

Abstract
Myocyte death occurs in many inherited and acquired cardiomyopathies, including arrhythmogenic cardiomyopathy (ACM), a genetic heart disease plagued by the prevalence of sudden cardiac death. Individuals with ACM and harboring pathogenic desmosomal variants, such as desmoglein-2 (DSG2), often show myocyte necrosis with progression to exercise-associated heart failure. Here, we showed that homozygous Dsg2 mutant mice (Dsg2mut/mut), a model of ACM, die prematurely during swimming and display myocardial dysfunction and necrosis. We detected calcium (Ca2+) overload in Dsg2mut/mut hearts, which induced calpain-1 (CAPN1) activation, association of CAPN1 with mitochondria, and CAPN1-induced cleavage of mitochondrial-bound apoptosis-inducing factor (AIF). Cleaved AIF translocated to the myocyte nucleus triggering large-scale DNA fragmentation and cell death, an effect potentiated by mitochondrial-driven AIF oxidation. Posttranslational oxidation of AIF cysteine residues was due, in part, to a depleted mitochondrial thioredoxin-2 redox system. Hearts from exercised Dsg2mut/mut mice were depleted of calpastatin (CAST), an endogenous CAPN1 inhibitor, and overexpressing CAST in myocytes protected against Ca2+ overload–induced necrosis. When cardiomyocytes differentiated from Dsg2mut/mut embryonic stem cells (ES-CMs) were challenged with β-adrenergic stimulation, CAPN1 inhibition attenuated CAPN1-induced AIF truncation. In addition, pretreatment of Dsg2mut/mut ES-CMs with an AIF-mimetic peptide, mirroring the cyclophilin-A (PPIA) binding site of AIF, blocked PPIA-mediated AIF-nuclear translocation, and reduced both apoptosis and necrosis. Thus, preventing CAPN1-induced AIF-truncation or barring binding of AIF to the nuclear chaperone, PPIA, may avert myocyte death and, ultimately, disease progression to heart failure in ACM and likely other forms of cardiomyopathies.
INTRODUCTION
Every year, about 400,000 to 460,000 people die from sudden cardiac death (SCD) (1). Arrhythmogenic cardiomyopathy (ACM) is one of the most arrhythmic forms of heart disease and a leading cause of SCD in young athletes (2). Clinical features of ACM include ventricular dysfunction and arrhythmias, whereas salient pathological traits encompass myocyte apoptosis and necrosis and replacement of the myocardium with fibrous and fatty (fibro-fatty) tissue (3). Despite myocyte death as one of the most reported characteristics, this is a poorly understood pathological feature of ACM. Myocyte death has a pivotal role in contributing to myocardial fibrosis and subsequent re-entrant ventricular arrhythmia. Furthermore, individuals with ACM are particularly at risk of increased disease progression and SCD in response to exercise (4, 5).
Desmoglein-2 (DSG2) is essential to the function and stability of the cardiac desmosome (6), and pathogenic variants in human DSG2 are the second most common cause of ACM (7). Homozygous Dsg2 mutant (Dsg2mut/mut) mice harbor a knock-in mutation in murine Dsg2, resulting in the loss of exons 4 and 5, generation of two stop codons in exon 6, and nonsense-mediated mRNA degradation, where recapitulation of ACM phenotypes is due to DSG2 loss of function (8). Our prior work demonstrated that sedentary Dsg2mut/mut mice recapitulate key ACM phenotypes by early adulthood (16 weeks of age), such as repolarization and depolarization abnormalities detected by electrocardiography (ECG), cardiac dysfunction, intercalated disc remodeling, myocyte injury characterized by inflammation and fibrosis (8), cardiac calcium (Ca2+) mishandling, and myocardial lipid accumulation via the presence of perilipin-1 immunofluorescence microscopy (9). Despite the presence of these functional and pathological cardiac phenotypes, including extensive biventricular fibrosis, sedentary Dsg2mut/mut mice live well into adulthood and their hearts have few or no apoptotic nuclei (8). However, in response to chronic physical effort, an increasing number of Dsg2mut/mut mice experience exercise-induced sudden death, and those that survive to the exercise end point exhibit many myocardial apoptotic nuclei (8). However, it remains unknown how environmental factors, such as exercise, promote myocyte cell death and pathological progression both in individuals with ACM and in this mouse model of ACM.
In this study, we report in vivo evidence that chronic endurance exercise in Dsg2mut/mut mice activated the Ca2+-dependent cysteine protease calpain-1 (CAPN1), leading to CAPN1-mediated truncation of mitochondrial-bound apoptosis-inducing factor (AIF). Excessive myocardial reactive oxygen species (ROS), in conjunction with a depleted mitochondrial thioredoxin-2 (TXN2) ROS buffering system, in the hearts of trained Dsg2mut/mut mice enhanced AIF oxidation. In turn, oxidized and truncated AIF migrated to the myocyte nucleus, triggering large-scale DNA fragmentation. In vitro, we showed that challenging HL-1 cells, an immortalized mouse cardiomyocyte (CM) cell line, with increased intracellular Ca2+ concentrations drives CAPN1 activation, mitochondrial membrane depolarization, movement of CAPN1 to the mitochondria, and necrotic cell death. Furthermore, hearts from exercised Dsg2mut/mut mice displayed a marked depletion in the amounts of the endogenous CAPN1 inhibitor, calpastatin (CAST). Overexpressing CAST or pretreatment with the cell-permeable CAPN1 inhibitor, calpeptin, protected HL-1 cells against Ca2+ overload–induced cell death. In addition, calpeptin blocked CAPN1-mediated truncation of AIF in CMs derived from embryonic stem cells (ES-CMs) generated from Dsg2mut/mut mouse embryos, even in mutant ES-CMs chronically stimulated with the β-adrenergic agonist isoproterenol (ISO) or increased intracellular Ca2+.
Last, we report the influence of peptidyl-prolyl cis-trans isomerase (PPIA; also known as cyclophilin-A) in the nuclear transport of AIF in CMs from the ACM model. Prior research demonstrated PPIA acts as an AIF nuclear chaperone, translocating AIF from the cytosol to the nucleus, leading to AIF-mediated DNA degradation (10–12), whereas heat shock protein–70 (HSP70) acts to prevent nuclear import of AIF (13, 14). In both the hearts from exercised mice and ES-CMs stimulated with ISO and Ca2+, Dsg2mut/mut CMs displayed reduced amounts of HSP70. We also detected reduced amounts of free, AIF-unbound PPIA and increased amounts of AIF-bound PPIA in the hearts of Dsg2mut/mut mice subjected to chronic exercise. Last, we showed that an AIF-mimetic peptide that disrupts the AIF-PPIA complex prevented PPIA-mediated nuclear import of AIF and reduced markers of apoptotic and necrotic death in Dsg2mut/mut ES-CMs chronically exposed to β-adrenergic and Ca2+ stimuli. Our study unravels a pathogenic role for a CAPN1-PPIA-AIF axis in exercise-induced or β-adrenergic–stimulated death of Dsg2mut/mut myocytes. Present findings offer new therapeutic avenues for preventing pathological progression to heart failure and cardiac dysfunction in individuals with ACM.
RESULTS
Exercise triggers extensive myocyte necrosis in the hearts of Dsg2mut/mut mice
Endurance exercise exacerbates left ventricular (LV) dysfunction in Dsg2mut/mut mice (8). Myocyte loss is a primary culprit of LV dysfunction in Dsg2mut/mut mice (8), yet right ventricular (RV) dysfunction is more prominent in patients with ACM (15). Therefore, we determined the impact of chronic swimming on both RV and LV function in Dsg2mut/mut mice, and the extent of myocyte loss contributing to cardiac dysfunction. We subjected 5-week-old wild-type (WT) and Dsg2mut/mut mice to an 11-week (90 min/day, 5 days/week) endurance swim protocol, as previously described (8). Only 56% (n = 15 of 27) of Dsg2mut/mut mice survived, whereas almost all WT mice survived (91% survival, n = 20 of 22) to the end of the 11-week protocol (Fig. 1A). Of the survivors, Dsg2mut/mut mice presented with both LV and RV dysfunction, represented by the grossly dilated RV and LV chambers (Fig. 1B) and impaired systolic function assessed by a considerable reduction in percent RV and LV ejection fraction (%RVEF and %LVEF, respectively; Fig. 1C and table S1), and aberrant ECG properties, such as reduced S-amplitude and increased Q-amplitude, indicative of repolarization and depolarization abnormalities (table S1 and fig. S1A). Furthermore, a robust correlative relationship between reduced LVEF and reduced RVEF was apparent in exercised Dsg2mut/mut mice (Fig. 1C).
Fig. 1. RV and LV dysfunction and myocardial necrosis in response to endurance exercise in Dsg2mut/mut mice.
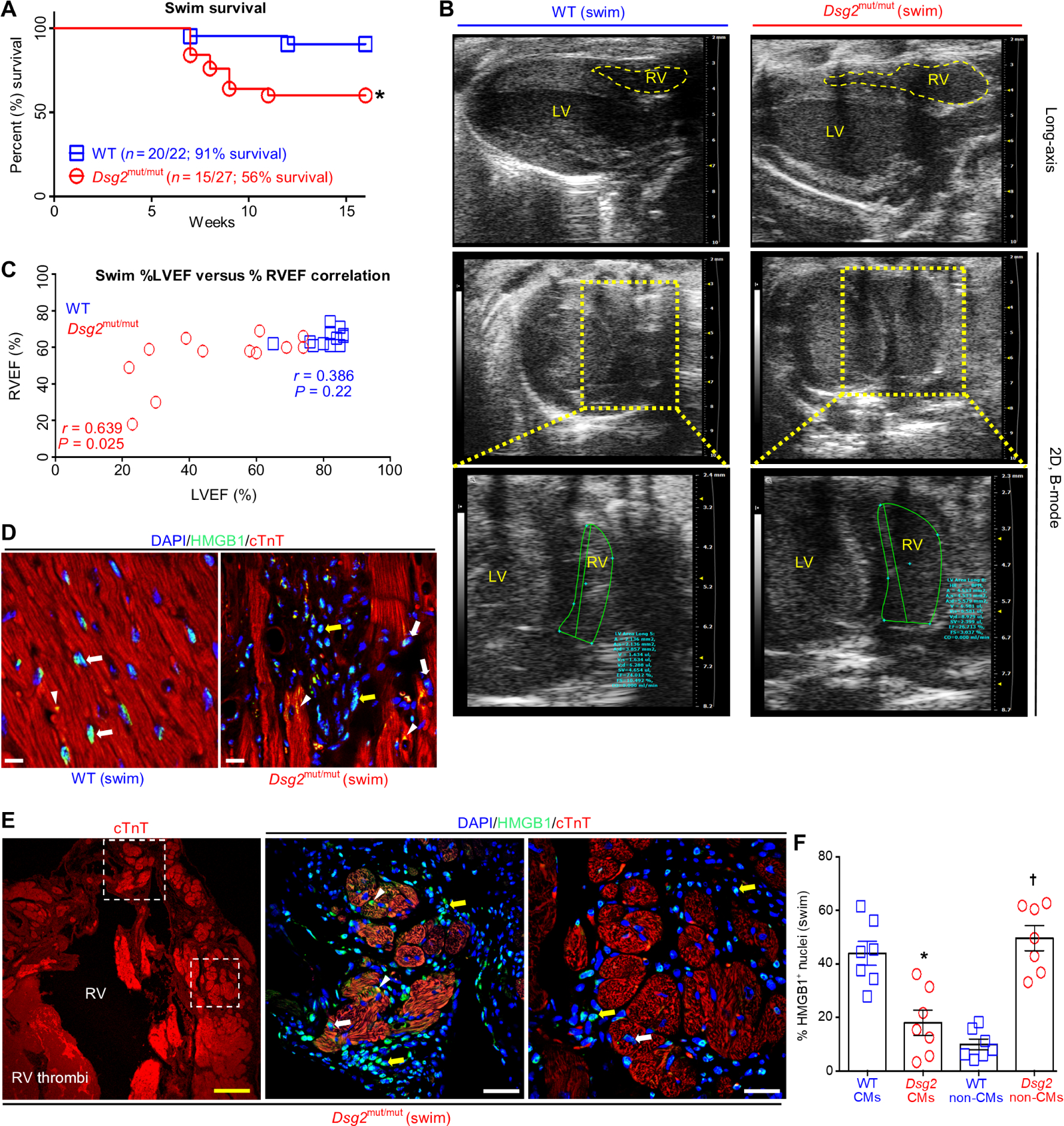
(A) Percent survival during swim. *P < 0.05 compared to WT, Mantel-Cox survival analysis. (B) Representative long-axis (top) and two-dimensional (2D), B-mode (bottom two panels) echocardiography from exercised WT and Dsg2mut/mut mice. Data are representative of n ≥ 15 mice per genotype. Yellow-dashed box indicates enlarged image of 2D, B-mode middle. (C) Comparison of the percent RV ejection fraction (%RVEF) with the percent left ventricle ejection fraction (%LVEF) using Pearson’s correlation analysis. P values represent Pearson’s correlation between %RVEF and %LVEF within each genotype (n ≥ 12 mice per genotype per parameter). (D and E) Representative HMGB1 immunostained myocardium from exercised WT and Dsg2mut/mut mice. White arrows, cardiomyocytes (CMs) positive (+) for HMGB1 nuclear localization; yellow arrows, non-CMs with HMGB1+ nuclei; white arrowheads, cytoplasmic HMGB1. In (E), dotted-line white boxes highlight enlarged areas in the right panels with the upper boxed area shown in the first panel to the right and the lower boxed area shown in the second panel to the right. Yellow scale bar in the first panel (E), 100 μm; white scale bars, 20 μm. (F) Quantification of CMs and non-CMs positive for nuclear HMGB1 in myocardium from exercised Dsg2mut/mut and WT mice. Data are presented as means ± SEM (n = 7 mice per genotype per parameter; *P < 0.05 Dsg2mut/mut CMs compared to WT CMs; †P < 0.05 Dsg2mut/mut non-CMs compared to WT non-CMs using paired t test).
Myocardial inflammation and fibrosis were both prominent in Dsg2mut/mut myocardium of exercised mice (fig. S1, B to D). We observed diffuse epicardial-to-endocardial fibrosis throughout the RV, with highly localized epicardial-to-endocardial fibrotic lesions within the LV free wall from exercised Dsg2mut/mut mice (fig. S1C). The fibrotic area within each ventricle of the exercised mutant mice was significantly greater than fibrotic areas in the ventricles of the WT mice (fig. S1D, P < 0.05 for both ventricles). These characteristics are more often associated with necrotic death than with apoptotic-induced cardiac remodeling (16). Thus, determining the extent and modality (that is, apoptosis or necrosis) by which exercise triggers myocyte cell death in Dsg2mut/mut myocardium is of pathological relevance. Previously, we showed that myocardium from exercised Dsg2mut/mut mice have increased numbers of apoptotic cells, as detected with the apoptotic marker, TUNEL (terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling) (8). Here, we evaluated myocardial tissue for necrosis by immunolabeling for high-mobility-group box-1 (HMGB1). Loss of nuclear HMGB1, a non-histone DNA binding protein, is a histological indicator of necrosis (17).
In hearts from exercised WT mice, HMGB1 was almost exclusively localized in the myocyte nucleus (Fig. 1D), denoting healthy myocytes. In contrast, Dsg2mut/mut hearts exhibited HMGB1-positive (HMGB1+) nuclei in myocytes, as well as HMGB1 localized in the perinuclear region and cytoplasm, indicating necrosis (Fig. 1, D and E). Release of nuclear HMGB1 from cells functions as a “danger signal,” acting as a chemotactic molecule for immune cells to sites of injury (17). Consistent with recruitment of immune cells to the myocardium of exercised Dsg2mut/mut mice, we additionally detected abundant HMGB1+ nuclei in non-CMs surrounding neighboring CMs (Fig. 1, E and F). Quantification of CMs with HMGB1+ nuclei revealed a significant decrease in the hearts of mutant mice compared with those in WT mice (Fig. 1F, P < 0.05). These results indicated that, in Dsg2mut/mut hearts from exercised mice, necrotic cells were primarily myocytes and the cells with HMGB1+ nuclear staining were primarily infiltrating immune cells (Fig. 1, E and F).
CAPN1 activation accounts for myocyte necrosis in exercised Dsg2mut/mut mice
Increased intracellular calcium (Ca2+) is well documented in individuals with ACM (18), and abnormal Ca2+ handling occurs in isolated Dsg2mut/mut myocytes (9). Ca2+ overload is a major cause of myocardial necrosis (19), and activation of the Ca2+-dependent cysteine proteases, CAPN1 and CAPN2, promotes Ca2+ overload–induced necrosis (20). Therefore, we assessed myocardial CAPN1 (Fig. 2, A to C) and CAPN2 abundance from the hearts of sedentary and exercised mice (fig. S2A). In vivo, native 80-kDa CAPNs undergo Ca2+-dependent autoproteolytic cleavage generating active 75-kDa CAPN peptides (21). Regardless of genotype, we detected a single CAPN2 protein in the myocardium of sedentary and exercised mice, indicating no regulated cleavage of this protein (fig. S2A). Conversely, hearts of exercised Dsg2mut/mut mice showed increased amounts of both native (80 kDa) CAPN1 and cleaved, active (75 kDa) CAPN1 compared to hearts from trained WT mice (Fig. 2, A to C).
Fig. 2. CAPN1 activation is associated with calcium overload and cell death, promoting myocyte necrosis.
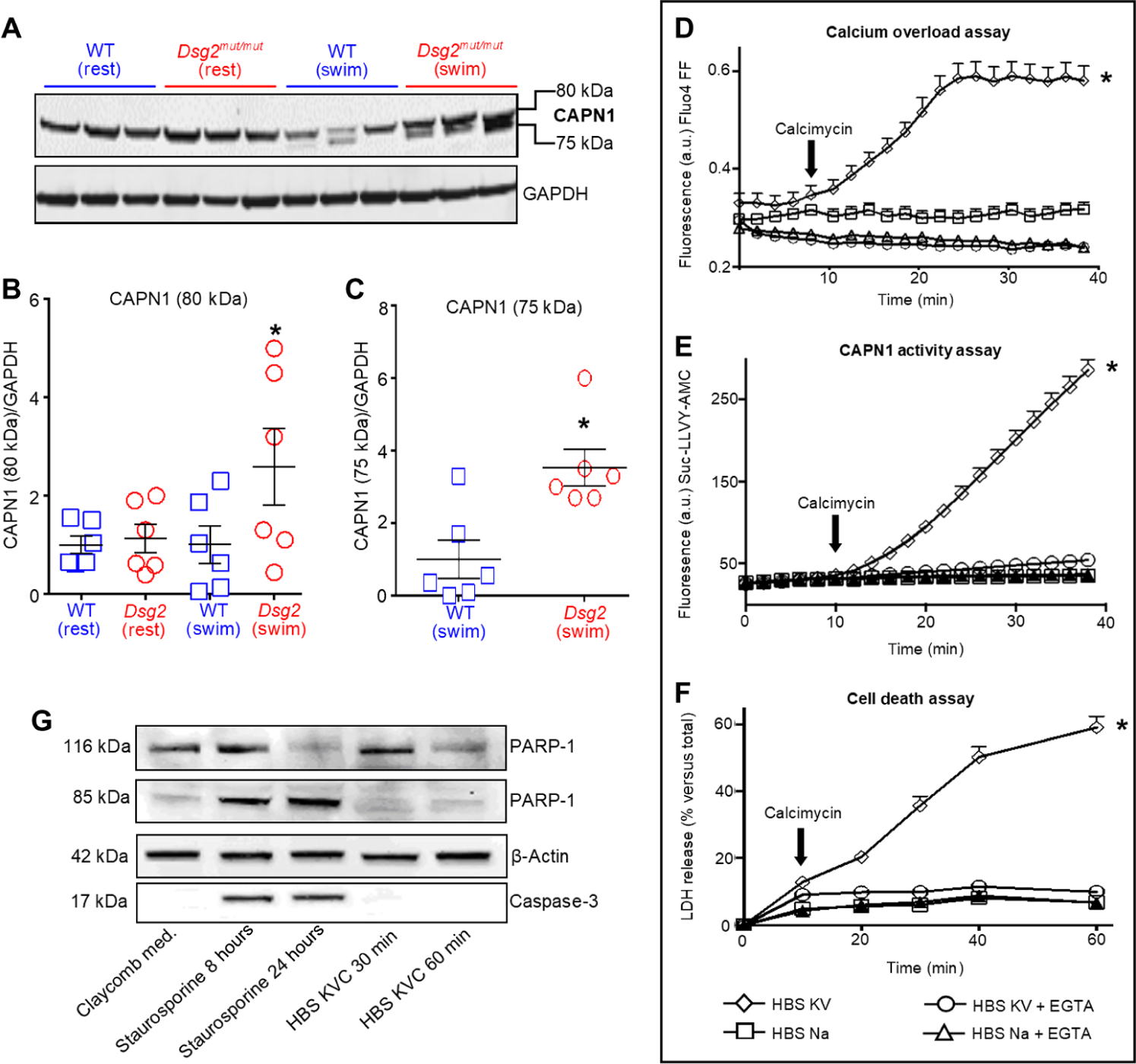
(A) Representative calpain-1 (CAPN1 recognizing domain IV) immunoblot of heart lysates from sedentary (rest) and exercised (swim) mice. (B and C) Quantification of CAPN1 in hearts from Dsg2mut/mut and WT mice. Data are presented as means ± SEM [n = 6 mice per genotype per cohort with *P < 0.05 Dsg2mut/mut (swim) compared to WT (swim) using one-way ANOVA in (B) and *P < 0.05 Dsg2mut/mut (swim) compared to WT (swim) using two-tailed, paired t test in (C)]. (D) HL-1 cells incubated in HBS sodium (HBS Na) medium or HBS with potassium and vanadate (HBS KV) medium in the absence or presence of 5 mM EGTA and calcimycin (1 μM, black arrow) to induce calcium (Ca2+) overload. a.u., arbitrary units. (E) CAPN1 activity, monitored by proteolytic cleavage of a synthetic peptide (Suc-LLVY-AMC) to produce fluorescence, in HL-1 cells exposed to the indicated media. (F) Death of HL-1 cells exposed to the indicated media, detected by lactose dehydrogenase (LDH) release into the media. (G) Representative immunoblot of poly (ADP-ribose) polymerase-1 (PARP-1) and caspase-3 and their cleaved product in HL-1 lysates. Cells were exposed to calcimycin in HBS KV medium (HBS KVC) or to staurosporin in the Claycomb medium (Sigma-Aldrich, no. 51800C) for the times indicated in the panel (8 or 24 hours for staurosporine; 30 or 60 min for HBS KVC). Data are representative of one of six experiments. For (D) to (F), data are presented as means ± SD (n = 6 independent experiments per cohort, with n = 3 cell culture replicates per condition; *P < 0.05 HBS KV compared to all other conditions using one-way ANOVA).
