

Abstract
The study focuses on identifying and screening natural products (NPs) based on their structural similarities with chemical drugs followed by their possible use in first-line treatment to COVID-19 infection. In the present study, the in-house natural product libraries, consisting of 26,311 structures, were screened against potential targets of SARS-CoV-2 based on their structural similarities with the prescribed chemical drugs. The comparison was based on molecular properties, 2 and 3-dimensional structural similarities, activity cliffs, and core fragments of NPs with chemical drugs. The screened NPs were evaluated for their therapeutic effects based on their predicted in-silico pharmacokinetic and pharmacodynamics properties, binding interactions with the appropriate targets, and structural stability of the bound complex using molecular dynamics simulations. The study yielded NPs with significant structural similarities to synthetic drugs currently used to treat COVID-19 infections. The study proposes the probable biological action of the selected NPs as Anti-retroviral protease inhibitors, RNA-dependent RNA polymerase inhibitors, and viral entry inhibitors.
Keywords: Drug design, Synthetic drugs, Structural diversity, SARS-CoV-2, Medicinal chemistry
1. Introduction
Viral infections play an important role in human diseases, and their regular outbreaks repeatedly underlined the need for their prevention in safeguarding public health [1]. The recent outbreak of COVID-19 disease was declared a ‘public health emergency of international concern' by World Health Organization (WHO) in view of its severity [2]. The Coronavirus disease (COVID-19), previously known as ‘2019 novel coronavirus' or ‘2019-nCoV′, is an infectious disease caused by a newly discovered coronavirus; severe acute respiratory syndrome coronavirus 2 or SARS-CoV-2 [3]. The SARS-CoV-2 is a member of the Coronavirinae family belonging to the Betacorona genus [4]. Structurally it is spherical or pleomorphic in shape, with a diameter of about 60–140 nm. All ages are susceptible to COVID-19 infection, and its clinical manifestations range from asymptomatic to mild to severe and even to death depending on the underlying health conditions of individuals [5,6]. The most commonly reported symptoms are fever, chills, headache, body aches, dry cough, fatigue, pneumonia, and complicated dyspnea. The virus transmits from person to person via the nasal, oral, eye, and mucosal secretions of the infected patient and direct transmission through the inhalation of droplets released during the patient's cough or sneeze [7,8].
Fig. 1 describes some of the most widely used vaccines currently developed against COVID-19. The other potential treatment strategies include inhibition of RNA-Dependent RNA Polymerase activity, viral protease inhibition, viral entry inhibition, immune modulation, monoclonal antibodies, janus kinase inhibitors, nutritional supplements, and conventional plasma therapy (Table S1) [9]. The developmental status of different antiviral drugs to treat COVID-19 conditions is shown in Fig. 2 .
Fig. 1.

Most widely used vaccines currently developed against COVID-19.
Fig. 2.

The broad-spectrum antiviral drugs currently being investigated to treat the COVID-19 condition.
Natural products and traditional medicines have been serving as the greatest source for modern drug discovery. Their derivatives have been recognized for many years as the source of therapeutic potential and structural diversity. There are over 200,000 compounds reported in the scientific literature. NPs are more often structurally complex, with well-organized structure and steric properties offering efficacy, efficiency, and selectivity of molecular targets [10]. However, their utilization on many health conditions is well documented; it is in the hands of existing traditional practitioners and herbologists to define their applications for newly emerging diseases. The biological activities reported from different plant extracts often narrow down to pre-reported molecules rather than novel compounds [11], creating a real challenge to medicinal chemists. In this avenue, the search for new therapeutic molecules is the need of the hour to combat new health challenges.
The biological activity of any molecule is attributed to its structural arrangements. If two molecules have a similar structure, they will probably have a similar biological effect [[12], [13], [14]] (Fig. 3 ). The computational chemists successfully exploit this principle for the construction of diverse compound libraries and select compounds for high-throughput screening experiments [12]. Computational advancements with the introduction of parallel processing clusters, cloud-based computing, and highly effective graphical processing units (GPUs), tremendous success has been achieved in the field of modern drug discovery [15]. The knowledge of natural products and ligands, earlier used as starting points for drug discovery, has greatly influenced computational biology techniques [16]. These advancements have been speeded up by the creation of new algorithms for more accurate predictions, simulations, and interpretations [[17], [18], [19], [20], [21], [22]]. The extensive molecular dynamics (MD) simulations can provide insights into the host-virus interactions, disease spread, and possible regulative/preventive mechanisms [23]. In this avenue, the present study proceeds to exploit these developments to identify natural products as potential drug leads targeting some of the most widely used antiviral drugs currently being used against SARS-CoV-2. The identified molecules envisage to be potential drug leads and, with critical screening and trials, could be used in the first-line treatment for COVID-19 infections.
Fig. 3.

