

ABSTRACT
Introduction
Coronavirus disease 2019 (COVID-19), which emerged as a major public health threat, has affected >400 million people globally leading to >5 million mortalities to date. Treatments of COVID-19 are still to be developed as the available therapeutic approaches are not able to combat the virus causing the disease (severe acute respiratory syndrome coronavirus-2; SARS-CoV-2) satisfactorily. However, antiviral peptides (AVPs) have demonstrated prophylactic and therapeutic effects against many coronaviruses (CoVs).
Areas covered
This review critically discusses various types of AVPs evaluated for the treatment of COVID-19 along with their mechanisms of action. Furthermore, the peptides inhibiting the entry of the virus by targeting its binding to angiotensin-converting enzyme 2 (ACE2) or integrins, fusion mechanism as well as activation of proteolytic enzymes (cathepsin L, transmembrane serine protease 2 (TMPRSS2), or furin) are also discussed.
Expert opinion
Although extensively investigated, successful treatment of COVID-19 is still a challenge due to emergence of virus mutants. Antiviral peptides are anticipated to be blockbuster drugs for the management of this serious infection because of their formulation and therapeutic advantages. Although they may act on different pathways, AVPs having a multi-targeted approach are considered to have the upper hand in the management of this infection.
KEYWORDS: SARS-CoV-2, COVID-19, peptides, coronavirus, antivirals
1. Introduction
Coronaviruses (CoVs) form the largest cluster of viruses that cause respiratory and gastrointestinal infections [1,2]. The seven CoVs that infect humans are HCoV-HKU1, HCoV-OC43, HCoV-NL63, HCoV-229E, MERS-CoV (Middle east respiratory syndrome-corona virus), SARS-CoV (severe acute respiratory syndrome-corona virus), and SARS-CoV-2 (Figure 1) [3].
Figure 1.
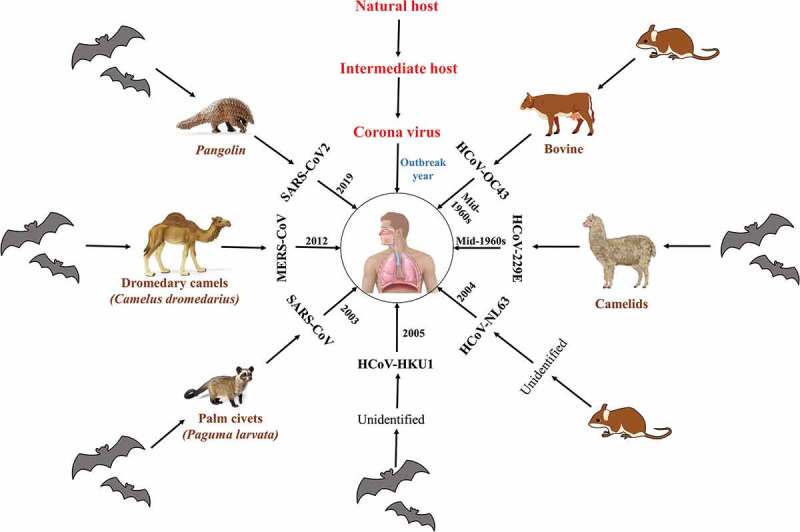
Classification of coronavirus diseases based on natural and intermediate hosts [4,5,18].
In December 2019, a mysterious pneumonia, characterized by fever, cough, chest discomfort, dyspnea, and bilateral lung infiltration was detected in China [6]. The causative agent of the disease was identified as the novel CoV, named later as ‘SARS-CoV-2,’ while the disease was given the name ‘COVID-19’ [7–9]. It was declared as a pandemic by WHO in March, 2020 [7].
Numerous clinical trials have either been conducted or are underway for repositioning the existing drugs to prevent and treat this disease. Some of these drugs are remdesivir, ivermectin, nelfinavir, cepharanthine, hydroxychloroquine, and dexamethasone [10].
Despite all the efforts, treatment of COVID-19 still remains elusive. Antimicrobial peptides (AMPs) have been reported to treat various viral infections such as AIDS, hepatitis, dengue, and influenza. AMPs, therefore, offer a potential treatment option of COVID-19 [11–13].
1.1. Search strategy and selection criteria
Articles for this review were identified through searches of PubMed for the period from January 2020, to July 2021, by use of the terms ‘COVID-19,’ and ‘SARS-CoV-2.’ Articles published in English only were included.
2. Structure assembly and replication of SARS-CoV-2 virus
SARS-CoV-2 virus is a large spherical albeit pleomorphic, enveloped virus, having positive sense RNA (with a length of 29.9 kb), encoding 9860 amino acids [14]. Proteins of SARS-CoV-2 include two polyproteins, i.e. ORF1a and ORF1b, which undergo proteolytic cleavage to form 16 nonstructural proteins, Nsp1-16, four structural proteins: nucleocapsid (N), membrane (M), envelope (E), and spike (S), and nine accessory proteins: Orf3a, Orf3b, Orf6, Orf7a, Orf7b, Orf8, Orf9b, Orf9c, and Orf10 [15,16.17]. The N protein forms the capsid outside the genome, while the remaining structural proteins participate in host cell infection and its replication [18]. Nsp1-16 facilitates viral replication and transcription (Figure 2) [19].
Figure 2.
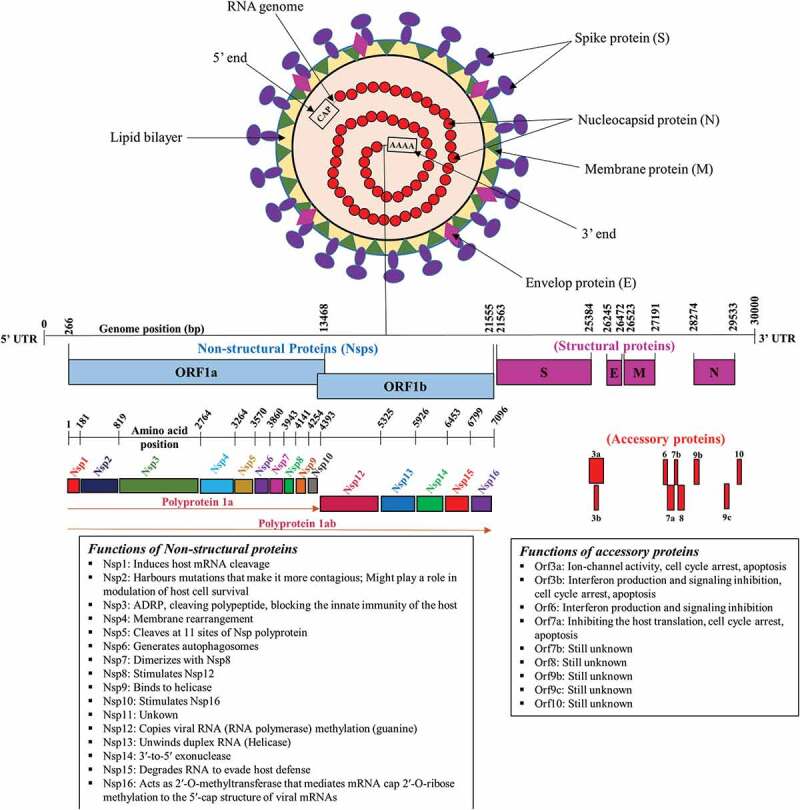
Genome organization of the SARS-CoV-2 and its genomic organization [16,20].
Virus entry to the host cells is facilitated by transmembrane S glycoprotein, consisting of S1 and S2 subunits, which are responsible for binding to host cell and fusion between the viral envelop and cellular membranes of host [21]. Receptor-binding domain (RBD), present in S1 subunit, recognizes the angiotensin-converting enzyme 2 (ACE2) peptidase domain of host cell and initiates direct binding with it, while membrane fusion is attributed to the basic elements present in S2 subunit [22]. ACE2 are considered as the target receptors of SARS-CoV-2 [23]. After binding, the host protease cleaves and activates S proteins to allow the insertion of viral fusion peptide into the host cell membrane. After that, the three pairs of heptad repeat region (HR1 and HR2) interact leading to formation of a six-helix coiled bundle, which pulls the viral membrane towards that of the host cell, leading to their fusion. This results in the entry of viral genome into the host cell [24]. Presence of 5ʹ methylated cap and a 3ʹ polyadenylated tail in viral genome permits the attachment of RNA to the ribosome of host cells for translation. Replication of viral genome and transcription of the negative-sense sub-genomic mRNA are mediated by RNA polymerase [25]. Host ribosomes translate these RNAs into structural and accessory proteins in the endoplasmic reticulum. Protein–protein interactions required for virus assembly are initiated by M proteins, leading to the formation of progeny viruses which are released into the extracellular space through exocytosis [26].
3. Peptides as drugs against COVID-19
Therapeutic peptides could not gain much popularity due to their limitations like susceptibility to degradation by enzymes, limitation of effective delivery methods, low membrane permeability, limited bioavailability on oral administration, rapid clearance from body, and low target specificity [27]. Recent advancement in processing technologies, however, has again generated interest in these therapeutic peptides. Properties of peptides, now, can be modulated to overcome the above-stated challenges by modification of amino acid sequences or addition of certain moieties. These modifications improve stability, transport, affinity profiles, and oral availability of peptides [28,29]. Peptides meant for treatment of COVID-19 are classified into the following categories:
3.1. Peptides preventing SARS-CoV-2 spike protein binding to ACE2 receptors
A crucial phase during the interaction of SARS-CoV-2 with the host cell involves the attachment of virus to ACE2 receptors, mediated by S glycoproteins present on virus cell surface. A plausible strategy for the prevention of infection is the inhibition of this interaction (Figure 3).
Figure 3.