We investigated whether increased cytosolic Ca2+ is necessary and sufficient to activate CAPNs and trigger CAPN-mediated myocyte cell death in vitro. We used HL-1 cells (an immortalized cardiac cell line), which have many features of an adult cardiac phenotype and are used frequently in ACM pathogenesis studies (22–25). HL-1 cells were incubated in sodium (Na+)–containing Hanks’ Balanced Salt (HBS) solution (HBS Na) or HBS in which Na+ was replaced by potassium (K+) to depolarize the plasma membrane and in the presence of vanadate to inhibit the plasma membrane Ca2+-ATPase (adenosine triphosphatase) (HBS KV). In HBS KV medium, calcimycin, a Ca2+ ionophore, triggered cytosolic Ca2+ overload in HL-1 cells (Fig. 2D). As controls, we showed that Ca2+ overload did not occur upon calcimycin addition in cells incubated in HBS Na medium alone or in HBS KV or HBS Na medium in the presence of the Ca2+-chelating agent EGTA (Fig. 2D).
To investigate Ca2+-mediated CAPN1 activation, cells were preloaded with a synthetic CAPN1 substrate, Suc-LLVY-AMC, that fluoresces upon cleavage. CAPN1-mediated substrate cleavage occurred only in cells subjected to Ca2+ overload (Fig. 2E). In addition, Ca2+ overload in HL-1 cells exposed to HBS KV medium resulted in cell death, which we measured by release of lactate dehydrogenase (LDH) into the medium (Fig. 2F). EGTA in the incubation media attenuated both CAPN1 activation (Fig. 2E) and cell death (Fig. 2F). Release of LDH does not discriminate between cell death modality; therefore, we investigated whether Ca2+ overload–induced cell death occurred through necrosis, apoptosis, or both. HL-1 cell culture lysates were assessed for cleavage products of caspase-3 (26) and poly [adenosine 5′-diphosphate (ADP)–ribose] polymerase-1 (PARP-1) (27), markers of apoptosis. Consistent with in vivo findings indicating that cell death was primarily due to necrosis (Fig. 1, D to F), cleavage of PARP-1 and caspase-3 was not detected in HL-1 cells subjected to Ca2+ overload (Fig. 2G). Conversely, these cleavage products were evident upon the exposure of HL-1 cells to staurosporine (Fig. 2G), an inducer of apoptosis (28).
The relationship between the extent of Ca2+ overload with CAPN1 activation and cell death was investigated by adding EGTA at different times to HL-1 cells subjected to Ca2+ overload (fig. S2, B to E). EGTA decreased in Fluo4 FF fluorescence, indicating a reduction in intracellular Ca2+ and thereby reducing the duration of intracellular Ca2+ overload (fig. S2B). Furthermore, EGTA addition to HL-1 cells 10 min after the addition of calcimycin resulted in minimal cell death, whereas the addition of EGTA 40 min after calcimycin resulted in the greatest extent of cell death, even more than resulted from calcimycin added to HBS KV media without EGTA (fig. S2C). In addition, CAPN1 activation displayed a strict dependence on the duration of Ca2+ overload (fig. S2D). Correlation analysis confirmed the positive relationship between the duration of Ca2+ overload and the extent of CAPN1 activity and cell death (fig. S2E).
Hearts from exercised Dsg2mut/mut mice display prominent CAST depletion
CAPN1 activation is endogenously inhibited by CAST (29), and CAST is a CAPN1 substrate (30). CAST exhibits tissue- and cell-specific isoforms. Skeletal (145/135-kDa doublet), cardiac (120/110-kDa doublet), T cell (70-kDa isoform), and erythrocyte (70-kDa isoform) CAST isoforms all contain CAPN1 binding domains (31). We tested whether any differences in endogenous CAST abundance and proteolytic CAST degradation product(s) occur in the hearts of Dsg2mut/mut mice compared to the hearts of WT mice, at rest and in response to swimming. The highest–molecular weight CAST protein detected in cardiac tissue, regardless of genotype or condition, migrated at 120 kDa (Fig. 3A). We additionally detected the 110- and 70-kDa CAST isoforms. Compared to exercised WT cohorts, both sedentary and exercised Dsg2mut/mut mice had reduced amounts of the 120-, 110-, and 70-kDa CAST isoforms (Fig. 3, A and B).
Fig. 3. CAST abundance is reduced in hearts of Dsg2mut/mut mice, and CAST overexpression or CAPN inhibition rescues HL-1 cells from Ca2+ overload–induced cell death.
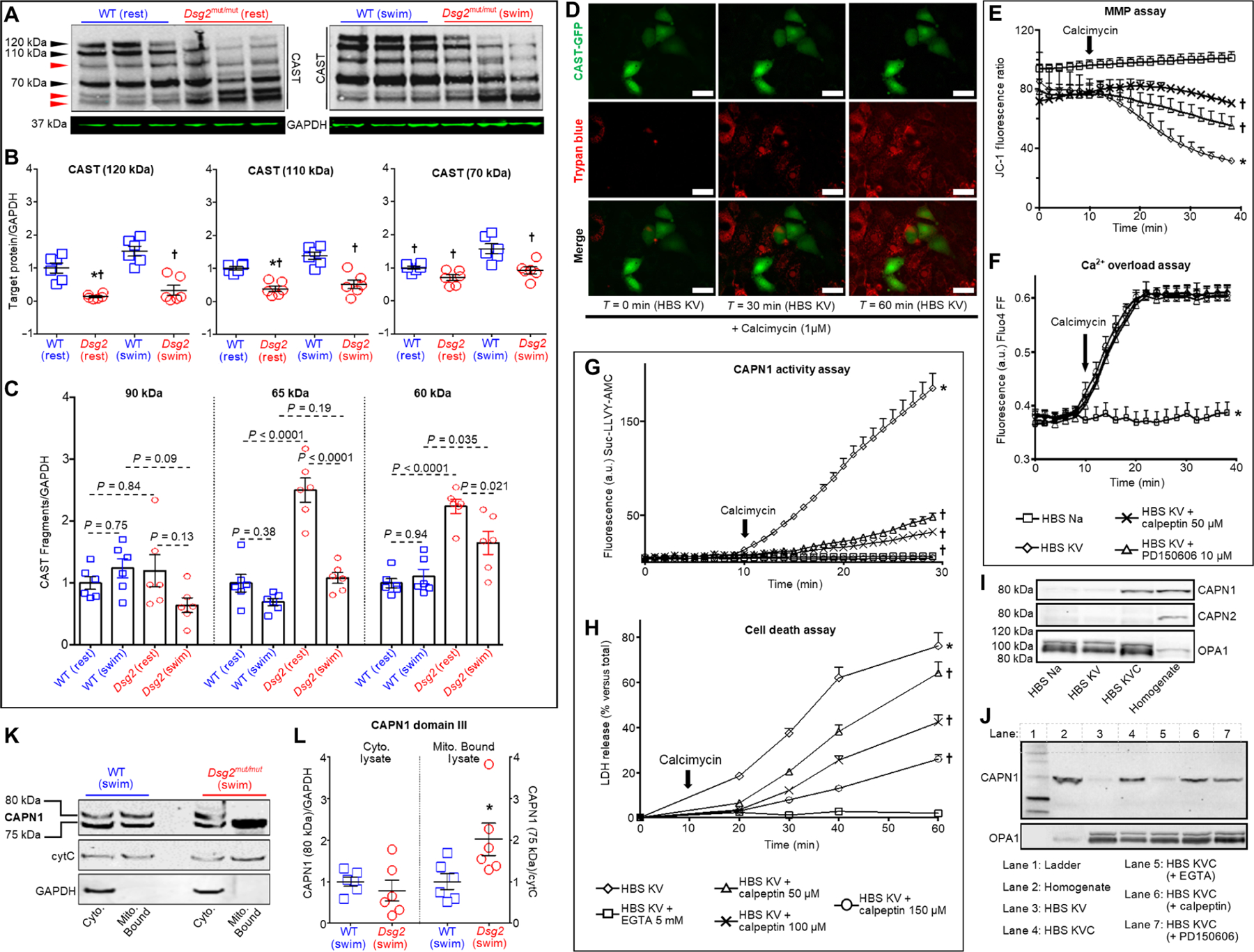
(A) Representative immunoblots probed for changes in calpastatin (CAST) from sedentary (left, rest) and exercised (right, swim) mice. Black arrowheads, CAST isoforms; red arrowheads, CAST proteolytic fragments. Immunoblots are representative of n = 6 mice per genotype per condition. GAPDH was used as a loading control. (B) Quantification of CAST isoforms in myocardial lysates from sedentary and exercised Dsg2mut/mut and WT mice. Data are presented as means ± SEM [n = 6 mice per genotype per condition; *P < 0.05 Dsg2mut/mut (rest) compared to WT (rest); †P < 0.05 for any group compared to WT (swim) using one-way ANOVA with Tukey’s post hoc test]. (C) Quantification of CAST fragments (90, 65, and 60 kDa) from sedentary and exercised cohorts. Data are presented as mean ± SEM (n = 6 mice per genotype per condition, indicated comparisons determined using one-way ANOVA with Tukey’s post hoc test). (D) Live-cell imaging of HL-1 cells transfected with a CAST-GFP (green) overexpression construct subjected to HBS KV medium in the absence and presence of calcimycin (1 μM), to induce Ca2+ overload. Trypan blue (red) admits red fluorescence via confocal microscopy and is only taken up in dead cells. Of note, HL-1 cells overexpressing CAST (green cells) are protected from Ca2+ overload–induced cell death (absence of red fluorescence in green cells). Images representative of n = 6 independent experiments, with n = 3 replicates per cell culture per condition. In (E) to (J), HL-1 cells were exposed to the conditions indicated in each panel. (E) Mitochondrial membrane potential (MMP) was monitored using the fluorescent ratiometric probe JC-1 (1.5 μM). *P < 0.05 for HBS KV compared to HBS Na; †P < 0.05 for HBS KV with either calpeptin or PD150606 compared to HBS KV using one-way ANOVA. (F) Intracellular calcium was monitored by means of Fluo4 FF fluorescence. *P < 0.05 for HBS Na compared to all other conditions using one-way ANOVA. (G) CAPN1 activity was monitored by proteolysis of the synthetic peptide Suc-LLVY-AMC (25 μM). (H) Cell death was detected as LDH release. In (G) and (H), *P < 0.05 for HBS KV compared to all conditions; †P < 0.05 for HBS KV with calpeptin (50, 100, or 150 μM) compared to HBS KV using one-way ANOVA. For (E) to (H), data are presented as means ± SD, n = 6 independent experiments per cohort, with n = 3 cell culture replicates/condition. (I) Representative immunoblots of CAPN1 or CAPN2 in purified mitochondria from HL-1 cells incubated under the indicated conditions for 2 min. Equal protein loading was indicated by staining with antibodies recognizing the mitochondrial protein OPA1. (J) Representative immunoblots of CAPN1 in purified mitochondria from HL-1 cells subjected to calcium overload (HBS KVC) in the absence or presence of EGTA (5 mM), calpeptin (50 μM), or PD150606 (10 μM). Cells were incubated under the indicated conditions for 2 min. For (I) and (J), immunoblots used CAPN1 antibody that recognizes domain IV and are representative of n = 6 independent experiments per cohort, with n = 3 cell culture replicates per condition. (K and L) Myocardial samples were analyzed from mice of the indicated genotype subjected to the swim protocol. Representative immunoblots of CAPN1 (using an antibody that recognizes domain III) in cytosolic (Cyto.) fractions and mitochondrial fractions (Mito. Bound). GAPDH was used as a loading control for cytosolic lysates; cytochrome C (cytC) was used as a loading control for mitochondria-bound samples. Quantification is shown and statistical differences [*P < 0.05 for 75-kDa CAPN1 Dsg2mut/mut (swim) compared to 75 kDa CAPN1 WT (swim)] were determined using one-way ANOVA.
We also analyzed CAST proteolytic fragments, as all full-length CAST isoforms are cleaved by CAPNs generating inactive CAST fragments (32). Regardless of genotype or condition, we observed myocardial CAST fragments at 90- and a 65/60-kDa doublet (Fig. 3, A and C). The abundance of the 60-kDa CAST fragment was higher in myocardial lysates from sedentary and exercised Dsg2mut/mut mice than from WT counterparts. The hearts from the sedentary Dsg2mut/mut mice had significantly higher amounts of the 65-kDa fragment than did the hearts from any other cohort of mice. These results suggested that a deficiency in CAST, associated with increased CAST degradation, contributes to the increase in Ca2+ and CAPN1-induced necrosis in the hearts of exercised Dsg2mut/mut mice.
To test our hypothesis that a deficiency in CAST contributes to Ca2+ and CAPN1-induced tissue damage, we evaluated the effect of overexpressing CAST on Ca2+ overload, CAPN1-induced death of HL-1 cells. We fused the CAPN1-inhibitor domain of CAST (29) with a green fluorescent protein (GFP) reporter (33) and overexpressed this CAST-GFP construct in HL-1 cells. The relatively low efficiency of the transfection generated a mixed population of CAST-GFP–positive cells and nontransfected (GFP-negative) cells. After transfection, HL-1 cells were cultured in HBS KV medium containing 0.4% trypan blue and then subjected to Ca2+ overload for 1 hour (Fig. 3D). Trypan blue emits red fluorescence when sequestered by dead or dying cells (34). Live-imaging, fluorescence-based cell viability was assessed at 30 and 60 min after the addition of calcimycin to induce Ca2+ overload in cells exposed to HBS KV medium. CAST-GFP–positive cells were protected from Ca2+ overload–induced cell death (Fig. 3D). Our in vivo and in vitro data collectively suggested that increasing CAST abundance delays Ca2+-mediated, CAPN1-induced necrosis in myocytes.
Mitochondrial dysfunction precedes Ca2+-mediated, CAPN1-induced necrosis
Mitochondrial perturbations are implicated in many cell death modalities (19, 35–37), and mitochondrial alterations and pathological intracellular Ca2+ concentrations ([Ca2+]i) occur in cardiac disorders, such as ischemia-reperfusion (I/R) (38). CMs derived from human pluripotent stem cells from an ACM patient display mitochondrial dysfunction and concomitant cell death (39). Hence, we monitored mitochondrial membrane potential (MMP) in HL-1 cells challenged with Ca2+ overload, in the presence or absence of two functionally distinct CAPN1 inhibitors (calpeptin and PD150606) (Fig. 3E). Calpeptin interacts with the catalytic site of CAPN1, whereas PD150606 interferes with the Ca2+-mediated activation of CAPN1 by affecting the EF-hand domain (Ca2+ binding site) of CAPN1 (40). Challenging HL-1 cells with Ca2+ overload decreased MMP, and calpeptin attenuated this effect (Fig. 3E). PD150606 was less effective in maintaining MMP than was calpeptin (Fig. 3E). Although both inhibitors attenuated Ca2+ overload–induced decrease in MMP (Fig. 3E), neither affected the kinetics of cytosolic Ca2+ accumulation (Fig. 3F).
Because both calpeptin and PD150606 (to a lesser extent) preserved MMP in Ca2+ overloaded cells, we interrogated whether these inhibitors attenuate Ca2+ overload–induced CAPN1 activity and cell death. Calpeptin displayed a dose-dependent reduction in Ca2+ overload–induced CAPN1 substrate hydrolysis (Fig. 3G) and cell death (Fig. 3H). Although PD150606 also showed a dose-dependent decrease in CAPN1 substrate hydrolysis (~60% reduction; fig. S3A), PD150606 did not attenuate CAPN1 activity to the same degree as did calpeptin (~75% reduction; Fig. 3G). Consistent with the less effective CAPN1 inhibition, PD150606 was less effective in reducing cell death (~15% reduction; fig. S3B) than was calpeptin (~40% reduction; Fig. 3H) when the responses to the highest concentrations tested of each drug were compared.
We examined the cytosolic and mitochondrial localization of CAPNs in HL-1 cells. Mitochondria were purified by density gradient, and purity was confirmed by the absence of proteins from other subcellular compartments and enrichment of both inner (optic atrophy 1, OPA1) and outer mitochondrial membrane (monoamine oxidase-A, MAO-A) markers (fig. S3C). CAPNs are polypeptide complexes, comprising an 80-kDa Ca2+-dependent peptide (domains I to IV) and 28-kDa regulatory peptide (domains V and VI) (21). Antibodies directed toward domain IV of CAPN1 and CAPN2 did not detect an 80-kDa band in purified mitochondrial extracts (lane 3 of both immunoblots; fig. S3D), indicating that neither CAPN1 nor CAPN2 are constitutively localized at mitochondria from HL-1 cells under physiological Ca2+ conditions.
Because we detected a decrease in MMP that was attenuated by CAPN inhibition, we investigated whether CAPNs translocate to mitochondria upon Ca2+ overload. In HL-1 cells, CAPN1, but not CAPN2, was abundantly localized in mitochondrial extracts within 2 min of Ca2+ overload (Fig. 3I). Both calpeptin and PD150606 failed to prevent mitochondrial CAPN1 translocation induced by Ca2+ overload (Fig. 3J), whereas Ca2+ chelation with EGTA reduced the amount of mitochondrial CAPN1 (Fig. 3J). Thus, our data indicated that the cytosolic-to-mitochondrial translocation of CAPN1 is Ca2+ dependent yet independent of CAPN1 enzymatic activity in HL-1 cells.
As a Ca2+-regulated protein, CAPN1 undergoes both conformational changes and autoproteolytic cleavage in response to increased [Ca2+]i. Within the 80-kDa peptide, domain IV binds Ca2+ to regulate CAPN1 autoproteolytic cleavage and activation, whereas domain III uses Ca2+ to control CAPN1 binding to lipid bilayers (32, 41, 42). Thus, antibodies directed against different domains can have different affinities and recognized inactive or active CAPN1 under different conditions. We used an antibody against domain IV to show that chronic exercise increased active, cleaved CAPN1 (75 kDa) in the hearts of WT and Dsg2mut/mut mice (Fig. 2, A to C). We also used this domain IV–targeted antibody to show that Ca2+ chelation prevented mitochondrial association of CAPN1 (Fig. 3, I and J, and fig. S3D). To verify these findings with another antibody (9A4H8D3 antibody, which specifically binds to domain III of CAPN1), we evaluated the subcellular localization of CAPN1 from heart lysates from exercised WT and mutant mice. We found both total (80 kDa) and active (75 kDa) CAPN1 in cytosolic and mitochondrial fractions from WT cardiac lysates (Fig. 3, K and L). However, mitochondrial fractions from hearts of exercised Dsg2mut/mut mice were enriched in the active, cleaved CAPN1 (75 kDa) peptide (Fig. 3, K and L). These data suggested that CAPN1 becomes activated at the mitochondria in ACM myocytes in response to exercise.
