

Abstract
Queuosine (Q) is a highly modified nucleoside of transfer RNA that is formed from guanosine triphosphate over the course of eight steps. The final step in this process, involving the conversion of epoxyqueuosine (oQ) to Q, is catalyzed by the enzyme QueG. A recent X-ray crystallographic study revealed that QueG possesses the same cofactors as reductive dehalogenases, including a base-off Co(II)cobalamin (Co(II)Cbl) species and two [4Fe-4S] clusters. While the initial step in the catalytic cycle of QueG likely involves the formation of a reduced Co(I)Cbl species, the mechanisms employed by this enzyme to accomplish the thermodynamically challenging reduction of base-off Co(II)Cbl to Co(I)Cbl and to convert oQ to Q remain unknown. In this study, we have used electronic paramagnetic resonance (EPR) and magnetic circular dichroism (MCD) spectroscopies in conjunction with whole-protein quantum mechanics/molecular mechanics (QM/MM) computations to further characterize wild type QueG and select variants. Our data indicate that the Co(II)Cbl cofactor remains five-coordinate upon substrate binding to QueG. Notably, during a QM/MM optimization of a putative QueG reaction intermediate featuring an alkyl–Co(III) species, the distance between the Co ion and coordinating C atom of oQ increased to >3.3 Å and the C–O bond of the epoxide reformed to regenerate the oQ-bound Co(I)Cbl reactant state of QueG. Thus, our computations indicate that the QueG mechanism likely involves single-electron transfer from the transient Co(I)Cbl species to oQ rather than direct Co–C bond formation, similar to the mechanism that has recently been proposed for the tetrachloroethylene reductive dehalogenase PceA.
Keywords: Epoxyqueuosine reductase, reductive dehalogenase, magnetic circular dichroism, electron paramagnetic resonance, quantum mechanics/molecular mechanics computations
Graphical Abstract
1. Introduction
To date, more than 140 different modifications have been identified in RNA molecules.1 These modified RNA molecules include intermediates along sequential, multistep syntheses of hypermodified nucleosides. Queuosine (Q) is one example of a highly modified nucleoside of transfer RNA (tRNA) that is formed from guanosine triphosphate (GTP) over the course of eight steps. The presence of Q in the wobble position of tRNA has been known for nearly 50 years;2,3 however, the final enzyme in the de novo biosynthetic pathway responsible for converting epoxyqueuosine (oQ) to Q, termed QueG (Figure 1), has only recently been identified.4,5 Q is incorporated at the wobble position, position 34, of the 5′-GUN-3′ anticodon, which decodes substrate codons with either a corresponding U or C in the 3’ position (encoding Asp, Asn, His, and Tyr).6 Q has an unclear biological role, but because it is conserved in the RNA of organisms in nearly all kingdoms of life, this tRNA modification may be of key importance.7 The presence of Q in tRNA has been shown to stimulate methylation of DNMT2, the most strongly conserved cytosine DNA methyltransferase in eukaryotes.8 Alternatively, the lack of Q in tRNA correlates with negative physiological phenomena such as cancer pathology9–13 and neurological disorders14. At the molecular level, the lack of Q has been implicated in the production of unfolded proteins that trigger endoplasmic reticulum stress15 and increased ribonuclease cleavage of cognate tRNA molecules.16
Figure 1.
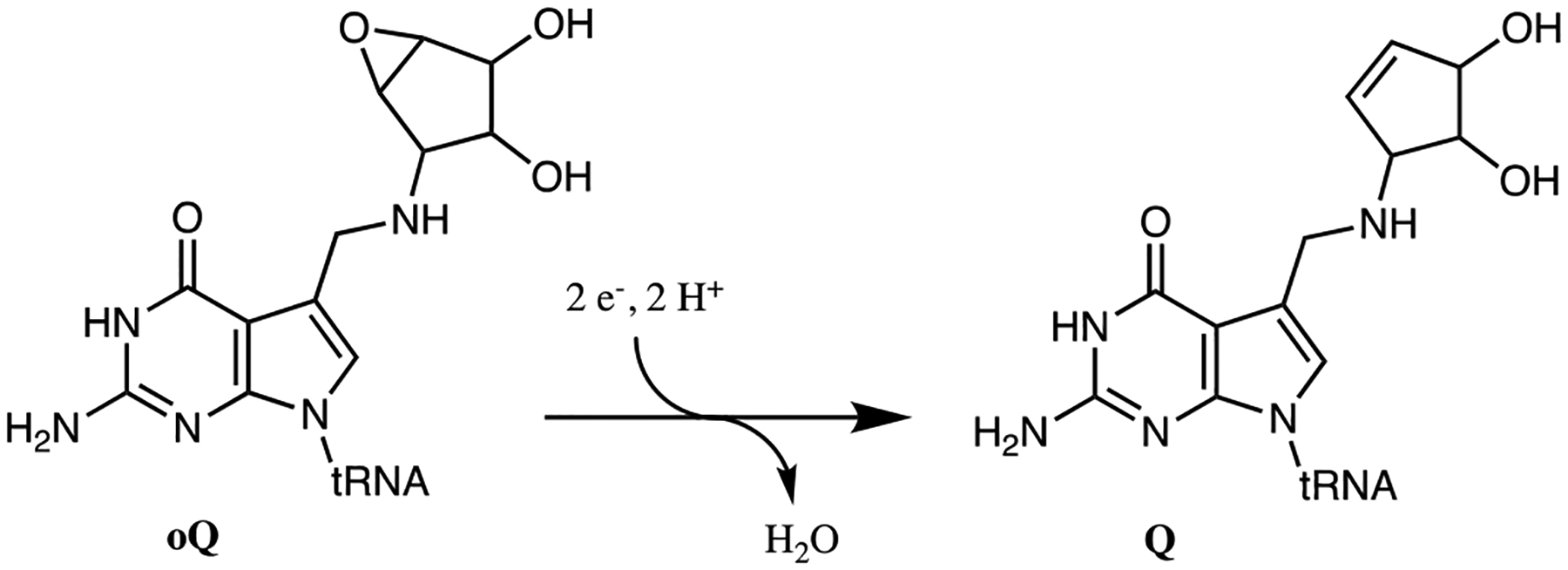
Conversion of oQ to Q as catalyzed by QueG.
Surprisingly, QueG shows little similarity with other tRNA modifying enzymes, even though oQ is already incorporated into the tRNA molecule before it is converted to Q. Instead, QueG was found to possess the same cofactors as, and share high sequence similarity with the reductive dehalogenases (RDases) PceA17 and NpRdhA,18 all of which contain a base-off Co(II)cobalamin (Co(II)Cbl) species and two [4Fe-4S] clusters.19 QueG and these RDases constitute a recently discovered, relatively poorly studied class of cobalamin-dependent enzymes that are isolated in the Co(II) state with an axial water molecule completing the five-coordinate, square pyramidal coordination environment of the cobalt center. This class differs from the extensively characterized adenosylcobalamin (AdoCbl) and methylcobalamin (MeCbl) dependent enzymes, which catalyze radical-mediated rearrangement reactions and methyl transfer reactions, respectively.20–28 While experimental and computational studies have provided evidence that RDases likely use an outer-sphere electron transfer from a reduced Co(I)Cbl species to initiate substrate dehalogenation reactions,17,29–31 the mechanism that QueG employs to convert oQ to Q remains unexplored. Also, it is not known how QueG facilitates the thermodynamically challenging reduction of base-off Co(II)Cbl to Co(I)Cbl to initiate substrate turnover.
Before an X-ray crystal structure of QueG had been solved, biochemical studies were performed to identify specific amino acid residues that important for enzymatic activity.32 These studies demonstrated that substitution of the conserved D104, Y105, H106, or D134 residues by an alanine residue had a particularly large negative effect on the rate of oQ reduction by QueG in vivo. Consistent with this finding, the electron paramagnetic resonance (EPR) spectrum obtained for the Y105A variant was dominated by contributions from a unique Co(II)Cbl species, while that of the D134A QueG variant could be simulated by adding the EPR spectra of the wild-type (WT) and Y105A proteins in a 1:1 ratio.32 In contrast, the EPR spectra of the H106A and D104A variants exhibited only minor differences from the WT QueG spectrum. X-ray crystal structures confirmed that the residues found to be critical to support enzymatic activity are in close proximity to the Co ion (D134 and Y105) or directly adjacent to the active site (H106 and D104).4 Additionally, it was observed that D134 is on a labile loop that points into the active site when substrate is bound (pdb entry 5d0b), but can be found pointing in (Asp-in state, pdb entry 5d08) or out (Asp-out state, pdb entry 5t8y) otherwise.4
In this study, we have used EPR and magnetic circular dichroism (MCD) spectroscopies to further characterize Co(II)Cbl bound to QueG. The effect of the hydrogen bond between Y105 and D134 was investigated by comparing data obtained for WT QueG as well as the Y105A and Y105F variants. Additionally, the first EPR spectra of QueG in the presence of oQ were collected to explore the effect of substrate binding to the active site on the Co(II)Cbl coordination environment. Whole-protein models of QueG in the absence and presence of substrate were generated via combined quantum mechanics/molecular mechanics (QM/MM) geometry optimizations and validated on the basis of published X-ray crystal structures and spectroscopic data.
2. Experimental Methods
Protein Production.
Protein was produced and reconstituted as previously described.32
Sample Preparation.
All chemicals were purchased from Sigma and used as received without further purification. Water was purified in a Millipore Milli-Q water purification system.
Diaquacob(III)inamide [(H2O)2Cbi2+] was prepared by adding the reductant NaBH4 to an aqueous solution of dicyanocob(III)inamide [(CN)2Cbi], loading the reaction mixture on a C18 SepPack column, washing with H2O, and eluting the product with methanol, as described in a previous report.33 (H2O)2Cbi2+ was then reduced to Co(II)Cbi+ in an anaerobic environment via dropwise addition of saturated potassium formate in buffer containing 50 mM TrisHCl, 100 mM KCl, and 2 mM DTT (pH 8).33 Absorption spectroscopy was used to verify the oxidation state of the product and determine its concentration using Beer’s Law via the reported molar extinction coefficient for Co(II)Cbi+ of 11 mM−1cm−1 at 470 nm.34
An oQ-containing stem loop substrate was generated as previously described.19 Dried oQ stem loop was dissolved in doubly distilled, RNA free water. QueG was incubated with oQ for 3 minutes prior to the addition of glycerol and freezing in liquid nitrogen.