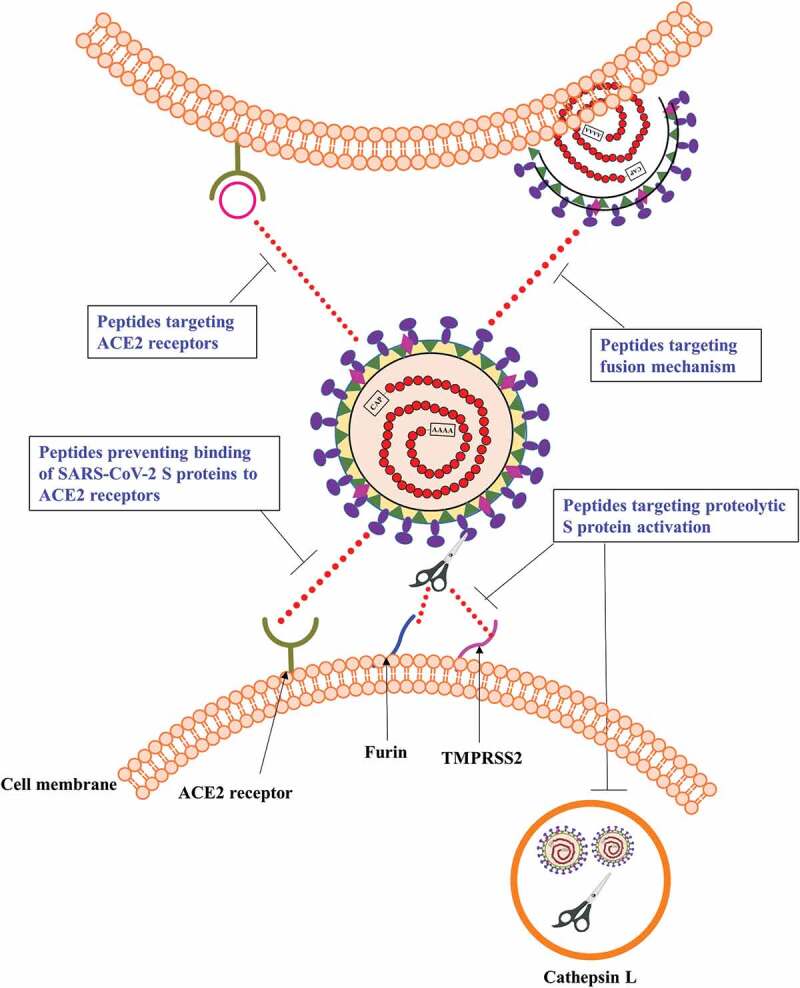
Targets of antiviral peptides against COVID-19.
Computational tools have been utilized to design novel peptides capable of inhibiting the interaction between ACE2 receptors and SARS-CoV-2 spike proteins, and thus preventing the virus entry into host cells. A portion consisting of 23 amino acids between Glu23 and Leu45 of the ACE2 PD N-terminal region is mainly involved in their interaction with spike protein. Computation alanine scanning further shortened the peptide chain containing 23 amino acids to 18 amino acids peptide (18aa peptide) that was found to be non-toxic in comparison with mutated peptides in the in silico toxicity evaluation [22]. In another study, protein–peptide docking revealed a stronger affinity of peptides toward ACE2 over RBD. Peptides generally associate with the residue binding motif (RBM), which, in turn, facilitates their binding to ACE2. On the other hand, peptides preferentially bind the core for ACE2. Among various peptides, AVP1244 and AVP0671 at ACE2 and RBD interfaces, respectively, decrease the binding affinity to a significant extent. They are also reported to alter the orientation of binding by RBD and ACE2 binding [30]. Chimeric peptide design strategy has been utilized for the designing of novel peptides, based on the analysis of key residues interacting with SARS-CoV-2-RBD and human ACE2, as well as screening of antibacterial peptides against SARS-CoV-2 spike RBD. Among them, peptides DBP6, AC23, AC20, and cnCoVP-1-cnCoVP-7 are expected to show potent activity against SARS-CoV-2 [31]. Badhe et al. created a series of peptides with a tendency to bind RBD of virus S protein and inhibit its binding on the ACE2 receptors. Two of the designed peptides exhibited both good RBD affinity as well as aqueous stability. However, for all the above studies, the peptides that have been designed by the in silico approach need to be synthesized and evaluated in in vitro and in vivo studies, before their use in COVID-19 therapy is proposed [32].
In addition to ACE2, glucose-regulating protein 78 (GRP78) receptor is another target assisting SARS-CoV-2 for its entry into the host cells. Allam et al. used in silico approach to identify bioactive peptides (satpdb12488, satpdb14438, satpdb18446, satpdb18674, and satpdb28899) which were found to act on regions III and IV of the viral S proteins and on its binding sites in GRP78. Bioassays, however, must be performed to ensure their inhibitory activity [33]. In another study, a protein inhibitor (ΔABP-D25Y) designed by computational approach has been found to exhibit exclusive binding at the ACE2 binding site of S protein in docking as well as molecular dynamic simulation studies. Higher binding affinity of the inhibitor as compared to that of ACE2 indicates its potential role in SARS-CoV-2 infection [34].
Han et al. prepared a number of peptide inhibitors after analyzing the interactions of amino acids at the ACE2 and RBD interface. The inhibitors consist of two sequential, self-supporting α-helices (bundle) from the protease domain (PD) of ACE2, which bind to the SARS-CoV-2 RBD. α-helical peptides are responsible for maintaining their secondary structure and specificity, as well as stable binding (blocking) to SARS-CoV-2. Enhancement of binding affinity can be achieved by multivalent binding of a number of peptides that are anchored onto the surfaces of various novel particulate delivery systems like dendrimers, nanoparticles, and clusters [35].
Scientists have also explored the use of hrsACE2, a human recombinant soluble ACE2. As ACE2 regulates many cellular functions including blood pressure and other cardiovascular parameters, its modulation with hrsACE2 could result in dysregulation of these key functions, leading eventually to serious side effects in patients. To overcome this, a series of peptides (22–44, 22–57, 22–44-g-351-357 and 351–357) were prepared and evaluated for their binding inhibition properties. All the synthesized peptides exhibited IC50 values in the μM range (1–10 μM). Peptide [22–44] was able to inhibit the formation of S1–ACE2 to the maximum extent (~76% reduction in binding probability) followed by [22–57] peptide (~74% reduction) and ~60% for the remaining peptides, pointing at their high therapeutic potential [21]. Three short peptides (pep1c, pep1d and pep1e), based on the ACE2 sequence/structure fragments, were designed and evaluated for their affinity to RBD of S protein. Two of these peptides (pep1c, pep1d) associate with the RBD of S protein. Dissociation constant for this binding was reported to be in nM range (<300 nM). In in vitro RBD-ACE2 interaction inhibition assay, IC50 value of 3 · 3 ± 0 · 8 mM was exhibited by pep1c. This is considered to be too high for any pharmacological application. Nevertheless, this may be taken as a lead molecule for further preparation of similar agents [36].
Cao et al. designed miniproteins by de novo sequencing utilizing ACE2 as scaffold, based on interaction motifs of RBD-binding. Out of these, two miniproteins i.e. AHB1 and AHB2 were found to exhibit IC50 of 35 and 15 · 5 nM, respectively, against SARS-CoV-2. Neutralization of SARS-CoV-2 of a much higher magnitude was observed with LCB1 and LCB3 (IC50 of 23 · 54 and 48 · 1 pM respectively) miniproteins designed by de novo approach. The designed miniproteins have many potential advantages over antibodies, such as a range of high-affinity binding modes, long term retention of activity even at high temperature, less molecular weight (5% of full antibody molecule), less scale up cost and amenability to formulation in the form of nasal gel or inhalational spray for respiratory tract [37]. Karoyan et al. designed a series of peptides resembling the N-terminal helix of human ACE2 protein and exhibiting high helical folding tendency in aqueous media, which is responsible for enhanced interactions with spike RBD and thereby, blocks SARS-CoV-2 pulmonary cell infection. These peptides were able to decrease SARS-CoV-2 viral titers while exhibiting complete efficacy at 1 μM in pulmonary cells. Also, these peptides were devoid of cell toxicity at effective concentrations, highlighting their safety [38].
A series of conformationally constrained peptidomimetics of ACE2 PD α1 helix were synthesized by solid-phase peptide synthesis approach and evaluated in a number of assays for antiviral activity. Interestingly, all of these failed to show any activity. This indicates the requirement of an enhanced binding interface in these peptides to display higher affinity for SARS-CoV-2 S-protein RBD binding than the membrane bound ACE2, thereby preventing virus entry [39].
3.2. Peptides targeting ACE2 and integrins
Blockage of the ACE2 receptors that act as entry point for the virus is another promising strategy for the management of COVID-19 (Figure 3) [40,41]. This proof-of-concept is supported by a number of clinically available molecules such as maraviroc (targeting HIV-1 co-receptor CCR5) [42], AMD3100 and EPI-X4 (HIV-1 co-receptor CXCR4 inhibitors) [43,44] and myrcludex B (that not only targets sodium taurocholate co-transporting polypeptide, but also the receptors for viral entry of hepatitis B and D) [45].
Defensins, a group of cytotoxic peptides, play a significant role in host defense mechanisms. Human defensin 5 (HD5) has much higher affinity to ACE2 receptors (39 · 3 nM) in comparison with that of SARS-CoV-2 (14 · 7 nM). It is also known to bind glycosylated Corona S1 protein, thus preventing the virus invasion due to its dual action [46]. Although pretreatment with HD5 has been shown to decrease the titers of SARS-CoV-2 pseudoparticles, its effect on SARS-CoV-2 isolates has not been established yet [47]. Moreover, blockage of ACE2 receptors may lead to deleterious consequences, as these receptors regulate some of the important cellular functions in the body. Further development of MLN-4760 (ACE2 blocker) was, therefore, stopped after phase Ib/IIa clinical trials due to toxicity concerns [48].
