

Abstract
Targeting the menin-MLL protein-protein interaction is being pursued as a new therapeutic strategy for the treatment of acute leukemia carrying MLL-rearrangements (MLLr leukemia). Herein, we report M-1121, a covalent and orally active inhibitor of the menin-MLL interaction capable of achieving complete and persistent tumor regression. M-1121 establishes covalent interactions with Cysteine 329 located in the MLL binding pocket of menin and potently inhibits growth of acute leukemia cell lines carrying MLL translocations with no activity in cell lines with wild-type MLL. Consistent with the mechanism of action, M-1121 drives dose-dependent down-regulation of HOXA9 and MEIS1 gene expression in the MLL-rearranged MV4;11 leukemia cell line. M-1121 is orally bioavailable and showed potent antitumor activity in vivo with tumor regressions observed at tolerated doses in the MV4;11 subcutaneous and disseminated models of MLL-rearranged leukemia. Together, our findings support development of an orally active covalent menin inhibitor as a new therapy for MLLr leukemia.
Graphical Abstract

Introduction
Chromosomal translocations of the mixed lineage leukemia 1 (MLL1, also known as MLL) are found in 5–10% of acute leukemias in adults and in approximately 70% of acute lymphoid leukemia (ALL) in infants1, 3. Acute myeloid leukemias (AML) carrying MLL rearrangements (MLLr leukemia) have poor clinical prognosis with a 5-year survival rate of about 35%1, 3. MLLr leukemias are resistant to current therapies, highlighting the need for developing new therapeutic strategies for this disease.
Upon chromosomal translocations, the MLL gene is fused with one of over 80 partner genes, resulting in chimeric genes that encode oncogenic MLL fusion proteins8. The protein-protein interaction between these MLL fusion proteins and the oncogenic co-factor menin is critical for overexpression of MEIS1 and HOXA genes that led to the development and maintenance of MLLr leukemia9–13. Thus, targeting the menin-MLL protein-protein interaction is being pursued as a new therapeutic strategy for MLLr leukemia.7, 12, 14–16 To date, potent small-molecule inhibitors of the menin-MLL protein-protein interaction (hereafter called menin inhibitors, Figure 1) have been reported.18–28 Two of those small-molecule inhibitors have been advanced into early phase clinical development and encouraging early clinical activity have been recently reported for both compounds 36–37.
Figure 1.

Representative menin nihibitors
In 2018, we published the structure-based discovery of M-525, the first-in-class, potent, covalent small-molecule menin inhibitor29. We demonstrated that M-525 is more potent than its non-covalent inhibitor counterparts in reducing the expression of HOXA9 and MEIS1 genes and in inhibiting growth of leukemia cells carrying MLL translocations. Optimization of M-525 yielded M-80833, a potent, covalent menin inhibitor with antitumor activity in in vitro and in in vivo models of MLLr leukemia (Figure 1). Despite its superior cellular potency, M-808 was discontinued for further development due to the low oral bioavailability demonstrated in mice. In this study, we describe our efforts to further improve upon M-808, that resulted in the discovery of M-1121 as the first, potent and orally active covalent menin inhibitor, capable of achieving complete and long-lasting tumor regression.
Results and Discussion
Design and synthesis of new non-covalent menin inhibitors.
As can be seen from the chemical structures of M-525 and M-808 in Figure 1, a covalent menin inhibitor consists of a non-covalent menin-binding scaffold and an electrophile for the formation of a covalent bond with a cysteine residue in menin. We reasoned that superior oral bioavailability for a covalent inhibitor could be achieved by improving the oral bioavailability of the non-covalent portion of the molecule. We modified the inhibitor portion of M-525 and M-808 lacking the electrophile group to first obtain a potent and orally bioavailable non-covalent inhibitor that could later be added an electrophile group for establishing covalent interactions.
Based upon M-525, compound 7 lacking an electrophile was designed and synthesized as a non-covalent menin inhibitor. Compound 7 binds to menin with an IC50 value of 6.9 nM as determined by fluorescence polarization (FP)-based, competitive binding assay32. This compound was then tested for its ability to inhibit cell proliferation of the acute leukemia cell lines MV4;11 and MOLM-13 carrying MLL-AF4 and MLL-AF9 fusion, respectively. Compound 7 showed moderate antiproliferative activity in MV4;11 and MOLM-13 cells with IC50 values of 798 nM and 840 nM, respectively.
Based on the co-crystal structure of M-808-menin complex, the positively charged amino group in M-808 establishes charge-charge interactions with two negatively charged carboxyl groups of Glu359 and Asp180 residues16. Accordingly, we have synthesized and evaluated a series of new menin inhibitors containing different positively charged groups, with the results summarized in Table 1.
Table 1.
Binding affinity and cell growth inhibition of menin inhibitors (7–13)

| ||||
|---|---|---|---|---|
|
| ||||
| Compd. | R1 | Binding Affinity to Menin IC50 (nM)a | Cell growth inhibition IC50 (nM)b |
|
| MV4;11 | MOLM13 | |||
|
| ||||
| 7 | -CN | 6.9 ± 0.4 | 798 ± 67 | 870 ± 224 |
| 8 |
|
4.0 ± 0.3 | 192 ± 23 | 553 ± 226 |
| 9 |

|
1.7 ± 0.4 | 178 ± 3 | 703 ± 150 |
| 10 |

|
3.5 ± 1.2 | 105 ± 11 | 274 ± 36 |
| 11 |

|
2.8 ± 0.2 | 516 ± 101 | 599 ± 119 |
| 12 |

|
3.4 ± 0.2 | 681 ± 165 | 675 ± 157 |
| 13 |

|
3.5 ± 0.8 | 421 ± 40 | 684 ± 240 |
IC50 values were determined using a FP-based competitive binding assay from at least three independent experiments.
Cell viability was determined using CellTiter-Glo® Luminescent Cell Viability Assay after 4 days of treatment for each compound, with average values and SD calculated from three independent experiments.
Compound 8 containing a primary amino group binds to menin with an IC50 value of 4.0 nM and inhibits cell growth with IC50 values of 192 nM and 553 nM in the MV4;11 and MOLM-13 cell lines, respectively. Dimethylation of the primary amine group in 8 led to compound 9, which binds to menin with an IC50 value of 1.7 nM and displays IC50 values of 178 nM for MV4;11 cells and 703 nM for MOLM-13 cells. Replacing the dimethylamino group with an azetidine group yielded compound 10, which binds to menin with an IC50 value of 3.5 nM and achieves IC50 values of 105 nM and 274 nM in the MV4;11 and MOLM-13 cell lines, respectively. Expanding the 4-membered ring in compound 10 to a 5-membered ring generated compound 11, that resulted in an IC50 value of 2.8 nM for menin binding and antiproliferative activity for MV4;11 and MOLM-13 cell lines with IC50 values of 516 nM and 599 nM, respectively. Next, we synthesized compounds 12 and 13 both of which contain a 6-membered ring. Compounds 12 and 13 have binding affinity and cell growth inhibitory activity similar to that of compound 11. Based upon the antiproliferative activity in both, MV4;11 and MOLM-13 cell lines, compound 10 is the most potent non-covalent inhibitor among the tested compounds shown in Table 1.
We then evaluated the pharmacokinetic (PK) properties of compound 10, when dosed orally at 25 mg/kg in mice (for details, see Supporting Information - SI). We found that compound 10 achieves an encouraging oral exposure with an average plasma concentration of 624, 735 and 928 ng/mL at 1, 3 and 6 h post-dosing, respectively.
We next modified the ‘linker’ region in compound 10 to further improve oral exposure, while retaining antiproliferative activity in the MV4;11 and MOLM-13 cell lines. We reasoned that different R2 groups on the bridge atom of the azetidine will affect the pKa of the nitrogen atom of the piperidine. This in turn could have a significant effect on the binding affinity to menin and antiproliferative activity in MLL cell lines, as well as on the PK of the resulting compounds. Accordingly, we synthesized and evaluated several analogues of compound 10 with different groups on the bridge atom of the azetidine. These results are summarized in Table 2.
Table 2.
Binding affinity and cell growth inhibition of menin inhibitors (14–18)a

| ||||
|---|---|---|---|---|
|
| ||||
| Compd. | R2 | Binding Affinity to Menin IC50 (nM) | Cell growth inhibition (nM) |
|
| MV4;11 | MOLM13 | |||
|
| ||||
| 10 | -H | 3.5 ± 1.2 | 105 ± 11 | 274 ± 36 |
| 14 | -F | 3.2 ± 0.2 | 222 ± 31 | 948 ± 305 |
| 15 | -Me | 2.9 ± 0.5 | 272 ± 17 | 544 ± 145 |
| 16 | -OH | 3.6 ± 0.8 | 379 ± 54 | 833 ± 179 |
| 17 | -OMe | 2.5 ± 0.3 | 166 ± 9 | 443 ± 120 |
| 18 | -COOMe | 42.4 ± 4.0 | >1000 | >10000 |
IC50 values are average of three independent experiments.
Introduction of a bridge fluorine atom in 10 yielded compound 14, that shows an IC50 value of 3.2 nM for binding to menin and inhibits cell growth in the MV4;11 and MOLM-13 cell lines with IC50 values of 222 nM and 948 nM, respectively. Adding a bridge methyl group in 10 generated compound 15, which binds to menin with an IC50 value of 2.9 nM. Compound 15 has antiproliferative activity in the MV4;11 and MOLM-13 cell lines with IC50 values of 272 nM and 544 nM, respectively. Introduction of a bridge hydroxyl or methoxyl group at the same bridge carbon in 10 resulted in compounds 16 and 17 which bind to menin with IC50 values of 3.6 nM and 2.5 nM, respectively. Compound 16 has IC50 values of 379 nM in MV4;11 cells and 833 nM in the MOLM-13 cell line, while compound 17 is about 2-fold more potent than 16. Addition of a bridging methyl ester group to 10 led to compound 18, which is more than10-fold less potent than 10 in its binding affinity to menin. Consistent with its lower binding affinity, compound 18 has a weak cell growth inhibitory activity in both the MV4;11 and MOML-13 cell lines. Hence, among these analogues with a substituted bridging atom, compound 17 with a bridging methoxyl group is the most potent compound based upon its cell growth inhibitory activity in both the MV4;11 and MOLM-13 cell lines.
We then evaluated the oral exposure of compound 17 in mice. A single oral administration of compound 17 at 25 mg/kg achieves average plasma compound concentrations of 8265, 5750 and 2195 ng/mL at 1, 3 and 6 h post-dosing, respectively. Hence, while compound 17 is slightly less potent than compound 10 in inhibiting cell growth in both the MV4;11 and MOLM-13 cell lines, 17 displays much improved oral plasma exposure when compared to 10 in mice.
Design of covalent inhibitors based upon compound 17
These promising cellular and oral exposure data for compound 17 promoted us to design and synthesize a series of covalent menin inhibitors based on this non-covalent inhibitor with the objective to improve cellular potency in MLLr cell lines. We tested these covalent inhibitors for their binding affinity to menin by FP-based assay and their antiproliferative activity in the MV4;11 and MOLM-13 cell lines. These results are summarized in Table 3.
Table 3.
Binding affinity and cell growth inhibition of menin inhibitors 19–26a

| ||||
|---|---|---|---|---|
|
| ||||
| Compd. | R3 | Binding affinity to menin (IC50 (nM)) | Cell growth inhibition (IC50 (nM)) |
|
| MV4;11 | MOLM13 | |||
|
| ||||
| 19 |

