

Abstract
We developed and validated a method for direct determination of per- and polyfluoroalkylated substances (PFASs) in environmental water samples without prior sample concentration. Samples are centrifuged and supernatants passed through an Acrodisc Filter (GXF/GHP 0.2 um, 25 mm diameter). After addition of ammonium acetate, samples are analyzed by UPLC-MS/MS using an AB Sciex 6500 plus Q-Trap mass spectrometer operated in negative multiple reaction-monitoring (MRM) mode. The instrument system incorporates a delay column between the pumps and autosampler to mitigate interference from background PFAS. The method monitors eight short-/long-chain PFAS which are identified by monitoring specific precursor product ion pairs and by their retention times and quantified using isotope mass-labeled internal standard based calibration plots. Average spiked recoveries (n = 8) of target analytes ranged from 84 to 110% with 4–9% relative standard deviation (RSD). The mean spiked recoveries (n = 8) of four surrogates were 94–106% with 3–8% RSD. For continuous calibration verification (CCV), average spiked recoveries (n = 8) for target analytes ranged from 88 to 114% with 4–11% RSD and for surrogates ranged from 104–112% with 3–11% RSD. The recoveries (n = 6) of matrix spike (MX), matrix spike duplicate (MXD), and field reagent blank (FRB) met our acceptance criteria. The limit of detection for the target analytes was between 0.007 and 0.04 ng/mL The method was used to measure PFAS in tap water and surface water.
Keywords: PFAS, Direct injection, Acrodisc filtration, Drinking and surface water, UPLC-MS/MS
1. Introduction
Per- and polyfluoroalkylated substances (PFASs) are a very large class of synthetic surfactant chemicals used in food packaging, water and stain resistant textiles fand aqueous firefighting foams [1]. Global production of two PFAS, perfluorooctane sulfonate (PFOS) and perfluorootanoic acid (PFOA) was ~4650 metric tons in 2000 but these have been supplanted by shorter chain compounds that continue to be produced in similar quantities [2]. The stability of PFAS combined with their widespread manufacturing and use have led to significant environmental contamination and human exposure. PFAS concentrations in surface and ground water are commonly in the parts per trillion (ppt) range and PFAS are commonly detectable in drinking water with an EPA established health advisory level of 70 ppt for the sum of PFOA and PFOS [3]. PFAS can be detected in the blood of almost all adult Americans at ng/L levels [4]. Circulating levels of PFAS associate with adverse human health effects in multiple epidemiological studies including elevated levels of circulating lipids (a risk factor for cardiovascular diseases), decreased immune function and increased risk for certain types of cancer [5], [6], [7], [8]. Studies using preclinical models identify potential mechanisms for these toxic effects of PFAS [9]. Accordingly, PFAS are now recognized as a significant public health concern [1].
Human biomonitoring measurements for PFAS at ng/mL levels in plasma and serum amenable to HPLC coupled electrospray ionization tandem mass spectrometry [10,11]. PFAS levels in surface and drinking water are generally 1000 times lower (ng/L) which requires pre-concentration of samples prior to analysis using solid-phase extraction as described in the widely used EPA 537 method [12,13]. This approach is time consuming, costly, and can introduce a significant source of technical variability into the analytical workflow. Rapid, sensitive reproducible methods for routine screening of water samples would be a useful advance for the field [14]. The latest triple quadrupole instruments have sufficient sensitivity to quantify PFAS at the ng/L levels present in surface and drinking water with mitigation of interference from background levels of PFAS. A method for PFAS analysis by direct sample injection would simplify the analytical workflow, speed up the screening process and result in significant cost savings by eliminating the need for costly solid-phase extraction supplies, multiple sample handling steps and addition of expensive internal standards and surrogates to the large starting sample volumes. Here, we report a direct injection method for analysis of PFAS in environmental water samples. The method employs centrifugation and membrane filtration of small volumes of samples which are then analyzed by an adaptation of our previously reported UHPLC ESI MS/MS method incorporating a delay column to mitigate interference from background PFAS contamination. The method employs surrogates and stable isotope labeled internal standards is technically robust and has sufficient sensitivity and reproducibility for use as a primary screening method to detect and quantify PFAS at commonly observed levels in surface water and drinking water. The method can accurately detect and quantify common PFAS species including PFOA and PFOS at levels below the commonly recommended screening level of 70 ng/L.
