

Abstract
Periodontitis is a complex inflammatory disease affecting the supporting structures of teeth and is associated with systemic inflammatory disorders. Regulator of G-protein signaling 12 (RGS12), the largest protein in the RGS protein family, plays a crucial role in the development of inflammation and bone remodeling. However, the role and mechanism(s) by which RGS12 may regulate periodontitis have not been elucidated. Here, we showed that ablation of RGS12 in Mx1+ hematopoietic cells blocked bone loss in the ligature-induced periodontitis model, as evidenced morphometrically and by micro–computed tomography analysis of the alveolar bone. Moreover, hematopoietic cell-specific deletion of RGS12 inhibited osteoclast formation and activity as well as the production of inflammatory cytokines such as IL1β, IL6, and TNFα in the diseased periodontal tissue. In the in vitro experiments, we found that the overexpression of RGS12 promoted the reprogramming of macrophages to the proinflammatory M1 type, but not the anti-inflammatory M2 type, and enhanced the ability of macrophages for migration. Conversely, knockdown of RGS12 in macrophages inhibited the production of inflammatory cytokines and migration of macrophages in response to lipopolysaccharide stimulation. Our results demonstrate for the first time that inhibition of RGS12 in macrophages is a promising therapeutic target for the treatment of periodontitis.
Keywords: bone loss, immunity, inflammation, innate immunity, periodontal disease(s), bone biology
Introduction
Periodontitis is a complex inflammatory disease that involves the supporting structures of teeth and is also associated with increased risk of systemic inflammatory disorders (Hajishengallis and Chavakis 2021). Chronic periodontitis usually results from untreated gingivitis that progresses to the loss of ligament and bone and eventually leads to tooth loss (Kononen et al. 2019). In the United States, periodontitis affects more than 50% of the population and mostly occurs in elderly individuals (Eke et al. 2020). The role of genetics in chronic periodontitis has been studied, but no single-nucleotide polymorphism has been consistently identified (Laine et al. 2010). Studies showed that the levels of inflammatory factors such as IL1β, IL17, TNFα, and COX2 are related to periodontal tissue damage and bone loss (Kayal 2013; Dutzan et al. 2018). Therefore, the dysfunctional host response caused by the immune system may be an important reason for periodontitis.
Macrophages, as one of the most important inflammatory cells, play critical roles in the pathogenesis of periodontitis (Hasturk and Kantarci 2015). Macrophages have a feature of highly plastic phenotypes in response to microenvironmental signals (Italiani and Boraschi 2014). Macrophages have 2 phenotypes: M1 macrophages (classically activated macrophages) are related to the proinflammatory factors’ activation and host defense, which can be induced by IL1β, IL6, and TNFα (Mosser and Edwards 2008). Conversely, M2 macrophages (nonclassically activated macrophages) are involved in stimulating immune cells to secrete anti-inflammatory cytokines such as IL4 and IL10 (Mosser and Edwards 2008), which can inhibit the inflammatory process and promote local tissue repair and wound healing (Wynn and Vannella 2016). Importantly, the major functions of macrophages are the elimination of bacteria, clearance of the extra neutrophils, and activation of the immune response (Navegantes et al. 2017). Macrophages first migrate to the microorganisms and engulf them through binding certain surface molecules of bacteria such as lipopolysaccharides (LPSs) (Hirayama et al. 2017). Inflammatory mediators such as cytokines and chemokines are released from activated immune cells, which can also stimulate the macrophages to migrate into the defective area to remove the necrotic tissues (Sokol and Luster 2015). Moreover, under an inflammatory environment, macrophages can differentiate into osteoclasts to dissolve the hard tissue such as alveolar bone. Thus, the dysregulation of macrophages during periodontitis contributes to inflammation and bone loss (Hasturk et al. 2012).
Regulators of G protein signaling (RGS proteins) regulate the G protein–dependent signals from the external environment (O’Brien et al. 2019). As the largest protein in the RGS family, RGS12 is involved in multiple signaling pathways, including the G protein–coupled receptors (GPCRs) and nuclear factor κ light chain enhancer of activated B-cell (NF-κB) pathways (Lambert et al. 2010; Yuan, Yang, Ng, et al. 2020). RGS12 regulates the occurrence and development of numerous diseases such as oral cancer, arthritis, and bone diseases (Fu et al. 2020; Yuan, Yang, Liu, et al. 2020; Yuan, Yang, Ng, et al. 2020). More important, RGS12 in macrophages was found as a key regulator in bone erosion and inflammation (Yang et al. 2013; Ng et al. 2019; Yuan, Yang, Ng, et al. 2020). Deletion of RGS12 in myeloid cell lineage using Mx1-Cre transgenic mice and LysM-Cre leads to a significant increase of bone mass (Yang et al. 2013; Ng et al. 2019). Moreover, RGS12 deficiency in macrophages can prevent the development of rheumatoid arthritis by regulating the nuclear translocation and activation of NF-κB (Yuan, Yang, Ng, et al. 2020). However, how RGS12 regulates the function of macrophages to affect periodontitis and bone erosion is still unclear.
In this study, we found that RGS12 plays a critical role in inflammatory bone erosion in periodontitis through controlling proinflammatory cytokines’ production, macrophage polarization, and migration, indicating that RGS12 is an important regulator of macrophages and a potential drug target for periodontitis and other inflammatory bone loss diseases.
