

SUMMARY
RAS guanosine triphosphatases (GTPases) are mutated in nearly 20% of human tumors, making them an attractive therapeutic target. Following our discovery that nucleotide-free RAS (apo RAS) regulates cell signaling, we selectively target this state as an approach to inhibit RAS function. Here, we describe the R15 monobody that exclusively binds the apo state of all three RAS isoforms in vitro, regardless of the mutation status, and captures RAS in the apo state in cells. R15 inhibits the signaling and transforming activity of a subset of RAS mutants with elevated intrinsic nucleotide exchange rates (i.e., fast exchange mutants). Intracellular expression of R15 reduces the tumor-forming capacity of cancer cell lines driven by select RAS mutants and KRAS(G12D)-mutant patient-derived xenografts (PDXs). Thus, our approach establishes an opportunity to selectively inhibit a subset of RAS mutants by targeting the apo state with drug-like molecules.
Graphical Abstract
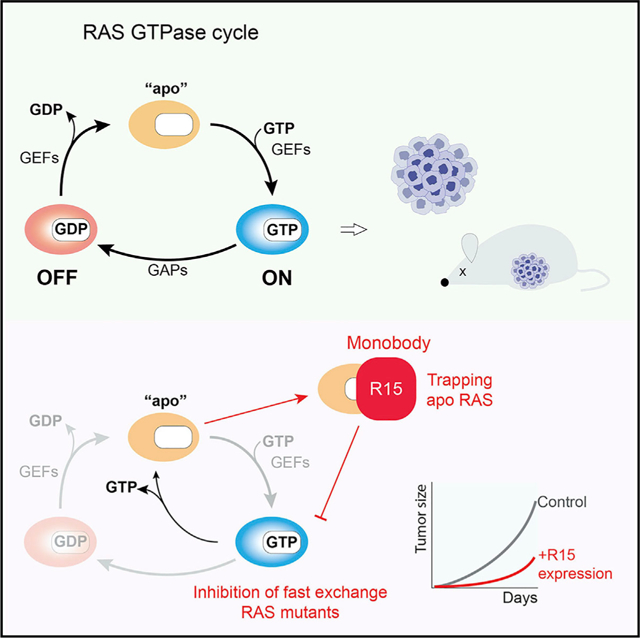
In brief
Khan et al. develop a high-affinity monobody to nucleotide-free RAS that, when expressed intracellularly, inhibits oncogenic RAS-mediated signaling and tumorigenesis. This study reveals the feasibility of targeting the nucleotide-free state to inhibit tumors driven by oncogenic RAS mutants that possess elevated nucleotide exchange activity.
INTRODUCTION
The RAS guanosine triphosphatase (GTPase) is a central hub in the regulation of cellular signaling. RAS cycles between inactive guanosine diphosphate (GDP)-bound and active GTP-bound states. This transition is mediated by guanine nucleotide exchange factors (GEFs) that promote the release of GDP to form a transient nucleotide-free state (apo RAS). Due to the picomolar affinity of apo RAS for nucleotide coupled with the higher concentration of GTP than GDP in cells (Traut, 1994), apo RAS subsequently binds GTP, leading to the activation of downstream effector pathways. Termination of RAS activation occurs upon hydrolysis of RAS-bound GTP, which is facilitated by the action of GTPase accelerating proteins (GAPs) that enhance the intrinsic GTPase activity by >100-fold (Karnoub and Weinberg, 2008; Simanshu et al., 2017; Spencer-Smith and O’Bryan, 2019).
Activating mutations in RAS are present in 19% of cancer patients (Prior et al., 2020; Zuberi et al., 2020). Oncogenic activation of RAS frequently occurs in human cancers predominantly through mutations at codons 12, 13, or 61. Mutations at these positions impair the intrinsic GTPase activity of RAS and/or disrupt interaction with GAPs, shifting RAS to the active, GTP-bound state, resulting in enhanced engagement of effector pathways and oncogenic transformation of cells.
Despite the prevalence of RAS mutations in cancer, pharmacological inhibition of mutant RAS has been challenging due in part to an apparent lack of deep hydrophobic pockets for binding small-molecule inhibitors. Further, the picomolar affinity for guanine nucleotides has precluded the successful development of nucleotide competitive inhibitors, an approach effectively utilized with kinases (Hanke et al., 1996; Lopez et al., 2014; Moore et al., 2020; O’Bryan, 2019; Papke and Der, 2017; Stalnecker and Der, 2020). However, the recent development of mutation-specific KRAS(G12C) inhibitors from preclinical tool compounds to a US Food and Drug Administration (FDA)-approved therapeutic has demonstrated that RAS is indeed druggable (Canon et al., 2019; Hallin et al., 2020; Janes et al., 2018; Lito et al., 2016; Ostrem et al., 2013). Although the KRAS(G12C) mutation occurs frequently in lung adenocarcinoma, this mutation is present in less than 3% of all human cancers (Zuberi et al., 2020). Thus, there remains a clear need for development of additional mutant-selective RAS therapeutics.
We have employed monobody (Mb) technology as a platform to identify vulnerabilities in RAS (Spencer-Smith et al., 2017a; Teng et al., 2021). The NS1 Mb binds the α4-β6-α5 allosteric interface of both HRAS and KRAS with low nanomolar affinity, but not to NRAS (Spencer-Smith et al., 2017a, 2017b). This interface has been implicated in dimerization and nanoclustering (Ambrogio et al., 2018; Guldenhaupt et al., 2012; Mysore et al., 2021; Packer et al., 2021; Spencer-Smith et al., 2017a), and NS1 expression decreases both HRAS and KRAS dimerization and nanoclustering in cells (Spencer-Smith et al., 2017a). Further, NS1 potently inhibits HRAS- and KRAS-mediated signaling, transformation, and tumorigenesis both in cellulo and in vivo (Khan et al., 2019, 2021; Spencer-Smith et al., 2017a) and has become a widely used tool in the RAS community (Alabi et al., 2021; Killoran and Smith, 2019; Lee et al., 2020; Lim et al., 2021; Roth et al., 2020). More recently, we reported the development of a mutation-specific Mb, 12VC1, that selectively inhibited the active state of KRAS(G12V) and KRAS(G12C) (Teng et al., 2021). 12VC1 directly recognized the mutant residues along with the switch 1 and switch 2 regions (SW1 and SW2, respectively) to inhibit KRAS-effector interaction (Teng et al., 2021). Thus, our approach using the Mb technology has led to the development of powerful tool biologics to further understand RAS biochemistry and define opportunities to disrupt the oncogenic activity of RAS mutants.
Although oncogenic RAS mutants are frequently locked in the GTP-loaded state, several observations call this premise into question. Recently described covalent KRAS inhibitors trap KRAS(G12C) in the GDP-bound state due to its intrinsic GTPase activity (Canon et al., 2019; Hallin et al., 2020; Janes et al., 2018; Lito et al., 2016). In addition, the fraction of the GTP-loaded state of RAS mutants is lower in cells than that observed in vitro, suggesting that RAS mutants continue to cycle despite impairment in their GTPase activity (Zhao et al., 2020). These findings are also consistent with the observation that certain RAS mutants, e.g., G12D (Bollag et al., 1996; Franken et al., 1993; Wey et al., 2013), G13D (Hunter et al., 2015; Smith et al., 2013; Wey et al., 2013), Q61L (Smith et al., 2013), and A146T (Wey et al., 2013), exhibit elevated spontaneous nucleotide release rates. Further, various oncogenic RAS mutants, particularly KRAS(G13D), have been reported to exist predominantly in the apo state in complex with SOS (Moghadamchargari et al., 2021). Together, these findings indicate that the nucleotide state of oncogenic RAS is more dynamic than previously appreciated.
To date, most efforts to inhibit RAS have focused on targeting the GDP- or GTP-bound states. Although conventional wisdom has been that the apo state exists only for a brief period before it re-binds a nucleotide, our prior discovery that apo RAS contributes to the regulation of cellular signaling pathways (Wong et al., 2012) raised the possibility that this state of RAS may represent a tractable therapeutic target. Indeed, the monoclonal antibody anti-p21Ser inhibited viral KRAS(G12S) transformation by blocking nucleotide loading, presumably by binding the nucleotide-free state of KRAS(G12S) (Clark et al., 1985; Feramisco et al., 1985; Stephen et al., 2014). Building on our success with Mbs directed to RAS, we developed a Mb, R15, that selectively binds apo RAS but lacks detectable binding to the GTP- or GDP-loaded states. Unexpectedly, when expressed in cells, R15 inhibited a subset of oncogenic RAS mutants, although biochemical binding assays showed no selectivity toward these mutants in vitro. Further, R15 trapped a subset of oncogenic RAS proteins in the apo state and inhibited their oncogenic activity. Thus, our studies demonstrate that it is indeed possible to target the nucleotide state of RAS and establish the possibility of selectively inhibiting a subset of oncogenic RAS mutants by targeting the apo state with drug-like molecules.
RESULTS
Monobodies selective for apo RAS
Using previously established methods (Spencer-Smith et al., 2017a; Teng et al., 2021), we developed the R15 Mb using EDTA-treated HRAS as a target. Clone characterization was performed using apo RAS samples that were confirmed to be predominantly free of bound nucleotides (Figures 1A and S1A). R15 exhibited high nanomolar affinity for the apo state of all three RAS isoforms but undetectable binding to GTP or GDP-loaded RAS (Figures 1B and C). When expressed in cells as a fusion with cyan fluorescent protein (CFP), R15 preferentially interacted with HRAS(K16N) over wild type (WT) and HRAS(G12V), consistent with HRAS(K16N) residing in the apo state (Sigal et al., 1986; Figure S2A). Furthermore, R15 bound HRAS(D119N), which has reduced affinity for nucleotide (Cool et al., 1999). Surprisingly, however, R15 also robustly interacted with oncogenic KRAS(G13D) and HRAS(Q61L) (Figure S2A). Although these mutants are transforming due to their shift to the GTP-bound state, biochemical analyses indicate that they exhibit elevated spontaneous nucleotide exchange activity (Smith et al., 2013), suggesting that R15 may trap these mutants in the apo state in cells. Given the pattern of RAS mutants that interacted with the apo-specific Mb, we sought to further characterize its activity both in vitro and in vivo.
Figure 1. Binding of apo-specific Mb to RAS.
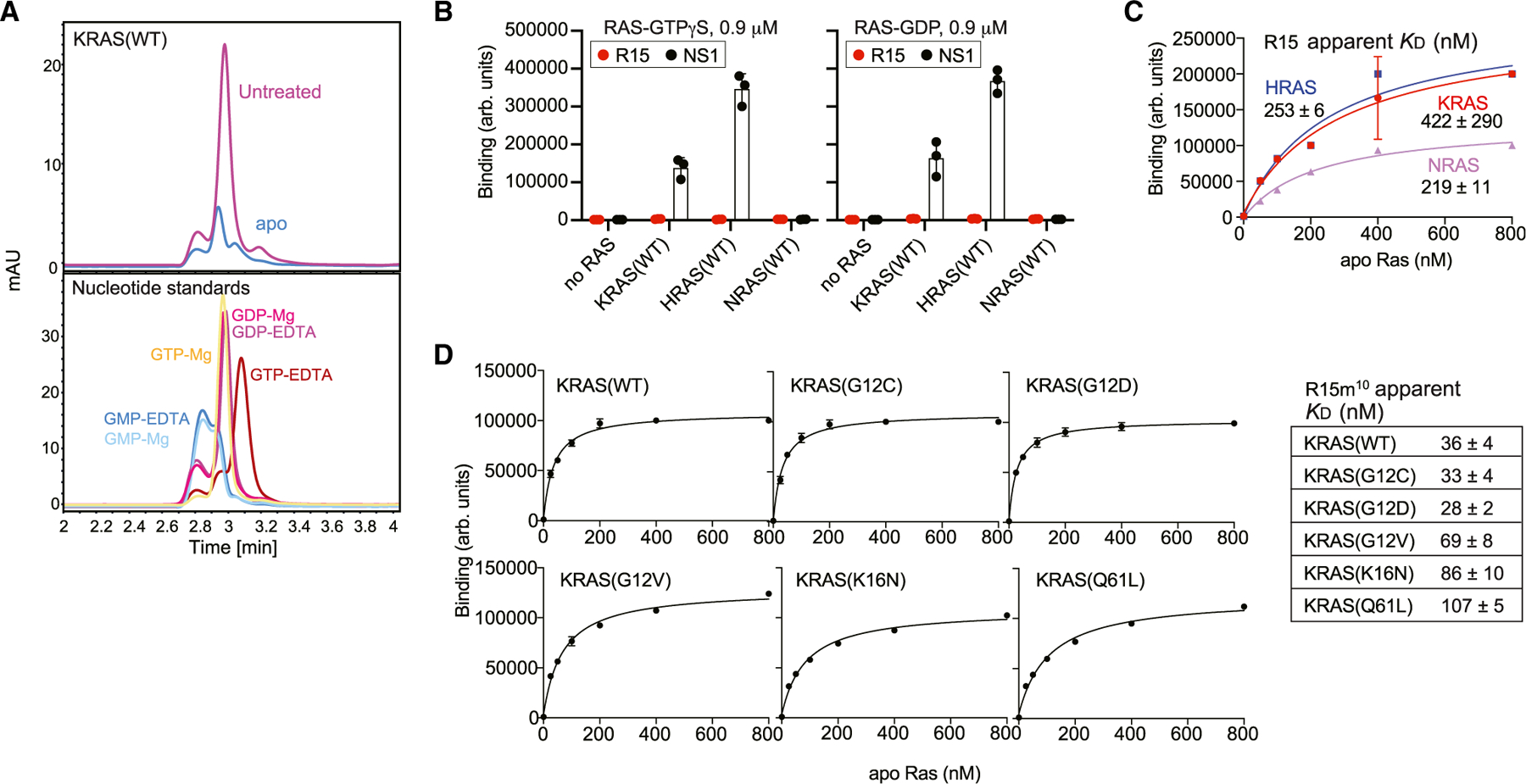
(A) Confirmation of the nucleotide-free nature of apo RAS preparations (Jeganathan et al., 2018). Chromatograms showing nucleotides released from RAS samples (top) and free nucleotide standards in the presence of 0.1 mM EDTA or 0.5 mM MgCl2.
(B) Binding of R15 Mb to GTP- or GDP-loaded RAS and to apo RAS, as measured using yeast surface display. NS1 Mb, which binds to GTP- and GDP-bound states of HRAS and KRAS, was used as a positive control.
(C) Binding titration of R15 Mb expressed on yeast cell surface to nucleotide-free RAS isoforms using flow cytometry is shown. Apparent KD values are shown (mean and SD; n = 3; technical replicates).
(D) Binding titration of R15m10 to various apo KRAS proteins. The table shows apparent KD values (mean and SD; n = 3; technical replicates).
See also Figure S1 for nucleotide-state specificity of R15m10.
