

Summary
The recruitment of signaling proteins into activated receptor tyrosine kinases (RTKs) to produce rapid, high-fidelity downstream response is exposed to the ambiguity of random diffusion to the target site. Liquid-liquid phase separation (LLPS) overcomes this by providing elevated, localized concentrations of the required proteins while impeding competitor ligands. Here, we show a subset of phosphorylation-dependent RTK-mediated LLPS states. We then investigate the formation of phase-separated droplets comprising a ternary complex including the RTK, (FGFR2); the phosphatase, SHP2; and the phospholipase, PLCγ1, which assembles in response to receptor phosphorylation. SHP2 and activated PLCγ1 interact through their tandem SH2 domains via a previously undescribed interface. The complex of FGFR2 and SHP2 combines kinase and phosphatase activities to control the phosphorylation state of the assembly while providing a scaffold for active PLCγ1 to facilitate access to its plasma membrane substrate. Thus, LLPS modulates RTK signaling, with potential consequences for therapeutic intervention.
Keywords: Liquid-liquid phase separation (LLPS), Receptor tyrosine kinases (RTKs), FGFR2, Shp2, Plcγ1, kinase activity, phosphatase activity, phospholipase activity
Graphical abstract
Highlights
-
•
Phosphorylated RTKs undergo phase separation with downstream effectors
-
•
Phosphorylation-dependent multivalent interaction drives FGFR2-SHP2 phase separation
-
•
The FGFR2-SHP2 complex colocalizes PLCγ1 to its plasma membrane substrate
-
•
Enzymatic activities are regulated within the FGFR2-SHP2-PLCγ1 membraneless droplets
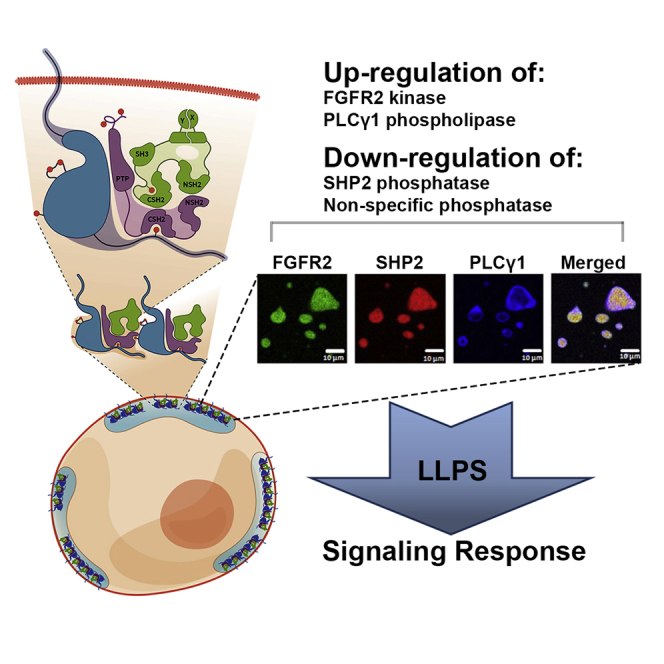
Lin et al. demonstrate that phosphorylated RTKs undergo liquid-liquid phase separation upon the recruitment of downstream proteins. Focusing on the RTK FGFR2, this process is shown to modulate enzymatic activities within the subcellular membraneless compartment.
Introduction
Receptor tyrosine kinases (RTKs) initiate signaling pathways, which regulate diverse cellular processes. On activation, multiple moderate affinity tyrosyl phosphate (pY) binding sites become available for the rapid recruitment of downstream effector proteins. However, the mechanism for expedient recruitment via random molecular diffusion through the cytoplasm is not fully understood. One way in which the probabilistic outcome associated with random diffusion could be alleviated is through the localized accumulation of high effective concentrations of signaling proteins in discrete pools in the cell (Cebecauer et al., 2010). The inclusion of interacting proteins into liquid-liquid phase-separated (LLPS) membraneless droplets maintains functionally relevant proteins at high concentrations in a liquid phase at the required point of action, enhancing equilibrium binding and enzyme activity (Banani et al., 2017; Bracha et al., 2018; Case et al., 2019a; Hyman et al., 2014; Wang et al., 2018). These LLPS states have been associated with a wide range of cellular functions including the regulation of signaling through, e.g., nephrin (Case et al., 2019a; Li et al., 2012), the T-cell receptor (Su et al., 2016), mTOR (Zhang et al., 2018), and RAS (Huang et al., 2019; Tulpule et al., 2021); however, whether LLPS extends to plasma membrane-bound RTK signal transduction has not been investigated. Here, we show that a subset of RTKs undergo LLPS with downstream effector proteins. We then demonstrate that one of these RTKs, fibroblast growth factor receptor 2 (FGFR2), forms a signaling-competent LLPS state with two downstream enzymes: a tandem Src homology 2 (SH2) domain-containing protein tyrosine phosphatase 2 (SHP2) and 1-phosphatidylinositol 4,5-bisphosphate phosphodiesterase gamma 1 (PLCγ1). We show that these proteins assemble into a ternary complex, which exploits LLPS condensation to simultaneously modulate kinase, phosphatase, and phospholipase activities. Therefore, LLPS formation ensures that the requirement for prolonged, high-fidelity signaling is achieved. Our work suggests that the formation of biological condensates might be a key organizing principle of RTK-mediated signaling, with broad implications for further mechanistic studies as well as therapeutic intervention.
Results
Phosphorylated RTKs form droplets with signaling proteins
To understand whether droplet formation could play a role in the regulation of RTK-containing signaling complexes, in the first instance, the intracellular domains of a subset of RTKs were investigated: pEGFRKinase−Tail (residues 712–1210), pHer2Kinase−Tail (720–1255), pHer4Kinase−Tail (718–1305), pFGFR1Kinase−Tail (478–822), pFGFR2Cyto (400–821), pVEGFR1Kinase−Tail (827–1338), and pVEGFR2Kinase−Tail (834–1356) (a schematic of all constructs used in this work is given in Figure 1A and purity in Figure S1A). We investigated the droplet formation with phosphorylated RTK intracellular domains with two known downstream effector proteins: phosphatase SHP2 (the inactive C459S mutant, SHP2C459S [Agazie and Hayman, 2003]) and adaptor protein SHC (the P52 isoform) (Figure S1A). Both effector proteins contain a pair of domains, which bind to pY-containing ligands. Visible droplets are apparent in an in vitro droplet formation assay using fluorescently labeled phosphorylated RTKs with SHP2C459S or SHC (Figure 1A) in 150-mM NaCl and using the optimum concentrations we obtained from the phase diagram analysis of different RTKs with SHP2C459S (Figure 1B) or SHC (Figure S1B). SHP2C459S and SHC were able to form droplets with all the RTKs we tested apart from the pVEGFR2Kinase-Tail-SHP2C459S pair, which could reflect differences in affinities between the RTK pY sequences and SHP2C459S or SHC.
Figure 1.

Phosphorylated RTK-mediated condensation of protein complexes
(A) (Above) Images of recombinant phosphorylated receptors from the EGFR, FGFR, and VEGFR families (Atto-488 labeled) droplet formation upon adding SHP2C459S (top panel) or SHC (middle panel); phosphorylated RTK proteins alone do not form droplets (third panel). Concentrations of each RTK-SHP2C459S or SHC pair were shown (x axis: RTK concentration; y axis: SHP2C459S or SHC concentration) and scale bars, 10 μm. (Below) Schematic diagram with residue numbers shows the defined boundaries of RTK intracellular regions of SHP2, SHC, and PLCγ1 proteins and polypeptides used in this study.
(B) Phase diagrams of phosphorylated EGFR, FGFR, and VEGFR family proteins (Atto-488 labeled) with concentrations shown in x axis and SHP2C459S (y axis) in 20 mM HEPES (pH7.5), 150 mM NaCl, and 1 mM TCEP. The sizes of the circles represent the average sizes of droplets (μm2), and the color scale bars represent the number of droplets in a 0.0256-mm2 area.
We focused our investigation on droplet formation involving FGFR2 (pFGFR2Cyto) and SHP2C459S (Chen et al., 2020). Active SHP2 resulted in lesser droplet formation than SHP2C459S, as did the use of unphosphorylated FGFR2Cyto or the “kinase-dead” K517I mutant (FGFR2Cyto,K517I). Thus, the appearance of droplets depends on prolonged RTK phosphorylation (Figure S1C). To mitigate against the rapid phosphorylation-dephosphorylation cycles in in vitro experiments, we adopted the SHP2C459S phosphatase-dead trapping mutant in the following experiments, except where we have stated that WT protein is used.
The droplets of FGFR2-SHP2 exhibit liquid-like features in cells
Using optimal protein concentrations derived from phase diagrams (Figure 1B), we found that pFGFR2Cyto-SHP2C459S droplets display the liquid-like features of mobilization and fusing upon encounter (Figure 2A), as well as in fluorescence recovery after the photobleaching (FRAP) experiments where the fluorescence of pFGFR2Cyto-SHP2C459S droplets recovered within minutes (Figure 2B). To determine whether the receptor forms droplets with SHP2C459S in fixed cells, we overexpressed full-length RFP-tagged FGFR2 and GFP-tagged SHP2C459S and stimulated with FGF9, a specific FGFR2 ligand, in HEK293T cells in which endogenous FGFR2 is negligible (Figure S2A). To remove the alternative mode of SHP2 recruitment to FGFR2 via FRS2, we adopted a FGFR2 mutant deleted for 428VT429 (FGFR2ΔVT: see STAR Methods). Compared with the basal state, the stimulation of FGFR2 results in the coalescence of micrometer-sized clusters at the plasma membrane of complexes containing SHP2C459S (Figure S2A).
Figure 2.

The dynamic LLPS properties of phosphorylated pFGFR2Cyto-SHP2C459S-pPLCγ1 condensates
(A) The dynamic LLPS properties of pFGFR2Cyto (10 μM)-SHP2C459S (60 μM) condensates was assessed by the fusion experiment. Images were taken every 5 min. Scale bars, 100 μm.
(B) Quantification of FRAP data (means ± SD, n = 2 experiments) for pFGFR2Cyto (10 μM)-SHP2C459S (60 μM) condensates.
(C) The dynamic LLPS property of full-length SHP2C459S-RFP condensates coexpressed with FGFR2ΔVT and stimulated with 10 ng/ml of FGF9 ligand in HEK293T cells. Images were taken every 30 s. Scale bars, 250 nm.
(D) Droplet formation observed between pFGFR2Cyto Atto-488 (10 μM), SHP2C459S Atto-550 (60 μM), and pPLCγ1 Atto-647 (12 μM). (i) Individual proteins showed no evidence of droplet formation. Droplet formation was observed after 1 min between different combinations of proteins: (ii) all three proteins; (iii) pFGFR2Cyto with SHP2C459S, and (v) SHP2C459S with pPLCγ1. (iv) No droplet formation was observed for pFGFR2Cyto with pPLCγ1. Droplet size was diminished with increasing concentrations of NaCl after incubation for 1 min (vi: 250 mM, vii: 500 mM) compared with (ii) and in the presence of 10% 1, 6-hexanediol for 1 min (viii). Addition of 20 μM of lipoamide (ix) or lipoic acid (x) (in 0.1% DMSO) also reduces droplet numbers and sizes while 0.1% DMSO has negligible effect. Scale bars, 10 μm.
(E) The dynamic LLPS properties of pFGFR2Cyto Atto-488 (10 μM)-SHP2C459S Atto-550 (60 μM)-pPLCγ1 Atto-647 (12 μM) condensates was assessed by the fusion experiment. Images were taken every 2 min. Scale bars, 10 μm.
To understand how the droplets affect signaling, another known substrate protein of FGFR2, PLCγ1 (Huang et al., 2016), was also investigated. Although it has been reported that the RTK-dependent phosphorylation of PLCγ1 on Y783 abrogates its interaction with active RTKs (Bunney et al., 2012; Gresset et al., 2010), the phospholipase was still seen to condense into droplets on plasma membrane upon the stimulation of cells (Figure S2B), whereas no such clusters were observed in the absence of the receptor where both SHP2C459S and PLCγ1 appear to be randomly diffusing in cells (Figure S2C). We also demonstrated that SHP2C459S-RFP exhibited condensate fluidity on the membrane when FGFR2ΔVT is expressed and activated by FGF9, as assessed by the fusion experiments in HEK293T cells (Figure 2C). These results indicate that the recruitment of SHP2C459S to active pFGFR2 promotes the formation of droplets that exhibit a dynamic liquid-like behavior.
Reconstituted FGFR2-SHP2C459S LLPS droplets promote the recruitment of active PLCγ1
PLCγ1 is recruited into active FGFR2 through binding to pY769 on the receptor (Bunney et al., 2012). The concomitant phosphorylation of PLCγ1 on Y783 causes an intramolecular interaction between pY783 and the CSH2 domain of the phospholipase; this intramolecular interaction induces a structural rearrangement and leads to the dissociation of phosphorylated PLCγ1 from the recruiting RTK (Bunney et al., 2012; DeBell et al., 2007; Poulin et al., 2005). However, active PLCγ1 needs to be retained proximal to the plasma membrane, where it can access and hydrolyze its substrate phosphatidylinositol 4,5-bisphosphate (PIP2). To explore a potential retention mechanism, we tested the ability of FGFR2-SHP2C459S LLPS droplets to recruit active PLCγ1 in vitro.
Using the concentrations of pFGFR2Cyto, SHP2C459S, and pPLCγ1 established from phase diagrams (Figure S2D), we saw no droplet formation with the individual fluorescently labeled proteins (Figure 2Di); however, submicrometer-sized droplets formed upon the addition of pPLCγ1 to pFGFR2Cyto-SHP2C459S (Figure 2Dii). Size exclusion chromatography revealed that these three proteins can form a high-molecular-weight complex (Figure S2E). The pairwise combination of pFGFR2Cyto/SHP2C459S formed droplets (Figure 2Diii), but the lack of droplet formation between pFGFR2 and pPLCγ1 is consistent with the reported abrogation of interaction upon Y783 PLCγ1 phosphorylation (Figure 2Div) (Bunney et al., 2012; Gresset et al., 2010). Interestingly, SHP2C459S/pPLCγ1 also exhibited droplet formation (Figure 2Dv). To provide a robust confirmation of LLPS formation, we employed two complementary methods that have emerged as current standards in the field (Alberti et al., 2019). First, increasing the concentration of NaCl from 150 mM to 250 mM inhibited the electrostatic interactions and, therefore, led to a decrease in the size of droplets (Figures 2Dii and 2Dvi). At 500-mM NaCl concentration, the droplets completely dispersed (Figure 2Dvii). Second, the addition of 10% 1,6-hexanediol disrupted condensates (Figure 2Dviii). As an additional test of LLPS, lipoamide and lipoic acid, which dissolve stress granules (Wheeler et al., 2019), appeared to do the same to pFGFR2Cyto-SHP2C459S-pPLCγ1 droplets (Figures 2Dix, 2Dx, and 2Dxi). Finally, the fusion experiment using the pFGFR2Cyto-SHP2C459S-pPLCγ1 condensates was consistent with a fluid state (Figure 2E).
Reconstituted pFGFR2Cyto-SHP2C459S-pPLCγ1 assemblies undergo LLPS on lipid membrane bilayers
To mimic FGFR2 signaling complex assembly on a membrane and confirm that a complex can form as a part of the LLPS, we prepared fluorescently labeled pFGFR2Cyto with an N-terminal 6x His tag that allowed attachment to the supported lipid bilayers containing 2% Ni-NTA (Banjade and Rosen, 2014; Case et al., 2019a; Huang et al., 2019; Su et al., 2017; Zeng et al., 2018). pFGFR2Cyto alone was uniformly distributed, as observed by fluorescence microscopy (Figure 3Ai)), and freely diffusing on the bilayers, as revealed by FRAP analysis (Figure S3A). To initiate the condensate formation, we added labeled, untagged SHP2C459S and incubated for 1 min. After buffer exchange to remove the excess protein, submicron-sized condensates were observed (Figure 3Aii). We added increasing concentrations of labeled, untagged pPLCγ1 (Figures 3Aiii and 3Aiv). This led to the appearance of robust pFGFR2Cyto-SHP2C459S-pPLCγ1 clusters. Confocal imaging confirmed that pFGFR2Cyto, SHP2C459S, and pPLCγ1 colocalized within the clusters. The concentrations of the three proteins used were based on our phase diagrams derived in the context of 3D droplet formation; however, it should be noted that condensates do form at lower concentrations on the 2D membrane. FRAP analysis showed that all three proteins in the condensed clusters exhibited a slow dynamic exchange with their counterparts into and out of the condensates (Figure 3B). Unlike the typical liquid-like behavior seen in pFGFR2Cyto-SHP2C459S condensates that show more substantial fluorescence recovery, the addition of pPLCγ1 may drive the condensate properties toward more gel-like ones. This in vitro experiment indicated that both SHP2C459S and pPLCγ1 can be recruited into membrane-bound pFGFR2 and form phase-separated clusters.