In addition to the ACE2 receptors, involvement of an interaction between the RGD (arginine-glycine-aspartate) motif in the spike protein and integrin α5β1 is also reported in the viral cell entry mechanism [49]. ATN-161, a non-RGD peptide obtained from fibronectin, has been reported to bind certain integrins, leading to inhibition of their activity [50,51]. ATN-161 was reported to reduce SARS-CoV-2 infection in renal cell line (VeroE6) of Chlorocebus atheiops. Pretreatment with ATN-161 increased cell viability, demonstrated by increased production of adenosine triphosphate [52]. Moreover, therapeutic efficacy of ATN-161 in in vivo study against closely related betacoronavirus (porcine hemagglutinating encephalomyelitis virus) [53] as well as in phase 2 clinical trial (NCT00352313) supported the further exploration of ATN-161 as a promising treatment for COVID-19.
3.3. Peptides targeting the fusion mechanism
The six-helix bundle created by interaction of HR1 with HR2, responsible for getting both viral and host cell membranes in each other<apos;>s vicinity for fusion, is another significant target for the development of antiviral drugs [54]. Certain lipopeptides obtained from EK1, a pan-coronavirus fusion inhibitor effective against five HCoVs, including SARS-CoV, MERS-CoV, and three SARS-related CoVs, were synthesized employing covalent attachment to cholesterol or palmitic acid using polyethylene glycol as a spacer to give corresponding lipopeptides EK1C and EK1P, respectively. These two peptides inhibited SARS-CoV2 mediated cell–cell fusion indicating that lipidation, especially cholesterol-modification is an attractive approach. To explore further, Xia et al. synthesized a number of derivatives of EK1C using multiple linkers containing 3–5 glycine/serine amino acid residues and polyethylene glycol-based spacer with variable arm length. Among the synthesized derivatives, EK1C4 containing GSGSG linker and PEG4 spacer was reported to be the most potent fusion inhibitor. The in vivo study in murine model also indicated the protective effect of EK1C4 against HCoV-OC43 infection, indicating its potential in prophylaxis as well as treatment of SARS-CoV-2 infection [55].
The S2 subunits of SARS-CoV and SARS-CoV-2 are quite strongly conserved in terms of their sequence alignment as they were found to be homologous to the extent of 92 · 6% and 100% in HR1 and HR2 domains, respectively [56]. Therefore, peptides obtained from SARS-CoV HR are anticipated to prevent infection. Working in this direction, Xia et al. prepared HR1- and HR2-derived peptides, named as 2019-nCoV-HR1P (aa924–965) and 2019-nCoV-HR2P (aa1168–1203). In cell–cell fusion assay, 2019-nCoV-HR2P demonstrated strong inhibition of fusion activity (IC50: 0 · 18 μM), pointing the possibility of 2019-nCoV HR1 region as a potential target site. 2019-nCoV-HR1P, however, failed to exhibit any considerable inhibitory effect (up to a strength of 40 μM), similar to the lack of activity of other coronavirus HR1-derived peptides, such as SARS-HR1P and MERS-HR1P [57]. 2019-nCoV-HR2P also inhibited the pseudovirus infection in a dose-dependent manner in ACE2-expressing 293 T cells [56]. Zhu et al. synthesized IPB01 peptide containing the HR2 sequence, which exhibited strong inhibition of SARS-CoV-2 S protein-mediated cell–cell fusion and pseudovirus transduction. For enhancing the antiviral activity as well as in vivo stability, IPB01 was conjugated with cholesterol group at its C terminus, leading to the formation of lipopeptide IPB02. Although IPB02 exhibited similar cell–cell fusion inhibitory activity (IC50: 0 · 25 μM), there was significant improvement in anti-SARS-CoV-2 pseudovirus activity (IC50: 0 · 08 μM) [58]. To further improve antiviral potency, de Varies et al. designed a series of lipophilic peptides by PEGylation, lipidation, and dimerization. Dimerization of both nonlipidated as well as lipidated peptides was reported to potentiate their effect. Among various peptides, SARSHRC-PEG24-chol (monomeric peptide) and [SARSHRC-PEG4]2-chol (dimeric peptide) were found to be the most potent lipopeptides. In biodistribution studies in humanized K18 hACE2 mice, both the peptides achieved similar lung concentration after intranasal administration in the first hour, followed by longer retention of dimeric peptide in lungs after 24 h as compared to that of monomeric peptide. On intranasal administration, this dimeric peptide was found to show thorough distribution throughout the lungs. Cellular toxicity assay in primary human airway epithelial cells, exhibited minimal toxicity even after 6 days at the highest concentrations tested while at its 90% maximal inhibitory concentration any kind of toxicity was absent. The dimeric peptide inhibited SARS-CoV-2 entry into VeroE6 cells or VeroE6 cells overexpressing transmembrane serine protease 2 (TMPRSS2). Intranasal administration of the dipeptide to ferrets for prophylactic use was able to protect them from transmission when they were co-housed with SARS-CoV-2-infected counterparts [59].
A series of short- and long-chain peptides, comprising 24 or 36 amino acids, were designed as SARS-CoV-2 fusion inhibitors. Although short chain peptide showed no effect on fusion activity, long-chain peptide exhibited potent inhibitory activity (IC50: 1 μM). It also suppressed the infectious activity of the pseudotyped viral particles by prevention of viral-cell fusion, through its association with the SARS-CoV-2 spike [60]. Sun and coworkers prepared four peptides from S protein of the virus. One of these peptides exhibited substantial inhibitory effects on cell–cell fusion and excellent neutralization activity against pseudotype SARS-CoV-2, and was able to inhibit the authentic viral infection to Vero E6 [61].
3.4. Peptides targeting proteolytic S protein activation
The steps of SARS-CoV-2 viral propagation including viral cell entry, its replication, polyprotein maturation as well as assemblage of the virions for further diffusion are regulated by various proteolytic enzymes of either host (serine-proteases) or virus (cysteine-proteases) origin. Binding and fusion of CoV S protein are initiated by cleavage events occurring at different sites, facilitated by a number of proteases at a number of sites in various cell compartments. Depending upon the viral strain and host cell type, S protein gets activated by one or more of the host protease enzymes, e.g. furin, trypsin, cathepsin L, transmembrane protease serine protease 2 (TMPRSS2), TMPRSS4, or human airway trypsin-like protease [62].
Furin protease is the most abundant host protease, which is expressed in a number of cells. Despite the role of furin to SARS-CoV-2 S activation still being ambiguous, it has been widely reported to be involved in other CoV infections as well. Furin is responsible for the cleavage of S protein at polybasic S1/S2 site (PRRAR), leading to two non-covalently linked distinct sub-units [63]. The second cleavage needed for SARS-CoV-2 activation is processed by TMPRSS2, leading to release of the fusion protein of S2 [64]. Cysteine protease, cathepsin L in lysosomes has also been reported to mediate activation of S protein without any involvement of furin-mediated priming [65]. Therefore, targeting these proteases is another promising approach for treatment of COVID-19 infection.
3.4.1. Peptides targeting furin
Two synthetic inhibitors targeting furin i.e. decanoyl-Arg-Val-Lys-Arg-chloromethylketone (dec-RVKR-cmk) and hexa-D-arginine (D6R) demonstrated antiviral activity against many viruses. They inhibit cleavage mediated by furin and the subsequent fusion of viral glycoproteins, thus exerting antiviral activity against viruses responsible for human immunodeficiency syndrome, Chikungunya, hepatitis B, influenza A, Ebola, and papilloma [66]. Recently, the role of dec-RVKR-cmk in COVID-19 has been explored. It has been reported to suppress the concentration of processed S protein fragments, cytopathic effects, and viral production in SARS-CoV-2-infected VeroE6 cells, indicating furin inhibitor activity [67,68]. D6R, another furin inhibitor, is safer and therapeutically more effective than the other members of this class, but its effectiveness in SARS-CoV-2 infection is yet to be explored [69]. MI-1851 is another synthetic furin inhibitor that has been reported to suppress SARS-CoV-2 replication in Calu-3 human airway cells, leading to a 30- to 190-fold reduction in virus titers at a dose of 10 μM [70,71]. All these studies suggest that furin inhibitors may lead to a plausible approach for the treatment of SARS-CoV-2 infection.
3.4.2. Peptides targeting TMPRSS2
An androgen-responsive enzyme, TMPRSS2, which is encoded by TMPRSS2 gene, is involved in various processes such as digestion, tissue remodeling, blood coagulation, fertility, inflammatory responses, tumour cell invasion, apoptosis, and pain [72]. It is expressed widely in epithelial cells of gastrointestinal, respiratory, and urogenital tracts [73]. This enzyme plays a significant role in cleavage of SARS-CoV-2 S glycoprotein, thus mediating entry of virus into the cell and its activation [74]. Inhibition of TMPRSS2 by camostat mesylate and nafamostat mesylate led to a reduction of SARS-CoV-2 infection in human pulmonary tissue in an ex vivo evaluation [75].