Due to difficulties in expressing R15 in cells, we generated derivatives with better expression and stability, and we focused subsequent studies on one clone, R15m10. Like the parental R15 clone, R15m10 bound selectively to the apo state of all three RAS isoforms with no detectable binding to GTP- or GDP-loaded states of any RAS isoform (Figures 1D and S1B). Given the preferential interaction of R15 for some RAS mutants in cells, we tested whether this selectivity reflected an in vitro preference for these mutants. R15m10 bound exclusively to the apo state of each KRAS mutant with only small differences in affinity, within 4-fold changes in the apparent KD value, for a specific mutation (Figures 1D and S1B).
When expressed in cells, R15m10 (hereafter referred to simply as R15) exhibited similar specificity for RAS mutants as the parental R15 Mb clone, binding preferentially to HRAS(K16N) and (Q61L) versus (G12V), whereas NS1 Mb bound all three HRAS mutants as expected (Figure 2A; Spencer-Smith et al., 2017a). Furthermore, R15 did not interact with the RAS-related GTPase RRAS2(Q71L), which is equivalent to HRAS(Q61L) (Figure 2A). Thus, R15 is selective for the apo state of KRAS, HRAS, and NRAS.
Figure 2. Selectivity of R15 binding to RAS mutants in cells.
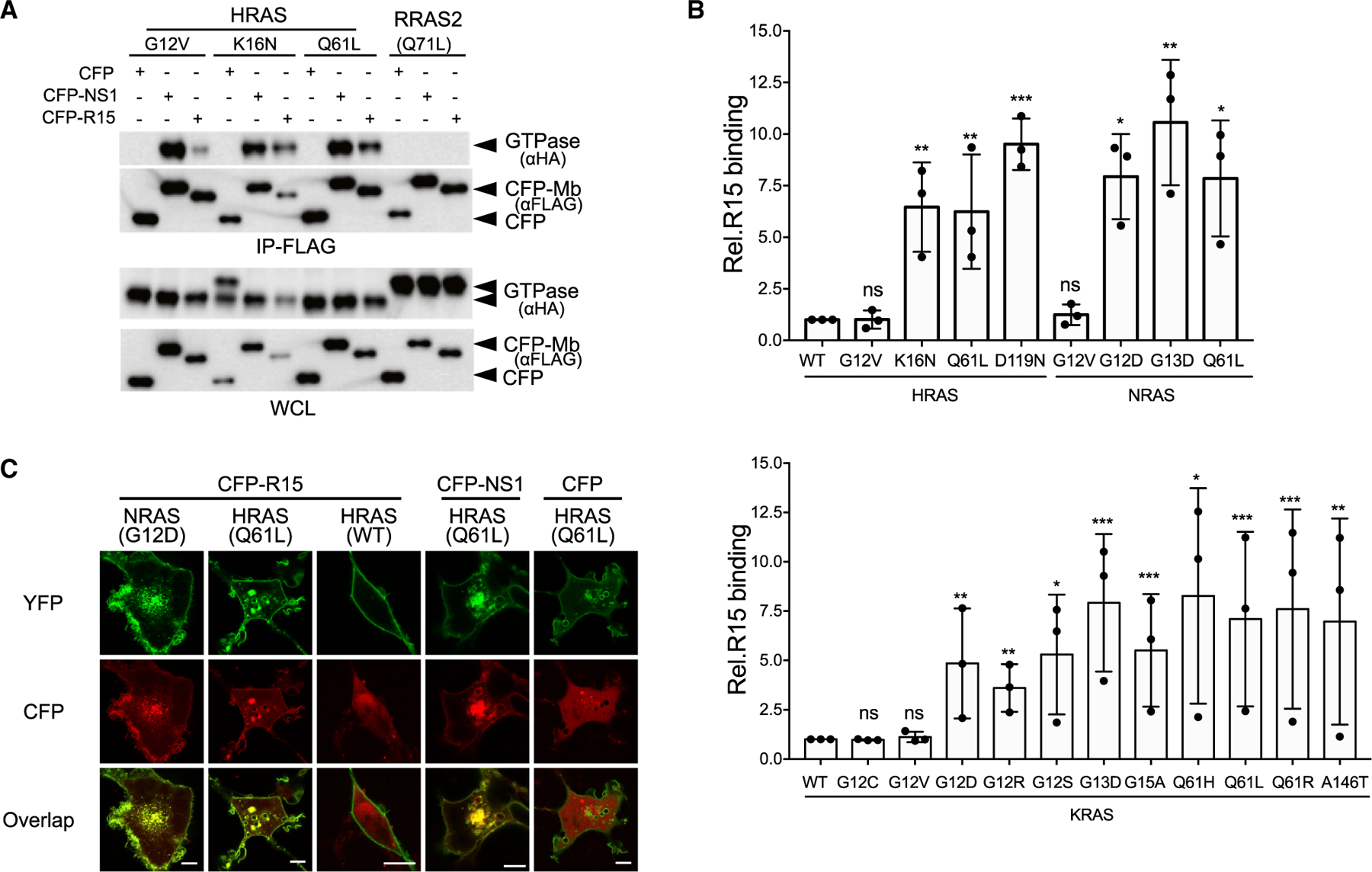
(A and B) Coimmunoprecipitation of hemagglutinin (HA)-tagged RAS isoforms with CFP-FLAG-R15.
(A) R15 interaction with HRAS and RRAS2 mutants. IP, immunoprecipitation; WCL, whole-cell lysate.
(B) Quantification of R15 interaction with various oncogenic mutant RAS proteins: HRAS and NRAS (top graph) and KRAS (bottom graph). Plotted values represent the relative binding of R15 to various isoforms of oncogenic mutants compared with wild-type RAS. For HRAS and NRAS mutants, binding is relative to HRAS(WT), and KRAS mutant binding is compared with KRAS(WT). The results represent the average of three biological replicates ± SD. ns, not significant. ***p < 0.0005, **p < 0.005, and *p < 0.05.
(C) Co-localization of CFP-R15, CFP-NS1, or CFP alone (pseudocolored red) with various YFP-tagged RAS proteins (pseudocolored green). Scale bars, 10 μm.
See also Figure S2.
To further examine the binding preference of R15 in cells, we examined the interaction of R15 with additional RAS mutants (Figures 2B, S2B, and S2C). R15 preferentially bound RAS mutants with reduced affinity for nucleotides, i.e., G15A, K16N, and D119N. Surprisingly, it also interacted with several oncogenic mutants, including G12D, G13D, Q61x (where x = L, H, R), and A146T. Many of these mutant RAS proteins exhibit elevated spontaneous nucleotide exchange activity (Bollag et al., 1996; Franken et al., 1993; Hunter et al., 2015; Smith et al., 2013; Wey et al., 2013), suggesting that R15 traps these mutants in the apo state upon release of nucleotide. In contrast, R15 bound weakly to G12V and G12C RAS mutants, consistent with their slower intrinsic exchange kinetics (Hunter et al., 2015; Smith et al., 2013; Wey et al., 2013). Intriguingly, R15 also bound to KRAS(G12R), which does not exhibit elevated spontaneous nucleotide exchange (Hunter et al., 2015; Moore et al., 2020), suggesting that R15 interaction is not solely dependent on the spontaneous exchange properties. Thus, in the cellular context, R15 is a pan-RAS but mutant-selective Mb that targets the oncoprotein branch of the RAS superfamily.
Cellular imaging analysis further supports the specificity of R15 (Figure 2C). CFP-tagged R15 co-localized with yellow fluorescent protein (YFP)-tagged NRAS(G12D) and HRAS(Q61L) at the plasma membrane and intracellular vesicles (Figure 2C). However, little co-localization of CFP-R15 was seen with YFP-HRAS(WT) (Figure 2C). These data indicate that R15 selectively binds and captures a subset of oncogenic RAS mutants at cellular membranes.
R15 traps RAS in the nucleotide-free state in cells
Based on the in vitro specificity of R15 for apo RAS and its preferential binding in cells to several RAS mutants with either accelerated nucleotide exchange or decreased nucleotide affinity, we next tested whether R15 traps RAS in the apo state in cells. We designed a strategy to determine the nucleotide state of various KRAS mutants bound to either NS1 or R15 Mb (Figure 3A). KRAS(Q61L) immunopurified with NS1 bound the glutathione S-transferase (GST)-RAF RBD independent of GTPγS addition (Figure 3B, lanes 3 and 4), consistent with the ability of NS1 to bind GTP-loaded oncogenic KRAS (Spencer-Smith et al., 2017a, 2017b). In contrast, KRAS(Q61L) immunopurified with R15 bound weakly to GST-RAF RBD. However, addition of GTPγS to the released R15-purified KRAS(Q61L) resulted in a significant increase in GST-RAF RBD binding, suggesting that R15-purified KRAS(Q61L) was nucleotide free (Figure 3B, lanes 5 and 6). Similar results were observed for additional KRAS mutants (Figures 3C and 3D). Although R15 only weakly bound the G12V mutant (Figure 2), the portion of RAS(G12V) immunopurified with R15 was also nucleotide free (Figures 3E and 3F). To rule out the possibility that Mb-trapped RAS proteins remained nucleotide loaded but spontaneously released nucleotide in vitro, we incubated the NS1-purified KRAS proteins with either GDP or GTPγS and tested for binding to GST-RAF RBD. The addition of GDP did not affect binding of immunopurified KRAS mutants to GST-RAF RBD, demonstrating that the addition of exogenous nucleotide to Mb-purified RAS proteins does not displace the bound nucleotide (Figure 3G). Together, these data indicate that R15 traps RAS in the apo state in cells.
Figure 3. R15 captures apo RAS in cells.
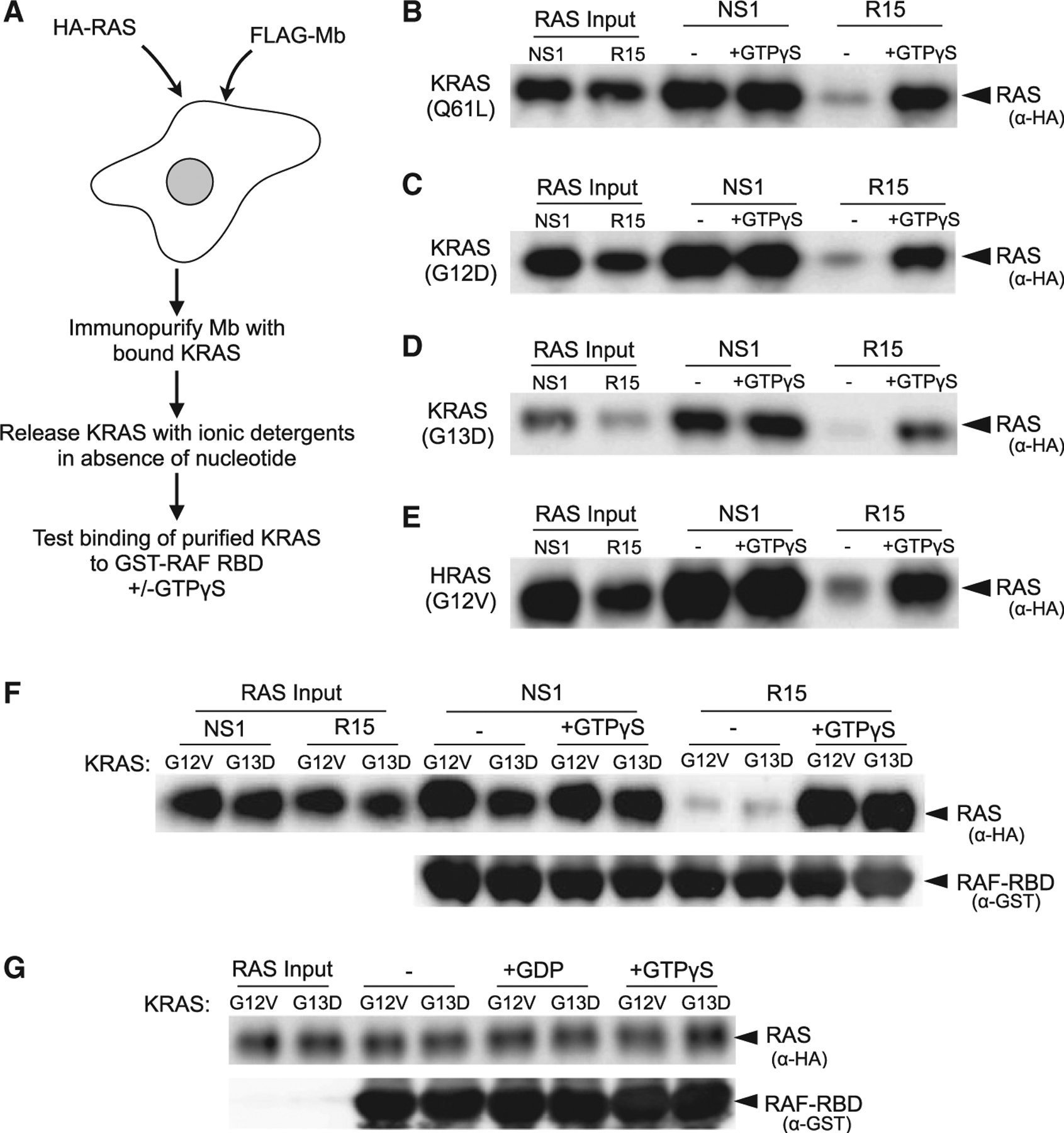
(A) Experimental strategy. HEK293 cells were co-transfected with constructs expressing the indicated CFP-FLAG-Mb and a selected HA-tagged oncogenic RAS mutant. Following a FLAG immunoprecipitation, Mb-bound RAS proteins were eluted from the immunocomplex (see STAR Methods) and then tested for binding to GST-RAF RBD in the absence (−) or presence (+) of added GTPγS.
(B–E) Different RAS mutant proteins were purified and then tested for binding to GST-RAF RBD in vitro. Lanes 1 and 2 represent the input of RAS protein eluted and purified from the indicated Mb. Lanes 3 and 4, binding of NS1-purified RAS protein to GST-RAF RBD in the absence (−) or presence (+) of added GTPγS; lanes 5 and 6, binding of R15-purified RAS protein to GST-RAF RBD in the absence (−) or presence (+) of added GTPγS. The specific RAS mutant protein examined is indicated on the left side of panel.
(F) Experiment was performed with either NS1 or R15 Mbs as in (A) using KRAS(G12V)- or KRAS(G13D)-expressing cells. −, absence of added GTPγS; +, presence of added GTPγS. The experiments were repeated at least two times for each RAS mutant.
(G) KRAS proteins immunopurified with NS1 and then incubated in the absence (−) or presence (+) of added GDP or GTPγS as indicated demonstrate that spontaneous nucleotide exchange in vitro is not responsible for binding to R15.
