

Abstract
A method for C–H bond amination via an electrophotocatalytic Ritter-type reaction is described. The reaction is catalyzed by a trisaminocyclopropenium (TAC) ion in an electrochemical cell under irradiation. These conditions convert benzylic C–H bonds to acetamides without the use of a stoichiometric chemical oxidant. A range of functionality is shown to be compatible with this transformation, and several complex substrates are demonstrated.
Graphical Abstract
The invention of methods to convert unactivated C–H bonds to C–N bonds has long been established as a desirable goal.1–2,3,4,5 In this regard, the classic Hoffmann-Löfler-Freitag reaction has been a mainstay for complex molecule synthesis,6–7,8,9 while modern incarnations of this strategy have recently been described using photoredox catalysis.10 – 11 12 Alternatively, a variety of reactions involving nitrene intermediates that can undergo C–H insertion or H-atom abstraction have proven exceptionally useful.13–14 15 16 17 18 Moreover, the use of designer transition metal complexes that can effect C–N couplings of C–H and N–H bonds have become increasingly popular, and a wide range of impressive applications of this strategy have been disclosed.1,19–20,21,22 An orthogonal strategy entails the oxidation of a C–H bond to the corresponding carbocation, which can then be trapped by a nitrile, usually as solvent, leading to amide products (Figure 1a). This process is in essence a Ritter reaction,23–24,25,26 with the difference being the means by which the carbocation is generated. Several catalytic methods for Ritter-type C–H amination have been reported, including work by Ishii, Baran, Kiyokawa and Minakata, and Liu and Chen (Figure 1b).27–28 29 30 While these are important advances, the oxidants employed are incompatible with many functional groups and in the case of Selectfluor are quite expensive. A potentially attractive alternative would be to use electrochemistry,31–32,33,34,35,36,37,38,39,40,41,42,43 which would obviate the need for the chemical oxidant altogether. While there have been examples of electrochemical Ritter-type C–H aminations,44–45,46,47 these processes tend to be low-yielding and incompatible with much native functionality due to the high anodic potentials required. However, very recently an electrochemical method for benzylic C–H amination with sulfonamides was recently reported that proceeded with good efficiency,48 although it required the use of HFIP solvent and the range of demonstrated functional group compatibility was narrow. Thus, there remains the need for an electrochemically-coupled version of this chemistry that has the generality needed to operate in the context of greater molecular complexity.
Figure 1.

(a) Ritter reaction of carbocation intermediates. (b) Selected examples of Ritter-type chemistry. (c) Electrophotocatalytic Ritter-type C–H amination.
In this regard, we recently reported49 an electrophotocatalytic50,51,52,53,54,55,56,57,58,59 – 60 61 62 vicinal C–H diamination reaction that achieved the conversion of alkylarene substrates, particularly those that were α-branched, to 3,4-dihydroimidazole products (Figure 1c). Although these products represent a valuable increase in molecular complexity, it would also be desirable to access the classic Ritter-type monoamination products as well. In this Communication, we report that unbranched substrates undergo efficient single site C–H amination under modified electrophotocatalytic conditions.
With the goal of developing a method that would be applicable to a wide variety of substrates, we chose the complex substrate 2, which is an analogue of the pharmaceutical agent celecoxib, for our optimization studies (Table 1). Under the optimal conditions we identified, TAC 1 catalyzed the conversion of this substrate to acetamide 3 by reaction in a divided cell (Ecell = 2.2 V, Eanode = 1.6 V vs SCE) under irradiation from a compact fluorescent light (CFL), in the presence of TFA, H2O, and n-Bu4NPF6 in acetonitrile solvent (entry 1). A small amount (4%) of aldehyde 4 was also observed. Table 1 illustrates the impact of variation from these conditions. We found that the identity of the electrolyte was important. Thus, the use of LiClO4 (entry 2) or n-Bu4NBF4 (entry 3) resulted in a significant decrease in yield of 3 and a small increase in the formation of the side product 4. Not surprisingly, without the application of the cell potential (entry 4) or in the absence of the catalyst (entry 5), no reaction occurred. When only the light was omitted, a small amount (9%) of background reaction was observed (entry 6). Changing the acid additive from TFA to AcOH was detrimental, with the yield dropping to only 10% (entry 7). Although product was observed without the addition of water (entry 8), the yield was significantly diminished. Notably, the use of a divided cell was critical to the success of this process, as the yield of product in an undivided cell was very low (entry 9). Finally, we also attempted to conduct this reaction via direct electrolysis, with potentials up to 3.5 V (entries 10 and 11). While some product was generated under these conditions, the yields were low, the reaction mixtures were complicated, and a large amount of aldehyde side product 4 was formed. The contrast between the direct electrolysis and electrophotocatalytic approach underscores the ability of the latter to effect potent yet selective oxidative chemistry.