Figure 3.

The formation of LLPS pFGFR2-SHP2C459S-pPLCγ1 condensates on supported lipid bilayers and plasma membranes
(A) pFGFR2Cyto-SHP2C459S-pPLCγ1 condensates on supported lipid bilayers. (i) Confocal images of homogeneously distributed pFGFR2Cyto Atto-488 (20 μM, 6xHis tagged) on membrane bilayers, (ii) pFGFR2Cyto Atto-488 gradually clustered upon the addition of SHP2C459S Atto-594 (60 μM, untagged), and (iii) pPLCγ1 Atto-647 (6 μM, untagged), followed by (iv) additional 36 μM of untagged pPLCγ1 Atto-647. Scale bars, 10 μm.
(B) FRAP analysis showing the dynamic nature of pFGFR2Cyto-SHP2C459S-pPLCγ1 condensates on supported lipid bilayers as all pFGFR2Cyto, SHP2C459S, and pPLCγ1 exchanged with their counterparts in the dilute phase. Data are presented as mean ± SD, n = 2 experiments.
(C) Immunofluorescence staining images showing colocalized SHP2-Alexa 488 and PLCγ1-Alexa 647 droplet formation on plasma membrane in FGF9-stimulated (10 ng/ml, 15 min) Caco-2 cells and Caco-2 FGFR2i cells. Inset image: magnification of regions shown to exemplify endogenous SHP2-PLCγ1 clusters on membranes. Graph (right of image): statistical analysis of droplet formation in parental Caco-2 cells and Caco-2 FGFR2i cells. Only the SHP2-Alexa 488 and PLCγ1-Alexa 647 colocalized droplets were counted. Knocking down FGFR2 reduces SHP2-Alexa 488 and PLCγ1-Alexa 647 colocalized droplets. Degree of colocalization of endogenous PLCγ1 and SHP2 determined by Pearson's R value. Sample numbers = 37 (wild type) or 38 (FGFR2i) from 2 independent experiments.
(D) Live cell images showing FGFR2ΔVT-SHP2C459S-PLCγ1 LLPS droplet formation on plasma membrane upon FGFR2ΔVT expression and activation in HEK293T SHP2 KO cells. FGFR2ΔVT, SHP2C459, and PLCγ1 were tagged with Neptune 2.5, mOrange, and mEGFP, respectively. Alexa 350-conjugated wheat germ agglutinin was used to stain the plasma membrane. (i) Serum-starved (-FGF9) cells show a low level of FGFR2ΔVT-SHP2C459S-PLCγ1 droplets colocalized on membrane; this could have been due to protein recruitment by the basally activated FGFR2. Most SHP2 and PLCγ1 proteins are diffused in cytosol. (ii) FGF9-stimulation (+FGF9, 10 ng/ml for 15 min) led to the activation of FGFR2 and enhanced FGFR2ΔVT-SHP2C459S-PLCγ1 LLPS droplet formation on the plasma membrane (see also Video S1). (iii) Expression of fluorescent tags alone does not initiate the droplet formation. (iv) In the absence of SHP2C459S-mOrange and PLCγ1-mEGFP, activated FGFR2ΔVT still forms droplets on the membrane with other endogenous cellular proteins. (v) SHP2C459S-mOrange does not form droplets in the absence of FGFR2ΔVT. (vi) PLCγ1-mEGFP does not form droplets in the absence of FGFR2ΔVT. (vii) FGFR2ΔVT cannot recruit active PLCγ1-mEGFP to the membrane in the absence of SHP2 expression, resulting in the random diffusion of PLCγ1-mEGFP.
(E) Statistical analysis of FGFR2ΔVT-SHP2C459S-PLCγ1 LLPS droplet formation in the absence (light peach) or presence (dark peach) of FGF9 stimulation. Sample numbers = 40 per condition from 3 independent experiments. Data are presented as mean ± SD.
Endogenous SHP2 and PLCγ1 form discrete membrane-bound puncta in cells
Next, we tested whether the formation of membrane-bound, phase-separated FGFR2-SHP2-PLCγ1 complex can occur with endogenous protein expression in cells. We used the human colorectal adenocarcinoma cell line Caco-2, which expresses all three proteins of interest. First, we confirmed that both endogenous SHP2 and PLCγ1 colocalized to punctate structures at the plasma membrane upon FGF9 stimulation by immunofluorescent (IF) staining (Figure 3C). Due to the lack of specific antibodies against FGFR2, we were unable to directly demonstrate the formation of endogenous FGFR2-SHP2-PLCγ1 puncta by IF. However, knocking down FGFR2 in Caco-2 cells (Caco-2 FGFR2i) (Figure S3B) resulted in decreased colocalization of endogenous SHP2 and PLCγ1 membrane-bound puncta (Figure 3C), indicating the involvement of FGFR2 in the formation of such puncta. The prevailing SHP2 and PLCγ1 puncta in the Caco-2 FGFR2i cells could be due to the incomplete knockdown or the expression of other membrane-bound RTKs, such as FGFR3, which is also highly endogenously expressed in Caco-2 cells (Erdem et al., 2017).
The puncta of FGFR2-SHP2-PLCγ1 exhibited liquid-like features in cells
We next assessed whether FGFR2-SHP2-PLCγ1 puncta displayed any liquid-like features in cells using live cell fluorescence microscopy. The results of this assessment suggest that the formation of FGFR2-SHP2-PLCγ1 condensates in vitro is highly dependent on SHP2 binding to FGFR2ΔVT. To test this, while ruling out the endogenous SHP2 forming puncta and affecting the imaging results, fluorescent protein-tagged FGFR2ΔVT, SHP2C459S, and PLCγ1 were expressed in HEK293T SHP2 KO cells (Figure S3B) and the average expression level of SHP2C459S was adjusted to approximately the endogenous SHP2 expression level (Figure S3C). Using live cell imaging with a plasma membrane marker, we observed ligand-dependent, membrane-localized puncta formation between independently fluorescent protein-tagged FGFR2ΔVT, SHP2C459S, and PLCγ1 (Figures 3Di and 3Dii). The expression of all three fluorescent protein tags alone does not promote the puncta formation (Figure 3Diii). The mobilization of the puncta containing the three proteins was visible on the plasma membrane (Video S1 of red box from Figure 3Dii). The formation of FGFR2ΔVT-EGFP or PLCγ1-EGFP droplets on the plasma membrane upon FGF9 stimulation, as well as their dynamic liquid-like fusion/fission behavior, was further supported by highly inclined and laminated optical sheet (HILO) imaging (Figures S3D and S3E; Videos S2–S4). This indicates that the FGFR2ΔVT-SHP2C459S-PLCγ1 puncta exhibit a dynamic liquid-like behavior. In the absence of SHP2C459S and PLCγ1, FGFR2ΔVT was still able to form puncta on the membrane; these are likely to include other cellular proteins, such as SHC, as we present in Figure 1A (Figure 3Div). By contrast, without FGFR2ΔVT expression and activation, neither SHP2C459S nor PLCγ1 can independently form puncta on the membrane (Figures 3Dv and 3Dvi); both proteins are randomly diffused in the cytosol. Importantly, without SHP2C459S expression, the FGFR2ΔVT is not able to recruit and retain PLCγ1 on the membrane, highlighting the role of SHP2 in controlling the membrane localization of PLCγ1 (Figure 3Dvii).
Dynamic FGFR2ΔVT-SHP2C459S-PLCγ1 droplets on plasma membrane. Confocal imaging revealed the dynamic property of FGFR2ΔVT-SHP2C459S-PLCγ1 droplets on plasma membrane upon FGF9 stimulation. FGFR2ΔVT was Neptune2.5 tagged (red), SHP2C459S was mOrange tagged (yellow), and PLCγ1 was mEGFP tagged (green). The video was recorded at a rate of 1 frame/30 sec. Scale bars, 1 μm
HILO imaging demonstrated a fusion event of FGFR2ΔVT-EGFP (coexpressed with untagged SHP2C459S and PLCγ1) on plasma membrane upon FGF9 stimulation. The video was recorded at a rate of 1 frame/1 sec. Scale bars, 10 μm
HILO imaging demonstrated a second fusion event of FGFR2ΔVT-EGFP (coexpressed with untagged SHP2C459S and PLCγ1) on plasma membrane upon FGF9 stimulation. The video was recorded at a rate of 1 frame/1 sec. Scale bars, 10 μm
HILO imaging demonstrated the fusion event of PLCγ1-EGFP (coexpressed with untagged FGFR2ΔVT and SHP2C459S) on plasma membrane upon FGF9 stimulation. The video was recorded at a rate of 1 frame/5 sec. Scale bars, 10 μm
SH2 domain interactions mediate the formation of complexes
We next examined the molecular features of the complex(es) formed by the three components of the droplets. First, we characterized the pairwise protein interactions involved in mediating the ternary complex formation. A direct interaction between FGFR2 and SHP2 has not been reported. To explore the nature of this, we used two constructs, GST-SHP2C459S and GST-SHP22SH2, in a pull-down assay to precipitate FGFR2 proteins from the HEK293T cells stably expressing FGFR2ΔVT or an enzymatically disabled version of FGFR2 (FGFR2ΔVT-KD: double Y/F mutants on the activation loop Y656/Y657 render FGFR2 kinase dead, KD) (Figure 4A). Cells were either serum-starved or stimulated with the FGF9 ligand to activate receptors. Both SHP2C459S and SHP22SH2 were able to bind FGFR2ΔVT, but not the unphosphorylatable KD mutant. Growth factor stimulation had a modest impact on the interactions observed (Figure 4A). This can be attributed to previously observed basal receptor phosphorylation (Ahmed et al., 2013; Lin et al., 2012) providing constitutive recruitment site(s) for the SH2 domain-containing SHP2 constructs (Figure 4A, input).
Figure 4.

Characterization of the interactions between FGFR2ΔVT-SHP2C459S droplets
(A) (Left) Pull-down experiments using GST-SHP2C459S or GST-SHP22SH2 (see schematic in Figure 1A) show that the binding of SHP2 requires phosphorylation of FGFR2ΔVT. FGFR2ΔVT or FGFR2ΔVT-KD (double mutant Y656/657F) stably expressing HEK293T cells were unstimulated or FGF9-stimulated (10 ng/ml, 15 min). Arrows highlight GST fusion as part of the SHP2 constructs. The lower level of interaction without FGF9 stimulation due to protein recruitment by the basally activated FGFR2 as shown in the pFGFR2 blot (Input). (Right) Densitometry analysis of GST pull down, n = 3. Data are presented as mean ± SD. Replicate data are shown in Data S1A.
(B) (Left) Pull-down experiments using different SHP2 constructs (see schematic in Figure 1A). GST-SHP2, GST-SHP2C459S, and GST-SHP22SH2 pull down pFGFR2Cyto and pFGFR2C58 while pFGFR2kinase was pulled down at a significantly lower level. Red arrows: input of recombinant pFGFR2Cyto, pFGFR2kinase, and pFGFR2C58. Long exposure was required to observe pFGFR2C58. Black arrows: fusion protein loading control indicating different SHP2 constructs. (Right) Densitometry analysis of GST pull down, n = 2. Data are presented as mean ± SD. Replicate data are shown in Data S1B.
(C) (Left) C-terminal 58 residues of FGFR2, GST-FGFR2C58 with individual Y to F substitutions, were phosphorylated and used to pull down recombinant SHP22SH2. The Y769F mutation abrogates binding. (Right) Densitometry analysis of GST pull-down level (salmon) and phosphorylation level (green) of different GST-FGFR2C58 Y/F mutants, n = 4. Data are presented as mean ± SD. Replicate data are shown in Data S1C.
(D) (Left) Pull-down experiment using GST-SHP2C459S. FGFR2ΔVT and FGFR2ΔVT-Y769F were transfected into HEK293T cells: unstimulated or FGF9-stimulated for 5 min, 15 min, or 60 min. GST-SHP2C459S binds to FGFR2ΔVT, but not to FGFR2ΔVT-Y769F, confirming that the interaction is mediated by pY769. The lower level of interaction in the absence of FGF9 stimulation is due to protein recruitment by the basally activated FGFR2 as shown in the pFGFR2 blot (Input). (Right) Densitometry analysis of GST pull down, n = 3. Data are presented as mean ± SD. Replicate data are shown in Data S1D.
To identify the pY binding site on FGFR2 and the recognition site on SHP2, we recombinantly produced and phosphorylated three different regions of the FGFR2: the intact cytoplasmic domain (pFGFR2Cyto; residues 400–821, contains the juxtamembrane region, the kinase domain, and the C-terminal tail), the kinase domain (pFGFR2kinase; 464–763), and the C-terminal tail (pFGFR2C58; 764–821). Using a GST pull-down assay with a variant of the SHP2 domains, we demonstrated the direct interaction of pFGFR2Cyto and pFGFR2C58 with full-length wild-type SHP2 (SHP2) or SHP2C459S (Figure 4B). The decrease in complex formation seen using GST-SHP2 compared with GST-SHP2C459S is likely due to the depletion of pY binding sites by phosphatase activity toward FGFR2. No interaction could be seen using the isolated PTP domains (GST-SHP2PTP and GST-SHP2PTP-C459S) or the SHP2 C-terminal tail (GST-SHP2C69) (Figure 4B). The tandem SHP2 SH2 domains (GST-SHP22SH2) also interacted with the pFGFR2Cyto and pFGFR2C58 (Figure 4B). The larger SHP2C459S construct was more efficient in precipitating FGFR2 proteins than GST-SHP22SH2. The pFGFR2Kinase interacted with SHP2, SHP2C459S, and SHP22SH2 with an apparent low affinity. The interactions of both SHP2C459S and SHP22SH2 with pFGFR2Cyto were confirmed by microscale thermophoresis (MST; Figure S4A; Table S1). Bio-layer interferometry (BLI) provided further evidence of the interaction between immobilized GST-SHP22SH2 and pFGFR2Cyto (5 μM; Figure S4B).
Having demonstrated that the C-terminal 58 residues of the receptor were sufficient to bind to SHP22SH2, a polypeptide containing these residues was used to investigate the interaction of pFGFR2 with SHP22SH2 using HSQC nuclear magnetic resonance (NMR) spectroscopy. In this case, pFGFR2C58 was titrated into 15N-SHP22SH2, and major changes in chemical shifts indicate the direct interaction (Figure S4C). Isothermal titration calorimetry (ITC) confirmed the binding and gave a stoichiometry of 1:1 for the complex (Kd∼8 μM; Figure S4D), indicating that only one SH2 domain of the tandem SH2 domains is able to bind to one pY in the C-terminal tail. To identify the pY residue(s) in GST-tagged FGFR2C58 responsible for the recruitment of SHP22SH2, each tyrosine residue was individually replaced by a phenylalanine residue. We prepared both tyrosine phosphorylated FGFR2C58 (GST-pC58 and its single Y/F mutants) and unphosphorylated FGFR2C58 (GST-C58 and its single Y/F mutants) for pulling down SHP22SH2. For the pFGFR2C58 mutants, only the Y769F mutant (GST-pC58Y769F) was unable to pull down SHP22SH2, even with a background of other pY residues (Figure 4C), indicating that Y769 is the major phosphorylation site on the FGFR2 C-terminal tail and serves as the binding site for SHP22SH2. This is the same pY residue that is required to recruit PLCγ1 to the receptor prior to the phosphorylation of the phospholipase. This previously unrecognized direct interaction involving pY769 was confirmed using a GST-SHP2C459S pull-down assay in the HEK293T cells transfected with FGFR2ΔVT or the FGFR2ΔVT-Y769F mutant (Figure 4D). As seen in Figure 4A, the FGFR2-SHP2 interaction can occur in the absence of growth factor, consistent with Y769 being highly phosphorylated even under basal conditions (Chen et al., 2008). This basal phosphorylation has been observed on equivalent tyrosine residues on other FGFRs (Huang et al., 2016; Kostas et al., 2018; Krick et al., 2018).