R15 competes with GEFs for binding RAS
Next, we used two approaches to determine the role of GEFs in generating the pool of apo RAS captured by R15. First, we tested whether disruption of RAS-SOS association altered R15-RAS interaction (Quilliam et al., 1996). Mutation of Asp69 to Asn, which disrupts GEF binding (Quilliam et al., 1996), enhanced the binding of R15 with both KRAS(Q61L) and (G12D) mutants (Figures 4A and 4B, lanes 1 and 2), Next, we measured the binding of R15 to KRAS mutants in the presence or absence of serum. Serum depletion should quiesce cells and reduce GEF association with RAS at the plasma membrane. Serum depletion enhanced the binding of R15 with KRAS(Q61L) and (G12D) (Figures 4A and 4B, compare lanes 1 versus 3). Furthermore, the combination of serum depletion and disruption of SOS binding (i.e., with the D69N mutation) further enhanced the binding of R15 with both KRAS mutants (Figures 4A and 4B). Similar results were seen with HRAS(G12V) and (Q61L) (Figure 4C). Different levels of enhanced binding of R15 were observed for different RAS mutations. Mutants with elevated intrinsic nucleotide exchange, e.g., Q61L, showed greater enhancement in R15 binding upon serum starvation and inhibition of GEF binding (Figure 4A). However, HRAS(G12V) binding to R15 was only moderately enhanced by disrupting GEF binding with the D69N mutation (Figure 4C). Mutation of Ala59 to Gly (A59G), which impairs GAP-stimulated hydrolysis of RAS (Lito et al., 2016; Lu et al., 2018), did not alter the binding of KRAS mutants to R15, indicating that RAS does not need to transition through the GDP-loaded state (followed by GEF-mediated GDP release) to bind R15 (Figure 4D). Thus, the improvements in binding of R15 to the oncogenic RAS mutants either by chronic serum starvation or by the introduction of the SOS-binding-defective D69N mutation are likely indicators that apo-RAS captured by R15 is mainly generated by spontaneous nucleotide exchange and independent of GEF-stimulated nucleotide exchange.
Figure 4. Exchange factors compete with R15 for binding to RAS.
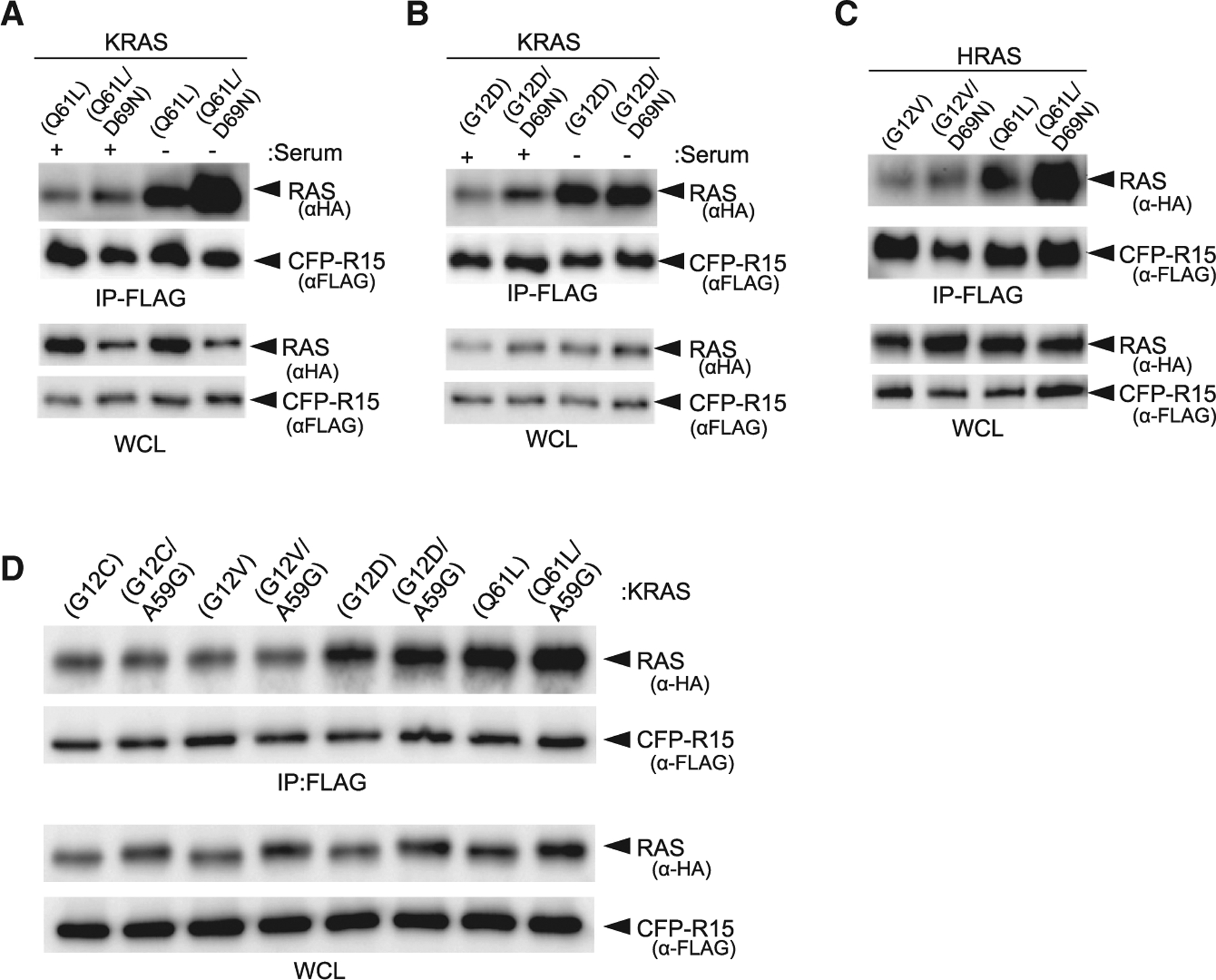
(A–C) Effects of impairing SOS binding either by serum starvation or D69N mutation on R15-RAS binding (A) KRAS(Q61L), (B) KRAS(G12D), and (C) HRAS(G12V) and HRAS(Q61L).
(D) Evaluation of A59G mutation on binding of R15 to various oncogenic RAS mutants.
R15 inhibits signaling and biological transformation mediated by a subset of oncogenic RAS mutants
Given the selective binding of R15 to a subset of RAS mutants, we next tested the impact of R15 on RAS-mediated signaling. When expressed as genetically encoded, intracellular reagents, both R15 and NS1 Mbs inhibited epidermal growth factor (EGF)-stimulated ERK-mitogen-activated protein kinase (MAPK) activation (Figure 5A). Furthermore, R15 selectively impaired ERK-MAPK activation by G12D, G12R, G13D, Q61L, Q61H, Q61R, and A146T mutants in all three RAS isoforms but had little effect on ERK-MAPK activation by either HRAS(G12V) or KRAS(G12V) (Figures 5B, S3A, and S3B). These results are consistent with the binding preference of R15 to RAS mutants (Figure 2). In contrast, NS1 inhibited ERK activation by all HRAS and KRAS mutants tested, but not NRAS, as previously described (Spencer-Smith et al., 2017a). Neither R15 nor NS1 inhibited ERK-MAPK activation by oncogenic kinases downstream of RAS, such as MEK(DD) and BRAF(V600E) (Figures 5B and S3B). Together, these results illustrate the selective inhibitory activity of R15 toward specific RAS mutants.
Figure 5. R15 inhibits RAS signaling and biological transformation.
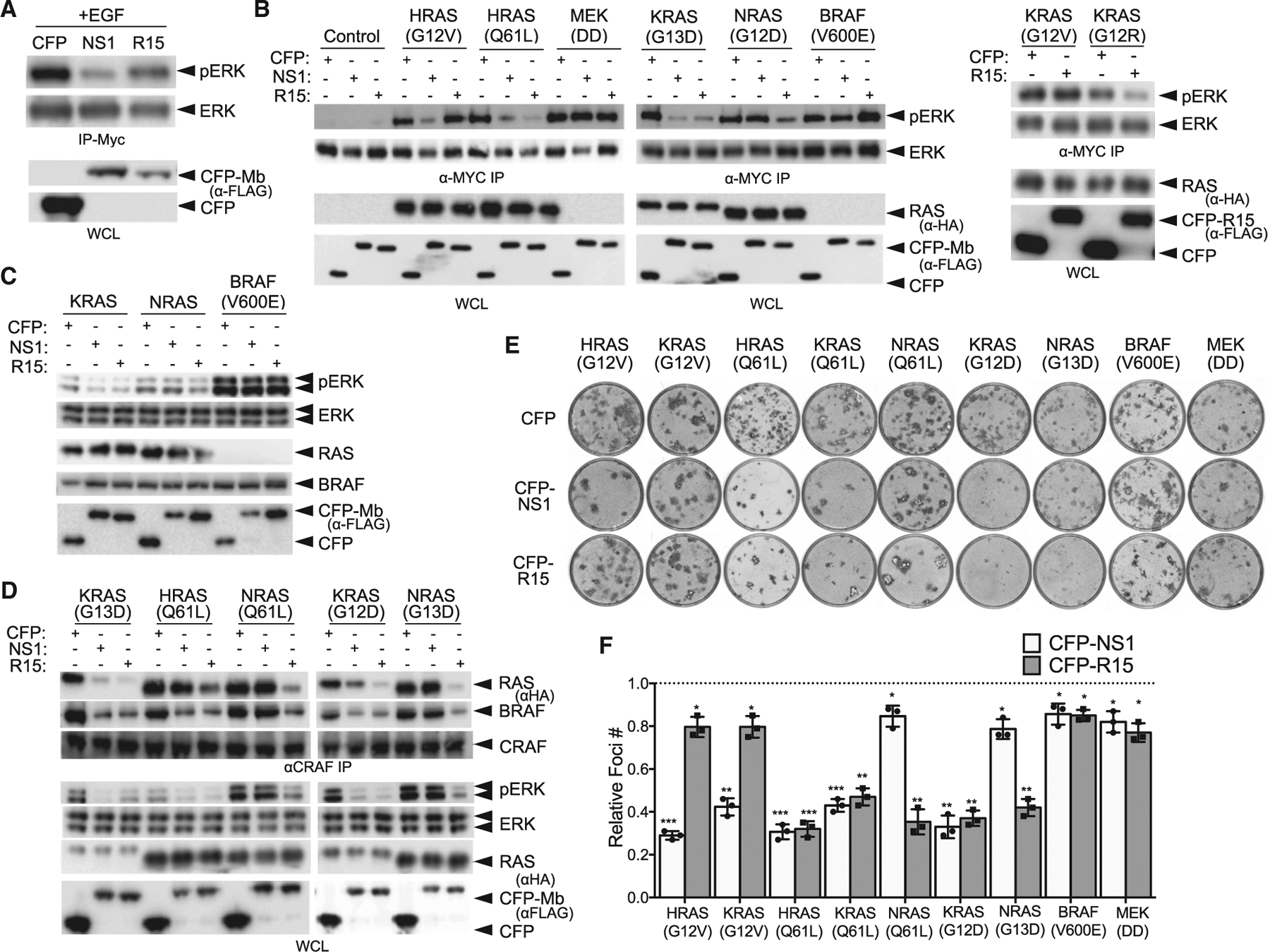
(A) Effect of CFP-R15 on EGF-stimulated ERK-MAPK activation in HEK293 cells. CFP, CFP-Mbs (NS1 or R15) and MYC-tagged ERK were co-expressed, and phosphorylation of MYC-tagged ERK was detected following MYC IP and western blot with phosphospecific ERK antibodies. CFP and CFP-NS1 were used as controls.
(B) Cells transfected with the indicated oncogene along with CFP or CFP-Mbs were analyzed for ERK activation as in (A). Quantification of results from (B) are presented in Figure S3B. The experiments were repeated three times for each mutant other than KRAS(G12R).
(C) Effects of R15 on ERK-MAPK signaling in isogenic MEFs expressing either a single RAS locus (KRAS or NRAS) or RASless MEFs rescued by expression of oncogenic BRAF(V600E). The experiments were repeated two times for each isogenic MEF cell line.
(D) Effect of R15 on heterodimerization of endogenous CRAF with RAS and BRAF is shown. HEK cells were co-transfected with the indicated expression constructs encoding oncogenic RAS and CFP or CFP-Mbs. After 48 h, cells were serum starved and cell lysates used to immunoprecipitate endogenous CRAF. The CRAF IPs were then examined for presence of HA-tagged RAS (top panel) and endogenous BRAF (middle panel). Levels of pERK were measured in WCLs to demonstrate efficacy of each Mb at inhibiting specific RAS mutants.
(E) NIH/3T3 cells were transfected with the indicated RAS mutants or oncogenic BRAF or MEK along with CFP or CFP-tagged Mb and allowed to sit at confluence for 2 to 3 weeks. Foci were stained with crystal violet and counted.
(F) Quantification of relative foci number from (E). Results represent the ratio of foci number in presence of CFP-Mb versus CFP alone and are the average of three independent biological experiments, each performed in technical triplicate ± SD. p values were determined by a Student’s t test between CFP and CFP-Mb for each oncogene. ***p < 0.001, **p < 0.01, and *p < 0.05.
See also Figure S3.
To further examine the specificity of R15, we tested the inhibitory activity of R15 in a panel of modified “RASless” mouse embryo fibroblasts (MEFs) (Drosten et al., 2010). Expression of CFP-R15, but not CFP alone, reduced pERK-MAPK levels in MEFs expressing only WT KRAS or NRAS but did not affect ERK activation in RASless MEFs rescued by expression of BRAF(V600E). In contrast, CFP-NS1 expression reduced pERK-MAPK levels in KRAS, but not NRAS or BRAF(V600E)-expressing MEFs (Figure 5C). These results demonstrate the selectivity of R15 at inhibiting RAS and a lack of off-target inhibition.
RAS activation results in the binding and heterodimerization of CRAF:BRAF that is necessary for RAF activation and subsequent MEK-MAPK activation (Freeman et al., 2013; Rajakulendran et al., 2009). Based on the ability of R15 to trap selected RAS mutants in the apo state, we anticipated that R15 would antagonize RAS-RAF interaction. Indeed, R15, but not CFP alone, reduced association of G12D, G13D, and Q61L RAS mutants with CRAF but did not affect the binding of the G12V mutant to CRAF (Figures 5D, S3C, and S3D). Further, R15 inhibited CRAF:BRAF interaction induced by these same oncogenic RAS mutants (Figures 5D, S3C, and S3E). These results are consistent with the expectation that R15 reduces the population of the GTP-bound state of RAS.
Given the inhibitory activity of R15 on RAS-mediated signaling, we next tested whether R15 impaired the biological activity of RAS. Consistent with the effects on signaling, R15 potently inhibited transformation of NIH/3T3 cells mediated by KRAS(G12D), H/K/NRAS(Q61L), and NRAS(G13D) with minimal effect on H/KRAS(G12V) (Figures 5E and 5F). In contrast, NS1 inhibited the focus-forming activity of all HRAS and KRAS mutants but was ineffective against NRAS, as expected (Spencer-Smith et al., 2017a). Further, neither R15 nor NS1 inhibited transformation by oncogenic BRAF(V600E) or MEK(DD) (Figures 5E and 5F), also as expected. These results highlight the selectivity of R15 for inhibiting a subset of oncogenic RAS mutants.
