

SUMMARY
The central nervous system has long been thought to regulate insulin secretion, an essential process in the maintenance of blood glucose levels. However, the anatomical and functional connections between the brain and insulin-producing pancreatic β-cells remain undefined. Here, we describe a functional transneuronal circuit connecting the hypothalamus to β-cells in mice. This circuit originates from a subpopulation of oxytocin neurons in the paraventricular hypothalamic nucleus (PVNOXT) and via the sympathetic autonomic branch it reaches the islets of the endocrine pancreas to innervate β-cells. Stimulation of PVNOXT neurons rapidly suppresses insulin secretion and causes hyperglycemia. Conversely, silencing of these neurons elevates insulin levels by dysregulating neuronal signaling and secretory pathways in β-cells and induces hypoglycemia. PVNOXT neuronal activity is triggered by glucoprivation. Our findings thus reveal that a subset of PVNOXT neurons form functional multisynaptic circuits with β-cells in mice to regulate insulin secretion and their function is necessary for the β-cell response to hypoglycemia.
Graphical Abstract
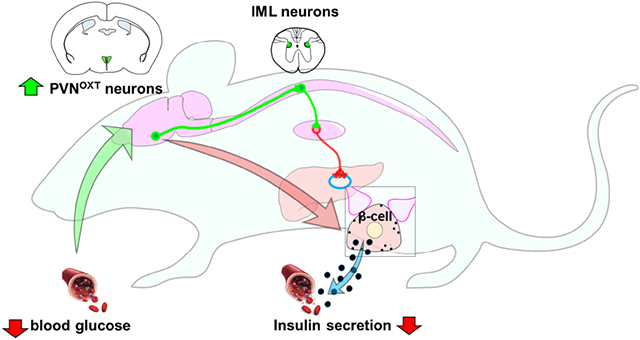
In Brief
The central nervous system has long been presumed to regulate insulin secretion. Papazoglou et al. utilize a multidisciplinary approach to investigate the anatomical and functional properties of a brain to β-cell connection. They identify and characterize a population of preautonomic neurons in the paraventricular hypothalamus that suppresses insulin secretion and prevents hypoglycemia.
INTRODUCTION
The central regulation of glycemia (Bernard, 1855) and pancreatic secretion (Pavlov and Thompson, 1902) have been known for more than a century. Innervation of pancreatic islets was also described decades ago (Langerhans and Morrison, 1937). Insulin, the glucose-lowering hormone synthesized and secreted by pancreatic islet β-cells, was isolated a century ago and proven to be effective in the treatment of diabetes (Banting et al., 1922). Insulin secretion was hypothesized to be centrally controlled, with the initial evidence derived from classical Pavlovian conditioning experiments in rodents (Woods et al., 1972). Subsequently, experiments in rats and dogs involving either lesions or electrical stimulation of the brain suggested a number of regions plausibly involved in the regulation of insulin levels (Berthoud et al., 1980; Berthoud and Jeanrenaud, 1979; Kaneto et al., 1975; Rohner-Jeanrenaud et al., 1983; Thorens, 2010). More recently, studies using multisynaptic viral tracers attempted to identify the CNS connections with the pancreas (Buijs et al., 2001; Rosario et al., 2016). However, the neuroanatomical and functional cell-to-cell communication between specific neuronal populations in the brain and pancreatic β-cells remained unknown.
Brain-glucose sensing and its role in metabolism has also been long hypothesized (Mayer, 1953). Indeed, glucose-sensing neurons located primarily in the hypothalamus respond to either hyper and hypoglycemia (Routh et al., 2014; Stanley et al., 2013) and are thought to trigger acute responses to normalize blood glucose levels, including regulation of insulin secretion (Pozo and Claret, 2018). Here, we describe a functional circuit connecting a subset of preautonomic PVN neurons (PVNOXT) to β-cells tasked with rapid suppression of insulin secretion. Furthermore, we show that these PVNOXT neurons are responsive to glucoprivation signals and are necessary for suppressing insulin secretion in response to the onset of hypoglycemia.
RESULTS
Hypothalamic neurons project to β-cells through sympathetic efferents
To identify neuronal populations in the brain synaptically connected to β-cells, we devised an in vivo tracing approach that combined a fluorescence-aided retrograde viral tracer, a β-cell specific transgenic mouse, and a reproducible viral delivery technique (Figures 1A and 1B). Specifically, the retrograde tracer Ba2017 (Figure 1A), an enhanced version of the Cre-dependent pseudorabies virus Ba2001 (DeFalco et al., 2001; Enquist and Card, 2003; Lo and Anderson, 2011; Schnutgen et al., 2003), was injected into the pancreatic duct of Ins1-Cre mice (Figure 1B), which express Cre-recombinase exclusively in β-cells (Thorens et al., 2015; Xiao et al., 2014). Viral EGFP+ fluorescence was detected in islets, specifically in β-cells, within 24 and 48 hours of Ba2017 injection (Figures 1C and 1D). During that time-period, EGFP+ signal was not detected in the exocrine pancreas (Figures S1A), spinal cord or brainstem (Figures S1B). By 72 hours, the majority of islets were infected (53.4%) in the Ins1-Cre mice, but not the control wildtype C57B16 mice (Figures 1E, 1F and S1C).
Figure 1. Infection of pancreatic islet β-cells with Ba2017 PRV.

(A) Structure of Cre-dependent Ba2017 pseudorabies virus. In the presence of Cre recombinase, the floxed sequence is inversed resulting in expression of EGFP and thymidine kinase (TK), a viral protein that promotes efficient replication.
(B) Schematic representation of Ba2017 delivery in the pancreas of Ins1-Cre mice. The recombination leads to activation of the virus (proliferation and retrograde transfer) specifically in insulin-expressing β-cells. The “active” form of Ba2017 retrogradely ascends neuronal afferents all the way to the central nervous system. The localization of the virus can be visualized by EGFP fluorescence.
(C-E) Representative images of β-cell-specific (insulin antibody staining, magenta) localization of Ba2017 (EGFP fluorescence, green) in Ins1-Cre mice at 24, 48 and 72h after administration (n = 2 mice per condition, 5-10 images per mouse).
(F) Representative images of β-cell-specific (insulin antibody staining, magenta) localization of Ba2017 (EGFP fluorescence, green) in C57B16 mice at 72h after Ba2017 administration (n = 2 mice, 5 images per mouse).
All sections were counterstained with nuclear marker DAPI (blue). (Scale bar = 50μm)
In the CNS, the first EGFP+ cells appeared in the intermediolateral nucleus of the spinal cord (IML) and the hypothalamus, where they were located almost exclusively in the PVN (PVN) and to a lower scale in the lateral hypothalamic area (LHA), 72 hours after injection (Figures 2A, 2B). A very small number of neurons were also observed in the pons and, more specifically, in the A5 (or subcoeruleus nucleus) (Figure 2C). In contrast to what has been reported previously (Rosario et al., 2016), EGFP+ neurons were not detected in the ventromedial hypothalamus (VMH), the arcuate nucleus (ARC), the dorsomedial hypothalamic nucleus (DMH), the central nucleus of the amygdala, periaqueductal gray (PAG) or the parabrachial nucleus (PBN) and the dorsal motor nucleus of the vagus (DMV) up to 3 days after viral injection (Figure 2D-2I). No EGFP+ cells were observed in the pancreas of control wild type mice 72 hours after Ba2017 injection although viral particles were distributed throughout the pancreas as visualized by immunostaining with anti-PRV antiserum (rb134) (Figures, 1F, S1D and S1E). To further test the possibility of retrograde travel of non-recombined Ba2017 virus in the control wild type mice, we looked at the spinal cord and PVN of these mice. Contrary to Ins1-Cre mice, viral particles were not detected in these locations confirming that Cre recombinase expression and recombination of the virus is necessary for its retrograde transfer capability (Figure S1F). Together, these findings demonstrate a transneuronal circuit that connects a subset of PVN neurons with pancreatic β-cells.
Figure 2. EGFP signal in the central nervous system 72h after Ba2017 administration.

(A) Representative images of EGFP fluorescent labeling (green) showing the localization of Ba2017 in autonomic preganglionic neurons within the intermediolateral nucleus of the spinal cord (IML) (T9-12) (left) and in preautonomic neurons within the paraventricular nucleus of the hypothalamus (PVN) (right). All sections were counterstained with nuclear marker DAPI (blue) and the inset is a higher magnification of the boxed area (n = 4, 4-8 images per region per mouse). (Scale bar = 200 μm, IML large; 50 μm IML higher magnification inset; 1 mm PVN large; 100 μm PVN higher magnification inset)
(B-I) Representative images of EGFP (green) labeled cells in various brain regions (n = 4, 4-8 images per region per mouse). Dashed areas represent the regions of interest in green identified in the brain section micrographs above each image. (Scale bar = 200 μm)
LH= Lateral hypothalamus, SubC= Subcoereleus nucleus, VMH= Ventromedial hypothalamic nucleus, ARC= Arcuate nucleus, DMH= Dorsomedial hypothalamic nucleus, CeA= Central nucleus of the amygdala, PAG= Periaqueductal grey, PBN= Parabrachial nucleus, DMV= Dorsal motor nucleus of the vagus, 3V= 3rd Ventricle, Aq= Aqueduct.
We also compared our findings to those reported with Ba2001 - an earlier version of the Ba2017 virus - by repeating our tracing experiment using the same incubation time (120 hours) as in the previous report (Rosario et al., 2016). Compared to the robust signal detected upon Ba2017 incubation, we failed to observe GFP+ cells in the spinal cord and regions of the brain of mice injected with Ba2001 (Figure S2), perhaps due to the inability of the Ba2001 virus to recombine in β-cells from Ins1-Cre mice. However, as expected we detected EGFP+ cells in additional brain regions of Ins1-Cre mice 120 hours after Ba2017 injection. Specifically, we observed EGFP+ cells in all the previously reported areas (VMH, ARC, DMH, CeA, PAG, DMV) with the exception of the PBN (Figure S2C) (Buijs et al., 2001; Rosario et al., 2016). The additional time taken by the Ba2107 EGFP+ signal to arrive at these regions argues against them being the primary connections with β-cells, although we cannot completely discount their role in β-cell function. This suggests that the PVN and to a lesser extent the LHA may serve as hubs that mediate the action of the DMH and possibly the VMH, ARC or other upstream neuronal populations with respect to communication with β-cells.
PVNOXT neurons suppress insulin secretion
As the PVN represented a primary brain region projecting to β-cells, as indicated by the high number of EGFP+ cells, we next sought to identify specific PVN neurons making anatomical connections with the β-cells. Two major subpopulations of PVN neurons projecting to the thoracic segments of the spinal cord are either oxytocinergic or vasopressinergic (Cechetto and Saper, 1988; Sawchenko and Swanson, 1982). Immunofluorescence assays revealed that a group of EGFP+ cells were also positive for the neuropeptide oxytocin (Figure 3A). EGFP+ co-labeling was not observed with vasopressin (AVP) under the same conditions (Figure S3A).
Figure 3. PVNOXT neurons communicate with β-cells and suppress insulin secretion.

(A) (left) Representative image of β-cell-projecting PVN neurons expressing EGFP (green) co-labeled with oxytocin-NP (magenta) 72 hours after Ba2017 administration White arrows indicate double-labeled cells. Dashed box in the upper left panel represents the area of the magnified image shown in the right panel, (right) Quantification of EGFP+ and EGFP+/OXT+ neurons in the PVN of Ins1-Cre mice 72 hours after Ba2017 administration (n = 4, 4 images per mouse). graph shows mean ± sem. (Scale bar = 50 μm)
(B) Schematic representation of DREADD virus injection site with representative image of viral mCherry expression in PVNOXT neurons.
(C) Schematic representation of mCherry control virus injection site with representative image of viral mCherry expression in PVNOXT neurons.
(D) Plasma insulin levels 10 minutes after administration of CNO and/or glucose in male (n = 7 mice) and female (n = 6 mice) PVNOXT:hM3Dq mice.