CAPN1 activation leads to AIF truncation
Our in vivo and in vitro findings are similar to studies showing a “mitochondriocentric” signal-transducer-effector (MSTE) pathway in nonischemic CM necrosis (19, 43). Thus, we hypothesized that in both HL-1 and ACM myocytes, the MSTE pathway is triggered by an increase in Ca2+ (the signal), which activates CAPN1 (the transducer), leading to myocyte necrosis (the end outcome). Missing is the identity of the mitochondrial effector responsible for myocyte necrosis in Dsg2mut/mut myocytes. Therefore, myocardium from both sedentary and exercised WT and Dsg2mut/mut mice was assessed for changes in cytochrome C (cytC) and AIF abundance and localization (Fig. 4, A to D, and fig. S4, A to C), two mitochondrial proteins released in response to MMP depolarization and implicated in either apoptosis, necrosis, or both (44). In addition, AIF is cleaved into a death-inducing truncated form (tAIF) that migrates to the nucleus, triggering large-scale DNA fragmentation and cell death (45, 46). CAPN cleavage of AIF can produce tAIF (fig. S4D).
Fig. 4. Exercise or β-adrenergic stimulation increases truncation of myocardial AIF in Dsg2mut/mut myocytes.
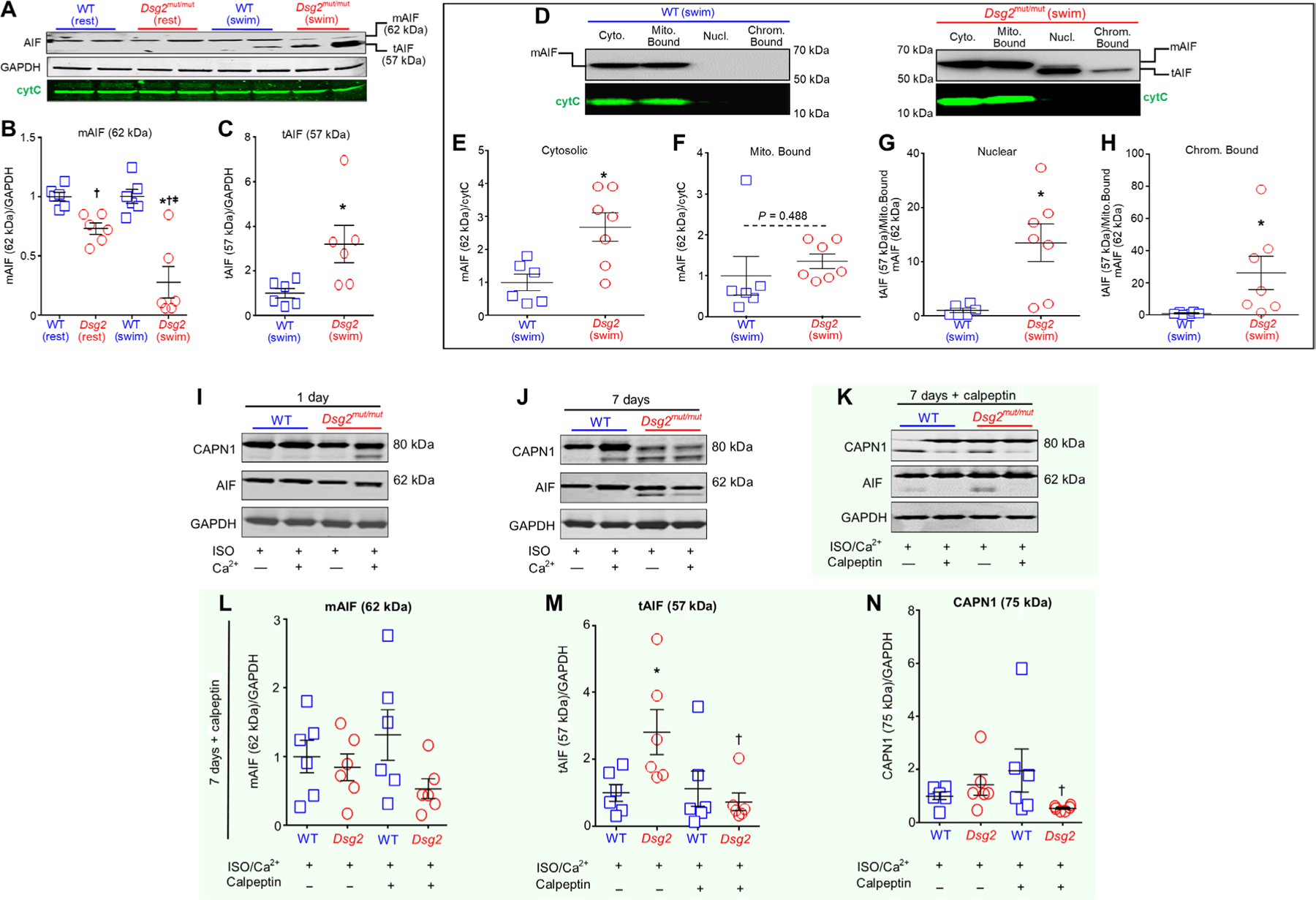
(A) Representative immunoblots of AIF with the mature AIF (mAIF) indicated at 62 kDa and truncated AIF (tAIF) at 57 kDa in hearts of mice of the indicated genotype under sedentary (rest) or exercised (swim) conditions. GADPH and cytC served as loading controls. (B and C) Quantification of mAIF and tAIF in hearts from the indicated mice. Data are presented as mean ± SEM [n = 6 per cohort per condition; *P < 0.05 compared to WT (rest); †P < 0.05 compared to WT (swim); and ǂP < 0.05 compared to Dsg2mut/mut (rest)] using one-way ANOVA in (B) and two-tailed t test in (C). (D) Representative immunoblots of AIF in subcellular fractions of ventricular lysates from exercised mice. Cyto., cytosolic extracts; Mito. Bound, mitochondrial fraction; Nucl., nuclear fraction; Chrom. Bound, chromatin-bound fraction. (E to H) Quantification of AIF in the indicated subcellular fraction. mAIF was quantified for cytosolic and mitochondrial fractions (E and F); tAIF was quantified for nuclear and chromatin-bound fractions (G and H). Data are presented as means ± SEM [n ≥ 6 per cohort per compartment; *P < 0.05 compared to WT (swim)] using two-tailed t test. (I and J) Representative immunoblots from WT and Dsg2mut/mut ES-CMs treated for 1 day or 7 days with either isoproterenol (ISO; 50 μM), calcium (Ca2+ 1 μM), or both (ISO/Ca2+). Data are representative of one of six experiments. (K) Representative immunoblots from WT and Dsg2mut/mut ES-CMs treated for 7 days with ISO/Ca2+, with or without calpeptin (50 μM) pretreatment. (L to N) Quantification of mAIF (62 kDa), tAIF (57 kDa), and active CAPN1 in ES-CMs of the indicated genotypes subjected to conditions as in (K). Data are presented as means ± SEM (n = 6 per genotype per parameter; *P < 0.05 untreated Dsg2mut/mut ES-CMs compared to untreated WT ES-CMs; †P < 0.05 calpeptin-pretreated Dsg2mut/mut ES-CMs compared to untreated Dsg2mut/mut ES-CMs using one-way ANOVA).
In total lysates, we found no changes in the abundance of cytC between genotypes, both at rest and after exercise (Fig. 4A and fig. S4A). Although we detected cytC in both mitochondrial and cytosolic extracts in exercised mice (Fig. 4D), the ratio between cytosolic:mitochondrial-bound or mitochondrial-bound:cytosolic cytC in exercised WT or Dsg2mut/mut mice was similar (fig. S4B). Conversely, in both genotypes, exercised myocardium showed two distinct AIF bands of 62 kDa [mature AIF (mAIF)] and 57 kDa (tAIF) (Fig. 4A and fig. S4D). We found that the hearts of Dsg2mut/mut mice had reduced amounts of mAIF compared to that in sedentary controls, and exercise exacerbated this phenomenon (Fig. 4B). Furthermore, myocardium from exercised Dsg2mut/mut mice showed increased amounts of tAIF compared to that in exercised controls (Fig. 4C).
Considering these results, we performed subcellular fractionation via stepwise, gradient centrifugation to isolate cellular compartments to determine the amounts of mAIF and tAIF in cytosolic, mitochondrial, nuclear, and chromatin-bound lysates (Fig. 4, D to H). Compared to hearts from exercised WT mice, cytosolic lysates from hearts of exercised Dsg2mut/mut mice showed elevated mAIF (Fig. 4E), whereas no differences in mitochondrial mAIF were observed between exercise cohorts (Fig. 4F). Only hearts from exercised Dsg2mut/mut mice showed the presence of nuclear and chromatin-bound tAIF; little or no tAIF was detectable in these fractions from WT counter parts (Fig. 4, G and H, and fig. S4C).
Building on our in vivo exercise findings, we assessed whether an exogenous β-adrenergic stimulus, alone or in the presence of increased [Ca2+]i, triggered autoproteolytic activation of CAPN1 and CAPN1-mediated truncation of AIF in ACM myocytes. We established embryonic stem cells (ESCs) from WT and Dsg2mut/mut mice and differentiated the ESCs into CMs (ES-CMs). The resulting ES-CMs were exposed to ISO (50 μM) in the absence or presence of Ca2+ (1 μM). One-day exposure to ISO alone or both ISO and Ca2+ (ISO/Ca2+) failed to induce the 75-kDa CAPN1 fragment in WT ES-CMs, although the 1-day exposure to ISO/Ca2+ was sufficient to generate activated CAPN1 in Dsg2mut/mut ES-CMs (Fig. 4I). In neither genotype was either 1-day stimulus sufficient to induce tAIF (Fig. 4I). Conversely, 7-day exposure (chronic) to ISO or ISO/Ca2+ was sufficient to induce tAIF in Dsg2mut/mut ES-CMs, an event accompanied by the cleavage of CAPN1 (Fig. 4J). In contrast, the 7-day exposure to ISO/Ca2+ induced CAPN1 activation, but not tAIF in WT ES-CMs (Fig. 4J).
Considering the higher efficacy of calpeptin over PD150606 on the attenuation of CAPN1 substrate hydrolysis in HL-1 cells (Fig. 3 and fig. S3), we assessed whether calpeptin attenuated CAPN1 activation and AIF truncation induced by chronic ISO/Ca2+ (Fig. 4K). The amount of mAIF (62 kDa) was similar in both ES-CMs of genotypes in the presence or absence of calpeptin upon chronic stimulation (Fig. 4L). However, pretreatment with calpeptin lowered the amounts of both tAIF (57 kDa) and cleaved (75 kDa) CAPN1 in Dsg2mut/mut ES-CMs (Fig. 4, M and N). Our findings suggested that ACM myocytes undergoing experimental (in vitro) and environmental (in vivo) exercise are more prone to Ca2+/CAPN1-mediated cleavage of AIF.
AIF translocates to the myocyte nucleus in Dsg2mut/mut mice and patients with ACM
Because tAIF translocates to the nucleus to induce chromatin condensation and cell death (45), we examined whether AIF nuclear translocation occurs in Dsg2mut/mut myocardium, as well as in the hearts of patients with ACM. Murine (Fig. 5, A to C) and human (Fig. 5, D to G) myocardia were analyzed for colocalization of AIF and 4′,6-diamidino-2-phenylindole (DAPI) (45). In mice, we observed increased AIF nuclear colocalization in the myocardium from exercised Dsg2mut/mut mice, when compared to either myocardium from exercised WT mice or to WT and ACM sedentary cohorts (Fig. 5, A to C).
Fig. 5. AIF nuclear localization is increased in ACM myocardium.
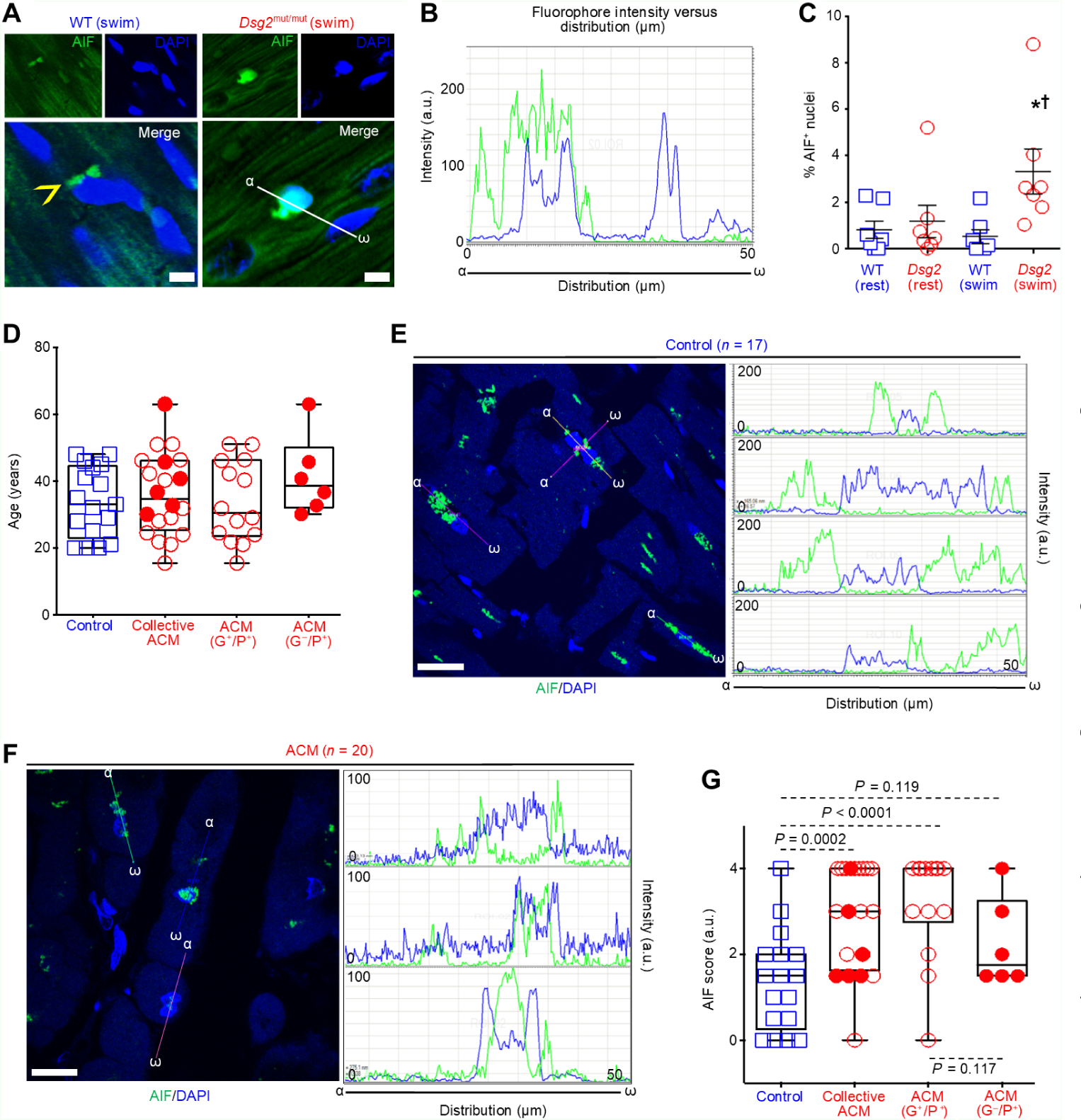
(A) Representative AIF-immunostained myocardium from exercised WT and Dsg2mut/mut mice. Yellow arrowhead, perinuclear-AIF. Scale bars, 20 μm. (B) AIF and DAPI fluorophore intensity versus fluorophore distribution (α-ω) (C) Quantification of AIF-positive (% AIF+) nuclei in myocardium from sedentary or exercised WT and Dsg2mut/mut mice. Data are presented as mean ± SEM [n = 7 per genotype per cohort; *P < 0.05 Dsg2mut/mut (swim) compared to WT (swim); †P < 0.05 Dsg2mut/mut (swim) compared to Dsg2mut/mut (rest), using one-way ANOVA]. (D) Age at biopsy collection. Data are presented as mean ± SEM [n = 17 controls, n = 6 ACM (G−/P+), and n = 14 ACM (G+/P+)]. (E and F) Representative AIF-immunostained myocardia and AIF and DAPI fluorophore intensity versus fluorophore distribution at three ROIs (α-ω). Scale bars, 20 μm. (G) AIF pathology scores from patients with ACM or healthy age-matched controls. AIF pathology score (AIF score) was determined from 3 to 10 ROIs by analysis of AIF and DAPI fluorophore intensity versus fluorophore distribution. Collective ACM denotes the total of G+/P+ and G−/P+ ACM patients. Data are presented as means ± SEM [n = 17 controls, n = 6 ACM (G−/P+), and n = 14 ACM (G+/P+) with significant differences determined by one-way ANOVA].
Myocardial samples from three age-matched patient cohorts were assessed for AIF localization (Fig. 5D). The first cohort included myocardial samples obtained at autopsy from individuals with no prior clinical history of heart disease (controls, n = 17). The second cohort consisted of age-matched myocardia from patients with ACM who had one of two known pathogenic desmosomal gene variants implicated in ACM [ACM G+/P+; DSG2 (n = 2) or plakophilin-2 (PKP2, n = 12); table S2]. The third cohort consisted of myocardia from patients with ACM in whom no pathogenic desmosomal gene variant had been identified but who met Task Force Criteria (TFC) (47) for ACM at the time of biopsy collection (G−/P+; n = 6). Following the methods by Daugas et al. (45), 3 to 10 regions of interest (ROIs) were labeled in each patient myocardial sample. Samples were then blinded and distributed to three reviewers, where each reviewer assigned a single AIF pathology score as nonpathological (grades 0 to 2; fig. S5, A to C) or pathological (grades 3 and 4; fig. S5D) based on the fluorophore intensity versus fluorophore distribution (that is the overlap between AIF and DAPI signals). All three reviewer AIF pathology scores were then averaged for a single patient. Myocardium from both control and patients with ACM had ROIs with a range of diffuse cytosolic AIF localization (grade 2), a 50:50 odds ratio of cytosolic and mitochondrial AIF localization (grade 1), and punctate mitochondrial or perinuclear localization of AIF (grade 0) (Fig. 5, E to G). However, significantly more patients with ACM (G+/P+ and G−/P+) had AIF-positive nuclei (grades 3 or 4; n = 13 of 20) compared to control myocardium (grades 3 or 4; n = 2 of 17) (P = 0.0002; Fig. 5G).
Considering the prominent AIF nuclear localization found in ACM myocardium compared to age-matched controls (Fig. 5G), we evaluated whether, and to what extent, AIF nuclear localization occurs in other cardiomyopathies using a tissue microarray (TMA) comprising age-matched (fig. S5E) LV tissue from cases of hypertrophic cardiomyopathy (HCM; n = 8), dilated cardiomyopathy (DCM; n = 28), ischemic heart disease (IHD; n = 25), and controls (n = 33). We observed no difference in AIF pathology scores for the TMA between the control and DCM, HCM, and IHD cohorts (fig. S5F). Blinded analysis of samples revealed that the average AIF pathology score for the TMA control cohort (1.49 ± 0.22, mean ± SEM, n = 33) was nearly identical to the score for the control cohort for the ACM analysis (1.35 ± 0.28, mean ± SEM, n = 17) (P > 0.999; fig. S5G). This indicated that AIF pathology scoring in control samples is independent of the number of samples assessed, consistent across the different times the evaluations were made, and reliable, considering three different blinded observers were used. We found increased AIF pathology scores in the combined ACM cohorts compared to either cohort of controls and in the G+/P+ ACM cohort compared to the scores of patients with DCM or IHD (fig. S5G).
We also investigated whether AIF pathology scores were differentially associated with the two cohorts of patients with ACM. Whereas collectively the two ACM cohorts (both G−/P+ and G+/P+ ACM patients) displayed higher AIF pathology scores compared to controls (Fig. 5G), we found that AIF pathological scores were only statistically significant between myocardial samples from the G+/P+ cohort compared to controls (P < 0.0005; Fig. 5G). No differences in myocardial AIF pathology scores were detected between controls and G−/P+ or between the two ACM cohorts (Fig. 5G). We also did not detect any significant differences among the clinical phenotypes between the G+/P+ and G−/P+ cohorts (table S3).