Targeting apo RAS inhibits tumor growth in vivo
Based on these results, we tested whether targeting apo RAS might be an effective approach to inhibit RAS-driven tumorigenesis in vivo. We established a chemical-genetic system to induce expression of R15 in a panel of human tumor lines expressing WT or mutant RAS proteins (Table S1). Consistent with the results from the cell signaling and focus formation assays, stable expression of R15 inhibited human tumor lines driven by specific RAS mutations (Figures 6, S4, and S5). DOX-induced expression of R15 reduced pERK-MAPK levels in Q61L, A146T, G12R, G12D, and G13D mutant RAS lines (Figures 6A–6D, S4A, and S4K). This effect was mirrored by a reduction in both proliferation (Figures 6F–6I) and anchorage-independent growth (Figures 6K–6N and S4F). In contrast, R15 did not inhibit ERK-MAPK activation (Figures 6E and S4E), proliferation, or anchorage-independent growth of G12V or G12C mutant lines (Figures 6J, 6O, and S4J), consistent with the inability of R15 to appreciably bind or inhibit these RAS mutants in cells (Figure 2). Although R15 did not reduce pERK-MAPK levels in Hec1AR15 and HCT-116R15 lines, there was a significant reduction in anchorage-independent growth of these lines, consistent with an apparent difference in signaling in 2D versus 3D growth conditions reported by others (Janes et al., 2018; Khan et al., 2019; Vartanian et al., 2013; Figures S4B, S4C, S4G, and S4H). Similar results were observed for H1299R15 (Figures S4D and S4I). In addition, A375R15 melanoma cells (BRAF(V600E)) were refractory to the inhibitory effects of R15 (Figure S4L).
Figure 6. Chemical induction of R15 expression inhibits signaling and growth of human tumor lines driven by select RAS mutations.
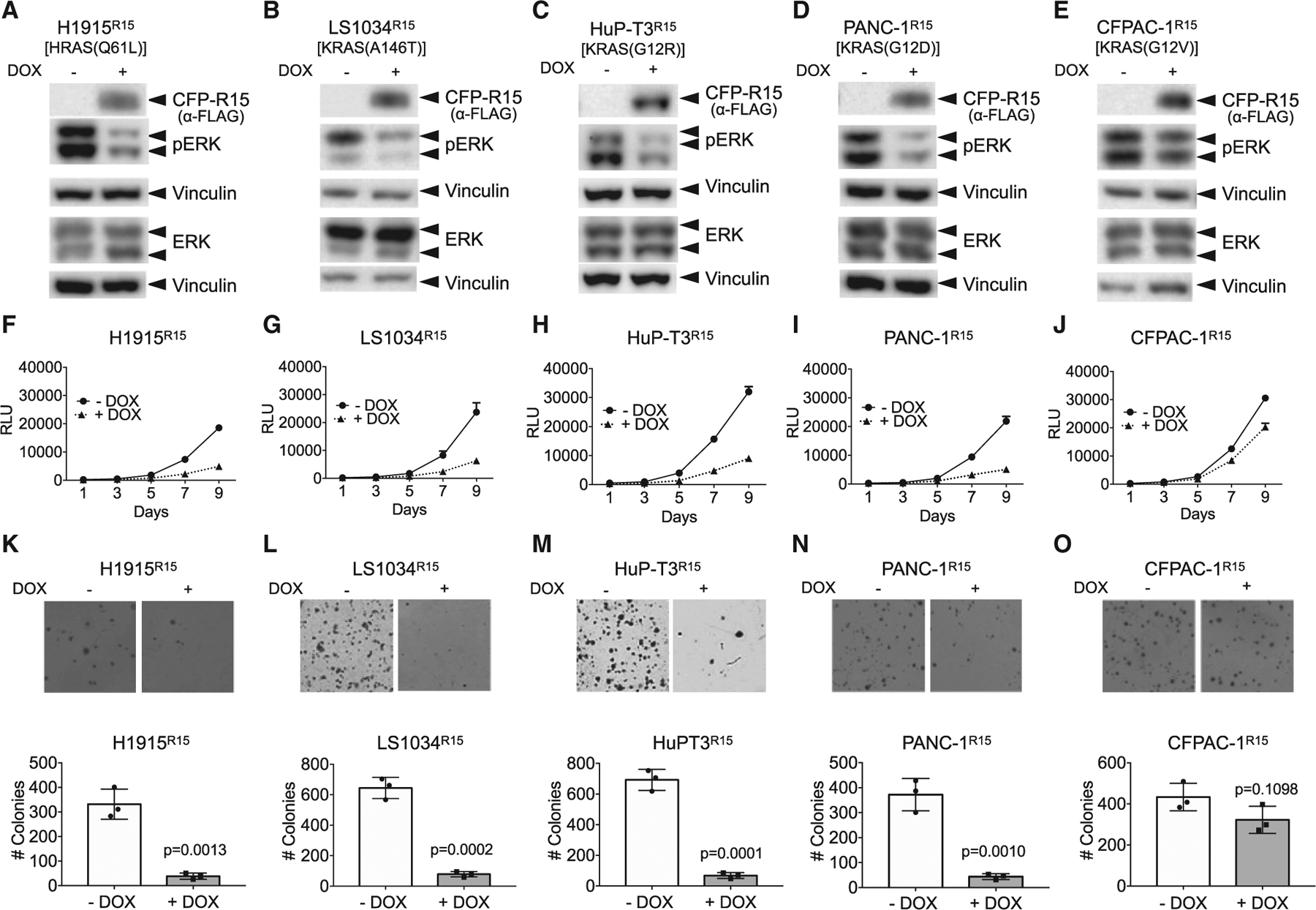
(A–E) Doxycycline (DOX)-inducible, R15-expressing stable lines were generated from RAS mutant tumor cells. ERK-MAPK activation was then measured ± DOX treatment by western blot analysis for pERK levels. The mutant RAS protein expressed in each tumor line is indicated above the panels. Vinculin expression was used as a control for loading.
(A, F, and K) H1915R15; (B, G, and L) LS1034R15; (C, H, and M) HuPT3R15; (D, I, and N) PANC-1R15; and (E, J, and O) CFPAC-1R15. Data from additional tumor lines are shown in Figure S4. The experiments were repeated three times for each cell line except H1944R15 and A375R15, which were repeated two times.
(F–J) R15 expression reduced the proliferation of a subset of RAS mutant human tumor cells. Results are the average of triplicate wells ± SEM shown by bars.
(K–O) R15 expression reduced anchorage-independent growth of a subset of RAS mutant human tumor cells. Engineered R15 cells were plated on soft agar in the absence (−) or presence (+) of DOX and allowed to grow for 3 to 4 weeks. Graphs represent the average colony number from three wells ± SD. Colonies were counted using NIH ImageJ software. Images are representative wells from each assay. The experiments were done in three technical replicates for each cell line. p values were determined by comparison of colony numbers between −DOX and +DOX conditions using a Student’s t test. Data on additional lines are shown in Figure S4. See Figure S5 for data on the effects of R15 on AKT activation.
The effects of R15 on phosphatidylinositol 3-kinase (PI3K)-AKT pathway varied in 2D growth conditions. Similar to its effects on RAS-MAPK signaling, the expression of R15 reduced the pAKT levels in H1915R15, LS1034R15, and PANC-1R15 cells (Figure S5) but enhanced pAKT levels in HCT-116R15 cells. This result is consistent with prior results in which the inhibition of RAS by NS1 resulted in increased pAKT levels in CFPAC-1R15 and Hec1aR15 cells (Khan et al., 2019). These data indicate that certain cells are more RAS dependent under 2D conditions, consistent with previously published studies (Janes et al., 2018; Khan et al., 2019; Vartanian et al., 2013). R15 expression did not impair the active AKT levels in KRAS(G12V) mutant CFPAC-1R15 cells (Figure S5E), consistent with the lack of effect of R15 on KRAS(G12V) in prior assays.
Next, we examined whether apo RAS might represent an effective therapeutic target to inhibit RAS-driven tumorigenesis in vivo. Doxycycline (DOX)-induced expression of R15 reduced tumor development in PANC-1R15, H1915R15, and HCT116R15 xenografts with only a moderate effect on CFPAC-1R15 xenografts, consistent with the selectivity of R15 for RAS(G12D), (G13D), and (Q61L) versus (G12V) mutants (Figures 7, S6, and S7). Interestingly, the DOX-treated cohort of H1915R15 did not form tumors by day 41 post-injection, at which point the control untreated (−DOX) groups reached endpoint criteria. Upon removal of DOX, tumors emerged in 50% of the animals; however, these tumors no longer responded to subsequent DOX treatment (Figure S6A). Analysis of these tumors revealed a loss of R15 expression and a lack of ERK inhibition (Figure S6B). DOX treatment of established tumors (50–100 mm3) in H1915R15-injected mice halted the tumor progression, and the average tumor sizes and tumor weights in this cohort (referred to as DOX(L)) were drastically lower than the untreated group (Figures S6D and S6F). Western blot analysis of tumor lysates revealed significant expression of R15 and a commensurate reduction in pERK-MAPK levels (Figure S6E). Although induction of R15 expression in HCT116R15 cells did not affect pERK-MAPK levels in 2D condition, there was a modest decrease in pERK-MAPK levels in tumors upon R15 expression (Figure S6H), consistent with our previous results observed following NS1 inhibition of KRAS in Hec1a cells (Khan et al., 2019).
Figure 7. R15 inhibits KRAS-driven tumor development.
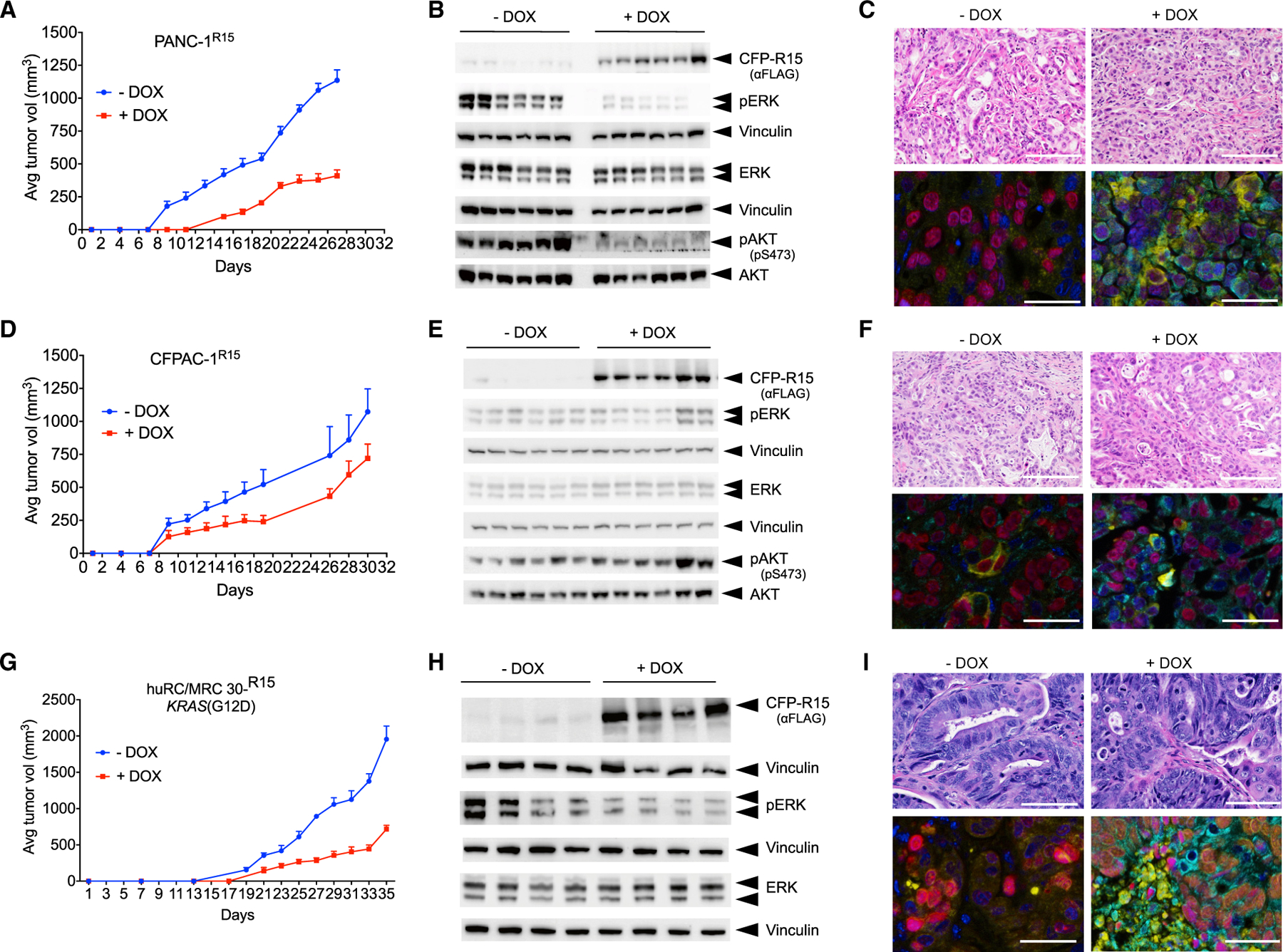
Athymic nude mice were injected subcutaneously in the flanks with PANC-1R15 (A) or CFPAC1R15 (D) cells. For PDXs, KRAS(G12D) mutant huRC/MRC30R15 colorectal (CRX) PDX was injected subcutaneously into the flanks of NSG mice (G). Mice were separated into two cohorts treated without (−) or with (+) DOX and monitored for tumor development.
(A, D, and G) Average tumor volume (n = 6 per condition for A and D and n = 4 per condition for G).
(B, E, and H) Effect of DOX-induced CFP-R15 expression on ERK-MAPK (B, E, and H) and AKT activation (B and E). Tumor lysates were probed for the indicated proteins by western blot. Vinculin was used as a loading control. Quantification of ERK activation from (B), (E), and (H), respectively, is shown in Figure S7.
(C, F, and I) H&E staining and multiplex immunohistochemical staining for various cohorts of PANC-1, CFPAC-1, and CRX PDX tumors. DAPI staining is in blue, GFP staining is in green, CC3 is in yellow, and Ki-67 is in magenta. The scale bars correspond to 50 μm. See Figure S6 for additional data on the effects of R15 on tumor development. Quantification of multiplex staining is shown in Figure S7.
