

Abstract
Novel psychoactive substances (NPS) continue to represent a threat to public health and safety. The number of new drugs in the latest emergent synthetic opioid class—the 2-benzylbenzimidazole analogs—also called the nitazenes—has begun to dominate the current new synthetic opioid (NSO) subclass of NPS. We describe a liquid chromatography–tandem quadrupole mass spectrometry method for the quantification of nine analogs and/or metabolites of drugs in this series: isotonitazene, metonitazene, protonitazene, etonitazene, clonitazene, flunitazene, N-desethyl isotonitazene, 5-amino isotonitazene and 4ʹ-hydroxy nitazene in human whole blood, urine, and tissue. Samples were prepared for analysis using a basic liquid–liquid extraction. Chromatographic separation was achieved using a C-18 analytical column. Multiple reaction monitoring mode was used for detection. The calibration range for the analytes was 0.5–50 ng/mL (except for 5-amino isotonitazene, which was 1.0–50 ng/mL). The limit of detection was 0.1 ng/mL, and the limit of quantitation was 0.5 ng/mL. The method had no carryover or interferences. Ionization enhancement was observed but did not affect quantitation. All analytes passed the method validation assessment. Authentic human samples suspected of containing NSOs were obtained from a medical examiner and coroner offices, as well as partnering forensic toxicology laboratories. Isotonitazene was confirmed in 92 blood samples, and its metabolites were confirmed across various matrices. Metonitazene (n = 35), flunitazene (n = 5), protonitazene (n = 3), etodesnitazene (n = 2) and butonitazene (n = 1) were also detected in cases. These newly emerging 2-benzylbenzimidazole analogs were commonly found in combination with NPS benzodiazepines and opioids (e.g., flualprazolam, fentanyl). Nitazene analogs are potent esoteric drugs that may not be identified during routine toxicological screening, and specialized assays based on sensitive instrumentation are needed to accurately characterize these NSOs.
Introduction
Since the onset of the opioid epidemic, new synthetic opioids (NSOs) have been reported as the largest contributors to drug overdose deaths in the USA (1, 2). Starting in 2017, NSOs outside of the fentanyl class began to proliferate, including drugs that retain opioid agonist activity but vary in structure and potency. The most recent NSO subclass to emerge and proliferate is the 2-benzylbenzimidazoles or the ‘nitazenes’ (Figure 1). Several analogs in this subclass have since been discussed in online drug use forums, offered for sale online and detected in drug materials. This subclass of analgesic drugs are structurally distinct from fentanyl, fentanyl analogs and other non-fentanyl analogs, such as U-47700, that were popular between 2016 and 2020 (3).
Figure 1.
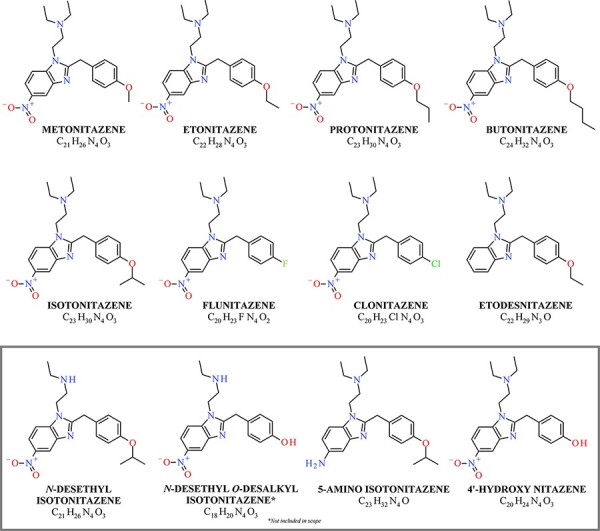
Structures of nitazene analogs included in analysis, as well as the metabolites of isotonitazene (box). Structures consist of the benzimidazole core, nitro group, alkoxy benzyl group and ethylamine component.
Isotonitazene marked the beginning of the current rise of nitazene analogs on the recreational drug market (4, 5). Isotonitazene is a structural analog of etonitazene, a potential therapeutic agent first synthesized in 1957 as an analgesic; however, etonitazene was never clinically approved for use in humans (6, 7). When first studied, etonitazene was the most potent drug of the 2-benzylbenzimidazole subclass with potency estimated at 100–1,000 times that of morphine in an animal model. From the same study, isotonitazene was reported to be the second most potent drug in the series, followed by protonitazene, metonitazene and clonitazene. Since this early work, additional nitazene analogs have been synthesized and studied, with potencies both greater than and less than etonitazene (8, 9).
Like other synthetic opioids, nitazene analogs bind to and activate μ-opioid receptors (MOR), yet little pharmacological information specific to these drugs is available regarding pharmacodynamics and pharmacokinetics. In vitro evaluation of isotonitazene by Blanckaert et al. demonstrated that isotonitazene is a strong opioid based on its high potency and efficacy (4). The half-maximal effective concentration (EC50) of isotonitazene was reported as 11.1 nM compared to 14.4 nM for fentanyl (8). Isotonitazene has a larger maximum response than fentanyl and hydromorphone, as the efficacy (Emax) of isotonitazene was determined to be 180% the Emax of hydromorphone, a value higher than the efficacy of fentanyl (163%) (4). In addition to isotonitazene, N-desethyl isotonitazene (EC50: 0.614 nM) and etonitazene (0.661 nM) also demonstrated high potency, closely followed by protonitazene (3.95 nM) and metonitazene (8.14 nM) (8). The potencies of other members of this series studied by Vandeputte et al. were lower than that of isotonitazene, including etodesnitazene (54.9 nM), clonitazene (140 nM), 4ʹ-hydroxy nitazene (176 nM), flunitazene (377 nM), and 5-amino isotonitazene (383 nM) (8).
Metabolism data for nitazene analogs are also limited; however, an early study involving isotonitazene provided insight into the likely metabolic pathways for newer members of the series. Four main metabolites of isotonitazene were reported by Krotulski et al. by examining in vivo metabolism following the ingestion of isotonitazene based on the presence of the parent drug (Figure 1) (10). Isotonitazene was metabolized through N-dealkylation and O-dealkylation to produce two primary urinary metabolites, N-desethyl isotonitazene and N-desethyl O-desalkyl isotonitazene (10). Of interest, N-desethyl isotonitazene was determined to exhibit higher in vitro potency than etonitazene and was identified in fatalities in which isotonitazene was present (8). A third metabolite, 5-amino isotonitazene, was produced by reduction of the nitro group. The fourth metabolite O-desalkyl isotonitazene, more commonly referred to as 4ʹ-hydroxy nitazene, was produced by O-dealkylation. 4ʹ-Hydroxy nitazene is a universal metabolite of nitazene analogs containing the 5-nitro group, N,N-diethylamine and an associated phenyl ether. This common metabolite may help simplify analytical toxicology screening methods; however, the identification of this metabolite in the absence of a parent drug could be indicative of the ingestion of any member of the series with the above common structural features. The metabolism of metonitazene is similar to that of isotonitazene (8).