A protein-based drug, aprotinin, popularly known as bovine pancreatic trypsin inhibitor, is a naturally occurring polypeptide (58 amino acids), which inhibits the activity of some serine proteases. Inhibition of SARS-CoV-2 replication by aprotinin, probably via inhibition of TMPRSS2 has also been reported. Aprotinin was reported to exert anti-SARS-CoV-2 effects in three models of cell culture [Caco2, Calu-3, and air–liquid interface (ALI) from primary bronchial epithelial cells] and against three SARS-CoV-2 strains (FFM1, FFM2, and FFM6). The IC50 values of aprotinin that reduce cytopathogenic effects formation, SARS-CoV-2 S levels, and SARS-CoV-2-induced caspase 3/7 activation were in the range of 0 · 8–1 · 0 μM, 0 · 8–1 · 65 μM, and 0 · 3–0 · 7 μM, respectively, for all the three tested SARS-strains. A targeted proteomics assay indicated the suppression of N and M proteins expression in SARS-CoV-2-infected ALI cultures by aprotinin. SARS-CoV-2 S expression in SARS-CoV-2/FFM7-infected Calu-3 lung adenocarcinoma cells were also found to be suppressed by aprotinin [76]. Bestle et al. also documented the suppression of SARS-CoV-2 multiplication in Calu-3 human airway cells by aprotinin treatment in a dose-dependent manner [70]. When aprotinin was added to broncho-alveolar lavage of COVID-19 patients, it led to inhibition of the viral entry into the host cells. The thrombo-inflammatory response was also found to decrease [77]. Aprotinin aerosol, approved for local treatment of influenza in Russia [78], may be particularly useful for the suppression of the viral replication cycle.
MI-432 and MI-1900 are synthetic peptidomimetic inhibitors of TMPRSS2 that prevent SARS-CoV-2 replication as well as cytopathogenic effects in Calu-3 cells in a dose-dependent manner, but with lower efficiency as compared to aprotinin. Treatment with MI-1900 were able to inhibit the multiplication of SARS-CoV-2 more effectively than MI-432. A combination of MI-1851 (furin inhibitor) with any of these i.e. aprotinin, MI-432 or MI-1900 was found to act synergistically leading to inhibition of both activation as well as replication of SARS-CoV-2. MI-432 and MI-1851 combination decreased the virus titers 10- to −32 folds in comparison with that by individual inhibitor. Low dose combination of MI-1900 and MI-1851 (10 μM each) failed to exhibit any increase in the antiviral activity as compared to that by individual drug treatments at 10 μM. There was, in fact, a fivefold decrease in viral titers in cells treated with 50 μM each of MI-1900 and MI-1851 in comparison to cells exposed to 50 μM of each inhibitor singly [70]. Thus, treatment modalities acting at different steps processing and activation of S protein by combining furin and TMPRSS2 inhibitors seems to be a plausible treatment strategy for this viral infection.
α1-Antitrypsin, the most widely present serine protease inhibitor in plasma, is reported to hinder SARS-CoV-2 viral infection by inhibition of TMPRSS2, disintegrin, and metalloproteinase 17 (ADAM17) activity [79,80]. Reduced ADAM17 activity helps to control the release of inflammatory mediators, and consequently ‘cytokine storm,’ a characteristic feature of COVID-19 that enhances the probability of vascular hyperpermeability, multi-organ failure, as well as mortality [81]. Currently, α1-antitrypsin along with other serine protease inhibitors is undergoing clinical trials for the treatment of COVID-19 (NCT04385836, NCT04547140, NCT04495101, EudraCT 2020–001391-15) [82–84].
3.4.3. Peptides targeting cathepsin L
Cathepsins are lysosomal enzymes that can be subdivided into three groups based on active site amino acid residue i.e. serine (cathepsins A and G), aspartyl (cathepsins D and E), and cysteine (cathepsins B, C/DPP1, F, H, K, L, O, S, W, V, and Z/X) [85]. Among them, cathepsin L is reported to be critical for entry of virus into the host cells by activating the viral S protein in the endosome or lysosome [86,87]. For optimum activity, cathepsins require acidic pH (between 4 · 5–5), such as found in the lysosome [88]. Inhibition of cathepsins L is expected to be a potential approach for the prevention of infection. A number of drugs already in the market for different indications such as clofazimine, rifampicin, saquinavir, chloroquine, astaxanthin, dexamethasone, clenbuterol, and heparin are reported to exhibit cathepsin inhibitory activity [89].
P9, a short peptide (30 amino acids) obtained from mouse β-defensin-4, showed potent and broad-spectrum activity against a number of respiratory viruses including influenza A virus, SARS-CoV and MERS-CoV. This is attributed to high-affinity binding to S protein along with the presence of abundant basic amino acids in its composition, which do not allow acidification in endosomes and thus, inhibit release of viral RNA [10,90]. In order to further improve the potency of P9, Zhao et al. developed P9R by substituting histidine and lysine that possess weak positive charge, with arginine at three positions i.e. 21, 23, and 28. This increases the net positive charge on the molecule. The developed peptide showed strong antiviral activity against pH-dependent viruses, including enveloped pandemic A(H1N1)pdm09 virus, avian influenza A(H7N9) virus, coronaviruses (SARS-CoV-2, MERS-CoV and SARS-CoV), and the non-enveloped rhinovirus. Mechanistic studies revealed that P9R shows potent antiviral activity via dual mechanisms i.e. direct binding to viruses and inhibition of virus-host endosomal acidification in contrast to PA1 (only binding to viruses) and P9RS (only inhibiting endosomal acidification) [91]. Recently, dual-functional cross-linking peptide 8P9R developed from P9 and P9R has been reported to inhibit virus entry mediated by TMPRSS2 surface pathway and endocytic pathway. Moreover, 8P9R is also demonstrated to enhance the antiviral efficacy of arbidol (a spike-ACE2 fusion inhibitor) at a strength lower than its normal IC50 [92]. However, further clinical trials are required for the assessment of the therapeutic efficacy of this combination therapy in COVID patients.
Teicoplanin, a glycopeptide antibiotic, has been reported to be effective against infections by Gram-positive bacteria. It has also exhibited antiviral activity against SARS-CoV and MERS-CoV [93]. Teicoplanin inhibits the activation of viral S protein by cathepsin L, thereby interfering with the virus replication cycle. The dual antibacterial and antiviral potential of teicoplanin has been explored in the treatment of S. aureus superinfection in highly morbid patients suffering from severe SARS-CoV-2 pneumonia [94].
Table 1.
Characteristic features of anti-viral peptides effective against SARS-CoV-2
| Target protein | Target domain | Name | Sequence derived from | Sequence/IUPAC name | IC50 | Reference |
|---|---|---|---|---|---|---|
| Spike protein | RBD | Inhibitor 1 | ACE2 (21–55) | IEEQAKTFLDKFNHEAEDLFYQSSLASWNYNTNIT | - | [35] |
| Inhibitor 2 | ACE2 (21–88 and 349–357) |
|
- | |||
| Inhibitor 3 | ACE2 (21–105 and 323–362) |
|
- | |||
| Inhibitor 4 | ACE2 (21–95 and 335–400) |
|
- | |||
| Spike protein | RBD | AVP0671 | Peptide library | TWLATRGLLRSPGRYVYFSPSASTWPVGIWTTGELVLGCDAAL | - | [30] |
| ACE2 | RBD-binding site | AVP1244 | Peptide library | GCASRCKAKCAGRRCKGWASAFRGRCYCKCFRC | - | [30] |
| Spike protein | RBD | 18aa | ACE (28–45) | FLDKFNHEAEDLFYQSSL | - | [22] |