Materials and Methods
The Appendix Methods section describes the following methods: the materials and reagents, the details of plasmid construction, migration assay, cell culture and osteoclastogenesis, real-time polymerase chain reaction (PCR), Western blot analysis, and immunofluorescence (IF).
Mice
All animal studies were performed in accordance with Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines and with approval by the Institutional Animal Care and Use Committee (IACUC) of the University of Pennsylvania. To generate the conditional RGS12-knockout (cKO) mice, RGS12fl/fl mice were randomly crossed with mice expressing Cre recombinase under the control of the Mx1 promoter (Mx1-Cre+). Cre control (8 wk old, n = 10) and cKO mice (8 wk old, n = 10) were littermates derived from the breeding of heterozygous animals. The mice were fed a chow diet and raised in a clean-grade room with 12-h light and 12-h dark cycles. All animals were of a BALB/c background.
Periodontitis Mouse Model
Experimental periodontitis and bone resorption were induced by insertion of ligature wire (5-0 silk ligature) between the left second molars for 10 d as described previously (Yuh et al. 2020). The ligature-induced periodontitis model generates a subgingival biofilm-retentive environment resulting in local inflammation and bone loss in conventional (but not germ-free) mice (Jiao et al. 2013; Kourtzelis et al. 2019). The mice were monitored and kept warm until recovery from the anesthesia. The presence of the ligature wire in each mouse was evaluated every 3 d. Mice in which the ligature wire was absent were excluded from the study.
The morphological changes in the alveolar bone and maxillae were analyzed by the micro–computed tomography (CT) system (Micro-CT 35; Scanco Medical AG). The distal coronal one-third area of the distal root and the mesial coronal one-third area were selected as the region of interest (ROI) for micro-CT analysis. Three-dimensional reconstruction images were used to determine bone volume over total volume (BV/TV) and bone mineral density (BMD) by using boneJ software (National Institutes of Health).
Maxillary samples were collected on day 10 following ligature insertion. For bone loss evaluation, the distance from the cement-enamel junction (CEJ) to the alveolar bone crest (ABC) for each mouse was measured on 6 predetermined points on the ligated second molar. The measurement of alveolar bone loss in the mice was performed by 3 experienced examiners (Yuh et al. 2020).
Histology
The maxillae were fixed in 10% formalin, decalcified, and embedded in paraffin. Tissues were cut into 5-µm sections and stained with hematoxylin and eosin to assess maxillae pathology. The degree of inflammation was evaluated on a scale from 0 to 3 scores: 0 = no inflammation, 1 = mild inflammation (5–25% inflammatory cells), 2 = moderate inflammation (25–50% inflammatory cells), and 3 = severe inflammation (>50% inflammatory cells) by 3 different experts as described previously (Gully et al. 2014).
For tartrate-resistant acid phosphatase (TRAP) staining, the maxillae were fixed in 4% formaldehyde and decalcified in 10% ethylenediaminetetraacetic acid (EDTA). The maxillae were stained with a TRAP activity kit (387A; Sigma) to measure the osteoclast activity according to the manufactural instructions. The TRAP-positive cells were quantified relative to the total trabecular bone surface by ImageJ software (National Institutes of Health).
Statistics
All statistical analyses were performed using Prism 7.0 software (GraphPad Software). The unpaired Student’s t test was used to compare the difference between 2 groups, and 1-way analysis of variance (ANOVA) followed by Tukey multiple comparison test was used to analyze the changes between multiple groups. Values are expressed as mean ± SEM, and P values <.05 were considered significant.
Results
Conditional Deletion of RGS12 in Mx1+ Cells Inhibits Ligature-Induced Bone Loss
RGS12 is a multi-functional protein that regulates the processes of inflammation and bone remodeling (Ng et al. 2019; Yuan, Yang, Ng, et al. 2020). Moreover, RGS12 plays a key role in regulating bone resorption (Yang and Li 2007). To identify the role of hematopoietic cell–derived RGS12 in periodontitis, RGS12 was deleted in hematopoietic lineage by mating RGS12 floxed mice with Mx1-Cre mice expressing Cre-recombinase under the control of the inducible Mx1 promoter (Kuhn et al. 1995; Yang et al. 2013). Poly I:C stimulates the production of IFNα, which leads to the activation of the Mx1 promoter and subsequent Cre expression. Thus, exon 2 of the floxed RGS12 gene was removed specifically in hematopoietic cells of RGS12-cKO mice by the Cre-recombinase induced by intraperitoneal (i.p.) injection of poly I:C (Fig. 1A, B). Loss of RGS12 protein in bone marrow macrophages (BMMs) of cKO animals was confirmed by immunoblotting (Fig. 1C). Mx1-Cre+ BMMs (control) showed a clear band indicating the RGS12 protein expression, whereas RGS12 protein was nearly absent in Mx1-Cre+;RGS12fl/fl BMMs (RGS12 cKO) after poly I:C injection (Fig. 1C), demonstrating effective RGS12 deletion.
Figure 1.