2. Materials and methods
2.1. Standard chemicals, solvents, and materials
Individual target analytes including perfluorooctane sulfonate (PFOS, >98%), perfluorootanoic acid (PFOA, >98%), perfluorononanoic acid (PFNA, > 98%), perfluorohexane sulfonate (PFHxS, > 98%), perfluoroheptanoic acid (PFHpA, > 98%), perfluorobutane sulfonates (PFBS, > 98%), hexafluoropropylene oxide dimer acid [2,3,3,3-tetrafluoro-2-(1,1,2,2,3,3,3-heptafluoropropanioc acid) (GenX, HFPO-DA, > 98%)], sodium 1H, 1H, 2H, 2H-PFHxS (4:2 FTS, >98%), surrogate standards (SS) 13C4–PFOA (> 98%), 13C5–PFNA (> 98%), 13C4–PFOS (> 98%), 13C2–4:2 FTS (>98%) and internal standard (IS) 13C4–PFHpA (< 98%) were from Wellington Laboratories (Guelph, ON, Canada). HPLC grade methanol, ammonium acetate, polypropylene bottles, luer lock disposable syringe were from Fisher Scientific, USA. Acrodisc filters (GXF/GHP, 0.2 µm, 25 mm diameter) were from Pall Corporation, GA, USA. Ultra-nanopure water was from our laboratory Milli Q water system.
2.2. Sample collection and storage
Six surface water samples obtained from a site of suspected PFAS contamination. About 250 mL surface water was collected in polypropylene bottle and 0.25 g ammonium acetate was added. The field reagent blank (FRB) was 250 mL of laboratory ultrapure water to which the same amount of ammonium acetate was added in the field. Samples were stored and shipped on ice and stored at ≤ 4°C until extraction and analysis.
2.3. Membrane filtration of water samples
A volume of 40 mL of the collected water samples was transferred to a polypropylene tube and centrifuged at 16,000 rpm for 45 min at 4 °C. The Acrodisc filter was pre-conditioned with 5.0 mL acetonitrile followed by 5 mL methanol at a flow rate of 1 mL/min using a Luer Lock disposable 10 mL syringe. The 13C labeled PFAS surrogates including 13C2–4:2 FTS, 13C4-PFOA, 13C5-PFNA, and 13C4-PFOS were added to 6.0 mL of the centrifuged water samples from stock solutions. The sample was passed through the acrodisc filter which was then washed with 1 mL methanol such that the total volume of the filtered sample was 7.0 mL. An aliquot of 0.985 ml of the filtered sample was taken and 1.0 ng of IS (13C4-PFHpA, 10 µl of 100 ng/mL) solution was added and the sample transferred to a polypropylene autosampler vial for analysis.
2.4. UPLC-MS/MS analysis of PFAS
We used an adaptation of our previously developed ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) [11]. In brief, this employs a Shimadzu Nexra X2 LC 30 AD UPLC and Nexra X2-SIL 30 AC autosampler coupled with an ABSciex 6500 plus QTRAP mass spectrometer and a Waters Atlantis T3 3 μm (50 mm length × 2.1 mm ID) column. The injection volume was 5.0 μL. To mitigate interference from PFASs contamination in the UHPLC solvents and instrument system, a delay column [Waters Atlantis T3 3 μm (100 mm length × 2.1 mm ID)] was installed between the solvent mixer and autosampler. Data were collected in negative multiple reaction monitoring (MRM) and processed using ABSciex Analyst software version 1.7/Multiquant version 3.02.