Table 1.

| |||
|---|---|---|---|
| entry | change from standard | yield 3 (%) | yield 4 (%) |
| 1 | none | 77 | 4 |
| 2 | LiCIO4 instead of n-Bu4NPF6 | 50 | 8 |
| 3 | n-Bu4NBF4 instead of n-Bu4NPF6 | 27 | 5 |
| 4 | no cell potential | 0 | 0 |
| 5 | no catalyst | 0 | trace |
| 6 | no light | 9 | trace |
| 7 | AcOH instead of TFA | 10 | trace |
| 8 | no H2O | 66 | trace |
| 9 | undivided cell | 14 | 4 |
| 10 | direct electrolysis (3.0 V) | 36 | 32 |
| 11 | direct electrolysis (3.5 V) | 33 | 20 |
| 12 | Pb as cathode | 28 | trace |
Reaction conditions: 2 (0.5 mmol), H2O (1.0 equiv), 1 (8 mol %), nBu4NPF6 (1.0 equiv), TFA (0.2 mL), MeCN (6.0 mL) [anode], and nBu4NPF6 (1.0 equiv), TFA (0.2 mL), MeCN (6.0 mL) [cathode] in H-type divided cell with carbon felt anode, Pt cathode. Reactions performed under constant cell potential conditions with irradiation for 48 h at rt under N2.
Yields are of isolated and purified products 3 and 4.
With these optimized conditions we explored the scope of this method (Table 2). Toluene and 4-halogenated toluenes gave rise to the corresponding benzylacetamides 5-9 in good yields. Notably, the position of halogenation had a significant impact on efficiency. Thus, whereas the 4-bromo product 8 was accessed in reasonable yield, the 2-bromo isomer 10 was formed less efficiently, and the 3-bromo isomer 11 was only produced in 31% yield. Also, in the latter two cases, a change of the electrolyte to LiClO4 proved necessary for maximal yield.63 On the other hand, 1-bromo-3,5-dimethylbenzene reacted with good efficiency to furnish acetamide 12 in 61% yield.
Table 2.
Electrophotocatalytic Ritter-type amination of benzylic C–H bonds.a

|
Reaction conditions: substrate (0.5 mmol), H2O (1.0 equiv), 1 (8 mol %), nBu4NPF6 (1.0 equiv), TFA (0.2 mL), MeCN (6.0 mL) [anode], and nBu4NPF6 (1.0 equiv), TFA (0.2 mL), MeCN (6.0 mL) [cathode] in H-type divided cell with carbon felt anode, Pt cathode. Reactions performed under constant voltage (CV) (2.2 V) conditions with light irradiation for 48 h at rt under N2, isolated yield.
LiClO4 instead of nBu4NPF6.
24 h.
2.6 V.
Worked up with 1M NaOH (aq).
Products resulting from functionalization of alkyl substituents other than methyl (e.g. 13 and 14), as long as the benzylic position was not branched. In the case of a branched substrate like cumene, the reaction gave rise to no acetamide product, but rather the dihydroimidazole product 15 as we previously described.49 In examining a substrate designed to probe the competition between methyl and methylene sites, we found that 16 was formed with a 6.2:1 preference over the alternative, methyl-functionalized site. Meanwhile, diphenylmethane was functionalized in good yield to furnish the α-diarylamine 17. Cyclic substrates such as tetralin, indane, and dibenzosuberone participated in the Ritter-type reaction as well, giving rise to acetamides 18–20 respectively.
