

Abstract
The administration of pan histone deacetylase (HDAC) inhibitors reduces ischemic damage to the CNS, both in vitro and in animal models of stroke, via mechanisms which we are beginning to understand. The acetylation of p53 is regulated by Class I HDACs and, because p53 appears to play a role in ischemic pathology, the purpose of this study was to discover, using an in vitro white matter ischemia model and an in vivo cerebral ischemia model, if neuroprotection mediated by HDAC inhibition depended on p53 expression. Optic nerves were excised from wild-type and p53-deficient mice, and then subjected to oxygen–glucose deprivation in the presence and absence of a specific inhibitor of Class I HDACs (MS-275, entinostat) while compound action potentials were recorded. Furthermore, transient focal ischemia was imposed on wild-type and p53-deficient mice, which were subsequently treated with MS-275. Interestingly, and in both scenarios, the beneficial effects of MS-275 were most pronounced when p53 was absent. These results suggest that modulation of p53 activity is not responsible for MS-275-mediated neuroprotection, and further illustrate how HDAC inhibitors variably influence p53 and associated apoptotic pathways.
Keywords: histone deacetylase, ischemia, mouse optic nerve, MS-275, neuroprotection, p53
Epigenetic mechanisms are integral to biological regulation across the life span and can enhance the intrinsic potential for repair within the CNS (Handel et al. 2009; Qureshi and Mehler 2010; Pearce 2011; Jakovcevski and Akbarian 2012; Schweizer et al. 2013). Perhaps most striking are the results obtained with histone deacetylase (HDAC) inhibitors (Chuang et al. 2009; Haberland et al. 2009; Thomas 2009; Fischer et al. 2010). For example, the acetylation status of the tumor suppressor p53 is regulated by specific HDACs, and increased acetylation and accumulation promotes growth arrest and apoptosis (Luo et al. 2000; Yao and Yang 2011). Hyper-acetylation, via the administration of HDAC inhibitors, has been clinically proven useful in a variety of cancer trials and these drugs are also effective in animal models of several inflammatory diseases (Shakespear et al. 2011). Recently, Brochier et al. (2013) identified the relevant p53 acetylation sites and this has provided a useful molecular understanding of the specific outcomes of HDAC inhibition.
While HDAC inhibitors promote the death of transformed, de-differentiated cells, these same drugs protect differentiated cells, such as neurons, from stressors that would normally lead to apoptosis (Uo et al. 2007, 2009). Indeed, the application of pan HDAC inhibitors (Dokmanovic et al. 2007) is a proven strategy with which to reduce experimental ischemic injury to the CNS, both in vitro (Langley et al. 2008; Baltan et al. 2011a; Baltan 2012) and in animal models of stroke (Chuang et al. 2009; Langley et al. 2009; Gibson and Murphy 2010).
As there appears to be a detrimental role for p53 in ischemic pathology (Crumrine et al. 1994; Culmsee et al. 2001; Leker et al. 2004; Endo et al. 2006; Vaseva et al. 2012), we set out to determine whether HDAC inhibitor-mediated neuroprotection required p53. We administered MS-275, reported to be a specific inhibitor of Class I HDACs (Hess-Stump et al. 2007), because Class 1 HDACs are implicated in p53 acetylation and we have shown previously that MS-275 is neuroprotective ex vivo (Baltan et al. 2011a; Baltan 2012). However, MS-275 has yet to be tested in a stroke model in vivo. Interestingly, we find that the neuroprotection elicited by MS-275, both in vivo and ex vivo, is most clearly apparent when p53 is absent.
Experimental procedures
Materials
The MS-275 was purchased from Selleck (Ontario, London). Sources of antibodies were as follows: cleaved caspase 3 (#9661, 1 : 1000; Cell Signaling Technology, Beverly, MA, USA), histone 3 (#9715, 1:1000; Cell Signaling), acetylated histone 3 (#06–599, final 1 : 5000; Millipore Corporation, Bedford, MA, USA), β-actin (AC-15, 1: 20 000; Sigma, St. Louis, MO, USA), neurofilament 160/200 (N2912, 1:100; Sigma), and donkey anti-mouse Cy3 (1 : 100; Jackson Immuno-Research, West Grove, PA, USA). The mitoCFP mice (Misgeld et al. 2007) were originally purchased from Jackson Laboratory and then crossed with p53−/− mice (Donehower et al. 1992) at the University of Washington. This study was in compliance with the ARRIVE guidelines.
Transient middle cerebral artery occlusion (MCAo)
This was performed as recently described (Jayadev et al. 2011), using male C57bl/6 mice (25–30 g), with approval from the Institutional Animal Care and Use Committee of the University of Washington. Anesthesia was induced by inhalation of 3% isoflurane and maintained by inhalation of 1.5% isoflurane. Body temperature was monitored throughout surgery (via a rectal probe) and maintained at 36–38°C using a heating blanket. Transient focal cerebral ischemia was induced by occlusion of the right middle cerebral artery for 60 min with a filament. Either MS-275 (30 mg/kg) or vehicle (0.05 N HCl and 0.1% Tween in phosphate buffered saline) was given ip immediately upon reperfusion, and again 24 and also 48 h later. Mice were killed 24 h after the final dose of MS-275, and the brain removed and cut into 1 mm sections using a mouse brain matrix (ASI Instruments, Warren, NJ, USA). To quantitate ischemic damage, brain slices were stained with 2,3,5-triphenyltetrazolium chloride (1 mg/mL saline). Digital photographs were taken and infarct areas calculated by at least two observers, who were blinded to genotype and treatment. All data are presented as mean of infarct (expressed as % hemisphere) ± SEM.
Recording techniques for optic nerve function and oxygen–glucose deprivation
This was performed as previously described (Baltan et al. 2011a). Mouse optic nerves (MONs) were gently freed from their dural sheaths and placed in a Haas top chamber superfused with artificial CSF (ACSF) at 37°C. Suction electrodes back-filled with ACSF were used for stimulation and recording the compound action potential (CAP). During experiments, a supramaximal CAP was elicited every 30 s. Oxygen–glucose deprivation (OGD) was induced by switching to glucose-free ACSF (replaced with equimolar sucrose to maintain osmolarity) and a gas mixture containing 95% N2/5% CO2. This was applied for 60 min, glucose-containing ACSF and O2 were restored, and CAP recorded for up to 5–6 h. Optic nerve function was assessed as the area under the supramaximal CAP. Irreversible injury was measured by determining residual CAP area, normalized to control CAP area, 5–6 h after the conclusion of OGD. Data were normalized by setting the mean of initial baseline values (measured over 20 min) to a value of 1.0. Results from several nerves were pooled, averaged, and plotted against time. Data are presented as mean ± SEM.
Immunohistochemistry
This was performed in perfusion-fixed (4% paraformaldehyde in phosphate-buffered saline) MONs. Cryoprotection was achieved in increasing sucrose concentrations (10–30% sucrose). Sections (16 μm) were blocked and permeabilized in 5% normal goat/donkey (50% by volume) serum (Sigma) and 0.3% Triton X-100 (Sigma) for 60 min at 23–25°C. The antibody against neurofilament was used at room temperature for 2 h. After a thorough wash in phosphate-buffered saline, the tissue was exposed to Donkey anti-mouse Cy3 in 2% normal goat serum for 2 h.