Conditional deletion of RGS12 in Mx1+ cells inhibits bone loss during ligature-induced periodontitis. (A) Diagrams of RGS12 conditional knockout (cKO) generation. Exons are depicted as blue boxes and are numbered. Cre, Cre recombinase coding sequence; loxP, loxP sites; neo, neomycin resistance gene. (B) Appearance of Mx1-Cre+ (Ctrl) and Mx1-Cre+;RGS12fl/fl (RGS12 cKO) after injection of poly I:C. (C) The bone marrow macrophages were extracted and the RGS12 deletion was confirmed by immunoblot after injection of poly I:C. Values are means ± SEMs. ***P < 0.01 versus Mx1-Cre+ control (Ctrl), n = 5. (D) Schematic diagram depicting the timing of injection of poly I:C and ligature-induced periodontitis model in mice. (E, F) The cementoenamel junction distance (CEJ-ABC) (white line) was to show the bone loss analysis of Mx1-Cre+ control (Ctrl) and RGS12 cKO mice in mock and ligation groups. Values are means ± SEMs. *P < 0.05 versus Ctrl, n = 10. (G–I) Micro–computed tomography analysis of bone density in Mx1-Cre+ control (Ctrl) and RGS12 cKO mice in mock and ligation groups. Quantitative analysis of bone volume fraction (BV/TV) (H) and bone mineral density (BMD) (I) are shown in (F). *P < 0.05, **P < 0.01, ***P < 0.001. Values are means ± SEMs (n = 10).
To determine the role of RGS12 in periodontitis, RGS12 cKO mice and Mx1-Cre+ controls were subjected to ligature-induced periodontitis. The expression of Cre recombinase was induced by i.p. administration of poly I:C on days 1, 3, and 5 before ligature (Fig. 1D).
To analyze alveolar bone changes upon the conditional deletion of RGS12, the distances between the cementoenamel junction (CEJ) and alveolar bone crest (ABC) in the palatal and buccal surfaces were measured at the second maxillary molar in three-dimensionally constructed images in the RGS12 cKO and Mx1-Cre+ control (Ctrl) mice with or without periodontitis (Fig. 1E–F). The CEJ-ABC distances were significantly decreased in the RGS12 cKO group in comparison to the control group (Fig. 1E–F). In periodontitis, the specimens from the Mx1-Cre+ control groups displayed aggravation of bone loss, but the bone loss was significantly reduced in RGS12 cKO groups (Fig. 1E–F). The average CEJ-ABC distance in the Mx1-Cre+ control group was 0.27 ± 0.01 mm, which was larger than that in the RGS12 cKO group (0.2 ± 0.03 mm) under the periodontitis condition (Fig. 1E–F).
To further determine if RGS12 affects alveolar bone erosion upon ligature-induced periodontitis using an independent approach, the morphology of the maxilla was reconstructed and analyzed by micro-CT in Mx1-Cre+ control and RGS12 cKO mice with or without ligature (10 d). We found that the alveolar bone BMD and the ratio of BV/TV increased in RGS12 cKO mice compared with those in Mx1-Cre+ control mice in the mock (no-ligature) groups (Fig. 1G–I). In the ligature-induced periodontitis groups, bone mass as indicated by BV/TV and BMD was decreased in both Mx1-Cre+ control and RGS12 cKO as compared to those in the mock condition. However, RGS12 cKO significantly inhibited bone loss compared to the Mx1-Cre+ controls in the ligature-induced periodontitis mice (Fig. 1G–I).
Ablation of RGS12 in Hematopoietic Lineage Prevents Inflammatory Bone Loss through Inhibiting Osteoclast Formation
Our previous studies showed that RGS12 cKO increases bone mass by decreasing osteoclastogenesis but not affecting the osteogenesis in long bone (Yang et al. 2013). Here, we further found RGS12 cKO mice also showed an increased bone mass in the skull bone in comparison with Mx1-Cre+ control mice (Appendix Fig. 1). To confirm whether RGS12 cKO affects bone formation or erosion, we detected the messenger RNA (mRNA) levels of osteoclast makers (Atp6v0d2 [adenosine triphosphatase V0] and Dcstamp [dendritic cell–specific transmembrane protein]) and osteoblast markers (ALP [alkaline phosphatase] and OC [osteocalcin]) in alveolar bone tissues. We found that only osteoclast markers were significantly reduced in RGS12 cKO mice with or without ligature but not the osteoblast markers (Appendix Fig. 2). These results suggested that RGS12 mainly regulates osteoclastogenesis in the alveolar bone.
Consistent with these findings, TRAP staining results showed that osteoclast numbers and osteoclast surface were significantly decreased in alveolar bone and especially in the junction areas of alveolar bone and cortical bone in RGS12 cKO mice in comparison to the Mx1-Cre+ control mice with or without ligature (Fig. 2A–C and Appendix Fig. 3). Furthermore, we found that osteoclast marker genes Nfatc1, Ctsk, and Acp5 were decreased in the alveolar bone of the RGS12 cKO group in comparison to the Mx1-Cre+ control group (Fig. 2D). To further confirm the RGS12 role in the osteoclast differentiation, BMMs from the Mx1-Cre+ control and RGS12 cKO mice were isolated and induced with macrophage colony stimulating factor (M-CSF)/receptor activation of NF-кB ligand (RANKL) and phosphate-buffered saline (PBS) or LPS for 4 d and stained by the TRAP activity kit (Fig. 2E). We found that RGS12 inhibited osteoclastogenesis in both conditions of M-CSF/RANKL and M-CSF/RANKL with LPS inductions (Fig. 2E). Collectively, these data establish that hematopoietic cell–specific RGS12 regulates bone resorption in ligature-induced periodontitis.
Figure 2.