Multi-panel immunohistochemical staining (IHC) coupled with image analysis algorithms of tumor tissue sections confirmed R15 expression (GFP) in DOX-treated cohorts of both CFPAC-1R15 and PANC-1R15 tumor sections (Figures 7C, 7F, S7C, and S7F). Concomitant with R15 expression, a robust decrease in Ki-67 and potent increase in caspase-3 cleavage was observed onlyin DOX-treated PANC-1R15, but not CFPAC-1R15, tumors (Figures 7C, 7F, S7C, and S7F).
To further examine the in vivo potency of R15, we transduced huRC/MRC 30 colorectal cancer PDX (KRAS(G12D)) with lentivirus encoding DOX-inducible CFP-R15 (Janakiraman et al., 2020). Induction of R15 expression with DOX treatment resulted in a delay in tumor development and a decrease in overall tumor burden (Figures 7G and S7G). Western blot analysis of tumor lysates confirmed R15 expression only in DOX-treated samples. R15 expression was accompanied by a decrease in ERK-MAPK activation (Figures 7H and S7H). H&E staining confirmed that the PDX tumor mirrored the architecture of human rectal cancers (Figure 7I). Further, multi-panel IHC staining demonstrated that R15 expression decreased proliferation and increased apoptosis, as seen by Ki-67 and cleaved caspase-3 profiling (Figures 7I and S7I). These results demonstrate that R15 selectively decreased proliferation (i.e., Ki-67 staining) and induced apoptosis (i.e., increased cleaved caspase 3) in multiple KRAS(G12D) xenograft models and highlight the potential feasibility and efficacy of targeting apo RAS as an approach to inhibit RAS-dependent tumorigenesis in vivo.
DISCUSSION
The success with covalent, allele-specific KRAS(G12C) inhibitors has been highly promising, as illustrated by the recent FDA approval of sotorasib/AMG510. However, non-KRAS(G12C) mutants lack readily tractable chemistries for direct covalent inhibition utilized with KRAS(G12C). Our findings open a new window of opportunity for pharmacological inhibition of a broad range of oncogenic RAS mutants. Despite the low picomolar affinity of RAS for guanine nucleotides in vitro and the high cellular concentrations of guanine nucleotides (Traut, 1994), R15 Mb captured apo RAS in cells and inhibited oncogenic RAS signaling and cellular transformation in vitro. Likewise, chemically regulated expression of R15 in select KRAS-mutant tumor and PDX models reduced tumor growth in vivo. Thus, our results reveal that it is possible to trap oncogenic RAS in the apo state and effectively inhibit its ability to couple to downstream effectors. Although R15 in its current form is not a suitable therapeutic, isolation of small-molecule mimetics of R15 may provide a viable approach to develop compounds that trap these KRAS mutants in the apo state in cells. This view is supported by a cell-permeable compound, BIM-46187, that trapped and inhibited heterotrimeric G-protein (Gαq) in a nucleotide-free conformation (Schmitz et al., 2014). Thus, our results provide a tractable approach to develop pharmacological inhibitors to a wider array of oncogenic KRAS mutants.
The activity of R15 toward specific oncogenic RAS mutants, i.e., G12D, Q61L, G13D, and A146T, can be rationalized in part by their elevated spontaneous exchange of GTP (Bollag et al., 1996; Franken et al., 1993; Hunter et al., 2015; Smith et al., 2013; Wey et al., 2013). Further, real-time nuclear magnetic resonance (NMR) studies demonstrated that the proportion of GTP-bound RAS in live cells is significantly lower than estimates made based on in vitro activity (Zhao et al., 2020). In addition, recent studies have highlighted that RAS-SOS complexes specifically for KRAS(G13D) are dominated by the nucleotide-free state, consistent with its elevated spontaneous nucleotide exchange rates (Moghadamchargari et al., 2021). Together, these studies suggest that RAS mutants, particularly “fast exchange” mutants, may transition through the apo state more frequently, thereby providing a potential opportunity to pharmacologically target this state of RAS.
While enhanced GTP dissociation explains the preference of R15 for a number of oncogenic RAS mutants, what accounts for the ability of R15 to target RAS mutants lacking fast exchange activity, such as G12R? One possibility is a difference in their association with GEFs. Disrupting GEF:RAS interaction (i.e., RAS(D69) mutation) increased RAS association with R15 (Figure 4), suggesting that R15 and GEFs either compete for overlapping binding sites on RAS or, alternatively, GEFs may allosterically inhibit RAS:R15 association. Indeed, SOS exhibited decreased catalytic activity toward KRAS(G12R) compared with WT and KRAS(G12V), suggesting a decrease in their interaction (Hobbs et al., 2020). Thus, decreasing the affinity of RAS for GEFs may enhance the probability of R15 capturing RAS proteins with slower spontaneous exchange rates.
Although a nucleotide (e.g., GTP) should rapidly bind apo RAS in the GEF:apo RAS complex, resulting in GEF dissociation and formation of nucleotide-bound RAS, R15 appears capable of inhibiting the binding of GTP and GDP to apo RAS. Similar results were seen with PIK3C2B, a target of apo HRAS (Wong et al., 2012). Given that the affinity of apo RAS for nucleotide is nearly 1,000-fold higher than for R15 (low pM versus nM) coupled with the sub-millimolar cellular concentrations of guanine nucleotides (Traut, 1994), we speculate that R15 allosterically inhibits nucleotide binding, i.e., reduces the affinity of RAS for nucleotide, rather than, or in addition to, directly competing for the nucleotide binding pocket. Thus, a combination of factors may determine the relative susceptibility of RAS mutants to R15-mediated binding and inhibition: nucleotide affinity (e.g., G15A, K16N, and D119N), exchange factor binding (e.g., G12R), and spontaneous nucleotide exchange rates (e.g., G12D, G13D, Q61L, and A146T). The inability of A59G mutation to affect the association of RAS with R15 (Figure 4D) further supports the premise that R15 binding is not dependent on RAS cycling through the GDP-bound state and subsequent GEF-mediated nucleotide dissociation. Future structural studies will address these questions.
Although the G12V and G12C mutants have lower intrinsic exchange rates and are less sensitive to inhibition by R15 (Figures 5, 6, and S3; Hunter et al., 2015; Killoran and Smith, 2019; Moore et al., 2020; Smith et al., 2013), the binding profile of R15 (Figures 2, 3, and S2) suggests that all RAS proteins have some degree of spontaneous nucleotide release activity and hence propensity to attain the apo state. Despite the reduced binding of R15 to RAS(G12V), (G12C), and WT relative to other RAS mutants, R15 nevertheless exhibited some inhibitory activity toward each of these RAS proteins (Figures 5, 6, and 7). We attribute this result to the longer duration of these experiments and the ability of R15 to bind and trap apo RAS, even though it forms more slowly in the context of these RAS proteins. Indeed, R15 immunopurified apo RAS from RAS(G12V)-transfected cells, although this required 5-fold more lysate to isolate levels of apo RAS comparable with that isolated from KRAS(Q61L)- or (G13D)-expressing cells (Figure 3).
Limitations of the study
Due to the inability to produce well-behaving purified protein samples of R15, we have been unable to determine the precise molecular interaction between R15 and apo RAS. Nevertheless, our data (Figure 4) suggest that R15 likely disrupts the switch regions to impair GTP loading and effector recruitment. Whether this is through direct binding of R15 to this region or allosteric regulation of the switch regions is not clear. Further studies will be necessary to distinguish these possibilities. Despite this limitation, our findings reveal the possibility for selective inhibition of a majority of oncogenic RAS mutants through targeting the apo state with “drug-like” molecules.
Our results with R15 further exemplify the power of Mbs as “tool biologics” to understand and explore vulnerabilities in RAS biochemistry, which can be exploited to inhibit RAS-dependent cancers. While RAS inhibitory Mbs are not viable therapeutics at this stage, due to their large size and poor cell penetrance, our studies nevertheless demonstrate the inhibitory activity of R15 as a genetically encodable reagent. Furthermore, rapidly advancing mRNA-delivery technologies currently employed for coronavirus disease 2019 (COVID-19) vaccines may enable effective delivery of mRNA-encoded Mb therapeutics. Indeed, a recent study delivered an mRNA-encoding, Mb-based RAS degrader into cells (Lim et al., 2021). In addition, development of small-molecule mimetics of R15 that target the apo state may provide a tractable approach to pharmacologically inhibit non-KRAS(G12C) mutant tumors. Furthermore, the ability of R15 to inhibit multiple RAS mutants provides an advantage over the exquisitely selective KRAS inhibitors, sotorasib and adagrasib. As anticipated, patients treated with these inhibitors develop resistance due in part to secondary mutations in RAS, either in cis or trans (Tanaka et al., 2021). However, the ability of R15 to target a wide range of RAS mutant proteins suggests that R15, or inhibitors that target apo RAS, would likely reduce the emergence of secondary RAS mutants as a mechanism of resistance. Thus, our studies provide a pathway forward for development of inhibitors that target a wider array of KRAS-mutant tumors than is currently possible with the available KRAS(G12C)-selective compounds.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and request for resources and reagents should be directed to and will be fulfilled by the lead contact, Prof. John P. O’Bryan (obryanjo@musc.edu).
Materials availability
All unique reagents (i.e., plasmids, cell lines, and monobodies) generated in this study are available from the lead contact with a completed materials transfer agreement.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This study did not generate any unique datasets or code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture and cell line authentication
HEK-293, HEK-293T and mouse embryonic fibroblasts (MEFs) were cultured in DMEM with 10% fetal bovine serum. NIH/3T3 cells were cultured in DMEM with 10% calf serum. Various oncogenic RAS mutant or non-RAS mutant cell lines were grown in the media as per ATCC recommendations using 10% tetracycline negative FBS. Media for culturing the cells was purchased commercially from Corning. The transient transfection in HEK-293, HEK-293T, MEFs and NIH/3T3 cells were done using polyethyleneimine (PEI). For HEK-293 and HEK-293T, typically, 3 μL of 1mg/ml PEI stock was used for each μg of DNA in Opti-MEM reduced serum media (Life Technologies). Briefly, Opti-MEM-PEI mixture was incubated for 10 minutes at room temperature. Next, DNA was added to Opti-MEM: PEI cocktail and incubated for at least 20 minutes at room temperature. The Opti-MEM: PEI-DNA mixture was added to cells in serum-free media and incubated for three hours after which the media was replaced with fresh complete media. Transfections in MEFs and NIH/3T3 cells were done with same procedures, however, 5 μL of 1mg/ml PEI stock was used for each μg of DNA and media was replenished after five hours. All human cell lines used in this study were authenticated by STR DNA profiling analysis at the Genome Research Core at University of Illinois at Chicago (UIC).
Animal models
For patient derived xenograft experiments, 4–8 week old NSG mice (both male and female) were used for subcutaneaous injection of cells. For nude mouse xenograft experiments, 5 week old athymic nude mice (both male and female) were used for subcutaneous injection of cells. All animal experiments were approved by the Institutional Animal Care and Use Committees of both the Ralph H. Johnson VA Medical Center and the Medical University of South Carolina prior to initiation. Mice were maintained in autoclaved, individually vented caging with sterilized corncob bedding and nesting material, irradiated chow and reverse osmosis water in bottles.
METHOD DETAILS
Development of monobodies
RAS proteins (residues 1–174) were produced as N-terminal fusions with a HIS-tag, Avi-tag and a TEV protease cleavage site as described previously (Teng et al., 2021). HRAS protein (residues 1–166) in the same format was produced as described previously (Spencer-Smith et al., 2017a). EDTA-treated RAS samples were produced by incubating the protein in 20 mM Tris-HCl buffer (pH 7.5) containing 10 mM EDTA and 0.5 mM DTT at 30°C for 30 min, followed by cooling on ice and diluted in 50 mM Tris-HCl buffer (pH 7.5) containing 150 mM NaCl, 10 mM EDTA and 0.5 mM DTT. Nucleotide exchange reactions to produce GTPγS- or GDP-bound RAS samples were performed by incubating the protein first in 20 mM Tris-HCl buffer (pH 7.5) containing 15 mM EDTA, 5 mM MgCl2, 0.5 mM DTT and 1 mM GTPγS or GDP at 30°C for 30 min, followed by cooling on ice and addition of MgCl2 to a final concentration of 20 mM. Nucleotide exchanged RAS protein was diluted in 50 mM Tris-HCl buffer (pH 7.5) containing 150 mM NaCl, 20 mM MgCl2 and 0.5 mM DTT.
Apo RAS was produced as follows. RAS protein was diluted 1:10 in nucleotide loading buffer (NL buffer; 25 mM HEPES buffer pH 8.0 containing 125 mM ammonium sulfate, 1 mM EDTA, 100 μM diethylenetriaminepentaacetic acid, and 0.2 mM TCEP). The sample was concentrated 10-fold followed by 10-fold dilution with NL buffer using an Amicon-0.5 concentrator (MilliporeSigma, catalog number UFC501024). This buffer exchange process was repeated a total of three times. Two micrograms of biotinylated alkaline phosphatase (ThermoFisher, catalog number 29339) was complexed with 400 μL Streptavidin MagneSphere® Paramagnetic Particles (Promega, catalog number Z5481). The beads were washed twice with TBS (50 mM TrisHCl, 150 mM NaCl, pH 7.5) containing 0.1 mM zinc sulfate. Two nanomole RAS protein in NL buffer was incubated with the immobilized alkaline phosphatase at 4°C overnight. The magnetic beads were removed using a magnetic stand, and the sample was buffer-exchanged using an Amicon-0.5 concentrator first with 25 mM HEPES buffer (pH 7.5) containing 100 mM NaCl, 1 mM EDTA, and 0.1 mM TCEP, followed with 25 mM HEPES buffer (pH 7.5) containing 100 mM NaCl, 0.1 mM EDTA, and 0.1 mM TCEP. The reduction of EDTA concentration was necessary for nucleotide detection (see below), because excess EDTA obscured signals from nucleotides. For a control, untreated RAS protein was diluted in 25 mM HEPES buffer (pH 7.5) containing 100 mM NaCl, 0.5 mM MgCl2, and 0.1 mM TCEP. Similar to EDTA, it was necessary to reduce the MgCl2 concentration to 0.5 mM, because the nucleotide peaks in the nucleotide detection assay shifted when higher concentrations of MgCl2 is added.
The nucleotide-free state of the apo RAS samples was confirmed by detecting nucleotides using a modified protocol from a published method (Jeganathan et al., Biochemistry 2018). An Eclipse C18 column (Agilent, 4.6 mm × 150 mm) was equilibrated in 50 mM potassium phosphate buffer pH6.6 containing 10 mM tetrabutylammonium bromide in 16% acetonitrile, and RAS proteins and control nucleotides were injected after heating at 98°C for 5 min and centrifugation for 5 min. Samples were buffer-exchanged using an Amicon concentrator to remove unbound nucleotide, if any, prior to the analysis.