Imaging and pixel-intensity measurements
Expression of NF160/200 was imaged using a Leica TCS SP5 upright confocal, whereas cyan fluorescent protein (CFP) was imaged using a Leica TCS SP511 upright multiphoton laser scanning microscope (Buffalo Grove, IL, USA). For NF160/200, sections were scanned with an argon and He–Ne laser for Cy3/543. Two to three adjacent sections from each MON were imaged for a total of five areas of interest. A total of 10–16 optical sections of 1-lm thickness at 512 9 512 pixel size were collected in the z-axis from a single microscopic field using 100x (HCX PLAN APO, oil immersion; numerical aperture 1.35; Leica) objective lens under fixed gain, laser power, pinhole, and photomultiplier tube (PMT) settings. For CFP, two to three adjacent sections for each MON were imaged with the wavelength set at 900 nm. A total of 10 optical sections of 1-lm thickness at 512 9 512 pixel size were collected in the z-axis from a single microscopic field using the 25x (HCX IRAPO, water immersion; numerical aperture, 0.95) objective lens under fixed gain, laser power, pinhole, and PMT settings. To compare pixel intensity and quantify immunohistochemical staining, all sections were processed concurrently with Leica imaging software (LAS-AF version 2.7; Buffalo Grove, IL, USA) using a single channel. The Z-stacks were projected into a single plane image before analysis and assessment of pixel intensity.
Cell death assay and western blotting
Immunofluorescence, preparation of protein extracts, and western blotting were carried out as described previously (Uo et al. 2005).
Statistical analysis
Data were summarized by means and standard errors. Mean values were compared by Student’s t-test and p-values < 0.05 were considered statistically significant.
Results
The functional integrity of optic nerve axons was monitored by quantifying the area under CAPs evoked by a supramaximal stimulus. After baseline recording, MS-275 was introduced for 30 min and the superfusion conditions were maintained during 60 min of OGD and the initial 30 min of reperfusion. We had reported previously that MS-275 does not affect basic axon function (Baltan et al. 2011a). The MON was then reperfused in ACSF for 5 h in the absence of the MS-275.
With the onset of OGD there was a rapid loss of axon function, and this partially recovered when oxygen and glucose were restored. While there was permanent CAP deficit in MONs from both wild-type and p53−/− mice, inclusion of MS-275 in the organ bath not only reduced the extent of CAP loss but also significantly improved recovery in both genotypes (Fig. 1). Interestingly, the effect of MS-275 on CAP recovery in p53−/− MONs was twofold that of MS-275 in wild-type mice (Fig. 2). As these mice express CFP in neuronal mitochondria we were able to image these organelles exclusively in axons of the optic nerve (Fig. 3). The increase in normalized CFP fluorescence intensity in sections taken from MONs functionally protected by MS-275 was elevated more than threefold in p53−/− as compared with wild type, implying enhanced preservation of mitochondrial viability. The neuroprotective effect of MS-275 was confirmed by assessing the expression of NF160/200 in adjacent sections (Fig. 4).
Fig. 1.
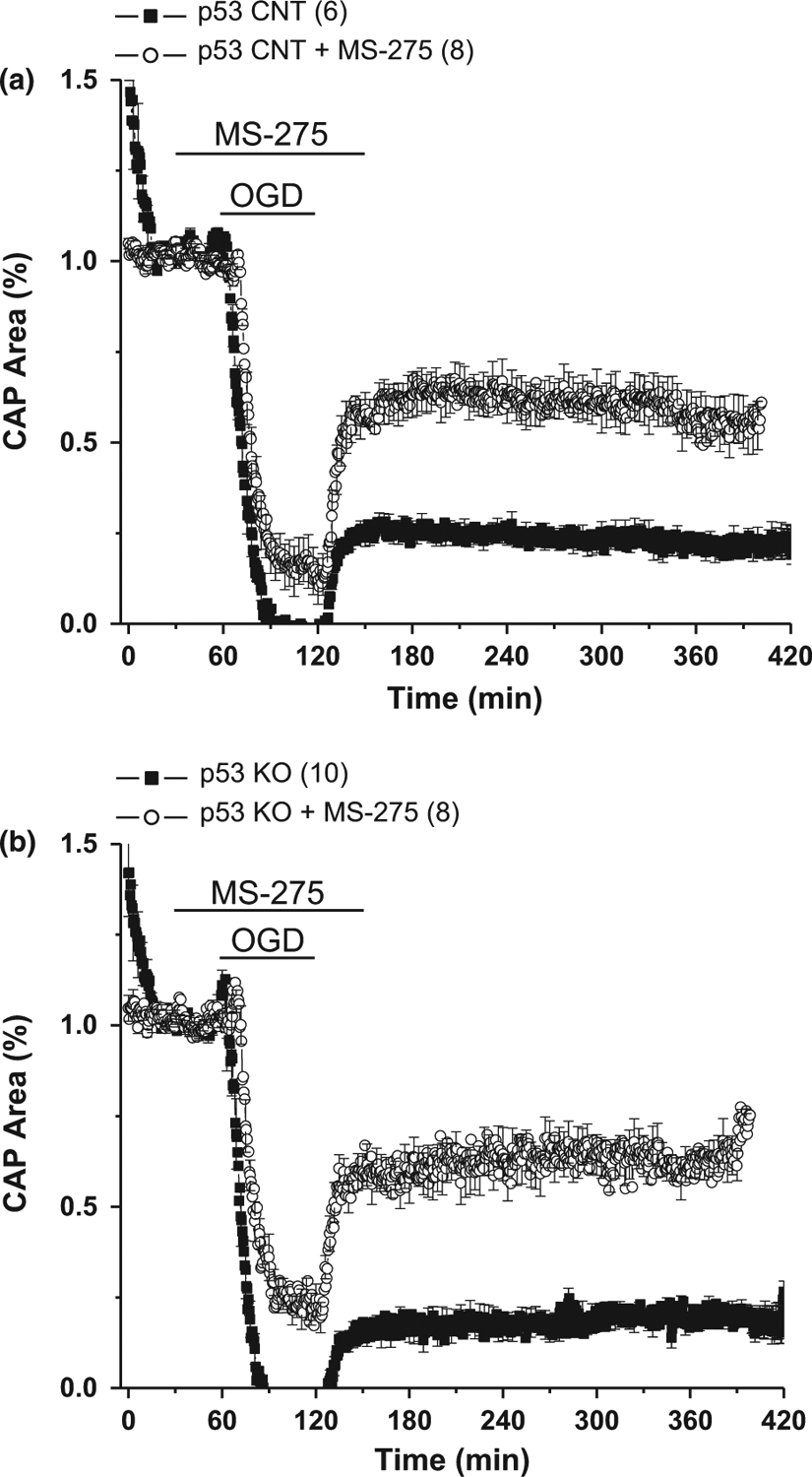
The onset of a period of ischemia [oxygen–glucose deprivation (OGD)] in mouse optic nerve caused immediate loss of axon function and incomplete recovery (filled squares) in both (a) wild-type (WT) and (b) p53 knockout (KO) mice. Addition of the histone deacetylase (HDAC) inhibitor MS-275 (1 μM) prior to oxygen–glucose deprivation (OGD) (open circles) somewhat ameliorated loss of function but dramatically improved compound action potential (CAP) recovery in both genotypes. Number of optic nerves in parentheses.