In terms of functional group compatibility, we found that an alcohol (21), carboxylic acid (22), ester (23 and 24), alkyl chloride (25), and tosylate groups (26) survived the amination procedure. While a terminal alkyne containing substrate furnished no product 27, the internal alkyne product 28 was obtained in 70% yield. Notably, products possessing nitrogen functionality of various types could be accessed, including carbazole (29) trifluoroacetamide (30 and 31), pyridine (32), and even free amine-containing structures (33 and 34).
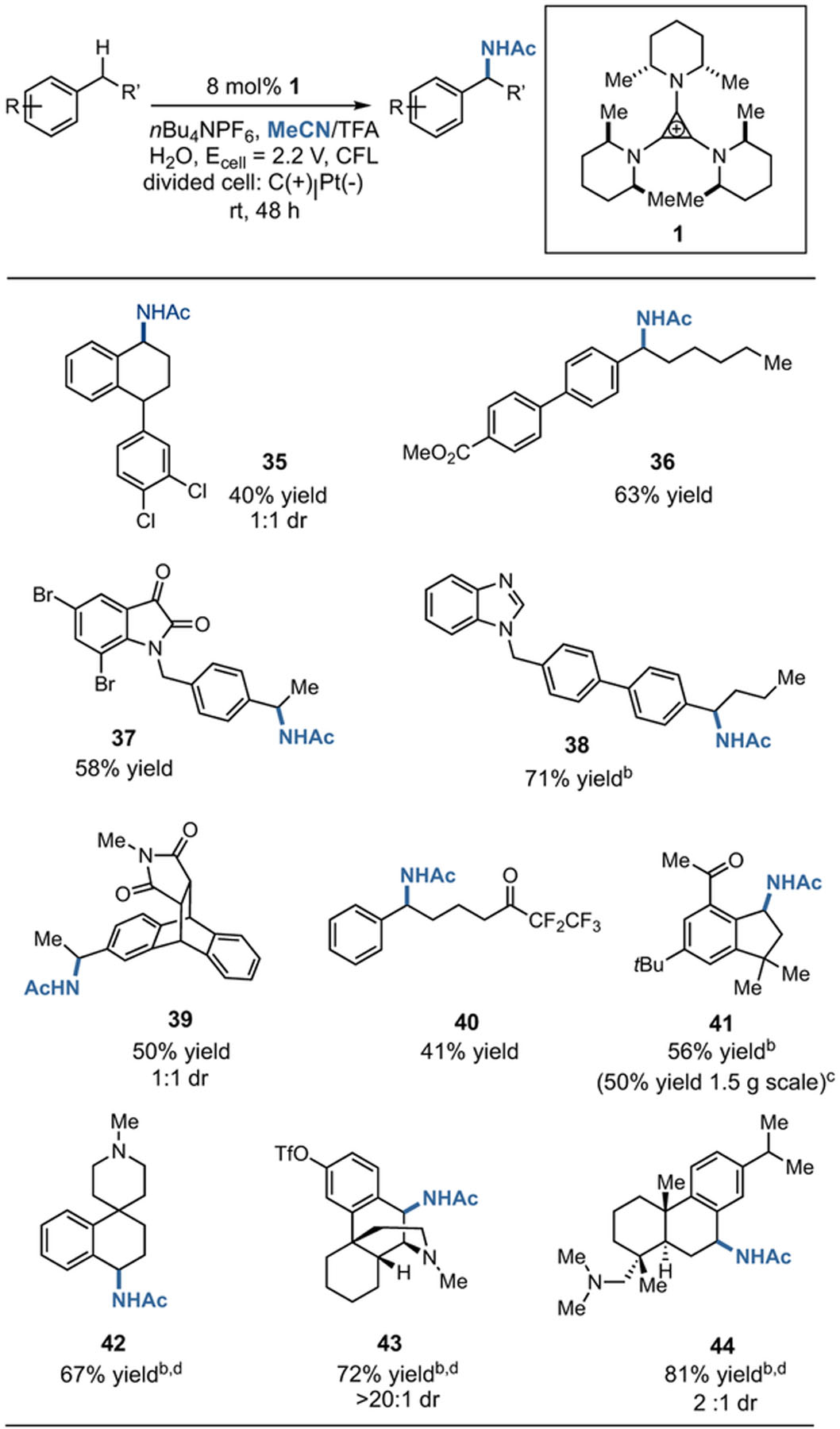
One of the most useful applications of C–H amination chemistry is the late-stage functionalization of complex molecules. For example, White has recently demonstrated efficient amination reactions on a number of bioactive molecules using a trichloroethylsulfoniminoiodinane using manganese catalysis.13 We decided to see whether some of these readily available compounds could be aminated using the electrophotocatalytic method, which has the benefits of using acetonitrile as the nitrogen source, requires no transition metal catalyst, and generates more desirable acetamide products (Table 3). Hence, the sertraline analogue 35 could be generated in 44% yield, while the retinoic acid receptor agonist derivative 3664 was produced in 63% yield. Meanwhile, an isatin derivative was selectively converted to compound 37 in 58% yield. Similarly, compound 38 derived from an inhibitor of the CYP11B165 was generated in good yield and with complete selectivity for the position shown. Other derivatives bearing either imide (39)66 or ketone functionalities (40 and 41) could also be accessed. Notably, products 42–4467 – 68 69 were generated with high efficiency despite the potential for undesired oxidation of the free amine functionality. To demonstrate the applicability of this method to preparative scale synthesis, we found that 41 could be generated on a 1.5 g scale in 50% yield by extending the reaction time to 72 h.
Table 3.
Electrophotocatalytic Ritter-type C–H amination of complex molecules.a
|
Reaction conditions: substrate (0.3–0.5 mmol, 1.0 equiv), H2O (1.0 equiv), 1 (8 mol %), nBu4NPF6 (1.0 equiv), TFA (0.2 mL), MeCN (6.0 mL) [anode], and nBu4NPF6 (1.0 equiv), TFA (0.2 mL), MeCN (6.0 mL) [cathode] in H-type divided cell with carbon felt anode, Pt cathode. Reactions performed under constant voltage (CV) conditions with light irradiation for 48 h at rt under N2, isolated yield.
