

Abstract
Tin-halide perovskites (THPs) have emerged as promising lead-free perovskites for photovoltaics and photocatalysis applications but still fall short in terms of stability and efficiency with respect to their lead-based counterpart. A detailed understanding of the degradation mechanism of THPs in a water environment is missing. This Letter presents ab initio molecular dynamics (AIMD) simulations to unravel atomistic details of THP/water interfaces comparing methylammonium tin iodide, MASnI3, with the lead-based MAPbI3. Our results reveal facile solvation of surface tin–iodine bonds in MASnI3, while MAPbI3 remains more robust to degradation despite a larger amount of adsorbed water molecules. Additional AIMD simulations on dimethylammonium tin bromide, DMASnBr3, investigate the origins of their unprecedented water stability. Our results indicate the presence of amorphous surface layers of hydrated zero-dimensional SnBr3 complexes which may protect the inner structure from degradation and explain their success as photocatalysts. We believe that the atomistic details of the mechanisms affecting THP (in-)stability may inspire new strategies to stabilize THPs.
Metal halide perovskites belong to the most promising semiconducting materials for energy conversion from renewable sources in the form of photovoltaics1 and photocatalysis2 thanks to their favorable optoelectronic properties, such as direct and tunable band gaps,3 fast charge transport,4 low nonradiative recombination,5 and low-temperature processing capabilities from solution.6 Thin film solar cells based on lead-halide perovskites (LHPs) have recently reached 25.2% efficiency with significant long-term stability,7 rivaling silicon solar cells.
A tremendous drawback which may limit the industrial market potential of state-of-the art perovskite photovoltaics is the presence of lead in the B-site of the ABX3 perovskite crystal structure. Recent reports8,9 have underlined the detrimental impact of lead on the environment and human health, and have further raised the need for lead-free alternatives. Tin-halide perovskites (THPs) have emerged as one of the most promising lead-free alternative for photovoltaics10−12 with record efficiencies of 14.6%13 in pure THPs and beyond 20% in mixed tin–lead perovskites.14−17 Despite having an optimal band gap (∼1.2–1.3 eV) for photovoltaic applications, THP performance and stability are still behind their lead-based counterpart. One of the critical aspects limiting the performance and the stability of THPs is the facile oxidation of Sn(II) to Sn(IV),18,19 which may induce a large self-p-doping in the perovskite bulk and deep electron trap states at the perovskite surface.20−22 Experimental reports demonstrate THPs degradation even in well-encapsulated thin films under exposure of light or under elevated temperatures.23−25
Understanding of the perovskite (in-)stability and associated degradation mechanisms is of utmost importance to achieve reliable and long-term stable THPs. Previous studies have shed light on the degradation of the lead-based counterpart, methylammonium lead iodide, MAPbI3. Ab initio molecular dynamics (AIMD) simulations26 were reported on the degradation of MAI-terminated surfaces by a rapid solvation process which occurs by the bonding of water molecules to the Pb atoms with subsequent release of surface iodine atoms. Metadynamics simulations showed that, after the initial Pb–I bond breaking, further hydration of iodine provides the required energy to overcome the I–-MA+ attraction.27 On the contrary, the PbI-terminated surface is more robust to degradation, while water molecules may enter into the MAPbI3 bulk and form intermediate hydrated phases,26,28,29 which is a key step of perovskite degradation upon water exposure.30−32 Water-intercalated MAPbI3 readily forms PbI2 vacancy complexes and induces electronic trap states.33 The presence of water can be both detrimental or beneficial to the excited state lifetime, depending on the level of humidity exposure.34 Light exposure can further drive perovskite degradation by dissociating water molecules to OH–, which may deprotonate the MA cation followed by desorption of CH3NH2.35−37 Interestingly, partial replacement of iodine with bromine can enhance perovskite stability toward water and oxygen.37,38
Recent studies connected the degradation of THPs with the release of SnO2 and SnI4, while in mixed tin-lead perovskites I2 formation also was observed.24 The presence of oxygen and moisture further accelerates THP degradation by formation of SnI4.39,40 Density functional theory (DFT) simulations suggest that adsorbed water or oxygen, by formation of H–I or Sn–O bonds, strongly affects the Sn–I bonds at the methylammonium tin iodide, MASnI3, surface,39,41 which is less significant in the case of MAPbI3.41 The presence of water molecules in THPs may further reduce light absorption and lead to faster charge relaxation.42 Several strategies such as metal doping,43 Lewis-base post-treatment,44 or capping 3D THP with analogue 2D large-cation perovskites45 were proposed to mitigate tin oxidation, which in turn may stabilize THPs.
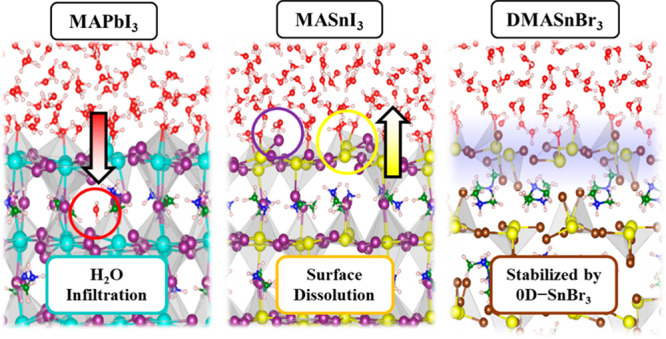
Despite recent efforts, a clear mechanistic picture of the factors affecting THP stability in a water environment is still missing. Here we report comparative AIMD simulations of MASnI3 with the prototypical lead-based MAPbI3 perovskite, Figure 1a,b, thus assessing the impact of the B-site metal on the interaction with water. A comparison with dimethylammonium tin bromide, DMASnBr3, Figure 1c, a water-stable perovskite catalyst,46 is also reported, which allows us to comparatively evaluate the concomitant impact of the A-site and X-site variation on the perovskite/water interface stability. Note that AIMD results on the water-induced degradation of MAPbI3 are partially reproduced from our previous analysis,26 together with additional statistical analysis, to provide a direct comparison with the prototypical MAPbI3/water interface as representative of the lead-based counterpart. Our results reveal a dissolution of the SnI2-terminated MASnI3 surface under water which is initiated by the formation of Sn–O bonds and the subsequent breaking of axial Sn–I bonds. The MAPbI3 surface, despite showing an increased amount of adsorbed water molecules, preserves its inorganic Pb–I framework without a substantial difference in the radial pair distribution between bulk and surface. However, water infiltration is observed, which presents the first step of MAPbI3 degradation. Our results indicate that the high water stability of DMASnBr3 is due to the presence of an amorphous surface layer made of disconnected, zero-dimensional SnBr3 complexes, stabilized by strong Sn–Br bonds, which strongly bind water molecules and protect the DMASnBr3 bulk.
