

Abstract
Whole-exome and whole-genome sequencing studies in autism spectrum disorder (ASD) have identified hundreds of thousands of exonic variants. Only a handful of them, primarily loss-of-function variants, have been shown to increase the risk for ASD, while the contributory roles of other variants, including most missense variants, remain unknown. New approaches that combine tissue-specific molecular profiles with patients’ genetic data can thus play an important role in elucidating the functional impact of exonic variation and improve understanding of ASD pathogenesis. Here, we integrate spatio-temporal gene co-expression networks from the developing human brain and protein-protein interaction networks to first reach accurate prioritization of ASD risk genes based on their connectivity patterns with previously known high-confidence ASD risk genes. We subsequently integrate these gene scores with variant pathogenicity predictions to further prioritize individual exonic variants based on the positive-unlabeled learning framework with gene- and variant-score calibration. We demonstrate that this approach discriminates among variants between cases and controls at the high end of the prediction range. Finally, we experimentally validate our top-scoring de novo mutation NP 001243143.1:p.Phe309Ser in the sodium/potassium-transporting ATPase ATP1A3 to disrupt protein binding with different partners.
1. Introduction
Autism spectrum disorder (ASD) is a group of complex neurodevelopmental disorders with a strong genetic component [1–4]. The field of psychiatric genetics has worked vigorously for more than a decade to discover genetic contributors to the risk for ASD. As a result, it is now understood that the genetic architecture of ASD represents a combination of high-risk rare copy number variants [5–8], rare coding variants detected through whole-exome sequencing of ASD families [9–12], and common variants identified in genome-wide association studies [3]. Recently, however, non-coding variants identified through the large whole-genome sequencing studies [13–15] have also begun to accumulate evidence for involvement in ASD. The genetic etiology of ASD is likely intermediate, with polygenic variation contributing additively in the presence of a strong de novo variant [1, 16]. In particular, pathogenic de novo variation shows potential to account for ASD occurrence in simplex families; i.e., those with a single affected child. ASD cases in such families have been found to harbor twice as many de novo loss-of-function (LoF) variants than expected by chance, although the recurrence of any particular variant is low [17]. Other types of variants, primarily missense variants, have subtler group signatures [17] and have recently attracted increased attention [18–21].
Due to the complex genetic architecture of ASD, identification of dysregulated signaling and regulatory pathways has remained challenging. Based on the biological functions of genes that carry recurrent de novo mutations, convergence on chromatin remodeling, synaptic and neuronal signaling, transcriptional and translational regulation have emerged [12, 17, 22, 23]. Collectively, mTor, MAPK and beta-catenin/Wnt signaling have all been implicated [24]. The integration of genetic data with other data types has further demonstrated that high-risk ASD genes are highly connected in co-expression and protein interaction networks, especially during late mid-fetal stages of brain development [25–29].
The abundance of available genetic and molecular data have led to the development of computational approaches to effectively identify new genes with association to ASD. For example, Mosca et al. [30] used a diffusion-based prioritization in a network to identify significantly connected gene modules associated with ASD. Krishnan et al. [31] performed a genome-wide prediction of ASD risk genes using a machine-learning approach based upon a brain-specific gene network, and used a case-control sequencing-study validation set to identify pathways and brain developmental stages to predict ASD risk genes with minimal or no prior genetic evidence. Similarly, Duda et al. [32] used a brain-specific functional relationship network for ASD risk gene prioritization. In the past several years, more comprehensive efforts have been made to integrate brain-specific gene expression data to further generate gene-level predictions for association with ASD [22, 33–38]. While these approaches have made strides in the identification of genes relevant to ASD, the challenge remains to incorporate this data with variant-level information to identify individual variants that significantly increase the risk for ASD.
Here, we seek to assess the utility of gene- and variant-scoring methods to prioritize impactful exonic de novo variation in individuals with ASD. We first quantify the strength of the relationship between a given gene and previously discovered high-confidence ASD risk genes by leveraging brain gene expression and protein-protein interaction data. We find that brain-specific co-expression networks improve model performance compared to the networks from other tissues, or to protein-protein interaction networks. Then, our approach integrates gene scores with variant pathogenicity predictions to prioritize individual exonic variants. The integration was carried out in a positive-unlabeled framework that allows for rigorous score calibration [39]. We apply this methodology to de novo variation derived from the Simons Foundation Collection families [9–11, 40, 41] and from other large-scale sequencing studies [12, 17, 42–57], and achieve effective discriminative case/control capacity on high-scoring variants. Finally, we validate one missense variant in an experimental follow-up study, confirming its putative contribution to ASD risk through the disruption of interactions with three protein-binding partners.
2. Materials and Methods
2.1. Systems data
To construct gene networks, we first integrated gene expression data and protein-protein interaction (PPI) data. We used the “RNA-Seq Gencode v10 summarized to genes” dataset from the BrainSpan atlas of the developing human brain [58, 59] to construct an expression matrix of 52,376 transcripts over 524 human brain samples derived from 57 postmortem brain specimens [58]. The 19,113 transcripts corresponding to protein-coding genes were subsequently grouped into 4 brain regions (Table 1) and 12 developmental periods (Table 2) as previously described [26, 28].
Table 1:
Brain region groupings for the BrainSpan dataset. Samples collected from various regions of the brain were separated into 4 groups, indexed from 1 to 4, as previously described [26, 28].
| Index | Brain Regions |
|---|---|
| 1 | Occipital neocortex, posterior (caudal) superior temporal cortex (area 22c), inferolateral temporal cortex (area TEv, area 20), posteroventral (inferior) parietal cortex, primary auditory cortex (core), primary visual cortex (striate cortex, area V1/17), temporal neocortex. |
| 2 | Anterior (rostral) cingulate (medial prefrontal) cortex, dorsolateral prefrontal cortex, orbital frontal cortex, parietal neocortex, primary motor cortex (area M1, area 4), primary motor-sensory cortex (samples), primary somatosensory cortex (area S1, area 3, 1, 2), ventrolateral prefrontal cortex. |
| 3 | Amygdaloid complex, caudal ganglionic eminence, hippocampus (hippocampal formation), lateral ganglionic eminence, medial ganglionic eminence, striatum. |
| 4 | Cerebellar cortex, cerebellum, dorsal thalamus, mediodorsal nucleus of thalamus, upper (rostral) rhombic lip. |
Table 2:
Time period groupings for the BrainSpan dataset. Samples collected at various stages of brain development were separated into 12 groups, indexed from 2 to 13, as previously described [26, 28].
| Index | Periods | Index | Periods |
|---|---|---|---|
| 2 | Early fetal | 8 | Neonatal and early infancy |
| 8–9 pcw | 0–5 mos | ||
| 3 | Early fetal | 9 | Late infancy |
| 10–12 pcw | 6–11 mos | ||
| 4 | Early mid-fetal | 10 | Early childhood |
| 13–15 pcw | 1–5 yrs | ||
| 5 | Early mid-fetal | 11 | Middle and late childhood |
| 16–18 pcw | 6–11 yrs | ||
| 6 | Late mid-fetal | 12 | Adolescence |
| 19–23 pcw | 12–19 yrs | ||
| 7 | Late fetal | 13 | Young adulthood |
| 24–37 pcw | 20–40 yrs |
Period abbreviations: pcw = post-conception weeks, mos = months, yrs = years.
Next, we assembled a dataset of 303,040 binary protein-protein interactions by combining physical PPIs from BioGRID v3.4.159 [60], gene-level interactions from the Autism Spliceform Interaction Network [27], the human interactome from [61] and gene-level PPIs from [62].
2.2. Rare de novo variants
We obtained 9,174 protein-coding de novo variants of three types; i.e., missense, in-frame insertion/deletion (indel) and loss-of-function (LoF; stop gain and frameshifting indels) from whole-exome sequencing studies of the Simons Foundation Collection families [9–11, 41] and other studies [12, 17, 42–57]. Variants present in the gnomAD database [63] as well as variants shared between cases and controls were filtered out from the case dataset because of their high likelihood of being non-pathogenic [64]. Our final set contained 3,608 variants (Table 3).
Table 3:
Breakdown of the de novo variants used in this study.
| Missense | LoF | Indel | Total | |
|---|---|---|---|---|
| Case (proband) | 2022 | 633 | 62 | 2717 |
| Control (sibling) | 737 | 132 | 22 | 891 |
| Total | 2759 | 765 | 84 | 3608 |
2.3. Network construction
We used gene expression data to build a correlation network for all brain regions (R), developmental periods (P) and their combinations (RP). Genes constituted nodes in these networks, whereas the links were constructed by reliably estimating the correlation of expression profiles across relevant samples for all pairs of genes. As criteria for noise filtering, we required more than five pairs of independent samples supporting the calculation of a Pearson’s correlation coefficient (ρ) as well as that at least one gene from each pair had at least 0.5 Transcripts Per Million (TPM) expression. The same network construction steps were taken for processing co-expression data from GTEx [65] to construct tissue-specific networks for our baseline approaches. GTEx data was available for multiple tissues, including adult human brain.
Gene co-expression networks were merged with PPI networks in two ways, referred to here as the “intersection” and “union” integration. In the “intersection” approach, the weight wij of the link between gene i and gene j was defined as
where Ic is an indicator function for the logical expression c, rij is the thresholded correlation coefficient ρij as described below, and | · | is an absolute value function. Similarly, the weight of each link in the “union” approach was defined as
In both approaches, we retained only co-expression edges with absolute values of at least 0.75; i.e., , where ρij is Pearson’s correlation between expressions of genes i and j over a set of samples in the BrainSpan dataset. In summary, three types of networks were built for each R, P, or RP for gene prioritization: (1) gene co-expression network without PPIs; (2) the intersection of co-expression and the PPI network; (3) the union of co-expression and the PPI network.
2.4. Gene and variant scoring
There may be hundreds or even thousands of genes involved in ASD [24, 66, 67], but the role of each gene and its contribution to the development of ASD is for the most part unknown. In the past decades, studies have identified about a hundred genes conferring high risk for ASD [4]. To prioritize the remaining genes, we used the functional flow network propagation approach [68] across spatio-temporal co-expression and PPI networks as described below.
The network seed genes (denoted as POS65; Supplementary Table S2) were derived from [11] and consisted of 65 highly confident genome-wide significant ASD risk genes. The performance of gene scoring was evaluated on an independent set of 63 genes (denoted as VAL63; Supplementary Table S2) assembled by removing POS65 genes from recently identified 102 high-risk autism genes [4]. As a negative control, additional evaluation datasets included 1000 lists of 63 genes, randomly sampled from BrainSpan to be similar in length and GC content (±10%) to the VAL63 genes.
The performance among methods with different parameter settings was compared along several dimensions: (i) edge weight normalization method in propagation: the original functional flow and its two variants (i.e., incoming and outgoing edge normalization); (ii) three different settings for edge cutoffs with controls of network sparsity; and (iii) number of propagation strides [68]. Through five-fold cross-validation, we identified the parameter settings with the best performance using 51 BrainSpan networks (4 regions, 12 periods and 35 region/period combinations) with and without PPI networks, and then used the best parameters for testing. Thirteen region/period combinations were omitted due to lack of samples.
The effect of missense variants was estimated with MutPred2 [19], loss-of-function variants with MutPred-LOF [69], and non-frameshifting indel variants and multi-residue substitutions with MutPred-Indel [70]. These predictors were selected based on the fact that they were all trained using similar protocols, their good performance in the prediction of both pathogenicity and protein function disruption [71], as well as that all report molecular mechanisms potentially causative of pathogenicity. High-scoring variants with “loss of protein binding” as an underlying mechanism were of primary interest for downstream experimental validation.
Although we were interested in variants that alter protein function, we note that exonic variants could lead to phenotypic changes via other molecular mechanisms, such as splicing disruption or impact on RNA stability and folding. Our approach has not directly considered such events.
2.5. Probabilistic model for autism-specific variant scoring
We propose a simple semi-supervised probabilistic model that combines the risk that a gene is involved in ASD with the probability that the variant disrupts the function of this gene. Before describing the model, we argue that both gene scoring and variant scoring can be approached through positive-unlabeled learning, a form of semi-supervised binary classification in which all labeled data is positive and unlabeled data is a mixture of positive and negative examples at unknown proportions [72]. In our problem, known ASD genes can be considered positive, whereas other genes can be considered to be unlabeled. The task of a gene prioritization model is then to identify remaining positive genes among unlabeled genes. Similarly, in variant scoring, we are given a set of disease-causing variants, such as those from the Human Gene Mutation Database [73], and a set of unlabeled variants from gnomAD. The task of a variant interpretation model is then to identify remaining disease-causing variants among unlabeled variants.
A common approach to positive-unlabeled learning is to develop classifiers by training positive against unlabeled data [74]. This approach was in fact shown to be optimal for a range of loss functions for model learning [75, 76], in the sense that minimizing the loss function from positive and unlabeled data (models referred to as non-traditional classifiers [74]), simultaneously minimizes the loss if one were to train a model from positive and negative data (models referred to as traditional classifiers [74]). Unfortunately, although ranking objectives such as area under the ROC curve fall under this scenario, the scores outputted by non-traditional classifiers are not calibrated to represent posterior probabilities [39]. As such, the outputs from gene prioritization tools and variant interpretation tools cannot be formally combined as probabilities. We will address the score calibration models after the model is introduced.
Let D (diagnosis) be a binary random variable indicating the diagnosis of ASD and v a single variant occurring in some gene g. We focus on a single variant at a time because the average number of de novo coding variants in an individual is around one [77]. Let now E (effect) and R (risk) denote binary random variables whether the function of a protein product of g is disrupted in the presence of v and whether g is an ASD risk gene, respectively. We can then use marginalization to write the probability of diagnosis d as
where d, e, and r are realizations of the random variables D, E, and R, respectively, variant v can be thought of as a realization of a random variable V, and p denotes an appropriate probability mass function; e.g., p(d|v) = P(D = d|v), etc. We are primarily interested in identifying new risk genes and variants and, thus, we focus on P(D = 1|v).