In the USA, isotonitazene was first reported in August 2019 but the drug was on the illicit drug market at least as early as April 2019 (9). By October 2020, isotonitazene was identified in more than 200 individual postmortem (PM) death investigation cases in the USA (11). NMS Labs (Horsham PA, USA) was receiving approximately 30–40 isotonitazene positive cases per month at the peak of its popularity (accounting for approximately 0.5% of all routine forensic toxicology casework screened via mass spectrometry per month) (11, 12). Isotonitazene was commonly found with novel psychoactive substance (NPS) benzodiazepines, such as flualprazolam and etizolam, a phenomenon that has become common for NSOs, including more recent nitazene analogs (11). In the USA, isotonitazene was reported in many states across the East and Midwest, including Illinois, Indiana, Ohio and Wisconsin (13, 14). In June 2020, the US Drug Enforcement Administration announced its intent to schedule isotonitazene under Schedule I of the Controlled Substances Act in an effort to curb increasing fatalities associated with this NSO; the temporary rule went into effect in August 2020 (15). This action placed isotonitazene alongside analogs etonitazene and clonitazene, which had been placed in Schedule I in 1961 (16). Federal scheduling of isotonitazene had an immediate, widespread impact on the prevalence of this drug, leading to a precipitous decline in its popularity, but clearing the way for the emergence and proliferation of other analogs. Isotonitazene has also been reported in Europe and Canada (3, 5).
Since the initial emergence of isotonitazene, several nitazene analogs have subsequently been identified in toxicology samples and/or drug materials linked to forensic investigations. Metonitazene, another one of the original analogs synthesized in 1957, was first reported in the USA in July 2020 (17). Butonitazene was reported in Ohio alongside metonitazene in January 2021, flunitazene was reported together with metonitazene and clonazolam in March 2021 and etodesnitazene (a nitazene analog without the nitro group) was reported in Oregon in February 2021 (18–20). N-Pyrrolidino etonitazene (or etonitazepyne) and protonitazene were reported in May 2021 in West Virginia and Iowa, respectively (21, 22). While most of these analogs were included in the early patent literature, the emergence of new nitazene analogs not found in the patents (e.g., N-pyrrolidino etonitazene) suggests that the increasing popularity of this subclass is beginning to drive creative clandestine synthesis for eventual proliferation on illicit drug markets. Forensic laboratories should be prepared for the continued emergence of new nitazene analogs with varying levels of potency and structural features.
In late 2019, the initial proliferation of isotonitazene led our laboratory to believe that the emergence of new nitazene analogs could become a high-level public health and safety concern. The prospect of new and emerging nitazenes in impairment cases and fatalities prompted the need for a proactive approach to anticipate future developments in drug design and pre-emptively create innovative analytical methods encompassing both current and potential future analogs. This forward-thinking approach is part of our broader strategy to have methods available to rapidly detect, quantify and report the appearance of new drugs to stakeholders in public health, public safety and forensic circles.
Methods
Materials
Standard reference materials were purchased from Cayman Chemical (Ann Arbor, MI) as powders and prepared at a concentration of 1 mg/mL in methanol, including isotonitazene, protonitazene, metonitazene, etonitazene, clonitazene, flunitazene, N-desethyl isotonitazene, 5-amino isotonitazene, 4ʹ-hydroxy nitazene, butonitazene, etodesnitazene and isotonitazene-d7. Drug-free human blood was purchased from BioIVT (Westbury, NY). Sodium borate decahydrate was purchased from Sigma-Aldrich (St. Louis, MO). Ethyl acetate, N-butyl chloride and liquid chromatography–mass spectrometry (LC–MS)-grade water and methanol were purchased from Honeywell Chemicals (Charlotte, NC). Formic acid was purchased from ThermoFisher Scientific (Waltham, MA).
Sample quantitation
For quantitative analysis, two spiking solutions containing nine initial drugs (isotonitazene, protonitazene, metonitazene, etonitazene, clonitazene, flunitazene, N-desethyl isotonitazene, 5-amino isotonitazene and 4ʹ-hydroxy nitazene) were prepared from the stock (1,000 ng/μL) by serial dilution in methanol at final concentrations 1.0 and 0.1 ng/μL. An internal standard (ISTD) spiking solution of isotonitazene-d7 was prepared at 0.1 ng/μL. The calibration range was evaluated from 0.5 to 50 ng/mL in blood. Low, mid and high control concentrations were 1.6, 8 and 40 ng/mL, respectively, in blood. Spiking solutions for butonitazene and etodesnitazene were prepared individually.
Sample preparation
A basic liquid–liquid extraction was used (10). Blood (0.5 mL) was aliquoted and spiked with the appropriate spiking solution. Internal standard (50 μL) was added to each sample at a final concentration of 10 ng/mL. Borax buffer (1 mL, 10 mM, pH 10.4) was added to each sample, followed by 3 mL of extraction solvent (70:30 N-butyl chloride, ethyl acetate). The samples were rotated for 15 min and centrifuged for 10 min at 4,600 rpm. The organic layer was transferred, and the samples were dried under nitrogen at 35°C for approximately 30 min. The samples were reconstituted in 200 μL of initial chromatographic conditions and transferred to autosampler vials. Analysis was performed using a Waters Xevo TQ-S Micro LC–tandem quadrupole mass spectrometer (LC–QQQ-MS) (Milford, MA). Data were processed using Waters MassLynx™ Software (Milford, MA).
Instrument method development
Each drug was directly infused individually into the mass spectrometer to determine optimal cone voltage and collision energy (Table I). Selected product ions are shown in the order of abundance. Dwell time (ms) was calculated by the software based on the number of precursor–product ion transitions. The two to three most abundant fragment ions were used to create a multiple reaction monitoring method. Chromatographic separation was achieved using an Agilent InfinityLab Poroshell C-18 120 (2.7 μm, 3.0 × 100 mm) analytical column. Mobile phase A (MPA) was 0.1% formic acid in water and mobile phase B (MPB) was 0.1% formic acid in methanol. The method used a linear reverse phase chromatographic gradient (60A:40B initial hold for 1 min, 70A:30B at 2 min, 40A:60B at 5.5 min, 60A:40B at 6 min, hold for 1 min). The flow rate was 0.4 mL/min. The injection volume was 5 μL. The column temperature was 30°C.