Figure 1.

Structural model of the (a) MAPbI3, (b) MASnI3, and (c) DMASnBr3 perovskite/water interfaces. The colors of the atoms are cyan: Pb, yellow: Sn, purple: I, orange: Br, green: C, white: H, blue: N, red: O. Further details on the structural models are provided in the Computational Details.
We start our analysis by comparing the interaction of the (001) MI2-terminated (M = Pb, Sn) tetragonal MAPbI3 and MASnI3 perovskite surfaces with the surrounding liquid water environment (see Computational Details for model setup and AIMD simulation details). The (001) surface belongs to the most stable perovskite surfaces47 and has been subject to several studies on the electronic properties of lead-26,28,32 and tin-based perovksites.21,22,39,45 The radial pair distribution function gM–O(r) (M = Sn, Pb), averaged over the AIMD trajectories, indicates a strong interaction of the Sn surface atoms with the water oxygen atoms, as shown by the peak located at 2.43 Å; see Figure 2a. The gPb–O(r) of the MAPbI3 surface comparatively shows a substantially broadened peak centered at around 2.53 Å. The interaction at the perovskite/water interface mainly occurs via the O lone pair electrons and the M2+ metal atoms forming M–O bonds (M= Sn, Pb), while the contribution of I–HW bonds between the surface iodine atoms and the water hydrogen atoms is less pronounced, Figure 2a. Fitting the tail of the gI–Hw(r) at low distances with a Gaussian distribution, we may estimate an average I–HW bond distance of 2.73 Å for MAPbI3 and 2.91 Å for MASnI3, indicating a stronger interaction at the MAPbI3/water interface. Both perovskite slabs show a lack of long-range correlation between the surface atoms and the water environment. Note that, within feasible AIMD time scales, the interaction of the perovskite slabs and the water environment mainly occurs at the surface, as visible in the M/O (M = Sn, Pb) g(r) in Figure S1, Supporting Information. Despite the pronounced Sn–O bond structure, the amount of water molecules adsorbed at the perovskite surface via M–O bonds is higher for MAPbI3, on average 0.88 ± 0.09 adsorbed water molecules per surface Pb compared to 0.69 ± 0.05 adsorbed water molecules per surface Sn atom, Figure 2b. Previous DFT studies48 showed a substantial increase in the ionic contribution to the M–O and I–HW bonds at the PbI2-terminated MAPbI3 surface compared to the SnI2-terminated MASnI3 surface. This should lead to an increased attraction of the water oxygen atoms at the MAPbI3 surface and may explain the gI–Hw(r) shift toward lower distances, Figure 2a, and finally in the increased number of adsorbed water molecules at the MAPbI3 surface.
Figure 2.

(a) M–O (M = Sn, Pb) and I–HW radial pair distribution, gM–O(r) and gI–Hw(r), and the integrated distribution, int[gM–O(r)] and int[gI–Hw(r)], for MAPbI3 (red) and MASnI3 (blue). (b) Time evolution of the number of adsorbed H2O molecules per surface metal atom, identified by M–O bonds below 3.0 Å.
We now move on to study the structural properties at the hydrated perovskite surfaces and bulk for MAPbI3 and MASnI3. We observe substantial differences in the surface structure when considering the time average for each atom over the AIMD trajectory, Figure 3a. Surprisingly, despite the substantial interaction of the MAPbI3 surface atoms with the water environment, the time-averaged MAPbI3 surface seems only weakly distorted. In contrast, the MASnI3 surface shows a strong structural distortion in the inorganic Sn–I framework and indicates broken Sn–I bonds in the surface planes and in between the layers. In the time-averaged MAPbI3/water interface, we observe a water molecule which has entered the MAPbI3 bulk being located in the first MAI layer, as reported in our previous study.26
Figure 3.

(a) Time-averaged structure for (left) MAPbI3/water and (right) MASnI3/water interface. Panels (b) and (c) give the Pb/I and Sn/I radial pair distribution, g(r) (solid), and the integrated distribution, int[g(r)] (dashed), for MAPbI3 and MASnI3, respectively. Panels (d) and (e) give the Pb/Pb and Sn/Sn g(r) (solid) and int[g(r)] (dashed) for MAPbI3 and MASnI3, respectively. In (c–e), we distinguish between the surface, defined by the outermost MI2 layers (M = Sn, Pb), and the bulk, defined by the inner layers. The notation, e.g., Sn/I (surf.), represents the g(r) of surface Sn atoms with all (surface and bulk) I atoms.
To quantify these visual observations, we compare the radial pair distributions for the individual components in both MAPbI3 and MASnI3. Note that we distinguish between surface atoms, given by the atoms located within the outer PbI2 and SnI2 layers, and identify the remaining atoms as bulk. The Pb/I bulk g(r), Figure 3b, shows a sharp peak at 3.17 Å, being near the experimental bond Pb–I length of ∼3.19 Å,49 and its integral reaches the expected coordination number of 6 within 3.8 Å. At the surface, the peak is shifted to 3.14 Å and slightly broadened; the Pb/I coordination number of surface Pb atoms reaches the expected value of 5 within 4.1 Å. The bulk and surface Pb/Pb g(r) both reach the maximum at 6.30 Å, while the surface Pb/Pb g(r) is slightly broadened, Figure 3d. Unlike for MAPbI3, we observe substantial differences between the surface and the bulk g(r) for MASnI3. The bulk Sn/I g(r), Figure 3c, is substantially broadened with a maximum at 3.07 Å, and the int[g(r)] reaches the expected Sn/I coordination number of 6 within 4.31 Å; surface Sn/I g(r) substantially differs from the bulk and shows a sharp peak at 2.99 Å, representing the equatorial Sn–I bonds, and a broadened contribution at large distances centered at around 3.48 Å, representing the axial Sn–I bonds between surface Sn and I from the first MAI layer. The Sn/I coordination number slowly increases with distance and reaches the expected value of 5 after 5.11 Å, which clearly points to elongated and potentially even broken bonds as observed in Figure 3a. The interaction of the water environment with the MASnI3 surface further broadens the surface Sn/Sn g(r), Figure 3e, while the bulk Sn/Sn g(r) remains at the expected Sn–Sn distance of 6.21 Å. Interestingly, the I/I g(r) do not show substantial differences between the hydrated surfaces and the bulk for both of the considered perovskites, Figure S2, Supporting Information.
