

Abstract
A plethora of experimental and epidemiological evidence supports a critical role for inflammation and adaptive immunity in the onset of cancer and in shaping its response to therapy. These data are particularly robust for gastrointestinal (GI) cancers, such as those affecting the GI tract, liver, and pancreas, on which this review is focused. We propose a unifying hypothesis according to which intestinal barrier disruption is the origin of tumor-promoting inflammation that acts in conjunction with tissue-specific cancer-initiating mutations. The gut microbiota and its products impact tissue-resident and recruited myeloid cells that promote tumorigenesis through secretion of growth- and survival-promoting cytokines that act on epithelial cells, as well as fibrogenic and immunosuppressive cytokines that interfere with the proper function of adaptive antitumor immunity. Understanding these relationships should improve our ability to prevent cancer development and stimulate the immune system to eliminate existing malignancies.
Keywords: immunity, inflammation, gut-liver axis, microbiome, immunotherapy
I must be cruel only to be kind; Thus bad begins, and worse remains behind.
—William Shakespeare, Hamlet
INTRODUCTION
A century and a half ago, the founding father of modern pathology, Rudolph Virchow, suggested that chronic irritation (inflammation) can lead to cancer. Very little was understood about cancer or inflammation at the time, but certain aspects of that prediction have been verified using the tools of modern molecular biology. In fact, inflammation has been recognized as one of the key enablers of the malignant state (1), contributing to cancer initiation, progression, and response to therapy (2). Oddly, Virchow’s academic nemesis was Robert Koch, a pioneer of modern microbiology who came up with the bacterial theory of disease, which Virchow was reluctant to acknowledge. Now we know that bacteria and their products are key drivers of inflammation, not only in the context of infectious disease but also in cancer and premalignant conditions. In this review, we cover recent advances in understanding the origin of tumor-elicited inflammation and the myriad of different mechanisms by which it affects cancer initiation and progression. As the topic of inflammation and cancer has been amply reviewed in the recent past (3, 4), we focus this review on gastrointestinal (GI) malignancies, those that arise along the GI tract, the liver, and the pancreas. Our decision to focus on GI cancer is based not only on our own research interests but also on the fact that the initial human epidemiological studies that provided important support for the role of inflammation were conducted on GI malignancies (5–8). Furthermore, the origin of tumor-promoting inflammation in GI cancer is fairly well understood, and much of it can be attributed to loss of epithelial barrier integrity. Once the barrier is compromised, commensal microbes and their products lead to activation of the major inflammation-activated transcription factor NF-κB, which through induction of IL-6, TNF, and other cytokines can accelerate the development of colon (7), liver (9), and pancreatic (10) cancers.
Although cancer is a disease of uncontrolled cell proliferation, it should be realized that the healthy human liver or pancreas hardly contains any proliferating cells, especially not at the age at which most cancers are detected. Also, the large intestine, encompassing the colon and rectum, the two sites at which most intestinal cancers are commonly initiated, exhibits lower rates of cell proliferation than the small intestine, at which primary cancer is hardly detected. In the adult liver, it is quite clear that cell proliferation only occurs as a response to necroinflammatory injuries triggered by chronic hepatitis B and C viral (HBV and HCV, respectively) infections, excessive alcohol consumption, fat accumulation, or exposure to certain toxicants. Such injuries trigger chronic inflammation (hepatitis) that stimulates proliferation of periportal hybrid hepatocytes, cells that hardly ever give rise to cancer (11), and fully differentiated zone 3 hepatocytes that have been implicated in the initiation of hepatocellular carcinoma (HCC), the major type of liver cancer (12). In the pancreas, chronic tissue damage gives rise to inflammation (pancreatitis), which converts fully differentiated acinar cells to ductal progenitors, which have higher proliferative capacity through acinar-to-ductal metaplasia (ADM) (13, 14). In colorectal cancer (CRC), however, the relationship between cell proliferation and tumor initiation is more complex, as the colon contains a population of true stem cells (whose existence in the mature liver remains controversial, although it is generally agreed that adult tissue stem cells are not part of the pancreas) that are in charge of maintaining mucosal layer integrity (15). However, the rate of cell division in the healthy colonic mucosa is considerably lower than that in the small intestinal mucosa. Since inflammatory bowel disease (IBD) affects either the large intestine (ulcerative colitis) or both small and large intestines (Crohn disease), the key difference in cancer rates between these two parts of the GI tract is unlikely to reside in their ability to develop inflammation. Instead, the colon and rectum host a much larger population of commensal microbes, with colonic bacteria accounting for 70% of all bacteria in the human body (16). As discussed below, loss of intestinal barrier integrity and the ensuing translocation of commensal microbes or their products is the key contributor to tumor-elicited inflammation in the colon (17). More surprisingly, loss of gut barrier integrity has also been implicated in some of the key inflammatory conditions that give rise to HCC, pancreatic ductal adenocarcinoma (PDAC), and bile duct cancer, which are some of the most aggressive and treatment-refractory malignancies (18), and for which an influential role of the microbiome has been recognized (19). Moreover, barrier disruption and the ensuing inflammatory response profoundly affect and modulate both natural immunosurveillance and drug-induced antitumor immunity.
THE GUT BARRIER AND ITS CONNECTION TO THE LIVER AND PANCREAS
Given the important role of gut barrier disruption in the initiation of most GI malignancies, we discuss key features of the intestinal epithelial barrier and its relationship with the mucosal immune system (Figure 1). The GI tract represents a unique challenge to the immune system, which must tolerate gut microbiota and maintain homeostatic conditions that prevent microbial dysbiosis and overproliferation of facultative pathogens. While not responding to food products and microbial components, the immune system needs to protect the host from invading bacteria and potentially harmful ingested antigens. The intestinal epithelium, composed of a single-cell layer, is crucial for preserving gut homeostasis and acts as not only a physical barrier but also a source for antimicrobial peptides and proteins, such as α-defensins, lysozyme, and different RegIII isoforms (20). Small intestinal villi and crypts and large intestinal crypts undergo constant cycles of intestinal epithelial cell (IEC) replenishment and renewal, and under homeostatic conditions, an entire crypt is replaced every four to five days. A crypt consists of enterocytes, the most prominent cell type of the intestinal epithelium that is responsible for nutrient and water absorption; mucin-secreting goblet cells; hormone-secreting enteroendocrine cells; Paneth cells that release antimicrobial factors/peptides (AMPs); and intestinal stem cells. Paneth cells are unique to the small intestine; they are the major sources of α-defensins, lysozymes, ribonucleases (like angiogenin 4), and secretory phospholipase A2. However, although Paneth cells are unequally distributed along the GI tract, enterocytes can also express different AMPs like REG3γ and REG3β, and in the large intestine β-defensin and cathelicidins are produced by IECs. Goblet cells, which are more abundant in the large intestine, express AMPs like RELMβ, ANG4, REG3γ, and REG3β (21). All of these cells express tight junction proteins (TJPs), including occludins, claudins, zonula occludens, and junctional adhesion molecules (JAMs) that provide a tight but highly regulated seal that allows passage of essential molecules but restricts entry of harmful substances and bacteria. Tight junctions are regulated by extracellular stimuli, including cytokines, via small GTPases and protein kinases, although the exact mechanism and function of individual TJPs remain elusive (22). Tight junctions form two types of barriers: the paracellular and intramembrane barriers, the first of which regulates selective paracellular permeability, which is the passive transport of molecules across the tissue and between distinct compartments of the body. The intramembrane barrier restricts the exchange of membrane components between the apical and basolateral cell surface domains (fence function) (22). IECs are in direct contact with the microbiota and express a variety of pattern recognition receptors (PRRs), like Toll-like receptors (TLRs) that signal via MyD88 and TRIF to activate NF-κB and IRF signaling and also promote NLRP3 inflammasome activation (23, 24).
Figure 1.

A schematic describing the interaction between the GI tract, liver, and pancreas in steady state. The gut-liver axis, which is based on the anatomical and physiological connection between the liver and the gut through the biliary tract and portal vein, allows delivery of liver-produced bile acids to the GI tract, while receiving lipid-rich and endotoxin-containing blood from the GI tract to be filtered and detoxified in the liver. The liver’s supply of blood has two sources: the hepatic artery and the portal vein. Some 70–75% of the liver’s blood supply is derived from the portal vein, which drains blood from mesenteric veins of the intestinal tract. In steady state, IgA+ cells, Tregs, myeloid cells, and ILCs regulate gut and liver homeostasis. Different populations of myeloid-derived antigen-presenting cells take up antigens, traffic to MLNs, and present them to naive T cells in the presence of TGF-β and RA, which favor the generation of food/bacterium-antigen-specific Tregs. IL-10 promotes Treg proliferation and IgA CSR. TGF-β, likely produced by Tregs or Tfhs, regulates B cell antibody class switching to IgA+ cells. Secretory IgA is transported across the epithelial barrier via pIgR into the intestinal lumen, where it regulates the entrance of luminal food- and bacterium-derived antigens and contributes to microbiome homeostasis. Tregs and IgA+ cells eventually exit the gut and enter the blood to regulate systemic tolerance to dietary and bacterial antigens. Healthy diet supports microbiome diversity and increased amounts of SCFA and tryptophan metabolites, which in turn suppress inflammation by inhibiting NF-κB and NLRP3 and increase TJP expression and IEC homeostasis. Abbreviations: CSR, class-switch recombination; ER, endoplasmic reticulum; GI, gastrointestinal; HSC, hepatic stellate cell; IEC, intestinal epithelial cell; ILC, innate lymphoid cell; KC, Kupffer cell; LC, lymphocyte; MLN, mesenteric lymph node; pIgR, polymeric immunoglobulin receptor; RA, retinoic acid; SCFA, short chain fatty acid; Tfh, follicular T helper cell; TJP, tight junction protein; Treg, regulatory T cell.