Ablation of RGS12 in hematopoietic lineage prevents inflammatory bone loss through inhibiting osteoclastogenesis. (A) The maxillae (ligature on day 10) were stained with the tartrate-resistant acid phosphatase (TRAP) kit (Sigma) (bar, 100 µm). (B) High-magnification image of (A) demonstrating the osteoclast in alveolar bone (bar, 25 µm). The RGS12 conditional knockout (cKO) showed fewer TRAP-positive osteoclasts compared to the Mx1-Cre+ control (Ctrl) group (red arrows) in ligature-induced periodontitis (day 10). (C) Histomorphometric analysis of the number of TRAP-positive osteoclasts (N.Oc/BS) and the percentage coverage of TRAP-positive osteoclasts on the alveolar bone surface (Oc.S/BS) from the Ctrl and RGS12 cKO after ligation for 10 d (values are means ± SEMs, ***P < 0.001 versus Ctrl, n = 5). (D) Relative messenger RNA expressions of Nfatc1 (nuclear factor of activated T cells 1), Ctsk (cathepsin K), and Acp5 (acid phosphatase 5) were measured by quantitative real-time polymerase chain reaction. *P < 0.05, **P < 0.01 versus Mx1-Cre+ control (Ctrl), n = 5. (E) Bone marrow macrophages (BMMs) from Ctrl and RGS12 cKO mice were incubated with macrophage colony stimulating factor (M-CSF) (20 ng/mL) and receptor activation of NF-kB ligand (RANKL) (100 ng/mL) and with and without lipopolysaccharide (LPS) (100 ng/mL) for 4 d. The cells were stained for TRAP. TRAP-positive cells with more than 3 nuclei were scored (bar, 50 µm). Note that BMMs from RGS12 cKO mice showed fewer TRAP-positive cells with or without LPS treatment. **P < 0.01, ***P < 0.001 versus Ctrl, n = 10.
Ablation of RGS12 in Mx1+ Cells Inhibits Periodontitis
To determine whether loss of RGS12 affects periodontitis, the maxillae pathology was analyzed by histology (Fig. 3A–B). The histological analysis showed higher inflammation scores in the Mx1-Cre+ control groups (2.4 ± 0.24 scores) in comparison to the RGS12 cKO groups (1.2 ± 0.2 scores) (Fig. 3D). We then stained the periodontal tissues with F4/80 antibody by immunofluorescence to further identify the infiltrated macrophages (Fig. 3C). As shown in Figure 3C and D, the F4/80+ cells were significantly decreased in RGS12 cKO groups in comparison with Mx1-Cre+ control groups during periodontitis. To further determine the regulation of RGS12 in inflammation, total RNA was extracted from the periodontal tissues of Mx1-Cre+ control and RGS12 cKO mice with or without ligature-induced periodontitis and analyzed for the expression of inflammation-related genes IL1β, IL6, IL12, TNFα, CCL2, and iNOS by a real-time quantitative polymerase chain reaction (qPCR) assay (Fig. 1G). The results showed that ablation of RGS12 significantly inhibited the expression of IL1β, IL6, CXCL1, and TNFα in periodontitis compared to the Mx1-Cre+ control group (Fig. 1G). These results suggest that the loss of RGS12 in macrophages inhibits ligature-induced periodontal inflammation.
Figure 3.

Ablation of RGS12 in Mx1+ cells inhibits periodontitis. (A) Histological analysis of periodontal tissues; arrow indicates increased infiltration of inflammatory cells (black box) (bar, 100 µm). (B) High-magnification image of (A) demonstrating infiltration of inflammatory cells. (C) F4/80 immunofluorescence to detect macrophages in periodontal tissues (besides the second molar) from Ctrl and RGS12 conditional knockout (cKO) mice (bar, 50 µm). (D) Infiltration of inflammatory cells as depicted in (A) and (B) was assessed as a histopathological score in the hematoxylin and eosin–stained section (top panel). The percentage of F4/80+ cells as depicted in (C) was analyzed by ImageJ software (National Institutes of Health). Note that RGS12 cKO mice showed less infiltration of inflammatory cells and F4/80-positive cells in comparison with Ctrl. Results are shown as mean ± SEM. **P < 0.01, ***P < 0.001 versus Mx1-Cre+ control (Ctrl), n = 5. (E–J) The control and inflamed periodontal tissues were collected from mock or ligature-induced periodontitis models on day 10. RNAs from Mx1-Cre+ control (Ctrl) and RGS12 cKO periodontal tissues were extracted by the Trizol kit (Sigma). Real-time polymerase chain reaction showing altered expression of IL1β (E), IL6 (F), CXCL1 (G), TNFα (H), CCL2 (I), and iNOS (J). *P < 0.05, **P < 0.01 versus Ctrl. Values are means ± SEMs (n = 5).
RGS12 Promotes the Migration of Macrophages
To investigate the effects of RGS12 on macrophage function, we first overexpressed RGS12 (pCMV-RGS12) or silenced RGS12 (psi-nU6.1-shRGS12) in RAW264.7 cells and confirmed the RGS12 protein changes by Western blot (Fig. 3A, B). To examine whether RGS12 plays a role in macrophage migration, we performed a Transwell migration assay in RGS12 overexpressed (OE) RAW264.7 cells, which were treated with or without LPS for 24 h. The results showed that the number of migrated macrophages in RGS12 OE group was much more than that in the control group after introducing the LPS (1 µg/mL) for 24 h (Fig. 3C, D). Moreover, without LPS induction, RGS12 OE also slightly increased macrophage migration (Fig. 3D). Consistently, we found that knockdown of RGS12 attenuated macrophage migration with and without LPS induction (Fig. 3E, F). To determine whether RGS12 affects the proliferation of macrophages, we performed the WST-1 assay (Appendix Fig. 4). RGS12 OE or knockdown did not affect macrophage proliferation (Appendix Fig. 4). These results indicate that RGS12 promotes macrophage migration but does not affect cell proliferation.