Spectroscopy.
Low temperature electronic absorption (Abs) and magnetic circular dichroism (MCD) spectra were collected with a Jasco J-715 spectropolarimeter in conjunction with an Oxford Instruments SM-4000 8 T super conducting magnetocryostat or 7 T SpectromagPT cryofree magnet. For low-temperature studies, samples were prepared with 55–60% (v/v) glycerol to ensure glass formation upon freezing in liquid nitrogen. Final protein concentrations ranged from 150 to 180 μM. CD background and glass-strain contributions to the MCD spectra were removed by taking the difference between spectra collected with the magnetic field oriented parallel and anti-parallel to the axis of light propagation.
X-Band EPR spectra were recorded on a Bruker ESP 300E spectrometer using an Oxford ESR 900 continuous-flow liquid helium cryostat and an Oxford ITC4 temperature controller. Final protein concentrations were 175 μM. Variable concentrations of glycerol were used to explore the effect of this glassing agent on the protein conformation. All spectra were collected with the following parameters: 2.0 mW (non-saturating) microwave power, 9.390 GHz microwave frequency, 87 ms conversion time, 327.68 ms time constant, 5 G modulation amplitude, 100 kHz modulation frequency, and 60 dB gain. Spectra were simulated with EasySpin.35
Generation of Computational Models.
All computational models were optimized by performing quantum mechanics/molecular mechanics (QM/MM) calculations with the ONIOM method as implemented in Gaussian09.36 Published crystallographic data for selenomethionine-labeled QueG (pdb entry 5d084) were used as the initial coordinates for the Asp-in model of WT QueG and served as the basis for the generation of all other models. Selenomethionine was reverted to methionine in silico. The D134 containing loop from pdb entry 5t8y4 was substituted for the corresponding amino acids from pdb entry 5d084 to generate the Asp-out model. To create the product-bound QueG model, the Q nucleoside from pdb entry 5d0b4 was truncated to queuine and added to the optimized model based on pdb entry 5d08.4 The substrate-bound QueG model was generated by in silico addition of the epoxide to the initial product-bound model. H atoms were added to the initial QueG models using the program PDBtoPQR37 for the protein, and manually for Co(II)Cbl, Q, and oQ. For the QM/MM geometry optimizations, the QM region included the sidechains of residues Y105, H106, D134, Q220, and W294, the Co ion and core of the corrin macrocycle (including the first carbon of each of the sidechains) of the Co(II)Cbl cofactor, water molecules within the active site, and substrate or product as applicable. The QM region was treated with the B3LYP38,39 functional and TZVP40 basis set (note that initial attempts to perform QM/MM geometry optimizations with the BP86 functional failed to converge). The remainder of the enzyme was modeled with the AMBER MM forcefield.36,41 Bonds that cross the boundary between the QM and MM regions were treated with a capping H atom, with the C–H bond lengths scaled to 0.709 of the original C–C bond lengths. AMBER parameters for the cobalamin were taken from the literature42 and were not needed for Q or oQ, as those molecules were included in the QM region. All optimizations were performed using the default ONIOM convergence criteria and a charge and spin multiplicity consistent with a low-spin Co(II)Cbl species for the resting and product/substrate bound states (total S=1/2), except for the model containing a Co(III)Cbl species formed via nucleophilic attack of the reduced Co(I) ion on one of the epoxide carbon atoms (total S=0). Figures for all models were created using PyMOL.43
The root mean square deviation (rmsd) of atomic positions between the optimized models and published crystal structures were determined using the align method in PyMOL.43 The Asp-in QueG model has a rmsd of 0.858 Å (for 2,410 of atoms) from the corresponding crystal structure (pdb entry 5d084). The Asp-out QueG model generated from a combination of pdb entries 5d084 and 5t8y4 has a rmsd of 1.048 Å (for 2,458 of atoms) from the Asp-out crystal structure (pdb entry 5t8y4). The substrate containing models with and without a lower axial water have rmsd values of 0.989 Å (2,397 atoms) and 0.946 Å (2,386 atoms), respectively, compared to the crystal structure of substrate-bound QueG (pdb entry 5d0b4). Similarly, the product containing models with and without a lower axial water have rmsd values of 0.985 Å (2,416 atoms) and 0.940 Å (2,387 atoms), respectively, when compared to the substrate-bound QueG crystal structure (pdb entry 5d0b4). These small rmsd values indicate that all of the QM/MM optimized QueG models are reasonable.
TD-DFT Calculations.
After convergence of the QM/MM geometry optimizations, the coordinates of the QM region, including those of the capping hydrogen atoms, were extracted for time-dependent density functional theory (TD-DFT) calculations using the ORCA4.044 software package. All TD-DFT calculations were performed with the cam-B3LYP45 functional. The def2-SVP46 and def2/J47 basis sets were employed for all atoms except for Co and all coordinating atoms, for which the TZVP40 basis set was employed. The RIJCOSX approximation as implemented in ORCA4.0 was used to speed up the calculation of the Hartree-Fock exchange term.
EPR Parameter Calculations.
EPR parameters were calculated with ORCA4.044 for the same active site models as those used for TD-DFT calculations. The B3LYP39,45 functional along with the CP(PPP)48 basis set was used for Co and IGLO-III49 for all ligating atoms. The SVP50 basis set was employed for all other atoms. The resolution of the identity (RI) approximation in conjunction with the SV/J auxiliary basis set were used to speed up the calculation of the Coulomb term.44,48,51 The EPR g values and A(59Co) hyperfine tensor were computed using coupled-perturbed self-consistent field (CP-SCF) theory, with a complete mean-field treatment of spin-orbit coupling and the IGLO gauge.49
3. Results
Characterization of QueG Resting State
Because the 4.5 K Abs spectrum of as-isolated QueG (Figure S1) has contributions from both the base-off Co(II)Cbl cofactor and the two [4Fe-4S]2+ clusters, we have used MCD spectroscopy to probe the electronic structure of QueG-bound Co(II)Cbl. MCD signal intensities of paramagnetic species increase with decreasing temperature; thus, low-temperature MCD spectra of QueG are dominated by contributions from Co(II)Cbl. An additional advantage of this technique is that the prominent features below ~22,000 cm−1 in the MCD spectra of Co(II)Cbl species are due to Co ligand field (LF) transitions, which are particularly sensitive to changes in the axial coordination environment of the cobalt center. Consistent with X-ray crystallographic and EPR data reported in the literature, our MCD spectrum of as-isolated QueG is similar to that of Co(II)cobinamide [Co(II)Cbi+] (Figure 2), a natural Co(II)Cbl precursor that lacks the intramolecular base and serves as a model of base-off Co(II)Cbl at neutral pH. Yet, the WT QueG MCD spectrum exhibits an additional weak, derivative-shaped feature centered at ~14,500 cm−1, and the features in the 17,000 – 22,000 cm−1 range have different relative intensities than their counterparts in the Co(II)Cbi+ spectrum, revealing small differences in the Co(II)–OH2 bonding interactions in these two species.
Figure 2.
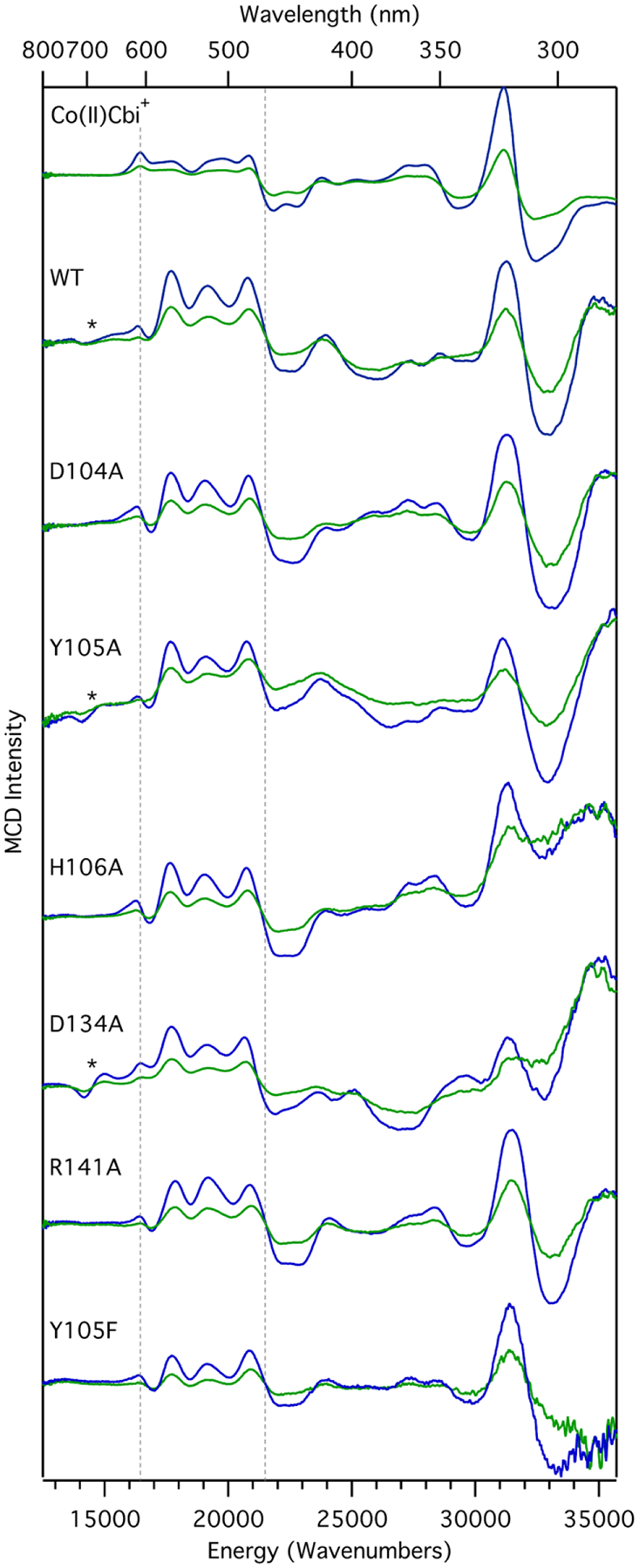
MCD spectra collected at 7 T and 4.5 K (blue) and 15 K (green) of Co(II)Cbi+, as well as WT QueG and select variants. The lower and upper limits of the region dominated by Co(II) LF transitions are indicated by vertical broken lines. The low energy, derivative-shaped feature centered at ~14,000 cm−1 is marked by an asterisk.