Under homeostatic conditions, RORγt-expressing lymphoid cells, including CD4+ T helper type 17 (Th17) cells, γδ T cells, and innate lymphoid cells (ILCs), support host defense and tissue repair (25, 26). IL-10-secreting T regulatory cells (Tregs) are abundantly represented in the healthy tissue and are responsible for preventing excessive inflammation. IgA-producing B cells (which are described in more detail below) maintain microbial homeostasis and prevent overproliferation of inflammation-causing bacteria (27, 28). Virulent pathogens, like Citrobacter rodentium, can induce an IgG response, which is not elicited by commensals or avirulent pathogens (29). Under inflammatory conditions (Figure 2), however, translocating bacteria or microbial products activate local myeloid cells to produce IL-23, promote IL-17-related inflammatory responses (30), and inhibit Treg generation (25, 31). These alterations not only lead to chronic gut inflammation but also influence systemic, liver, and pancreatic inflammation. The so-called gut-liver axis, which is based on the anatomical and physiological connection between the liver and gut through the biliary tract and portal vein, allows delivery of liver-produced bile acids to the GI tract, while lipid-rich and endotoxin-containing blood is received from the small intestine to be filtered in the liver. Since most bacterial metabolites and food antigens are cleared in the liver, the liver and intestine share some common immunological features. Given that the GI tract contains a larger mass of immune cells than any other organ system, many of these cells also reach the liver. Importantly, both tissues need to work together to maintain a delicate balance between immune reactivity and tolerance, which probably makes them more susceptible to chronic inflammatory diseases (32).
Figure 2.
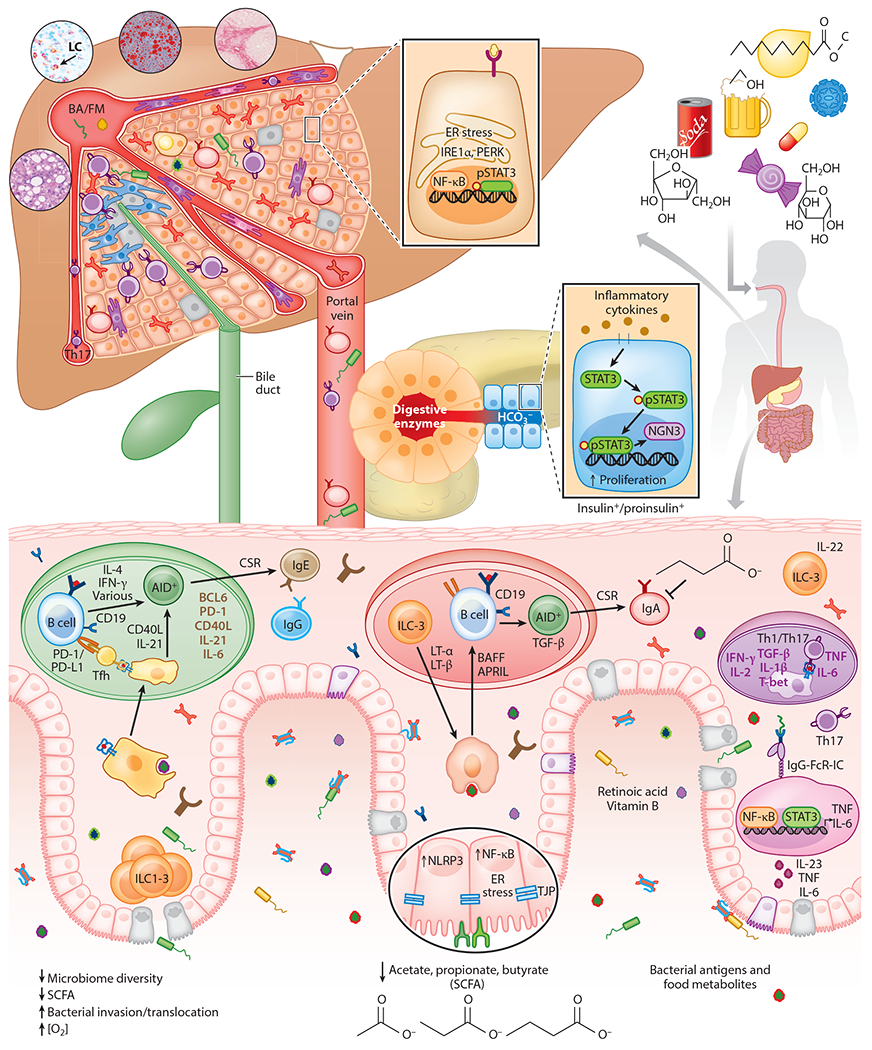
A schematic describing the interaction between the GI tract, liver, and pancreas during inflammatory conditions. If the gut barrier is disrupted and its contents enter the submucosa, the liver is the first organ in the body to encounter microbial products, toxins, and microorganisms (such as bacteria and fungi) from the intestine. The liver, therefore, serves as a receptacle and sensor for compounds and substances originating from the intestine to which it must respond. Moreover, hypernutrition, alcohol abuse, and other factors that can disrupt the gut barrier induce chronic intestinal inflammation and also affect the liver to support NASH and ASH development. Hypernutrition and alcohol decrease microbiome diversity and SCFAs and induce inflammation through activation of NF-κB and NLRP3. IL-1β and IL-23-induced IL-17 also contribute to intestinal inflammation and the likely induction of IgG and IgE. Abbreviations: ASH, alcoholic steatohepatitis; BA, bacterial antigen; CSR, class-switch recombination; ER, endoplasmic reticulum; FM, food metabolite; GI, gastrointestinal; IgG-FcR-IC, IgG Fc receptor immune complex; LC, lymphocyte; NASH, nonalcoholic steatohepatitis; SCFA, short chain fatty acid; Tfh, follicular T helper cell; Th, helper T cell; TJP, tight junction protein.
Nonparenchymal liver cells are thought to be responsible for establishment of tolerance, including resident dendritic cells (DCs), plasmacytoid DCs, liver sinusoidal endothelial cells, and Kupffer cells, as well as hepatic stellate cells (HSCs), Tregs, and IgA+ plasmocytes (33, 34). These cells mediate immunosuppression via expression of anti-inflammatory cytokines such as IL-10 and TGF-β, as well as the negative costimulatory molecule programmed cell death ligand 1 (PD-L1). The close interaction between the liver and gut enables them to influence and modulate each other’s fate during disease conditions. For example, a relatively common extraintestinal manifestation of IBD is hepatic steatosis. Furthermore, hypernutrition, fructose consumption, and alcohol abuse promote the development of steatohepatitis, in part through barrier disruption, dysbiosis, and perturbation of gut immune homeostasis. Interestingly, nonalcoholic fatty liver disease (NAFLD) is often associated with presence of colorectal adenomas (35, 36). Although the detailed mechanisms remain elusive, the balance between inflammation and tolerance induction seems to contribute to all of these pathologies, with inflammation due to barrier disruption playing a cardinal role, as discussed below.
INFLAMMATION IN COLORECTAL CANCER
Early studies of inflammation and its role in colorectal tumorigenesis were mostly focused on colitis-associated cancer (CAC), a specific form of CRC that is likely to appear in IBD patients. CAC accounts for only 2% of all CRCs, although it is responsible for 15% of all causes of death among IBD patients; but unlike sporadic CRC, its connection with inflammation was obvious, hence the early interest in this particular malignancy (37–41). Comparison between CRC and CAC is provided in Table 1. Of note, CAC risk increases with the length and severity of the disease. In ulcerative colitis, around 2% of patients develop cancer after 10 years, 8% after 20 years, and 18% after 30 years. Approximately 8% of Crohn disease patients develop precancerous lesions or CAC after 30 years (42, 43). One of the most popular mouse models of CRC—the azoxymethane (AOM) plus dextran sulfate sodium (DSS) model—is based on a combination of DSS-induced experimental colitis with carcinogen (AOM) exposure. The key differences in the mutational mechanisms that give rise to CAC on one hand and sporadic and familial CRC on the other have been amply discussed (41, 44–48). Although loss or inactivation of the tumor suppressor Adenomatous polyposis coli (APC) gene is the first genetic event in sporadic and familial CRC, followed by SMAD and TP53 mutations, next-generation sequencing has identified TP53 loss-of-function mutations and recurrent loss- or gain-of-function mutations in KRAS, APC, EGFR2, MLL, and EP300 in CAC (49–51). Most importantly, recent evidence also implicates inflammation in the initiation of sporadic and familial CRC (52, 53), making them less different from APC-independent CAC than previously thought. Life style factors, including obesity, cigarette smoking, and sedentary lifestyle, have been described to support CRC, probably by inducing or contributing to inflammation (8, 54). We, for instance, found that activation of NF-κB in murine IECs, which also takes place in both CAC and sporadic/familial CRC, results in induction of NO synthase (iNOS), which gives rise to nitrosatively damaged DNA (55). When this takes place in mouse IECs that harbor a single Apc allele, iNOS-induced DNA damage accelerates loss of the remaining Apc allele, giving rise to aberrant crypt foci (ACFs), the preneoplastic lesions that precede colonic adenomas. Exactly how DNA damage accelerates loss of heterozygosity is not entirely clear, but one plausible mechanism is recombination-based DNA repair. However, nitrosative DNA damage in IECs that harbor wild-type APC may result in acquisition of different oncogenic mutations.
Table 1.