Fig. 2.
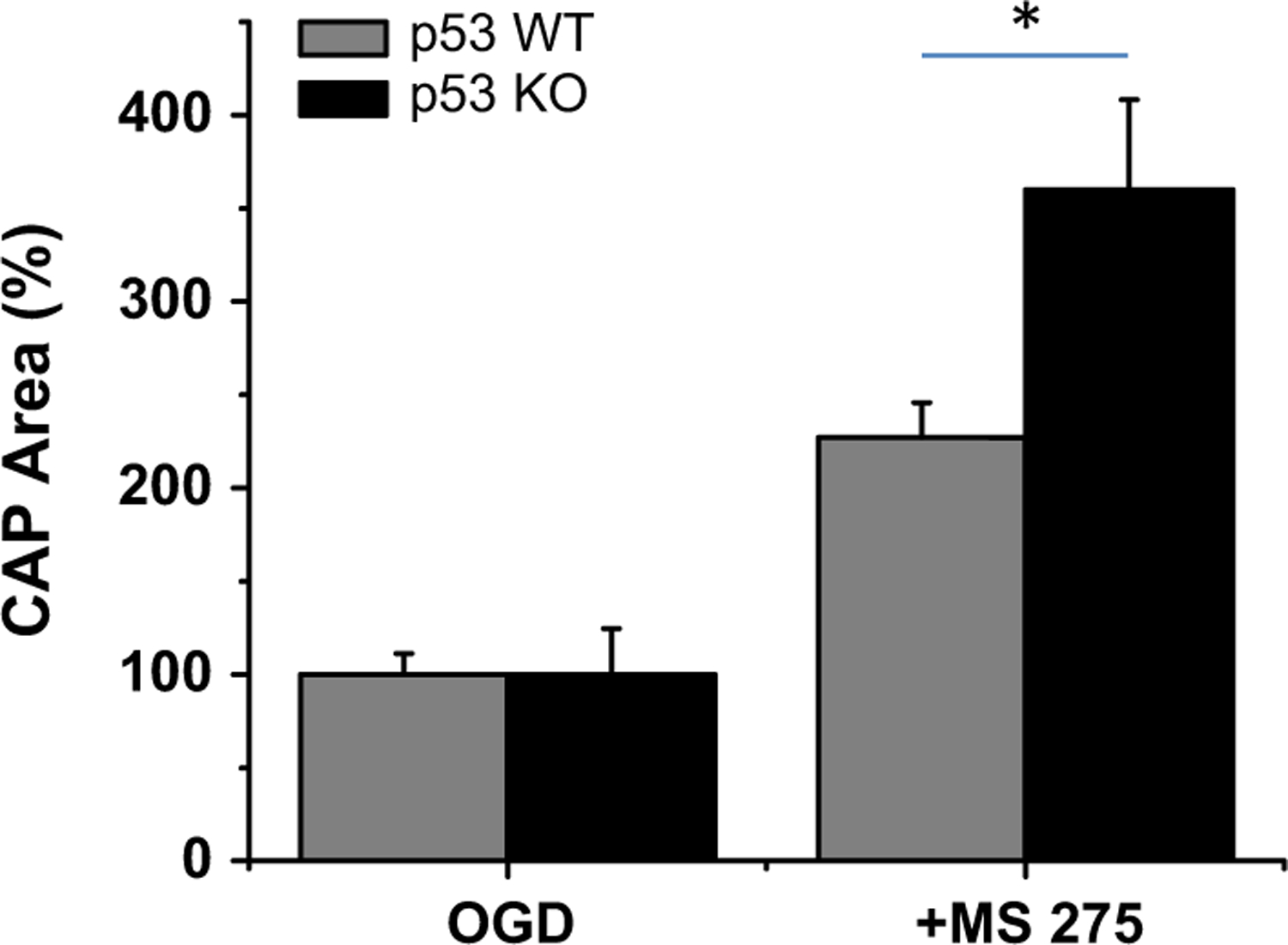
Recovery of compound action potential (CAP) area after oxygen–glucose deprivation (OGD) in the presence of MS-275 was significantly greater in optic nerves from p53−/− as compared with wild-type mice. *p < 0.02.
Fig. 3.
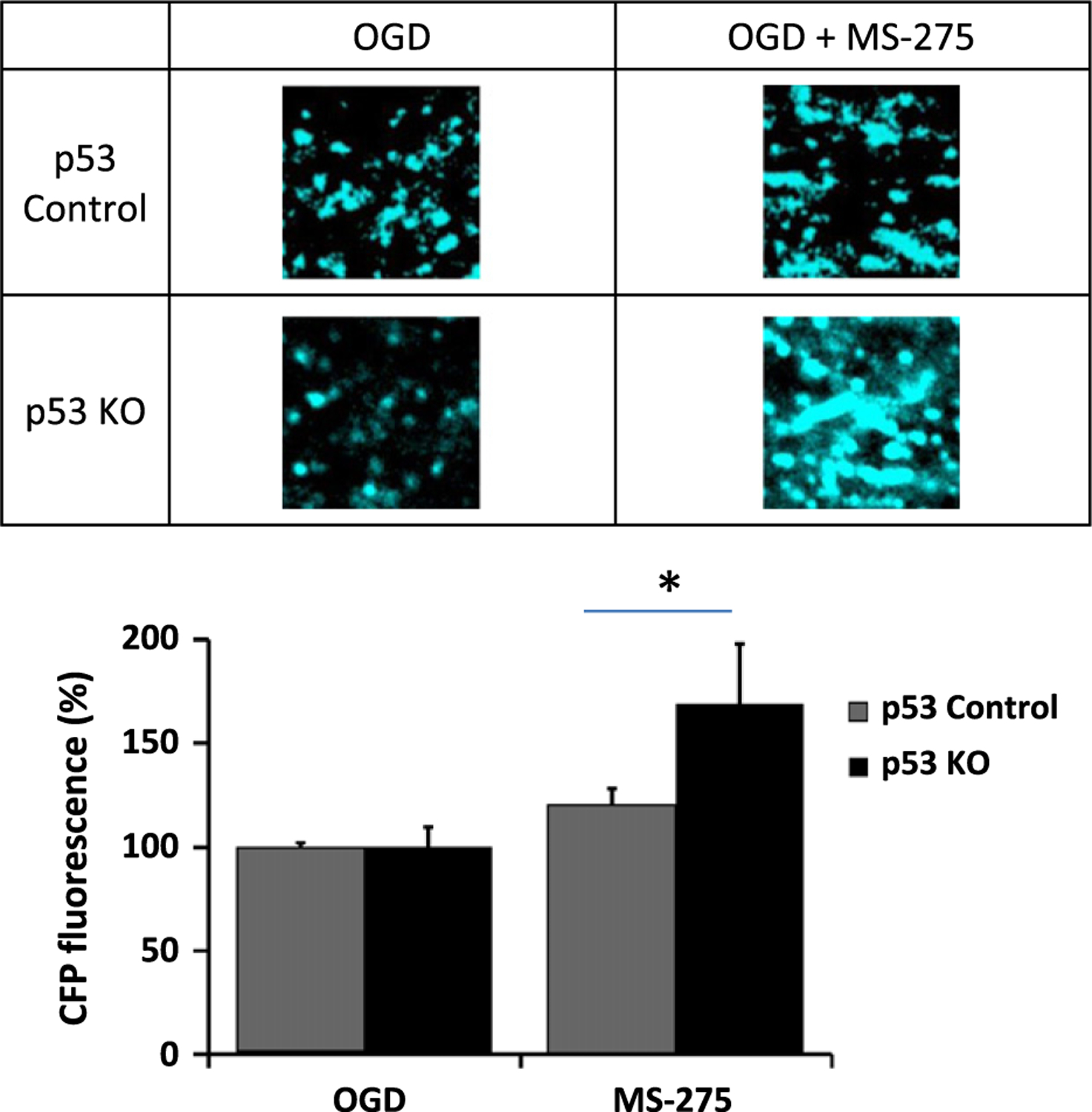
Images of neuronal [cyan fluorescent protein (CFP+)] mitochondria in mouse optic nerve (MON) from wt and p53 ko mice after oxygen–glucose deprivation (OGD), and in the absence or presence of MS-275. The histogram summarizes data from a minimum of three mice (± SEM) for each group. *p < 0.05.