| 23aa | ACE (23–45) | EQAKTFLDKFNHEAEDLFYQSSL | - | |||
| Spike protein | RBD | Peptide 1 | EQEERIQQDKRKNEQEDKRYQRYGRGKGHQP | - | [32] | |
| Peptide 2 | EQQQRIQEDQYRNDWEDEEYQRKGRGKGHQP | - | ||||
| Peptide 3 | EQQQRIQQDQDSNDREDKEYQKRGKGKGHNH | - | ||||
| Peptide 4 | QEEQKIQEDQRRNDKEHKRKQRYGRGCGKQN | - | ||||
| Peptide 5 | EQEERIQRDKRKNEKEHEEKQRRGRGCGKQN | - | ||||
| Peptide 6 | EQEERIQQDKRKNENEDKRYQRYGRGKGHQP | - | ||||
| Peptide 7 | EQEERIQQDKRKNEWEDQYYQKYGQGKGHQP | - | ||||
| Peptide 8 | EQEERIQRDQYKNDYEDEEYQRKGRGKGHQP | - | ||||
| Peptide 9 | EQEQRIQQDKRSNEQEDKRYQREGKGKGHNN | - | ||||
| Peptide 10 | EQEERIQQDQRKNDKEDQRYQREGKGKGHNH | - | ||||
| Peptide 11 | ACE (22–53) | EEQAKTFLDKFNHEAEDLFYQSSGLGKGDFR | - | |||
| Peptide 12 | ACE (21–43) | IEEQAKTFLDKFNHEAEDLFYQS | - | |||
| Spike protein | RBD | satpdb12488 | PTTFMLKYDENGTITDAVDC | - | [33] | |
| satpdb14438 | SNNTIAIPTNFSISITTEVM | - | ||||
| satpdb28899 | RDVSDFTDSVRDPKTSEILD | - | ||||
| satpdb18674 | QYGSFCTQLNRALSGIAAEQ | - | ||||
| satpdb18446 | VLYNSTFFSTFKCYGVSATK | - | ||||
| Spike protein | RBD | [22–44] | ACE (22–44) | EEQAKTFLDKFNHEAEDLFYQSS | - | [21] |
| [22–57] | ACE (22–57) | EEQAKTFLDKFNHEAEDLFYQSSLASWNYNTNITEE | - | |||
| [22–44-g-351-357] | ACE (22–44 and 351–357) | EEQAKTFLDKFNHEAEDLFYQSS G LGKGDFR | - | |||
| [351–357] | ACE (351–357) | LGKGDFR | - | |||
| Spike protein | RBD | pep1c | ACE (30–38) | DKFNHEAED | 3 · 3 ± 0 · 8 mM | [36] |
| pep1d | ACE (30–35) | DKFNHE | - | |||
| pep1e | ACE (37–42) | EDLFYQ | - | |||
| Spike protein | RBD | AHB1 | ACE2 | DEDLEELERLYRKAEEVAKEAKDASRRGDDERAKEQMERAMRLFDQVFELAQELQEKQTDGNRQKATHLDKAVKEAADELYQRVR | 35 nM | [37] |
| AHB2 | ACE2 | ELEEQVMHVLDQVSELAHELLHKLTGEELERAAYFNWWATEMMLELIKSDDEREIREIEEEARRILEHLEELARK | 15 · 5 nM | |||
| LCB1 | de novo design | DKEWILQKIYEIMRLLDELGHAEASMRVSDLIYEFMKKGDERLLEEAERLLEEVER | 23 · 54 pM | |||
| LCB2 | de novo design | SDDEDSVRYLLYMAELRYEQGNPEKAKKILEMAEFIAKRNNNEELERLVREVKKRL | - | |||
| LCB3 | de novo design | NDDELHMLMTDLVYEALHFAKDEEIKKRVFQLFELADKAYKNNDRQKLEKVVEELKELLERLLS | 48 · 1 pM | |||
| LCB4 | de novo design | QREKRLKQLEMLLEYAIERNDPYLMFDVAVEMLRLAEENNDERIIERAKRILEEYE | - | |||
| LCB5 | de novo design | SLEELKEQVKELKKELSPEMRRLIEEALRFLEEGNPAMAMMVLSDLVYQLGDPRVIDLYMLVTKT | - | |||
| LCB6 | de novo design | DREQRLVRFLVRLASKFNLSPEQILQLFEVLEELLERGVSEEEIRKQLEEVAKELG | - | |||
| LCB7 | de novo design | DDDIRYLIYMAKLRLEQGNPEEAEKVLEMARFLAERLGMEELLKEVRELLRKIEELR | - | |||
| LCB8 | de novo design | PIIELLREAKEKNDEFAISDALYLVNELLQRTGDPRLEEVLYLIWRALKEKDPRLLDRAIELFER | - | |||
| Spike protein | RBD | P1 | ACE | STIEE QAKTF LDKFN HEAED LFYQS SL-NH2 | - | [38] |
| P1scr | AHLFS YLTTK EEQDN DAIFL QEFSK ES-NH2 | - | ||||
| Ppen | IEE QAKTF LDKFN HEAED LFYQS-NH2 | - | ||||
| P2 | SALEE QLKTF LDKFL HELED LLYQL SL-NH2 | - | ||||
| P3 | SALEE QLKTF LDKFL HELED LLYQL AL-NH2 | - | ||||
| P4 | Ac-SALEE QLKTF LDKFL HELED LLYQL AL-NH2 | - | ||||
| P5 | SALEE QLKTF LDKFL HELED PLYQL AL-NH2 | - | ||||
| P6 | SALEE QLKTF LDKFL HELED LLYQL ALAL-NH2 | - | ||||
| P7 | SALEE QYKTF LDKFL HELED LLYQL ALAL-NH2 | >1 μM | ||||
| P8 | SALEE QLKTF LDKFM HELED LLYQL AL-NH2 | 46 nM | ||||
| P9 | SALEE QYKTF LDKFM HELED LLYQL SL-NH2 | 53 nM | ||||
| P10 | SALEE QYKTF LDKFM HELED LLYQL AL-NH2 | 42 nM | ||||
| Spike protein | RBD | Native (1) | ACE | IEEQAKTFLDKFNHEAEDLFYQS | - | |
| 2 | IR8EQAKTFS5DKFNHEAEDLFYQS | - | ||||
| 3 | IEEQR8KTFLDKS5NHEAEDLFYQS | - | ||||
| 4 | IEEQAR8TFLDKFS5HEAEDLFYQS | - | ||||
| 5 | IEEQAKTFR8DKFNHES5EDLFYQS | - | ||||
| 6 | IEEQAKTFLDKR8NHEAEDS5FYQS | - | ||||
| 7 | IEEQAKTFLDKFR8HEAEDLS5YQS | - | ||||
| 8 | IEEQAKTFLDKFNHER8EDLFYQS5 | - | ||||
| 9 | HEAEDLFYQS | - | ||||
| 10 | HES5EDLS5YQS | - | ||||
| 11 | IEEQAKTFLDKFNHE | - | ||||
| 12 | IEEQR8KTFLDKS5NHE | - | ||||
| 13 | TFLDKFNHEAEDL | - | ||||
| 14 | TFR8DKFNHES5EDL | - | ||||
| 15 | TS5LDKS5NHEAEDL | - | ||||
| α5β1 integrin | ACE2 and RBD-binding domain | ATN-161 | Fibronectin | Ac-PHSCN-NH2 | 3 · 16 nM | [52] |
| Spike S2 | HR1 | EK1 | HR2 (OC43) | SLDQINVTFLDLEYEMKKLEEAIKKLEESYIDLKEL | 286 · 7 nM | [55] |
| EK1P | SLDQINVTFLDLEYEMKKLEEAIKKLEESYIDLKEL-PEG4-Palm | 69 · 2 nM | ||||
| EK1C | SLDQINVTFLDLEYEMKKLEEAIKKLEESYIDLKEL-PEG4-Chol | 48 · 1 nM | ||||
| EK1C1 | SLDQINVTFLDLEYEMKKLEEAIKKLEESYIDLKEL-Chol | 56 · 8 nM | ||||
| EK1C2 | SLDQINVTFLDLEYEMKKLEEAIKKLEESYIDLKEL-GSG-Chol | 48 · 2 nM | ||||
| EK1C3 | SLDQINVTFLDLEYEMKKLEEAIKKLEESYIDLKEL-GSG-PEG4-Chol | 10 · 6 nM | ||||
| EK1C4 | SLDQINVTFLDLEYEMKKLEEAIKKLEESYIDLKEL-GSGSG-PEG4-Chol | 1 · 3 nM | ||||
| EK1C5 | SLDQINVTFLDLEYEMKKLEEAIKKLEESYIDLKEL-GSGSG-PEG8-Chol | 3 · 1 nM | ||||
| EK1C6 | SLDQINVTFLDLEYEMKKLEEAIKKLEESYIDLKEL-GSGSG-PEG12-Chol | 3 · 9 nM | ||||
| EK1C7 | SLDQINVTFLDLEYEMKKLEEAIKKLEESYIDLKEL-GSGSG-PEG24-Chol | 3 · 9 nM | ||||
| Spike S2 | HR1 | 2019-nCoV-HR2P | HR2 (1168–1203) | DISGINASVVNIQKEIDRLNEVAKNLNESLIDLQEL | 0 · 18 μM | [56] |
| Spike S2 | HR1 | IPB01 | HR2 (1162–1205) | ISGINASVVNIQKEIDRLNEVAKNLNESLIDLQEL | 0 · 022 μM | [58] |
| IPB02 | ISGINASVVNIQKEIDRLNEVAKNLNESLIDLQELK (Chol) | 0 · 025 μM | ||||
| IPB03 | INASVVNIQKEIDRLNEVAKNLNESLIDLQELGK (Chol) | 0 · 015 μM | ||||
| IPB04 | SVVNIQKEIDRLNEVAKNLNESLIDLQELGK (Chol) | 0 · 033 μM | ||||
| IPB05 | IQKEIDRLNEVAKNLNESLIDLQELGK (Chol) | >5 μM | ||||
| IPB06 | IDRLNEVAKNLNESLIDLQELGK (Chol) | >5 μM | ||||
| IPB07 | IQKEIDRLNEVAKNLNESLIDLQELGKYEQYIK (Chol) | 0 · 017 μM | ||||
| IPB08 | ISGINASVVNIQKEIDRLNEVAKNLNESLIK (Chol) | 4 · 66 μM | ||||
| IPB09 | SVVNIQKEIDRLNEVAKNLNESLIK (Chol) | >5 μM | ||||
| Spike S2 | HR1 | SARSHRC | HR2 (1168–1203) | DISGINASVVNIQKEIDRLNEVAKNLNESLIDLQEL | - | [59] |
| SARSHRC-chol | DISGINASVVNIQKEIDRLNEVAKNLNESLIDLQEL-chol | - | ||||
| SARSHRC-PEG4-chol | DISGINASVVNIQKEIDRLNEVAKNLNESLIDLQEL-PEG4-chol | - | ||||
| SARSHRC-PEG24-chol | DISGINASVVNIQKEIDRLNEVAKNLNESLIDLQEL-PEG24-chol | - | ||||
| [SARSHRC]2-PEG11 | [DISGINASVVNIQKEIDRLNEVAKNLNESLIDLQEL]2-PEG11 | - | ||||
| [SARSHRC-PEG4]2-chol | [DISGINASVVNIQKEIDRLNEVAKNLNESLIDLQEL-PEG4]2-chol | - | ||||
| Spike S2 | HR1 | Peptide #1 | HR2 (1164–1202 | VVNIQKEIDRLNEVAKNLNESLID | - | [60] |
| Peptide #2 | DISGINASVVNIQKEIDRLNEVAKNLNESLIDLQEL | 1 μM | ||||
| Peptide #3 | DISGINASVVNIQKEIDRLNEVAKNLNESLIELQEL | 7 · 2 μM | ||||
| Peptide #4 | DISGINASVVNIQKEIDRLNEVAKNLNESLIKLQEL | - | ||||
| Peptide #5 | DISGINASVVNIQKEIDRLNEVAKNLNESLIGLQEL | 4 · 7 μM | ||||
| Peptide #6 | DISGINASVVNIQKEIDRLNEVAKNLGESLIGLQEL | - | ||||
| Peptide #7 | DISGINASVVNIQKEIDRLNEVAKNLPESLIGLQEL | - | ||||
| Peptide #8 | DISGINASVVNIQKEIDRLDEVAKNLNESLIDLQEL | - | ||||
| Peptide #9 | DISGINASVVNIQKEIDRLGEVAKNLNESLIDLQEL | - | ||||
| Peptide #10 | DISGINASVENIQKEIDRLNEVAKNLNESLIDLQEL | 8 · 9 μM | ||||
| Peptide #11 | DISGINASVVNIQKEIDRLNEVAKNLGESLIELQEL | - | ||||
| Peptide #12 | DISGINASVVNIQKEIDRLDEVAKNLPESLIELQEL | - | ||||
| Spike S2 | HR1 | Peptide 1 | HR2 (1163–1203) | GYHLMSFPQSAPHGVVFLHVTW | - | [61] |
| Peptide 2 | GVFVSNGTHWFVTQRNFYE | - | ||||
| Peptide 3 | ISGINASVVNIQKEIDRLNEVAKNLNESLIDLQEL | 0 · 72 μM | ||||
| Peptide 4 | IQKEIDRLNEVAKNLNESLIDLQELGK | - | ||||
| Furin | Catalytic domain | dec-RVKR-cmk | PCSK target motif | N-(1,20-diamino-9-(4-aminobutyl)-6-(2-chloroacetyl)-1,20-diimino-12-isopropyl-8,11,14-trioxo-2,7,10,13,19-pentaazaicosan-15-yl)decanamide | 0 · 057 μM | [67] |