(E) Grouped % of basal changes of basal plasma insulin levels. All graphs show mean ± sem. **P < 0.01; ratio paired Student’s t-test.
(F) Plasma insulin levels 10 minutes after administration of CNO and/or glucose in male (n = 3) or female (n = 5) PVNOXT:mCherry mice.
(G) Grouped % of basal changes of basal plasma insulin levels. All graphs show mean ±sem. **P <0.01; ratio paired Student’s t-test.
(H) Representative EM images of β-cells in islets within pancreas sections from PVNOXT:mCherry (left; n = 4, 25-40 images per mouse) and PVNOXT:hM3Dq mice (right; n = 4, 25-40 images per mouse) 10 min after co-administration of glucose and CNO. Docked granules: located within 0.2 μm of the plasma membrane. Cyan lines indicate the cell membrane and yellow arrows point to the docked granules.
(I) Quantification of number of docked insulin granules per μm of plasma membrane. (n = 4 mice per group). Graph shows mean ± sem. **P < 0.01; two-tailed, unpaired Student’s t-test.
(J-K) Glycemic level changes (J) during GTT with or without chemogenetic stimulation of PVNOXT neurons with CNO (shaded areas represent error bars). Baseline glucose at time of injection (0 min): 89.75 ± 8.797 and 81.50 ± 3.571 respectively, and, (K) Glycemic change area under curve (left) and Δ peak glycemia (right) during GTT after PVNOXT stimulation via CNO. (n = 8 mice) *P < 0.05, **P < 0.01; paired Student’s t-test.
PVNOXT neurons have been suggested to play a role in glucose homeostasis, but so far this hypothesis has not been tested (Leng and Sabatier, 2017). We thus investigated this connection further by stimulating the PVNOXT neurons using chemogenetics. When stimulating PVNOXT neurons, we needed a system where the inhibitory effect of PVNOXT neurons on insulin secretion would be reproducibly quantifiable. For this reason, we decided to stimulate PVNOXT neurons during a glucose-stimulated insulin secretion (GSIS) challenge. This low baseline scenario is similar to AGRP stimulation in fed mice to demonstrate their role in inducing food intake (Krashes et al., 2011). We stereotactically injected AAV-DIO-hM3Dq or AAV-DIO-mCherry virus in the PVN of Oxt-ires-Cre mice (Wu et al., 2012) to induce expression of the hM3Dq receptor or the mCherry fluorescent reporter exclusively in PVNOXT neurons (PVNOXT:hM3Dq and PVNOXT:mCherry respectively) (Figures 3B and 3C). Mice were injected intraperitoneally (i.p) with either glucose alone (2 g/kg) or with glucose and the hM3Dq receptor agonist clozapine-N-oxide (CNO, 0.5 mg/kg). Due to sex differences in islet biology (Gannon et al., 2018) and PVNOXT-related behaviors (Pisansky et al., 2017), we performed these experiments in both sexes. The 10-minute time point to measure insulin levels was chosen for two reasons: (i) it was the earliest time point where we could see the effect of neuronal stimulation, considering the time taken by CNO to reach the CNS, and, (ii) a pre-stimulation or stimulation for longer periods would likely preclude an accurate interpretation due to indirect effects via parallel yet non-specific pathways. As shown in Figures 3D and 3E, GSIS was rapidly suppressed within 10 minutes of PVNOXT neurons stimulation in both male and female mice, respectively. No changes in insulin secretion were observed upon a similar injection (glucose +/− CNO) in control mice that express the fluorescent protein mCherry in PVNOXT neurons, but not the hM3Dq receptor (Figures 3F-G). Further, no significant effects on insulin levels were observed upon chemogenetic stimulation of PVNAVP neurons (Figure S3B). Stimulation of total PVNOXT neurons had no effect on levels of plasma glucagon and plasma adrenaline (Figures S4A-D). Similarly, stimulation of SONOXT neurons (SON = supraoptic nucleus; contains only OXT neurons that project to the posterior pituitary) had no effect on levels of plasma insulin (Figure S4E). Taken together, these observations negate any substantial contribution of these pathways on the suppression of glucose-stimulated insulin secretion.
Next, we wanted to demonstrate by means other than measuring plasma levels of insulin that the changes in activation of PVNOXT neurons are quickly transmitted to β cells. The observed time kinetics of insulin suppression led us to hypothesize that the neuronal effects are transmitted via changes in insulin granule trafficking, a highly synchronized and orchestrated process of insulin secretion (Rorsman and Renstrom, 2003). If so, such effect could be studied via changes in insulin granule movement by electron microscopy (EM). However, traditional EM of β cells to observe intracellular organelles and insulin granule trafficking typically utilizes isolated islets from harvested pancreas. EM on isolated islets, while convenient, would have defeated the purpose of demonstrating a neuronal effect on β cell granule movement, due to the extended mechanical and chemical islet isolation process. Thus, we utilized a recent protocol on intact pancreas sections (Fulgenzi et al., 2020). Here, pancreata are harvested at 10 minutes after CNO stimulation and immediately processed for EM. Typically, approximately 10% of all granules are docked on the plasma membrane and only a small fraction can be released immediately upon stimulation (Orci, 1985). Moreover, insulin granule docking is a rate limiting step for insulin secretion, and granule docking is impaired in human T2D (Gandasi et al., 2018). Here, our EM analysis revealed that stimulation of PVNOXT neurons resulted in a significant reduction in the number of docked insulin granules during glucose-stimulated insulin secretion (Figures 3H-I). Taken together, these findings suggest that PVNOXT neuron activation communicates a signal to β-cells to suppress insulin release via targeting granule trafficking.
PVNOXT stimulation elevates blood glucose levels
The effect of alterations in insulin secretion are manifested as changes in blood glucose levels. Thus, we investigated the effect of activating PVNOXT neurons on this parameter. Considering the rapid suppression of insulin secretion by PVNOXT neurons, we anticipated that the fluctuations in glycemia would be similarly quick and thus escape detection by traditional static blood glucose measurements at random times and might also be impacted by the stress induced by tail sampling. Thus, we employed an in vivo glucose monitoring telemetry system (Korstanje et al., 2017) which allows minute-to-minute glucose monitoring without handling of the mice. Chemogenetic stimulation of PVNOXT neurons during hyperglycemic challenge resulted in significant elevation of blood glucose levels - an effect that was sustained for at least the first 30 minutes after glucose administration (Figures 3J-K).
Next, we sought to investigate the role of this neurocircuit during feeding which reflects a physiological trigger for insulin secretion. To achieve this, mice were monitored during a fasting-refeeding regimen that generates a reproducible rise in blood glucose levels during the refeeding phase (Figure S5A). Stimulation of PVNOXT neurons resulted in significantly higher prandial blood glucose levels that persisted during the first hour of refeeding (Figures S5B-D). Feeding amounts did not differ between experiments (Figure S5E), which is in agreement with data from Li et al. (Li et al., 2019), showing that PVNOXT stimulation does not affect feeding and that PVNOXT neuronal activity is not affected by fasting/feeding states. Taken together, these observations are consistent with a role for PVNOXT neurons in suppression of insulin secretion, leading to elevation of blood glucose levels.
PVNOXT neurons suppress insulin secretion via sympathetic afferents
PVN neurons communicate with β-cells via autonomic neurons located in the spinal cord. In addition to PVNOXT neurons that project to autonomic areas, several PVN neurons project to other brain regions and the pituitary (Liao et al., 2020). To determine whether the circuit we uncovered involves sympathetic preganglionic neurons, we injected a retrograde Cre-dependent AAV-hM3Dq virus (rgAAV-hsyn-DIO-hM3Dq-mCherry) in the thoracic segments 9-13 of the spinal cord (Fasanella et al., 2008; Kohro et al., 2015) of OXT-ires-Cre mice (Figure 4A). PVN is the only brain region that contains oxytocin neurons projecting to the spinal cord (Hallbeck et al., 2001; Swanson and McKellar, 1979), so there is no possibility that this receptor gets expressed elsewhere. This approach induces expression of the DREADD receptor hM3Dq specifically in PVNOXT neurons that project to sympathetic neurons within this part of the spinal cord (Figure 4B), which includes the preganglionic neurons that are connected to the ganglia innervating the pancreas and excludes the ones connected to the adrenal glands (Hosoya et al., 1995). We refer to these targeted neurons as spPVNOXT neurons, with “sp” referring to both Spinal cord-Projecting and Sympathetic-Projecting. In addition, the targeted group of oxytocin neurons exclude those projecting to other brain areas as well as the pituitary. Stimulation of spPVNOXT neurons via CNO injection, similar to chemogenetic activation of PVNOXT neurons, resulted in suppression of glucose-stimulated insulin secretion (Figure 4C-D). Further, stimulation of spPVNOXT neurons during a fasting-refeeding regimen resulted in significantly higher prandial blood glucose levels that persisted during the first hour of refeeding (Figures S5F-H), wherein feeding amounts did not differ between experiments (Figure S5I). These observations strongly suggest that the PVNOXT neurons innervate the pancreas via a circuit that involves the sympathetic preganglionic neurons.
Figure 4. PVNOXT neurons that project to the spinal cord (spPVNOXT) segments 9-13 suppress insulin secretion.

(A) Schematic representation of retrograde DREADD virus injection in the spinal cord of Oxt-ires-Cxe mice.
(B) Representative image of viral mCherry expression (magenta) in preautonomic PVNOXT neurons (green) (n=9, 1-6 images per mouse). (Scale bar = 200 μm).
(C) Plasma insulin levels 10 minutes after administration of CNO and/or glucose in mice expressing the hM3Dq receptor in spPVNOXT neurons (left males, n = 4; right females, n = 5).
(D) Grouped % of basal changes of basal plasma insulin levels. All graphs show mean ± sem. *P < 0.05; ratio paired Student’s t-test.
PVNOXT neurons are required for fasting-induced insulin suppression
Insulin secretion by β-cells needs to be tightly regulated by glucose metabolism, hormonally or neuronally (Noguchi and Huising, 2019). During fasting, insulin secretion needs to be suppressed to prevent hypoglycemia and harmful neuroglycopenic effects (Shah et al., 2017). In mice, a 24-hour fast significantly lowers plasma insulin levels (Feldman and Lebovitz, 1970). To evaluate their role in fasting-induced insulin suppression, we silenced PVNOXT neurons by injecting AAV-DJ-CMV-DIO-eGFP-2A-TeNT virus, which transduces tetanus toxin (TeNT) and neutralizes neurotransmitter release by cleaving synaptobrevin (VAMP2) (Sweeney et al., 1995), in the PVN of Oxt-ires-Cre mice (PVNOXT:TeNT mice) (Figure 5A). As a control group we used Oxt-ires-Cre mice injected with AAV5-EF1a-DIO-EYFP-WPRE-pA virus (PVNOXT:YFP mice). After a 24-hour fast, PVNOXT:TeNT mice had significantly higher plasma insulin levels than PVNOXT:YFP mice (Figure 5B). Further, insulinemia in PVNOXT:TeNT mice was similar to that observed in the fed state, whereas insulin levels in the PVNOXT:YFP mice were suppressed as expected. The higher levels of plasma insulin were also reflected in the significantly lower blood glucose levels in PVNOXT:TeNT mice (Figure 5B). These findings suggest that functional PVNOXT neurons are necessary for suppression of insulin secretion and prevention of hypoglycemia during chronic fasting.
Figure 5. PVNOXT neuron silencing dysregulates insulin secretory apparatus.

(A) Schematic representation of YFP and TeNT virus injection site with representative images of viral YFP (n=7, 4 images per mouse) or EGFP (n=7, 4 images per mouse) expression in PVNOXT neurons, respectively.
(B) 24h-fasted and fed plasma insulin (left) and blood glucose (right) levels in PVNOXT:YFP (n = 7) and PVNOXT:TeNT (n = 7) mice. Graph shows mean ± sem. *P < 0.05, ***P < 0.001, ns: non-significant; two-tailed, unpaired Student’s t-test.
