

Abstract
Beta cells of the pancreatic islet express many different types of ion channels. These channels reside in the β-cell plasma membrane as well as subcellular organelles and their coordinated activity and sensitivity to metabolism regulate glucose-dependent insulin secretion. Here, we review the molecular nature, expression patterns, and functional roles of many β-cell channels, with an eye toward explaining the ionic basis of glucose-induced insulin secretion. Our primary focus is on KATP and voltage-gated Ca2+ channels as these primarily regulate insulin secretion; other channels in our view primarily help to sculpt the electrical patterns generated by activated β-cells or indirectly regulate metabolism. Lastly, we discuss why understanding the physiological roles played by ion channels is important for understanding the secretory defects that occur in type 2 diabetes.
Introduction
The islets of Langerhans are endocrine microorgans that are embedded in the larger exocrine pancreas (211). The approximately one million islets that reside within a typical human pancreas constitute a very small percentage of the total volume of the organ [~1%; (102)], while most of the organ is made of acinar and ductal tissue. Within an islet are several different types of endocrine cells. While insulin-secreting β-cells make up the majority (~80% in mice and ~50% in humans), islets also possess glucagon-secreting α-cells (~15% in mice and ~40% in humans), somatostatin-secreting δ-cells (~5% in mice and ~10% in human), ε-cells and PP-cells, which secrete ghrelin and pancreatic polypeptide, respectively (41, 44, 119, 120, 198). Mouse islets, which are a standard experimental model have a β-cell-rich core surrounded by a mantle of mainly α-cells (44, 139, 265). In contrast, human islets have a more random distribution of cells throughout the islet mantle and core (44, 139, 265). As the focus of this article is the pancreatic β-cell, interested readers may wish to consult recent reviews that focus on other islet cell types (95, 229, 297). To be clear, at the onset the focus of this article is not on all β-cell ion channels, but only those which have been shown to play a clear functional role. A list of the ion channels known to be involved in β-cell electrical activity and potentially in the control of insulin secretion can be found in Table 1. We also wish to apologize in advance to colleagues whose work is not fully discussed in the present article due to limitations of space.
Table 1.
Ion Channels in Pancreatic β-cells Involved in Insulin
| Family | Channel | Gene | Function in the β-cell | Activators | Blockers | References |
|---|---|---|---|---|---|---|
| Inward-rectifying K+ channels | Kir2.1 | Kcnj2/KCNJ2 | Spike repolarization? | Zacopride | ML133, Ba2+ | (221, 304, 305) |
| Kir3.2 (GIRK2) | Kcnj6 | Spike repolarization in response to hormones and neurotransmitters | Noradrenaline, somatostatin, galanin | Atomoxetine, Tertiapin, Ba2+ | (80, 144) | |
| Kir6.2/SUR1 (KATP) | Kcnj11/KCNJ11 Abcc8/ABCC8 | Mediates membrane potential and its coupling to metabolism | Diazoxide | Sulfonylureas, ATP/ADP ratio, glinides | (103, 163, 174) | |
| Voltage-gated Ca2+ channels | Cav1.2 (L-type) | Cacna1c/CACNA1C | Mediates Ca2+ influx during electrical activity | (S)-(-)-Bay K8644 | Dihydropyridines, benzothiazepines, phenylalkylamines | (153, 186, 251, 298) |
| Cav1.3 (L-type) | Cacna1d/CACNA1D | Mediates Ca2+ influx during electrical activity | (S)-(-)-Bay K8644 | Dihydropyridines, benzothiazepines, phenylalkylamines | (153, 186, 251, 298) | |
| Cav2.1 (P/Q-type) | Cacna1a/CACNA1A | Triggering exocytosis of insulin granules | – | ω-Agatoxin IVA | (36, 161) | |
| Cav2.2 (N-type) | Cacna1b/CACNA1B | Spiking? | – | ω-Conotoxin GVIA | (282) | |
| Cav2.3 (R-type) | Cacna1e | Spiking? | – | SNX482 | (188, 288) | |
| Cav3.1 (T-type) | Cacna1g/CACNA1G | Low threshold spiking | – | Amiloride, NiCl2, NNC55-3096 | (36, 154, 275, 282) | |
| Cav3.2 (T-Type) | Cacna1h/CACNA1H | Low threshold spiking | – | Amiloride, NiCl2, NNC55-3096 | (36, 154, 275, 282) | |
| Voltage-gated Na+ channels | Nav1.3 | Scn3a/SCN3A | Upstroke of action potential | Batrachotoxin, veratridine | Tetrodotoxin, saxitoxin | (18, 30, 36, 189) |
| Nav1.6 | Scn8a/SCN8A | Upstroke of action potential | Batrachotoxin, veratridine | Tetrodotoxin, saxitoxin | (18, 30, 36, 189) | |
| Nav1.7 | Scn9a/SCN9A | Upstroke of action potential | Batrachotoxin, veratridine | Tetrodotoxin, Saxitoxin | (18, 30, 36, 189) | |
| Volume-regulated Cl− channels | VRAC/SWELL1 | Lrrc8a/LRRC8A | Membrane depolarization and response to swelling; volume regulation | Cell swelling, cAMP | DCPIB, DIDS, NPPB, niflumic acid, glyburide | (131, 141) |
| Ca2+-activated Cl− channels | Anoctamin-1 | Ano1/ANO1 | Membrane depolarization | Ca2+ | T-AO1, tannic acid, niflumic acid | (61, 78, 204) |
| Anoctamin-2 | Ano2/ANO2 | Membrane depolarization | Ca2+ | T-AO1, tannic acid, niflumic acid | (61, 78, 204) | |
| Other Cl− channels | CFTR | ABCC7 | Acidification of insulin-containing granules | PKA phosphorylation | NPPB, niflumic acid | (65, 77) |
| Calcium-activated K+ (Kca) channels | BK (KCa1.1) | Kcnma1/KCNMA1 | Spike repolarization | Ca2+, membrane depolarization | Charybdotoxin, iberiotoxin, paxillin, TEA | (58, 116, 150, 187) |
| SK1 (KCa2.1) | Kcnn1/KCNN1 | Modulates Ca2+ oscillations, repolarization of the plateau potential | Ca2+, 1-EBIO, GW542573 | Apamin, UCL 1684 | (29, 112, 273, 307) | |
| SK2 (KCa2.2) | Kcnn2/KCNN2 | Modulates Ca2+ oscillations, repolarization of the plateau potential | Ca2+, 1-EBIO, CyPPA | Apamin, UCL 1684 | (29, 112, 273, 307) | |
| SK3 (KCa2.3) | Kcnn3/KCNN3 | Modulates Ca2+ oscillations, repolarization of the plateau potential | Ca2+, 1-EBIO, CyPPA | Apamin, UCL 1684 | (29, 112, 273, 307) | |
| SK4 (IK, KCa3.1) | Kcnn4/KCNN4 | Modulates Ca2+ oscillations, repolarization of the plateau potential | Ca2+, 1-EBIO, SKA-111 | TRAM-34 | (29, 72, 112, 273, 307) | |
| Voltage-dependent K+ channels | Kv2.1 | Kcnb1/KCNB1 | Spike repolarization | – | TEA, quinidine, aminopyridines, and dendrotoxin | (32, 117, 164, 222) |
| TRP channels | TRPC1 | Trpc1/TRPC1 | Mediation of store-operated Ca2+ channels | – | La3+, Gd3+, 2-APB | (166, 234, 267, 311) |
| TRPC3 | Trpc3 | β-Cell development and GPR40-induced membrane depolarization | GSK1702934A, artemisinin | Pyr3 | (143, 287, 300) | |
| TRPC4 | Trpc4 | KATP trafficking | leptin, (−)-englerin | M084, ML204 | (3, 181, 257, 264) | |
| TRPM2 | Trpm2/TRPM2 | Mediates Ca2+ influx induced by glucose and GLP-1 | Ca2+, Adenosine 5′-diphosphate ribose, forskolin, exendin-4 | N-(p-amylcinnamoyl) anthranilic acid, curcumin, JNJ-28583113 | (17, 82, 138, 172, 201, 280, 283) | |
| TRPM5 | Trpm5/TRPM5 | Mediates glucose-induced oscillation | Arachidonic acid, steviol glycoside, GLP-1 | Flufenamic acid, TPPO | (55, 200, 209, 259, 286) | |
| TRPV2 | Trpv2 | Membrane depolarization | Cell swelling, cannabidiol | SET2 | (51, 215, 246) | |
| Two-pore-domain K+ channels | TALK1 | Knck16/KNCK16 | Resting “leak” conductance | pH (alkaline), nitric oxide | pH (acidic) | (74, 132, 291) |
| TASK1 | Kcnk3/KCNK3 | Resting “leak” conductance | pH (alkaline) | pH (acidic), A1899, ML365 | (63, 74, 292) |
The β-cell synthesizes and secretes insulin in response to a glucose challenge. Thus, when plasma glucose concentration rises after a meal, for instance, glucose is first transported into the β-cell via specialized glucose transporter(s) molecules [GLUT1 and GLUT2 in humans, GLUT2 exclusively in mice (100, 171, 278)]. Once in the cytosol, glucose is phosphorylated by glucokinase and glycolysis and cellular respiration occur, leading to increased ATP production and, concomitantly, a rise in cytoplasmic ATP/ADP (1, 9, 70, 135). Increased ATP/ADP leads to the closure of ATP-sensitive K+ channels (KATP) and subsequently, membrane depolarization, as the conductance of the β-cell plasma membrane to K+ ions is dominated by the resting K+ conductance of these channels, gKATP. Closing KATP channels, in turn, leads to the opening of voltage-gated Ca2+ channels as the membrane potential of the β-cells depolarizes toward −40 mV. The opening of these channels in turn mediates an increase in Ca2+ influx due to Ca2+’s strong electrochemical gradient, resulting in a rise in the cytosolic Ca2+ concentration. As the exocytosis of insulin granules is Ca2+-dependent, the secretion of insulin results (237, 238).
Insulin is stored within the β-cell in large dense-core secretory granules, in complex with zinc, and there are estimated to be approximately 15,000 granules in a typical β-cell (67, 197). Somatostatin, a hormone which is secreted by islet δ-cells inhibits insulin secretion, while glucagon, which is synthesized and secreted by islet α-cells potentiates insulin secretion by raising cytosolic cAMP (235, 250, 266, 269).
Before discussing the critical role played by different β-cell ion channels in the regulation of insulin secretion, we will digress for a moment to consider some unique aspects of the β-cell as compared to other cells.
In most cells, glucose is metabolized to supply the cell with sufficient chemical energy (in the form of ATP) to fuel general as well as more cell-specific functions. While this also occurs in β-cells, the β-cell not only uses glucose as a fuel but as a specific signal to trigger insulin secretion. This is very different from the situation in neurons, for instance, where the neuron is stimulated not by fuels but by the release of excitatory neurotransmitters from the presynaptic terminals of neighboring neurons. While β-cells also have cell surface receptors for acetylcholine, ATP, glutamate, and other signaling molecules (these all being “excitatory”), the occupation of these receptors by their ligands is not required for glucose-induced insulin secretion, as glucose itself is the primary β-cell secretagogue. Indeed, the actions of nearly all β-cell modulators are potentiated by elevated glucose (242).
KATP Channels Are the Primary Ion Channel Regulators of β-cell Electrical Activity
The modulation of ATP-sensitive K+ channel (KATP) activity by fuel metabolism is the primary means by which β-cell electrical activity is coupled to glucose-induced changes in intracellular ATP/ADP (5, 190). This is because changes in KATP conductance, balanced against the cell’s resting ionic leak conductance determines the β-cell membrane potential (59).
When measured in excised membrane patches from β-cells in symmetrical K+ concentrations, KATP was shown to be a 50 pS, weakly inwardly rectifying K+ channel (7, 281). KATP channel inhibition by increased ATP/ADP is manifested by an increase in mean channel closed time as ATP rises, and a decrease in mean open time as ADP falls (190, 231); there is also a decrease in channel opening frequency as ATP/ADP increases. It is trivial to show that as mean open time (MOT) decreases and mean closed time (MCT) increases, the steady state open probability (Po) is reduced, as Po is equal to MOT/[MCT+MOT]. Thus, reducing Po depolarizes the β-cell away from EK [e.g. ≈ −70 mV, as set by Nernst; (244)] toward the value of the leak potential, which is more depolarized; it is to this new battery potential that the membrane potential heads when KATP closes (for full discussion, see Ref. 59). The β-cell leak conductance may in aggregate include open voltage-gated Ca2+ channels, activated nonselective cation channels such as TRP or SOCE channels, a resting sodium leak (270), and electrogenic pump currents, as long as they are net depolarizing (5, 20, 59, 84, 104, 141). The depolarizing reversal potential of islet Cl− channels could also contribute to the overall leak and depolarization to the voltage of the plateau phase (141). Our long experience studying mathematical models of β-cell electrophysiology clearly shows that β-cells are very sensitive to the value of the leak conductance that is chosen. Studying the leak conductance experimentally, however, is difficult because given the high input resistance (or low input conductance) of β-cells, which occurs when KATP channels are maximally inhibited or closed (11, 219), leak currents can be very small, on the order of 1 pA or less, which is near the limit of resolution of the patch clamp.
Two other considerations are worth noting when discussing the KATP channels of β-cells. First, calculations made over 30 years ago by Cook and colleagues showed that there are so many KATP channels in the β-cell plasma membrane that the membrane potential would not be predicted to even begin to depolarize (as glucose and ATP/ADP rise) until >90% of the KATP channels are shut, assuming that a typical β-cell has 15,000 pS of KATP conductance and 10 pS of leak; these authors called this the “Spare Channel Model” (59). While this simple model did not incorporate ADP activation of KATP channels (73, 130) or the possibility of ATP subdomains just under the plasma membrane (135), the calculation successfully explained why β-cells would not be persistently depolarized even assuming KATP channels have an IC50 of 15 μM in excised outside-out patches and are exposed to bulk cytosolic ATP levels in the range of 3–5 mM. The main point of the model was that even when most of the β-cell’s KATP channels were bound by ATP (and thus closed), the cell membrane would still be relatively hyperpolarized nonetheless and that only after closing the last few percent of open KATP channels would the cells depolarize significantly in response to glucose (59). Barg et al. (15) estimated there are about 7000 KATP channels in a single β-cell, yielding a maximum whole-cell conductance of about 140,000 pS (assuming a single-channel conductance of 20 pS). This suggests that the probability of opening (Po) of KATP rarely exceeds 0.1.
Subsequent to studies functionally characterizing the channel, biochemical and cloning work revealed that two protein subunits constituted the channel: an inward rectifying Kir6.2 subunit (KCNJ11) and an ATP-binding cassette transporter family member, called SUR1 for sulfonylurea receptor 1 (ABCC8). Structural characterization of the channel showed that it is an octameric complex made up of four pore-forming Kir6.2 subunits and four regulatory SUR1 subunits arranged around a central pore (54, 260). Each SUR1 protein interacts with the first transmembrane domain of Kir6.2 via its amino-terminal transmembrane domain (TMD0), which is connected to the ABC transporter core structure of SUR1. This consists of two transmembrane domains and two nucleotide-binding domains (NBDs) that are connected via an intracellular loop (L0). Recent work from two different groups using cryo-EM has produced an elegant three-dimensional reconstruction of the channel, as described in Refs 157, 169.
Intracellular adenine nucleotides regulate both Kir6.2 and SUR1 activity. In the absence of Mg2+, nucleotides inhibit the KATP channel activity by binding to a nucleotide-binding pocket formed between the amino and carboxy-terminal domains of two adjacent Kir6.2 subunits (190). In addition, ADP activation of the channel occurs via its interactions with the two Walker consensus domains of SUR1, as well as phosphorylation sites that in turn regulate KATP activity. Readers interested in a more detailed discussion of KATP channel gating are referred to Refs 6, 190. Furthermore, both gain and loss of function mutations in KATP have been linked to persistent hyperinsulinemic hypoglycemia of infancy [or PHHI; (48)] and neonatal diabetes [or NDM; (13, 218)]; details of these mutations are beyond the scope of the present article. However, the impact of these mutations in either SUR1 or Kir6.2 highlights that KATP is a critical regulator of insulin secretion, as gain of function mutations lead to insufficient insulin secretion and neonatal diabetes, and loss of function mutations produce excessive secretion and hypoglycemia (87, 277). In terms of the clinical significance of KATP pharmacology to type 2 diabetes, KATP was shown decades ago to be blocked by sulfonylureas [ligands to the sulfonylurea receptor; (103, 174)] as well as glinides (163), two different classes of oral medications used historically in the case of first-generation sulfonylureas to treat type 2 diabetes because of their powerful actions as insulin secretagogues.
More recent work in the field suggests that besides the acute regulation of KATP channel gating by changes in cytosolic ATP/ADP due to fuel metabolism, hormones such as leptin (53, 202) or insulin (137) as well as chronic glucose (88) can regulate the number of KATP channels resident on the β-cell surface. These perturbations have been shown to result from modulation of KATP trafficking from endosomal sites within the cell to the plasma membrane where they are electrophysiologically active (168). This is an entirely new form of KATP channel regulation and while it is doubtful that glucose-induced changes in KATP trafficking supersede in importance the modulation of KATP channel gating by glucose metabolism and ATP/ADP (98), this is a fascinating new area of KATP research, and it well justifies more research.
Voltage-gated Ca2+ Channels (VGCCs)
In response to β-cell depolarization due to KATP closure, VGCCs open and electrical bursting commences (68). Bursting consists of spontaneous slow plateau depolarizations with superimposed trains of rapid spikes that are believed to mediate the patterns of Ca2+ entry that drive pulsatile insulin exocytosis. This basic pulsatility is necessary for proper insulin action (238). VGCCs mediate the upstroke of the β-cell spikes and the regularly occurring and Ca-dependent slow plateau depolarizations (57, 60). Furthermore, blocking VGCCs pharmacologically inhibits insulin secretion under most cases (243).
VGCCs can be broadly separated into two different functional classes: high voltage-activated (HVA) channels that open in response to a large membrane depolarization, and low voltage-activated (LVA) channels that are activated by smaller voltage excursions, typically depolarizations not far from the β-cell silent phase potential (47, 97). HVA Ca2+ channels can undergo Ca2+- and/or voltage-dependent inactivation, where the channel can enter a refractory gating state wherein it is unable to pass Ca2+. For L-type Ca2+ channels, the most typical type of inactivation is where Ca2+ entering the cell through the open channel binds to calmodulin residues on the carboxy-terminal of the α subunit, mediating inactivation (105, 123). This type of inactivation occurs over 10s of milliseconds and is strikingly slowed by the replacement of Ca2+ with Ba2+ as the charge carrier (241).
While the L-type Ca2+ channel was the first VGCC to be functionally characterized, later studies using careful pharmacological as well as biophysical dissection of HVA current revealed not only L-type channels, the subtype modified by dihydropyridine drugs such as nifedipine; but N-type channels, which are blocked by omega conotoxin from the snail Conus (282); and P/Q-type, which are blocked by the funnel-web spider toxin, agatoxin-V (161); and an R-type (for “residual”) for which no clear selective blockers have been identified (217), although R-type can be inhibited by SNX-482 along with other Ca2+ channels (4, 188). While all of these channels can coexist in the same cell, sometimes in different proportions, and while all of them can bring Ca2+ into the cell, the extent of their coupling to insulin granule exocytosis may differ based on their proximity to hormone granule release sites or due to differences in their channel kinetics (243).
In Cav3.2 channels (referred to as LVA or T-type Ca2+ channels) replacing Ca2+ with Ba2+ does not alter the fast inactivation kinetics observed (282), as T-type channel inactivation is a purely voltage-activated process, resembling the inactivation of voltage-gated Na+ channels. Here fast inactivation occurs largely because of interactions of charged residues into the pore to occlude channel gating from the inside face of the membrane (107).