Similarities and differences between colorectal cancer and colitis-associated cancer
| Disease | Colorectal cancer | ||
|---|---|---|---|
| Sporadic/familial CRC | CAC | ||
| Origins | Mutational accumulation or family history (30% of CRC) (47) | Chronic inflammation (e.g., IBD) CAC accounts for 1–2% of all CRCs (40) |
|
| Presusceptibility and risk factors | History of CRC in first-degree relatives, obesity, red meat consumption, cigarette smoking, sedentary lifestyle, and low fruit and vegetable consumption (54) | IBD: UC (colon) and CD (SI & colon) | |
| Genetic predisposition mutations | Precancerous a | Adenomatous polyps (46): Lynch syndrome (MLH1, MSH2, MSH6, PMS2, Tacstd1/EpCAM—70%b) FAP (APC—90%b) Hamartomatous polyps (46): Peutz—Jeghers syndrome (STK11—50–70%b) Juvenile polyposis syndrome (SMAD4, BMPR1A—<50%b) Cowden syndrome (PTEN—65–80%b) BRCA1 (45) |
47 risk loci identified with UC and 71 for CD. 30% shared between both (41)c GNA12d, HNF4A, CDH1, ERRFI1, MUC19, ITLN1d, REL, PTGER4, NKX2-3, STAT3, PLA2G2A/E, SLC9A4, SLC22A5, SLC22A4d, AQP12A/B, SLC9A3, SLC26A3, NOD2d, ATG16L1d, XBP1d, CARD9d, SLC11A1, FCGR2Ad/B, CCL11/CCL2/CCL7/CCL8, CCR6, IL8RA/IL8RB, MST1d, ERAP2d, LNPEP, DENND1B, IL23Rd, JAK2, TYK2d, ICOSLG, IL21, TNFSF15d, NDFIP1, TNFSF8, TAGAP, IL2, TNFRSF9, PIM3, IL7Rd, IL12B, PRDM1, IFNG, IL5, IKZF1, BACH2, IRF5, IL10, IL27d, SBNO2, CREM, IL1R1/IL1R2, etc. (41) |
| Cancerous | Early APC (81%e), late TP53 (60%e), KRAS (43%e), SMAD4 (10%e), MSI, EGFR2, etc. (50, 51) | Late APC (13%e), early TP53 (63%e), KRAS (20%e), SMAD4 (13%e), EGFR2, MLL, EP300, MSI, RNF43, etc. (50, 51) | |
| Signaling pathways | WNT/β-catenin, K-Ras, p53, TGF-β, NF-κB, STAT3, etc. (44) | ||
| Percentage of cancer occurrence within individual patient | Lynch syndrome: 2–4% of CRC cases (46) FAP: 1% of new CRC cases (46) Hamartomatous polyposis syndromes: <0.5% of CRC cases (46) |
1.5- to 2.4-higher risk to develop CAC (40) CAC-UC: 18% after 30 years of disease (43) CAC-CD: 8% after 30 years of disease (43) |
|
| Liver metastasis | >50% of advanced CRC patients will develop liver metastases during their lifetime (139) | ||
| Cytokine involvement | Dysregulated production of TNF, IFN-γ, IL-17, IL-12, IL-23, etc. | ||
| Treatments | IBD related | Anti-α4β7 (e.g., vedolizumab), anti-TNF (e.g., infliximab, adalimumab), anti-IL-12/IL-23 (e.g., ustekinumab), anti-IL-6 (e.g., tocilizumab), anti-IL-2 receptor (e.g., basiliximab), anti-CD20 (e.g., rituximab), anti-IFN-γ (e.g., fontolizumab), JAK inhibitor (e.g., tofacitinib), etc. (44) | |
| Cancer related | Chemotherapy: combination therapy of 5-fluourouracil, fluoropyrimidine, oxaliplatin (e.g., FOLFOX, CAPEOX, FOLFIRI); targeted therapy: EGFR inhibitors (e.g., panitumumab, cetuximab), VEGF inhibitors (e.g., bevacizumab, ramucirumab); patients with MSI: PD-(L)1 inhibitors (e.g., nivolumab, pembrolizumab) (44, 48) | ||
Although a family history of CRC is reported in 30% of CRC patients, only 5–6% have germline mutations in familial cancer syndrome–associated genes (46).
Percentage of patients with specified mutations in genes associated with familial CRC (46).
Green font represents mutations specific to UC, purple font represents mutations specific to CD, and red font represents mutations related to both UC and CD.
Coding mutation.
Mutation frequency of commonly mutated genes in nonhypermutated sporadic CRC and CAC (51).
Abbreviations: CAC, colitis-associated cancer; CAPEOX, capecitabine; CD, Crohn disease; CRC, colorectal cancer; FAP, familial adenomatous polyposis; FOLFIRI, fluorouracil and irinotecan; FOLFOX, fluorouracil and oxaliplatin; IBD, inflammatory bowel disease; JAK, Janus kinase; MSI, microsatellite instability; SI, small intestine; UC, ulcerative colitis.
The origin of inflammation in human CAC can be either autoimmunity, as is the case of ulcerative colitis or Crohn disease, which, although not a typical autoimmune disease, is characterized by uncontrolled activation of immune cells, particularly T cells. Both diseases are associated with dysregulated production of TNF and other inflammatory cytokines, such as IFN-γ and IL-17 (26, 56–58). In DSS-treated mice, however, inflammation is initiated by barrier disruption and enhanced translocation of microbes or microbial products, such as endotoxins and nucleic acids, that activate TLR signaling in mucosal macrophages (53, 59). On NF-κB and NLRP3 inflammasome activation, these macrophages produce several tumor-promoting cytokines, including TNF, IL-6, and IL-1β (44). Beyond endotoxin and nucleic acids, commensal-IgG immune complexes also activate NF-κB and NLRP3 inflammasomes via Fcγ receptors (FcγRs) (31). Moreover, IgG immune complex cross-linking of neonatal Fc receptor (FcRn) in DCs promotes Th1/Tc1 cytokine secretion and shows protective immunity against CRC in mice (60). Much of the early work on the DSS-AOM model had focused on the role of IL-6, which activates STAT3 to increase the survival and proliferation of IECs that harbor AOM-induced oncogenic mutations (7, 61). Another cytokine that contributes to STAT3 activation in CAC is IL-11, which unlike IL-6 is mainly produced by activated stromal fibroblasts (62). Another model used for deliberate induction of oncogenic colonic inflammation is infection with Helicobacter felis (63). In addition to macrophages, both DSS and H. felis activate ILCs that produce IL-22 (64). IL-22 is a regeneration-inducing cytokine that activates intestinal stem cells (65). IL-22, however, is not the only regeneration-inducing cytokine, as IL-6 appears to be even more critical for postinjury barrier restoration (61). In addition to STAT3, the regenerative activity of IL-6, and probably IL-22 as well, depends on activation of the key regenerative and reparative transcriptional activator YAP (66). Whereas IL-6-induced STAT3 activation depends on the JAK subgroup of tyrosine kinases, YAP activation depends on Src tyrosine kinases (66). Notably, STAT3, JAK, YAP, and Src/Yes activation is not restricted to mouse models of CAC but was found to take place in 70% of human CRCs (67).
The common occurrence of STAT3 and YAP activation in sporadic CRC indicates that inflammatory processes are also activated in the major form of CRC. Indeed, large-scale epidemiological studies had shown that the incidence of CRC is substantially lower in individuals who take nonsteroidal anti-inflammatory drugs, including aspirin (68). However, it took some time to understand how inflammation is initiated in sporadic and familial CRC. As mentioned above, the key oncogenic event in sporadic and familial CRC is loss of the APC tumor suppressor. Using the CPC-APC mouse model of CRC, in which formation of colonic adenomas depends on Apc loss of heterozygosity (69), we found that in addition to activation of β-catenin, loss of APC compromises barrier integrity (17). Reduced expression of the junctional adhesion proteins JAM-A, B, and C, as well as mucin 2 (Muc2), was observed in early ACF lesions. Moreover, using an inducible biallelic loss of Apc, we detected JAM-A, JAM-B, and Muc2 downregulation at both the RNA and protein levels within three weeks after APC loss (17). Barrier disruption resulted in translocation of endotoxin and other microbial products that led to TLR2/4/9-dependent induction of IL-23 in lamina propria myeloid cells. Importantly, elevated expression of IL-23 was also observed in human CRC, where it correlates with a worse prognostic outcome (70). In CPC-APC mice, ablation of the Il23p19 or the Il23r gene attenuated development of colonic adenomas and reduced their histopathological grade (17). IL-23 receptor (IL-23R), however, is not expressed in either normal or transformed epithelial cells, and its effect on tumor growth is mediated through expansion of IL-17-producing T cells, including Th17 and γδ T cells. Correspondingly, IL-17 receptor A (IL-17RA) ablation attenuated colonic tumor growth in CPC-APC mice (17). Unlike IL-23R, IL-17RA is expressed in IECs, in which it mediates NF-κB and ERK activation and stimulates cell proliferation within ACF lesions, thereby leading to formation of colonic adenomas (71). Similar findings regarding the importance of IL-17A and IL-17RA signaling were made in Apc mice infected with Bacteroides fragilis (72–74). Elevated expression of IL-17A was also observed in early human CRC, and high IL-17A expressers were found to be at a much higher risk of lethal metastatic disease than low IL-17A expressers (75). Likewise, an IL-17A gene polymorphism (rs2275913, G198) was linked to increased CRC risk and worse prognostic outcome (76). Moreover, T cell—specific ablation of IL-1R1 decreased tumor-elicited inflammation dependent upon IL-17A and IL-22, thereby reducing CRC progression (77). Curiously, however, in patients with advanced CRC, high IL-17A expression correlated with improved outcome (78), suggesting that at some point IL-17A may stimulate antitumor immunity. These observations, however, are at odds with the general observation that most CRCs are refractory to checkpoint inhibitor therapy, whose success requires activation of tumor-resident T cells (79, 80). Another IL-17 family member, IL-17C, was also found to be upregulated in mouse CRC through microbial dysbiosis (81). Although its mechanistic basis remains obscure, barrier disruption also seems to be the inflammatory driver that accounts for development of serrated adenomas in mice with IEC-specific disruption of the atypical protein kinase C ζ (PKCζ) and PKCλ/ι (82). Just as first found in CPC-APC mice (17), adenoma development in Prkcifl/flPrkczfl/flVillin-Cre mice is inhibited by antibiotics (82). As aPKC downregulation and inflammation were also detected in human serrated tumors, it appears that barrier loss is a general and key event in all forms of CRC. Barrier loss may precede microbial dysbiosis, but microbial factors can further enhance barrier loss (Figure 3).
Figure 3.