|
2.7±0.5 | 2.3 ± 0.7 | 49.7 ± 7.2 |
| 20 |

|
2.8±0.3 | 112 ± 33 | 402 ± 246 |
| 21 |

|
3.8 ± 0.5 | 156 ± 30 | 594 ± 195 |
| 22 |

|
3.0 ± 0.4 | 77 ± 15 | 196 ± 44 |
| 23 |
|
3.7 ± 0.3 | 74 ± 24 | 188 ± 8 |
| 24 (M-1121) |

|
2.7 ± 0.1 | 10.3 ± 2.9 | 51.5 ± 13.5 |
| 25 |

|
4.6 ± 1.7 | 436 ± 89 | 504 ± 42 |
| 26 |

|
4.2 ± 0.7 | 289 ± 82 | 630 ± 192 |
IC50 values are average of three independent experiments.
We replaced the cyclopropyl group in 17 with the Michael acceptor used in M-525 to obtain compound 19. Compound 19 shows improved antiproliferative activity with IC50 values of 2.3 nM and 49 nM in MV4;11 and MOLM-13 cell lines, respectively. Compound 19 thus is 72- and 9- fold more potent than compound 17 in inhibiting proliferation of MV4;11 and MOLM-13 cell lines, respectively, suggesting that compound 19 behaves as a covalent menin inhibitor in cells.
In both M-525 and M-808, a positively charged group was attached to the Michael acceptor. Our data show that the positively charged group in M-525 and M-808 is critical in increasing the reactivity of the Michael acceptor for rapid formation of a covalent bond with menin, and such enhanced reactivity leads to improved antiproliferative activity in MLLr leukemia cells30, 33. However, we hypothesized that the positively charged group attached to the Michael acceptor in M-525 and M-808 molecules diminishes the oral bioavailability of these compounds. Therefore, we decided to synthesize and test a series of potential covalent menin inhibitors lacking the positively charged group attached to the Michael acceptor.
Removal of the positively charged group attached to the Michael acceptor in compound 19 yielded compound 20. As expected and consistent with our previous data, compound 20 is 49- and 8-fold less potent than compound 19 in inhibiting growth of MV4;11 and MOLM-13 cell lines, respectively. Therefore, while compounds 19 and 20 contains the same the acrylamide Michael acceptor, compound 20 has a much weaker cell growth inhibitory potency in both the MV4;11 and MOLM-13 cell lines, indicating that compound 20 probably does not form a covalent bond with menin very efficiently in cells.
We reasoned that the Michael acceptor in compound 20 may not able to adopt an optimal position and/or orientation for efficient formation of a covalent bond with menin upon binding. Therefore, we synthesized compounds 21-23 with different conformationally constrained linking groups between the SO2 group and the acrylamide to determine if other linkers would place the Michael acceptor in a more optimal position and orientation for efficient covalent bond formation with menin protein.
Compound 21 employing a 6-membered ring piperidine linker is slightly less potent than compound 20 in cell growth inhibition in the MV4;11 and MOLM-13 cell lines. Changing the acrylamide from 4-position in compound 21 to 3-position in the piperidine linker with either a (S)- or (R)-configuration generated compounds 22 and 23, respectively. Compounds 22 and 23 are both 2-times more potent than compound 21 in the MV4;11 and MOLM-13 cell lines and marginally more potent than compound 20 in both cell lines. Since compounds 21-23 are all much less potent than compound 19, these compounds are still likely incapable of efficiently forming a covalent bond with menin in cells.
We hypothesized that further conformational restriction of the linker may lock the acrylamide Michael acceptor group in an optimal position and orientation for efficient formation of a covalent bond with the thiol group of Cys329 in menin. Computational modeling suggested that a highly conformationally constrained (1S,4S)-2,5-diazabicyclo[2.2.1]heptane linker would lock the acrylamide in an optimal position and orientation for efficient formation of a covalent bond with the thiol group of Cys 329 (Supporting Information). Interestingly, the enantiomer (1R,4R)-2,5- diazabicyclo[2.2.1]heptane linker was predicted to place the acrylamide much further away (4.7 Å) from the thiol group of Cys 329 residue, suggesting that a formation of a covalent bond is unlikely.
To test our predictions, we synthesized compounds 24 and 25 using these two enantiomeric 2,5- diazabicyclo[2.2.1]heptane linkers. Consistent with our predictions, compound 24 containing the (1S,4S)-2,5-diazabicyclo[2.2.1]heptane linker is a very potent inhibitor with IC50 values of 10.3 nM and 51.5 nM in the MV4;11 and MOLM-13 cell lines. In comparison, the stereoisomer compound 25 containing the (1R,4R)-2,5-diazabicyclo[2.2.1]heptane is a much weaker inhibitor and displays IC50 values of 436 nM and 504 nM in the MV4;11 and MOLM-13 cell lines, respectively. Hence, compound 24, which was named M-1121, is 40- and 9-fold more potent than its stereoisomer compound 25 in the MV4;11 and MOLM-13 cell lines, respectively. These data show that the stereochemistry of the linker in M-1121 is critically important for its potent activity in MLLr leukemia cells. M-1121 is also 8–10 fold more potent than compound 20 in both MV4;11 and MOLM-13 cell lines.
We synthesized compound 26 by converting the acrylamide in compound 24 into propionamide to further test the importance of covalent bond formation for cell growth inhibition (Table 3). Compound 26 is >10-times less potent than 24 in inhibition of cell growth in MV4;11 and MOLM-13 cell lines (Table 3), highlighting the importance of covalent formation for achieving high cellular potency in MLLr leukemia cell lines.
To gain further insights into their mode of action and cellular activity, we analyzed compounds 19–26 for covalent complex formation with recombinant human menin protein by mass spectrometry using four different incubation times (10-, 30-, 60-minutes and overnight) and obtained the data summarized in Table 4.
Table 4.
Analysis of covalent complex formation of menin inhibitors with recombinant human menin protein by mass spectrometry.
| Compound | Incubation time with menin protein | |||
|---|---|---|---|---|
|
| ||||
| 10 minutes | 30 minutes | 1 hr | overnight | |
|
| ||||
| % of menin covalent complex | ||||
|
| ||||
| 19 | 36.9% | 83.4% | 85.4% | 90.9% |
| 20 | 24.1% | 40.9% | 59.4% | 81.5% |
| 21 | 0% | 0% | 0% | 27.0% |
| 22 | 8.8% | 27.9% | 37.4% | 93.1% |
| 23 | 9.8% | 22.6% | 40.5% | 90.0% |
| 24 (M-1121) | 27% | 59% | 66% | 100% |
| 25 | 0% | 0% | 0% | 45.1% |
| 26 | 0% | 0% | 0% | 0% |
In general, the reaction kinetic data for compounds 19-26 with menin protein (Table 4) correlate nicely with their potencies in inhibition of cell growth in both MV4;11 and MOLM-13 cell lines (Table 3). Compound 19 is the most potent inhibitor in cell growth inhibition and has the fastest kinetics in formation of a covalent complex with menin protein among compounds 19-26. With 10-, 30-, 60-minutes and overnight incubation time, 36.9%, 83.4%, 85.4% and 90.9% of menin protein forms a covalent complex with compound 19, respectively. M-1121 has the second fastest kinetics in formation of a covalent complex with menin protein and is also the second most potent inhibitor. This is followed by compound 20 as the third most potent compound with the third fastest kinetics. Compounds 22 and 23 have a slower reaction kinetics than compounds 19, 20 and M-1121, consistent with their weaker cellular activity than compounds 19, 20 and M-1121. Compounds 21 and 25 have a very slow reaction kinetics and no covalent complex was detected within 1 h incubation time. Consistent with its lack of a Michael acceptor, compound 26 does not form a covalent complex with menin even with overnight incubation. Of note, the stoichiometry for each inhibitor:menin covalent complex was 1:1.
A glutathione (GSH) reactivity assay was employed to test intrinsic reactivity of M-1121 toward glutathione (GSH). Incubation of M-1121 under conditions mimicking intracellular glutathione e levels (4.5 mM GSH, 37 °C, pH 7.4) revealed a GSH conjugation half-life of 89 min. These data indicated that M-1121 has only mild reactivity with GSH.
To further understand the precise binding mode, we determined the co-crystal structure for M-1121 in complex with menin at 2.74 Å resolution (Figure 2.). Consistent with our design and mass spectrometry data, M-1121 forms a covalent bond between the acrylamide Michael acceptor and the thiol group of Cys329 in menin. The 2,5-diazabicyclo [2.2.1] ring orients and places the acrylamide group in a position that is optimal for the formation of a covalent bond with the sulfur atom in Cys329 of menin. In addition, the 2,5-diazabicyclo [2.2.1] group establishes hydrophobic interactions with menin, specifically with Val371, Ala325 and Gly326. The nitrogen atom of azetidine head group is 4.1 Å from the negatively charged carboxylic acid group of the Asp180 side chain in menin, indicating a charge-charge interaction. The methoxy group on the azetidine linker in M-1121 forms a hydrogen bond with the phenol group of Tyr323.
Figure 2.

Co-crystal structure of compound 24 (M-1121) complexed with menin at 2.74 Å resolution (PDB code: 7M4T). Side chains of menin residues within 4 Å from compound are shown as sticks. Hydrogen bonds are shown as dashed lines.
M-1121 was evaluated for its antiproliferative activity in 4 MLLr and 3 MLL wild-type cell lines to further define its cellular activity and selectivity. We found that M-1121 potently inhibits proliferation of MLL rearranged but not MLL wild-type cell lines up to 10 μM, highlighting the selective activity of M-1121 for cells carrying MLL translocations (Table 5).
Table 5.
Cell growth inhibitory activity of M-1121 in a panel of leukemia cell lines after 4 days of treatment determined by CellTiter-Glo®
| Cell line | MLL status | Disease Indication | Cell growth inhibition IC50 (nM) |
|---|---|---|---|
|
| |||
| MV4;11 | MLL/AF4 | ALL | 10 |
| MOLM-13 | MLL/AF9 | AML | 52 |
| OCI-AML4 | MLL-ENL | AML | 3 |
| SEM | MLL/AF4 | ALL | 51 |
| KOPN8 | MLL/ENL | ALL | 110 |
| RS4;11 | MLL/AF4 | ALL | 1,000 |
| HL-60 | MLLwt | AML | >10,000 |
| K562 | MLLwt | CML | >10,000 |
| MEG-1 | MLLwt | CML | >10,000 |
AML: Acute Myeloid Leukemia; ALL: Acute Lymphocytic Leukemia; CML: Chronic Myeloid Leukemia.
In our previous studies, we showed that menin inhibitors suppress the expression of HOXA9 and MEIS1 in MLLr leukemia cells28, 33. We thus evaluated M-1121 for its effect on the expression of HOXA9 and MEIS1 in MV4;11 cells by qRT-PCR. Our data showed that M-1121 suppresses HOXA9 and MEIS1 gene transcription in a dose-dependent manner and effectively modulates the expression of HOXA9 and MEIS1 gene at concentrations as low as 10 nM and 30 nM, respectively (Figure 3). Hence, M-1121 is potent in inhibiting the expression of HOXA9 and MEIS1 gene transcription in the M4;11 cell line consistent with the expected mechanism of action of a menin inhibitor.
Figure 3.

Gene expression changes induced by M-1121. Quantitative real-time polymerase chain reaction (qRT-PCR) analysis of the effect of M-1121 on the mRNA levels of HOXA9 and MEIS1genes in MV4;11 cells after 24h of treatment.
Next, we evaluated the oral exposure of M-1121 in mice. A single oral administration of M-1121 at 25 mg/kg achieves an average plasma concentration of 3797, 4640 and 2055 ng/mL at 1, 3 and 6 h post-dosing, respectively, indicating excellent oral exposure. Subsequently, a PK study with both intravenous and oral administrations was performed with M-1121. The PK data showed that M-1121 has a low clearance and a moderate volume of distribution (Table 6). M-1121 dosed orally at 5 mg/kg achieves a Cmax value of 4153 ng/mL, and AUC0−∞ of 43567 ng/mL*h, respectively. Together, M-1121 has an acceptable PK profile in mice with 49.4% oral bioavailability.
Table 6.
Pharmacokinetic of M-1121 in mice. Female C57BL/6 mice were dosed either intravenously with a solution in 10% NMP, 10% Solutol® HS15 and 80% saline, or orally at 5 mg/kg in 0.5% methyl cellulose + 0.2% Tween 80 (w/w).
| 1 mg/kg (IV) |
5 mg/kg (PO) |
||||||
|---|---|---|---|---|---|---|---|
| Compd. | CL (L/h/kg) | Vss (L/kg) | AUC0-∞ (h·ng/mL) | tmax (h) | Cmax (ng/mL) | AUC0-∞ (h·ng/mL) | F% |
|
| |||||||
| M-1121 | 0.0567 | 0.330 | 17644 | 2.00 | 4153 | 43567 | 49.4 |
We determined the plasma protein binding data for M-1121 and found that M-1121 has 90.7%, 87.5% and 99.3% binding in human, rat and mouse respectively. The PPB data showed that while M-1121 has an excellent PPB in human and rat plasma, it has a very high PPB in mouse plasma, suggesting that high doses may be needed to achieve strong antitumor activity in mice.
We next evaluated M-1121 for its in vivo antitumor activity in SCID mice harboring MV4;11 subcutaneous tumors. In the first experiment, when xenograft tumors reached an average volume of 100 mm3, mice were treated with M-1121 at 100 mg/kg daily for 26 days via oral gavage (Figure 4.a and 4.b). M-1121 reduced the average tumor volume from 157 mm3 at the beginning of the treatment to 106 mm3 on day 26 of the treatment, a reduction of tumor volume of 32%. Significantly, M-1121 caused no animal weight loss or other signs of toxicity during and after the treatment.
Figure 4.

M-1121 exhibits potent anti-tumer efficacy in the MV4;11 (Mll rearranged) subcutaneous tumor xenograft model. The compound was administrated orally at the indicated dose schedules.
Because of the high PPB (99.3%) of M-1121 in mouse plasma and importantly its lack of any signs of toxicity in mice at 100 mg/kg, we further tested its antitumor activity in the MV4;11 subcutaneous tumor model at a higher dose to determine if M-1121 can achieve even stronger antitumor activity. In the second experiment, when tumors reached an average volume of 200 mm3, mice were treated with M-1121 at 300 mg/kg once daily for 15 days via oral gavage (Figure 4.c and 4.d). M-1121 led to complete tumor regression in 10 out of 10 mice with no tumor regrowth detected up to a month after last treatment (day 45 after treatment start) (Figure 4.c and Figure S2. in supporting information). Treatment with M-1121 was well tolerated with no significant body weight loss (Figure 4.d) or other signs of toxicity.
Since MLLr leukemias are bone marrow diseases, we next investigated whether M-1121 has activity in this compartment by evaluating the effect of M-1121 in bone marrow CD45+ leukemic cells in NCG mice engrafted with the Luciferase-tagged, MV4;11 disseminated model. Mice were dosed with M-1121 at 150 mg/kg once daily by oral gavage for 4 days to achieve steady-state drug levels and evaluated the effect at 48 h time-point post-last dose. We found that M-1121 suppresses the expression of MEIS1 gene to less than 2% compared to vehicle group with concomitant induction of the cell differentiation marker ITGAM (~ 67-fold) and CD11b protein in human CD45+ cells isolated from bone marrow (Figure 5.a, and b). It is interesting to note that even though mice were treated for only 4 days, M-1121 was able to exert anti-tumor activity with reduction in the number of human CD45+ CD33+ leukemic cells in bone marrow as detected by flow cytometry (Figure 5.c) and decrease in the intensity of whole-body bioluminescence signal (Figure 5.d). No significant changes in body weight were observed (data not shown).
Figure 5.

M-1121 shows activity in the disseminated Luciferase-tagged MV4;11 (MLL rearranged) xenograft model. M-1121 was administered via oral gavage at 150 mg/kg once daily for 4 days and data presented as mean ± SEM, five mice were evaluated per group. Readouts in a, b and c were measured 48h after last dose. MEIS1 and ITGAM gene expression in human CD45+ cells isolated from bone marrow evaluated by RT-qPCR (a), CD11b Mean Fluorescence Intensity (MFI) measured in human CD45+ CD33+ cells from bone marrow by flow cytometry (b), number of human CD45+ CD33+ cells measured by flow cytometry (c) and whole-body bioluminescence intensity determined in vehicle or M-1121 treated mice (d). Dotted line indicates last day of dosing.
Chemistry
The synthetic routes to these compounds are shown below (Scheme 1.). Compounds 8-13 were synthesized in a convergent manner. The Boc group in the known compound 2733 was deprotected with TFA and the resulting amine was converted into a methyl carbamate 28. A relay reduction of 28 with DIBAL-H followed by NaBH4 was employed to give primary amine 29, which was subjected to a Boc protection. Removal of the benzyl protecting group followed by substitution and removal of Boc group afforded compound 8, whose subsequent functionalization gave 9-13. Intermediate 36 was synthesized in 5 steps starting from 4-fluorobenzenethiol.
Scheme 1.