We now observe that nonfunctional variants cannot contribute to the positive diagnosis and neither can variants outside of the group of the ASD risk genes; i.e., P(D = 1|E = e, R = r, v) = 0 unless both e = 1 and r = 1. Hence, we can write the probability of the ASD diagnosis given variant v as
since all other terms reduce to 0. We now make an assumption that any variant disrupting the function of an ASD risk gene causes the phenotype with certainty. Then, by applying conditional independence between a variant disrupting gene function and that gene being an ASD risk gene, we obtain a probabilistic model of ASD diagnosis in the presence of a de novo variant v as
| (1) |
The last two terms on the right-hand side of Eq. (1) correspond to a variant-level score and a gene-level score, respectively. We have further replaced the probability P(R = 1|v) with P(R = 1|g) to clarify that the probability that g is a risk gene is strictly a gene property, as long as variant v is within g. Probabilities P(E = 1|v) and P(R = 1|g) are first obtained by applying a dedicated variant- or gene-prediction tool, which are then calibrated to be proper probabilities, as described in Section 2.6. To avoid multiplying small probabilities, we have scored each variant using a logarithm transform
| (2) |
This model described above is appropriate for phenotype-specific prioritization of highly penetrant variants. However, even in complex phenotypes such as ASD, it has been observed that the polygenic effect can be modulated in the presence of a strong de novo variant [1]. Therefore, although we expect a lower performance levels compared to Mendelian disorders, we believe that a useful diagnostic signal can still emerge. Polygenic scores corresponding to the set of individuals considered here were not available for the development of more sophisticated models.
2.6. Score calibration in the positive-unlabeled setting
According to the above derivation, the gene score P(R = 1|g) and variant score P(E = 1|v) can be simply multiplied to yield the probability that the variant v in gene g leads to ASD. However, the outputs of the gene and variant scoring tools require calibration before they can be considered good approximations of the posteriors and multiplied together. To illustrate this seeming subtlety, we will digress to discuss model development in a positive-unlabeled setting.
Consider a binary classification problem of mapping inputs into outputs on the dataset drawn i.i.d. from a fixed but unknown probability distribution p(x, y), where (x, y) is a realization of a random vector (X,Y) of inputs (X) and outputs (Y). In a traditional supervised setting, we are given a set of positive examples obtained from p(x|Y = 1) and a set of negative examples obtained from p(x|Y = 0), roughly available at proportions P(Y = 1) and P(Y = 0), respectively. In contrast, a positive-unlabeled setting considers a training data obtained through a selection process to contain a set of positive examples drawn from p(x|Y = 1) and a set of unlabeled examples drawn from the marginal distribution .
Using S to represent a binary random variable that a data point is labeled (S = 1 indicates labeled and S = 0 unlabeled), we can train a classifier to approximate the posterior distribution between labeled and unlabeled data as P(S = 1|x). Jain et al. [78] derived a formula to then convert P(S = 1|x) into P(Y = 1|x) as
| (3) |
where P(S = 1) = 1 − P(S = 0) is the probability of observing a (positively) labeled example in the training data and P(Y = 1) is the probability of observing a positive example in the unlabeled data. Therefore, to estimate the posterior probability of the positive output given some input x, two conditions must be fulfilled: (1) we must train a non-traditional classifier that estimates P(S = 1|x), and (2) we must estimate P(Y = 1).
The first condition can be reasonably achieved by training models that approximate the posterior distributions in a binary classification setting. Posterior approximation has been covered in the literature; e.g., Rojas [79] demonstrated it for neural networks, whereas Platt [80] gave a post-processing algorithm for learners such as support vector machines. The second condition requires a complex step of nonparametric estimation of class priors in unlabeled data [39, 78]. The class prior P(Y = 1) in this work was estimated using the AlphaMax algorithm [39, 78] and the fraction P(S = 0)/P(S = 1) was estimated as the fraction of unlabeled and labeled training examples. The uncalibrated probability P(S = 1|x) was the output of a dedicated tool in either gene prioritization or variant interpretation, as applicable. Finally, we note that errors in estimating P(S = 1|v) and P(Y = 1) can lead to undesired situations that the calibrated probability is greater than 1. The (monotonic) logarithmic transform from Eq. (2) allowed us to disregard such problems.
2.7. Clinical significance of variant scoring
Evaluation of variant interpretation for complex clinical phenotypes is a difficult task owing to the mostly low-to-moderate penetrance of pathogenic variants. Even when penetrance is high, de novo variation, compound heterozygosity, or structural variation could all be contributing factors for different subsets of individuals [24], which presents evaluation problems for de novo variation because the ground truth is unavailable. Therefore, standard machine learning approaches that include ROC curves, precision-recall curves and their derivatives (e.g., area under the curve) cannot be effectively used to evaluate the quality of predictive models.
To evaluate potential clinical impact of our scoring of de novo variation, we use the positive likelihood ratio (LR+), defined as the ratio of posterior and prior odds of pathogenicity [81]. Let P(Y = 1) be the fraction of pathogenic variants in the population of interest and P(Y = 1|f(x) = 1) be the fraction of pathogenic variants in the same population but when a computational model gives a positive prediction. Then, the likelihood ratio for the positive prediction f(x) = 1 is defined as
| (4) |
where the prior odds are defined as the ratio of P(Y = 1) and P(Y = 0), and the posterior odds are defined as the ratio of P(Y = 1|f(x) = 1) and P(Y = 0|f(x) = 1). The likelihood ratio is therefore the increase in odds of pathogenicity due to the positive prediction. It can be shown that LR+ is independent of the class prior P(Y = 1) and can be computed as the ratio of the true positive rate and false positive rate [81].
The positive likelihood ratio is related to the Diagnostic Odds Ratio (DOR) that has been often used for risk assessment, particularly in cancer studies [82]. The relationship is expressed as
where LR− is defined as the ratio of the false negative rate and true negative rate [81]. Since LR− is limited to a [0, 1] interval for predictors whose ROC curve never drops below the identity line, LR+ is generally lower than DOR and thus gives a more conservative view on clinical utility.
2.8. Experimental validation
As a proof of principle, we selected one missense variant for experimental validation. This variant was selected based on the following criteria: (1) the gene was not in the list of POS65 or 102 genes from [4]; (2) the gene was highly scored by top-performing BrainSpan networks to suggest that it was likely an ASD risk gene; (3) the mutation was scored with a high MutPred score to suggest pathogenicity; (4) the mutation was not present in either the Human Gene Mutation Database [73] or ClinVar [83]; (5) no variants from controls were found in the gene.
2.8.1. Plasmid cloning and cell transfection
The ORF clones of the gene of interest, ATP1A3, and its interacting partners were obtained from the ORFeome Collaboration [84]. The genes were transferred from the donor plasmid pDONR223 to destination plasmids, pDEST40 and pDEST47, using LR Gateway reaction (Invitrogen) following manufacturer’s instructions. The gene of interest was introduced into pDEST40 to obtain ATP1A3-V5 tagged, and the partners were transferred to pDEST47, to obtain GFP-tagged proteins.
HEK 293T cells were seeded at 5 × 105 cells per well in 60 mm plates (Genesee Scientific). After 24h, cells were transfected using Lypofectamine 3000 (Invitrogen) following manufacturer’s instructions and then harvested after additional 48h.
2.8.2. Co-Immunoprecipitation and Western Blot
HEK 293T cells were harvested and rinsed once with ice-cold 1xPBS, pH 7.2, and lysed in immunoprecipitation lysis buffer (20 mM Tris, pH 7.4, 140 mM NaCl, 10% glycerol, and 1% Triton X-100) supplemented with 1xEDTA-free complete protease inhibitor mixture (Roche) and phosphatase inhibitor cocktails-I, II (Sigma Aldrich). The cells were centrifuged at 16000 × g at 4°C for 30 min, and the supernatants were collected. Protein concentration was quantified by modified Lowry assay (DC protein assay; Bio-Rad). The cell lysates were resolved by SDS-PAGE and transferred onto PVDF Immobilon-P membranes (Millipore). After blocking with 5% nonfat dry milk in TBS containing 0.1% Tween 20 for 1hr at room temperature, membranes were probed overnight with the appropriate primary antibodies. They were then incubated for 1h with the species-specific peroxidase-conjugated secondary antibody. Membranes were developed using the Pierce-ECL Western Blotting Substrate Kit (Thermo Scientific).
For immunoprecipitation experiments, samples were lysed and quantified as described above. Then, 1mg of total protein was diluted with immunoprecipitation buffer to achieve a concentration of 1 mg/ml. A total of 30 μl of anti-V5-magnetic beads-coupled antibody (MBL) was add-ed to each sample and incubated for 4h at 4°C in tube rotator. Beads were then washed twice with immunoprecipitation buffer and three more times with ice cold 1xPBS. The proteins were then eluted with 40 μl of 2xLaemli buffer. After a short spin, supernatants were carefully removed, and SDS-PAGE was performed. The following primary antibodies were used: anti-V5 (1:1000; Invitrogen), anti-GFP (1:1000; Cell Signaling), anti-GAPDH (1:5000; Cell Signaling).
3. Results
3.1. Brain-specific co-expression networks improve ASD gene prioritization
To assess the extent to which brain-specific gene co-expression networks and protein-protein interaction (PPI) networks help in autism gene prioritization, we ran a label propagation algorithm with POS65 as the seed genes on all gene networks described in the Methods section. We assessed the quality of the predicted gene scores of the algorithm with various parameters using stratified 5-fold cross-validation for the three network types (i.e., co-expression networks and the “union” and “intersection” with PPI). The final parameter set included ρ = 0.75 for co-expression network construction and 5 strides with outgoing weight normalization for the functional flow procedure [68].
Figure 1A shows the area under the Receiver Operating Characteristic (ROC) curve (AUC) evaluated over all region-, period-, and region/period-specific networks with or without the PPI data; for detailed results see Supplementary Table S3. We observed that in most cases, networks constructed using the union of co-expression and PPI networks performed better than co-expression networks without the PPI information. Similarly, many of the networks using region- and period-specific brain co-expression data outperformed the PPI-only network. Region-wise, all brain regions performed similarly well, with R1 and R2 displaying a slightly better performance than other regions. Period-wise, P5 (16–18 pcw) and P10 (1–5 yrs) performed best. With regard to region/period networks, P10 in combination with any region, but especially R2-P10 and R4-P10 combinations, had superior performance. Top region/period-specific networks with the union of PPI outperformed region- and period-specific networks as well as the PPI-only network.
Figure 1:
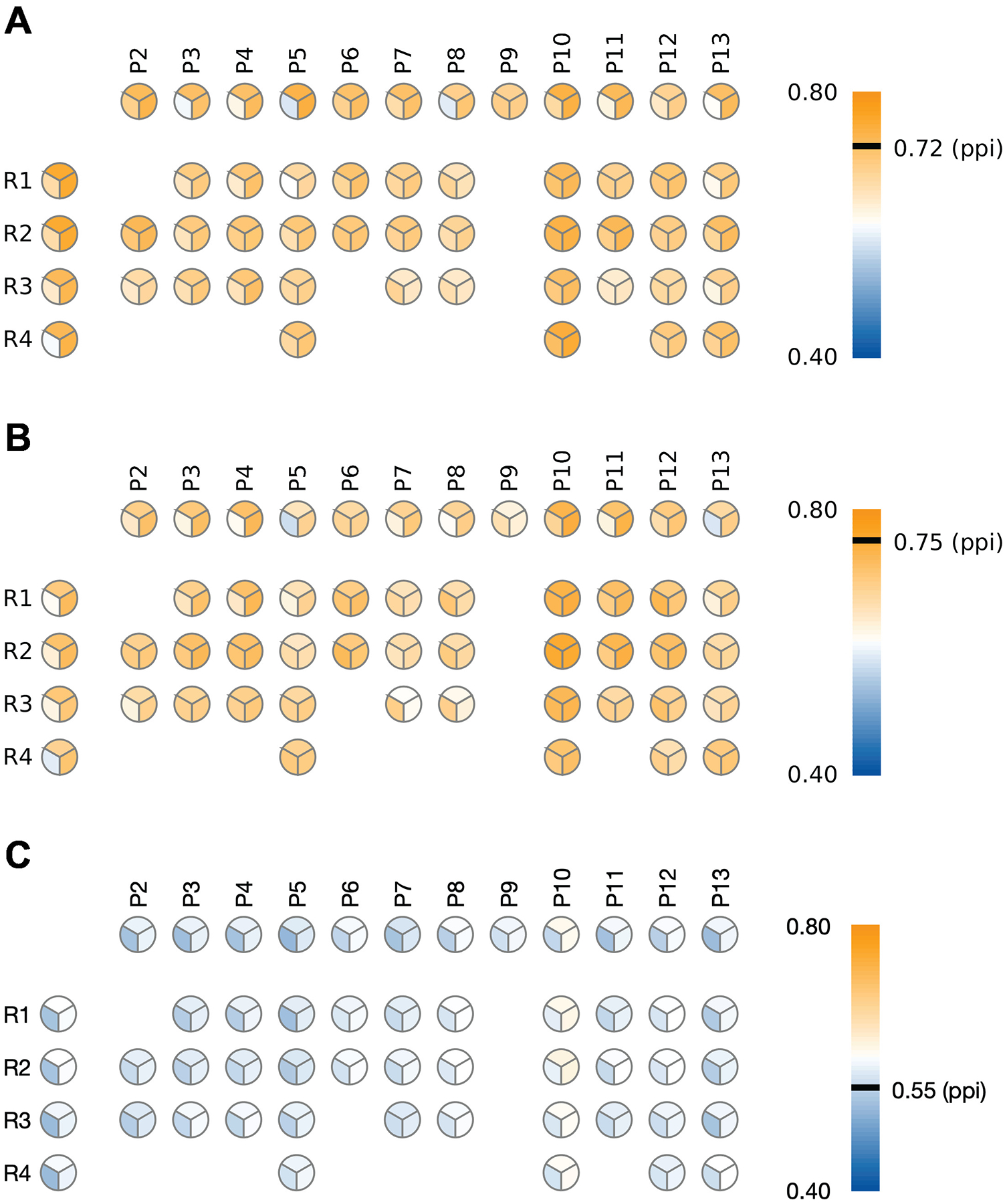
Heatmap plot of the performance of gene scoring of BrainSpan networks with POS65 as seed genes. (A) cross-validated performance with POS65 as seed genes. (B) Using POS65 as seed genes but evaluated on the VAL63 genes. (C) Using POS65 as seed genes but evaluated on the 1000 lists of simulated genes with similar length and GC content as VAL63. Each pie chart represents the estimated AUC on one BrainSpan region, period or region/period combination. The three patches in each pie chart represent: (top) the original BrainSpan network; (lower-left) the intersection of BrainSpan and PPI network; (lower-right) the union of BrainSpan and PPI network. More detailed results are shown in Supplementary Table S3
Next, we evaluated the quality of gene scoring obtained by a one-time propagation of POS65 but on an independent validation dataset VAL63. The classification performance was only slightly lower than POS65 cross-validation performance (Figure 1B; Supplementary Table S3). However, in agreement with cross-validation results, period P10 by itself, or P10 in combination with various regions remained the best performing networks on the out-of-sample VAL63 dataset. Again, the addition of PPI data improved the performance for the majority of datasets. As a negative control, we also generated 1000 simulated gene lists, each of which consisted of 63 brain-expressed genes with similar length and GC content as VAL63. The performance of gene scoring on these control networks (Figure 1C) was considerably lower than on the VAL63 gene set.
