

Abstract
Obesity has become a global epidemic in the modern world with the numbers of obese individuals having risen at alarming rates in the last decades. Obesity represents a serious medical condition that can lead to multiple complications, such as diabetes, dyslipidemia, cardiovascular disease including hypertension and atherosclerosis, stroke and increases in the risk of many types of cancer. Very few effective options exist to treat obesity, with many removed from the market due to associated complications. Obesity and metabolic syndrome display a sexual dichotomy, with (premenopausal) females displaying protection from weight gain and metabolic dysfunction compared to men. These beneficial effects are generally attributed to a class of female ovarian hormone, estrogens, which exert pleiotropic effects in multiple metabolic tissues, such as adipose, skeletal muscle, liver and pancreas. Multiple receptors mediate the actions of estrogens, including the classical nuclear estrogen receptors (ER α and ER β) and the G protein-coupled estrogen receptor (GPER). While the roles of nuclear ERs are more established, evidence of GPER function in metabolic homeostasis is still emerging. In this review, we will discuss the latest advances concerning the contributions of GPER towards obesity and metabolism utilizing GPER-selective pharmacological (agonists or antagonists) or genetic (GPER knock out mice or cells) tools. We present evidence that GPER regulates body weight, fat distribution, inflammation and glucose and lipid homeostasis via effects on metabolic tissues. Selective agonism of GPER by its agonist G-1 can alleviate symptoms of obesity and metabolic dysfunction in multiple murine models, thereby limiting weight gain, reducing insulin resistance and inflammation and improving glucose and lipid homeostasis in vivo. Thus, GPER represents a novel therapeutic target, with G-1 a first-in-class therapeutic agent, to treat obesity and its associated comorbidities, including diabetes.
Keywords: Estrogen, GPER, metabolism, obesity, diabetes, inflammation
1. Introduction: Obesity, metabolism and sex differences
Obesity and its pathophysiological consequences are increasing at alarming rates worldwide and pose a serious public health challenge (World Health Organization, 2020c; Flegal et al., 2010; Guh et al., 2009). Current estimates reveal that in United States alone more than 65% of population is either obese or overweight (Centers for Disease Control and Prevention, 2020b). Obesity is a major risk factor for metabolic syndrome, which is characterized by high blood sugar, visceral adiposity, high blood pressure and abnormal cholesterol/triglyceride levels (Alberti et al., 2006; Pantalone et al., 2017). Modern day obesity results from multiple factors ranging from genetic, behavioral and environmental, the primary ones being high calorie diets coupled with a sedentary life style (Centers for Disease Control and Prevention, 2020a; McAllister et al., 2009). Obesity leads to inflammation, insulin resistance and ectopic fat deposition and interferes with normal functions of metabolic tissues (Catoi et al., 2015; Mittendorfer, 2011; Shulman, 2014). These conditions can lead to increased incidences of diabetes, heart disease, stroke, as well as certain forms of cancer (Basen-Engquist and Chang, 2011; Eckel and Krauss, 1998; Kernan and Dearborn, 2015; Poirier et al., 2006). The estimated economic burden of obesity and its comorbidities exceeds 200 billion dollars per year (Apovian, 2016; Hammond and Levine, 2010). Thus, it is important to determine the underlying mechanisms involved in the development of obesity and its associated complications and identify novel molecular targets and therapeutic agents that can prevent or limit these metabolic abnormalities.
Metabolic homeostasis is differentially regulated in males and females as evidenced by both murine models and human observations and studies. Premenopausal women display lower incidences of obesity, diabetes and cardiovascular disease compared to age-matched men, with this protection lost in females following menopause (Garaulet et al., 2002; Kotani et al., 1994; Kozakowski et al., 2017; Nakhjavani et al., 2014; Regitz-Zagrosek et al., 2006; Tandon et al., 2010). After menopause, women exhibit increased propensities to gain weight (specifically in the visceral area), develop insulin resistance and display dysfunctional glucose and lipid homeostasis. Furthermore, after the onset of menopause, besides an increase in overall fat deposition, there is also a shift in the primary site of fat deposition (Proietto, 2017; Stefan, 2020). Premenopausal women display “gynoid” or subcutaneous fat deposition in the lower body whereas following menopause, fat deposition in women is more like that of men, namely an “android” distribution, primarily in the central body around the abdomen (Blaak, 2001; Regitz-Zagrosek et al., 2006). Android pattern fat preferentially promotes inflammation and insulin resistance, thereby increasing the risk of metabolic and cardiovascular disease with age (Fujioka et al., 1987; Lee et al., 2009; Schmidt et al., 2015; Stefan et al., 2017). Mouse models of metabolic dysfunction, such as ovariectomy (OVX) and diet-induced obesity (DIO), further confirm the metabolic disparities observed between males and females. When fed a high fat diet (HFD), male mice display rapid weight gain whereas female mice are protected against this weight gain (Hong et al., 2009; Ingvorsen et al., 2017). However, following ovariectomy (mimicking human menopause), female mice also exhibit central weight gain and develop metabolic dysfunction (Hong et al., 2009; Ingvorsen et al., 2017). Hormone replacement therapy in postmenopausal women, as well as in ovariectomized female mice, can alleviate weight gain and its deleterious metabolic effects (Bonds et al., 2006; Gurney et al., 2014; Stubbins et al., 2012a; Stubbins et al., 2012b). In human and rodent females, these protective effects on weight and metabolism are primarily attributed to estrogens. Role(s) of estrogens in metabolism are still poorly understood and therefore dissecting the underlying mechanisms of estrogenic action on metabolism and identifying novel therapeutic agents that mimic its actions are of considerable importance.
2. Estrogens, estrogen receptors and GPER
The female sex hormone estrogen plays a key role in the sexual and reproductive development of females (Deroo and Korach, 2006; Hewitt et al., 2016; Prossnitz et al., 2008). Of the various forms of estrogens (e.g estrone, estradiol, estriol), 17 β-estradiol (E2) is the most potent. Besides its classic roles in reproductive functions, it exerts protective effects on metabolism via central and peripheral effects in premenopausal females (Deroo and Korach, 2006; Mauvais-Jarvis, 2011; Wang and Xu, 2019). E2 can modulate food intake and energy expenditure through the central nervous system and also directly affect the physiology and function of peripheral metabolic tissues, such as adipose, skeletal muscle, liver, pancreas, as well as immune cells (Mauvais-Jarvis, 2011; 2015; Meyer et al., 2011). E2 regulates the amount and site of fat distribution in the body, promoting subcutaneous or gynoid fat deposition and suppressing lipogenesis in visceral organs (Eaton and Sethi, 2019; Mauvais-Jarvis, 2015). In addition, E2 maintains glucose and lipid homeostasis by promoting an anti-inflammatory phenotype, which enhances glucose-induced insulin secretion, increases insulin sensitivity and lowers systemic lipid levels (Gupte et al., 2015; Shen et al., 2014; Stubbins et al., 2012a; Stubbins et al., 2012b). As discussed above, postmenopausal women and OVX mice exhibit common attributes under E2-deficient conditions, leading to multiple aspects of metabolic dysfunction (Stubbins et al., 2012a). E2 supplementation attenuates these conditions both in humans and mice (Lundholm et al., 2008; Mauvais-Jarvis, 2015). Surprisingly, the sexual dimorphism of male mice, with an increased propensity to gain weight on calorie-rich diet, is sensitive to E2 an its mimetics (Hong et al., 2009; Mauvais-Jarvis, 2015; Murata et al., 2002).