It is also interesting to investigate the structural properties of the MA cations in terms of the metal/MA g(r) and the orientation of the MA within the inorganic cages. The apparent stability of the MAPbI3 surface is also reflected in the Pb/MA g(r), Figure S2c, Supporting Information. We observe a broad distribution for both the bulk and the surface, which, however, follow the same shape. In contrast, the Sn/MA g(r) of the surface Sn atoms varies from the bulk and shows a flattened distribution, which indicates a lack of correlation between the surface Sn atoms and the MA cations. Interestingly, the MA cations in the MAI layers near the SnI2-terminated surface are highly oriented at ±30° (layer 1 and layer 4 in Figure S3b, Supporting Information), while MA orientation in the hydrated MAPbI3 is strongly perturbed in comparison to the bare MAPbI3 slabs.26 Despite the strong structural disorder in the hydrated MASnI3 surface, the I/MA g(r) of surface I atoms follows the bulk g(r) closely, Figure S2f, Supporting Information, which may explain the high orientation of the MA cations in MASnI3.
To establish the main water-induced degradation mechanism of the MASnI3 surface, we now analyze the temporal evolution of the surface bonds in terms of the Sn/I coordination number in comparison to MAPbI3, Figure 4a. The Pb/I coordination number in the MAPbI3 bulk remains close to 6 throughout the trajectory; at the surface, it remains approximately 5 until 5.2 ps and then drops to ∼3.5 for a short time. This is caused by a breaking of equatorial Pb–I bonds at the hydrated surface, while the axial Pb–I bonds between the surface and the first MAI layer remain stable, Figure S4, Supporting Information. The equatorial Pb–I bonds, however, are restored after a short time period such that the coordination number approaches a value of 5 again. As shown in our previous study, the fundamental degradation mechanism of the MAPbI3 surface is initiated by the infiltration of the water molecule,26Figure 4b. Recent classical molecular dynamics simulations underlined that water infiltration into the perovskite bulk is a crucial step to initiate the degradation of PbI2-terminated surfaces.32 Within the AIMD trajectory, we did not observe the infiltration of water molecules for the MASnI3 surface. The Sn/I coordination number of the surface Sn decreases slowly in time from ∼4.5 after the thermalization period (first ∼2 ps) to ∼3.5 at 8 ps, Figure 4a. The origin of this change in coordination number is the breaking of axial bonds between the surface Sn atoms and the I atoms in the first MAI layer. The average Sn–I interlayer distance increases from ∼3.5 Å at 3.7 ps to a maximum of ∼4.0 Å at 6.8 ps, Figure 4c, while individual Sn–I distances above 5.4 Å can be observed, Figure 4d. In contrast, the Pb–I interlayer distance remains stable at around 3.2 Å as the main effect of the water is on the equatorial Pb–I bonds, without strongly changing the axial Pb–I bonds.
Figure 4.

(a) Time evolution of the (left) Pb/I and (right) Sn/I coordination number in the bulk and at the surface. (b) Key steps of the infiltration of the water molecule into the MAPbI3 slab. (c) Time evolution of the average interlayer distance between the surface metal atoms and the iodine atoms in the first MAI layer. (d) Structure visualizing the dissolution of the MASnI3 surface; interlayer distances are given for several Sn–I pairs, in units of Angstrom.
The presented observations give clear indication of a facile solvation of the SnI2-terminated surface by water. As a consequence, we may expect an enhanced surface defect density in humid conditions which can introduce deep trap states and strongly suppresses the device performance of THP solar cells.21 On a longer time scale, water may easily enter the MASnI3 bulk through the dissolved surface to accelerate the degradation32 and hamper the electronic properties.42 This emphasizes the need for perovskite synthesis under a controlled moisture-free environment and additionally may require passivation of MASnI3 using hydrophobic protection layers. Note that we previously observed the rapid solvation of the MAI-terminated MAPbI3 surface from AIMD simulations, which occurs by the release of I atoms under the attack of water molecules at Pb sites.26 Further DFT studies reported comparable water adsorption energies on MAI-terminated MAPbI3 and MASnI3 surfaces.48 We thus are confident that the degradation process of the MAI-terminated MASnI3 will follow a similar mechanism as in MAPbI3.
We now analyze the results on the (001) SnBr2-terminated orthorhombic DMASnBr3/water interface to gain a deeper understanding of the origin of the high water stability of DMASnBr3.46,50 The gSn–O(r) of the DMASnBr3 surface shows a maximum at 2.37 Å, close to the Sn–O peak of MASnI3 (2.43 Å), while the gBr–Hw(r) is substantially shifted with respect to MASnI3 to shorter distances, Figure 5a. Additionally, the intensity of the DMASnBr3 gSn–O(r) is less pronounced, which we may attribute to Sn–Br bonds being stronger than Sn–I ones. The overall amount of adsorbed water molecules per surface Sn atom does not substantially vary between the considered THPs, Figure S5, Supporting Information. Furthermore, the DMASnBr3 int[g(r)] follows closely the one of MASnI3, suggesting a comparable interaction of the perovskite surface and water environment. In contrast to the substantial distortion of the Sn–I bonds at the MASnI3 surface, however, the Sn/Br g(r) indicates highly stable metal-halide bonds at the DMASnBr3 surface, Figure 5b. Interestingly, the short-range correlation, given by the first peak in the Sn/Br g(r), is nicely preserved, but the second peak is flattened, which points to a rather disordered surface structure. Also, the Br/Br g(r) is preserved at the surfaces, Figure S6, Supporting Information. The Sn/Br coordination number does not strongly change with time, with an average of 3.61 and 2.99 of the bulk and of the surface Sn atoms, respectively, Figure S7, Supporting Information. In contrast to MASnI3, the interlayer distance between the surface Sn atoms and the Br atoms in the first DMABr layer remains relatively stable but shows larger fluctuations, Figure S8, Supporting Information. The most significant difference to MASnI3 is the highly disordered Sn/Sn g(r) at the DMASnBr3 surface, Figure 5c. While the bulk Sn/Sn distribution shows a well-pronounced peak at 5.97 Å, the surface Sn/Sn g(r) lacks both short- and long-range order. Such a distribution is atypical for a crystalline solid and points to a rather amorphous phase.