To understand whether region- and period-specific brain expression data was indeed important for the ASD risk gene prioritization, we repeated the experiment with the same network construction procedures for gene scoring, only using tissue-specific gene expression data from the GTEx database v.7 instead of BrainSpan. We found that all GTEx networks, including those from the brain, showed inferior performance even to the protein-protein interaction network (Figure 2; Supplementary Table S3). Of note, GTEx brain datasets are derived from adult brain samples. This suggests that spatio-temporal developmental brain transcriptome from BrainSpan, and especially from fetal and early postnatal periods, significantly improves ASD gene prioritization.
Figure 2:
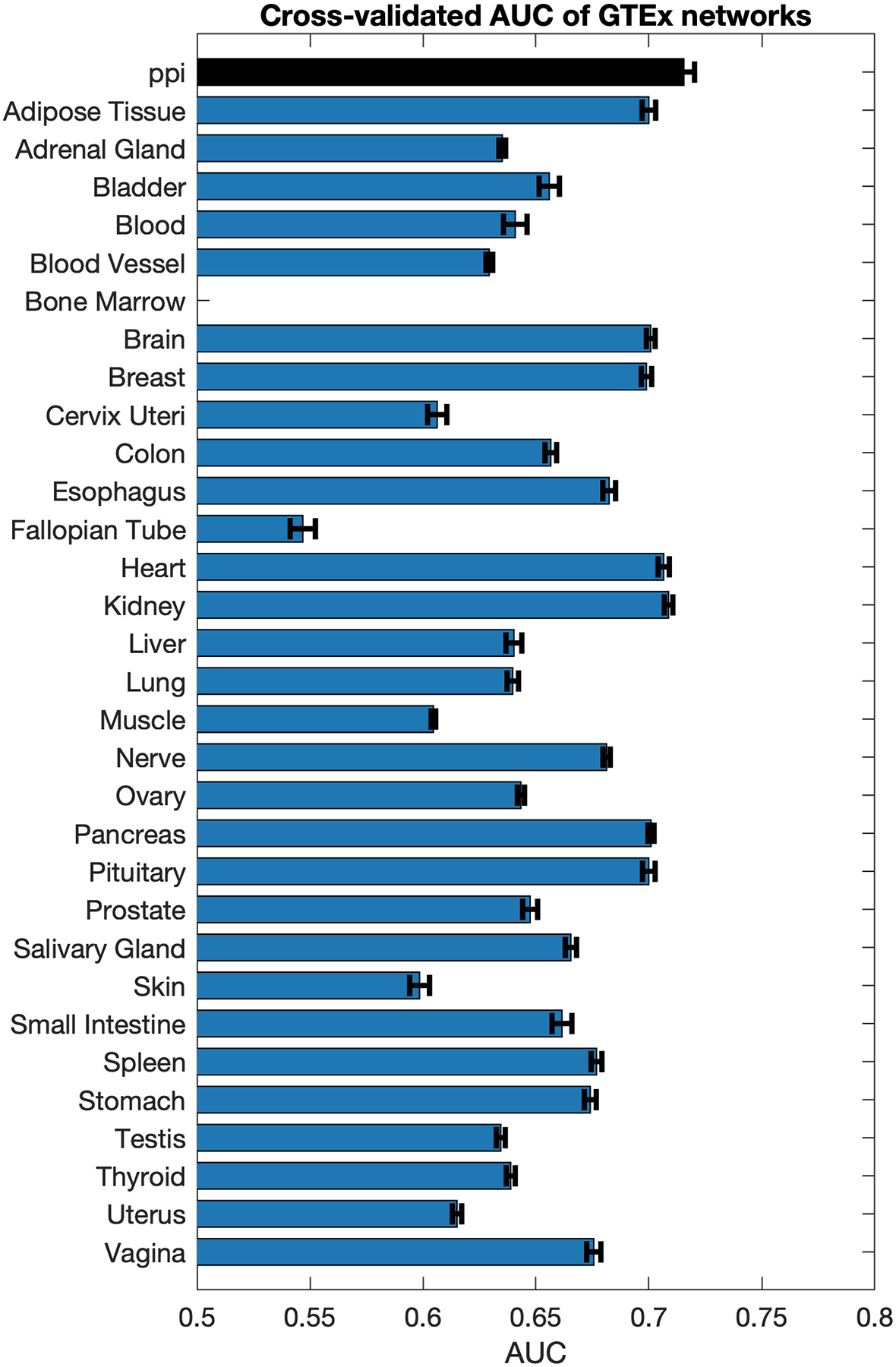
Performance of gene scoring on networks constructed from 31 GTEx tissues. Black bar corresponds to the performance of the PPI network. The bars show average AUC and standard error over 100 restarts of network propagation through cross-validation.
3.2. Estimating prior probabilities
To further prioritize individual exonic variants, the variant scores were calculated by an appropriate tool from the MutPred family; i.e., MutPred2 [19] was used on missense variants, MutPred-LOF [69] on frameshifting indels and stop variants (LOF variants), and MutPred-Indel [70] on non-frameshifting indels and multiresidue substitutions. The gene scores were calculated by gene prioritization tools described in Section 2.
After non-traditional scores were obtained, we used the AlphaMax algorithm [39, 78] to estimate the prior probability of pathogenicity caused by different types of variants; i.e., missense, loss-of-function (LoF) and indel to be 1.5%, 2.5% and 5%, respectively, while the prior probability for a gene being categorized as an ASD risk gene was estimated at 10%. All raw prediction scores were then re-scaled according to Eq. (3) to acquire calibrated scores.
3.3. Gene scoring improves discriminatory power of highly scored variants
We next demonstrate that the integration of gene-scoring with variant-scoring from Eq. (1) increases the discriminatory power of highly scored variants between autism cases and controls. After re-weighting with gene scores, we defined high-risk variants as those whose final scores were larger than 90% of control variants. This score corresponds to the variants with the one-sided p-value below 0.1 given an empirical null distribution defined by the control variants. We then applied Fisher’s exact test to determine whether the proportion of high-risk de novo variants is higher among probands than in their healthy siblings. A more stringent threshold corresponding to the 95% showed similar results, although we considered it less reliable due to the relatively small sample sizes of LoF and indel variants at this threshold.
We benchmarked the discriminating power of our gene prioritization with brain-specific networks against several baseline gene scoring methods: (i) POS65 – the known 65 risk seed genes were assigned the probability of 1, while all other genes were assigned the probability of 0; (ii) MutPred – this baseline scoring scheme does not use any gene prioritization and scores variants simply based on the outputs of MutPred; (iii) Krishnan – the gene probabilities were obtained from [31]; (iv) Duda – the gene probabilities were obtained from [32]; (v) PPI – the genes were scored by propagating over the PPI network. The blue dashed line indicates the significance value corresponding to the p-value of 0.05 in each plot, whereas the gray dashed line indicates the Bonferroni-corrected p-value (Figures 3–5).
Figure 3:
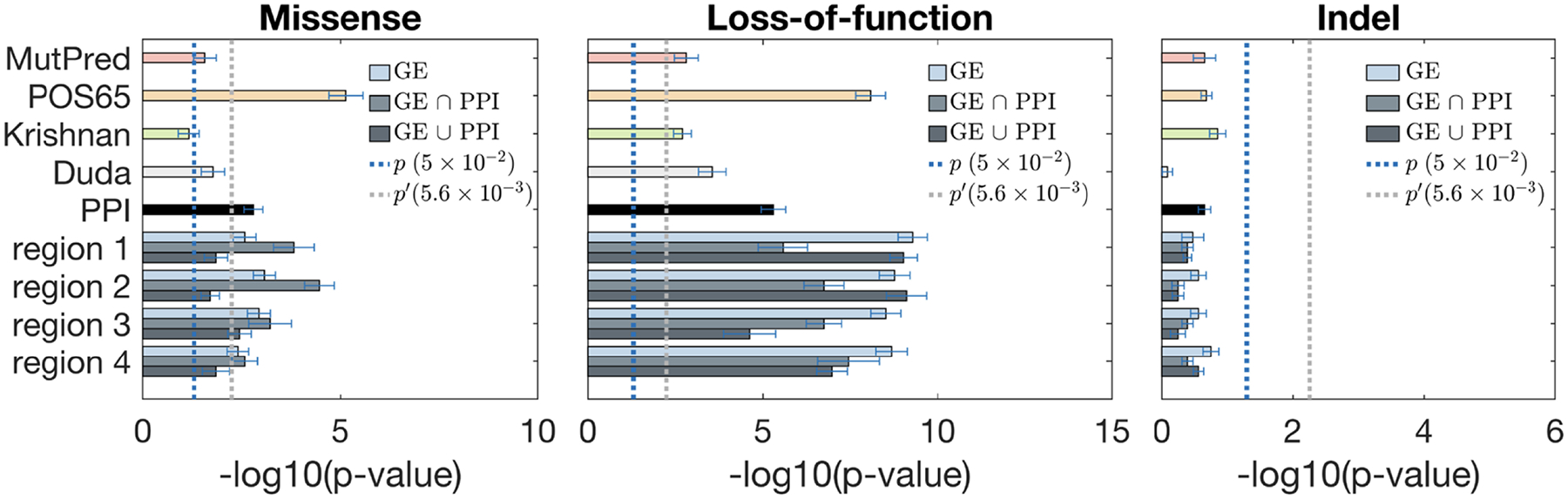
Fisher’s exact test for discriminating case and control exonic de novo variants by using gene scores from various brain region networks. From left to right: missense, LoF, and indel. Each region has three bars (color coded from light to dark) corresponding to the co-expression network, and merged networks with PPI using the “intersection” and the “union” methods, respectively. Dotted lines show the thresholds for statistical significance, with p′ being the Bonferroni-corrected value.
Figure 5:
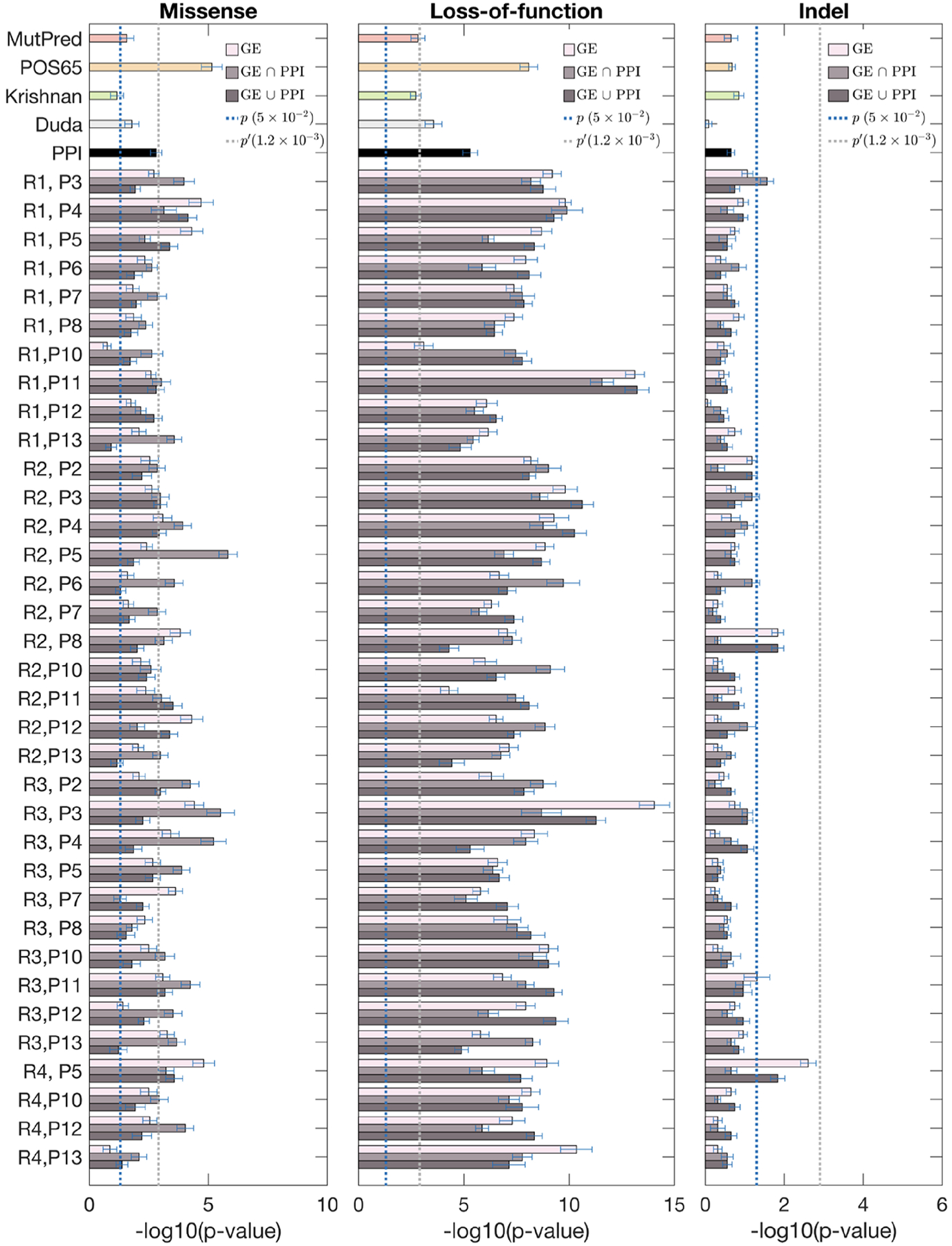
Fisher’s exact test for discriminating case and control exonic de novo variants by using gene scores from various region/period combination networks. From left to right: missense, LoF, and indel. Each combination has three bars (color coded from light to dark) corresponding to the co-expression network, and merged networks with PPI using the “intersection” and the “union” methods, respectively. Dotted lines show the thresholds for statistical significance, with p′ being the Bonferroni-corrected value.