Fig. 4.
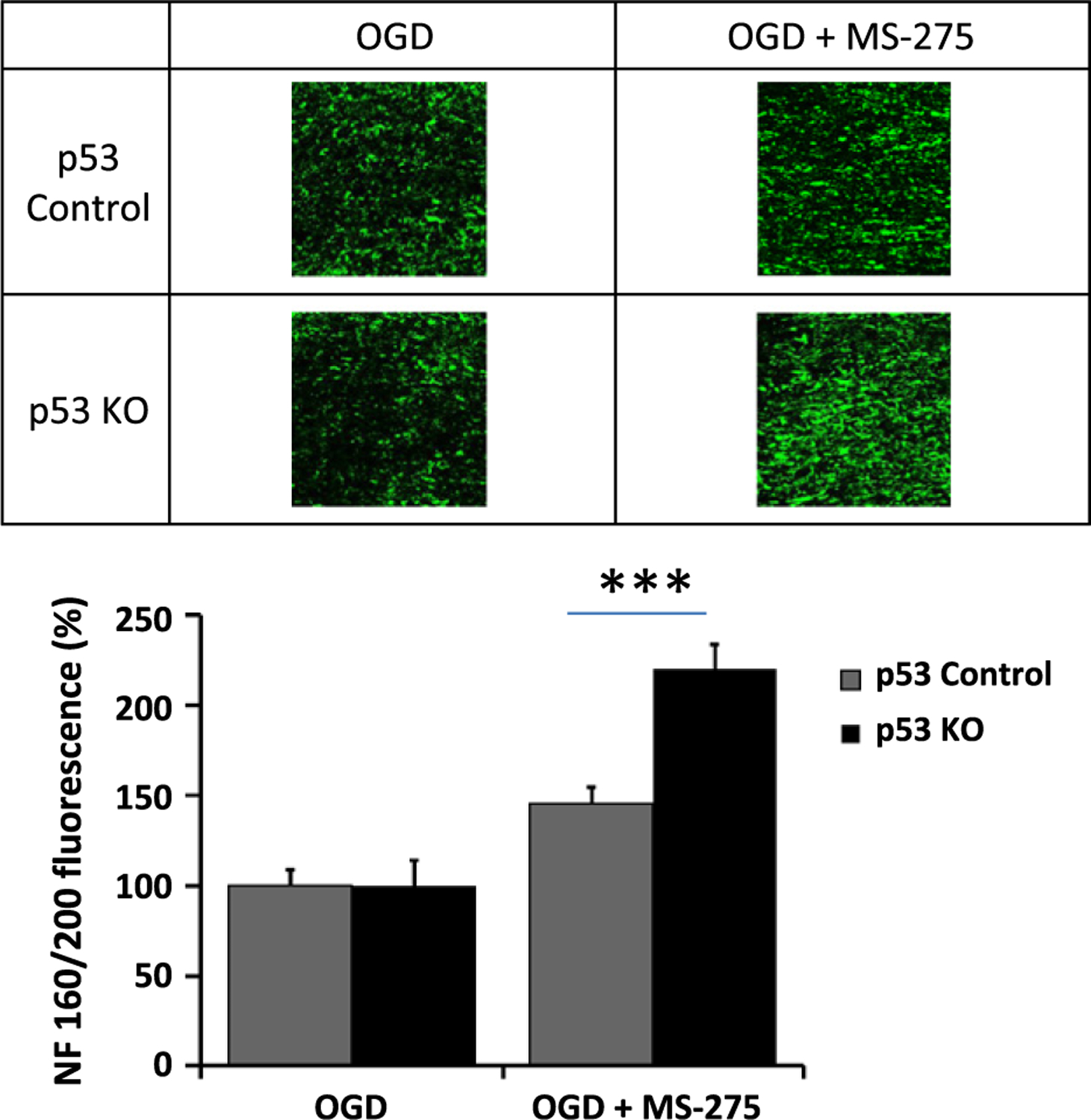
Images of NF160/200 in axons of mouse optic nerve (MON) from wt and p53 ko mice after oxygen–glucose deprivation (OGD), and in the absence or presence of MS-275. The histogram summarizes data from a minimum of three mice (± SEM) for each group. ***p < 0.01.
To validate the central effects of MS-275, we first delivered 30 mg/kg ip to mice after stroke, solubilized brain, and ran western blots for acetylated histone 3, total H3, and actin. It is clear that MS-275 accessed the brain to cause HDAC inhibition, as acetylated histone 3 was more abundant in these brains compared to those of untreated mice (Fig. 5). Then we looked at the effects of MS-275 and p53 expression on anatomic outcome after MCAo. Three days after occlusion of the right MCA for 60 min, there was an infarct that involved approximately 50% of the right hemisphere in wild-type mice (Fig. 6). As previously observed, mice devoid of p53 generally had smaller infarcts, though this failed to reach significance. While the administration of MS-275 post-stroke did not affect infarct size in wild-type mice, the p53−/− mice receiving MS-275 had consistently smaller infarcts.
Fig. 5.