Estrogens regulate metabolism through a complex interplay between multiple tissues and its actions through multiple receptors. Traditionally, effects of E2 have largely been attributed to the nuclear estrogen receptors (ER α and β), which act as ligand-activated transcription factors to facilitate changes in gene expression (Hewitt and Korach, 2018; Prossnitz et al., 2008; Prossnitz and Barton, 2011). In addition to these genomic responses, E2 can activate rapid non-genomic responses through extra-nuclear ERs and the G protein-coupled estrogen receptor (GPER, previously known as GPR30). GPER is a 7-transmembrane G protein-coupled receptor (GPCR), located predominantly in intracellular membranes, particularly those of the endoplasmic reticulum and Golgi apparatus, as well as the plasma membrane. In addition to activating classical GPCR-mediated pathways (e.g. adenylyl cyclase, kinases and ion channels), GPER can also mediate long term effects by indirectly regulating expression of target genes, secondary to rapid signaling (Prossnitz and Hathaway, 2015). The culmination of evidence over the last 15–20 years has revealed that GPER is a novel estrogen receptor that mediates both rapid and genomic responses. GPER is expressed in a variety of cell and tissue types, such as reproductive and metabolic organs and tissues, the CNS, cardiovascular and immune cells, and is thus functionally implicated in numerous physiological and pathophysiological conditions, including cancer, metabolism, immune regulation, and cardiovascular, reproductive and nervous systems (Prossnitz and Barton, 2014; Prossnitz and Hathaway, 2015). Although ERs have well described roles in metabolic homeostasis, the significance of GPER-mediated effects has only been uncovered more recently (Barros and Gustafsson, 2011; Prossnitz and Hathaway, 2015). Interestingly, in murine models, loss of ER α or GPER leads to similar pathophysiological consequences such as obesity, abnormal glucose/lipid homeostasis and inflammation, although with differential severities, suggesting a potential partial overlap in the mechanisms of action of both the receptors (Barros and Gustafsson, 2011; Prossnitz and Hathaway, 2015; Sharma et al., 2018; Sharma and Prossnitz, 2016; 2017). An important distinction, however, exists in reproductive capacity as ER α-deficient female mice are sterile, whereas GPER-deficient female mice are fertile. Nuclear ERs and GPER may activate differential pathways in a synergistic or antagonistic manner to regulate metabolic signaling and in the absence of one receptor, the other may partially compensate for its physiological role. Thus, dissecting the mechanisms of action of these different receptors to address the individual contribution of a given receptor remains of great significance.
Because E2 binds non-selectively to all its receptors, both genetic and pharmacologic tools have been developed to differentiate between the activity of GPER and classical ERs. Discovery of GPER-selective pharmacological agents, the agonist G-1 (Bologa et al., 2006) and antagonists G15 (Dennis et al., 2009) and G36 (Dennis et al., 2011), has made it possible to probe GPER function with high specificity, as these compounds have very low affinity for nuclear ERs (Prossnitz and Barton, 2014; Prossnitz and Hathaway, 2015). These ligands have been used for both in vitro as well as in vivo studies, ranging from acute to chronic exposures. The importance of ligand specificity and selectivity in experimental interpretation, as well as potential clinical applications, is exemplified by selective ER modulators (SERMs), such as tamoxifen or raloxifene, and selective ER downregulators (SERDs, e.g. fulvestrant), that have been shown to act as GPER agonists (Prossnitz and Arterburn, 2015; Sharma and Prossnitz, 2017).
To complement pharmacological approaches, genetically modified mice lacking GPER (GPER KO mice) have been developed to investigate the physiological or pathophysiological significance of GPER in vivo. Genetic and pharmacological approaches are often complementary, with the former revealing roles of the protein under normal conditions or in disease models, and the latter testing the consequence of continued activation of the endogenous protein (Prossnitz and Barton, 2014; Prossnitz and Hathaway, 2015). GPER KO mice, or cells and tissues obtained from them, lack responsiveness to the GPER-selective agonist G-1, highlighting its specificity and lack of off-target effects, as well as to a subset of E2-mediated effects, further confirming the role of GPER as an E2 receptor in vivo. In addition, the physiological effects of E2 in ER αβ double KO mice to maintain glucose homeostasis further support an integral role of GPER in estrogen biology (Liu et al., 2009). Taken together, these experimental systems have revealed much regarding the complex roles of these multiple receptors in physiology and disease.
3. Roles of GPER in obesity, fat deposition and energy homeostasis
Studies from our group and others have highlighted the importance of GPER in maintaining overall body weight and energy balance, as well as overall body fat content and the site of fat deposition (Sharma and Prossnitz, 2017). The first studies of body weight regulation by GPER were reported in 2009 by two independent groups. Whereas Haas et al. observed increased body weight and visceral adiposity in both male and female GPER-deficient mice compared to WT mice (Haas et al., 2009), Mårtensson et al. observed lower body weights in female GPER KO mice vs. WT mice, with no body weight changes in males (Mårtensson et al., 2009). Differences in the phenotype of GPER KO mice could result from the method used to create the knockout mouse, differences in chow, bedding or environmental factors (Prossnitz and Hathaway, 2015). In 2013, we reported that male GPER KO mice were heavier at ages ranging from 6 to 24 months (Sharma et al., 2013). Male GPER KO mice also exhibited increased overall fat content, both in the visceral as well as subcutaneous depots, and higher circulating lipids without any changes in food intake or locomotor activity (Sharma et al., 2013).
Subsequently, Clegg and colleagues reported adiposity in both the sexes of GPER KO mice (Davis et al., 2014). Surprisingly, they observed sex-specific differences in the onset of adiposity as well as satiety. Whereas GPER KO males were significantly heavier at 8 weeks, females displayed adiposity relative to WT controls only by 16 weeks. They further confirmed our observations of increased body weight in GPER KO mice in the absence of changes in food intake or activity and observed decreased energy expenditure in GPER KO mice (Davis et al., 2014; Sharma et al., 2013). Expression of the thermogenic genes uncoupling protein 1 (Ucp1) and β3 -adrenergic receptor was reduced in the brown adipose tissue (BAT) from GPER KO mice. Furthermore, female GPER KO mice exhibited a lower sensitivity to leptin-mediated food intake and cholecystokinin-mediated satiety signals, whereas males revealed no differences (Davis et al., 2014). Investigation into the mechanisms of these differences revealed diminished ERK activation in female hypothalamus of GPER KO mice compared to WT controls. ERK activation in hypothalamus is suggested to be a positive regulator of energy balance (Davis et al., 2014). Contrary to these multiple reports, a single study has reported a decrease in the body weights and increased energy expenditure in GPER KO mice fed a HFD (Wang et al., 2016). The reasons for these latter contrasting results remain unclear. In summary however, the majority of studies suggest that GPER deficiency increases body weight and decreases energy expenditure.
Contrary to determining the physiological role(s) of GPER through genetic approaches, we recently employed a pharmacological strategy to investigate the role of GPER in weight regulation and energy balance. We utilized multiple murine mouse models of obesity, including DIO, where mice are fed a HFD, and OVX mice, where surgical removal of the ovaries produces a significant reduction in endogenous E2 levels. Both models result in obesity and metabolic dysfunction in mice. Our hypothesis that chronic activation of GPER by G-1 in these models might mitigate obesity and improve metabolic function was validated as treatment of obese OVX and DIO mice with the GPER-selective agonist G-1 attenuated adiposity both in males and females (Sharma et al., 2020). However, differential effects on weight patterns of DIO males and females were observed. Male mice, upon G-1 treatment, stopped gaining weight compared to vehicle controls but female mice (on both normal chow and HFD) lost weight following G-1 treatment. In line with previous results (Dennis et al., 2009), unlike E2, G-1 treatment in OVX mice did not yield feminizing effects as it did not increase uterine wet weights. Furthermore, levels of circulating lipids were lowered in both male and female G-1-treated mice (Sharma et al., 2020).
OVX mice exhibited an increase in the weight of visceral fat depots, including gonadal, perirenal and mesenteric fat pads, all of which were reduced following G-1 treatment (Sharma et al., 2020). Analysis of body composition by dual-energy X-ray absorptiometry (DEXA) demonstrated a decrease in overall body fat content as well as body fat percentage in OVX mice treated with G-1, without any changes in lean mass or bone density and mineral content (Sharma et al., 2020). Both ovariectomy and HFD resulted in a decrease in energy expenditure, an effect that was reversed by G-1, during both the light and dark cycles. No changes in food intake or activity levels were observed. Investigations into the mechanisms of G-1-mediated actions on weight and energy balance revealed an increase in the expression of thermogenic gene Ucp1 and mitochondrial genes, such as peroxisome proliferator-activated receptor-gamma coactivator 1 α (Ppargc1), acetyl co-A carboxylase (Acaca), nuclear respiratory factor (Nrf1) and transcriptional factor A mitochondrial (Tfam1) in BAT from OVX mice. These genes regulate oxidative metabolism and maintain glucose and lipid homeostasis and energy balance, suggesting increased fuel utilization, in particular through β-oxidation, upon G-1 treatment. In addition, expression of tyrosine hydroxylase (Th), an important enzyme in norepinephrine synthesis, and thus a marker for sympathetic innervation was higher in the BAT from OVX mice treated with G-1 compared to vehicle-treated controls. These results suggest that GPER activation may regulate BAT thermogenesis in part through mechanisms involving the central nervous system (Sharma et al., 2020). Interestingly, a recent study by Butler et al. reported an acute decrease in food intake in OVX rats treated with PPT and G-1, as selective activators of ER α and GPER, respectively (Butler et al., 2018). Within 1 hour of treatment with PPT or G-1, food intake decreased, with PPT reducing feeding for a longer time compared to G-1. Furthermore, the authors reported that GPER activation appeared necessary for the rapid anorexiogenic effects of PPT, suggesting a possible synergy in the function of ER α and GPER (Butler et al., 2018) or the fact that PPT has also been shown to act as an agonist of GPER (Petrie et al., 2013). These observations indicate that GPER may counteract obesity by increasing BAT thermogenesis and energy expenditure as well as modulating short term food intake.