LiClO4 instead of nBu4NPF6.
72 h reaction time.
Run at 2.6 V.
A mechanistic rationale for this electrophotocatalytic Ritter-type reaction is shown in Figure 2. The TAC 1 can be oxidized to generate the stable radical dication 45 (Ep/2 = 1.12 V in 30:1 MeCN:TFA vs SCE). Irradiation of 45 then leads to the photoexcited intermediate 46, which engages in single-electron oxidation of the arene substrate 47 to generate radical cation 48. Deprotonation of this intermediate results in benzylic radical 49, which then undergoes a second oxidation event, either by the TAC radical dication 45 or directly at the anode, leading to carbocation 50. The carbocation then proceeds through the classic Ritter steps by reaction with acetonitrile solvent to form nitrilium 51 followed by hydrolysis (either by reaction with adventitious water or with trifluoroacetic acid) to furnish the amide product 52. Meanwhile, the redox reaction is balanced by cathodic reduction of protons to produce dihydrogen. A key aspect of this approach is that the cell potential is insufficient to oxidize the substrate directly, leading to selective single electron oxidations by the electrophotocatalyst, which minimizes the risk of side reactions.
Figure 2.

Mechanistic rationale for electrophotocatalytic Ritter-type reaction.
An important distinction should be made between the monoaminations described in this work and the diaminations we recently reported under similar conditions.49 Our hypothesis for the diamination reaction is that initially formed benzylic acetamides 52 undergo an acid-catalyzed E1 elimination reaction to furnish styrenes 53, which then undergo further oxidation and Ritter-type trapping reactions to eventually form one or both of the regiosiomeric dihydroimidazoles 54a,b. In the case of benzylic-branched substrates, the elimination reaction is facile with TFA, and thus monoamination products 52 are not observed. With unbranched substrates, the elimination reaction requires a stronger acid like triflic acid. Thus, under the current conditions using TFA, the unbranched substrates furnish only the monoamination products 52.