| Probably catalytic domain | MI-1851 | Peptidomimetic | N-(1-((4-carbamimidoylbenzyl)amino)-4-(guanidinooxy)-1-oxobutan-2-yl)-2-(2-(2-(4-(guanidinomethyl)phenyl)acetamido)-3-guanidinooxy)propanamido)-3,3-dimethylbutanamide) | - | [70] | |
| TMPRSS2 | Catalytic site | Aprotinin | Natural polypeptide | RPDFC LEPPY TGPCK ARIIR YFYNA KAGLC QTFVY GGCRA KRNNF KSAED CMRTC GGA | - | [70,76] |
| MI-432 | Synthetic peptidomimetic | 3-(3-(4-(2-Aminoethyl)piperidin-1-yl)-2-((2’,4’-dichloro-[1,1’-biphenyl])-3-sulfonamido)-3-oxopropyl)benzimidamide | - | [70] | ||
| MI-1900 | Synthetic peptidomimetic | 4-(3-(3-Carbamimidoylphenyl)-2-((2’,4’-dimethoxy-[1,1’-biphenyl])-3-sulfonamido)propanoyl)-N-cyclohexylpiperazine-1-carboxamide | - | [70] | ||
| α1-Antitrypsin | Natural glycoprotein | Polypeptide (394 amino acids with three carbohydrate side chains) | 10–20 μM | [79,95] | ||
| Cathepsin L | Acidification | P9 | Mouse-β-defensin-4 derived | NGAICWGPCPTAFRQIGNCGHFKVRCCKIR | 2 · 4 μg/mL | [91] |
| P9R | Mouse-β-defensin-4 derived | NGAICWGPCPTAFRQIGNCGRFRVRCCRIR | 0 · 9 μg/mL | [91] | ||
| PA1 | Mouse-β-defensin-4 derived | NGAICWGPCPTAFRQIGNCGHFKVRCCKIRDED | - | [91] | ||
| P9RS | Mouse-β-defensin-4 derived | NGAHSWHPNETHFRQIHNSGRHRVRSHRIR | - | [91] | ||
| 8P9R | Mouse-β-defensin-4 derived | NGAICWGPCPTAFRQIGNCGRFRVRCCRIR | 0 · 3 μg/mL | [92] |
PCSK: Pro-protein convertase subtilisin/kexin
4. Conclusions
Despite the extensive investigations on SARS-CoV-2, the prevention and treatment of COVID-19 still remain a challenge. Bioactive peptides, because of their satisfactory bioavailability, therapeutic efficacy, and negligible off-target activity seem to be strong candidates for mitigation of SARS-CoV-2 etiopathology at different steps of viral infection. Moreover, rapid and large-scale cost-effective synthesis (depending on peptide composition), high target specificity and better tolerability makes them an attractive alternative to small synthetic molecules. A number of peptide-based drugs have been rationally designed and few to them have been synthesized and tested in in vitro and in vivo studies. These peptides act on various processes from virus entry to replication in host such as binding to ACE2, fusion mechanism, and proteolytic activation of S protein by targeting furin, TMPRSS2, and cathepsins L. However, emergence of virus mutants and reduced antibodies titers after recovery, enhance the probability of COVID-19 infection in healthy individuals as well as patients already recovered from this infection. Antiviral peptides having multi-target approach is considered to have upper-hand over the drugs acting on single target.
5. Expert opinion
Since the emergence of COVID-19 pandemic, extensive studies have been conducted to understand its etiopathogenesis, viral protein structure and composition, and the viral-host interaction to develop effective direct-acting antiviral and host-directed agents. However, none of the treatment strategies developed hitherto has been able to exhibit the optimum clinical efficacy. This has been attributed to a number of factors like development of viral resistance, co-infections, and the emergence of new viral strains [96]. Development of novel antiviral agents that are effective either in combination with the currently used drugs or in a stand-alone manner is, therefore, the need of the hour.
A number of peptide-based therapeutics have been developed and evaluated for the management of different diseases. Along with more than 100 peptide-based drugs already in the global market, a number of peptide drugs are undergoing clinical trials [11,97]. Approval of peptide-based antiviral therapeutics for the Human immunodeficiency virus (Enfuvirtide and Maraviroc) [98], and Hepatitis virus (Myrcludex b) [99] indicates their potential in the treatment of life-threatening viral infections. The extensive investigation of SARS-CoV-2 structural proteins and their cellular targets resulted in rational designing and optimizing of AVP against this deadly virus. These peptides inhibit SARS-CoV-2 entry to the host cells virus by targeting its binding to ACE2 or integrins, fusion mechanism as well as activation of proteolytic enzymes (cathepsin L, TMPRSS2, or furin). It is pertinent to mention here that targeting the virus entry is the most pragmatic approach for combating the infection as it does not require cell-penetration property. Moreover, it minimizes the detrimental effects of infection because it is able to inhibit the viral replication cycle in its early stages. Notably, the S-protein-targeting peptides EK1C4, SARSHRC-PEG24-chol (monomeric peptide), [SARSHRC-PEG4]2-chol (dimeric peptide), LCB1, and LCB3 exhibited promising antiviral potency against SARS-CoV-2 without affecting the function of the host protein. Therefore, application of these peptides via oral, intranasal, or inhalational routes directly at the site of replication may obviate the systemic administration and therefore, the need for optimization of their properties such as tissue-penetration, plasma stability, or half-life.
Despite their strong potential, the use of peptide-based drugs is limited by bottlenecks like instability, short half-life, less potency, inability to cross membrane barriers and poor bioavailability due to protease degradation. For the successful development of peptide-based drugs, these challenges need to be overcome. For this, a number of approaches have been utilized which include their conjugation with polymers like polyethylene glycol to enhance chemical stability [100], replacement of redundant hydrophobic amino acids to charged/polar residues for improving oral bioavailability [101,102], linkage of peptides to the cell penetrating peptides to enhance their cell permeability [100], and encapsulation in nanoparticles for efficient delivery [103]. Although these modifications help to address the challenges of poor pharmacokinetic properties of non-modified peptides, the systematic designing and appropriate formulation development need to be focused for the successful commercialization of peptide-based therapeutics.
Declaration of Interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.Pal M, Berhanu G, Desalegn C, et al. Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2): an update. Cureus. 2020;12(3):e7423–e7423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li FS. Function, and evolution of coronavirus spike proteins. Annu Rev Virol. 2016;3(1):237–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen B, Tian E-K, He B, et al. Overview of lethal human coronaviruses. Signal Transduct Target Ther. 2020. Jun 10;5(1):89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Corman VM, Muth D, Niemeyer D, et al. Hosts and Sources of Endemic Human Coronaviruses. Adv Virus Res. 2018;100:163–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Swelum AA, Shafi ME, Albaqami NM, et al. COVID-19 in human, animal, and environment: a review [review]. Front Vet Sci. 2020. September 04;7(578). doi: 10.3389/fvets.2020.00578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Y-c W, Chen C-S, Chan Y-J. The outbreak of COVID-19: an overview. J Chin Med Assoc. 2020;83(3): 217–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.https://www.euro.who.int/en/health-topics/health-emergencies/coronavirus-covid-19/novel-coronavirus-2019-ncov (Assessed on February 22, 2022).