Our scoring method based on BrainSpan networks was more powerful in discriminating high-risk variants between case and control groups than PPI networks and two other published methods [31, 32]. Interestingly, all scoring approaches performed better on LoF mutations compared to missense and indel variants. This is consistent with the notion that LoF mutations are generally more pathogenic, and that ASD patients have an excess of LoF variants compared to controls. Across region-based networks, R1 and R2 cortical regions generally outperformed other regions, which is consistent with previous observations from the literature [25, 26, 28, 29]. Across period-based networks, P3 (early fetal) for all mutation types, and P11 (middle and late childhood) for LoF generally outperformed other periods. This is a surprising finding since previously P6 (late mid-fetal) period has been strongly implicated in ASD based on the gene network-level [26, 28]. This suggests that adding variant scoring to the networks may pick up additional signals that were not present in the gene-based models. Furthermore, predictions using region-period combination networks (Figure 5) generally performed better than region-based (Figure 3) or period-based (Figure 4) did individually. Some of the region/period network-based predictions significantly outperformed the existing methods; i.e., R2-P5 and R3-P3 for missense, R1-P11 and R3-P3 for LoFs, and R2-P8 and R4-P5 for indels (Figure 5). Note that the addition of gene scoring increased predictive performance compared to just pure variant scoring by MutPred. In most cases, combination of gene and variant scoring on certain region/period combinations improved upon POS65. This suggests that our method improves the predictions of novel ASD risk genes and variants. We note that the statistical signal also holds when POS65 were completely removed from the networks (Supplementary Materials).
Figure 4:
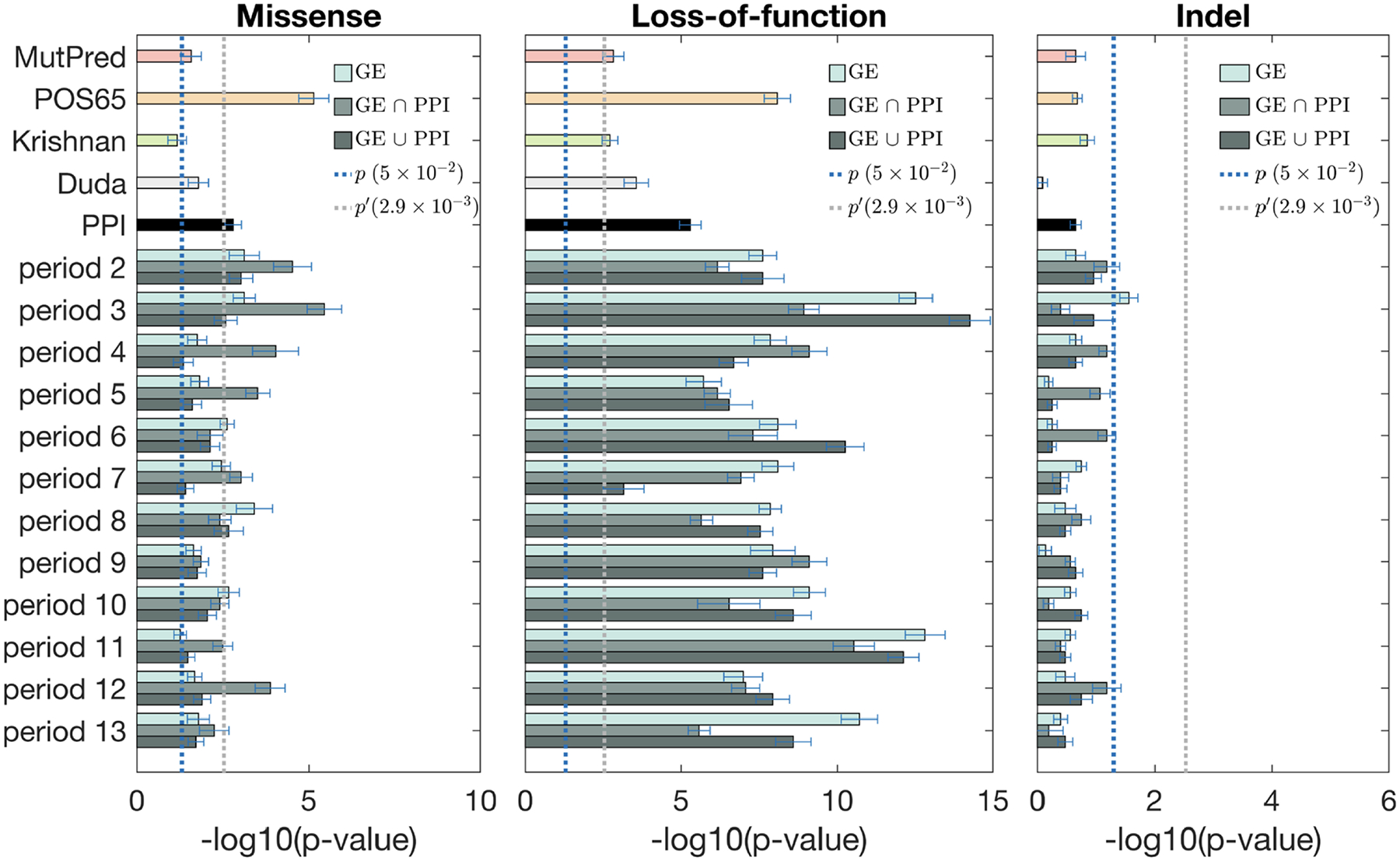
Fisher’s exact test for discriminating case and control exonic de novo variants by using gene scores from various period networks. From left to right: missense, LoF, and indel. Each period has three bars (color coded from light to dark) corresponding to the co-expression network, and merged networks with PPI using the “intersection” and the “union” methods, respectively. Dotted lines show the thresholds for statistical significance, with p′ being the Bonferroni-corrected value.
3.4. Assessing clinical significance
Recent guidelines on variant interpretation in the clinic allow for the use of computational models [85] with concrete likelihood ratio values proposed by Tavtigian et al. [86]. While these recommendations are relatively new and, for now, mostly apply to known Mendelian genes, the numerical values of likelihood ratios on the strength of clinical support can be seen as a form of guidance. In this light, we computed positive likelihood ratios (Eq. 4; Section 2.7) for our method when the decision threshold for the raw scores of the predictor was set at the level of the top 10th (and top 5th) percentile of the empirical null distribution defined by the scores from control variants. For easier interpretation, we also report the estimates of the diagnostic odds ratio (DOR). Areas under the ROC curve are reported in Supplementary Table S5.
The averaged and maximum values of LR+ and DOR are shown in Table 4. These results indicate that the expected increase in odds of pathogenicity is around 1.5 for missensse variants, around 4.5 for loss-of-function variants, and around 2 for indels, with their maximum values being higher, depending on the best-performing network. Following Tavtigian et al. [86], we can classify the increase in odds of pathogenicity as informative for clinical decision-making. We observe, however, that LR+ values do not completely reflect the results from Section 3.3 because of the discrepancy in the number of variants from each group. That is, we found useful statistical signal for the missense variants but their diagnostic value is the lowest, whereas the small dataset size of indel variation resulted in a loss of statistical signal despite a generally informative diagnostic value. The most trustworthy results in interpreting variation are therefore provided by our scoring of the LoF variants, where both statistical significance and moderate diagnostic signal were found.
Table 4:
Aggregated positive likelihood ratio (LR+) and diagnostic odds ratio (DOR) over the BrainSpan datasets. Each entry shows the average LR+ and DOR over all networks in a category and the maximum LR+ and DOR in parentheses. The cutoff represents the percentile of scores over control variants used to define pathogenic variants in cases.
| Metric | Cutoff | Networks | Missense | LoF | Indel |
|---|---|---|---|---|---|
| LR+ | 90% | Regions | 1.42 (1.59) | 3.25 (3.58) | 1.67 (2.31) |
| Periods | 1.38 (1.67) | 3.35 (4.41) | 2.00 (3.55) | ||
| Combinations | 1.41 (1.70) | 3.26 (4.38) | 2.11 (4.79) | ||
| 95% | Regions | 1.64 (1.93) | 4.44 (5.24) | 2.04 (2.48) | |
| Periods | 1.64 (2.02) | 4.84 (6.20) | 2.62 (4.97) | ||
| Combinations | 1.71 (2.11) | 4.61 (6.91) | 3.02 (7.81) | ||
| DOR | 90% | Regions | 1.49 (1.70) | 4.33 (4.98) | 1.80 (2.65) |
| Periods | 1.45 (1.81) | 4.56 (7.03) | 2.29 (4.76) | ||
| Combinations | 1.48 (1.85) | 4.36 (6.94) | 2.45 (7.71) | ||
| 95% | Regions | 1.87 (2.19) | 5.54 (6.88) | 2.15 (2.67) | |
| Periods | 1.67 (2.24) | 6.24 (8.74) | 2.90 (6.13) | ||
| Combinations | 1.77 (2.23) | 5.86 (10.33) | 3.48 (11.55) |
3.5. Experimental validation
To validate functional effect of missense mutations predicted by our ASD variant effect predictor, we selected one highly ranked mutation (Supplementary Table S4), and investigated its impact on protein-protein interactions using co-immunoprecipitation. We selected the mutation NP_001243143.1:p.Phe309Ser in the sodium/potassium-transporting ATPase ATP1A3 gene, which had a score of 0.92 and was annotated by our model as “altered PPI hotspot”. This mutation was initially identified by the exome sequencing of ASD families [12]. The ATP1A3 gene carries several more de novo missense mutations from other ASD or developmental delay sequencing studies [87–89]; however, the pathogenicity of F309S or other mutations in ATP1A3 is unknown.
We tested the interaction of the wild type (WT) and F309S mutant (MT) ATP1A3 proteins for interaction with its three interacting protein partners, TOMM22, VDAC1 and TGFβ1 (Figure 6B). All these PPIs had highly confident interaction scores in the BioGRID database. To investigate the impact of the mutation on PPIs with these three partners, we tagged the WT and MT forms of ATP1A3 with the V5-tag, and the interacting partners with the GFP tag (Section 2.8). We then co-transfected the WT or MT forms with one of its partners into HEK 293T cells, and assessed the strength of interaction by performing a V5 immunoprecipitation and blotting against GFP.
Figure 6:
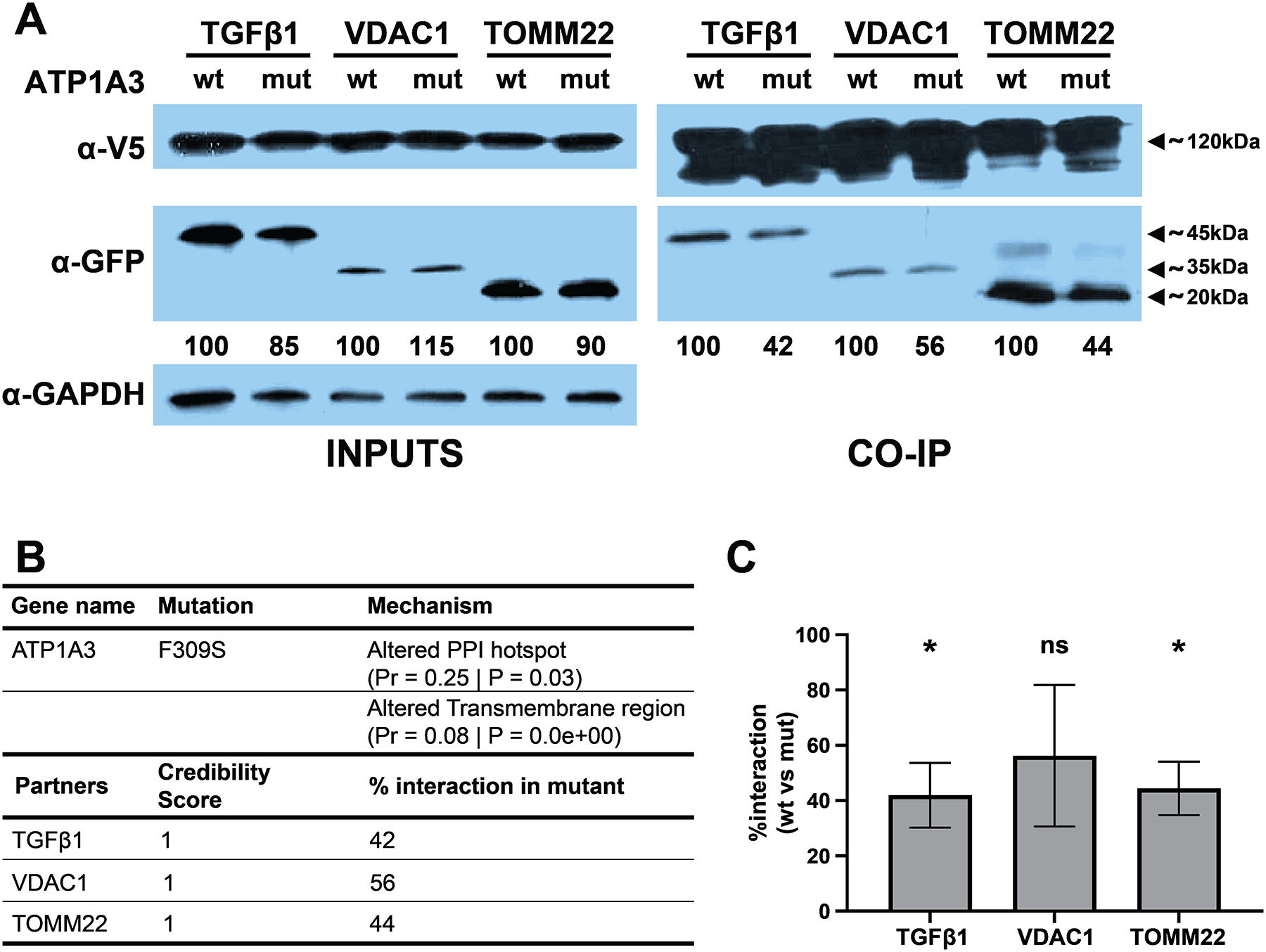
(A) Representative images of Western Blot for ATP1A3 interaction with its selected partners. Numbers below the anti-GFP blot represent the percentage of densitometry intensity for each mutant partner compared to its WT counterpart. (B) Table representing different relevant parameters for ATP1A3 and the selected partners. (C) Graph representing the percentage of interaction for each partner, comparing the F309S mutant against its WT counterpart (n = 3, paired ratio t-test, *p < 0.05).