To complement our spectroscopic data of as-isolated QueG, a whole-protein computational model was generated via QM/MM geometry optimization starting from the crystal structure of WT QueG in the Asp-in conformation (pdb entry 5d084). The optimized model is in excellent agreement with the crystallographically determined structure, with the most notable difference being a ~15° rotation of W294 into the active site (Figure S2). The computational model was validated further by comparing the TD-DFT computed Abs spectrum for the QM region, which included the core of the Co(II)Cbl cofactor and relevant active site residues, to our spectroscopic data. Consistent with our MCD spectra, the TD-DFT computed Co(II) LF transition energies and Abs intensities for QueG and Co(II)Cbi+ are very similar (Figure S3). The small differences in energies and relative MCD intensities of these transitions observed experimentally can be attributed to the distinct hydrogen-bonding interactions involving the axial water molecule of the base-off Co(II)Cbl cofactor in the QueG active site and of free Co(II)Cbi+ in aqueous solution. Notably, the TD-DFT computation for the WT QueG model does not predict the appearance of a new transition near 14,500 cm−1, where a derivative-shaped feature with variable relative intensity is observed in some of the MCD spectra obtained for the different QueG species investigated (Figure 2).
Analysis of Active Site Variants
Previous EPR spectroscopic studies of QueG32 demonstrated that the D104A, H106A, and R141A substitutions perturbed the QueG-bound Co(II)Cbl cofactor minimally. On the other hand, the D134A and Y105A QueG variants displayed EPR spectra with unique features that had no counterparts in the WT spectrum. It is thus surprising that our MCD spectra obtained for the Y105A and D134A variants are very similar to that of WT QueG (Figure 2). As the MCD features in the 15,000–21,000 cm−1 region are particularly sensitive to changes in the axial ligand environment,52 the energies and relative intensities of these features would be expected to vary much more dramatically, especially between the MCD spectra of the Y105A variant and the WT enzyme.
Because different relative amounts of glycerol were used to prepare our MCD samples (55% v/v) and the EPR samples by Britt and coworkers32 (15% v/v), it seemed possible that the enzyme conformation is affected by the addition of glycerol. Indeed, while the EPR spectrum of a Y105A QueG sample containing no glycerol (Figure 3) is similar to that previously reported for this variant,32 the spectrum obtained for a Y105A QueG sample containing 55% (v/v) glycerol is much more similar to that of the WT enzyme (Figure 3). The striking similarities between the MCD spectra of WT QueG and the different variants investigated can then be attributed to the high concentration of glycerol needed to prepare these samples. As glycerol is known to cause proteins to favor of more compact states,53 it is possible that the catalytic D134 residue, located on a labile loop, is stabilized in the Asp-in configuration in samples containing 55% glycerol (v/v).
Figure 3.
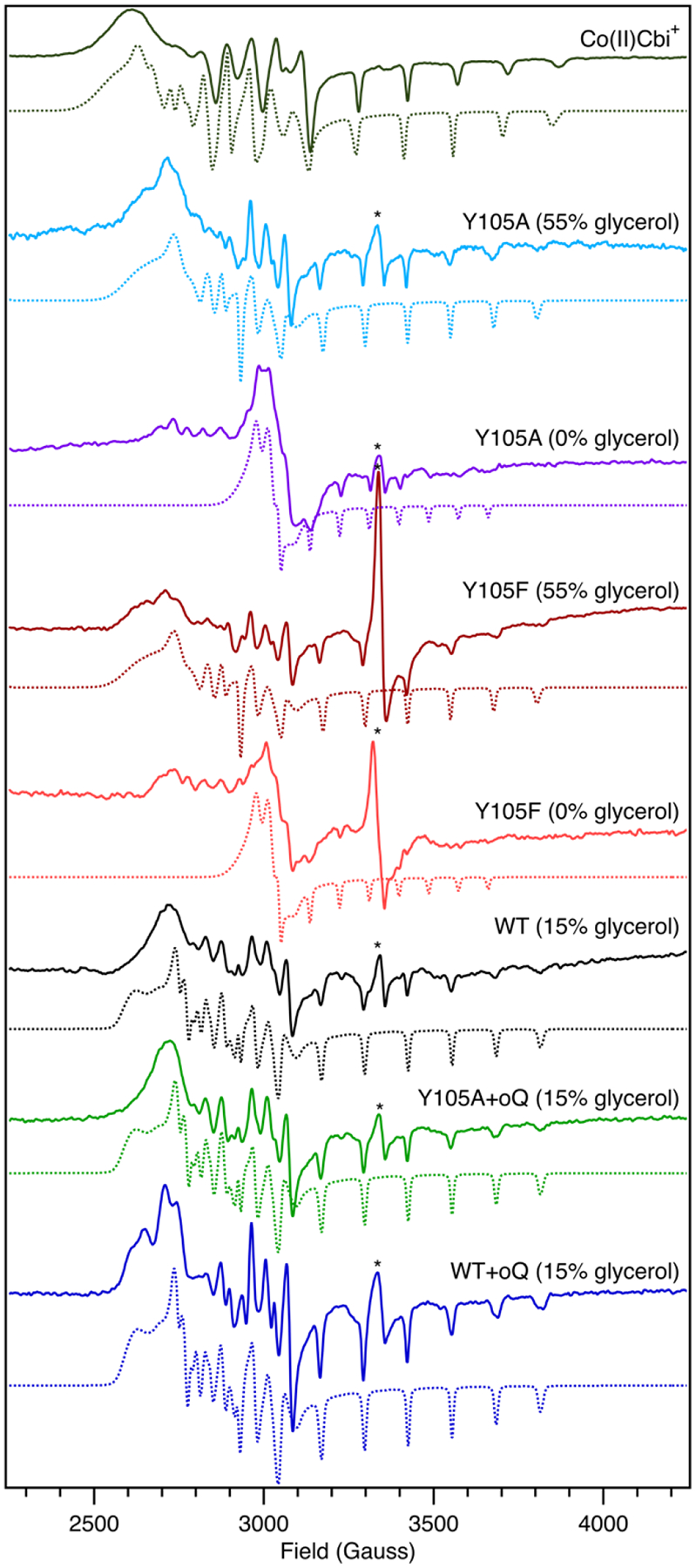
EPR data collected at 20 K of Co(II)Cbi+, as well as WT QueG and select variants in the absence and presence of oQ and variable amounts of glycerol. EPR simulation parameters are provided in Table 1. The asterisk (*) denotes contributions from [4Fe-4S] cluster degradation products.32
To test the hypothesis that the Y105A QueG EPR spectrum is unique because this variant favors the Asp-out configuration, a computational model of QueG with the loop containing D134 swung out of the active site (as captured in pdb entry 5t8y4) was generated. Inspection of this model reveals that when D134 is unavailable for hydrogen bonding, H106 and Y105 shift but maintain their relative positioning (Figure 4). Residues across the active site, such as W294 and Q220, do not move in response to the Asp-in to Asp-out transition. DFT-computed EPR g values and cobalt hyperfine coupling constants for Co(II)Cbi+ and the Co(II)Cbl species in the WT QueG Asp-out and Asp-in models are shown in Table 1. The calculated and observed g values and 59Co hyperfine tensors for Co(II)Cbi+ serve to assess the accuracy of these EPR calculations. While the magnitude of the z-component of the computed 59Co hyperfine tensor is overestimated and the computed gx value is underestimated compared to the experimentally determined parameters, the computational results are consistent with those previously reported for Co(II)Cbi+.54
Figure 4.