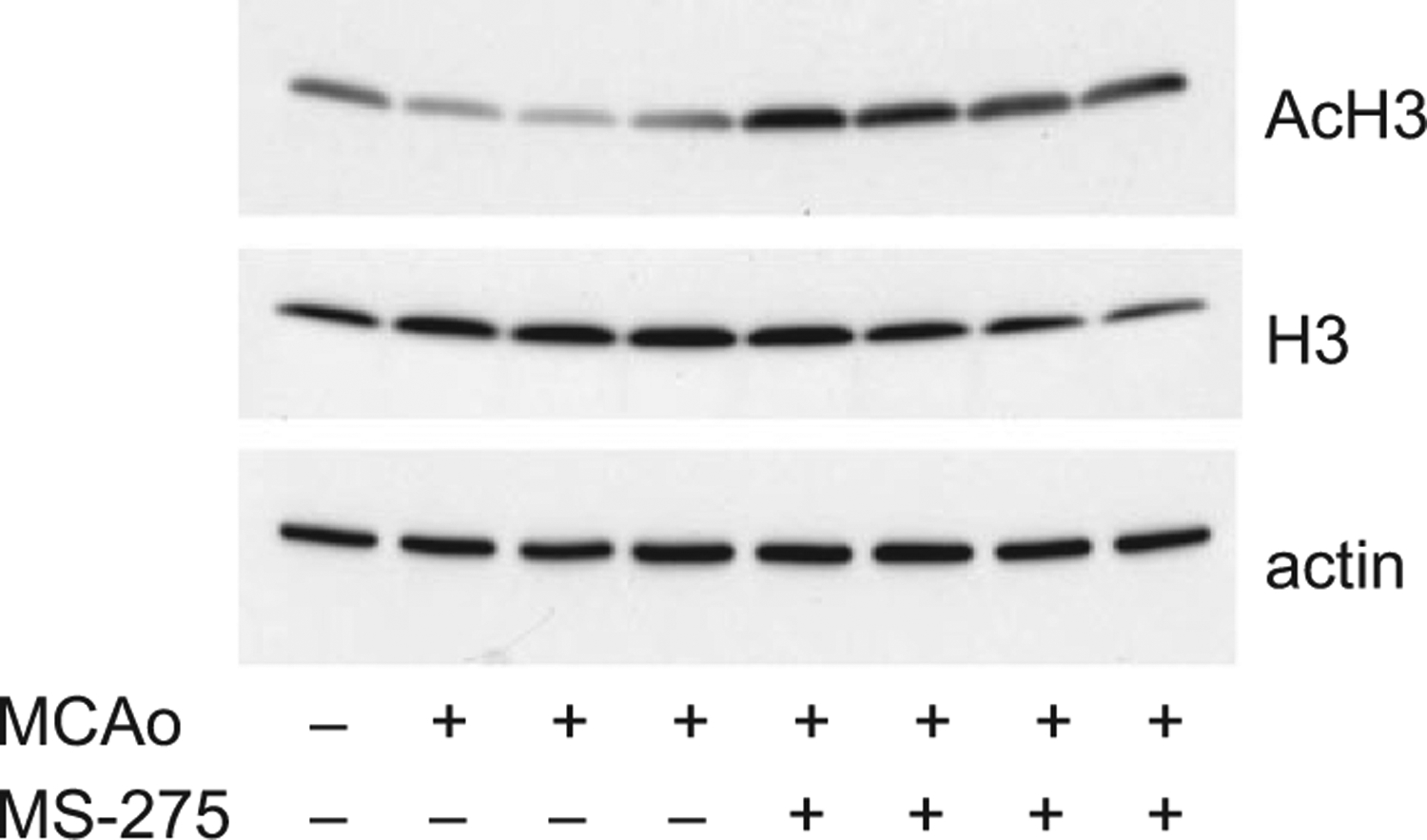
Intraperitoneal injection of MS-275 after middle cerebral artery occlusion (MCAo) resulted in the accumulation of acetylated histone protein 3 (AcH3) in mouse brain, as revealed by western blotting. Each lane comprises protein extracted from a different animal. Total histone protein 3 (H3) and β-actin served as controls.
Fig. 6.
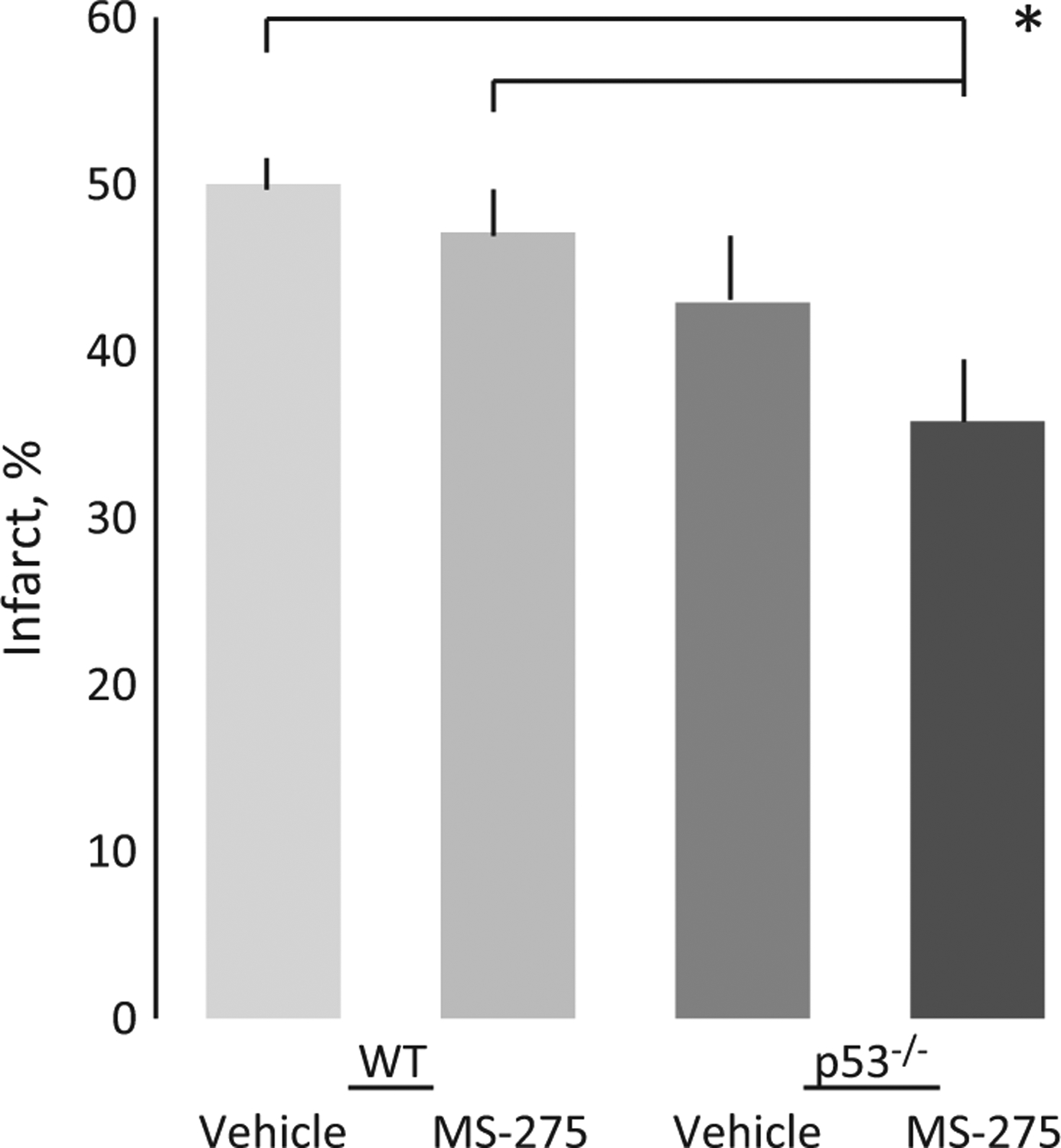
The ip administration of MS-275 after 60 min middle cerebral artery occlusion (MCAo) resulted in smaller infarcts (% hemisphere) in p53−/− as compared with wild-type mice. n = 10 ± SEM for each group. *p < 0.05.