Finally, for mutations that disrupt the binding of pY residues in the SH2 domains (R32A in the NSH2 and R138A in CSH2), we identified that the CSH2 domain is required for the interaction with the receptor (Figures S4E and S4F); the NSH2 domain interacts with pFGFR2 with a low affinity. The recruitment of the CSH2 domain to pY769, therefore, leaves the NSH2 available for other pY-mediated interactions.
Direct binding of SHP2 and PLCγ1 is mediated by their tandem SH2 domains in a phosphorylation-independent manner
To characterize the interaction of SHP2 and PLCγ1, we first focused on the pY sites on PLCγ1 (771YGAL774, 775YEGR778, and 783YVEN786). However, both MST and ITC studies showed that SHP22SH2 fails to interact with any of the three tyrosyl phosphopeptides derived from PLCγ1 (Figures S5A and S5B; Table S1). Because the dimerization of SH2 domains has been reported (Stein et al., 2003; Hu et al., 2003; Depetris et al., 2005; Frese et al, 2006; Huculeci et al., 2015), we tested the possibility of the SHP2-PLCγ1 interaction being mediated by the heterodimerization of their tandem SH2 domains. Because the action of PLCγ1 is modulated by the intramolecular interaction with pY783, we used an extended tandem SH2 construct (residues 545–791) that includes three C-terminal tyrosine residues, namely Y771, Y775, and Y783 (schematic, Figure 1A). This polypeptide permits the added assessment of the impact of the phosphorylation state of Y783 on complex formation. Moderate affinity binding was shown to occur between the tandem SH2 domains of SHP2 and PLCγ1 (SHP22SH2-PLCγ12SH2: Kd∼1.16 ± 0.09 μM) and also when PLCγ1 is phosphorylated (SHP22SH2-pPLCγ12SH2: Kd∼0.48 ± 0.04 μM; Figure S5C; Table S1). This interaction is independent of the pY residues that are normally required to bind to SH2 domains because the SHP2 with pY recognition site mutants SHP22SH2 R32/138A still retains the ability to interact with pPLCγ12SH2 (Kd∼0.48 ± 0.03 μM; Figure S5D; Table S1).
Comparison of the NMR spectra of the isolated 15N-PLCγ12SH2 with those of the 15N-PLCγ12SH2-SHP22SH2 complex showed mostly minor, widely distributed changes (Figures S5E–S5G), consistent with small structural/dynamic changes. However, a limited number of residues showed pronounced chemical shift perturbations (CSPs) indicative of a specific binding event (Figure 5A). The mapping of the CSPs onto the structure of PLCγ12SH2 revealed that they localized to a potential binding region involving both SH2 domains (Figure 5B). The binding of SHP2 to this site would occlude the N-terminal pY binding pocket abrogating the binding of PLCγ1 to pY769 on FGFR2, thereby removing competition for this site with SHP2. The CSPs of residues connecting the CSH2 domain with the Y783 peptide region on PLCγ1 are known to be sensitive to any direct or allosteric interference because of the fast exchange equilibria between bound and unbound states (Koss et al., 2018). The absence of any corresponding CSPs indicates that the previously observed intramolecular interactions of the CSH2 domain on PLCγ1 with the pY783 binding pocket remain largely unperturbed by SHP2 binding and, hence, can preserve the active state of the phospholipase (Figure S5H; Table S1). These data lead to the assumption that when both proteins are present, only SHP2 can engage the receptor.
Figure 5.

Interactions between SHP2 and PLCγ1 droplets and the formation of ternary complexes
(A) Plot of the chemical shift changes (ppm) of the backbone amide peaks of 1H, 15N-labeled PLCγ12SH2 (200 μM) upon the addition of 3 mol L−1 equivalent of SHP22SH2. The residue numbers are indicated on the x axis.
(B) CSP of residues mapped on to the crystal structure of the PLCγ12SH2 (PDB code: 4FBN). The gradient indicates the strength of the perturbation. The pY binding pockets for NSH2 and CSH2 are shown in cyan (R562, R586, S588, E589, T590, and T596) and green (R675, R694, R696 and A703), respectively. Left hand image shows putative binding region (highlighted by increasing CSP). Right hand image shows the structure rotated into plane by 180° to show the comparatively negligible CSP on the ‘non-binding’ surface.
(C) Plot of the chemical shift changes (ppm) of the backbone amide peaks of 1H, 15N-labeled SHP22SH2 (100 μM) upon the addition of 6 mol L−1 equivalent of PLCγ12SH2. The residue numbers are indicated on the x axis.
(D) CSP of residues mapped on to the crystal structure of the SHP22SH2 (PDB code: 2SHP). The gradient indicates the strength of the perturbation. The critical pY binding residue for NSH2 and CSH2 are shown in cyan (R32) and green (R138), respectively. Left hand image shows mild CSP on the “non-binding” surface. Right hand image shows the structure rotated into plane by 180° to show the putative binding region (highlighted by increasing CSP).
(E) Formation of ternary complex revealed by GST pull-down assay. Upper panel blot: unphosphorylated GST-FGFR2Cyto does not interact with SHP22SH2 (lane 4), PLCγ12SH2 (lane 5), or pPLCγ12SH2 (lane 6). Phosphorylated GST-pFGFR2Cyto can precipitate SHP22SH2 (lane 7), PLCγ12SH2 (Lane 8), and coprecipitated SHP22SH2 and PLCγ12SH2 (lane 9). The addition of ATP/MgCl2 (5 mM) does not affect GST-pFGFR2Cyto precipitating SHP22SH2 (lane 10), whereas the phosphorylation of PLCγ12SH2 (pPLCγ12SH2) by GST-pFGFR2Cyto abolishes the interaction (lane 11). However, pPLCγ12SH2 can be precipitated by GST-pFGFR2Cyto when SHP22SH2 is present (lane 12), suggesting an adaptor function of SHP22SH2 in the ternary complex formation. Second panel blot: the phosphorylation state of PLCγ12SH2. Third panel blot: the phosphorylation state of FGFR2Cyto. Lower panel blot: total GST-FGFR2Cyto protein loading control. The figure represents 3 independent experiments. Replicate data are shown in Data S2A.
(F) The ternary complex was constituted using BLI. Upper panel: GST-pFGFR2Cyto captured on an anti-GST sensor was exposed sequentially to excess SHP22SH2 (200 μM, 0 s) and pPLCγ12SH2 (200 μM, 240 s) in the presence of 5 mM ATP/Mg2+. After each binding equilibrium was reached, the sensor was washed (120 s and 360 s). Lower panel: GST-pPLCγ12SH2 was captured on an anti-GST sensor. Then sequential binding of SHP22SH2 (200 μM, 0 s) and pFGFR2Cyto (200 μM, 240 s) was measured in the presence of 5 mM ATP/Mg2+. Dotted red lines mark the equilibrium binding of each complex. Figures are representative sensorgrams from 3 independent experiments.
(G) BiFC was used to study the formation of ternary complex. Inset: schematic depicting the interaction: pFGFR2Cyto (blue) and pPLCγ12SH2 (yellow) with split CFP tag (green). 100 nM of CN173-pFGFR2Cyto and CC173-pPLCγ12SH2 were used for the assay. The addition of increasing concentration (x axis on graph) of SHP22SH2 (purple) produces fluorescent signal (cyan), n = 3. Data are presented as mean ± SD.
(H) Time evolution of the ternary complexes simulated from the deterministic mathematical model; out of the four possible ternary complexes (pF·S·P, pF·P·S, pF·S·pP, and pF·S·pP), only pF·S·pP prevails. F, FGFR2Cyto; S, SHP22SH2; P, PLCγ12SH2.
Next, NMR was used to map the pPLCγ12SH2 binding interface on 15N-SHP22SH2; similar to the PLCγ12SH2 example, the 15N-SHP22SH2-pPLCγ12SH2 complex showed mostly minor, widely distributed changes (Figures 5C and 5D). The specific binding event is indicated by a limited number of residues that showed CSPs (F41, F71, E83, H84, S165, V170, and L212; Figures S5I and S5J). The binding of pPLCγ12SH2 appears to have no effect on either SHP22SH2 pY binding pockets that are capable of binding to pFGFR2 (i.e., CSH2 binding to pY769 and NSH2 binding to other potential pY sites, e.g., on the kinase domain).
FGFR2, SHP2, and PLCγ1 form a stable ternary complex in the LLPS state
The direct pairwise interactions between pFGFR2/SHP2 and SHP2/pPLCγ1 maintained through mutually exclusive interfaces suggests the formation of a ternary complex, which we postulate is the minimal complex required for the droplet formation involving the three interacting components. To investigate this, we conjugated FGFR2Cyto or pFGFR2Cyto on agarose beads and performed pull-down experiments using recombinant SHP22SH2, PLCγ12SH2, and pPLCγ12SH2. Unphosphorylated FGFR2Cyto failed to recruit any protein as the pairwise interactions are pY-SH2 domain-dependent (Figure 5E, lanes 4, 5, and 6). Consistent with the reported data (Bunney et al., 2012; Gresset et al., 2010), we observed an initial binding between Pfgfr2Cyto and PLCγ12SH2 (Figure 5E, lane 8) that declined as the phospholipase was phosphorylated by the receptor in the presence of ATP/MgCl2 (Figure 5E, lane 11). We also observed SHP22SH2 binding to Pfgfr2Cyto (Figure 5E, lane 7). The addition of ATP/MgCl2 to the pFGFR2Cyto and SHP22SH2 mixture does not affect the binding (Figure 5E, lane 10). Importantly, when SHP22SH2 and PLCγ12SH2 were mixed with pFGFR2, both were found to bind the receptor concurrently (Figure 5E, lane 9), possibly due to the competition between SHP22SH2 and PLCγ12SH2 bindings to pY769 on pFGFR2Cyto and/or the recruitment of a complex including both SHP22SH2 and PLCγ12SH2 into the receptor. In the presence of ATP/MgCl2, SHP22SH2 and the phosphorylated pPLCγ12SH2 can be concomitantly precipitated by pFGFR2Cyto (Figure 5E, lane 12). Because pPLCγ12SH2 does not bind to pFGFR2, our data reveal that the phospholipase must be recruited into the receptor via SHP22SH2, thus inferring the existence of a ternary complex including pFGFR2, SHP2, and pPLCγ1.
The mechanism of formation of the ternary complex was elucidated by reconstituting the interactions on a BLI sensor with immobilized GST-pFGFR2Cyto (Figure 5F, upper panel). To observe binding, the concentrations of the added proteins were typically 10-fold greater than the previously measured Kds (Table S1). Saturating the sensor with SHP22SH2 in the presence of ATP/MgCl2 showed binary complex formation. Subsequent exposure of the GST-pFGFR2Cyto/SHP22SH2 complex to excess pPLCγ12SH2, which was phosphorylated by the receptor in situ, resulted in an increase in signal, indicating the recruitment of the phospholipase to the binary complex. The reconstitution of the ternary complex on immobilized GST-tagged pPLCγ12SH2 on the sensor followed by exposure to SHP22SH2 and subsequently pFGFR2Cyto confirmed the role of SHP2 in mediating the interaction between the receptor and pPLCγ12SH2 (Figure 5F, lower panel). Additional confirmation of ternary complex formation was provided using bimolecular fluorescence complementation (BiFC) where pFGFR2Cyto and pPLCγ12SH2 were N-terminally tagged with a split cyan fluorescent protein (CFP; CN173-pFGFR2Cyto and CC173-pPLCγ12SH2, respectively; CN173: CFP residue 1–173 and CC173: CFP residue 174–238 [Kodama and Hu, 2012]). As expected, pPLCγ12SH2 was unable to bind to FGFR2, hence the absence of fluorescent response (Figure 5G). However, the addition of increasing concentrations of SHP22SH2 resulted in an increase in the fluorescent signals as pFGFR2Cyto and pPLCγ12SH2 were brought into proximity through the formation of the ternary complex. A deterministic mathematical model supports our mechanistic proposal, whereby ternary complex formation is based on the pFGFR2-SHP2 complex, which recruits pPLCγ1 (Figure 5H). In this model, the known affinities, estimated rates, and interaction constraints between pFGFR2 and pPLCγ1 were included under a range of different concentration regimes of the three interacting components of the ternary complex. In all cases, after a time course, the prevailing complex included pFGFR2, SHP2, and pPLCγ1 (see STAR Methods).
SHP2 tandem SH2 domains drive FGFR2-SHP2-PLCγ1 LLPS
Having identified the interactions that sustain the ternary complex, we sought to reconstitute the features of LLPS using SHP22SH2 and pPLCγ12SH2 constructs that represent the minimal binding regions of the intact proteins. As with the intact proteins, SHP22SH2 was able to drive LLPS with pFGFR2Cyto (Figure 6Ai); the optimum concentrations are revealed by a phase diagram (Figure S6A), and the LLPS property was confirmed by FRAP (Figure S6B). As the pY769 of FGFR2 provides the binding site for the CSH2 domain of SHP2, the addition of a pY769-mimicking peptide, which occludes the SHP2 SH2 binding site, and a general SH2 domain pY-containing peptide inhibitor resulted in the shrinking of the droplet number and size in pFGFR2Cyto–SHP22SH2 LLPS condensates (Figures 6Aii, 6Aiii and S6C). This is mirrored in condensates formed using full-length SHP2C459S (Figure S6D), thus confirming that the pY-SH2 domain interaction(s) helps to sustain the LLPS. Finally, we used the pY-binding incompetent SH2 domain mutations in SHP22SH2 (SHP22SH2 R32A, SHP22SH2 R138A, and SHP22SH2 R32/138A) to show that the blocking of the interaction of the CSH2 domain with pY769 on the C-terminal tail of pFGFR2Cyto was able to abrogate LLPS (SHP22SH2 R138A and SHP22SH2 R32/138A; Figure 6B). Importantly, SHP22SH2 R32A also abrogated phase separation, suggesting that an NSH2 domain-mediated, nonspecific, weak, pY-dependent interaction(s) is (are) necessary for phase separation. Thus, our combined binding data reveal multiple potential interactions between SHP2 and pFGFR2; i.e., CSH2 domain interacts with the phosphorylated FGFR2 C-terminal tail (Figure S4D), and also weak interactions between NSH2 from the SHP22SH2 R138A mutant and FGFR2 were observed (Figures S4E and S4F). The kinase domain of FGFR2 has six available tyrosine residues for phosphorylation. MST assays confirmed that SHP22SH2 R138A can interact with any of these six pY residues (Figure S6E; Table S1). As a result, the interactions of SHP2 SH2 domains with pFGFR2Cyto can sustain the multivalent features of phase-separated molecules.
Figure 6.

Characterization of the ternary complex formation
(A) (i) In vitro phase separation assay using Atto-labeled pFGFR2Cyto (10 μM) and truncated SHP22SH2 (30 μM). The addition of a pY769 peptide (ii) or a general pY peptide (ii) to compete SH2 domain binding reduces droplet formation. Scale bars, 10 μm.
(B) R to A mutation of residues 32 or/and 138 in the pY binding sites show that both wild-type SH2 domains of SHP2 are required (30 μM of each mutant) for LLPS with pFGFR2Cyto (10 μM). (i) Wild-type SHP22SH2. (ii) SHP22SH2 R32A. (iii) SHP22SH2 R138A. (iv) SHP22SH2 R32/138A. Scale bars, 10 μm.
(C) In vitro phase separation assay using Atto-labeled pFGFR2Cyto (10 μM), SHP22SH2 (30 μM), and pPLCγ12SH2 (12 μM). (i) Individual proteins showed no evidence of droplet formation. Droplet formation was observed after 1 min: (ii) with all three proteins; (iii) with pFGFR2Cyto and SHP22SH2, not with pFGFR2Cyto with pPLCγ12SH2 and (iv) with SHP22SH2 with pPLCγ12SH2. (v) Droplet size was diminished with increasing concentration of NaCl after incubation for 1 min and (vi) (250 mM compared with 150 mM in (ii)) in the presence of 10% 1, 6-hexanediol for 1 min (vii). Scale bars, 10 μm.