Chronic inflammation and its role in colorectal and pancreatic cancer development and metastasis to the liver. Bacterial translocation due to barrier disruption induces myeloid cell expression of TNF, IL-6, and IL-23, which in turn activate and expand Th17 cells, support NF-κB and STAT3 activation in intraepithelial cells, and accelerate tumor development and metastatic spread. Abbreviations: ER, endoplasmic reticulum; FcR, fragment crystallizable receptor; LPS, lipopolysaccharide; Th17, T helper type 17; TLR, Toll-like receptor.
MICROBES IN GASTROINTESTINAL CANCER
Dietary and various environmental factors modulate the composition and metabolic activity of the gut microbiota, which in turn can impact health. Furthermore, dietary factors also modulate barrier function (83–85). Particularly, high-fat diet (HFD) induces barrier dysfunction and dysbiosis and alters the gut metabolome (34, 86–90). HFD-induced dysbiosis is thought to promote obesity by increasing the capacity for energy harvest and storage (20), but it can also operate through inflammation-dependent mechanisms (91, 92). In particular, dysbiosis-induced inflammation was proposed to contribute to nonalcoholic steatohepatitis (NASH) development and thereby increase HCC risk (34, 93, 94), but as mentioned above, dysbiosis can be secondary to barrier disruption (95). Regardless of this classic chicken-or-egg question, dysbiosis can trigger intestinal inflammation and barrier impairment, but barrier impairment is also likely to precede dysbiosis (56, 89). A summary regarding the role of the most abundant bacterial phyla and their metabolites and corresponding physiological impact on health and cancer has been provided in Supplemental Table 1. Either way, microbial products can reach the liver via the portal circulation to induce hepatic inflammation and contribute to NASH and its progression to HCC (96). Although the main inducer of steatohepatitis remains to be identified, it could be none other than endotoxin (97). Although correlative studies suggest a disconnect between gram-negative bacteria, endotoxin, and NASH, these factors were also elevated in healthy obese individuals not suffering from NASH (98). The gut microbiome was also proposed to modulate gut and liver immunity through short-chain fatty acids (SCFAs), tryptophan metabolites that bind to the aryl hydrocarbon receptor (AhR), and retinoic acid (99–101). SCFAs (acetate, propionate, and butyrate) can be detected by the intracellular receptor PPARγ and the G protein—coupled receptors (GPRs) GPR41, GPR43, and GPR109a, the last of which also binds butyrate. Paradoxically, however, SCFAs enhance barrier function and immune tolerance and promote gut homeostasis through increased mucus production by goblet cells (100). SCFAs inhibit NF-κB, activate inflammasomes and induce IL-18 production, increase IgA secretion by B cells, reduce expression of T cell—activating molecules on antigen-presenting cells, and increase the number and function of colonic Tregs. Commensal Lactobacillus uses tryptophan to produce AhR ligands, such as indole-3-aldehyde (102). AhR activation is critical for the organogenesis of intestinal lymphoid follicles (ILFs) and AhR-expressing immune cells, including RORγt+ ILC3s that are involved in ILF genesis (103). In addition, AhR-induced IL-22 production by ILCs drives secretion of the antimicrobial peptides lipocalin-2, S100A8, and S100A9, which provide protection from translocating microbes (103, 104), as well as Candida albicans (102). Furthermore, IL-22 is a potent regenerative cytokine that restores barrier integrity (105). Several bacterial factors that engage noncanonical PRRs have also been identified, including polysaccharide A (PSA), formyl peptides, and D-glycero-β-D-manno-heptose-1,7-biphosphate (HBP). PSA from Bacteroides fragilis can alter the Th1–Th2 cell balance (100, 101). Formyl peptides released by all bacteria bind formyl peptide receptors (FPRs), which are GPRs found on neutrophils and other immune cells (100–103). Staphylococcus aureus formyl peptides can signal through FPR1 and contribute to activation of nociceptor-driven mechanical pain and release of immunosuppressive neuropeptides (106). At high nanomolar concentrations, S. aureus formyl peptides, also known as phenol-soluble modulins (PSMs), stimulate massive neutrophil influx to infection sites by binding FPR2 (100). Induced neutrophil activation leads to an oxidative burst. PSMs affect the adaptive immune system by inducing a tolerogenic phenotype in DCs and inhibiting Th1 differentiation (107). These are just a few examples of how bacterial products and metabolites can regulate adaptive immune cells and therefore modulate GI cancer development (23, 100), both positively via TLRs and negatively via tolerogenic GPRs.
Another way in which the intestinal microbiota and barrier disruption can affect HCC development in mice and humans is through modulation of IgA plasmocyte development. The term gut-liver axis was first described when researchers reported production of IgA antibodies to dietary antigens in patients with liver cirrhosis, indicating interactions between the gut and liver (108). IgA can also be derived from naive B cells recruited into the liver tumor microenvironment (TME) by CAF-generated chemokines (109, 110). After binding specific antigens that could be bacterium, food, or tumor derived to B cell receptors (BCRs) and further exposure to TGF-β and other cytokines, including IL-21 from circulatory follicular T helper (Tfh) cells, lymphotoxin β (LTβ), IL-33, and IL-10, naive B cells undergo class-switch recombination (CSR) to produce IgA (34, 110, 111). While the initial inflammatory response in the liver entails induction of B cell—recruiting chemokines, such as CXCL12 and CXCL13, chronic hepatitis eventually results in production of high amounts of TGF-β, an anti-inflammatory cytokine that is typical of the healing phase of the inflammatory response (112–114). In addition, both NASH and alcoholic steatohepatitis (ASH) result in recruitment of circulating Tfh cells that produce IL-21 (34). Together, TGF-β and Tfh-expressed IL-21 initiate T cell—dependent CSR that converts IgM-expressing naive B cells to IgA+ plasmablasts and plasma cells (collectively referred to as plasmocytes) that also express high amounts of the immunosuppressive molecules IL-10 and PD-L1 (34). Correspondingly, both human and mouse NASH-afflicted livers contain numerous IgA+ plasmocytes that express high amounts of IL-10 and PD-L1 (34). Although these cells are most likely generated within the liver through a T cell—dependent mechanism, gut sterilization results in their disappearance and inhibition of NASH to HCC progression. IgA ablation also inhibits NASH to HCC progression, which is supported by IgA+ plasmocytes that induce the exhaustion of HCC-directed cytotoxic T lymphocytes (CTLs). By contrast, depletion of polymeric immunoglobulin receptor gene (Pigr) enhances HCC development, indicating different functions of secretory IgA (sIgA) and IgA+ plasmocytes in HCC (34). In addition to inducing CTL exhaustion, IgA+ plasmocytes can also regulate and dampen neuroinflammation (115). These anti-inflammatory and immunosuppressive effects are in line with the well-established homeostatic and regulatory role of IgA+ plasmocytes in mucosal immunity (28, 116, 117). Given that the primary role of IgA is to maintain bacterial homeostasis, IgA+ plasmocytes may suppress the growth of tumors that depend on microbial signals, like CRC. Accordingly, IL-33-deficient mice showed decreased IgA level and dysbiosis, which support IL-1α-dependent colitis and CAC (118). The gut microbiome can also affect the liver immune system through bile acid regulation of chemokine-dependent accumulation of natural killer T (NKT) cells (119).
Barrier disruption as an early event in CRC development places the gut microbiota as the key instigator of tumor-elicited inflammation in both APC-dependent and APC-independent CRC. Treatment of CPC-APC or a PKC-deficient mice with broad-spectrum antibiotics, which get rid of approximately 99.5% of the colonic microbiota, attenuates development of both tubular (17) and serrated adenomas (82). A similar effect has been observed after triple ablation of TLR2, 4, and 9 in bone marrow—derived cells, suggesting that most colonic bacteria act nonspecifically through pathogen-associated molecular pattern (PAMP)-mediated activation of TLR signaling. Other studies, however, have implicated specific microbes in CRC development. Toxigenic Escherichia coli and B. fragilis were reported to invade polyps and shown to encode colibactin and B. fragilis cancer-promoting oncotoxins, respectively (120, 121). Colibactin can alkylate DNA to cause damage that contributes to genomic instability, mutations, and cancer (121). However, it is questionable whether the number of colibactin-producing E. coli is sufficiently high to induce such damage. Likewise, there is little evidence that B. fragilis populates premalignant lesions. Only one bacterium, Helicobacter pylori, has been confirmed to act as a carcinogen in the stomach (6, 122). However, even in this case, only 2% of H. pylori—infected individuals ever develop gastric cancer. Moreover, the carcinogenic activity of H. pylori depends on a particular variant of its cytotoxin-associated gene A (CagA) locus, which codes for a scaffold protein that modulates Ras and Wnt signaling (123). Furthermore, even the oncogenic CagA variant does not act alone, as it only leads to induction of gastric metaplasia, whose further progression to dysplasia requires epithelial injury and inflammation brought about by secondary factors, such as foods with high salt content or high levels of nitrates (124). An etiologic relationship between gut bacteria and immune-suppressive inflammation in PDAC has also been described, showing that distinct and abundant microbes like Bifidobacterium pseudolongum drive suppressive monocytic cellular differentiation via selective TLR engagement that leads to T cell anergy and enhanced tumor growth (125).