2.5. Assay performance and quality control
To evaluate the technical reproducibility and sensitivity of the method, 18 quality control (QC) samples were prepared from the collected surface water and field reagent blank (FRB) samples. These encompassed 12 of these were matrix spike (MX) and matrix spike duplicate (MXD) prepared from the surface water samples and an additional 6 matrix spike samples were from the FRB. All QC samples were fortified with known concentration of target analytes and surrogates. The QC sample spiking concentrations and volumes of these are shown in Table 1. The QC samples were processed using the centrifugation/filtration method described above. Analyte recoveries were calculated for all QC samples. LODs of target analytes were evaluated using additional standard samples that were prepared at the indicated lower concentrations (Table 1) and injected multiple times. The limit of detection (LOD) was determined using the signal to noise (S/N) ratio of the response of individual target analyte ion and noise immediately preceding and after the peaks using the ABSciex Analyst software S/N calculation protocol.
Table 1.
Analytical method performance: LODs, LDR with QC recoveries from tap water, MX, MXD, FRB and CCV for PFAS compounds.
| Compounds | LOD, (ng/mL) | LDR, (ng/mL) | Spiking* (ng/mL) | Spike RecŦ (n = 8) (%) | MX ± RSD (n = 6) (%) | MXD ± RSD (n = 6) (%) | FRB ± RSD (n = 6) (%) | CCV ± RSD (n = 8) (%) |
|---|---|---|---|---|---|---|---|---|
| Short-chain | ||||||||
| PFBS | 0.007 | 0.02–0.56 | 0.42 | 85.8 ± 6.0 | 87.4 ± 5.7 | 89.5 ± 4.1 | 87.9 ± ±4.9 | 94.8 ± 4.0 |
| GenX | 0.04 | 0.08–2.40 | 1.8 | 84.8 ± 8.9 | 88.1 ± 5.1 | 86.8 ± 4.2 | 88.9 ± 7.1 | 87.8 ± 5.6 |
| 4.2 FTS | 0.007 | 0.02–0.56 | 0.42 | 99.8 ± 4.6 | 85.8 ± 7.6 | 85.6 ± 4.7 | 94.1 ± 5.8 | 105.3 ± 5.0 |
| Long-chain | ||||||||
| PFHpA | 0.021 | 0.05–1.60 | 1.2 | 94.6 ± 4.2 | 91.1 ± 5.6 | 87.1 ± 4.5 | 90.0 ± 3.4 | 103.1 ± 8.4 |
| PFHxS | 0.019 | 0.05–1.52 | 1.14 | 83.9 ± 5.7 | 91.7 ± 4.8 | 94.8 ± 2.4 | 93.4 ± 5.5 | 98.4 ± 3.9 |
| PFOA | 0.015 | 0.04–1.20 | 0.9 | 101.8 ± 4.3 | 88.6 ± 6.8 | 91.0 ± 2.5 | 95.7 ± 4.9 | 111.2 ± 4.4 |
| PFOS | 0.02 | 0.05–1.60 | 1.2 | 110.1 ± 7.8 | 84.5 ± 17.7 | 95.9 ± 25.9 | 104.3 ± 11.9 | 113.5 ± 10.6 |
| PFNA | 0.02 | 0.05–1.60 | 1.2 | 99.3 ± 6.9 | 97.2 ± 5.3 | 106.0 ± 9.9 | 107.8 ± 6.7 | 110.1 ± 9.1 |
| Surrogates | ||||||||
| 13C2–4:2 FTS | NM | 0.04–1.80 | 0.96 | 94.3 ± 7.5 | 94.5 ± 20.0 | 83.9 ± 4.9 | 90.9 ± 4.1 | 104.3 ± 3.5 |
| 13C4-PFOA | NM | 0.06–1.92 | 1.44 | 97.6 ± 3.0 | 90.7 ± 6.2 | 90.9 ± 2.2 | 100.4 ± 4.0 | 104.4 ± 5.2 |
| 13C4-PFOS | NM | 0.10–3.20 | 2.4 | 106.0 ± 8.2 | 83.9 ± 17.6 | 97.1 ± 25.7 | 103.8 ± 9.7 | 111.6 ± 9.9 |
| 13C5-PFNA | NM | 0.06–1.92 | 1.44 | 97.1 ± 8.3 | 97.7 ± 4.6 | 99.6 ± 11.5 | 112.7 ± 8.2 | 107.3 ± 6.0 |
LODs were calculated at S/N = ~ 3 – 5 for all compounds, LDR - linear dynamic range
MX - surface water matrix spike, MXD - surface water matrix spike duplicate