RGS12 Promotes the Polarization of Macrophages toward the M1 Phenotype
Numerous pharmacological agents have been identified to regulate the inflammatory pathways of M1 and M2 macrophages (Gensel et al. 2017). Recent studies showed that the inflammatory setting can be diverted from proinflammation to anti-inflammation by using different natural analogs (Amantea et al. 2016). However, the regulatory mechanism by which these molecules control the M1–M2 phenotype remains unknown. To determine whether RGS12 affects macrophage polarization, RAW264.7 cells were transfected with pCMV-empty (control) or pCMV-RGS12 (RGS12 OE) plasmids for 48 h, and then M1 macrophage marker CD86 and M2 macrophage marker CD163 were detected by performing the immunofluorescence staining (Fig. 4A). We found that fluorescence intensity of M1 macrophage marker CD86, but not of M2 marker CD163, was significantly increased in the RGS12 OE group compared with the control group (Fig. 4A, B). To further detect the cytokine markers of the M1 and M2 macrophages, RAW264.7 cells were transfected with pCMV-RGS12 plasmids for 48 h, and then the phenotype marker genes for M1 (IL1β, IL6, and TNFα) and M2 macrophages (IL4, IL1RA, and IL10) were determined by the real-time PCR as described (Jablonski et al. 2015). The results showed that RGS12 markedly upregulated the expression of M1 macrophage marker genes (Fig. 4C–E) but did not affect the expression of M2 macrophage marker genes (Fig. 4F–H), suggesting that RGS12 can promote the M1 macrophage formation.
Figure 4.

RGS12 promotes the migration of macrophages. (A) RAW264.7 cells were transfected with pCMV-empty or pCMV-RGS12 plasmids for 24 h by using Lipofectamine 3000 (ThermoFisher), and lysates were extracted to perform immunoblots. The right panels show the quantitative data of relative intensity from immunoblots (means ± SEMs, ***P < 0.001, n = 3). (B) RAW264.7 cells were transfected with shCtrl or shRGS12 for 24 h as described in (A), and lysates were analyzed by immunoblots. The right panels show the quantitative data of relative intensity. The data were normalized to β-actin. ***P < 0.001 versus the shCtrl group (n = 3). (C, D) Representative images from Transwell assays in RAW264.7 macrophages transfected with pCMV-RGS12 for 24 h with lipopolysaccharide (LPS) or phosphate-buffered saline (PBS) (1 µg/mL). The migrated average cell number was measured and calculated under random 20× fields (C). Bar charts showed the quantification of migrated cells per field (D). **P < 0.01, ***P < 0.001 versus the vector group, n = 5. (E, F) RAW264.7 macrophages were transfected with shRGS12 or shCtrl for 24 h with LPS or PBS and analyzed by Transwell assays (bar, 25 µm). Quantification of migrated cells as described in (C) and (D). Values are means ± SEMs (***P < 0.001 versus the shCtrl group, n = 5).
Discussion
The pathological consequences of periodontitis result from the dynamic and polymicrobial oral microbiome and thereby inflammatory cell infiltration (Lamont et al. 2018). Phagocytosis is a basic process for nutrition in unicellular organisms, and it is also found in professional phagocytes such as macrophages and neutrophils (Uribe-Querol and Rosales 2020). These cells are critical in eliminating dead cells and maintaining homeostasis (Gordon 2016; Uribe-Querol and Rosales 2020). By analyzing the protein database in human peripheral blood mononuclear cells, we found that RGS12 showed the highest expression level in monocytes and the lowest expression level in neutrophils compared to other immune cells. Its expression level in monocytes was 8-fold higher than that in neutrophils (Appendix Fig. 5). Thus, these findings suggest that the knockout of RGS12 may dominantly affect the monocytes and have a limited impact on the activation of neutrophils (Appendix Fig. 5). M1 macrophages could transfer to M2 macrophages under specific conditions. M1 macrophages exposed to the IL13 cytokine showed the M2 phenotype and gained phagocytic activity, whereas M2 macrophages that switched to M1 macrophages lost their endocytic activity (Tarique et al. 2015). A previous study also found M2 macrophages have higher phagocytic activity than M1 macrophages, so the M2 macrophages play a major role in phagocytosis (Jaggi et al. 2020). In our study, we found RGS12 promotes the M1 macrophage polarization and its marker gene expression and does not alter M2 macrophage marker gene expression. These findings suggest that phagocytosis may not be a major factor regulated by RGS12. However, we cannot neglect the phagocytosis ability since it could maintain homeostasis. Therefore, in future studies, we will focus on whether RGS12 can regulate the macrophage phagocytosis ability in different environments.