(D) (Top panel) An excess of inactive FGFR2Cyto K517I (FGFR2Cyto K517I [2000 μM]:pFGFR2Cyto [10 μM] = 200:1) was used as the substrate to monitor kinase activity. 500 μM of ATP/MgCl2 was added and incubated at room temperature for 15 min. In the context of phase-separated droplets (by adding SHP2C459S [60 μM] and pPLCγ1 [12 μM]; lane 5), the kinase activity of pFGFR2Cyto was enhanced (compare lanes 3 and 5). The addition of 10% 1,6-hexanediol (lane 9) results in the reduction of kinase activity by dissolving the phase-separated droplets (compare lane 5 and lane 9). (Bottom panel) Densitometry analysis of kinase assay, n = 5. Data are presented as mean ± SD. Replicate data are shown in Data S2C.
(E) (Top panel) A synthesized GST-phospho-substrate (600 μM, lane 1) was used to measure SHP2 (60 μM) activity in the context of phase-separated droplets. Upon the addition of FGFR2Cyto and pPLCγ1 (10 μM and 12 μM, respectively) and incubation for 15 min, the SHP2 activity is reduced in the droplets compared with the isolated phosphatase (compare lane 2 and lane 4). The addition of 10% 1,6-hexanediol (lane 5) results in the upregulation of activity by dissolving the droplets (compare lane 4 and lane 5). (Bottom panel) Densitometry analysis of phosphatase assay, n = 2. Data are presented as mean ± SD. Replicate data are shown in Data S2C.
(F) Confocal images of the effect of external phosphatase CIP (10 μM) on pFGFR2Cyto-SHP2C459S-pPLCγ1 LLPS formation. Top panel: droplet formation without CIP. Middle panel: the addition of CIP after droplet formation has limited effect on the dephosphorylation of proteins, hence droplets are still present. Lower panel: the addition of CIP to pFGFR2Cyto before droplet formation (before the addition of SHP2C459S and pPLCγ1) efficiently dephosphorylates pFGFR2Cyto; therefore, no droplet can form at the unphosphorylated state.
Western blots (below left) confirmed that pFGFR2Cyto-SHP2C459S-pPLCγ1 (10 μM, 60 μM, and 12 μM, respectively) droplet formation prevents the dephosphorylation of pPLCγ1 and pFGFR2Cyto by 10 μM of CIP (exposure 0.5 h). (Below right) Densitometry analysis of the phosphorylation state of pPLCγ1 (salmon) and pFGFR2Cyto (dark green), n = 2. Data are presented as mean ± SD. Replicate data are shown in Data S2D.
(G) The lipase activity of PLCγ1 (50 μM) was dramatically enhanced in the phase-separated environment (by adding pFGFR2Cyto [10μM] and SHP2C459S [60 μM]; green curve) compared with PLCγ1 alone (magenta curve). Sample sizes n = 4. Data are presented as mean ± SD.
pPLCγ12SH2 was introduced to determine whether the interactions between SHP2 and pPLCγ1 tandem SH2 domains are sufficient for the LLPS droplet formation. Consistent with the full-length proteins, submicrometer-sized droplets were formed upon the addition of pPLCγ12SH2 into pFGFR2Cyto-SHP22SH2, as revealed by confocal images (Figures 6Ci, 6Cii, and 6Ciii). Again, no droplet formation was observed upon adding pPLCγ12SH2 to pFGFR2Cyto because of the inability of these two molecules to interact (Figure 6Civ). The addition of salt or 10% 1,6 hexanediol supported LLPS characteristics by the shrinkage of droplet size (Figures 6Cvi and 6Cvii). The observation of droplet fusion underscored the existence of pFGFR2Cyto-SHP22SH2-pPLCγ12SH2 in an LLPS state (Figure S6F).
The LLPS affects enzyme activities in the ternary complex
To demonstrate a potential functional outcome of LLPS formation in the context of FGFR2 signaling, we employed in vitro enzymatic assays. Increased tyrosine kinase activity was demonstrated through mixing an excess of the inactive K517I-mutated FGFR2Cyto, as the substrate (FGFR2Cyto K517I; Figure 6D, lane 1), with an LLPS containing the unlabeled pFGFR2Cyto-SHP2C459S-pPLCγ1 complex in the presence of ATP/MgCl2. The formation of LLPS droplets was confirmed using a light microscope (Figure S6G). The active pFGFR2Cyto in the solution efficiently phosphorylated the FGFR2Cyto K517I substrate (Figure 6D, third panel, lane 3). Phosphorylation activity appears to be more efficient in the presence of all three components of the ternary complex in the droplet environment (Figure 6D, third panel, lane 5). The dissolution of the droplet by addition of 10% 1,6-hexanediol led to a reduction in kinase activity (Figure 6D, lane 9). Note that hexanediol does not affect the kinase activity of FGFR2 in this system (Figures 6D, lane 7 and S6H).
Replacing SHP2C459S with wild-type SHP2, we were also able to show that within the context of the pFGFR2Cyto-SHP2-pPLCγ1 LLPS, the phosphatase activity of wild-type SHP2 toward a GST-tagged phosphopeptide substrate was lowered (Figure 6E, lanes 1, 2, and 4). Again, the dissolution of the droplet by hexanediol increases the efficiency of phosphatase activity (Figure 6E lane 5) without itself affecting interactions of the phosphatase within the LLPS (Figures 6E, lane 3 and S6I). Moreover, condensed droplet formation may help to inhibit the effects of nonspecific phosphatases (alkaline phosphatase, calf intestinal [CIP]) on the activated receptor and pPLCγ1 (Figure 6F).
Finally, using an artificial substrate 4-methylumbelliferyl myo-inositol-1-phosphate, N-methyl-morpholine salt in an in vitro assay (White et al., 2014), the lipolytic activity of pPLCγ1 was shown to be greatly enhanced when it is in the phase-separated pFGFR2Cyto-SHP2C459S-pPLCγ1 complex (Figures 6G and S6J). Thus, within the LLPS state, the functional output of the ternary complex is enhanced through increased kinase and phospholipase activities and the downregulation of the phosphatase activity.
SHP2-mediated assembly of the ternary complex on the membrane provides a scaffold for PLCγ1 downstream signaling
To confirm that our in vitro functional studies extend to the in cellulo context, we stably knocked out the SHP2 expression in MCF7 cells (MCF7 SHP2 KO) (Figure 7A) or the SHP2 expression in A431 cells (A431 SHP2i) (Figure 7A) and Caco-2 cells (Caco-2 SHP2i) (Figure S7A). All three cell lines endogenously express FGFR2. In the absence of SHP2, FGF9-stimulated cells displayed impaired dephosphorylation of PLCγ1 (Figures 7A and S7A). Although the knockdown of SHP2 should result in prolonged activation of the receptor, we observed that when the phospholipase is unable to bind to SHP2, phosphorylation of the PLCγ1 downstream effectors PKCβII and AKT is significantly reduced in all cell lines (Figures 7A and S7A). The downregulation of PKCβII, a downstream protein phosphorylated on serine 660 (pS660) in response to diacyl glycerol production through the turnover of PIP2 by PLCγ1, suggests that PLCγ1 function is compromised; i.e., despite being in an activated state, PLCγ1 is unable to access its membrane-localized substrate in the absence of the scaffolding function of the FGFR2-SHP2 complex. The decoupling of the phosphatase activity of SHP2 and PLCγ1-mediated signaling is further exemplified with the addition of the SHP2 inhibitor NSC87877 (50 μM) to A431 cells (Figure S7B). Here, the level of phosphorylated PKCβII remained unaffected by the inhibition of SHP2, underscoring the idea that it is the presence, rather than the activity of SHP2, that is required for PLCγ1 downstream signaling.
Figure 7.

Phase transition of FGFR2-SHP2-PLCγ1 upregulates downstream signaling
(A) (Left) Depletion of SHP2 upregulates PLCγ1 through phosphorylation of Y783 but downregulates its downstream effectors (shown by reduced phosphorylation of PKCβII-S660 and AKT-S473) in FGF9-stimulated (10 ng/ml) MCF7 cells and A431 cells. (Right) Densitometry analysis of SHP2 expression and the activation levels of various signaling proteins (dark green: parental cells; light green: SHP2 depletion cells). MCF7 cells: n = 3; A431 cells: n = 2. Data are presented as mean ± SD. Replicate data are shown in Data S3A.
(B) Inhibition of calcium response in MCF7 SHP2 KO cells (sample size = 8) and A431 SHP2i cells (sample size = 16) (light cyan) upon FGF9 stimulation (10 ng/ml) for 1 h compared with the parental cells (dark cyan).
(C) (Left) Cell motility is reduced in the SHP2 depletion cells upon FGF9 stimulation (10 ng/ml) (MCF7 SHP2 KO cells and A431 SHP2i cells) compared with control the parental cells. (Right) Graphical representation of percentage recovery; (dark cyan) parental cells, (light cyan) SHP2 depletion cells (n = 3).
(D) (Left) Knockin SHP2 constructs restore PKCβII activity upon FGF9 stimulation (10 ng/ml) in A431 SHP2i cells. Knockin wild-type SHP2 has lower effect on restoring PKCβII activity, which could have been due to the rapid phospho-turnover mediated by overexpressed SHP2. Both SHP2C459S and SHP22SH2 greatly increase PKCβII activity, indicating that the phosphatase activity is dispensable and only the tandem SH2 domains are required. (Right) Densitometry analysis of PKCβII activity upon knock in of various SHP2 constructs (dark cyan: parental cells; light cyan: SHP2 depletion cells), n = 3. Data are presented as mean ± SD. Replicate data are shown in Data S3B.
(E) (Top) Mutations of the PLCγ1 binding interface residues on SHP2 tandem SH2 domains (SHP2C459S NMR) abolish the ability of SHP2C459S to restore PKCβII activity in MCF7 SHP2 KO cells. (Bottom) Densitometry analysis of PKCβII activity upon knockin of various SHP2 constructs (dark cyan: parental cells; light cyan: SHP2 depleted cells), n = 3. Data are presented as mean ± SD. Replicate data are shown in Data S4C.
(F) Schematic diagrams of the domains of FGFR2Cyto, SHP2, and PLCγ1. (i) Upon ligand stimulation (orange), membrane-localized FGFR2 (blue) recruits the NSH2 domain of PLCγ1 (green) into pY769 on its C terminus. This results in the phosphorylation (pY783), activation dissociation from FGFR2 of PLCγ1. FGFR2 can also recruit SHP2 (purple) CSH2 domain into pY769 (ii). The FGFR2-SHP2 complex is then available to recruit the active PLCγ1 through the tandem SH2 domains of SHP2 and PLCγ1. The “secondary interaction” mediated by the SHP2 NSH2 domain and pYs on the FGFR2 kinase domain further provide the multivalency for the phase separation of the ternary complexes on cellular membrane (iii).
Further corroboration of this scaffolding role for SHP2 was shown by the depletion of SHP2 in MCF7 SHP2 KO cells and A431 SHP2i cells, which led to depressed Ca2+ concentrations as a result of the downregulation of PLCγ1-mediated signaling (Figure 7B). In agreement with this, we also observed a reduction in the functional output of cell motility in MCF7 SHP2 KO cells and A431 SHP2i cells (Figure 7C), concomitant with the loss of PLCγ1 function.
To confirm whether the SHP2 scaffold or enzymatic function is responsible for impaired PLCγ1 signaling, we transiently transfected three different SHP2 constructs (SHP2, SHP2C459S, and SHP22SH2) into A431 SHP2i cells. The expression of wild-type SHP2 in A431 SHP2i cells results in the rescue of PKCβII activity as we expected; however, the expression of phosphatase-dead mutant (SHP2C459S) or just the tandem SH2 domains (SHP22SH2) is also sufficient to restore the activity of PKCβII (Figure 7D).
Downstream signaling outputs of the FGFR2-SHP2-PLCγ1 LLPS complex reveal a distinct membraneless subcellular platform for RTK FGFR2 signaling
We reasoned that if a higher-order assembly is critical for robust PLCγ1-PKCβII signaling, then blocking the formation of FGFR2-SHP2-PLCγ1 LLPS complexes should decrease downstream signaling outputs. We first focused on the FGFR2-SHP2 interaction. As we have shown in Figure 6A, the addition of pY peptides disrupts the formation of FGFR2-SHP2 LLPS condensates. Indeed, the overexpression of the RFP-tagged FGFR2 C-terminal tail resulted in the downregulation of PKCβII activity in MCF7 cells in the presence of FGF9 stimulation (Figure S7C). We also selected a shortened peptide from the 58-residue C-terminal tail (764TTNEEpYLDLSQP775) that included pY769. The overexpression and phosphorylation of this peptide in Caco-2 cells upon FGF9-induced FGFR2 activation (Chen et al., 2008) also result in the downregulation of PKCβII activity (Figure S7D). These data further demonstrate the importance of the FGFR2-SHP2 interaction in maintaining PLCγ1-PKCβII signaling.
Finally, we disrupted the SHP2-PLCγ1 binding interface on SHP2 to validate the role of SHP2 in connecting FGFR2 and PLCγ1 signaling. Based on our NMR study, w=4e mutated a number of residues on the binding interface of SHP22SH2 (F41A, F71A, E83A, H84A, S165A, V170A, and L212A; Figures S5I and S5J). These mutations on SHP2 reduce its affinity for pPLCγ12SH2 but have no effect on its ability to bind to pFGFR2Cyto (Figure S7E; Table 1). Importantly, knocking in the SHP2C459S construct shows the expected PKCβII phosphorylation in response to PLCγ1 activation at the membrane. However, the SHP2C459S construct containing the interface mutations (SHP2C459S NMR) fails to rescue the PKCβII phosphorylation in MCF7 SHP2 KO cells compared with SHP2C459S (Figure 7E), suggesting a potential targeting area for treatment.
Discussion
RTK function depends on the ability to recruit downstream, cytoplasmic signaling components to the plasma membrane. In the cytoplasm, proteins diffuse randomly to their receptor targets largely unaided energetically. Thus, it is expected that the propagation of a mutually exclusive signal needs to be supported by mechanisms that ensure timely delivery of the correct proteins to the RTK with greater reliability than the probability-dependent diffusion process.
Here, we report the formation of LLPS condensates sustained by phosphorylated RTKs upon the recruitment of the signaling protein SHP2 or SHC. We honed in on a functional plasma membrane-localized LLPS, the basic element of which is a ternary protein complex composed of active pFGFR2, SHP2, and pPLCγ1 (Figures 7Fi, 7Fii, and 7Fiii). The assembly of the three proteins to form the condensate requires the phosphorylation-dependent interactions of FGFR2 and the tandem SH2 domains of SHP2, which provide multivalency and drive the higher-order FGFR2-SHP2 complex assembly. The FGFR2-SHP2 condensate further recruits activated pPLCγ1 through the tandem SH2 domain heterodimerization; this process ensures the membrane localization of the activated PLCγ1 facilitating access to the phospholipid substrate.
A unique aspect of the RTK-LLPS complex observed in this work is that, unlike previously reported membraneless organelles or particles (Amaya et al., 2018; Brangwynne et al., 2009; Delarue et al., 2018; Sheu-Gruttadauria and MacRae, 2018; Shin et al., 2018), it is sustained by a plasma membrane-bound receptor. This membrane association limits the extent of the expansion of the droplet into the cytoplasm (Su et al., 2016; Case et al., 2019b), resembling a droplet of condensed protein on the inner leaflet of the membrane ( Su et al., 2016; Huang et al., 2019).
The idea that LLPS can function as a switch for enzyme activity (Li et al., 2012) in RTK-mediated pathways is observed here in controlling RTK FGFR2 signaling. The restriction of SHP2 phosphatase and amplification of both FGFR2 and PLCγ1 activities drive PKCβII signaling with a potential cancer outcome (El-Gamal et al., 2014; Sledge and Gökmen-Polar, 2006; Teicher, 2006). Alongside the elevated apparent local concentrations associated with the condensed state, it is likely that within the highly charged, multivalent milieu of a globule, catalytically favorable conformations are more easily accessible. Indeed, LLPS-component mutant forms of SHP2 have been seen to show elevated activity in vitro based on conformational perturbation (Zhu et al, 2020).