Synthesis of compounds 8–13
Reagents and conditions: (a) TFA, DCM, rt; (b) dimethyl dicarbonate, Et3N, DCM, rt; (c) DIBAL-H, toluene, 0 °C; (d) NaBH4, MeOH, 0 °C to rt; (e) Boc2O, DCM, rt; (f) H2, Pd/C, MeOH, rt; (g) 36, K2CO3, KI, MeCN, 80 °C; (h) TFA, DCM, rt; (i) formaldehyde, NaBH(OAc)3, MeOH, rt (9); 1,4-dibromobutane, K2CO3, MeCN, 80 °C (10); 1,5-dibromopentane, K2CO3, MeCN, 80 °C (11); 1-bromo-2-(2-bromoethoxy)ethane, K2CO3, MeCN, 80 °C (12); methallyltrimethylsilane, formaldehyde, H2O (13); (j) bromocyclopropane, t-BuONa, DMSO, 80 °C; (k) m-CPBA, DCM, rt; (l) methyl azetidine-3-carboxylate hydrochloride, K2CO3, DMSO, 80 °C; (m) LiAlH4, THF, 0 °C; (n) MsCl, Et3N, DCM, rt;
Synthesis of compounds 14–18 (Scheme 2) started with compound 29. A ring-closure/substitution reaction gave an azetidine compound 37. Removal of the benzyl protecting group followed by substitution with 41 afforded compound 39. Then the Boc group was cleaved and the resulting product was submitted to a nucleophilic aromatic substitution reaction with 34 to give 14-18 as the final compounds.
Scheme 2.

Synthesis of compounds 14–18
Reagents and conditions: (a) 1,3-dibromopropane, K2CO3, KI, MeCN, 80 °C; (b) H2, Pd/C, MeOH, rt; (c) 41, K2CO3, KI, MeCN, 80 °C; (d) TFA, DCM, rt; (e) 34, K2CO3, DMSO, 80 °C;
Starting from 39d, compounds 19–26 were synthesized in a similar manner (Scheme 3.). Removal of Boc protection from 39d followed by aromatic substitution with 42 gave compound 40. Removal of the Boc protection from 40 produced the corresponding amine intermediates, which reacted with diverse carbonyl chlorides or anhydrides to afford the final products 19-26.
Scheme 3.