Last, we evaluated whether exercise contributed to the higher AIF pathology scores in ACM patient myocardium. We reviewed retrospective exercise participation reports, as previously described (48), from six patients with ACM for whom we had both myocardial samples immunostained for AIF and exercise histories. These six patients were from G+/P+ patients and all harbored a PKP2 variant (n = 4 deletion variants, n = 1 amino acid substitution, and n = 1 mutant splice product). We analyzed metabolic equivalent of task hours (METhrs) against AIF pathology scores and found a positive, albeit not significant, trend between AIF pathology score and METhrs (Pearson’s coefficient r = 0.54 and P = 0.267; fig. S5H).
Dsg2mut/mut myocardium displays lower antioxidant capacity of the TXN system
Physical effort increases ROS production (49). If not adequately scavenged, ROS accumulation in the heart can lead to myocardial inflammation, fibrosis, and, ultimately, cell death. The mitochondrial TXN2 system is essential for cell viability and is a critical regulator of H2O2 accumulation by the mitochondria (50). We examined whether endurance exercise contributes to the destabilization of mitochondria in hearts of Dsg2mut/mut mice, stemming from ROS accumulation. Furthermore, we hypothesized that ROS accumulation was due, at least in part, to a deficient TXN2 system. Using electron paramagnetic resonance (EPR) spectroscopy (Fig. 6A), we found that sedentary mice, regardless of genotype, displayed similar amounts of ROS accumulation at rest (Fig. 6, B and C). Conversely, swimming augmented ROS accumulation in Dsg2mut/mut myocardium, an effect not detected in exercised WT mice (Fig. 6C). Thus, the hearts of Dsg2mut/mut mice were deficient in scavenging ROS accumulation in response to exercise.
Fig. 6. Exercise fails to up-regulate the mitochondrial TXN2 system in Dsg2mut/mut hearts.
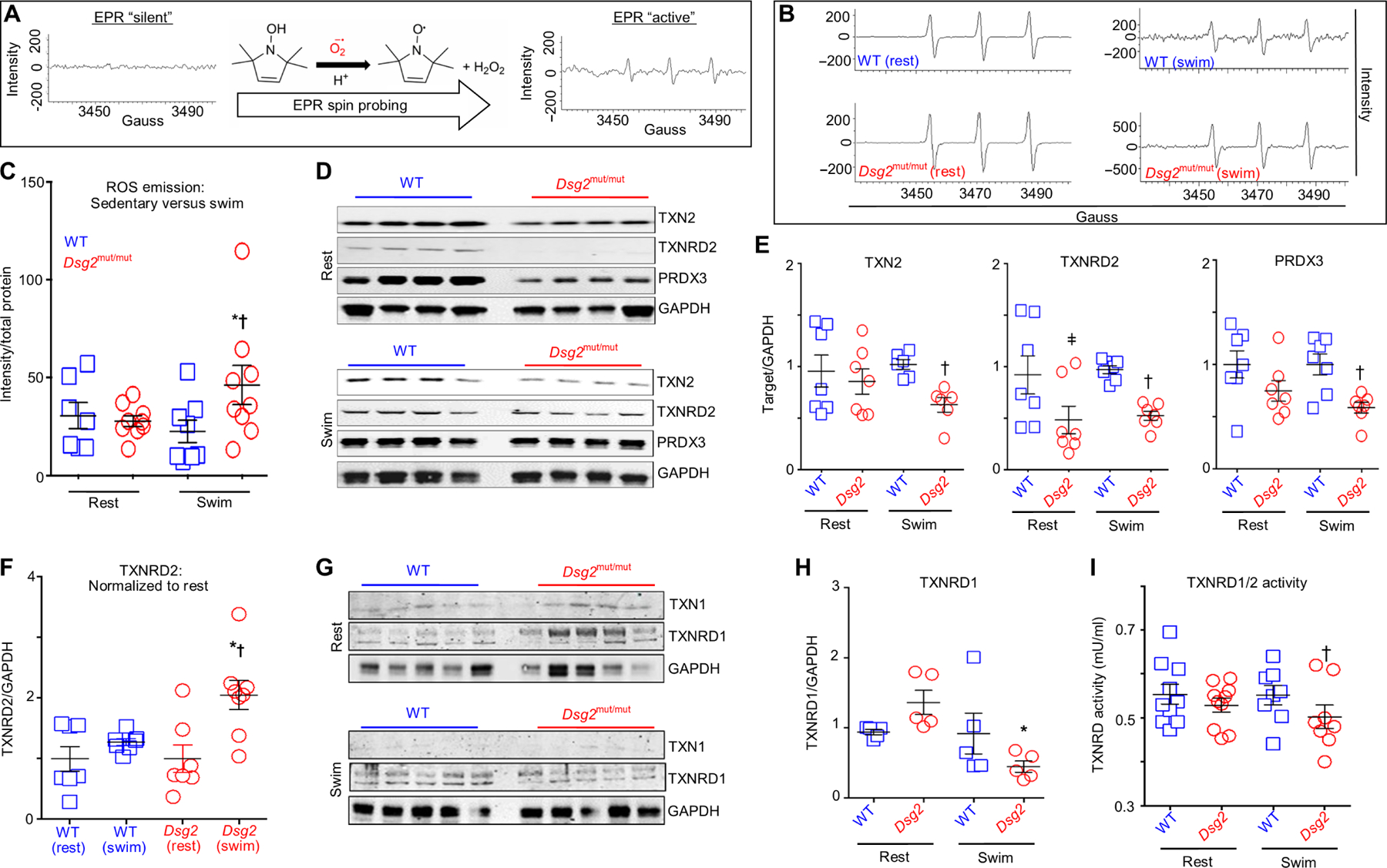
(A) Schematic of ROS emission recordings made with electron paramagnetic resonance (EPR) spectroscopy. (B) Representative EPR tracings from sedentary (rest) and exercised (swim) cohorts. (C) ROS emission in sedentary (rest) and exercised (swim) WT and Dsg2mut/mut mice. Data are presented as means ± SEM [n ≥ 7 genotype per cohort; *P < 0.05 Dsg2mut/mut (swim) compared to Dsg2mut/mut (rest); †P < 0.05 Dsg2mut/mut (swim) compared to WT (swim), using one-way ANOVA]. (D and E) Immunoblots and quantification from sedentary and exercised cohorts probed for thioredoxin-2 (TXN2), TXN2 reductase (TXNRD2), and peroxiredoxin-3 (PRXD3) with GAPDH as the loading control. Quantified data are presented as means ± SEM [n ≥ 6 mice per genotype per parameter; ǂP < 0.05 Dsg2mut/mut (rest) compared to WT (rest); †P < 0.05 Dsg2mut/mut (swim) compared to WT (swim), using one-way ANOVA]. (F) Exercised TXNRD2 levels normalized to sedentary TXNRD2 levels, within genotype. Note the increased TXNRD2 levels from exercised Dsg2mut/mut mice compared to TXNRD2 levels from sedentary Dsg2mut/mut mice. Data presented as means ± SEM, n ≥ 7 genotype per cohort, *P < 0.05 Dsg2mut/mut (swim) versus Dsg2mut/mut (rest); †P < 0.05 Dsg2mut/mut (swim) versus WT (swim) using one-way ANOVA. (G) Western immunoblots from sedentary and exercised cohorts probed for thioredoxin-1 (TXN1) and TXN1-reductase (TXNRD1), normalized to GAPDH. (H) Sedentary Dsg2mut/mut mice showed an increased trend toward elevated TXNRD1 levels, which was markedly down-regulated in response to swimming. Data presented as mean ± SEM, n = 5 genotype per cohort. *P < 0.05 Dsg2mut/mut (swim) versus Dsg2mut/mut (rest) using one-way ANOVA. (I) TXNRD1/2 activity was assessed in myocardial homogenates. Of note, myocardial lysates from exercised Dsg2mut/mut mice showed reduced TXNRD1/2 activity compared to exercised WT mice. Data presented as means ± SEM, n ≥ 8 genotype per cohort, †P < 0.05 Dsg2mut/mut (swim) versus WT (swim) using one-way ANOVA.
To determine whether this deficiency in ROS scavenging is related to the TXN2 system, we evaluated the abundance of components of the TXN2 system in the hearts from WT and Dsg2mut/mut mice under resting or exercised conditions. Compared to hearts from WT mice, hearts from sedentary Dsg2mut/mut mice displayed a trend toward reduced amounts of TXN2 and peroxiredoxin-3 (PRDX3), a mitochondria-specific peroxidase (51), albeit not significant (Fig. 6, D and E). Myocardial TXN2 reductase (TXNRD2) was nearly undetectable in sedentary mutants (Fig. 6, D and E). In healthy rodents, myocardial TXNRD2 is up-regulated in response to physical training (52). Therefore, we tested whether swimming increased TXNRD2 abundance in exercised cohorts. Although TXNRD2 abundance remained considerably lower in the myocardium from exercised Dsg2mut/mut mice compared to that in the myocardium of exercised WT mice (Fig. 6, D and E), TXNRD2 abundance increased in response to exercise (Fig. 6, D and F). When normalized to the amount in hearts from sedentary Dsg2mut/mut mice, myocardial samples from exercised Dsg2mut/mut mice displayed increased TXNRD2 abundance (Fig. 6F). In contrast to TXNRD2, exercise led to a further decrease in TXN2 and PRDX3 content in myocardium from ACM mice (Fig. 6, D and E).
Mitochondrial ROS can induce ROS release from cytosolic sources (53), and inhibition of cytosolic TXN1 leads to myocardial oxidative damage (54). Therefore, we evaluated the status of cytosolic TXN1 and TXN1 reductase (TXNRD1). Regardless of genotype, both sedentary and exercise cohorts displayed similar TXN1 content (Fig. 6G). Although TXNRD1 abundance was up-regulated in hearts from sedentary ACM mice, swimming reduced TXNRD1 abundance (Fig. 6, G and H). To determine whether there were functional differences in TXN reductase activity, we incubated myocardial homogenates with NADPH (reduced form of nicotinamide adenine dinucleotide phosphate) and a disulfide substrate, either in the presence or in the absence of an inhibitor, auranofin, of both TXNRD1 and TXNRD2 (50). Only hearts from exercised Dsg2mut/mut mice had a marked decline in TXNRD1/2 activity compared to exercised controls (Fig. 6I). Thus, these results indicated that, in response to exercise, inadequate function of the mitochondrial and cytosolic TXN systems leads to augmented ROS accumulation in Dsg2mut/mut hearts, which likely contribute to mitochondrial dysfunction, increased MMP, and access of CAPN1 to the mitochondria and AIF.
Impaired AIF function in the mitochondria augments mitochondrial ROS accumulation and reduces cytC oxidase (COX) IV abundance (55–57). We expected that with the production of tAIF and reduction in mAIF (Fig. 4, A and C) and the increase in ROS accumulation in the hearts of exercised Dsg2mut/mut mice (Fig. 6C), COX IV abundance would be lower in the hearts of these mice. Therefore, we assessed COX IV abundance by immunoblotting of myocardial lysates and immunohistochemistry of myocardial tissue in both genotypes under rest and exercise conditions. In contrast to prior reports showing exercise-induced increase in COX IV (58–60), we found a reduction in COX IV, regardless of genotype (fig. S6, A and B). This unexpected finding prompted us to examine COX IV by immunohistochemistry. We observed a drastic reduction in COX IV within the myocardium in exercised Dsg2mut/mut mice, particularly in areas with extensive myocardial damage and infiltration of inflammatory cells (fig. S6C). Considering that COX IV is encoded by nuclear DNA (61), decreased COX IV abundance in Dsg2mut/mut hearts may stem from a nuclear insult. Alternatively, and a simpler explanation, continued loss in Dsg2mut/mut myocardium would ultimately result in reduced mitochondria and thus reduced COX IV abundance.
Oxidized truncated AIF binds DNA
Whether in its mature or truncated form, AIF contains three cysteinyl residues (C256, C317, and C441) (Fig. 7A) (46). Previous studies showed that AIF undergoes oxidative modification before CAPN1 proteolytic processing and mitochondrial release (62). Considering only exercised myocardium displayed tAIF (Fig. 4, A and D), we tested the redox status of AIF cysteines in mitochondrial fractions of hearts from sedentary or exercised mice. Specifically, mitochondrial fractions from myocardial lysates were exposed to a 5-kDa cysteine-labeling agent, methoxypolyethylene glycol maleimide (mPEG) (Fig. 7A). Covalent binding of mPEG with the cysteine-SH group (i.e., the reduced state) generates a 5-kDa shift from the original molecular weight of any protein with each mPEG-bound cysteine adding another 5 kDa (Fig. 7A and fig. S7A). In addition to evaluating size shifts for AIF, we included glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and cytC in this analysis. We detected a shift indicating that mPEG bound to all three cysteines in a fraction of mAIF, as well as to cysteines in a fraction of GAPDH and cytC, from sedentary cohorts, regardless of genotype (Fig. 7B). Quantification of the ratio of unmodified mAIF (62 kDa) and reduced mAIF (77 kDa, mPEGged) indicated that the proportions were about 60 and 40%, respectively, in sedentary mice of each genotype (Fig. 7C). Conversely, in exercised mutants, heart mitochondrial fractions had a large proportion of the tAIF and oxidized form of tAIF (ox-tAIF), whereas heart mitochondrial fractions from WT exercised mice had less ox-tAIF as a fraction of the total AIF (Fig. 7, D and E).
Fig. 7. Exercise promotes AIF-oxidation and DNA fragmentation.
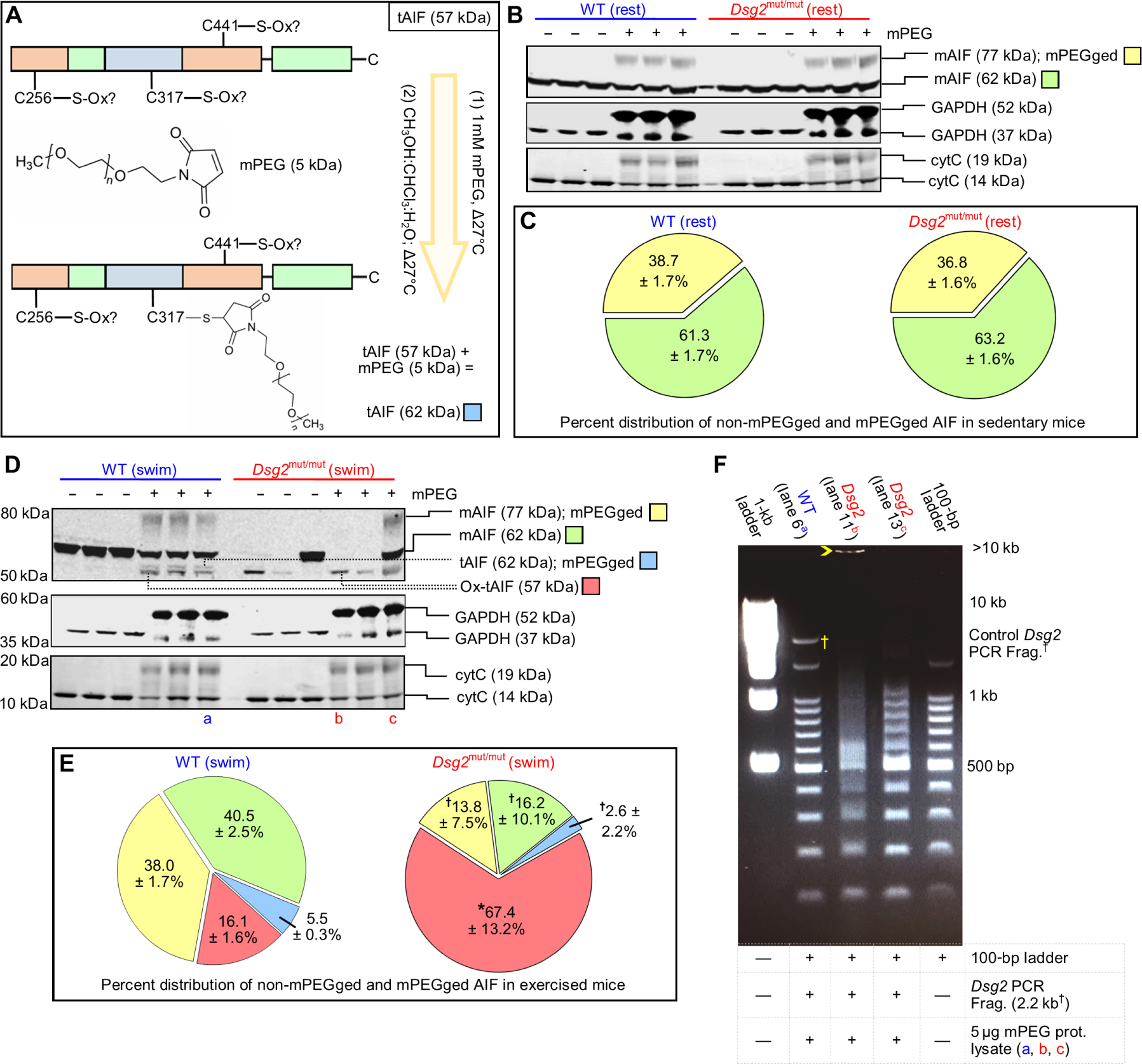
(A) Representative schematic of methoxypolyethylene glycol maleimide (mPEG) modification of AIF. Any nonoxidized cysteine (C256, C317, or C441) binds mPEG, adding 5 kDa to AIF per mPEG-modified cysteine. (B) Representative immunoblots from untreated (−) and mPEG-treated (+) lysates from sedentary (rest) cohorts. (C) Percent (%) distribution of unmodified (non-mPEGged) and mPEG-modified (mPEGged) AIF from sedentary mice. Data are presented as mean ± SEM. No significant differences in the distribution within and between sedentary cohorts were detected by one-way ANOVA (n = 6 per genotype per parameter). (D and E) Representative immunoblots and percent distribution of non-mPEGged and mPEGged mAIF and truncated AIF (tAIF) from exercised (swim) cohorts. Data are presented with means ± SEM [n = 6 WT mice per parameter, n = 7 Dsg2mut/mut mice per parameter; *P < 0.05 for Dsg2mut/mut compared to WT within each respective AIF condition; for example, Dsg2mut/mut mAIF 77-kDa mPEGged (yellow) versus WT mAIF 77-kDa mPEGged (yellow)], using one-way ANOVA with Tukey’s post hoc test. †P < 0.05 for any AIF condition (yellow, green, or blue) compared to ox-tAIF (57 kDa, red) using one-way ANOVA with Tukey’s post hoc test. For (C) and (E), percent distribution was calculated as the amount of each mPEGged or non-mPEGged AIF form divided by the sum of all AIF forms times 100. (F) Representative DNA retardation assay. Five micrograms of mPEG-treated protein lysates [from the samples shown in (D): lane 6 (a), lane 11 (b), and lane 13 (c)] was incubated with a 2.2-kb DNA fragment (†Frag.) generated via polymerase chain reaction (PCR) from the WT form of murine Dsg2 and a 100-bp DNA ladder. Yellow open arrowhead marks most retarded fragment. Data are representative of n ≥ 6 mice per genotype per lane.
Prior studies indicate that AIF must bind DNA to induce cell death (63), and large-scale DNA fragmentation is a hallmark of AIF-driven cell death (64). Therefore, we investigated whether myocardial proteins in lysates from exercised Dsg2mut/mut mice bound DNA, using a previously described in vitro DNA retardation assay (63). We used both a DNA ladder ranging from 100 base pairs (bp) to 1.5 kb and a 2.2-kb fragment of the WT Dsg2 gene (8) to evaluate the binding of proteins from control myocardial protein lysates and mPEG-modified lysates. Binding of proteins to the DNA fragments retards their movement through the gel, resulting in either a discrete band or smearing of the band, depending on the amount of the DNA binding protein in the sample. We used aliquots of the same samples from those evaluated in Fig. 7D: an mPEG-treated WT (swim) sample (marked with the blue “a”) and two mPEG-treated Dsg2mut/mut (swim) samples (marked with red “b” and “c”). All of the bands detected in the sample from the WT mouse were at the same size as those of the 100-bp ladder or of the 2.2-kb Dsg2 fragment, indicating no evidence of DNA binding (Fig. 7F, lane 2), which is consistent with the low proportions of tAIF and ox-tAIF in that sample. The sample from the mutant mouse with predominantly nonoxidized tAIF exhibited smearing of the DNA ladder starting at ~500 bp (Fig. 7F, lane 4), indicating that proteins in that sample bound DNA. The sample from the mutant mouse with only ox-tAIF as the detected form of AIF showed smearing of the DNA ladder starting below 500 bp without distinct DNA laddering after 500 bp and had a band above 10 kb (Fig. 7F, lane 3). Thus, these results suggested that the amount of ox-tAIF determines the amount of DNA retardation and that ox-tAIF has a stronger interaction with DNA than does nonoxidized tAIF.