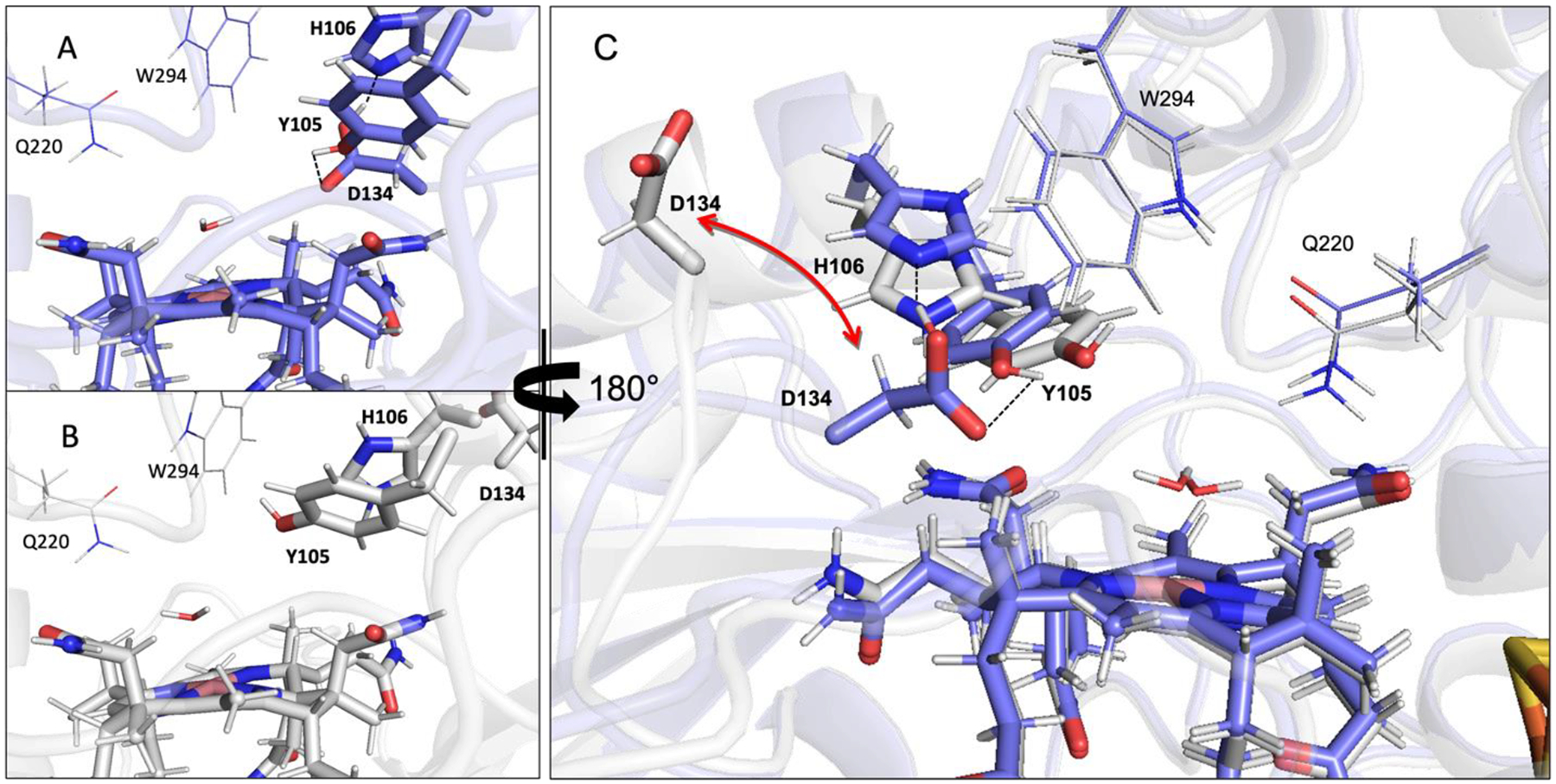
Comparison between the active site regions of (A) the Asp-in and (B) the Asp-out QueG computational models. These models are shown together in panel C, rotated by 180° from their orientations in panels A and B. The dashed lines in panel A and C indicate the hydrogen bond between D134 and Y105 that exists in the Asp-in state.
Table 1.
Computational and Experimental EPR g Values and Hyperfine Coupling Constants (MHz)
| gx | gy | gz | Ax (Co) | Ay (Co) | Az (Co) | |
|---|---|---|---|---|---|---|
| Computational Models | ||||||
| Co(II)Cbi+ | 2.252 | 2.231 | 2.008 | 218 | 222 | 584 |
| Asp-In | 2.228 | 2.212 | 2.008 | 184 | 179 | 560 |
| Asp-Out | 2.244 | 2.204 | 1.998 | 6 | 23 | 552 |
| oQ, no wat | 2.247 | 2.239 | 2.009 | 223 | 232 | 581 |
| oQ, ax wat | 2.260 | 2.227 | 2.009 | 227 | 251 | 602 |
| Experimental from This Study (no glycerol unless indicated otherwise) | ||||||
| Co(II)Cbi+ | 2.42 | 2.32 | 2.00 | 226 | 210 | 402 |
| Y105A (55% glycerol) | 2.422 | 2.290 | 1.922 | 123 | 146 | 358 |
| Y105A | 2.234 | 2.190 | 2.000 | 6 | 23 | 245 |
| Y105F (55% glycerol) | 2.422 | 2.290 | 1.922 | 123 | 146 | 358 |
| Y105F | 2.234 | 2.190 | 2.000 | 6 | 23 | 245 |
| WT (15% glycerol) | 2.415 | 2.293 | 1.992 | 162 | 144 | 358 |
| WT+oQ (15% glycerol) | 2.415 | 2.293 | 1.992 | 162 | 144 | 358 |
| Y105A+oQ (15% glycerol) | 2.411 | 2.295 | 1.992 | 163 | 146 | 358 |
| Experimental reported Previously (15% glycerol present)32 | ||||||
| WT | 2.437 | 2.355 | 2.061 | 186 | 133 | 340 |
| H106A | 2.435 | 2.354 | 2.064 | 186 | 130 | 330 |
| D104A | 2.396 | 2.349 | 2.065 | 125 | 125 | 333 |
| Y105A | 2.282 | 2.250 | 2.063 | 9 | 11 | 235 |
The most notable difference between the computed EPR parameters for the Asp-in and Asp-out QueG models in Table 1 is the larger g spread predicted for the first model. Thus, the decrease in g spread observed experimentally from WT QueG to its Y105A variant is consistent with the former adopting the Asp-in conformation and the latter adopting the Asp-out conformation, as predicted on the basis of the EPR results obtained for the Y105A variant in the absence and presence of glycerol. Because the upper axial water is bound at nearly the same distance for the Asp-in and Asp-out models (2.33 Å and 2.36 Å respectively), the difference in the computed EPR parameters underscores the importance of including second sphere residues in these calculations.
The Y105F QueG variant, in which the –OH functional group of the original Tyr residue is missing, was prepared to test the hypothesis that the key role of Y105 is to control the positioning of the axial water molecule of the Co(II)Cbl cofactor via its hydroxyl group. Indeed, the EPR spectrum of Y105F QueG is nearly identical to that observed for the Y105A variant both in the absence and presence of glycerol (Figure 3). These data suggest that the Y105A substitution leads to the appearance of a unique EPR spectrum primarily due to the loss of a hydrogen bond donor rather than a substantial active site reorganization caused by the replacement of the bulky Tyr residue by Ala.
Notably, the relative intensity of the weak, derivative-shaped MCD feature centered at ~14,500 cm−1 (Figure 2) correlates with the relative intensity of the EPR signals arising from [4Fe-4S] degradation products (Figure 3 and EPR data provided in ref32). For this reason and the fact that our TD-DFT computations do not predict the appearance of a new low-energy transition for QueG-bound Co(II)Cbl, we attribute the MCD feature at ~14,500 cm−1 to [4Fe-4S] degradation products.
Effects of Substrate Binding to the QueG Active Site
EPR spectroscopy was used to probe the effect that substrate (oQ) binding to the QueG active site has on the Co(II)Cbl cofactor. The EPR spectrum of WT QueG incubated with oQ is nearly superimposable on that of resting WT QueG, indicating that the Co(II) ion retains an axial ligand in the presence of substrate (Figure 3). Interestingly, binding of substrate has a much larger effect on the Y105A variant, with the EPR spectrum of this variant in the presence oQ more closely resembling that of WT QueG than of the Y105A variant in the absence of substrate and confirming that substrate does in fact bind to the active site. These data indicate that the binding of substrate oQ to QueG causes the enzyme to favor the Asp-in conformation, even in the case of the Y105A variant.
X-ray crystal structures of QueG revealed the presence of a water molecule 2.6 Å away from the Co(II) ion, serving as the upper axial ligand of Co(II)Cbl prior to substrate binding.4 No electron density attributable to a water molecule was found near the lower face of the corrin ring,4 and R141 is properly positioned to restrict accessibility of the Co(II) ion from the lower face. While these crystal structures could be interpreted to suggest that substrate binding to QueG leads to the formation of a four-coordinate Co(II)Cbl species, our EPR data clearly indicate that the Co(II) ion remains five-coordinate in this process, rather than four-coordinate as seemingly captured in the X-ray crystal structure of product bound QueG (pdb entry 5d0b4). To establish the coordination environment of the Co(II)Cbl species in substrate bound QueG, two computational models were considered, one with a water molecule added to the pocket between R141 and the lower face of the Co(II)Cbl cofactor to serve as the lower axial ligand, and one without water molecule to explore if oQ could coordinate to the Co(II) ion in the upper axial position (Figure 5). For both models the upper axial water molecule present in the crystal structure and computational model of substrate free QueG was removed prior to QM/MM optimization, as stipulated by the crystal structure of product bound QueG.
Figure 5.