Obesity also results in insulin resistance and dyslipidemia, which in turn adversely affect the functions of metabolic tissues, including decreased insulin-stimulated glucose uptake in skeletal muscle, increased hepatic glucose production, increased triglyceride and non-esterified fatty acid release from adipose tissue and reduced insulin secretion from pancreatic β-cells, resulting in hyperglycemia (Bays et al., 2013; Consitt et al., 2009; Shoelson et al., 2007; Shulman, 2014; Tchernof and Despres, 2013). GPER is widely expressed in these metabolically active tissues. In adipose tissue, GPER expression is higher in female vs. male mice (Davis et al., 2014). GPER is also expressed in 3T3-L1 preadipocytes, where its expression increases with differentiation (Zhu et al., 2013). Activation of GPER by E2 or G-1 in 3T3-L1 cells during adipogenesis blocked lipid accumulation. During adipocyte differentiation, preadipocytes first undergo mitotic clonal expansion, then arrest at the G 1 growth phase of cell cycle and initiate the process of adipogenesis. However, following GPER activation, 3T3-L1 cells continued to divide even after 2 days of initiation of differentiation, failing to arrest in G 0 /G 1 state and increasing the expression of cell-cycle regulating factors such as CDK4,CDK6 and cyclinD. Thus, GPER inhibited lipid accumulation in adipocytes in part by preventing cell cycle arrest and subsequent differentiation (Zhu et al., 2013). Interestingly, with adipocyte differentiation, GPER expression increases but expression of classical ERs decreases, leading to the speculation that classical ERs may play a role during differentiation whereas GPER may have a more predominant role in mature adipocytes.
Mice lacking GPER display increased adiposity, as revealed by larger visceral fat pads and larger adipocytes compared to WT mice (Davis et al., 2014; Haas et al., 2009; Sharma et al., 2013). Lipid-rich visceral adipocytes are associated with the secretion of pro-inflammatory cytokines, hypoxia and mitochondrial dysfunction, conditions that are mitigated with decreased adipocyte size resulting in an anti-inflammatory state and improved insulin sensitivity (Woo et al., 2019; Ye, 2009). Our studies on G-1 treated OVX mice revealed decreased gonadal fat pad weight with a concomitant decrease in systemic inflammatory markers (Sharma et al., 2020). Furthermore, gonadal WAT demonstrated an increase in the expression of genes involved in mitochondrial biogenesis and fatty acid oxidation and a reduction in the genes involved in hypoxia and angiogenesis. We also observed an increase in the expression of mitochondrial markers and fatty acid oxidation in skeletal muscle and BAT obtained from G-1-treated OVX mice. In addition, real-time cellular metabolic studies revealed an increase in both basal and maximal respiration in brown preadipocytes upon acute (24 hour) G-1 treatment. In OVX mice, G-1 treatment may promote fatty acid oxidation in WAT resulting in smaller adipocytes, abrogation of hypoxia, a decrease in the secretion of pro-inflammatory cytokines and the metabolic hormones resistin and leptin (Balistreri et al., 2010; Sharma et al., 2020; Ye, 2009). An anti-inflammatory environment can improve insulin sensitivity, increase energy expenditure and improve glucose homeostasis as observed in OVX mice treated with G-1 (de Luca and Olefsky, 2008; Rogers et al., 2009). Thus, our results demonstrate that GPER re-programs WAT to alleviate hypoxia, promote anti-inflammatory phenotype and fatty acid oxidation to decrease obesity and improve metabolic phenotype.
G-1 also mimics E2 in upregulating the expression of mitochondrial genes to promote fat oxidation in WAT (D’Eon et al., 2005; Sharma et al., 2020). Our observations are consistent with reports in which postmenopausal women displayed reduced fat oxidation while hormone replacement resulted in lower serum leptin levels (Abildgaard et al., 2013; Di Carlo et al., 2000). Similarly, treatment of DIO mice with E2 increased lipid oxidation to reduce obesity (D’Eon et al., 2005). Cell-based studies revealed that in 3T3-L1 adipocytes treated with IL6 to induce inflammation-mediated mitochondrial dysfunction, treatment with E2 or G-1 attenuated the adverse effects of inflammation on mitochondrial function through activation of protein kinase A by ER α and GPER, thereby increasing mitochondrial biogenesis and oxygen consumption (Bauza-Thorbrugge et al., 2019). Interestingly, in another model of GPER KO mice, GPER-lacZ mice (mutant mouse that harbors a β-galactosidase (lacZ) reporter within the Gper locus, disrupting Gper expression), female but not male GPER KO mice exhibited increased lipid accumulation in the liver along with decreased circulating HDL levels compared to WT mice (Meoli et al., 2014). Consistent with this, in a human cohort of the Northern European descent, individuals carrying a hypofunctional P16L genetic variant of GPER have increased plasma LDL cholesterol. Finally, in HepG2 liver cells, expression of the LDL receptor was increased upon activation of GPER with G-1, an effect blocked by both GPER antagonist G15 and knockdown of GPER expression (Hussain et al., 2015). In conclusion, GPER regulates body weight, energy expenditure and fat deposition in various tissues to maintain metabolic health.
4. GPER functions in diabetes and glucose homeostasis
Emerging evidence supports extensive roles for GPER in glucose homeostasis in vivo. Mårtensson et al. showed disparate characteristics in glucose management between female and male GPER KO mice in 2009 (Mårtensson et al., 2009). Six-month-old female GPER KO mice exhibited higher plasma glucose, impaired glucose tolerance and defective glucose-stimulated insulin secretion compared to WT controls. Male mice however did not reveal any differences between genotypes. Furthermore, E2-stimulated increases in serum insulin levels in OVX mice were abrogated in GPER KO mice (Mårtensson et al., 2009). Mauvais-Jarvis et al. reported that in a model of type I diabetes, female GPER KO mice exposed to streptozotocin (STZ) displayed decreased pancreatic insulin content, a loss of pancreatic β-cells and higher blood glucose levels compared to their WT counterparts, whereas male GPER KO mice revealed no such differences (Liu and Mauvais-Jarvis, 2009). In addition, they demonstrated that the severity of diabetes worsened in ER αβ double KO mice following ovariectomy but that E2 supplementation could still mitigate these symptoms. These observations suggested that E2 could manifest its antidiabetic effects in the absence of ER α and ER β, perhaps through another receptor, such as GPER (Liu and Mauvais-Jarvis, 2009). In 2013, we reported the characterization of male GPER KO mice from 6–24 months of age (Sharma et al., 2013). At 6 months of age, male GPER KO mice were heavier than their WT type counterparts but displayed normal glucose tolerance despite being insulin resistant. With increasing age, their insulin resistance worsened and male GPER KO mice exhibited glucose intolerance only after 12 months of age. Although fasting glucose levels were normal in GPER KO mice, they did exhibit higher fasting insulin levels than WT mice, clearly demonstrating the presence of insulin resistance, requiring elevated insulin levels to maintain normal blood glucose levels. We also observed higher levels of systemic inflammatory markers (IL6, TNF α and IL1 β), higher plasma lipids (cholesterol and triglycerides) and lower adiponectin levels, which cumulatively, along with insulin resistance, could result in abnormal glucose homeostasis (Sharma et al., 2013). In 2014, Davis et al. confirmed these results and reported increased systemic levels of the inflammatory marker SAA3 and a decrease in adiponectin levels in both sexes of GPER KO mice (Davis et al., 2014). Furthermore, they demonstrated that while WT OVX mice responded to chronic E2 supplementation and displayed improved glucose homeostasis, GPER KO mice failed to do so. E2-treated OVX GPER KO mice exhibited delayed glucose clearance from blood during glucose tolerance tests compared to the WT mice, suggesting that GPER plays a key role in E2-mediated glucose homeostasis in vivo (Davis et al., 2014).