It might be stressed that the risk of overoxidation presents a significant challenge for C–H amination chemistry, particularly if the installed nitrogen is not sufficiently deactivated (e.g. by a tosyl protecting group). We have measured the redox potentials of several of the products shown in Table 2 using cyclic voltammetry and have found that they are very close to those of the corresponding starting materials (see Supporting Information for details). It seems that the introduction of an acetamide moiety slows the kinetics of single-electron oxidation by TAC 1, which may be why the electrophotocatalytic process is more efficient than the direct electrochemical one. However, we have found that prolonged exposure to the standard reaction conditions (48 h) does result in some acetamide decomposition (7–30% in the compounds tested).
The direct amination of C–H bonds via Ritter-type reactions offers a potentially simple and scalable approach to molecular upgrading. The current work demonstrates that electrophotocatalysis can help achieve these transformations without the need for stoichiometric chemical oxidants and with enough selectivity to operate with a reasonable range of native functionality. As such, this method should be a useful addition to C–H functionalization toolbox.
Supplementary Material
Acknowledgement:
Financial support for this work was provided by NIGMS (R35 GM127135).
Footnotes
Supporting Information Available: Experimental procedures and product characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Davies HML; Manning JR Catalytic C-H functionalization by metal carbenoid and nitrenoid insertion. Nature 2008, 451, 417–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Park Y; Kim Y; Chang S Transition metal-catalyzed C-H amination: scope, mechanism, and applications. Chem. Rev 2017, 117, 9247–9301. [DOI] [PubMed] [Google Scholar]
- 3.Che C-M; Lo VK-Y; Zhou C-Y; Huang J-S Selective functionalization of saturated C-H bonds with metalloporphyrin catalysts. Chem. Soc. Rev 2011, 40, 1950–1975. [DOI] [PubMed] [Google Scholar]
- 4.Roizen JL; Harvey ME; Du Bois J Metal-catalyzed nitrogen-atom transfer methods for the oxidation of aliphatic C-H bonds. Acc. Chem. Res 2012, 45, 911–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Louillat M-L; Patureau FW Oxidative C-H amination reactions. Chem. Soc. Rev 2014, 43, 901–910. [DOI] [PubMed] [Google Scholar]
- 6.Wolff ME Cyclization of N-halogenated amines (The Hofmann-Löffler reaction). Chem. Rev 1963, 63, 55–64. [Google Scholar]
- 7.Stateman LM; Nakafuku KM; Nagib DA Remote C–H functionalization via selective hydrogen atom transfer. Synthesis 2018, 50, 1569–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hofmann AW Piperidine and pyridine. Ber. Dtsch. Chem. Ges 1879, 12, 984–990. [Google Scholar]
- 9.ffler K; Freytag C New method for the formation of N-alkylated pyrrolidines. Ber. Dtsch. Chem. Ges 1909, 42, 3427–3431. [Google Scholar]
- 10.Wappes EA; Fosu SC; Chopko TC; Nagib DA Triiodide-mediated delta-amination of secondary C-H bonds. Angew. Chem. Int. Ed 2016, 55, 9974–9978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martinez C; Muñiz K An iodine-catalyzed Hofmann-Loffler reaction. Angew. Chem. Int. Ed 2015, 54, 8287–8291. [DOI] [PubMed] [Google Scholar]