Synthesis of compounds 19–26
Reagents and conditions: (a) TFA, DCM, rt; (b) 42, K2CO3, DMSO, 80 °C; (c) TFA, DCM, rt; (d) (2E)-4-(dimethylamino)-2-butenoyl chloride hydrochloride, DIPEA, DCM, 0 °C to rt (19); acrylic anhydride, DIPEA, DCM, rt (20-25); propionic anhydride, DIPEA, DCM, rt (26);
EXPERIMENTAL SECTION
General Methods for Chemistry.
Unless otherwise noted, commercial solvents and reagents were used without further purification. Proton nuclear magnetic resonance (1H NMR) spectra were recorded on a Bruker Advance 400 MHz spectrometer and are reported in parts per million (ppm) downfield from tetramethylsilane (TMS). In the spectral data reported, the format (δ) chemical shift (multiplicity, J values in Hz, integration) was used with the following abbreviations: s = singlet, d =doublet, t = triplet, q = quartet, and m = multiplet. Mass spectrometric (MS) analysis was carried out with a Waters ultraperformance liquid chromatography (UPLC) – mass spectrometer. The final compounds were all purified by a C18 reverse-phase preparative high-performance liquid chromatography (HPLC) column with solvent A (0.1% TFA in H2O) and solvent B (0.1% TFA in MeCN). The purity of all the final compounds was confirmed to be >95% by UPLC analysis (10 – 100% MeCN in H2O containing 0.1% TFA in 10 min).
Methyl ((1S,2R)-2-(cyano(1-((1-(4-(cyclopropylsulfonyl)phenyl)azetidin-3-yl)methyl)piperidin-4-yl)(3-fluorophenyl)methyl)cyclopentyl)carbamate (7).
Compound 7 was prepared from compounds 28 and 36 with the procedure that was used to produce compound 31. 1H NMR (400 MHz, MeOH-d4) δ 7.49 – 7.44 (m, 2H), 7.25 (td, J = 8.1, 6.1 Hz, 1H), 7.15 (d, J = 7.9 Hz, 1H), 7.07 (d, J = 10.2 Hz, 1H), 6.98 – 6.92 (m, 1H), 6.36 – 6.26 (m, 2H), 3.95 (td, J = 8.0, 2.0 Hz, 2H), 3.68 (q, J = 6.8 Hz, 1H), 3.53 (dd, J = 8.1, 5.6 Hz, 2H), 3.38 (t, J = 11.6 Hz, 1H), 3.25 (s, 1H), 3.23 (s, 3H), 3.13 (p, J = 1.6 Hz, 1H), 3.08 – 2.94 (m, 1H), 2.92 – 2.79 (m, 2H), 2.67 (q, J = 7.9 Hz, 1H), 2.37 (tt, J = 7.9, 4.8 Hz, 1H), 2.26 (t, J = 12.1 Hz, 1H), 2.10 – 1.91 (m, 2H), 1.83 – 1.75 (m, 1H), 1.70 – 1.59 (m, 1H), 1.58 – 1.31 (m, 5H), 1.29 – 1.13 (m, 1H), 1.00 – 0.93 (m, 2H), 0.84 – 0.77 (m, 2H); ESI-MS calcd for C33H42FN4O4S [M + H]+ = 609.29, found: 609.14.
Methyl ((1S,2R)-2-(2-amino-1-(1-((1-(4-(cyclopropylsulfonyl)phenyl)azetidin-3-yl)methyl)piperidin-4-yl)-1-(3-fluorophenyl)ethyl)cyclopentyl)carbamate (8).
Compound 31 (680 mg, 0.954 mmol) was dissolved in DCM (3 mL) and TFA (1.5 mL) was added slowly. After stirring for 3 h at room temperature (rt), the reaction mixture was concentrated under vacuum. The residue was purified with preparative HPLC to give the title compound (495 mg, 85%). 1H NMR (400 MHz, MeOH-d4) δ 7.64 – 7.59 (m, 2H), 7.44 (q, J = 7.6 Hz, 1H), 7.24 – 7.16 (m, 2H), 7.13 (t, J = 8.8 Hz, 1H), 6.77 – 6.71 (m, 2H), 3.81 – 3.66 (m, 4H), 3.66 – 3.53 (m, 2H), 3.53 – 3.45 (m, 1H), 3.31 – 3.20 (m, 4H), 3.20 – 3.09 (m, 3H), 3.02 (t, J = 12.3 Hz, 1H), 2.93 (t, J = 12.5 Hz, 1H), 2.89 – 2.70 (m, 2H), 2.57 (m, 1H), 2.41 (s, 1H), 2.18 – 2.08 (m, 1H), 2.04 – 1.94 (m, 3H), 1.94 – 1.82 (m, 1H), 1.82 – 1.67 (m, 2H), 1.67 – 1.54 (m, 3H), 1.21 – 1.12 (m, 2H), 1.06 – 0.97 (m, 2H); ESI-MS calcd for C33H46FN4O4S [M + H]+ = 613.32, found: 613.17.
Methyl ((1S,2R)-2-(1-(1-((1-(4-(cyclopropylsulfonyl)phenyl)azetidin-3-yl)methyl)piperidin-4yl)-2-(dimethylamino)-1-(3-fluorophenyl)ethyl)cyclopentyl)carbamate (9).
Triethylamine (23 03CL, 0.163 mmol), acetic acid (15 μL, 0.261 mmol) and formaldehyde (3 μL, 0.196 mmol) were added to a solution of compound 8 (40 mg, 0.06 53 mmol) in MeCN (30 mL). After 3 h, sodium triacetoxyborohydride (42 mg, 0.196 mmol) was added. The mixture was stirred overnight, quenched with H2O and concentrated under vacuum. The residue was purified by reverse phase preparative HPLC to give the title compound (22 mg, 53%). 1H NMR (400 MHz, MeOH-d4) δ 7.71 – 7.66 (m, 2H), 7.56 – 7.48 (m, 1H), 7.46 – 7.37 (m, 2H), 7.21 – 7.14 (m, 1H), 6.58 – 6.51 (m, 2H), 4.21 (t, J = 8.0 Hz, 2H), 4.01 – 3.83 (m, 3H), 3.79 (ddd, J = 8.3, 5.6, 2.9 Hz, 2H), 3.72 – 3.59 (m, 2H), 3.57 – 3.43 (m, 4H), 3.31 – 3.19 (m, 2H), 3.16 – 3.01 (m, 3H), 3.01 – 2.82 (m, 4H), 2.69 (d, J = 7.4 Hz, 1H), 2.58 (tt, J = 8.0, 4.8 Hz, 1H), 2.44 (s, 1H), 2.28 – 2.11 (m, 2H), 2.06 – 1.93 (m, 1H), 1.70 (m, 3H), 1.60 (m, 3H), 1.42 (m, 1H), 1.33 (t, J = 7.3 Hz, 1H), 1.21 – 1.14 (m, 2H), 1.05 – 0.98 (m, 2H); ESI-MS calcd for C35H50FN4O4S [M + H]+ = 641.35, found: 641.20.
Methyl ((1S,2R)-2-(2-(azetidin-1-yl)-1-(1-((1-(4-(cyclopropylsulfonyl)phenyl)azetidin-3-yl)methyl)piperidin-4-yl)-1-(3-fluorophenyl)ethyl)cyclopentyl)carbamate (10).
1,3-Dibromopropane (8 μL, 0.0784 mmol), K2CO3 (27 mg, 0.196 mmol) and KI (1 mg, 0.0053 mmol) were added to a solution of compound 8 (40 mg, 0.0653 mmol) in MeCN (1.5 mL). The mixture was stirred at 80 °C overnight, quenched with H2O and concentrated under vacuum. The residue was purified by reverse phase preparative HPLC to give the title compound (26 mg, 61%). 1H NMR (400 MHz, MeOH-d4) δ 7.69 – 7.63 (m, 2H), 7.47 (td, J = 8.1, 6.2 Hz, 1H), 7.19 – 7.11 (m, 2H), 7.06 (d, J = 8.0 Hz, 1H), 6.55 – 6.49 (m, 2H), 4.60 – 4.46 (m, 2H), 4.42 – 4.28 (m, 2H), 4.21 – 4.10 (m, 3H), 3.79 (d, J = 15.6 Hz, 1H), 3.72 (ddd, J = 8.0, 5.6, 2.4 Hz, 2H), 3.61 – 3.44 (m, 3H), 3.41 (d, J = 7.1 Hz, 2H), 3.26 – 3.16 (m, 1H), 2.99 (dt, J = 24.9, 12.4 Hz, 2H), 2.79 (d, J = 9.4 Hz, 1H), 2.57 (tt, J = 7.9, 4.8 Hz, 2H), 2.50 – 2.39 (m, 1H), 2.13 – 1.96 (m, 5H), 1.93 – 1.83 (m, 1H), 1.83 – 1.74 (m, 1H), 1.73 – 1. 57 (m, 3H), 1.47 (q, J = 12.9 Hz, 1H), 1.19 – 1.12 (m, 2H), 1.04 – 0.96 (m, 2H); ESI-MS calcd for C36H50FN4O4S [M + H]+ = 653.35, found: 653.10.
Methyl ((1S,2R)-2-(1-(1-((1-(4-(cyclopropylsulfonyl)phenyl)azetidin-3-yl)methyl)piperidin-4-yl)-1-(3-fluorophenyl)-2-(pyrrolidin-1-yl)ethyl)cyclopentyl)carbamate (11).
Compound 11 was prepared from compound 8 and 1,4-dibromobutane according to the procedure used to produce compound 10 (26 mg, 60%). 1H NMR (400 MHz, MeOH-d4) δ 7.71 – 7.65 (m, 2H), 7.49 (td, J = 8.0, 6.3 Hz, 1H), 7.39 – 7.30 (m, 2H), 7.20 – 7.12 (m, 1H), 6.56 – 6.50 (m, 2H), 4.17 (t, J = 8.0 Hz, 2H), 3.91 – 3.67 (m, 6H), 3.64 – 3.50 (m, 3H), 3.49 – 3.40 (m, 3H), 3.38 – 3.35 (m, 2H), 3.29 – 3.18 (m, 2H), 3.05 (t, J = 12.4 Hz, 1H), 2.96 (t, J = 12.5 Hz, 1H), 2.90 – 2.77 (m, 1H), 2.58 (tt, J = 7.9, 4.8 Hz, 1H), 2.32 – 1.98 (m, 8H), 1.98 – 1.86 (m, 1H), 1.86 – 1.49 (m, 6H), 1.44 – 1.25 (m, 1H), 1.20 – 1.13 (m, 2H), 1.06 – 0.97 (m, 2H); ESI-MS calcd for C37H52FN4O4S [M + H]+ = 667.37, found: 667.25.
Methyl ((1S,2R)-2-(1-(1-((1-(4-(cyclopropylsulfonyl)phenyl)azetidin-3-yl)methyl)piperidin-4yl)-1-(3-fluorophenyl)-2-morpholinoethyl)cyclopentyl)carbamate (12).
Compound 12 was prepared from compound 8 and bis(2-bromoethyl) ether according to the procedure used to prepare compound 10 (20 mg, 45%). 1H NMR (400 MHz, MeOH-d4) δ 7.70 – 7.64 (m, 2H), 7.45 (q, J = 7.6 Hz, 1H), 7.39 (d, J = 9.6 Hz, 2H), 7.17 – 7.06 (m, 1H), 6.56 – 6.49 (m, 2H), 4.18 (t, J = 8.0 Hz, 2H), 3.92 (s, 4H), 3.80 – 3.68 (m, 4H), 3.64 – 3.52 (m, 4H), 3.49 – 3.36 (m, 6H), 3.29 – 3.10 (m, 2H), 3.04 (t, J = 12.5 Hz, 1H), 2.92 (t, J = 12.4 Hz, 1H), 2.81 (s, 1H), 2.58 (tt, J = 7.9, 4.8 Hz, 1H), 2.24 – 2.07 (m, 3H), 2.07 – 1.93 (m, 2H), 1.70 (s, 7H), 1.20 – 1.13 (m, 2H), 1.05 – 0.97 (m, 2H); ESI-MS calcd for C37H52FN4O5S [M + H]+ = 683.36, found: 683.23.
Methyl ((1S,2R)-2-(1-(1-((1-(4-(cyclopropylsulfonyl)phenyl)azetidin-3-yl)methyl)piperidin-4-yl)-1-(3-fluorophenyl)-2-(4-hydroxy-4-methylpiperidin-1-yl)ethyl)cyclopentyl)carbamate (13)
Compound 13 was prepared according to a reported procedure34. Methallyltrimethylsilane (32 μL, 0.184 mmol) and formal aldehyde (41 μL, 0.488 mmol) were added to a solution of compound 8 (75 mg, 0.1223 mmol) in MeCN (1 mL) and H2O (1 mL). The mixture was stirred at 45 °C overnight and then was purified by reverse phase preparative HPLC to give the title compound (60 mg, 69%). 1H NMR (400 MHz, MeOH-d4) δ 7.71 – 7.64 (m, 2H), 7.52 – 7.44 (m, 1H), 7.44 – 7.33 (m, 2H), 7.18 – 7.10 (m, 1H), 6.55 – 6.47 (m, 2H), 4.15 (t, J = 8.0 Hz, 2H), 4.03 (s, 1H), 3.87 – 3.71 (m, 3H), 3.71 – 3.61 (m, 3H), 3.61 – 3.48 (m, 3H), 3.48 – 3.38 (m, 3H), 3.38 – 3.35 (m, 1H), 3.31 – 3.14 (m, 3H), 3.04 (t, J = 12.4 Hz, 1H), 2.98 – 2.79 (m, 2H), 2.58 (tt, J = 7.9, 4.8 Hz, 1H), 2.33 – 1.98 (m, 5H), 1.98 – 1.86 (m, 2H), 1.85 – 1.45 (m, 8H), 1.25 (s, 3H), 1.20 – 1.12 (m, 2H), 1.05 – 0.96 (m, 2H); ESI-MS calcd for C39H56FN4O5S [M + H]+ = 711.40, found: 711.20.
Methyl ((1S,2R)-2-(2-(azetidin-1-yl)-1-(1-((1-(4-(cyclopropylsulfonyl)phenyl)-3-fluoroazetidin-3-yl)methyl)piperidin-4-yl)-1-(3-fluorophenyl)ethyl)cyclopentyl)carbamate (14).
Compound 14 was prepared from compounds 37, 41a and 34 according to the procedure used to prepare compound 17. 1H NMR (400 MHz, MeOH-d4) δ 7.74 – 7.68 (m, 2H), 7.52 – 7.42 (m, 1H), 7.20 – 7.11 (m, 2H), 7.11 – 7.02 (m, 1H), 6.63 – 6.58 (m, 2H), 4.61 – 4.45 (m, 2H), 4.42 – 4.30 (m, 2H), 4.30 – 4.10 (m, 6H), 3.85 – 3.73 (m, 4H), 3.60 (t, J = 12.1 Hz, 2H), 3.55 – 3.42 (m, 1H), 3.16 (dt, J = 24.0, 12.4 Hz, 2H), 2.87 – 2.73 (m, 1H), 2.63 – 2.51 (m, 2H), 2.51 – 2.39 (m, 1H), 2.14 – 1.95 (m, 5H), 1.95 – 1.85 (m, 1H), 1.85 – 1.74 (m, 1H), 1.73 – 1.48 (m, 5H), 1.20 – 1.13 (m, 2H), 1.05 – 0.98 (m, 2H); ESI-MS calcd for C36H49F2N4O4S [M + H]+ = 671.34, found: 671.26.
Methyl ((1S,2R)-2-(2-(azetidin-1-yl)-1-(1-((1-(4-(cyclopropylsulfonyl)phenyl)-3-methylazetidin-3-yl)methyl)piperidin-4-yl)-1-(3-fluorophenyl)ethyl)cyclopentyl)carbamate (15).
Compound 15 was prepared from compounds 37, 41b and 34 according to the method used to produce compound 17. 1H NMR (400 MHz, MeOH-d4) δ 7.69 – 7.62 (m, 2H), 7.46 (td, J = 8.3, 6.3 Hz, 1H), 7.19 – 7.10 (m, 2H), 7.10 – 7.00 (m, 1H), 6.56 – 6.49 (m, 2H), 4.58 – 4.42 (m, 2H), 4.41 – 4.21 (m, 2H), 4.12 (d, J = 15.6 Hz, 1H), 3.92 – 3.71 (m, 6H), 3.59 – 3.42 (m, 3H), 3.38 (s, 3H), 3.18 – 2.98 (m, 2H), 2.87 – 2.72 (m, 1H), 2.56 (tt, J = 7.9, 4.8 Hz, 2H), 2.49 – 2.35 (m, 1H), 2.10 – 1.93 (m, 5H), 1.93 – 1.81 (m, 1H), 1.81 – 1.72 (m, 1H), 1.72 – 1.52 (m, 5H), 1.49 (s, 3H), 1.18 – 1.12 (m, 2H), 1.03 – 0.97 (m, 2H); ESI-MS calcd for C37H52FN4O4S [M + H]+ = 667.37, found: 667.27.
Methyl ((1S,2R)-2-(2-(azetidin-1-yl)-1-(1-((1-(4-(cyclopropylsulfonyl)phenyl)-3-hydroxy- azetidin-3-yl)methyl)piperidin-4-yl)-1-(3-fluorophenyl)ethyl)cyclopentyl)carbamate (16).
Compound 16 was prepared from compounds 37, 41c and 34 according to the procedure used to prepare compound 17. 1H NMR (400 MHz, MeOH-d4) δ 7.72 – 7.66 (m, 2H), 7.49 (td, J = 8.2, 6.2 Hz, 1H), 7.20 – 7.13 (m, 2H), 7.11 – 7.02 (m, 1H), 6.61 – 6.55 (m, 2H), 4.61 – 4.44 (m, 2H), 4.44 – 4.25 (m, 2H), 4.24 – 4.05 (m, 3H), 3.94 (d, J = 8.8 Hz, 2H), 3.80 (d, J = 15.6 Hz, 1H), 3.62 (t, J = 12.8 Hz, 2H), 3.49 (s, 3H), 3.22 – 3.02 (m, 2H), 2.90 – 2.75 (m, 1H), 2.58 (tt, J = 8.0, 4.8 Hz, 2H), 2.53 – 2.39 (m, 1H), 2.15 – 1.94 (m, 5H), 1.85 (d, J = 24.0 Hz, 3H), 1.69 (d, J = 12.5 Hz, 4H), 1.55 (d, J = 12.7 Hz, 1H), 1.22 – 1.12 (m, 2H), 1.06 – 0.97 (m, 2H); ESI-MS calcd for C36H50FN4O5S [M + H]+ = 669.35, found: 669.11.
Methyl ((1S,2R)-2-(2-(azetidin-1-yl)-1-(1-((1-(4-(cyclopropylsulfonyl)phenyl)-3-methoxy-azetidin-3-yl)methyl)piperidin-4-yl)-1-(3-fluorophenyl)ethyl)cyclopentyl)carbamate (17).