FRB - field reagent blank, Spike RecŦ -Tap water spike recovery
RSD-relative standard deviation, NM-not monitored
Spiking conc for Spike RecŦ, MX, MXD, FRB and CCV
2.6. Calibration curve, calibration verification and quality assurance (QA)
A series of samples containing a range of calibration of analytes and mass labeled surrogates and mass-labeled internal standards (IS) were prepared. The analyte versus IS peak areas generated using the above methods were linear unweighted regression plots, forced through the origin for each analyte and surrogate. Correlation coefficients (R2) were between 0.9992 and 0.9999 for all target analytes and surrogates. A separate continuous calibration verification (CCV) sample was prepared with standards and surrogates following the US EPA method 537 procedure, with a concentration at the mid-level calibration point. The CCV samples were injected to monitor instrument response/calibration and recovery of surrogates within each sample batch. These CCV standards were analyzed using calibration plots before the beginning analysis of data from each batch of extracts.
The U.S. EPA method-537.1 approved CCV acceptance criteria are 70 −130% surrogate recovery and MX, MXD and FRB acceptance criteria are 50 to 150% analytes and surrogates recoveries. For the method presented here, average surrogate recoveries were 104–111% for CCV; 84–98% for MX; 84–100% for MXD and 91–113% for FRB. The mean analytes recoveries were 88 to 114% for CCV; 85 to 97% for MX; 86 to 106% for MXD and 88 to 108% for FRB. If CCV surrogate recoveries were outside of approved criteria such as < 70% or > 130%, the problem was addressed, and the sample batch was reanalyzed. For each sample batch, samples were injected in the order of solvent blank, IS-blank, calibration standards, CCV samples, solvent blank, QC samples, IS-blank, sample batch, CCV samples and solvent blank. Before processing the surface water data, CCV standards that bracketed the sample and QC extracts were verified to conform compliance with the EPA 537 method criteria. If target compounds were detected in FRBs these values were subtracted from the levels reported for the analytical samples.
3. Results and discussion
The purpose of this study was to develop and validate a method for measurement of a panel of common PFAS in water at sub ng/L levels without use of sample pre-concentration by solid-phase extraction. The method reported here involves centrifugation, pre-conditioning of an Acrodisc membrane filter, sample filtration and direct analysis of filtered sample by UPLC-MS/MS. To validate the method, we used the EPA method 537 sampling protocols and assay performance recommendations including samples for continuous calibration verification (CCV), matrix spike (MX), matrix spike duplicate (MXD), field reagent blank (FRB) and IS-blank (IS-BLK). Unweighted isotope-mass-labeled-IS calibration plots were used to calculate all QCs recoveries and PFAS quantification.