(C) KEGG pathways significantly altered upon PVNOXT silencing in islets from 24h-fasted mice (n = 4 per group).
(D) Heat map of expression patterns of significantly down- or upregulated genes implicated in granule docking (n = 4 per group).
(E) Fold change in mRNA levels of genes implicated in regulation of insulin secretion (n = 4 per group).
(F) Map of gene products involved in insulin secretion significantly altered in PVNOXT:TeNT mice (affected genes highlighted in blue; unaffected genes in grey) (n = 4 per group).
All data from the RNASeq were selected with a cutoff at *P < 0.05.
We next reasoned that the effects of the functional arrest of the PVNOXT neurons would be transmitted to β-cells in the PVNOXT:TeNT mice and would be manifested in changes to the islet transcriptome. Thereby, to gain more insight into the impact of PVNOXT silencing, we performed RNA sequencing to analyze gene expression in islets from PVNOXT:TeNT and PVNOXT:GFP mice after a 24-hour fast. Pathway analysis revealed that a select group of neurotransmission-related signaling pathways were significantly affected (Figures 5C-F). Additionally, genes representing calcium signaling, GPCR signaling, cAMP signaling, gap junction signaling, vesicle trafficking and granule fusion, that are directly implicated in insulin secretion (Perelis et al., 2015; Rorsman and Ashcroft, 2018), were significantly modulated. Finally, expression of genes associated with insulin granule docking (Gandasi et al., 2018) were selectively affected in the PVNOXT:TeNT mice (Figure 5D). This is consistent with the granule docking defects observed upon stimulation of PVNOXT neurons (Figures 3H and I). Together, these observations further demonstrate the functional role of PVNOXT neurons in regulating insulin release, plausibly via targeting key nodes in insulin granule trafficking.
PVNOXT neurons respond to glucoprivation
Central sensing of hypoglycemia largely occurs in the hypothalamus and is thought to orchestrate counter-regulatory responses (CRR) that include suppression of insulin secretion (Graveling and Frier, 2010; Pozo and Claret, 2018). Also, the PVN has been previously reported to contain preautonomic neurons that respond strongly to low glucose levels (Melnick et al., 2011). Having demonstrated the ability of PVNOXT neurons to prevent hypoglycemia by suppressing insulin secretion and raising glucose levels, we next sought to examine if these neurons are part of a central sensing mechanism to counter hypoglycemia. To test this physiological response, we utilized 2-deoxy-D-glucose (2-DG), a glucose analog that cannot be metabolized and provokes a glucoprivic/hypoglycemic response (Wick et al., 1957). Neuronal activity of PVNOXT neurons in response to 2-DG injection was monitored by (i) Fos immunoreactivity (Figures 6A and B) and (ii) in vivo fiber photometry (Figures 6C-E). The total Fos staining in the PVN remained unchanged between the two groups. Importantly, the number of neurons double-positive for oxytocin and Fos immunostaining nearly tripled in the 2-DG group compared to vehicle (Figure 6B). Next, we used Oxt-Ires-Cre mice and virally-induced Cre-dependent expression of the calcium indicator GCaMP6s selectively in PVNOXT neurons (Figure 6C). PVNOXT neurons responded to 2-DG injection by increasing their activity within 20 minutes of administration, which corresponds to the time when 2-DG is known to reach the brain (Hom et al., 1984; Sokoloff et al., 1977; Wong et al., 2011) (Figures 6D and E). Interestingly, 2-DG-induced glucoprivation is known to induce centrally-mediated sympathetic activity in the pancreas of dogs within a similar time frame (Havel et al., 1988). As expected, the 2-DG-induced glucoprivation signal was associated with increased food intake (Figure 6F). These observations are indicative of a distinctive role for a subpopulation of PVNOXT neurons in responding to glucoprivation as part of a brain-sensing mechanism to counter hypoglycemia. Taken together, these findings suggest that PVNOXT neurons are activated by hypoglycemic stimuli as a mechanism to suppress insulin release.
Figure 6. PVNOXT neurons respond to glucoprivation.

(A) Representative images of PVN sections showing c-fos (green) and oxytocin-neurophysin (magenta) immunostaining after i.p. injection of saline (top; n=4, 4 images per mouse) or 2-DG (400 mg/kg; bottom; n=5, 4 images per mouse). Dashed box in the left panels represents the area of the magnified image shown in the right panels. White arrows indicate double-labeled cells.
(B) Quantification of the number cells with double c-fos and OXT labeling in the PVN after saline (n = 4) or 2-DG injection (n = 5) (top). Quantification of the number of cells positive for c-fos staining in the entire PVN after saline (n = 4) or 2-DG injection (n = 5) (bottom). Graphs show mean ± sem. ***P < 0.001; two-tailed, unpaired Student’s t-test
(C) Schematic representation and representative image of AAV1-CAG-FLEX-GCaMP6s-WPRE-SV40 virus injection site and fiber implantation, shown as a dashed region (n=8, 4 images per mouse).
(D and E) Plot of calcium signals from PVNOXT neurons (D) and quantification of fluorescence changes in 15-minute increments (E) after i.p. injection of saline or 2-DG (200 mg/kg). (n = 8) Graph shows mean ± sem. **P < 0.01 two-tailed, paired Student’s t-test
(F) 1h food intake after the end of GCaMP recording in mice injected with saline or 2-DG (200 mg/kg). (n = 8) ***P <0.001 two-tailed, paired Student’s t-test
DISCUSSION
Our findings reveal a functional brain-to-β-cell transneuronal circuit that regulates insulin secretion. Although a role for the central nervous system in regulation of insulin levels has long been suggested (Rohner-Jeanrenaud et al., 1983), precise anatomical and functional validation of its existence was not achievable due to limitations in the tools and experimental techniques. Using a novel Ba2017 pseudorabies virus in combination with a β-cell-specific transgenic Ins1-Cre mouse model (Thorens et al., 2015), we uncover that the region more closely and robustly projecting to β-cells via sympathetic innervation is the PVN. Challenges of functionally characterizing connections between the brain and β-cells were overcome with the use of (i) retrograde rabies viral tracing using pancreatic ductal delivery, (ii) chemogenetics to target neuronal subsets in the PVN and the spinal cord, (iii) automated in vivo glucose telemetry, and (iv) in vivo fiber photometry to monitor neuronal activity in real time. These integrated approaches allowed us to (i) reliably identify and manipulate distinct subpopulations of hypothalamic neurons while monitoring systemic glycemia and changes in insulin secretion, and (ii) enabled robust monitoring of specific neuronal activity during extreme glycemic conditions. Thus, we demonstrate that a distally located group of hypothalamic neurons are connected to β-cells via sympathetic efferents to fine tune insulin release as a response to dynamic glycemic changes.
It is worth noting that our retrograde tracing results yield a different pattern of innervation of β-cells as compared to previously published studies (Rosario et al., 2016). There happen to be multiple areas where the two studies vary fundamentally that might explain the different findings. The histological analysis in Rosario et al., was done on sections from mice sacrificed 120 hours after viral administration whereas in our study the primary tracing data was analyzed after 72 hours. The 72-hour time-frame takes into account the highly infectious nature and rapid propagation of such viruses in the brain (slower in peripheral nerves) (Card and Enquist, 2014). With longer incubation times, the cell-specificity of projecting cells is rendered ambiguous due to recombined viral particles released from dying β-cells that could potentially infect neuronal or other islet cells, locally. Indeed, longer incubation periods result in more profound viral EGFP labeling in many brain and hypothalamic regions compared to the earlier 3-day timepoint. Further, we provide careful analyses of the virus originating in β-cells of the islets and the virus tracking upstream and also show that exocrine cells were not infected in our pancreatic ductal delivery approach. Moreover, we utilized a delicate but reproducible technique to deliver the tracing virus to the entire pancreatic tissue. In contrast to injecting the tracing virus in the pancreatic parenchyma (Rosario et al., 2016), we injected the virus in the pancreatic duct of Ins1-cre mice that allowed for a consistent delivery route and β-cell specificity from experiment to experiment, in every mouse used in the tracing study. Taken together, we demonstrate that our tracing results are robust and reproducible in detecting neuronal connections between pancreatic islet β-cells and the central nervous system.
Our findings unravel a transneuronal circuit that connects a subpopulation of PVN neurons with pancreatic β-cells. Having said that, we emphasize that our results describe the role of a subset of PVN neurons communicating with β-cells and the role of the remaining PVN neurons in this process is unclear. After three days of incubation, a limited fraction of the PVN neurons connected to the β-cells express oxytocin and most oxytocin neurons are not labelled by retrograde EGFP expression. The retrograde tracing method with PRV viruses and, in general, most tracing methods are qualitative and not quantitative, including a recent study using pancreatic injection of non-Cre-dependent PRV (Duncan et al., 2019). While a small fraction of the PVN neurons closely connected to the β-cells express oxytocin and most oxytocin neurons are not labelled by retrograde EGFP expression during the selected time window used for retrograde tracing, majority of the PVNOXT neurons were targeted during the physiological studies. Using present day neuroscience tools, it is not feasible to perform the physiological experiments in a way that selectively targets the β-cell projecting neurons identified via tracing. Thus, we are limited to conducting these studies via stimulating all PVNOXT neurons or by examining their activity during an experimental challenge.
Also, we cannot discount the role of additional hypothalamic regions - such as the VMH and the ARC - in the regulation of insulin secretion (Rosario et al., 2016; Stanley et al., 2016). Our tracing results demonstrate a lack of direct connectivity of VMH or ARC to the sympathetic autonomic areas that innervate β-cells. However, we cannot exclude the possibility that neurons within these regions could regulate insulin secretion via longer neurocircuits and, as such, further tracing studies are needed to investigate the order of connectivity of these areas to β-cells. Tracing data at the 72h timepoint showed that the PVN and LHA are the forebrain structures with close synaptic proximity to β-cells. EGFP+ neurons were not detected in the VMH, ARC, DMH, CeA, PBN or PAG, and the DMV. This suggests that the PVN and to a lesser extent the LHA may serve as hubs that mediate the action of the DMH and possibly the VMH, ARC or other upstream neuronal populations with respect to regulating β-cell function. While EGFP+ cells were limited to the PVN and LHA at the 3-day timepoint, extended Ba2017 virus incubation (120h/5 days) identified a number of EGFP+ cells in previously reported areas (VMH, ARC, DMH, CeA, PAG, DMV) with the exception of the PBN. As these regions were not positive for EGFP+ cells at the 72h incubation timepoint, we deduce that these regions are not the primary connections to β-cells. However, not having individually characterized regions besides the PVN precludes us from discounting their role in insulin secretion. Further, our tracing methods did not detect evidence of an intra-pancreatic neuronal network as monosynaptic tracing studies would enable better characterization of such circuits. Establishing a functional circuit required us to manipulate the identified pancreas-projecting neurons in the PVN while also manipulating the proximal projections to the pancreas. To achieve this, we chemogenetically stimulated PVN oxytocin neurons that project to preganglionic sympathetic neurons in the spinal cord. We found that, similar to that observed upon stimulation of the total PVNOXT neuronal population, stimulation of spPVNOXT neurons that project to the spinal cord (T9-13) also led to suppression of insulin secretion. Glucose telemetry data revealed no effects upon chemogenetic stimulation of spPVNOXT neurons (data not shown) supporting the notion that there exists a network of interconnected neurons that not only modulate β-cell function but also modify the function of other peripheral organs involved in glucose homeostasis. Taken together, these findings underscore the existence of more complex processes than we have described here.
We complemented the insulin level determinations by identification of islet-centric pathways affected by disruption of the PVNOXT neuron-β-cell circuit. Unbiased gene expression studies provided a window into the islet-specific targets of the neuronal circuitry we describe here. In addition to changes in neurotransmission-related signaling pathway, genes representing calcium signaling, GPCR signaling, cAMP signaling, gap junction signaling, vesicle trafficking and granule exocytosis that are implicated in insulin secretion were substantially modulated. Finally, expression of genes associated with insulin granule docking were selectively affected, which is consistent with the granule docking defects observed upon stimulation of PVNOXT neurons. Having said that, these gene expression changes are mere signposts and detailed studies will reveal their functional roles in this circuit.