The pancreas, specifically β-cells in pancreatic islets, are critical to maintaining physiological glucose levels. Numerous studies have reported GPER-mediated regulation of islet function, including hormone release and cell survival, specifically that of β-cells (Liu and Mauvais-Jarvis, 2010). GPER is expressed in pancreatic islets with females exhibiting higher expression than males (Kumar et al., 2011). Islets from both male and female GPER KO mice exhibited reduced basal and E2-induced insulin secretion compared to their WT counterparts (Mårtensson et al., 2009). Furthermore, compared to WT controls, islets from female GPER KO mice exhibited a greater reduction in insulin secretion compared to islets obtained from male GPER KO mice. E2 treatment in OVX GPER KO mice did not increase serum insulin levels but islets from these mice exhibited higher pancreatic insulin content (presumably via ER α), suggesting that GPER may have a more prominent role in insulin secretion (Mårtensson et al., 2009). Stimulation of murine pancreatic islets as well as those from Type 2 diabetic patients with the GPER-selective agonist G-1 and E2 exerted antidiabetic effects by improving glucose-stimulated insulin release while suppressing glucagon and somatostatin secretion (Balhuizen et al., 2010; Kumar et al., 2011). Our previous studies in MIN6 cells (a widely used model to study β-cell function) showed that E2- or G-1-mediated insulin secretion was inhibited by the GPER-selective antagonist G15. Furthermore, insulin secretion in response to E2 or G-1 stimulation in WT islets was abolished in islets from GPER KO mice (Sharma and Prossnitz, 2011).
Insulin resistance, obesity and ectopic fat deposition can interfere with pancreatic function, leading to hyperinsulinemia due to compensation in islets (Chadt et al., 2000). Eventually, the toxic milieu, a result of inflammation, glucolipotoxicity and oxidative stress, leads to β-cell death and diabetes (Chadt et al., 2000). Stimulation of GPER by E2 or G-1 activates pro-survival pathways and protects β-cells from apoptosis in murine and human islets, as well as in MIN6 cells (Balhuizen et al., 2010; Kumar et al., 2011; Liu and Mauvais-Jarvis, 2009). GPER activates multiple signaling pathways to mediate insulin release and β-cell survival. With regard to insulin secretion, GPER activates both cAMP/PKA and PLC/IP3 pathways, with G-1 being more potent for IP3 production, and E2 for cAMP generation (Balhuizen et al., 2010; Kumar et al., 2011). Furthermore, our studies in MIN6 cells revealed that GPER stimulates insulin release through intracellular calcium release and activation of ERK and PI3K pathways (Sharma and Prossnitz, 2011), with EGFR transactivation required for ERK activation, as reported in many cancer cells (Filardo et al., 2000; Sharma and Prossnitz, 2011). GPER also promotes islet cell survival by phosphorylation of pro-survival genes such as CREB, Akt, and ERK1/2, while simultaneously suppressing the activity of stress proteins, such as SAPK/JNK and p38 (Balhuizen et al., 2010; Kumar et al., 2011; Liu et al., 2009). Interestingly, islets isolated from ER αβ KO mice, upon challenge with STZ, still exhibited protection against cell death by E2, suggesting the involvement of GPER or another unknown ER (Liu et al., 2009). Activation of GPER further protects islets and cultured β-cells from lipotoxicity by downregulating important transcription factors involved in lipogenesis, such as chSREBP and SREBP1 via the STAT3 pathway (Liu and Mauvais-Jarvis, 2010). Finally, GPER has an unexpected role in maintaining β-cell mass during pregnancy via downregulation of islet-specific microRNA mi-338–3p, thereby promoting proliferation and survival of β-cells (Jacovetti et al., 2012).
Our recent study sought to determine whether G-1 also elicits antidiabetic effects in murine models of metabolic dysfunction employing OVX and DIO models (Sharma et al., 2020). Our results demonstrate that chronic G-1 treatment improved glucose clearance from the blood of OVX or DIO mice (both males and females) as revealed by glucose tolerance tests. In addition, both fasting blood glucose and insulin levels were lower in both models, suggesting more effective insulin function in peripheral tissues in G-1-treated mice. This was confirmed by an improved HOMA-IR, an index used to assess insulin resistance. It is well known that E2 counteracts visceral obesity and can have direct as well as indirect effects on glucose homeostasis (Liu et al., 2009; Liu and Mauvais-Jarvis, 2009; 2010; Stubbins et al., 2012a). In our studies, OVX female mice (both on normal chow and HFD) displayed weight loss and reduced hyperglycemia following G-1 treatment, whereas DIO male mice did not lose weight but still exhibited improved glucose homeostasis, supporting the concept that G-1 exerts direct effects on glucose homeostasis even in the absence of weight loss (Sharma et al., 2020). However, OVX models pose a challenge in ascertaining the direct vs indirect effects of GPER activation since they display both weight loss as well as improved glucose homeostasis. Two key events that regulate blood glucose levels are insulin production from pancreatic β-cells and effective insulin action in peripheral or central tissues. GPER may improve glucose homeostasis by exerting direct effects through both these modes. These results suggest that GPER, through its activation by G-1, manifests a critical role in glucose metabolism in vivo by acting at multiple levels and that activation of GPER presents a novel therapeutic approach to mitigate metabolic dysfunctions associated with diabetes.
5. GPER roles in inflammation
Obesity initiates a complex cycle involving various pathophysiological events in the body (Heymsfield and Wadden, 2017). Weight gain resulting from loss of E2, not only leads to insulin resistance, ectopic fat deposition and hyperglycemia, but also low grade chronic inflammation (Eaton and Sethi, 2019). These effects involve a multitude of metabolically relevant tissues, such as skeletal muscle, liver, adipose and pancreas, interfering with their normal function. We and others have reported that GPER KO mice display higher levels of circulating pro-inflammatory cytokines, with a concomitant decrease in adiponectin levels (Davis et al., 2014; Sharma et al., 2013). Conversely in our OVX and DIO models, long term treatment with G-1 lowered circulating levels of TNF α, MCP1 and IL6 and also reduced the expression of inflammatory genes in multiple metabolic tissues (Sharma et al., 2020). Our results are supported by previous studies that have highlighted the anti-inflammatory role of GPER both in vivo as well as in vitro. In a murine experimental autoimmune encephalomyelitis (EAE) model of multiple sclerosis, GPER KO mice displayed impaired E2-mediated protection against disease and further abrogated the protective effects of E2 on white matter damage (Blasko et al., 2009; Wang et al., 2009). Conversely, GPER activation by G-1 reduced the severity of disease as supported by clinical and histological manifestations of EAE. The lack of effects of G-1 in GPER KO mice demonstrated the selectivity of G-1 and GPER action. GPER-mediated mechanisms on immune cells have revealed inhibition of inflammatory cytokine production from macrophages and enhanced suppressive activity of T regulatory cells via upregulation of programmed cell death (Wang et al., 2009). Furthermore, previous studies from our lab have demonstrated that G-1 treatment promotes de novo production of the anti-inflammatory cytokine IL10 in pro-inflammatory Th 17 cells (Brunsing et al., 2013; Brunsing and Prossnitz, 2011).
GPER is also involved in mediating the anti-inflammatory effects of genistein in microglia (Du et al., 2018). In adipocytes, treatment of differentiated 3T3-L1 cells with E2 reduced pro-inflammatory gene expression in spite of ER α knockdown, suggesting an alternative pathway, possibly via GPER, was involved (Santos et al., 2017). A role for GPER is also indicated in the pathophysiology of liver cancer, where mice lacking GPER displayed increased inflammation, fibrosis and increased tumorigenesis in a diethylnitrosamine-induced mouse liver tumor model (Wei et al., 2016). Consistent with this, cell-based studies demonstrate that activation of GPER downregulates the expression of fibrosis markers in hepatic stellate cell line, LX2. In this study, the authors conclude that GPER may protect against liver cancer by counteracting inflammation (Wei et al., 2016). These observations support a potent anti-inflammatory role for GPER with its agonists having potential therapeutic benefits as anti-inflammatory agents.