3.1. Optimization of sample filtration
Centrifugation and membrane filtration were used to remove particulate material from samples. Previous reports indicated that PFAS readily absorb to nitrocellulose filters but are less readily absorbed to cellulose or polyethersulphone filters [15]. The acrodisc filters we selected for our study are polypropylene and incorporate a glass fiber pre filter with a hydrophobic polytetrafluoroethylene (PTFE) membrane. We evaluated recoveries of PFAS and added surrogates and internal standards using laboratory tap water which contains several common PFAS at levels that are typically reported in municipal water. Pre-conditioning of the filter was essential for efficient recovery of PFAS compounds. We evaluated pre-rinsing the filter with water, methanol and acetonitrile and mixtures of these solvents and examined effects of processing different volumes of tap water samples using these pre-conditioning protocols (Fig. 1). Initially (Trial 1), we pre-conditioned the Acrodisc with 1.0 mL water, and extracted 2.0 to 3.0 mL of spiked tap water. The spiked analyte recovery was between 9.8 and 70% and surrogate recoveries ranged from 8.8 to 62.9%. The lowest recovery for PFOS was 9.8% and for 13C4-PFOS surrogate was 8.8%. To improve the recovery of PFOS and 13C4-PFOS, in the Trial 2, we pre-conditioned Acrodisc filter with 5 mL acetonitrile followed by 5 mL methanol and then loaded 5.0, 7.0 and 10.0 mL of analyte and surrogate spiked tap water. With this approach the average analyte recovery ranged from 34.6 to 90.1% and surrogate recoveries were 36.4 to 87.8%. PFOS and 13C4-PFOS surrogate recoveries were between 34.6 and 36.4%, respectively. In Trial 3, we maintained the same Trial 2 pre-conditioning protocol but loaded 6 mL of our spiked tap water sample and then rinsed the Acrodisc filter with 1.0 mL methanol. With this approach, average analyte recoveries were between 83.9 and 110.1% with 4.3 to 8.9% relative standard deviation (RSD) and surrogate recoveries ranged from 94.3 to 106% with 3.0 to 8.3% RSD. We used this method for subsequent studies.
Fig. 1.
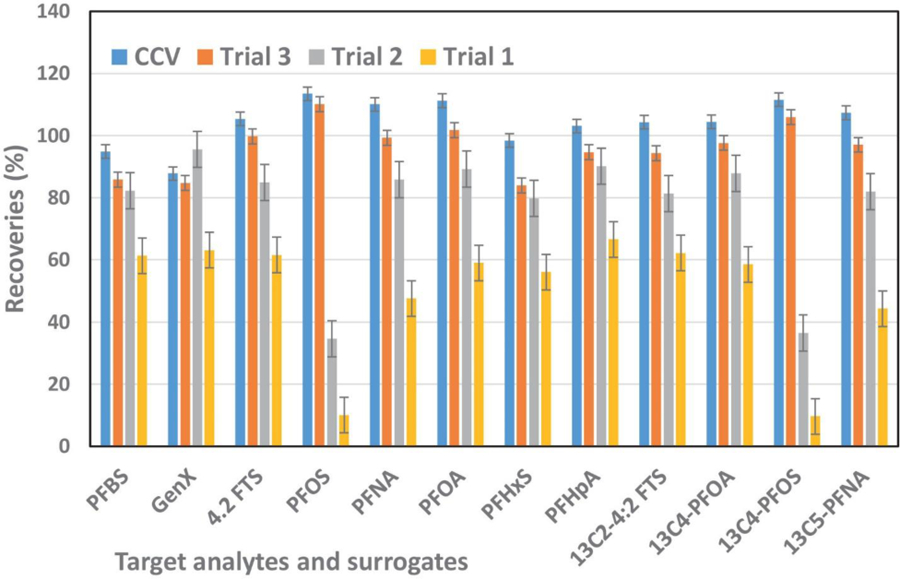
Acrodisc filter recovery optimization for PFAS analytes and surrogates. The isotope mass labeled 13C2–4:2 FTS, 13C4-PFOA, 13C4-PFOS and 13C5-PFNA, are surrogates and others are target analytes. Trial 1, Acrodisc filter conditioned with 1.0 mL nanopure water; Trial 2, Acrodisc filter conditioned with 5.0 mL acetonitrile following 5.0 mL methanol; Trial 3, Acrodisc filter conditioned with 5.0 mL acetonitrile following 5.0 mL methanol and after loading the sample filter was rinsed with 1.0 mL methanol; CCV, continuous calibration verification or laboratory control sample used to calibration check through the analysis.