Figure 5.

(a) Sn–O and X–HW (X = Br, I) radial pair distribution and the integrated distribution for MASnI3 (blue) and DMASnBr3 (green). (b) Sn/Br and (c) Sn/Sn g(r) of DMASnBr3 in the bulk (blue) and at the surface (red).
Further inspection reveals, indeed, that the DMASnBr3 surface consists of disconnected, zero-dimensional SnBr3complexes which form a surface layer possibly protecting the inner bulk structure. Surface Sn atoms form three stable Sn–Br bonds with well-preserved Sn/Br and Br/Br g(r) and lack of Sn/Sn correlations, which are strong signatures of such 0D-SnBr3 complexes, Figure S9, Supporting Information. A comparison of the crystal structures of DMASnBr3 and MASnBr3 (Figure S10, Supporting Information) shows equal Sn–Br bond lengths along the (001) direction for MASnBr3 and alternating short (∼2.74 Å) and long Sn–Br bonds (∼3.45 Å) for DMASnBr3. Consequently, the presence of SnBr3 complexes within DMASnBr3 can be attributed to the large DMA cation. Recent AIMD simulations also observed the presence of partially decoupled SnBr3 complexes within formamidinium tin bromide, FASnBr3,51 which form instantaneously caused by the large size of the FA cation. Water molecules form strong Sn–O bonds with such surface complexes, which is supported by DFT calculations of the water adsorption energy, with Eads(DMASnBr3) = −0.72 eV to Eads(MASnI3) = −0.55 eV, Figure S11, Supporting Information. Relaxing the DMASnBr3 slab at t = 8 ps after removal of the water molecules does not lead to a more stable structure, Figure S12, Supporting Information, which indicates that the amorphous surface layer is formed upon interaction with water molecules at finite temperatures. Within the simulated time scales, water molecules neither enter into the perovskite nor are able to dissolve the surface. The 0D-SnBr3 complexes can easily rearrange at the surface to bond with water molecules and consequently to prohibit water from entering the surface. Moreover, the water environment cannot easily break the strong Sn–Br bonds, which limits the solubility of the DMASnBr3 surface by water. As a final remark, DMASnBr3 is further stabilized by the relatively strong hydrophobic nature of the DMA cation with respect to MA.46 As a consequence, higher stability of DMABr-terminated surfaces compared to MASnI3 may be expected due a reduced solvation of DMA cations, and water infiltration into the SnBr2-terminated DMASnBr3 surface may be further suppressed. The high band gap (∼2.8 eV50) in combination with the amorphous surface layer may prevent the applicability of DMASnBr3 in photovoltaics. On the contrary, this inherent surface stability can be the crucial aspect for exploitation of DMASnBr3 as water-stable THP in photocatalytic applications.
In summary, we have reported ab initio molecular dynamics simulations to investigate the surface degradation of tin-halide perovskites using two prototypical systems, i.e., MASnI3 and DMASnBr3, and provided a comparison with the prototypical lead-based counterpart MAPbI3. Our results show a facile dissolution of the SnI2-terminated MASnI3 surface under water. Water molecules bond with the surface Sn atoms and induce the breaking of axial Sn–I bonds with the I atoms of the adjacent MAI layer. Our AIMD results predict a higher water stability of the PbI2-terminated MAPbI3 surface. Despite showing an increased number of adsorbed water molecules on the MAPbI3 surface, the lead iodine bonds remain stable throughout the trajectory without showing any substantial difference in the radial pair distribution between the bulk and the surface. Still, water molecules may enter the MAPbI3 bulk where they may initiate the solvation of the perovskite.26,32 Finally, we reported AIMD simulations to analyze the origin of the experimentally reported46,50 surface stability of DMASnBr3 in a liquid water environment. We observed highly stable Sn–Br bonds at the surface and an increased stability of the inorganic framework along the c-axis. Our results show that the DMASnBr3 surface is made of disconnected, zero-dimensional SnBr3 complexes which form an amorphous layer that can easily reorient within the surface and hereby protect the DMASnBr3 bulk. Such amorphous surface structures typically are believed to be detrimental in terms of photovoltaics but may be key for the applicability of THPs in photocatalysis. Overall, we believe that the reported atomistic details of the THP/water interface in this work will inspire novel surface modification and device engineering strategies to enhance the stability of THPs for lead-free perovskite photovoltaics and photocatalysis.
Computational Details. The models of the THP–water interfaces are constructed by five SnX2 (X = I, Br) layers such that the perovskite slabs are terminated by SnX2 layers, Figure 1. The cell parameters in the a and b axes are fixed to the experimental values: MASnI3a = b = 17.5154 Å,52 DMASnBr3a = 12.274 Å, b = 12.071 Å,46 representing 2 × 2 (001) slabs of the tetragonal MASnI3 and of the orthorhombic DMASnBr3 phases. A vacuum region of 15 Å was added on top of the perovskite slabs, such that MASnI3c = 47.709 Å and DMASnBr3c = 47.5 Å. The vacuum region was filled by 155 and 72 water molecules for the MASnI3 and the DMASnBr3 model, respectively, thus giving rise to a liquid water environment characterized by the experimental density of liquid water. We reproduce part of our previous AIMD analysis on the water-induced degradation of MAPbI3,26 together with additional statistical analysis, to provide a direct comparison with the prototypical MAPbI3/water interface as representative of the lead-based counterpart.
We carried out AIMD simulations on the THP–water interface models using the Quickstep module in the CP2K software package.53−56 We used a double-ζ basis set (DZVP-MOLOPT)57 combined with the norm-conserving Goedecker–Teter–Hutter (GTH) pseudopotentials58 and a CUTOFF = 800 Ry, and REL_CUTOFF = 50 Ry for the expansion of the electron density. The revised PBE (revPBE) parametrization of the PBE functional was used to account for the exchange and correlation potentials. van der Waals corrections are included by means of the rVV10 functional to account for nonlocal electron correlations.59 The temperature was controlled by a Nosé–Hoover thermostat60,61 with a target temperature of 350 K and a time constant of 16.68 fs, and the volume was kept constant by applying an NVT ensemble. The integration time step of the dynamics was set to 0.48 fs. The AIMD simulations were run for at least 8 ps for both the MASnI3/water and the DMASnBr3/water interface model.