We developed Mb R15 following a previously published method that combines phage display and yeast display technologies (Koide et al., 2012). Four rounds of phage display library sorting using 100 nM apo HRAS (residues 1–166) and one round of yeast display library sorting using 200 nM apo HRAS (residues 1–166) were performed to enrich apo RAS binding Mbs, using the buffer supplemented with 1 mM EDTA, then negative sorting using 500 nM each of the HRAS(residues 1–166)-GDP and HRAS(residues 1–166)-GTPγS complexes was used to eliminate Mbs that were not specific to apo RAS, using the buffer supplemented with 20 mM MgCl2. R15m10 was identified from a library based on R15 in which residues 75, 77, 79, 81 and 83 in the FG loop were randomized to all 20 amino acids. Two rounds of yeast-display library sorting were performed using 500 and 100 nM apo HRAS (residues 1–174) for the first and second rounds, respectively.
Affinity measurements were performed using the yeast display format using previously published method (Koide et al., 2012) with RAS proteins (residues 1–174), with modifications of using 10 mM EDTA-supplemented buffer for apo RAS and 20 mM MgCl2-supplemented buffer for RAS/GTPγS and RAS/GDP. Fluorescence intensity was measured using an iQue Screener Plus instrument (Sartorius).
Generation of mammalian cell expression construct for R15 Mb
R15 Mb was cloned either into CFP-tagged, pECFP-C1 for transient expression and pCW5.1 for stable cell expression using Gibson assembly. Briefly, R15 was PCR amplified from the from bacterial expression vector pHBT by two separate PCRs using 5′ Mb Gibson primer (GACGATGACGACAAGGGATCCGTTTCTTCTGTTCC) and a 3′ Mb Gibson primer (TCAGTTATCTAGATCCGGTGGATCCCTAGGTACGGTAGTT AATCGAGATTGG) in one reaction and 5′ Mb Gibson primer and 3′ pCW Mb Gibson primer (GGCGCAACCCCAACCCCGGCCTAGGTACGGTAGTTAATCGAGATTGG) in the other. The two resulting amplicons (325 and 320 bp respectively) were mixed with BamH1 digested vectors (pECFP-NS1 and pCW5.1-CFP-NS1) and ligated using Gibson Assembly Mix (New England Biolabs) and then used to transform E. coli. The resulting transformants were screened for inserts and sequenced to confirm mutants.
Confocal Microscopy
To study the co-localization of RAS and Monobodies,COS-1 cells were co-transfected with vector encoding CFP-Mb (NS1 or R15)along with indicated YFP-RAS mutant using PEI transfection. The vector encoding CFP alone serves as negative control for co-localization studies. Forty-eight hours post transfection, cells were imaged live, and images captured on Zeiss LSM 880 Confocal Microscope.
NIH/3T3 focus formation assays
Freshly revived NIH/3T3 cells were passaged no more than twice and then seeded in 60 mm dishes to a density of 2.5 × 105 cells in complete media. Cells were co-transfected with the indicated RAS mutant or downstream oncogenic kinases, negative control CFP-alone vector, positive control CFP-NS1 vector and test CFP-R15 vector using PEI. Cells were replenished with fresh media every two days. For oncogenic RAS, foci begin to emerge approximately 10 days post-transfection and assay is usually terminated by 2 weeks due to the size and number of foci. However, the foci for downstream oncogenic kinases usually appear after two weeks and were terminated after three weeks. Foci were stained with 0.1% crystal violet and counted. All assays were performed in triplicate and repeated three times.
Stable cell generation of inducible R15
The viral particles for DOX-inducible R15 expression were generated in HEK-293T cells. The packaging HEK-293T cells were transfected with pCW57.1-CFP-R15 (transfer plasmid) along with a plasmid encoding packaging plasmid (pCMVdR8.74) and the viral envelope (pMD2.G) in 4:3:1 ratio using calcium phosphate. Next day packaging cells were placed in fresh media and on day 2 post-transfection, conditioned media from the HEK-293T cells were collected, filtered using 0.45μm syringe filters. The filtered supernatant was used to infect cells and cells were selected using puromycin. The concentration of puromycin for selection was cell line dependent. Following selection, colonies were pooled to generate a polyclonal cell line that was used for all subsequent analyses.
Immunoblotting and antibodies
For transient transfections, 48 hours post transfection and for stable cells 48 hours after DOX administration, cells were serum starved O/N. Following day, cell lysates were made by washing cells once in cold PBS followed by lysis using PLC buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 10% glycerol, 1% Triton X-100, 1 mM EGTA, 1.5 mM magnesium chloride, 100 mM sodium fluoride supplemented with 1 mM vanadate, 10μg/ml leupeptin and 10μg/ml aprotinin). To generate tumor lysates, tumors were harvested, transferred to microfuge tubes, and snap-frozen by immersing in liquid nitrogen. Tumor tissue (40–50 mg) was then homogenized in ~1 mL of cold PLC buffer on ice. Homogenates were passed through 70μm cell strainer to clear the lysates then centrifuged at 13,000 rpm for 20 min at 4°C. Supernatant was collected and transferred to fresh tubes. The lysates were directly used for protein estimation and analysis or stored at −80°C for later use. The following antibodies were used: monoclonal HA (clone 16B12, Biolegend #90154), polyclonal rabbit HA (Poly9023, Biolegend #923502), monoclonal FLAG (Clone M2, Sigma #F1804), polyclonal rabbit FLAG (Sigma #F7425), phospho-ERK (Thr202/Tyr204, CST #9101), total ERK (CST #9102), phosphor-AKT (Ser473, CST #9271), total AKT (CST #9272S), phospho-AKT (T308, CST #9275S), Vinculin (SC #73614), Anti-MYC (Clone A46, Millipore-Sigma #05–724), cleaved Caspase-3 (Asp-175, CST #9661), CRAF (BD Biosciences # 610151), BRAF (Santa Cruz #sc-9002), Anti-GST (Santa Cruz # sc-459).
R15 binding and cell signaling assays
For the in-cell binding assays, FLAG tagged CFP-R15 (CFP-R15) was co-transfected with the indicated GTPase mutant (RAS and RRAS2) into HEK293 cells using PEI. FLAG tagged CFP alone (CFP) and FLAG tagged CFP-NS1 (CFP-NS1) were used as negative and positive controls respectively. Forty-eight hours post transfection, cells were replenished with fresh serum containing media or serum starved O/N depending on the experimental purpose. Lysates were collected and analyzed by Western blot for the expression of RAS or RRAS2 and Mb (Mb). Following this, lysates were immunoprecipitated using FLAG antibody followed by Western blot for co-precipitation of the HA-tagged RAS or RRAS2. Western blots of R15 immunoprecipitates were quantified by Image Studio Lite (v.5.2.5, LICOR Biosciences). Ratio for IP fraction of RAS to R15 was determined for each condition. The resulting value was divided by the value of the RAS in the whole cell lysate (WCL) fraction for each oncoprotein. Binding of R15 to wild type RAS was arbitrarily set to 1. The relative binding of R15 for H/N-RAS mutants was compared to HRAS(WT) and for KRAS mutants to KRAS(WT).
For transient MAPK cell signaling assays, HEK293 cells were transfected with CFP-R15 or indicated HA-tagged RAS mutant or downstream oncogenic kinases [(MEKDD), BRAF(V600E)]. Again, CFP and CFP-NS1 were used as controls. Forty-eight hours after transfection, cells were serum starved overnight in DMEM alone then stimulated with EGF (100 ng/mL for 10 min) where indicated. Oncogene-transfected cells were not stimulated with growth factor. Cells were then lysed in PLC buffer and analyzed for effects on MAPK signaling as previously described (Spencer-Smith et al., 2017a). Western blots were quantified with Image Studio Lite (v.5.2.5, LI-COR Biosciences) using the Analysis function. Ratio of pERK divided by total ERK was determined for each condition. The resulting ratios were divided by ratio for CFP alone for each protein. Dotted line represents the level of ERK-MAPK activation by each RAS mutant in the presence of CFP and was arbitrarily set to 1.
The effects of R15 on RAS interaction with RAF and CRAF and BRAF association were also measured by co-immunoprecipitation (co-IP). Briefly, HEK293 cells were transfected with indicated constructs. After immunoprecipitation of endogenous CRAF, samples were analyzed by Western blot for coprecipitation of BRAF and HA-tagged RAS. Ratio for BRAF to CRAF was determined for each condition. The resulting ratio was divided by the ratio for the CFP alone sample for each oncoprotein. Dotted line represents the level of BRAF/CRAF activation by each RAS mutant in the presence of CFP and was arbitrarily set to 1. For determining the effects of R15 on RAS-RAF association, the ratio of IP fraction HA-RAS and CRAF was evaluated for each condition. Again, the values of each oncoprotein for NS1 and R15 were divided by CFP alone. HA-RAS/CRAF for each condition for CFP was arbitrarily set to 1.
For stable cell signaling assays in oncogenic cells, 2–4 μg/mL of DOX was added for 48 hours and then cells were serum starved overnight in the presence of DOX. Lysates were prepared the following day and analyzed by Western blot to determine expression of R15. The samples were then examined for effects of R15 expression on ERK-MAPK and PI3K-AKT signaling using phosphospecific ERK (pERK) or AKT [pAKT(S473)] antibodies along with the total protein antibodies (ERK or AKT).
Nucleotide status of RAS bound to Mbs
HEK293 cells were co-transfected with HA-tagged RAS mutant and FLAG-tagged Mbs (NS1 or R15). Forty-eight hours post transfection, lysates were collected and evaluated for expression of RAS (α-HA) and Mbs (α-FLAG). Once uniform expression was confirmed, Mbs were immunopurified using FLAG antibody. We used 3-fold more lysate for immunoprecipitation of RAS from R15 transfected lysates vs NS1 transfected cells as R15 preferentially binds apo-RAS which represents only a fraction of total RAS protein. Following immunopurification, Mb-bound RAS was dissociated by treating the immune complex with buffer containing 1% Deoxycholate and 0.1% SDS on ice for 5 min. Samples were then spun at 3500 RPM for 5 min and the supernatant containing eluted RAS saved. Treatment was repeated with detergent 3x, and eluted fractions combined. A fraction of total eluate was saved for input control and the remaining sample diluted 10-fold with PLC buffer. The diluted fractions were divided into two tubes with addition of MgCl2 to a final concentration of 15 mM. GTPγS was added to one tube and incubated on ice for 30 min. Next, samples were incubated with bead-bound GST-RAF RBD at 4°C for 30 min. RAF RBD-bound samples were collected by centrifugation at 3500 RPM for 5 min. Beads were then washed 3x with 1 mL PLC buffer then resuspend in 2x sample loading buffer. Samples were then analyzed by Western blot.
Proliferation assays
The effects of R15 on the growth inhibition of various human tumor lines in 2D tissue culture growth conditions were evaluated by proliferation assays. Briefly, cells were plated on 24-well plates in complete medium (DMEM or RPM1 with 10% FBS plus optimized dose of puromycin) and divided into −DOX and +DOX wells in triplicate for each condition. 2– 4μg/ml DOX was used to induce expression of R15 for the indicated number of days. On the indicated day, medium was removed and replaced with 100μL of serum-free DMEM, cells were harvested after 30 min at 37°C. Viability was assayed using CellTiter-Glo (Promega) and luminescence was quantified using Clariostar (BMG, Labtech) 96-well microtiter plate luminometer following the manufacturer’s instructions.
Soft agar colony formation assays
Soft agar colony formation assays were performed essentially as described elsewhere (Clark et al., 1995). A solidified base agar layer (0.5%) was topped with cell suspension in 0.33%soft agar and allowed to set. Cells were fed 1–2x per week by careful drop wise addition of growth media to top layer. Doxycycline (DOX; 2–4 ug/ml) was added to induce expression of R15. Two weeks after platting, cells were stained using MTT (100μL of 2mg/ml solution of MTT per well). Colony number and average colony size were quantified using ImageJ.
Multiplex immunohistochemistry and image analysis
Tumors harvested from mice were fixed in neutral-buffered formalin for 24 hours, washed in 70% ethanol and then processed and embedded in paraffin using standard techniques. Subsequently, 4–5 μm sections of FFPE tissue on Histobond plus slides were deparaffinized and stained using the Ventana Discovery Ultra automated immuno-stainer (Roche Diagnostics Corp. Indianapolis, IN) and Akoya OPAL™ reagents (Akoya Biosciences, Marlborough, MA). Heat-induced epitope retrieval (HIER) was performed in EDTA buffer pH 9 (Cat. #S2367 Agilent/Dako Santa Clara, CA) for 32 minutes at 95°C and endogenous peroxidase was blocked with a solution of hydrogen peroxide after incubation of the first primary antibody. Antibodies used included GFP (Cell Signaling Technologies, #2956), Ki-67 (Abcam, #16667) and cleaved caspase (Cell Signaling Technologies, #9661). After incubation with primary and secondary antibodies, the Akoya Opal tyramide signal amplification reagents were used for fluorescence detection. The following fluorophores were used: OPAL 480, OPAL 520, and OPAL 620. DAPI was used for nuclear counterstaining. Between each sequential antibody staining step, slides were incubated in citrate buffer pH 6 (Cell Conditioning Solution (CC2) Cat. #980–223, Roche Diagnostics) at 90°C for 8 min to remove the previous primary and secondary antibody complexes. Stained slides are mounted with ProLong™ Gold Antifade Reagent (Cat. #P36934, ThermoFisher) and imaged using the Akoya Vectra® Polaris™ Automated Imaging system (Akoya Biosciences, Marlborough, MA). Whole slide scans were done at 20X magnification and subsequently 8 regions of interest (ROIs) where chosen at random across each tumor for further analysis. Spectral unmixing and removal of autofluorescence was performed using the inForm® Software v2.4.10 (Akoya Biosciences, Marlborough, MA) and resulting images were exported in TIFF format for further analysis.
For imaging analysis, multispectral images were acquired at 320 magnification using the Akoya Vectra® Polaris™ Automated Quantitative Pathology Imaging System, 200 slides (Akoya Biosciences, Marlborough, MA). Standard settings were used for multispectral image acquisition. Multispectral image analysis of multiplex IHC stains was performed using inForm Image Analysis Software (Akoya Biosciences, Marlborough, MA). A representative set of training images were first loaded and spectrally unmixed by using spectral libraries generated from the library stains for each fluorophore and the autofluorescence slide. Next, a machine learning algorithm was trained by user-specified tissue annotations aided by the signal from the epithelial markers to accurately segment tumor tissue versus stromal tissue and background, as well as individual cells using the nuclear DAPI signal. All images were reviewed after batch processing; necrotic tissue, tissue folds, and other technical artefacts were excluded from further image analyses. Percentage of cells positive for the protein expression was calculated in segmented tumor tissue as the mean signal intensity within the respective cellular compartment.