3.2. Analytical method performance
Technical method performance and reproducibility of the entire analytical procedure was evaluated for reproducibility, limits of detection (LODs) linearity and dynamic range (LDR) using surface and drinking water samples. Table 1 presents the analytical method performance showing LODs, LDR, and mean analyte recoveries with relative standard deviation (RSD). The average spike analyte recoveries (n = 8) from tap water were 84–−10% with 4–9% RSD, surrogate recoveries 94–106% with 3–8% RSD. For CCV, the mean spike analytes recoveries (n = 8) corresponded to 88–114% with 4–11% RSD, and for surrogates to 104–112% with 3–11% RSD. In the case of MX, average spiked recoveries (n = 6) 85–97% with 5–17% RSD for analytes, 84–98% with 5–20% RSD for surrogates were achieved. For MXD, the mean spiked recoveries (n = 6) were found to be 86–106% with 4–26% RSD for analytes, 84–100% with 2–26% RSD for surrogates. The spiked recoveries (n = 6) from FRB for analytes were 88 to 108% with 3 to 12% RSD, for surrogates 91–113% with 4–10% RSD. Our method meets or exceeds the acceptance criteria for EPA method-537, which are 70 −130% for CCV surrogates recoveries and 50 to 150% for MX, MXD and FRB for analytes and surrogate recoveries. The target compound LODs were estimated at S/N = 3 against blank / background signal. The LODs of the targeted analytes were assessed at between 0.007 and 0.04 ng/mL, as low as 0.007 ng/mL for PFBS (Table 1). The limit of quantitation was estimated 3 fold above the LODs which meets our requirement for a method that can measure PFAS at commonly used screening levels in surface water or drinking water. Thus, we developed a robust, simple and reproducible analytical method has been developed that could be used for PFAS analysis in groundwater by UPLC-MS/MS maintaining the acceptance criteria for EPA method 537.
3.3. Identification and measurement of PFAS in tap and surface water
PFAS were identified from their precursor/product ion signals and retention times as determined using authentic synthetic standards. When the ratio of quantifier to qualifier ion abundance was within ± 20% of the expected value the identity of the target compound was confirmed. A typical chromatogram shown in Fig. 2 B indicates the characteristic mass signals and retention time of targeted long-chain PFOA and PFOS analytes detected in surface water samples collected from a suspected site of PFAS contamination. The PFOA eluted from analytical column at 8.08 min and was detected using its quantifier ion 368.7 m/z and confirmation ion 168.7 m/z. In the case of PFOS, the analyte RT had 8.81 min and the quantifier ion was 79.9 m/z and confirmation ion was 98.8 m/z. Peaks eluting at around 9.8 min in Fig. 2 (A) and (B) are interfering PFAS compounds that are present in the instrument system but separated from PFAS in the analytical samples using the delay column.
Fig. 2.
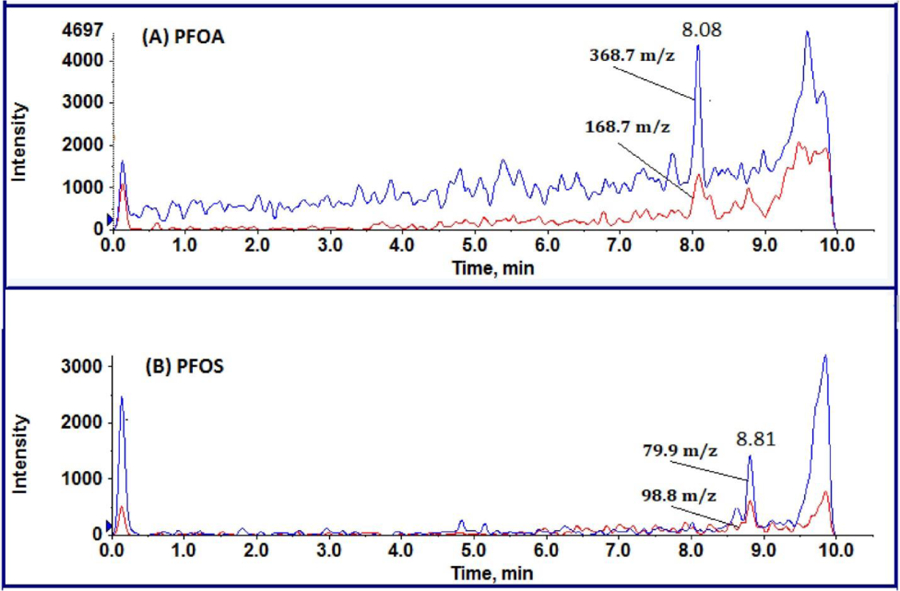
Typical detection and characterization of PFAS compounds protocol surface water collected from suspected. Extracted ion chromatogram for (A) PFOA quantitation ion 368.7 m/z, confirmation ion 168.7 m/z, and retention time 8.08 min, (B) PFOS quantitation ion 79.9 m/z, confirmation ion 98.8 m/z and retention time 8.81 min.