RGS proteins were reported to play critical roles in regulating cell functions (De Vries and Gist Farquhar 1999). Our previous studies showed that RGS12 promotes osteoclast differentiation through regulating RANKL signaling (Yang and Li 2007), but whether RGS12 also regulates osteoclastogenesis in alveolar bone during periodontitis is unknown. Osteoclasts are multinucleated giant cells that differentiate from myeloid precursors under the influence of the M-CSF and RANKL (Feng and Teitelbaum 2013). Thus, study of hematopoietic lineage cells will help us better understand the different stages for RGS12 regulating osteoclastogenesis and inflammatory response during periodontitis. Macrophages have different functional properties in response to environment-derived signals (Mantovani et al. 2004). Gosselin et al. (2007) reported that macrophages transferred to a different environment can upregulate or downregulate the expression of a vast array of genes, such as preferentially decreasing the expression of microglia-specific genes such as RUNX (RUNX1 and RUNX2) and SMAD3 (Gosselin et al. 2017). In an inflammatory environment, RGS12 is activated and translocated into the nucleus to further promote inflammation (Yuan, Yang, Ng, et al. 2020). In this study, we found overexpression of RGS12 in macrophages showed a feature similar to macrophage exposure to LPS, which promotes M1 macrophage polarization and function. These findings suggest that RGS12 may trigger the potentiated cytotoxic properties of macrophages via promoting an inflammatory setting.
Taken together, this study for the first time uncovers that RGS12 activates the innate immune response through promoting the macrophage polarization toward M1 phenotype, migration, and osteoclastogenesis (Fig. 5I). Importantly, the lack of RGS12 in macrophages can inhibit both bone loss and inflammatory cell infiltration in periodontitis. Thus, our study provides a promising drug target that may facilitate the clinical treatments for periodontitis.
Figure 5.
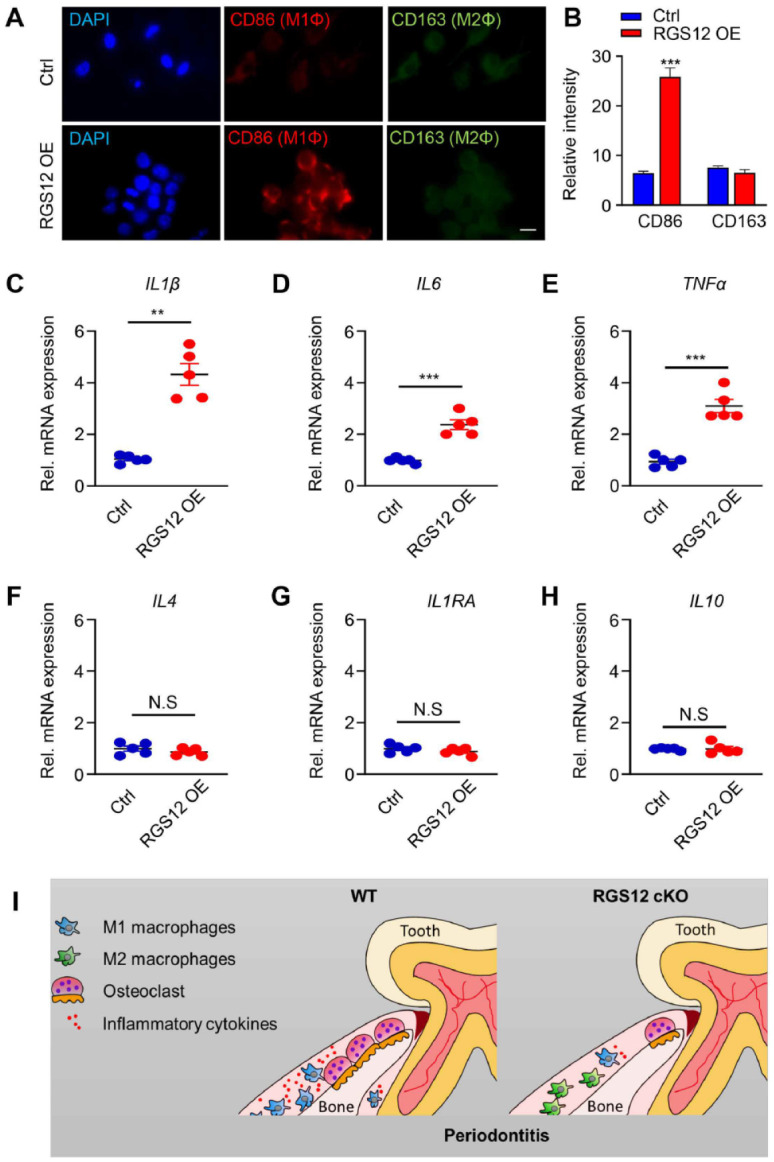
RGS12 promotes the polarization of macrophages toward the M1 phenotype. (A, B) RAW264.7 cells were transfected with pCMV-empty or pCMV-RGS12 plasmids for 48 h, and the M1 macrophage marker CD86 and M2 macrophage marker CD163 were detected by the immunofluorescence staining assay. (A) RAW264.7 cells were incubated with the anti-CD86 or anti-CD163 antibody followed by incubation with fluorescein isothiocyanate–conjugated secondary antibody, and the nuclei were stained with DAPI (blue). Images show CD86 in red and CD163 in green. (B) The expression of CD86 and CD163 in each cell was analyzed by the ImageJ densitometry method. Ten views in each well were analyzed. The relative intensity of the fluorescence was determined by comparing each intensity value to the average intensity. The blue bar represents pCMV-empty (Ctrl) and the red bar represents the pCMV-RGS12 overexpression (RGS12 OE) (bar, 10 µm). n = 5, ***P < 0.001. (C–E) RAW264.7 cells were transfected with pCMV-RGS12 or pCMV-empty (Ctrl) plasmids for 48 h, and the M1 macrophage markers were detected by real-time polymerase chain reaction (PCR) assay. Note that overexpression of RGS12 can increase the IL1β, IL6, and TNFα levels. **P < 0.01, ***P < 0.001 versus the control group, n = 5. (F–H) RAW264.7 cells were treated as described in (C) to (E), and the M2 macrophage markers (IL4, IL1RA, and IL10) were detected by real-time PCR assay. Values are means ± SEMs (P > 0.05 versus the control group, n = 5). (I) Schematic for RGS12 role in periodontitis. RGS12 promotes the polarization of M1 macrophages, the migration of macrophages, and the expression of proinflammatory factors. Moreover, RGS12 enhances osteoclast formation under inflammatory conditions. Finally, the conditional knockout of RGS12 prevents inflammation and alveolar bone erosion during periodontitis.