Targeting PPIA prevents AIF nuclear import and reduces markers of cell death in ACM myocytes challenged with sustained β-adrenergic stimulation and Ca2+ overload
An inhibitor targeting the AIF putative deoxyribonuclease activity is potentially an ideal approach to preventing AIF-mediated DNA fragmentation and cell death; however, the site of this activity has yet to be identified. HSP70 is an endogenous inhibitor of AIF nuclear import (13, 14), whereas PPIA (also known as cyclophilin-A) binds AIF in the cytosol, translocating AIF into the cell nucleus (12). Therefore, we evaluated the amount of HSP70 and PPIA in the hearts of sedentary and exercised mice and in ES-CMs subjected to chronic ISO and Ca2+ stimulation. We used dual-color fluorophore histogram crossover to determine whether HSP70 and/or PPIA formed a complex with AIF (mAIF or tAIF) (fig. S8A). Although HSP70 was significantly reduced in sedentary (fig. S8, B and C) and exercised (Fig. 8, A and B) Dsg2mut/mut mice and ISO/Ca2+-treated Dsg2mut/mut ES-CMs (fig. S8D, E) compared to the respective controls, HSP70 did not comigrate with mAIF or tAIF (fig. S8B and Fig. 8A).
Fig. 8. Targeting the PPIA binding site of AIF prevents nuclear import of tAIF, nuclear loss of HMGB1, and AIF-mediated necroptosis.
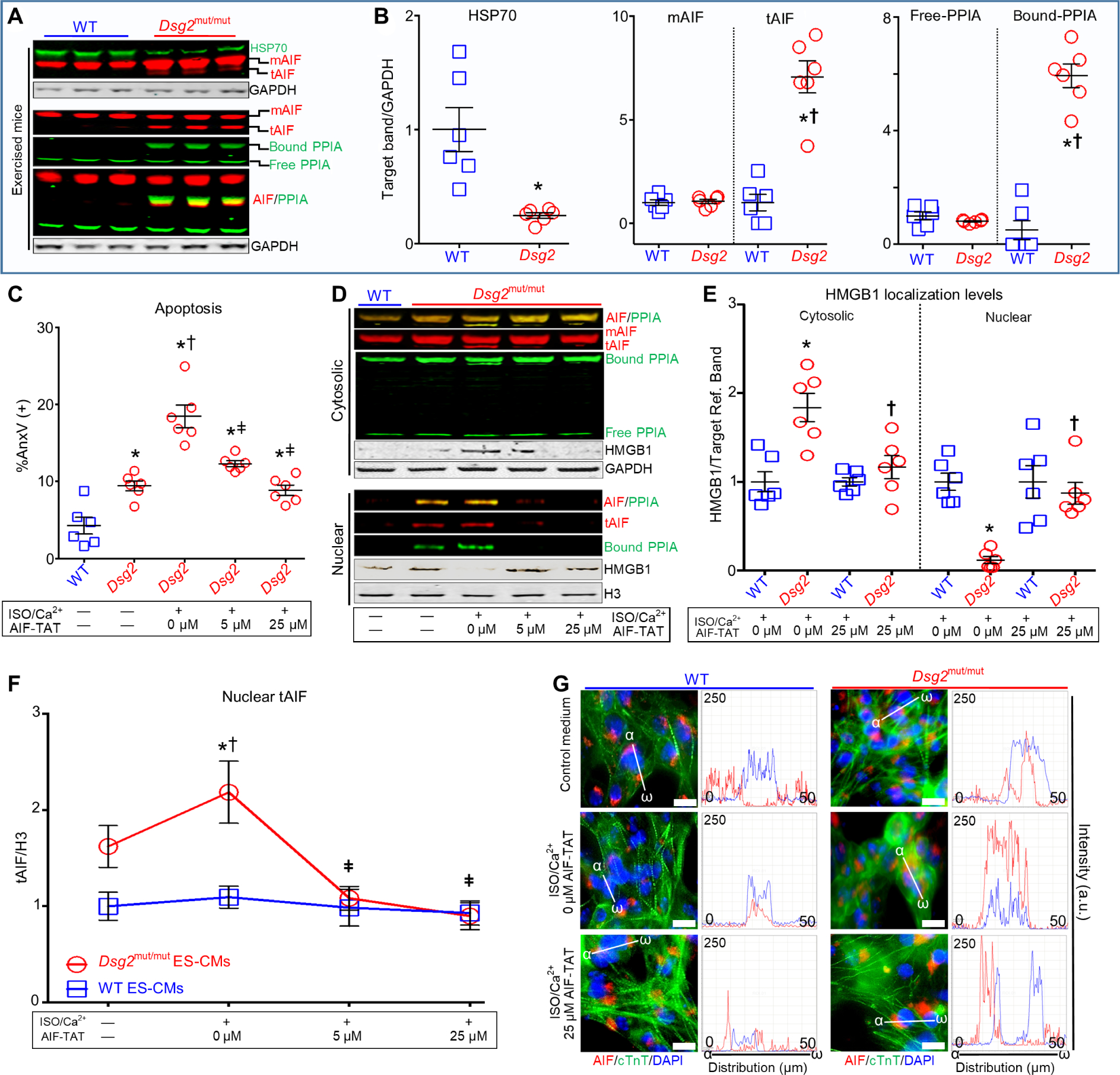
(A) Representative myocardial immunoblots from exercised WT and Dsg2mut/mut mice probed for heat shock protein-70 (HSP70), cyclophilin-A (PPIA), and AIF, with GAPDH as loading control. (B) Quantification of HSP70, mAIF, tAIF, and free and bound PPIA. Data are presented as means ± SEM [*P < 0.05 Dsg2mut/mut (HSP70) compared to WT (HSP70) using two-tailed paired t test; *P < 0.05 Dsg2mut/mut (tAIF or bound PPIA) compared to WT (tAIF or bound PPIA); †P < 0.05 Dsg2mut/mut (tAIF or bound PPIA) compared to Dsg2mut/mut (mAIF or free PPIA)], using one-way ANOVA. For (C) to (G), ES-CMs were treated for 7 days with 50 μM ISO and 1 μM calcium (Ca2+), in the presence or absence of 0, 5, or 25 μM AIF-TAT mimetic peptide (AIF-TAT). (C) Flow cytometry analyses of annexin V (AnxV)–positive cells. *P < 0.05 any group compared to WT (no ISO/Ca2+; no AIF-TAT); †P < 0.05 any group compared to Dsg2mut/mut (no ISO/Ca2+; no AIF-TAT); ǂP < 0.05 Dsg2mut/mut (5 or 25 μM AIF-TAT) compared to Dsg2mut/mut (ISO/Ca2+; 0 μM AIF-TAT). (D and E) Representative immunoblots of AIF, PPIA, mAIF, tAIF, AIF/PPIA complexes, and HMGB1 in cytosolic and nuclear fractions. Nuclear and cytosolic HMGB1 was quantified. *P < 0.05 Dsg2mut/mut (ISO/Ca2+; 0 μM AIF-TAT) compared to WT (ISO/Ca2+; 0 μM AIF-TAT) within each subcellular compartment; †P < 0.005 Dsg2mut/mut (ISO/Ca2+; 25 μM AIF-TAT) compared to WT (ISO/Ca2+; 25 μM AIF-TAT) within each subcellular compartment, using one-way ANOVA. (F) Effect of AIF-TAT on tAIF nuclear localization. *P < 0.005 Dsg2mut/mut (ISO/Ca2+; 0 μM AIF-TAT) compared to Dsg2mut/mut (no ISO/Ca2+; no AIF-TAT); †P < 0.005 Dsg2mut/mut (ISO/Ca2+; 0 μM AIF-TAT) compared to WT (ISO/Ca2+; 0 μM AIF-TAT); ǂP < 0.005 Dsg2mut/mut (ISO/Ca2+; 5 or 25 μM AIF-TAT) compared to Dsg2mut/mut (ISO/Ca2+; 0 μM AIF-TAT). All data are presented as means ± SEM (n = 6 per genotype per cohort per treatment using one-way ANOVA with Tukey’s post hoc analysis). (G) Representative ES-CMs immunostained for DAPI/AIF/cTnT with corresponding AIF/DAPI fluorophore intensity versus fluorophore distribution (α-ω) histograms. Images are representative of n = 6 independent experiments per cohort, with n = 3 cell culture replicates per condition. White scale bars, 25 μM.
Next, we assayed the amounts of free and AIF-bound PPIA in CMs. Regardless of genotype, sedentary mice showed no changes in free-PPIA and neither tAIF nor AIF-bound PPIA was detected in myocardial lysates (fig. S8, B and C). Myocardium from exercised Dsg2mut/mut mice showed significantly increased amounts of bound-PPIA levels that comigrated with tAIF, whereas this complex was not apparent in myocardium from exercised WT mice (Fig. 8, A and B). Unexpectedly, the amounts of free PPIA were similar in exercised cohorts of either genotype (Fig. 8, A and B), but the amount of free-PPIA was reduced in ISO/Ca2+-challenged ACM ES-CMs (fig. S8, D and E). Although both WT and Dsg2mut/mut ES-CMs had increased amounts of bound-PPIA that comigrated with both mAIF and tAIF, the difference was only statistically significant for Dsg2mut/mut ES-CMs and appeared to result from increased binding to tAIF (fig. S8, D and E).
To test whether blocking the interaction with PPIA could prevent ACM-related cell death, we used an AIF mimetic peptide (amino acids 370 to 394), representing most of the PPIA binding domain of AIF (amino acids 367 to 399). This AIF peptide binds PPIA with a KD of 1.2 × 10−5 and sequesters PPIA in the cytosol (12), disrupting the formation of the PPIA/AIF complex (65). To increase cellular uptake, the AIF-mimetic peptide was fused to the cell-penetrating HIV transactivator of transcription (TAT) fragment at its N terminus (henceforth called “AIF-TAT”) (66). In unstimulated cell cultures, Dsg2mut/mut ES-CMs exhibited increased apoptosis, indicated by cells positive for annexin V, compared to WT ES-CMs, and apoptosis was exacerbated in ISO/Ca2+-stimulated ES-CM cultures (Fig. 8C and fig. S8F). We used two assays for necrosis. By flow cytometry, we did not detect any difference between the genotypes in necrotic cell death in ES-CM cultures at baseline or in response to ISO/Ca2+ stimulation (fig. S8G). However, by immunoblot analysis of subcellular fractions from ES-CM lysates, we detected a loss of nuclear HMGB1 that was associated with accumulation of cytosolic HMGB1 in Dsg2mut/mut ES-CMs (Fig. 8, D and E), indicating active necrosis. In the presence of sustained β-adrenergic stimulation/Ca2+ overload, AIF-TAT reduced apoptosis (Fig. 8C and fig. S8F), PPIA/tAIF complexes in the nucleus (Fig. 8D), tAIF nuclear localization (Fig. 8F), and cytosolic HMGB1 (Fig. 8E) in Dsg2mut/mut ES-CMs.
To gain additional insight, we performed immunofluorescent analysis of the ES-CMs and the effect of AIF-TAT on ISO/Ca2+ stimulation. Using identical methods to those performed in mouse and patient myocardial samples (Fig. 5), the overlap of AIF and DAPI fluorophore intensity versus fluorophore distribution was used to confirm the localization of AIF in the cell nucleus (Fig. 8G). Although we found numerous AIF+ nuclei in Dsg2mut/mut ES-CMs either treated for 7 days with control media (Fig. 8G, top) or stimulated with ISO/Ca2+ (Fig. 8G, middle, and fig. S9A), the latter cohort additionally showed loss of cardiac troponin striation and cell membrane swelling with enlarged nuclei, indicative of necrosis (fig. S9A). In contrast, Dsg2mut/mut ES-CMs treated with ISO/Ca2+ for 7 days in the presence of AIF-TAT were devoid of AIF and DAPI nuclear colocalization with normal cardiac troponin striation (Fig. 8G, bottom).
We additionally found HMGB1+ nuclei in Dsg2mut/mut ES-CMs treated for 7 days with ISO/Ca2+, suggesting healthy CMs, yet these were also swollen with enlarged nuclei (fig. S9B), suggesting that these cells were in the early stages of necrosis. These results are consistent with the lack of a change in the proportion of necrotic cells based on positivity for propidium iodide and negativity for annexin V, which are indicators of later stages of necrosis (fig. S8G) (17). We also detected Dsg2mut/mut ES-CMs stimulated with ISO/Ca2+ that had morphologies consistent with apoptosis (fig. S9A). Conversely, WT ES-CMs subjected to the same conditions exhibited little to null AIF and DAPI colocalization, prominent HMGB1 nuclear localization, and normal striation of cardiac troponin (fig. S9, A and B), indicative of healthy cells.
Collectively, these results indicated that AIF-TAT prevented the interaction of AIF with PPIA and nuclear translocation of AIF. Consequently, AIF-TAT reduced both necrotic cells death (HMGB1 release from the nucleus) and apoptotic cell death. These data indicated that AIF plays a role in calcium-induced, caspase-independent cell death.
DISCUSSION
In all cardiomyopathies, regardless of etiology, myocyte death is a major determinant of cardiac dysfunction and heart failure (67). In ACM, exercise can increase the risk of pathological progression and SCD. Our study uncovered four previously unrecognized mechanisms accounting for exercise-triggered myocyte cell death in ACM: Ca2+ overload or low CAST abundance is associated with (i) increased total and active CAPN1 amounts, (ii) perturbed MMP and redox regulation, (iii) CAPN1-dependent cleavage of AIF and oxidization of tAIF, and (iv) PPIA-mediated translocation of tAIF to the myocyte nucleus, resulting in apoptosis and necrotic HMGB1 release.
Very low chronic levels of apoptosis are sufficient to cause lethal dilated cardiomyopathy (67), and myocyte cell death is a common pathological hallmark in ACM (68–70). Mounting evidence confirms that caspase-independent, AIF-mediated necrosis can be a regulated process (71, 72). Often, apoptosis and necrosis intersect in cardiac disease and remodeling (44). The fact that Dsg2mut/mut mice harbor extensive biventricular fibrosis prompted us to determine whether regulated necrosis also contributes to myocyte loss in Dsg2mut/mut hearts in response to endurance exercise.
CAPN1: A new pathogenic factor in ACM
CAPNs mediate the proteolytic cleavage of a wide array of proteins involved in many physiological processes. Dysregulation of CAPN1 activity has been associated with sarcomere protein degradation leading to LV decompensation (73), the development of hypertension (74), atherosclerosis (75), myocardial infarction (76), and pressure overload (77), yet never investigated in ACM. When CAPN1 activation is potentiated, pathological phenotypes associated with the loss of Ca2+ control are exacerbated, as in the case of I/R injury (20). Here, we add ACM to this palette of pathological cardiac conditions associated with CAPN1 activation. We demonstrated that CAPN1 is activated in response to exercise in Dsg2mut/mut hearts, by increased Ca2+ load and reduced CAST abundance. Our data indicated a cause-effect nexus among prolonged increases in intracellular Ca2+, CAPN1 activation, CAPN1 migration to the mitochondria, and, ultimately, necrotic cell death in HL-1 cells—events that were prevented by the cell-permeable CAPN1 inhibitor, calpeptin. In Dsg2mut/mut ES-CMs, calpeptin not only prevented CAPN1 activation but also blocked the formation of tAIF even in the presence of β-adrenergic stimuli and increased [Ca2+]i. Thus, we propose that, in addition to conferring protection against I/R injury and cardiac hypertrophy (74), CAPN1 inhibitors may serve as therapeutic agents in ACM.
Mitochondrial dysfunction plays a pivotal role in cell death, and mitochondrial alterations are related to Ca2+ overload during I/R injury (78). In isolated mitochondria, Ca2+ addition directly impairs mitochondrial function by Ca2+-dependent permeability transition pore opening (79). However, these alterations occur at [Ca2+] in the millimolar range, thus far from physiological conditions in viable CMs. Therefore, the link between increased [Ca2+]i and mitochondrial dysfunction is likely the result of Ca2+-dependent processes. Here, we revealed that CAPN1 activation is one causal linchpin between persistently increased [Ca2+]i and mitochondrial dysfunction in Dsg2mut/mut myocytes.
CAPN1 and CAPN2 were undetectable in mitochondria isolated from HL-1 cells exposed to physiological Ca2+ concentrations, yet only 2 min of exposure to Ca2+ overload was sufficient to induce a substantial mitochondrial fraction of CAPN1 but not CAPN2. This preceded the fall in MMP, which occurred after ~10 min of Ca2+ overload in HL-1 cells. Thus, intracellular CAPN1 redistribution could contribute to the fall in MMP. Others have described the mitochondrial localization of CAPN members (80). However, we showed that mitochondrial CAPN1 translocation in response to Ca2+ overload occurs independently of CAPN1 activity, as indicated by the inability of calpeptin to prevent CAPN1 mitochondrial localization. Previous studies conducted in re-perfused myocardium show that CAPN binding to the sarcolemma also occurs in the presence of the CAPN inhibitor, MDL-28170 (81). Additional studies are needed to determine the mechanisms underlying CAPN1 translocation into mitochondria; however, it is tempting to speculate that the relationship between the increase in [Ca2+]i and CAPN1 redistribution relates to covalent changes of the protease. In particular, the increase in [Ca2+]i might cause CAPN1 dephosphorylation by means of calcineurin activation.
CAST is both an endogenous inhibitor and substrate of CAPN1; thus, degradation of CAST isoforms promotes CAPN1 release from CAST (31, 32). There are intriguing physiological and cell-specific implications of the different CAST isoforms observed in Dsg2mut/mut mice. We previously reported that innate nuclear factor κB signaling and infiltrating T cells in the myocardium of Dsg2mut/mut mice lead to a cytokine storm, resulting in myocardial inflammation (82). Our findings here showing an increase in the 70-kDa CAST isoform, which is found in T cells (31), may shed light on a myocyte-extrinsic (that T cells) role of CAPN1 activation in cardiac injury in ACM. Prior research shows that T cells constitutively transcribe CAPN1, CAPN2, and CAST genes even during quiescence (83). In response to antigenic stimuli or increased Ca2+, they can proliferate and promote neighboring cell death. Inhibiting CAPN1 in T cells not only reduces T cell proliferation but also inhibits their ability to mount a pro-inflammatory cytokine surge (83). Infiltrating immune cells contribute to the pathogenesis of ACM: Hearts from Dsg2mut/mut mice showed infiltrating macrophages and T cells and increased inflammatory cytokine production (82). Although we cannot rule out that 70-kDa CAST isoforms originate from erythrocytes, we also advance the possibility that they arise from infiltrating T cells. Last, increased CAST in the hearts of WT and Dsg2mut/mut mice subjected to the swim protocol could be an exercise-induced adaptation, because CAST is increased in skeletal and cardiac tissues in response to exercise or β-adrenergic stimulation, or both (31).
CAPN1-PPIA-AIF–mediated myocyte cell death in ACM
Mitochondrial perturbations can be a central step leading to a programmed necrotic pathway (PNP), such as caspase-independent AIF-mediated parthanatos (84–86) or caspase-independent AIF-mediated necroptosis (71, 72). Mechanistically, these two PNPs differ in how AIF-mediated cell death occurs. In AIF-mediated parthanatos, release of nontruncated mAIF (62 kDa) from mitochondria occurs via PARylation of proteins by PAR polymers generated by PARP-1, leading to mitochondria depolarization (84, 85, 87). In AIF-mediated necroptosis, AIF truncation (57 kDa) and mitochondrial release are mediated by CAPN1 (62, 72, 88). Thus, our study suggests that exercise-induced myocardial damage in ACM involves AIF necroptosis, because both activated CAPN1 and tAIF were present in Dsg2mut/mut myocardium and inhibition of CAPN1 in Dsg2mut/mut ES-CMs reduced active CAPN1 that was accompanied by nearly undetectable tAIF. This new evidence may provide a mechanistic explanation for the findings reported by Pilichou et al. (16), who showed that myocyte necrosis, and not apoptosis, underlies cardiac dysfunction in another Dsg2-mutant mouse model (Dsg2-N271S; homolog of human DSG2-N266S).