Table I.
Mass Spectrometer Detection Parameters
| Compound | RT (min) | Precursor ion (m/z) | Product ions (m/z) | Dwell (s) | Cone (V) | Collision (V) |
|---|---|---|---|---|---|---|
| 4ʹ-Hydroxy nitazene | 1.79 | 369.2 | 100.0a | 0.011 | 46 | 24 |
| 72.0 | 32 | |||||
| Flunitazene | 4.74 | 371.2 | 100.0a | 0.011 | 60 | 26 |
| 109.0 | 46 | |||||
| 72.0 | 34 | |||||
| 5-Amino isotonitazene | 1.21 | 381.2 | 100.0a | 0.011 | 48 | 22 |
| 72.0 | 42 | |||||
| Metonitazene | 4.57 | 383.2 | 100.0a | 0.011 | 48 | 22 |
| 121.0 | 34 | |||||
| 72.0 | 20 | |||||
| N-Desethyl isotonitazene | 6.45 | 383.2 | 72.0a | 0.011 | 48 | 20 |
| 311.9 | 18 | |||||
| 130.0 | 56 | |||||
| Clonitazene | 5.98 | 387.1 | 100.0a | 0.011 | 66 | 24 |
| 124.9 | 36 | |||||
| 72.0 | 36 | |||||
| Etonitazene | 5.76 | 397.2 | 100.0a | 0.011 | 52 | 22 |
| 106.9 | 52 | |||||
| 72.0 | 42 | |||||
| Protonitazene | 6.69 | 411.2 | 100.0a | 0.011 | 46 | 20 |
| 106.9 | 52 | |||||
| 72.0 | 42 | |||||
| Isotonitazene | 6.34 | 411.2 | 100.0a | 0.011 | 46 | 20 |
| 106.9 | 52 | |||||
| 72.0 | 42 | |||||
| Isotonitazene-d7 | 6.34 | 418.2 | 100.0a | 0.011 | 50 | 24 |
| 72.0 | 46 | |||||
| 107.9 | 52 | |||||
| Butonitazeneb | 7.63 | 425.2 | 100.0a | 0.011 | 46 | 20 |
| 106.9 | 52 | |||||
| 72.0 | 42 | |||||
| Etodesnitazeneb | 2.81 | 352.2 | 100.0a | 0.011 | 52 | 18 |
| 106.9 | 40 | |||||
| 72.0 | 42 |
Quantification ion.
Not included in the scope of full validation—evaluated using standard addition only.
Method validation
The described method was validated according to a standard set forth by the AAFS Standards Board (23). Only the initial nine analogs (isotonitazene, protonitazene, metonitazene, etonitazene, clonitazene, flunitazene, N-desethyl isotonitazene, 5-amino-isotonitazene and 4ʹ-hydroxynitazene) were included in this validation. Validation experiments included calibration model, bias/accuracy, precision, interferences, recovery, matrix effects (ion suppression/enhancement), dilution integrity, carryover, limit of detection (LOD), limit of quantitation (LOQ), processed sample stability and matrix matching. An additional assessment of standard addition was added.
A calibration model of seven non-zero calibration points was evaluated in individual runs over 5 days. R2 values and calibrator back-calculations were used to assess the performance. Passing performance included an R2 value greater than 0.98 and back-calculations less than ±20% deviation. Bias and precision studies were assessed concurrently in individual runs over 5 days by running control samples (e.g., low, mid and high) in triplicate. Bias was acceptable within ±20% of target concentration. Precision was evaluated within-run and between-run and was acceptable at a threshold of less than 20% for each concentration and run.
Interference studies were performed by assessing four factors: matrix, internal standard, analyte and commonly encountered drugs. Matrix interferences were evaluated using 10 sources of blank blood. Analyte and internal standard interferences were determined by monitoring each individually without the other. Commonly encountered drug interferences were evaluated against more than 250 traditional, therapeutic, abused and NPS drugs commonly detected in forensic toxicology casework to determine whether any interfered with the analytes of interest.
Recovery, matrix effects and process efficiency were evaluated by preparing three sets of samples: unextracted, pre-spiked and post-spiked (10 ng/mL). These sample sets help determine the efficiency of the sample preparation procedure, the effects that the matrix have on the analytes (if any), and the recovery of each analyte. Matrix effects (also referred to as ion suppression or enhancement) were acceptable if the average signal deviation was less than ±20% when comparing post-spike and unextracted samples. If the value exceeded what was deemed acceptable, the laboratory needs to determine that the suppression or enhancement does not affect any critical validation parameters (e.g., LOD, LOQ) (23).
The LOD was determined by fortifying drug-free blood with decreasing concentrations of the analytes and analyzing these samples in individual runs over 3 days. A passing LOD exhibits a signal-to-noise (S/N) ratio greater than three, but also must pass all criteria set forth in the method for acceptable identification (e.g., retention time, ion ratio, peak shape, etc.). A decision point LOQ was determined by analyzing the lowest calibrator in individual runs over 3 days. A passing LOQ must pass all criteria set forth in the method for quantitation (e.g., accuracy, retention time, ion ratio, peak shape, etc.) and exhibit a S/N ratio greater than 10.
Carryover was evaluated in individual runs over 5 days by analyzing blank samples directly after the highest calibrator (50 ng/mL). Blank samples must be free of signal (i.e., chromatographic peak) or absent signal that could affect the assay performance. Dilution integrity was assessed in individual runs over 5 days in triplicate by diluting the middle calibrator (10 ng/mL) with deionized water at 1:2 and 1:4. Diluted samples must still pass all criteria for identification and quantitation, as well as criteria for bias (±20%) and precision (20%) across runs.
Processed sample stability was evaluated over four non-consecutive days by re-analyzing control samples from the initial validation run and comparing the response over time. The controls were stored in the autosampler until the next validation run was prepared. The analytes were considered stable if they passed all quantitative criteria and/or did not change significantly (±20%) in response.
A matrix matching study was performed by preparing control samples in quintuplicate in urine. These controls in urine were compared against the blood calibration curve. The assay was considered ‘matrix matched’ if the quantitative values were within ±20% that of the target value.
Although not required, standard addition studies were added as part of this validation to allow for the analysis of other matrices. Standard addition can help alleviate issues arising from matrix effects and provides an avenue for quantitation when typical full validation experiments (as described above) are not performed. This process can be especially useful for infrequently encountered alternate matrices, such as the liver and bile, when the blank authentic matrix is not available for validation studies. Standard addition was performed by preparing two pools of blood at different concentrations (5 and 10 ng/mL). These pools were aliquoted (0.5 mL) in quadruplicate and up-spiked with a known drug (sample 1: no drug, sample 2: 0.5 ng/mL, sample 3: 5 ng/mL and sample 4: 50 ng/mL). The peak area ratio from each aliquot was plotted against the up-spiked concentration. The R2 value was expected to be at least 0.98. The sample was expected to quantitate within 20% of the prepared concentration.