The state of current knowledge provides convincing evidence that GPER regulates body weight and multiple aspects of metabolism in various metabolically active tissues; however, the mechanisms through which these effects are mediated are still not yet fully understood and merit further study. Based on our results and published reports, we propose a model for the anti-obesity and anti-diabetic effects of G-1 and GPER in vivo in Fig. 1. As discussed above, GPER exerts direct and/or indirect effects on multiple metabolic tissues such as WAT, BAT, skeletal muscle, pancreas and liver to reduce obesity and improve glucose homeostasis. Body weight and glucose homeostasis in turn regulate each other in a positive feedback loop. Finally, although the regulation of energy metabolism and glucose homeostasis by E2 through actions in the central nervous system is well known, the effects of GPER on centrally mediated pathways remain largely unknown.
Figure 1.
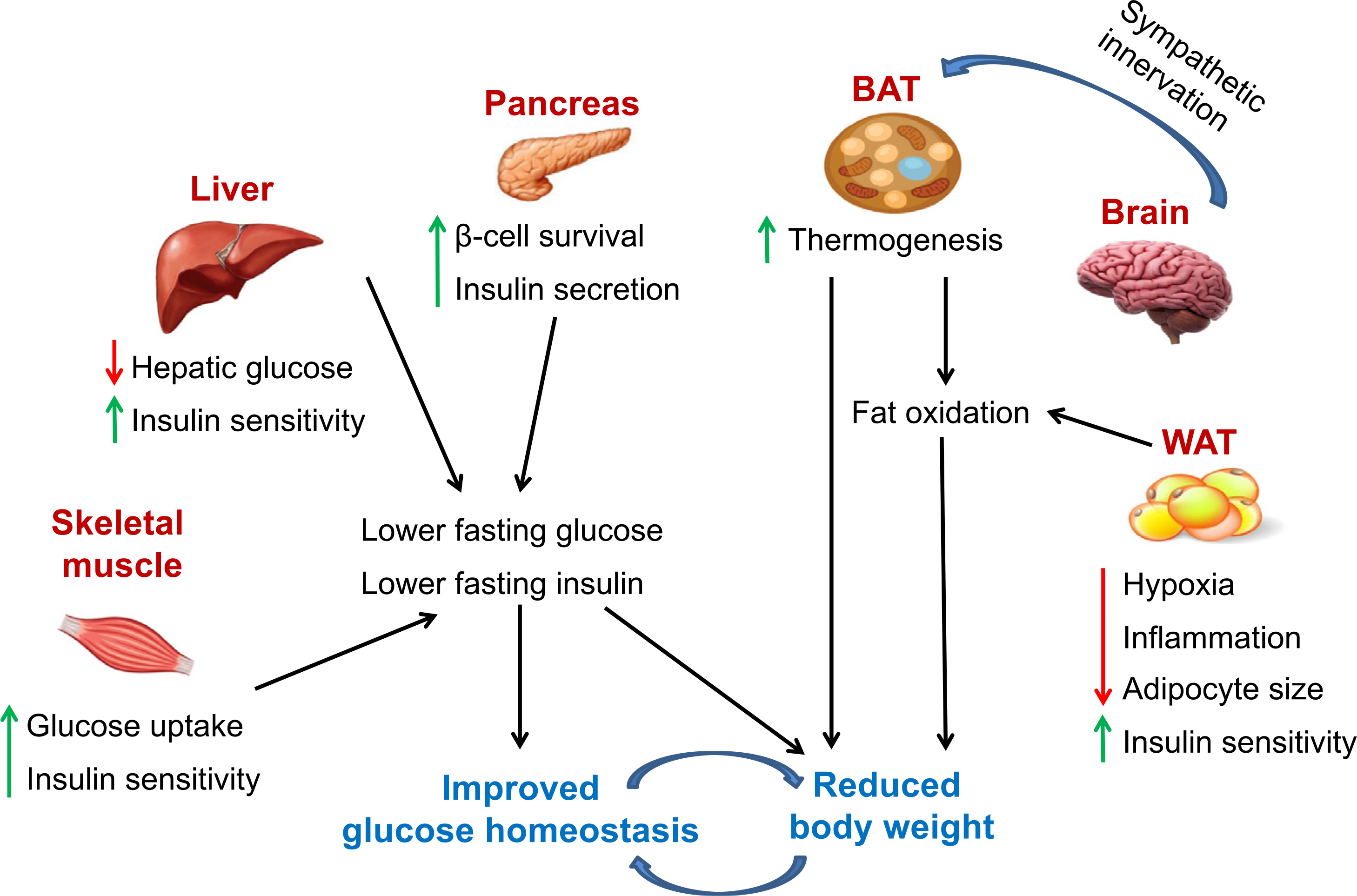
Schematic representation of the proposed effects of GPER activation on body weight and glucose homeostasis. In this model, GPER activation promotes thermogenesis in BAT and fatty acid oxidation in BAT and WAT. BAT stimulated by the sympathetic nervous system promotes thermogenesis. With GPER activation, WAT exhibits smaller adipocytes, likely due to decreased hypoxia and inflammation, resulting in improved insulin sensitivity. Increased fatty acid oxidation coupled with attenuated inflammation upon stimulation of GPER improves skeletal muscle, pancreatic and liver function with improved pancreatic β-cell survival and insulin secretion, resulting in lower fasting glucose and insulin levels, thereby restoring glucose homeostasis. Increased thermogenesis and fat oxidation, with a concomitant decrease in glucose and insulin levels, result in a reduction in body weight. Body weight and glucose homeostasis in turn regulate by each other. Red arrows denote reductions, whereas green arrows depict increases upon GPER stimulation. See text for additional details
6. Conclusions and future directions
Much of the current knowledge regarding the physiological effects of GPER action is derived from global GPER KO mice. While investigating the mechanisms of GPER-mediated effects in global GPER KO mice, care should be taken to account for the possibility of compensatory effects. As studies on metabolism are further confounded by the extensive cross-talk between metabolic tissues, results from global GPER KO mice should be carefully interpreted. To determine the individual contributions of respective metabolic tissues, the use of a tissue-specific conditional knockout mice may represent an attractive alternative. Furthermore, most of the evidence of metabolic regulation via GPER comes from peripheral effects, with few central effects described, both of which should be the subject of future studies.
The global epidemics of obesity and diabetes call for new therapeutic drugs that can effectively ameliorate these diseases. Despite E2’s potent effects on body weight and metabolism, it is not a viable therapy in men and premenopausal women. As described in this review, GPER exerts multiple beneficial effects on various tissues to promote a healthy weight and metabolism. Thus, the GPER-selective agonist G-1 represents a promising first-in-class drug for the treatment of obesity and diabetes. Furthrmore, because G-1 lacks the feminizing effects of E2 as defined by classical uterine imbibition, it represents a viable option for the treatment of metabolic disorders in men as well as women.
Acknowledgments
The authors are supported by grants from the National Institutes of Health (NIH R01 CA163890 and CA194496 to E.R.P.) from Dialysis Clinic, Inc. (E.R.P.), and from the University of New Mexico School of Medicine Research Allocation Committee (G.S.) and by the UNM Comprehensive Cancer Center (P30 CA118100), and the Autophagy, Inflammation and Metabolism (AIM) Center of Biomedical Research Excellence (CoBRE, NIH P20 GM121176).
Footnotes
Declaration of interests
The authors declare that they have competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Competing interests
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests.
E.R.P. is an inventor on U.S. Patent Nos. 10,251,870, 10,561,648 and 10,682,341 and G.S. and E.R.P. are inventors on U.S. patent No. 10,471,047, all for the therapeutic use of compounds targeting GPER (“Method for treating obesity, diabetes, cardiovascular and kidney diseases by regulating GPR30/GPER”). E.R.P. is an inventor on U.S. Patent Nos. 7,875,721 and 8,487,100 for GPER-selective ligands and imaging agents (“Compounds for binding to ER α/ β and GPR30, methods of treating disease states and conditions mediated through these receptors and identification thereof”).
References
- Centers for Disease Control and Prevention, Adult Obesity Causes & Consequences (2020a) https://www.cdc.gov/obesity/adult/causes.html.
- Centers for Disease Control and Prevention, Overweight and Obesity, Data and Statistics (2020b) https://www.cdc.gov/obesity/data/index.html.