The types of Ca2+ channels expressed in β-cells depends on the species
In mouse islets, one of the most well-studied experimental preparations, the major high voltage-activated Ca2+ channel of β-cells is the L-type channel (237, 258). The L-type channel is closely tied to insulin secretion, as its blockade by dihydropyridines such as nifedipine or nimodipine profoundly suppresses GSIS (106, 192) and blocks or at least largely curtails islet bursting. The L-type channel is also functionally important in rat (83) and human β-cells (36, 227), but besides this channel, rat β-cells express T-type LVA channels. T-type channel activity in the β-cells of these preparations not only bring in Ca2+ but also augment the amplitude of membrane depolarization to facilitate the opening of L-type channels (27), likely because of their fast rate of voltage-dependent activation (however, they inactivate more quickly, too) and low activation threshold. However, progress in understanding the detailed role of T-type channels has been hampered by the lack of sufficiently specific drugs or toxins to target T-type channels). The available evidence suggests that interfering with T-type channels reduces insulin secretion in human and rat β-cells (64, 183).
Molecular structures
L-type channels exist as a complex of several different subunits: a pore-forming α1 subunit, a disulfide-linked α2δ dimer, an intracellular β subunit, and a transmembrane γ subunit. The α1 subunit is composed of four repeating homologous domains (I to IV) each containing six transmembrane segments (S1 to S6), a pore loop (P-loop) between the S5 and S6 segments, and three intracellular linkers (303). The P-loops form the lining of the aqueous pore and contain either four glutamic acid residues (in high voltage-activated VGCCs) or two glutamic acid and two aspartic acid residues (in low voltage-activated VGCCs); these constitute the Ca2+ channel selectivity filter (236, 271). The voltage sensors of the VGCCs are made up of charged amino acid residues, primarily within the S4 segment which create charge movement and lead to channel opening in response to depolarization of the membrane potential (303). The auxiliary subunits of the channel are involved in channel trafficking to the cell surface and also modify the biophysical and pharmacological properties of the channel (50). In neurons, the α2δ subunit has been shown to regulate Ca2+ channel density at the synapse and it also helps promote assembly of the channel (114). Genetic ablation of α2δ−1, the dominant α2δ subunit in pancreatic β-cells, causes a 50% reduction in Ca2+ current amplitude, reduced insulin secretion, glucose intolerance, and diabetes suggesting an important role for α2δ in β-cells (170).
The various HVA VGCCs that play significant roles in GSIS by mediating Ca2+ entry include L-type channels (Cav1.2/CACNA1C and Cav1.3/CACNA1D), P/Q-type (Cav2.1/CACNA1A), N-type (Cav2.2/CACNA1B), and R-type (Cav2.3/Cacna1e). For LVA or T-type VGCCs, Cav3.1/CACNA1G and Cav3.2/CACNA1H are expressed (303). The relative levels of expression of each of the channel isoforms, and the activity of each varies depending on the species (303). The different VGCCs can be identified and classified by their different biophysical properties and the effects of differential pharmacological inhibitors on their activity.
L-type Ca2+ channels activate near −50 mV and reach maximal activation near 0 mV. They exhibit slow inactivation and are modified by dihydropyridines drugs (DHPs) that alter their gating modes (106, 192), or by phenylalkylamine or benzothiazepine blockers that cause use-dependent channel blockade (111). In β-cells, whole-cell L-type VGCC currents were first reported in cultured neonatal rat pancreatic β-cells (239). Since their initial characterization, VGCCs have been studied in a wide range of insulin-secreting cell lines (115, 243) and primary β-cells from many species (18, 113, 142, 184, 225). The L-type VGCC generally comprises the majority of VGCC current in most of the species where it has been quantitatively examined (237, 258).
Mouse β-cells express two L-type subtypes Cav1.2 and Cav1.3 (186, 251, 298). Cav1.2 has been reported to be the primary L-type isoform for VGCC current as its genetic ablation results in the near complete loss of DHP-sensitive L-type Ca2+ current (251). Furthermore, mutant mouse β-cells expressing a DHP-resistant variant of Cav1.2 had no residual, DHP-sensitive Ca2+ current (159, 160). It has been estimated that approximately 60% to 80% of glucose-induced insulin secretion can be attributed to the activation of L-channels (64, 194, 251). Cav1.2 plays an especially prominent role in triggering first phase insulin secretion in mouse islets, as its inhibition drastically reduces first phase secretion (251).
The role of Cav1.3 in β-cells has been more controversial. Some groups have reported that L-type channel blockers have no effect on the residual Ca2+ current observed in mouse β-cells lacking Cav1.2, and additionally, had difficulty confirming Cav1.3 immunoreactivity (16, 251). However, in contrast, other groups found that mouse β-cells express Cav1.3 mRNA and protein and that knocking out Cav1.3 resulted in a compensatory overexpression of Cav1.2 and reduced basal insulin secretion (186) that Cav1.3 plays a role in mediating basal insulin secretion, although more work needs to be done to verify this hypothesis. However, other groups have shown that there are more Cav1.3 than Cav1.2 mRNA in human β-cells (36) which, combined with the reported shift in total Ca2+ current activation toward more negative voltages (34, 147), suggests that Cav1.3 likely plays a more significant role in human β-cells.
P/Q-type VGCC in β-cells
As stated above, L-type channels carry the majority of Ca2+ current in β-cells. However, DHPs and other CCBs do not fully block total Ca2+ current in most β-cell preparations (212, 232, 251, 261). P/Q-type channels were reported in mouse β-cells following pharmacological inhibition of competing HVA currents (251). Thus, using a mixture of isradipine, a DHP that blocks L-type VGCCs, and the R-type blocker SNX482, Cav current was reduced by about 80% but was still not fully eliminated. The subsequent addition of ω-agatoxin, a specific inhibitor of P/Q-type channels isolated from the funnel-web spider, blocked nearly all of the remaining Cav current (251). P/Q-type channels rapidly activate near −30 mV, reach a peak amplitude at about +10 mV and exhibit slow inactivation (158). P/Q-type channels are encoded by the CACNA1A gene (50).
Rat β-cells and rat-derived insulin-secreting cell lines were found to express functional P/Q-type VGCCs (158) and Cav2.1 is also expressed in human β-cells. P/Q is manifested as a residuum of HVA Cav current in solutions containing both nifedipine and ω-conotoxin GVIA (64, 258). About 25% of human β-cell Cav current appears to be of the P/Q-type, based on its sensitivity to ω-agatoxin (258). The Cav2.1 blocker ω-agatoxin also partially blocks Cav currents in rat β-cells and inhibits the DHP-resistant component of GSIS by about 30% (158). In human β-cells, in contrast, ω-agatoxin inhibits about 65% of human GSIS (258).
N-type VGCC in β-cells
The role of N-type VGCCs encoded by Cav2.2 has been somewhat controversial in β-cells. While it has been reported that mouse and human β-cells do not express functional Cav2.2 as ω-conotoxin GVIA, an N-channel specific neurotoxin from the marine snail Conus (195) does not affect β-cell Cav currents (86), there is evidence showing ω-conotoxin GVIA decreases arachidonic acid-induced Ca2+ influx in β-cells (216). N-type VGCCs have also been detected in several rat-derived insulin-secreting cells, including hamster insulinoma tumor (HIT) (243). However, other groups have reported that Ca2+ signaling in these lines were insensitive to the actions of ω-conotoxin GVIA (115, 167, 247).
Several studies have shown that ω-conotoxin GVIA has no effect on GSIS in human islets (64, 214), while others reported that ω-conotoxin GVIA reduced HVA Cav current in human β-cells and reduced second phase insulin secretion in rat islets (145). The reduction in second phase secretion, however, has been attributed to a ω-conotoxin-induced reduction of β-cell ATP/ADP, rather than via a direct effect on Ca2+ channels (194). This suggests that Cav2.2 could conceivably play a role in regulating glucose metabolism and ATP generation, but the mechanism involved remains obscure (2, 230).
R-type VGCC in β-cells
In other cell preparations, even blocking L, N, and P/Q channels does not completely eliminate all HVA current, suggesting that a possible residual (or “R” current) current persists that is mediated by a separate type of Ca2+ channel (217). An R-type VGCC was reported in mouse β-cells and could be inhibited by SNX-482, a high affinity Cav2.3 channel blocking peptide (188). Thus, SNX-482 was observed to reduce isradipine-resistant Cav current by 60% in mouse β-cells (251). SNX-482 sensitive-current could also be detected in INS-1 cells (288). Cav2.3 knockout mice had reduced Cav current, as elicited by depolarizations to −10 mV or more (128), again supporting the existence of R channels in β-cells. Functional R channel current was shown to be due to Cav2.3 gene expression (50).
R-type channels were shown to regulate insulin secretion in both INS-1 cells and mouse β-cells (206, 207, 288, 302). In terms of whole-body responses, Cav2.3 knockout mice had reduced glucose tolerance, reduced insulin secretion, and were hyperglycemic (206). Further studies with Cav2.3 knockout mice and SNX-482 blocking peptide demonstrated that Ca2+ entry through Cav2.3 channels appeared to be most strongly coupled to second phase rather than first phase insulin secretion (128). While L-type Ca2+ channels are localized close to the exocytotic machinery of the β-cell (31, 243), Cav2.3 appears to be more distant from the exocytosis sites and may instead help recruit insulin-containing granules from the large reserve pool to the more readily releasable pool (128, 302).
T-type VGCC in β-cells
Unlike the other VGCCs, the T-type Ca2+ channels are low-voltage-activating, meaning they activate at more negative membrane potentials close to the silent phase potential of oscillating β-cells. T-type channels are rapidly inactivating and have activation thresholds as negative as −60 mV (208, 310). They mediate maximal current at about −30 mV and they are reversibly blocked by amiloride (275), Ni2+ (154), and NNC55–3096 (36). The inactivation of T-type Ca2+ channels is a voltage-dependent process, exhibiting half-maximal inactivation at −65 mV. T-type channels are expressed in rat and human, but not mouse β-cells (8, 18, 108, 240, 310). However, T-type channels have been reported in β-cells from nonobese diabetic (NOD) mice (295), suggesting that the genes that encode these channels might be disallowed in normal mouse β-cells. In islet α-cells, however, T-type channels are crucial for the secretion of glucagon when the cells are exposed to low glucose concentrations (94, 223). T-type Ca2+ channels have been identified in α-cells of guinea pigs (223) and mice (40), although mouse delta cells may express more T-type channels than α-cells (40).
The Na+ Channels of β-cells
Na+ channels mediate the classic, fast activating, and inactivating inward current first identified by Alan Hodgkin and Andrew Huxley in the squid giant axon, where they mediate the rapid upstroke of the sodium spike (109, 110). Na+ channels are expressed in human, rat, and mouse β-cells (36, 71, 213, 308). However, there is a consensus that they play only a minor role in mouse β-cell excitability because their steady state inactivation(or h-infinity curve) is quite left-shifted in mouse β-cells (93, 213); Na+ current can thus only be elicited by de-inactivating the channels using very negative holding potentials. In mouse islets, the Na+ channel specific toxin, tetrodotoxin (TTX) clearly blocks Na+ current elicited from very negative prepotentials but under physiological conditions, TTX is without effect on GSIS (308). However, this is not the case for human or rat β-cells where the channels are more active physiologically (36, 71)
Voltage-gated Na+ channels (VGSCs) are composed of an α-subunit (Nav1.1-Nav1.9/SCN1A-SCN5A, SCN8A SCN11A), which makes up the voltage-dependent pore, and an auxiliary beta-subunit (beta1–4/SCN1B-SCN4B) which regulates channel trafficking and gating (90, 203). As in VGCCs, the α-subunit is made up of four repeating domains (termed I to IV), each containing six transmembrane segments (S1 to S6), with the S4 segment acting as a voltage sensor. The P-loop between S5 and S6 is the ionic selectivity filter (101).
Mouse β-cells predominantly express Nav1.7 (Scn9a), some Nav1.3 (Scn3a), and very little Nav1.6 (Scn8a) (224). Of the β-subunits, beta1 (Scn1b) is expressed at higher levels than beta3 (Scn3b) (308). While there is some evidence to support the idea that Na+ currents may be important for rodent β-cells (308), the topic is controversial, and more work needs to be done to fully understand if Na+ current affects rodent β-cells and if so, under what conditions.
Human β-cells mainly express Nav1.6 (SCN8A) with Nav1.7 (SCN9A), Nav1.3 (SCN3A), and Nav1.2 (SCN2A) making up the remainder (30, 189, 224). Of the beta-subunits, beta1 (SCN1B) and beta3 (SCN3B) are expressed about equally. Unlike mouse β-cells, human β-cells have large TTX-sensitive Na+ currents, having half-maximal inactivation at −40 mV, much more positive than mouse (36). Because of this characteristic, Na+ channels play a more important role in the upstroke of the action potential, and several studies have shown that TTX reduces human β-cell action potential amplitude, as well as glucose-stimulated insulin secretion, although this action is reduced at higher glucose concentrations (10–20 mM) (18, 36).
Transient Receptor Potential (TRP) Channels
Over the years, increased attention has been paid to the possible roles of Transient Receptor Potential channels in islets (reviewed recently in Ref. 122). TRP channels, first identified in the photoreceptors of Drosophila, where they are involved in the generation of light-induced receptor potentials (182) are a superfamily consisting of a number of different TRP families such as TRPC, TRPM, TRPV, TRPML, TRPP, and TRPA channels. Accumulating evidence gathered through molecular as well as functional studies has provided support for TRPC, TRPM, TRPP, and other TRP channels in β-cells, along with various mRNA splice variants (122).
TRP channels are broadly speaking nonselective cation channels, but their Ca2+-permeability in particular and their activation by Ca2+ vary by channel type, as do their functional roles. TRP channels that have been shown to have constitutive activity appear well suited to contribute to the “leakage conductance” of the β-cell, whose reversal potential drives cell depolarization when gKATP is reduced by (say) glucose metabolism. But a further interesting role of TRP channels is their sensitivity to environmental factors such as pH, mechanical perturbation or swelling, odorants and taste stimuli, temperature, and the like. Additionally, these channels are also subject to regulation by cAMP dependent as well as PKC-dependent phosphorylation and activators such as intracellular Ca2+, ADP-ribose, cyclic ADP-ribose, and others (290). We note, however, that the specific types of TRP channels that are expressed in mouse, rat, and human primary β-cells and those present in the range of insulinoma β-cell lines can be controversial and is quite heterogeneous. Thus, caution has to be exerted before assuming that a particular functional attribute of an insulin-secreting cell must necessarily reflect the participation of a TRP channel in the process under study. This situation was not helped, especially in the early days of TRP channel research when the pharmacological agents being employed were limited and often quite nonspecific.
TRP channels have been proposed to play an essential role in β-cell electrophysiology in a number of ways. TRPC1, for instance, has been proposed to be part of the SOCE current along with Orai-1 which activates following ER Ca2+ depletion and concomitantly, STIM1 puncta formation (234). Or TRP as has been proposed for TRPC3 channels have been proposed to couple to GCPRs such as the fatty acid receptor GPR40, which can activate the channel due to PKC activation (300). In INS1 cells, TRPC4 may be involved in leptin-induced KATP channel translocation to the plasma membrane (264).
Relevant to studies of islet electrical oscillations was a study where the role of TRPM5 was examined by genetically deleting its gene in a mouse and studying its isolated islets. Unlike wild type islets that in this study tended to display either fast (period <1 min.), slow (period <5 min.), or mixed fast/slow oscillations in suprathreshold glucose, the islets from the knockout mice tended to show mostly slow oscillations at the expense of fast ones (55). The authors proposed that during the nadir of the bursting phase, TRPM5 channels contribute to a rise in Ca2+ that facilitates cell depolarization to the next active phase, increasing burst frequency (55). Consistent with this, TRPM5 null mice were found to be glucose intolerant and had displayed reduced insulin secretion. While these are intriguing results, a complexity of the study was that even the wild-type islets had slow, as well as fast oscillating islets, and the defective secretion that was seen in isolated islets could be due to more direct effects of the TRPM5 gene (43). It also would have been more convincing if the TRPM5 deletion was β-cell-specific and not a global one (55).
Due to limitations of space, we cannot discuss in detail many of the other roles that have been proposed for β-cell TRP channels in any detail. However, TRP channels have been implicated in GLP-1 mediated insulin secretion (136, 201, 285), β-cell proliferation (262) and apoptosis (49, 121), the sensitivity of insulin secretion to changes in temperature (134, 284), and responses to cell swelling (245, 246). The contribution of TRP channels to zinc transport in β-cells (294), and a possible role for them in intracellular Ca2+ release (151) have also been suggested by experimental work.
Cl− Channels
The role of Cl− channels (and anion channels, more generally) in pancreatic β-cell physiology has not been as thoroughly studied as have cation channels. Originally thought to play a passive role, Cl− was long ago shown to actively accumulate against its electrochemical gradient in rat β-cells and that its channels can therefore mediate Cl− efflux-induced membrane depolarization (254). A few years later, several studies showed that Cl− flux could modulate insulin secretion from rat β-cells in a glucose-dependent manner (263, 272). In 1988, Sehlin and Meissner reported that changes in [Cl−]i in mouse β-cells could alter voltage-gated Ca2+ channel activity, providing the first link between glucose metabolism, altered Cl− fluxes, and insulin secretion (255).
Since those early studies, several different chloride channels have been reported in pancreatic β-cells, most notably a volume-regulated anion channel (VRAC/SWELL1) (131, 141), the cystic fibrosis transmembrane conductance regulator (CFTR) (78, 96), and Ca2+-activated Cl− channels (Ano1/2) (78, 148).
VRAC/SWELL1
Volume-regulated anion channels (VRAC) were first described in β-cells by Kinard and Satin (141) and by Best et al (26). These authors found that rodent β-cells exhibit swelling-regulated, outwardly rectifying Cl− currents that were sensitive to anion channel blockers, although only Satin’s group reported that the current was also cAMP activated (141). VRAC activation led to membrane depolarization due to increased Cl− efflux that in turn could increase insulin secretion (25, 42). In determining whether VRAC was activated by glucose, it was found that increasing glucose similarly activated depolarizing Cl− currents (22) and it was proposed that this resulted from β-cell swelling in response to increased glucose metabolism; the finding of ATP-dependence is concordant with the glucose sensitivity of the channels (178, 179).
As mentioned earlier, KATP closure alone is insufficient to depolarize the β-cell membrane because a depolarizing “leak conductance” must also be part of the prevailing circuit. As the reversal potential for Cl− channels (about −35 mV) is close to the plateau potential (141, 220), Cl− channels are likely a component for this leak conductance. A VRAC model, by Best and colleagues, incorporated VRAC into the consensus model of insulin secretion as a carrier of the depolarizing leak current (23). In this model, glucose decreases outward KATP current by raising ATP/ADP while also increasing inward current, which together promote β-cell depolarization and consequently insulin secretion. Best and colleagues proposed that glucose metabolism can regulate VRAC, depolarizing the β-cell and facilitating insulin secretion (24).
The molecular identity of VRAC was recently determined. VRAC is composed of a hexameric complex of leucine-rich repeat-containing protein 8 (LRRC8A-E), with LRRC8A (commonly called SWELL1) being essential for channel function (69, 146). Knocking down SWELL1 in MIN6 cells and primary mouse β-cells using CRISPR/Cas9 reduces swelling- and glucose-induced Cl− current and blunts insulin secretion (131, 268). Stuhlmann et al. have shown that all five VRAC subunits are expressed in mouse islets, with Lrrc8a, Lrrc8c, and Lrrc8d attaining the highest expression levels (268). Interestingly, VRAC containing LRRC8D channels are also permeable to taurine, myo-inositol, GABA, and lysine, which can also affect insulin secretion (162). Indeed, when cells expressing VRAC are exposed to hypotonic solutions that activate VRAC by causing cell swelling, taurine, inositol, and other molecules can be shown to efflux the cells (118, 249).
CFTR
Cystic fibrosis transmembrane conductance regulator (CFTR) is a member of the ATP-binding cassette superfamily of proteins (ABCC7), whose open/closed gating allows anions to flow down their electrochemical gradient. CFTR is made up of two sets of transmembrane domains, each containing six membrane-spanning α-helices and two cytoplasmic nucleotide-binding domains (309). CFTR also contains an important regulatory (or R) domain that allows for PKA-mediated phosphorylation that regulates channel gating. Functional studies have demonstrated that the open probability of CFTR is increased by cAMP-dependent phosphorylation. In terms of its electrophysiological properties, CFTR exhibits a linear IV relationship, with a single-channel conductance of approximately 6 to 11 pS (19).
The presence of CFTR in β-cells has been very controversial as the very low levels of expression cast doubt on their functional relevance for islets. A lack of signal found with RNAseq has been interpreted as evidence that Cftr is not expressed in mouse β-cells (224). However, other studies have detected expression of Cftr (30), possibly due to contamination by high levels of CFTR expression from residual exocrine tissue (252).