Among the mitochondrial assets that limit myocyte cell death is the TXN2/TXNRD2 ROS buffering system (89). This major antioxidant system is under the transcriptional regulation of the Wnt/β-catenin signaling pathway, a pathway down-regulated in the ACM heart (8, 24). CAPN activity can promote β-catenin degradation in human colon cancer cells and mouse embryo fibroblasts, even in the absence of the negative regulator of Wnt/β-catenin signaling, glycogen synthase kinase 3β (GSK3β) (90). Thus, our study revealed a mechanism of CAPN1 activation that clarifies why suppression of the Wnt/β-catenin pathway, and consequentially TXN2/TXNRD2 down-regulation in Dsg2mut/mut mice, contributes to ACM pathogenesis. CAPN1 can also truncate GSK3β at its N-terminal inhibitory domain [serine-9 (S9)], thereby constitutively activating GSK3β (91). Our prior work in mouse (Dsg2mut/mut and JUP2157del2) and in vitro models (neonatal rat ventricular myocytes expressing JUP2157del2 or PKP21851del123) of ACM and myocardium of patients with ACM demonstrates that GSK3β plays a central role in ACM pathogenesis (8). Thus, finding increased CAPN1 activity in Dsg2mut/mut mice provides an additional mechanistic explanation for our discovery that Dsg2mut/mut mice expressing constitutively active GSK3β (S9 to alanine-9 mutation) exhibit a worse functional and pathological phenotype compared to Dsg2mut/mut mice expressing nonmutated GSK3β (8). Furthermore, in its active form (nonphosphorylated S9), GSK3β phosphorylates proline-rich serine/threonine (PP[S/T]xP) motifs and/or (S/T)xxxx(S/T) motifs on target substrates (where x is any amino acid) (92). AIF contains both motifs in its C-terminal nuclear localization signal domains (46). Thus, AIF may undergo GSK3β-mediated phosphorylation at these domains, thus affecting AIF nuclear translocation in ACM myocytes. The current lack of publicly available phosphorylated AIF antibodies targeting such motifs defers answering this intriguing possibility to future studies. Therefore, in addition to the beneficial effects exerted by GSK3β inhibitors [see, for instance, their anti-ischemic properties (93)], preventing GSK3β-mediated AIF posttranslational modifications (94) could uncover details of the relationship between GSK3β and AIF in ACM.
Our study revealed that other oxidation is another posttranslational modification of AIF. Finding that AIF was not only truncated but also oxidized in the hearts of Dsg2mut/mut mice after exercise dovetails with a study of non–small cell lung carcinoma cells showing that increased Ca2+ activates mitochondrial CAPN1 and ROS production, the latter event resulting in oxidative modifications of AIF (62). Presumably, the oxidative modification of AIF augments accessibility to AIF’s CAPN1 cleavage site (62). For clinical application, genetic ablation or irreversible pharmacological inhibition of AIF is an impractical avenue for preventing AIF-mediated cell death, because AIF is required for mitochondrial oxidative respiration (46, 95). Muscle-specific loss of AIF results in mitochondrial dysfunction, muscular atrophy, and dilated cardiomyopathy (96). Additional in-depth studies shall determine the factors that limit mitochondrial AIF export or its posttranslational modifications. Here, we showed (i) that CAPN1 inhibition is necessary and sufficient to prevent AIF truncation in ACM myocytes, and (ii) that prevention of PPIA binding to AIF using an AIF-TAT mimetic peptide is efficacious in preventing apoptosis, nuclear localization of tAIF, and nuclear loss of HMGB1 (a marker of cell necrosis).
Limitations and studies in perspective
The TXN system does not fully portray the antioxidant armamentarium, either in mitochondria or in the cytosol. We suspect that glutathione (GSH) is also affected in ACM CMs at rest and, even more so, after chronic exercise. We defer a thorough investigation of the GSH system status to future studies to assess whether rescuing the TXN or the GSH system or both shields the ACM heart against exercise-induced mitochondrial dysfunction and AIF-mediated cell death. Second, CAPNs can disrupt ATP synthase inducing superoxide production (97) or participate in mitochondrial permeability transition pore induction. These events can also contribute to ACM pathogenesis and deserve future, in-depth investigation.
There are inherent physiological and technical limits when using an ES-CM system as a substitute for primary adult myocytes or as an in vivo surrogate. We chose this approach for three main reasons. First, isolation of adult CMs primarily results in single-cell isolation with infrequent paired myocytes (<1 to 2% of isolation culture); thus, primary adult CMs are not the ideal in vitro system for studying a disease involving cell-cell mechanical and electrical contact dependent on desmosomal integrity. Using cell monolayers provided a system for assessing the impact of ISO/Ca2+ on apoptotic and necrotic levels and cellular morphology. Second, infiltrating CD68+ macrophages and CD3+ T cells are present in the hearts of Dsg2mut/mut mice (82). Thus, the presence of immune cells isolated from adult Dsg2mut/mut cardiac tissue would have confounded the analysis, particularly in light of previous studies that showed the regulatory role of CAPN1 in T cell proliferation (83). Use of ES-CM cultures provided an ideal surrogate, because this system guarantees ≥95 to 99% CM purity in the population and is devoid of immune cells. Third, using primary adult myocytes longer than 72 hours was unfeasible; this limitation would have precluded the possibility of testing the impact of sustained ISO/Ca2+ challenge up to 7 days. We previously demonstrated that the amount and intensity of exercise increase disease progression in patients with ACM (4). Thus, it was paramount for us to compare the impact of acute (1 day) versus chronic (7 days) β-adrenergic/Ca2+ challenge in ACM ES-CMs.
Evidence of AIF+ nuclei in the myocardium of DCM, HCM, and IHD subjects, albeit to a lower extent than ACM samples, suggested that AIF nuclear translocation (and the consequent adverse effects) may contribute to myocyte cell death in other forms of cardiomyopathies. On the other hand, this evidence reiterates the necessity of correlating histopathological findings with clinical history, such as exercise training or other forms of physiological or pathological stressors. We do not have exercise history data available from controls or DCM, HCM, and IHD cohorts for the present study. However, evidence obtained in a small group (six) of G+/P+ ACM individuals, all of whom harbor a pathogenic PKP2 mutation, showed a trend for a correlation between AIF scores with exercise history. Future, dedicated studies should include more retrospective exercise participation reports from ACM patients for which myocardial samples have been collected. Despite this limitation, the data support the current mechanistic notion that AIF-mediated myocyte death is more likely to occur in the presence of a pathological “substrate” (for example, a desmosomal variant) and an environmental “trigger” (for example, exercise or hemodynamic stress).
Last, it is tempting to extend a similar pathogenic scenario (Ca2+/CAPN1/TXN2/PPIA/AIF) to other cell types, including cardiac sympathetic neurons or mesenchymal cells. Such cells may contribute to arrhythmias associated with ACM. Therefore, heart rhythm disorders may arise from a combination of abnormal Ca2+/CAPN1/TXN2/PPIA/AIF handling, the loss of myocardium, neuronal or mesenchymal cell death, or sympathetic nerve hyperinnervation.
Conclusions
Our study revealed a means by which exercise induces cell death in ACM that is mediated by a system involving Ca2+, CAPN1, TXN2, PPIA, and AIF (fig. S10), offering unprecedented opportunities to deepen our understanding of ACM pathophysiology. Although the present mechanism unlikely exhausts all the possibilities underpinning exercise-instigated myocyte loss, ACM appears an example of AIF-induced myocyte necroptosis (45, 98). Moreover, the adverse consequences of CAPN1-mediated AIF truncation and PPIA-tAIF complex formation could be at play in other forms of cardiomyopathies characterized by high levels of myocardial cell death and fibrotic remodeling, such as myocarditis, and a clinical history of either sustained physiological or pathological stressors. Thus, the current findings have the potential to expand beyond existing paradigms of disease mechanisms triggered in ACM and may provide a novel therapeutic approach in the prevention of disease onset and progression in ACM individuals and subjects affected by other forms of cardiomyopathies, of an inherited or acquired nature.
MATERIALS AND METHODS
Study design
Using a murine model of ACM, two different myocyte cell lines (ACM ES-CMs and HL-1 cells), and myocardial samples from patients with ACM, we used a randomized study design to determine the pathological role of AIF in myocyte cell death in ACM. All procedures conformed to the Guide for the Care and Use of Laboratory Animals from the National Institutes of Health (NIH publication no. 85–23, revised 1996) and followed adherence to the Johns Hopkins School of Medicine Animal Care and Use Committee. Studies were conducted in male WT and Dsg2mut/mut mice at rest and in response to 11 weeks of endurance exercise (5 to 16 weeks of age, 15 to 25 g). Mice were given access to water and standard mouse chow ad libitum. A full phenotypic description of the WT and Dsg2mut/mut mice used in these experiments, the protocol of homologous recombination that generated the Dsg2mut/mut mouse line, and polymerase chain reaction (PCR) primers and conditions for genotyping can be found in Chelko et al. (8). Mice have a C57bl background and the following numbers were used: exercised mice, n = 22 for WT mice and n = 27 for Dsg2mut/mut mice; sedentary mice, n = 7 for both WT and Dsg2mut/mut mice. All experiments were performed in myocardial samples from age-matched mice (16 weeks of age; sedentary and exercised), and a minimum of n = 5 mice, myocardial lysates, and/or histological samples were used for all studies. Any mouse that died during exercise was used strictly for survival analysis; no myocardial samples were used from these mice. The Johns Hopkins University (JHU) Echocardiography and Electrocardiography Physiology Core (Director, D. Kass) were blinded to genotype and environmental condition (i.e., rest or swim).
In addition, we used age-matched myocardial samples from individuals with no history of heart disease and patients with ACM, DCM, HCM, and IHD. The main study was approved by the Johns Hopkins School of Medicine institutional review board (IRB; study no. NA_00041248 to H.C.). All participants provided written informed consent. Anonymized, nonconsented tissues were obtained for the TMA under IRB study no. IRB00183127 to M.K.H. All histology experiments were immunostained and coded (S.P.C.) and then distributed to three independent reviewers (M.K.H., C.A.J., and P.A.) to allow for blinded AIF pathological scoring. After averaging of blinded reviewer scores, the final averaged score for each patient was compiled in an Excel file, recoded, and sent to the JHU Arrhythmogenic Right Ventricular Dysplasia (ARVD) Program (Director, H. Calkins) for input of clinical phenotypes by genetic counselors (B.M. and C.T.) not familiar with study design.
Initial in vitro studies in HL-1 cells used pharmacological agents to determine the prerequisites needed in ES-CMs for CAPN1-mediated truncation of AIF, which was dependent on both ISO and Ca2+ stimulation and time (7 days of treatment). These initial HL-1 experiments additionally determined the most appropriate inhibitor for CAPN1, calpeptin, and the PPIA-AIF complex inhibitor, AIF-TAT, in ES-CMs. For all experiments, the number of myocardial samples and lysates, mice, and/or cell cultures used, along with the number of replicates, can be found in the figure legends.
Swim protocol
All mice followed a previously described endurance swim protocol (8). In brief, mice underwent 2 weeks of swim training beginning at 5 weeks of age, where mice swam 10 min on training day 1 (Monday), and then each following day, 10-min increments were added. By the end of training day 10 (second Friday), mice swam 90 min/day, 5 days/week until swim end point (16 weeks of age). No mouse swam on Saturday or Sunday.
Cardiac function assessment by echocardiography and ECG
All exercised and age-matched sedentary mice were assessed for cardiac function with transthoracic echocardiography and ECG telemetry, as previously described (8). Transthoracic echocardiography was assessed in nonsedated mice using a Vevo 2100 Visualsonic imaging system, and measurements were obtained according to the American Society of Echocardiography (99). Parasternal long-axis views of the LV, at the level of the papillary muscles, were acquired at a sweep speed of 200 mm/s (M-mode) (8). Three to five measurements were acquired from each mouse and averaged.
ECG telemetry was obtained as previously described (8). In brief, mice underwent nose cone anesthesia (2% isoflurane, in 100% O2), and an ECG wireless telemeter (model ETA-F10, DSI PhysioTel) was implanted (subcutaneous, ventral) and sutured in place (7–0 Ethicon), following the manufacturer’s protocol (DSI PhysioTel) to obtain Lead I ECG recordings. All sedentary and exercised mice became fully ambulatory within 2 to 3 min after implantation and were given 3 days for recovery. After implantation recovery, ECGs were recorded for either 24 hours in sedentary cohorts or 90 min in exercised cohorts during a 90-min swim as previously described (8). All ECG parameters were analyzed following previously described guidelines (100), using the LabChart Pro ECG Analysis Add-on Software (LabChart Pro 8, MLS360/8, ADInstruments). After functional data collection (echocardiography and ECG telemetry), all exercised mice and sedentary age-matched cohorts (16 weeks of age) were euthanized, and hearts were excised for the following experimental protocols listed below.
Mouse ES-CM cell differentiation and treatment
Mouse ESCs were maintained and differentiated as previously described (101), and the absence of mycoplasma contamination was confirmed before experimental studies. Briefly, mouse ESCs from both WT and Dsg2mut/mut mice were derived from embryonic day 3.5 embryos and maintained on gelatin-coated dishes in maintenance medium [Glasgow minimum essential medium (MEM) supplemented with 10% fetal bovine serum and 3 μM Chir99021 and 1 μM PD98059 or ESGRO (1000 U/ml); Millipore], GlutaMAX, sodium pyruvate, and MEM nonessential amino acids (Thermo Fisher Scientific). For CM differentiation, ESCs were plated in Iscove’s Modified Dulbecco’s Medium (Thermo Fisher Scientific) and Ham’s F12 Medium (Cellgro) at 3:1, supplemented with N2, B27, penicillin/streptomycin, 2 mM GlutaMAX, 0.05% bovine serum albumin (BSA), l-ascorbic acid (5 ng/ml; Sigma-Aldrich), and α-monothioglycerol (Sigma-Aldrich) at a final density of 100,000 cells/ml to allow spheroid formation. After 48 hours, embryoid bodies were collected and transferred to ultralow attachment plastic surface and induced for 40 hours with activin A and Bmp4 (R&D Systems). Cells were then dissociated and plated as monolayers for 48 hours in the presence of 10 μM XAV (3,5,7,8-Tetrahydro-2-[4-(trifluoromethyl)phenyl]-4H-thiopyrano [4,3-d]pyrimidin-4-one; R&D Systems) to allow CM differentiation. The resulting ES-CMs were cultured 45 days before experimental procedures were performed.
Four ES-CM experimental protocols were performed. (i) For acute (1 day) versus chronic (7 days) β-adrenergic and calcium overload experiments, WT and Dsg2mut/mut ES-CMs were treated for 1 or 7 days with media partially (50%) changed every day containing either (a) control media, (b) media containing 50 μM ISO alone, or (c) media containing 50 μM ISO and 1 μM Ca2+. (ii) For chronic CAPN1 inhibition experiments, WT and Dsg2mut/mut ES-CM were incubated in medium with 30 μM calpeptin (CAPN1 inhibitor, catalog no. 0448, Tocris Bioscience) for 2 hours before the addition of 50 μM ISO/1 μM Ca2+ on day 1 and each consecutive day for 7 days and 30 μM calpeptin was re-administered each following morning for 2 hours before partial (50%) 50 μM ISO/1 μM Ca2+ media change. (iii) For chronic AIF-TAT mimetic peptide inhibition experiments, WT and Dsg2mut/mut ES-CMs were incubated in medium with either 0, 5, or 25 μM AIF-TAT for 2 hours before the addition of 50 μM ISO/1 μM Ca2+ on day 1 and each consecutive day for 7 days, where 0, 5, or 25 μM AIF-TAT was re-administered every other morning for 2 hours before partial (50%) 50 μM ISO/1 μM Ca2+ media change. (iv) After 7 days of treatment, ES-CMs were incubated with fluorescent-conjugated annexin V in a buffer containing 10 mM Hepes (pH 7.4), 140 mM NaCl, and 2.5 mM CaCl2, followed by the addition of propidium iodide (1 mg/ml) and analyzed by flow cytometry.
HL-1 cell culture and treatment
HL-1 cells are an immortalized cell line derived from mouse atria cardiac myocytes, which maintains a differentiated adult cardiac phenotype, and were grown as previously described (102). Briefly, cells were cultured as a monolayer (37°C, 5% CO2) in plates precoated with 1 μg/cm2 fibronectin and 0.02% (w/v) gelatin solution. HL-1 cells were maintained in the Claycomb medium (Sigma-Aldrich) supplemented with 10% fetal bovine serum, penicillin (100 U/ml), streptomycin (0.1 mg/ml), 0.1 mM norepinephrine, and 4 mM l-glutamine. Each experiment was performed 48 hours after cell plating.
HL-1 cells were incubated for 10 min in HBS (Sigma-Aldrich) KV medium (1.26 mM CaCl2, 0.81 mM MgSO4, 0.44 mM KH2PO4, 142 mM KCl, 0.27 mM K2HPO4, 5.55 mM D-glucose, 1 mM Na3VO4, and 20 mM Hepes, pH 7.4), and then rapid and sustained intracellular Ca2+ overload was obtained by adding the Ca2+ ionophore, calcimycin (1 μM). To monitor intracellular Ca2+ variations, HL-1 cells were loaded with 5 μM Fluo-4 FF AM (Molecular Probes) in HBS Na (Sigma-Aldrich) for 30 min before the medium was replaced with HBS KV. The fluorescence was recorded using a fluorimeter plate reader (Fluoroskan Ascent Labsystem) at wavelength settings of 485/530 nm for excitation/emission.
CAPN activity was measured as proteolytic cleavage of the fluorescent CAPN substrate, Suc-LLVY-AMC (Suc-Leu-Leu-Val-Tyr-7-Amino-4-Methyl-Coumarin, Calbiochem). HL-1 cells were grown in 24-well plates and incubated for 30 min in HBS Na (Sigma-Aldrich) with 25 μM Suc-LLVY-AMC and 0.01% pluronic F127 (Molecular Probes) before treatment protocols. The fluorescence was recorded using a fluorimeter plate reader (Fluoroskan Ascent Labsystem) at wavelength settings of 390/460 nm for excitation/emission. CAPN activity was inhibited by the addition of either calpeptin (Tocris Bioscience) or PD150606 (Calbiochem) in the incubation medium, as previously described (80, 103). Concentrations of calpeptin or PD150606 used in HL-1 cell studies are provided in the figures or legends.
HL-1 plasma membrane integrity was assessed by measuring the release of LDH into the culture supernatant. LDH activity was evaluated by an enzymatic assay. Samples of the cell-free culture supernatant were collected at indicated times in the figures or legends. Cells were lysed in 1% (w/v) Triton X-100 at the end of each experiment to estimate the remaining cellular content of LDH. The LDH activity at each time point was expressed as a percentage of total LDH activity (cellular + culture supernatant).
MMP was determined using the potentiometric probe, JC-1 (5,5′,6,6′-Tetrachloro-1,1′,3,3′-tetraethyl-imidacarbocyanine iodide), as described previously (104). JC-1 (Molecular Probes) is a cationic carbocyanine dye that selectively accumulates in polarized mitochondria, where it forms red fluorescent aggregates (excitation 485/emission 590 nm). Upon mitochondrial depolarization, JC-1 is released into the cytosol, whereas a monomer emits green fluorescence (excitation 485 nm/emission 520 nm). Therefore, MMP variations were monitored as the ratio of fluorescent emissions at 590/520 nm using a fluorimeter plate reader (Fluoroskan Ascent Labsystem). HL-1 cells were incubated with JC-1 (1.5 μM) for 30 min at 37°C in HBS Na before treatment protocols.
For cell line transfection, both coverslips and cell culture plate surfaces were pretreated with the following coating solutions: 100 to 200 μl of Opti-MEM reagent was combined with CAST-overexpression plasmid (0.66 μg/cm2) containing a GFP reporter and incubated at room temperature for 5 min. Then, 100 to 200 μl of Opti-MEM reagent was combined with 0.22 μl/cm2 of cationic lipid reagent and incubated at room temperature for 5 min. These two solutions were then integrated and complexed for 20 min at room temperature. The transfection solution was distributed over the cell seeding surface and left to dry at room temperature for 30 to 45 min. Cells were counted and resuspended in 2 ml of serum-free and antibiotic-free medium and then directly spread over the surface. Cells were incubated at 37°C with 95% O2 and 5% CO2 for 30 min, and then medium was supplemented with the appropriate serum and antibiotic concentrations. Treatments were conducted 24 hours after cell transfection. After transfection and treatment, cell cultures were subjected to calcium overload in the presence of HBS KV medium containing 0.4% trypan blue. Live-imaging, fluorescence-based cell viability was detected at 30 and 60 min after Ca2+ overload.