A full drug stability assessment was included for these nitazene analogs. A pool of blood was fortified with all analytes at a concentration of 10 ng/mL. This pool was aliquoted (0.5 mL), and the individual aliquots were stored at varying temperatures: freezer (−20°C), refrigerator (4°C) and room temperature (∼20°C). Aliquots were extracted and analyzed in triplicate at varying time points: 0 h, 24 h, 48 h, 7 days, 14 days, 28 days and 60 days. A fresh calibration curve and controls were prepared for each individual analysis. Analytes were considered stable until there was a loss of >20% concentration or the criteria needed for identification and quantitation were not met.
Authentic samples
Various biological samples (n = 171) were received from individual cases (n = 114) for the analysis for the presence of nitazene analogs. PM (n = 47) and driving-under-the-influence-of-drugs (DUID) (n = 67) cases were submitted between 2019 and 2021 and analyzed on a rolling basis as cases were triaged for quantitation. Cases originated from medical examiner and coroners’ offices as well as partnering forensic toxicology laboratories. PM cases included 31 peripheral blood samples, 15 central blood samples, 6 unspecified blood samples, 1 serum sample, 26 urine samples, 3 bile samples, 3 liver samples, 2 brain samples and 16 vitreous fluid samples. DUID cases included 67 antemortem blood samples. Blood, serum, urine and vitreous samples were stored refrigerated (approx. 4°C) for their life cycle. Bile, liver and brain samples were stored in the freezer (approx. −20°C) prior to shipment for analysis where they were thawed, prepared and stored refrigerated thereafter. The total sample storage time ranged from less than 1 month to up to 1 year and varied with respect to an individual case.
These authentic samples were generally suspected to contain isotonitazene, metonitazene and/or other new synthetic opioids. The authentic samples were prepared via the validated workflow described above and analyzed via the Waters Xevo TQ-S Micro LC–QQQ-MS. For matrices other than blood and urine, quantitative analysis was performed using standard addition. Cases involving nitazene analogs outside the scope of the fully validated method (e.g., butonitazene, etodesnitazene) were screened by liquid chromatography quadrupole time-of-flight mass spectrometry (LC–QTOF-MS) and subsequently quantitated using standard addition with analysis by LC–QQQ-MS, after verification of the standard addition process for each drug (24).
Results and Discussion
Method development and validation
Chromatographic separation (Figure 2) of the nitazene analogs was observed within this 7-min method. Special attention was given to isotonitazene and protonitazene—two very closely related positional isomers (Figure 1) that cannot be distinguished based on mass spectral analysis alone.
Figure 2.
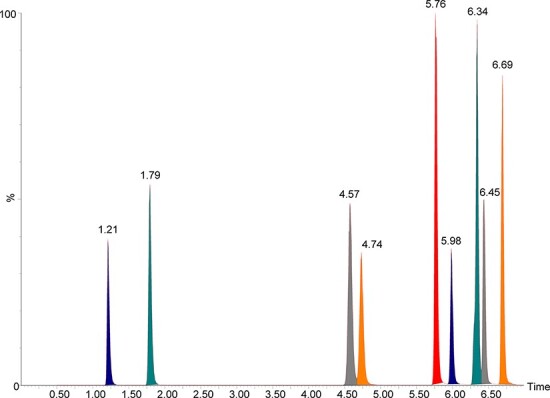
Chromatographic separation achieved (at 50 ng/mL). From left to right: 5-amino isotonitazene (1.21 min), 4ʹ-hydroxy nitazene (1.79 min), metonitazene (4.57 min), flunitazene (4.74 min), etonitazene (5.76 min), clonitazene (5.98 min), isotonitazene (6.34 min), N-desethyl isotonitazene (6.45 min) and protonitazene (6.69 min).
Table II shows the quantitative parameters for each nitazene analog assessed via full validation. The calibration model for all drugs was linear with 1/x weighting and the origin excluded. The final calibration range was 0.5–50 ng/mL for all analytes except for 5-amino isotonitazene, which had an amended calibration range of 1.0–50 ng/mL due to the method’s inability to accurately quantitate down to 0.5 ng/mL. All drugs passed the calibration model studies with R2 values at or above 0.98. The LOQ for all drugs was 0.5 ng/mL, with the exception of 5-amino isotonitazene, which had an LOQ of 1.0 ng/mL. The LOD for all drugs was 0.1 ng/mL.
Table II.
Quantitation Parameters and Calibration Results
| Compound | Calibration rangea (ng/mL) | Model | Weighting | LOQ (ng/mL) | LOD (ng/mL) | Correlation (R2) | y-Intercept |
|---|---|---|---|---|---|---|---|
| Isotonitazene | 0.5–50 | Linear | 1/x | 0.5 | 0.1 | 0.999 | 0.16 |
| Protonitazene | 0.5–50 | Linear | 1/x | 0.5 | 0.1 | 0.999 | 0.13 |
| Etonitazene | 0.5–50 | Linear | 1/x | 0.5 | 0.1 | 0.999 | 0.17 |
| Metonitazene | 0.5–50 | Linear | 1/x | 0.5 | 0.1 | 0.998 | 0.23 |
| Clonitazene | 0.5–50 | Linear | 1/x | 0.5 | 0.1 | 0.999 | 0.20 |
| Flunitazene | 0.5–50 | Linear | 1/x | 0.5 | 0.1 | 0.998 | 0.30 |
| N-Desethyl isotonitazene | 0.5–50 | Linear | 1/x | 0.5 | 0.1 | 0.998 | 0.11 |
| 5-Amino isotonitazene | 1.0–50 | Linear | 1/x | 1.0 | 0.1 | 0.998 | 0.27 |
| 4ʹ-Hydroxy nitazene | 0.5–50 | Linear | 1/x | 0.5 | 0.1 | 0.998 | 0.12 |
Calibrator values were 0.5, 1, 2, 5, 10, 20 and 50 ng/mL.