3.4. PFAS levels in tap and surface water
Tap water and surface water samples were analyzed using our validated method described in above. Table 2 presents the concentration of targeted short- and long-chain PFAS compounds. The PFOS and PFOA concentrations were 0.016 – 0.069ng/ml and 0.019 – 0.052 ng/ml, respectively. PFNA was observed in some water samples ranged from 0.007 to 0.015 ng/ml. The short-chain PFBS detected at 0.007 to 0.012 ng/mL in three samples. Other compounds such as PFHxS, PFHpA, GenX and 4:2 FTS were not detected in either the tap or surface water samples.
Table 2.
PFAS concentration (ng/mL) in tap and suspected site surface water samples.
| Sample ID | PFBS | GenX | 4.2 FTS | PFOS | PFNA | PFOA | PFHxS | PFHpA |
|---|---|---|---|---|---|---|---|---|
| Site 1 | 0.012 | ND | ND | 0.037 | 0.007 | 0.043 | ND | ND |
| Site 2 | 0.007 | ND | ND | 0.04 | ND | 0.032 | ND | ND |
| Site 3 | ND | ND | ND | 0.038 | ND | 0.052 | ND | ND |
| Site 4 | ND | ND | ND | 0.05 | ND | 0.042 | ND | ND |
| Site 5 | 0.007 | ND | ND | 0.016 | 0.015 | 0.048 | ND | ND |
| Site 6 | ND | ND | ND | 0.017 | 0.011 | 0.019 | ND | ND |
| Tap water | ND | ND | ND | 0.069 | 0.008 | 0.037 | ND | ND |
4. Conclusions
A reproducible and robust UPLC-MS/MS has been developed and validated for direct injection analysis of PFAS in water samples. The method is faster and less expensive than solid phase extraction based approaches (the acrodisc filters cost < 20% of the cost of solid phase extraction columns). The method is suitable for routine screening of common PFAS in surface or drinking water.
Highlights.
PFAS are widely distributed persistent environmental chemicals.
PFAS exposure is associated with adverse human health effects.
Measurement of PFAS in surface and drinking water is required.
This is feasible using sample filtration/liquid chromatography mass spectrometry.
Acknowledgments
This research was supported by NIH/NIEHS Grants P42ES007380 and P30ES026529 and resources provided by the Lexington Veterans Affairs Medical Center.