Acknowledgments
The Ministero dell’Istruzione dell’Università e della Ricerca (MIUR) and Università degli Studi di Perugia are acknowledged for financial support through the program “Dipartimenti di Eccellenza 2018–2022” (Grant AMIS) to F.D.A. This research was funded by PON Project “Tecnologia per celle solari bifacciali ad alta Efficienza a 4 terminali per utility scale” (BEST-4U) of the Italian Ministry MIUR (CUP B88D19000160005) and by project Ricerca@CNR PHOTOCAT (CUP B93C21000060006). F.A. acknowledges support from by the European Union’s Horizon 2020 research and innovation program under Grant Agreement No 771528 of the SOPHY project. E.M. and A.A.A. wish to thank the Distinguished Scientist Fellowship Program (DSFP) of King Saud University, Riyadh, Saudi Arabia.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpclett.2c00273.
Radial pair distribution functions for MAPbI3 and MASnI3; methylammonium orientation in the individual MAI layers; snapshot visualizing the equatorial Pb–I bond breaking at the hydrated MAPbI3 surface; time evolution of the adsorbed water molecules at the MASnI3 and the DMASnBr3 surface; Sn/DMA and Br/Br radial pair distribution functions for DMASnBr3; AIMD snapshot of the DMASnBr3–water interface visualizing SnBr3 complexes; crystal structures of MASnI3, MASnBr3, and DMASnBr3; DFT calculations of the adsorption energy of a single water molecule on the perovskite surfaces; optimized DMASnBr3 slabs before and after the interaction with water (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Snaith H. J. Perovskites: The Emergence of a New Era for Low-Cost, High-Efficiency Solar Cells. J. Phys. Chem. Lett. 2013, 4 (21), 3623–3630. 10.1021/jz4020162. [DOI] [Google Scholar]
- Bian Z.; Wang Z.; Jiang B.; Hongmanorom P.; Zhong W.; Kawi S. A Review on Perovskite Catalysts for Reforming of Methane to Hydrogen Production. Renewable Sustainable Energy Rev. 2020, 134, 110291. 10.1016/j.rser.2020.110291. [DOI] [Google Scholar]
- Umari P.; Mosconi E.; De Angelis F. Relativistic GW Calculations on CH3NH3PbI3 and CH3NH3SnI3 Perovskites for Solar Cell Applications. Sci. Rep. 2015, 4, 4467. 10.1038/srep04467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranks S. D.; Eperon G. E.; Grancini G.; Menelaou C.; Alcocer M. J. P.; Leijtens T.; Herz L. M.; Petrozza A.; Snaith H. J. Electron-Hole Diffusion Lengths Exceeding 1 Micrometer in an Organometal Trihalide Perovskite Absorber. Science 2013, 342 (6156), 341–344. 10.1126/science.1243982. [DOI] [PubMed] [Google Scholar]
- Johnston M. B.; Herz L. M. Hybrid Perovskites for Photovoltaics: Charge-Carrier Recombination, Diffusion, and Radiative Efficiencies. Acc. Chem. Res. 2016, 49 (1), 146–154. 10.1021/acs.accounts.5b00411. [DOI] [PubMed] [Google Scholar]
- Jung M.; Ji S.-G.; Kim G.; Seok S. I. Perovskite Precursor Solution Chemistry: From Fundamentals to Photovoltaic Applications. Chem. Soc. Rev. 2019, 48 (7), 2011–2038. 10.1039/C8CS00656C. [DOI] [PubMed] [Google Scholar]
- Jeong J.; Kim M.; Seo J.; Lu H.; Ahlawat P.; Mishra A.; Yang Y.; Hope M. A.; Eickemeyer F. T.; Kim M.; Yoon Y. J.; Choi I. W.; Darwich B. P.; Choi S. J.; Jo Y.; Lee J. H.; Walker B.; Zakeeruddin S. M.; Emsley L.; Rothlisberger U.; Hagfeldt A.; Kim D. S.; Grätzel M.; Kim J. Y. Pseudo-Halide Anion Engineering for α-FAPbI3 Perovskite Solar Cells. Nature 2021, 592 (7854), 381–385. 10.1038/s41586-021-03406-5. [DOI] [PubMed] [Google Scholar]
- Li J.; Cao H.-L.; Jiao W.-B.; Wang Q.; Wei M.; Cantone I.; Lü J.; Abate A. Biological Impact of Lead from Halide Perovskites Reveals the Risk of Introducing a Safe Threshold. Nat. Commun. 2020, 11 (1), 310. 10.1038/s41467-019-13910-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schileo G.; Grancini G. Lead or No Lead? Availability, Toxicity, Sustainability and Environmental Impact of Lead-Free Perovskite Solar Cells. J. Mater. Chem. C 2021, 9 (1), 67–76. 10.1039/D0TC04552G. [DOI] [Google Scholar]
- Di Girolamo D.; Pascual J.; Aldamasy M. H.; Iqbal Z.; Li G.; Radicchi E.; Li M.; Turren-Cruz S.-H.; Nasti G.; Dallmann A.; De Angelis F.; Abate A. Solvents for Processing Stable Tin Halide Perovskites. ACS Energy Lett. 2021, 6, 959–968. 10.1021/acsenergylett.0c02656. [DOI] [Google Scholar]
- Hao F.; Stoumpos C. C.; Cao D. H.; Chang R. P. H.; Kanatzidis M. G. Lead-Free Solid-State Organic–Inorganic Halide Perovskite Solar Cells. Nat. Photonics 2014, 8 (6), 489–494. 10.1038/nphoton.2014.82. [DOI] [Google Scholar]
- Nasti G.; Abate A. Tin Halide Perovskite (ASnX3) Solar Cells: A Comprehensive Guide toward the Highest Power Conversion Efficiency. Adv. Energy Mater. 2020, 10 (13), 1902467. 10.1002/aenm.201902467. [DOI] [Google Scholar]