10% Pd/C (344 mg, 10%wt) was added to a solution of compound 37 (1.6 g, 3.24 mmol) in MeOH (50 mL) under an N2 atmosphere. The solution was briefly vacuumed to remove the N2 atmosphere then put under a H2 atmosphere, this was repeated three times. The mixture was then stirred for 2 h at rt under a H2 atmosphere. The Pd/C catalyst was then filtered off, the solvent was removed by rotary evaporation to give compound 38 which was not purified further. 1H NMR (400 MHz, MeOH-d4) δ 7.48–7.43 (m, 1H), 7.16–7.06 (m, 3H), 4.51–4.45 (m, 2H), 4.38–4.27 (m, 2H), 4.10 (d, J = 15.6 Hz, 1H), 3.77 (d, J = 15.2 Hz, 1H), 3.55–3.52 (m, 1H), 3.40–3.33 (m, 2H), 3.31 (s, 3H), 3.01–2.89 (m, 2H), 2.78–2.72 (m, 1H), 2.58–2.48 (m, 1H), 2.46–2.39 (m, 1H), 2.05–1.93 (m, 5H), 1.78–1.70 (m, 1H), 1.68–1.54 (m, 3H), 1.39–1.30 (m, 1H), 1.08–1.02 (m, 1H); ESI-MS calculated for C23H34FN3O2 [M + H]+ = 404.26, found: 404.42.
Compound 38 (200 mg, 0.496 mmol), K2CO3 (206 mg, 1.49 mmol), and KI (8 mg, 0.0496 mmol) were added to a solution of the compound 41d (176 mg, 0.595 mmol) in MeCN (4 mL). The mixture was refluxed overnight. Then, the mixture was filtered through celite, concentrated, and purified with preparative HPLC to give 39d (155 mg, 52%) as a white solid.
Compound 39d (155 mg, 0.257 mmol) was dissolved in DCM (4 mL), then TFA (380 μL, 5.14 mmol) was added. After stirring for 2 h at rt, the reaction mixture was then evaporated. The crude product was dissolved in DMSO (3 mL). Compound 34 (62 mg, 0.308 mmol) and K2CO3 (142 mg, 1.03 mmol) were added and the mixture was stirred at 80 °C overnight. The mixture was quenched with H2O and purified by reverse phase preparative HPLC to give compound 17 (112 mg, 64%). 1H NMR (400 MHz, MeOH-d4) δ 7.69 – 7.62 (m, 2H), 7.48 – 7.39 (m, 1H), 7.16 – 7.08 (m, 2H), 7.04 (d, J = 8.0 Hz, 1H), 6.59 – 6.53 (m, 2H), 4.57 – 4.39 (m, 2H), 4.38 – 4.20 (m, 4H), 4.11 (d, J = 15.6 Hz, 1H), 3.96 – 3.87 (m, 2H), 3.75 (d, J = 15.6 Hz, 1H), 3.61 – 3.50 (m, 4H), 3.50 – 3.39 (m, 5H), 3.15 – 2.97 (m, 2H), 2.84 – 2.70 (m, 1H), 2.59 – 2.46 (m, 2H), 2.46 – 2.33 (m, 1H), 2.10 – 1.90 (m, 5H), 1.90 – 1.71 (m, 3H), 1.71 – 1.57 (m, 4H), 1.56 – 1.44 (m, 1H), 1.18 – 1.08 (m, 2H), 1.03 – 0.93 (m, 2H); ESI-MS calculated for C37H52FN4O5S [M + H]+ = 683.36, found: 683.16.
Methyl 3-((4-(2-(azetidin-1-yl)-1-(3-fluorophenyl)-1-((1R,2S)-2-((methoxycarbonyl)amino)-cyclopentyl)ethyl)piperidin-1-yl)methyl)-1-(4-(cyclopropylsulfonyl)phenyl)azetidine-3-car-boxylate (18).
Compound 18 was prepared from compounds 37, 41e and 34 according to the procedure used to prepare compound 17. 1H NMR (400 MHz, MeOH-d4) δ 7.73 – 7.67 (m, 2H), 7.52 – 7.41 (m, 1H), 7.20 – 7.10 (m, 2H), 7.06 (d, J = 7.9 Hz, 1H), 6.62 – 6.53 (m, 2H), 4.60 – 4.43 (m, 2H), 4.41 – 4.32 (m, 2H), 4.28 (dd, J = 8.3, 2.3 Hz, 2H), 4.13 (d, J = 15.6 Hz, 1H), 4.01 (dd, J = 8.4, 6.5 Hz, 2H), 3.88 – 3.79 (m, 4H), 3.79 – 3.72 (m, 3H), 3.69 – 3.60 (m, 2H), 3.55 – 3.40 (m, 1H), 3.25 – 3.08 (m, 2H), 2.83 – 2.72 (m, 1H), 2.58 (tt, J = 8.0, 4.8 Hz, 2H), 2.49 – 2.38 (m, 1H), 2.12 – 1.94 (m, 5H), 1.89 (s, 1H), 1.84 – 1.72 (m, 1H), 1.72 – 1.59 (m, 4H), 1.59 – 1.42 (m, 1H), 1.21 – 1.11 (m, 2H), 1.06 – 0.97 (m, 2H). ESI-MS calcd for C38H52FN4O6S [M + H]+ = 711.36, found: 711.15.
Methyl ((1S,2R)-2-(2-(azetidin-1-yl)-1-(1-((1-(4-((1-((E)-4-(dimethylamino)but-2-enoyl)-azetidin-3-yl)sulfonyl)phenyl)-3-methoxyazetidin-3-yl)methyl)piperidin-4-yl)-1-(3-fluorophenyl)ethyl)cyclopentyl)carbamate (19).
Compound 19 was prepared from compounds 39d, 42a and (2E)-4-(dimethylamino)-2-butenoyl chloride hydrochloride instead of acrylic anhydride according to the method used to produce compound 24. 1H NMR (400 MHz, MeOH-d4) δ 7.70 – 7.64 (m, 2H), 7.44 – 7.36 (m, 1H), 7.12 – 7.04 (m, 2H), 7.00 (d, J = 8.0 Hz, 1H), 6.68 (dt, J = 15.3, 7.1 Hz, 1H), 6.58 – 6.52 (m, 2H), 6.41 (dt, J = 15.2, 1.3 Hz, 1H), 4.53 – 4.37 (m, 4H), 4.35 – 4.20 (m, 6H), 4.21 – 4.02 (m, 4H), 3.96 – 3.85 (m, 4H), 3.76 – 3.67 (m, 1H), 3.49 (s, 4H), 3.46 – 3.38 (m, 5H), 3.11 – 2.95 (m, 2H), 2.84 (s, 6H), 2.73 (d, J = 9.2 Hz, 1H), 2.48 (dt, J = 19.0, 9.5 Hz, 1H), 2.42 – 2.30 (m, 1H), 2.06 – 1.87 (m, 5H), 1.87 – 1.76 (m, 1H), 1.76 – 1.67 (m, 1H), 1.67 – 1.54 (m, 3H), 1.54 – 1.40 (m, 1H), 1.20 – 1.00 (m, 1H); ESI-MS calcd for C43H62FN6O6S [M + H]+ = 809.44, found: 809.28.
Methyl ((1S,2R)-2-(1-(1-((1-(4-((1-acryloylazetidin-3-yl)sulfonyl)phenyl)-3-methoxyazetidin-3-yl)methyl)piperidin-4-yl)-2-(azetidin-1-yl)-1-(3-fluorophenyl)ethyl)cyclopentyl)carbamate (20).
Compound 20 was prepared from compounds 39d and 42a according to the procedure used to produce compound 24. 1H NMR (400 MHz, MeOH-d4) δ 7.79 – 7.73 (m, 2H), 7.54 – 7.43 (m, 1H), 7.21 – 7.13 (m, 2H), 7.08 (d, J = 8.0 Hz, 1H), 6.32 (dd, J = 17.0, 9.5 Hz, 1H), 6.25 (dd, J = 17.0, 2.7 Hz, 1H), 5.77 (dd, J = 9.5, 2.7 Hz, 1H), 4.61 – 4.44 (m, 4H), 4.42 – 4.34 (m, 1H), 4.34 – 4.23 (m, 4H), 4.23 – 4.10 (m, 3H), 3.98 (dd, J = 9.7, 5.8 Hz, 2H), 3.79 (d, J = 15.6 Hz, 1H), 3.57 (s, 4H), 3.54 – 3.42 (m, 5H), 3.19 – 3.01 (m, 2H), 2.87 – 2.75 (m, 1H), 2.62 – 2.51 (m, 1H), 2.51 – 2.39 (m, 1H), 2.13 – 1.95 (m, 4H), 1.93 – 1.75 (m, 2H), 1.75 – 1.61 (m, 4H), 1.61 – 1.46 (m, 1H), 1.27 – 1.04 (m, 1H); ESI-MS calcd for C40H55FN5O6S [M + H]+ = 752.39, found: 752.21.
Methyl ((1S,2R)-2-(1-(1-((1-(4-((1-acryloylpiperidin-4-yl)sulfonyl)phenyl)-3-methoxyazetidin-3-yl)methyl)piperidin-4-yl)-2-(azetidin-1-yl)-1-(3-fluorophenyl)ethyl)cyclopentyl)carbamate (21).
Compound 21 was prepared from compounds 39d and 42b according to the procedure used to produce compound 24. 1H NMR (400 MHz, MeOH-d4 ) δ 7.65 – 7.59 (m, 2H), 7.43 (td, J = 8.3, 6.2 Hz, 1H), 7.17 – 7.07 (m, 2H), 7.02 (d, J = 8.0 Hz, 1H), 6.69 (dd, J = 16.8, 10.7 Hz, 1H), 6.60 – 6.53 (m, 2H), 6.13 (dd, J = 16.8, 1.9 Hz, 1H), 5.69 (dd, J = 10.7, 1.9 Hz, 1H), 4.58 (d, J = 13.5 Hz, 1H), 4.47 (s, 2H), 4.38 – 4.22 (m, 5H), 4.22 – 4.04 (m, 2H), 3.92 (dd, J = 9.7, 5.9 Hz, 2H), 3.74 (d, J = 15.5 Hz, 1H), 3.60 – 3.49 (m, 4H), 3.45 (s, 5H), 3.37 – 3.31 (m, 1H), 3.16 – 2.95 (m, 3H), 2.83 – 2.72 (m, 1H), 2.72 – 2.61 (m, 1H), 2.57 – 2.46 (m, 1H), 2.45 – 2.29 (m, 1H), 2.11 – 1.89 (m, 6H), 1.88 – 1.79 (m, 1H), 1.78 – 1.70 (m, 1H), 1.70 – 1.54 (m, 4H), 1.54 – 1.35 (m, 3H), 1.13 (s, 1H); ESI-MS calcd for C42H59FN5O6S [M + H]+ = 780.42, found:780.33.
Methyl ((1S,2R)-2-(1-(1-((1-(4-(((S)-1-acryloylpiperidin-3-yl)sulfonyl)phenyl)-3-methoxy-azetidin-3-yl)methyl)piperidin-4-yl)-2-(azetidin-1-yl)-1-(3-fluorophenyl)ethyl)-cyclopentyl)carbamate (22).
Compound 22 was prepared from compounds 39d and 42c according to the procedure used to produce compound 24. 1H NMR (400 MHz, MeOH-d4) δ 7.73 – 7.67 (m, 2H), 7.53 – 7.43 (m, 1H), 7.21 – 7.13 (m, 2H), 7.08 (d, J = 8.0 Hz, 1H), 6.73 (dd, J = 16.8, 10.7 Hz, 1H), 6.66 – 6.59 (m, 2H), 6.16 (d, J = 16.8 Hz, 1H), 5.79 – 5.69 (m, 1H), 4.64 (d, J = 12.8 Hz, 1H), 4.53 (s, 2H), 4.44 – 4.22 (m, 5H), 4.16 (d, J = 15.6 Hz, 1H), 3.98 (dd, J = 9.6, 6.7 Hz, 3H), 3.80 (d, J = 15.6 Hz, 1H), 3.65 – 3.54 (m, 4H), 3.51 (s, 5H), 3.19 – 3.02 (m, 3H), 2.96 (t, J = 11.7 Hz, 1H), 2.91 – 2.76 (m, 1H), 2.64 – 2.50 (m, 1H), 2.50 – 2.38 (m, 1H), 2.22 – 2.10 (m, 1H), 2.10 – 1.96 (m, 5H), 1.96 – 1.85 (m, 2H), 1.85 – 1.75 (m, 2H), 1.75 – 1.59 (m, 4H), 1.59 – 1.42 (m, 2H), 1.17 (s, 1H); ESI-MS calcd for C42H59FN5O6S [M + H]+ = 780.42, found: 780.23.
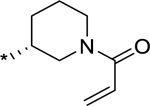
Methyl ((1S,2R)-2-(1-(1-((1-(4-(((R)-1-acryloylpiperidin-3-yl)sulfonyl)phenyl)-3-methoxy-azetidin-3-yl)methyl)piperidin-4-yl)-2-(azetidin-1-yl)-1-(3-fluorophenyl)ethyl)cyclopentyl)-carbamate (23).
Compound 23 was prepared from compounds 39d and 42d according to the procedure used to produce compound 24. 1H NMR (400 MHz, MeOH-d4) δ 7.74 – 7.66 (m, 2H), 7.49 (td, J = 8.3, 6.4 Hz, 1H), 7.21 – 7.13 (m, 2H), 7.08 (d, J = 8.0 Hz, 1H), 6.73 (dd, J = 16.9, 10.8 Hz, 1H), 6.67 – 6.59 (m, 2H), 6.16 (d, J = 16.7 Hz, 1H), 5.79 – 5.70 (m, 1H), 4.64 (d, J = 12.8 Hz, 1H), 4.53 (s, 2H), 4.44 – 4.21 (m, 5H), 4.16 (d, J = 15.6 Hz, 1H), 3.98 (dd, J = 9.7, 6.6 Hz, 3H), 3.80 (d, J = 15.6 Hz, 1H), 3.57 (s, 4H), 3.52 (s, 5H), 3.19 – 3.02 (m, 3H), 2.96 (t, J = 11.6 Hz, 1H), 2.89 – 2.75 (m, 1H), 2.65 – 2.51 (m, 1H), 2.51 – 2.40 (m, 1H), 2.24 – 2.11 (m, 1H), 2.11 – 1.98 (m, 5H), 1.97 – 1.75 (m, 4H), 1.75 – 1.59 (m, 4H), 1.59 – 1.43 (m, 2H), 1.17 (s, 1H); ESI-MS calcd for C42H59FN5O6S [M + H]+ = 780.42, found: 780.19.
Methyl ((1S,2R)-2-(1-(1-((1-(4-(((1S,4S)-5-acryloyl-2,5-diazabicyclo[2.2.1]heptan-2-yl)-sulfonyl)phenyl)-3-methoxyazetidin-3-yl)methyl)piperidin-4-yl)-2-(azetidin-1-yl)-1-(3-fluoro-phenyl)ethyl)cyclopentyl)carbamate (24).
Compound 39d (2.20 g, 3.48 mmol) was dissolved in DCM (50 mL), then trifluoroacetic acid (5.0 mL, 73.1 mmol) was added. After stirring for 2 h at rt, the reaction mixture was evaporated. The crude product was dissolved in DMSO (30 mL), compound 42e (1.49 g, 4.18 mmol) and K2CO3 (1.92 g, 13.9 mmol) were added. The mixture was stirred at 80 °C overnight. The mixture was quenched with H2O and purified by reverse phase preparative HPLC to give 40e (1.66 g, 59%).
Compound 40e (1.28 g, 1.50 mmol) was dissolved in DCM (25 mL), then TFA (2.2 mL, 30.0 mmol) was added slowly. After stirring for 2 h at rt, the reaction mixture was evaporated to give the crude product without further purification. Acrylic anhydride (206 μL, 1.80 mmol) and Et3N (623 μL, 4.50 mmol) were added to a solution of this crude product in DCM (20 mL). After stirring for 3 h at rt, the reaction mixture was evaporated and the residue was purified by reverse phase preparative HPLC to give 24 (927 mg, 78%). 1H NMR (400 MHz, MeOH-d4) δ 7.69 – 7.62 (m, 2H), 7.45 (q, J = 7.6 Hz, 1H), 7.18 – 7.08 (m, 2H), 7.07 – 6.98 (m, 1H), 6.66 – 6.59 (m, 0.5H), 6.59 – 6.52 (m, 2H), 6.32 (dd, J = 16.8, 10.0 Hz, 0.5H), 6.23 (ddd, J = 16.8, 2.1, 1.2 Hz, 1H), 5.72 (ddd, J = 10.0, 3.4, 2.1 Hz, 1H), 4.73 (s, 1H), 4.58 – 4.41 (m, 2H), 4.40 – 4.29 (m, 2H), 4.29 – 4.21 (m, 2H), 4.13 (d, J = 15.6 Hz, 1H), 3.91 (t, J = 8.7 Hz, 2H), 3.76 (d, J = 15.6 Hz, 1H), 3.63 – 3.54 (m, 2H), 3.54 – 3.50 (m, 3H), 3.50 – 3.45 (m, 4H), 3.44 – 3.38 (m, 1H), 3.34 (dd, J = 11.6, 2.2 Hz, 0.5H), 3.27 (s, 3H), 3.18 (dd, J = 9.6, 2.1 Hz, 0.5H), 3.14 – 2.97 (m, 2H), 2.85 – 2.71 (m, 1H), 2.61 – 2.47 (m, 1H), 2.47 – 2.34 (m, 1H), 2.13 – 1.90 (m, 4H), 1.89 – 1.72 (m, 4H), 1.72 – 1.57 (m, 4H), 1.49 (d, J = 12.8 Hz, 1H), 1.41 – 1.23 (m, 1H), 1.18 – 0.98 (m, 1H); 13C NMR (101 MHz, MeOH-d4) δ 165.14, 162.70, 159.82, 154.38 (d, J = 4.4 Hz), 131.20 (d, J = 8.4 Hz), 130.26 (d, J = 6.2 Hz), 129.25, 129.10, 129.05, 128.76, 126.37 (d, J = 3.9 Hz), 125.56, 117.05 (d, J = 23.1 Hz), 115.61 (d, J = 21.2 Hz), 112.28, 74.16, 61.38, 60.53, 59.72, 58.61 (d, J = 6.7 Hz), 57.53, 56.72, 55.74, 55.06, 54.84, 52.96, 51.73, 50.94, 41.32, 37.54, 35.90, 33.72, 26.90, 26.20, 25.77, 21.21, 17.08; ESI-MS calcd for C42H58FN6O6S [M + H]+ = 793.41, found: 793.16.
Methyl ((1S,2R)-2-(1-(1-((1-(4-(((1R,4R)-5-acryloyl-2,5-diazabicyclo[2.2.1]heptan-2-yl)-sulfonyl)phenyl)-3-methoxyazetidin-3-yl)methyl)piperidin-4-yl)-2-(azetidin-1-yl)-1-(3-fluoro-phenyl)ethyl)cyclopentyl)carbamate (25).
Compound 25 was prepared from compounds 39d and 42f according to the procedure used to produce compound 24. 1H NMR (400 MHz, MeOH-d4) δ 7.72 – 7.67 (m, 2H), 7.49 (q, J = 7.7 Hz, 1H), 7.21 – 7.12 (m, 2H), 7.07 (d, J = 8.0 Hz, 1H), 6.69 – 6.62 (m, 0.5H), 6.62 – 6.57 (m, 2H),, 6.36 (dd, J = 16.8, 10.0 Hz, 0.5H), 6.30 – 6.23 (m, 1H), 5.75 (ddd, J = 10.0, 3.8, 2.1 Hz, 1H), 4.77 (s, 1H), 4.53 (s, 2H), 4.36 (s, 2H), 4.32 – 4.24 (m, 2H), 4.16 (d, J = 15.7 Hz, 1H), 3.95 (dd, J = 9.5, 4.8 Hz, 2H), 3.84 – 3.75 (m, 1H), 3.67 – 3.57 (m, 2H), 3.57 – 3.54 (m, 3H), 3.54 – 3.49 (m, 4H), 3.48 – 3.42 (m, 1H), 3.38 (dd, J = 11.7, 2.1 Hz, 0.5H), 3.31 – 3.27 (m, 3H), 3.22 (dd, J = 9.5, 2.1 Hz, 0.5H), 3.18 – 3.00 (m, 2H), 2.81 (d, J = 9.5 Hz, 1H), 2.62 – 2.38 (m, 2H), 2.13 – 1.95 (m, 4H), 1.93 – 1.76 (m, 3H), 1.76 – 1.61 (m, 4H), 1.54 (d, J = 12.8 Hz, 1H), 1.46 – 1.35 (m, 1H), 1.35 – 1.27 (m, 1H), 1.15 (s, 1H); ESI-MS calcd for C42H58FN6O6S [M + H]+ = 793.41, found: 793.12.