Limitations of the study
The assembly of membraneless RTK-LLPS complexes may be a general phosphorylation- and concentration-dependent mechanism for activating RTKs and recruiting signal proteins. Additional work is needed to ascertain the biological roles of LLPS in RTK signaling, as well as to investigate its potential links to the manifold disease states associated with the perturbations of RTK function. It will also be of interest how the LLPS formation affects the efficacy of therapeutic agents that target RTKs or their effectors.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-FGFR Rabbit Polyclonal | Santa Cruz Biotechnology | Cat#: sc-122; RRID:AB_631509 |
| Anti-Phospho-FGF Receptor (Tyr653/654) Rabbit Polyclonal | Cell Signaling Technology | Cat#: 3471; RRID:AB_331072 |
| Anti-SHP22SH2 Mouse Monoclonal | Santa Cruz Biotechnology | Cat#: 271053; RRID:AB_10612217 |
| Anti-SHP2 Rabbit Polyclonal | Sigma-Aldrich | Cat#: SAB1300500; RRID:AB_10610174 |
| Anti-SHP2 Goat Polyclonal, knockdown validated | Invitrogen | Cat#: PA5-17956; RRID:AB_10984971 |
| Anti-Phospho-p44/42 MAPK (Erk1/2) Rabbit Polyclonal | Cell Signaling Technology | Cat#: 9101; RRID:AB_331646 |
| Anti-p44/42 MAPK (Erk1/2) Rabbit Monoclonal | Cell Signaling Technology | Cat#: 4695; RRID:AB_390779 |
| Anti-PLCγ1 Rabbit Polyclonal | Santa Cruz Biotechnology | Cat#: SC-81; RRID:AB_632202 |
| Anti-PLCγ1 Rabbit Polyclonal - KO Validated | Abcam | Cat#: ab107455; RRID:AB_11156766 |
| Anti-Phospho-PLCγ1 (Tyr783) Rabbit Polyclonal | Cell Signaling Technology | Cat#: 2821; RRID:AB_330855 |
| Anti-PLCγ1 Rabbit Polyclonal, KO validated | Cell Signaling Technology | Cat#: 2822; RRID:AB_2163702 |
| Anti-Phospho-PKC (pan) (βII Ser660) Rabbit Polyclonal | Cell Signaling Technology | Cat#: 9371; RRID:AB_2168219 |
| Anti-PKC (pan) Mouse Monoclonal | Santa Cruz Biotechnology | Cat#: SC-13149; RRID:AB_628144 |
| Anti-Phospho-Akt (Ser473) Rabbit Monoclonal | Cell Signaling Technology | Cat#: 3787; RRID:AB_331170 |
| Anti-Akt Rabbit Polyclonal | Cell Signaling Technology | Cat#: 9272; RRID:AB_329827 |
| Anti- α-Tubulin Rabbit Monoclonall | Cell Signaling Technology | Cat#: 2125; RRID:AB_2619646 |
| Anti-GST Rabbit Polyclonal | Cell Signaling Technology | Cat#: 2622; RRID:AB_331670 |
| Anti-p-Tyr Mouse Monoclonal | Santa Cruz Biotechnology | Cat#: 7020; RRID:AB_628123 |
| Anti-6xHis Mouse Monoclonal | Clontech | Cat#: 631212; RRIB:AB_2721905 |
| Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 488 | Invitrogen | Cat#: A32731; RRID:AB_2633280 |
| Donkey anti-Goat IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 594 | Invitrogen | Cat#: A32758; RRID:AB_2762828 |
| Bacterial and virus strains | ||
| BL21 (DE3) | New England Biolabs | Cat#: C2527H |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant Human FGF-9 Protein, CF | R&D Systems | Cat#: 273-F9/CF |
| Protease Inhibitor Cocktail Set III, EDTA-Free | Merck | Cat#: 539134 |
| Metafectene reagent | Biontex | Cat#: T020-1.0 |
| TransIT-2020 reagent | Mirus | Cat#: MIR 5404 |
| X-tremeGENE HP DNA transfection reagent | SIGMA | Cat#: 6366236001 |
| TransfeX transfection reagent | ATCC | Cat#: ATCC ACS-4005 |
| NSC 87877 | Santa Cruz Biotechnology | Cat#: sc-204139 |
| DMSO | SIGMA | Cat#: D2650 |
| 1, 6-Hexanediol | SIGMA | Cat#: 240117 |
| Lipoic acid | SIGMA | Cat#: 62320 |
| Lipoamide | Santa Cruz Biotechnology | Cat#: sc-239160 |
| 4-methylumbelliferyl myo-inositol-1-phosphate, N-methyl-morpholine salt |
Carbosynth Limited | Cat#: M-5717 |
| Alkaline Phosphatase, Calf Intestinal (CIP) | New England Biolabs | Cat#: M0290 |
| DOPC | Avanti | Cat#: 850375 |
| Ni2+-NTA DOGS | Avanti | Cat#: 790404 |
| PEG5000 PE | Avanti | Cat#: 880230 |
| Atto-488 NHS ester | SIGMA | Cat#: 41698 |
| Atto-594 NHS ester | SIGMA | Cat#: 79636 |
| Atto-647 NHS ester | SIGMA | Cat#: 07376 |
| NTA-Atto 550 | SIGMA | Cat#: 94159 |
| Ammonium Chloride (15N, 99%) | Goss Scientific | Cat#: NLM-467 |
| D-Glucose (U-13C6, 99%; 1,2,3,4,5,6,6-D7, 97-98%) | Goss Scientific | Cat#: CDLM-3813 |
| FGFR2 pY769 peptide: TTNEE{pY}LDLSQP | Genscript | Customized |
| PLCγ1 pY771 peptide: TAEPD{pY}GALYEG | Genscript | Customized |
| PLCγ1 pY775 peptide: DYGAL{pY}EGRNPG | Genscript | Customized |
| PLCγ1 pY783 peptide: RNPGF{pY}VEANPM | Genscript | Customized |
| Experimental models: Cell lines | ||
| HEK293T | ATCC | CRL-1573; RRID:CVCL_0045 |
| HEK293T SHP2 KO | This manuscript | N/A |
| A431 SHP2i | Ahmed et al., 2013 | N/A |
| A431 | ATCC | CRL-1555; RRID:CVCL_0037 |
| Caco-2 | ATCC | HTB-37; RRID:CVCL_0025 |
| Caco-2 SHP2i | This manuscript | N/A |
| MCF7 | ATCC | HTB-22; RRID:CVCL_0031 |
| MCF7 SHP2 KO | This manuscript | N/A |
| Recombinant DNA | ||
| EGFRKinase-Tail | This manuscript, residue 712-1210, cloned in pET28b | N/A |
| Her2Kinase-Tail | This manuscript, residue 720-1255, cloned in pET28b | N/A |
| Her4Kinase-Tail | This manuscript, residue 718-1308, cloned in pET28b | N/A |
| FGFR1Kinase-Tail | This manuscript, residue 478-822, cloned in pET28b | N/A |
| FGFR2Cyto | This manuscript, residue 400-821, cloned in pET28b | N/A |
| VEGFR1Kinase-Tail | This manuscript, residue 827-1338, cloned in pET28b | N/A |
| VEGFR2Kinase-Tail | This manuscript, residue 834-1356, cloned in pMAL-c5X | N/A |
| SHP2 | This manuscript, residue 1-593, cloned in pET28b | N/A |
| SHP2C459S | This manuscript, residue 1-593, with C459S mutant, cloned in pET28b | N/A |
| SHC (p52) | Suen et al., 2013 | N/A |
| PLCγ1 | This manuscript, residue 1-1291, cloned in pET28b | N/A |
| FGFR2Cyto K517I | This manuscript, residue 00-821, with K517I mutant, cloned in pET28b | N/A |
| FGFR2Kinase | This manuscript, residue 464-763, cloned in pET28b | N/A |
| FGFR2C58 | This manuscript, residue 764-821, cloned in pET28b | N/A |
| SHP22SH2 | This manuscript, residue 1-221, cloned in pET28b | N/A |
| SHP22SH2 R32A | This manuscript, residue 1-221, with R32A mutant, cloned in pET28b | N/A |
| SHP22SH2 R138A | This manuscript, residue 1-221, with R138A mutant, cloned in pET28b | N/A |
| SHP22SH2 R32/138A | This manuscript, residue 1-221, with R32/138A mutant, cloned in pET28b | N/A |
| PLCγ12SH2 | This manuscript, residue 545-791, cloned in pET28b | N/A |
| GST-SHP2 | This manuscript, residue 1-593, cloned in pGEX-4T1 | N/A |
| GST-SHP2C459S | This manuscript, residue 1-593, with C459S mutant, cloned in pGEX-4T1 | N/A |
| GST-SHP22SH2 | This manuscript, residue 1-221, cloned in pGEX-4T1 | N/A |
| GST-SHP22SH2 R32A | This manuscript, residue 1-221, with R32A mutant, cloned in pGEX-4T1 | N/A |
| GST-SHP22SH2 R138A | This manuscript, residue 1-221, with R138A mutant, cloned in pGEX-4T1 | N/A |
| GST-SHP22SH2,R32/138A | This manuscript, residue 1-221, with R32/138A mutant, cloned in pGEX-4T1 | N/A |
| GST-SHP2PTP | This manuscript, residue 247-525, cloned in pGEX-4T1 | N/A |
| GST-SHP2PTP C459S | This manuscript, residue247-525, with C459S mutant, cloned in pGEX-4T1 | N/A |
| GST-SHP2C69 | This manuscript, residue 526-593, cloned in pGEX-4T1 | N/A |
| GST-PLCγ12SH2 | This manuscript, residue 545-791, cloned in pGEX-4T1 | N/A |
| GST-FGFR2C58 | This manuscript, residue 763-821, cloned in pGEX-4T1 | N/A |
| GST-FGFR2C58 Y769F | This manuscript, residue763-821, with Y769F mutant, cloned in pGEX-4T1 | N/A |
| GST-FGFR2C58 Y779F | This manuscript, residue 763-821, with Y799F mutant, cloned in pGEX-4T1 | N/A |
| GST-FGFR2C58 Y783F | This manuscript, residue 763-821, with Y783F mutant, cloned in pGEX-4T1 | N/A |
| GST-FGFR2C58 Y805F | This manuscript, residue 763-821, with Y805F mutant, cloned in pGEX-4T1 | N/A |
| GST-FGFR2C58 Y812F | This manuscript, residue 763-821, with Y812F mutant, cloned in pGEX-4T1 | N/A |
| GST-FGFR2Cyto | This manuscript, residue 400-821, cloned in pGEX-4T1 | N/A |
| GST-substrate | This manuscript, gene fragment encodes 4xDADEYLIPQQG, cloned in pGEX-4T1 | N/A |
| CN173-FGFR2Cyto | This manuscript, residue 400-821, N-terminally fused with residue 1-173 of CFP, cloned in pET28b | N/A |
| CC173-PLCγ12SH2 | This manuscript, residue 545-791, N-terminally fused with residue 174-238 of CFP, cloned in pET28b | N/A |
| SHP2C459S-RFP | This manuscript, residue 1-593, with C459S mutant, cloned in pRFP-C | N/A |
| pcDNA6_FGFR2ΔVT | This manuscript, residue 1-821, cloned in pcDNA6 | N/A |
| pcDNA6_FGFR2ΔVT Y769F | This manuscript, residue 1-821, with Y769F mutant, cloned in pcDNA6 | N/A |
| pcDNA6_FGFR2ΔVT-Y656/657F | This manuscript, residue 1-821, with Y656/657F mutant, cloned in pcDNA6 | N/A |
| FGFR2ΔVT-Neptune 2.5 | This manuscript, residue 1-821, cloned in Neptune 2.5 | N/A |
| SHP2C459S-mOrange | This manuscript, residue 1-593, cloned in mOrange-N1 | N/A |
| PLCγ1-mEGFP | Bunney et al., 2012 | N/A |
| Deposited data | ||
| Original western data and microscopy data for figures | This paper | DOI:10.17632/hpm8ccskgh.1 |
| Software and algorithms | ||
| Origin | OriginLab | Version 9.1; RRID:SCR_014212 |
| ImageJ (Fiji) | NIH | https://imagej.nih.gov/ij/; RRID:SCR_002285 |
| PyMOL | PyMOL | https://www.pymol.org; RRID:SCR_000305 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, John E. Ladbury (j.e.ladbury@leeds.ac.uk)
Materials availability
Plasmids generated in this study will be available upon request.
Experimental model and subject details
Mammalian cell culture
HEK293T, A431 and MCF7 cells were maintained in DMEM (Dulbecco’s modified Eagle’s high glucose medium) supplemented with 10% (v/v) FBS (foetal bovine serum) (Caco-2 cells were maintained in EMEM (Eagle’s minimum essential medium with 20% FBS) and 1% antibiotic/antimycotic (Lonza) in a humidified incubator with 10% CO2. shRNA control cells (A431 Ci) and SHP2 knockdown cells (A431 SHP2i) were maintained as described previously (Ahmed et al., 2013).
Escherichia coli strains
E. coli BL21(DE3) cells were used in this study for the production of recombinant proteins. Cells were cultured in 2x YT medium.
Method details
Cloning, expression and purification of recombinant proteins
SHP2 is known to be recruited to FGFRs through the scaffold protein FRS2 upon receptor activation (Hadari et al., 1998; Ong et al., 1997; Ong et al., 2000). To mitigate this in our cell-based assays the FGFR2ΔVT variant, which lacks the critical 428VT429 motif was adopted (Burgar et al., 2002). The full length SHP2, EGFR, Her2, and Her4 plasmid templates were obtained from Addgene (SHP2: #8381, EGFR: #81926, Her2: #16257, Her4: #29527). The full-length PLCγ1 mEGFP expression vector was a kind gift from Dr. Matilda Katan (University College London, UK). The full-length VEGFR1 and VEGFR2 mEGFP expression vector was a kind gift from Dr. Sreenivasan Ponnambalam (University of Leeds, UK). Gene fragments that encode different regions of SHP2 or RTK proteins as we described in the text and key resources table were amplified using standard PCR and cloned into prokaryotic or eukaryotic expression vectors as designed.
All His-tagged, GST-tagged, or MBP-tagged recombinant proteins were purified from BL21(DE3) cells. A single colony was used to transform 100 ml of 2xYT which was grown overnight at 37°C. 1L of 2xYT were inoculated with 10 ml of this overnight culture and were allowed to grow at 37°C until the OD600=0.8 at which point the culture was cooled down to 20°C and expression was induced with 1 mM IPTG. Cultures were allowed to grow for a further 12 hours before harvesting by centrifugation. Cells were re-suspended in 20 mM Tris, 150 mM NaCl, 10% glycerol, pH 8.0 in the presence of protease inhibitors and lysed by sonication. Insoluble material was removed by centrifugation (40,000 g at 4°C for 60 min).
For the purification of His-tagged proteins, the soluble fraction was applied to a Talon column. Following a wash with 10 times column volume of washing buffer (20 mM Tris, pH 8.0, 150 mM NaCl, and 1mM ß-mercaptoethanol) protein was eluted from the column with elution buffer ((20 mM Tris, pH 8.0, 150 mM NaCl, 150 mM imidazole, and 1mM ß-mercaptoethanol). Eluted proteins were concentrated to 5 ml and applied to a Superdex75 gel filtration column in buffer containing 20 mM HEPES, 150 mM NaCl and 1 mM TCEP pH 7.5. Analysis of pure proteins on SDS-PAGE showed greater than 98% purity. For the preparation of untagged SHP2C459S and PLCγ1 proteins, the purified proteins were incubated with 1 ml of talon beads for the cleavage procedure as described below.
For the purification of GST-tagged proteins, the soluble fraction was applied to a GST column. Following a wash with 10 times column volume of washing buffer (20 mM Tris, pH 8.0, 150 mM NaCl, and 1mM ß-mercaptoethanol) protein was eluted from the column with elution buffer ((20 mM Tris, pH 8.0, 150 mM NaCl, 20 mM glutathione, and 1mM ß-mercaptoethanol). Eluted proteins were concentrated to 5 mL and applied to a Superdex75 gel filtration column in buffer containing 20 mM HEPES, 150 mM NaCl and 1 mM TCEP pH 7.5. Analysis of pure proteins on SDS-PAGE showed greater than 98% purity.