Author Contributions
G. Yuan, C. Fu, S. Yang, contributed to conception, design, data acquisition, analysis, and interpretation, drafted and critically revised the manuscript; S.T. Yang, D.Y. Yuh, contributed to conception and data acquisition, drafted and critically revised the manuscript; G. Hajishengallis, contributed to conception and data analysis, drafted and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Supplemental Material
Supplemental material, sj-docx-1-jdr-10.1177_00220345211045303 for RGS12 Drives Macrophage Activation and Osteoclastogenesis in Periodontitis by G. Yuan, C. Fu, S.T. Yang, D.Y. Yuh, G. Hajishengallis and S. Yang in Journal of Dental Research
Footnotes
A supplemental appendix to this article is available online.
Declaration of Conflicting Interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the National Institute on Aging (AG048388), National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) (AR066101), and National Institute of Dental and Craniofacial Research (DE026152) to S. Yang. This work was supported by grants from the Penn Center for Musculoskeletal Disorders, National Institutes of Health/NIAMS P30-AR069619.
ORCID iDs: C. Fu  https://orcid.org/0000-0001-5921-559X
https://orcid.org/0000-0001-5921-559X
G. Hajishengallis
https://orcid.org/0000-0001-7392-8852
References
- Amantea D, Certo M, Petrelli F, Tassorelli C, Micieli G, Corasaniti MT, Puccetti P, Fallarino F, Bagetta G. 2016. Azithromycin protects mice against ischemic stroke injury by promoting macrophage transition towards M2 phenotype. Exp Neurol. 275(Pt 1):116–125. [DOI] [PubMed] [Google Scholar]
- De Vries L, Gist Farquhar M. 1999. Rgs proteins: more than just gaps for heterotrimeric g proteins. Trends Cell Biol. 9(4):138–144. [DOI] [PubMed] [Google Scholar]
- Dutzan N, Kajikawa T, Abusleme L, Greenwell-Wild T, Zuazo CE, Ikeuchi T, Brenchley L, Abe T, Hurabielle C, Martin D, et al. 2018. A dysbiotic microbiome triggers TH17 cells to mediate oral mucosal immunopathology in mice and humans. Sci Transl Med. 10(463):eaat0797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eke PI, Borgnakke WS, Genco RJ. 2020. Recent epidemiologic trends in periodontitis in the USA. Periodontology. 2000. 82(1):257–267. [DOI] [PubMed] [Google Scholar]
- Feng X, Teitelbaum SL. 2013. Osteoclasts: new insights. Bone Res. 1(1):11–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu C, Yuan G, Yang ST, Zhang D, Yang S. 2020. RGS12 represses oral cancer via the phosphorylation and SUMOylation of PTEN. J Dent Res. 100(5):522–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gensel JC, Kopper TJ, Zhang B, Orr MB, Bailey WM. 2017. Predictive screening of M1 and M2 macrophages reveals the immunomodulatory effectiveness of post spinal cord injury azithromycin treatment. Sci Rep. 7:40144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S. 2016. Phagocytosis: an immunobiologic process. Immunity. 44(3):463–475. [DOI] [PubMed] [Google Scholar]
- Gosselin D, Skola D, Coufal NG, Holtman IR, Schlachetzki JCM, Sajti E, Jaeger BN, O’Connor C, Fitzpatrick C, Pasillas MP, et al. 2017. An environment-dependent transcriptional network specifies human microglia identity. Science. 356(6344):eaal3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gully N, Bright R, Marino V, Marchant C, Cantley M, Haynes D, Butler C, Dashper S, Reynolds E, Bartold M. 2014. Porphyromonas gingivalis peptidylarginine deiminase, a key contributor in the pathogenesis of experimental periodontal disease and experimental arthritis. PLoS One. 9(6):e100838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajishengallis G, Chavakis T. 2021. Local and systemic mechanisms linking periodontal disease and inflammatory comorbidities. Nat Rev Immunol. 21(7):426–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasturk H, Kantarci A. 2015. Activation and resolution of periodontal inflammation and its systemic impact. Periodontology 2000. 69(1):255–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasturk H, Kantarci A, Van Dyke TE. 2012. Oral inflammatory diseases and systemic inflammation: role of the macrophage. Front Immunol. 3:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirayama D, Iida T, Nakase H. 2017. The phagocytic function of macrophage-enforcing innate immunity and tissue homeostasis. Int J Mol Sci. 19(1):92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Italiani P, Boraschi D. 2014. From monocytes to M1/M2 macrophages: phenotypical vs. functional differentiation. Front Immunol. 5:514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablonski KA, Amici SA, Webb LM, Ruiz-Rosado Jde D, Popovich PG, Partida-Sanchez S, Guerau-de-Arellano M. 2015. Novel markers to delineate murine M1 and M2 macrophages. PLoS One. 10(12):e0145342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaggi U, Yang M, Matundan HH, Hirose S, Shah PK, Sharifi BG, Ghiasi H. 2020. Increased phagocytosis in the presence of enhanced M2-like macrophage responses correlates with increased primary and latent HSV-1 infection. PLoS Pathog. 