To investigate how the stimulation of PVNOXT neurons rapidly suppresses insulin release, we studied insulin granule dynamics using a novel electron microscopy protocol on pancreas sections (Fulgenzi et al., 2020). Electron microscopy analyses showed that stimulation of PVNOXT neurons during GSIS leads to a reduction in the number of docked granules on the plasma membrane of β-cells within 10 minutes. Adrenergic signaling and, by extension, the sympathetic input to the β-cell suppresses insulin release, via changes in insulin granule docking, and increases T2D susceptibility (Rosengren et al., 2010). Further, insulin granule docking is a rate limiting step for insulin secretion, and granule docking is impaired in human T2D (Gandasi et al., 2018). How the PVNOXT neuronal circuit precisely relays the information to the granule trafficking machinery is unclear. Although the EM experiments were designed to study acute effects of neuronal stimulation and the RNAseq analyses enabled investigation into pathways affected upon chronic ablation of the neuronal signals, collectively the data indicate that the PVNOXT neuron input to the β-cell has quite marked and widespread effects on the cell, i.e., that the reduced granule-docking may simply be the endpoint of a number of different processes.
Sympathetic inhibition of insulin secretion (Ahren, 2000; Gilliam et al., 2007; Havel and Taborsky, 1989; Porte and Williams, 1966) is one of the first native physiologic responses to hypoglycemia, being part of the counter-regulatory response to protect against declining glucose levels (Graveling and Frier, 2010; Porte, 1969). Failure to trigger this response as a result of impaired awareness of hypoglycemia (IAH) can be fatal, due to the risk of neuroglycopenia. Both CRR and IAH are known to involve neuronal mechanisms (Arbelaez et al., 2008; Cryer, 2013; Gerich et al., 1991; Graveling and Frier, 2010; Meek et al., 2016; Mitrakou et al., 1991). The PVNOXT-β-cell circuit we identified sheds light on the organization of such neuronal processes with physiological and pathological outcomes. More specifically, our study provides anatomical and functional evidence of a well-defined neuronal circuit that links the central response to severe hypoglycemia and the sympathetic inhibition of insulin secretion. Thus, we show that preautonomic PVNOXT neurons respond to glucoprivation and trigger sympathetic efferents which inhibit insulin secretion. Conversely, functional silencing of PVNOXT neurons prevents fasting-induced insulin suppression leading to extreme hypoglycemia which is suggestive of a potential role for these neurons in IAH. Glucose counter regulation in response to acute hypoglycemia in humans is mediated principally by glucagon and epinephrine (Cryer and Gerich, 1985). We did not detect appreciable changes in the levels of either of the two hormones upon stimulation of PVNOXT neurons under the conditions utilized in this study. It is plausible that the circuit described here accounts solely for the suppression of insulin secretion, which is considered to be the earliest event in the response to hypoglycemia (Fanelli et al., 1994; Zammitt and Frier, 2005). Having said that, these data do not imply that sympathetic innervation at the level of the islet has dedicated fibers for β cells that do not target α cells or vascular cells (and blood flow) simultaneously. Distinct from what we show here, oxytocin release is modulated in response to hypoglycemia and oxytocin concentrations after hypoglycemia were significantly higher in diabetic subjects compared with normal subjects (Fisher et al., 1989). Further, insulin can stimulate oxytocin neurons and induce its release into the circulation (Zhang et al., 2018). As we have excluded the involvement of the neuroendocrine system, the adrenal glands and α cells, it seems very likely that that the suppression of insulin secretion via sympathetic innervation is the main mechanism by which PVN oxytocin neurons contribute to the CRR and IAH.
Although prior studies have elegantly described intra-islet innervation in rodent and human islets (Rodriguez-Diaz et al., 2011a; Rodriguez-Diaz et al., 2011b), these studies did not address central regulation of insulin secretion. Moreover, to our knowledge, no studies have characterized the central control of pancreatic endocrine cells in any organism with the exception of neurons in the Drosophila brain that are synaptically connected to insulin-producing cells to regulate insulin secretion (Oh et al., 2019). Our study is the first to provide such evidence in mammals. Having said that, how the findings presented here relate to humans remain to be seen considering the neuroanatomical differences between mice and humans with respect to innervation of the endocrine pancreas as well as islet organization (Cabrera et al., 2006; Dybala and Hara, 2019; Rodriguez-Diaz et al., 2011a). While brain modulation of pancreatic β-cell function plays a role in certain situations, the fact that in vitro/ex vivo and transplanted β-cells function optimally argues that neuronal inputs are not 'necessary' for normal β-cell function. Thus, we believe that the neural input we describe is part of a multisystem response to a life-threatening event like hypoglycemia. The notion that specialized neurons would influence β-cell function and, by extension, monitor the levels of insulin to preclude extreme hypoglycemia is consistent with the teleological role of glucose as the primary fuel source for the brain. Overall, we propose that transneuronal circuits such as the one described here are essential nodes of an integrated multiorgan functional network tasked with the maintenance of normal insulin and glucose levels in mammals. Identification and characterization of similar circuits will enable better understanding of the role of the brain in glucose homeostasis in physiology and disease.
LIMITATIONS OF STUDY
While selectively targeting β-cell projecting neurons identified via tracing would be the best way to characterize their role, it is not possible to do so using existing tools. Moreover, our report focuses on the role of PVNOXT neurons in insulin secretion and we cannot exclude a role of the remaining PVN neurons, as well as of neurons in other brain regions, in the process. Also, 2-DG is a limited model of glucoprivation because of its impact on systemic glucose levels and effects in the cell beyond disruptions of glucose sensing. However, similar to high fat diet-feeding typically used in metabolic studies, 2-DG is the most well-studied glucoprivic agent. The relevance of the findings presented here to humans remains to be explored.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to the lead contact, Sushil G. Rane (ranes@nih.gov).
Data and Code Availability
RNA-seq data have been deposited at GEO and are publicly available. The accession number is GSE188856. Microscopy data reported in this paper will be shared by the lead contact upon request.
This paper does not report any original codes.
Data S1 represents an Excel file containing the values that were used to create all graphs in the paper. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse Models
All animal care and experimental procedures were approved by the National Institutes of Health Institutional Animal Care and Use Committee (protocol K014-DEOB-18). Mice were housed at 22° C with a 12/12-hour light/dark cycle (lights on at 06:00). Humidity was set between 40-60% and mice were fed a standard mouse chow (Teklad F6 Rodent Diet 8664, Harlan Teklad, Madison, WI) ad libitum. Mice used in the various studies were between 8-32 weeks old. All mice were purchased from the Jackson Laboratory (Bar Harbor, ME) and maintained on a C57BL/6 background. All experimental mice used in the study were derived upon breeding’s in the local animal facility and no additional acclimatization was needed. General health of mice was good with all surgical procedures followed as per approved guidelines for monitoring of post-surgery mice. Two mice were excluded from the spinal cord experiment due to lower limb paralysis.
For the retrograde tracing experiments with intraductal injection of Ba2017 or Ba2001 8- to 12-week-old male and female mice were used. Mice were single housed after surgery until euthanized. Lines used: C57BL/6, Ins1-Cre.
For all DREADD experiments 8- to 32-week-old male and female mice were used and data were pooled. Mice were group-housed throughout the experiment. Littermates of the same sex were randomly assigned to experimental groups. Lines used: Oxt-ires-Cre, Avp-ires-Cre.
For the Fos experiment 8– to 12-week-old male mice were used. Littermates of the same sex were randomly assigned to experimental groups. Line used: C57BL/6.
For the photometry experiments 8– to 12-week-old male and female were used and data were pooled. Mice were single housed throughout the duration of the experiment until euthanized. Littermates of the same sex were randomly assigned to experimental groups. Line used: Oxt-ires-Cre.
For the tetanus toxin experiments 8- to 12-week-old female mice were used. Mice were group-housed throughout the experiment. Littermates of the same sex were randomly assigned to experimental groups. Line used: Oxt-ires-Cre.
METHOD DETAILS
Ba2017 Virus Production
Ba2017 is a novel pseudorabies virus (PRV; suid herpesvirus 1) recombinant for Cre-dependent multisynaptic retrograde neurotracing. Ba2017 is derived from the attenuated retrograde tracing strain, PRV Bartha (Enquist and Card, 2003), and exhibits improved genetic stability and fluorescent protein expression compared to the previous-generation Cre-dependent retrograde tracer, Ba2001 (DeFalco et al., 2001). Ba2017 contains a transgene expression cassette, which expresses PRV thymidine kinase (TK) and monomeric EGFP dependent on Cre-mediated recombination events, inserted into and disrupting the native TK gene. The Cre-dependent cassette consists of a CAG promoter, LoxP site, SV40 transcription termination and polyadenylation signal (polyA), Lox2272 site, inverted codon-optimized PRV thymidine kinase coding sequence, inverted FMDV 2A polyprotein cleavage sequence (F2A), inverted monomeric EGFP coding sequence, inverted LoxP and Lox2722 sites, WPRE post-transcriptional regulatory sequence, and BGH polyA signal. To reduce leaky transgene expression, the Cre-dependent cassette in Ba2017 combines the “Lox-stop-Lox” approach used in Ba2001 (DeFalco et al., 2001), where the transgene is interrupted by a transcription termination signal flanked by Lox sites, and the double-floxed inverted ORF (DIO)/flip-excision (FLEX) approach (Schnutgen et al., 2003), where transgene coding sequences are inverted relative to the promoter. Prior to Cre-mediated recombination, Ba2017 is phenotypically TK-null, which limits viral replication in neurons, and does not express the EGFP marker. After two Cre-mediated recombination events, to revert transgene orientation and excise the SV40 polyA sequence, Ba2017 expresses TK, enabling robust replication in neurons, and strongly expresses the EGFP fluorescent marker. The CAG promoter, SV40 polyA flanked by Lox sites, and WPRE element were derived from the H129ΔTK-TT plasmid from Lo and Anderson (Lo and Anderson, 2011), and the remaining sequences were assembled by de novo gene synthesis (Genscript, Inc.). The resulting shuttle plasmid was co-transfected with purified PRV Bartha nucleocapsid DNA into PK15 porcine kidney epithelial cells, selected for loss of TK enzymatic activity by treatment with 1-beta-D-Arabinofuranosylthymine (AraT), cloned through three rounds of plaque purification, and screened for Cre-dependent EGFP expression (Staniszewska Goraczniak, Hogue, and Enquist, unpublished data).
Intraductal Injection
Procedure was adapted from Xiao et al. (Xiao et al., 2014). Mice are anesthetized with 2-2.5% (wt/vol) isoflurane by inhalation and placed under a microscope with warming pad over 3 layers of paper/gauze. Breathing and heart rate were monitored during the surgery and isoflurane concentration adapted accordingly. A midline abdominal incision is made, and the abdominal muscle is cut along the linea alba to create a laparotomy incision of 1.5-2.0 cm. Stomach is pulled out and the duodenum is rotated and stretched to expose the biliary-pancreatic duct and its junction with the duodenum (the sphincter of Oddi) using Q-tips. A microclamp is placed on the bile duct above the branching of the pancreatic duct to block infusion towards the liver. Two low-pressure microclamps are closely placed on the duodenum on both sides of the sphincter of Oddi to prevent backflow of the virus to the duodenum. A small hole is made with a 30-gauge needle opposite to the sphincter of Oddi, 1–2 mm away from the ampulla of Vater. A 10 mm-long 31-gauge blunt-ended cannula (PlasticsOne, Roanoke, VA USA; C316I/SPC) is inserted into the biliary-pancreatic duct through the sphincter of Oddi in the duodenum with the tip reaching the entry of the pancreatic duct. The cannula is connected via a P50 tube (PlasticsOne, Roanoke, VA USA; C313CT) to a 1 ml syringe (BD, Temse Belgium; 309628), fixed on to a microinfusion pump. 75 μl of virus solution (Ba2017, titer: 2.58-2.76 x 10^9 pfu/ml) are infused in the pancreatic duct at a steady flow of 6 ul/min. 5 minutes after the end of the infusion, the clamps and cannula are removed and tissue adhesive (Vetbond; 3M, St. Paul, MN USA) is applied on the hole created by the needle to avoid leakage. The stomach and duodenum are placed back to the abdomen. Abdominal muscles and skin are sutured (CP Medical, Portland, OR USA; CP-S682S-02). The mouse receives a single subcutaneous injection of Buprenorphine SR® (1 mg/kg; ZooPharm, Laramy, WY USA) and an intraperitoneal injection of 0.5 ml of sterile 0.9% saline. After that the mouse is put back to its cage, placed on a hand warmer. Post-operationally mice are injected with Buprenorphine SR® (1 mg/kg) every 48-72 hours until they are euthanized.