We observed a reduction in the interaction of F309S MT form of ATP1A3 with all three partners compared to the WT ATP1A3 (Figure 6A). We did not observe significant reduction in the expression of MT ATP1A3 or its partners after transfection, as evident from full lysate inputs before the immunoprecipitation (Figure 6A). Thus, the observed reduction in interaction strength is not due to lower expression levels of the MT protein or the partners. The reduction of the interaction was around 50% for all interacting partners (Figure 6C). These results suggest that the F309S mutation weakens or disrupts the interaction of ATP1A3 with its partners, in agreement with our prediction. In addition to ASD, heterozygous mutations in ATP1A3 gene are also implicated in other neurological disorders including alternating hemiplegia of childhood 2 [OMIM 614820], CAPOS syndrome [OMIM 601338], and dystonia-12 [OMIM 128235]. This example demonstrates the utility of our combined gene- and variant-scoring model for formulating and validating testable hypothesis with regards to the functional impact of missense mutations in ASD.
4. Discussion
As a result of whole-exome and whole-genome sequencing of affected families, the number of genes and variants potentially implicated in complex neurodevelopmental diseases will continue to grow. It is therefore increasingly important to be able to interpret the significance of the newly found variants in the disease context and identify molecular mechanisms, in the form of specific alterations of molecular function [90, 91], underlying the development of the phenotype. In this work, we proposed a probabilistic framework to prioritize exonic de novo variation and evaluated the usefulness of gene- and variant-scoring methods to discriminate between individuals with and without ASD. We found that the higher-resolution systems data was beneficial to prioritization; i.e., that brain region-specific and developmental period-specific gene co-expression networks provide a valuable source of information for prioritizing ASD genes. We have also shown that gene scoring based on a network propagation method using a combination of co-expression and protein-protein interaction networks outperforms each single source of information, suggesting complementarity between the two types of data. Furthermore, co-expression networks focusing on particular brain regions and developmental periods, especially with inclusion of fetal and early postnatal brain development, were more powerful in scoring ASD genes than general tissue-specific networks, including adult brain networks from GTEx.
A novel aspect of this study is that formal integration of gene and variant scoring was based on formulating the inference in a positive-unlabeled setting through which we were able to convert general-purpose variant- and gene-scoring methods into a disease-specific variant interpretation method. We have shown that the final variant-level scores were accurate in distinguishing high-risk exonic de novo variants of all types between case and control individuals. A combination of these scoring methods is advantageous for predicting molecular mechanisms of pathogenicity in ASD risk genes and, more broadly, offers a probabilistic model for comparing the impact of multiple variants in an individual’s genome. Although we have only evaluated the impact of exonic de novo variants in the context of a pre-specified phenotype, we anticipate that this formulation has the potential to be incorporated into polygenic risk scoring schemes that combine common and rare variation [92].
While our results are generally positive, there are also limitations that merit discussion. First, from a technical perspective, the calibration method used in this work depends on the ability of the underlying methods to estimate prior and posterior distributions in the positive-unlabeled setting. Both problems remain open and actively researched in machine learning [93, 94]. Second, our probabilistic framework for scoring variants in a disease-specific context relied on simplifying assumptions; e.g., any variant that disrupts gene function in a known disease gene was automatically assumed to be disease-causing. This limitation will be difficult to overcome until such a time as gene function can be predicted at the level of protein domains or, optimistically, in a residue-specific manner for all aspects of protein function. Third, we relied on the MutPred family of tools to capture disease-causing variants based on our familiarity with these models, their good performance, and their ability to predict specific types of functional alteration. These tools, however, have not been benchmarked against others in this project and thus a higher performance may be achievable. Fourth, our main evaluation strategies were based on our ability to discriminate cases and controls for high-scoring variants. We have selected this evaluation because only a small fraction of cases may be caused by exonic de novo variation. Since this fraction is unknown and difficult to estimate, we could not apply the available correction strategies to give robust performance estimates [95, 96]. Consideration of the top 5–10% of high-scoring variants to evaluate the accuracy of our models was simply pragmatic. Finally, as recent studies have demonstrated [97], bulk gene expression data could be confounded by cell type-specific gene expression signals. Thus, using single-cell transcriptomic data could be beneficial in the context of the current study, although it is not yet available across multiple brain developmental periods and regions.
The probabilistic framework and findings from this study have led to encouraging results in prioritizing de novo variation in a disease-specific context. They have also lead to candidates that could be experimentally evaluated and thus contribute to the knowledge of complex neurodevelopmental disorders. The F309S variant in ATP1A3 is one such candidate that significantly reduces protein-protein interaction propensity and is therefore a candidate for further studies of causality.
Supplementary Material
Acknowledgements
We thank Nicholas Chew, Jasmine Le, Yssis Davis, and Andres Frayde for help with the experiments, and Kevin Chau, Pan Zhang, and Patricia Moran Losada for help with dataset assembly. We also thank four anonymous reviewers for their constructive critiques that helped us to improve the quality of this paper.
Funding.
This study was supported by the National Institutes of Health award MH105524 (LMI, PR), the Precision Health Initiative of Indiana University (PR) and, in part, by the Simons Foundation for Autism Research grant 345469 (LMI), MH104766 (LMI), and MH109885 (LMI).
Footnotes
Conflicts of interests. On behalf of all authors, the corresponding author states that there is no conflict of interest.
Code availability
The code is available at https://github.com/yuxjiang/ASD_Hum_Genet.
References
- [1].Weiner DJ, Wigdor EM, Ripke S, Walters RK, Kosmicki JA, Grove J, Samocha KE, Goldstein JI, Okbay A, Bybjerg-Grauholm J, Werge T, Hougaard DM, Taylor J, Skuse D, Devlin B, Anney R, Sanders SJ, Bishop S, Mortensen PB, Børglum AD, Smith GD, Daly MJ, and Robinson EB. Polygenic transmission disequilibrium confirms that common and rare variation act additively to create risk for autism spectrum disorders. Nat Genet, 49(7):978–985, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Bai D, Yip BHK, Windham GC, Sourander A, Francis R, Yoffe R, Glasson E, Mahjani B, Suominen A, Leonard H, Gissler M, Buxbaum JD, Wong K, Schendel D, Kodesh A, Breshnahan M, Levine SZ, Parner ET, Hansen SN, Hultman C, Reichenberg A, and Sandin S. Association of genetic and environmental factors with autism in a 5-country cohort. JAMA Psychiatry, 76(10):1035–1043, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Grove J, Ripke S, Als TD, Mattheisen M, Walters RK, Won H, Pallesen J, Agerbo E, Andreassen OA, Anney R, Awashti S, Belliveau R, Bettella F, Buxbaum JD, Bybjerg-Grauholm J, Bækvad-Hansen M, Cerrato F, Chambert K, Christensen JH, Churchhouse C, Dellenvall K, Demontis D, De Rubeis S, Devlin B, Djurovic S, Dumont AL, Goldstein JI, Hansen CS, Hauberg ME, Hollegaard MV, Hope S, Howrigan DP, Huang H, Hultman CM, Klei L, Maller J, Martin J, Martin AR, Moran JL, Nyegaard M, Nærland T, Palmer DS, Palotie A, Pedersen CB, Pedersen MG, dPoterba T, Poulsen JB, Pourcain BS, Qvist P, Rehnström K, Reichenberg A, Reichert J, Robinson EB, Roeder K, Roussos P, Saemundsen E, Sandin S, Satterstrom FK, Davey Smith G, Stefansson H, Steinberg S, Stevens CR, Sullivan PF, Turley P, Walters GB, Xu X, Stefansson K, Geschwind DH, Nordentoft M, Hougaard DM, Werge T, Mors O, Mortensen PB, Neale BM, Daly MJ, and Børglum AD. Identification of common genetic risk variants for autism spectrum disorder. Nat Genet, 51(3):431–444, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Satterstrom FK, Kosmicki JA, Wang J, Breen MS, De Rubeis S, An J-Y, Peng M, Collins R, Grove J, Klei L, Stevens C, Reichert J, Mulhern MS, Artomov M, Gerges S, Sheppard B, Xu X, Bhaduri A, Norman U, Brand H, Schwartz G, Nguyen R, Guerrero EE, Dias C, Betancur C, Cook EH, Gallagher L, Gill M, Sutcliffe JS, Thurm A, Zwick ME, Børglum AD, State MW, Cicek AE, Talkowski ME, Cutler DJ, Devlin B, Sanders SJ, Roeder K, Daly MJ, Buxbaum JD, Aleksic B, Anney R, Barbosa M, Bishop S, Brusco A, Bybjerg-Grauholm J, Carracedo A, Chan MC, Chiocchetti AG, Chung BH, Coon H, Cuccaro ML, Curró A, Dalla Bernardina B, Doan R, Domenici E, Dong S, Fallerini C, Fernández-Prieto M, Ferrero GB, Freitag CM, Fromer M, Gargus JJ, Geschwind D, Giorgio E, González-Peñas J, Guter S, Halpern D, Hansen-Kiss E, He X, Herman GE, Hertz-Picciotto I, Hougaard DM, Hultman CM, Ionita-Laza I, Jacob S, Jamison J, Jugessur A, Kaartinen M, Knudsen GP, Kolevzon A, Kushima I, Lee SL, Lehtimäki T, Lim ET, Lintas C, Lipkin WI, Lopergolo D, Lopes F, Ludena Y, Maciel P, Magnus P, Mahjani B, Maltman N, Manoach DS, Meiri G, Menashe I, Miller J, Minshew N, Montenegro EM, Moreira D, Morrow EM, Mors O, Mortensen PB, Mosconi M, Muglia P, Neale BM, Nordentoft M, Ozaki N, Palotie A, Parellada M, Passos-Bueno MR, Pericak-Vance M, Persico AM, Pessah I, Puura K, Reichenberg A, Renieri A, Riberi E, Robinson EB, Samocha KE, Sandin S, Santangelo SL, Schellenberg G, Scherer SW, Schlitt S, Schmidt R, Schmitt L, Silva IM, Singh T, Siper PM, Smith M, Soares G, Stoltenberg C, Suren P, Susser E, Sweeney J, Szatmari P, Tang L, Tassone F, Teufel K, Trabetti E, Walsh CA, Weiss LA, Werge T, Werling DM, Wigdor EM, Wilkinson E, Willsey AJ, Yu TW, Yu MH, Yuen R, Zachi E, Agerbo E, Als TD, Appadurai V, Bækvad-Hansen M, Belliveau R, Buil A, Carey CE, Cerrato F, Chambert K, Churchhouse C, Dalsgaard S, Demontis D, Dumont A, Goldstein J, Hansen CS, Hauberg ME, Hollegaard MV, Howrigan DP, Huang H, Maller J, Martin AR, Martin J, Mattheisen M, Moran J, Pallesen J, Palmer DS, Pedersen CB, Pedersen MG, Poterba T, Poulsen JB, Ripke S, Schork AJ, Thompson WK, Turley P, and Walters RK. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell, 180(3):568–584.e23, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, Leotta A, Pai D, Zhang R, Lee YH, Hicks J, Spence SJ, Lee AT, Puura K, Lehtimaki T, Ledbetter D, Gregersen PK, Bregman J, Sutcliffe JS, Jobanputra V, Chung W, Warburton D, King MC, Skuse D, Geschwind DH, Gilliam TC, Ye K, and Wigler M. Strong association of de novo copy number mutations with autism. Science, 316(5823):445–449, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, Shago M, Moessner R, Pinto D, Ren Y, Thiruvahindrapduram B, Fiebig A, Schreiber S, Friedman J, Ketelaars CE, Vos YJ, Ficicioglu C, Kirkpatrick S, Nicolson R, Sloman L, Summers A, Gibbons CA, Teebi A, Chitayat D, Weksberg R, Thompson A, Vardy C, Crosbie V, Luscombe S, Baatjes R, Zwaigenbaum L, Roberts W, Fernandez B, Szatmari P, and Scherer SW. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet, 82(2):477–488, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, Conroy J, Magalhaes TR, Correia C, Abrahams BS, Almeida J, Bacchelli E, Bader GD, Bailey AJ, Baird G, Battaglia A, Berney T, Bolshakova N, Bolte S, Bolton PF, Bourgeron T, Brennan S, Brian J, Bryson SE, Carson AR, Casallo G, Casey J, Chung BH, Cochrane L, Corsello C, Crawford EL, Crossett A, Cytrynbaum C, Dawson G, de Jonge M, Delorme R, Drmic I, Duketis E, Duque F, Estes A, Farrar P, Fernandez BA, Folstein SE, Fombonne E, Freitag CM, Gilbert J, Gillberg C, Glessner JT, Goldberg J, Green A, Green J, Guter SJ, Hakonarson H, Heron EA, Hill M, Holt R, Howe JL, Hughes G, Hus V, Igliozzi R, Kim C, Klauck SM, Kolevzon A, Korvatska O, Kustanovich V, Lajonchere CM, Lamb JA, Laskawiec M, Leboyer M, Le Couteur A, Leventhal BL, Lionel AC, Liu XQ, Lord C, Lotspeich L, Lund SC, Maestrini E, Mahoney W, Mantoulan C, Marshall CR, McConachie H, McDougle CJ, McGrath J, McMahon WM, Merikangas A, Migita O, Minshew NJ, Mirza GK, Munson J, Nelson SF, Noakes C, Noor A, Nygren G, Oliveira G, Papanikolaou K, Parr JR, Parrini B, Paton T, Pickles A, Pilorge M, Piven J, Ponting CP, Posey DJ, Poustka A, Poustka F, Prasad A, Ragoussis J, Renshaw K, Rickaby J, Roberts W, Roeder K, Roge B, Rutter ML, Bierut LJ, Rice JP, Salt J, Sansom K, Sato D, Segurado R, Sequeira AF, Senman L, Shah N, Sheffield VC, Soorya L, Sousa I, Stein O, Sykes N, Stoppioni V, Strawbridge C, Tancredi R, Tansey K, Thiruvahindrapduram B, Thompson AP, Thomson S, Tryfon A, Tsiantis J, Van Engeland H, Vincent JB, Volkmar F, Wallace S, Wang K, Wang Z, Wassink TH, Webber C, Weksberg R, Wing K, Wittemeyer K, Wood S, Wu J, Yaspan BL, Zurawiecki D, Zwaigenbaum L, Buxbaum JD, Cantor RM, Cook EH, Coon H, Cuccaro ML, Devlin B, Ennis S, Gallagher L, Geschwind DH, Gill M, Haines JL, Hallmayer J, Miller J, Monaco AP, Nurnberger J, I. J, Paterson AD, Pericak-Vance MA, Schellenberg GD, Szatmari P, Vicente AM, Vieland VJ, Wijsman EM, Scherer SW, Sutcliffe JS, and Betancur C. Functional impact of global rare copy number variation in autism spectrum disorders. Nature, 466(7304):368–372, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Malhotra D and Sebat J. CNVs: harbingers of a rare variant revolution in psychiatric genetics. Cell, 148(6):1223–1241, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, Yamrom B, Lee Y.-h., Narzisi G, Leotta A, Kendall J, Grabowska E, Ma B, Marks S, Rodgers L, Stepansky A, Troge J, Andrews P, Bekritsky M, Pradhan K, Ghiban E, Kramer M, Parla J, Demeter R, Fulton LL, Fulton RS, Magrini VJ, Ye K, Darnell JC, Darnell RB, Mardis ER, Wilson RK, Schatz MC, McCombie WR, and Wigler M. De novo gene disruptions in children on the autistic spectrum. Neuron, 74(2):285–299, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, Turner EH, Stanaway IB, Vernot B, Malig M, Baker C, Reilly B, Akey JM, Borenstein E, Rieder MJ, Nickerson DA, Bernier R, Shendure J, and Eichler EE. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature, 485(7397):246–250, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, Walker MF, Ober GT, Teran NA, Song Y, El-Fishawy P, Murtha RC, Choi M, Overton JD, Bjornson RD, Carriero NJ, Meyer KA, Bilguvar K, Mane SM, Šestan N, Lifton RP, Günel M, Roeder K, Geschwind DH, Devlin B, and State MW. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature, 485(7397):237–241, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Ercument Cicek A, Kou Y, Liu L, Fromer M, Walker S, Singh T, Klei L, Kosmicki J, Fu S-C, Aleksic B, Biscaldi M, Bolton PF, Brownfeld JM, Cai J, Campbell NG, Carracedo A, Chahrour MH, Chiocchetti AG, Coon H, Crawford EL, Crooks L, Curran SR, Dawson G, Duketis E, Fernandez BA, Gallagher L, Geller E, Guter SJ, Sean Hill R, Ionita-Laza I, Jimenez Gonzalez P, Kilpinen H, Klauck SM, Kolevzon A, Lee I, Lei J, Lehtimäki T, Lin C-F, Ma’ayan A, Marshall CR, McInnes AL, Neale B, Owen MJ, Ozaki N, Parellada M, Parr JR, Purcell S, Puura K, Rajagopalan D, Rehnström K, Reichenberg A, Sabo A, Sachse M, Sanders SJ, Schafer C, Schulte-Rüther M, Skuse D, Stevens C, Szatmari P, Tammimies K, Valladares O, Voran A, Wang L-S, Weiss LA, Willsey AJ, Yu TW, Yuen RKC, Cook EH, Freitag CM, Gill M, Hultman CM, Lehner T, Palotie A, Schellenberg GD, Sklar P, State MW, Sutcliffe JS, Walsh CA, Scherer SW, Zwick ME, Barrett JC, Cutler DJ, Roeder K, Devlin B, Daly MJ, and Buxbaum JD. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature, 515(7526):209–215, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Yuen RK, Thiruvahindrapuram B, Merico D, Walker S, Tammimies K, Hoang N, Chrysler C, Nalpathamkalam T, Pellecchia G, Liu Y, Gazzellone MJ, D’Abate L, Deneault E, Howe JL, Liu RS, Thompson A, Zarrei M, Uddin M, Marshall CR, Ring RH, Zwaigenbaum L, Ray PN, Weksberg R, Carter MT, Fernandez BA, Roberts W, Szatmari P, and Scherer SW. Whole-genome sequencing of quartet families with autism spectrum disorder. Nat Med, 21(2):185–191, 2015. [DOI] [PubMed] [Google Scholar]
- [14].An J-Y, Lin K, Zhu L, Werling DM, Dong S, Brand H, Wang HZ, Zhao X, Schwartz GB, Collins RL, Currall BB, Dastmalchi C, Dea J, Duhn C, Gilson MC, Klei L, Liang L, Markenscoff-Papadimitriou E, Pochareddy S, Ahituv N, Buxbaum JD, Coon H, Daly MJ, Kim YS, Marth GT, Neale BM, Quinlan AR, Rubenstein JL, Sestan N, State MW, Willsey AJ, Talkowski ME, Devlin B, Roeder K, and Sanders SJ. Genome-wide de novo risk score implicates promoter variation in autism spectrum disorder. Science, 362(6420):eaat6576, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Brandler WM, Antaki D, Gujral M, Kleiber ML, Whitney J, Maile MS, Hong O, Chapman TR, Tan S, Tandon P, Pang T, Tang SC, Vaux KK, Yang Y, Harrington E, Juul S, Turner DJ, Thiruvahindrapuram B, Kaur G, Wang Z, Kingsmore SF, Gleeson JG, Bisson D, Kakaradov B, Telenti A, Venter JC, Corominas R, Toma C, Cormand B, Rueda I, Guijarro S, Messer KS, Nievergelt CM, Arranz MJ, Courchesne E, Pierce K, Muotri AR, Iakoucheva LM, Hervas A, Scherer SW, Corsello C, and Sebat J. Paternally inherited cis-regulatory structural variants are associated with autism. Science, 360(6386):327–331, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Leblond CS, Cliquet F, Carton C, Huguet G, Mathieu A, Kergrohen T, Buratti J, Lemiere N, Cuisset L, Bienvenu T, Boland A, Deleuze JF, Stora T, Biskupstoe R, Halling J, Andorsdottir G, Billstedt E, Gillberg C, and Bourgeron T. Both rare and common genetic variants contribute to autism in the Faroe Islands. NPJ Genom Med, 4:1, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, Stessman HA, Witherspoon KT, Vives L, Patterson KE, Smith JD, Paeper B, Nickerson DA, Dea J, Dong S, Gonzalez LE, Mandell JD, Mane SM, Murtha MT, Sullivan CA, Walker MF, Waqar Z, Wei L, Willsey AJ, Yamrom B, Lee Y.-h., Grabowska E, Dalkic E, Wang Z, Marks S, Andrews P, Leotta A, Kendall J, Hakker I, Rosenbaum J, Ma B, Rodgers L, Troge J, Narzisi G, Yoon S, Schatz MC, Ye K, McCombie WR, Shendure J, Eichler EE, State MW, and Wigler M. The contribution of de novo coding mutations to autism spectrum disorder. Nature, 515(7526):216–221, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Chen S, Fragoza R, Klei L, Liu Y, Wang J, Roeder K, Devlin B, and Yu H. An interactome perturbation framework prioritizes damaging missense mutations for developmental disorders. Nat Genet, 50(7):1032–1040, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Pejaver V, Urresti J, Lugo-Martinez J, Pagel KA, Lin GN, Nam HJ, Mort M, Cooper DN, Sebat J, Iakoucheva LM, Mooney SD, and Radivojac P. Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat Comm, (11):5918, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Chen S, Wang J, Cicek E, Roeder K, Yu H, and Devlin B. De novo missense variants disrupting protein–protein interactions affect risk for autism through gene co-expression and protein networks in neuronal cell types. Mol Autism, 11(1):76, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Koire A, Katsonis P, Kim YW, Buchovecky C, Wilson SJ, and Lichtarge O. A method to delineate de novo missense variants across pathways prioritizes genes linked to autism. Sci Transl Med, 13(594):eabc1739, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Gilman SR, Iossifov I, Levy D, Ronemus M, Wigler M, and Vitkup D. Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron, 70(5):898–907, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Pinto D, Delaby E, Merico D, Barbosa M, Merikangas A, Klei L, Thiruvahindrapuram B, Xu X, Ziman R, Wang Z, Vorstman JA, Thompson A, Regan R, Pilorge M, Pellecchia G, Pagnamenta AT, Oliveira B, Marshall CR, Magalhaes TR, Lowe JK, Howe JL, Griswold AJ, Gilbert J, Duketis E, Dombroski BA, De Jonge MV, Cuccaro M, Crawford EL, Correia CT, Conroy J, Conceiçáo IC, Chiocchetti AG, Casey JP, Cai G, Cabrol C, Bolshakova N, Bacchelli E, Anney R, Gallinger S, Cotterchio M, Casey G, Zwaigenbaum L, Wittemeyer K, Wing K, Wallace S, van Engeland H, Tryfon A, Thomson S, Soorya L, Rogé B, Roberts W, Poustka F, Mouga S, Minshew N, McInnes LA, McGrew SG, Lord C, Leboyer M, Le Couteur AS, Kolevzon A, Jiménez González P, Jacob S, Holt R, Guter S, Green J, Green A, Gillberg C, Fernandez BA, Duque F, Delorme R, Dawson G, Chaste P, Café C, Brennan S, Bourgeron T, Bolton PF, Bölte S, Bernier R, Baird G, Bailey AJ, Anagnostou E, Almeida J, Wijsman EM, Vieland VJ, Vicente AM, Schellenberg GD, Pericak-Vance M, Paterson AD, Parr JR, Oliveira G, Nurnberger JI, Monaco AP, Maestrini E, Klauck SM, Hakonarson H, Haines JL, Geschwind DH, Freitag CM, Folstein SE, Ennis S, Coon H, Battaglia A, Szatmari P, Sutcliffe JS, Hallmayer J, Gill M, Cook EH, Buxbaum JD, Devlin B, Gallagher L, Betancur C, and Scherer SW. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am J Hum Genet, 94(5):677–694, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Iakoucheva LM, Muotri AR, and Sebat J. Getting to the cores of autism. Cell, 178(6):1287–1298, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Parikshak NN, Luo R, Zhang A, Won H, Lowe JK, Chandran V, Horvath S, and Geschwind DH. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell, 155(5):1008–1021, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Willsey AJ, Sanders SJ, Li M, Dong S, Tebbenkamp AT, Muhle RA, Reilly SK, Lin L, Fertuzinhos S, Miller JA, Murtha MT, Bichsel C, Niu W, Cotney J, Ercan-Sencicek AG, Gockley J, Gupta AR, Han W, He X, Hoffman EJ, Klei L, Lei J, Liu W, Liu L, Lu C, Xu X, Zhu Y, Mane SM, Lein ES, Wei L, Noonan JP, Roeder K, Devlin B, Sestan N, and State MW. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell, 155(5):997–1007, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Corominas R, Yang X, Lin GN, Kang S, Shen Y, Ghamsari L, Broly M, Rodriguez M, Tam S, Trigg SA, Fan C, Yi S, Tasan M, Lemmens I, Kuang X, Zhao N, Malhotra D, Michaelson JJ, Vacic V, Calderwood MA, Roth FP, Tavernier J, Horvath S, Salehi-Ashtiani K, Korkin D, Sebat J, Hill DE, Hao T, Vidal M, and Iakoucheva LM. Protein interaction network of alternatively spliced isoforms from brain links genetic risk factors for autism. Nat Comm, 5(1):3650, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lin GN, Corominas R, Lemmens I, Yang X, Tavernier J, Hill DE, Vidal M, Sebat J, and Iakoucheva LM. Spatiotemporal 16p11.2 protein network implicates cortical late mid-fetal brain development and KCTD13-Cul3-RhoA pathway in psychiatric diseases. Neuron, 85(4):742–754, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lin GN, Corominas R, Nam HJ, Urresti J, and Iakoucheva LM. Comprehensive analyses of tissue-specific networks with implications to psychiatric diseases. Methods Mol Biol, 1613:371–402, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Mosca E, Bersanelli M, Gnocchi M, Moscatelli M, Castellani G, Milanesi L, and Mezzelani A. Network diffusion-based prioritization of autism risk genes identifies significantly connected gene modules. Front Genet, 8:129, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Krishnan A, Zhang R, Yao V, Theesfeld CL, Wong AK, Tadych A, Volfovsky N, Packer A, Lash A, and Troyanskaya OG. Genome-wide prediction and functional characterization of the genetic basis of autism spectrum disorder. Nat Neurosci, 19(11):1454–1462, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Duda M, Zhang H, Li H-D, Wall DP, Burmeister M, and Guan Y. Brain-specific functional relationship networks inform autism spectrum disorder gene prediction. Transl Psychiatry, 8(1):56, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Liu L, Lei J, Sanders SJ, Willsey AJ, Kou Y, Cicek AE, Klei L, Lu C, He X, Li M, Muhle RA, Ma’ayan A, Noonan JP, Sestan N, McFadden KA, State MW, Buxbaum JD, Devlin B, and Roeder K. DAWN: a framework to identify autism genes and subnetworks using gene expression and genetics. Mol Autism, 5(1):22, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhang C and Shen Y. A cell type-specific expression signature predicts haploinsufficient autism-susceptibility genes. Hum Mutat, 38(2):204–215, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Norman U and Cicek AE. ST-Steiner: a spatio-temporal gene discovery algorithm. Bioinformatics, 35(18):3433–3440, 2019. [DOI] [PubMed] [Google Scholar]