Patient derived xenografts (PDXs) and nude mouse xenografts
For PDX generation, single cell suspensions were isolated from established PDX rectal tumor models endogenously harboring KRAS(G12D) mutation [(huRC/MRC 30)] (Janakiraman et al., 2020). The cells were resuspended in a media containing virus with MOI of 2 or higher. The cocktail of cells/virus with added polybrene was incubated 37°C for 4 hrs with gentle shaking at 50–70 RPM. Cells were centrifuged at 1500 RPM for 5 minutes; cell pellet was washed 3X with PBS and resuspended in 50% Matrigel with RPMI. Resuspended cell solution (100 ul with 0.5 × 106) was injected s.c. into the flanks of 4–8 weeks NSG mice. Two days after injections, mice were divided into −DOX and +DOX cohorts. The tumor progression in both cohorts was recorded my measuring tumor dimensions by digital caliper three times a week. Tumor dimensions were noted every two days with a digital caliper and the tumor volume was estimated as V(mm3) = π/6(length × width2).
For the xenograft tumor assays, five-week-old male or female athymic nude mice were purchased from Charles River Laboratories (CRL) or Taconic Biosciences and acclimatized for one week. A 100 μL suspension of 10 × 106 cells in a 1:1 solution (v/v) of serum-free RPMI/Matrigel basement membrane matrix was injected subcutaneously (s.c) into the flanks of mice. Two days after cell inoculation, mice were randomly assigned to control (−DOX) or treatment (+DOX) cohorts. Cells were assessed for their ability to form tumors in the absence (−DOX) or presence of Mb expression (+DOX). DOX was provided at 2 mg/mL in water supplemented with sucrose. For H1915R15 experiments, tumors were allowed to reach 50–75mm3, upon which mice were randomized into −DOX and +DOX groups (D15). All animal experiments described herein were in compliance with protocols approved by the Institutional Animal Care and Use Committees (IACUC) at the Medical University of South Carolina and the Ralph H Johnson VA Medical Center.
QUANTIFICATION AND STATISTICAL ANALYSIS
The statistical details for each experiment can be found in the figure legends, figures, and results sections throughout the main text. All experiments were repeated independently at least three times and the data (bar graphs), unless otherwise specified, are presented as mean ± standard deviation. In animal experiments, “N” represents number of animals utilized in each treatment group. The investigators were blinded during evaluation of tumor size variations following treatments and imaging analysis for multiplex staining. All statistical analysis was performed using GraphPad Prism v9 software for Mac. Significance between two groups was assessed by the student’s two-tailed t test.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-HA(Mouse) | BioLegend | Cat. #901515; RRID:AB_2565334 |
| Anti-HA(Rabbit) | BioLegend | Cat. #923502; RRID:AB_2565438 |
| Anti-FLAG(Mouse) | Sigma | Cat. #F1804; RRID:AB_262044 |
| Anti-FLAG(Rabbit) | Sigma | Cat. #F7425; RRID:AB_439687 |
| Anti-Phospho-ERK | Cell Signaling Technology (CST) | Cat.#9101; RRID:AB_331646 |
| Anti-ERK | CST | Cat. #9102; RRID:AB_330744 |
| Anti-Phospho-AKT(S473) | CST | Cat. #9271; RRID:AB_329825 |
| Anti-Phospho-AKT(T308) | CST | Cat. #9275; RRID:AB_329828 |
| Anti-AKT | CST | Cat. #9272; RRID:AB_329827 |
| Anti-MYC | Millipore-Sigma | Cat. #05–724; RRID:AB_309938 |
| Anti-Cleaved Caspase-3 | CST | Cat. #9661; RRID:AB_2341188 |
| Anti-CRAF | BD Biosciences | Cat. #610151; RRID:AB_397552 |
| Anti-BRAF | Santa Cruz | Cat. #sc-9002; RRID:AB_2067494 |
| Anti-GST | Santa Cruz | Cat. #sc-459; RRID:AB_631586 |
| Anti- Vinculin | Santa Cruz | Cat. #sc-73614; RRID:AB_1131294 |
| Anti-Ki-67 | Abcam | Cat. #16667; RRID:AB_302459 |
| Anti-GFP | CST | Cat. #2956; RRID:AB_1196615 |
| Anti-V5 | ThermoFisher | Cat. #MA5–15253; RRID:AB_10977225 |
| Cell Conditioning Solution (CC2) | Roche Diagnostics | Cat. #980–223 |
| DAPI | Akoya Biosciences | Cat. #FP1490 |
| Doxycycline (DOX) | Sigma | Cat. #D9891–100G |
| GTPγS | Sigma | Cat. #G8634 |
| 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) | Fisher | Cat. #M6494 |
| Opal Polaris 480 | Akoya Biosciences | Cat. #FP1500001KT |
| Opal 520 | Akoya Biosciences | Cat. #FP1487001KT |
| Opal 620 | Akoya Biosciences | Cat. #FP1495001KT |
| ProLong™ Gold Antifade Reagent | ThermoFisher | Cat. #P36934 |
| Bacterial and virus strains | ||
| E. coli BL21(DE3) | ThermoFisher | Cat. #694504 |
| S. cerevisiae EBY100 | Invitrogen | Discontinued |
| Biological samples | ||
| Patient Derived Xenografts (PDXs) | Ramsay Camp’s Lab, MUSC | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| His-Avi-TEV-HRAS (1–166) | Koide Lab, NYUSoM | N/A |
| His-Avi-TEV-HRAS (1–174) | Koide Lab, NYUSoM | N/A |
| His-Avi-TEV-KRAS (1–174) | Koide Lab, NYUSoM | N/A |
| His-Avi-TEV-NRAS (1–174) | Koide Lab, NYUSoM | N/A |
| His-Avi-TEV-KRAS(G12C) (1–174) | Koide Lab, NYUSoM | N/A |
| His-Avi-TEV-KRAS(G12D) (1–174) | Koide Lab, NYUSoM | N/A |
| His-Avi-TEV-KRAS(G12V) (1–174) | Koide Lab, NYUSoM | N/A |
| His-Avi-TEV-KRAS(K16N) (1–174) | Koide Lab, NYUSoM | N/A |
| His-Avi-TEV-KRAS(Q61L) (1–174) | Koide Lab, NYUSoM | N/A |
| GST-RAF-RBD | O’Bryan Lab, MUSC | N/A |
| Critical commercial assays | ||
| Gibson Assembly Master Mix | NEB | Cat. #E2611 |
| CellTiter-Glo Luminescent Cell Viability Assay | Promega | Cat. #G7571 |
| CloneAmp™ HiFi PCR Premix | Clontech | Cat. #639298 |
| Experimental models: Cell lines | ||
| HEK-293 | O’Bryan Lab, MUSC | N/A |
| HEK-293T | O’Bryan Lab, MUSC | N/A |
| COS-1 | O’Bryan Lab, MUSC | N/A |
| NIH/3T3 | O’Bryan Lab, MUSC | N/A |
| RASless MEFs | NCI-RAS Initiative | N/A |
| NCI-H1915 | ATCC | Cat. #CRL-5904 |
| LS1034 | NCI-RAS Initiative | N/A |
| HuP-T3 | Channing Der Lab, UNC, NC | N/A |
| PANC-1 | Gregory Thatcher Lab, UIC, IL | N/A |
| CF-PAC-1 | Channing Der Lab, UNC, NC | N/A |
| PSN-1 | Channing Der Lab, UNC, NC | N/A |
| HCT 116 | NCI-RAS Initiative | N/A |
| Hec1-A | Todd Waldman Lab, Georgetown University, Washington, DC | N/A |
| NCI-H1299 | Robert Winn Lab, UIC, IL | N/A |
| NCI-H1792 | Robert Winn Lab, UIC, IL | |
| NCI-H1944 | NCI-RAS Initiative | N/A |
| A375 | Andrew Aplin Lab, Thomas Jefferson University, PA | N/A |
| Experimental models: Organisms/strains | ||
| Athymic NU/NU nude mice | Charles River | Cat. #CR1 athymic nude mice |
| Athymic NU/NU nude mice | Taconic | Cat. #NCRNU |
| NSG (NOD-SCID gamma) mice | Jackson Laboratory | Cat. #005557 |
| Oligonucleotides | ||
| Monobody Gibson Forward Primer 5’→3’ (GACGATGACGACAAGGGATCCGTTTCTTCTGTTCC) | Eurofins | Custom |
| Monobody Gibson Reverse Primer 5’→3’ (TCAGTTATCTAGATCCGGTGGATCCCTAGGTACGGTAGTTAATCGAGATTGG) | Eurofins | Custom |
| KRAS Gibson Forward Primer 5’→3’ GGAGGACCTTCTAGCGGATCCATGACTGAATATAAACTTGTGGTAGTTGGAGCT | Eurofins | Custom |
| KRAS Gibson Reverse Primer 5’→3’ TCACCCTGAAGTTCTCAGGATCCTTACATAATTACACACTTTGTCTTTGAC | Eurofins | Custom |
| HRAS Gibson Forward Primer 5’→3’ GGAGGACCTTCTAGCGGATCCATGACAGAATAC | Eurofins | Custom |
| HRAS Gibson Reverse Primer 5’→3’ TCACCCTGAAGTTCTCAGGATCCTCAGGAGAGCAC | Eurofins | Custom |
| NRAS Gibson Forward Primer 5’→3’ GGAGGACCTTCTAGCGGATCCATGACTGAGTACAAACTGGTGGTG | Eurofins | Custom |
| NRAS Gibson Reverse Primer 5’→3’ TCACCCTGAAGTTCTCAGGATCCTTACATCACCACACATGGCAATCCC | Eurofins | Custom |
| Recombinant DNA | ||
| pCGN-HA tagged human KRAS WT | O’Bryan Lab, MUSC | N/A |
| pCGN-HA tagged human KRAS (G12V) | O’Bryan Lab, MUSC | N/A |
| pCGN-HA tagged human KRAS (G13D) | This Paper | N/A |
| pCGN-HA tagged human KRAS (G12C) | This Paper | N/A |
| pCGN-HA tagged human KRAS (G12D) | This Paper | N/A |
| pCGN-HA tagged human KRAS (G12R) | This Paper | N/A |
| pCGN-HA tagged human KRAS (G12S) | This Paper | N/A |
| pCGN-HA tagged human KRAS (G15A) | This Paper | N/A |
| pCGN-HA tagged human KRAS (Q61L) | This Paper | N/A |
| pCGN-HA tagged human KRAS (Q61H) | This Paper | N/A |
| pCGN-HA tagged human KRAS (Q61R) | This Paper | N/A |
| pCGN-HA tagged human KRAS (A146T) | This Paper | N/A |
| pCGN-HA tagged human KRAS [G12D(D69N)] | This Paper | N/A |
| pCGN-HA tagged human KRAS [G12V(D69N)] | This Paper | N/A |
| pCGN-HA tagged human KRAS [Q61L(D69N)] | This Paper | N/A |
| pCGN-HA tagged human KRAS [G12C(A59G)] | This Paper | N/A |
| pCGN-HA tagged human KRAS [G12D(A59G)] | This Paper | N/A |
| pCGN-HA tagged human KRAS [G12V(A59G)] | This Paper | N/A |
| pCGN-HA tagged human KRAS [Q61L(A59G)] | This Paper | N/A |
| pCGN-HA tagged human HRAS WT | O’Bryan Lab, MUSC | N/A |
| pCGN-HA tagged human HRAS(G12V) | O’Bryan Lab, MUSC | N/A |
| pCGN-HA tagged human HRAS(Q61L) | O’Bryan Lab, MUSC | N/A |
| pCGN-HA tagged human HRAS(K16N) | O’Bryan Lab, MUSC | N/A |
| pCGN-HA tagged human HRAS(D119N) | O’Bryan Lab, MUSC | N/A |
| pCGN-HA tagged human HRAS [G12V(D69N)] | This Paper | N/A |
| pCGN-HA tagged human HRAS [Q61L(D69N)] | This Paper | N/A |
| pCGN-HA tagged human NRAS(G12V) | This Paper | N/A |
| pCGN-HA tagged human NRAS(G12D) | O’Bryan Lab, MUSC | N/A |
| pCGN-HA tagged human NRAS(G13D) | O’Bryan Lab, MUSC | N/A |
| pCGN-HA tagged human NRAS(Q61L) | This Paper | N/A |
| pCGN-HA tagged human MEK(DD) | O’Bryan Lab, MUSC | N/A |
| pBabe MYC tagged human BRAF(V600E) | O’Bryan Lab, MUSC | N/A |
| pECFP | O’Bryan Lab, MUSC | N/A |
| pECFP-FLAG tagged NS1 | O’Bryan Lab, MUSC | N/A |
| pECFP-FLAG tagged R15 | This Paper | N/A |
| pECFP-FLAG tagged R15m10 | This Paper | N/A |
| pEYFP-HRAS WT | O’Bryan Lab, MUSC | N/A |
| pEYFP-HRAS (Q61L) | O’Bryan Lab, MUSC | N/A |
| pEYFP-NRAS (G12D) | O’Bryan Lab, MUSC | N/A |
| pCW57.1-CFP-FLAG tagged NS1 | This Paper | N/A |
| pCW57.1-CFP-FLAG tagged R15m10 | This Paper | N/A |
| pMD2.G VSV-G | Dider Trono Lab, | N/A |
| pCMVdR8.74 | Dider Trono Lab, University of Geneva | N/A |
| Software and algorithms | ||
| Prism | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| NIH-ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Image Studio Lite | LICOR Biosciences | https://www.licor.com/bio/image-studio-lite/ |
| inForm® | Akoya Biosciences | https://www.akoyabio.com/phenoptics/software/inform-tissue-finder/ |
| Vectra Polaris | Akoya Biosciences | https://www.akoyabio.com/phenoptics/mantra-vectra-instruments/vectra-polaris/ |
| Other | ||
| Monobody sequence, R15: VSSVPTKLEVVAATPTSLLISWDASSSSVSYYRITYG ETGGNSPVQEFTVPGYYSTATISGLKPGVDYTITVYAYWYGYWSYISPISINYRT | Koide Lab, NYUSoM | N/A |
| Monobody sequence, R15m10: VSSVPTKLEVVAATPTSLLISWDASSSSVSYYRIT YGETGGNSPVQEFTVPGYYSTATISGLKPGVDYTITVYAVWQGVWRYVSPISINYRT | Koide Lab, NYUSoM | N/A |
Highlights.