- Jiang X.; Li H.; Zhou Q.; Wei Q.; Wei M.; Jiang L.; Wang Z.; Peng Z.; Wang F.; Zang Z.; Xu K.; Hou Y.; Teale S.; Zhou W.; Si R.; Gao X.; Sargent E. H.; Ning Z. One-Step Synthesis of SnI2·(DMSO)x Adducts for High-Performance Tin Perovskite Solar Cells. J. Am. Chem. Soc. 2021, 143 (29), 10970–10976. 10.1021/jacs.1c03032. [DOI] [PubMed] [Google Scholar]
- Lin R.; Xiao K.; Qin Z.; Han Q.; Zhang C.; Wei M.; Saidaminov M. I.; Gao Y.; Xu J.; Xiao M.; Li A.; Zhu J.; Sargent E. H.; Tan H. Monolithic All-Perovskite Tandem Solar Cells with 24.8% Efficiency Exploiting Comproportionation to Suppress Sn(II) Oxidation in Precursor Ink. Nat. Energy 2019, 4 (10), 864–873. 10.1038/s41560-019-0466-3. [DOI] [Google Scholar]
- Yang Z.; Yu Z.; Wei H.; Xiao X.; Ni Z.; Chen B.; Deng Y.; Habisreutinger S. N.; Chen X.; Wang K.; Zhao J.; Rudd P. N.; Berry J. J.; Beard M. C.; Huang J. Enhancing Electron Diffusion Length in Narrow-Bandgap Perovskites for Efficient Monolithic Perovskite Tandem Solar Cells. Nat. Commun. 2019, 10 (1), 4498. 10.1038/s41467-019-12513-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C.; Song Z.; Chen C.; Xiao C.; Subedi B.; Harvey S. P.; Shrestha N.; Subedi K. K.; Chen L.; Liu D.; Li Y.; Kim Y.-W.; Jiang C.; Heben M. J.; Zhao D.; Ellingson R. J.; Podraza N. J.; Al-Jassim M.; Yan Y. Low-Bandgap Mixed Tin–Lead Iodide Perovskites with Reduced Methylammonium for Simultaneous Enhancement of Solar Cell Efficiency and Stability. Nat. Energy 2020, 5 (10), 768–776. 10.1038/s41560-020-00692-7. [DOI] [Google Scholar]
- Kapil G.; Bessho T.; Maekawa T.; Baranwal A. K.; Zhang Y.; Kamarudin M. A.; Hirotani D.; Shen Q.; Segawa H.; Hayase S. Tin-Lead Perovskite Fabricated via Ethylenediamine Interlayer Guides to the Solar Cell Efficiency of 21.74. Adv. Energy Mater. 2021, 11 (25), 2101069. 10.1002/aenm.202101069. [DOI] [Google Scholar]
- Noel N. K.; Stranks S. D.; Abate A.; Wehrenfennig C.; Guarnera S.; Haghighirad A.-A.; Sadhanala A.; Eperon G. E.; Pathak S. K.; Johnston M. B.; Petrozza A.; Herz L. M.; Snaith H. J. Lead-Free Organic–Inorganic Tin Halide Perovskites for Photovoltaic Applications. Energy Environ. Sci. 2014, 7 (9), 3061–3068. 10.1039/C4EE01076K. [DOI] [Google Scholar]
- Pascual J.; Nasti G.; Aldamasy M. H.; Smith J. A.; Flatken M.; Phung N.; Di Girolamo D.; Turren-Cruz S.-H.; Li M.; Dallmann A.; Avolio R.; Abate A. Origin of Sn(II) Oxidation in Tin Halide Perovskites. Mater. Adv. 2020, 1 (5), 1066–1070. 10.1039/D0MA00245C. [DOI] [Google Scholar]
- Meggiolaro D.; Ricciarelli D.; Alasmari A. A.; Alasmary F. A. S.; De Angelis F. Tin versus Lead Redox Chemistry Modulates Charge Trapping and Self-Doping in Tin/Lead Iodide Perovskites. J. Phys. Chem. Lett. 2020, 11 (9), 3546–3556. 10.1021/acs.jpclett.0c00725. [DOI] [PubMed] [Google Scholar]
- Ricciarelli D.; Meggiolaro D.; Ambrosio F.; De Angelis F. Instability of Tin Iodide Perovskites: Bulk p-Doping versus Surface Tin Oxidation. ACS Energy Lett. 2020, 5 (9), 2787–2795. 10.1021/acsenergylett.0c01174. [DOI] [Google Scholar]
- Ambrosio F.; Meggiolaro D.; Almutairi T. M.; De Angelis F. Composition-Dependent Struggle between Iodine and Tin Chemistry at the Surface of Mixed Tin/Lead Perovskites. ACS Energy Lett. 2021, 6 (3), 969–976. 10.1021/acsenergylett.1c00111. [DOI] [Google Scholar]
- Akbulatov A. F.; Tsarev S. A.; Elshobaki M.; Luchkin S. Y.; Zhidkov I. S.; Kurmaev E. Z.; Aldoshin S. M.; Stevenson K. J.; Troshin P. A. Comparative Intrinsic Thermal and Photochemical Stability of Sn(II) Complex Halides as Next-Generation Materials for Lead-Free Perovskite Solar Cells. J. Phys. Chem. C 2019, 123 (44), 26862–26869. 10.1021/acs.jpcc.9b09200. [DOI] [Google Scholar]
- Leijtens T.; Prasanna R.; Gold-Parker A.; Toney M. F.; McGehee M. D. Mechanism of Tin Oxidation and Stabilization by Lead Substitution in Tin Halide Perovskites. ACS Energy Lett. 2017, 2 (9), 2159–2165. 10.1021/acsenergylett.7b00636. [DOI] [Google Scholar]
- Mundt L. E.; Tong J.; Palmstrom A. F.; Dunfield S. P.; Zhu K.; Berry J. J.; Schelhas L. T.; Ratcliff E. L. Surface-Activated Corrosion in Tin–Lead Halide Perovskite Solar Cells. ACS Energy Lett. 2020, 5 (11), 3344–3351. 10.1021/acsenergylett.0c01445. [DOI] [Google Scholar]
- Mosconi E.; Azpiroz J. M.; De Angelis F. Ab Initio Molecular Dynamics Simulations of Methylammonium Lead Iodide Perovskite Degradation by Water. Chem. Mater. 2015, 27 (13), 4885–4892. 10.1021/acs.chemmater.5b01991. [DOI] [Google Scholar]
- Zheng C.; Rubel O. Unraveling the Water Degradation Mechanism of CH3NH3PbI3. J. Phys. Chem. C 2019, 123 (32), 19385–19394. 10.1021/acs.jpcc.9b05516. [DOI] [Google Scholar]