All nine nitazene analogs fell within the acceptable criteria for bias and precision at all three control concentrations (Table III). Recovery was greater than 85% for all drugs. Matrix effects were greater than 100%, indicating ionization enhancement for all drugs; however, there was no impact on assay performance (e.g., accuracy, precision, peak shape, etc.). Process efficiency was greater than 100% for all drugs; however, this is not a metric commonly assessed in forensic toxicology validations and therefore does not associate acceptance criteria in the standard document—like matrix effects, there was no impact on assay performance. Isotonitazene, protonitazene, etonitazene, clonitazene, flunitazene and N-desethyl isotonitazene all were stable in the autosampler for up to 216 h (9 days). 5-Amino isotonitazene was stable for up to 192 h (8 days), and metonitazene and 4ʹ-hydroxy nitazene were stable for less than 120 h (5 days).
Table III.
Validation Results by Nitazene Analog
| Drug | Accuracy (%) | Precision (%) | Recovery (%) | Matrix effect (%) | Process efficiency (%) | Processed sample stability (hours) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Low | Mid | High | Low | Mid | High | |||||
| Isotonitazene | 2.5 | −4.1 | −4.6 | 5.1 | 8.8 | 7.1 | 87.7 | 175 | 153 | 216 |
| Protonitazene | 1.5 | −3.1 | −1.6 | 4.2 | 7.4 | 6.2 | 85.5 | 174 | 149 | 216 |
| Etonitazene | 2.4 | −4.8 | −4.6 | 3.6 | 8.2 | 6.8 | 92.5 | 177 | 163 | 216 |
| Metonitazene | 2.4 | −4.7 | −4.4 | 5.6 | 9.0 | 7.9 | 97.3 | 174 | 169 | 120 |
| Clonitazene | 1.4 | −5.4 | −4.1 | 3.8 | 8.5 | 8.2 | 90.9 | 174 | 158 | 216 |
| Flunitazene | 0.7 | −7.5 | −6.2 | 4.2 | 8.9 | 7.6 | 95.5 | 171 | 163 | 216 |
| N-Desethyl isotonitazene | 2.9 | −6.0 | −5.1 | 4.6 | 8.3 | 5.7 | 94.5 | 150 | 141 | 216 |
| 5-Amino isotonitazene | 8.6 | −0.7 | −1.0 | 7.5 | 10.6 | 14.2 | 96.4 | 234 | 225 | 192 |
| 4ʹ-Hydroxy nitazene | 5.0 | −2.5 | −5.5 | 5.5 | 9.9 | 7.4 | 98.1 | 189 | 186 | <120 |
No interferences and no carryover were observed for this assay. All analytes passed dilution integrity studies except for 5-amino isotonitazene, which showed unacceptable bias and variation for both the 1:2 and 1:4 dilutions. 5-Amino isotonitazene was also the only analyte that failed matrix matching studies; all others met the criteria. As a result, it was determined that this method is only valid for 5-amino isotonitazene if the matrix of the sample is blood and undiluted.
All nine analogs met the necessary criteria for the two standard addition experiments performed. All drugs exhibited R2 values greater than 0.99. The percent deviation was less than 10% from the target concentration for both the 5 and 10 ng/mL samples for all drugs.
Overall, our laboratory was able to develop an LC–QQQ-MS assay for the simultaneous quantitation of nine nitazene analogs, including two isomers (protonitazene and isotonitazene) and parent drugs and metabolites. The assay passed validation with exceptional performance. Ultimately, the method was able to identify this drug subclass with significant sensitivity in sub-ng/mL concentrations.
Stability assessment
A stability assessment was conducted over 60 days in blood at three storage conditions: freezer (−20°C), refrigerator (4°C) and room temperature (∼20°C). All nitazene analogs were observed to be stable under refrigerated conditions except for 5-amino isotonitazene (Figure 3). During the quantitation of stability samples, 5-amino isotonitazene failed criteria needed for quantitation (i.e., accuracy of control values). This was anecdotally attributed to the poor stability of this metabolite; however, further research is warranted. 5-Amino isotonitazene is included in Figure 3 for reference only.
Figure 3.
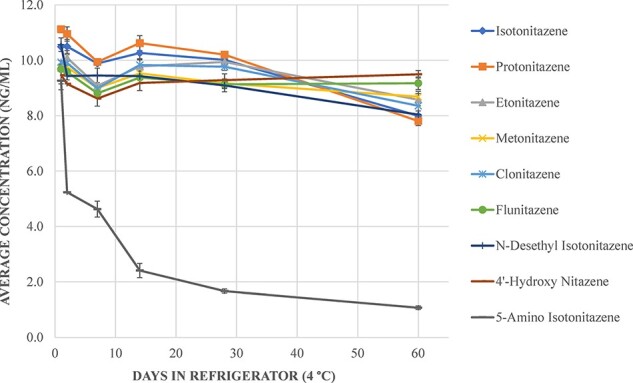
Stability of nitazene analog parent drugs and isotonitazene metabolites in the refrigerator over 60 days. 5-Amino isotonitazene failed criteria for calibration during quantitation, however, was included for reference.
At room temperature, all nitazene analogs were stable for up to 60 days except for isotonitazene (≥14 days), protonitazene (≥14 days), N-desethyl isotonitazene (≥7 days) and 4ʹ-hydroxy nitazene (≥14 days). Stability results for nitazene analogs in the freezer were inconsistent, as most parent drugs were observed to be unstable after 28 days—further assessment is warranted. The Supplementary File contains figures for stability at room temperature (Figure S1) and frozen (Figure S2). For reliable results, blood samples suspected to contain a nitazene analog can generally be stored in the refrigerator (4°C) for at least 28 days before analysis based on the data generated from this study.
Method additions and verification
Following validation, subsequent iterations of the method were developed to included newly emergent nitazene analogs butonitazene and etodesnitazene, which were assessed via standard addition rather than traditional quantitation by the calibration curve. Etodesnitazene was added to the initial method without the need for chromatographic changes as it eluted (2.81 min) within the original run time. For butonitazene, the chromatographic gradient was adjusted due to this drug eluting after the full run time of the initial method. The final organic hold (40A:60B) in the gradient was lengthened from 0.5 to 1.5 min. This allowed butonitazene to elute (7.63 min) properly for detection and quantitation.
The standard addition verification process was acceptable for both butonitazene and etodesnitazene. Both assays were determined to be linear between 0.2 and 50 ng/mL. The LOD values for both analytes were determined to be less than 0.1 ng/mL. No interferences from the matrix, analyte, internal standard or commonly encountered drugs were discovered. Simulated scenarios of standard addition were acceptable: for butonitazene, the control prepared at 5 ng/mL resulted in quantitation at 5.9 ng/mL (+18%) and the control prepared at 10 ng/mL resulted in quantitation at 9.8 ng/mL (−2%) and for etodesnitazene, the control prepared at 5 ng/mL resulted in quantitation at 5.2 ng/mL (+4%) and the control prepared at 10 ng/mL resulted in quantitation at 8.9 ng/mL (−11%). Recovery for butonitazene was 88% and the recovery for etodesnitazene was 80%.