Development of a high-affinity monobody, R15, selective for apo RAS
R15 inhibits RAS mutants with elevated spontaneous nucleotide release rates
>50% of oncogenic RAS mutants may be susceptible to inhibitors binding to apo RAS
Targeting apo RAS represents a viable approach for inhibiting RAS-driven tumors
ACKNOWLEDGMENTS
We wish to thank Negin Martin, Director of the Viral Vector Core, National Institute of Environmental Health Sciences, NIH, Research Triangle Park, NC, for the lentiviral packaging of pCW5.1 CFP-FLAG-NS1 and R15 constructs; the Translational Sciences Laboratory of the Hollings Cancer Center for the multiplex imaging analysis of samples; and Dr. David Lewin for examining the pathology of the CRC PDX samples. J.P.O. was supported by grants from the Department of Veterans Affairs Biomedical Laboratory Research and Development Service MERIT Award (1I01BX002095) and the NCI (P30 CA138313). J.P.O. and S.K. were supported by a grant from the NCI (R01CA212608). J.P.O. and G.A.H. were supported by a grant from the NIGMS (P20 GM103542). K.W.T. was supported by an NIH fellowship (F32 CA225131) and ACS fellowship (PF-18-180-01-TBE). S.K. was supported by a grant from the NCI (R01 CA194864). E.R.C. was supported by a grant from the Department of Veterans Affairs Clinical Science Research and Development MERIT Award (1I01CX001880-01A1). The contents of this article do not represent the views of the US Department of Veterans Affairs or the United States government.
DECLARATION OF INTERESTS
J.P.O., A.K., and S.K. are listed as inventors on a patent application on monobodies targeting the nucleotide-free state of RAS files by the Medical University of South Carolina and New York University (no. 62/862,924). K.W.T., A.K., and S.K. are listed as inventors on a patent application on mutant RAS targeting monobodies filed by New York University (application no. 63/121,903). A.K. and S.K. are listed as inventors on issued and pending patents on monobody technology filed by The University of Chicago (US patent 9,512,199 B2 and related pending applications). S.K. is an SAB member, holds equity in and receives consulting fees from Black Diamond Therapeutics, and receives research funding from Puretech Health and Argenx BVBA. The other authors declare no competing interests.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110322.
REFERENCES
- Alabi S, Jaime-Figueroa S, Yao Z, Gao Y, Hines J, Samarasinghe KTG, Vogt L, Rosen N, and Crews CM (2021). Mutant-selective degradation by BRAF-targeting PROTACs. Nat. Commun 12, 920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrogio C, Kohler J, Zhou ZW, Wang H, Paranal R, Li J, Capelletti M, Caffarra C, Li S, Lv Q, et al. (2018). KRAS dimerization impacts MEK inhibitor sensitivity and oncogenic activity of mutant KRAS. Cell 172, 857–868.e815. [DOI] [PubMed] [Google Scholar]
- Bollag G, Adler F, elMasry N, McCabe PC, Conner E Jr., Thompson P, McCormick F, and Shannon K (1996). Biochemical characterization of a novel KRAS insertion mutation from a human leukemia. J. Biol. Chem 271, 32491–32494. [DOI] [PubMed] [Google Scholar]
- Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, Gaida K, Holt T, Knutson CG, Koppada N, et al. (2019). The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 575, 217–223. [DOI] [PubMed] [Google Scholar]
- Clark G, Cox AD, Graham SM, and Der CJ (1995). Biological assays for Ras transformation. Methods Enzymol 255, 395–412. [DOI] [PubMed] [Google Scholar]
- Clark R, Wong G, Arnheim N, Nitecki D, and McCormick F (1985). Antibodies specific for amino acid 12 of the ras oncogene product inhibit GTP binding. Proc. Natl. Acad. Sci. U S A 82, 5280–5284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cool RH, Schmidt G, Lenzen CU, Prinz H, Vogt D, and Wittinghofer A (1999). The Ras mutant D119N is both dominant negative and activated. Mol. Cell Biol 19, 6297–6305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosten M, Dhawahir A, Sum EY, Urosevic J, Lechuga CG, Esteban LM, Castellano E, Guerra C, Santos E, and Barbacid M (2010). Genetic analysis of Ras signalling pathways in cell proliferation, migration and survival. EMBO J 29, 1091–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feramisco JR, Clark R, Wong G, Arnheim N, Milley R, and McCormick F (1985). Transient reversion of ras oncogene-induced cell transformation by antibodies specific for amino acid 12 of ras protein. Nature 314, 639–642. [DOI] [PubMed] [Google Scholar]
- Franken SM, Scheidig AJ, Krengel U, Rensland H, Lautwein A, Geyer M, Scheffzek K, Goody RS, Kalbitzer HR, Pai EF, et al. (1993). Three-dimensional structures and properties of a transforming and a nontransforming glycine-12 mutant of p21H-ras. Biochemistry 32, 8411–8420. [DOI] [PubMed] [Google Scholar]
- Freeman AK, Ritt DA, and Morrison DK (2013). The importance of Raf dimerization in cell signaling. Small GTPases 4, 180–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guldenhaupt J, Rudack T, Bachler P, Mann D, Triola G, Waldmann H, Kotting C, and Gerwert K (2012). N-Ras forms dimers at POPC membranes. Biophys. J 103, 1585–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallin J, Engstrom LD, Hargis L, Calinisan A, Aranda R, Briere DM, Sudhakar N, Bowcut V, Baer BR, Ballard JA, et al. (2020). The KRAS(G12C) inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discov 10, 54–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, Pollok BA, and Connelly PA (1996). Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J. Biol. Chem 271, 695–701. [DOI] [PubMed] [Google Scholar]
- Hobbs GA, Baker NM, Miermont AM, Thurman RD, Pierobon M, Tran TH, Anderson AO, Waters AM, Diehl JN, Papke B, et al. (2020). Atypical KRAS(G12R) mutant is impaired in PI3K signaling and macropinocytosis in pancreatic cancer. Cancer Discov 10, 104–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter JC, Manandhar A, Carrasco MA, Gurbani D, Gondi S, and Westover KD (2015). Biochemical and structural analysis of common cancer-associated KRAS mutations. Mol. Cancer Res 13, 1325–1335. [DOI] [PubMed] [Google Scholar]
- Janakiraman H, Zhu Y, Becker SA, Wang C, Cross A, Curl E, Lewin D, Hoffman BJ, Warren GW, Hill EG, et al. (2020). Modeling rectal cancer to advance neoadjuvant precision therapy. Int. J. Cancer 147, 1405–1418. [DOI] [PubMed] [Google Scholar]
- Janes MR, Zhang J, Li LS, Hansen R, Peters U, Guo X, Chen Y, Babbar A, Firdaus SJ, Darjania L, et al. (2018). Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell 172, 578–589 e517. [DOI] [PubMed] [Google Scholar]
- Jeganathan S, Muller MP, Ali I, and Goody RS (2018). Assays for nucleotide competitive reversible and irreversible inhibitors of ras GTPases. Biochem 57, 4690–4699. [DOI] [PubMed] [Google Scholar]
- Karnoub AE, and Weinberg RA (2008). Ras oncogenes: split personalities. Nat. Rev. Mol. Cell Biol 9, 517–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan I, MarElia-Bennet C, Lefler J, Zuberi M, Denbaum E, Koide A, Connor DM, Broome AM, Pécot T, Timmers C, et al. (2021). Targeting the KRAS α4–α5 allosteric interface inhibits pancreatic cancer tumorigenesis. Small GTPases, 1–14. 10.1080/21541248.2021.1906621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan I, Spencer-Smith R, and O’Bryan JP (2019). Targeting the alpha4–alpha5 dimerization interface of K-RAS inhibits tumor formation in vivo. Oncogene 38, 2984–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killoran RC, and Smith MJ (2019). Conformational resolution of nucleotide cycling and effector interactions for multiple small GTPases determined in parallel. J. Biol. Chem 294, 9937–9948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koide A, Wojcik J, Gilbreth RN, Hoey RJ, and Koide S (2012). Teaching an old scaffold new tricks: monobodies constructed using alternative surfaces of the FN3 scaffold. J. Mol. Biol 415, 393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KY, Fang Z, Enomoto M, Gasmi-Seabrook G, Zheng L, Koide S, Ikura M, and Marshall CB (2020). Two distinct structures of membrane-associated homodimers of GTP- and GDP-bound KRAS4B revealed by paramagnetic relaxation enhancement. Angew. Chem 59, 11037–11045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S, Khoo R, Juang YC, Gopal P, Zhang H, Yeo C, Peh KM, Teo J, Ng S, Henry B, et al. (2021). Exquisitely specific anti-KRAS biodegraders inform on the cellular prevalence of nucleotide-loaded states. ACS Cent. Sci 7, 274–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lito P, Solomon M, Li LS, Hansen R, and Rosen N (2016). Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science 351, 604–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez MS, Kliegman JI, and Shokat KM (2014). The logic and design of analog-sensitive kinases and their small molecule inhibitors. Methods Enzymol 548, 189–213. [DOI] [PubMed] [Google Scholar]
- Lu J, Bera AK, Gondi S, and Westover KD (2018). KRAS switch mutants D33E and A59G crystallize in the state 1 conformation. Biochem 57, 324–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghadamchargari Z, Shirzadeh M, Liu C, Schrecke S, Packianathan C, Russell DH, Zhao M, and Laganowsky A (2021). Molecular assemblies of the catalytic domain of SOS with KRas and oncogenic mutants. Proc. Natl. Acad. Sci. U S A 118, e2022403118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore AR, Rosenberg SC, McCormick F, and Malek S (2020). RAS-targeted therapies: is the undruggable drugged? Nat. Rev. Drug Discov 19, 533–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mysore VP, Zhou ZW, Ambrogio C, Li L, Kapp JN, Lu C, Wang Q, Tucker MR, Okoro JJ, Nagy-Davidescu G, et al. (2021). A structural model of a Ras-Raf signalosome. Nat. Struct. Mol. Biol 28, 847–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Bryan JP (2019). Pharmacological targeting of RAS: recent success with direct inhibitors. Pharmacol. Res 139, 503–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrem JM, Peters U, Sos ML, Wells JA, and Shokat KM (2013). K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503, 548–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packer MR, Parker JA, Chung JK, Li Z, Lee YK, Cookis T, Guterres H, Alvarez S, Hossain MA, Donnelly DP, et al. (2021). Raf promotes dimerization of the Ras G-domain with increased allosteric connections. Proc. Natl. Acad. Sci. U S A 118, e201564811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papke B, and Der CJ (2017). Drugging RAS: know the enemy. Science 355, 1158–1163. [DOI] [PubMed] [Google Scholar]
- Prior IA, Hood FE, and Hartley JL (2020). The frequency of ras mutations in cancer. Cancer Res 80, 2969–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quilliam LA, Hisaka MM, Zhong S, Lowry A, Mosteller RD, Han J, Drugan JK, Broek D, Campbell SL, and Der CJ (1996). Involvement of the switch 2 domain of Ras in its interaction with guanine nucleotide exchange factors. J. Biol. Chem 271, 11076–11082. [DOI] [PubMed] [Google Scholar]
- Rajakulendran T, Sahmi M, Lefrancois M, Sicheri F, and Therrien M (2009). A dimerization-dependent mechanism drives RAF catalytic activation. Nature 461, 542–545. [DOI] [PubMed] [Google Scholar]
- Roth S, Macartney TJ, Konopacka A, Chan KH, Zhou H, Queisser MA, and Sapkota GP (2020). Targeting endogenous K-RAS for degradation through the affinity-directed protein missile system. Cell Chem. Biol 27, 1151–1163.e1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz AL, Schrage R, Gaffal E, Charpentier TH, Wiest J, Hiltensperger G, Morschel J, Hennen S, Haussler D, Horn V, et al. (2014). A cell-permeable inhibitor to trap Galphaq proteins in the empty pocket conformation. Chem. Biol 21, 890–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigal IS, Gibbs JB, D’Alonzo JS, Temeles GL, Wolanski BS, Socher SH, and Scolnick EM (1986). Mutant ras-encoded proteins with altered nucleotide binding exert dominant biological effects. Proc. Natl. Acad. Sci. U S A 83, 952–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simanshu DK, Nissley DV, and McCormick F (2017). RAS proteins and their regulators in human disease. Cell 170, 17–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MJ, Neel BG, and Ikura M (2013). NMR-based functional profiling of RASopathies and oncogenic RAS mutations. Proc. Natl. Acad. Sci. U S A 110, 4574–4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer-Smith R, Koide A, Zhou Y, Eguchi RR, Sha F, Gajwani P, Santana D, Gupta A, Jacobs M, Herrero-Garcia E, et al. (2017a). Inhibition of RAS function through targeting an allosteric regulatory site. Nat. Chem. Biol 13, 62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer-Smith R, Li L, Prasad S, Koide A, Koide S, and O’Bryan JP (2017b). Targeting the alpha4-alpha5 interface of RAS results in multiple levels of inhibition. Small GTPases 10, 378–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer-Smith R, and O’Bryan JP (2019). Direct inhibition of RAS: quest for the holy grail? Semin. Cancer Biol 54, 138–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stalnecker CA, and Der CJ (2020). RAS, wanted dead or alive: advances in targeting RAS mutant cancers. Sci. Signal 13, eaay6013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephen AG, Esposito D, Bagni RK, and McCormick F (2014). Dragging ras back in the ring. Cancer Cell 25, 272–281. [DOI] [PubMed] [Google Scholar]
- Tanaka N, Lin JJ, Li C, Ryan MB, Zhang J, Kiedrowski LA, Michel AG, Syed MU, Fella KA, Sakhi M, et al. (2021). Clinical acquired resistance to KRASG12C inhibition through a novel KRAS switch-II pocket mutation and polyclonal alterations converging on RAS-MAPK reactivation. Cancer Discov 11, 1913–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng KW, Tsai ST, Hattori T, Fedele C, Koide A, Yang C, Hou X, Zhang Y, Neel BG, O’Bryan JP, et al. (2021). Selective and noncovalent targeting of RAS mutants for inhibition and degradation. Nat. Commun 12, 2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traut TW (1994). Physiological concentrations of purines and pyrimidines. Mol. Cell Biochem 140, 1–22. [DOI] [PubMed] [Google Scholar]
- Vartanian S, Bentley C, Brauer MJ, Li L, Shirasawa S, Sasazuki T, Kim JS, Haverty P, Stawiski E, Modrusan Z, et al. (2013). Identification of mutant K-Ras-dependent phenotypes using a panel of isogenic cell lines. J. Biol. Chem 288, 2403–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wey M, Lee J, Jeong SS, Kim J, and Heo J (2013). Kinetic mechanisms of mutation-dependent Harvey Ras activation and their relevance for the development of Costello syndrome. Biochemistry 52, 8465–8479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong KA, Russo A, Wang X, Chen Y-J, Lavie A, and O’Bryan JP (2012). A new dimension to Ras function: a novel role for nucleotide-free Ras in Class II phosphatidylinositol 3-kinase beta (PI3K-C2b) regulation. PLoS One 7, e45360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Fujimiya R, Kubo S, Marshall CB, Ikura M, Shimada I, and Nishida N (2020). Real-time in-cell NMR reveals the intracellular modulation of GTP-bound levels of RAS. Cell Rep 32, 108074. [DOI] [PubMed] [Google Scholar]
- Zuberi M, Khan I, and O’Bryan JP (2020). Inhibition of RAS: proven and potential vulnerabilities. Biochem. Soc. Trans 48, 1831–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This study did not generate any unique datasets or code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.