Mitochondria isolation and subcellular fractionation
Mitochondria were isolated from HL-1 cells in Percoll density gradient (105). Briefly, 30 × 106 adherent cells were harvested with trypsin/EDTA, centrifuged at 500g for 10 min, and washed in buffer A [100 mM sucrose, 1 mM EGTA, 20 mM Mops (pH 7.4), and BSA (1 mg/ml)] before cell disruption with a Dounce homogenizer. The suspension was centrifuged twice at 500g for 5 min, and the resulting supernatant was removed, then centrifuged at 10,000g for 10 min at 4°C. The pellet was resuspended in buffer B [300 mM sucrose, 1 mM EGTA, 20 mM Mops (pH 7.4), and BSA (1 mg/ml)], and the homogenate was layered on a two-phase (25 and 40%) Percoll density gradient. After centrifugation at 30,000g for 15 min at 4°C, mitochondria (layered at the interface) were removed, washed with buffer A, and resuspended in RIPA buffer.
Subcellular fractionation was performed in mouse ventricular myocardium and ES-CMs lysed with compartment-specific isolation buffers according to the manufacturer’s protocol (Subcellular Fractionation Kit for Tissues, Thermo Fisher Scientific, 87790). Centrifugation speeds, spin times, temperatures, and resulting subcellular compartment (supernatant) were acquired in sequential order and are as follows: (i) 500g, 5 min at 4°C, cytosolic extract; (ii) 3000g, 5 min at 4°C, mitochondrial-bound extract; (iii) 5000g, 5 min at 4°C, nuclear extract; and (iv) 16,000g, 5 min at 4°C, chromatin-bound extract.
DNA retardation assay
A DNA retardation assay was performed, as previously described (63), to examine the effects of oxidized or reduced AIF lysates on the migration of double-stranded DNA. Specifically, a 10-μl sample was prepared to contain (i) 5 μg of protein lysate, (ii) 4 μl of a 100-bp DNA ladder (ranging from 100 bp to 1.5 kb; Promega, G2101), and (iii) 1 μl (400 ng/μl) of the WT Dsg2 2.2-kb double-stranded DNA fragment generated by PCR (8), and then incubated at 37°C for 30 min. After incubation, 5 μl of sample mixture was run on a 3% agarose gel at 150 V. A 1-kb DNA ladder (New England BioLabs, N3232L) was used as a reference band.
Tissue microarray
A TMA composed of 94 LV tissues was generated consisting of 33 control tissues (noncardiac deaths), and three cohorts with failing hearts: 28 DCM tissues, 8 HCM tissues, and 25 IHD tissues. All tissue cores were 1.5 mm in diameter, embedded in a single paraffin block, and cut at 5 μm thickness.
AIF subcellular localization analysis and determination of AIF pathology score
After immunostaining, AIF fluorescence and nuclear fluorescence (DAPI) were detected by laser scanning microscopy (Zeiss LSM 510 Meta) in a sequential fashion to avoid cross-talk between fluorophores, as previously described (45). Merged images were obtained using Zeiss LSM software, and about 3 to 10 ROIs were assessed per condition (mouse myocardium and patient myocardial biopsies). Colocalization analyses were determined by the overlap of fluorophore distribution (x axis) versus fluorophore intensity (y axis) between AIF and DAPI at each ROI, as previously described (45). Anonymized, immunostained samples were distributed to three independent reviewers for blinded AIF pathological scoring. All patient-immunostained slides received a single AIF pathology score from each blinded reviewer ranging from 0 to 4 as follows: grade 0 (mitochondrial AIF), grade 1 (mitochondrial and cytosolic AIF, 50:50 odds ratio), grade 2 (diffuse cytosolic AIF, rare mitochondrial localization), grade 3 (presence of 2 or 3 AIF-positive nuclei, determined by the overlap of fluorophore intensity and fluorophore distribution), or grade 4 (presence of ≥4 AIF-positive nuclei, determined by the overlap of fluorophore intensity and fluorophore distribution). Blinded reviewer scores were then averaged (reviewers 1 to 3) for each patient, compiled and recoded, and sent to the JHU ARVD Program for input of clinical phenotypes for assessment of AIF pathology scores and TFC phenotypes. AIF-to-DAPI colocalization pathological scores (grades 0 to 4) were only performed in the patient myocardium due to the availability of myocardial biopsy size. The amount of total myocardial AIF-positive (AIF+) nuclei was performed in mouse myocardium because of the availability of tissue and presented as percent AIF+ nuclei over total nuclei.
Statistical analysis
For all mouse and patient ACM findings, data are presented as means ± SEM, and n values are provided in the figure, table, or legend. For all HL-1 cell findings, data are presented as means ± SD from six independent experiments/conditions. P < 0.05 was deemed statistically significant. As appropriate, associations between continuous dependent variables were tested using Student’s paired/unpaired t test (binary independent variables) or using one-way or two-way analysis of variance (ANOVA; two or more variables). Specifics of statistical analyses performed are included in each figure legend or table. Raw data are provided in data file S1.
Supplementary Material
Data file S1. Raw data.
Fig. S1. Dsg2mut/mut mice display ECG abnormalities and myocardial injury in response to endurance exercise.
Fig. S2. CAPN1 activation and cell death in response to Ca2+ overload.
Fig. S3. CAPN1 inhibition in HL-1 cells by PD150606.
Fig. S4. CytC in Dsg2mut/mut mice and posttranslational modifications of AIF.
Fig. S5. AIF localization in ACM patient myocardium.
Fig. S6. Dsg2mut/mut mice display reduced COX IV abundance and aberrant myocardial localization.
Fig. S7. Labeled cysteines and DNA gel electrophoresis.
Fig. S8. HSP70 and PPIA abundance in ACM myocytes.
Fig. S9. Increased AIF nuclear localization and nuclear loss of HMGB1 in ACM ES-CMs.
Fig. S10. Graphical abstract of exercise-induced, CAPN1/PPIA-mediated AIF-nuclear import in ACM.
Table S1. Echocardiographic and electrocardiographic indices from exercised WT and Dsg2mut/mut mice.
Table S2. Desmosomal gene variants in patients with ACM.
Table S3. Clinical characteristics and AIF pathology scores of patients with ACM.
Acknowledgments:
We are grateful to the ARVD/C patients and families who have made this work possible. We are grateful to C. Culmsee (Philipps-Universität Marburg) for the very helpful advice. Editorial services were provided by N. R. Gough (BioSerendipity, LLC, Elkridge, MD). We thank the IBB-CNR (Naples, Italy) for the construction and production of AIF-TAT mimetic peptide and W. Claycomb (Louisiana State University Medical Center, New Orleans, LA) for providing HL-1 cells.
Funding:
This work was supported by the NIH [R01 HL136918 and R01 HL063030 (to N.P.)]; R01 HL137259 (to B.O.); R01 HL138475 (to R.N.K.); T32 AG058527 (G.K.); R01 HD086026 (to C.K.) and TEDCO [2015-MSCRFI-1622 to (P.A.)]; American Heart Association Career Development Award [19CDA34760185 (to S.P.C.) and 19CDA34760161 (to P.A.)], Innovative Project Award [18IPA34170446 to (P.A.)], and Grant-in-Aid [17GRNT33670405 to (M.H.)]; the Department of Defense [PR191593 (to R.N.K.)]; Leducq Transatlantic Network of Excellence [16CVD04 (to F.D.L.)]; Leducq [RA15CVD04 (to R.N.K.)]; University of Padova Strategico Grant “DYCENDI” and CNR Neuroscience Institute (to F.D.L.); the Magic That Matters Fund [internal JHU award (to N.P.)]; Heart Rhythm Society Cardiac Pacing and Electrophysiology Fellowship Award sponsored by an educational grant from St. Jude Medical (to S.P.C.); and the Gilead Research Scholars in Cardiovascular Disease Fund (to A.A.). The Johns Hopkins ARVD Program is supported by the Dr. Francis P. Chiaramonte Private Foundation; Foundation Leducq [16CVD02 (to H.C.)]; the Leyla Erkan Family Fund for ARVD Research; the Dr. Satish, Rupal, and Robin Shah ARVD Fund at Johns Hopkins; the Bogle Foundation; the Healing Hearts Foundation; the Campanella Family; the Patrick J. Harrison Family; the Peter French Memorial Foundation; and the Wilmerding Endowments.
Footnotes
Competing interests: H.C. is a consultant for Medtronic Inc. and St. Jude Medical/Abbott. H.C. receives research support from Boston Scientific Corp. C.T. and C.A.J. receive salary support from the Boston Scientific Corp research grant. C.A.J. received a lecture fee from Abbott. All other authors declare that they have no competing interests.
Data and materials availability: All data associated with this study are present in the paper or the Supplementary Materials.
SUPPLEMENTARY MATERIALS
stm.sciencemag.org/cgi/content/full/13/581/eabf0891/DC1
Materials and Methods
View/request a protocol for this paper from Bio-protocol.
REFERENCES AND NOTES
- 1.Lloyd-Jones D, Adams R, Carnethon M, De Simone G, Ferguson TB, Flegal K, Ford E, Furie K, Go A, Greenlund K, Haase N, Hailpern S, Ho M, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, Dermott MM, Meigs J, Mozaffarian D, Nichol G, O’Donnell C, Roger V, Rosamond W, Sacco R, Sorlie P, Stafford R, Steinberger J, Thom T, Wasserthiel-Smoller S, Wong N, Wylie-Rosett J, Hong Y; American Heart Association Statistics Committee and Stroke Statistics Subcommittee, Heart disease and stroke statistics—2009 update. Circulation 119, 480–486 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Maron BJ, Doerer JJ, Haas TS, Tierney DM, Mueller FO, Sudden deaths in young competitive athletes: Analysis of 1866 deaths in the United States, 1980–2006. Circulation 119, 1085–1092 (2009). [DOI] [PubMed] [Google Scholar]
- 3.Saffitz JE, Asimaki A, Huang H, Arrhythmogenic right ventricular cardiomyopathy: New insights into mechanisms of disease. Cardiovasc. Pathol 19, 166–170 (2010). [DOI] [PubMed] [Google Scholar]
- 4.James CA, Bhonsale A, Tichnell C, Murray B, Russell SD, Tandri H, Tedford RJ, Judge DP, Calkins H, Exercise increases age-related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. J. Am. Coll. Cardiol 62, 1290–1297 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sawant AC, Bhonsale A, te Riele ASJM, Tichnell C, Murray B, Russell SD, Tandri H, Tedford RJ, Judge DP, Calkins H, James CA, Exercise has a disproportionate role in the pathogenesis of arrhythmogenic right ventricular dysplasia/cardiomyopathy in patients without desmosomal mutations. J. Am. Heart Assoc 3, e001471 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Awad MM, Calkins H, Judge DP, Mechanisms of disease: Molecular genetics of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Nat. Clin. Pract. Cardiovasc. Med 5, 258–267 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Groeneweg JA, Bhonsale A, James CA, te Riele AS, Dooijes D, Tichnell C, Murray B, Wiesfeld ACP, Sawant AC, Kassamali B, Atsma DE, Volders PG, de Groot NM, de Boer K, Zimmerman SL, Kamel IR, van der Heijden JF, Russell SD, Cramer MJ, Tedford RJ, Doevendans PA, van Veen TA, Tandri H, Wilde AA, Judge DP, van Tintelen JP, Hauer RN, Calkins H, Clinical presentation, long-term follow-up, and outcomes of 1001 arrhythmogenic right ventricular dysplasia/cardiomyopathy patients and family members. Circ. Cardiovasc. Genet 8, 437–446 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Chelko SP, Asimaki A, Andersen P, Bedja D, Amat-Alarcon N, De Mazumder D, Jasti R, MacRae CA, Leber R, Kleber AG, Saffitz JE, Judge DP, Central role for GSK3β in the pathogenesis of arrhythmogenic cardiomyopathy. JCI Insight 1, e85923 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cho G-S, Lee DI, Tampakakis E, Murphy S, Andersen P, Uosaki H, Chelko S, Chakir K, Hong I, Seo K, Chen H-SV, Chen X, Basso C, Houser SR, Tomaselli GF, O’Rourke B, Judge DP, Kass DA, Kwon C, Neonatal transplantation confers maturation of PSC-derived cardiomyocytes conducive to modeling cardiomyopathy. Cell Rep. 18, 571–582 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Candé C, Vahsen N, Kouranti I, Schmitt E, Daugas E, Spahr C, Luban J, Kroemer RT, Giordanetto F, Garrido C, Penninger JM, Kroemer G, AIF and cyclophilin A cooperate in apoptosis-associated chromatinolysis. Oncogene 23, 1514–1521 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Zhu C, Wang X, Deinum J, Huang Z, Gao J, Modjtahedi N, Neagu MR, Nilsson M, Eriksson PS, Hagberg H, Luban J, Kroemer G, Blomgren K, Cyclophilin A participates in the nuclear translocation of apoptosis-inducing factor in neurons after cerebral hypoxia-ischemia. J. Exp. Med 204, 1741–1748 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Doti N, Reuther C, Scognamiglio PL, Dolga AM, Plesnila N, Ruvo M, Culmsee C, Inhibition of the AIF/CypA complex protects against intrinsic death pathways induced by oxidative stress. Cell Death Dis. 5, e993 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ravagnan L, Gurbuxani S, Susin SA, Maisse C, Daugas E, Zamzami N, Mak T, Jäättelä M, Penninger JM, Garrido C, Kroemer G, Heat-shock protein 70 antagonizes apoptosis-inducing factor. Nat. Cell Biol 3, 839–843 (2001). [DOI] [PubMed] [Google Scholar]
- 14.Gurbuxani S, Schmitt E, Cande C, Parcellier A, Hammann A, Daugas E, Kouranti I, Spahr C, Pance A, Kroemer G, Garrido C, Heat shock protein 70 binding inhibits the nuclear import of apoptosis-inducing factor. Oncogene 22, 6669–6678 (2003). [DOI] [PubMed] [Google Scholar]
- 15.Fressart V, Duthoit G, Donal E, Probst V, Deharo J-C, Chevalier P, Klug D, Dubourg O, Delacretaz E, Cosnay P, Scanu P, Extramiana F, Keller D, Hidden-Lucet F, Simon F, Bessirard V, Roux-Buisson N, Hebert J-L, Azarine A, Casset-Senon D, Rouzet F, Lecarpentier Y, Fontaine G, Coirault C, Frank R, Hainque B, Charron P, Desmosomal gene analysis in arrhythmogenic right ventricular dysplasia/cardiomyopathy: Spectrum of mutations and clinical impact in practice. Europace 12, 861–868 (2010). [DOI] [PubMed] [Google Scholar]
- 16.Pilichou K, Nava A, Basso C, Beffagna G, Bauce B, Lorenzon A, Frigo G, Vettori A, Valente M, Towbin J, Thiene G, Danieli GA, Rampazzo A, Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation 113, 1171–1179 (2006). [DOI] [PubMed] [Google Scholar]
- 17.Klune JR, Dhupar R, Cardinal J, Billiar TR, Tsung A, HMGB1: Endogenous danger signaling. Mol. Med 14, 476–484 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Opbergen CJM, Delmar M, van Veen TAB, Potential new mechanisms of pro-arrhythmia in arrhythmogenic cardiomyopathy: Focus on calcium sensitive pathways. Neth. Heart J 25, 157–169 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakayama H, Chen X, Baines CP, Klevitsky R, Zhang X, Zhang H, Jaleel N, Chua BHL, Hewett TE, Robbins J, Houser SR, Molkentin JD, Ca2+- and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. J. Clin. Invest 117, 2431–2444 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Inserte J, Hernando V, Garcia-Dorado D, Contribution of calpains to myocardial ischaemia/reperfusion injury. Cardiovasc. Res 96, 23–31 (2012). [DOI] [PubMed] [Google Scholar]
- 21.Ono Y, Sorimachi H, Calpains –– An elaborate proteolytic system. Biochim. Biophys. Acta 1824, 224–236 (2012). [DOI] [PubMed] [Google Scholar]
- 22.Cerrone M, Lin X, Zhang M, Agullo-Pascual E, Pfenniger A, Gusky HC, Novelli V, Kim C, Tirasawadichai T, Judge DP, Rothenberg E, Chen H-SV, Napolitano C, Priori SG, Delmar M, Missense mutations in plakophilin-2 cause sodium current deficit and associate with a Brugada syndrome phenotype. Circulation 129, 1092–1103 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen SN, Gurha P, Lombardi R, Ruggiero A, Willerson JT, Marian AJ, The hippo pathway is activated and is a causal mechanism for adipogenesis in arrhythmogenic cardiomyopathy. Circ. Res 114, 454–468 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garcia-Gras E, Lombardi R, Giocondo MJ, Willerson JT, Schneider MD, Khoury DS, Marian AJ, Suppression of canonical Wnt/β-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J. Clin. Invest 116, 2012–2021 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Q, Deng C, Rao F, Modi RM, Zhu J, Liu X, Mai L, Tan H, Yu X, Lin Q, Xiao D, Kuang S, Wu S, Silencing of desmoplakin decreases connexin43/Nav1.5 expression and sodium current in HL1 cardiomyocytes. Mol. Med. Rep 8, 780–786 (2013). [DOI] [PubMed] [Google Scholar]
- 26.Laugwitz KL, Moretti A, Weig HJ, Gillitzer A, Pinkernell K, Ott T, Pragst I, Stadele C, Seyfarth M, Schomig A, Ungerer M, Blocking caspase-activated apoptosis improves contractility in failing myocardium. Hum. Gene Ther 12, 2051–2063 (2001). [DOI] [PubMed] [Google Scholar]
- 27.Zingarelli B, Hake PW, O’Connor M, Denenberg A, Kong S, Aronow BJ, Absence of poly(ADP-ribose)polymerase-1 alters nuclear factor-kappa B activation and gene expression of apoptosis regulators after reperfusion injury. Mol. Med 9, 143–153 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yue TL, Wang C, Romanic AM, Kikly K, Keller P, De Wolf WE Jr., Hart TK, Thomas HC, Storer B, Gu JL, Wang X, Feuerstein GZ, Staurosporine-induced apoptosis in cardiomyocytes: A potential role of caspase-3. J. Mol. Cell. Cardiol 30, 495–507 (1998). [DOI] [PubMed] [Google Scholar]
- 29.Melloni E, Michetti M, Salamino F, Minafra R, Pontremoli S, Modulation of the calpain autoproteolysis by calpastatin and phospholipids. Biochem. Biophys. Res. Commun 229, 193–197 (1996). [DOI] [PubMed] [Google Scholar]
- 30.Blomgren K, Hallin U, Andersson AL, Puka-Sundvall M, Bahr BA, McRae A, Saido TC, Kawashima S, Hagberg H, Calpastatin is up-regulated in response to hypoxia and is a suicide substrate to calpain after neonatal cerebral hypoxia-ischemia. J. Biol. Chem 274, 14046–14052 (1999). [DOI] [PubMed] [Google Scholar]
- 31.Parr T, Sensky PL, Bardsley RG, Buttery PJ, Calpastatin expression in porcine cardiac and skeletal muscle and partial gene structure. Arch. Biochem. Biophys 395, 1–13 (2001). [DOI] [PubMed] [Google Scholar]
- 32.Goll DE, Thompson VF, Li H, Wei W, Cong J, The calpain system. Physiol. Rev 83, 731–801 (2003). [DOI] [PubMed] [Google Scholar]
- 33.De Tullio R, Averna M, Stifanese R, Parr T, Bardsley RG, Pontremoli S, Melloni E, Multiple rat brain calpastatin forms are produced by distinct starting points and alternative splicing of the N-terminal exons. Arch. Biochem. Biophys 465, 148–156 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Kumar T, Majumdar A, Das P, Sarafis V, Ghose M, Trypan blue as a fluorochrome for confocal laser scanning microscopy of arbuscular mycorrhizae in three mangroves. Biotech. Histochem 83, 153–159 (2008). [DOI] [PubMed] [Google Scholar]
- 35.Ricci J-E, Muñoz-Pinedo C, Fitzgerald P, Bailly-Maitre B, Perkins GA, Yadava N, Scheffler IE, Ellisman MH, Green DR, Disruption of mitochondrial function during apoptosis is mediated by caspase cleavage of the p75 subunit of complex I of the electron transport chain. Cell 117, 773–786 (2004). [DOI] [PubMed] [Google Scholar]