Tissue Preparation
Brain:
Mice were anesthetized by intraperitoneal injection of Ketamine-Xylazine solution (80 mg/kg-10 mg/kg) and intracardially perfused with ice cold 0.9% saline followed by ice cold 4% Paraformaldehyde in 0.1M PBS. The brain was removed and postfixed (4% PFA, 4°C, overnight). After that, the brain is incubated in 30% sucrose in 0.1M PBS (48h or until they sink to the bottom of the vial) and then coronally sectioned into 40 μm sections using a sliding microtome (SM2010 R, Leica). Sections were collected in 0.1M PBS or PBT azide for short- or long-term analysis respectively.
Spinal cord and pancreas:
Mice were anesthetized by intraperitoneal injection of Ketamine-Xylazine solution (80 mg/kg-10 mg/kg) and intracardially perfused with ice cold 0.9% saline followed by ice cold 4% Paraformaldehyde in 0.1M PBS. Spinal cord (segments T6-15) and whole pancreas were dissected and postfixed (4% PFA, 4°C, overnight). After that, the tissues were successively incubated for 24h in 10, 20 and 30% sucrose in 0.1 M PBS and then sectioned into 15 μm and 40 μm sections using a cryostat (Leica, Buffalo Grove, IL) and mounted on microscopy slides (Superfrost Plus, Fisher, Waltham, MA) and stored at −20°C or 80°C.
Immunohistochemistry/Immunofluorescence
Brain sections were washed in 0.1 M PBS for 10 min, incubated in blocking buffer (5% normal donkey serum; 0.5% Triton X-100) for 2h and then incubated with primary antibody in blocking buffer (4°C, overnight). Next day the sections were washed 3 × 10 min in 0.1 M PBS, incubated with secondary antibody solution and then washed again 3 × 10min in 0.1 M PBS, mounted on microscopy slides, and coverslipped (Vectashield Hardset mounting medium with DAPI; Vector, Burlingame, CA). Spinal cord and pancreas sections were left at RT for 10 min to air dry, washed in 0.1M PBS for 10 min, incubated in blocking buffer (5% normal donkey serum; 0.5% Triton X-100 in PBS) for 2h and then incubated with primary antibody in blocking buffer (4°C, overnight). Next day the sections were washed 3 × 10 min in 0.1 M PBS, incubated with secondary antibody solution, washed again 3 × 10 min in 0.1 M PBS and then coverslipped (Vectashield Hardset mounting medium with DAPI; Vector, Burlingame, CA). Images were captured using Olympus VS120 (Olympus, Center Valley, PA) slide scanner microscope and/or Zeiss Observer Z1 confocal microscope (Zeiss, Peabody, MA). Antibody details are provided in the KEY RESOURCES TABLE section. We used the following primary antibodies: rabbit anti-cFos (1:1000; Santa Cruz, Sc-52), mouse anti-oxytocin-NP (1:1000; PS 38), mouse anti-vasopressin-NP (1:1000; PS 41), guinea-pig anti-insulin (1:1000; Dako, A0564). rabbit anti-rb134 (1:2000; rb134). We used the following secondary antibodies: Alexa 568 donkey anti-rabbit (1:1000; Invitrogen, A10042), Alexa 488 donkey anti-rabbit (1:1000; Jackson Immunoresearch, 711-545-152), Alexa 594 donkey anti-mouse (1:1000; Jackson Immunoresearch, 715-585-150), Alexa 594 donkey anti-guineapig (1:1000; Jackson Immunoresearch, 706-585-148).
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-Fos | Santa Cruz Biotechnology | Cat# sc-52; RRID:AB_2106783 |
| Mouse anti-oxytocin-neurophysin | H. Gainer, NINDS | Cat# PS-38; RRID:AB_2315026 |
| Mouse anti-vasopressin-neurophysin | H. Gainer, NINDS | Cat# PS-41; RRID:AB_2313960 |
| Guinea pig anti-insulin | Agilent (Dako) | Cat# A0564; RRID:AB_10013624 |
| Rabbit DsRed | ||
| Rabbit anti-rb134 | L.W. Enquist, Princeton University | Cat# Rb133/Rb134, RRID:AB_2315209 |
| Alexa Fluor 488 donkey anti-rabbit | Jackson ImmunoResearch Labs | Cat# 711-545-152, RRID: AB_2313584 |
| Alexa Fluor 568 donkey anti-rabbit | Invitrogen | Cat# A10042, RRID:AB_2534017 |
| Alexa Fluor 594 donkey anti-mouse | Jackson ImmunoResearch Labs | Cat# 715-585-150, RRID:AB_2340854 |
| Alexa Fluor 594 donkey anti-guinea-pig | Jackson ImmunoResearch Labs | Cat# 706-585-148, RRID:AB_2340474 |
| Bacterial and virus strains | ||
| Ba2017 | this manuscript | N/A |
| Ba2001 | CNNV | http://www.cnnv.pitt.edu/ |
| AAV8-hSyn-DIO-hM3D(Gq)-mCherry | Addgene | RRID:Addgene_44361 |
| AAV8-hSyn-DIO-mCherry | Addgene | RRID:Addgene_50459 |
| AAV5-EF1a-DIO-EYFP-WPRE-pA | UNC Vector Core | https://www.med.unc.edu/genetherapy/vectorcore |
| AAV-DJ-CMV-DIO-eGFP-2A-TeNT | Stanford Viral Core | Cat# GVVC-AAV-71 |
| AAV1-CAG-FLEX-GCaMP6s-WPRE-SV40 | UPenn Vector Core | Cat# AV-1-PV2818 |
| Chemicals, peptides, and recombinant proteins | ||
| Glucose | Sigma | Cat# G7021 |
| 2-Deoxy-D-Glucose | Sigma | D6134 |
| Clozapine-N-Oxide | MedChemExpress | Cat# HY-17366 |
| Collagenase Type1A | Sigma | C9891 |
| Normal donkey serum | Jackson ImmunoResearch Labs | Cat# 017-000-121, RRID:AB_2337258 |
| Critical commercial assays | ||
| Mouse Ultrasensitive Insulin ELISA | Alpco Diagnostics | Cat# 80-INSMSU-E01, RRID:AB_2792981 |
| Glucagon ELISA - 10 μL | Mercodia | Cat# 10-1281-01, RRID:AB_2783839 |
| Noradrenaline high sensitive ELISA | DLD Diagnostika | Cat# EA633/96 |
| RNAqueous™-Micro Total RNA Isolation Kit | ThermoFisher | Cat# AM1931 |
| NEBNext® Ultra™ II RNA Library Prep Kit for Illumina® | NEB | Cat# E7770S |
| Deposited data | ||
| RNA sequencing data | GEO | GEO: GSE188856 |
| Data S1 | N/A | Excel file containing values used to create all graphs in the paper. |
| Experimental models: Organisms/strains | ||
| C57BL/6 mice | The Jackson Laboratory | RRID:IMSR_JAX:000664 |
| Ins1-Cre | The Jackson Laboratory | RRID:IMSR_JAX:026801 |
| Oxt-ires-Cre | The Jackson Laboratory | RRID:IMSR_JAX:024234 |
| Avp-ires-Cre | The Jackson Laboratory | RRID:IMSR_JAX:023530 |
| Software and algorithms | ||
| Ponemah v6.30 | DSI | https://www.datasci.com/ |
| VitalView software 5.0 | Starr Life Sciences | https://www.starrlifesciences.com/ |
| Olyvia 2.9 | Olympus | https://www.olympus-lifescience.com/ |
| Zen 2012 | Zeiss | https://www.zeiss.com/microscopy/ |
| Synapse, Browser | TDT | https://www.tdt.com/component/ |
| Prism 8.2.0 | GraphPad | https://www.graphpad.com/ |
| ImageJ-Fiji | NIH | https://imagej.net/ |
| Partek Flow | Partek | https://www.partek.com/ |
| Ingenuity Pathway Analysis | Qiagen | https://digitalinsights.qiagen.com/ |
| DAVID | LHRI | https://david.ncifcrf.gov/ |
| Other | ||
| VECTASHIELD HardSet Mounting Medium with DAPI | Vector Laboratories | Cat# H-1500, RRID:AB_2336788 |
| Meloxicam SR® | ZooPharm | N/A |
| Buprenorphine SR® | ZooPharm | N/A |
Stereotaxic AAV Injections
Mice were anaesthetized with 0.5–1.5% isoflurane and placed into a stereotaxic apparatus (Stoelting, Wood Dale, IL). After exposing and disinfecting the skin area above the skull, a small incision was made to expose the skull. Two small holes were drilled bilaterally with a small burr drill over the appropriate stereotaxic coordinates. A 30-gauge blunt end stainless steel needle attached to a 5-μ1 Hamilton syringe was lowered into the burr hole and 50-200 nl of the viral solution was administered bilaterally in the PVN (bregma: AP: −0.80 mm, DV: −4.70 mm, L: +/−0.25 mm) or SON (bregma: AP: −0.58 mm, DV: −5.40 mm, L: +/−1.13 mm) over 3-5 min per side. The needle was left in place for 5 min after each injection. Following removal of the needle, the skin was closed with wound clips and sealed with Vetbond (3M, St. Paul, MN) surgical glue. Post-operationally the mice were placed on a warm surface and were administered 0.3-0.5 ml of warm sterile 0.9% saline intraperitoneally and Meloxicam SR® subcutaneously (4 mg/kg for 72-hour action; ZooPharm, Laramy, WY USA). We performed stereotaxic injection of the following viruses: AAV-DJ-CMV-DIO-eGFP-2A-TeNT (#GVVCAAV-71, Stanford Viral Core; Stanford, CA), AAV8-hSyn-DIO-hM3D(Gq)-mCherry (44361, Addgene Viral Core; Watertown, MA). AAV8-hSyn-DIO-mCherry (50459, Addgene Viral Core; Watertown, MA), AAV5-EF1a-DIO-EYFP-WPRE-pA (UNC Vector Core, Chapel Hill, NC).