Whether CFTR plays a functional role in insulin secretion is similarly controversial, as several studies support (78, 96) while others dispute (99, 196) such a role for the channel. Despite its low level of expression, CFTR has been demonstrated to play a role in regulating [Cl−]i in neurons (185, 199), supporting the idea that even low levels of CFTR expression may be sufficient. As a significant percentage of cystic fibrosis patients develop CF-related diabetes (96), this remains an important and clinically relevant question.
However, CFTR may not necessarily influence insulin secretion and β-cell function directly via channels in the β-cell plasma membrane. In several cell types, CFTR is active in the trans-Golgi network (14), endosomes and lysosomes (28), and in acidic organelles and granules (149). Several studies suggest that CFTR may play a role in insulin processing and the exocytosis of insulin-containing granules via the acidification of the insulin granule, and hence not directly through an action on the electrophysiology of the β-cell (65, 77). The classic interpretation of the role of CFTR in CF diabetes attributes its action in acinar cell function whereby defective CFTR regulation leads to poor mucus secretion from the pancreatic acini, ductal malfunction, and concomitant inflammation, and islet damage (81, 99).
Ca2+-activated Cl− channels (CaCCs)
CaCCs were first described in guinea pig β-cells as mediating a current which reverses at −22 mV, assuming an internal Cl− concentration of 65 mM, which is higher than predicted from passive distribution of the anion (148). This current is functionally similar to that mediated by the anoctamin channels Ano1 and Ano2 (204), which have been suggested to play a role in insulin secretion (61, 78). Ano1 is expressed in mouse and rat β-cells and its inhibition by T-AO1, tannic acid, or Ano1-specific antibodies, reduces glucose-stimulated insulin secretion, the rate of action potential firing, and results in the partial repolarization of the β-cell membrane potential (61). Ano1 is expressed as well in human β-cells and siRNA-mediated silencing of Ano1 or T-AO1 inhibits insulin secretion (78, 127, 299). While the above studies support Ano1’s involvement in insulin secretion, other studies have failed to find expression of Ano1 or Ano2 in mouse or human β-cells and have concluded that they play a minimal role (224). Clearly, this issue needs to be reexamined more thoroughly.
Role of β-cell Cl− channels in autocrine regulation
Besides insulin, pancreatic β-cells contain several other substances which are co-released with insulin in response to glucose. Several of these substances have been proposed to be autocrine and/or paracrine regulators of islet cell electrical or secretory activity (35, 301). Two substances which could regulate Cl− channel activity, and therefore β-cell excitability, are GABA and glycine.
Pancreatic β-cells contain two types of GABA receptors: ionotropic GABAA receptors and metabotropic GABAB receptors. GABAA receptor activation in human β-cells depolarizes the cell membrane toward the Cl− equilibrium potential [ca. −35 mV (35)] due to the prevailing intracellular Cl− concentration and can increase insulin secretion (35). Inhibiting GABAA receptors using a GABAA receptor antagonist such as SR95531 prevents insulin secretion (35). Activation of the metabotropic GABAB receptor, however, has been shown to inhibit insulin secretion when activated (33, 38) while GABAB antagonists such as CGP55485 stimulate secretion (274), suggesting that GABA can both stimulate and inhibit insulin secretion depending on the receptor type engaged. Because pancreatic β-cells contain high levels of GABA (276), it is possible that GABA could act as an autocrine and/or paracrine signal to neighboring cells. GABA has been shown to be released from both rodent (85) and human (37) β-cells via exocytosis (35, 39) or via nonvesicular release, possibly through bestrophin-1 (BEST1) channels (155) although more work needs to be done to understand the details of GABA release.
Glycine is also stored inside the β-cell secretory granule and is co-released with insulin (301). Glycine receptors are found on the membranes of both rodent and human β-cells and have been shown to result in membrane depolarization in response to glycine (156, 296, 301). Antagonizing the glycine receptor with strychnine has been shown to reduce glucose-sensitive insulin secretion in human islets (301). Yan-Do et al. (301) proposed that β-cells use glycine as an autocrine positive feedback signal to stimulate insulin secretion.
Ca2+-dependent K+ Channels
Potassium channels that are activated by a rise in intracellular Ca2+ concentration or KCa channels have been of interest in the β-cell field for a long time, from even before the advent of the patch clamp and the subsequent discovery of KATP channels. This was the case because the presence of KCa channels in β-cells was a logical way in which to link voltage-dependent Ca2+ influx into the β-cell by VGCCs to subsequent membrane repolarization during bursting, a simple type of negative feedback mechanism. The identification of potential KCa channel blockers increased the attractiveness and testability of this hypothesis. Thus, the idea took hold that the cyclic activation of KCa following a phase of Ca2+ influx could periodically terminate bursts of spikes in β-cells by repolarizing the membrane potential (10, 12). We now know that there are two main types of KCa channels in β-cells: a voltage-insensitive, small conductance K+ channel named SK (273, 307) and a larger, highly voltage- and Ca2+-sensitive K channel, called BK (58, 150).
Along with KATP, the KCa channels play specific roles in β-cell physiology. For SK channels, there are three different SK channel isoforms and all three are expressed in β-cells (72, 126, 273). SK channels slowly activate as cytosolic Ca2+ rises during spiking or bursting, they have a unitary conductance of 9 to 10 pS (293) and they have a distinctive pharmacology; the classic SK inhibitor is apamin, a component of bee venom (307). A major step forward in our understanding of slowly activating KCa (likely SK) was the development of a burst waveform voltage-clamp protocol by Göpel and Rorsman (92) who found they could isolate slow KCa current by subjecting the β-cell membrane to this physiological clamp command. In response to the imposed waveform, KCa slowly rises following the rise in Ca2+ creating an “envelope current” that reaches a peak by the end of the pulse burst, a phase corresponding to the voltage plateau of a burst. Once the spike-like voltage command pulses subside, a slow tail current of KCa occurs as Ca2+ falls to a low value corresponding to the holding phase potential. While the envelope and slow tail currents evoked by this protocol may include contributions from multiple types of KCa currents, there is also evidence that KATP channels can also contribute to the current (133). We note that the burst wave protocol described here has been used productively by other labs, including our own in other studies (89, 307).
A large amount of work has been done in neurons to identify the role of SK and other K+ channels mediating slow K+ currents like those underlying spike after-hyperpolarization of hippocampal neurons (205), and a number of compounds have been identified as putative SK selective blockers (29). Work with these compounds applied to β-cells suggests that the slow KCa tail current is likely carried by SK3 (307) or possibly SK4 (72) or a combination thereof. Regardless, the KCa current due to SK likely is important in regulating slow bursting when KATP is not mediating the majority of negative feedback (20). We will discuss this point further in the Modeling Section below.
Besides SK channels, large conductance (e.g. 200 pS), voltage-, and Ca2+-activated “BK” channels are a second class of KCa channel in β-cells, as first described by Cook and Hales in isolated inside-out patches (58). Unlike the KCa channels of red blood cells or SK channels, the steep voltage dependence of these channels, their Ca2+ sensitivity, and their rapid speed of activation and deactivation are hallmarks (129, 152). The gene encoding the BK channels is known as KCNMA1 (56, 91).
Because of its dynamics and voltage dependence, BK channels are rather poorly equipped to mediate islet bursting (unlike KATP or SK) and it is believed by most workers in the field that their main role is to contribute to the repolarization of fast action potentials in the β-cell (116, 126, 241), likely in concert with Kv channels (see below). BK channels are quite sensitive to blockade by toxins such as charybdotoxin (248), iberiotoxin (187), or paxillin (116), as well low dose tetraethylammonium (TEA). In response to BK blockade, the amplitude of the β-cell action potentials is potentiated, as is insulin secretion (116). However, inhibiting these channels has no pronounced effect on the pattern or kinetics of islet bursting (116).
Voltage-dependent K+ Channels
Pulse depolarizations applied in voltage clamp not only activate BK current as the membrane depolarizes and Ca2+ enters the cell, but also a voltage-dependent delayed rectifier K+ current, which activates with a delay and usually inactivates slowly, if at all (227, 228). In β-cells, the current carried by these channels strongly overlaps current mediated by BK channels; this delayed activating current is referred to as Kdr (delayed rectifier) or Kv current, and channel expression is encoded by the gene KCNB1 (Kv2.1) (164, 165). Progressively depolarizing the β-cell membrane from −30 to +50 mV results in an outward current that turns on with a characteristic delay and then smoothly activates further; with progressive depolarization, the current grows in amplitude and activates more rapidly. Blockers of the current include the classic inhibitors such as TEA, quinidine, and aminopyridines but also dendrotoxin (32, 117, 164, 222). We believe that the main role of this current is to repolarize the action potential in concert with BK current, although overexpression of Kv2.1 does reduce insulin secretion, rendering the mice glucose intolerant (210).
Inward Rectifying K+ Channels Besides Kir6.2
KIR channels are expressed in a variety of tissues, including β-cells. Their role is to help maintain the resting membrane potential near their reversal potential and to regulate the intrinsic excitability of electrically active cells. As their name suggests, KIR channels display inward rectification, with conductance that increases with hyperpolarization. Under physiological conditions, KIR channels mediate a large inward current at potentials negative to EK and small outward current at potentials positive to it (97). This rectification is due to blockade of the channel by polyamines or intracellular cations such as Mg2+ when the cell membrane is depolarized; the block is relieved by hyperpolarization and so conductance increases (191). The most important KIR channel in the β-cell is Kir6.2, the pore-forming subunit of KATP. However, other KIR channels have also been suggested to play a role in regulating insulin secretion.
Using sulfonylureas to inhibit KATP currents allows other KIR channels to be studied. In human β-cells, hyperpolarizing voltage steps applied from −70 mV cause progressively increasing, tolbutamide-resistant inward current that showed inward rectification (221, 227). Blockade of the current using Ba2+ or Cs+ (both of which inhibit KIR channels) resulted in membrane depolarization. However, this type of functional study was unable to identify which KIR channel likely was contributing to the current observed.
G protein-coupled inward-rectifying K+ (GIRK) channels, a type of inward rectifier that is activated in a G-protein-dependent manner have been shown to be present in mouse β-cells, where they mediate epinephrine- and somatostatin-induced hyperpolarization (124, 226).
Another type of KIR channel that has recently been studied in β-cells is Kir2.1, which is encoded by KCNJ11. Interestingly, in SUR1 knockout mice lacking functional KATP channels, islets still exhibit electrical bursting, although the bursting seen is no longer glucose- or tolbutamide-sensitive (253). Moreover, mice lacking KATP exhibit relatively normal blood glucose levels unless they are metabolically stressed or age (177, 253). A very similar phenotype is seen in Kir6.2 knockout mice (256). We proposed that to compensate for their loss of KATP, SUR1−/− β-cells upregulate Kir2.1 (304). Our model is based on the report that Kir2.1 contains several phosphorylation sites for protein kinase A (PKA) and that phosphorylation of these ser/thr sites results in potentiation of Kir2.1 current (79, 233, 305). Recent studies using Förster resonance energy transfer (FRET) and total internal reflection fluorescence (TIRF) microscopy show that elevated glucose induces oscillations in cAMP in mouse β-cells (76, 279). As PKA activity is cAMP-dependent, we proposed that changes in intracellular cAMP concentrations may regulate Kir2.1 channel activity and give rise to electrical oscillations in the absence of KATP (304). However, more work needs to be done to more completely prove that this occurs.
Two-pore-domain K+ (K2P) Channels
Two-pore-domain K+ channels (K2P) are small conductance K+ channels that are active at all membrane potentials and can serve as resting “leak” channels. K2P channels are formed as dimers having two α-subunits, each containing four transmembrane domains and two pore domains (180). They have been proposed to primarily serve to stabilize the plateau potential during glucose-induced electrical bursting (63). However, the conductance of these channels is significantly less than that of KATP (62, 291). Thus, it is believed that K2P channels have a more prominent role in the regulation of membrane potential when KATP is largely inhibited.
The existence of several K2Ps has been reported in mouse and human β-cells, most notably TALK1 (encoded by KNCK16), TWIK1 (encoded by KCNK1), and TASK1 (encoded by KCNK3) (125). TALK1 is an outwardly rectifying, and very small (about 1pA/pF) K+ current that is detectable at voltages above −45 mV and which has a reversal potential near −50 mV (291). Islets isolated from TALK1-deficient mice have more depolarized β-cells, reduced interburst duration, and elevated second phase GSIS (291). How TALK1 is regulated is not completely clear as yet. Interestingly, a GOF polymorphism in TALK1 (A277E) has been linked to increased risk of T2D (289). This is a gain-of-function polymorphism that increases TALK1 channel open probability, resulting in membrane hyperpolarization, decreased β-cell Ca2+ influx, and reduced in insulin secretion. TALK1 is also pH sensitive, with increased channel activity observed under alkaline conditions and decreased activity under acidic conditions (132). TALK1 has also been shown to be activatable by nitric oxide (74).
The two-pore-domain acid-sensitive K+ channel called TASK-1 is highly expressed in human and rodent β-cells and localizes to the plasma membrane, where it resembles an outwardly rectifying, noninactivating K+ current that is active at all physiological membrane potentials (66, 75, 140). In both human and mouse β-cells, blockade of TASK-1 using the inhibitor A1899 depolarizes the membrane in high glucose (14 mM) and has been shown to increase GSIS (63).
Modeling β-cells
As islet oscillations result from coordinated and highly complex interactions between β-cell ion channels, mechanisms for handling cytosolic Ca2+, and cell fuel metabolism such complexities require the construction of predictive quantitative models to probe the underlying mechanisms of islet oscillations. Our group has thus worked for many years to develop and test increasingly sophisticated and powerful β-cell models, culminating in the Integrated Oscillation Model or IOM (as reviewed in Ref. 20). The IOM, a variant of our previous Dual Oscillator Model [DOM; (21)] predicts that both autonomous glycolytic oscillations (AGOs), resulting from the autocatalytic activity of phosphofructokinase, and passive glycolytic oscillations (PGOs) due to Ca2+-driven ATP consumption leading to KATP activation can predominate during different conditions. The model is uniquely suited for determining islet glucose responses to a range of conditions, and most importantly how islets can account for fast oscillations, slow oscillations, and mixed or compound oscillations (20). While a full discussion of the mathematical details of these models is beyond the scope of the present article, all of the models to date going back to the seminal model of Chay and Keizer (52) involve two key subsystems: a fast feedback loop involving Ca2+ and K+ channels of the voltage-dependent and/or Ca2+-activated variety, and a slower feedback loop where a slow variable drives the slow plateau depolarizations that are observed in response to suprathreshold glucose. By plotting the behaviors of the model with its fast and slow components on the phase plane, a representation of the solutions of the many equations that compose the model one can readily predict the expected dynamics of the oscillations and/or stability of the complete model.
In the DOM, KATP oscillations that are driven by pulses of FBP produce oscillations in pyruvate that, once amplified by mitochondria, drive slow oscillations in ATP/ADP, Vm and Ca2+. Fast oscillations largely result from negative feedback of Ca2+ on K+ channels interacting with the slow oscillator (21). In the IOM, a rise in Ca2+ facilitates glycolytic flux by activating mitochondrial PDH and depleting the F6P pool while increasing mitochondrial ATP production (173). The ATP rise, in turn, increases Ca2+ pumping, which hydrolyzes ATP.
To help understand how each of the main ion channels participates in β-cell bursting, Figure 1 shows the predictions of the IOM model for each current that activates in islets under steady state conditions and 11 mM glucose. The left-hand column corresponds to slow bursting, while the one on the right shows the case for fast bursting.
Figure 1.
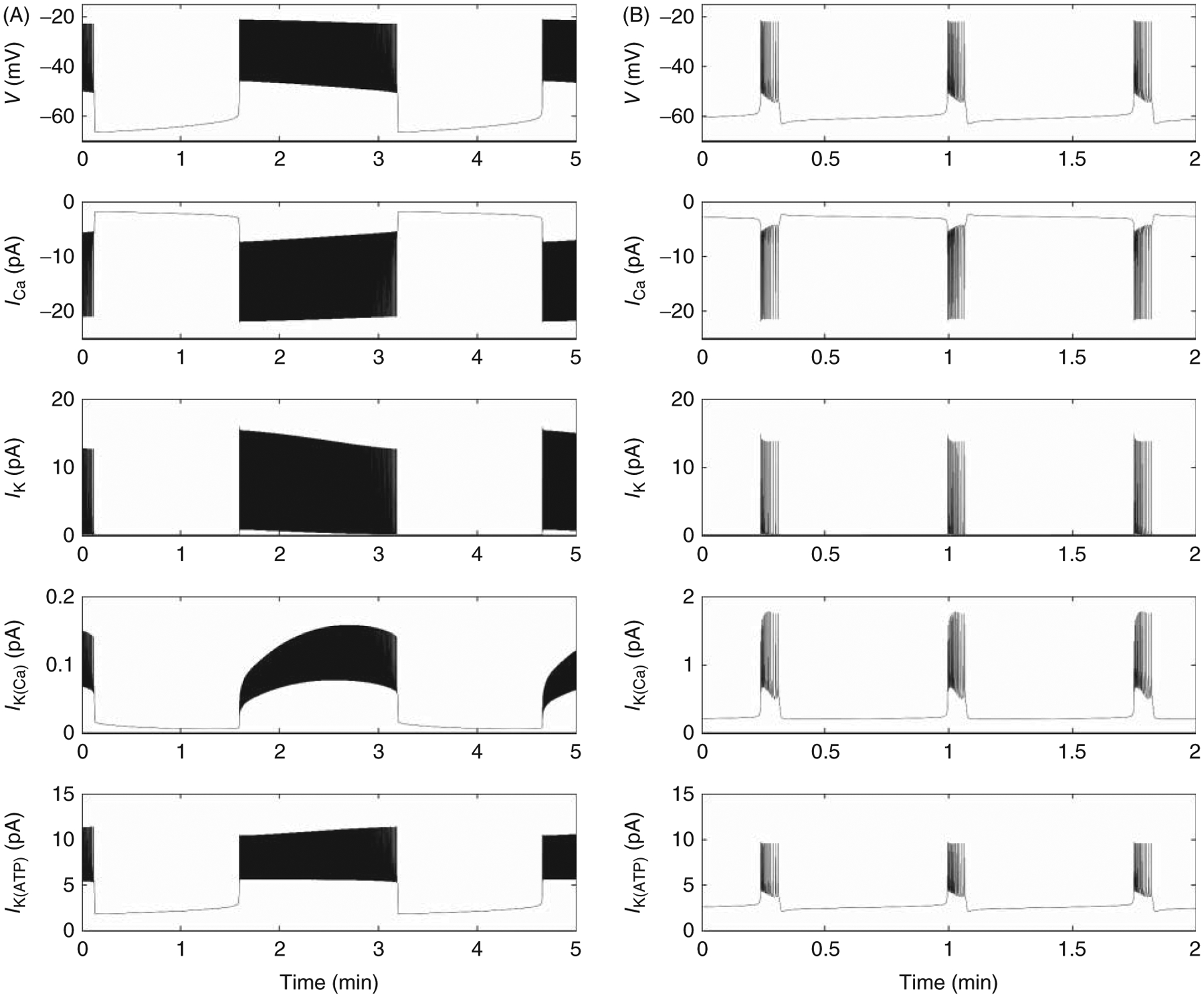
Shows a simulation performed using the Integrated Oscillator Model [IOM; (20)] of β-cell bursting in the steady state (e.g. in 11 mM glucose) to show the respective contributions of voltage-gated Ca2+ channels (ICa), voltage-dependent K+ channels (IK), slow Ca2+-activated K+ channels (IK(Ca)) and ATP-sensitive K+ channels (IK(ATP)) to slow (A) versus fast (B) bursting electrical activity (Vm). Simulations were performed by Dr. Isabella Marinelli, to whom we are grateful.
During slow bursting, L-type Ca2+ current activates quickly at the beginning of the burst plateau and spikes of inward current correspond to each of the fast action potentials triggered during the plateau. A small amount of Ca2+ current decline occurs during the train of action potentials, corresponding to the decline in activation voltage, and the Ca2+ current shuts off promptly after plateau repolarization.