THE MANY FACES OF INFLAMMATION IN HEPATOCELLULAR CARCINOMA
HCC, the main type of liver cancer and the second leading cause of cancer-related death, is the prototypical inflammation-dependent cancer (Figure 4). The main cause of HCC is viral hepatitis caused by HBV or HCV However, whereas HBV is a DNA virus that integrates into the host genome and leads to induction of oncogenic mutations by insertion mutagenesis (126), HCV is a nonintegrating RNA virus that exerts its oncogenic effect via induction of inflammation and endoplasmic reticulum (ER) stress (127, 128). HBV infection results in CD8+ T cell activation, but these cells quickly assume an exhausted phenotype (129). By contrast, the immune/inflammatory response evoked by HCV is mainly mediated by innate cells, since HCV does not sustain an effective T cell response. Like HCV, whose importance as an HCC initiator is declining due to the recent introduction of highly effective anti-HCV drugs, the two major nonviral causes of HCC, NASH and ASH, also operate through inflammation and ER stress (130). The availability of reasonably good mouse models of NASH-induced HCC (131) revealed that the two main tumor-promoting cytokines in the liver are TNF and IL-6 (132–136). Whereas TNF exerts its oncogenic effect through NF-κB (134), IL-6 mainly functions through STAT3 activation (132, 133). Nonetheless, hepatocyte-specific ablation of either IKKβ, the protein kinase responsible for NF-κB activation, or STAT3 does not always lead to inhibition of HCC development. Under certain circumstances, particularly in models that are associated with induction of hepatocyte necrosis or repetitive liver injury, IKKβ/NF-κB or STAT3 ablation can actually accelerate HCC development (137). Moreover, damaged hepatocytes coordinate myeloid cell accumulation and fibrosis within the liver and support metastasis. Interestingly, during early pancreatic tumorigenesis in mice, hepatocytes show activation of STAT3 signaling, increased production of serum amyloid A1 and A2 (SAA), and release of IL-6, which supports the establishment of a prometastatic niche in the liver (138). Gut inflammation may also contribute to the growth of metastatic CRC in the liver (139), but the mechanisms remain to be elucidated. The involvement of certain microbiota strains to support CRC metastasis has also been recently described (140) (Supplemental Table 1).
Figure 4.

Hepatocellular carcinoma and the effect of barrier disruption on its development. Chronic inflammation and sustained production of the two main tumor-promoting cytokines, TNF and IL-6, support HCC development. TNF exerts its oncogenic effect through NF-κB, whereas IL-6 mainly functions through activation of STAT3. However, release of tumor, bacterial, or food antigens, in combination with cytokines and chemokines produced by different types of monocytes and activated HSCs, can activate humoral and cellular immune cells, like tumor-promoting Th17 cells, Tregs, and IgA+ cells that compromise the antitumorigenic activity of different immune cells such as CTLs (cytotoxic CD8+ T cells). Abbreviations: BA, bacterial antigen; BCR, B cell receptor; CD, cell death; CSR, class-switch recombination; CTL, cytotoxic T lymphocyte; DAMP, damage-associated molecular pattern; ER, endoplasmic reticulum; FcR, fragment crystallizable receptor; FM, food metabolite; HCC, hepatocellular carcinoma; HSC, hepatic stellate cell; KC, Kupffer cell; LPS, lipopolysaccharide; NK, natural killer; NOS, nitric oxide synthase; ROS, reactive oxygen species; Tfh, follicular T helper cell; TLR, Toll-like receptor.
NASH-induced HCC is also supported by IL-17A produced by Th17 cells (141). IL-17A is likely to act through activation of NF-κB in epithelial cells (71). Consistent with its ability to stimulate the proliferation of HCC progenitors, expression of constitutively active IKKβ in mouse hepatocytes accelerates HCC development (142). However, in addition to stimulating hepatocyte proliferation, IKKβ/NF-κB activation results in production of organotypic cytokines that orchestrate the generation of ectopic lymphoid clusters (ELCs), composed of T cells, B cells, and stromal fibroblasts/HSCs, that provide niches that support the growth of HCC progenitors. These niches may also protect HCC progenitors from killing by activated CTLs.
Another immune related mechanism through which chronic hepatitis supports HCC development involves the generation of IgA+ plasmocytes, which as discussed above suppress HCC-directed CTLs that otherwise prevent growth and expansion of HCC progenitors (34). While HCC-specific antigens that are recognized by HCC-targeting T cells remain to be identified, T cell receptor (TCR) sequencing indicates that the HCC-elicited immune response is oligoclonal in nature, encompassing the selection and amplification of approximately 20–30 immunodominant clones. Given the presence of exhausted but HCC-directed CD8+ T cells, treatment of HCC-bearing mice with a blocking PD-L1 antibody that induces reinvigoration of the latter results in regression of 60–70% of large liver tumors (34). In accordance with the important role of the physical interaction between PD-L1-expressing B cells and PD-1-expressing Tfh cells in sustaining the CSR reaction (143), anti-PD-L1 treatment also results in a substantial reduction in the number of liver-resident IgA+ plasmocytes, further facilitating CTL reinvigoration (34). In fact, circulating IgA was also described to be elevated in ASH and other chronic liver diseases, and its amounts show a direct correlation with the extent of liver fibrosis (34), which is not all that surprising given the profibrogenic function of TGF-β (144, 145), the main inducer of IgA CSR. Most likely, the PD-L1- and IL-10-expressing IgA+ plasmocytes in human liver carry out the same immunosuppressive and HCC-promoting functions ascribed to their mouse counterparts. Furthermore, the likely involvement of these cells in HCC development explains the surprisingly good response of human nonviral HCC to PD-1 blockade (146). By contrast to HCC, the majority of CRC is refractory to PD-1 blockade, with the exception of mismatch repair—deficient and microsatellite instability—high tumors (80, 147, 148), an effect that was correlated with the number of somatic mutations (149). Another key difference in the immunobiology of CRC versus that of HCC lies in their relationship with IgA+ plasmocytes, as also mentioned above. Although ER stress in hepatocytes drives NASH development (134), in IECs ER stress orchestrates a protective IgA response (150).
CONCLUSIONS AND FUTURE PROSPECTS
While the tumor-promoting function of chronic inflammation has been recognized and amply reviewed (2, 4, 151, 152), more recent work highlighted above indicates that most tumor-promoting inflammation, at least in cancers of the GI system, originates in barrier disruption. Initially described as a source of tumor-elicited inflammation (17), barrier disruption can precede tumor development and contribute to pathological conditions that greatly increase cancer risk. Such pathologies include IBD, steatohepatitis, alcoholic hepatitis, and chronic pancreatitis. So far, the protumorigenic function of barrier disruption is mainly supported by association studies and by the ability of broad-spectrum antibiotics to inhibit CRC, HCC, and PDAC in mouse models (17, 34, 125). Pending development of the right experimental tools, we hope that future work will allow clinical investigators to determine the prevalence of barrier disruption in human GI malignancies and their associated premalignant states—IBD, hepatitis, and pancreatitis. Just as important is the development of specific experimental and pharmacological tools that can prevent barrier deterioration or enhance barrier restoration. In addition to establishing the importance of barrier disruption, such tools can be used to prevent oncogenic progression of chronic GI-related inflammatory conditions and reduce the enormous toll levied by highly aggressive malignancies such as HCC, PDAC, and liver-metastatic CRC.
Although translocating gut microbes and microbial products can trigger tumor-promoting inflammation through general means such as TLR engagement, we should not discount the possible existence of cancer-promoting bacterial toxins and metabolites. If the carcinogenic activity of such substances is validated in human disease, future research should be directed at identification and development of specific antidotes and protective interventions.
In summary, tumor-promoting inflammation is initiated as a protective response to translocating commensals. In addition to stimulating the proliferation and survival of tumor progenitors, chronic tumor-promoting inflammation also acts via immune mechanisms, such as the TGF-β—and Tfh-driven generation of immunosuppressive IgA+ plasmocytes. Much more effort is needed to better understand the immune mechanisms through which chronic inflammation supports the growth and progression of colonic and pancreatic adenomas and adenocarcinomas. Such understanding will improve our ability to successfully use immunotherapeutics, such as checkpoint inhibitors, in cancers where they have not been proven effective.
Supplementary Material
ACKNOWLEDGMENTS
We thank C.R. Lichtenstern for writing and editorial assistance and R.K. Ngu for illustration and figure preparation. S.S. was supported by a PCF Young Investigator Award and by SCRC for ALPD & Cirrhosis, funded by the NIAAA (P50 AA011999). Work in M.K.’s laboratory was supported by grants from the NIH (R01 AI043477, R01 CA211794) and Tower Cancer Research Foundation. Additional support came from U01 AA027681 to S.S. and M.K., P01 CA128814 to M.K./Ze’ev Ronai, and Padres Pedal the Cause C3 award #PTC2018.