- 12.Becker P; Duhamel T; Stein CJ; Reiher M; Muñiz K Cooperative light activated iodine and photoredox catalysis for the amination of Csp3-H bonds. Angew. Chem., Int. Ed 2017, 56, 8004–8008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clark JR; Feng K; Sookezian A; White MC Manganese-catalysed benzylic C(sp3)-H amination for late-stage functionalization. Nat. Chem 2018, 10, 583–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang M; Yang T; Paretsky JD; Berry JF; Schomaker JM Inverting steric effects: Using “attractive” noncovalent interactions to direct silver-catalyzed nitrene transfer. J. Am. Chem. Soc 2017, 139, 17376–17386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nasrallah A; Boquet V; Hecker A; Retailleau P; Darses B; Dauban P Catalytic enantioselective intermolecular benzylic C(sp3)–H amination. Angew. Chem. Int. Ed 2019, 58, 8192–8196. [DOI] [PubMed] [Google Scholar]
- 16.Bess EN; DeLuca RJ; Tindall DJ; Oderinde MS; Roizen JL; Du Bois J; Sigman MS Analyzing site selectivity in Rh2(esp)2-catalyzed intermolecular C–H amination reactions. J. Am. Chem. Soc 2014, 136, 5783–5789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prier CK; Zhang RK; Buller AR; Brinkmann-Chen S; Arnold FH Enantioselective, intermolecular benzylic C-H amination catalysed by an engineered iron-haem enzyme. Nat. Chem 2017, 9, 629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bakhoda A; Jiang Q; Bertke JA; Cundari TR; Warren TH Elusive terminal copper arylnitrene intermediates. Angew. Chem. Int. Ed 2017, 56, 6426–6430. [DOI] [PubMed] [Google Scholar]
- 19.Collet F; Lescot C; Dauban P Catalytic C–H amination: the stereoselectivity issue. Chem. Soc. Rev 2011, 40, 1926–1936. [DOI] [PubMed] [Google Scholar]
- 20.Ramirez TA; Zhao B; Shi Y Recent advances in transition metal-catalyzed sp3 C–H amination adjacent to double bonds and carbonyl groups. Chem. Soc. Rev 2012, 41, 931–942. [DOI] [PubMed] [Google Scholar]
- 21.Collet F; Dodd RH; Dauban P Catalytic C–H amination: recent progress and future directions. Chem. Commun 2009, 5061–5074. [DOI] [PubMed] [Google Scholar]
- 22.Cho SH; Kim JY; Kwak J; Chang S Recent advances in the transition metal-catalyzed twofold oxidative C–H bond activation strategy for C–C and C–N bond formation. Chem. Soc. Rev 2011, 40, 5068–5083. [DOI] [PubMed] [Google Scholar]
- 23.Ritter JJ; Minieri PP A new reaction of nitriles; amides from alkenes and mononitriles. J. Am. Chem. Soc 1948, 70, 4045–4048. [DOI] [PubMed] [Google Scholar]
- 24.Ritter JJ; Kalish J A new reaction of nitriles; synthesis of t-carbinamines. J. Am. Chem. Soc 1948, 70, 4048–4050. [DOI] [PubMed] [Google Scholar]
- 25.Jiang D; He T; Ma L; Wang Z Recent developments in Ritter reaction. RSC Adv., 2014, 4, 64936–64946. [Google Scholar]
- 26.Guérinot A; Reymond S; Cossy J Ritter reaction: recent catalytic developments. Eur. J. Org. Chem 2012, 19–28. [Google Scholar]
- 27.Michaudel Q; Thevenet D; Baran PS Intermolecular Ritter-type C-H amination of unactivated sp3 carbons. J. Am. Chem. Soc 2012, 134, 2547–2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kiyokawa K; Takemoto K; Minakata S Ritter-type amination of C-H bonds at tertiary carbon centers using iodic acid as an oxidant. Chem. Commun 2016, 52, 13082–13085. [DOI] [PubMed] [Google Scholar]
- 29.Sakaguchi S; Hirabayashia T; Ishii Y First Ritter-type reaction of alkylbenzenes using N-hydroxyphthalimide as a key catalyst. Chem. Commun 2002, 5, 516–517. [DOI] [PubMed] [Google Scholar]
- 30.Li G-X; Morales-Rivera CA; Gao F; Wang Y; He G; Liu P; Chen G A unified photoredox-catalysis strategy for C(sp3)–H hydroxylation and amidation using hypervalent iodine. Chem. Sci, 2017, 8, 7180–7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Milton RD; Minteer SD Nitrogenase bioelectrochemistry for synthesis applications. Acc. Chem. Res 2019, 52, 3351–3360. [DOI] [PubMed] [Google Scholar]
- 32.Shi S-H; Liang Y; Jiao N Electrochemical oxidation induced selective C–C bond cleavage. Chem. Rev 2021, 121, 485–505. [DOI] [PubMed] [Google Scholar]