Voltage-dependent K+ current plays a minor role during the interburst period but then activates rapidly during each action potential to help repolarize them. In contrast, IK(Ca) has much slower kinetics, as these channels (most likely mediated by SK type isoform) follow changes in cytosolic Ca2+, IK(Ca) slowly ramps up during the burst and mainly reaches its peak by the end of the active phase. Notably, a slow KCa tail current is clearly visible during the interburst period.
The KATP current grows slowly during the burst active phase due to an increase in the total channel conductance brought about by a slow decline in the ATP:ADP ratio. It quickly falls at the end of an active phase due to a rapid decline in the K+ driving force, but slowly rises as the membrane depolarizes throughout the interburst period.
Qualitatively similar patterns are seen for most of the currents during fast bursting (right-hand column). A particularly notable exception to this is IK(Ca), which more rapidly activates at the initiation of the active phase and then declines instead of continually growing.
Thus, the model can be used to understand how ion currents can contribute to the bursting process and the predictions of these models can be tested in a laboratory. A particularly interesting and important aspect of our models has been their ability to predict unexpected islet behaviors that were subsequently verified by experiment. Three examples will be discussed here.
First, modeling predicted that increasing the cytoplasmic concentration of fructose2,6 bisphosphate (FBP2), an allosteric activator of PFK1 should increase the frequency while decreasing the amplitude of islet Ca2+ oscillations. We tested this experimentally by manipulating the expression of PFK2/FBPase2 mutants, which allowed us to either increase or decrease FBP2 levels. In both cases, the model predictions were verified (175).
As a second example, the model predicted that the abolition of islet metabolic oscillations seen following the application of diazoxide, which hyperpolarizes the β-cell membrane by opening KATP channels should be reversed by the addition of KCl (in the continual presence of diazoxide) to depolarize the membrane and restore Ca2+ influx. This novel prediction was verified experimentally by measuring endogenous NAD(P)H fluorescence (193) or more recently measuring FBP with our PKAR probe (176).
As a last example, it was predicted that individual mouse β-cells exhibiting repetitive spiking but not bursting should be capable of being converted to a more islet-like bursting pattern by increasing their voltage-dependent Ca2+ conductance. Using the dynamic clamp technique, whereby a computer-generated ion conductance is calculated in an iterative manner in real time and then injected into the cell in response to changes in the cell’s own membrane potential we confirmed that injection of such a conductance indeed was able to convert the cell to bursting (306). These examples, and others, have convinced us over many years that models are not only useful for accounting for the interactions between many variables in a complex dynamical system but that they can make novel yet experimentally testable predictions.
Conclusion and Future Directions
The plethora of ion channels identified in pancreatic β-cells has provided investigators with a robust palette of elements that can be combined in expected as well as unexpected ways to mediate glucose-induced islet electrical bursting and its modulation by not only metabolizable fuels but also neurohormonal modulators and mediators to produce a wide spectrum of electrical behaviors and, in turn, patterns of secretion. The more well-understood roles that classic ion channels play in other cell types typically serve as starting points in our understanding of β-cells, as once these channels are modified by metabolism, or Ca2+, a much wider range of behaviors become possible. Hence, our work has emphasized the importance of mathematical models in understanding how ion channels interact with one another and with metabolism to mediate the range of oscillatory phenomena observed in islets.
Future endeavors in this field will need to build on new work describing the importance of the heterogeneity in channel expression that has been observed, and the recent use of RNAseq combined with patch clamp electrophysiology (45, 46) to decipher the molecular basis of β-cell ion channel heterogeneity is encouraging. Furthermore, extending these studies into understanding human islets, a relatively new development, as well as islet behavior in vivo, should be even more illuminating and will likely lead to increasingly clinically relevant findings.
Didactic Synopsis.
Major teaching points
Understanding the physiology of pancreatic β-cell ion channels is important for understanding how glucose metabolism is coupled to insulin secretion.
The KATP channel is the primary regulator of β-cell electrical activity by coupling metabolism to changes in membrane potential.
Voltage-gated Ca2+ channels (VGCCs) are responsible for triggering and sustaining insulin secretion by regulating Ca2+ influx across the plasma membrane in response to changes in membrane potential.
While KATP and VGCCs are the main ion channels which control insulin secretion, other channels also play a role in modulating secretion.
Understanding the role of different ion channels in regulating insulin secretion allows for the better design of complex dynamical models to account for β-cell oscillations and hence pulsatile insulin secretion.
Acknowledgements
Work in the Satin Lab is supported by NIH RO1 DK46409, NIH HIRN U01 DK127747; funding from the Juvenile Diabetes Research Foundation (JDRF), CDA-2016-189 and support from the FastForward Initiative of the UM Medical School. The authors would like to thank Drs. Arthur Sherman and Richard Bertram for their helpful comments on an earlier version of the manuscript and Dr. Isabella Marinelli of the University of Birmingham, UK for providing a figure depicting the role of different simulated ion currents in a bursting β-cell (Figure 1).
Footnotes
Financial Conflicts
None.
References
- 1.Ainscow EK, Rutter GA. Mitochondrial priming modifies Ca2+ oscillations and insulin secretion in pancreatic islets. Biochem J 353: 175–180, 2001. DOI: 10.1042/bj3530175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ainscow EK, Rutter GA. Glucose-stimulated oscillations in free cytosolic ATP concentration imaged in single islet B-cells: Evidence for a Ca2+-dependent mechanism. Diabetes 51: S162–S170, 2002. DOI: 10.2337/diabetes.51.2007.s162. [DOI] [PubMed] [Google Scholar]
- 3.Akbulut Y, Gaunt HJ, Muraki K, Ludlow MJ, Amer MS, Bruns A, Vasudev NS, Radtke L, Willot M, Hahn S, Seitz T, Ziegler S, Christ-mann M, Beech DJ, Waldmann H. (−)-Englerin A is a potent and selective activator of TRPC4 and TRPC5 calcium channels. Angew Chem Int Ed 54: 3787–3791, 2015. DOI: 10.1002/anie.201411511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arroyo G, Aldea M, Fuentealba J, Albillos A, Garcia AG. SNX482 selectively blocks P/Q Ca2+ channels and delays the inactivation of Na+ channels of chromaffin cells. Eur J Pharmacol 475: 11–18, 2003. DOI: 10.1016/s0014-2999(03)02084-3. [DOI] [PubMed] [Google Scholar]
- 5.Ashcroft F, Rorsman P. Electrophysiology of the pancreatic beta-cell. Prog Biophys Mol Biol 54: 87–143, 1989. DOI: 10.1016/0079-6107(89)90013-8. [DOI] [PubMed] [Google Scholar]
- 6.Ashcroft FM. ATP-sensitive potassium channelopathies: Focus on insulin secretion. J Clin Investig 115: 2047–2058, 2005. DOI: 10.1172/JCI25495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ashcroft FM, Harrison DE, Ashcroft SJH. Glucose induces closure of single potassium channels in isolated rat pancreatic β-cells. Nature 312: 446–448, 1984. DOI: 10.1038/312446a0. [DOI] [PubMed] [Google Scholar]
- 8.Ashcroft FM, Kelly RP, Smith PA. Two types of Ca channel in rat pancreatic β-cells. Pflugers Arch 415: 504–506, 1990. DOI: 10.1007/bf00373633. [DOI] [PubMed] [Google Scholar]
- 9.Ashcroft FM, Rorsman P. ATP-sensitive K+ channels: A link between B-cell metabolism and insulin secretion. Biochem Soc T 18: 109–111, 1990. DOI: 10.1042/bst0180109. [DOI] [PubMed] [Google Scholar]
- 10.Atwater I, Dawson CM, Ribalet B, Rojas E. Potassium permeability activated by intracellular calcium ion concentration in the pancreatic beta-cell. J Physiol 288: 575–588, 1979. DOI: 10.1113/jphysiol.1979.sp012714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Atwater I, Ribalet B, Rojas E. Cyclic changes in potential and resistance of the beta-cell membrane induced by glucose in islets of Langerhans from mouse. J Physiol 278: 117–139, 1978. DOI: 10.1113/jphysiol.1978.sp012296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Atwater I, Rosario L, Rojas E. Properties of the Ca-activated K+ channel in pancreatic β-cells. Cell Calcium 4: 451–461, 1983. DOI: 10.1016/0143-4160(83)90021-0. [DOI] [PubMed] [Google Scholar]
- 13.Balamurugan K, Kavitha B, Yang Z, Mohan V, Radha V, Shyng S. Functional characterization of activating mutations in the sulfonylurea receptor 1 (ABCC8) causing neonatal diabetes mellitus in Asian Indian children. Pediatr Diabetes 20: 397–407, 2019. DOI: 10.1111/pedi.12843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barasch J, Kiss B, Prince A, Saiman L, Gruenert D, AI-Awqati Q. Defective acidification of intracellular organelles in cystic fibrosis. Nature 352: 70–73, 1991. DOI: 10.1038/352070a0. [DOI] [PubMed] [Google Scholar]
- 15.Barg S, Galvanovskis J, Gopel SO, Rorsman P, Eliasson L. Tight coupling between electrical activity and exocytosis in mouse glucagon-secreting alpha-cells. Diabetes 49: 1500–1510, 2000. DOI: 10.2337/diabetes.49.9.1500. [DOI] [PubMed] [Google Scholar]
- 16.Barg S, Ma X, Eliasson L, Galvanovskis J, Göpel SO, Obermüller S, Platzer J, Renström E, Trus M, Atlas D, Striessnig J, Rorsman P. Fast exocytosis with few Ca2+ channels in insulin-secreting mouse pancreatic B cells. Biophys J 81: 3308, 3323, 2001. DOI: 10.1016/s0006-3495(01)75964-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bari MR, Akbar S, Eweida M, Kühn FJP, Gustafsson AJ, Lückhoff A, Islam MdS. H2O2-induced Ca2+ influx and its inhibition by N-(p-amylcinnamoyl) anthranilic acid in the β-cells: Involvement of TRPM2 channels. J Cell Mol Med 13: 3260–3267, 2009. DOI: 10.1111/j.1582-4934.2009.00737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barnett DW, Pressel DM, Misler S. Voltage-dependent Na+ and Ca2+ currents in human pancreatic islet β-cells: Evidence for roles in the generation of action potentials and insulin secretion. Pflugers Arch 431: 272–282, 1995. DOI: 10.1007/bf00410201. [DOI] [PubMed] [Google Scholar]
- 19.Berger HA, Anderson MP, Gregory RJ, Thompson S, Howard PW, Maurer RA, Mulligan R, Smith AE, Welsh MJ. Identification and regulation of the cystic fibrosis transmembrane conductance regulator-generated chloride channel. J Clin Invest 88: 1422–1431, 1991. DOI: 10.1172/jci115450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bertram R, Satin LS, Sherman AS. Closing in on the mechanisms of pulsatile insulin secretion. Diabetes 67: 351–359, 2018. DOI: 10.2337/dbi17-0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bertram R, Sherman A, Satin LS. Metabolic and electrical oscillations: Partners in controlling pulsatile insulin secretion. Am J Physiol Endocrinol Metab 293: E890–E900, 2007. DOI: 10.1152/ajpendo.00359.2007. [DOI] [PubMed] [Google Scholar]
- 22.Best L Glucose and a-ketoisocaproate induce transient inward currents in rat pancreatic beta cells. Diabetologia 40: 1–6, 1997. DOI: 10.1007/s001250050635. [DOI] [PubMed] [Google Scholar]
- 23.Best L, Brown P, Tomlinson S. Anion fluxes, volume regulation and electrical activity in the mammalian pancreatic beta-cell. Exp Physiol 82: 957–966, 1997. DOI: 10.1113/expphysiol.1997.sp004081. [DOI] [PubMed] [Google Scholar]
- 24.Best L, Brown PD, Sener A, Malaisse WJ. Electrical activity in pancreatic islet cells: The VRAC hypothesis. Islets 2: 59–64, 2010. DOI: 10.4161/isl.2.2.11171. [DOI] [PubMed] [Google Scholar]
- 25.Best L, Miley H, Yates A. Activation of an anion conductance and beta-cell depolarization during hypotonically induced insulin release. Exp Physiol 81: 927–933, 1996. DOI: 10.1113/expphysiol.1996.sp003993. [DOI] [PubMed] [Google Scholar]
- 26.Best L, Sheader E, Brown P. A volume-activated anion conductance in insulin-secreting cells. Pflugers Arch - Eur J Physiol 431: 363–370, 1996. DOI: 10.1007/bf02207273. [DOI] [PubMed] [Google Scholar]
- 27.Bhattacharjee A, Whitehurst RM, Zhang M, Wang L, Li M. T-type calcium channels facilitate insulin secretion by enhancing general excitability in the insulin-secreting β-cell line, INS-1. Endocrinology 138: 3735–3740, 1997. DOI: 10.1210/endo.138.9.5390. [DOI] [PubMed] [Google Scholar]
- 28.Biwersi J, Verkman AS. Functional CFTR in endosomal compartment of CFTR-expressing fibroblasts and T84 cells. Am J Physiol-Cell Ph 266: C149–C156, 1994. DOI: 10.1152/ajpcell.1994.266.1.c149. [DOI] [PubMed] [Google Scholar]
- 29.Blank T, Nijholt I, Kye M-J, Spiess J. Small conductance ca2+-activated k+ channels as targets of CNS drug development. Curr Drug Target -CNS Neurological Disord 3: 161–167, 2004. DOI: 10.2174/1568007043337472. [DOI] [PubMed] [Google Scholar]
- 30.Blodgett DM, Nowosielska A, Afik S, Pechhold S, Cura AJ, Kennedy NJ, Kim S, Kucukural A, Davis RJ, Kent SC, Greiner DL, Garber MG, Harlan DM, diIorio P. Novel observations from next-generation RNA sequencing of highly purified human adult and fetal islet cell subsets. Diabetes 64: 3172–3181, 2015. DOI: 10.2337/db15-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bokvist K, Eliasson L, Ammälä C, Renström E, Rorsman P. Co-localization of L-type Ca2+ channels and insulin-containing secretory granules and its significance for the initiation of exocytosis in mouse pancreatic B-cells. EMBO J 14: 50–57, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bokvist K, Rorsman P, Smith PA. Effects of external tetraethylammonium ions and quinine on delayed rectifying K+ channels in mouse pancreatic beta-cells. J Physiol 423: 311–325, 1990. DOI: 10.1113/jphysiol.1990.sp018024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bonaventura MM, Crivello M, Ferreira ML, Repetto M, Cymeryng C, Libertun C, Lux-Lantos VA. Effects of GABAB receptor agonists and antagonists on glycemia regulation in mice. Eur J Pharmacol 677: 188–196, 2012. DOI: 10.1016/j.ejphar.2011.12.013. [DOI] [PubMed] [Google Scholar]
- 34.Braun M The αβδ of ion channels in human islet cells. Islets 1: 160–162, 2009. DOI: 10.4161/isl.1.2.9405. [DOI] [PubMed] [Google Scholar]
- 35.Braun M, Ramracheya R, Bengtsson M, Clark A, Walker JN, Johnson PR, Rorsman P. γ-Aminobutyric acid (GABA) is an autocrine excitatory transmitter in human pancreatic β-cells. Diabetes 59: 1694–1701, 2010. DOI: 10.2337/db09-0797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Braun M, Ramracheya R, Bengtsson M, Zhang Q, Karanauskaite J, Partridge C, Johnson PR, Rorsman P. Voltage-gated ion channels in human pancreatic beta-cells: Electrophysiological characterization and role in insulin secretion. Diabetes 57: 1618–1628, 2008. DOI: 10.2337/db07-0991. [DOI] [PubMed] [Google Scholar]
- 37.Braun M, Wendt A, Birnir B, Broman J, Eliasson L, Galvanovskis J, Gromada J, Mulder H, Rorsman P. Regulated exocytosis of GABA-containing synaptic-like microvesicles in pancreatic β-cells. J Gen Physiol 123: 191–204, 2004. DOI: 10.1085/jgp.200308966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Braun M, Wendt A, Buschard K, Salehi A, Sewing S, Gromada J, Rorsman P. GABAB receptor activation inhibits exocytosis in rat pancreatic β-cells by G-protein-dependent activation of calcineurin. J Physiol 559: 397–409, 2004. DOI: 10.1113/jphysiol.2004.066563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Braun M, Wendt A, Karanauskaite J, Galvanovskis J, Clark A, MacDonald PE, Rorsman P. Corelease and differential exit via the fusion pore of GABA, serotonin, and ATP from LDCV in rat pancreatic β cells. J Gen Physiol 129: 221–231, 2007. DOI: 10.1085/jgp.200609658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Briant LJ, Zhang Q, Vergari E, Kellard JA, Rodriguez B, Ashcroft FM, Rorsman P. Functional identification of islet cell types by electrophysiological fingerprinting. J Roy Soc Interface 14: 20160999, 2017. DOI: 10.1098/rsif.2016.0999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brissova M, Fowler MJ, Nicholson WE, Chu A, Hirshberg B, Harlan DM, Powers AC. Assessment of human pancreatic islet architecture and composition by laser scanning confocal microscopy. J Histochem Cytochem 53: 1087–1097, 2005. DOI: 10.1369/jhc.5c6684.2005. [DOI] [PubMed] [Google Scholar]
- 42.Britsch S, Krippeitdrews P, Gregor M, Lang F, Drews G. Effects of osmotic changes in extracellular solution on electrical activity of mouse pancreatic B-cells. Biochem Bioph Res Co 204: 641–645, 1994. DOI: 10.1006/bbrc.1994.2507. [DOI] [PubMed] [Google Scholar]