Authentic casework samples
Comprehensive confirmation results for PM and DUID samples are shown in Table IV. Isotonitazene (n = 25), N-desethyl isotonitazene (n = 13), metonitazene (n = 35), protonitazene (n = 4), butonitazene (n = 3), etodesnitazene (n = 1), flunitazene (n = 4) and 4ʹ-hydroxy nitazene (n = 10) were confirmed quantitatively in PM samples analyzed using this validated nitazene analog method. Only isotonitazene (n = 50) and N-desethyl isotonitazene (n = 12) were confirmed in the DUID samples. Isotonitazene and metonitazene were identified with the highest frequency. Isotonitazene positive samples were collected between October 2019 and January 2020, while metonitazene positive samples were collected between November 2020 and February 2021. This illustrates the quick turnover of these NSOs on recreational drug markets. Brorphine, a non-nitazene NSO, was prevalent in the months in between.
Table IV.
Overall Results for Nitazene Analogs Confirmed (Including Both Quantitative and Qualitative)
| Case type | Drug | Matrix | N (total) | N (qualitative, positive, <0.5 ng/mL) | N (quantitative, >0.5 ng/mL) | Mean (±SD) | Median | Min. | Max. |
|---|---|---|---|---|---|---|---|---|---|
| PM | Isotonitazene | Central blood | 8 | 2 | 6 | 1.4 (±0.6) | 1.5 | 0.6 | 2 |
| Peripheral blood | 13 | 3 | 10 | 2 (±2.5) | 1.3 | 0.5 | 8.6 | ||
| Unspecified blood | 4 | 2 | 2 | 0.85 (±0.1) | 0.85 | 0.8 | 0.9 | ||
| Urine | 9 | 5 | 4 | 1.4 (±0.9) | 1.2 | 0.6 | 2.5 | ||
| Bile | 3 | 0 | 3 | 2 (±1.4) | 1.6 | 0.9 | 3.5 | ||
| Brain | 1 | 1 | 0 | - | - | - | - | ||
| N-Desethyl isotonitazene | Central blood | 8 | 7 | 1 | 3 | N/A | N/A | N/A | |
| Peripheral blood | 12 | 9 | 3 | 1.1 (±1.0) | 0.6 | 0.5 | 2.2 | ||
| Unspecified blood | 4 | 4 | 0 | - | - | - | - | ||
| Urine | 9 | 3 | 6 | 4.5 (±6.1) | 2.5 | 0.6 | 16 | ||
| Bile | 3 | 0 | 3 | 90 (±39) | 70 | 65 | 136 | ||
| Brain | 1 | 1 | 0 | - | - | - | - | ||
| 5-Amino isotonitazene | Central blood | 4 | 4 | 0 | - | - | - | - | |
| Peripheral blood | 7 | 7 | 0 | - | - | - | - | ||
| Unspecified blood | 1 | 1 | 0 | - | - | - | - | ||
| Urine | 3 | 3 | 0 | - | - | - | - | ||
| Bile | 1 | 1 | 0 | - | - | - | - | ||
| Metonitazene | Central blood | 5 | 1 | 4 | 5.5 (±4.7) | 4.2 | 1.6 | 12 | |
| Peripheral blood | 17 | 1 | 16 | 5.5 (±3.5) | 4.5 | 0.5 | 13 | ||
| Unspecified blood | 2 | 0 | 2 | 17 (±22) | 17 | 1.4 | 33 | ||
| Serum | 1 | 0 | 1 | 18 | N/A | N/A | N/A | ||
| Urine | 13 | 1 | 12 | 14 (±13) | 9.2 | 0.58 | 31 | ||
| Vitreous | 12 | 12 | 0 | - | - | - | - | ||
| Protonitazene | Central blood | 1 | 0 | 1 | 5 | N/A | N/A | N/A | |
| Peripheral blood | 2 | 0 | 2 | 14 (±16) | 14 | 3.1 | 25 | ||
| Urine | 1 | 0 | 1 | 1 | N/A | N/A | N/A | ||
| Butonitazene | Unspecified blood | 1 | 0 | 1 | 3.2 | N/A | N/A | N/A | |
| Serum | 1 | 0 | 1 | 2.4 | N/A | N/A | N/A | ||
| Urine | 1 | 0 | 1 | 10 | N/A | N/A | N/A | ||
| Etodesnitazene | Central blood | 1 | 0 | 1 | 30 | N/A | N/A | N/A | |
| Peripheral blood | 1 | 1 | 0 | - | - | - | - | ||
| Flunitazene | Central blood | 3 | 1 | 2 | 2.7 (±2.9) | 2.7 | 0.6 | 4.8 | |
| Peripheral blood | 2 | 1 | 1 | 2.1 | N/A | N/A | N/A | ||
| Urine | 4 | 3 | 1 | 0.5 | N/A | N/A | N/A | ||
| Vitreous | 1 | 1 | 0 | - | - | - | - | ||
| 4ʹ-Hydroxy nitazene | Central blood | 2 | 2 | 0 | - | - | - | - | |
| Peripheral blood | 5 | 5 | 0 | - | - | - | - | ||
| Unspecified blood | 2 | 2 | 0 | - | - | - | - | ||
| Urine | 10 | 3 | 7 | 5.7 (±5.9) | 2.9 | 0.7 | 14 | ||
| Bile | 3 | 0 | 3 | 64 (±29) | 52 | 43 | 98 | ||
| DUID | Isotonitazene | Blood | 67 | 17 | 50 | 1.6 (±1.9) | 1 | 0.5 | 9 |
| N-Desethyl isotonitazene | Blood | 64 | 52 | 12 | 0.8 (±0.5) | 0.6 | 0.5 | 2.2 | |
| 4ʹ-Hydroxy nitazene | Blood | 47 | 47 | 0 | - | - | - | - |
In total, isotonitazene was confirmed in 75 samples (44%), including qualitative confirmations (<0.5 ng/mL) in 30 samples (18%): 13 PM and 17 DUID. Mean, median and range concentrations are shown in Table IV. The mean and median concentrations in blood were lowest for isotonitazene compared to the other analogs confirmed, likely owing to its higher degree of potency. There was only one instance in a DUID sample where isotonitazene was identified without the presence of metabolites; all PM samples contained at least one metabolite.
For DUID cases, N-desethyl isotonitazene was present in 96% (n = 64) of samples containing isotonitazene and 4ʹ-hydroxy nitazene was present in 70% (n = 47). N-Desethyl isotonitazene was found with isotonitazene in 24 PM samples (96%), while 4ʹ-hydroxy nitazene was present with isotonitazene in 13 samples (52%). 5-Amino isotonitazene was identified in PM samples with isotonitazene (n = 16, 64%) but was not identified in DUID samples.