Finally, as other HDAC inhibitors show protection against MCAo in wild-type mice (Gibson and Murphy 2010), we looked at the effects of MS-275 on p53 expression in neurons in culture (Fig. 7). In previous studies, addition of suberoylanilide hydroxamate and TSA were shown to dramatically increase p53 expression at the same time as suppressing p53-mediated transcriptional activity and cell death (Uo et al. 2009). As expected, both camptothecin and nutlin exposure increased p53 expression but, in contrast to other HDAC inhibitors, MS-275 suppressed p53 expression.
Fig. 7.
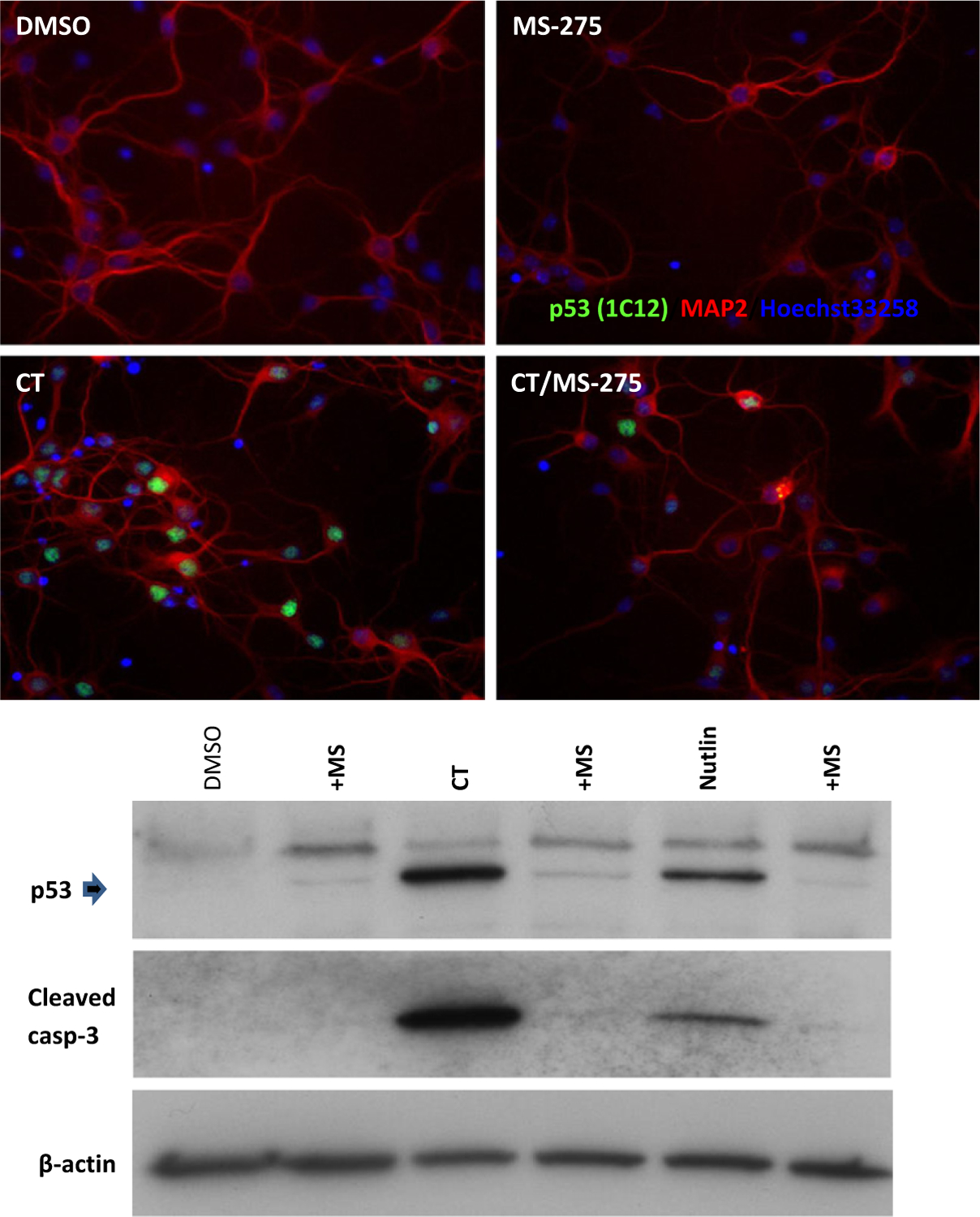
Administration of MS-275 to cortical neurons in culture prevented the accumulation of p53 and cleaved caspase 3 invoked by exposure to camptothecin (CT) and nutlin. Neurons were treated with CT (2.5 μM) in the presence or absence of MS-275 (10 μM) for 12 h (DMSO as a vehicle control), fixed, and stained for p53 and a neuronal marker, MAP2, with Hoechst 33258 staining of nuclei. Protein samples were prepared and subjected to western blotting for p53 and cleaved caspase 3 expression. β-actin was the control for sample loading.
Discussion
Our previous results from ex vivo (optic nerve) and in vitro (cultured neuron) studies support the notion that HDAC inhibitors engage a diverse array of targets involved in the maintenance of cell function and cell survival in the face of injury (Haberland et al. 2009). Potential explanations for the beneficial effects of compounds such as MS-275 are numerous (Baltan et al. 2013). For example, HDAC inhibitors can promote neuronal survival by blocking the action of p53 (Uo et al. 2009; Brochier et al. 2013), which has been implicated in acute CNS injury (Morrison et al. 2003). The apoptotic activity of p53 in post-natal cortical neurons is exclusively mediated through transcription-dependent functions (Uo et al. 2007) and HDAC inhibitors protect cultured post-natal cortical neurons from p53-dependent cell death by suppressing PUMA expression, a critical signaling intermediate linking p53 to Bax activation (Uo et al. 2009). Although many HDAC inhibitors (such as suberoylanilide hydroxamate, TSA) cause p53 to accumulate in neurons, despite blocking p53 transcriptional activity, we observed that MS-275 treatment reduced p53 accumulation after DNA damage and this may reflect a different mechanism of action (Uo et al. 2009). It is conceivable that MS-275, in contrast to other HDAC inhibitors, does not directly affect p53 acetylation, or its stability and activity. This drug might indirectly reduce p53 levels, without causing a significant decline in p53 activity, and this could explain why MS-275 is hardly effective in the presence of p53.