- 8.Keni R, Alexander A, Nayak PG, et al. COVID-19: emergence, spread, possible treatments, and global burden [mini review]. Front Public Health. 2020. May 28;8(216). doi: 10.3389/fpubh.2020.00216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kapoor B, Kochhar RS, Gulati M, et al. Triumvirate to treat mucormycosis: interplay of pH, metal ions and antifungal drugs. Med Hypotheses. 2022. Feb 01;159:110748. [Google Scholar]
- 10.Mahendran ASK, Lim YS, Fang C-M, et al. The potential of antiviral peptides as COVID-19 therapeutics. Front Pharmacol. 2020;11:575444.*This article describes the potential use of AVPs against COVID19 based on the documented evidence against SARS.*This article describes the potential use of AVPs against COVID19 based on the documented evidence against SARSCoV2, SARS SARSCoV, MERSCoV, SARS MERSCoV, SARSrelated CoVs, and other respiratory viruses. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Agarwal G, Gabrani R. Antiviral peptides: identification and validation. Int J Pept Res Ther. 2020;271–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marcocci ME, Amatore D, Villa S, et al. The amphibian antimicrobial peptide temporin b inhibits in vitro herpes simplex virus 1 infection. Antimicrob Agents Chemother. 2018. May;62(5). doi: 10.1128/AAC.02367-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Angelis M, Casciaro B, Genovese A, et al. Temporin G, an amphibian antimicrobial peptide against influenza and parainfluenza respiratory viruses: insights into biological activity and mechanism of action. Faseb J. 2021. Feb;35(2):e21358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang Y, Yang C, X-f X, et al. Structural and functional properties of SARS-CoV-2 spike protein: potential antivirus drug development for COVID-19. Acta Pharmacol Sin. 2020;41(9):1141–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This article highlghts recent research advance in the structure, function and development of antivirus drugs targeting the S protein
- 15.Mariano G, Farthing RJ, Lale-Farjat SLM, et al. Structural characterization of SARS-CoV-2: where we are, and where we need to be [review]. Front Mol Biosci. 2020. December 17;7(344). doi: 10.3389/fmolb.2020.605236 [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This article provides molecular details of the SARS-COV-2 proteins and the basis of their biological functions during the viral infection cycle
- 16.Al-Qaaneh AM, Alshammari T, Aldahhan R, et al. Genome composition and genetic characterization of SARS-CoV-2. Saudi J Biol Sci. 2021. Mar 01;28(3):1978–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This review summarizes the genome composition and genetic characterization of the SARS-CoV-2
- 17.Awasthi A, Vishwas S, Corrie L, et al. OUTBREAK of novel corona virus disease (COVID-19): antecedence and aftermath. Eur J Pharmacol. 2020. 10 05;884:173381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mittal A, Manjunath K, Ranjan RK, et al. COVID-19 pandemic: insights into structure, function, and hACE2 receptor recognition by SARS-CoV-2. PLoS Pathog. 2020;16(8):e1008762. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This article describes origin and transmission of pathogenic human CoVs
- 19.Wang M-Y, Zhao R, Gao L-J, et al. SARS-CoV-2: structure, biology, and structure-based therapeutics development [review]. Front Cell Infect Microbiol. 2020. November 25;10(724). doi: 10.3389/fcimb.2020.587269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arya R, Kumari S, Pandey B, et al. Structural insights into SARS-CoV-2 proteins. J Mol Biol. 2021. 01 22; 4332:166725. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This article summarizes the important structural features of different SARS-CoV-2 proteins with their function, and their potential as a target for therapeutic interventions
- 21.Yang J, Petitjean SJL, Koehler M, et al. Molecular interaction and inhibition of SARS-CoV-2 binding to the ACE2 receptor. Nat Commun. 2020. Sep 11;11(1):4541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baig MS, Alagumuthu M, Rajpoot S, et al. Identification of a potential peptide inhibitor of sars-Cov-2 targeting its entry into the host cells. Drugs R D. 2020. 09 01; 20:161–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bhalla V, Blish CA, South AM. A historical perspective on ACE2 in the COVID-19 era. J Hum Hypertens. 2020. 12 14;35:935–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yesudhas D, Srivastava A, Gromiha MM. COVID-19 outbreak: history, mechanism, transmission, structural studies and therapeutics. Infection. 2021. Apr;49(2):199–213. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This article highligths the mechanism of entry of SARS-CoV-2 along with structural study of spike-ACE2 complex that provide insights to understand disease pathogenesis and development of vaccines and drug
- 25.Ghanbari R, Teimoori A, Sadeghi A, et al. Existing antiviral options against SARS-CoV-2 replication in COVID-19 patients. Future Microbiol. 2020;15(18):1747–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Habas K, Nganwuchu C, Shahzad F, et al. Resolution of coronavirus disease 2019 (COVID-19). Expert Rev Anti Infect Ther. 2020. Dec;18(12):1201–1211. [DOI] [PubMed] [Google Scholar]
- 27.Bruno BJ, Miller GD, Lim CS. Basics and recent advances in peptide and protein drug delivery. Ther Deliv. 2013. Nov;4(11):1443–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Recio C, Maione F, Iqbal AJ, et al. The potential therapeutic application of peptides and peptidomimetics in cardiovascular disease [review]. Front Pharmacol. 2017. January 06;7(526). doi: 10.3389/fphar.2016.00526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schütz D, Ruiz-Blanco YB, Münch J, et al. Peptide and peptide-based inhibitors of SARS-CoV-2 entry. Adv Drug Deliv Rev. 2020. Dec 01;167:47–65. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This article summarizes the current status of peptides inhibiting SARS-CoV-2 entry and outline the strategies used to design peptides targeting the ACE2 receptor or the viral spike protein and its activating proteases
- 30.Rathod SB, Prajapati PB, Punjabi LB, et al. Peptide modelling and screening against human ACE2 and spike glycoprotein RBD of SARS-CoV-2. Silico Pharmacol. 2020. Nov 09;8(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barh D, Tiwari S, Silva Andrade B, et al. Potential chimeric peptides to block the SARS-CoV-2 spike receptor-binding domain. F1000Res. 2020;9:576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Badhe Y, Gupta R, Rai B. In silico design of peptides with binding to the receptor binding domain (RBD) of the SARS-CoV-2 and their utility in bio-sensor development for SARS-CoV-2 detection [ 10.1039/D0RA09123E]. RSC Adv. 2021;11(7):3816–3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Allam L, Ghrifi F, Mohammed H, et al. Targeting the GRP78-dependant SARS-CoV-2 cell entry by peptides and small molecules. Bioinform Biol Insights. 2020;14:1177932220965505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jaiswal G, Kumar V, Abel SM. In-silico design of a potential inhibitor of SARS-CoV-2 S protein. PloS one. 2020;15(10):e0240004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Han Y, Král P. Computational design of ACE2-based peptide inhibitors of SARS-CoV-2. ACS Nano. 2020. Apr 28;14(4):5143–5147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Odolczyk N, Marzec E, Winiewska-Szajewska M, et al. Native structure-based peptides as potential protein–protein interaction inhibitors of SARS-CoV-2 spike protein and human ACE2 receptor. Molecules. 2021;26(8):2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cao L, Goreshnik I, Coventry B, et al. De novo design of picomolar SARS-CoV-2 miniprotein inhibitors. Science (New York, NY). 2020;370(6515):426–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Karoyan P, Vieillard V, Gómez-Morales L, et al. Human ACE2 peptide-mimics block SARS-CoV-2 pulmonary cells infection. Commun Biol. 2021. Feb 12;4(1):197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morgan DC, Morris C, Mahindra A, et al. Stapled ACE2 peptidomimetics designed to target the SARS-CoV-2 spike protein do not prevent virus internalization.Pept Sci.2021;113:e24217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Belouzard S, Millet JK, Licitra BN, et al. Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses. 2012;4(6):1011–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shang J, Wan Y, Luo C, et al. Cell entry mechanisms of SARS-CoV-2. Proceedings of the National Academy of Sciences. 2020;117:11727–11734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hunt JS, Romanelli F. Maraviroc, a CCR5 coreceptor antagonist that blocks entry of human immunodeficiency virus type 1. Pharmacother. 2009. Mar;29(3):295–304. [DOI] [PubMed] [Google Scholar]
- 43.Donzella GA, Schols D, Lin SW, et al. AMD3100, a small molecule inhibitor of HIV-1 entry via the CXCR4 co-receptor. Nat Med. 1998. Jan;4(1):72–77. [DOI] [PubMed] [Google Scholar]
- 44.Buske C, Kirchhoff F, Münch J. EPI-X4, a novel endogenous antagonist of CXCR4. Oncotarget. 2015;6(34):35137–35138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao K, Liu S, Chen Y, et al. Upregulation of HBV transcription by sodium taurocholate cotransporting polypeptide at the postentry step is inhibited by the entry inhibitor myrcludex B. Emerg Microbes Infect. 2018 Nov 21 7(1):186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Niv Y. Defensin 5 for prevention of SARS-CoV-2 invasion and Covid-19 disease. Med Hypotheses. 2020;143:110244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang C, Wang S, Li D, et al. Human intestinal defensin 5 inhibits SARS-CoV-2 invasion by cloaking ACE2. Gastroenterology. 2020;159(3):1145–1147.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Teralı K, Baddal B, Gülcan HO. Prioritizing potential ACE2 inhibitors in the COVID-19 pandemic: insights from a molecular mechanics-assisted structure-based virtual screening experiment. J Mol Graph Model. 2020;100:107697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Makowski L, Olson-Sidford W, W. Weisel J. Biological and clinical consequences of integrin binding via a rogue rgd motif in the SARS CoV-2 spike protein. Viruses. 2021;13(2):146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Doñate F, Parry GC, Shaked Y, et al. Pharmacology of the novel antiangiogenic peptide ATN-161 (Ac-PHSCN-NH2): observation of a U-shaped dose-response curve in several preclinical models of angiogenesis and tumor growth. Clin Cancer Res off J Am Assoc Cancer Res. 2008 Apr 1 14(7):2137–2144. [DOI] [PubMed] [Google Scholar]