- World Health Organization, Obesity and Overweight (2020c) https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight.
- Abildgaard J, Pedersen AT, Green CJ, Harder-Lauridsen NM, Solomon TP, Thomsen C, Juul A, Pedersen M, Pedersen JT, Mortensen OH, Pilegaard H, Pedersen BK, Lindegaard B, 2013. Menopause is associated with decreased whole body fat oxidation during exercise. Am J Physiol Endocrinol Metab 304, E1227–E1236. [DOI] [PubMed] [Google Scholar]
- Alberti KG, Zimmet P, Shaw J, 2006. Metabolic syndrome–a new world-wide definition. A Consensus Statement from the International Diabetes Federation. Diabet Med 23, 469–480. [DOI] [PubMed] [Google Scholar]
- Apovian CM, 2016. Obesity: definition, comorbidities, causes, and burden. Am J Manag Care 22, s176–s185. [PubMed] [Google Scholar]
- Balhuizen A, Kumar R, Amisten S, Lundquist I, Salehi A, 2010. Activation of G protein–coupled receptor 30 modulates hormone secretion and counteracts cytokine-induced apoptosis in pancreatic islets of female mice. Mol Cell Endocrinol 320, 16–24. [DOI] [PubMed] [Google Scholar]
- Balistreri CR, Caruso C, Candore G, 2010. The role of adipose tissue and adipokines in obesity-related inflammatory diseases. Mediators Inflamm 2010, 802078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barros RP, Gustafsson JA, 2011. Estrogen receptors and the metabolic network. Cell Metab 14, 289–299. [DOI] [PubMed] [Google Scholar]
- Basen-Engquist K, Chang M, 2011. Obesity and cancer risk: recent review and evidence. Curr Oncol Rep 13, 71–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauza-Thorbrugge M, Rodriguez-Cuenca S, Vidal-Puig A, Galmes-Pascual BM, Sbert-Roig M, Gianotti M, Llado I, Proenza AM, 2019. GPER and ERalpha mediate estradiol enhancement of mitochondrial function in inflamed adipocytes through a PKA dependent mechanism. J Steroid Biochem Mol Biol 185, 256–267. [DOI] [PubMed] [Google Scholar]
- Bays HE, Toth PP, Kris-Etherton PM, Abate N, Aronne LJ, Brown WV, Gonzalez-Campoy JM, Jones SR, Kumar R, La Forge R, Samuel VT, 2013. Obesity, adiposity, and dyslipidemia: a consensus statement from the National Lipid Association. J Clin Lipidol 7, 304–383. [DOI] [PubMed] [Google Scholar]
- Blaak E, 2001. Gender differences in fat metabolism. Curr Opin Clin Nutr Metab Care 4, 499–502. [DOI] [PubMed] [Google Scholar]
- Blasko E, Haskell CA, Leung S, Gualtieri G, Halks-Miller M, Mahmoudi M, Dennis MK, Prossnitz ER, Karpus WJ, Horuk R, 2009. Beneficial role of the GPR30 agonist G-1 in an animal model of multiple sclerosis. J Neuroimmunol 214, 67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bologa CG, Revankar CM, Young SM, Edwards BS, Arterburn JB, Kiselyov AS, Parker MA, Tkachenko SE, Savchuck NP, Sklar LA, Oprea TI, Prossnitz ER, 2006. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat Chem Biol 2, 207–212. [DOI] [PubMed] [Google Scholar]
- Bonds DE, Lasser N, Qi L, Brzyski R, Caan B, Heiss G, Limacher MC, Liu JH, Mason E, Oberman A, O’Sullivan MJ, Phillips LS, Prineas RJ, Tinker L, 2006. The effect of conjugated equine oestrogen on diabetes incidence: the Women’s Health Initiative randomised trial. Diabetologia 49, 459–468. [DOI] [PubMed] [Google Scholar]
- Brunsing RL, Owens KS, Prossnitz ER, 2013. The G protein-coupled estrogen receptor (GPER) agonist G-1 expands the regulatory T-cell population under TH17-polarizing conditions. J Immunother 36, 190–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunsing RL, Prossnitz ER, 2011. Induction of interleukin-10 in the T helper type 17 effector population by the G protein coupled estrogen receptor (GPER) agonist G-1. Immunology 134, 93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler MJ, Hildebrandt RP, Eckel LA, 2018. Selective activation of estrogen receptors, ERalpha and GPER-1, rapidly decreases food intake in female rats. Horm Behav 103, 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catoi AF, Parvu A, Muresan A, Busetto L, 2015. Metabolic Mechanisms in Obesity and Type 2 Diabetes: Insights from Bariatric/Metabolic Surgery. Obes Facts 8, 350–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadt A, Scherneck S, Joost HG and Al-Hasani H (2000) Molecular links between Obesity and Diabetes: “Diabesity”, in Endotext (Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dungan K, Grossman A, Hershman JM, Hofland HJ, Kaltsas G, Koch C, Kopp P, Korbonits M, McLachlan R, Morley JE, New M, Purnell J, Singer F, Stratakis CA, Trence DL and Wilson DP eds), South Dartmouth (MA). [Google Scholar]
- Consitt LA, Bell JA, Houmard JA, 2009. Intramuscular lipid metabolism, insulin action, and obesity. IUBMB Life 61, 47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Eon TM, Souza SC, Aronovitz M, Obin MS, Fried SK, Greenberg AS, 2005. Estrogen regulation of adiposity and fuel partitioning. Evidence of genomic and non-genomic regulation of lipogenic and oxidative pathways. J Biol Chem 280, 35983–35991. [DOI] [PubMed] [Google Scholar]
- Davis KE, Carstens EJ, Irani BG, Gent LM, Hahner LM, Clegg DJ, 2014. Sexually dimorphic role of G protein-coupled estrogen receptor (GPER) in modulating energy homeostasis. Horm Behav 66, 196–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Luca C, Olefsky JM, 2008. Inflammation and insulin resistance. FEBS Lett 582, 97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis MK, Burai R, Ramesh C, Petrie WK, Alcon SN, Nayak TK, Bologa CG, Leitao A, Brailoiu E, Deliu E, Dun NJ, Sklar LA, Hathaway HJ, Arterburn JB, Oprea TI, Prossnitz ER, 2009. In vivo effects of a GPR30 antagonist. Nat Chem Biol 5, 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis MK, Field AS, Burai R, Ramesh C, Petrie WK, Bologa CG, Oprea TI, Yamaguchi Y, Hayashi S, Sklar LA, Hathaway HJ, Arterburn JB, Prossnitz ER, 2011. Identification of a GPER/GPR30 antagonist with improved estrogen receptor counter-selectivity. J Steroid Biochem Mol Biol 127, 358–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deroo BJ, Korach KS, 2006. Estrogen receptors and human disease. J Clin Invest 116, 561–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Carlo C, Tommaselli GA, Pisano G, Nasti A, Rossi V, Palomba S, Nappi C, 2000. Serum leptin levels in postmenopausal women: effects of transdermal hormone replacement therapy. Menopause 7, 36–41. [DOI] [PubMed] [Google Scholar]
- Du ZR, Feng XQ, Li N, Qu JX, Feng L, Chen L, Chen WF, 2018. G protein-coupled estrogen receptor is involved in the anti-inflammatory effects of genistein in microglia. Phytomedicine 43, 11–20. [DOI] [PubMed] [Google Scholar]
- Eaton SA, Sethi JK, 2019. Immunometabolic Links between Estrogen, Adipose Tissue and Female Reproductive Metabolism. Biology (Basel) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckel RH, Krauss RM, 1998. American Heart Association call to action: obesity as a major risk factor for coronary heart disease. AHA Nutrition Committee. Circulation 97, 2099–2100. [DOI] [PubMed] [Google Scholar]
- Filardo EJ, Quinn JA, Bland KI, Frackelton AR Jr., 2000. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol 14, 1649–1660. [DOI] [PubMed] [Google Scholar]
- Flegal KM, Carroll MD, Ogden CL, Curtin LR, 2010. Prevalencea and trends in obesity among US adults, 1999–2008. JAMA 303, 235–241. [DOI] [PubMed] [Google Scholar]
- Fujioka S, Matsuzawa Y, Tokunaga K, Tarui S, 1987. Contribution of intra-abdominal fat accumulation to the impairment of glucose and lipid metabolism in human obesity. Metabolism 36, 54–59. [DOI] [PubMed] [Google Scholar]
- Garaulet M, Perez-Llamas F, Baraza JC, Garcia-Prieto MD, Fardy PS, Tebar FJ, Zamora S, 2002. Body fat distribution in pre-and post-menopausal women: metabolic and anthropometric variables. J Nutr Health Aging 6, 123–126. [PubMed] [Google Scholar]