- 33.Siu JC; Fu N; Lin S Catalyzing electrosynthesis: a homogeneous electrocatalytic approach to reaction discovery. Acc. Chem. Res 2020, 53, 547–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiao KJ; Xing YK; Yang QL; Qiu H; Mei TS Site-selective C-H functionalization via synergistic use of electrochemistry and transition metal catalysis. Acc. Chem. Res 2020, 53, 300–310. [DOI] [PubMed] [Google Scholar]
- 35.Wang F; Stahl SS Electrochemical oxidation of organic molecules at lower overpotential: accessing broader functional group compatibility with electron-proton transfer mediators. Acc. Chem. Res 2020, 53, 561–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kingston C; Palkowitz MD; Takahira Y; Vantourout JC; Peters BK; Kawamata Y; Baran PS A survival guide for the “electro-curious”. Acc. Chem. Res 2020, 53, 72–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yan M; Kawamata Y; Baran PS Synthetic organic electrochemical methods since 2000: On the verge of a renaissance. Chem. Rev 2017, 117, 13230–13319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ackermann L Metalla-electrocatalyzed C-H activation by earth-abundant 3d metals and beyond. Acc. Chem. Res 2020, 53, 84–104. [DOI] [PubMed] [Google Scholar]
- 39.Tang S; Liu Y; Lei A Electrochemical oxidative cross-coupling with hydrogen evolution: a green and sustainable way for bond formation. Chem. 2018, 4, 27–45. [Google Scholar]
- 40.Sperry JB; Wright DL The application of cathodic reductions and anodic oxidations in the synthesis of complex molecules. Chem. Soc. Rev 2006, 35, 605–621. [DOI] [PubMed] [Google Scholar]
- 41.Xiong P; Xu H-C Chemistry with electrochemically generated N-centered radicals. Acc. Chem. Res 2019, 52, 3339–3350. [DOI] [PubMed] [Google Scholar]
- 42.Jing Q; Moeller KD From molecules to molecular surfaces. exploiting the interplay between organic synthesis and electrochemistry. Acc. Chem. Res 2020, 53, 135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yoshida J; Shimizu A; Hayashi R Electrogenerated cationic reactive intermediates: The pool method and further advances. Chem. Rev 2018, 118, 4702–4730. [DOI] [PubMed] [Google Scholar]
- 44.Becker JY; Byrd LR; Miller LL; So Y-H Remote anodic substitution of ketones. J. Am. Chem. Soc 1975, 97, 853–856. [Google Scholar]
- 45.Becker JY; Byrd LR; Miller LL Remote, anodic rearrangement-substitution reaction of aliphatic-ketones. J. Am. Chem. Soc 1974, 96, 4718–4719. [Google Scholar]
- 46.Becker JY; Miller LL; Siegel TM Anodic α-cleavage of ketones. J. Am. Chem. Soc 1975, 97, 849–853. [Google Scholar]
- 47.Tajima T; Ishii H; Fuchigami T Anodic benzylic fluorination of toluene, ethylbenzene, and cumene derivatives. Electrochem. Commun 2002, 4, 589–592. [Google Scholar]
- 48.Hou Z-W; Liu D-J; Xiong P; Lai X-L; Song J; Xu H-C Site-selective electrochemical benzylic C–H amination. Angew. Chem. Int. Ed 2021, 60, 2943–2947. [DOI] [PubMed] [Google Scholar]
- 49.Shen T; Lambert TH Electrophotocatalytic diamination of vicinal C–H bonds. Science 2021, 371, 620–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moutet J-C; Reverdy G Phototochemistry of cation radicals in solution; photoinduced electron-transfer reactions between alcohols and the N, N, N’, N’,-tetraphenyl-p-phenylenediamine cation radical. J. Chem. Soc., Chem. Commun 1982, 654–655. [Google Scholar]
- 51.Scheffold R; Orlinski R Carbon-carbon bond formation by light-assisted B12 catalysis. nucleophilic acylation of Michael olefins. J. Am. Chem. Soc 1983, 105, 7200–7202. [Google Scholar]
- 52.Yan H; Hou Z-W; Xu H-C Photoelectrochemical C-H alkylation of heteroarenes with organotrifluoroborates. Angew. Chem. Int. Ed 2019, 58, 4592–4595. [DOI] [PubMed] [Google Scholar]