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
Footnotes
The Annual Review of Immunology is online at immunol.annualreviews.org
LITERATURE CITED
- 1.Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: the next generation. Cell 144(5):646–74 [DOI] [PubMed] [Google Scholar]
- 2.Grivennikov SI, Greten FR, Karin M. 2010. Immunity, inflammation, and cancer. Cell 140(6):883–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shalapour S, Karin M. 2015. Immunity, inflammation, and cancer: an eternal fight between good and evil. J. Clin. Investig 125(9):3347–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ruffell B, Coussens LM. 2015. Macrophages and therapeutic resistance in cancer. Cancer Cell 27(4):462–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Warren JR, Marshall B. 1983. Unidentified curved bacilli on gastric epithelium in active chronic gastritis. Lancet 321(8336):1273–75 [PubMed] [Google Scholar]
- 6.Suerbaum S, Michetti P. 2002. Helicobacter pylori infection. N. Engl. J. Med 347:1175–86 [DOI] [PubMed] [Google Scholar]
- 7.Greten FR, Eckmann L, Greten TF, Park JM, Li Z-W, et al. 2004. IKKβ links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 118(3):285–96 [DOI] [PubMed] [Google Scholar]
- 8.Willett WC, Stampfer MJ, Colditz GA, Rosner BA, Speizer FE. 1990. Relation of meat, fat, and fiber intake to the risk of colon cancer in a prospective study among women. N. Engl. J. Med 323(24):1664–72 [DOI] [PubMed] [Google Scholar]
- 9.Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, et al. 2004. NF-κB functions as a tumour promoter in inflammation-associated cancer. Nature 431(7007):461–66 [DOI] [PubMed] [Google Scholar]
- 10.Zhang Y, Yan W, Collins MA, Bednar F, Rakshit S, et al. 2013. Interleukin-6 is required for pancreatic cancer progression by promoting MAPK signaling activation and oxidative stress resistance. Cancer Res. 73(20):6359–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Font-Burgada J, Shalapour S, Ramaswamy S, Hsueh B, Rossell D, et al. 2015. Hybrid periportal hepatocytes regenerate the injured liver without giving rise to cancer. Cell 162(4):766–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McLoughlin MR, Orlicky DJ, Prigge JR, Krishna P, Talago EA, et al. 2019. TrxR1, Gsr, and oxidative stress determine hepatocellular carcinoma malignancy. PNAS 116(23):11408–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zambirinis CP, Pushalkar S, Saxena D, Miller G. 2014. Pancreatic cancer, inflammation and microbiome. Cancer J. 20(3):195–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee PJ, Papachristou GI. 2019. New insights into acute pancreatitis. Nat. Rev. Gastroenterol. Hepatol 16(8):479–96 [DOI] [PubMed] [Google Scholar]
- 15.de Sousa e Melo F, de Sauvage FJ. 2019. Cellular plasticity in intestinal homeostasis and disease. Cell Stem Cell 24(1):54–64 [DOI] [PubMed] [Google Scholar]
- 16.Hillman ET, Lu H, Yao T, Nakatsu CH. 2017. Microbial ecology along the gastrointestinal tract. Microbes Environ. 32(4):300–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, et al. 2012. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 491(7423):254–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Siegel RL, Miller KD, Jemal A. 2018. Cancer statistics, 2018. CA Cancer J. Clin 68(1):7–30 [DOI] [PubMed] [Google Scholar]
- 19.Wei M-Y, Shi S, Liang C, Meng Q-C, Hua J, et al. 2019. The microbiota and microbiome in pancreatic cancer: more influential than expected. Mol. Cancer 18:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Conlon MA, Bird AR. 2014. The impact of diet and lifestyle on gut microbiota and human health. Nutrients 7(1):17–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bowcutt R, Forman R, Glymenaki M, Carding SR, Else KJ, Cruickshank SM. 2014. Heterogeneity across the murine small and large intestine. World J. Gastroenterol 20(41):15216–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zihni C, Mills C, Matter K, Balda MS. 2016. Tight junctions: from simple barriers to multifunctional molecular gates. Nat. Rev. Mol. Cell Biol 17(9):564–80 [DOI] [PubMed] [Google Scholar]
- 23.Dzutsev A, Badger JH, Perez-Chanona E, Roy S, Salcedo R, et al. 2017. Microbes and cancer. Annu. Rev. Immunol 35(1):199–228 [DOI] [PubMed] [Google Scholar]
- 24.Lavelle E, Murphy C, O’Neill L, Creagh E. 2010. The role of TLRs, NLRs, and RLRs in mucosal innate immunity and homeostasis. Mucosal Immunol. 3(1):17–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harrison OJ, Powrie FM. 2013. Regulatory T cells and immune tolerance in the intestine. Cold Spnng Harb. Perspect. Biol 5(7):a018341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Friedrich M, Pohin M, Powrie F. 2019. Cytokine networks in the pathophysiology of inflammatory bowel disease. Immunity 50(4):992–1006 [DOI] [PubMed] [Google Scholar]
- 27.Nakajima A, Vogelzang A, Maruya M, Miyajima M, Murata M, et al. 2018. IgA regulates the composition and metabolic function of gut microbiota by promoting symbiosis between bacteria. J. Exp. Med 215(8):2019–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Macpherson AJ, Yilmaz B, Limenitakis JP, Ganal-Vonarburg SC. 2018. IgA function in relation to the intestinal microbiota. Annu. Rev. Immunol 36(1):359–81 [DOI] [PubMed] [Google Scholar]
- 29.Kamada N, Sakamoto K, Seo S-U, Zeng MY, Kim Y-G, et al. 2015. Humoral immunity in the gut selectively targets phenotypically virulent attaching-and-effacing bacteria for intraluminal elimination. Cell Host Microbe 17(5):617–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fatkhullina AR, Peshkova IO, Dzutsev A, Aghayev T, McCulloch JA, et al. 2018. An interleukin-23-interleukin-22 axis regulates intestinal microbial homeostasis to protect from diet-induced atherosclerosis. Immunity 49(5):943–957.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Castro-Dopico T, Dennison TW, Ferdinand JR, Mathews RJ, Fleming A, et al. 2019. Anti-commensal IgG drives intestinal inflammation and type 17 immunity in ulcerative colitis. Immunity 50(4):1099–114.e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mannon PJ 2019. Immunological diseases of the gastrointestinal. tract. In Clinical Immunology: Principles and Practice, ed. Rich RR, Fleisher TA, Shearer WT, Schroeder HW Jr., Frew AJ, et al. , pp. 1005–19.e1. London: Elsevier. 5th ed. [Google Scholar]
- 33.Tiegs G, Lohse AW. 2010. Immune tolerance: what is unique about the liver. J. Autoimmun 34(1):1–6 [DOI] [PubMed] [Google Scholar]
- 34.Shalapour S, Lin X-J, Bastian IN, Brain J, Burt AD, et al. 2017. Inflammation-induced IgA+ cells dismantle anti-liver cancer immunity. Nature 551(7680):340–45. Erratum. 2017. Nature 552(7685):430. Correction. 2018. Nature 561(7721):E1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shen H, Lipka S, Kumar A, Mustacchia P. 2014. Association between nonalcoholic fatty liver disease and colorectal adenoma: a systemic review and meta-analysis. J. Gastrointest. Oncol 5(6):440–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mantovani A, Dauriz M, Byrne CD, Lonardo A, Zoppini G, et al. 2018. Association between non-alcoholic fatty liver disease and colorectal tumours in asymptomatic adults undergoing screening colonoscopy: a systematic review and meta-analysis. Metabolism 87:1–12 [DOI] [PubMed] [Google Scholar]
- 37.Baker A-M, Cross W, Curtius K, Bakir IA, Choi C-HR, et al. 2019. Evolutionary history of human colitis-associated colorectal cancer. Gut 68(6):985–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Al Bakir I, Curtius K, Graham TA. 2018. From colitis to cancer: an evolutionary trajectory that merges maths and biology. Front. Immunol 9:2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rubin DC, Shaker A, Levin MS. 2012. Chronic intestinal inflammation: inflammatory bowel disease and colitis-associated colon cancer. Front. Immunol 3:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhen Y, Luo C, Zhang H. 2018. Early detection of ulcerative colitis-associated colorectal cancer. Gastroenterol. Rep 6(2):83–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Khor B, Gardet A, Xavier RJ. 2011. Genetics and pathogenesis of inflammatory bowel disease. Nature 474(7351):307–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Triantafillidis JK, Nasioulas G, Kosmidis PA. 2009. Colorectal cancer and inflammatory bowel disease: epidemiology, risk factors, mechanisms of carcinogenesis and prevention strategies. Anticancer Res. 29(7):2727–37 [PubMed] [Google Scholar]
- 43.Waldner MJ, Neurath MF 2015. Mechanisms of immune signaling in colitis-associated cancer. Cell Mol. Gastroenterol. Hepatol 1(1):6–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Terzić J, Grivennikov S, Karin E, Karin M. 2010. Inflammation and colon cancer. Gastroenterology 138(6):2101–14.e5 [DOI] [PubMed] [Google Scholar]
- 45.Oh M, McBride A, Yun S, Bhattacharjee S, Slack M, et al. 2018. BRCA1 and BRCA2 gene mutations and colorectal cancer risk: systematic review and meta-analysis. J. Natl. Cancer Inst 110(11):1178–89 [DOI] [PubMed] [Google Scholar]
- 46.Stoffel EM, Kastrinos F. 2014. Familial colorectal cancer, beyond Lynch syndrome. Clin. Gastroenterol. Hepatol 12(7):1059–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lowery JT, Ahnen DJ, Schroy PC, Hampel H, Baxter N, et al. 2016. Understanding the contribution of family history to colorectal cancer risk and its clinical implications: a state-of-the-science review. Cancer 122(17):2633–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cunningham D, Atkin W, Lenz H-J, Lynch HT, Minsky B, et al. 2010. Colorectal cancer. Lancet 375(9719):1030–47 [DOI] [PubMed] [Google Scholar]
- 49.Fujita M, Matsubara N, Matsuda I, Maejima K, Oosawa A, et al. 2017. Genomic landscape of colitis-associated cancer indicates the impact of chronic inflammation and its stratification by mutations in the Wnt signaling. Oncotarget 9(1):969–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kameyama H, Nagahashi M, Shimada Y, Tajima Y, Ichikawa H, et al. 2018. Genomic characterization of colitis-associated colorectal cancer. World J. Surg. Oncol 16(1):121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Robles AI, Traverso G, Zhang M, Roberts NJ, Khan MA, et al. 2016. Whole-exome sequencing analyses of inflammatory bowel disease—associated colorectal cancers. Gastroenterology 150(4):931–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Canli Ö, Nicolas AM, Gupta J, Finkelmeier F, Goncharova O, et al. 2017. Myeloid cell-derived reactive oxygen species induce epithelial mutagenesis. Cancer Cell 32(6):869–83.e5 [DOI] [PubMed] [Google Scholar]