N-Desethyl isotonitazene was the primary metabolite found with isotonitazene. N-Desethyl isotonitazene concentrations (Table IV) were slightly lower than isotonitazene; N-desethyl isotonitazene is reported to have greater potency than the parent drug, reinforcing the need to consider metabolites within the analytical scope (8). N-Desethyl isotonitazene was found in urine and bile and at much higher concentrations in bile than isotonitazene itself. The detection of N-desethyl isotonitazene could be crucial to the interpretation of drug concentrations following ingestion.
Metonitazene was found in combination with other nitazene analogs. Metonitazene was discovered with flunitazene (n = 5), butonitazene (n = 3) and protonitazene (n = 2) in blood, serum and urine. Metonitazene was confirmed in 35 (20%) PM samples, primarily blood (central, peripheral and unspecified) and urine. The mean concentration of metonitazene in PM blood (Table IV) was observed to be higher than that for isotonitazene. One serum sample contained 18 ng/mL of metonitazene.
Butonitazene was confirmed in three (1.8%) PM samples: unspecified blood (3.2 ng/mL), serum (2.4 ng/mL) and urine (10 ng/mL). Flunitazene, one of the lesser potent nitazene analogs, was confirmed in four (2.3%) PM samples: central and peripheral blood (0.6–4.8 ng/mL) and urine (0.5 ng/mL). In more recent months, protonitazene was confirmed in four (2.3%) PM samples: central and peripheral blood (3.1–25 ng/mL) and urine (1.0 ng/mL). As mentioned previously, protonitazene was found along with metonitazene, and protonitazene was also discovered in combination with etodesnitazene (qualitative only). Etodesnitazene was quantified in a separate central blood sample (30 ng/mL).
4ʹ-Hydroxy nitazene, the universal metabolite of the nitazene analogs, was confirmed with isotonitazene (n = 6), metonitazene (n = 2), protonitazene (n = 2) and butonitazene (n = 1) in the PM samples. One sample contained both metonitazene and butonitazene and in this instance the source of the 4ʹ-hydroxy nitazene could not be attributed, although it is likely that both parent drugs could each have produced the metabolite. Mean and median bile concentrations were elevated for this metabolite (mean: 64 ± 30 ng/mL, median: 52 ng/mL).
Other drugs commonly detected alongside isotonitazene were flualprazolam, fentanyl and quinine (Figure 4). It is clear from this case series that the nitazene analogs can be often used with NPS benzodiazepines and other opioids, including NPS opioids. These drugs are generally found at relatively low concentrations (<10 ng/mL). N-Desethyl isotonitazene, however, was found at highly variable concentrations depending on the matrix source (i.e., blood vs. bile).
Figure 4.
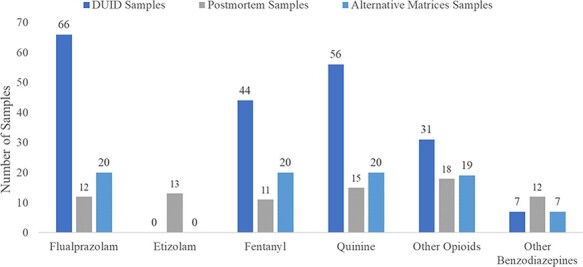
Most common drugs found alongside isotonitazene in authentic PM and DUID samples.
Conclusion
Our laboratory developed and validated a proactive workflow for the detection and quantification of nitazene analogs using a Waters Xevo TQ-S Micro LC–QQQ-MS. The assay included isotonitazene, protonitazene, metonitazene, etonitazene, clonitazene, flunitazene, N-desethyl isotonitazene, 5-amino isotonitazene and 4ʹ-hydroxy nitazene. This method was readily adapted to include newly emerging nitazene analogs (e.g., butonitazene and etodesnitazene) and exhibited an appropriate level of sensitivity. Nitazene analogs and their metabolites were identified, confirmed and quantitated in 171 biological samples from 114 distinct cases suspected of involving NSOs. This manuscript provides comprehensive quantitative data for this NSO series and is the first to compare concentrations in blood, urine and other matrices; isotonitazene (the most potent analog detected) was found at the lowest concentrations. The most prevalent nitazene analog in late 2019 was isotonitazene, while the most prevalent nitazene in early 2021 was metonitazene. The detection of N-desethyl isotonitazene remains a cause for concern due to its increased potency over the parent drug and potential to be sold as a parent drug itself. This NSO subclass of NPS remains a threat to public health and sensitive instrumentation is required to appropriately assess toxicologically significant levels in authentic cases.
Supplementary Material
Acknowledgments
The authors of this manuscript would like to acknowledge the following individuals for their contributions to the project: Donna Papsun and Kyle Miller from NMS Labs; Eric Lavins and Szabi Sofalvi from Cuyahoga County Medical Examiner’s Office; Amy Miles and Aaron Zane from the Wisconsin State Lab of Hygiene; and Judith Rodriguez Salas from the Center for Forensic Science Research and Education, as well as additional scientists at the CFSRE and NMS Labs.
Contributor Information
Sara E Walton, Center for Forensic Science Research and Education, Fredric Rieders Family Foundation, 2300 Stratford Avenue, Willow Grove, PA 19090, USA; Jefferson College of Life Sciences, Thomas Jefferson University, 1020 Locust Street, Philadelphia, PA 19107, USA.
Alex J Krotulski, Center for Forensic Science Research and Education, Fredric Rieders Family Foundation, 2300 Stratford Avenue, Willow Grove, PA 19090, USA; Jefferson College of Life Sciences, Thomas Jefferson University, 1020 Locust Street, Philadelphia, PA 19107, USA.
Barry K Logan, Center for Forensic Science Research and Education, Fredric Rieders Family Foundation, 2300 Stratford Avenue, Willow Grove, PA 19090, USA; Jefferson College of Life Sciences, Thomas Jefferson University, 1020 Locust Street, Philadelphia, PA 19107, USA; NMS Labs, 200 Welsh Road, Horsham, PA 19044, USA.
Supplementary Data
Supplementary Data is available at Journal of Analytical Toxicology online.
Funding
This project was supported by the Fredric Rieders Family Foundation and Thomas Jefferson University. A portion of this project was funded by the National Institute of Justice, Office of Justice Programs, U.S. Department of Justice (Award Number 2020-DQ-BX-0007, “Real-Time Sample-Mining and Data-Mining Approaches for the Discovery of Novel Psychoactive Substances (NPSs)”). The opinions, findings, conclusions and/or recommendations expressed in this publication are those of the author(s) and do not necessarily reflect those of the Department of Justice.