Methyl ((1S,2R)-2-(2-(azetidin-1-yl)-1-(3-fluorophenyl)-1-(1-((3-methoxy-1-(4-(((1S,4S)-5-propionyl-2,5-diazabicyclo[2.2.1]heptan-2-yl)sulfonyl)phenyl)azetidin-3-yl)methyl)piperidin-4-yl)ethyl)cyclopentyl)carbamate (26).
Compound 26 was prepared from compounds 39d, 42e and propionic anhydride instead of acrylic anhydride according to the procedure used to produce compound 24. 1H NMR (400 MHz, MeOH-d4) δ 7.72 – 7.66 (m, 2H), 7. 53 – 7.44 (m, 1H), 7.20 – 7.12 (m, 2H), 7.08 (d, J = 8.0 Hz, 1H), 6.64 – 6.55 (m, 2H), 4.74 – 4.43 (m, 4H), 4.43 – 4.31 (m, 2H), 4.31 – 4.24 (m, 2H), 4.21 – 4.10 (m, 1H), 3.96 (t, J = 8.5 Hz, 2H), 3.84 – 3.75 (m, 1H), 3.56 (s, 3H), 3.54 – 3.44 (m, 6H), 3.42 – 3.35 (m, 0.5H), 3.30 – 3.25 (m, 2H), 3.19 (dd, J = 9.5, 2.0 Hz, 0.5H), 3.17 – 3.02 (m, 2H), 2.81 (d, J = 9.4 Hz, 1H), 2.61 – 2.49 (m, 1H), 2.50 – 2.40 (m, 1H), 2.40 – 2.29 (m, 1H), 2.22 – 2.08 (m, 2H), 2.08 – 1.95 (m, 5H), 1.94 – 1.84 (m, 1H), 1.84 – 1.73 (m, 2H), 1.73 – 1.59 (m, 4H), 1.59 – 1.48 (m, 1H), 1.37 – 1.26 (m, 1H), 1.11 (t, J = 7.5 Hz, 2H), 1.05 (t, J = 7.5 Hz, 2H); ESI-MS calcd for C42H60FN6O6S [M + H]+ = 795.43, found: 827.32.
Methyl ((1S,2R)-2-((S)-(1-benzylpiperidin-4-yl)(cyano)(3-fluorophenyl)methyl)cyclopentyl)-carbamate (28).
Compound 27 (2.28 g, 4.64 mmol) was dissolved in dichloromethane (14 mL) and TFA (6.9 mL, 92.9 mmol) was added slowly. After stirring for 2 h at rt, the reaction mixture was concentrated under vacuum. The crude product was dissolved in dry dichloromethane (50 mL). Then, Et3N (5.2 mL, 37.1 mmol) and dimethyl dicarbonate (1.24 mg, 9.28 mmol) were added at 0 °C. After stirring for 2 h at rt, the reaction mixture was concentrated under vacuum. The residue was purified column chromatography (silica gel, DCM/MeOH 20:1 to 10:1) to give 28 (1.86 g, 89%). 1H NMR (400 MHz, MeOH-d4) δ 7.43 (td, J = 8.0, 6.0 Hz, 1H), 7.38 – 7.23 (m, 7H), 7.11 (tdd, J = 8.3, 2.5, 1.0 Hz, 1H), 3.88 (q, J = 7.0 Hz, 1H), 3.49 (s, 2H), 3.46 (s, 3H), 2.99 – 2.89 (m, 2H), 2.84 (td, J = 8.3, 6.9 Hz, 1H), 2.15 – 2.04 (m, 2H), 2.04 – 1.94 (m, 3H), 1.88 – 1.76 (m, 1H), 1.72 – 1.60 (m, 3H), 1.60 – 1.48 (m, 2H), 1.43 – 1.34 (m, 1H), 1.26 (td, J = 12.7, 4.0 Hz, 1H); ESI-MS calcd for C27H33FN3O2 [M + H]+ = 450.26, found: 450.08.
Methyl ((1S,2R)-2-((S)-2-amino-1-(1-benzylpiperidin-4-yl)-1-(3-fluorophenyl)ethyl)cyclo-pentyl)carbamate (29).
Compound 28 (7.62 g, 16.9 mmol) was added to a dry RB-flask then covered with a kimwipe and put in a desiccator that was put under vacuum for 1–2 days. After the vacuuming step, the flask was removed from the desiccator and quickly capped with a septum and the system was vacuumed under an N2 atmosphere. The anhydrous toluene (150 ml) was added to the flask, then was cooled to 0 °C in the ice-bath. Diisobutylaluminiumhydride (25% in toluene, 45.2 mL, 67.8 mmol) was injected into the reaction mixture slowly at 0 °C with a syringe with stirring. After 4 h, the reaction was quenched with potassium sodium tartrate(aq). The mixture was stirred vigorously overnight and was extracted with EtOAc three times. The organic solvent was washed with brine and dried with Na2SO4, filtered, and concentrated under rotatory vacuum. The residue was redissolved in MeOH (200 mL), NaBH4 (2.56 g, 67.8 mmol) was added slowly at 0 °C, the reaction mixture was stirred at rt, overnight. The reaction mixture was concentrated and diluted with water and extracted with DCM three times. The organic solvent was washed with brine, dried with Na2SO4, filter, and concentrated under rotatory vacuum to give crude product 29 (6.52 g, 85%) with was used without further purification. 1H NMR (400 MHz, MeOH-d4) δ 7.37 – 7.24 (m, 6H), 7.08 – 7.00 (m, 2H), 6.99 – 6.92 (m, 1H), 3.64 – 3.51 (m, 2H), 3.39 – 3.25 (m, 2H), 3.23 (s, 3H), 3.08 (s, 2H), 2.93 – 2.81 (m, 1H), 2.81 – 2.72 (m, 1H), 2.60 (q, J = 9.4 Hz, 1H), 2.00 – 1.92 (m, 1H), 1.90 (s, 2H), 1.89 – 1.68 (m, 4H), 1.64 – 1.52 (m, 2H), 1.52 – 1.41 (m, 2H), 1.40 – 1.30 (m, 1H); ESI-MS calcd for C27H37FN3O2 [M + H]+ = 454.29, found: 454.24.
Methyl ((1S,2R)-2-((S)-1-(1-benzylpiperidin-4-yl)-2-((tert-butoxycarbonyl)amino)-1-(3-fluoro-phenyl)ethyl)cyclopentyl)carbamate (30).
Compound 29 (2.68 g, 5.91 mmol) was dissolved in dry dichloromethane (50 mL). Then, Et3N (1.2 mL, 8.87 mmol) and di-tert-butyl dicarbonate (2.71 mL, 11.8 mmol) were added. After stirring for 2 h at rt, the reaction mixture was concentrated under vacuum. The residue was purified column chromatography (silica gel, DCM/MeOH 20:1 to 8:1) to give 30 (2.30 g, 70%). 1H NMR (400 MHz, MeOH-d4) δ 7.42 – 7.18 (m, 8H), 6.99 (td, J = 8.1, 2.3 Hz, 1H), 4.10 (q, J = 6.8 Hz, 1H), 3.84 – 3.73 (m, 1H), 3.70 – 3.60 (m, 1H), 3.54 (s, 3H), 3.49 (s, 2H), 3.00 – 2.85 (m, 2H), 2.45 (q, J = 8.4 Hz, 1H), 2.05 – 1.92 (m, 3H), 1.92 – 1.78 (m, 2H), 1.67 – 1.51 (m, 4H), 1.47 (s, 9H), 1.42 – 1.30 (m, 3H), 1.25 – 1.15 (m, 1H); ESI-MS calcd for C32H45FN3O4 [M + H]+ = 554.34, found: 554.09.
tert-butyl (2-(1-((1-(4-(cyclopropylsulfonyl)phenyl)azetidin-3-yl)methyl)piperidin-4-yl)-2-(3-fluorophenyl)-2-((1R,2S)-2-((methoxycarbonyl)amino)cyclopentyl)ethyl)carbamate (31).
Palladium on carbon (150 mg, 10% wt) was added to a solution of compound 30 (500 mg, 0.903 mmol) in MeOH (10 mL) under an N2 atmosphere. The solution was briefly vacuumed to remove the N2 atmosphere then put under a H2 atmosphere, this was repeated three times. The mixture was then stirred for 6h at rt under a H2 atmosphere. The Pd/C catalyst was then filtered off, the solvent was removed by rotary evaporation. The residue was then redissolved in acetonitrile (5 mL). K2CO3 (374 mg, 2.71 mmol) and KI (15 mg, 0.0903 mmol) and 36 (374 mg, 1.08 mmol) were added to the solution. The mixture was stirred at 80 °C overnight. Then, the mixture was concentrated in vacuum and purified by reverse phase preparative HPLC to give 31 (399 mg, 62%). ESI-MS calcd for C38H54FN4O6S [M + H]+ = 713.37, found: 713.15.
Cyclopropyl(4-fluorophenyl)sulfane (33).
To a stirred solution of 32 (2.0 mL, 18.7 mmol) in DMSO (50 mL) under an N2 atmosphere were added cyclopropyl bromide (1.6 mL, 20.6 mmol) and t-BuONa (4.49 g, 46.8 mmol). Then the reaction mixture was heated at 80° C for 24 h. After cooling to rt, the mixture was poured into H2O (250 mL) and extracted with Et2O three times. The combined organic phases were washed with brine, dried over Na2SO4 and concentrated in vacuum. The residue was purified column chromatography (silica gel, hexane/EtOAc 100:1 to 15:1) to give 33 (1.32 g, 42%). Spectral data was identical to literature compound35.
1-(cyclopropylsulfonyl)-4-fluorobenzene (34).
m-CPBA (4.28 g, 17.4 mmol, 70%) was added to a stirred solution of 33 (1.46 g, 8.68 mmol) in DCM (80 mL) at 0 °C. After 2h, the reaction mixture was quenched with 1M NaOH (aq.), extracted with DCM three times. The combined organic phases were washed with brine, dried over Na2SO4 and concentrated in vacuum. The residue was purified by column chromatography (silica gel, hexane/EtOAc 10:1 to 1:1) to give 34 (1.58 g, 91%). 1H NMR (400 MHz, CDCl3) δ 7.97 – 7.91 (m, 2H), 7.28 – 7.22 (m, 2H), 2.47 (tt, J = 8.0, 4.8 Hz, 1H), 1.40 – 1.34 (m, 2H), 1.11 – 1.03 (m, 2H).
Methyl 1-(4-(cyclopropylsulfonyl)phenyl)azetidine-3-carboxylate (35).
Compound 34 (819 mg, 4.09 mmol) was dissolved in DMSO (20 mL), methyl azetidine-3-carboxylate hydrochloride (620 mg, 4.09 mmol) and K2CO3 (2.26 g, 16.4 mmol) were added. The mixture was quenched with water, extracted with EA three times. The combined organic phases were washed with brine, dried over Na2SO4 and concentrated in vacuum. The residue was purified column chromatography (silica gel, hexane/EtOAc 2:1 to 1:2) to give 35 (729 mg, 60%). 1H NMR (400 MHz, CDCl3) δ 7.71 – 7.66 (m, 2H), 6.46 – 6.41 (m, 2H), 4.21 – 4.10 (m, 4H), 3.76 (s, 3H), 3.60 (tt, J = 8.6, 6.0 Hz, 1H), 2.40 (tt, J = 8.0, 4.8 Hz, 1H), 1.32 – 1.22 (m, 2H), 1.01 – 0.90 (m, 2H); ESI-MS calcd for C14H18NO4S [M + H]+ = 296.10, found: 259.82.
(1-(4-(Cyclopropylsulfonyl)phenyl)azetidin-3-yl)methyl methanesulfonate (36).
Compound 35 (729 mg, 2.47 mmol) was dissolved in THF (30 mL) and LiAlH4 (4.9 mL, 1 M in THF) was added at 0 °C. After 4 h, the reaction was quenched with potassium sodium tartrate (aq). The mixture was stirred vigorously overnight and was extracted with EtOAc three times. The organic solvent was washed with brine and dried with Na2SO4, filtered, and concentrated under rotatory vacuum. The residue was redissolved in DCM (25 mL), Et3N (1.0 mL, 7.41 mmol) and methanesulfonyl chloride (230 μL, 2.96 mmol) were added. After stirring under rt for 1h, the reaction was quenched with water, extracted with DCM three times. The combined organic phases were washed with brine, dried over Na2SO4 and concentrated in vacuum. The residue was purified by column chromatography (silica gel, DCM/MeOH 20:1 to 15:1) to give 36 (699 mg, 82%). 1H NMR (400 MHz, CDCl3) δ 7.73 – 7.67 (m, 2H), 6.46 – 6.40 (m, 2H), 4.46 (d, J = 6.7 Hz, 2H), 4.10 (t, J = 8.1 Hz, 2H), 3.81 (dd, J = 8.0, 5.0 Hz, 2H), 3.25 – 3.13 (m, 1H), 3.06 (s, 3H), 2.41 (tt, J = 8.0, 4.8 Hz, 1H), 1.32 – 1.25 (m, 2H), 1.00 – 0.93 (m, 2H); ESI-MS calcd for C14H20NO5S2 [M + H]+ = 346.08, found: 345.92.
tert-Butyl (1S,4S)-5-((4-fluorophenyl)sulfonyl)-2,5-diazabicyclo[2.2.1]heptane-2-carboxylate (42e).
(1S,4S)-2-Boc-2,5-diazabicyclo[2.2.1]heptane (1.0 g, 5.05 mmol) was added to a solution of 4-fluorobenzenesulfonyl chloride (1.08 g, 5.55 mmol) and Et3N (2.1 mL, 15.2 mmol) in 40 mL of DCM. After 5 h, H2O was added and the reaction was extracted three times with DCM, concentrated and purified by column (silica gel, hexane/EtOAc 3:1 to 1:2) to give 42e as white solid. 1H NMR (400 MHz, CDCl3) δ 7.93 – 7.80 (m, 2H), 7.21 (td, J = 8.7, 2.3 Hz, 2H), 4.50 – 4.30 (m, 2H), 3.40 (ddd, J = 23.1, 14.1, 9.2 Hz, 2H), 3.29 – 3.13 (m, 2H), 1.71 (dd, J = 15.1, 10.1 Hz, 1H), 1.41 (d, J = 12.2 Hz, 9H), 1.33 (t, J = 8.0 Hz, 1H). ESI-MS calcd for C16H22FN2O4S [M + H]+ = 357.13, found: 357.01.
Fluorescence Polarization (FP)-Based Binding Assay.
Compound binding was measured using an FP assay as described previously32. Briefly, 5 μL of compounds at various concentrations in dimethyl sulfoxide (DMSO) solution was added to 195 μL of a mixture of menin and the fluorescein-labeled tracer (compound 37 in our previous publication32) in the assay buffer (phosphate-buffered saline, 100 μ g/mL bovine γ -globulin, with 0.01% Triton X-100), and the mixture was incubated for 1 h at rt. Final concentrations of the menin protein and the fluorescein-labeled tracer were 4 and 2 nM, respectively. FP values were measured using an In finite M-1000 plate reader (Tecan, Morrisville, NC). The IC50 values were determined by nonlinear regression fitting of the sigmoidal dose-dependent FP decreases as a function of total compound concentrations using the GraphPad Prism 5.0 software.
Cell Growth Evaluation.
MLLr and MLL wild-type leukemia cell lines were plated in 96-well tissue culture plates in triplicates. Compounds were added in a 9-point dose response curve starting from 10 μM top concentration with 1:3 serial dilutions. Compounds were diluted in DMSO to a final concentration of 0.1% DMSO in media. One column on each plate was designated for the 0.1% DMSO vehicle control. Total cellular ATP levels were measured using CellTiter-Glo (Promega) reagent on the day of cell plating (day 0 readout). The cells were then incubated at 37°C in RPMI 1640 media (Gibco) and 5% CO2 for 4 days, total cellular ATP levels were measured again using the CellTiter-Glo® reagent. ATP standard curves were generated on both day 0 and day 4. IC50 was calculated using the GraphPad Prism data analysis software. The catalog numbers for each cell lines are MV4;11 (CRL-9591, ATCC), OCI-AML4 (ACC-729, DSMZ), MOLM-13 (C0003003, AddexBio), SEM (ACC-546, DSMZ), KOPN8 (ACC-552, DSMZ), RS4;11(CRL-1873, ATCC), HL-60 (CCL-240, ATCC), K562 (CCL-243, ATCC) and MEG-1 (CRL-2021, ATCC).