- [36].Brueggeman L, Koomar T, and Michaelson JJ. Forecasting risk gene discovery in autism with machine learning and genome-scale data. Sci Rep, 10(1):4569, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Beyreli I, Karakahya O, and Cicek AE. Deep multitask learning of gene risk for comorbid neurodevelopmental disorders. bioRxiv, 2020.06.13.150201, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Schaaf CP, Betancur C, Yuen RKC, Parr JR, Skuse DH, Gallagher L, Bernier RA, Buchanan JA, Buxbaum JD, Chen C-A, Dies KA, Elsabbagh M, Firth HV, Frazier T, Hoang N, Howe J, Marshall CR, Michaud JL, Rennie O, Szatmari P, Chung WK, Bolton PF, Cook EH, Scherer SW, and Vorstman JAS. A framework for an evidence-based gene list relevant to autism spectrum disorder. Nat Rev Genet, 21(6):367–376, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Jain S, White M, and Radivojac P. Estimating the class prior and posterior from noisy positives and unlabeled data. Advances in Neural Information Processing Systems, pages 2693–2701, 2016. [Google Scholar]
- [40].Fischbach GD and Lord C. The Simons Simplex Collection: a resource for identification of autism genetic risk factors. Neuron, 68(2):192–195, 2010. [DOI] [PubMed] [Google Scholar]
- [41].Neale BM, Kou Y, Liu L, Ma’ayan A, Samocha KE, Sabo A, Lin C-F, Stevens C, Wang L-S, Makarov V, Polak P, Yoon S, Maguire J, Crawford EL, Campbell NG, Geller ET, Valladares O, Schafer C, Liu H, Zhao T, Cai G, Lihm J, Dannenfelser R, Jabado O, Peralta Z, Nagaswamy U, Muzny D, Reid JG, Newsham I, Wu Y, Lewis L, Han Y, Voight BF, Lim E, Rossin E, Kirby A, Flannick J, Fromer M, Shakir K, Fennell T, Garimella K, Banks E, Poplin R, Gabriel S, DePristo M, Wimbish JR, Boone BE, Levy SE, Betancur C, Sunyaev S, Boerwinkle E, Buxbaum JD, Cook EH Jr, Devlin B, Gibbs RA, Roeder K, Schellenberg GD, Sutcliffe JS, and Daly MJ. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature, 485(7397):242–245, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].O’Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S, Karakoc E, Mackenzie AP, Ng SB, Baker C, Rieder MJ, Nickerson DA, Bernier R, Fisher SE, Shendure J, and Eichler EE. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet, 43(6):585–589, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Xu B, Roos JL, Dexheimer P, Boone B, Plummer B, Levy S, Gogos JA, and Karayiorgou M. Exome sequencing supports a de novo mutational paradigm for schizophrenia. Nat Genet, 43(9):864–868, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Michaelson JJ, Shi Y, Gujral M, Zheng H, Malhotra D, Jin X, Jian M, Liu G, Greer D, Bhandari A, Wu W, Corominas R, Peoples A, Koren A, Gore A, Kang S, Lin GN, Estabillo J, Gadomski T, Singh B, Zhang K, Akshoomoff N, Corsello C, McCarroll S, Iakoucheva LM, Li Y, Wang J, and Sebat J. Whole-genome sequencing in autism identifies hot spots for de novo germline mutation. Cell, 151(7):1431–1442, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Rauch A, Wieczorek D, Graf E, Wieland T, Endele S, Schwarzmayr T, Albrecht B, Bartholdi D, Beygo J, Di Donato N, Dufke A, Cremer K, Hempel M, Horn D, Hoyer J, Joset P, Röpke A, Moog U, Riess A, Thiel CT, Tzschach A, Wiesener A, Wohlleber E, Zweier C, Ekici AB, Zink AM, Rump A, Meisinger C, Grallert H, Sticht H, Schenck A, Engels H, Rappold G, Schröck E, Wieacker P, Riess O, Meitinger T, Reis A, and Strom TM. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet, 380(9854):1674–1682, 2012. [DOI] [PubMed] [Google Scholar]
- [46].Xu B, Ionita-Laza I, Roos JL, Boone B, Woodrick S, Sun Y, Levy S, Gogos JA, and Karayiorgou M. De novo gene mutations highlight patterns of genetic and neural complexity in schizophrenia. Nat Genet, 44(12):1365–1369, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Gulsuner S, Walsh T, Watts AC, Lee MK, Thornton AM, Casadei S, Rippey C, Shahin H, Nimgaonkar VL, Go RCP, Savage RM, Swerdlow NR, Gur RE, Braff DL, King M-C, and McClellan JM. Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell, 154(3):518–529, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Jiang Y.-h., Yuen RKC, Jin X, Wang M, Chen N, Wu X, Ju J, Mei J, Shi Y, He M, Wang G, Liang J, Wang Z, Cao D, Carter MT, Chrysler C, Drmic IE, Howe JL, Lau L, Marshall CR, Merico D, Nalpathamkalam T, Thiruvahindrapuram B, Thompson A, Uddin M, Walker S, Luo J, Anagnostou E, Zwaigenbaum L, Ring RH, Wang J, Lajonchere C, Wang J, Shih A, Szatmari P, Yang H, Dawson G, Li Y, and Scherer SW. Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. Am J Hum Genet, 93(2):249–263, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].O’Roak BJ, Stessman HA, Boyle EA, Witherspoon KT, Martin B, Lee C, Vives L, Baker C, Hiatt JB, Nickerson DA, Bernier R, Shendure J, and Eichler EE. Recurrent de novo mutations implicate novel genes underlying simplex autism risk. Nat Commun, 5:5595, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Krumm N, Turner TN, Baker C, Vives L, Mohajeri K, Witherspoon K, Raja A, Coe BP, Stessman HA, He Z-X, Leal SM, Bernier R, and Eichler EE. Excess of rare, inherited truncating mutations in autism. Nat Genet, 47(6):582–588, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Brandler WM, Antaki D, Gujral M, Noor A, Rosanio G, Chapman TR, Barrera DJ, Lin GN, Malhotra D, Watts AC, Wong LC, Estabillo JA, Gadomski TE, Hong O, Fajardo KVF, Bhandari A, Owen R, Baughn M, Yuan J, Solomon T, Moyzis AG, Maile MS, Sanders SJ, Reiner GE, Vaux KK, Strom CM, Zhang K, Muotri AR, Akshoomoff N, Leal SM, Pierce K, Courchesne E, Iakoucheva LM, Corsello C, and Sebat J. Frequency and complexity of de novo structural mutation in autism. Am J Hum Genet, 98(4):667–679, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Hashimoto R, Nakazawa T, Tsurusaki Y, Yasuda Y, Nagayasu K, Matsumura K, Kawashima H, Yamamori H, Fujimoto M, Ohi K, Umeda-Yano S, Fukunaga M, Fujino H, Kasai A, Hayata-Takano A, Shintani N, Takeda M, Matsumoto N, and Hashimoto H. Whole-exome sequencing and neurite outgrowth analysis in autism spectrum disorder. J Hum Genet, 61(3):199–206, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Turner TN, Hormozdiari F, Duyzend MH, McClymont SA, Hook PW, Iossifov I, Raja A, Baker C, Hoekzema K, Stessman HA, Zody MC, Nelson BJ, Huddleston J, Sandstrom R, Smith JD, Hanna D, Swanson JM, Faustman EM, Bamshad MJ, Stamatoyannopoulos J, Nickerson DA, McCallion AS, Darnell R, and Eichler EE. Genome sequencing of autism-affected families reveals disruption of putative noncoding regulatory dna. Am J Hum Genet, 98(1):58–74, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Yuen RKC, Merico D, Cao H, Pellecchia G, Alipanahi B, Thiruvahindrapuram B, Tong X, Sun Y, Cao D, Zhang T, Wu X, Jin X, Zhou Z, Liu X, Nalpathamkalam T, Walker S, Howe JL, Wang Z, MacDonald JR, Chan A, D’Abate L, Deneault E, Siu MT, Tammimies K, Uddin M, Zarrei M, Wang M, Li Y, Wang J, Wang J, Yang H, Bookman M, Bingham J, Gross SS, Loy D, Pletcher M, Marshall CR, Anagnostou E, Zwaigenbaum L, Weksberg R, Fernandez BA, Roberts W, Szatmari P, Glazer D, Frey BJ, Ring RH, Xu X, and Scherer SW. Genome-wide characteristics of de novo mutations in autism. NPJ Genom Med, 1:160271–1602710, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].van Bon BWM, Coe BP, Bernier R, Green C, Gerdts J, Witherspoon K, Kleefstra T, Willemsen MH, Kumar R, Bosco P, Fichera M, Li D, Amaral D, Cristofoli F, Peeters H, Haan E, Romano C, Mefford HC, Scheffer I, Gecz J, de Vries BBA, and Eichler EE. Disruptive de novo mutations of DYRK1A lead to a syndromic form of autism and ID. Mol Psychiatry, 21(1):126–132, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Stessman HAF, Xiong B, Coe BP, Wang T, Hoekzema K, Fenckova M, Kvarnung M, Gerdts J, Trinh S, Cosemans N, Vives L, Lin J, Turner TN, Santen G, Ruivenkamp C, Kriek M, van Haeringen A, Aten E, Friend K, Liebelt J, Barnett C, Haan E, Shaw M, Gecz J, Anderlid B-M, Nordgren A, Lindstrand A, Schwartz C, Kooy RF, Vandeweyer G, Helsmoortel C, Romano C, Alberti A, Vinci M, Avola E, Giusto S, Courchesne E, Pramparo T, Pierce K, Nalabolu S, Amaral DG, Scheffer IE, Delatycki MB, Lockhart PJ, Hormozdiari F, Harich B, Castells-Nobau A, Xia K, Peeters H, Nordenskjöld M, Schenck A, Bernier RA, and Eichler EE. Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat Genet, 49(4):515–526, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].C Yuen RK, Merico D, Bookman M, L Howe J, Thiruvahindrapuram B, Patel RV, Whitney J, Deflaux N, Bingham J, Wang Z, Pellecchia G, Buchanan JA, Walker S, Marshall CR, Uddin M, Zarrei M, Deneault E, D’Abate L, Chan AJS, Koyanagi S, Paton T, Pereira SL, Hoang N, Engchuan W, Higginbotham EJ, Ho K, Lamoureux S, Li W, MacDonald JR, Nalpathamkalam T, Sung WWL, Tsoi FJ, Wei J, Xu L, Tasse A-M, Kirby E, Van Etten W, Twigger S, Roberts W, Drmic I, Jilderda S, Modi BM, Kellam B, Szego M, Cytrynbaum C, Weksberg R, Zwaigenbaum L, Woodbury-Smith M, Brian J, Senman L, Iaboni A, Doyle-Thomas K, Thompson A, Chrysler C, Leef J, Savion-Lemieux T, Smith IM, Liu X, Nicolson R, Seifer V, Fedele A, Cook EH, Dager S, Estes A, Gallagher L, Malow BA, Parr JR, Spence SJ, Vorstman J, Frey BJ, Robinson JT, Strug LJ, Fernandez BA, Elsabbagh M, Carter MT, Hallmayer J, Knoppers BM, Anagnostou E, Szatmari P, Ring RH, Glazer D, Pletcher MT, and Scherer SW. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat Neurosci, 20(4):602–611, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Kang HJ, Kawasawa YI, Cheng F, Zhu Y, Xu X, Li M, Sousa AMM, Pletikos M, Meyer KA, Sedmak G, Guennel T, Shin Y, Johnson MB, Krsnik Ž, Mayer S, Fertuzinhos S, Umlauf S, Lisgo SN, Vortmeyer A, Weinberger DR, Mane S, Hyde TM, Huttner A, Reimers M, Kleinman JE, and Šestan N. Spatio-temporal transcriptome of the human brain. Nature, 478(7370):483–489, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Li M, Santpere G, Imamura Kawasawa Y, Evgrafov OV, Gulden FO, Pochareddy S, Sunkin SM, Li Z, Shin Y, Zhu Y, Sousa AMM, Werling DM, Kitchen RR, Kang HJ, Pletikos M, Choi J, Muchnik S, Xu X, Wang D, Lorente-Galdos B, Liu S, Giusti-Rodríguez P, Won H, de Leeuw CA, Pardiñas AF, Hu M, Jin F, Li Y, Owen MJ, O’Donovan MC, Walters JTR, Posthuma D, Reimers MA, Levitt P, Weinberger DR, Hyde TM, Kleinman JE, Geschwind DH, Hawrylycz MJ, State MW, Sanders SJ, Sullivan PF, Gerstein MB, Lein ES, Knowles JA, and Sestan N. Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science, 362(6420):eaat7615, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Chatr-Aryamontri A, Oughtred R, Boucher L, Rust J, Chang C, Kolas NK, O’Donnell L, Oster S, Theesfeld C, Sellam A, Stark C, Breitkreutz B-J, Dolinski K, and Tyers M. The BioGRID interaction database: 2017 update. Nucleic Acids Res, 45(D1):D369–D379, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Rolland T, Taşan M, Charloteaux B, Pevzner SJ, Zhong Q, Sahni N, Yi S, Lemmens I, Fontanillo C, Mosca R, Kamburov A, Ghiassian SD, Yang X, Ghamsari L, Balcha D, Begg BE, Braun P, Brehme M, Broly MP, Carvunis A-R, Convery-Zupan D, Corominas R, Coulombe-Huntington J, Dann E, Dreze M, Dricot A, Fan C, Franzosa E, Gebreab F, Gutierrez BJ, Hardy MF, Jin M, Kang S, Kiros R, Lin GN, Luck K, MacWilliams A, Menche J, Murray RR, Palagi A, Poulin MM, Rambout X, Rasla J, Reichert P, Romero V, Ruyssinck E, Sahalie JM, Scholz A, Shah AA, Sharma A, Shen Y, Spirohn K, Tam S, Tejeda AO, Trigg SA, Twizere J-C, Vega K, Walsh J, Cusick ME, Xia Y, Barabási A-L, Iakoucheva LM, Aloy P, De Las Rivas J, Tavernier J, Calderwood MA, Hill DE, Hao T, Roth FP, and Vidal M. A proteome-scale map of the human interactome network. Cell, 159(5):1212–1226, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Yang X, Coulombe-Huntington J, Kang S, Sheynkman GM, Hao T, Richardson A, Sun S, Yang F, Shen YA, Murray RR, Spirohn K, Begg BE, Duran-Frigola M, MacWilliams A, Pevzner SJ, Zhong Q, Trigg SA, Tam S, Ghamsari L, Sahni N, Yi S, Rodriguez MD, Balcha D, Tan G, Costanzo M, Andrews B, Boone C, Zhou XJ, Salehi-Ashtiani K, Charloteaux B, Chen AA, Calderwood MA, Aloy P, Roth FP, Hill DE, Iakoucheva LM, Xia Y, and Vidal M. Widespread expansion of protein interaction capabilities by alternative splicing. Cell, 164(4):805–817, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, Gauthier LD, Brand H, Solomonson M, Watts NA, Rhodes D, Singer-Berk M, England EM, Seaby EG, Kosmicki JA, Walters RK, Tashman K, Farjoun Y, Banks E, Poterba T, Wang A, Seed C, Whiffin N, Chong JX, Samocha KE, Pierce-Hoffman E, Zappala Z, O’Donnell-Luria AH, Minikel EV, Weisburd B, Lek M, Ware JS, Vittal C, Armean IM, Bergelson L, Cibulskis K, Connolly KM, Covarrubias M, Donnelly S, Ferriera S, Gabriel S, Gentry J, Gupta N, Jeandet T, Kaplan D, Llanwarne C, Munshi R, Novod S, Petrillo N, Roazen D, Ruano-Rubio V, Saltzman A, Schleicher M, Soto J, Tibbetts K, Tolonen C, Wade G, Talkowski ME, Neale BM, Daly MJ, and MacArthur DG. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581(7809):434–443, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Kosmicki JA, Samocha KE, Howrigan DP, Sanders SJ, Slowikowski K, Lek M, Karczewski KJ, Cutler DJ, Devlin B, Roeder K, Buxbaum JD, Neale BM, MacArthur DG, Wall DP, Robinson EB, and Daly MJ. Refining the role of de novo protein-truncating variants in neurodevelopmental disorders by using population reference samples. Nat Genet, 49(4):504–510, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Mele M, Ferreira PG, Reverter F, DeLuca DS, Monlong J, Sammeth M, Young TR, Goldmann JM, Pervouchine DD, Sullivan TJ, Johnson R, Segre AV, Djebali S, Niarchou A, Consortium TG, Wright FA, Lappalainen T, Calvo M, Getz G, Dermitzakis ET, Ardlie KG, and Guigo R. The human transcriptome across tissues and individuals. Science, 348(6235):660–665, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Brandler WM and Sebat J. From de novo mutations to personalized therapeutic interventions in autism. Annu Rev Med, 66:487–507, 2015. [DOI] [PubMed] [Google Scholar]
- [67].de la Torre-Ubieta L, Won H, Stein JL, and Geschwind DH. Advancing the understanding of autism disease mechanisms through genetics. Nat Med, 22(4):345–361, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Nabieva E, Jim K, Agarwal A, Chazelle B, and Singh M. Whole-proteome prediction of protein function via graph-theoretic analysis of interaction maps. Bioinformatics, 21(Suppl 1):i302–i310, 2005. [DOI] [PubMed] [Google Scholar]
- [69].Pagel KA, Pejaver V, Lin GN, Nam HJ, Mort M, Cooper DN, Sebat J, Iakoucheva LM, Mooney SD, and Radivojac P. When loss-of-function is loss of function: assessing mutational signatures and impact of loss-of-function genetic variants. Bioinformatics, 33(14):i389–i398, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Pagel KA, Antaki D, Lian A, Mort M, Cooper DN, Sebat J, Iakoucheva LM, Mooney SD, and Radivojac P. Pathogenicity and functional impact of non-frameshifting insertion/deletion variation in the human genome. PLoS Comput Biol, 15(6):e1007112, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Pejaver V, Mooney SD, and Radivojac P. Missense variant pathogenicity predictors generalize well across a range of function-specific prediction challenges. Hum Mutat, 38(9):1092–1108, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Denis F, Gilleron R, and Letouzey F. Learning from positive and unlabeled examples. Theor Comput Sci, 348(1):70–83, 2005. [Google Scholar]
- [73].Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, Hussain M, Phillips AD, and Cooper DN. The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet, 136(6):665–677, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Elkan C and Noto K. Learning classifiers from only positive and unlabeled data. ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, pages 213–220, 2008. [Google Scholar]
- [75].Blanchard G, Lee G, and Scott C. Semi-supervised novelty detection. J Mach Learn Res, 11:2973–3009, 2010. [Google Scholar]
- [76].Reid MD and Williamson RC. Composite binary losses. J Mach Learn Res, 11:2387–2422, 2010. [Google Scholar]
- [77].Acuna-Hidalgo R, Veltman JA, and Hoischen A. New insights into the generation and role of de novo mutations in health and disease. Genome Biol, 17(1):241, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Jain S, White M, Trosset MW, and Radivojac P. Nonparametric semi-supervised learning of class proportions. arXiv preprint:1601.01944, 2016. [Google Scholar]
- [79].Rojas R. A short proof of the posterior probability property of classifier neural networks. Neural Comput, 8(1):41–43, 1996. [Google Scholar]
- [80].Platt JC. Probabilistic outputs for support vector machines and comparison to regularized likelihood methods, pages 61–74. MIT Press, 1999. [Google Scholar]
- [81].Glas AS, Lijmer JG, Prins MH, Bonsel GJ, and Bossuyt PM. The diagnostic odds ratio: a single indicator of test performance. J Clin Epidemiol, 56(11):1129–1135, 2003. [DOI] [PubMed] [Google Scholar]
- [82].Breast Cancer Association Consortium, Dorling L, Carvalho S, Allen J, Gonzalez-Neira A, Luccarini C, Wahlstrom C, Pooley KA, Parsons MT, Fortuno C, Wang Q, Bolla MK, Dennis J, Keeman R, Alonso MR, Alvarez N, Herraez B, Fernandez V, Nunez-Torres R, Osorio A, Valcich J, Li M, Torngren T, Harrington PA, Baynes C, Conroy DM, Decker B, Fachal L, Mavaddat N, Ahearn T, Aittomaki K, Antonenkova NN, Arnold N, Arveux P, Ausems M, Auvinen P, Becher H, Beckmann MW, Behrens S, Bermisheva M, Bialkowska K, Blomqvist C, Bogdanova NV, Bogdanova-Markov N, Bojesen SE, Bonanni B, Borresen-Dale AL, Brauch H, Bremer M, Briceno I, Bruning T, Burwinkel B, Cameron DA, Camp NJ, Campbell A, Carracedo A, Castelao JE, Cessna MH, Chanock SJ, Christiansen H, Collee JM, Cordina-Duverger E, Cornelissen S, Czene K, Dork T, Ekici AB, Engel C, Eriksson M, Fasching PA, Figueroa J, Flyger H, Forsti A, Gabrielson M, Gago-Dominguez M, Georgoulias V, Gil F, Giles GG, Glendon G, Garcia EBG, Alnaes GIG, Guenel P, Hadjisavvas A, Haeberle L, Hahnen E, Hall P, Hamann U, Harkness EF, Hartikainen JM, Hartman M, He W, Heemskerk-Gerritsen BAM, Hillemanns P, Hogervorst FBL, Hollestelle A, Ho WK, Hooning MJ, Howell A, Humphreys K, Idris F, Jakubowska A, Jung A, Kapoor PM, Kerin MJ, Khusnutdinova E, Kim SW, Ko YD, Kosma VM, Kristensen VN, Kyriacou K, Lakeman IMM, Lee JW, Lee MH, Li J, Lindblom A, Lo WY, Loizidou MA, Lophatananon A, Lubinski J, MacInnis RJ, Madsen MJ, Mannermaa A, Manoochehri M, Manoukian S, Margolin S, Martinez ME, Maurer T, Mavroudis D, McLean C, Meindl A, Mensenkamp AR, Michailidou K, Miller N, Mohd Taib NA, Muir K, Mulligan AM, Nevanlinna H, Newman WG, Nordestgaard BG, Ng PS, Oosterwijk JC, Park SK, Park-Simon TW, Perez JIA, Peterlongo P, Porteous DJ, Prajzendanc K, Prokofyeva D, Radice P, Rashid MU, Rhenius V, Rookus MA, Rudiger T, Saloustros E, Sawyer EJ, Schmutzler RK, Schneeweiss A, Schurmann P, Shah M, Sohn C, Southey MC, Surowy H, Suvanto M, Thanasitthichai S, Tomlinson I, Torres D, Truong T, Tzardi M, Valova Y, van Asperen CJ, Van Dam RM, van den Ouweland AMW, van der Kolk LE, van Veen EM, Wendt C, Williams JA, Yang XR, Yoon SY, Zamora MP, Evans DG, de la Hoya M, Simard J, Antoniou AC, Borg A, Andrulis IL, Chang-Claude J, Garcia-Closas M, Chenevix-Trench G, Milne RL, Pharoah PDP, Schmidt MK, Spurdle AB, Vreeswijk MPG, Benitez J, Dunning AM, Kvist A, Teo SH, Devilee P, and Easton DF. Breast cancer risk genes - association analysis in more than 113,000 women. N Engl J Med, 384(5):428–439, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Landrum MJ, Chitipiralla S, Brown GR, Chen C, Gu B, Hart J, Hoffman D, Jang W, Kaur K, Liu C, Lyoshin V, Maddipatla Z, Maiti R, Mitchell J, O’Leary N, Riley GR, Shi W, Zhou G, Schneider V, Maglott D, Holmes JB, and Kattman BL. ClinVar: improvements to accessing data. Nucleic Acids Res, 48(D1):D835–D844, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].The ORFeome Collaboration. The ORFeome Collaboration: a genome-scale human ORF-clone resource. Nat Methods, 13(3):191–192, 2016. [DOI] [PubMed] [Google Scholar]
- [85].Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, and ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med, 17(5):405–424, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Tavtigian SV, Greenblatt MS, Harrison SM, Nussbaum RL, Prabhu SA, Boucher KM, Biesecker LG, and ClinGen Sequence Variant Interpretation Working Group. Modeling the ACMG/AMP variant classification guidelines as a Bayesian classification framework. Genet Med, 20(9):1054–1060, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, Gudjonsson SA, Sigurdsson A, Jonasdottir A, Jonasdottir A, Wong WS, Sigurdsson G, Walters GB, Steinberg S, Helgason H, Thorleifsson G, Gudbjartsson DF, Helgason A, Magnusson OT, Thorsteinsdottir U, and Stefansson K. Rate of de novo mutations and the importance of father’s age to disease risk. Nature, 488(7412):471–475, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Deciphering Developmental Disorders Study. Large-scale discovery of novel genetic causes of developmental disorders. Nature, 519(7542):223–228, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Takata A, Miyake N, Tsurusaki Y, Fukai R, Miyatake S, Koshimizu E, Kushima I, Okada T, Morikawa M, Uno Y, Ishizuka K, Nakamura K, Tsujii M, Yoshikawa T, Toyota T, Okamoto N, Hiraki Y, Hashimoto R, Yasuda Y, Saitoh S, Ohashi K, Sakai Y, Ohga S, Hara T, Kato M, Nakamura K, Ito A, Seiwa C, Shirahata E, Osaka H, Matsumoto A, Takeshita S, Tohyama J, Saikusa T, Matsuishi T, Nakamura T, Tsuboi T, Kato T, Suzuki T, Saitsu H, Nakashima M, Mizuguchi T, Tanaka F, Mori N, Ozaki N, and Matsumoto N. Integrative analyses of de novo mutations provide deeper biological insights into autism spectrum disorder. Cell Rep, 22(3):734–747, 2018. [DOI] [PubMed] [Google Scholar]
- [90].Rost B, Radivojac P, and Bromberg Y. Protein function in precision medicine: deep understanding with machine learning. FEBS Lett, 590(15):2327–2341, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Lugo-Martinez J, Pejaver V, Pagel KA, Jain S, Mort M, Cooper DN, Mooney SD, and Radivojac P. The loss and gain of functional amino acid residues is a common mechanism causing human inherited disease. PLoS Comput Biol, 12(8):e1005091, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Torkamani A, Wineinger NE, and Topol EJ. The personal and clinical utility of polygenic risk scores. Nat Rev Genet, 19(9):581–590, 2018. [DOI] [PubMed] [Google Scholar]
- [93].Zeiberg D, Jain S, and Radivojac P. Fast nonparametric estimation of class proportions in the positive-unlabeled classification setting. AAAI Conference on Artificial Intelligence, pages 6729–6736, 2020. [Google Scholar]
- [94].Kiryo R, Niu G, du Plessis MC, and Sugiyama M. Positive-unlabeled learning with non-negative risk estimator. Advances in Neural Information Processing Systems, 2017. [Google Scholar]
- [95].Jain S, White M, and Radivojac P. Recovering true classifier performance in positive-unlabeled learning. AAAI Conference on Artificial Intelligence, pages 2066–2072, 2017. [Google Scholar]
- [96].Ramola R, Jain S, and Radivojac P. Estimating classification accuracy in positive-unlabeled learning: characterization and correction strategies. Pac Symp Biocomput, 24:124–135, 2019. [PMC free article] [PubMed] [Google Scholar]
- [97].Farahbod M and Pavlidis P. Untangling the effects of cellular composition on coexpression analysis. Genome Res, 30(6):849–859, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.