For the purification of MBP-VEGFR2Kinase-Tail protein, the soluble fraction was applied to 1 ml of Amylose agarosed. Following a wash with 20 times column volume of washing buffer (20 mM Tris, pH 8.0, 150 mM NaCl, and 1mM ß-mercaptoethanol), the purity of MBP-VEGFR2Kinase-Tail on beads was analysed on SDS-PAGE and showed greater than 98% purity.
Transformed E. coli for the expression of 1H, 15N, 13C-labelled protein for backbone resonance assignment was initially grown in normal LB broth overnight at 37°C. The next day cells were harvested and transferred to 100 ml M9 media containing deuterated D-glucose (U-13C6, 99%, 1, 2, 3, 4, 5, 6, 6-D7, 97-98%), 15N-labeled ammonium chloride (15NH4Cl), and grew overnight at 30°C. The next day the pre-culture was transferred to 1000 ml of labelled M9 media (starting OD600∼0.1) and incubated at 37°C, 220rpm. When the OD600 was reached ∼0.8, IPTG was added to the final concentration of 1 mM. The protein was expressed for 16 hr at 20°C before harvesting.
Cleavage of affinity tags
Purified proteins were rebound to their affinity agarose beads for the cleavage of tags. Briefly, protein on agarose beads was prepared as a 50% slurry. Thrombin (1 unit for 1mg of protein) was added to the slurry and rotate gently at 4°C overnight. On the following day, thrombin was removed by passing the solution from the slurry through 1 ml of benzamidine agarose beads and the untagged target protein was collected from the benzamidine beads flowthrough. The untagged proteins were further purified using a Superdex 75 gel filtration column as described above.
Plasmids transfection and viral infection
Transfection of plasmids into HEK293T cells was performed using Metafectene transfection reagent. Transfection of plasmids into MCF7 cells was performed using TransIT transfection reagent. Transfection of plasmids into A431 cells was performed using X-tremeGENE HP DNA transfection reagent. Transfection of plasmids into Caco-2 cells was performed using TransfeX transfection reagent. Transfections were carried out according to manufacture’s protocols and all plasmid DNA was prepared using QIAprep Spin Miniprep Kit (QIAGEN).
CRISPR knockdown/knockout
FGFR2 or SHP2 expression in different cell lines was knockdown or knockout using CRISPR. Briefly, pLentiCRISPR v2 plasmid containing FGFR2 gRNA target sequence (GTACCGTAACCATGGTCAGC) of SHP2 gRNA target sequence (GAGACTTCACACTTTCCGTT) were purchased from GenScript. pLentiCRISPR v2 was co-transfected with the packaging plasmids pMD2.G and psPAX2 (2:1:1 ratio) into HEK293T cells. Collect virus-containing medium 48 hours after transfection and pass viral media through a 0.45uM low protein-binding filter. The viral supernatant can be used to infect cells or frozen at -80°C. For the infection, cells were treated with 10 μg/ml polybrene and 1 ml of virus solution was used to infect cells in one well of a 6-well plate with 1 ml of culture medium. Infected cells were incubated for 72 hours, selection was performed by changing the medium containing puromycin (1 μg/ml for HEK293T, 2 μg/ml for A431, MCF7, and Caco-2 cells) every 2-3 days. Protein expression levels were confirmed using western blotting.
In vitro phosphorylation of purified proteins
Purified RTK proteins were autophosphorylated by incubating 10μM of protein with 5mM of ATP/MgCl2 at room temperature for 2 hours. Reactions were quenched by adding 50mM EDTA. Phoshorylated RTK proteins were further purified using a Superdex 75 gel filtration column (in 20 mM HEPES, pH 7.5, 150 mM NaCl, and 1 mM TCEP) as described above.
Phosphorylated PLCγ1 proteins were prepared by incubating with recombinant FGFR2pCyto conjugated on agarose beads with 5 mM ATP and 5 mM MgCl2 for 2 hours. The phosphorylation reactions were quenched by adding EDTA (prepared in 20 mM HEPES, pH 7.5) to a final concentration of 50mM, and FGFR2pCyto protein (on beads) was removed after the phosphorylation and quenching reactions by centrifugation. The supernatant solution that contains phosphorylated PLCγ1 proteins was further purified using a Superdex 75 gel filtration column (in 20 mM HEPES, pH 7.5, 150 mM NaCl, and 1 mM TCEP) as described above.
The phosphorylation states of proteins were analysed by gel-shift assays (Lee et al., 2019) on SDS-PAGE and immunoblotting using an anti-phosphotyrosine antibody and showed high degrees of homologues.
Protein fluorescent labelling
Highly purified FGFR2 and PLCγ1 proteins (in 20 mM HEPES, pH 7.5, 150 mM NaCl, and 1 mM TCEP) were prepared in 100 mM NaHCO3 buffer (pH 8.3) at 2 mg/ml and labelled with Atto-488 NHS ester or Atto-647 NHS ester (Sigma) respectively and incubated at room temperature for 1 hr (fluorophore to protein molar ratio was 1:1). Highly purified SHP2 proteins (in 20 mM HEPES, pH 7.5, 150 mM NaCl, and 1 mM TCEP) were directly labelled with NTA Atto-550 at room temperature for 10 minutes (fluorophore to protein molar ratio was 1:1). For untagged SHP2 labelling (used in support lipid bilayers experiment) highly purified SHP2 proteins (in 20 mM HEPES, pH 7.5, 150 mM NaCl, and 1 mM TCEP) were prepared in 100 mM NaHCO3 buffer (pH 8.3) at 0.5 mg/ml and labelled with Atto-594 NHS ester and incubated at room temperature for 20 minutes to reduce non-specific labelling that affect the SH2 domain binding ability. Excess dye was removed using G-15 desalting chromatography. Proteins were concentrated and labelling efficiency was measured by Nanodrop 2000 (ThermoFisher).
Confocal microscopy for in vitro droplet formation
Purified proteins were mixed or diluted with buffer (20 mM HEPES, pH 7.5, 150 mM NaCl, and 1 mM TCEP) to desired combinations and concentrations, and incubated at room temperature for 1 min before imaging. To monitor the effect of NaCl, 1, 6-hexanediol, lipoic acid, or lipoamide in the formation of droplets, mixtures were prepared by adding compounds at last and incubated for 1 minutes. Finally, 4μl of each sample was pipetted onto a 3-well chambered cover glass slides (Thermo Scientific, ER-303B-CE24). Images were acquired on either Leica SP8 or Zeiss LSM880 microscopes. Super-resolution imaging was performed on Zeiss LSM880 using the AiryScan settings.
Supported lipid bilayer assay
Methods for preparing supported lipid bilayers have been described (Banjade and Rosen, 2014; Case et al., 2019a; Huang et al., 2019; Su et al., 2017; Zeng et al., 2018). In general, phospholipids containing 98% POPC (Avantilipids), 2% DGS-NTA-Ni (Avantilipids) and 0.1% PEG 5000 PE (Avantilipids) were mixed and dried under a stream of nitrogen and resuspended in PBS. Finally, the vesicle solution was sonicated for 90 seconds in an ice-water bath to make small unilamellar vesicles (SUVs) and the solution was subjected to a centrifugation at 33,500 g for 45 min at 4°C. Supernatant containing SUVs was collected. The membrane reconstitution system was prepared on a homemade chambered cover glass (cleaned with Hellmanex III (Helllma Analytics) overnight, thoroughly rinsed with H2O followed by 15 minutes incubation in 1:1 IPA/H2O, followed by excessive rinsing of H2O. Supported lipid bilayers were form on the cleaned glass slides by incubating the SUVs mixed with PBS for at least 30 min. The chambers were then rinsed with PBS buffer, followed by incubation of 1 mg/mL BSA in PBS for 10 min to block defects in supported membranes. Supported lipid membranes were further washed with the protein buffer for twenty times.
pFGFR2Cyto (20μM; His-tagged) was added to the membrane and incubated for 5 minutes, solution was buffer exchanged into protein buffer (20 mM HEPES, pH 7.5, 150 mM NaCl, and 1 mM TCEP) to remove excess protein. After imaging, untagged SHP2 and PLCγ1 proteins were sequential added for confocal imaging. Between all incubation steps, the chamber were washed with protein buffer (20 mM HEPES, pH 7.5, 150 mM NaCl, and 1 mM TCEP) thoroughly to remove unbound proteins. All preparations were done at room temperature.
Fluorescence recovery after photobleaching (FRAP) assay
FRAP assay was performed on a Zeiss LSM 880 inverted confocal microscope at room temperature. Fluoroscent signals were bleached using the appropriate corresponding laser beam. Fluorescence intensity was recorded in two regions: a region that was bleached and a region of an equal size that was not bleached. The unbleached region was used as a control for the stability of fluorescence signal throughout the FRAP experiment. Fluorescence signal from the bleached region was normalised to the unbleached region, which was then expressed as a fraction of the normalised signal prior to bleaching. The fluorescence intensity difference between pre-bleaching and at time 0 (the time point immediately after photobleaching pulse) was normalized to 1. The experimental control was based on quantification of fluorescence intensities of similar droplet/membrane regions without photobleaching.
Immunofluorescence
HEk293T cells expressing fluorescent tagged FGFR2, SHP2, or PLCγ1 and Caco-2 cells that endogenously express all three proteins were seeded on coverslips in a 24-well plate. After serum-starvation for 16 hours, cells were stimulated with 10 ng/ml FGF9 for 15 minutes or remained untreated. Cells were fixed for 15 minutes with 4%paraformaldehyde, washed, and incubated in blocking buffer for 1 hour (1X PBS with 1% BSA and 0.5% Triton X-100). Blocking buffer was aspirated and cells were incubated with primary antibody overnight in the dark at 4°C. The following day, cells were washed, incubated with fluorophore-conjugated secondary antibodies for 1 hour at room temperature in the dark, washed, and then mounted. Slides were analyzed using a Zeiss LSM 880 inverted confocal microscope and images were analyzed using ImageJ software.
Plasma membrane staining and live cell imaging
HEK293T SHP2 KO cells expressing FGFR2ΔVT-Neptune 2.5, SHP2C459S-mOrange, and PLCγ1-mEGFP or expressing fluorescent proteins as control experiments were seeded in Ibidi μ-Dish (Ibidi, 81156) coated with Poly-D-Lysine (Sigma, P4832). Cells were serum-starved for 16 hours. For plasma membrane staining, wheat germ agglutinin (Alexa 350 conjugated, Invitrogen, W11263) was prepared in PBS as a 1 mg/ml stock. Before imaging experiment, cells were washed 3 times with PBS and labelling was performed using 0.01 mg/ml wheat germ agglutinin at 37°C for 10 min. When the labelling is complete, labelling solution was removed and cells were washed 3 times with PBS and starvation medium was added. Cells were direct subjected to imaging experiment or stimulated with FGF9 (10 ng/ml) for 15 minutes before performing confocal imaging.
Highly inclined and laminated optical sheet (HILO) microscopy
HILO microscopy (Tokunaga et al., 2008) was performed on a home-built system based on an open stage for mounting the sample and fluorescence microscopy optics (RM21, Mad City Labs, with XYZ-nanopositioner). The illumination beam (488 nm laser, LBX-488-150-CSB-PP, Oxxius) was expanded 14x (aspheric lens A220TM-A, pinhole P20D, collimator AC254-150-A-ML, Thorlabs), and translated across and focussed on the back focal plane of the objective (60X, NA 1.5: UPLAPO60XOHR, Olympus) using a motorised mirror and lens stage (TIRF Module, Mad City Labs). A spectral filter (Di01-405/488/561/635-25x36, Semrock) separated excitation and emission light. Images were formed with a tube lens (TTL180-A, f = 180 mm, Thorlabs) and captured on a cooled sCMOS camera (Prime BSI, Photometrics Teledyne). The system was controlled with Micro-Manager 2.0 (Edelstein et al., 2010).
Image processing was carried out in FIJI (Schindelin et al., 2012). The Z-stack (depth step: 0.25 μm) was depth-coded (colour map: “Ice”) with a macro based on K_TimeRGBcolorcode.ijm by Kota Miura. Maximum intensity projections are shown. Contrast stretching aids the visibility of the protein clusters and cellular area. For the XZ view, the image was scaled in Z so that the scale bar applies isotropically to both the XY and XZ images (XY pixels: 0.108 μm). Images were acquired after stimulation of FGFR2 with FGF9 ligand.
Pulldown and western blots
GST-tagged proteins immobilized on Glutathione Sepharose (GE Healthcare Life Science) as a 50% slurry. For pulldown experiments using mammalian cells, cells were grown in 10cm dishes, serum starved overnight and stimulated with 10 ng/ml FGF9 for the indicated time period. Cells were lysed with buffer containing 50 mM HEPES (pH 7.5), 0.1% (v/v) NP-40, 10 mM NaF, 1 mM sodium orthovanadate, 10% (v/v) glycerol, 150 mM NaCl, 1mM PMSF and Protease Inhibitor Cocktail Set III (Calbiochem). The cell debris was removed by centrifugation at 13,000 rpm for 20 minutes. Cell lysates were prepared at a concentration of 1mg/ml in the lysis buffer (0.5mg per vial). 50 μl of the slurry beads were added to the lysates and incubated at 4 °C overnight with gentle rotation. For pulldown experiments using purified proteins, 1 μg of purified proteins were prepared in a 500 μl volume (in 20 mM HEPES, pH 7.5, 150 mM NaCl, and 1mM TCEP) and 50 μl of the slurry beads were added to the sloutions and incubated at 4 °C overnight with gentle rotation.
The beads were then spun down at 4,000 rpm for 3 minutes, supernatant was removed and the beads were washed with 1 ml lysis buffer. This washing procedure was repeated five times in order to remove non-specific binding. After the last wash, 50 μl of 2x Laemmli sample buffer were added, the sample was boiled and subjected to SDS-PAGE and western blot assay using specific antibodies. Immune complexes were detected with horseradish peroxidase conjugated secondary antibodies and visualized by enhanced chemiluminescence reagent according to the manufacturer’s instructions (Pierce).
Bimolecular fluorescence complementation (BiFC)
The sequences encoding resides 1-173 (CN173) and 174-238 (CC173) of CFP were fused upstream of sequences encoding FGFR2Cyto and PLCγ12SH2 in pET28b vectors for expression in E. coli (Kodama and Hu, 2012). Both proteins were purified and phosphorylated as described above in the presence of FGFR2Cyto, ATP and MgCl2. The fluorescence complementation was measured using the Monolith NT.115 (NanoTemper Technologies, GmbH) as described above. Protein samples were filled into capillaries and the change in fluorescence intensity upon the adding of SHP22SH2 domain was monitored. Data were plotted using Origin 7.0.
Bio-layer interferometry (BLI)
BLI experiments were performed using a FortéBio Octet Red 384 using Anti-GST biosensors (18-5096). Assays were done in 384 well plates at 25 °C. Association was measured by dipping sensors into solutions of analyte protein for a designed time period as indicated in each figure, and was followed by moving sensors to wash buffer for a designed time period as indicated in each figure to monitor the dissociation process. Raw data shows a rise in signal associated with binding followed by a diminished signal after application of wash buffer. In experiments in which the ternary complex was reconstituted on the sensor ATP/Mg2+ was added as described in the Results.
Microscale thermophoresis (MST)
The binding affinities were measured using the Monolith NT.115 (NanoTemper Technologies, GmbH). Proteins were fluorescently labelled with Atto488 according to the manufacturer’s protocol. Labelling efficiency was determined to be 1:1 (protein;dye) by measuring the absorbance at 280 and 488 nm. A 16 step dilution series of the unlabelled binding partner was prepared and mixed with the labelled protein at 1:1 ratio and loaded into capillaries. Measurements were performed at 25 °C in 20 mM HEPES, 150 mM NaCl and 1 mM TCEP pH 7.5 buffer containing 0.01% Tween 20. Data analyses were performed using Nanotemper Analysis software, v.1.2.101 and were plotted using Origin 7.0. All measurements were conducted as triplicates and the errors were presented as the standard error of the triplicates. Equilibrium dissociation constants (Kd’s) are reported as ‘apparent’ values because for the interaction of FGFR2pCyto with SHP2 and PLCγ1 because the MST binding profiles appear to include two independent binding sites. The second of which is much weaker and is likely to be due to non-specific effects. Nonetheless the possibility of competing equilibria is flagged by the use of the term apparent. Reported data fits are based only on the initial tight binding event. Where the Kd is reported without the superscript ‘app’ the data has been fit to the standard 1:1 binding model.