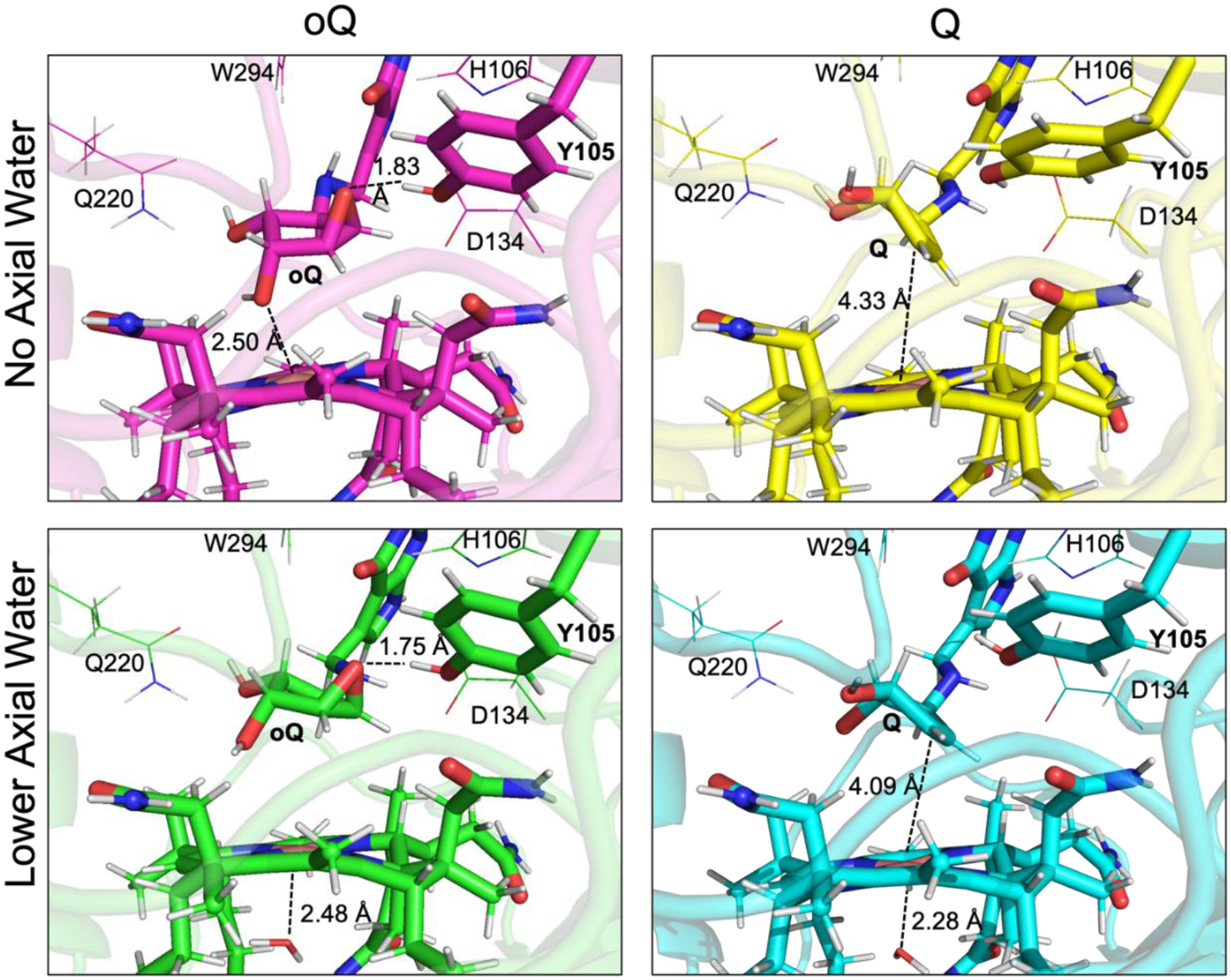
Substrate (left) and product (right) containing models. The models in the bottom row contain an axial water on the lower face of the Co(II)Cbl, which is absent in the models shown in the top row.
In the QM/MM optimized model of oQ-bound QueG that includes water molecule in the lower axial position, the epoxide-containing cyclopentanediol moiety rotated to place the epoxide oxygen 1.75 Å from the hydroxyl group of Y105 (Figure 5, bottom left), supporting a role of Y105 as the immediate proton donor to the substrate-derived anion intermediate during oQ turnover. Interestingly, D134, which has also been suggested to fulfill this role,4,5 is situated 4 Å from the epoxide oxygen. One of the hydroxyl groups of the cyclopentanediol moiety assists in proper substrate positioning through hydrogen bonding with Q220 (at a distance of 1.85 Å). Additionally, H106 and D134, which were identified as necessary for enzyme turnover,32 engage in a hydrogen-bonding network that can shuttle a proton to the transiently formed Y105 anion. In all models with D134 oriented into the active site (Asp-in conformation), H106 was initially modeled as an imidazolium cation. During geometry optimization, the extra proton migrated to the carboxylate group of D134, further supporting a role of these residues in proton shuttling.
Surprisingly, during QM/MM optimization of the oQ-bound QueG model lacking a water molecule in the lower axial position, a five-coordinate Co(II) environment was restored (Figure 5, top left). While the cylopentanediol moiety again rotated to place the epoxide oxygen 1.83 Å from the Y105 hydroxyl group, the ring additionally torqued to place the oxygen atom of one of the hydroxyl groups of to within bonding distance (2.50 Å) of the Co(II) ion. The description of this model as containing a five-coordinate Co(II) center is supported by EPR parameter calculations, which yielded similar results for this model and that of oQ-bound QueG with a lower axial water ligand (Table 1). Both substrate-bound models show hydrogen-bonding interactions between several active site residues and oQ, supporting a role of substrate in stabilizing the Asp-in QueG configuration. On the basis of these computational models and our EPR data, the diminished activity of the Y105A variant32 can then be attributed to the loss of the necessary proton donor rather than an Asp-out enzyme conformation in the presence of substrate.
Corresponding models with and without the lower axial water ligand were also generated with product Q bound (Figure 5, right). While the orientation of the cyclopentanediol moiety is largely unchanged between the substrate and product bound QueG models containing an axial water ligand, it is very different in the models lacking this ligand. Specifically, in the Q-bound QueG model lacking an axial water ligand, the Co(II) center no longer engages in a bonding interaction with the oxygen atom of one of the alcohol groups and thus resides in a four-coordinate ligand environment. However, during QueG turnover, this four-coordinate Co(II)Cbl species could be stabilized by coordination of the water molecule that is formed upon oQ reduction.
Computational Insights into the QueG Reaction Mechanism
Thus far, mechanistic proposals for QueG have been limited to a nucleophilic attack of the reduced Co(I) ion on one of the epoxide carbon atoms, coupled with protonation by either Y105 or D134 (B. subtilis numbering).4,5 However, due to striking structural similarities between QueG and RDases, the possibility of outer sphere single electron transfer (SET) from Co(I)Cbl to the substrate cannot be discounted. While the Co(II) substrate containing computational models indicate a Co–substrate distance that is incompatible with Co–C bond formation, it is possible that the active site reorganizes after reduction to Co(I)Cbl. To evaluate the possibly that an alkyl–Co(III) intermediate is formed in the QueG catalytic cycle, a computational model with the distance between the epoxide C and Co shortened and the C–O bond broken was prepared (model of the first intermediate for the nucleophilic attack pathway in Figure 6). During the optimization, the cobalamin reverted to the geometry expected for Co(I)Cbl, with the distance between the Co ion and C atom increasing to >3.3 Å, well beyond the ~2 Å bond length expected for an organometallic bond with cobalamin (Figure S6). Additionally, the C–O bond of the epoxide that was cleaved in the input geometry reformed in the course of the optimization. Thus, our computations indicate that Co–C bond formation is highly unlikely to occur during the QueG-catalyzed reduction of oQ to Q. Rather, this reaction likely proceeds through SET, as has recently been proposed for PceA.31
Figure 6.
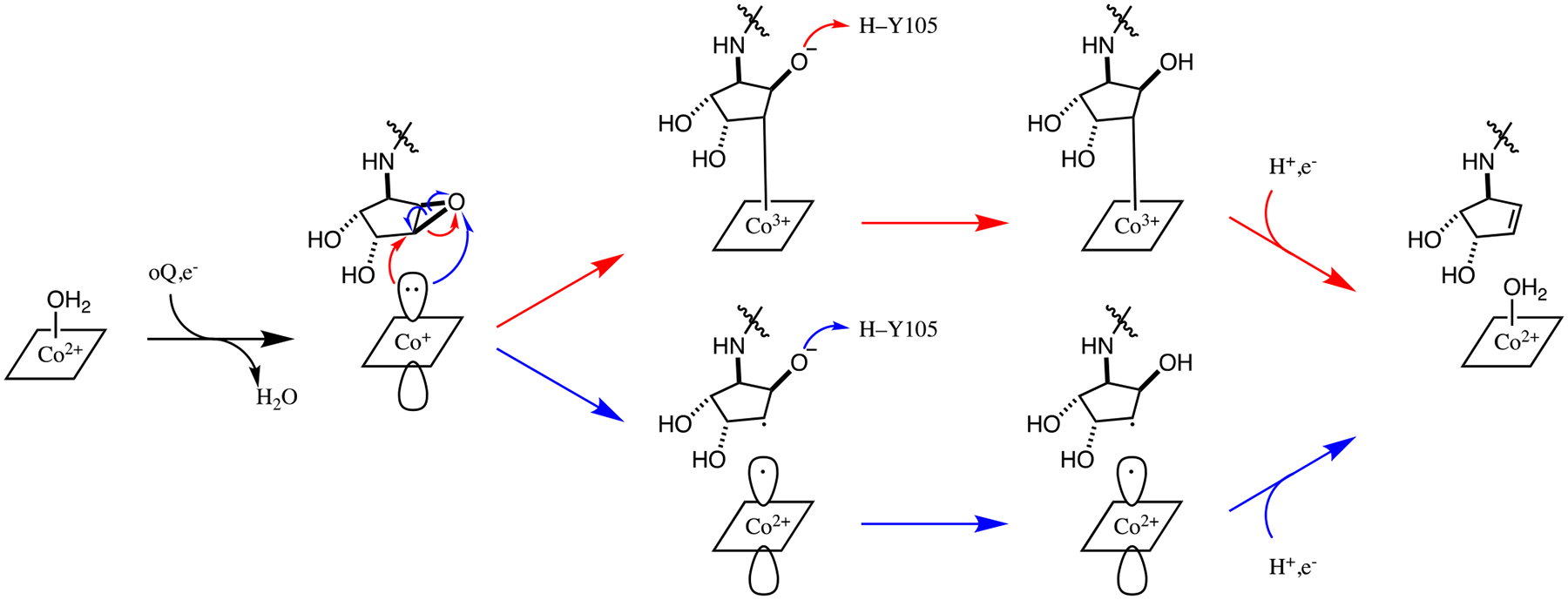
Potential reaction mechanisms for the reduction of oQ to produce Q. Red arrows: nucleophilic attack of the Co(I) ion on an epoxide C of the substrate. Blue arrows: SET from the Co(I) ion to the substrate.
4. Discussion
The EPR data obtained in this study provide compelling evidence that Co(II)Cbl remains five-coordinate upon substrate binding to QueG. Our computations reveal two ways by which the Co(II) center can remain five-coordinate; i.e., either a water molecule migrates into the pocket between R141 and the cobalamin to serve as the lower axial ligand, or the cyclopentanediol moiety on the substrate rotates such that one of the alcohol groups can become the upper axial ligand. In the latter case, the substrate could simply swing up upon Co(II) → Co(I)Cbl reduction to form a four-coordinate Co(I)Cbl species. Interestingly, during the QM/MM optimization of the model of product-bound QueG lacking a lower axial water, no Co–O(product) bond formed. The four-coordinate Co(II)Cbl species present in this model could be stabilized through coordination of the water molecule released in the course of the reaction (Figure 6).