- 53.Wang F; Stahl SS Merging photochemistry with electrochemistry: functional-group tolerant electrochemical amination of C(sp3)-H bonds. Angew. Chem. Int. Ed 2019, 58, 6385–6390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang L; Liardet L; Luo J; Ren D; Grätzel M; Hu X Photoelectrocatalytic arene C-H amination. Nat. Catal 2019, 2, 366–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang W; Carpenter K; Lin S Electrochemistry broadens the scope of flavin photocatalysis: photoelectrocatalytic oxidation of unactivated alcohols. Angew. Chem. Int. Ed 2020, 59, 409–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Niu L; Jiang C; Liang Y; Liu D; Bu F; Shi R; Chen H; Dutta Chowdhury A; Lei A Manganese-catalyzed oxidative azidation of C(sp3)–H bonds under electrophotocatalytic conditions. J. Am. Chem. Soc 2020, 142, 17693–17702. [DOI] [PubMed] [Google Scholar]
- 57.Liu J; Lu L; Wood D; Lin S New redox strategies in organic synthesis by means of electrochemistry and photochemistry. ACS Cent. Sci 2020, 6, 1317–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yu Y; Guo P; Zhong J-S; Yuan Y; Ye K-Y Merging photochemistry with electrochemistry in organic synthesis. Org. Chem. Front 2020, 7, 131–135. [Google Scholar]
- 59.Huang H; Strater ZM; Rauch M; Shee J; Sisto TJ; Nuckolls C; Lambert TH Electrophotocatalysis with a trisaminocyclopropenium radical dication. Angew. Chem. Int. Ed 2019, 58, 13318–13322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huang H; Strater ZM; Lambert TH Electrophotocatalytic C-H functionalization of ethers with high regioselectivity. J. Am. Chem. Soc 2020, 142, 1698–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim H; Kim H; Lambert TH; Lin S Reductive Electrophotocatalysis: Merging Electricity and Light To Achieve Extreme Reduction Potentials. J. Am. Chem. Soc 2020, 142, 2087–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cowper NGW; Chernowsky CP; Williams OP; Wickens ZK Potent Reductants via Electron-Primed Photoredox Catalysis: Unlocking Aryl Chlorides for Radical Coupling. J. Am. Chem. Soc 2020, 142, 2093–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.We found that changing the electrolyte to LiClO4 often increased conversion and reaction rate. In addition, using LiClO4 in many cases reduced the difficulty of isolating the pure products because TBAPF6 often has very similar polarity as some of the products, making them difficult to purify by chromatography.
- 64.Lund BW; Piu F; Gauthier NK; Eeg A; Currier E; Sherbukhin V; Brann MR; Hacksell U; Olsson R Discovery of a potent, orally available, and isoform-selective retinoic acid β2 receptor agonist. J. Med. Chem 2005, 48, 7517–7519. [DOI] [PubMed] [Google Scholar]
- 65.Hille UE; Zimmer C; Vock CA; Hartmann RW First selective CYP11B1 inhibitors for the treatment of cortisol-dependent diseases. ACS. Med. Chem. Lett 2011, 2, 2–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Alibert S; Santelli-Rouvier C; Castaing M; Berthelot M; Spengler G; Molnar J; Barbe J Effects of a series of dihydroanthracene derivatives on drug efflux in multidrug resistant cancer cells. Eur. J. Med. Chem 2003, 38, 253–263. [DOI] [PubMed] [Google Scholar]
- 67.Chambers MS; Baker R; Billington DC; Knight AK; Middlemiss DN; Wonget EHF Spiropiperidines as high-affinity, selective σ ligands. J. Med. Chem 1992, 35, 2033–2039. [DOI] [PubMed] [Google Scholar]
- 68.Taylor CP;Traynelis SF; Siffert J; Pope LE; Matsumoto RR Pharmacology of dextromethorphan: relevance to dextromethorphan/quinidine (Nuedexta®) clinical use. Pharmacology & Therapeutics 2016, 164, 170–182. [DOI] [PubMed] [Google Scholar]
- 69.Merachi M; Jung YY; Fan L; Sethi G; Ahn KS A brief overview of the antitumoral actions of leelamine. Biomedicines 2019, 7, 53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.