- 43.Brixel LR, Monteilh-Zoller MK, Ingenbrandt CS, Fleig A, Penner R, Enklaar T, Zabel BU, Prawitt D. TRPM5 regulates glucose-stimulated insulin secretion. Pflugers Arch - Eur J Physiol 460: 69–76, 2010. DOI: 10.1007/s00424-010-0835-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cabrera O, Berman DM, Kenyon NS, Ricordi C, Berggren P-O, Caicedo A. The unique cytoarchitecture of human pancreatic islets has implications for islet cell function. P Natl Acad Sci USA 103: 2334–2339, 2006. DOI: 10.1073/pnas.0510790103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Camunas-Soler J, Dai X, Hang Y, Bautista A, Lyon J, Suzuki K, Kim SK, Quake SR, MacDonald PE. Pancreas patch-seq links physiologic dysfunction in diabetes to single-cell transcriptomic phenotypes. BioRxiv: 555110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Camunas-Soler J, Dai X-Q, Hang Y, Bautista A, Lyon J, Suzuki K, Kim SK, Quake SR, MacDonald PE. Patch-seq links single-cell transcriptomes to human islet dysfunction in diabetes. Cell Metab 31: 1017–1031.e4, 2020. DOI: 10.1016/j.cmet.2020.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carbone E, Lux HD. A low voltage-activated, fully inactivating Ca channel in vertebrate sensory neurones. Nature 310: 501–502, 1984. DOI: 10.1038/310501a0. [DOI] [PubMed] [Google Scholar]
- 48.Cartier EA, Conti LR, Vandenberg CA, Shyng S-L. Defective trafficking and function of KATP channels caused by a sulfonylurea receptor 1 mutation associated with persistent hyperinsulinemic hypoglycemia of infancy. Proc Natl Acad Sci 98: 2882–2887, 2001. DOI: 10.1073/pnas.051499698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Casas S, Novials A, Reimann F, Gomis R, Gribble FM. Calcium elevation in mouse pancreatic beta cells evoked by extracellular human islet amyloid polypeptide involves activation of the mechanosensitive ion channel TRPV4. Diabetologia 51: 2252–2262, 2008. DOI: 10.1007/s00125-008-1111-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Bi 16: 521–555, 2000. DOI: 10.1146/annurev.cellbio.16.1.521. [DOI] [PubMed] [Google Scholar]
- 51.Chai H, Cheng X, Zhou B, Zhao L, Lin X, Huang D, Lu W, Lv H, Tang F, Zhang Q, Huang W, Li Y, Yang H. Structure-based discovery of a subtype-selective inhibitor targeting a transient receptor potential vanilloid channel. J Med Chem 62: 1373–1384, 2019. DOI: 10.1021/acs.jmedchem.8b01496. [DOI] [PubMed] [Google Scholar]
- 52.Chay TR, Keizer J. Minimal model for membrane oscillations in the pancreatic beta-cell. Biophys J 42: 181–189, 1983. DOI: 10.1016/s0006-3495(83)84384-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen P-C, Kryukova YN, Shyng S-L. Leptin regulates KATP channel trafficking in pancreatic β-cells by a signaling mechanism involving AMP-activated protein kinase (AMPK) and cAMP-dependent protein kinase (PKA). J Biol Chem 288: 34098–34109, 2013. DOI: 10.1074/jbc.m113.516880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Clement JP, Kunjilwar K, Gonzalez G, Schwanstecher M, Panten U, Aguilar-Bryan L, Bryan J. Association and stoichiometry of KATP channel subunits. Neuron 18: 827–838, 1997. DOI: 10.1016/s0896-6273(00)80321-9. [DOI] [PubMed] [Google Scholar]
- 55.Colsoul B, Schraenen A, Lemaire K, Quintens R, Lommel LV, Segal A, Owsianik G, Talavera K, Voets T, Margolskee RF, Kokrashvili Z, Gilon P, Nilius B, Schuit FC, Vennekens R. Loss of high-frequency glucose-induced Ca2+ oscillations in pancreatic islets correlates with impaired glucose tolerance in Trpm5−/− mice. Proc National Acad Sci 107: 5208–5213, 2010. DOI: 10.1073/pnas.0913107107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Contreras GF, Castillo K, Enrique N, Carrasquel-Ursulaez W, Castillo JP, Milesi V, Neely A, Alvarez O, Ferreira G, González C, Latorre R. A BK (Slo1) channel journey from molecule to physiology. Channels 7: 442–458, 2013. DOI: 10.4161/chan.26242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cook DL. Electrical pacemaker mechanisms of pancreatic islet cells. Fed Proc 43: 2368–2372, 1984. [PubMed] [Google Scholar]
- 58.Cook DL, Ikeuchi M, Fujimoto WY. Lowering of pHi inhibits Ca2+-activated K+ channels in pancreatic B-cells. Nature 311: 269–271, 1984. DOI: 10.1038/311269a0. [DOI] [PubMed] [Google Scholar]
- 59.Cook DL, Satin LS, Ashford ML, Hales NC. ATP-sensitive k+ channels in pancreatic β-cells: Spare-channel hypothesis. Diabetes 37: 495–498, 1988. DOI: 10.2337/diab.37.5.495. [DOI] [PubMed] [Google Scholar]
- 60.Cook DL, Satin LS, Hopkins WF. Pancreatic B cells are bursting, but how? Trends Neurosci 14: 411–414, 1991. DOI: 10.1016/0166-2236(91)90033-q. [DOI] [PubMed] [Google Scholar]
- 61.Crutzen R, Virreira M, Markadieu N, Shlyonsky V, Sener A, Malaisse WJ, Beauwens R, Boom A, Golstein PE. Anoctamin 1 (Ano1) is required for glucose-induced membrane potential oscillations and insulin secretion by murine β-cells. Pflugers Arch - Eur J Physiol 468: 573–591, 2016. DOI: 10.1007/s00424-015-1758-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dadi PK, Luo B, Vierra NC, Jacobson DA. TASK-1 potassium channels limit pancreatic α-cell calcium influx and glucagon secretion. Mol Endocrinol 29: 777–787, 2015. DOI: 10.1210/me.2014-1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dadi PK, Vierra NC, Jacobson DA. Pancreatic β-cell-specific ablation of TASK-1 channels augments glucose-stimulated calcium entry and insulin secretion, improving glucose tolerance. Endocrinology 155: 3757–3768, 2014. DOI: 10.1210/en.2013-2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Davalli AM, Biancardi E, Pollo A, Socci C, Pontiroli AE, Pozza G, Clementi F, Sher E, Carbone E. Dihydropyridine-sensitive and -insensitive voltage-operated calcium channels participate in the control of glucose-induced insulin release from human pancreatic β cells. J Endocrinol 150: 195–203, 1996. DOI: 10.1677/joe.0.1500195. [DOI] [PubMed] [Google Scholar]
- 65.Davidson HW, Rhodes CJ, Hutton JC. Intraorganellar calcium and pH control proinsulin cleavage in the pancreatic β cell via two distinct site-specific endopeptidases. Nature 333: 93–96, 1988. DOI: 10.1038/333093a0. [DOI] [PubMed] [Google Scholar]
- 66.Davies LA, Hu C, Guagliardo NA, Sen N, Chen X, Talley EM, Carey RM, Bayliss DA, Barrett PQ. TASK channel deletion in mice causes primary hyperaldosteronism. Proc National Acad Sci 105: 2203–2208, 2008. DOI: 10.1073/pnas.0712000105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dean PM. Ultrastructural morphometry of the pancreatic β-cell. Diabetologia 9: 115–119, 1973. DOI: 10.1007/bf01230690. [DOI] [PubMed] [Google Scholar]
- 68.Dean PM, Matthews EK. Glucose-induced electrical activity in pancreatic islet cells. J Physiol 210: 255–264, 1970. DOI: 10.1113/jphysiol.1970.sp009207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Deneka D, Sawicka M, Lam AKM, Paulino C, Dutzler R. Structure of a volume-regulated anion channel of the LRRC8 family. Nature 558: 254–259, 2018. DOI: 10.1038/s41586-018-0134-y. [DOI] [PubMed] [Google Scholar]
- 70.Detimary P, Gilon P, Henquin J-C. Interplay between cytoplasmic Ca2+ and the ATP/ADP ratio: A feedback control mechanism in mouse pancreatic islets. Biochem J 333: 269–274, 1998. DOI: 10.1042/bj3330269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Donatsch P, Lowe DA, Richardson BP, Taylor P. The functional significance of sodium channels in pancreatic beta-cell membranes. J Physiol 267: 357–376, 1977. DOI: 10.1113/jphysiol.1977.sp011817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Düfer M, Gier B, Wolpers D, Krippeit-Drews P, Ruth P, Drews G. Enhanced glucose tolerance by SK4 channel inhibition in pancreatic β-cells. Diabetes 58: 1835–1843, 2009. DOI: 10.2337/db08-1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dunne MJ, Petersen OH. Intracellular ADP activates K+ channels that are inhibited by ATP in an insulin-secreting cell line. FEBS Lett 208: 59–62, 1986. DOI: 10.1016/0014-5793(86)81532-0. [DOI] [PubMed] [Google Scholar]
- 74.Duprat F, Girard C, Jarretou G, Lazdunski M. Pancreatic two P domain K+ channels TALK-1 and TALK-2 are activated by nitric oxide and reactive oxygen species. J Physiol 562: 235–244, 2005. DOI: 10.1113/jphysiol.2004.071266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Duprat F, Lesage F, Fink M, Reyes R, Heurteaux C, Lazdunski M. TASK, a human background K+ channel to sense external pH variations near physiological pH. EMBO J 16: 5464–5471, 1997. DOI: 10.1093/emboj/16.17.5464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dyachok O, Idevall-Hagren O, Sågetorp J, Tian G, Wuttke A, Arrieumerlou C, Akusjärvi G, Gylfe E, Tengholm A. Glucose-induced cyclic AMP oscillations regulate pulsatile insulin secretion. Cell Metab 8: 26–37, 2008. DOI: 10.1016/j.cmet.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 77.Edlund A, Barghouth M, Huhn M, Abels M, Esguerra J, Mollet I, Svedin E, Wendt A, Renstrom E, Zhang E, Wierup N, Scholte BJ, Flodström-Tullberg M, Eliasson L. Defective exocytosis and processing of insulin in a cystic fibrosis mouse model. J Endocrinol 1: 45–57, 2019. DOI: 10.1530/joe-18-0570. [DOI] [PubMed] [Google Scholar]
- 78.Edlund A, Esguerra JL, Wendt A, Flodström-Tullberg M, Eliasson L. CFTR and Anoctamin 1 (ANO1) contribute to cAMP amplified exocytosis and insulin secretion in human and murine pancreatic beta-cells. BMC Med 12: 87, 2014. DOI: 10.1186/1741-7015-12-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fakler B, Brändle U, Glowatzki E, Zenner H, Ruppersberg J. Kir2.1 inward rectifier K+ channels are regulated independently by protein kinases and ATP hydrolysis. Neuron 13: 1413–1420, 1994. DOI: 10.1016/0896-6273(94)90426-X. [DOI] [PubMed] [Google Scholar]
- 80.Ferrer J, Nichols C, Makhina E, Salkoff L, Bernstein J, Gerhard D, Wasson J, Ramanadham S, Permutt A. Pancreatic islet cells express a family of inwardly rectifying K+ channel subunits which interact to form G-protein-activated channels. J Biol Chem 270: 26086–26091, 1995. [DOI] [PubMed] [Google Scholar]
- 81.Fontés G, Ghislain J, Benterki I, Zarrouki B, Trudel D, Berthiaume Y, Poitout V. The ΔF508 mutation in the cystic fibrosis transmembrane conductance regulator is associated with progressive insulin resistance and decreased functional β-cell mass in mice. Diabetes 64: 4112–4122, 2015. DOI: 10.2337/db14-0810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fourgeaud L, Dvorak C, Faouzi M, Starkus J, Sahdeo S, Wang Q, Lord B, Coate H, Taylor N, He Y, Qin N, Wickenden A, Carruthers N, Lovenberg TW, Penner R, Bhattacharya A. Pharmacology of JNJ-28583113: A novel TRPM2 antagonist. Eur J Pharmacol 853: 299–307, 2019. DOI: 10.1016/j.ejphar.2019.03.043. [DOI] [PubMed] [Google Scholar]
- 83.Fox JE, Gyulkhandanyan AV, Satin LS, Wheeler MB. Oscillatory membrane potential response to glucose in islet β-cells: A comparison of islet-cell electrical activity in mouse and rat. Endocrinology 147: 4655–4663, 2006. DOI: 10.1210/en.2006-0424. [DOI] [PubMed] [Google Scholar]
- 84.Gall D, Gromada J, Susa I, Rorsman P, Herchuelz A, Bokvist K. Significance of Na/Ca exchange for Ca2+ buffering and electrical activity in mouse pancreatic β-cells. Biophys J 76: 2018–2028, 1999. DOI: 10.1016/s0006-3495(99)77359-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gaskins HR, Baldeón ME, Selassie L, Beverly JL. Glucose modulates γ-aminobutyric acid release from the pancreatic βTC6 cell line (*). J Biol Chem 270: 30286–30289, 1995. DOI: 10.1074/jbc.270.51.30286. [DOI] [PubMed] [Google Scholar]
- 86.Gilon P, Yakel J, Gromada J, Zhu Y, Henquin JC, Rorsman P. G protein-dependent inhibition of L-type Ca2+ currents by acetylcholine in mouse pancreatic B-cells. J Physiol 499: 65–76, 1997. DOI: 10.1113/jphysiol.1997.sp021911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gloyn AL, Pearson ER, Antcliff JF, Proks P, Bruining G, Slinger-land AS, Howard N, Srinivasan S, Silva JM, Molnes J, Edghill EL, Frayling TM, Temple I, Mackay D, Shield JP, Sumnik Z, van Rhijn A, Wales JK, Clark P, Gorman S, Aisenberg J, Ellard S, Njølstad PR, Ashcroft FM, Hattersley AT. Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N Engl J Med 350: 1838–1849, 2004. DOI: 10.1056/NEJMoa032922. [DOI] [PubMed] [Google Scholar]
- 88.Glynn E, Thompson B, Vadrevu S, Lu S, Kennedy RT, Ha J, Sherman A, Satin LS. Chronic glucose exposure systematically shifts the oscillatory threshold of mouse islets: Experimental evidence for an early intrinsic mechanism of compensation for hyperglycemia. Endocrinology 157: 611–623, 2016. DOI: 10.1210/en.2015-1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Goforth PB, Bertram R, Khan FA, Zhang M, Sherman A, Satin LS. Calcium-activated K+ channels of mouse β-cells are controlled by both store and cytoplasmic Ca2+ experimental and theoretical studies. J Gen Physiol 120: 307–322, 2002. DOI: 10.1085/jgp.20028581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Goldin AL. Resurgence of sodium channel research. Annu Rev Physiol 63: 871–894, 2001. DOI: 10.1146/annurev.physiol.63.1.871. [DOI] [PubMed] [Google Scholar]
- 91.Gonzalez-Perez V, Lingle CJ. Regulation of BK channels by beta and gamma subunits. Annu Rev Physiol 81: 113–137, 2016. DOI: 10.1146/annurev-physiol-022516-034038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Göpel S, Kanno T, Barg S, Eliasson L, Galvanovskis J, Renström E, Rorsman P. Activation of Ca(2+)-dependent K(+) channels contributes to rhythmic firing of action potentials in mouse pancreatic beta cells. J Gen Physiol 114: 759–770, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Göpel S, Kanno T, Barg S, Galvanovskis J, Rorsman P. Voltage-gated and resting membrane currents recorded from B-cells in intact mouse pancreatic islets. J Physiol 521: 717–728, 1999. DOI: 10.1111/j.1469-7793.1999.00717.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Göpel SO, Kanno T, Barg S, Weng X-G, Gromada J, Rorsman P. Regulation of glucagon release in mouse α-cells by KATP channels and inactivation of TTX-sensitive Na+ channels. J Physiol 528: 509–520, 2000. DOI: 10.1111/j.1469-7793.2000.00509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gromada J, Chabosseau P, Rutter GA. The α-cell in diabetes mellitus. Nat Rev Endocrinol 14: 1, 2018. DOI: 10.1038/s41574-018-0097-y. [DOI] [PubMed] [Google Scholar]
- 96.Guo JH, Chen H, Ruan YC, Zhang XL, Zhang XH, Fok KL, Tsang LL, Yu MK, Huang WQ, Sun X, Chung YW, Jiang X, Sohma Y, Chan HC. Glucose-induced electrical activities and insulin secretion in pancreatic islet β-cells are modulated by CFTR. Nat Commun 5: 4420, 2014. DOI: 10.1038/ncomms5420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hagiwara S, Ozawa S, Sand O. Voltage clamp analysis of two inward current mechanisms in the egg cell membrane of a starfish. J Gen Physiol 65: 617–644, 1975. DOI: 10.1085/jgp.65.5.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Han Y-EE, Chun JN, Kwon MJ, Ji Y-SS, Jeong M-HH, Kim H-HH, Park S-HH, Rah JC, Kang J, Lee S-HH, Ho W-KK. Endocytosis of KATP channels drives glucose-stimulated excitation of pancreatic β cells. Cell Rep 22: 471–481, 2018. DOI: 10.1016/j.celrep.2017.12.049. [DOI] [PubMed] [Google Scholar]
- 99.Hart NJ, Aramandla R, Poffenberger G, Fayolle C, Thames AH, Bautista A, Spigelman AF, Babon JAB, DeNicola ME, Dadi PK, Bush WS, Balamurugan AN, Brissova M, Dai C, Prasad N, Bottino R, Jacobson DA, Drumm ML, Kent SC, MacDonald PE, Powers AC. Cystic fibrosis–related diabetes is caused by islet loss and inflammation. Jci Insight 3: e98240, 2018. DOI: 10.1172/jci.insight.98240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Heimberg H, Vos AD, Pipeleers D, Thorens B, Schuit F. Differences in glucose transporter gene expression between rat pancreatic α-and β-cells are correlated to differences in glucose transport but not in glucose utilization. J Biol Chem 270: 8971–8975, 1995. DOI: 10.1074/jbc.270.15.8971. [DOI] [PubMed] [Google Scholar]
- 101.Heinemann SH, Terlau H, Stühmer W, Imoto K, Numa S. Calcium channel characteristics conferred on the sodium channel by single mutations. Nature 356: 441–443, 1992. DOI: 10.1038/356441a0. [DOI] [PubMed] [Google Scholar]
- 102.Hellman B The frequency distribution of the number and volume of the islets Langerhans in man. I. Studies on non-diabetic adults. Acta Soc Med Ups 64: 432–460, 1959. [PubMed] [Google Scholar]
- 103.Henquin J-C, Meissner HP. Opposite effects of tolbutamide and diazoxide on 86Rb+ fluxes and membrane potential in pancreatic B cells. Biochem Pharmacol 31: 1407–1415, 1982. DOI: 10.1016/0006-2952(82)90036-3. [DOI] [PubMed] [Google Scholar]
- 104.Herchuelz A, Diaz-Horta O, Eylen F. Na/Ca exchange in function, growth, and demise of β-cells. Ann N Y Acad Sci 976: 315–324, 2002. DOI: 10.1111/j.1749-6632.2002.tb04754.x. [DOI] [PubMed] [Google Scholar]
- 105.Hering S, Berjukow S, Sokolov S, Marksteiner R, Weiß RG, Kraus R, Timin EN. Molecular determinants of inactivation in voltage-gated Ca2+ channels. J Physiol 528: 237–249, 2000. DOI: 10.1111/j.1469-7793.2000.t01-1-00237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hess P, Lansman JB, Tsien RW. Different modes of Ca channel gating behaviour favoured by dihydropyridine Ca agonists and antagonists. Nature 311: 538–544, 1984. DOI: 10.1038/311538a0. [DOI] [PubMed] [Google Scholar]
- 107.Hille B Ion Channels of Excitable Membranes (3rd ed). Sinauer, 2001. [Google Scholar]
- 108.Hiriart M, Matteson DR. Na channels and two types of Ca channels in rat pancreatic B cells identified with the reverse hemolytic plaque assay. J Gen Physiol 91: 617–639, 1988. DOI: 10.1085/jgp.91.5.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hodgkin AL, Huxley AF. Currents carried by sodium and potassium ions through the membrane of the giant axon of Loligo. J Physiol 116: 449–472, 1952. DOI: 10.1113/jphysiol.1952.sp004717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hodgkin AL, Huxley AF. The dual effect of membrane potential on sodium conductance in the giant axon of Loligo. J Physiol 116: 497–506, 1952. DOI: 10.1113/jphysiol.1952.sp004719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hondeghem LM, Katzung BG. Antiarrhythmic agents: The modulated receptor mechanism of action of sodium and calcium channel-blocking drugs. Annu Rev Pharmacol 24: 387–423, 1984. DOI: 10.1146/annurev.pa.24.040184.002131. [DOI] [PubMed] [Google Scholar]