Isothermal titration calorimetry (ITC)
ITC experiments were carried out using a MicroCal iTC200 (Malvern) at 25 °C. 20 15 μl injections of 100 μM FGFR2pC58 were made into 10 μM SHP22SH2 in the calorimeter cell. A control experiment involving the injection of 100 μM FGFR2pC58 into buffer was performed. The heat per injection was determined and subtracted from the binding data. Data was analysed using a single independent site model using Origin software.
Nuclear magnetic resonance (NMR)
All NMR spectroscopic experiments were carried out on Bruker Avance III (700, 800 and 950 MHz) NMR spectrometers equipped with cryogenically cooled triple resonance probes with a z-axis pulse field gradient coil were used. Resonance assignment spectra were recorded in 25 mM Na2HPO4/NaH2PO4, pH 6.5, 50 mM NaCl, 5 mM DTT, 1 mM EDTA. For the 1H,15N,13C labelled construct, a standard set of 3D backbone resonance assignment experiments (HNCA, HNCOCA, HNCACB, CACBCONH, HNCO and HNCACO) using standard Bruker library pulse sequences (with Watergate water suppression) or BEST versions (Lescop et al., 2007; Schulte-Herbrüggen and Sørensen, 2000) of amide transverse relaxation optimized spectroscopy (TROSY) (Pervushin et al., 1997) pulse sequences applying Non-Uniform Sampling (17-25%) were used to obtain a high-resolution spectra.
The NMR titration of SHP22SH2 into PLCγ12SH2 experiments were recorded at 25 °C using 200 μM uniformly 15N-labelled PLCγ12SH2 in 20 mM HEPES (pH 7.5) containing 150 mM NaCl, 1mM TCEP and 10% (v/v) D2O. 0, 150, 300, 450 600 and 900 μM unlabelled SHP22SH2 were added and an amide BEST TROSY pulse sequence recorded.
The NMR titration of PLCγ12SH2 into SHP22SH2 experiments were recorded at 25 °C using 100 μM uniformly 15N-labelled SHP22SH2 in 20 mM HEPES (pH 7.5) containing 150 mM NaCl, 1mM TCEP and 10% (v/v) D2O. 600 μM unlabelled PLCγ12SH2 were added and an amide BEST TROSY pulse sequence recorded.
All NMR data was processed with NMRPipe (Delaglio et al., 1995) and analyzed with CcpNmr Analysis software package (Skinner et al., 2016; Vranken et al., 2005) available in-house and on NMRBox platform (Maciejewski et al., 2017). Chemical shift perturbations (CSPs) for individual residues were calculated from the chemical shift for the backbone amide 1H (ΔωH) and 15N (ΔωN) using the following equation: CSP = (Evenäs et al., 2001).
In vitro FGFR2 kinase assay
A kinase-dead mutant FGFR2Cyto K517I was expressed and purified as a substrate for active FGFR2Cyto kinase assay (in 20 mM HEPES, pH 7.5, 150 mM NaCl, and 1 mM TCEP). To accurately measure the kinase activity in a LLPS environment as we described in the text, 200-time access of FGFR2Cyto K517I substrate was mixed with 10 μM of the active FGFR2Cyto in the solution state, in the LLPS state, or in other control conditions. 500 μM of ATP/MgCl2 was added and incubated at room temperature for 15 minutes. The phosphorylation states of substrate under various conditions were analysed using immunoblotting.
In vitro SHP2 phosphatase assay
A 4xDADEYLIPQQG peptide was cloned as a GST-fusion (GST-substrate) and used for in vitro SHP2 phosphatase assay. Briefly, GST-substrate on GST beads was phosphorylated by FGFR2Cyto in the presence of 5mM ATP/MgCl2 for 2 hours. After washing with PBS to remove FGFR2Cyto and ATP/MgCl2, GST-substrate was eluted from GST beads using 20 mM glutathione. Eluted phosphorylated GST-substrate was passed through a Superdex 75 column for further purification and buffer exchange (in 20 mM HEPES, pH 7.5, 150 mM NaCl, and 1 mM TCEP). 10-time access of phosphorylated GST-substrate was mixed with 60 μM of wild type SHP2 in the solution state, in the LLPS state, or in other control conditions at room temperature for 15 minutes. The phosphorylation states of substrate under various conditions were analysed using immunoblotting.
In vitro phosphatase assay
The effect of 10% 1, 6-hexanediol on SHP2 activity (60 μM) was measured using the non-radioactive phosphatase assay system (Promega V2471) according to the manufacture’s protocol.
Dephosphorylation by CIP
10 μM of phosphatase CIP (NEB, M0290) was incubated with individual protein (10 μM of pFGFR2Cyto, 60 μM of SHP2C459S, or 12 μM of pPLCγ1) for 30 minutes before droplet formation, or added after the pFGFR2Cyto, SHP2C459S and PLCγ1 droplets were formed (incubation with CIP for 15 minutes). The effects of CIP in the dephosphorylation of individual protein and pFGFR2Cyto-SHP2C459S-PLCγ1 droplets were examined using immunoblotting.
In vitro PLCγ1 activity assay
The lipolytic activity of pPLCγ1 was determined using the artificial substrate 4-methylumbelliferyl myo-inositol-1-phosphate, N-methyl-morpholine salt (Biosynth). Briefly, reaction mixtures consisted of 10 mM Tris, pH 6.8, 0.8 mM substrate, and 50% of protein mixtures (50 μM of pPLCγ1 alone or other protein mixtures as we described in the text). The reaction mixtures were placed in a 96-well plate, and fluorescence was measured using 350 nm excitation/450 nm emission filters on a plate reader. Reactions were allowed to proceed for 45 min at room temperature, with measurements taken every 5 min.
Wound-healing assay
Cells were seeded in a 96 well plate. After overnight serum starvation the cells were scratched using IncuCyte® WoundMaker and stimulated with 10 ng/ml FGF9, then incubated for further 24 hr (MCF7 cells; 16 hr for A431 cells). Images were taken at 0 hr and after incubation by a microscope gantry inside a cell incubator (Incucyte, Essen Bioscience, Ann Arbor, MI, USA). The identical experiment was also performed using HEK293T cells stably transfected with FGFR2ΔVT.
Calcium concentration assay
The calcium assay was perform using Fluo-4 NW Calcium Assay Kit (F36206, ThermoFisher Scientific) according to the manufactural manual. Briefly, cells were seeded in a 96 well plate. After FGF9 stimulation, medium was removed and cells were exposed to the dye loading solution and incubated for 30 minutes at 37°C then 30 minutes at 25°C. The Ca2+ level (fluorescence) was measured using instrument settings for excitation at 490 nm and emission at 510 nm.
Mathematical model
We have developed a deterministic mathematical model to describe the in vitro interactions between FGFR2Cyto (F), PLC12SH2 (P) and SHP22SH2 (S). The model has been formulated to include the reactions depicted in Reaction Figure, which involve the following chemical species
-
•
(or ) = unphosphorylated FGFR2,
-
•
(or ) = phosphorylated FGFR2,
-
•
(or ) = SHP2,
-
•
(or ) = phosphorylated FGFR2-SHP2 complex,
-
•
(or ) = unphosphorylated PLC,
-
•
(or ) = phosphorylated FGFR2-PLC complex,
-
•
(or ) = phosphorylated FGFR2-phosphorylated PLC complex,
-
•
(or ) = phosphorylated PLC,
-
•
(or ) = SHP2-unphosphorylated PLC complex,
-
•
(or ) = SHP2-phosphorylated PLC complex,
-
•
(or ) = phosphorylated FGFR2-SHP2-unphosphorylated PLC complex,
-
•
(or ) = phosphorylated FGFR2-unphosphorylated PLC-SHP2 complex,
-
•
(or ) = phosphorylated FGFR2-SHP2-phosphorylated PLC complex, and
-
•
(or ) = phosphorylated FGFR2-phosphorylated PLC-SHP2 complex,
where in every row above the first symbol is the abbreviated species name provided in Reaction Figure and the symbol in parentheses is used in the differential equations describing the variables of the mathematical model. We have assumed that there are no allosteric binding effects between SHP2, PLC, or the phosphorylated receptor. From the reactions in Reaction Figure and assuming mass-action kinetics, a set of ordinary differential equations (ODEs) for the concentrations (in units of ) for each molecular species can be written as follows:
| (Equation 1) |
| (Equation 2) |
| (Equation 3) |
| (Equation 4) |
| (Equation 5) |
| (Equation 6) |
| (Equation 7) |
| (Equation 8) |
| (Equation 9) |
| (Equation 10) |
| (Equation 11) |
| (Equation 12) |
| (Equation 13) |
| (Equation 14) |
We note that in the previous set of equations for . Rate constants corresponding to phosphorylation events (, ) or molecular dissociation events
(, , , , ) have dimensions of inverse time, and thus, units of s-1. Rate constants corresponding to molecular association events (, , , ) have dimensions of inverse concentration and time, and thus, units of .
We have assumed that at time , the initial time for the system under consideration, only , and are present. Thus, the initial concentrations for all other chemical species vanish. Given the timescales to be studied in the experimental model, the mathematical model does not include protein synthesis, degradation or trafficking of any of the molecular species and hence, the total number of molecules of , and are constant in time. We can write down conservation expressions for the total concentration of (), () and (), since we assume the experimental volume of the system does not change with time. We, therefore, can write
| (Equation 15) |
| (Equation 16) |
| (Equation 17) |
We note that in the previous set of constraints for . The above equations hold since the total concentration of a molecule at any time t is the sum of the concentrations of all species containing this molecule at that time point.
It is of interest to see which ternary complex species will prevail in the system for sufficiently late times, and thus, we have analysed the steady states of the mathematical model. Steady states can be found by setting the right hand sides of the differential equations to zero and simultaneously solving the resulting equations for the different molecular species in the system. In this case, due to the complexity of the equations, we cannot find explicit expressions for the species at steady state but we find the following implicit equations for a stable steady state solution (denoted by )
| (Equation 18) |
| (Equation 19) |
| (Equation 20) |
| (Equation 21) |
| (Equation 22) |
| (Equation 23) |
Constraints (15), (16) and (17), together with equations (18), (19), (20), (21), (23) and (23) provide a set of implicit polynomial equations for , and . It is interesting to observe that the stable steady state defined by the previous equations only provides a non-vanishing value for the ternary complex (or ), and that the other ternary complexes, , and , are such that . This is in agreement with the experimental results presented in this manuscript.
The parameters k+2, k+3, k+6, k+7 μM.s-1 were fixed by the experimental kd values where, kd,2 = 25.1 μM, kd,3 = 0.223 μM, kd,6 = 1.16 μM, kd,7 = 0.48 μM.
;, and the molecular association rates , , and with units of are fixed by the experimental values where, , , and .
Reaction Figure:
Quantification and statistical analysis
Data for quantification analyses are presented as mean ± standard deviation (SD). The protein levels in the western blot images were quantified by ImageJ (Fiji) and the number of replicates is shown in the figure legends. In vitro condensates were quantified using the Analyse Particle function in ImageJ (Fiji). Average of the number of condensates from 4 independent areas are shown. Colocalisation of cellular condensates were determined by using the "Coloc 2" function in ImageJ (Fiji). Degree of colocalization of endogenous PLCγ1 and SHP2 in Caco-2 cells is determined by Pearson's R value.
Acknowledgments
We thank A. Zhuravleva, A. Kalverda (bioNMR Facility, University of Leeds), and M. Peckham (Bio-imaging Facility, University of Leeds). This work was funded by Cancer Research UK (grant C57233/A22356), MRC, UK (grant MR/K015613/1), and BBSRC, UK (grant BB/S015787/1).
Author contributions
C.-C.L., K.M.S., L.W., J.A.L., P.B., A.P.C., A.S., C.S., H.K., E.M., S.D.E., Z.A., and J.E.L. conceived and/or performed experiments and analyzed data. P.-A.J. and C.M.-P. performed the mathematical analysis. C.-C.L., K.M.S., and J.E.L. wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: February 28, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.molcel.2022.02.005.
Contributor Information
Chi-Chuan Lin, Email: c.c.lin@leeds.ac.uk.
John E. Ladbury, Email: j.e.ladbury@leeds.ac.uk.