- 53.Rakoff-Nahoum S, Medzhitov R. 2007. Regulation of spontaneous intestinal tumorigenesis through the adaptor protein MyD88. Science 317(5834):124–27 [DOI] [PubMed] [Google Scholar]
- 54.Johnson CM, Wei C, Ensor JE, Smolenski DJ, Amos CI, et al. 2013. Meta-analyses of colorectal cancer risk factors. Cancer Causes Control 24(6):1207–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shaked H, Hofseth LJ, Chumanevich A, Chumanevich AA, Wang J, et al. 2012. Chronic epithelial NF-κB activation accelerates APC loss and intestinal tumor initiation through iNOS up-regulation. PNAS 109(35):14007–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Neurath MF. 2019. Targeting immune cell circuits and trafficking in inflammatory bowel disease. Nat. Immunol 20(8):970–79 [DOI] [PubMed] [Google Scholar]
- 57.West NR, Hegazy AN, Owens BMJ, Bullers SJ, Linggi B, et al. 2017. Oncostatin M drives intestinal inflammation and predicts response to tumor necrosis factor-neutralizing therapy in patients with inflammatory bowel disease. Nat. Med 23(5):579–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Danne C, Powrie F. 2018. Helicobacter hepaticus polysaccharide induces an anti-inflammatory response in intestinal macrophages. Microb. Cell 5(4):208–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. 2004. Recognition of commensal microflora by Toll-like receptors is required for intestinal homeostasis. Cell 118(2):229–41 [DOI] [PubMed] [Google Scholar]
- 60.Baker K, Rath T, Flak MB, Arthur JC, Chen Z, et al. 2013. Neonatal Fc receptor expression in dendritic cells mediates protective immunity against colorectal cancer. Immunity 39(6):1095–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Grivennikov S, Karin E, Terzic J, Mucida D, Yu G-Y, et al. 2009. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 15(2):103–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Putoczki TL, Thiem S, Loving A, Busuttil RA, Wilson NJ, et al. 2013. Interleukin-11 is the dominant IL-6 family cytokine during gastrointestinal tumorigenesis and can be targeted therapeutically. Cancer Cell 24(2):257–71 [DOI] [PubMed] [Google Scholar]
- 63.Cai X, Carlson J, Stoicov C, Li H, Wang TC, Houghton J. 2005. Helicobacter felis eradication restores normal architecture and inhibits gastric cancer progression in C57BL/6 mice. Gastroenterology 128(7):1937–52 [DOI] [PubMed] [Google Scholar]
- 64.Kirchberger S, Royston DJ, Boulard O, Thornton E, Franchini F, et al. 2013. Innate lymphoid cells sustain colon cancer through production of interleukin-22 in a mouse model. J. Exp. Med 210(5):917–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lindemans CA, Calafiore M, Mertelsmann AM, O’Connor MH, Dudakov JA, et al. 2015. Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature 528(7583):560–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Taniguchi K, Wu L-W, Grivennikov SI, de Jong PR, Lian I, et al. 2015. A gp130-Src-YAP module links inflammation to epithelial regeneration. Nature 519(7541):57–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Taniguchi K, Moroishi T, de Jong PR, Krawczyk M, Grebbin BM, et al. 2017. YAP-IL-6ST autoregulatory loop activated on APC loss controls colonic tumorigenesis. PNAS 114(7):1643–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ruder EH, Laiyemo AO, Graubard BI, Hollenbeck AR, Schatzkin A, Cross AJ. 2011. Non-steroidal anti-inflammatory drugs and colorectal cancer risk in a large, prospective cohort. Am. J. Gastroenterol 106(7):1340–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hinoi T, Akyol A, Theisen BK, Ferguson DO, Greenson JK, et al. 2007. Mouse model of colonic adenoma-carcinoma progression based on somatic Apc inactivation. Cancer Res. 67(20):9721–30 [DOI] [PubMed] [Google Scholar]
- 70.Hu W-H, Chen H-H, Yen S-L, Huang H-Y, Hsiao C-C, Chuang J-H. 2017. Increased expression of interleukin-23 associated with progression of colorectal cancer. J. Surg. Oncol 115(2):208–12 [DOI] [PubMed] [Google Scholar]
- 71.Wang K, Kim MK, Di Caro G, Wong J, Shalapour S, et al. 2014. Interleukin-17 receptor A signaling in transformed enterocytes promotes early colorectal tumorigenesis. Immunity 41(6):1052–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Housseau F, Wu S, Wick EC, Fan H, Wu X, et al. 2016. Redundant innate and adaptive sources of IL-17 production drive colon tumorigenesis. Cancer Res. 76(8):2115–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hurtado CG, Wan F, Housseau F, Sears CL. 2018. Roles for interleukin 17 and adaptive immunity in pathogenesis of colorectal cancer. Gastroenterology 155(6):1706–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wu S, Rhee K-J, Albesiano E, Rabizadeh S, Wu X, et al. 2009. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med 15(9):1016–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tosolini M, Kirilovsky A, Mlecnik B, Fredriksen T, Mauger S, et al. 2011. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, Th2, Treg, Th17) in patients with colorectal cancer. Cancer Res. 71(4):1263–71 [DOI] [PubMed] [Google Scholar]
- 76.Omrane I, Marrakchi R, Baroudi O, Mezlini A, Ayari H, et al. 2014. Significant association between interleukin-17A polymorphism and colorectal cancer. Tumor Biol. 35(7):6627–32 [DOI] [PubMed] [Google Scholar]
- 77.Dmitrieva-Posocco O, Dzutsev A, Posocco DF, Hou V, Yuan W, et al. 2019. Cell-type-specific responses to interleukin-1 control microbial invasion and tumor-elicited inflammation in colorectal cancer. Immunity 50(1):166–80.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Amicarella F, Muraro MG, Hirt C, Cremonesi E, Padovan E, et al. 2017. Dual role of tumour-infiltrating T helper 17 cells in human colorectal cancer. Gut 66(4):692–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Routy B, Chatelier EL, Derosa L, Duong CPM, Alou MT, et al. 2018. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 359(6371):91–97 [DOI] [PubMed] [Google Scholar]
- 80.Ganesh K, Stadler ZK, Cercek A, Mendelsohn RB, Shia J, et al. 2019. Immunotherapy in colorectal cancer: rationale, challenges and potential. Nat. Rev. Gastroenterol. Hepatol 16(6):361–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Song X, Gao H, Lin Y, Yao Y, Zhu S, et al. 2014. Alterations in the microbiota drive interleukin-17C production from intestinal epithelial cells to promote tumorigenesis. Immunity 40(1):140–52 [DOI] [PubMed] [Google Scholar]
- 82.Nakanishi Y, Duran A, L’Hermitte A, Shelton PM, Nakanishi N, et al. 2018. Simultaneous loss of both atypical protein kinase C genes in the intestinal epithelium drives serrated intestinal cancer by impairing immunosurveillance. Immunity 49(6):1132–47.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sellmann C, Priebs J, Landmann M, Degen C, Engstler AJ, et al. 2015. Diets rich in fructose, fat or fructose and fat alter intestinal barrier function and lead to the development of nonalcoholic fatty liver disease over time. J. Nutr. Biochem 26(11):1183–92 [DOI] [PubMed] [Google Scholar]
- 84.Hamilton MK, Boudry G, Lemay DG, Raybould HE. 2015. Changes in intestinal barrier function and gut microbiota in high-fat diet-fed rats are dynamic and region dependent. Am. J. Physiol. Gastrointest. Liver Physiol 308(10):G840–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bach Knudsen KE, Lærke HN, Hedemann MS, Nielsen TS, Ingerslev AK, et al. 2018. Impact of diet-modulated butyrate production on intestinal barrier function and inflammation. Nutrients 10(10):1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fabbiano S, Suárez-Zamorano N, Chevalier C, Lazarević V, Kieser S, et al. 2018. Functional gut microbiota remodeling contributes to the caloric restriction-induced metabolic improvements. Cell Metab. 28(6):907–21.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Parks BW, Nam E, Org E, Kostem E, Norheim F, et al. 2013. Genetic control of obesity and gut microbiota composition in response to high-fat, high-sucrose diet in mice. Cell Metab. 17(1):141–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schulz MD, Atay C, Heringer J, Romrig FK, Schwitalla S, et al. 2014. High-fat-diet-mediated dysbiosis promotes intestinal carcinogenesis independently of obesity. Nature 514(7523):508–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Arias-Jayo N, Abecia L, Alonso-Sáez L, Ramirez-Garcia A, Rodriguez A, Pardo MA. 2018. High-fat diet consumption induces microbiota dysbiosis and intestinal inflammation in zebrafish. Microb. Ecol 76(4):1089–101 [DOI] [PubMed] [Google Scholar]
- 90.Hildebrandt MA, Hoffmann C, Sherrill-Mix SA, Keilbaugh SA, Hamady M, et al. 2009. High-fat diet determines the composition of the murine gut microbiome independently of obesity. Gastroenterology 137(5):1716–24.e2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Brown K, DeCoffe D, Molcan E, Gibson DL. 2012. Diet-induced dysbiosis of the intestinal microbiota and the effects on immunity and disease. Nutrients 4(8):1095–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Saltzman ET, Palacios T, Thomsen M, Vitetta L. 2018. Intestinal microbiome shifts, dysbiosis, inflammation, and non-alcoholic fatty liver disease. Front. Microbiol 9:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Brandl K, Schnabl B. 2017. The intestinal microbiota and NASH. Curr. Opin. Gastroenterol 33(3):128–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yu L-X, Schwabe RF. 2017. The gut microbiome and liver cancer: mechanisms and clinical translation. Nat. Rev. Gastroenterol. Hepatol 14(9):527–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fukui H 2016. Increased intestinal permeability and decreased barrier function: Does it really influence the risk of inflammation? Inflamm. Intest. Dis 1(3):135–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tripathi A, Debelius J, Brenner DA, Karin M, Loomba R, et al. 2018. The gut-liver axis and the intersection with the microbiome. Nat. Rev. Gastroenterol. Hepatol 15(7):397–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rahman K, Desai C, Iyer SS, Thorn NE, Kumar P, et al. 2016. Loss of junctional adhesion molecule A promotes severe steatohepatitis in mice on a diet high in saturated fat, fructose, and cholesterol. Gastroenterology 151(4):733–46.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yuan J, Baker SS, Liu W, Alkhouri R, Baker RD, et al. 2014. Endotoxemia unrequired in the pathogenesis of pediatric nonalcoholic steatohepatitis. J. Gastroenterol. Hepatol 29(6):1292–98 [DOI] [PubMed] [Google Scholar]