- 112.Honrath B, Krabbendam IE, Culmsee C, Dolga AM. Small conductance Ca2+-activated K+ channels in the plasma membrane, mitochondria and the ER: Pharmacology and implications in neuronal diseases. Neurochem Int 109: 13–23, 2017. DOI: 10.1016/j.neuint.2017.05.005. [DOI] [PubMed] [Google Scholar]
- 113.Hopkins WF, Satin LS, Cook DL. Inactivation kinetics and pharmacology distinguish two calcium currents in mouse pancreatic B-cells. J Membr Biol 119: 229–239, 1991. DOI: 10.1007/bf01868728. [DOI] [PubMed] [Google Scholar]
- 114.Hoppa MB, Lana B, Margas W, Dolphin AC, Ryan TA. α2δ expression sets presynaptic calcium channel abundance and release probability. Nature 486: 122–125, 2012. DOI: 10.1038/nature11033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Horváth A, Szabadkai G, Várnai P, Arányi T, Wollheim CB, Spät A, Enyedi P. Voltage dependent calcium channels in adrenal glomerulosa cells and in insulin producing cells. Cell Calcium 23: 33–42, 1998. DOI: 10.1016/s0143-4160(98)90072-0. [DOI] [PubMed] [Google Scholar]
- 116.Houamed KM, Sweet IR, Satin LS. BK channels mediate a novel ionic mechanism that regulates glucose-dependent electrical activity and insulin secretion in mouse pancreatic β-cells. J Physiol 588: 3511–3523, 2010. DOI: 10.1113/jphysiol.2009.184341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hurst RS, Busch AE, Kavanaugh MP, Osborne PB, North RA, Adelman JP. Identification of amino acid residues involved in dendrotoxin block of rat voltage-dependent potassium channels. Mol Pharmacol 40: 572–576, 1991. [PubMed] [Google Scholar]
- 118.Hyzinski-García MC, Rudkouskaya A, Mongin AA. LRRC8A protein is indispensable for swelling-activated and ATP-induced release of excitatory amino acids in rat astrocytes. J Physiol 592: 4855–4862, 2014. DOI: 10.1113/jphysiol.2014.278887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ichii H, Inverardi L, Pileggi A, Molano RD, Cabrera O, Caicedo A, Messinger S, Kuroda Y, Berggren P, Ricordi CA. Novel method for the assessment of cellular composition and beta-cell viability in human islet preparations. Am J Transplant 5: 1635–1645, 2005. DOI: 10.1111/j.1600-6143.2005.00913.x. [DOI] [PubMed] [Google Scholar]
- 120.Ionescu-Tirgoviste C, Gagniuc PA, Gubceac E, Mardare L, Popescu I, Dima S, Militaru M. A 3D map of the islet routes throughout the healthy human pancreas. Sci Rep 5: 14634, 2015. DOI: 10.1038/srep14634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ishii M, Hagiwara T, Mori Y, Shimizu S. Involvement of TRPM2 and L-type Ca2+ channels in Ca2+ entry and cell death induced by hydrogen peroxide in rat β-cell line RIN-5F. J Toxicol Sci 39: 199–209, 2014. DOI: 10.2131/jts.39.199. [DOI] [PubMed] [Google Scholar]
- 122.Islam MdS. Molecular regulations and functions of the transient receptor potential channels of the islets of langerhans and insulinoma cells. Cell 9: 685, 2020. DOI: 10.3390/cells9030685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ivanina T, Blumenstein Y, Shistik E, Barzilai R, Dascal N. Modulation of L-type Ca2+ channels by Gβγ and calmodulin via Interactions with N and C termini of α1C. J Biol Chem 275: 39846–39854, 2000. DOI: 10.1074/jbc.m005881200. [DOI] [PubMed] [Google Scholar]
- 124.Iwanir S, Reuveny E. Adrenaline-induced hyperpolarization of mouse pancreatic islet cells is mediated by G protein-gated inwardly rectifying potassium (GIRK) channels. Pflugers Arch - Eur J Physiol 456: 1097–1108, 2008. DOI: 10.1007/s00424-008-0479-4. [DOI] [PubMed] [Google Scholar]
- 125.Jacobson D, Shyng S-L. Ion channels of the islets in type 2 diabetes. J Mol Biol 432: 1326–1346, 2019. DOI: 10.1016/j.jmb.2019.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Jacobson DA, Mendez F, Thompson M, Torres J, Cochet O, Philipson LH. Calcium-activated and voltage-gated potassium channels of the pancreatic islet impart distinct and complementary roles during secret-agogue induced electrical responses. J Physiol 588: 3525–3537, 2010. DOI: 10.1113/jphysiol.2010.190207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Jian X, Felsenfeld G. Insulin promoter in human pancreatic β cells contacts diabetes susceptibility loci and regulates genes affecting insulin metabolism. Proc National Acad Sci 115: 201803146, 2018. DOI: 10.1073/pnas.1803146115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Jing X, Li D-Q, Olofsson CS, Salehi A, Surve VV, Caballero J, Ivarsson R, Lundquist I, Pereverzev A, Schneider T, Rorsman P, Renström E. CaV2.3 calcium channels control second-phase insulin release. J Clin Invest 115: 146–154, 2005. DOI: 10.1172/jci22518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kaczorowski GJ, Knaus H-G, Leonard RJ, McManus OB, Garcia ML. High-conductance calcium-activated potassium channels; Structure, pharmacology, and function. J Bioenerg Biomembr 28: 255–267, 1996. DOI: 10.1007/bf02110699. [DOI] [PubMed] [Google Scholar]
- 130.Kakei M, Kelly RP, Ashcroft SJH, Ashcroft FM. The ATP-sensitivity of K+ channels in rat pancreatic B-cells is modulated by ADP. FEBS Lett 208: 63–66, 1986. DOI: 10.1016/0014-5793(86)81533-2. [DOI] [PubMed] [Google Scholar]
- 131.Kang C, Xie L, Gunasekar SK, Mishra A, Zhang Y, Pai S, Gao Y, Kumar A, Norris AW, Stephens SB, Sah R. SWELL1 is a glucose sensor regulating β-cell excitability and systemic glycaemia. Nat Commun 9: 367, 2018. DOI: 10.1038/s41467-017-02664-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kang D, Kim D. Single-channel properties and pH sensitivity of two-pore domain K+ channels of the TALK family. Biochem Bioph Res Co 315: 836–844, 2004. DOI: 10.1016/j.bbrc.2004.01.137. [DOI] [PubMed] [Google Scholar]
- 133.Kanno T, Rorsman P, Göpel SO. Glucose-dependent regulation of rhythmic action potential firing in pancreatic beta-cells by K(ATP)-channel modulation. J Physiol 545: 501–507, 2002. DOI: 10.1113/jphysiol.2002.031344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kashio M, Tominaga M. Redox signal-mediated enhancement of the temperature sensitivity of transient receptor potential melastatin 2 (TRPM2) elevates glucose-induced insulin secretion from pancreatic islets. J Biol Chem 290: 12435–12442, 2015. DOI: 10.1074/jbc.m115.649913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kennedy HJ, Pouli AE, Ainscow EK, Jouaville LS, Rizzuto R, Rutter GA. Glucose generates sub-plasma membrane ATP microdomains in single islet β-cells potential role for strategically located mitochondria. J Biol Chem 274: 13281–13291, 1999. DOI: 10.1074/jbc.274.19.13281. [DOI] [PubMed] [Google Scholar]
- 136.Khajavi N, Finan B, Kluth O, Müller TD, Mergler S, Schulz A, Kleinau G, Scheerer P, Schürmann A, Gudermann T, Tschöp MH, Krude H, DiMarchi RD, Biebermann H. An incretin-based tri-agonist promotes superior insulin secretion from murine pancreatic islets via PLC activation. Cell Signal 51: 13–22, 2018. DOI: 10.1016/j.cellsig.2018.07.006. [DOI] [PubMed] [Google Scholar]
- 137.Khan FA, Goforth PB, Zhang M, Satin LS. Insulin activates ATP-sensitive K+ channels in pancreatic -cells through a phosphatidylinositol 3-kinase-dependent pathway. Diabetes 50: 2192–2198, 2001. DOI: 10.2337/diabetes.50.10.2192. [DOI] [PubMed] [Google Scholar]
- 138.Kheradpezhouh E, Barritt GJ, Rychkov GY. Curcumin inhibits activation of TRPM2 channels in rat hepatocytes. Redox Biol 7: 1–7, 2016. DOI: 10.1016/j.redox.2015.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kim A, Miller K, Jo J, Kilimnik G, Wojcik P, Hara M. Islet architecture: A comparative study. Islets 1: 129–136, 2009. DOI: 10.4161/isl.1.2.9480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kim Y, Bang H, Kim D. TBAK-1 and TASK-1, two-pore K+ channel subunits: Kinetic properties and expression in rat heart. Am J Physiol-heart C 277: H1669–H1678, 1999. DOI: 10.1152/ajp-heart.1999.277.5.h1669. [DOI] [PubMed] [Google Scholar]
- 141.Kinard TA, Satin LS. An ATP-sensitive Cl− channel current that is activated by cell swelling, cAMP, and glyburide in insulin-secreting cells. Diabetes 44: 1461–1466, 1995. DOI: 10.2337/diab.44.12.1461. [DOI] [PubMed] [Google Scholar]
- 142.Kinard TA, Satin LS. Temperature modulates the Ca2+ current of HIT T15 and mouse pancreatic β-cells. Cell Calcium 20: 475–482, 1996. DOI: 10.1016/s0143-4160(96)90089-5. [DOI] [PubMed] [Google Scholar]
- 143.Kiyonaka S, Kato K, Nishida M, Mio K, Numaga T, Sawaguchi Y, Yoshida T, Wakamori M, Mori E, Numata T, Ishii M, Takemoto H, Ojida A, Watanabe K, Uemura A, Kurose H, Morii T, Kobayashi T, Sato Y, Sato C, Hamachi I, Mori Y. Selective and direct inhibition of TRPC3 channels underlies biological activities of a pyrazole compound. Proc National Acad Sci 106: 5400–5405, 2009. DOI: 10.1073/pnas.0808793106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kobayashi T, Washiyama K, Ikeda K. Inhibition of G-protein-activated inwardly rectifying K+ channels by the selective norepinephrine reup-take inhibitors atomoxetine and reboxetine. Neuropsychopharmacology 35: 1560–1569, 2010. DOI: 10.1038/npp.2010.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Komatsu M, Yokokawa N, Takeda T, Nagasawa Y, Aizawa T, Yamada T. Pharmacological characterization of the voltage-dependent calcium channel of pancreatic B-cell. Endocrinology 125: 2008–2014, 1989. DOI: 10.1210/endo-125-4-2008. [DOI] [PubMed] [Google Scholar]
- 146.König B, Stauber T. Biophysics and structure-function relationships of LRRC8-formed volume-regulated anion channels. Biophys J 116: 1185–1193, 2019. DOI: 10.1016/j.bpj.2019.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Koschak A, Reimer D, Huber I, Grabner M, Glossmann H, Engel J, Striessnig J. α1d (cav1.3) subunits can form l-type ca2+ channels activating at negative voltages. J Biol Chem 276: 22100–22106, 2001. DOI: 10.1074/jbc.m101469200. [DOI] [PubMed] [Google Scholar]
- 148.Kozak JA, Logothetis DE. A calcium-dependent chloride current in insulin-secreting βTC-3 cells. Pflugers Arch 433: 679–690, 1997. DOI: 10.1007/s004240050332. [DOI] [PubMed] [Google Scholar]
- 149.Krishnan V, Maddox JW, Rodriguez T, Gleason E. A role for the cystic fibrosis transmembrane conductance regulator in the nitric oxide-dependent release of Cl − from acidic organelles in amacrine cells. J Neurophysiol 118: 2842–2852, 2017. DOI: 10.1152/jn.00511.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kukuljan M, Goncalves AA, Atwater I. Charybdotoxin-sensitive K(Ca) channel is not involved in glucose-induced electrical activity in pancreatic β-cells. J Membr Biol 119: 187–195, 1991. DOI: 10.1007/bf01871418. [DOI] [PubMed] [Google Scholar]
- 151.Lange I, Yamamoto S, Partida-Sanchez S, Mori Y, Fleig A, Penner R. TRPM2 functions as a lysosomal Ca2+-release channel in β cells. Sci Signal 2: ra23–ra23, 2009. DOI: 10.1126/scisignal.2000278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Latorre R, Castillo K, Carrasquel-Ursulaez W, Sepulveda RV, Gonzalez-Nilo F, Gonzalez C, Alvarez O. Molecular determinants of BK channel functional diversity and functioning. Physiol Rev 97: 39–87, 2017. DOI: 10.1152/physrev.00001.2016. [DOI] [PubMed] [Google Scholar]
- 153.Lebrun P, Atwater I. Effects of the calcium channel agonist, BAY K 8644, on electrical activity in mouse pancreatic B-cells. Biophys J 48: 919–930, 1985. DOI: 10.1016/s0006-3495(85)83855-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lee J-H, Gomora JC, Cribbs LL, Perez-Reyes E. Nickel block of three cloned T-type calcium channels: Low concentrations selectively block α1H. Biophys J 77: 3034–3042, 1999. DOI: 10.1016/s0006-3495(99)77134-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Lee S, Yoon B-E, Berglund K, Oh S-J, Park H, Shin H-S, Augustine GJ, Lee CJ. Channel-mediated tonic GABA release from glia. Science 330: 790–796, 2010. DOI: 10.1126/science.1184334. [DOI] [PubMed] [Google Scholar]
- 156.Li C, Liu C, Nissim I, Chen J, Chen P, Doliba N, Zhang T, Nissim I, Daikhin Y, Stokes D, Yudkoff M, Bennett MJ, Stanley CA, Matschinsky FM, Naji A. Regulation of glucagon secretion in normal and diabetic human islets by γ-hydroxybutyrate and glycine. J Biol Chem 288: 3938–3951, 2013. DOI: 10.1074/jbc.m112.385682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Li N, Wu J-X, Ding D, Cheng J, Gao N, Chen L. Structure of a pancreatic ATP-sensitive potassium channel. Cell 168: 101–110.e10, 2017. DOI: 10.1016/j.cell.2016.12.028. [DOI] [PubMed] [Google Scholar]
- 158.Ligon B, Boyd AE, Dunlap K. Class a calcium channel variants in pancreatic islets and their role in insulin secretion. J Biol Chem 273: 13905–13911, 1998. DOI: 10.1074/jbc.273.22.13905. [DOI] [PubMed] [Google Scholar]
- 159.Lin M, Aladejebi O, Hockerman GH. Distinct properties of amlodipine and nicardipine block of the voltage-dependent Ca2+ channels Cav1.2 and Cav2.1 and the mutant channels Cav1.2/Dihydropyridine insensitive and Cav2.1/Dihydropyridine sensitive. Eur J Pharmacol 670: 105–113, 2011. DOI: 10.1016/j.ejphar.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Liu G, Hilliard N, Hockerman GH. Cav1.3 is preferentially coupled to glucose-induced [Ca2+]i oscillations in the pancreatic β Cell Line INS-1. Mol Pharmacol 65: 1269–1277, 2004. DOI: 10.1124/mol.65.5.1269. [DOI] [PubMed] [Google Scholar]
- 161.Llinás R, Sugimori M, Lin JW, Cherksey B. Blocking and isolation of a calcium channel from neurons in mammals and cephalopods utilizing a toxin fraction (FTX) from funnel-web spider poison. Proc National Acad Sci 86: 1689–1693, 1989. DOI: 10.1073/pnas.86.5.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lutter D, Ullrich F, Lueck JC, Kempa S, Jentsch TJ. Selective transport of neurotransmitters and –modulators by distinct volume-regulated LRRC8 anion channels. J Cell Sci 130: 1122–1133, 2017. DOI: 10.1242/jcs.196253. [DOI] [PubMed] [Google Scholar]
- 163.Lv W, Wang X, Xu Q, Lu W. Mechanisms and characteristics of sulfonylureas and glinides. Curr Top Med Chem 20: 37–56, 2019. DOI: 10.2174/1568026620666191224141617. [DOI] [PubMed] [Google Scholar]
- 164.MacDonald PE, Ha XF, Wang J, Smukler SR, Sun AM, Gaisano HY, Salapatek AMF, Backx PH, Wheeler MB. Members of the Kv1 and Kv2 voltage-dependent K+ channel families regulate insulin secretion. Mol Endocrinol 15: 1423–1435, 2001. DOI: 10.1210/mend.15.8.0685. [DOI] [PubMed] [Google Scholar]
- 165.MacDonald PE, Sewing S, Wang J, Joseph JW, Smukler SR, Sakellaropoulos G, Wang J, Saleh MC, Chan CB, Tsushima RG, Salapatek AMF, Wheeler MB. Inhibition of Kv2.1 voltage-dependent K+channels in pancreatic β-cells enhances glucose-dependent insulin secretion. J Biol Chem 277: 44938–44945, 2002. DOI: 10.1074/jbc.m205532200. [DOI] [PubMed] [Google Scholar]
- 166.Marabita F, MdS I. Expression of transient receptor potential channels in the purified human pancreatic &bgr;-cells. Pancreas 46: 97–101, 2017. DOI: 10.1097/mpa.0000000000000685. [DOI] [PubMed] [Google Scholar]
- 167.Marchetti C, Amico C, Podestà D, Robello M. Inactivation of voltage-dependent calcium current in an insulinoma cell line. Eur Biophys J 23: 51–58, 1994. DOI: 10.1007/bf00192205. [DOI] [PubMed] [Google Scholar]
- 168.Martin GM, Chen P-C, Devaraneni P, Shyng S-L. Pharmacological rescue of trafficking-impaired ATP-sensitive potassium channels. Front Physiol 4: 386, 2013. DOI: 10.3389/fphys.2013.00386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Martin GM, Yoshioka C, Rex EA, Fay JF, Xie Q, Whorton MR, Chen JZ, Shyng S-L. Cryo-EM structure of the ATP-sensitive potassium channel illuminates mechanisms of assembly and gating. elife 6: e24149, 2017. DOI: 10.7554/eLife.24149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Mastrolia V, Flucher SM, Obermair GJ, Drach M, Hofer H, Renström E, Schwartz A, Striessnig J, Flucher BE, Tuluc P. Loss of α 2 δ−1 calcium channel subunit function increases the susceptibility for diabetes. Diabetes 66: 897–907, 2017. DOI: 10.2337/db16-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.McCulloch LJ, van de Bunt M, Braun M, Frayn KN, Clark A, Gloyn AL. GLUT2 (SLC2A2) is not the principal glucose transporter in human pancreatic beta cells: Implications for understanding genetic association signals at this locus. Mol Genet Metab 104: 648–653, 2011. DOI: 10.1016/j.ymgme.2011.08.026. [DOI] [PubMed] [Google Scholar]
- 172.McHugh D, Flemming R, Xu S-Z, Perraud A-L, Beech DJ. Critical intracellular Ca2+ dependence of transient receptor potential melastatin 2 (TRPM2) cation channel activation. J Biol Chem 278: 11002–11006, 2003. DOI: 10.1074/jbc.m210810200. [DOI] [PubMed] [Google Scholar]
- 173.McKenna JP, Bertram R. Fast-slow analysis of the integrated oscillator model for pancreatic β-cells. J Theor Biol: 152-162, 2018. DOI: 10.1016/j.jtbi.2018.08.029. [DOI] [PubMed] [Google Scholar]
- 174.Meissner H, Atwater I. The kinetics of electrical activity of beta cells in response to a “square wave” stimulation with glucose or glibenclamide. Horm Metab Res 8: 11–16, 1976. DOI: 10.1055/s-0028-1093685. [DOI] [PubMed] [Google Scholar]
- 175.Merrins MJ, Bertram R, Sherman A, Satin LS. Phosphofructo-2-kinase/fructose-2,6-bisphosphatase modulates oscillations of pancreatic islet metabolism. PLoS ONE 7: e34036, 2012. DOI: 10.1371/journal.pone.0034036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Merrins MJ, Dyke AR, Mapp AK, Rizzo MA, Satin LS. Direct measurements of oscillatory glycolysis in pancreatic islet β-cells using novel fluorescence resonance energy transfer (FRET) biosensors for pyruvate kinase m2 activity. J Biol Chem 288: 33312–33322, 2013. DOI: 10.1074/jbc.M113.508127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Miki T, Nagashima K, Tashiro F, Kotake K, Yoshitomi H, Tamamoto A, Gonoi T, Iwanaga T, Miyazaki J, Seino S. Defective insulin secretion and enhanced insulin action in KATP channel-deficient mice. Proc Natl Acad Sci 95: 10402–10406, 1998. DOI: 10.1073/pnas.95.18.10402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Miley HE, Holden D, Grint R, Best L, Brown PD. Regulatory volume increase in rat pancreatic β-cells. Pflugers Arch 435: 227–230, 1997. DOI: 10.1007/s004240050505. [DOI] [PubMed] [Google Scholar]