- 51.Edwards DN, Salmeron K, Lukins DE, et al. Integrin α5β1 inhibition by ATN-161 reduces neuroinflammation and is neuroprotective in ischemic stroke. J Cereb Blood Flow and Metab. 2020. Aug;40(8):1695–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Beddingfield BJ, Iwanaga N, Chapagain PP, et al. The integrin binding peptide, ATN-161, as a novel therapy for SARS-CoV-2 infection. Jacc. 2021. Jan 01;6(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lv X, Li Z, Guan J, et al. ATN-161 reduces virus proliferation in PHEV-infected mice by inhibiting the integrin α5β1-FAK signaling pathway. Vet Microbiol. 2019. Jun;233:147–153. [DOI] [PubMed] [Google Scholar]
- 54.Wang X, Xia S, Zhu Y, et al. Pan-coronavirus fusion inhibitors as the hope for today and tomorrow. Protein Cell. 2021. Feb 01;12(2):84–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xia S, Liu M, Wang C, et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020. Nov 01;30(4):343–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xia S, Zhu Y, Liu M, et al. Fusion mechanism of 2019-nCoV and fusion inhibitors targeting HR1 domain in spike protein. Cell Mol Immunol. 2020. Jul 01;17(7):765–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lu L, Liu Q, Zhu Y, et al. Structure-based discovery of middle east respiratory syndrome coronavirus fusion inhibitor. Nat Commun. 2014;5:3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhu Y, Yu D, Yan H, et al. Design of potent membrane fusion inhibitors against SARS-CoV-2, an emerging coronavirus with high fusogenic activity. J Virol. 2020;94(14):e00635–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.de Vries Rd, Schmitz KS, Bovier FT, et al. Intranasal fusion inhibitory lipopeptide prevents direct-contact SARS-CoV-2 transmission in ferrets. Science (New York, NY). 2021;371(6536):1379–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kandeel M, Yamamoto M, Tani H, et al. Discovery of new fusion inhibitor peptides against SARS-CoV-2 by targeting the spike S2 subunit. Biomol Ther (Seoul). 2021 May 1;29(3):282–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sun H, Li Y, Liu P, et al. Structural basis of HCoV-19 fusion core and an effective inhibition peptide against virus entry. Emerg Microbes Infect. 2020. Jan 01;9(1):1238–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gioia M, Ciaccio C, Calligari P, et al. Role of proteolytic enzymes in the COVID-19 infection and promising therapeutic approaches. Biochem Pharmacol. 2020. Dec 01;182:114225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xia S, Lan Q, Su S, et al. The role of furin cleavage site in SARS-CoV-2 spike protein-mediated membrane fusion in the presence or absence of trypsin. Signal Transduct Target Ther. 2020. Jun 12;5(1):92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mollica V, Rizzo A, Massari F. The pivotal role of TMPRSS2 in coronavirus disease 2019 and prostate cancer. Future Oncol. 2020;16(27):2029–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xiu S, Dick A, Ju H, et al. Inhibitors of SARS-CoV-2 entry: current and future opportunities. J Med Chem. 2020. Nov 12;63(21):12256–12274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Imran I, Saleemi S, Chen C, et al. Decanoyl-Arg-Val-Lys-Arg-chloromethylketone: an antiviral compound that acts against flaviviruses through the inhibition of furin-mediated prM cleavage. Viruses. 2019;11(11):1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cheng Y-W, Chao T-L, Li C-L, et al. Furin inhibitors block SARS-CoV-2 spike protein cleavage to suppress virus production and cytopathic effects. Cell Rep. 2020. Oct 13;33(2):108254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tang T, Jaimes JA, Bidon MK, et al. Proteolytic activation of SARS-CoV-2 spike at the S1/S2 boundary: potential role of proteases beyond furin. ACS Infect Dis. 2021. Feb 12;7(2):264–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhou M, Zhang Y, Wei H, et al. Furin inhibitor D6R suppresses epithelial-mesenchymal transition in SW1990 and PaTu8988 cells via the Hippo-YAP signaling pathway. Oncol Lett. 2018;15(3):3192–3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bestle D, Heindl MR, Limburg H, et al. TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life Sci Alliance. 2020;3(9):e202000786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Faheem F, Kumar BK, Sekhar KVGC, et al. Druggable targets of SARS-CoV-2 and treatment opportunities for COVID-19. Bioorg Chem. 2020. Nov 01;104:104269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Thunders M, Delahunt B. Gene of the month: TMPRSS2 (transmembrane serine protease 2). J Clin Pathol. 2020;73(12):773–776. [DOI] [PubMed] [Google Scholar]
- 73.Iwata-Yoshikawa N, Okamura T, Shimizu Y, et al. TMPRSS2 contributes to virus spread and immunopathology in the airways of murine models after coronavirus infection. J Virol. 2019;93(6):e01815–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Strope JD, PharmD CHC, Figg WD. TMPRSS2: potential biomarker for COVID-19 outcomes. J Clin Pharmacol. 2020;60(7):801–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hoffmann M, Hofmann-Winkler H, Smith JC, et al. Camostat mesylate inhibits SARS-CoV-2 activation by TMPRSS2-related proteases and its metabolite GBPA exerts antiviral activity. EBioMedicine. 2021. Mar 01;65:103255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bojkova D, Bechtel M, McLaughlin K-M, et al. Aprotinin inhibits SARS-CoV-2 replication. Cells. 2020;9(11):2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vanassche T, Engelen MM, Van Thillo Q, et al. A randomized, open-label, adaptive, proof-of-concept clinical trial of modulation of host thromboinflammatory response in patients with COVID-19: the dawn-antico study. Trials. 2020. Dec 09;21(1):1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhirnov OP, Klenk HD, Wright PF. Aprotinin and similar protease inhibitors as drugs against influenza. Antiviral Res. 2011. Oct 01;92(1):27–36. [DOI] [PubMed] [Google Scholar]
- 79.Yang C, Chapman KR, Wong A, et al. α1-antitrypsin deficiency and the risk of COVID-19: an urgent call to action. Lancet Respir Med. 2021;9(4):337–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dutta AK, Goswami K. Host genomics of COVID-19: evidence point towards alpha 1 antitrypsin deficiency as a putative risk factor for higher mortality rate. Med Hypotheses. 2021;147:110485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.de Loyola Mb, Dos Reis TTA, de Oliveira Gxlm, et al. Alpha-1-antitrypsin: a possible host protective factor against Covid-19. Rev Med Virol. 2021;31(2):e2157–e2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.https://clinicaltrials.gov/ct2/show/NCT04547140?term=NCT04547140&draw=2&rank=1 (Assessed on February 22, 2022).
- 83.https://clinicaltrials.gov/ct2/show/NCT04495101?term=NCT04495101&draw=2&rank=1 (Assessed on February 22, 2022).
- 84.https://clinicaltrials.gov/ct2/show/NCT04385836?term=NCT04385836&draw=2&rank=1 (Assessed on February 22, 2022).
- 85.Pišlar A, Mitrović A, Sabotič J, et al. The role of cysteine peptidases in coronavirus cell entry and replication: the therapeutic potential of cathepsin inhibitors. PLoS Pathog. 2020;16(11):e1009013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ou X, Liu Y, Lei X, et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat Commun. 2020. Mar 27;11(1):1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sacco MD, Ma C, Lagarias P, et al. Structure and inhibition of the SARS-CoV-2 main protease reveal strategy for developing dual inhibitors against Mpro and cathepsin L. Sci Adv. 2020;6(50):eabe0751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gomes CP, Fernandes DE, Casimiro F, et al. Cathepsin L in COVID-19: from pharmacological evidences to genetics [review]. Front Cell Infect Microbiol. 2020. December 08;10(777). doi: 10.3389/fcimb.2020.589505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Liu T, Luo S, Libby P, et al. Cathepsin L-selective inhibitors: a potentially promising treatment for COVID-19 patients. Pharmacol Ther. 2020;213:107587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhao H, Zhou J, Zhang K, et al. A novel peptide with potent and broad-spectrum antiviral activities against multiple respiratory viruses. Sci Rep. 2016. Feb 25;6(1):22008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhao H, KKW T, Sze K-H, et al. A broad-spectrum virus- and host-targeting peptide against respiratory viruses including influenza virus and SARS-CoV-2. Nat Commun. 2020;11(1):4252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhao H, KKW T, Lam H, et al. Cross-linking peptide and repurposed drugs inhibit both entry pathways of SARS-CoV-2. Nat Commun. 2021. Mar 09;12(1):1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Baron SA, Devaux C, Colson P, et al. Teicoplanin: an alternative drug for the treatment of COVID-19? Int J Antimicrob Agents. 2020;55(4):105944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ceccarelli G, Alessandri F, d’Ettorre G, et al. Is teicoplanin a complementary treatment option for COVID-19? The question remains. Int J Antimicrob Agents. 2020;56(2):106029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wettstein L, Weil T, Conzelmann C, et al. Alpha-1 antitrypsin inhibits TMPRSS2 protease activity and SARS-CoV-2 infection. Nat Commun. 2021. Mar 19;12(1):1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Heydari H, Golmohammadi R, Mirnejad R, et al. Antiviral peptides against Coronaviridae family: a review. Peptides. 2021. May;139:170526.**This article summarizes data relating to antiviral peptides against Coronaviridae family particularly with respect to their applicability for development as novel treatments. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Badani H, Garry RF, Wimley WC. Peptide entry inhibitors of enveloped viruses: the importance of interfacial hydrophobicity. Biochim Biophys Acta - Biomembr. 2014. Sep 01;1838(9):2180–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shi S, Nguyen PK, Cabral HJ, et al. Development of peptide inhibitors of HIV transmission. Bioact Mater. 2016. Dec 01;1(2):109–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Turon-Lagot V, Saviano A, Schuster C, et al. Targeting the host for new therapeutic perspectives in hepatitis D. J Clin Med. 2020;9(1):222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chew M-F, Poh K-S, Poh C-L. Peptides as therapeutic agents for dengue virus. Int J Med Sci. 2017;14(13):1342–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mant CT, Kovacs JM, Kim HM, et al. Intrinsic amino acid side-chain hydrophilicity/hydrophobicity coefficients determined by reversed-phase high-performance liquid chromatography of model peptides: comparison with other hydrophilicity/hydrophobicity scales. Biopolymers. 2009;92(6):573–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wu SJ, Luo J, O’Neil KT, et al. Structure-based engineering of a monoclonal antibody for improved solubility. Protein Eng Des Sel. 2010. Aug;23(8):643–651. [DOI] [PubMed] [Google Scholar]
- 103.Lee AC, Harris JL, Khanna KK, et al. A comprehensive review on current advances in peptide drug development and design. Int J Mol Sci. 2019 May 14;20(10). doi: 10.3390/ijms20102383 [DOI] [PMC free article] [PubMed] [Google Scholar]