Located on the lower face of the Cbl inside the QueG active site, R141 is oriented roughly parallel to the plane of the corrin ring (Figure 7). This residue, in conjunction with the propionamide side chains on the periphery of the corrin ring, limits accessibility to the lower face of the Cbl cofactor. Recently, an X-ray crystal structure of TsrM was published revealing an analogous Arg residue, R69, oriented with the guanidinium group perpendicular to the face of the Cbl (Figure 7).55 In TsrM, R69 is thought to weakly coordinate to the Co ion, destabilizing methylcobalamin [i.e., the Co(III) state] relative to Co(I)Cbl and thus promoting transfer of the methyl group to the substrate tryptophan. In part, R69 accomplishes this destabilization by preventing a water molecule from coordinating to the lower face of the Cbl cofactor, which would stabilize the Co(III) state. While in QueG the side chain of R141 is too far from the Co ion to invoke a direct interaction, this residue may serve a similar role in preventing a water molecule from coordinating to the Co ion. However, this residue is not strictly conserved in QueG enzymes32 and so is unlikely to play a critical mechanistic role.
Figure 7.
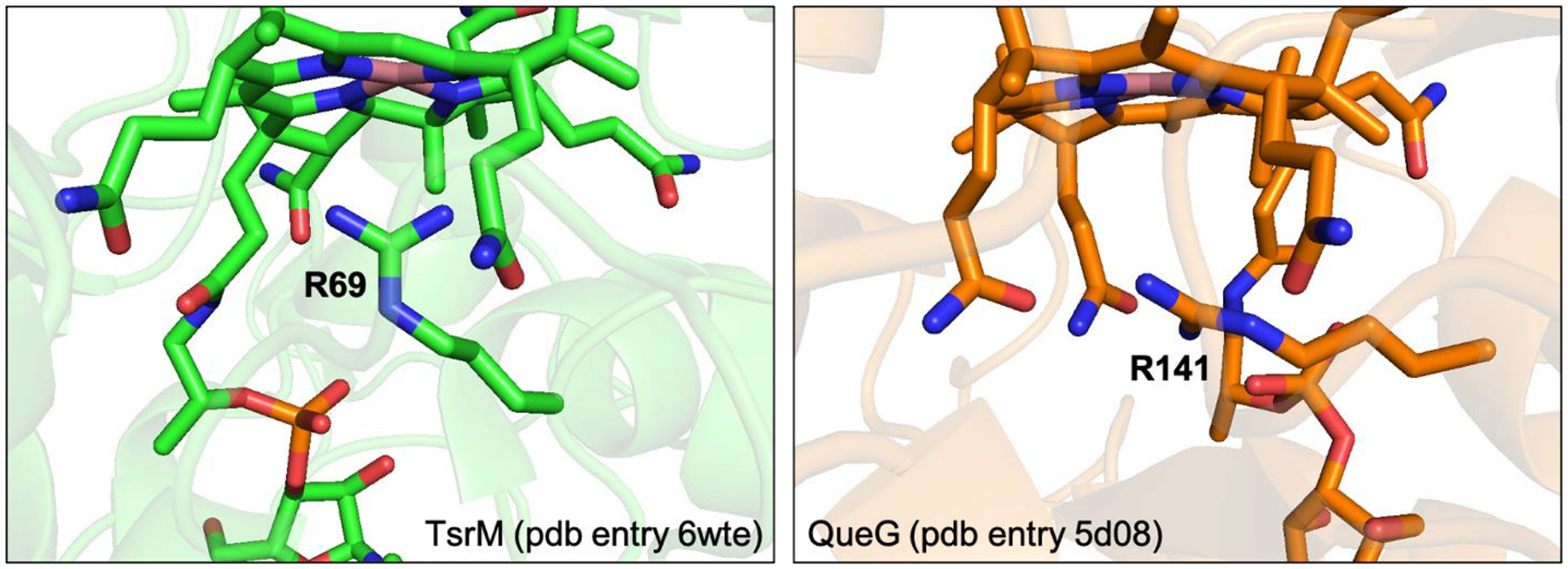
Comparison between the active sites of TsrM55 and QueG4. Note the different positioning of the Arg residue that is present below the face of the cobalamin in both enzymes.
Because Co(II)Cbl remains five-coordinate upon binding of the oQ substrate to QueG, the mechanism used by QueG to perform the thermodynamically challenging Co(II)→Co(I)Cbl reduction differs fundamentally from that employed by adenosyltransferases (ATRs), which catalyze the Co–C bond formation step during adenosylcobalamin biosynthesis.56–58 All ATRs studied to date generate a four-coordinate Co(II)Cbl species upon addition of co-substrate ATP, lowering the thermodynamic barrier for Co(II) ion reduction by destabilizing the Co(II) state. Contrastingly, a low potential [4Fe-4S] cluster in the corrinoid/FeS protein facilitates the reduction of the catalytically inactive Co(II)corrinoid species that is formed via off-cycle oxidation of the Co(I) intermediate.59 Similarly, the RDase PceA maintains a five-coordinate Co(II) coordination environment following substrate binding and utilizes low potential [4Fe-4S] clusters to perform the Co(II)→Co(I)Cbl reduction.31 As the [4Fe-4S] clusters in QueG have been determined to possess redox potentials below that of the Co(II/I)Cbl redox couple,4 it is unsurprising that in this enzyme Co(II)Cbl also remains five-coordinate upon substrate binding.
In addition to using similar mechanisms for the Co(II)→Co(I)Cbl reduction, QueG and PceA also share common structural features. In both enzymes the cobalamin and [4Fe-4S] cofactors are positioned in nearly the same way and located within similar binding domains.4,17 Moreover, in each case, the substrate is bound with the closest atom more than 4 Å from Co ion and an active site Tyr residue likely serves as the proton donor for the respective reduction reactions (Figure 8). Moreover, calculations on QueG and PceA disfavor mechanisms invoking Co–C bond formation and instead support an outer sphere electron transfer mechanism.
Figure 8.
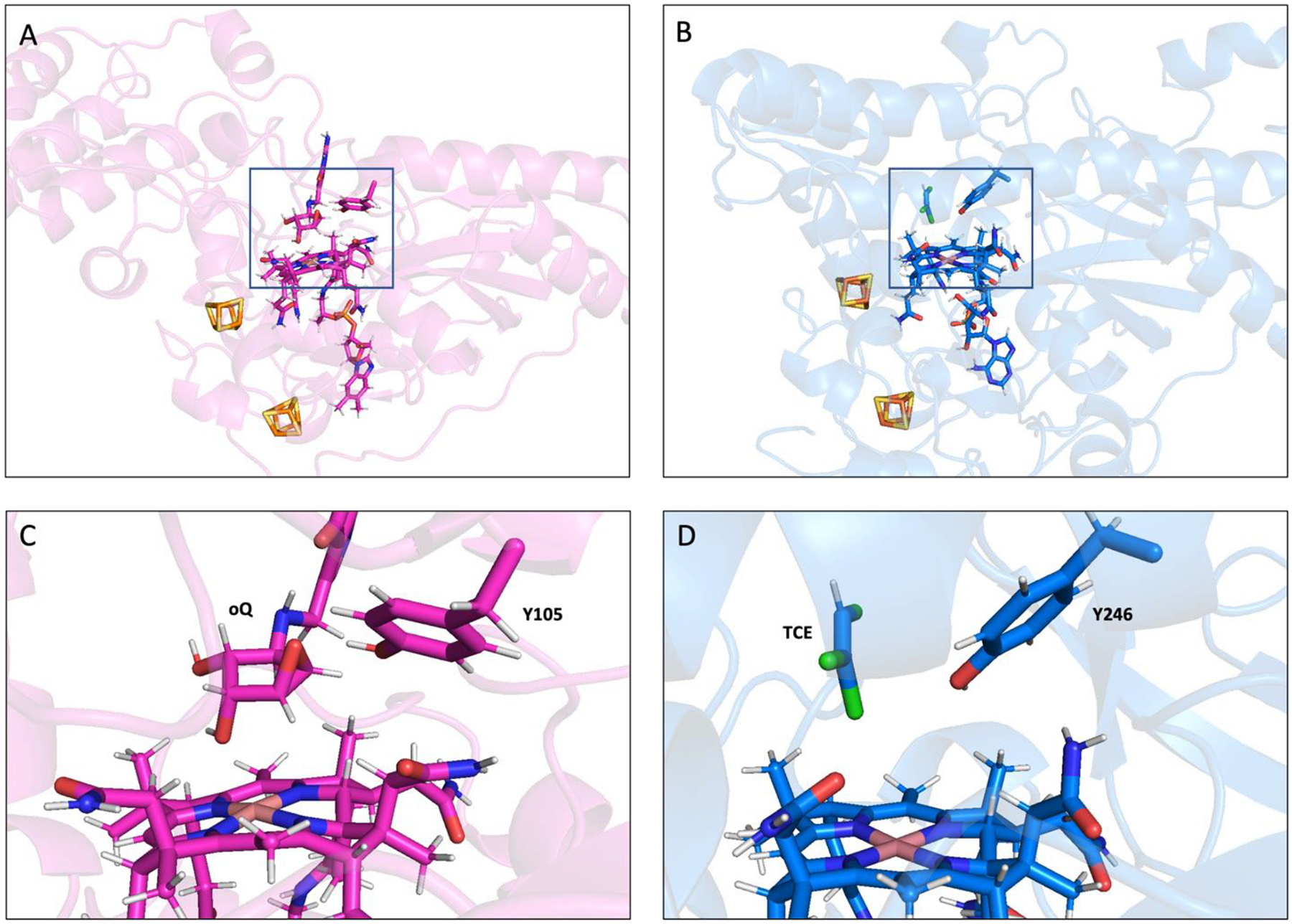
Comparison between the computational models of substrate bound QueG and PceA at the enzyme level (A and B respectively) and in the active site region (C and D respectively). The PceA substrate shown is trichloroethylene (TCE).