Crystallization and Structure determination.
Menin (residues 2–610 containing a deletion from 460–519) was purified as previously described 30. For crystallization, Menin (25 mg/mL in 25 mM Tris 8.0, 150 mM NaCl and 5mM DTT) was mixed with M-1121 in a protein to compound ratio of 1:1.2 then immediately setup for crystallization at 4 °C. All crystals grew in drops containing 1 0075L of complex and 1 uL of well solution (1.96 M NaCl, 89 mM Bis-Tris pH 6.8, 0.178 M MgCl2 and 10.7 mM Pr Acetate). Crystals were cryoprotected by progressively soaking crystals in well solution with increasingly higher amounts of sodium formate (1 M - 5 M in 1M steps). Diffraction data were collected on a Eiger 9M detector at the Advanced Photon Source LS-CAT 21-ID-D beamline at Argonne National Laboratory. All data were processed with HKL2000 38. The structure of the menin-1121 was solved by molecular replacement (Molrep 39) using the protein structure from PDB ID 6WNH as the search model. The structure went through iterative rounds of electron density fitting and structural refinement using Coot 40 and Buster 41, respectively. The coordinates and restraint files for the ligands were created from smiles in Grade 42 with the mogul+qm option. The initial Fo-Fc electron density map showed the presence of one compound covalently bound to C329 (Figure S1). The following regions were disordered in the structures: 71–73, 386–401, 528–547, 582–610 in menin:M-1121. Data collection and structural refinement statistics are shown in Table S1.
Mass Spectroscopic Analysis of the Human Menin Protein Incubated with Menin Inhibitors.
Samples of menin (25 mg/mL in 25 mM Tris 8.0, 150 mM NaCl, and 5 mM DTT) were incubated with compounds in a protein-to-compound molar ratio of 1:1.2 for 1 h or overnight at 4 °C. Following incubation, the sample was diluted to 1 mg/mL with H2O. Each sample (0.1 mL) was subjected to a reverse-phase HPLC column (Phenomenex Aeris widepore C4 column 3.6 μM, 50 mm × 2.10 mm) at a flow rate of 0.5 mL/min in H2O with 0.2% (v/v) HCOOH. The protein was eluted using a gradient of 5 − 100% MeCN with 0.2% (v/v) HCOOH over 4 min. The liquid chromatography − mass spectrometry (LC−MS) experiment (Agilent Q-TOF 6545) was carried out under the following conditions: fragmentor voltage, 300 V; skimmer voltage, 75 V; nozzle voltage, 100 V; sheath gas temperature, 350 °C; and drying gas temperature, 325 °C. The MassHunter Qualitative Analysis software (Agilent) was used to analyze the data. Intact protein masses were obtained using the maximum entropy deconvolution algorithm.
Real-Time PCR.
Total RNA was isolated from either cell grown in vitro or bone marrow samples enriched for human CD45+ cells by EasySep Human CD45 Depletion Kit (STEMCELL Technologies) using a RNeasy kit (QIAGEN) according to the manufacturer’s protocol. The cDNA was generated using a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Real-time PCR amplifications of HOXA9, MEIS1, ITGAM, GAPDH and HPRT1genes were carried out with primers specific for each gene, using TaqMan gene expression assays (Applied Biosystems). Relative quantification of each gene transcript was calculated by a comparative cycle threshold (Ct) method. The results were presented as relative expression to vehicle treatment after normalizing to an internal loading control GAPDH or HPRT1. The catalog numbers for primers of each genes are HOXA9 (Hs00365956_m1), MEIS1 (Hs00180020_m1), ITGAM (Hs00167304_m1), GAPDH (Hs99999905_m1) and HPRT1 (Hs99999909_m1).
Plasma Protein Binding.
To determine plasma protein binding using a dialysis method, dialysis buffer was loaded into the receiver side of a dialysis chamber and plasma or dialysis buffer spiked with M-1121 (1μM), or plasma spiked with warfarin (1 μM) or quinidine (1μM) were loaded into the donor side of the dialysis chambers and the dialysis block was shaken at 37°C for 5 hours. After incubation, samples from both the donor and receiver sides of the dialysis apparatus were added to a 96-well plate and mixed with same volume of opposite matrices (blank buffer to plasma and blank plasma to buffer). To prepare 0-hour samples, plasma or dialysis buffer spiked with M-1121 (1 μM), or 100 μL plasma spiked with warfarin (1μM) or quinidine (1μM) were added to a 96-well plate and mixed with same volume of blank buffer. Zero-hour samples were stored at ‑20°C until analysis. At the time of analysis, all samples were quenched with acetonitrile containing the internal standard imipramine. After quenching, assay plates were shaken followed by centrifugation. Supernatants were removed and added to a new 96-well plate then diluted with Mill-Q® water before being analyzed by liquid chromatography with tandem mass spectrometry. The peak area ratio between the sample and the internal standard was used to calculate percent bound, percent unbound, and recovery.
Flow cytometry.
Bone marrow cells were collected, and cell suspensions were filtered through 70μM cell strainer and washed with PBS. Red blood cells were removed by using 1 × RBC lysis buffer (Sigma, cat#R7757.) Samples were washed twice with pre-cold PBS and stained with fixable viability stain (BD, cat# 565388) followed by human FcR blocker treatment (BD, cat#564219). Next, cells were stained with Anti-human CD45 (BD, cat#563204), Anti-human CD33 (BD, cat#555626) and Anti-human CD11b (BD, cat#562721) antibodies mixture at 4°C for 40 minutes in the dark. Samples were suspended with cell staining buffer and analyzed on Thermo Attune NxT flow cytometer.
Animal Experiments.
Animal experiments were performed under the guidelines of the University of Michigan Committee for Use and Care of Animals and using an approved animal protocol (PI, Shaomeng Wang) or in accordance with ChemPartner approved Institutional Animal Care and Use Committee (IACUC) protocols.
In Vivo Efficacy Studies in an MV4;11 Human AML Xenograft Model in Mice.
Five-six-week-old female C.B.−17 SCID mice were purchased from Charles River and implanted subcutaneously in the right flank with MV4;11 cells at an inoculum of 5×106 cells/mouse (cell suspension and Matrigel® at 1:1 ratio.) Once tumors reached 200mm3 mice were randomized into vehicle (0.5%MC (400cP) + 0.2% tween 80 (w/w)) or M-1121 treated groups. Treatment was administered daily by oral gavage (QD) at 10ml/kg.
Body weights were measured daily, and tumor volume measured three times a week using Vernier calipers. Tumor volume was calculated using the formula 0.5xWxWxL with W being tumor width and L tumor length. Results graphed as mean ± standard error (SEM). Graphing and statistical analysis were performed using GraphPad Prism 8.00 (GraphPad Software).
In Vivo Studies in a Luciferase-tagged MV4;11 AML Xenograft Model.
Five-six-week-old female NCG mice obtained from Charles River Laboratories were implanted systemically via tail vein injection with Luciferase-tagged-MV;411 cells at an inoculum level of 5×106 cells/mouse in serum free media. Severity of disease was monitored by IVIS® SpectrumCT Imaging System twice weekly as recommended by manufacturer (PerkinElmer) until disease burden reached log phase (27 days post implant.) Mice were randomized into two groups, vehicle and M-1121, that were formulated and administered as mentioned above. Mice were treated for 4 days and bone marrow samples were collected 48 hours after the last dose and subjected to either staining for flow cytometry evaluation or human CD45+ cell enrichment for downstream gene expression evaluation by RT-qPCR.
Bioluminescence imaging.
Mice implanted with Luciferase-tagged MV4;11 cells were peritoneally injected with luciferin potassium salt (PerkinElmer, cat#122799–5) at a dose of 150mg/kg before undergoing anesthesia with 3% isoflurane (Patterson Veterinary Supply, cat#07–806-3204) in air in anesthesia induction chamber. Bioluminescence imaging was performed using IVIS imaging system and mice were imaged approximately 5 minutes after substrate injection. Acquisition setting (binning and duration) were set up depending upon tumor activity at the time of acquisition.
Supplementary Material
ACKNOWLEDGMENT
We thank G.W. A. Milne for critical reading and editing of the manuscript.
Funding
This work is supported in part by the National Institutes of Health/National Cancer Institute (R01 CA208267 to SW), the Prostate Cancer Foundation (to SW) and the Rogel Cancer Center Core Grant from the National Cancer Institutes, NIH (P30 CA046592) and a research contract from Agios Pharmaceuticals (to S.W.). Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817).
ABBREVIATIONS
- ALL
acute lymphoblastic leukemia
- AML
acute myeloid leukemia
- DCM
dichloromethane
- DIBAL-H
diisobutylaluminium hydride
- DIPEA
N,N-Diisopropylethylamine
- ESI-MS
electrospray ionization mass spectrometry
- EtOAc
ethyl acetate
- FP
fluorescence polarization
- MEN1
multiple endocrine neoplasia 1
- MLL
mixed lineage leukemia protein
- GSH
glutathione
- PBS
phosphate buffered saline
- PD
pharmacodynamics
- RT
room temperature
- SCID
severe combined immunodeficient
- TFA
trifluoroacetic acid
- UPLC
ultra-performance liquid chromatography
Footnotes
Disclosure of Conflicts of Interest
The authors declare the following competing financial interest(s): The University of Michigan has filed a number of patent applications on this class of menin inhibitors, which have been licensed to Medsyn Biopharma, LLC. S. Wang, M. Zhang, A. Aguilar, S. Xu, L. Huang, T. Xu, K. Zheng, D. McEachern, and J. Stuckey are co-inventors on one or more of these patents and receive royalties on these patents from the University of Michigan. S. Wang is a co-founder of Medsyn Biopharma LLC. The University of Michigan and S. Wang own stock in Medsyn. T.S, B.W, S.G., B.N.N., S.R., A.E.T and T.L are current or former employees and shareholders of Agios Pharmaceuticals, Inc.
ASSOCIATED CONTENT
Supporting Information.
The following Supporting Information is available free of charge on the ACS Publications website at DOI:
Crystallography data collection and refinement statistics. In vivo pharmacokinetics and efficacy data of compounds 10, 17, 24 (M-1121) in mice. Purity and spectral data of M-1121 (PDF).
A molecular string file for all the final target compounds (CSV). Method for computational modeling for compounds 24 and 25, and modeled structures of compounds 24 and 25 in complex with menin (PDB).
Accession Codes. The coordinates for M-1121 complexed with menin have been deposited into the Protein Data Bank with the PDB code: 7M4T. Authors will release the atomic coordinates upon publication of this article.
REFERENCES
- 1.Dimartino JF; Cleary ML Mll rearrangements in haematological malignancies: lessons from clinical and biological studies. Br. J. Haematol. 1999, 106, 614–26. [DOI] [PubMed] [Google Scholar]
- 2.Ernst P; Wang J; Korsmeyer SJ The role of MLL in hematopoiesis and leukemia. Curr. Opin. Hematol. 2002, 9, 282–7. [DOI] [PubMed] [Google Scholar]
- 3.Marschalek R Mechanisms of leukemogenesis by MLL fusion proteins. Br. J. Haematol. 2011, 152, 141–54. [DOI] [PubMed] [Google Scholar]
- 4.Popovic R; Zeleznik-Le NJ MLL: how complex does it get? J. Cell. Biochem. 2005, 95, 234–42. [DOI] [PubMed] [Google Scholar]