If the target for MS-275 is not p53, the drug could modulate neuronal survival by modifying the activity of other transcription factors. For example, nuclear erythroid 2-related factor 2 promotes multiple pathways of cellular protection by coordinating the transcription of genes that confer protection against oxidative stress and inflammation (Martín-Montalvo et al. 2011; Wang et al. 2012). Activation of the nuclear erythroid 2-related factor 2 pathway has been shown to increase the resistance of neurons to oxidative stress and metabolic insults in vitro, and to ischemic stroke in vivo (Son et al. 2010).
During ischemic injury, and consistent with increased fission and the loss of ATP, morphological changes in mitochondria are induced in the mouse optic nerve. However, HDAC inhibitors like MS-275 preserve mitochondria and maintain ATP (current studies; Baltan et al. 2011a). We have also found that a period of OGD depletes the glutamate transporter 1 transporter in optic nerve, but that the administration of HDAC inhibitors robustly up-regulates expression of this pump and prominently delays and attenuates glutamate release (Baltan et al. 2011a).
Which HDACs are implicated in stroke pathology? Based on the outcome of shRNA knockdown in cultured primary neurons (RS Morrison, unpublished observation), our focus has been on those of Class 1. Recently, we mapped the cell- and region-specific expression of HDACs 1, 2, and 3 in mouse brain, and reported that a period of ischemia up-regulates expression of these enzymes (Baltan et al. 2011b). Deletion of each of the Class 1 HDACs is embryonically lethal, mainly through effects on cardiac development, and for HDAC 3 the issue is defective repair of DNA double-stranded breaks (Haberland et al. 2009). There appear to be distinct roles for Class 1 HDACs in neuronal differentiation. The activity of HDAC 1 is required for differentiation to neurons, whereas HDAC 2 blocks differentiation to oligodendrocytes and inhibits differentiation to astrocytes (Humphrey et al. 2008). Both HDACs 1 and 2 are also involved in precursor cell maturation (Montgomery et al. 2009). Although it is not at all clear if the notable amplification of neurogenesis post-stroke contributes to subsequent restoration of function, the ability of HDAC inhibitors to promote migration and ‘neuronal’ differentiation could be exploited, therapeutically (Kahle and Bix 2013).
Although MS-275 selectively inhibits Class 1 HDACs (Hess-Stump et al. 2007; Khan et al. 2008), this drug does not discriminate between HDACs 1, 2, and 3 (Lauffer et al. 2013), and there is good evidence to suggest that inhibition of HDAC 1 in the CNS could be detrimental (Kim et al. 2008). Activity of HDAC 2 counteracts memory formation and over-expression impairs synaptic plasticity (Guan et al. 2009). HDAC 2 modulates nuclear factor kappa B and contributes to the signaling linked to cell proliferation, survival and differentiation, and pharmacological intervention to precisely target HDAC 2 is considered to be a valid therapeutic option (Kramer 2009). Recently, the neurotoxicity associated with mutant Huntingtin was shown to be much reduced in the absence of HDAC 3 (Bardai et al. 2013). The search for more selective inhibitors of HDACs 2 and 3 continues (Thomas 2009; Fischer et al. 2010; Durham 2012).
In summary, the Class 1 HDAC inhibitor, MS-275, ameliorates ischemic pathology in the CNS more robustly in the absence of p53, distinguishing its activity from other HDAC inhibitors. The acute (preserving white matter) and longer term effects of HDAC inhibitors (reducing cell death, promoting progenitor cell differentiation) suggest these drugs to be potential stroke therapeutics. Differences between broadly effective HDAC inhibitors, as shown in this study, further underscore the need to develop more selective inhibitors.
Acknowledgements
Thanks are due to Yoshito and Chizuru Kinoshita for generating wild-type and p53 knockout mice expressing CFP, and for western blotting, respectively. Support for this work was from the American Heart Association (RSM, SB) and from National Institutes for Health grants NS035533 and NS056031 (RSM), NS065319 (SPM and RSM) and AG033720 (SB). The authors declare no conflicts of interest.
Abbreviations used:
- AcH3
acetylated histone 3
- ACSF
artificial CSF
- CAP
compound action potential
- CFP
cyan fluorescent protein
- CT
camptothecin
- GFAP
glial fibrillary acidic protein
- HDAC
histone deactylase
- MCAo
middle cerebral artery occlusion
- MON
mouse optic nerve
- MS-275
N-(2-aminophenyl)-4-[N-(pyridine-3yl-methoxy-carbonyl) aminomethyl]benzamide
- NF160/200
neurofilament
- OGD
oxygen–glucose deprivation
- SAHA
suberoylanilide hydroxamate
- TTC
2,3,5-triphenyltetrazolium chloride
- WM
white matter
References
- Baltan S (2012) Histone deacetylase inhibitors preserve function in aging axons. J. Neurochem 12(Suppl. 2), 108–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltan S, Murphy SP, Danilov CA, Bachleda A and Morrison RS (2011a) Histone deacetylase inhibitors preserve white matter structure and function during ischemia by conserving ATP and reducing excitotoxicity. J. Neurosci 31, 3990–3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltan S, Bachleda A, Morrison RS and Murphy SP (2011b) Expression of histone deacetylases in cellular compartments of the mouse brain and the effects of ischemia. Transl. Stroke Res 2, 411–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltan S, Morrison RS and Murphy SP (2013) Novel protective effects of histone deacetylase inhibition on stroke and white matter ischemic injury. Neurotherapeutics 10, 798–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardai FH, Verma P, Smith C, Rawat V, Wang L and D’Mello SR (2013) Disassociation of HDAC 3 from normal Huntingtin underlies mutant Huntingtin neurotoxicity. J. Neurosci 33, 11833–11838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brochier C, Dennis G, Rivieccio MA et al. (2013) Specific acetylation of p53 by HDAC inhibition prevents DNA damage-induced apoptosis in neurons. J. Neurosci 33, 8621–8623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang D-W, Leng Y, Marinova Z, Kim H-J and Chiu C-T (2009) Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci. 32, 591–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crumrine RC, Thomas AL and Morgan PF (1994) Attenuation of p53 expression protects against focal ischemic damage in transgenic mice. J. Cereb. Blood Flow Metab 14, 887–891. [DOI] [PubMed] [Google Scholar]
- Culmsee C, Zhu X, Yu QS et al. (2001) A synthetic inhibitor of p53 protects neurons against death induced by ischemic and excitotoxic insults, and amyloid beta-peptide. J. Neurochem 77, 220–228. [DOI] [PubMed] [Google Scholar]
- Dokmanovic M, Clarke C and Marks PA (2007) Histone deacetylase inhibitors: overview and perspectives. Mol. Cancer Res 5, 981–989. [DOI] [PubMed] [Google Scholar]
- Donehower LA, Harvey M, Slagle BL et al. (1992) Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 356, 215–221. [DOI] [PubMed] [Google Scholar]