- Koocher N. Z.; Saldana-Greco D.; Wang F.; Liu S.; Rappe A. M. Polarization Dependence of Water Adsorption to CH3NH3PbI3 (001) Surfaces. J. Phys. Chem. Lett. 2015, 6 (21), 4371–4378. 10.1021/acs.jpclett.5b01797. [DOI] [PubMed] [Google Scholar]
- Müller C.; Glaser T.; Plogmeyer M.; Sendner M.; Döring S.; Bakulin A. A.; Brzuska C.; Scheer R.; Pshenichnikov M. S.; Kowalsky W.; Pucci A.; Lovrinčić R. Water Infiltration in Methylammonium Lead Iodide Perovskite: Fast and Inconspicuous. Chem. Mater. 2015, 27 (22), 7835–7841. 10.1021/acs.chemmater.5b03883. [DOI] [Google Scholar]
- Christians J. A.; Miranda Herrera P. A.; Kamat P. V. Transformation of the Excited State and Photovoltaic Efficiency of CH3NH3PbI3 Perovskite Upon Controlled Exposure to Humidified Air. J. Am. Chem. Soc. 2015, 137 (4), 1530–1538. 10.1021/ja511132a. [DOI] [PubMed] [Google Scholar]
- Kakekhani A.; Katti R. N.; Rappe A. M. Water in Hybrid Perovskites: Bulk MAPbI3 Degradation via Super-Hydrous State. APL Mater. 2019, 7 (4), 041112. 10.1063/1.5087290. [DOI] [Google Scholar]
- Caddeo C.; Saba M. I.; Meloni S.; Filippetti A.; Mattoni A. Collective Molecular Mechanisms in the CH3NH3PbI3 Dissolution by Liquid Water. ACS Nano 2017, 11 (9), 9183–9190. 10.1021/acsnano.7b04116. [DOI] [PubMed] [Google Scholar]
- Kye Y.-H.; Yu C.-J.; Jong U.-G.; Chen Y.; Walsh A. Critical Role of Water in Defect Aggregation and Chemical Degradation of Perovskite Solar Cells. J. Phys. Chem. Lett. 2018, 9 (9), 2196–2201. 10.1021/acs.jpclett.8b00406. [DOI] [PubMed] [Google Scholar]
- Long R.; Fang W.; Prezhdo O. V. Moderate Humidity Delays Electron-Hole Recombination in Hybrid Organic-Inorganic Perovskites: Time-Domain Ab Initio Simulations Rationalize Experiments. J. Phys. Chem. Lett. 2016, 7 (16), 3215–3222. 10.1021/acs.jpclett.6b01412. [DOI] [PubMed] [Google Scholar]
- Aristidou N.; Eames C.; Islam M. S.; Haque S. A. Insights into the Increased Degradation Rate of CH3NH3PbI3 Solar Cells in Combined Water and O2 Environments. J. Mater. Chem. A 2017, 5 (48), 25469–25475. 10.1039/C7TA06841G. [DOI] [Google Scholar]
- Peng C.; Chen J.; Wang H.; Hu P. First-Principles Insight into the Degradation Mechanism of CH3NH3PbI3 Perovskite: Light-Induced Defect Formation and Water Dissociation. J. Phys. Chem. C 2018, 122 (48), 27340–27349. 10.1021/acs.jpcc.8b07294. [DOI] [Google Scholar]
- Zhang L.; Sit P. H.-L. Ab Initio Study of Interaction of Water, Hydroxyl Radicals, and Hydroxide Ions with CH3NH3PbI3 and CH3NH3PbBr3 Surfaces. J. Phys. Chem. C 2015, 119 (39), 22370–22378. 10.1021/acs.jpcc.5b07000. [DOI] [Google Scholar]
- Aziz A.; Aristidou N.; Bu X.; Westbrook R. J. E.; Haque S. A.; Islam M. S. Understanding the Enhanced Stability of Bromide Substitution in Lead Iodide Perovskites. Chem. Mater. 2020, 32 (1), 400–409. 10.1021/acs.chemmater.9b04000. [DOI] [Google Scholar]
- Lanzetta L.; Webb T.; Zibouche N.; Liang X.; Ding D.; Min G.; Westbrook R. J. E.; Gaggio B.; Macdonald T. J.; Islam M. S.; Haque S. A. Degradation Mechanism of Hybrid Tin-Based Perovskite Solar Cells and the Critical Role of Tin (IV) Iodide. Nat. Commun. 2021, 12 (1), 2853. 10.1038/s41467-021-22864-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanzetta L.; Aristidou N.; Haque S. A. Stability of Lead and Tin Halide Perovskites: The Link between Defects and Degradation. J. Phys. Chem. Lett. 2020, 11 (2), 574–585. 10.1021/acs.jpclett.9b02191. [DOI] [PubMed] [Google Scholar]
- Xie G.; Xu L.; Sun L.; Xiong Y.; Wu P.; Hu B. Insight into the Reaction Mechanism of Water, Oxygen and Nitrogen Molecules on a Tin Iodine Perovskite Surface. J. Mater. Chem. A 2019, 7 (10), 5779–5793. 10.1039/C8TA11705E. [DOI] [Google Scholar]
- Kachmar A.; Berdiyorov G.; Madjet M. E.-A. Effect of Water on the Structural, Optical, and Hot-Carrier Cooling Properties of the Perovskite Material MASnI3. J. Phys. Chem. C 2019, 123 (7), 4056–4063. 10.1021/acs.jpcc.8b11651. [DOI] [Google Scholar]
- Park C.; Choi J.; Min J.; Cho K. Suppression of Oxidative Degradation of Tin–Lead Hybrid Organometal Halide Perovskite Solar Cells by Ag Doping. ACS Energy Lett. 2020, 5 (10), 3285–3294. 10.1021/acsenergylett.0c01648. [DOI] [Google Scholar]
- Kamarudin M. A.; Hirotani D.; Wang Z.; Hamada K.; Nishimura K.; Shen Q.; Toyoda T.; Iikubo S.; Minemoto T.; Yoshino K.; Hayase S. Suppression of Charge Carrier Recombination in Lead-Free Tin Halide Perovskite via Lewis Base Post-treatment. J. Phys. Chem. Lett. 2019, 10 (17), 5277–5283. 10.1021/acs.jpclett.9b02024. [DOI] [PubMed] [Google Scholar]