- 5.Slany RK When epigenetics kills: MLL fusion proteins in leukemia. Hematol. Oncol. 2005, 23, 1–9. [DOI] [PubMed] [Google Scholar]
- 6.Tomizawa D; Koh K; Sato T; Kinukawa N; Morimoto A; Isoyama K; Kosaka Y; Oda T; Oda M; Hayashi Y; Eguchi M; Horibe K; Nakahata T; Mizutani S; Ishii E Outcome of risk-based therapy for infant acute lymphoblastic leukemia with or without an MLL gene rearrangement, with emphasis on late effects: a final report of two consecutive studies, MLL96 and MLL98, of the Japan Infant Leukemia Study Group. Leukemia 2007, 21, 2258–2263. [DOI] [PubMed] [Google Scholar]
- 7.Slany RK The molecular biology of mixed lineage leukemia. Haematologica 2009, 94, 984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meyer C; Burmeister T; Gröger D; Tsaur G; Fechina L; Renneville A; Sutton R; Venn NC; Emerenciano M; Pombo-de-Oliveira MS; Barbieri Blunck C; Almeida Lopes B; Zuna J; Trka J; Ballerini P; Lapillonne H; De Braekeleer M; Cazzaniga G; Corral Abascal L; van der Velden VHJ; Delabesse E; Park TS; Oh SH; Silva MLM; LundAho T; Juvonen V; Moore AS; Heidenreich O; Vormoor J; Zerkalenkova E; Olshanskaya Y; Bueno C; Menendez P; Teigler-Schlegel A; zur Stadt U; Lentes J; Göhring G; Kustanovich A; Aleinikova O; Schäfer BW; Kubetzko S; Madsen HO; Gruhn B; Duarte X; Gameiro P; Lippert E; Bidet A; Cayuela JM; Clappier E; Alonso CN; Zwaan CM; van den Heuvel-Eibrink MM; Izraeli S; Trakhtenbrot L; Archer P; Hancock J; Möricke A; Alten J; Schrappe M; Stanulla M; Strehl S; Attarbaschi A; Dworzak M; Haas OA; Panzer-Grümayer R; Sedék L; Szczepański T; Caye A; Suarez L; Cavé H; Marschalek R The MLL recombinome of acute leukemias in 2017. Leukemia 2018, 32, 273–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Faber J; Krivtsov AV; Stubbs MC; Wright R; Davis TN; van den Heuvel-Eibrink M; Zwaan CM; Kung AL; Armstrong SA HOXA9 is required for survival in human MLL-rearranged acute leukemias. Blood 2009, 113, 2375–2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kumar AR; Sarver AL; Wu B; Kersey JH Meis1 maintains stemness signature in MLL-AF9 leukemia. Blood 2010, 115, 3642–3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caslini C; Yang Z; El-Osta M; Milne TA; Slany RK; Hess JL Interaction of MLL Amino Terminal Sequences with Menin Is Required for Transformation. Cancer Research 2007, 67, 7275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yokoyama A; Somervaille TCP; Smith KS; Rozenblatt-Rosen O; Meyerson M; Cleary ML The Menin Tumor Suppressor Protein Is an Essential Oncogenic Cofactor for MLLAssociated Leukemogenesis. Cell 2005, 123, 207–218. [DOI] [PubMed] [Google Scholar]
- 13.Chen Y-X; Yan J; Keeshan K; Tubbs AT; Wang H; Silva A; Brown EJ; Hess JL; Pear WS; Hua X The tumor suppressor menin regulates hematopoiesis and myeloid transformation by influencing Hox gene expression. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yokoyama A; Wang Z; Wysocka J; Sanyal M; Aufiero DJ; Kitabayashi I; Herr W; Cleary ML Leukemia Proto-Oncoprotein MLL Forms a SET1-Like Histone Methyltransferase Complex with Menin To Regulate <em>Hox</em> Gene Expression. Mol. Cell. Biol. 2004, 24, 5639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shi A; Murai MJ; He S; Lund G; Hartley T; Purohit T; Reddy G; Chruszcz M; Grembecka J; Cierpicki T Structural insights into inhibition of the bivalent menin-MLL interaction by small molecules in leukemia. Blood 2012, 120, 4461–4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murai MJ; Chruszcz M; Reddy G; Grembecka J; Cierpicki T Crystal structure of menin reveals binding site for mixed lineage leukemia (MLL) protein. J. Biol. Chem 2011, 286, 31742–31748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cierpicki T; Grembecka J Challenges and opportunities in targeting the menin–MLL interaction. Future Med Chem 2014, 6, 447–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grembecka J; He S; Shi A; Purohit T; Muntean AG; Sorenson RJ; Showalter HD; Murai MJ; Belcher AM; Hartley T; Hess JL; Cierpicki T Menin-MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nat. Chem. Biol. 2012, 8, 277–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Borkin D; Pollock J; Kempinska K; Purohit T; Li X; Wen B; Zhao T; Miao H; Shukla S; He M; Sun D; Cierpicki T; Grembecka J Property Focused Structure-Based Optimization of Small Molecule Inhibitors of the Protein–Protein Interaction between Menin and Mixed Lineage Leukemia (MLL). J. Med. Chem. 2016, 59, 892–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He S; Malik B; Borkin D; Miao H; Shukla S; Kempinska K; Purohit T; Wang J; Chen L; Parkin B; Malek SN; Danet-Desnoyers G; Muntean AG; Cierpicki T; Grembecka J Menin-MLL inhibitors block oncogenic transformation by MLL-fusion proteins in a fusion partner-independent manner. Leukemia 2016, 30, 508–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu Y; Yue L; Wang Y; Xing J; Chen Z; Shi Z; Liu R; Liu Y-C; Luo X; Jiang H; Chen K; Luo C; Zheng M Discovery of Novel Inhibitors Targeting the Menin-Mixed Lineage Leukemia Interface Using Pharmacophore- and Docking-Based Virtual Screening. J. Chem. Inf. Model. 2016, 56, 1847–1855. [DOI] [PubMed] [Google Scholar]
- 22.He S; Senter TJ; Pollock J; Han C; Upadhyay SK; Purohit T; Gogliotti RD; Lindsley CW; Cierpicki T; Stauffer SR; Grembecka J High-Affinity Small-Molecule Inhibitors of the Menin-Mixed Lineage Leukemia (MLL) Interaction Closely Mimic a Natural Protein–Protein Interaction. J. Med. Chem. 2014, 57, 1543–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Borkin D; Klossowski S; Pollock J; Miao H; Linhares BM; Kempinska K; Jin Z; Purohit T; Wen B; He M; Sun D; Cierpicki T; Grembecka J Complexity of Blocking Bivalent Protein–Protein Interactions: Development of a Highly Potent Inhibitor of the Menin– Mixed-Lineage Leukemia Interaction. J. Med. Chem. 2018, 61, 4832–4850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Borkin D; He S; Miao H; Kempinska K; Pollock J; Chase J; Purohit T; Malik B; Zhao T; Wang J; Wen B; Zong H; Jones M; Danet-Desnoyers G; Guzman Monica L.; Talpaz M; Bixby Dale L.; Sun D; Hess Jay L.; Muntean Andrew G.; Maillard I; Cierpicki T; Grembecka J Pharmacologic Inhibition of the Menin-MLL Interaction Blocks Progression of MLL Leukemia In Vivo. Cancer Cell 2015, 27, 589–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brzezinka K; Nevedomskaya E; Lesche R; Haegebarth A; ter Laak A; FernándezMontalván AE; Eberspaecher U; Werbeck ND; Moenning U; Siegel S; Haendler B; Eheim AL; Stresemann C Characterization of the Menin-MLL Interaction as Therapeutic Cancer Target. Cancers 2020, 12, 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krivtsov AV; Evans K; Gadrey JY; Eschle BK; Hatton C; Uckelmann HJ; Ross KN; Perner F; Olsen SN; Pritchard T; McDermott L; Jones CD; Jing D; Braytee A; Chacon D; Earley E; McKeever BM; Claremon D; Gifford AJ; Lee HJ; Teicher BA; Pimanda JE; Beck D; Perry JA; Smith MA; McGeehan GM; Lock RB; Armstrong SA A Menin-MLL Inhibitor Induces Specific Chromatin Changes and Eradicates Disease in Models of MLL-Rearranged Leukemia. Cancer Cell 2019, 36, 660–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klossowski S; Miao H; Kempinska K; Wu T; Purohit T; Kim E; Linhares BM; Chen D; Jih G; Perkey E; Huang H; He M; Wen B; Wang Y; Yu K; Lee SC-W; Danet-Desnoyers G; Trotman W; Kandarpa M; Cotton A; Abdel-Wahab O; Lei H; Dou Y; Guzman M; Peterson L; Gruber T; Choi S; Sun D; Ren P; Li L-S; Liu Y; Burrows F; Maillard I; Cierpicki T; Grembecka J Menin inhibitor MI-3454 induces remission in MLL1rearranged and NPM1-mutated models of leukemia. J. Clin. Investig. 2020, 130, 981–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aguilar A; Zheng K; Xu T; Xu S; Huang L; Fernandez-Salas E; Liu L; Bernard D; Harvey KP; Foster C; McEachern D; Stuckey J; Chinnaswamy K; Delproposto J; Kampf JW; Wang S Structure-Based Discovery of M-89 as a Highly Potent Inhibitor of the Menin-Mixed Lineage Leukemia (Menin-MLL) Protein–Protein Interaction. J. Med. Chem. 2019, 62, 6015–6034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lonsdale R; Ward RA Structure-based design of targeted covalent inhibitors. Chem. Soc. Rev. 2018, 47, 3816–3830. [DOI] [PubMed] [Google Scholar]
- 30.Xu S; Aguilar A; Xu T; Zheng K; Huang L; Stuckey J; Chinnaswamy K; Bernard D; Fernández-Salas E; Liu L; Wang M; McEachern D; Przybranowski S; Foster C; Wang S Design of the First-in-Class, Highly Potent Irreversible Inhibitor Targeting the Menin-MLL Protein–Protein Interaction. Angew. Chem. Int. Ed. 2018, 57, 1601–1605. [DOI] [PubMed] [Google Scholar]
- 31.Strelow JM A Perspective on the Kinetics of Covalent and Irreversible Inhibition. SLAS Discov. 2017, 22, 3–20. [DOI] [PubMed] [Google Scholar]
- 32.Zhou H; Liu L; Huang J; Bernard D; Karatas H; Navarro A; Lei M; Wang S Structure-Based Design of High-Affinity Macrocyclic Peptidomimetics to Block the Menin-Mixed Lineage Leukemia 1 (MLL1) Protein–Protein Interaction. J. Med. Chem. 2013, 56, 1113–1123. [DOI] [PubMed] [Google Scholar]
- 33.Xu S; Aguilar A; Huang L; Xu T; Zheng K; McEachern D; Przybranowski S; Foster C; Zawacki K; Liu Z; Chinnaswamy K; Stuckey J; Wang S Discovery of M-808 as a Highly Potent, Covalent, Small-Molecule Inhibitor of the Menin-MLL Interaction with Strong In Vivo Antitumor Activity. J. Med. Chem. 2020, 63, 4997–5010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Larsen SD; Grieco PA; Fobare WF Reactions of allylsilanes with simple iminium salts in water: A facile route to piperidines via a aminomethano desilylation-cyclization process. J. Am. Chem. Soc. 1986, 108, 3512–3513. [Google Scholar]
- 35.Benoit E; Bueno B; Choinière C; Gagnon A First use of an organobismuth reagent in C(sp3)–S bond formation: Access to aryl cyclopropyl sulfides via copper-catalyzed S– Cyclopropylation of thiophenols using tricyclopropylbismuth. J. Organomet. Chem. 2019, 893, 72–77. [Google Scholar]
- 36.Wang S; Altman J; Pettit K; Botton S; Walter R; Fenaux P; Burrows F; Tomkinson B; Martell B; Fathi A Preliminary data on a phase 1/2A first in human study of the menin-KMT2A (MLL) inhibitor KO-539 in patients with relapsed or refractory acute myeloid leukemia. Presented at: 2020 American Society of Hematology Annual Meeting and Exposition. December 5–8, 2020; Virtual. Abstract 115. https://ash.confex.com/ash/2020/webprogram/Paper134942.html. [Google Scholar]
- 37.McGeehan J A first-in-class Menin-MLL1 antagonist for the treatment of MLL-r and NPM1 mutant leukemias. Presented at: the 2020 AACR Virtual Annual Meeting I; April 27–28, 2020. Abstract DDT01–01. https://www.abstractsonline.com/pp8/#!/9045/presentation/6808. [Google Scholar]
- 38.Otwinowski Z; Minor W, [20] Processing of X-ray diffraction data collected in oscillation mode. In Meth. Enzymol, Carter CW, Ed. Academic Press: San Diego, USA, 1997; Vol. 276, pp 307–326. [DOI] [PubMed] [Google Scholar]
- 39.Vagin A; Teplyakov A, MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 1997, 30 (6), 1022–1025. [Google Scholar]
- 40.Emsley P; Cowtan K, Coot: model-building tools for molecular graphics. Acta Cryst. D 2004, 60 (12 Part 1), 2126–2132. [DOI] [PubMed] [Google Scholar]
- 41.Roversi P; Sharff A; Smart OS; Vonrhein C; Womack TO BUSTER version 2.11.2 Cambridge, United Kingdom: Global Phasing Ltd., 2011. [Google Scholar]
- 42.Smart OS; Womack TO; Sharff A; Flensburg C; Keller P; Paciorek W; Vonrhein C; Bricogne G GRADE, 1.2.13, Cambridge, United Kingdom: Global Phasing Ltd., 2017. [Google Scholar]
- 43.Molecular Operating Environment (MOE); Chemical Computing Group: Montreal, Canada, 2019.0102. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.