- Durham B (2012) Novel HDAC inhbiitors with improved selectivity for HDAC 2 and 3 protect against cell death. Biosci. Horiz 5, 1–7. [Google Scholar]
- Endo H, Kamada H, Nito C, Nishi T and Chan PH (2006) Mitochondrial translocation of p53 mediates release of cytochrome c and hippocampal CA1 neuronal death after transient global cerebral ischemia in rats. J. Neurosci 26, 7974–7983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A, Sananbenesi F, Mungenast A and Tsai L-H (2010) Targeting the correct HDAC(s) to treat cognitive disorders. Trends Pharmacol. Sci 31, 605–617. [DOI] [PubMed] [Google Scholar]
- Gibson C and Murphy SP (2010) Benefits of histone deacetylase inhibitors for acute brain injury; a systematic review of animal studies. J. Neurochem 115, 806–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan J-S, Haggarty SJ, Giacometti E et al. (2009) HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 459, 55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberland M, Montgomery RL and Olson EN (2009) The many roles for HDACs in development and physiology. Nat. Rev. Gen 10, 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handel AE, Ebers GC and Ramagoplan SV (2009) Epigenetics: molecular mechanisms and implications for disease. Trends Mol. Med 16, 7–16. [DOI] [PubMed] [Google Scholar]
- Hess-Stump H, Bracker TU, Henderson D and Politz O (2007) MS-275, a potent orally available inhibitor of HDACs. Int. J. Biochem. Cell Biol 39, 1388–1405. [DOI] [PubMed] [Google Scholar]
- Humphrey GW, Wang Y-H, Hirai T et al. (2008) Complementary roles for HDACs 1, 2, and 3 in differentiation of pluripotent stem cells. Differentiation 76, 348–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakovcevski M and Akbarian S (2012) Epigenetic mechanisms in neurological disease. Nat. Med 18, 1194–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayadev S, Nesser NK, Hopkins S et al. (2011) The transcription factor p53 influences microglia activation phenotype. Glia 59, 1402–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahle MP and Bix GJ (2013) Neuronal restoration following ischemic stroke: influences, barriers, and therapeutic potential. Neurorehabil. Neural Repair 27, 469–478. [DOI] [PubMed] [Google Scholar]
- Khan N, Jeffers M, Kumar S et al. (2008) Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem. J 409, 581–589. [DOI] [PubMed] [Google Scholar]
- Kim D, Frank CL, Dobbin MM et al. (2008) Deregulation of HDAC1 by p25/Cdk5 in neurotoxicity. Neuron 60, 803–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer OH (2009) HDAC2: a critical factor in health and disease. Trends Pharmacol. Sci 30, 647–655. [DOI] [PubMed] [Google Scholar]
- Langley B, D’Annibale MA, Suh K et al. (2008) Pulse inhibition of HDACs induces complete resistance to oxidative death in cortical neurons without toxicity and reveals a role for cytoplasmic p21 waf1/cip1 in cell cycle-independent neuroprotection. J. Neurosci 28, 163–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley B, Brochier C and Rivieccio MA (2009) Targeting HDACs as a multifaceted approach to treat the diverse outcomes of stroke. Stroke 40, 2899–2905. [DOI] [PubMed] [Google Scholar]
- Lauffer BEL, Mintzer R, Fong R et al. (2013) Histone deacetylase (HDAC) inhibitor kinetic rate constants correlate with cellular histone acetylation but not transcription and cell viability. J. Biol. Chem 288, 26926–26943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leker RR, Aharonowiz M, Greig NH and Ovadia H (2004) The role of p53-induced apoptosis in cerebral ischemia: effects of the p53 inhibitor pifithrin alpha. Exp. Neurol 187, 478–486. [DOI] [PubMed] [Google Scholar]
- Luo J, Su F, Chen D, Shiloh A and Gu W (2000) Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature 408, 377–381. [DOI] [PubMed] [Google Scholar]
- Martín-Montalvo A, Villalba JM, Navas P and de Cabo R (2011) NRF2, cancer and calorie restriction. Oncogene 30, 505–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misgeld T, Kerschensteiner M, Bareyre FM, Burgess RW and Lichtman JW (2007) Imaging axonal transport of mitochondria in vivo. Nat. Methods 4, 559–561. [DOI] [PubMed] [Google Scholar]
- Montgomery RL, Hsieh J, Barbosa AC, Richardson JA and Olson EN (2009) HDACs 1 and 2 control the progression of neural precursors to neurons during brain development. Proc. Natl Acad. Sci. USA 106, 7876–7881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison R, Kinoshita Y, Johnson MD, Guo W and Garden GA (2003) p53-dependent cell death signaling in neurons. Neurochem. Res 28, 15–27. [DOI] [PubMed] [Google Scholar]
- Pearce WJ (2011) Epigenetics: an expanding new piece of the stroke puzzle. Transl. Stroke Res 2, 243–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qureshi IA and Mehler MF (2010) Emerging role of epigenetics in stroke. Part 1: DNA methylation and chromatin modifications. Arch. Neurol 67, 1316–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweizer S, Meisel A and Marschenz S (2013) Epigenetic mechanisms in cerebral ischemia. J. Cereb. Blood Flow Metab 33, 1335–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakespear MR, Halili MA, Irvine KM, Fairlie DP and Sweet MJ (2011) Histone deacetylases as regulators of inflammation and immunity. Trends Immunol. 32, 335–343. [DOI] [PubMed] [Google Scholar]
- Son TG, Camandola S, Arumugam TV et al. (2010) Plumbagin, a novel Nrf2/ARE activator, protects against cerebral ischemia. J. Neurochem 112, 1316–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas EA (2009) Focal nature of neurological disorders necessitates isotype-selective HDAC inhibitors. Mol. Neurobiol 40, 33–45. [DOI] [PubMed] [Google Scholar]
- Uo T, Kinoshita Y and Morrison RS (2005) Neurons exclusively express N-Bak, a BH3 domain-only Bak isoform that promotes neuronal apoptosis. J. Biol. Chem 280, 9065–9073. [DOI] [PubMed] [Google Scholar]
- Uo T, Kinoshita Y and Morrison RS (2007) Apoptotic actions of p53 require transcriptional activation of PUMA and do not involve a direct mitochondrial/cytoplasmic site of action in postnatal cortical neurons. J. Neurosci 27, 12198–12210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uo T, Veenstra TD and Morrison RS (2009) HDAC inhibitors prevent p53-dependent and -independent Bax-mediate neuronal apoptosis through two distinct mechanisms. J. Neurosci 29, 2824–2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaseva AV, Marchenko ND, Ji K, Tsirka SE, Holzmann S and Moll UM (2012) p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell 149, 1536–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Zhu X, Kim YT et al. (2012) HDAC inhibition activates transcription factor Nrf2 and protects against cerebral ischemic damage. Free Radic. Biol. Med 52, 928–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y-L and Yang W-M (2011) Beyond histone and deacetylase: an overview of cytoplasmic HDACs and their non-histone substrates. J. Biomed. Biotech 2011, 146493. [DOI] [PMC free article] [PubMed] [Google Scholar]