16(10):e1008971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Y, Darzi Y, Tawaratsumida K, Marchesan JT, Hasegawa M, Moon H, Chen GY, Nunez G, Giannobile WV, Raes J, et al. 2013. Induction of bone loss by pathobiont-mediated Nod1 signaling in the oral cavity. Cell Host Microbe. 13(5):595–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayal RA. 2013. The role of osteoimmunology in periodontal disease. Biomed Res Int. 2013:639368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kononen E, Gursoy M, Gursoy UK. 2019. Periodontitis: a multifaceted disease of tooth-supporting tissues. J Clin Med. 8(8):1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourtzelis I, Li X, Mitroulis I, Grosser D, Kajikawa T, Wang B, Grzybek M, von Renesse J, Czogalla A, Troullinaki M, et al. 2019. Del-1 promotes macrophage efferocytosis and clearance of inflammation. Nat Immunol. 20(1):40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn R, Schwenk F, Aguet M, Rajewsky K. 1995. Inducible gene targeting in mice. Science. 269(5229):1427–1429. [DOI] [PubMed] [Google Scholar]
- Laine ML, Loos BG, Crielaard W. 2010. Gene polymorphisms in chronic periodontitis. Int J Dent. 2010:324719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert NA, Johnston CA, Cappell SD, Kuravi S, Kimple AJ, Willard FS, Siderovski DP. 2010. Regulators of G-protein signaling accelerate GPCR signaling kinetics and govern sensitivity solely by accelerating GTPase activity. Proc Natl Acad Sci U S A. 107(15):7066–7071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamont RJ, Koo H, Hajishengallis G. 2018. The oral microbiota: dynamic communities and host interactions. Nat Rev Microbiol. 16(12):745–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. 2004. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 25(12):677–686. [DOI] [PubMed] [Google Scholar]
- Mosser DM, Edwards JP. 2008. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 8(12):958–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navegantes KC, de Souza Gomes R, Pereira PAT, Czaikoski PG, Azevedo CHM, Monteiro MC. 2017. Immune modulation of some autoimmune diseases: the critical role of macrophages and neutrophils in the innate and adaptive immunity. J Transl Med. 15(1):36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng AYH, Li Z, Jones MM, Yang S, Li C, Fu C, Tu C, Oursler MJ, Qu J, Yang S. 2019. Regulator of G protein signaling 12 enhances osteoclastogenesis by suppressing Nrf2-dependent antioxidant proteins to promote the generation of reactive oxygen species. Elife. 8:e42951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien JB, Wilkinson JC, Roman DL. 2019. Regulator of G-protein signaling (RGS) proteins as drug targets: progress and future potentials. J Biol Chem. 294(49):18571–18585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokol CL, Luster AD. 2015. The chemokine system in innate immunity. Cold Spring Harb Perspect Biol. 7(5):a016303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarique AA, Logan J, Thomas E, Holt PG, Sly PD, Fantino E. 2015. Phenotypic, functional, and plasticity features of classical and alternatively activated human macrophages. Am J Respir Cell Mol Biol. 53(5):676–688. [DOI] [PubMed] [Google Scholar]
- Uribe-Querol E, Rosales C. 2020. Phagocytosis: our current understanding of a universal biological process. Front Immunol. 11:1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn TA, Vannella KM. 2016. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. 44(3):450–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Li YP. 2007. RGS12 is essential for RANKL-evoked signaling for terminal differentiation of osteoclasts in vitro. J Bone Miner Res. 22(1):45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Li YP, Liu T, He X, Yuan X, Li C, Cao J, Kim Y. 2013. Mx1-cre mediated rgs12 conditional knockout mice exhibit increased bone mass phenotype. Genesis. 51(3):201–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan G, Yang S, Liu M, Yang S. 2020. RGS12 is required for the maintenance of mitochondrial function during skeletal development. Cell Discov. 6:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan G, Yang S, Ng A, Fu C, Oursler MJ, Xing L, Yang S. 2020. RGS12 is a novel critical NF-κB activator in inflammatory arthritis. iScience. 23(6):101172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuh DY, Maekawa T, Li X, Kajikawa T, Bdeir K, Chavakis T, Hajishengallis G. 2020. The secreted protein DEL-1 activates a β3 integrin-FAK-ERK1/2-RUNX2 pathway and promotes osteogenic differentiation and bone regeneration. J Biol Chem. 295(21):7261–7273. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, sj-docx-1-jdr-10.1177_00220345211045303 for RGS12 Drives Macrophage Activation and Osteoclastogenesis in Periodontitis by G. Yuan, C. Fu, S.T. Yang, D.Y. Yuh, G. Hajishengallis and S. Yang in Journal of Dental Research