In principle, the second electron necessary for product formation (see Figure 6) could either be directly transferred to the substrate radical intermediate from the [4Fe-4S] clusters or shuttled through the transient Co(II)Cbl species (generated via initial outer-sphere single electron transfer to the substrate) in both the QueG and PceA reaction mechanisms. For the computational models of QueG and PceA with their respective substrates bound, the shortest S…C(substrate) distance between the proximal [4Fe-4S] cluster and the substrate ranges from 10.6 Å (QueG) to 8.5 Å (PceA).31 While these distances are not unreasonable for a direct electron transfer, the ~5 Å distance from the proximal cluster to the edge of the corrin ring in both enzymes suggests that the second electron is more likely passed through the Co(II)Cbl intermediate to regenerate a transient Co(I)Cbl species. The absence of a water molecule on the lower face of the Co(II)Cbl intermediate so as to maintain a four-coordinate Co(II) coordination environment (Figure 6) would lower the thermodynamic barrier for Co(II)→Co(I)Cbl reduction, as discussed above.
5. Conclusion
To further characterize the QueG-bound Co(II)Cbl cofactor, we have collected MCD data of the WT enzyme and validated a corresponding computational model on the basis of spectroscopic and X-ray crystallographic data. Enzyme variants that had previously been studied biochemically and with EPR spectroscopy,32 as well as the new variant, Y105F, were characterized using MCD and EPR spectroscopies. Minimal differences among the MCD data sets led to us to investigate the effect of glycerol concentration on enzyme state. On the basis of our EPR and computational studies, we propose that high glycerol concentrations favor the more compact Asp-in state, while the Asp-out state is favored by some variants when the glycerol concentration is low. Further, the transition from the Asp-in to the Asp-out state primarily affects the residues involved in proton transfer (Y105, H106, and D134), while the portion of the active site remote from D134 is largely unaffected, supporting a role for the Asp-out state in substrate recognition and orientation for binding and a role for the Asp-in state in catalysis. EPR data and computational models of QueG in the presence of oQ indicate that the Co(II)Cbl cofactor remains five-coordinate upon substrate binding. Importantly, substrate binding favors the catalytically relevant Asp-in state, even for the Y105A QueG variant, demonstrating the importance of hydrogen-bonding interaction between oQ and other active site residues.
Supplementary Material
Funding Sources
This work was supported by the National Science Foundation (Grant CHE-1710339 to T.C.B) and a National Institute of General Medical Sciences of the National Institutes of Health (Grant R35 GM126956 to V.B.).
ABBREVIATIONS
- QueG
epoxyqueuosine reductase
- RDases
reductive dehalogneases
- Cbl
cobalamin
- Cbi
cobinamide
- oQ
epoxyqueuosine
- Q
queuosine
- TCE
trichloroethylene
- SET
single electron transfer
- EPR
electron paramagnetic resonance
- MCD
magnetic circular dichroism
- QM/MM
quantum mechanics/molecular mechanics
- Abs
electronic absorption
- CD
circular dichroism
- rmsd
root mean squared deviation
- TD-DFT
time-dependent density functional theory
- ATRs
adenosyltransferases
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Experimental Abs spectra of WT QueG and Co(II)Cbi+, comparison between the active site regions of the crystal structure of resting QueG and the corresponding QM/MM optimized computational model, TD-DFT computed Abs spectra, EPR data of substrate-bound WT and Y105A QueG with differing relative amounts of glycerol, MCD data of WT and Y105A QueG and Y105A in the absence and presence of oQ, input and optimized geometries for the model of the putative alkyl–Co(III)Cbl intermediate, and computed Co spin densities for spin unrestricted QM/MM models (PDF), and as well as coordinates for the QM region of all models (XLS).
Accession Code
QueG from Bacillus subtilis: UniProt P97030.
The authors declare no competing financial interest.
References
- 1.Machnicka MA, Milanowska K, Oglou OO, Purta E, Kurkowska M, Olchowik A, Januszewski W, Kalinowski S, Dunin-Horkawicz S, Rother KM, Helm M, Bujnicki JM & Grosjean H (2013) MODOMICS: A database of RNA modification pathways - 2013 update. Nucleic Acids Res 41, D262–D267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harada F & Nishimura S (1970) Possible Anticodon Sequences of tRNAHIS, tRNAAsn, and tRNAAsp from Escherichia coli B. Universal Presence of Nucleoside Q in the First Position of the Anticondons of These Transfer Ribonucleic Acids. Biochem. Biophys. Acta 187, 3047. [DOI] [PubMed] [Google Scholar]
- 3.Goodman HM, Abelson J, Landy A, Brenner S & Smith JD (1968) Amber suppression: A Nucleotide change in the anticodon of a tyrosine transfer RNA. Nature 217, 1019–1024. [DOI] [PubMed] [Google Scholar]
- 4.Dowling DP, Miles ZD, Köhrer C, Maiocco SJ, Elliott SJ, Bandarian V & Drennan CL (2016) Molecular basis of cobalamin-dependent RNA modification. Nucleic Acids Res 44, 9965–9976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Payne KAP, Fisher K, Sjuts H, Dunstan MS, Bellina B, Johannissen L, Barran P, Hay S, Rigby SEJ & Leys D (2015) Epoxyqueuosine Reductase Structure Suggests a Mechanism for Cobalamin-Dependent tRNA Modification. J. Biol. Chem 290, 27572–27581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nishimura S (1983) Structure, Biosynthesis, and Function of Queuosine in Transfer RNA. Prog. Nucleic Acid Res. Mol. Biol 28, 49–73. [DOI] [PubMed] [Google Scholar]
- 7.Katze, Basile B & McCloskey J (1982) Queuine, a Modified Base Incorporated Posttranscriptionally Into Eukaryotic Transfer RNA: Wide Distribution in Nature. Science 216, 55–56. [DOI] [PubMed] [Google Scholar]
- 8.Johannsson S, Neumann P, Wulf A, Welp LM, Gerber HD, Krull M, Diederichsen U, Urlaub H & Ficner R (2018) Structural insights into the stimulation of S. pombe Dnmt2 catalytic efficiency by the tRNA nucleoside queuosine. Sci. Rep 8, 8880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aytac U & Gunduz U (1994) Q-modification of tRNAs in human brain tumors. Cancer Biochem. Biophys 14, 93–98. [PubMed] [Google Scholar]
- 10.Baranowski W, Dirheimer G, Jakowicki JA & Keith G (1994) Deficiency of Queuine, a Highly Modified Purine Base, in Transfer RNAs from Primary and Metastatic Ovarian Malignant Tumors in Women. Cancer Res 54, 4468–4471. [PubMed] [Google Scholar]
- 11.Emmerich B, Zubrod E, Weber H, Maubach PA, Kersten H & Kersten W (1985) Relationship of Queuine-lacking Transfer RNAs to the Grade of Malignancy in Human Leukemias and Lymphomas. Cancer Res 45, 4308–4314. [PubMed] [Google Scholar]
- 12.Huang B-S, Wu R-T & Chien K-Y (1992) Relationship of the Queuine Content of Transfer Ribonucleic Acids to Histopathological Grading and Survival in Human Lung Cancer. Cancer Res 52, 4696–4700. [PubMed] [Google Scholar]
- 13.Katze JR & Beck WT (1980) Administration of queuine to mice relieves modified nucleoside queuosine deficiency in Ehrlich ascites tumor tRNA. Top. Catal 96, 313–319. [DOI] [PubMed] [Google Scholar]
- 14.Marks T & Farkas WR (1997) Effects of a diet deficient in tyrosine and queuine on germfree mice. Biochem. Biophys. Res. Commun 230, 233–237. [DOI] [PubMed] [Google Scholar]
- 15.Tuorto F, Legrand C, Cirzi C, Federico G, Liebers R, Müller M, Ehrenhofer‐Murray AE, Dittmar G, Gröne H & Lyko F (2018) Queuosine‐modified tRNAs confer nutritional control of protein translation. EMBO J 37, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang X, Matuszek Z, Huang Y, Parisien M, Dai Q, Clark W, Schwartz MH & Pan T (2018) Queuosine modification protects cognate tRNAs against ribonuclease cleavage. RNA 24, 1305–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bommer M, Kunze C, Fesseler J, Schubert T, Diekert G & Dobbek H (2014) Structural Basis for Organohalide Respiration. Science 346, 455–458. [DOI] [PubMed] [Google Scholar]
- 18.Payne KAP, Quezada CP, Fisher K, Dunstan MS, Collins FA, Sjuts H, Levy C, Hay S, Rigby SEJ & Leys D (2015) Reductive Dehalogenase Structure Suggests a Mechanism for B12-Dependent Dehalogenation. Nature 517, 513–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miles ZD, Mccarty RM, Molnar G & Bandarian V (2011) Discovery of Epoxyqueuosine (oQ) Reductase Reveals Parallels Between Halorespiration and tRNA Modification. PNAS 108, 7368–7372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reitzer R, Gruber K, Jogl G, Wagner UG, Bothe H, Buckel W & Kratky C (1999) Glutamate Mutase from Clostridium cochlearium: The Structure of a Coenzyme B12-Dependent Enzyme Provides New Mechanistic Insights. Structure 7, 891–902. [DOI] [PubMed] [Google Scholar]