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].E. National Academies of Sciences Medicine, E. division on, s. life, i. environmental health matters, the national academies collection: reports funded by national institutes of health Mantus E, Shelton-Davenport M, Johnson A (Eds.), Understanding, Controlling, and Preventing Exposure to PFAS: Proceedings of a Workshop-in Brief, National Academies Press, Washington (DC) (2020), 10.17226/25856 [DOI] [PubMed] [Google Scholar]
- [2].Glüge J, Scheringer M, Cousins IT, DeWitt JC, Goldenman G, Herzke D, Lohmann R, Ng CA, Trier X, Wang Z An overview of the uses of per- and polyfluoroalkyl substances (PFAS), environmental science Process. Impacts, 22 (12) (2020), pp. 2345–2373, 10.1039/d0em00291g [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Domingo JL, Nadal M Human exposure to per- and polyfluoroalkyl substances (PFAS) through drinking water: a review of the recent scientific literature Environ. Res, 177 (2019), Article 108648, 10.1016/j.envres.2019.108648 [DOI] [PubMed] [Google Scholar]
- [4].Calafat AM, Wong LY, Kuklenyik Z, Reidy JA, Needham LL Polyfluoroalkyl chemicals in the U.S. population: data from the national health and nutrition examination survey (NHANES) 2003–2004 and comparisons with NHANES 1999–2000 Environ. Health Perspect, 115 (11) (2007), pp. 1596–1602, 10.1289/ehp.10598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Averina M, Brox J, Huber S, Furberg AS Exposure to perfluoroalkyl substances (PFAS) and dyslipidemia, hypertension and obesity in adolescents. the fit futures study Environ. Res, 195 (2021), Article 110740, 10.1016/j.envres.2021.110740 [DOI] [PubMed] [Google Scholar]
- [6].Gardener H, Sun Q, Grandjean P PFAS concentration during pregnancy in relation to cardiometabolic health and birth outcomes Environ. Res, 192 (2021), Article 110287, 10.1016/j.envres.2020.110287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Geiger SD, Yao P, Vaughn MG, Qian Z PFAS exposure and overweight/obesity among children in a nationally representative sample Chemosphere, 268 (2021), Article 128852, 10.1016/j.chemosphere.2020.128852 [DOI] [PubMed] [Google Scholar]
- [8].Steenland K, Winquist A PFAS and cancer, a scoping review of the epidemiologic evidence Environ. Res, 194 (2021), Article 110690, 10.1016/j.envres.2020.110690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Szilagyi JT, Avula V, Fry RC Perfluoroalkyl substances (PFAS) and their effects on the placenta, pregnancy, and child development: a potential mechanistic role for placental peroxisome proliferator-activated receptors (PPARs) Curr. Environ. Health Rep, 7 (3) (2020), pp. 222–230, 10.1007/s40572-020-00279-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Berger U, Kaiser MA, Karrman A, Barber JL, van SP Leeuwen Recent developments in trace analysis of poly- and perfluoroalkyl substances Anal. Bioanalyt. Chem, 400 (6) (2011), pp. 1625–1635, 10.1007/s00216-011-4823-8 [DOI] [PubMed] [Google Scholar]
- [11].Mottaleb MA, Petriello MC, Morris AJ High throughput UHPLC MS/MS measurement of per and poly fluorinated alkyl substances (PFAS) in human serum J. Anal. Toxicol, 44 (4) (2020), pp. 339–347, 10.1093/jat/bkz097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Shoemaker JA, Boutin B, Grimmett P Development of a U.S. EPA drinking water method for the analysis of selected perfluoroalkyl acids by solid-phase extraction and LC-MS-MS J. Chromatogr. Sci, 47 (1) (2009), pp. 3–11, 10.1093/chromsci/47.1.3 [DOI] [PubMed] [Google Scholar]
- [13].Shoemaker JA, Tettenhorst D, Method 537.1: determination of selected per- and polyfluorinated alkyl substances in drinking water by solid-phase extraction and liquid chromatography/tandem mass spectrometry (LC/MS/MS), (2018).
- [14].Meegoda JN, Kewalramani JA, Li B, Marsh RW A review of the applications, environmental release, and remediation technologies of per- and polyfluoroalkyl substances Int. J. Environ. Res. Public Health, 17 (21) (2020), 10.3390/ijerph17218117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sörengård M, Franke V, Tröger R R, Ahrens L Losses of poly- and perfluoroalkyl substances to syringe filter materials J. Chromatogr. A, 1609 (2020), Article 460430, 10.1016/j.chroma.2019.460430 Jan 4 [DOI] [PubMed] [Google Scholar]