Supplemental information
Data and code availability
-
•
Original western data and microscopy data for figures in this paper have been deposited at Mendeley Data and are publicly available as of the date of publication. The DOI is in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Agazie Y.M., Hayman M.J. Development of an efficient “substrate-trapping” mutant of Src homology phosphotyrosine phosphatase 2 and identification of the epidermal growth factor receptor, Gab1, and three other proteins as target substrates. J. Biol. Chem. 2003;278:13952–13958. doi: 10.1074/jbc.M210670200. [DOI] [PubMed] [Google Scholar]
- Ahmed Z., Lin C.C., Suen K.M., Melo F.A., Levitt J.A., Suhling K., Ladbury J.E. Grb2 controls phosphorylation of FGFR2 by inhibiting receptor kinase and SHP2 phosphatase activity. J. Cell Biol. 2013;200:493–504. doi: 10.1083/jcb.201204106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberti S., Gladfelter A., Mittag T. Considerations and challenges in studying liquid-liquid phase separation and biomolecular condensates. Cell. 2019;176:419–434. doi: 10.1016/j.cell.2018.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaya J., Ryan V.H., Fawzi N.L. The SH3 domain of Fyn kinase interacts with and induces liquid–liquid phase separation of the low-complexity domain of hnRNPA2. J. Biol. Chem. 2018;293:19522–19531. doi: 10.1074/jbc.RA118.005120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banani S.F., Lee H.O., Hyman A.A., Rosen M.K. Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017;18:285–298. doi: 10.1038/nrm.2017.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banjade S., Rosen M.K. Phase transitions of multivalent proteins can promote clustering of membrane receptors. eLife. 2014;3 doi: 10.7554/eLife.04123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracha D., Walls M.T., Wei M.T., Zhu L., Kurian M., Avalos J.L., Toettcher J.E., Brangwynne C.P. Mapping local and gobal liquid phase behavior in living cells using photo-oligomerizable seeds. Cell. 2018;175:1467–1480. doi: 10.1016/j.cell.2018.10.048. e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brangwynne C.P., Eckmann C.R., Courson D.S., Rybarska A., Hoege C., Gharakhani J., Jülicher F., Hyman A.A. Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science. 2009;324:1729–1732. doi: 10.1126/science.1172046. [DOI] [PubMed] [Google Scholar]
- Bunney T.D., Esposito D., Mas-Droux C., Lamber E., Baxendale R.W., Martins M., Cole A., Svergun D., Driscoll P.C., Katan M. Structural and functional integration of the PLCγ interaction domains critical for regulatory mechanisms and signaling deregulation. Structure. 2012;20:2062–2075. doi: 10.1016/j.str.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgar H.R., Burns H.D., Elsden J.L., Lalioti M.D., Heath J.K. Association of the signaling adaptor FRS2 with fibroblast growth factor receptor 1 (Fgfr1) Is mediated by alternative splicing of the juxtamembrane domain. J. Biol. Chem. 2002;277:4018–4023. doi: 10.1074/jbc.M107785200. [DOI] [PubMed] [Google Scholar]
- Case L.B., Ditlev J.A., Rosen M.K. Regulation of transmembrane signaling by phase separation. Annu. Rev. Biophys. 2019;48:465–494. doi: 10.1146/annurev-biophys-052118-115534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case L.B., Zhang X., Ditlev J.A., Rosen M.K. Stoichiometry controls activity of phase-separated clusters of actin signaling proteins. Science. 2019;363:1093–1097. doi: 10.1126/science.aau6313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cebecauer M., Spitaler M., Sergé A., Magee A.I. Signalling complexes and clusters: functional advantages and methodological hurdles. J. Cell Sci. 2010;123:309–320. doi: 10.1242/jcs.061739. [DOI] [PubMed] [Google Scholar]
- Chen H., Libring S., Ruddraraju K.V., Miao J., Solorio L., Zhang Z.Y., Wendt M.K. SHP2 is a multifunctional therapeutic target in drug resistant metastatic breast cancer. Oncogene. 2020;39:7166–7180. doi: 10.1038/s41388-020-01488-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Xu C.F., Ma J., Eliseenkova A.V., Li W., Pollock P.M., Pitteloud N., Miller W.T., Neubert T.A., Mohammadi M. A crystallographic snapshot of tyrosine trans-phosphorylation in action. Proc. Natl. Acad. Sci. USA. 2008;105:19660–19665. doi: 10.1073/pnas.0807752105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBell K., Graham L., Reischl I., Serrano C., Bonvini E., Rellahan B. Intramolecular regulation of phospholipase C-γ1 by its C-terminal Src homology 2 domain. Mol. Cell. Biol. 2007;27:854–863. doi: 10.1128/MCB.01400-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio F., Grzesiek S., Vuister G.W., Zhu G., Pfeifer J., Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Delarue M., Brittingham G.P., Pfeffer S., Surovtsev I.V., Pinglay S., Kennedy K.J., Schaffer M., Gutierrez J.I., Sang D., Poterewicz G., et al. mTORC1 controls phase separation and the biophysical properties of the cytoplasm by tuning crowding. Cell. 2018;174:338–349. doi: 10.1016/j.cell.2018.05.042. e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depetris R.S., Hu J., Gimpelevich I., Holt L.J., Daly R.J., Hubbard S.R. Structural basis for inhibition of the insulin receptor by the adaptor protein Grb14. Mol. Cell. 2005;20:325–333. doi: 10.1016/j.molcel.2005.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelstein A., Amodaj N., Hoover K., Vale R., Stuurman N. Computer control of microscopes using μManager. Curr. Protoc. Mol. Biol. 2010;92:14–20. doi: 10.1002/0471142727.mb1420s92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Gamal D., Williams K., LaFollette T.D., Cannon M., Blachly J.S., Zhong Y., Woyach J.A., Williams E., Awan F.T., Jones J., et al. PKC-β as a therapeutic target in CLL: PKC inhibitor AEB071 demonstrates preclinical activity in CLL. Blood. 2014;124:1481–1491. doi: 10.1182/blood-2014-05-574830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdem Z.N., Schwarz S., Drev D., Heinzle C., Reti A., Heffeter P., Hudec X., Holzmann K., Grasl-Kraupp B., Berger W., et al. Irinotecan upregulates fibroblast growth factor receptor 3 expression in colorectal cancer cells, which mitigates irinotecan-induced apoptosis. Transl. Oncol. 2017;10:332–339. doi: 10.1016/j.tranon.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evenäs J., Tugarinov V., Skrynnikov N.R., Goto N.K., Muhandiram R., Kay L.E. Ligand-induced structural changes to maltodextrin-binding protein as studied by solution NMR spectroscopy. J. Mol. Biol. 2001;309:961–974. doi: 10.1006/jmbi.2001.4695. [DOI] [PubMed] [Google Scholar]
- Frese S., Schubert W.D., Findeis A.C., Marquardt T., Roske Y.S., Stradal T.E.B., Heinz D.W. The phosphotyrosine peptide binding specificity of Nck1 and Nck2 Src homology 2 domains. J. Biol. Chem. 2006;281:18236–18245. doi: 10.1074/jbc.M512917200. [DOI] [PubMed] [Google Scholar]
- Gresset A., Hicks S.N., Harden T.K., Sondek J. Mechanism of phosphorylation-induced activation of phospholipase C-γ isozymes. J. Biol. Chem. 2010;285:35836–35847. doi: 10.1074/jbc.M110.166512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadari Y.R., Kouhara H., Lax I., Schlessinger J. Binding of SHP2 tyrosine phosphatase to FRS2 is essential for fibroblast growth factor-induced PC12 cell differentiation. Mol. Cell. Biol. 1998;18:3966–3973. doi: 10.1128/mcb.18.7.3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J., Liu J., Ghirlando R., Saltiel A.R., Hubbard S.R. Structural basis for recruitment of the adaptor protein APS to the activated insulin receptor. Mol. Cell. 2003;12:1379–1389. doi: 10.1016/s1097-2765(03)00487-8. [DOI] [PubMed] [Google Scholar]
- Huang W.Y.C., Alvarez S., Kondo Y., Lee Y.K., Chung J.K., Lam H.Y.M., Biswas K.H., Kuriyan J., Groves J.T. A molecular assembly phase transition and kinetic proofreading modulate Ras activation by SOS. Science. 2019;363:1098–1103. doi: 10.1126/science.aau5721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z., Marsiglia W.M., Basu Roy U., Rahimi N., Ilghari D., Wang H., Chen H., Gai W., Blais S., Neubert T.A., et al. Two FGF receptor kinase molecules act in concert to recruit and transphosphorylate phospholipase Cγ. Mol. Cell. 2016;61:98–110. doi: 10.1016/j.molcel.2015.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huculeci R., Garcia-Pino A., Buts L., Lenaerts T., van Nuland N. Structural insights into the intertwined dimer of fyn SH2. Protein Sci. 2015;24:1964–1978. doi: 10.1002/pro.2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman A.A., Weber C.A., Jülicher F. Liquid-liquid phase separation in biology. Annu. Rev. Cell Dev. Biol. 2014;30:39–58. doi: 10.1146/annurev-cellbio-100913-013325. [DOI] [PubMed] [Google Scholar]
- Kodama Y., Hu C.D. Bimolecular fluorescence complementation (BiFC): a 5-year update and future perspectives. Biotechniques. 2012;53:285–298. doi: 10.2144/000113943. [DOI] [PubMed] [Google Scholar]
- Koss H., Bunney T.D., Esposito D., Martins M., Katan M., Driscoll P.C. Dynamic allostery in PLCγ1 and Its modulation by a cancer mutation revealed by MD simulation and NMR. Biophys. J. 2018;115:31–45. doi: 10.1016/j.bpj.2018.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostas M., Haugsten E.M., Zhen Y., Sørensen V., Szybowska P., Fiorito E., Lorenz S., Jones N., de Souza G.A., Wiedlocha A., Wesche J. Protein tyrosine phosphatase receptor type G (PTPRG) controls fibroblast growth factor receptor (FGFR) 1 activity and influences sensitivity to FGFR kinase inhibitors. Mol. Cell. Proteomics. 2018;17:850–870. doi: 10.1074/mcp.RA117.000538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krick S., Helton E.S., Hutcheson S.B., Blumhof S., Garth J.M., Denson R.S., Zaharias R.S., Wickham H., Barnes J.W. FGF23 induction of O-linked N-acetylglucosamine regulates IL-6 secretion in human bronchial epithelial cells. Front. Endocrinol. 2018;9:708. doi: 10.3389/fendo.2018.00708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.R., Park Y.H., Min H., Kim Y.R., Seok Y.J. Determination of protein phosphorylation by polyacrylamide gel electrophoresis. J. Microbiol. 2019;57:93–100. doi: 10.1007/s12275-019-9021-y. [DOI] [PubMed] [Google Scholar]
- Lescop E., Schanda P., Brutscher B. A set of BEST triple-resonance experiments for time-optimized protein resonance assignment. J. Magn. Reson. 2007;187:163–169. doi: 10.1016/j.jmr.2007.04.002. [DOI] [PubMed] [Google Scholar]
- Li P., Banjade S., Cheng H.C., Kim S., Chen B., Guo L., Llaguno M., Hollingsworth J.V., King D.S., Banani S.F., et al. Phase transitions in the assembly of multivalent signalling proteins. Nature. 2012;483:336–340. doi: 10.1038/nature10879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C.C., Melo F.A., Ghosh R., Suen K.M., Stagg L.J., Kirkpatrick J., Arold S.T., Ahmed Z., Ladbury J.E. Inhibition of basal FGF receptor signaling by dimeric Grb2. Cell. 2012;149:1514–1524. doi: 10.1016/j.cell.2012.04.033. [DOI] [PubMed] [Google Scholar]
- Maciejewski M.W., Schuyler A.D., Gryk M.R., Moraru I.I., Romero P.R., Ulrich E.L., Eghbalnia H.R., Livny M., Delaglio F., Hoch J.C. NMRbox: a resource for biomolecular NMR computation. Biophys. J. 2017;112:1529–1534. doi: 10.1016/j.bpj.2017.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong S.H., Guy G.R., Hadari Y.R., Laks S., Gotoh N., Schlessinger J., Lax I. FRS2 proteins recruit intracellular signaling pathways by binding to diverse targets on fibroblast growth factor and nerve growth factor receptors. Mol. Cell. Biol. 2000;20:979–989. doi: 10.1128/mcb.20.3.979-989.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong S.H., Lim Y.P., Low B.C., Guy G.R. SHP2 associates directly with tyrosine phosphorylated p90 (SNT) protein in FGF-stimulated cells. Biochem. Biophys. Res. Commun. 1997;238:261–266. doi: 10.1006/bbrc.1997.7272. [DOI] [PubMed] [Google Scholar]
- Pervushin K., Riek R., Wider G., Wüthrich K. Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc. Natl. Acad. Sci. USA. 1997;94:12366–12371. doi: 10.1073/pnas.94.23.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulin B., Sekiya F., Rhee S.G. Intramolecular interaction between phosphorylated tyrosine-783 and the C-terminal Src homology 2 domain activates phospholipase C-γ1. Proc. Natl. Acad. Sci. USA. 2005;102:4276–4281. doi: 10.1073/pnas.0409590102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte-Herbrüggen T., Sørensen O.W. Clean TROSY: compensation for relaxation-induced artifacts. J. Magn. Reson. 2000;144:123–128. doi: 10.1006/jmre.2000.2020. [DOI] [PubMed] [Google Scholar]
- Sheu-Gruttadauria J., MacRae I.J. Phase transitions in the assembly and function of human miRISC. Cell. 2018;173:946–957. doi: 10.1016/j.cell.2018.02.051. e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin Y., Chang Y.-C., Lee D.S.W., Berry J., Sanders D.W., Ronceray P., Wingreen N.S., Haataja M., Brangwynne C.P. Liquid nuclear condensates mechanically sense and restructure the genome. Cell. 2018;175:1481–1491. doi: 10.1016/j.cell.2018.10.057. e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner S.P., Fogh R.H., Boucher W., Ragan T.J., Mureddu L.G., Vuister G.W. CcpNmr AnalysisAssign: a flexible platform for integrated NMR analysis. J. Biomol. NMR. 2016;66:111–124. doi: 10.1007/s10858-016-0060-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sledge G.W., Jr., Gökmen-Polar Y. Protein kinase C-beta as a therapeutic target in breast cancer. Semin. Oncol. 2006;33(Supplement 9) doi: 10.1053/j.seminoncol.2006.03.019. S15–S18. [DOI] [PubMed] [Google Scholar]
- Stein E.G., Ghirlando R., Hubbard S.R. Structural basis for dimerization of the Grb10 Src homology 2 domain. Implications for ligand specificity. J. Biol. Chem. 2003;278:13257–13264. doi: 10.1074/jbc.M212026200. [DOI] [PubMed] [Google Scholar]
- Su X., Ditlev J.A., Hui E., Xing W., Banjade S., Okrut J., King D.S., Taunton J., Rosen M.K., Vale R.D. Phase separation of signaling molecules promotes T cell receptor signal transduction. Science. 2016;352:595–599. doi: 10.1126/science.aad9964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su X., Ditlev J.A., Rosen M.K., Vale R.D. Reconstitution of TCR signaling using supported lipid bilayers. Methods Mol. Biol. 2017;1584:65–76. doi: 10.1007/978-1-4939-6881-7_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suen K.M., Lin C.C., George R., Melo F.A., Biggs E.R., Ahmed Z., Drake M.N., Arur S., Arold S.T., Ladbury J.E. Interaction with Shc prevents aberrant Erk activation in the absence of extracellular stimuli. Nat. Struct. Mol. Biol. 2013;20:620–627. doi: 10.1038/nsmb.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teicher B.A. Protein kinase C as a therapeutic target. Clin. Cancer Res. 2006;12:5336–5345. doi: 10.1158/1078-0432.CCR-06-0945. [DOI] [PubMed] [Google Scholar]
- Tokunaga M., Imamoto N., Sakata-Sogawa K. Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat. Methods. 2008;5:159–161. doi: 10.1038/nmeth1171. [DOI] [PubMed] [Google Scholar]
- Tulpule A., Guan J., Neel D.S., Allegakoen H.R., Lin Y.P., Brown D., Chou Y.T., Heslin A., Chatterjee N., Perati S., et al. Kinase-mediated RAS signaling via membraneless cytoplasmic protein granules. Cell. 2021;184:2649–2664. doi: 10.1016/j.cell.2021.03.031. e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vranken W.F., Boucher W., Stevens T.J., Fogh R.H., Pajon A., Llinas M., Ulrich E.L., Markley J.L., Ionides J., Laue E.D. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins. 2005;59:687–696. doi: 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- Wang Z., Ma J., Miyoshi C., Li Y., Sato M., Ogawa Y., Lou T., Ma C., Gao X., Lee C., et al. Quantitative phosphoproteomic analysis of the molecular substrates of sleep need. Nature. 2018;558:435–439. doi: 10.1038/s41586-018-0218-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler R.J., Lee H., Poser I., Pal A., Doeleman T., Kishigami S., Kour S., Anderson E.N., Marrone L., Murthy A.C., Jahnel M. Small molecules for modulating protein driven liquid-liquid phase separation in treating neurodegenerative disease. Preprint at bioRxiv. 2019 doi: 10.1101/721001. [DOI] [Google Scholar]
- White M.J., Boyd J.M., Horswill A.R., Nauseef W.M. Phosphatidylinositol-specific phospholipase C contributes to survival of Staphylococcus aureus USA300 in human blood and neutrophils. Infect. Immun. 2014;82:1559–1571. doi: 10.1128/IAI.01168-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng M., Chen X., Guan D., Xu J., Wu H., Tong P., Zhang M. Reconstituted postsynaptic density as a molecular platform for understanding synapse formation and plasticity. Cell. 2018;174:1172–1187. doi: 10.1016/j.cell.2018.06.047. e16. [DOI] [PubMed] [Google Scholar]
- Zhang G., Wang Z., Du Z., Zhang H. mTOR regulates phase separation of PGL granules to modulate their autophagic degradation. Cell. 2018;174:1492–1506. doi: 10.1016/j.cell.2018.08.006. e22. [DOI] [PubMed] [Google Scholar]
- Zhu G., Xie J., Kong W., Xie J., Li Y., Du L., Zheng Q., Sun L., Guan M., Li H., et al. Phase separation of disease-associated SHP2 mutants underlies MAPK hyperactivation. Cell. 2020;183:490–502. doi: 10.1016/j.cell.2020.09.002. e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Dynamic FGFR2ΔVT-SHP2C459S-PLCγ1 droplets on plasma membrane. Confocal imaging revealed the dynamic property of FGFR2ΔVT-SHP2C459S-PLCγ1 droplets on plasma membrane upon FGF9 stimulation. FGFR2ΔVT was Neptune2.5 tagged (red), SHP2C459S was mOrange tagged (yellow), and PLCγ1 was mEGFP tagged (green). The video was recorded at a rate of 1 frame/30 sec. Scale bars, 1 μm
HILO imaging demonstrated a fusion event of FGFR2ΔVT-EGFP (coexpressed with untagged SHP2C459S and PLCγ1) on plasma membrane upon FGF9 stimulation. The video was recorded at a rate of 1 frame/1 sec. Scale bars, 10 μm
HILO imaging demonstrated a second fusion event of FGFR2ΔVT-EGFP (coexpressed with untagged SHP2C459S and PLCγ1) on plasma membrane upon FGF9 stimulation. The video was recorded at a rate of 1 frame/1 sec. Scale bars, 10 μm
HILO imaging demonstrated the fusion event of PLCγ1-EGFP (coexpressed with untagged FGFR2ΔVT and SHP2C459S) on plasma membrane upon FGF9 stimulation. The video was recorded at a rate of 1 frame/5 sec. Scale bars, 10 μm
Data Availability Statement
-
•
Original western data and microscopy data for figures in this paper have been deposited at Mendeley Data and are publicly available as of the date of publication. The DOI is in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.