- 21.Zelder O, Beatrix B, Kroll F & Buckel W (1995) Coordination of a Histidine Residue of the Protein-Component S to the Cobalt Atom in Coenzyme B12-Dependent Glutamate Mutase from Clostridium cochlearium. FEBS Lett 369, 252–254. [DOI] [PubMed] [Google Scholar]
- 22.Shibata N, Masuda J, Tobimatsu T, Toraya T, Suto K, Morimoto Y & Yasuoka N (1999) A New Mode of B12 Binding and the Direct Participation of a Potassium Ion in Enzyme Catalysis: X-Ray Structure of Diol Dehydratase. Structure 7, 997–1008. [DOI] [PubMed] [Google Scholar]
- 23.Brown KL (2005) Chemistry and Enzymology of Vitamin B12. Chem. Rev 105, 2075–2149. [DOI] [PubMed] [Google Scholar]
- 24.Banerjee R & Ragsdale SW (2003) The Many Faces of Vitamin B12: Catalysis by Cobalamin-Dependent Enzymes. Annu. Rev. Biochem 72, 209–247. [DOI] [PubMed] [Google Scholar]
- 25.Ludwig and ML & Matthews RG (1997) Structure-Based Perspectives on B12 Dependent Enzymes. Annu. Rev. Biochem 66, 269–313. [DOI] [PubMed] [Google Scholar]
- 26.Bauerle MR, Schwalm EL & Booker SJ (2015) Mechanistic Diversity of Radical S-Adenosylmethionine (SAM)-Dependent Methylation. J. Biol. Chem 290, 3995–4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matthews RG (2001) Cobalamin-Dependent Methyltransferases. Acc. Chem. Res 34, 681–689. [DOI] [PubMed] [Google Scholar]
- 28.Svetlitchnaia T, Svetlitchnyi V, Meyer O & Dobbek H (2006) Structural Insights Into Methyltransfer Reactions of a Corrinoid Iron-Sulfur Protein Involved in Acetyl-CoA Synthesis. Proc. Natl. Acad. Sci 103, 14331–14336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kunze C, Bommer M, Hagen WR, Uksa M, Dobbek H, Schubert T & Diekert G (2017) Cobamide-Mediated Enzymatic Reductive Dehalogenation via Long-Range Electron Transfer. Nat. Commun 8, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Diekert G, Boland W, Svatos A, Ploss K, Neumann A, Habel A, Wolf J, Schmidtz RPH, Schmitz RPH, Wolf J, Habel A, Neumann A, Ploss K, Svatos A, Boland W & Diekert G (2007) Evidence for a Radical Mechanism of the Dechlorination of Chlorinated Propenes Mediated by the Tetrachloroethene Reductive Dehalogenase of Sulfurospirillum multivorans. Environ. Sci. Technol 41, 7370–7375. [DOI] [PubMed] [Google Scholar]
- 31.Greenhalgh ED, Kunze C, Schubert T, Diekert G & Brunold TC (2021) A Spectroscopically Validated Computational Investigation of Viable Reaction Intermediates in the Catalytic Cycle of the Reductive Dehalogenase PceA. Biochemistry 2022–2032 doi: 10.1021/acs.biochem.1c00271. [DOI] [PubMed] [Google Scholar]
- 32.Miles ZD, Myers WK, Kincannon WM, Britt RD & Bandarian V (2015) Biochemical and Spectroscopic Studies of Epoxyqueuosine Reductase: A Novel Iron–Sulfur Cluster- and Cobalamin-Containing Protein Involved in the Biosynthesis of Queuosine. Biochemistry 54, 4927–4935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stich TA, Buan NR, Escalante-Semerena JC & Brunold TC (2005) Spectroscopic and Computational Studies of the ATP:Corrinoid Adenosyltransferase (CobA) from Salmonella enterica: Insights into the Mechanism of Adenosylcobalamin Biosynthesis. J. Am. Chem. Soc 127, 8710–8719. [DOI] [PubMed] [Google Scholar]
- 34.Hay BP & Finke RG (1987) Thermolysis of the Co-C Bond in Adenosylcorrins. 3. Quantification of the Axial Base Effect in Adenosylcobalamin by the Synthesis and Thermolysis of Axial Base-Free Adenosylcobinamide. Insights into the Energetics of Enzyme-Assisted Cobalt-Carbon Bond Hom. J. Am. Chem. Soc 109, 8012–8018. [Google Scholar]
- 35.Stoll S & Schweiger A (2005) EasySpin, a comprehensive software package for spectral simulation and analysis in EPR doi: 10.1016/j.jmr.2005.08.013. [DOI] [PubMed]
- 36.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery J, J. A, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyez O, Austin AJ, Cammi R, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowksi J & Fox DJ Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT. [Google Scholar]
- 37.Dolinsky TJ, Nielsen JE, McCammon JA & Baker NA (2004) PDB2PQR: an Automated Pipeline for the Setup, Execution, and Analysis of Poisson-Boltzmann Electrostatics Calculations. Nucleic Acids Res 32, W665–W667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Becke AD (1993) Density-Function Thermochemistry 3. The Role of Exact Exchange. J. Chem. Phys 98, 5648–5652. [Google Scholar]
- 39.Lee CT, Yang WT & Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron-density. Phys. Rev. B 37, 785–789. [DOI] [PubMed] [Google Scholar]
- 40.Schäfer A, Huber C & Ahlrichs R (1994) Fully Optimized Contracted Gaussian Basis Sets of Triple Zeta Valence Quality for Atoms Li to Kr. J. Chem. Phys 100, 5829–5835. [Google Scholar]
- 41.Case DA, Belfon K, Ben-Shalom IY, Brozell SR, Cerutti DS, Cheatham TE III, C. VWD, Darden TA, Duke RE, Giambasu G, Gilson MK, Gohlke H, Goetz AW, Harris R, Izadi S, Izmailov SA, Kasavajhala K, Kovalenko A, Krasny R, Kurtzman T, Lee TS, LeGrand S, Li P, Lin C, L. J, Luchko T, Luo R, Man V, Merz KM, Miao Y, Mikhailovskii O, Monard G, Nguyen H, Onufriev A, Pan F, Pantano S, Qi R, Roe DR, Roitberg A, Sagui C, Schott-Verdugo S, Shen J, Simmerling CL, Skrynnikov NR, Smith J, Swails J, Walker RC, Wang J, Wilson L, Wolf RM, Wu X, Xiong Y, X. Y & Kollman DMY and A. P (2020) AMBER 2020 University of California, San Francisco. [Google Scholar]
- 42.Marques HM, Ngoma B, Egan TJ & Brown KL (2001) Parameters for the AMBER Force Field for the Molecular Mechanics Modeling of the Cobalt Corrinoids. J. Mol. Struct 561, 71–91. [Google Scholar]
- 43.Schrodinger L The PyMOL Molecular Graphics System, 1.3 ed. [Google Scholar]
- 44.Neese F (2012) The ORCA Program System. Wiley Interdiscip. Rev. Comput. Mol. Sci 2 73–78. [Google Scholar]
- 45.Yanai T, Tew DP & Handy NC (2004) A New Hybrid Exchange-Correlation Functional Using the Coulomb-Attenuating Method (CAM-B3LYP). Chem. Phys. Lett 393, 51–57. [Google Scholar]
- 46.Weingend F & Ahlrichs R (2005) Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys 7, 3297–3305. [DOI] [PubMed] [Google Scholar]
- 47.Weingend F (2006) Accurate Coulomb-Fitting Basis Sets for H to Rn. Phys. Chem. Chem. Phys 8, 1057–1065. [DOI] [PubMed] [Google Scholar]
- 48.Neese F (2002) Prediction and interpretation of the 57Fe isomer shift in Mössbauer spectra by density functional theory. Inorganica Chim. Acta 337, 181–192. [Google Scholar]
- 49.Godbout N, Salahub DR, Andzelm J & Wimmer E (1992) Optimization of Gaussian-type basis sets for local spin density functional calculations. Part I. Boron through neon, optimization technique and validation. Can. J. Chem 70, 560–571. [Google Scholar]
- 50.Schaefer A, Horn H & Achlrichs R (1992) Fully Optimized Contracted Gaussian Basis Sets for Atoms Lithiium to Krypton. J. Chem. Phys 97, 2571–2577. [Google Scholar]
- 51.Neese F (2003) An Improvement of the Resolution of the Identity Approximation for the Formation of the Coulomb Matrix. J. Comput. Chem 24, 1740–1747. [DOI] [PubMed] [Google Scholar]
- 52.Stich TA, Buan NR & Brunold TC (2004) Spectroscopic and Computational Studies of Co2+Corrinoids: Spectral and Electronic Properties of the Biologically Relevant Base-On and Base-Off Forms of Co2+Cobalamin. J. Am. Chem. Soc 126, 9735–9749. [DOI] [PubMed] [Google Scholar]
- 53.Vagenende V, Yap MGS & Trout BL (2009) Mechanisms of protein stabilization and prevention of protein aggregation by glycerol. Biochemistry 48, 11084–11096. [DOI] [PubMed] [Google Scholar]
- 54.Stich TA, Buan NR & Brunold TC (2004) Spectroscopic and computational studies of Co2+corrinoids: Spectral and electronic properties of the biologically relevant base-on and base-off forms of Co2+cobalamin. J. Am. Chem. Soc 126, 9735–9749. [DOI] [PubMed] [Google Scholar]
- 55.Knox HL, Chen PYT, Blaszczyk AJ, Mukherjee A, Grove TL, Schwalm EL, Wang B, Drennan CL & Booker SJ (2021) Structural basis for non-radical catalysis by TsrM, a radical SAM methylase. Nat. Chem. Biol 17, 485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stich TA, Yamanishi M, Banerjee R & Brunold TC (2005) Spectroscopic Evidence for the Formation of a Four-Coordinate Co2+Cobalamin Species upon Binding to the Human ATP:Cobalamin Adenosyltransferase. J. Am. Chem. Soc 127, 7660–7661. [DOI] [PubMed] [Google Scholar]
- 57.Fonseca MV & Escalante-Semerena JC (2001) An in Vitro Reducing System for the Enzymic Conversion of Cobalamin to Adenosylcobalamin. J. Biol. Chem 276, 32101–32108. [DOI] [PubMed] [Google Scholar]
- 58.Lexa D & Saveant J-M (1983) The Electrochemistry of Vitamin B12. Acc. Chem. Res 16, 235–243. [Google Scholar]
- 59.Stich TA, Seravalli J, Venkateshrao S, Spiro TG, Ragsdale SW & Brunold TC (2006) Spectroscopic Studies of the Corrinoid/Iron-Sulfur Protein from Moorella thermoacetica. J. Am. Chem. Soc 128, 5010–5020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.