- 179.Miley HE, Sheader EA, Brown PD, Best L. Glucose-induced swelling in rat pancreatic β-cells. J Physiol 504: 191–198, 1997. DOI: 10.1111/j.1469-7793.1997.00191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Miller AN, Long SB. Crystal structure of the human two–pore domain potassium channel K2P1. Science 335: 432–436, 2012. DOI: 10.1126/science.1213274. [DOI] [PubMed] [Google Scholar]
- 181.Miller M, Shi J, Zhu Y, Kustov M, Tian J, Stevens A, Wu M, Xu J, Long S, Yang P, Zholos AV, Salovich JM, Weaver CD, Hopkins CR, Lindsley CW, McManus O, Li M, Zhu MX. Identification of ML204, a novel potent antagonist that selectively modulates native TRPC4/C5 ion channels. J Biol Chem 286: 33436–33446, 2011. DOI: 10.1074/jbc.m111.274167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Minke B The history of the Drosophila TRP channel: The birth of a new channel superfamily. J Neurogenet 24: 216–233, 2010. DOI: 10.3109/01677063.2010.514369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Misler S, Barnett DW, Gillis KD, Pressel DM. Electrophysiology of stimulus-secretion coupling in human-cells. Diabetes 41: 1221–1228, 1992. DOI: 10.2337/diab.41.10.1221. [DOI] [PubMed] [Google Scholar]
- 184.Misler S, Barnett DW, Pressel DM, Gillis KD, Scharp DW, Falke LC. Stimulus-secretion coupling in -cells of transplantable human islets of langerhans: Evidence for a critical role for Ca2+ entry. Diabetes 41: 662–670, 1992. DOI: 10.2337/diab.41.6.662. [DOI] [PubMed] [Google Scholar]
- 185.Morales FR, Silveira V, Damián A, Higgie R, Pose I. The possible additional role of the cystic fibrosis transmembrane regulator to motoneuron inhibition produced by glycine effects. Neuroscience 177: 138–147, 2011. DOI: 10.1016/j.neuroscience.2010.12.034. [DOI] [PubMed] [Google Scholar]
- 186.Namkung Y, Skrypnyk N, Jeong M-J, Lee T, Lee M-S, Kim H-L, Chin H, Suh P-G, Kim S-S, Shin H-S. Requirement for the L-type Ca2+ channel α1D subunit in postnatal pancreatic β cell generation. J Clin Invest 108: 1015–1022, 2001. DOI: 10.1172/jci13310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Nardi A, Olesen S-P. BK Channel modulators: A comprehensive overview. Curr Med Chem 15: 1126, 1146, 2008. DOI: 10.2174/092986708784221412. [DOI] [PubMed] [Google Scholar]
- 188.Newcomb R, Szoke B, Palma A, Wang G, Chen X, Hopkins W, Cong R, Miller J, Urge L, Tarczy-Hornoch K, Loo JA, Dooley DJ, Nadasdi L, Tsien RW, Lemos J, Miljanich G. Selective peptide antagonist of the class E calcium channel from the venom of the tarantula hysterocrates gigas †. Biochemistry 37: 15353–15362, 1998. DOI: 10.1021/bi981255g. [DOI] [PubMed] [Google Scholar]
- 189.Nica AC, Ongen H, Irminger J-C, Bosco D, Berney T, Antonarakis SE, Halban PA, Dermitzakis ET. Cell-type, allelic, and genetic signatures in the human pancreatic beta cell transcriptome. Genome Res 23: 1554–1562, 2013. DOI: 10.1101/gr.150706.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Nichols CG. KATP channels as molecular sensors of cellular metabolism. Nature 440: 470–476, 2006. DOI: 10.1038/nature04711. [DOI] [PubMed] [Google Scholar]
- 191.Nichols CG, Lopatin AN. Inward rectifier potassium channels. Annu Rev Physiol 59: 171–191, 1997. DOI: 10.1146/annurev.physiol.59.1.171. [DOI] [PubMed] [Google Scholar]
- 192.Nowycky MC, Fox AP, Tsien RW. Three types of neuronal calcium channel with different calcium agonist sensitivity. Nature 316: 440–443, 1985. DOI: 10.1038/316440a0. [DOI] [PubMed] [Google Scholar]
- 193.Nunemaker CS, Bertram R, Sherman A, Tsaneva-Atanasova K, Daniel CR, Satin LS. Glucose modulates [Ca2+]i oscillations in pancreatic islets via ionic and glycolytic mechanisms. Biophys J 91: 2082–2096, 2006. DOI: 10.1529/biophysj.106.087296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Ohta M, Nelson J, Nelson D, Meglasson MD, Erecińska M. Effect of Ca++ channel blockers on energy level and stimulated insulin secretion in isolated rat islets of Langerhans. J Pharmacol Exp Ther 264: 35–40, 1993. [PubMed] [Google Scholar]
- 195.Olivera BM, McIntosh JM, Curz LJ, Luque FA, Gray WR. Purification and sequence of a presynaptic peptide toxin from Conus geographus venom. Biochemistry 23: 5087–5090, 1984. DOI: 10.1021/bi00317a001. [DOI] [PubMed] [Google Scholar]
- 196.Olivier AK, Yi Y, Sun X, Sui H, Liang B, Hu S, Xie W, Fisher JT, Keiser NW, Lei D, Zhou W, Yan Z, Li G, Evans TIA, Meyerholz DK, Wang K, Stewart ZA, Norris AW, Engelhardt JF. Abnormal endocrine pancreas function at birth in cystic fibrosis ferrets. J Clin Invest 122: 3755–3768, 2012. DOI: 10.1172/jci60610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Olofsson CS, Göpel SO, Barg S, Galvanovskis J, Ma X, Salehi A, Rorsman P, Eliasson L. Fast insulin secretion reflects exocytosis of docked granules in mouse pancreatic B-cells. Pflugers Arch 444: 43–51, 2002. DOI: 10.1007/s00424-002-0781-5. [DOI] [PubMed] [Google Scholar]
- 198.Orci L, Baetens D, Ravazzola M, Stefan Y, Malaisse-Lagae F. Pancreatic polypeptide and glucagon: Non-random distribution in pancreatic islets. Life Sci 19: 1811–1816, 1976. DOI: 10.1016/0024-3205(76)90112-0. [DOI] [PubMed] [Google Scholar]
- 199.Ostroumov A, Simonetti M, Nistri A. Cystic fibrosis transmembrane conductance regulator modulates synaptic chloride homeostasis in motoneurons of the rat spinal cord during neonatal development. Dev Neurobiol 71: 253–268, 2011. DOI: 10.1002/dneu.20855. [DOI] [PubMed] [Google Scholar]
- 200.Palmer KR, Atwal K, Bakaj I, Carlucci-Derbyshire S, Buber TM, Cerne R, Cortés RY, Devantier HR, Jorgensen V, Pawlyk A, Lee PS, Sprous DG, Zhang Z, Bryant R. Triphenylphosphine oxide is a potent and selective inhibitor of the transient receptor potential melastatin-5 ion channel. Assay Drug Dev Technol 8: 703–713, 2010. DOI: 10.1089/adt.2010.0334. [DOI] [PubMed] [Google Scholar]
- 201.Pang B, Kim S, Li D, Ma Z, Sun B, Zhang X, Wu Z, Chen L. Glucagon-like peptide-1 potentiates glucose-stimulated insulin secretion via the transient receptor potential melastatin 2 channel. Exp Ther Med 14: 5219–5227, 2017. DOI: 10.3892/etm.2017.5136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Park S-H, Ryu S-Y, Yu W-J, Han Y, Ji Y-S, Oh K, Sohn J-W, Lim A, Jeon J-P, Lee H, Lee K-H, Lee S-H, Berggren P-O, Jeon J-H, Ho WK. Leptin promotes KATP channel trafficking by AMPK signaling in pancreatic β-cells. Proc Natl Acad Sci 110: 12673–12678, 2013. DOI: 10.1073/pnas.1216351110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Payandeh J, Scheuer T, Zheng N, Catterall WA. The crystal structure of a voltage-gated sodium channel. Nature 475: 353–358, 2011. DOI: 10.1038/nature10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Pedemonte N, Galietta LJV. Structure and function of TMEM16 proteins (Anoctamins). Physiol Rev 94: 419–459, 2014. DOI: 10.1152/physrev.00039.2011. [DOI] [PubMed] [Google Scholar]
- 205.Pennefather P, Lancaster B, Adams PR, Nicoll RA. Two distinct Ca-dependent K currents in bullfrog sympathetic ganglion cells. Proc National Acad Sci 82: 3040–3044, 1985. DOI: 10.1073/pnas.82.9.3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Pereverzev A, Mikhna M, Vajna R, Gissel C, Henry M, Weiergräber M, Hescheler J, Smyth N, Schneider T. Disturbances in glucose-tolerance, insulin-release, and stress-induced hyperglycemia upon disruption of the Ca v 2.3 (α1E) subunit of voltage-gated Ca 2+ channels. Mol Endocrinol 16: 884–895, 2002. DOI: 10.1210/mend.16.4.0801. [DOI] [PubMed] [Google Scholar]
- 207.Pereverzev A, Vajna R, Pfitzer G, Hescheler J, Klockner U, Schneider T. Reduction of insulin secretion in the insulinoma cell line INS-1 by overexpression of a Ca(v)2.3 (alpha1E) calcium channel antisense cassette. Eur J Endocrinol 146: 881–889, 2002. DOI: 10.1530/eje.0.1460881. [DOI] [PubMed] [Google Scholar]
- 208.Perez-Reyes E, Cribbs LL, Daud A, Lacerda AE, Barclay J, Williamson MP, Fox M, Rees M, Lee J-H. Molecular characterization of a neuronal low-voltage-activated T-type calcium channel. Nature 391: 896–900, 1998. DOI: 10.1038/36110. [DOI] [PubMed] [Google Scholar]
- 209.Philippaert K, Pironet A, Mesuere M, Sones W, Vermeiren L, Kerselaers S, Pinto S, Segal A, Antoine N, Gysemans C, Laureys J, Lemaire K, Gilon P, Cuypers E, Tytgat J, Mathieu C, Schuit F, Rorsman P, Talavera K, Voets T, Vennekens R. Steviol glycosides enhance pancreatic beta-cell function and taste sensation by potentiation of TRPM5 channel activity. Nat Commun 8: 14733, 2017. DOI: 10.1038/ncomms14733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Philipson LH, Rosenberg MP, Kuznetsov A, Lancaster ME, Worley JF, Roe MW, Dukes ID. Delayed rectifier K+ channel overexpression in transgenic islets and beta-cells associated with impaired glucose responsiveness. J Biol Chem 269: 27787–27790, 1994. [PubMed] [Google Scholar]
- 211.Pipeleers D, Kiekens R, Veld PI. Morphology of the pancreatic β-cell. In: Ashcroft FM, Ashcroft SJH, editors. Insulin: Molecular Biology to Pathology. IRL Press at Oxford University Press, 1992, p. 5–31. [Google Scholar]
- 212.Plant TD. Properties and calcium-dependent inactivation of calcium currents in cultured mouse pancreatic B-cells. J Physiol 404: 731–747, 1988. DOI: 10.1113/jphysiol.1988.sp017316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Plant TD. Na+ currents in cultured mouse pancreatic B-cells. Pflugers Arch 411: 429–435, 1988. DOI: 10.1007/bf00587723. [DOI] [PubMed] [Google Scholar]
- 214.Pollo A, Lovallo M, Biancardi E, Sher E, Socci C, Carbone E. Sensitivity to dihydropyridines, ω-conotoxin and noradrenaline reveals multiple high-voltage-activated Ca2+ channels in rat insulinoma and human pancreatic β-cells. Pflugers Arch 423: 462–471, 1993. DOI: 10.1007/bf00374942. [DOI] [PubMed] [Google Scholar]
- 215.Qin N, Neeper MP, Liu Y, Hutchinson TL, Lubin ML, Flores CM. TRPV2 is activated by cannabidiol and mediates CGRP release in cultured rat dorsal root ganglion neurons. J Neurosci 28: 6231–6238, 2008. DOI: 10.1523/jneurosci.0504-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Ramanadham S, Turk J. ω-Conotoxin inhibits glucose- and arachidonic acid-induced rises in intracellular [Ca2+] in rat pancreatic islet β-cells. Cell Calcium 15: 259–264, 1994. DOI: 10.1016/0143-4160(94)90065-5. [DOI] [PubMed] [Google Scholar]
- 217.Randall A, Tsien R. Pharmacological dissection of multiple types of Ca2+ channel currents in rat cerebellar granule neurons. J Neurosci 15: 2995–3012, 1995. DOI: 10.1523/jneurosci.15-04-02995.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Remedi MS, Friedman JB, Nichols CG. Diabetes induced by gain-of-function mutations in the Kir6.1 subunit of the KATP channel. J Gen Physiol 149: jgp.201611653, 2016. DOI: 10.1085/jgp.201611653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Ren J, Sherman A, Bertram R, Goforth PB, Nunemaker CS, Waters CD, Satin LS. Slow oscillations of KATP conductance in mouse pancreatic islets provide support for electrical bursting driven by metabolic oscillations. Am J Physiol Endocrinol Metab 305: E805–E817, 2013. DOI: 10.1152/ajpendo.00046.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Ribalet B, Beigelman PM. Calcium action potentials and potassium permeability activation in pancreatic beta-cells. Am J Physiol-cell Ph 239: C124–C133, 1980. DOI: 10.1152/ajpcell.1980.239.3.c124. [DOI] [PubMed] [Google Scholar]
- 221.Riz M, Braun M, Wu X, Pedersen MG. Inwardly rectifying Kir2.1 currents in human β-cells control electrical activity: Characterisation and mathematical modelling. Biochem Biophys Res Commun 459: 284–287, 2015. DOI: 10.1016/j.bbrc.2015.02.099. [DOI] [PubMed] [Google Scholar]
- 222.Roe MWM, Worley JF, Mittal AA, Kuznetsov A, DasGupta S, Mertz RJ, Witherspoon SM, Blair N, Lancaster ME, McIntyre MS, Shehee WR, Dukes ID, Philipson LH. Expression and function of pancreatic β-cell delayed rectifier k+ channels role in stimulus-secretion coupling. J Biol Chem 271: 32241–32246, 1996. DOI: 10.1074/jbc.271.50.32241. [DOI] [PubMed] [Google Scholar]
- 223.Rorsman P Two types of Ca2+ currents with different sensitivities to organic Ca2+ channel antagonists in guinea pig pancreatic alpha 2 cells. J Gen Physiol 91: 243–254, 1988. DOI: 10.1085/jgp.91.2.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Rorsman P, Ashcroft FM. Pancreatic β-cell electrical activity and insulin secretion: Of mice and men. Physiol Rev 98: 117–214, 2018. DOI: 10.1152/physrev.00008.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Rorsman P, Ashcroft FM, Trube G. Single Ca channel currents in mouse pancreatic B-cells. Pflugers Arch 412: 597–603, 1988. DOI: 10.1007/bf00583760. [DOI] [PubMed] [Google Scholar]
- 226.Rorsman P, Bokvist K, Ämmälä C, Arkhammar P, Berggren P-O, Lars-son O, Wåhlander K. Activation by adrenaline of a low-conductance G protein-dependent K+ channel in mouse pancreatic B cells. Nature 349: 77–79, 1991. DOI: 10.1038/349077a0. [DOI] [PubMed] [Google Scholar]
- 227.Rorsman P, Braun M. Regulation of insulin secretion in human pancreatic islets. Physiology 75: 155–179, 2013. DOI: 10.1146/annurevphysiol-030212-183754. [DOI] [PubMed] [Google Scholar]
- 228.Rorsman P, Eliasson L, Kanno T, Zhang Q, Gopel S. Electrophysiology of pancreatic β-cells in intact mouse islets of Langerhans. Prog Biophys Mol Biol 107: 224–235, 2011. DOI: 10.1016/j.pbiomolbio.2011.06.009. [DOI] [PubMed] [Google Scholar]
- 229.Rorsman P, Huising MO. The somatostatin-secreting pancreatic δ-cell in health and disease. Nat Rev Endocrinol 14: 404–414, 2018. DOI: 10.1038/s41574-018-0020-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Rorsman P, Renström E. Insulin granule dynamics in pancreatic beta cells. Diabetologia 46: 1029–1045, 2003. DOI: 10.1007/s00125-003-1153-1. [DOI] [PubMed] [Google Scholar]
- 231.Rorsman P, Trube G. Glucose dependent K+-channels in pancreaticβ-cells are regulated by intracellular ATP. Pflugers Arch 405: 305–309, 1985. DOI: 10.1007/bf00595682. [DOI] [PubMed] [Google Scholar]
- 232.Rorsman P, Trube G. Calcium and delayed potassium currents in mouse pancreatic beta-cells under voltage-clamp conditions. J Physiol 374: 531–550, 1986. DOI: 10.1113/jphysiol.1986.sp016096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Ruppersberg JP, Fakler B. Complexity of the regulation of Kir2.1 K+ channels. Neuropharmacology 35: 887–893, 1996. DOI: 10.1016/0028-3908(96)00092-5. [DOI] [PubMed] [Google Scholar]
- 234.Sabourin J, Gal L, Saurwein L, Haefliger J-AA, Raddatz E, Allagnat F. Store-operated Ca2+ entry mediated by Orai1 and TRPC1 participates to insulin secretion in rat β-cells. J Biol Chem 290: 30530–30539, 2015. DOI: 10.1074/jbc.M115.682583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Samols E, Marri G, Marks V. Promotion of insulin secretion by glucagon. Lancet 286: 415–416, 1965. DOI: 10.1016/s0140-6736(65)90761-0. [DOI] [PubMed] [Google Scholar]
- 236.Sather WA, McCleskey EW. Permeation and selectivity in calcium channels. Annu Rev Physiol 65: 133–159, 2003. DOI: 10.1146/annurev.physiol.65.092101.142345. [DOI] [PubMed] [Google Scholar]
- 237.Satin LS. Localized calcium influx in pancreatic β-cells. Endocrine 13: 251–262, 2000. DOI: 10.1385/endo:13:3:251. [DOI] [PubMed] [Google Scholar]
- 238.Satin LS, Butler PC, Ha J, Sherman AS. Pulsatile insulin secretion, impaired glucose tolerance and type 2 diabetes. Mol Asp Med 42: 61–77, 2015. DOI: 10.1016/j.mam.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Satin LS, Cook DL. Voltage-gated Ca2+ current in pancreatic B-cells. Pflugers Arch 404: 385–387, 1985. DOI: 10.1007/bf00585354. [DOI] [PubMed] [Google Scholar]
- 240.Satin LS, Cook DL. Evidence for two calcium currents in insulin-secreting cells. Pflugers Arch 411: 401–409, 1988. DOI: 10.1007/bf00587719. [DOI] [PubMed] [Google Scholar]
- 241.Satin LS, Cook DL. Calcium current inactivation in insulin-secreting cells is mediated by calcium influx and membrane depolarization. Pflugers Arch 414: 1–10, 1989. DOI: 10.1007/bf00585619. [DOI] [PubMed] [Google Scholar]
- 242.Satin LS, Kinard TA. Neurotransmitters and their receptors in the islets of langerhans of the pancreas. Endocrine 8: 213–223, 1998. DOI: 10.1385/endo:8:3:213. [DOI] [PubMed] [Google Scholar]
- 243.Satin LS, Tavalin SJ, Kinard TA, Teague J. Contribution of L- and non-L-type calcium channels to voltage-gated calcium current and glucose-dependent insulin secretion in HIT-T15 cells. Endocrinology 136: 4589–4601, 1995. DOI: 10.1210/endo.136.10.7545106. [DOI] [PubMed] [Google Scholar]
- 244.Satin LS, Watts M, Sherman AS. An introduction to beta cell electrophysiology and modeling. In: Khadra A, editor. Diabetes Systems Biology. IOP Publishing, 2020, p. 2–1 to 2–46. [Google Scholar]
- 245.Sawatani T, Kaneko Y, Doutsu I, Ogawa A, Ishikawa T. TRPV2 channels mediate cell swelling-induced insulin secretion in mouse pancreatic β-cells. Proc Annu Meet Jpn Pharmacol Soc WCP2018: PO2–7–33, 2020. DOI: 10.1254/jpssuppl.wcp2018.0_po2-7-33. [DOI] [Google Scholar]
- 246.Sawatani T, Kaneko YK, Doutsu I, Ogawa A, Ishikawa T. TRPV2 channels mediate insulin secretion induced by cell swelling in mouse pancreatic β-cells. Am J Physiol-cell Ph 316: C434–C443, 2019. DOI: 10.1152/ajpcell.00210.2017. [DOI] [PubMed] [Google Scholar]
- 247.Schmidt A, Hescheler J, Offermanns S, Spicher K, Hinsch KD, Klinz FJ, Codina J, Birnbaumer L, Gausepohl H, Frank R. Involvement of pertussis toxin-sensitive G-proteins in the hormonal inhibition of dihydropyridine-sensitive Ca2+ currents in an insulin-secreting cell line (RINm5F). J Biol Chem 266: 18025–18033, 1991. [PubMed] [Google Scholar]