Blood Glucose Telemetry
Mice were subjected to surgical implantation of HD-XG blood glucose implant (Data Sciences International, St. Paul., MN). The body weight of the mice were between 25 and 30 g. All animals were anesthetized using 1-3% Isoflurane. Once general anesthesia is achieved, hair was removed from the ventral surface of the neck, from the chest and the abdomen and Buprenorphine SR® (1 mg/kg; ZooPharm, Laramy, WY USA) was administered subcutaneously. The mouse was placed on an operating table in dorsal recumbency on a heated water blanket. Surgical preparation (3x alternating chlorhexadine, followed by alcohol) was applied to all areas where hair has been removed and the surgical site was prepared using aseptic technique. A midline incision was made in the ventral surface of the neck, extending from the mandible to the level of the sternum. Using blunt dissection, the skin was separated from the underlying muscle, to create a subcutaneous pouch and a tunnel that begins at the cervical incision and extends to the lateral chest and flank. After irrigation of the tunnel with warm sterile saline, the transmitter was placed through the incision and moved through the tunnel to the subcutaneous pouch. Once the transmitter body is in place, the glands in the neck were retracted to expose the muscles along the trachea and the carotid artery, the latter of which was dissected (either bluntly or hydraulically) isolated from the vagus nerve. Three silk suture ties (6-0) were placed under a non-branching segment of the vessel at 6-7 mm apart. The two proximal ties were used to secure a catheter in the vessel and the distal one, near the bifurcation of the internal and external carotid arteries, was used to ligate the artery. A 25-27 gauge needle was used to puncture the vessel between the two ties to create an arteriotomy for placement of the catheter. Using a catheter introducer and vessel cannulation forceps, the transmitter catheter (PA-C10) was placed into the vessel and advanced until the catheter tip was in the aortic arch. The catheter was secured in the vessel using silk ties. The skin incision was closed with 5-0 or 6-0 Vicryl suture. Following placement of the transmitter and catheter, Bupivacaine was dripped on the skin incision(s) and the mouse was given 1 ml sterile saline SQ and was placed in a heated, oxygenated chamber to recover. The animals were monitored multiple times a day - incision site, signs of pain, food, and water intake, etc. and were provided pain medication (Buprenorphine SR® 1 mg/kg every 2 to 3 days) and 1-2 mL of subcutaneous fluids (neck skinfold) as needed for 24-48 hours. At that time, they were single housed in a micro-isolator cage and put on a heated water blanket inside an oxygenated intensive care cage in ICU. They were continually monitored until they are moving around freely and fully recovered. The telemetry implant was switched on 24 hours after the implantation and remained on throughout the recovery and experimental period an until the mice were euthanized. During all the telemetry experiments the animals remained single-housed in temperature-controlled chambers at 22° C with a 12-hour light/12-hour dark cycle (lights on at 06:00) in their home cages placed on top of ER4000 energizer/receivers (Starr Life Sciences). Blood glucose was continuously measured by telemetry, and 1-min means were collected with Ponemah software (Starr Life Sciences). The telemetry setup for blood glucose was calibrated once for each mouse 24 hours after the animals were placed on the chamber with the receivers. Calibrations were repeated once per week. Animals with inaccurate viral injection were excluded.
Chemogenetic Experiment 1 (GSIS)
Mice were fasted overnight (16h) and at 4h in the light phase (10:00 a.m.) half of them received a single i.p. injection of glucose (2 g/kg; Sigma, G7021) solution and the other half received a solution of glucose (2 g/kg) plus CNO (0.5 mg/kg; MedChemExpress. HY-17366) (all solutions in sterile 0.9% saline). Blood was collected through tail bleeding before the injection (time 0) and at 10 minutes (time 10) post-injection and kept on ice. Mice were placed in a restraining apparatus during blood collection. Blood samples were later spun down at 4000xg for 15 minutes and plasma was collected which was later analyzed for insulin content by ELISA (ALPCO, 80-INSMSU-E01; Salem, NH USA). This procedure was repeated at least 4 times for each mouse, twice with glucose and twice with glucose+CNO injection in a crossover design with one-week gaps to ensure recovery of blood volume and dissipation of the effects of CNO. Experiments with similar basal fasting insulin levels for both injection types were used for analysis. All chemogenetic experiments were pseudo-randomized by crossover design where half of the mice received injections with CNO and half with vehicle.
Chemogenetic Experiment 2 (GTT Telemetry)
Like experiment 1, the mice were fasted overnight (16h) and at 4h in the light phase (10:00 a.m.) half of them received a single i.p. injection of glucose (2 g/kg) solution and the other half received a solution of glucose (2 g/kg) plus CNO (0.5 mg/kg) (all solutions in sterile 0.9% saline). After injection mice returned to their cage and there was no more handling. Baseline blood glucose at time zero was subtracted from data that were collected for the first 30 minutes, and these data were used for comparison analysis of area under curve and peak values. This procedure was repeated in a crossover design with 3-4 day intervals to ensure dissipation of the effects of CNO and recovery from fasting.
Chemogenetic Experiment 3 (Refeeding Telemetry)
Food was removed at 8h in the light phase (2:00 p.m.) and was added again 4h later (6:00 p.m.). One hour before adding the food back (5:00 p.m.) the mice received a single injection of either vehicle or CNO (1 mg/kg) solution. The average glycemic levels of the period between injection and feeding were used as a baseline to calculate the delta of glucose change after feeding. This procedure was repeated in a crossover design with 3-4 day intervals to ensure dissipation of the effects of CNO.
Plasma Insulin, Glucagon and Noradrenaline Measurements
Plasma insulin was measured with mouse ultrasensitive insulin ELISA (ALPCO). Plasma glucagon was measured with glucagon ELISA - 10 μL (Mercodia). Plasma noradrenaline was measured with noradrenaline high sensitive ELISA (DLD Diagnostika).
Electron Microscopy
Protocol used for the transmission electron microscope (TEM) analysis of mouse islets is described elsewhere (Fulgenzi et al., 2020). In brief, pvNOXT:hM3Dq and pvNOXT:mCherry mice (n=4 per group) were fasted overnight (16h) and at 4h in the light phase (10:00 a.m.) (similar to chemogenetic experiment 1) they all received a single i.p. injection of glucose (2 g/kg) plus CNO (1 mg/kg). 10 minutes later they were euthanized by cervical dislocation and were perfused for 30 seconds with PBS followed by 5 minutes with fixative solution (2.5% glutaraldehyde, 0.5% tannic acid, 30 mM sucrose in 0.1 M cacodylate buffer). The pancreata were removed and post-fixed for 2 hrs. in the same fixative followed by 1 hour in 1% osmium tetroxide buffer. Small, trimmed tissue blocks of about 1 mm3 were washed 3 times in buffer and stained with 2% uranil acetate in 50% ethyl alcohol for 1 hr. dehydrated and embedded in Epon-Araldite resin per standard protocol. 60 nm ultrathin sections were stained with lead citrate and imaged with TEM, (Technai T12 FEI at 1900x magnification). Analysis of insulin granule content was performed using ImageJ software. Between 10-20 EM images (24 x 16 μm) were analyzed from each islet (total 20-40 from 2 islets combined) for quantification of docked insulin granules. Each image contained 2-8 beta cells which accounted for 135 μm of membrane length and 138 docked insulin granules on average (total ~4000 docked granules per animal). All docked granules from all beta cells were quantified. There was no area selection to avoid bias.
Spinal Cord Viral Injection
This surgical approach was adapted from Kohro et al (Kohro et al., 2015). Mice were anaesthetized with isoflurane and placed into a stereotaxic apparatus (Stoelting). The skin was incised at T8–L1 segments and the spinal cord immobilized to a fixed position. The dura mater and the arachnoid membrane between the bones are carefully incised using the tip of a 30G needle to create a hole bilaterally between segments T9-T10 and T11-T12. A 30G blunt end stainless steel needle attached to a 5-μl Hamilton syringe was lowered into each hole directly in the lateral horn (approximately 500μm lateral from the midline and 550 μm in depth from the surface of the dorsal root entry) and 500 nl of the viral solution was administered over 3-5 min per site. The needle was left in place for 5 min after the injection. Following removal of the needle, the skin was closed with wound clips and sealed with Vetbond surgical glue. Post-operationally the mice were placed on a warm surface and administered 0.3-0.5 ml of warm sterile 0.9% saline i.p., and Meloxicam SR® s.c. Mice were left to recover for 6 weeks before the GSIS and GTT experiments. For the automated glucose telemetry experiments, mice underwent a second surgery for implantation of glucose monitoring device 4 weeks after the spinal cord viral injection and were left to recover for 2 weeks.
RNA Sequencing
PVNOXT:TeNT and PVNOXT:GFP mice were fasted for 24 hours (2 p.m. to 2 p.m.) and euthanized in a CO2 chamber. Mouse pancreatic islets were isolated via bile duct collagenase digestion with Collagenase type 1A (1 mg/ml, Sigma, St. Louis, MO, USA) using Histopaque (Sigma) gradient, followed by hand-picking under a stereomicroscope and collected islets were used for RNA-seq analysis. Total RNA was extracted from isolated mouse pancreatic islet cells, using RNAqueous Micro Total RNA isolation Kit (ThermoFisher) according to manufacturer's instructions. The quality of the input RNA was assessed using Bioanalyzer 2100 and the RNA nano chip (Agilent Technologies, Santa Clara, CA). After enrichment of mRNA by NEBNext Poly(A) mRNA Magnetic Isolation Module (NEB), processes of cDNA synthesis and sequencing library generation were performed using NEBNext Ultra DNA Prep Kit (NEB) with NEBNext Multiplex Oligos, following the manufacturer’s protocol. Library fragment-size distribution was assessed using the Bioanalyzer 2100 and the DNA high-sensitivity chip (Agilent Technologies). Quantification of libraries was performed using Qubit 2.0 (ThermoFisher) and Qubit dsDNA HS Quantification Kit (ThermoFisher) before sequencing. 12 samples per lane were multiplexed in HiSeq3000, single-end 50. Downstream analysis of demultiplexed fastq files was executed using Partek Flow (Partek, St. Louis, MO): alignment to the reference genome (mm10), and quantification to annotation model (mm10_refseq_v91_19_08_01_v2). STAR aligner was used for sequence alignment and the genomic alignments that map uniquely to the set of known Refseq were used as raw input. TPM normalized count matrix was further visualized. DESeq2 un-normalized transcript count matrix was used as input, low abundant features were removed by strict pre-filtering, DESeq2 comparison analysis data were used for selection of significantly changed gene expression. Cut-off values were set at p<0.05. KEGG Pathway analysis was performed using three independent analysis software: Ingenuity Pathway Analysis (Qiagen), DAVID Bioinformatics Resources 6.8 (LHRI) and Gene Ontology (Panther). Corroborating results were collected as well as results that were only available on DAVID but not on the other data bases (DAVID has more comprehensive listing of KEGG pathways). Z-score heatmap was generated using TPM normalized count values.
Fos Experiment
On the day of the experiment, food was removed 2h prior to injection and mice were placed in a clean cage. (~8am). C57BL/6 mice were injected i.p. with 0.9% saline or 2-DG (400 mg/kg). 2h post-injection mice were anesthetized by intraperitoneal injection of Ketamine-Xylazine solution (80 mg/kg-10 mg/kg) and intracardially perfused with ice cold 0.9% saline followed by ice cold 4% Paraformaldehyde in 0.1M PBS. The brain was removed and postfixed (4% PFA, 4°C, overnight) prior to processing for immunofluorescence assays.