- Guh DP, Zhang W, Bansback N, Amarsi Z, Birmingham CL, Anis AH, 2009. The incidence of co-morbidities related to obesity and overweight: a systematic review and meta-analysis. BMC Public Health 9, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupte AA, Pownall HJ, Hamilton DJ, 2015. Estrogen: an emerging regulator of insulin action and mitochondrial function. J Diabetes Res 2015, 916585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney EP, Nachtigall MJ, Nachtigall LE, Naftolin F, 2014. The Women’s Health Initiative trial and related studies: 10 years later: a clinician’s view. J Steroid Biochem Mol Biol 142, 4–11. [DOI] [PubMed] [Google Scholar]
- Haas E, Bhattacharya I, Brailoiu E, Damjanovic M, Brailoiu GC, Gao X, Mueller-Guerre L, Marjon NA, Gut A, Minotti R, Meyer MR, Amann K, Ammann E, Perez-Dominguez A, Genoni M, Clegg DJ, Dun NJ, Resta TC, Prossnitz ER, Barton M, 2009. Regulatory role of G protein-coupled estrogen receptor for vascular function and obesity. Circ Res 104, 288–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond RA, Levine R, 2010. The economic impact of obesity in the United States. Diabetes Metab Syndr Obes 3, 285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt SC, Korach KS, 2018. Estrogen Receptors: New Directions in the New Millennium. Endocr Rev 39, 664–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt SC, Winuthayanon W, Korach KS, 2016. What’s new in estrogen receptor action in the female reproductive tract. J Mol Endocrinol 56, R55–R71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heymsfield SB, Wadden TA, 2017. Mechanisms, Pathophysiology, and Management of Obesity. N Engl J Med 376, 254–266. [DOI] [PubMed] [Google Scholar]
- Hong J, Stubbins RE, Smith RR, Harvey AE, Nunez NP, 2009. Differential susceptibility to obesity between male, female and ovariectomized female mice. Nutr J 8, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain Y, Ding Q, Connelly PW, Brunt JH, Ban MR, McIntyre AD, Huff MW, Gros R, Hegele RA, Feldman RD, 2015. G-protein estrogen receptor as a regulator of low–density lipoprotein cholesterol metabolism: cellular and population genetic studies. Arterioscler Thromb Vasc Biol 35, 213–221. [DOI] [PubMed] [Google Scholar]
- Ingvorsen C, Karp NA, Lelliott CJ, 2017. The role of sex and body weight on the metabolic effects of high-fat diet in C57BL/6N mice. Nutr Diabetes 7, e261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacovetti C, Abderrahmani A, Parnaud G, Jonas JC, Peyot ML, Cornu M, Laybutt R, Meugnier E, Rome S, Thorens B, Prentki M, Bosco D, Regazzi R, 2012. MicroRNAs contribute to compensatory beta cell expansion during pregnancy and obesity. J Clin Invest 122, 3541–3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kernan WN, Dearborn JL, 2015. Obesity increases stroke risk in young adults: opportunity for prevention. Stroke 46, 1435–1436. [DOI] [PubMed] [Google Scholar]
- Kotani K, Tokunaga K, Fujioka S, Kobatake T, Keno Y, Yoshida S, Shimomura I, Tarui S, Matsuzawa Y, 1994. Sexual dimorphism of age-related changes in whole–body fat distribution in the obese. Int J Obes Relat Metab Disord 18 207–202. [PubMed] [Google Scholar]
- Kozakowski J, Gietka-Czernel M, Leszczynska D, Majos A, 2017. Obesity in menopause - our negligence or an unfortunate inevitability? Prz Menopauzalny 16, 61–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R, Balhuizen A, Amisten S, Lundquist I, Salehi A, 2011. Insulinotropic and antidiabetic effects of 17beta-estradiol and the GPR30 agonist G-1 on human pancreatic islets. Endocrinology 152, 2568–2579. [DOI] [PubMed] [Google Scholar]
- Lee CG, Carr MC, Murdoch SJ, Mitchell E, Woods NF, Wener MH, Chandler WL, Boyko EJ, Brunzell JD, 2009. Adipokines, inflammation, and visceral adiposity across the menopausal transition: a prospective study. J Clin Endocrinol Metab 94, 1104–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Le May C, Wong WP, Ward RD, Clegg DJ, Marcelli M, Korach KS, Mauvais-Jarvis F, 2009. Importance of extranuclear estrogen receptor-alpha and membrane G protein-coupled estrogen receptor in pancreatic islet survival. Diabetes 58, 2292–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Mauvais-Jarvis F, 2009. Rapid, nongenomic estrogen actions protect pancreatic islet survival. Islets 1, 273–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Mauvais-Jarvis F, 2010. Minireview: Estrogenic protection of beta-cell failure in metabolic diseases. Endocrinology 151, 859–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundholm L, Bryzgalova G, Gao H, Portwood N, Falt S, Berndt KD, Dicker A, Galuska D, Zierath JR, Gustafsson JA, Efendic S, Dahlman-Wright K, Khan A, 2008. The estrogen receptor {alpha}-selective agonist propyl pyrazole triol improves glucose tolerance in ob/ob mice; potential molecular mechanisms. J Endocrinol 199, 275–286. [DOI] [PubMed] [Google Scholar]
- Mårtensson UE, Salehi SA, Windahl S, Gomez MF, Sward K, Daszkiewicz-Nilsson J, Wendt A, Andersson N, Hellstrand P, Grande PO, Owman C, Rosen CJ, Adamo ML, Lundquist I, Rorsman P, Nilsson BO, Ohlsson C, Olde B, Leeb-Lundberg LM, 2009. Deletion of the G protein-coupled receptor 30 impairs glucose tolerance, reduces bone growth, increases blood pressure, and eliminates estradiol-stimulated insulin release in female mice. Endocrinology 150, 687–698. [DOI] [PubMed] [Google Scholar]
- Mauvais-Jarvis F, 2011. Estrogen and androgen receptors: regulators of fuel homeostasis and emerging targets for diabetes and obesity. Trends Endocrinol Metab 22, 24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauvais-Jarvis F, 2015. Sex differences in metabolic homeostasis, diabetes, and obesity. Biol Sex Differ 6, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister EJ, Dhurandhar NV, Keith SW, Aronne LJ, Barger J, Baskin M, Benca RM, Biggio J, Boggiano MM, Eisenmann JC, Elobeid M, Fontaine KR, Gluckman P, Hanlon EC, Katzmarzyk P, Pietrobelli A, Redden DT, Ruden DM, Wang C, Waterland RA, Wright SM, DB Allison, 2009. Ten putative contributors to the obesity epidemic. Crit Rev Food Sci Nutr 49, 868–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meoli L, Isensee J, Zazzu V, Nabzdyk CS, Soewarto D, Witt H, Foryst-Ludwig A, Kintscher U, Noppinger PR, 2014. Sex- and age-dependent effects of Gpr30 genetic deletion on the metabolic and cardiovascular profiles of diet-induced obese mice. Gene 540, 210–216. [DOI] [PubMed] [Google Scholar]
- Meyer MR, Clegg DJ, Prossnitz ER, Barton M, 2011. Obesity, insulin resistance and diabetes: sex differences and role of oestrogen receptors. Acta Physiol (Oxf) 203, 259–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittendorfer B, 2011. Origins of metabolic complications in obesity: adipose tissue and free fatty acid trafficking. Curr Opin Clin Nutr Metab Care 14, 535–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata Y, Robertson KM, Jones ME, Simpson ER, 2002. Effect of estrogen deficiency in the male: the ArKO mouse model. Mol Cell Endocrinol 193, 7–12. [DOI] [PubMed] [Google Scholar]