References
- 1. CDC Injury Center . (2021) Drug Overdose. Drug Overdose Deaths. https://www.cdc.gov/drugoverdose/deaths/index.html (accessed Jul 28, 2021).
- 2. Mattson C.L., Tanz L.J., Quinn K., Kariisa M., Patel P., Davis N.L. (2021) Trends and geographic patterns in drug and synthetic opioid overdose deaths—United States, 2013–2019. MMWR Morbidity and Mortality Weekly Report, 70, 202–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Krotulski A.J. (2019) Public Alert Isotonitazene NPS Discovery. https://www.npsdiscovery.org/wp-content/uploads/2019/11/Public-Alert_Isotonitazene_NPS-Discovery_111919-1.pdf (accessed Mar 28, 2020).
- 4. Blanckaert P., Cannaert A., Van Uytfanghe K., Hulpia F., Deconinck E., Van Calenbergh S., et al. (2019) Report on a novel emerging class of highly potent benzimidazole NPS opioids: chemical and in vitro functional characterization of isotonitazene. Drug Testing and Analysis, 12, 422–430. doi: 10.1002/dta.2738 [DOI] [PubMed] [Google Scholar]
- 5. Krotulski A. J., Papsun D. M., Kacinko S. L., Logan B. K. (2020) Isotonitazene quantitation and metabolite discovery in authentic forensic casework. Journal of Analytical Toxicology, 44, 521–530. [DOI] [PubMed] [Google Scholar]
- 6. Hoffman K., Hunger A., Kebrle J., Rossi A. (1960) United States Patent Office - Benzimidazoles. https://patentimages.storage.googleapis.com/c4/25/6b/003a7dcea52ce3/US2935514.pdf(accessed May 3, 1960). [Google Scholar]
- 7. Hunger A., Kebrle J., Rossi A., Hoffmann K. (1957) Synthese basisch substituierter, analgetisch wirksamer Benzimidazol-Derivate. Experientia, 13, 400–401. [DOI] [PubMed] [Google Scholar]
- 8. Vandeputte M., Uytfanghe K.V., Layle N., St. Germaine D., Iula D.M., Stove C. (2020) Synthesis, chemical characterization, and mu-opioid receptor activity assessment of the emerging group of nitazene new synthetic opioids. ACS Chemical Neuroscience, 12, 1241–1251. [DOI] [PubMed] [Google Scholar]
- 9.EMCDDA (2020), EMCDDA technical report on the new psychoactive substance N,N-diethyl-2-[[4-(1-methylethoxy)phenyl]methyl]-5-nitro-1H-benzimidazole-1-ethanamine (isotonitazene), EMCDDA, Lisbon.
- 10. Krotulski A.J., Papsun D.M., Kacinko S.L., Logan B.K. (2020) Isotonitazene quantitation and metabolite discovery in authentic forensic casework. Journal of Analytical Toxicology, 44, 521–530. [DOI] [PubMed] [Google Scholar]
- 11. Krotulski A.J., Papsun D.M., Noble C., Kacinko S.L., Logan B.K. (2021) Brorphine—investigation and quantitation of a new potent synthetic opioid in forensic toxicology casework using liquid chromatography-mass spectrometry. Journal of Forensic Sciences, 6, 664–676. [DOI] [PubMed] [Google Scholar]
- 12. Papsun D.M., Krotulski A.J., Homan J., Temporal K.D.H., Logan B.K. (2020) Flualprazolam blood concentrations in 197 forensic investigation cases. Journal of Analytical Toxicology, 45, 226–232. [DOI] [PubMed] [Google Scholar]
- 13. Shover C.L., Falasinnu T.O., Freedman R.B., Humphreys K. (2021) Emerging characteristics of isotonitazene-involved overdose deaths: a case-control study. Journal of Addiction Medicine, 15, 429–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. The Woods at Parkside . (2021) Isotonitazene Found in Growing Ohio Overdose Deaths. Woods at Parkside. https://www.thewoodsatparkside.com/isotonitazene-found-in-growing-ohio-overdose-deaths/ (accessed Mar 8, 2021).
- 15. Drug Enforcement Administration . (2020) Temporary Placement of Isotonitazene in ScheduleI. https://www.deadiversion.usdoj.gov/fed_regs/rules/2020/fr0618.htm (accessed Nov 2, 2020).
- 16. Controlled Substances - Alphabetical Order Alphabetical Order.
- 17.NPS Discovery. (2020) Metonitazene Monograph. https://www.npsdiscovery.org/wp-content/uploads/2020/07/Metonitazene_073020_NMSLabs_Report.pdf (accessed Mar 8, 2021).
- 18. NPS Discovery . (2021) Butonitazene Monograph. https://www.npsdiscovery.org/wp-content/uploads/2021/01/Butonitazene_011521_ToxicologyAnalyticalReport.pdf (accessed Mar 8, 2021).
- 19. NPS Discovery . (2021) Etodesnitazene Monograph. https://www.npsdiscovery.org/wp-content/uploads/2021/02/Etodesnitazene_022321_ToxicologyAnalyticalReport.pdf (accessed Mar 8, 2021).
- 20. NPS Discovery . (2021) Flunitazene Monograph. https://www.npsdiscovery.org/wp-content/uploads/2021/03/Flunitazene_032621_ToxicologyAnalyticalReport.pdf?mc_cid=47c602c143&mc_eid=0872426f82 (accessed Apr 19, 2021).
- 21. NPS Discovery . (2021) Protonitazene Monograph. https://www.npsdiscovery.org/wp-content/uploads/2021/05/Protonitazene_052621_ToxicologyAnalyticalReport.pdf (accessed Jul 28, 2021).
- 22. NPS Discovery . (2021) N-Pyrrolidino-Etonitazene Monograph. https://www.npsdiscovery.org/wp-content/uploads/2021/05/N-Pyrrolidino-Etonitazene_051321_ToxicologyAnalyticalReport.pdf (accessed Jul 28, 2021).
- 23. AAFS Standards Board . (2019) Standard PracticesforMethod Validation in Forensic Toxicology. http://www.asbstandardsboard.org/wp-content/uploads/2019/11/036_Std_e1.pdf (accessed May 15, 2020).
- 24. Krotulski A.J., Papsun D.M., Walton S.E., Logan B.K. (2021) Metonitazene in the United States—forensic toxicology assessment of a potent new synthetic opioid using liquid chromatography mass spectrometry. Drug Testing and Analysis, 13, 1697–1711. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.