In Vivo Fiber Photometry
Stereotaxic injections, optical fiber implantation and in vivo fiber photometry were performed as previously described (Li et al., 2019). Mice were anaesthetized with isoflurane and placed into a stereotaxic apparatus (Stoelting Just for Mice). For postoperative care, mice were injected intraperitoneally with Meloxicam SR® (4 mg/kg). After exposing the skull via small incision, a small hole was drilled for injection. A pulled-glass pipette with 20–40 mm tip diameter was inserted into the brain and virus was injected by an air pressure system. A micromanipulator (Grass Technologies, Model S58 Stimulator) was used to control injection speed at 25 nl/min and the pipette was withdrawn 5 min after injection. AAV1-CAG-FLEX-GCaMP6s-WPRE- SV40 (#AV-1-PV2818, Penn Vector Core; Philadelphia, PA), was unilaterally injected into the PVN (25-50 nl, bregma: AP: −0.80 mm, DV: −4.70 mm, L: +/−0.20 mm). Optical fibers (fiber: core = 400 mm; 0.48 NA; M3 thread titanium receptacle; Doric Lenses) were implanted unilaterally over the PVN (25-50 nl, bregma: AP: −0.80 mm, DV: −4.60/−4.65 mm, L: +/−0.20 mm). Fibers were fixed to the skull using C&B Metabond Quick Cement and dental acrylic and mice were allowed 2 weeks for recovery before acclimatization. Recordings started 4-6 weeks after cranial surgeries to allow for adequate recovery and viral expression. During the last 2 weeks of that period, mice were habituated to handling by the investigator. All recordings were done in the home cage of the individually housed experimental animal near the beginning of the light cycle. Mice were allowed to adapt to the tethered patchcord for 2 days prior to experiments (core = 400 mm; 0.48 NA; M3 connector; Doric Lenses) and given 10 minutes to acclimate to the tethered patchcord prior to any recording. Continuous <20 Wblue LED at 470 nm and UV LED at 405 nm served as excitation light sources, driven by a multichannel hub (Thorlabs), modulated at 211hz and 511hz respectively, and delivered to a filtered minicube (FMC5, Doric Lenses) before connecting through optic fibers to a rotary joint (FRJ 1 3 1, Doric Lenses) to allow for movement. GCaMP6s calcium GFP signals and UV autofluorescent signals were collected through the same fibers back to the dichroic ports of the minicube into a femtowatt silicon photoreceiver (2151, Newport). Digital signals were then demodulated, amplified, and collected through a lock-in amplifier (RZ5P, Tucker-Davis Technologies). Data was collected through the software Synapse (TDT), exported via Browser (TDT), and analyzed in Microsoft Excel. Synchronized multi-angled high-definition videos were recorded for time-locked data analysis in Synapse. On the day of the experiment, food was removed 2h prior to injection and mice were placed in a clean cage (~8am). Mice were plugged to the fiber and baseline signal was recorded for 5 min. After that, mice were injected i.p. with 0.9% saline or 2-DG (200 mg/kg) and fluorescent signal was recorded for 35 more minutes. After the end of the recording, food was introduced into the cage and 1h later the remaining food was weighed, and food consumption was calculated. Animals with inaccurate viral injection or fiber placement were excluded.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data for group analysis are presented as dot plots that include each datapoint, mean and SEM. Statistical significance for these data was calculated using unpaired two-tailed Student’s t-test. Data for paired analysis are presented as dot plots that include each datapoint. Statistical significance for these data was calculated using paired Student’s t-test. Statistical significance for RNA-seq data was calculated with DESeq analysis.
Quantification of islet infection, docked granules and c-fos positivity were done in blinded fashion.
No statistical methods were used to predetermine sample size. Samples sizes were chosen based on previous reports of experiments of the same type in the published literature.
Chemogenetic experiments were pseudo-randomized by crossover design where half of the mice received injections with CNO and half with vehicle. All viral injections were pseudo-randomized by randomly injecting mice of the same litter for chemogenetics (AAV-hsyn-DIO-hM3Dq-mCherry or AAV-hsyn-DIO-mCherry) and tetanus toxin (AAV-CMV-DIO-TeNT or AAV-EF1A-DIO-EYFP) experiments. Littermate mice were randomly assigned to different groups without being separated.
Data excluded:
Based on immunofluorescence analyses, animals with inaccurate stereotaxic viral injection (4 mice) or fiber placement (5 mice) were excluded from chemogenetic and fiber photometry data analyses, respectively. One animal with glucose intolerance was excluded from tetanus toxin data analyses. One mouse was excluded from tracing analyses for lack of viral signal. Two mice were excluded from the spinal cord experiment due to lower limb paralysis.
All data follow normal distribution.
Supplementary Material
Data S1. Unprocessed data for all graphs. Related to Figures 3, 4, 5, 6 and Supplemental Figures 1, 3, 4, 5.
Highlights.
Hypothalamic neurons are multisynaptically connected to pancreatic islet β-cells
Stimulation of PVNOXT neurons suppresses insulin secretion and increases glycemia
Functional silencing of PVNOXT neurons induces hypoglycemia
Hypoglycemia stimulates PVNOXT neuronal activity to suppress insulin secretion
Acknowledgements
We thank Jens B. Bosse and Esteban Engel for assistance with production of Ba2017, Harold Gainer for providing antibodies, Alice Franks for assistance with mouse colony maintenance, Audrey Noguchi and Danielle Springer of the NHLBI Murine Phenotyping Core for glucose telemetry implantation surgeries and the NHLBI Genomics Core for RNAseq analyses support. We thank Marc Reitman, Clifton Bogardus, Kevin Hall, Aaron Cypess, Claire-Marie Vacher and Ramana V. Tantravahi for feedback and critical reading of the manuscript. This research was supported by the Intramural Research Program (DK055105 SGR, DK055107 SGR, DK075073 SGR and DK075087 MJK) of the NIDDK, NIH.
Footnotes
Inclusion and Diversity
We worked to ensure sex balance in the selection of non-human subjects. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science.
Declaration of Interests
The authors declare no competing interests
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahren B (2000). Autonomic regulation of islet hormone secretion--implications for health and disease. Diabetologia 43, 393–410. [DOI] [PubMed] [Google Scholar]
- Arbelaez AM, Powers WJ, Videen TO, Price JL, and Cryer PE (2008). Attenuation of counterregulatory responses to recurrent hypoglycemia by active thalamic inhibition: a mechanism for hypoglycemia-associated autonomic failure. Diabetes 57, 470–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banting FG, Best CH, Collip JB, Campbell WR, and Fletcher AA (1922). Pancreatic Extracts in the Treatment of Diabetes Mellitus. Can Med Assoc J 12, 141–146. [PMC free article] [PubMed] [Google Scholar]
- Bernard C (1855). Leçons de physiologie expérimental appliquée à la médecine, faites au Collège de France. (Paris,: J.B. Baillière et fils; etc.). [Google Scholar]
- Berthoud HR, Bereiter DA, and Jeanrenaud B (1980). Role of the autonomic nervous system in the mediation of LHA electrical stimulation-induced effects on insulinemia and glycemia. J Auton Nerv Syst 2, 183–198. [DOI] [PubMed] [Google Scholar]
- Berthoud HR, and Jeanrenaud B (1979). Acute hyperinsulinemia and its reversal by vagotomy after lesions of the ventromedial hypothalamus in anesthetized rats. Endocrinology 105, 146–151. [DOI] [PubMed] [Google Scholar]
- Buijs RM, Chun SJ, Niijima A, Romijn HJ, and Nagai K (2001). Parasympathetic and sympathetic control of the pancreas: a role for the suprachiasmatic nucleus and other hypothalamic centers that are involved in the regulation of food intake. J Comp Neurol 431, 405–423. [DOI] [PubMed] [Google Scholar]
- Cabrera O, Berman DM, Kenyon NS, Ricordi C, Berggren PO, and Caicedo A (2006). The unique cytoarchitecture of human pancreatic islets has implications for islet cell function. Proc Natl Acad Sci U S A 103, 2334–2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Card JP, and Enquist LW (2014). Transneuronal circuit analysis with pseudorabies viruses. Curr Protoc Neurosci 68, 1 5 1–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cechetto DF, and Saper CB (1988). Neurochemical organization of the hypothalamic projection to the spinal cord in the rat. J Comp Neurol 272, 579–604. [DOI] [PubMed] [Google Scholar]
- Cryer PE (2013). Mechanisms of hypoglycemia-associated autonomic failure in diabetes. N Engl J Med 369, 362–372. [DOI] [PubMed] [Google Scholar]
- Cryer PE, and Gerich JE (1985). Glucose counterregulation, hypoglycemia, and intensive insulin therapy in diabetes mellitus. N Engl J Med 313, 232–241. [DOI] [PubMed] [Google Scholar]
- DeFalco J, Tomishima M, Liu H, Zhao C, Cai X, Marth JD, Enquist L, and Friedman JM (2001). Virus-assisted mapping of neural inputs to a feeding center in the hypothalamus. Science 291, 2608–2613. [DOI] [PubMed] [Google Scholar]
- Duncan A, Heyer MP, Ishikawa M, Caligiuri SPB, Liu XA, Chen Z, Micioni Di Bonaventura MV, Elayouby KS, Abies JL, Howe WM, et al. (2019). Habenular TCF7L2 links nicotine addiction to diabetes. Nature 574, 372–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dybala MP, and Hara M (2019). Heterogeneity of the Human Pancreatic Islet. Diabetes 68, 1230–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enquist LW, and Card JP (2003). Recent advances in the use of neurotropic viruses for circuit analysis. Curr Opin Neurobiol 13, 603–606. [DOI] [PubMed] [Google Scholar]
- Fanelli C, Pampanelli S, Epifano L, Rambotti AM, Ciofetta M, Modarelli F, Di Vincenzo A, Annibale B, Lepore M, Lalli C, et al. (1994). Relative roles of insulin and hypoglycaemia on induction of neuroendocrine responses to, symptoms of, and deterioration of cognitive function in hypoglycaemia in male and female humans. Diabetologia 37, 797–807. [DOI] [PubMed] [Google Scholar]
- Fasanella KE, Christianson JA, Chanthaphavong RS, and Davis BM (2008). Distribution and neurochemical identification of pancreatic afferents in the mouse. J Comp Neurol 509, 42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman JM, and Lebovitz HE (1970). Effect of fasting on insulin secretion and action in mice. Endocrinology 86, 313–321. [DOI] [PubMed] [Google Scholar]
- Fisher BM, Baylis PH, Thornton S, and Frier BM (1989). Arginine vasopressin and oxytocin responses to insulin-induced hypoglycemia in type 1 (insulin-dependent) diabetes. J Clin Endocrinol Metab 68, 688–692. [DOI] [PubMed] [Google Scholar]
- Fulgenzi G, Hong Z, Tomassoni-Ardori F, Barella LF, Becker J, Barrick C, Swing D, Yanpallewar S, Croix BS, Wess J, et al. (2020). Novel metabolic role for BDNF in pancreatic beta-cell insulin secretion. Nat Commun 11, 1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandasi NR, Yin P, Omar-Hmeadi M, Ottosson Laakso E, Vikman P, and Barg S (2018). Glucose-Dependent Granule Docking Limits Insulin Secretion and Is Decreased in Human Type 2 Diabetes. Cell Metab 27, 470–478 e474. [DOI] [PubMed] [Google Scholar]
- Gannon M, Kulkarni RN, Tse HM, and Mauvais-Jarvis F (2018). Sex differences underlying pancreatic islet biology and its dysfunction. Mol Metab 15, 82–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerich JE, Mokan M, Veneman T, Korytkowski M, and Mitrakou A (1991). Hypoglycemia unawareness. Endocr Rev 12, 356–371. [DOI] [PubMed] [Google Scholar]
- Gilliam LK, Palmer JP, and Taborsky GJ Jr. (2007). Tyramine-mediated activation of sympathetic nerves inhibits insulin secretion in humans. J Clin Endocrinol Metab 92, 4035–4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graveling AJ, and Frier BM (2010). Impaired awareness of hypoglycaemia: a review. Diabetes Metab 36 Suppl 3, S64–74. [DOI] [PubMed] [Google Scholar]
- Hallbeck M, Larhammar D, and Blomqvist A (2001). Neuropeptide expression in rat paraventricular hypothalamic neurons that project to the spinal cord. J Comp Neurol 433, 222–238. [DOI] [PubMed] [Google Scholar]