- Nakhjavani M, Imani M, Larry M, Aghajani-Nargesi A, Morteza A, Esteghamati A, 2014. Metabolic syndrome in premenopausal and postmenopausal women with type 2 diabetes: loss of protective effects of premenopausal status. J Diabetes Metab Disord 13, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantalone KM, Hobbs TM, Chagin KM, Kong SX, Wells BJ, Kattan MW, Bouchard J, Sakurada B, Milinovich A, Weng W, Bauman J, Misra-Hebert AD, Zimmerman RS, Burguera B, 2017. Prevalence and recognition of obesity and its associated comorbidities: cross-sectional analysis of electronic health record data from a large US integrated health system. BMJ Open 7, e017583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrie WK, Dennis MK, Hu C, Dai D, Arterburn JB, Smith HO, Hathaway HJ, Prossnitz ER, 2013. G protein-coupled estrogen receptor-selective ligands modulate endometrial tumor growth. Obstet Gynecol Int 2013, 472720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier P, Giles TD, Bray GA, Hong Y, Stern JS, Pi-Sunyer FX, Eckel RH, American Heart, AObesity Committee of the Council on Nutrition PA and Metabolism, 2006. Obesity and cardiovascular disease: pathophysiology, evaluation, and effect of weight loss: an update of the 1997 American Heart Association Scientific Statement on Obesity and Heart Disease from the Obesity Committee of the Council on Nutrition, Physical Activity, and Metabolism. Circulation 113, 898–918. [DOI] [PubMed] [Google Scholar]
- Proietto J, 2017. Obesity and weight management at menopause. Aust Fam Physician 46, 368–370. [PubMed] [Google Scholar]
- Prossnitz ER, Arterburn JB, 2015. International Union of Basic and Clinical Pharmacology. XCVII. G Protein-Coupled Estrogen Receptor and Its Pharmacologic Modulators. Pharmacol Rev 67, 505–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prossnitz ER, Arterburn JB, Smith HO, Oprea TI, Sklar LA, Hathaway HJ, 2008. Estrogen signaling through the transmembrane G protein-coupled receptor GPR30. Annu Rev Physiol 70, 165–190. [DOI] [PubMed] [Google Scholar]
- Prossnitz ER, Barton M, 2011. The G-protein-coupled estrogen receptor GPER in health and disease. Nat Rev Endocrinol 7, 715–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prossnitz ER, Barton M, 2014. Estrogen biology: New Insights into GPER function and clinical opportunities. Mol Cell Endocrinol 389, 71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prossnitz ER, Hathaway HJ, 2015. What have we learned about GPER function in physiology and disease from knockout mice? J Steroid Biochem Mol Biol 153, 114–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regitz-Zagrosek V, Lehmkuhl E, Weickert MO, 2006. Gender differences in the metabolic syndrome and their role for cardiovascular disease. Clin Res Cardiol 95, 136–147. [DOI] [PubMed] [Google Scholar]
- Rogers NH, Perfield JW 2nd, Strissel KJ, Obin MS, Greenberg AS, 2009. Reduced energy expenditure and increased inflammation are early events in the development of ovariectomy-induced obesity. Endocrinology 150, 2161–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos RS, de Fatima LA, Frank AP, Carneiro EM, Clegg DJ, 2017. The effects of 17 alpha-estradiol to inhibit inflammation in vitro. Biol Sex Differ 8, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt FM, Weschenfelder J, Sander C, Minkwitz J, Thormann J, Chittka T, Mergl R, Kirkby KC, Fasshauer M, Stumvoll M, Holdt LM, Teupser D, Hegerl U, Himmerich H, 2015. Inflammatory cytokines in general and central obesity and modulating effects of physical activity. PLoS One 10, e0121971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma G, Hu C, Brigman JL, Zhu G, Hathaway HJ, Prossnitz ER, 2013. GPER deficiency in male mice results in insulin resistance, dyslipidemia, and a proinflammatory state. Endocrinology 154, 4136–4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma G, Hu C, Staquicini DI, Brigman JL, Liu M, Mauvais-Jarvis F, Pasqualini R, Arap W, Arterburn JB, Hathaway HJ, Prossnitz ER, 2020. Preclinical efficacy of the GPER-selective agonist G-1 in mouse models of obesity and diabetes. Sci Transl Med 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma G, Mauvais-Jarvis F, Prossnitz ER, 2018. Roles of G protein-coupled estrogen receptor GPER in metabolic regulation. J Steroid Biochem Mol Biol 176, 31–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma G, Prossnitz ER, 2011. Mechanisms of estradiol-induced insulin secretion by the G protein-coupled estrogen receptor GPR30/GPER in pancreatic beta-cells. Endocrinology 152, 3030–3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma G, Prossnitz ER, 2016. GPER/GPR30 Knockout Mice: Effects of GPER on Metabolism. Methods Mol Biol 1366, 489–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma G, Prossnitz ER, 2017. G-Protein-Coupled Estrogen Receptor (GPER) and Sex-Specific Metabolic Homeostasis. Adv Exp Med Biol 1043, 427–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen M, Kumar SP, Shi H, 2014. Estradiol regulates insulin signaling and inflammation in adipose tissue. Horm Mol Biol Clin Investig 17, 99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoelson SE, Herrero L, Naaz A, 2007. Obesity, inflammation, and insulin resistance. Gastroenterology 132, 2169–2180. [DOI] [PubMed] [Google Scholar]
- Shulman GI, 2014. Ectopic fat in insulin resistance, dyslipidemia, and cardiometabolic disease. N Engl J Med 371, 2237–2238. [DOI] [PubMed] [Google Scholar]
- Stefan N, 2020. Causes, consequences, and treatment of metabolically unhealthy fat distribution. Lancet Diabetes Endocrinol 8, 616–627. [DOI] [PubMed] [Google Scholar]
- Stefan N, Schick F, Haring HU, 2017. Causes, Characteristics, and Consequences of Metabolically Unhealthy Normal Weight in Humans. Cell Metab 26, 292–300. [DOI] [PubMed] [Google Scholar]
- Stubbins RE, Holcomb VB, Hong J, Nunez NP, 2012a. Estrogen modulates abdominal adiposity and protects female mice from obesity and impaired glucose tolerance. Eur J Nutr 51, 861–870. [DOI] [PubMed] [Google Scholar]
- Stubbins RE, Najjar K, Holcomb VB, Hong J, Nunez NP, 2012b. Oestrogen alters adipocyte biology and protects female mice from adipocyte inflammation and insulin resistance. Diabetes Obes Metab 14, 58–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tandon VR, Mahajan A, Sharma S, Sharma A, 2010. Prevalence of cardiovascular risk factors in postmenopausal women: A rural study. J Midlife Health 1, 26–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchernof A, Despres JP, 2013. Pathophysiology of human visceral obesity: an update. Physiol Rev 93, 359–404. [DOI] [PubMed] [Google Scholar]
- Wang A, Luo J, Moore W, Alkhalidy H, Wu L, Zhang J, Zhen W, Wang Y, Clegg DJ, Bin X, Cheng Z, McMillan RP, Hulver MW, Liu D, 2016. GPR30 regulates diet-induced adiposity in female mice and adipogenesis in vitro. Sci Rep 6, 34302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Dehghani B, Li Y, Kaler LJ, Proctor T, Vandenbark AA, Offner H, 2009. Membrane estrogen receptor regulates experimental autoimmune encephalomyelitis through up-regulation of programmed death 1. J Immunol 182, 3294–3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Xu Y, 2019. Mechanisms for Sex Differences in Energy Homeostasis. J Mol Endocrinol 62, R129–R143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei T, Chen W, Wen L, Zhang J, Zhang Q, Yang J, Liu H, Chen BW, Zhou Y, Feng X, Yang Q, Bai X, Liang T, 2016. G protein-coupled estrogen receptor deficiency accelerates liver tumorigenesis by enhancing inflammation and fibrosis. Cancer Lett 382, 195–202. [DOI] [PubMed] [Google Scholar]
- Woo CY, Jang JE, Lee SE, Koh EH, Lee KU, 2019. Mitochondrial Dysfunction in Adipocytes as a Primary Cause of Adipose Tissue Inflammation. Diabetes Metab J 43, 247–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, 2009. Emerging role of adipose tissue hypoxia in obesity and insulin resistance. Int J Obes (Lond) 33, 54–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu P, Yuen JM, Sham KW, Cheng CH, 2013. GPER mediates the inhibitory actions of estrogen on adipogenesis in 3T3-L1 cells through perturbation of mitotic clonal expansion. Gen Comp Endocrinol 193, 19–26. [DOI] [PubMed] [Google Scholar]