- Mahata A.; Meggiolaro D.; Gregori L.; De Angelis F. Suppression of Tin Oxidation by 3D/2D Perovskite Interfacing. J. Phys. Chem. C 2021, 125 (20), 10901–10908. 10.1021/acs.jpcc.1c02686. [DOI] [Google Scholar]
- Romani L.; Speltini A.; Ambrosio F.; Mosconi E.; Profumo A.; Marelli M.; Margadonna S.; Milella A.; Fracassi F.; Listorti A.; De Angelis F.; Malavasi L. Water-Stable DMASnBr3 Lead-Free Perovskite for Effective Solar-Driven Photocatalysis. Angew. Chem., Int. Ed. 2021, 60 (7), 3611–3618. 10.1002/anie.202007584. [DOI] [PubMed] [Google Scholar]
- Haruyama J.; Sodeyama K.; Han L.; Tateyama Y. Termination Dependence of Tetragonal CH3NH3PbI3 Surfaces for Perovskite Solar Cells. J. Phys. Chem. Lett. 2014, 5 (16), 2903–2909. 10.1021/jz501510v. [DOI] [PubMed] [Google Scholar]
- Li Q.; Chen Z.; Tranca I.; Gaastra-Nedea S.; Smeulders D.; Tao S. Compositional Effect on Water Adsorption on Metal Halide Perovskites. Appl. Surf. Sci. 2021, 538, 148058. 10.1016/j.apsusc.2020.148058. [DOI] [Google Scholar]
- Weller M. T.; Weber O. J.; Henry P. F.; Di Pumpo A. M.; Hansen T. C. Complete Structure and Cation Orientation in the Perovskite Photovoltaic Methylammonium Lead Iodide Between 100 and 352 K. Chem. Commun. 2015, 51 (20), 4180–4183. 10.1039/C4CC09944C. [DOI] [PubMed] [Google Scholar]
- Pisanu A.; Speltini A.; Quadrelli P.; Drera G.; Sangaletti L.; Malavasi L. Enhanced Air-Stability of Sn-Based Hybrid Perovskites Induced by Dimethylammonium (DMA): Synthesis, Characterization, Aging and Hydrogen Photogeneration of the MA1-xDMAxSnBr3 System. J. Mater. Chem. C 2019, 7 (23), 7020–7026. 10.1039/C9TC01743G. [DOI] [Google Scholar]
- Pisanu A.; Mahata A.; Mosconi E.; Patrini M.; Quadrelli P.; Milanese C.; De Angelis F.; Malavasi L. Exploring the Limits of Three-Dimensional Perovskites: The Case of FAPb1-xSnxBr3. ACS Energy Lett. 2018, 3 (6), 1353–1359. 10.1021/acsenergylett.8b00615. [DOI] [Google Scholar]
- Stoumpos C. C.; Malliakas C. D.; Kanatzidis M. G. Semiconducting Tin and Lead Iodide Perovskites with Organic Cations: Phase Transitions, High Mobilities, and Near-Infrared Photoluminescent Properties. Inorg. Chem. 2013, 52 (15), 9019–9038. 10.1021/ic401215x. [DOI] [PubMed] [Google Scholar]
- Hutter J.; Iannuzzi M.; Schiffmann F.; VandeVondele J. CP2K: Atomistic Simulations of Condensed Matter Systems. WIREs Comput. Mol. Sci. 2014, 4 (1), 15–25. 10.1002/wcms.1159. [DOI] [Google Scholar]
- Kühne T. D.; Iannuzzi M.; Del Ben M.; Rybkin V. V.; Seewald P.; Stein F.; Laino T.; Khaliullin R. Z.; Schütt O.; Schiffmann F.; Golze D.; Wilhelm J.; Chulkov S.; Bani-Hashemian M. H.; Weber V.; Borstnik U.; Taillefumier M.; Jakobovits A. S.; Lazzaro A.; Pabst H.; Müller T.; Schade R.; Guidon M.; Andermatt S.; Holmberg N.; Schenter G. K.; Hehn A.; Bussy A.; Belleflamme F.; Tabacchi G.; Glöß A.; Lass M.; Bethune I.; Mundy C. J.; Plessl C.; Watkins M.; VandeVondele J.; Krack M.; Hutter J. CP2K: An Electronic Structure and Molecular Dynamics Software Package - Quickstep: Efficient and Accurate Electronic Structure Calculations. J. Chem. Phys. 2020, 152 (19), 194103. 10.1063/5.0007045. [DOI] [PubMed] [Google Scholar]
- VandeVondele J.; Krack M.; Mohamed F.; Parrinello M.; Chassaing T.; Hutter J. Quickstep: Fast and Accurate Density Functional Calculations Using a Mixed Gaussian and Plane Waves Approach. Comput. Phys. Commun. 2005, 167 (2), 103–128. 10.1016/j.cpc.2004.12.014. [DOI] [Google Scholar]
- Kaiser W.; Carignano M.; Alothman A. A.; Mosconi E.; Kachmar A.; Goddard W. A.; De Angelis F. First-Principles Molecular Dynamics in Metal-Halide Perovskites: Contrasting Generalized Gradient Approximation and Hybrid Functionals. J. Phys. Chem. Lett. 2021, 12 (49), 11886–11893. 10.1021/acs.jpclett.1c03428. [DOI] [PubMed] [Google Scholar]
- VandeVondele J.; Hutter J. Gaussian Basis Sets for Accurate Calculations on Molecular Systems in Gas and Condensed Phases. J. Chem. Phys. 2007, 127 (11), 114105. 10.1063/1.2770708. [DOI] [PubMed] [Google Scholar]
- Goedecker S.; Teter M.; Hutter J. Separable Dual-Space Gaussian Pseudopotentials. Phys. Rev. B 1996, 54 (3), 1703–1710. 10.1103/PhysRevB.54.1703. [DOI] [PubMed] [Google Scholar]
- Vydrov O. A.; van Voorhis T. Nonlocal van der Waals Density Functional: The Simpler the Better. J. Chem. Phys. 2010, 133 (24), 244103. 10.1063/1.3521275. [DOI] [PubMed] [Google Scholar]
- Nosé S. A Unified Formulation of the Constant Temperature Molecular Dynamics Methods. J. Chem. Phys. 1984, 81 (1), 511–519. 10.1063/1.447334. [DOI] [Google Scholar]
- Hoover W. G. Constant-Pressure Equations of Motion. Phys. Rev. A 1986, 34 (3), 2499–2500. 10.1103/PhysRevA.34.2499. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.