- 248.Schneider MJ, Rogowski RS, Krueger BK, Blaustein MP. Charybdotoxin blocks both Ca-activated K channels and Ca-independent voltage-gated K channels in rat brain synaptosomes. FEBS Lett 250: 433–436, 1989. DOI: 10.1016/0014-5793(89)80771-9. [DOI] [PubMed] [Google Scholar]
- 249.Schober AL, Wilson CS, Mongin AA. Molecular composition and heterogeneity of the LRRC8-containing swelling-activated osmolyte channels in primary rat astrocytes. J Physiol 595: 6939–6951, 2017. DOI: 10.1113/jp275053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Schuit FC, Derde M-P, Pipeleers DG. Sensitivity of rat pancreatic A and B cells to somatostatin. Diabetologia 32: 207–212, 1989. DOI: 10.1007/bf00265096. [DOI] [PubMed] [Google Scholar]
- 251.Schulla V, Renström E, Feil R, Feil S, Franklin I, Gjinovci A, Jing X, Laux D, Lundquist I, Magnuson MA, Obermüller S, Olofsson CS, Salehi A, Wendt A, Klugbauer N, Wollheim CB, Rorsman P, Hofmann F. Impaired insulin secretion and glucose tolerance in β cell-selective CaV1.2 Ca2+ channel null mice. EMBO J 22: 3844–3854, 2003. DOI: 10.1093/emboj/cdg389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Segerstolpe Å, Palasantza A, Eliasson P, Andersson E-M, Andréasson A-C, Sun X, Picelli S, Sabirsh A, Clausen M, Bjursell MK, Smith DM, Kasper M, Ämmälä C, Sandberg R. Single-cell transcriptome profiling of human pancreatic islets in health and type 2 diabetes. Cell Metab 24: 593–607, 2016. DOI: 10.1016/j.cmet.2016.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Seghers V, Nakazaki M, DeMayo F, Aguilar-Bryan L, Bryan J. Sur1 knockout mice. A model for K(ATP) channel-independent regulation of insulin secretion. J Biol Chem 275: 9270–9277, 2000. [DOI] [PubMed] [Google Scholar]
- 254.Sehlin J Interrelationship between chloride fluxes in pancreatic islets and insulin release. Am J Physiol-endoc M 235: E501, 1978. DOI: 10.1152/ajpendo.1978.235.5.e501. [DOI] [PubMed] [Google Scholar]
- 255.Sehlin J, Meissner HP. Effects of Cl− deficiency on the membrane potential in mouse pancreatic β-cells. Biochimica Et Biophysica Acta Bba - Biomembr 937: 309–318, 1988. DOI: 10.1016/0005-2736(88)90253-2. [DOI] [PubMed] [Google Scholar]
- 256.Seino S, Iwanaga T, Nagashima K, Miki T. Diverse roles of K(ATP) channels learned from Kir6.2 genetically engineered mice. Diabetes 49: 311–318, 2000. DOI: 10.2337/diabetes.49.3.311. [DOI] [PubMed] [Google Scholar]
- 257.Sharma S, Hopkins CR. Review of transient receptor potential canonical (TRPC5) channel modulators and diseases: Miniperspective. J Med Chem 62: 7589–7602, 2019. DOI: 10.1021/acs.jmedchem.8b01954. [DOI] [PubMed] [Google Scholar]
- 258.Sher E, Giovannini F, Codignola A, Passafaro M, Giorgi-Rossi P, Volsen S, Craig P, Davalli A, Carrera P. Voltage-operated calcium channel heterogeneity in pancreatic cells: Physiopathological implications. J Bioenerg Biomembr 35: 687–696, 2003. DOI: 10.1023/b:jobb.0000008032.49504.48. [DOI] [PubMed] [Google Scholar]
- 259.Shigeto M, Ramracheya R, Tarasov AI, Cha CY, Chibalina MV, Hastoy B, Philippaert K, Reinbothe T, Rorsman N, Salehi A, Sones WR, Vergari E, Weston C, Gorelik J, Katsura M, Nikolaev VO, Vennekens R, Zaccolo M, Galione A, Johnson PR, Kaku K, Ladds G, Rorsman P. GLP-1 stimulates insulin secretion by PKC-dependent TRPM4 and TRPM5 activation. J Clin Invest 125: 4714–4728, 2015. DOI: 10.1172/JCI81975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Shyng S-L, Nichols CG. Octameric stoichiometry of the KATP channel complex. J Gen Physiol 110: 655–664, 1997. DOI: 10.1085/jgp.110.6.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Sinnegger-Brauns MJ, Hetzenauer A, Huber IG, Renström E, Wietzorrek G, Berjukov S, Cavalli M, Walter D, Koschak A, Waldschütz R, Hering S, Bova S, Rorsman P, Pongs O, Singewald N, Striessnig J. Isoform-specific regulation of mood behavior and pancreatic β cell and cardiovascular function by L-type Ca2+ channels. J Clin Invest 113: 1430–1439, 2004. DOI: 10.1172/jci20208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Skrzypski M, Khajavi N, Mergler S, Billert M, Szczepankiewicz D, Wojciechowicz T, Nowak KW, Strowski MZ. Orexin A modulates INS-1E cell proliferation and insulin secretion via extracellular signal-regulated kinase and transient receptor potential channels. J Physiology Pharmacol Official J Pol Physiological Soc 67: 643–652, 2016. [PubMed] [Google Scholar]
- 263.Somers G, Somer A, Devis G, Malaisse WJ. The stimulus-secretion coupling of glucose-induced insulin release. Pflugers Arch 388: 249–253, 1980. DOI: 10.1007/bf00658490. [DOI] [PubMed] [Google Scholar]
- 264.Srivastava S, Li Z, Soomro I, Sun Y, Wang J, Bao L, Coetzee WA, Stanley CA, Li C, Skolnik EY. Regulation of KATP channel trafficking in pancreatic β cells by protein histidine phosphorylation. Diabetes 67: db171433, 2018. DOI: 10.2337/db17-1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Steiner DJ, Kim A, Miller K, Hara M. Pancreatic islet plasticity: Inter-species comparison of islet architecture and composition. Islets 2: 135–145, 2010. DOI: 10.4161/isl.2.3.11815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Strowski M, Parmar R, Blake A, Schaeffer J. Somatostatin inhibits insulin and glucagon secretion via two receptors subtypes: An in vitro study of pancreatic islets from somatostatin receptor 2 knockout mice. Endocrinology 141: 111–117, 2000. DOI: 10.1210/endo.141.1.7263. [DOI] [PubMed] [Google Scholar]
- 267.Strübing C, Krapivinsky G, Krapivinsky L, Clapham DE. TRPC1 and TRPC5 form a novel cation channel in mammalian brain. Neuron 29: 645–655, 2001. DOI: 10.1016/s0896-6273(01)00240-9. [DOI] [PubMed] [Google Scholar]
- 268.Stuhlmann T, Planells-Cases R, Jentsch TJ. LRRC8/VRAC anion channels enhance β-cell glucose sensing and insulin secretion. Nat Commun 9: 1974, 2018. DOI: 10.1038/s41467-018-04353-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Sussman KE, Vaughan GD. Insulin release after ACTH, glucagon and adenosine-3′−5′-phosphate (Cyclic AMP) in the perfused isolated rat pancreas. Diabetes 16: 449–454, 1967. DOI: 10.2337/diab.16.7.449. [DOI] [PubMed] [Google Scholar]
- 270.Swayne LA, Mezghrani A, Lory P, Nargeot J, Monteil A. The NALCN ion channel is a new actor in pancreatic β-cell physiology. Islets 2: 54–56, 2010. DOI: 10.4161/isl.2.1.10522. [DOI] [PubMed] [Google Scholar]
- 271.Talavera K, Staes M, Janssens A, Klugbauer N, Droogmans G, Hofmann F, Nilius B. Aspartate residues of the Glu-Glu-Asp-Asp (EEDD) pore locus control selectivity and permeation of the t-type Ca2+channel α1G. J Biol Chem 276: 45628–45635, 2001. DOI: 10.1074/jbc.m103047200. [DOI] [PubMed] [Google Scholar]
- 272.Tamagawa T, Henquin J-C. Chloride modulation of insulin release, 86Rb+ Efflux, and 45Ca2+ fluxes in rat islets stimulated by various secretagogues. Diabetes 32: 416–423, 1983. DOI: 10.2337/diab.32.5.416. [DOI] [PubMed] [Google Scholar]
- 273.Tamarina NA, Wang Y, Mariotto L, Kuznetsov A, Bond C, Adelman J, Philipson LH. Small-conductance calcium-activated K+ channels are expressed in pancreatic islets and regulate glucose responses. Diabetes 52: 2000–2006, 2003. DOI: 10.2337/diabetes.52.8.2000. [DOI] [PubMed] [Google Scholar]
- 274.Taneera J, Jin Z, Jin Y, Muhammed SJ, Zhang E, Lang S, Salehi A, Korsgren O, Renström E, Groop L, Birnir B. γ-Aminobutyric acid (GABA) signalling in human pancreatic islets is altered in type 2 diabetes. Diabetologia 55: 1985–1994, 2012. DOI: 10.1007/s00125-012-2548-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Tang C, Presser F, Morad M. Amiloride selectively blocks the low threshold (T) calcium channel. Science 240: 213–215, 1988. DOI: 10.1126/science.2451291. [DOI] [PubMed] [Google Scholar]
- 276.Taniguchi H, Okada Y, Shimada C, Baba S. GABA in pancreatic islets. Arch Histol Japon 40: 87–97, 1977. DOI: 10.1679/aohc1950.40.supplement_87. [DOI] [PubMed] [Google Scholar]
- 277.Thomas P, Cote G, Wohllk N, Haddad B, Mathew P, Rabl W, Aguilar-Bryan L, Gagel R, Bryan J. Mutations in the sulfonylurea receptor gene in familial persistent hyperinsulinemic hypoglycemia of infancy. Science 268: 426–429, 1995. DOI: 10.1126/science.7716548. [DOI] [PubMed] [Google Scholar]
- 278.Thorens B GLUT2, glucose sensing and glucose homeostasis. Diabetologia 58: 221–232, 2015. DOI: 10.1007/s00125-014-3451-1. [DOI] [PubMed] [Google Scholar]
- 279.Tian G, Sandler S, Gylfe E, Tengholm A. Glucose- and hormone-induced cAMP oscillations in α- and β-cells within intact pancreatic islets. Diabetes 60: 1535–1543, 2011. DOI: 10.2337/db10-1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Togashi K, Hara Y, Tominaga T, Higashi T, Konishi Y, Mori Y, Tominaga M. TRPM2 activation by cyclic ADP-ribose at body temperature is involved in insulin secretion. EMBO J 25: 1804–1815, 2006. DOI: 10.1038/sj.emboj.7601083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Trube G, Rorsman P, Ohno-Shosaku T. Opposite effects of tolbutamide and diazoxide on the ATP-dependent K+ channel in mouse pancreatic β-cells. Pflugers Arch 407: 493–499, 1986. DOI: 10.1007/bf00657506. [DOI] [PubMed] [Google Scholar]
- 282.Tsien RW, Lipscombe D, Madison DV, Bley KR, Fox AP. Multiple types of neuronal calcium channels and their selective modulation. Trends Neurosci 11: 431–438, 1988. DOI: 10.1016/0166-2236(88)90194-4. [DOI] [PubMed] [Google Scholar]
- 283.Uchida K, Dezaki K, Damdindorj B, Inada H, Shiuchi T, Mori Y, Yada T, Minokoshi Y, Tominaga M. Lack of TRPM2 impaired insulin secretion and glucose metabolisms in mice. Diabetes 60: 119–126, 2011. DOI: 10.2337/db10-0276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Uchida K, Tominaga M. The role of thermosensitive TRP (transient receptor potential) channels in insulin secretion [Review]. Endocr J 58: 1021–1028, 2011. DOI: 10.1507/endocrj.ej11-0130. [DOI] [PubMed] [Google Scholar]
- 285.Uchida K, Tominaga M. The role of TRPM2 in pancreatic β-cells and the development of diabetes. Cell Calcium 56: 332–339, 2014. DOI: 10.1016/j.ceca.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 286.Ullrich ND, Voets T, Prenen J, Vennekens R, Talavera K, Droogmans G, Nilius B. Comparison of functional properties of the Ca2+-activated cation channels TRPM4 and TRPM5 from mice. Cell Calcium 37: 267–278, 2005. DOI: 10.1016/j.ceca.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 287.Urban N, Schaefer M. Direct activation of TRPC3 channels by the antimalarial agent artemisinin. Cell 9: 202, 2020. DOI: 10.3390/cells9010202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Vajna R, Klöckner U, Pereverzev A, Weiergräber M, Chen X, Miljanich G, Klugbauer N, Hescheler J, Perez-Reyes E, Schneider T. Functional coupling between ‘R-type’ Ca2+ channels and insulin secretion in the insulinoma cell line INS-1. Eur J Biochem 268: 1066–1075, 2001. DOI: 10.1046/j.1432-1327.2001.01969.x. [DOI] [PubMed] [Google Scholar]
- 289.Varshney A, Scott LJ, Welch RP, Erdos MR, Chines PS, Narisu N, Albanus RD, Orchard P, Wolford BN, Kursawe R, Vadlamudi S, Cannon ME, Didion JP, Hensley J, Kirilusha A, Program NCS, Bonnycastle LL, Taylor DL, Watanabe R, Mohlke KL, Boehnke M, Collins FS, Parker SCJ, Stitzel ML, Barnabas BB, Bouffard GG, Brooks SY, Coleman H, Dekhtyar L, Guan X, Han J, Ho S, Legaspi R, Maduro QL, Masiello CA, McDowell JC, Montemayor C, Mullikin JC, Park M, Riebow NL, Rosarda J, Schandler K, Schmidt B, Sison C, Smith R, Stantripop S, Thomas JW, Thomas PJ, Vemulapalli M, Young AC. Genetic regulatory signatures underlying islet gene expression and type 2 diabetes. Proc National Acad Sci 114: 2301–2306, 2017. DOI: 10.1073/pnas.1621192114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Venkatachalam K, Montell C. TRP channels. Annu Rev Biochem 76: 387–417, 2007. DOI: 10.1146/annurev.biochem.75.103004.142819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Vierra NC, Dadi PK, Jeong I, Dickerson M, Powell DR, Jacobson DA. Type 2 diabetes–associated K+ channel TALK-1 modulates β-cell electrical excitability, second-phase insulin secretion, and glucose homeostasis. Diabetes 64: 3818–3828, 2015. DOI: 10.2337/db15-0280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Vierra NC, Dadi PK, Milian SC, Dickerson MT, Jordan KL, Gilon P, Jacobson DA. TALK-1 channels control β cell endoplasmic reticulum Ca2+ homeostasis. Sci Signal 10: eaan2883, 2017. DOI: 10.1126/scisignal.aan2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Vogalis F, Storm JF, Lancaster B. SK channels and the varieties of slow after-hyperpolarizations in neurons. Eur J Neurosci 18: 3155–3166, 2003. DOI: 10.1111/j.1460-9568.2003.03040.x. [DOI] [PubMed] [Google Scholar]
- 294.Wagner TFJ, Drews A, Loch S, Mohr F, Philipp SE, Lambert S, Oberwinkler J. TRPM3 channels provide a regulated influx pathway for zinc in pancreatic beta cells. Pflügers Archiv Eur J Physiol 460: 755–765, 2010. DOI: 10.1007/s00424-010-0838-9. [DOI] [PubMed] [Google Scholar]
- 295.Wang L, Bhattacharjee A, Fu J, Li M. Abnormally expressed low-voltage-activated calcium channels in -cells from NOD mice and a related clonal cell line. Diabetes 45: 1678–1683, 1996. DOI: 10.2337/diab.45.12.1678. [DOI] [PubMed] [Google Scholar]
- 296.Weaver CD, Partridge JG, Yao TL, Moates JM, Magnuson MA, Verdoorn TA. Activation of glycine and glutamate receptors increases intracellular calcium in cells derived from the endocrine pancreas. Mol Pharmacol 54: 639–646, 1998. [PubMed] [Google Scholar]
- 297.Wendt A, Eliasson L. Pancreatic α-cells – The unsung heroes in islet function. Semin Cell Dev Biol 103: 41–50, 2020. DOI: 10.1016/j.semcdb.2020.01.006. [DOI] [PubMed] [Google Scholar]
- 298.Wiser O, Trus M, Hernández A, Renström E, Barg S, Rorsman P, Atlas D. The voltage sensitive Lc-type Ca2+ channel is functionally coupled to the exocytotic machinery. Proc National Acad Sci 96: 248–253, 1999. DOI: 10.1073/pnas.96.1.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Xu Z, Lefevre GM, Gavrilova O, Claire MBFS, Riddick G, Felsenfeld G. Mapping of long-range INS promoter interactions reveals a role for calcium-activated chloride channel ANO1 in insulin secretion. Proc National Acad Sci 111: 16760–16765, 2014. DOI: 10.1073/pnas.1419240111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Yamada H, Yoshida M, Ito K, Dezaki K, Yada T, Ishikawa S, Kakei M. Potentiation of glucose-stimulated insulin secretion by the GPR40–PLC–TRPC pathway in pancreatic β-cells. Sci Rep 6: 25912, 2016. DOI: 10.1038/srep25912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Yan-Do R, Duong E, Fox JEM, Dai X, Suzuki K, Khan S, Bautista A, Ferdaoussi M, Lyon J, Wu X, Cheley S, MacDonald PE, Braun M. A glycine-insulin autocrine feedback loop enhances insulin secretion from human β-cells and is impaired in type 2 diabetes. Diabetes 65: 2311–2321, 2016. DOI: 10.2337/db15-1272. [DOI] [PubMed] [Google Scholar]
- 302.Yang S-N, Berggren P-O. CaV2.3 channel and PKCλ: New players in insulin secretion. J Clin Invest 115: 16–20, 2005. DOI: 10.1172/jci23970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Yang S-N, Berggren P-O. The role of voltage-gated calcium channels in pancreatic β-cell physiology and pathophysiology. Endocr Rev 27: 621–676, 2006. DOI: 10.1210/er.2005-0888. [DOI] [PubMed] [Google Scholar]
- 304.Yildirim V, Vadrevu S, Thompson B, Satin LS, Bertram R. Upregulation of an inward rectifying K+ channel can rescue slow Ca2+ oscillations in K(ATP) channel deficient pancreatic islets. PLoS Comput Biol 13: e1005686, 2017. DOI: 10.1371/journal.pcbi.1005686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Zhang L, Liu Q, Liu C, Zhai X, Feng Q, Xu R, Cui X, Zhao Z, Cao J, Wu B. Zacopride selectively activates the Kir2.1 channel via a PKA signaling pathway in rat cardiomyocytes. Sci China Life Sci 56: 788–796, 2013. DOI: 10.1007/s11427-013-4531-z. [DOI] [PubMed] [Google Scholar]
- 306.Zhang M, Goforth P, Bertram R, Sherman A, Satin L. The Ca2+ dynamics of isolated mouse beta-cells and islets: Implications for mathematical models. Biophys J 84: 2852–2870, 2003. DOI: 10.1016/S0006-3495(03)70014-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Zhang M, Houamed K, Kupershmidt S, Roden D, Satin LS. Pharmacological properties and functional role of Kslow current in mouse pancreatic β-cells SK channels contribute to Kslow tail current and modulate insulin secretion. J Gen Physiol 126: 353–363, 2005. DOI: 10.1085/jgp.200509312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Zhang Q, Chibalina MV, Bengtsson M, Groschner LN, Ramracheya R, Rorsman NJG, Leiss V, Nassar MA, Welling A, Gribble FM, Reimann F, Hofmann F, Wood JN, Ashcroft FM, Rorsman P. Na+ current properties in islet α- and β-cells reflect cell-specific Scn3a and Scn9a expression. J Physiol 592: 4677–4696, 2014. DOI: 10.1113/jphysiol.2014.274209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Zhang Z, Liu F, Chen J. Molecular structure of the ATP-bound, phosphorylated human CFTR. Proc National Acad Sci 115: 201815287, 2018. DOI: 10.1073/pnas.1815287115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Zhuang H, Bhattacharjee A, Hu F, Zhang M, Goswami T, Wang L, Wu S, Berggren PO, Li M. Cloning of a T-type Ca2+ channel isoform in insulin-secreting cells. Diabetes 49: 59–64, 2000. DOI: 10.2337/diabetes.49.1.59. [DOI] [PubMed] [Google Scholar]
- 311.Zitt C, Zobel A, Obukhov AG, Harteneck C, Kalkbrenner F, Lückhoff A, Schultz G. Cloning and functional expression of a human Ca2+-permeable cation channel activated by calcium store depletion. Neuron 16: 1189–1196, 1996. DOI: 10.1016/s0896-6273(00)80145-2. [DOI] [PubMed] [Google Scholar]