- 36.Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y, Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 434, 652–658 (2005). [DOI] [PubMed] [Google Scholar]
- 37.Chouchani ET, Pell VR, James AM, Work LM, Saeb-Parsy K, Frezza C, Krieg T, Murphy MP, A unifying mechanism for mitochondrial superoxide production during ischemia-reperfusion injury. Cell Metab. 23, 254–263 (2016). [DOI] [PubMed] [Google Scholar]
- 38.De J García-Rivas G, Carvajal K, Correa F, Zazueta C, Ru360, a specific mitochondrial calcium uptake inhibitor, improves cardiac post-ischaemic functional recovery in rats in vivo. Br. J. Pharmacol 149, 829–837 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim C, Wong J, Wen J, Wang S, Wang C, Spiering S, Kan NG, Forcales S, Puri PL, Leone TC, Marine JE, Calkins H, Kelly DP, Judge DP, Chen H-SV, Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature 494, 105–110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Del Bello B, Moretti D, Gamberucci A, Maellaro E, Cross-talk between calpain and caspase-3/−7 in cisplatin-induced apoptosis of melanoma cells: A major role of calpain inhibition in cell death protection and p53 status. Oncogene 26, 2717–2726 (2007). [DOI] [PubMed] [Google Scholar]
- 41.Khorchid A, Ikura M, How calpain is activated by calcium. Nat. Struct. Biol 9, 239–241 (2002). [DOI] [PubMed] [Google Scholar]
- 42.Wanichawan P, Hafver TL, Hodne K, Aronsen JM, Lunde IG, Dalhus B, Lunde M, Kvaløy H, Louch WE, Tønnessen T, Sjaastad I, Sejersted OM, Carlson CR, Molecular basis of calpain cleavage and inactivation of the sodium-calcium exchanger 1 in heart failure. J. Biol. Chem 289, 33984–33998 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Borkowski BJ, Cheema Y, Shahbaz AU, Bhattacharya SK, Weber KT, Cation dyshomeostasis and cardiomyocyte necrosis: The Fleckenstein hypothesis revisited. Eur. Heart J 32, 1846–1853 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kung G, Konstantinidis K, Kitsis RN, Programmed necrosis, not apoptosis, in the heart. Circ. Res 108, 1017–1036 (2011). [DOI] [PubMed] [Google Scholar]
- 45.Daugas E, Susin SA, Zamzami N, Ferri KF, Irinopoulou T, Larochette N, Prévost MC, Leber B, Andrews D, Penninger J, Kroemer G, Mitochondrio-nuclear translocation of AIF in apoptosis and necrosis. FASEB J. 14, 729–739 (2000). [PubMed] [Google Scholar]
- 46.Sevrioukova IF, Apoptosis-inducing factor: Structure, function, and redox regulation. Antioxid. Redox Signal 14, 2545–2579 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Richardson P, McKenna W, Bristow M, Maisch B, Mautner B, O’Connell J, Olsen E, Thiene G, Goodwin J, Gyarfas I, Martin I, Nordet P, Report of the 1995 World Health Organization/International society and federation of cardiology task force on the definition and classification of cardiomyopathies. Circulation 93, 841–842 (1996). [DOI] [PubMed] [Google Scholar]
- 48.Wang W, Orgeron G, Tichnell C, Murray B, Crosson J, Monfredi O, Cadrin-Tourigny J, Tandri H, Calkins H, James CA, Impact of exercise restriction on arrhythmic risk among patients with arrhythmogenic right ventricular cardiomyopathy. J. Am. Heart Assoc 7, e008843 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Powers SK, Jackson MJ, Exercise-induced oxidative stress: Cellular mechanisms and impact on muscle force production. Physiol. Rev 88, 1243–1276 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stanley BA, Sivakumaran V, Shi S, Donald IM, Lloyd D, Watson WH, Aon MA, Paolocci N, Thioredoxin reductase-2 is essential for keeping low levels of H2O2 emission from isolated heart mitochondria. J. Biol. Chem 286, 33669–33677 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Matsushima S, Ide T, Yamato M, Matsusaka H, Hattori F, Ikeuchi M, Kubota T, Sunagawa K, Hasegawa Y, Kurihara T, Oikawa S, Kinugawa S, Tsutsui H, Overexpression of mitochondrial peroxiredoxin-3 prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation 113, 1779–1786 (2006). [DOI] [PubMed] [Google Scholar]
- 52.Fisher-Wellman KH, Mattox TA, Thayne K, Katunga LA, La Favor JD, Neufer PD, Hickner RC, Wingard CJ, Anderson EJ, Novel role for thioredoxin reductase-2 in mitochondrial redox adaptations to obesogenic diet and exercise in heart and skeletal muscle. J. Physiol 591, 3471–3486 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ, Reactive oxygen species (ROS)-induced ROS release: A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med 192, 1001–1014 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yamamoto M, Yang G, Hong C, Liu J, Holle E, Yu X, Wagner T, Vatner SF, Sadoshima J, Inhibition of endogenous thioredoxin in the heart increases oxidative stress and cardiac hypertrophy. J. Clin. Invest 112, 1395–1406 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ghezzi D, Sevrioukova I, Invernizzi F, Lamperti C, Mora M, D’Adamo P, Novara F, Zuffardi O, Uziel G, Zeviani M, Severe X-linked mitochondrial encephalomyopathy associated with a mutation in apoptosis-inducing factor. Am. J. Hum. Genet 86, 639–649 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bano D, Prehn JHM, Apoptosis-Inducing Factor (AIF) in physiology and disease: The tale of a repented natural born killer. EBioMedicine 30, 29–37 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Delavallée L, Mathiah N, Cabon L, Mazeraud A, Brunelle-Navas M-N, Lerner LK, Tannoury M, Prola A, Moreno-Loshuertos R, Baritaud M, Vela L, Garbin K, Garnier D, Lemaire C, Langa-Vives F, Cohen-Salmon M, Fernández-Silva P, Chrétien F, Migeotte I, Susin SA, Mitochondrial AIF loss causes metabolic reprogramming, caspase-independent cell death blockade, embryonic lethality, and perinatal hydrocephalus. Mol Metab. 40, 101027 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Garcia-Roves PM, Huss J, Holloszy JO, Role of calcineurin in exercise-induced mitochondrial biogenesis. Am. J. Physiol. Endocrinol. Metab 290, E1172–E1179 (2006). [DOI] [PubMed] [Google Scholar]
- 59.Sun Y, Qi Z, He Q, Cui D, Qian S, Ji L, Ding S, The effect of treadmill training and N-acetyl-l-cysteine intervention on biogenesis of cytochrome c oxidase (COX). Free Radic. Biol. Med 87, 326–335 (2015). [DOI] [PubMed] [Google Scholar]
- 60.Li F-H, Li T, Ai J-Y, Sun L, Min Z, Duan R, Zhu L, Liu Y-Y, Liu TC-Y, Beneficial autophagic activities, mitochondrial function, and metabolic phenotype adaptations promoted by high-intensity interval training in a rat model. Front. Physiol 9, 571 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kadenbach B, Huttemann M, The subunit composition and function of mammalian cytochrome c oxidase. Mitochondrion 24, 64–76 (2015). [DOI] [PubMed] [Google Scholar]
- 62.Norberg E, Gogvadze V, Vakifahmetoglu H, Orrenius S, Zhivotovsky B, Oxidative modification sensitizes mitochondrial apoptosis-inducing factor to calpain-mediated processing. Free Radic. Biol. Med 48, 791–797 (2010). [DOI] [PubMed] [Google Scholar]
- 63.Ye H, Cande C, Stephanou NC, Jiang S, Gurbuxani S, Larochette N, Daugas E, Garrido C, Kroemer G, Wu H, DNA binding is required for the apoptogenic action of apoptosis inducing factor. Nat. Struct. Biol 9, 680–684 (2002). [DOI] [PubMed] [Google Scholar]
- 64.Zhang X, Chen J, Graham SH, Du L, Kochanek PM, Draviam R, Guo F, Nathaniel PD, Szabó C, Watkins SC, Clark RSB, Intranuclear localization of apoptosis-inducing factor (AIF) and large scale DNA fragmentation after traumatic brain injury in rats and in neuronal cultures exposed to peroxynitrite. J. Neurochem 82, 181–191 (2002). [DOI] [PubMed] [Google Scholar]
- 65.Farina B, Sturlese M, Mascanzoni F, Caporale A, Monti A, Sorbo GD, Fattorusso R, Ruvo M, Doti N, Binding mode of AIF(370–394) peptide to CypA: Insights from NMR, label-free and molecular docking studies. Biochem. J 475, 2377–2393 (2018). [DOI] [PubMed] [Google Scholar]
- 66.Rodriguez J, Xie C, Li T, Sun Y, Wang Y, Xu Y, Li K, Zhang S, Zhou K, Wang Y, Mallard C, Hagberg H, Doti N, Wang X, Zhu C, Inhibiting the interaction between apoptosis-inducing factor and cyclophilin A prevents brain injury in neonatal mice after hypoxia-ischemia. Neuropharmacology 171, 108088 (2020). [DOI] [PubMed] [Google Scholar]
- 67.Wencker D, Chandra M, Nguyen K, Miao W, Garantziotis S, Factor SM, Shirani J, Armstrong RC, Kitsis RN, A mechanistic role for cardiac myocyte apoptosis in heart failure. J. Clin. Invest 111, 1497–1504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mallat Z, Tedgui A, Fontaliran F, Frank R, Durigon M, Fontaine G, Evidence of apoptosis in arrhythmogenic right ventricular dysplasia. N. Engl. J. Med 335, 1190–1197 (1996). [DOI] [PubMed] [Google Scholar]
- 69.Runge MS, Stouffer GA, Sheahan RG, Yamamoto S, Tsyplenkova VG, James TN, Morphological patterns of death by myocytes in arrhythmogenic right ventricular dysplasia. Am. J. Med. Sci 320, 310–319 (2000). [DOI] [PubMed] [Google Scholar]
- 70.Saffitz JE, The pathobiology of arrhythmogenic cardiomyopathy. Annu. Rev. Pathol 6, 299–321 (2011). [DOI] [PubMed] [Google Scholar]
- 71.Candé C, Cohen I, Daugas E, Ravagnan L, Larochette N, Zamzami N, Kroemer G, Apoptosis-inducing factor (AIF): A novel caspase-independent death effector released from mitochondria. Biochimie 84, 215–222 (2002). [DOI] [PubMed] [Google Scholar]
- 72.Polster BM, Basañez G, Etxebarria A, Hardwick JM, Nicholls DG, Calpain I induces cleavage and release of apoptosis-inducing factor from isolated mitochondria. J. Biol. Chem 280, 6447–6454 (2005). [DOI] [PubMed] [Google Scholar]
- 73.Galvez AS, Diwan A, Odley AM, Hahn HS, Osinska H, Melendez JG, Robbins J, Lynch RA, Marreez Y, Dorn GW 2nd, Cardiomyocyte degeneration with calpain deficiency reveals a critical role in protein homeostasis. Circ. Res. 100, 1071–1078 (2007). [DOI] [PubMed] [Google Scholar]
- 74.Letavernier E, Perez J, Bellocq A, Mesnard L, de Castro Keller A, Haymann J-P, Baud L, Targeting the calpain/calpastatin system as a new strategy to prevent cardiovascular remodeling in angiotensin II-induced hypertension. Circ. Res 102, 720–728 (2008). [DOI] [PubMed] [Google Scholar]
- 75.Miyazaki T, Koya T, Kigawa Y, Oguchi T, Lei X-F, J.-r. Kim-Kaneyama, A. Miyazaki, Calpain and atherosclerosis. J. Atheroscler. Thromb 20, 228–237 (2013). [DOI] [PubMed] [Google Scholar]
- 76.Mani SK, Balasubramanian S, Zavadzkas JA, Jeffords LB, Rivers WT, Zile MR, Mukherjee R, Spinale FG, Kuppuswamy D, Calpain inhibition preserves myocardial structure and function following myocardial infarction. Am. J. Physiol. Heart Circ. Physiol 297, H1744–H1751 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mani SK, Shiraishi H, Balasubramanian S, Yamane K, Chellaiah M, Cooper G, Banik N, Zile MR, Kuppuswamy D, In vivo administration of calpeptin attenuates calpain activation and cardiomyocyte loss in pressure-overloaded feline myocardium. Am. J. Physiol. Heart Circ. Physiol 295, H314–H326 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Di Lisa F, Bernardi P, Mitochondria and ischemia-reperfusion injury of the heart: Fixing a hole. Cardiovasc. Res 70, 191–199 (2006). [DOI] [PubMed] [Google Scholar]
- 79.Bernardi P, Krauskopf A, Basso E, Petronilli V, Blachly-Dyson E, Di Lisa F, Forte MA, The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 273, 2077–2099 (2006). [DOI] [PubMed] [Google Scholar]
- 80.Guttmann RP, Johnson GV, Oxidative stress inhibits calpain activity in situ. J. Biol. Chem 273, 13331–13338 (1998). [DOI] [PubMed] [Google Scholar]
- 81.Hernando V, Inserte J, Sartório CL, Parra VM, Poncelas-Nozal M, Garcia-Dorado D, Calpain translocation and activation as pharmacological targets during myocardial ischemia/reperfusion. J. Mol. Cell. Cardiol 49, 271–279 (2010). [DOI] [PubMed] [Google Scholar]
- 82.Chelko SP, Asimaki A, Lowenthal J, Bueno-Beti C, Bedja D, Scalco A, Amat-Alarcon N, Andersen P, Judge DP, Tung L, Saffitz JE, Therapeutic modulation of the immune response in arrhythmogenic cardiomyopathy. Circulation 140, 1491–1505 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mikosik A, Jasiulewicz A, Daca A, Henc I, Frąckowiak JE, Ruckemann-Dziurdzińska K, Foerster J, Page AL, Bryl E, Fulop T, Witkowski JM, Roles of calpain-calpastatin system (CCS) in human T cell activation. Oncotarget 7, 76479–76495 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G, Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 397, 441–446 (1999). [DOI] [PubMed] [Google Scholar]
- 85.Wang Y, Kim NS, Li X, Greer PA, Koehler RC, Dawson VL, Dawson TM, Calpain activation is not required for AIF translocation in PARP-1-dependent cell death (parthanatos). J. Neurochem 110, 687–696 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yu S-W, Andrabi SA, Wang H, Kim NS, Poirier GG, Dawson TM, Dawson VL, Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc. Natl. Acad. Sci. U.S.A 103, 18314–18319 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yuste VJ, Moubarak RS, Delettre C, Bras M, Sancho P, Robert N, d’Alayer J, Susin SA, Cysteine protease inhibition prevents mitochondrial apoptosis-inducing factor (AIF) release. Cell Death Differ. 12, 1445–1448 (2005). [DOI] [PubMed] [Google Scholar]
- 88.Chen Q, Paillard M, Gomez L, Ross T, Hu Y, Xu A, Lesnefsky EJ, Activation of mitochondrial μ-calpain increases AIF cleavage in cardiac mitochondria during ischemia-reperfusion. Biochem. Biophys. Res. Commun 415, 533–538 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tanaka T, Hosoi F, Yamaguchi-Iwai Y, Nakamura H, Masutani H, Ueda S, Nishiyama A, Takeda S, Wada H, Spyrou G, Yodoi J, Thioredoxin-2 (TRX-2) is an essential gene regulating mitochondria-dependent apoptosis. EMBO J. 21, 1695–1703 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Benetti R, Copetti T, Dell’Orso S, Melloni E, Brancolini C, Monte M, Schneider C, The calpain system is involved in the constitutive regulation of β-catenin signaling functions. J. Biol. Chem 280, 22070–22080 (2005). [DOI] [PubMed] [Google Scholar]
- 91.Goñi-Oliver P, Lucas JJ, Avila J, Hernández F, N-terminal cleavage of GSK-3 by calpain: A new form of GSK-3 regulation. J. Biol. Chem 282, 22406–22413 (2007). [DOI] [PubMed] [Google Scholar]
- 92.Kimura T, Sharma G, Ishiguro K, S.-i. Hisanaga, Phospho-tau bar code: Analysis of phosphoisotypes of tau and its application to tauopathy. Front. Neurosci 12, 44 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW, Ziman BD, Wang S, Ytrehus K, Antos CL, Olson EN, Sollott SJ, Glycogen synthase kinase-3β mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J. Clin. Invest 113, 1535–1549 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yeste-Velasco M, Folch J, Jiménez A, Rimbau V, Pallàs M, Camins A, GSK-3β inhibition and prevention of mitochondrial apoptosis inducing factor release are not involved in the antioxidant properties of SB-415286. Eur. J. Pharmacol 588, 239–243 (2008). [DOI] [PubMed] [Google Scholar]
- 95.Bénit P, Goncalves S, Dassa EP, Brière J-J, Rustin P, The variability of the Harlequin mouse phenotype resembles that of human mitochondrial-complex I-deficiency syndromes. PLOS ONE 3, e3208 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Joza N, Oudit GY, Brown D, Bénit P, Kassiri Z, Vahsen N, Benoit L, Patel MM, Nowikovsky K, Vassault A, Backx PH, Wada T, Kroemer G, Rustin P, Penninger JM, Muscle-specific loss of apoptosis-inducing factor leads to mitochondrial dysfunction, skeletal muscle atrophy, and dilated cardiomyopathy. Mol. Cell. Biol 25, 10261–10272 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ni R, Zheng D, Xiong S, Hill DJ, Sun T, Gardiner RB, Fan GC, Lu Y, Abel ED, Greer PA, Peng T, Mitochondrial calpain-1 disrupts ATP synthase and induces superoxide generation in type 1 diabetic hearts: A novel mechanism contributing to diabetic cardiomyopathy. Diabetes 65, 255–268 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Galluzzi L, Kepp O, Chan FK-M, Kroemer G, Necroptosis: Mechanisms and relevance to disease. Annu. Rev. Pathol 12, 103–130 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Syed F, Diwan A, Hahn HS, Murine echocardiography: A practical approach for phenotyping genetically manipulated and surgically modeled mice. J. Am. Soc. Echocardiogr 18, 982–990 (2005). [DOI] [PubMed] [Google Scholar]
- 100.Boukens BJ, Rivaud MR, Rentschler S, Coronel R, Misinterpretation of the mouse ECG: ‘Musing the waves of Mus musculus’. J. Physiol 592, 4613–4626 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Shenje LT, Andersen P, Uosaki H, Fernandez L, Rainer PP, Cho G.-s., Lee D.-i., Zhong W, Harvey RP, Kass DA, Kwon C, Precardiac deletion of Numb and Numblike reveals renewal of cardiac progenitors. eLife 3, e02164 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Claycomb WC, Lanson NA Jr., Stallworth BS, Egeland DB, Delcarpio JB, Bahinski A, Izzo NJ Jr., HL-1 cells: A cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc. Natl. Acad. Sci. U.S.A 95, 2979–2984 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Johnson GVW, Guttmann RP, Calpains: Intact and active? Bioessays 19, 1011–1018 (1997). [DOI] [PubMed] [Google Scholar]
- 104.Di Lisa F, Blank PS, Colonna R, Gambassi G, Silverman HS, Stern MD, Hansford RG, Mitochondrial membrane potential in single living adult rat cardiac myocytes exposed to anoxia or metabolic inhibition. J. Physiol 486, 1–13 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sims NR, Rapid isolation of metabolically active mitochondria from rat brain and subregions using Percoll density gradient centrifugation. J. Neurochem 55, 698–707 (1990). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data file S1. Raw data.
Fig. S1. Dsg2mut/mut mice display ECG abnormalities and myocardial injury in response to endurance exercise.
Fig. S2. CAPN1 activation and cell death in response to Ca2+ overload.
Fig. S3. CAPN1 inhibition in HL-1 cells by PD150606.
Fig. S4. CytC in Dsg2mut/mut mice and posttranslational modifications of AIF.
Fig. S5. AIF localization in ACM patient myocardium.
Fig. S6. Dsg2mut/mut mice display reduced COX IV abundance and aberrant myocardial localization.
Fig. S7. Labeled cysteines and DNA gel electrophoresis.
Fig. S8. HSP70 and PPIA abundance in ACM myocytes.
Fig. S9. Increased AIF nuclear localization and nuclear loss of HMGB1 in ACM ES-CMs.
Fig. S10. Graphical abstract of exercise-induced, CAPN1/PPIA-mediated AIF-nuclear import in ACM.
Table S1. Echocardiographic and electrocardiographic indices from exercised WT and Dsg2mut/mut mice.
Table S2. Desmosomal gene variants in patients with ACM.
Table S3. Clinical characteristics and AIF pathology scores of patients with ACM.