- 99.Levy M, Thaiss CA, Elinav E. 2016. Metabolites: messengers between the microbiota and the immune system. Genes Dev. 30(14):1589–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rooks MG, Garrett WS. 2016. Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol 16(6):341–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Shibata N, Kunisawa J, Kiyono H. 2017. Dietary and microbial metabolites in the regulation of host immunity. Front. Microbiol 8:2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zelante T, Iannitti RG, Cunha C, De Luca A, Giovannini G, et al. 2013. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 39(2):372–85 [DOI] [PubMed] [Google Scholar]
- 103.Kiss EA, Diefenbach A. 2012. Role of the aryl hydrocarbon receptor in controlling maintenance and functional programs of RORγt+ innate lymphoid cells and intraepithelial lymphocytes. Front. Immunol 3:124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lee JS, Cella M, Colonna M. 2012. AHR and the transcriptional regulation of type-17/22 ILC. Front. Immunol 3:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sonnenberg GF, Fouser LA, Artis D. 2011. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat. Immunol 12(5):383–90 [DOI] [PubMed] [Google Scholar]
- 106.Haas P-J, de Haas CJC, Kleibeuker W, Poppelier MJJG, van Kessel KPM, et al. 2004. N-terminal residues of the chemotaxis inhibitory protein of Staphylococcus aureus are essential for blocking formylated peptide receptor but not C5a receptor. J. Immunol 173(9):5704–11 [DOI] [PubMed] [Google Scholar]
- 107.Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL. 2005. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell 122(1):107–18 [DOI] [PubMed] [Google Scholar]
- 108.Ash M 2018. The gut-liver axis. Clinical Education. https://www.clinicaleducation.org/resources/reviews/the-gut-liver-axis/ [Google Scholar]
- 109.Ammirante M, Shalapour S, Kang Y, Jamieson CAM, Karin M. 2014. Tissue injury and hypoxia promote malignant progression of prostate cancer by inducing CXCL13 expression in tumor myofibroblasts. PNAS 111(41):14776–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Shalapour S, Font-Burgada J, Di Caro G, Zhong Z, Sanchez-Lopez E, et al. 2015. Immunosuppressive plasma cells impede T-cell-dependent immunogenic chemotherapy. Nature 521(7550):94–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cerutti A 2008. The regulation of IgA class switching. Nat. Rev. Immunol 8(6):421–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wynn TA, Ramalingam TR. 2012. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat. Med 18(7):1028–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kim KK, Sheppard D, Chapman HA. 2018. TGF-β1 signaling and tissue fibrosis. Cold Spnng Harb. Perspect. Biol 10(4):a022293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Batlle E, Massagué J. 2019. Transforming growth factor-β signaling in immunity and cancer. Immunity 50(4):924–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Rojas OL, Pröbstel A-K, Porfilio EA, Wang AA, Charabati M, et al. 2019. Recirculating intestinal IgA-producing cells regulate neuroinflammation via IL-10. Cell 176(3):610–24.e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gutzeit C, Magri G, Cerutti A. 2014. Intestinal IgA production and its role in host-microbe interaction. Immunol. Rev 260(1):76–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mantis NJ, Rol N, Corthésy B. 2011. Secretory IgA’s complex roles in immunity and mucosal homeostasis in the gut. Mucosal Immunol. 4(6):603–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Malik A, Sharma D, Zhu Q, Karki R, Guy CS, et al. 2016. IL-33 regulates the IgA-microbiota axis to restrain IL-1α-dependent colitis and tumorigenesis. J. Clin. Investig 126(12):4469–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ma C, Han M, Heinrich B, Fu Q, Zhang Q, et al. 2018. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science 360(6391):eaan5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dejea CM, Fathi P, Craig JM, Boleij A, Taddese R, et al. 2018. Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science 359(6375):592–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wilson MR, Jiang Y, Villalta PW, Stornetta A, Boudreau PD, et al. 2019. The human gut bacterial genotoxin colibactin alkylates DNA. Science 363(6428):eaar7785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wang TC, Dangler CA, Chen D, Goldenring JR, Koh T, et al. 2000. Synergistic interaction between hypergastrinemia and Helicobacter infection in a mouse model of gastric cancer. Gastroenterology 118(1):36–47 [DOI] [PubMed] [Google Scholar]
- 123.Hatakeyama M 2014. Helicobacter pylori CagA and gastric cancer: a paradigm for hit-and-run carcinogenesis. Cell Host Microbe 15(3):306–16 [DOI] [PubMed] [Google Scholar]
- 124.Yordanov D, Boyanova L, Markovska R, Ilieva J, Andreev N, et al. 2017. Influence of dietary factors on Helicobacter pylori and CagA seroprevalence in Bulgaria. Gastroenterol. Res. Pract 2017:9212143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Pushalkar S, Hundeyin M, Daley D, Zambirinis CP, Kurz E, et al. 2018. The pancreatic cancer microbiome promotes oncogenesis by induction of innate and adaptive immune suppression. Cancer Discov. 8(4):403–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tarocchi M, Polvani S, Marroncini G, Galli A. 2014. Molecular mechanism of hepatitis B virus-induced hepatocarcinogenesis. World J. Gastroenterol 20(33):11630–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chusri P, Kumthip K, Hong J, Zhu C, Duan X, et al. 2016. HCV induces transforming growth factor β1 through activation of endoplasmic reticulum stress and the unfolded protein response. Sci. Rep 6:22487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Waris G, Tardif KD, Siddiqui A. 2002. Endoplasmic reticulum (ER) stress: hepatitis C virus induces an ER-nucleus signal transduction pathway and activates NF-κB and STAT-3. Biochem. Pharmacol 64(10):1425–30 [DOI] [PubMed] [Google Scholar]
- 129.Wang Q, Pan W, Liu Y, Luo J, Zhu D, et al. 2018. Hepatitis B virus-specific CD8+ T cells maintain functional exhaustion after antigen reexposure in an acute activation immune environment. Front. Immunol 9:219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Baiceanu A, Mesdom P, Lagouge M, Foufelle F. 2016. Endoplasmic reticulum proteostasis in hepatic steatosis. Nat. Rev. Endocrinol 12(12):710–22 [DOI] [PubMed] [Google Scholar]
- 131.Febbraio MA, Reibe S, Shalapour S, Ooi GJ, Watt MJ, Karin M. 2019. Preclinical models for studying NASH-driven HCC: How useful are they? Cell Metab. 29(1):18–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.He G, Yu G-Y, Temkin V, Ogata H, Kuntzen C, et al. 2010. Hepatocyte IKKβ/NF-κB inhibits tumor promotion and progression by preventing oxidative stress-driven STAT3 activation. Cancer Cell 17(3):286–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.He G, Dhar D, Nakagawa H, Font-Burgada J, Ogata H, et al. 2013. Identification of liver cancer progenitors whose malignant progression depends on autocrine IL-6 signaling. Cell 155(2):384–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Nakagawa H, Umemura A, Taniguchi K, Font-Burgada J, Dhar D, et al. 2014. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 26(3):331–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, et al. 2007. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science 317(5834):121–24 [DOI] [PubMed] [Google Scholar]
- 136.Park EJ, Lee JH, Yu G-Y, He G, Ali SR, et al. 2010. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 140(2):197–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Maeda S, Kamata H, Luo J-L, Leffert H, Karin M. 2005. IKKβ couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell 121(7):977–90 [DOI] [PubMed] [Google Scholar]
- 138.Lee JW, Stone ML, Porrett PM, Thomas SK, Komar CA, et al. 2019. Hepatocytes direct the formation of a pro-metastatic niche in the liver. Nature 567(7747):249–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zarour LR, Anand S, Billingsley KG, Bisson WH, Cercek A, et al. 2017. Colorectal cancer liver metastasis: evolving paradigms and future directions. Cell. Mol. Gastroenterol. Hepatol 3(2):163–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Bullman S, Pedamallu CS, Sicinska E, Clancy TE, Zhang X, et al. 2017. Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science. 358(6369):1443–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Gomes AL, Teijeiro A, Burén S, Tummala KS, Yilmaz M, et al. 2016. Metabolic inflammation-associated IL-17A causes non-alcoholic steatohepatitis and hepatocellular carcinoma. Cancer Cell 30(1):161–75 [DOI] [PubMed] [Google Scholar]
- 142.Finkin S, Yuan D, Stein I, Taniguchi K, Weber A, et al. 2015. Ectopic lymphoid structures function as microniches for tumor progenitor cells in hepatocellular carcinoma. Nat. Immunol 16(12):1235–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Good-Jacobson KL, Szumilas CG, Chen L, Sharpe AH, Tomayko MM, Shlomchik MJ. 2010. PD-1 regulates germinal center B cell survival and the formation and affinity of long-lived plasma cells. Nat. Immunol 11(6):535–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Meng X, Nikolic-Paterson DJ, Lan HY. 2016. TGF-β: the master regulator of fibrosis. Nat. Rev. Nephrol 12(6):325–38 [DOI] [PubMed] [Google Scholar]
- 145.Györfi AH, Matei A-E, Distler JHW. 2018. Targeting TGF-β signaling for the treatment of fibrosis. Matrix Biol. 68–69:8–27 [DOI] [PubMed] [Google Scholar]
- 146.El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, et al. 2017. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 389(10088):2492–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Overman MJ, Ernstoff MS, Morse MA. 2018. Where we stand with immunotherapy in colorectal cancer: deficient mismatch repair, proficient mismatch repair, and toxicity management. Am. Soc. Clin. Oncol. Educ. Book 38:239–47 [DOI] [PubMed] [Google Scholar]
- 148.Ciardiello D, Vitiello PP, Cardone C, Martini G, Troiani T, et al. 2019. Immunotherapy of colorectal cancer: challenges for therapeutic efficacy. Cancer Treat. Rev 76:22–32 [DOI] [PubMed] [Google Scholar]
- 149.Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, et al. 2017. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357(6349):409–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Grootjans J, Krupka N, Hosomi S, Matute JD, Hanley T, et al. 2019. Epithelial endoplasmic reticulum stress orchestrates a protective IgA response. Science 363(6430):993–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Galdiero MR, Marone G, Mantovani A. 2018. Cancer inflammation and cytokines. Cold Spring Harb. Perspect. Biol 10(8):a028662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. 2017. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol 14(7):399–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.