- Havel PJ, and Taborsky GJ Jr. (1989). The contribution of the autonomic nervous system to changes of glucagon and insulin secretion during hypoglycemic stress. Endocr Rev 10, 332–350. [DOI] [PubMed] [Google Scholar]
- Havel PJ, Veith RC, Dunning BE, and Taborsky GJ Jr. (1988). Pancreatic noradrenergic nerves are activated by neuroglucopenia but not by hypotension or hypoxia in the dog. Evidence for stress-specific and regionally selective activation of the sympathetic nervous system. J Clin Invest 82, 1538–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hom FG, Goodner CJ, and Berrie MA (1984). A [3H]2-deoxyglucose method for comparing rates of glucose metabolism and insulin responses among rat tissues in vivo. Validation of the model and the absence of an insulin effect on brain. Diabetes 33, 141–152. [DOI] [PubMed] [Google Scholar]
- Hosoya Y, Matsukawa, Okado N, Sugiura Y, and Kohno K (1995). Oxytocinergic innervation to the upper thoracic sympathetic preganglionic neurons in the rat. A light and electron microscopical study using a combined retrograde transport and immunocytochemical technique. Exp Brain Res 107, 9–16. [DOI] [PubMed] [Google Scholar]
- Kaneto A, Kajinuma H, and Kosaka K (1975). Effect of splanchnic nerve stimulation on glucagon and insulin output in the dog. Endocrinology 96, 143–150. [DOI] [PubMed] [Google Scholar]
- Kohro Y, Sakaguchi E, Tashima R, Tozaki-Saitoh H, Okano H, Inoue K, and Tsuda M (2015). A new minimally-invasive method for microinjection into the mouse spinal dorsal horn. Sci Rep 5, 14306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korstanje R, Ryan JL, Savage HS, Lyons BL, Kane KG, and Sukoff Rizzo SJ (2017). Continuous Glucose Monitoring in Female NOD Mice Reveals Daily Rhythms and a Negative Correlation With Body Temperature. Endocrinology 158, 2707–2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krashes MJ, Koda S, Ye C, Rogan SC, Adams AC, Cusher DS, Maratos-Flier E, Roth BL, and Lowell BB (2011). Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J Clin Invest 121, 1424–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langerhans P, and Morrison H (1937). Contributions to the microscopic anatomy of the pancreas. (Baltimore: John Hopkins Press; ). [Google Scholar]
- Leng G, and Sabatier N (2017). Oxytocin - The Sweet Hormone? Trends Endocrinol Metab 28, 365–376. [DOI] [PubMed] [Google Scholar]
- Li C, Navarrete J, Liang-Guallpa J, Lu C, Funderburk SC, Chang RB, Liberles SD, Olson DP, and Krashes MJ (2019). Defined Paraventricular Hypothalamic Populations Exhibit Differential Responses to Food Contingent on Caloric State. Cell Metab 29, 681–694 e685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao PY, Chiu YM, Yu JH, and Chen SK (2020). Mapping Central Projection of Oxytocin Neurons in Unmated Mice Using Cre and Alkaline Phosphatase Reporter. Front Neuroanat 14, 559402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo L, and Anderson DJ (2011). A Cre-dependent, anterograde transsynaptic viral tracer for mapping output pathways of genetically marked neurons. Neuron 72, 938–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer J (1953). Glucostatic mechanism of regulation of food intake. N Engl J Med 249, 13–16. [DOI] [PubMed] [Google Scholar]
- Meek TH, Nelson JT, Matsen ME, Dorfman MD, Guyenet SJ, Damian V, Allison MB, Scarlett JM, Nguyen HT, Thaler JP, et al. (2016). Functional identification of a neurocircuit regulating blood glucose. Proc Natl Acad Sci U S A 113, E2073–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnick IV, Price CJ, and Colmers WF (2011). Glucosensing in parvocellular neurons of the rat hypothalamic paraventricular nucleus. Eur J Neurosci 34, 272–282. [DOI] [PubMed] [Google Scholar]
- Mitrakou A, Ryan C, Veneman T, Mokan M, Jenssen T, Kiss I, Durrant J, Cryer P, and Gerich J (1991). Hierarchy of glycemic thresholds for counterregulatory hormone secretion, symptoms, and cerebral dysfunction. Am J Physiol 260, E67–74. [DOI] [PubMed] [Google Scholar]
- Noguchi GM, and Huising MO (2019). Integrating the inputs that shape pancreatic islet hormone release. Nature Metabolism 1, 1189–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh Y, Lai JS, Mills HJ, Erdjument-Bromage H, Giammarinaro B, Saadipour K, Wang JG, Abu F, Neubert TA, and Suh GSB (2019). A glucose-sensing neuron pair regulates insulin and glucagon in Drosophila. Nature 574, 559–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orci L (1985). The insulin factory: a tour of the plant surroundings and a visit to the assembly line. The Minkowski lecture 1973 revisited. Diabetologia 28, 528–546. [DOI] [PubMed] [Google Scholar]
- Pavlov IP, and Thompson WH (1902). The work of the digestive glands, lectures by Professor J. P. Pawlow. (London,: C. Griffin & Company, limited; ). [Google Scholar]
- Perelis M, Marcheva B, Ramsey KM, Schipma MJ, Hutchison AL, Taguchi A, Peek CB, Hong H, Huang W, Omura C, et al. (2015). Pancreatic beta cell enhancers regulate rhythmic transcription of genes controlling insulin secretion. Science 350, aac4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisansky MT, Hanson LR, Gottesman II, and Gewirtz JC (2017). Oxytocin enhances observational fear in mice. Nat Commun 8, 2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porte D Jr. (1969). Sympathetic regulation of insulin secretion. Its relation to diabetes mellitus. Arch Intern Med 123, 252–260. [PubMed] [Google Scholar]
- Porte D Jr., and Williams RH (1966). Inhibition of insulin release by norepinephrine in man. Science 152, 1248–1250. [DOI] [PubMed] [Google Scholar]
- Pozo M, and Claret M (2018). Hypothalamic Control of Systemic Glucose Homeostasis: The Pancreas Connection. Trends Endocrinol Metab 29, 581–594. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Diaz R, Abdulreda MH, Formoso AL, Gans I, Ricordi C, Berggren PO, and Caicedo A (2011a). Innervation patterns of autonomic axons in the human endocrine pancreas. Cell Metab 14, 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Diaz R, Dando R, Jacques-Silva MC, Fachado A, Molina J, Abdulreda MH, Ricordi C, Roper SD, Berggren PO, and Caicedo A (2011b). Alpha cells secrete acetylcholine as a non-neuronal paracrine signal priming beta cell function in humans. Nat Med 17, 888–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohner-Jeanrenaud F, Bobbioni E, Ionescu E, Sauter JF, and Jeanrenaud B (1983). Central nervous system regulation of insulin secretion. Adv Metab Disord 10, 193–220. [DOI] [PubMed] [Google Scholar]
- Rorsman P, and Ashcroft FM (2018). Pancreatic beta-Cell Electrical Activity and Insulin Secretion: Of Mice and Men. Physiol Rev 98, 117–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rorsman P, and Renstrom E (2003). Insulin granule dynamics in pancreatic beta cells. Diabetologia 46, 1029–1045. [DOI] [PubMed] [Google Scholar]
- Rosario W, Singh I, Wautlet A, Patterson C, Flak J, Becker TC, Ali A, Tamarina N, Philipson LH, Enquist LW, et al. (2016). The Brain-to-Pancreatic Islet Neuronal Map Reveals Differential Glucose Regulation From Distinct Hypothalamic Regions. Diabetes 65, 2711–2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosengren AH, Jokubka R, Tojjar D, Granhall C, Hansson O, Li DQ, Nagaraj V, Reinbothe TM, Tuncel J, Eliasson L, et al. (2010). Overexpression of alpha2A-adrenergic receptors contributes to type 2 diabetes. Science 327, 217–220. [DOI] [PubMed] [Google Scholar]
- Routh VH, Hao L, Santiago AM, Sheng Z, and Zhou C (2014). Hypothalamic glucose sensing: making ends meet. Front Syst Neurosci 8, 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawchenko PE, and Swanson LW (1982). Immunohistochemical identification of neurons in the paraventricular nucleus of the hypothalamus that project to the medulla or to the spinal cord in the rat. J Comp Neurol 205, 260–272. [DOI] [PubMed] [Google Scholar]
- Schnutgen F, Doerflinger N, Calleja C, Wendling O, Chambon P, and Ghyselinck NB (2003). A directional strategy for monitoring Cre-mediated recombination at the cellular level in the mouse. Nat Biotechnol 21, 562–565. [DOI] [PubMed] [Google Scholar]
- Shah P, Rahman SA, Demirbilek H, Guemes M, and Hussain K (2017). Hyperinsulinaemic hypoglycaemia in children and adults. Lancet Diabetes Endocrinol 5, 729–742. [DOI] [PubMed] [Google Scholar]
- Sokoloff L, Reivich M, Kennedy C, Des Rosiers MH, Patlak CS, Pettigrew KD, Sakurada O, and Shinohara M (1977). The [14C]deoxyglucose method for the measurement of local cerebral glucose utilization: theory, procedure, and normal values in the conscious and anesthetized albino rat. J Neurochem 28, 897–916. [DOI] [PubMed] [Google Scholar]
- Stanley S, Domingos AI, Kelly L, Garfield A, Damanpour S, Heisler L, and Friedman J (2013). Profiling of Glucose-Sensing Neurons Reveals that GHRH Neurons Are Activated by Hypoglycemia. Cell Metab 18, 596–607. [DOI] [PubMed] [Google Scholar]
- Stanley SA, Kelly L, Latcha KN, Schmidt SF, Yu X, Nectow AR, Sauer J, Dyke JP, Dordick JS, and Friedman JM (2016). Bidirectional electromagnetic control of the hypothalamus regulates feeding and metabolism. Nature 531, 647–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson LW, and McKellar S (1979). The distribution of oxytocin- and neurophysin-stained fibers in the spinal cord of the rat and monkey. J Comp Neurol 188, 87–106. [DOI] [PubMed] [Google Scholar]
- Sweeney ST, Broadie K, Keane J, Niemann H, and O'Kane CJ (1995). Targeted expression of tetanus toxin light chain in Drosophila specifically eliminates synaptic transmission and causes behavioral defects. Neuron 14, 341–351. [DOI] [PubMed] [Google Scholar]
- Thorens B (2010). Central control of glucose homeostasis: the brain--endocrine pancreas axis. Diabetes Metab 36 Suppl 3, S45–49. [DOI] [PubMed] [Google Scholar]
- Thorens B, Tarussio D, Maestro MA, Rovira M, Heikkila E, and Ferrer J (2015). Ins1(Cre) knock-in mice for beta cell-specific gene recombination. Diabetologia 58, 558–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wick AN, Drury DR, Nakada HI, and Wolfe JB (1957). Localization of the primary metabolic block produced by 2-deoxyglucose. J Biol Chem 224, 963–969. [PubMed] [Google Scholar]
- Wong KP, Sha W, Zhang X, and Huang SC (2011). Effects of administration route, dietary condition, and blood glucose level on kinetics and uptake of 18F-FDG in mice. J Nucl Med 52, 800–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods SC, Alexander KR, and Porte D Jr. (1972). Conditioned insulin secretion and hypoglycemia following repeated injections of tolbutamide in rats. Endocrinology 90, 227–231. [DOI] [PubMed] [Google Scholar]
- Wu Z, Xu Y, Zhu Y, Sutton AK, Zhao R, Lowell BB, Olson DP, and Tong Q (2012). An obligate role of oxytocin neurons in diet induced energy expenditure. PLoS One 7, e45167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X, Guo P, Prasadan K, Shiota C, Peirish L, Fischbach S, Song Z, Gaffar I, Wiersch J, El-Gohary Y, et al. (2014). Pancreatic cell tracing, lineage tagging and targeted genetic manipulations in multiple cell types using pancreatic ductal infusion of adeno-associated viral vectors and/or cell-tagging dyes. Nat Protoc 9, 2719–2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zammitt NN, and Frier BM (2005). Hypoglycemia in type 2 diabetes: pathophysiology, frequency, and effects of different treatment modalities. Diabetes Care 28, 2948–2961. [DOI] [PubMed] [Google Scholar]
- Zhang B, Nakata M, Nakae J, Ogawa W, and Yada T (2018). Central insulin action induces activation of paraventricular oxytocin neurons to release oxytocin into circulation. Sci Rep 8, 10415. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Unprocessed data for all graphs. Related to Figures 3, 4, 5, 6 and Supplemental Figures 1, 3, 4, 5.
Data Availability Statement
RNA-seq data have been deposited at GEO and are publicly available. The accession number is GSE188856. Microscopy data reported in this paper will be shared by the lead contact upon request.
This paper does not report any original codes.
Data S1 represents an Excel file containing the values that were used to create all graphs in the paper. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.