A comparison describing the structural similarities with variations highlighted inside the ellipse between thymidine, a naturally occurring nucleotide base, and zidovudine, a synthetic drug used to treat HIV patients. Structurally both the molecules share >93% identity.
2. Material and method
2.1. Dataset collection and library construction
An in-house natural product library consisting of 26,311 natural product structures was constructed using natural products information from different databases like Dr. Duke's database (https://phytochem.nal.usda.gov/phytochem/search) [24], Phytochemical Interactions Database (http://www.genome.jp/db/pcidb), and Natural product activity and species source database (NPASS) (http://bidd.group/NPASS/index.php) [25]. The plant's secondary metabolites are classified into various classes according to their chemical structures [26]. Their classification as phenolics, alkaloids, Flavonoids, Tannins, Coumarins, terpenes, Lignans, etc., with polyphenols further classified into flavans, flavones, and isoflavonoids are of particular interest to medicinal chemists due to their diverse therapeutic effects [27,28]. Therefore, the polyphenols from the natural product library were further categorized as flavans (339), flavones (193), and isoflavonoids (457), and the rest of the molecules as a general group. The broad-spectrum antiviral drugs currently under investigation to treat COVID-19 conditions were collected from the drugvirus.info server (https://drugvirus.info). For comparison, the small molecule synthetic drugs were categorized into molecules present in Pubchem COVID-19 portal (306) [29] (https://pubchem.ncbi.nlm.nih.gov/#query=covid-19), and molecules present at different stages of clinical trials (138) (As of 31st August 2020) based on the available information from ClinicalTrials.gov database [30] (https://www.clinicaltrials.gov/ct2/home). Further, the study was extended to compare the most promising investigational drugs like Remdesivir, Arbidol, Lopinavir, and Ritonavir. The top 10 structures with structurally most similar to investigational drugs were selected for in-silico PK/PD analysis and HTVS (high throughput virtual screening) studies.
2.2. Structure-based screening of natural products
The non-redundant natural product libraries were compared against chemical drugs currently under prescription/study to treat COVID-19 infection. The comparison was based on 2 and 3-dimensional structural similarities, activity cliffs (ACs), and core fragments (CFs). The structural similarities were assessed based on the number of fragments that both molecules have to the number of fragments found in any two structures [31]. The structural scaffolds (SSs) were analyzed based on plane ring system to determine the sub-structures. ACs, CFs, and SSs were determined employing Osiris DataWarrior V.4.4.3 software [31].
-
a
Pharmacophore based comparison of natural products with their synthetic drugs counterparts:
To find structurally similar compounds rather than compounds sharing a common sub-structure, core fragment-based SAR analysis was performed by considering the most central ring structure. The similarities between the fragments were assessed based on the number of fragments that both molecules have in common, divided by the number of fragments being found in any of the two structures [[31], [32], [33]]. The structures were further analyzed for structural scaffolds based on plane ring system to determine the substructures and to define the similarity cut-off during the structure comparison. The molecular properties, activity cliff, core fragments, and structural scaffolds were predicted using Osiris DataWarrior V.4.4.3 software [31,34].
-
b
Similarity score cut-off limit:
Natural products exist in many of their stable analogs forms in nature. Even with minor structural variations, the analogous forms of natural products can exert unique biological effects [35]. Therefore, there was a need to set an appropriate limit to filter natural products and their analogous structures. Due to their complex structures, at higher cut-off limits, such derivatives are expected to exclude, while at lower cut-off limits, the analogous structures become inclusive (Table S2). By considering such variations, a similarity cut-off limit of 60% was fixed for the comparison [34].
2.3. Molecular properties based PK/PD analysis
Natural products are the major source of oral drugs ‘beyond Lipinski's rule of five' [[36], [37], [38]]. The drug-likeness assessment, pharmacokinetic (PK), and pharmacodynamics (PD) of NPs were determined based on their molecular properties like molecular weight, cLogP, hydrogen atom donors, hydrogen atom acceptors, and rotatable hydrogen bonds. These properties are used as filtering parameters to estimate the oral bioavailability, solubility, and permeability of new drug candidates [36,38,39]. The natural products obtained from structural comparison were considered as hits for in-silico PK/PD assessment to analyze mutagenicity, tumorigenicity, reproductive effectiveness, irritant properties, and drug likeliness. Molecular properties were predicted using Osiris Data warrior V.4.4.3 software [31]. The admetSAR server [40] was used to predict solubility, permeability, GPCR ligand, ion channel modulator, protease inhibitor, kinase inhibitor, enzyme inhibitor, nuclear receptor inhibitor, aqueous solubility, TPSA (Topological polar surface area), blood-brain barrier penetration, human intestinal absorption, Caco-2 permeability, AMES toxicity, carcinogenicity and acute oral toxicity of the selected molecules [38,21].
2.4. Molecular interactions studies using automated docking
Automated docking was performed to deduce the binding interactions of selected natural products with appropriate target proteins. Broyden-Fletcher-Goldfarb-Shanno algorithm implemented in the AutoDockVina was employed to study proper binding modes of the selected natural products in different conformations [42]. The antiviral drugs currently being prescribed for COVID-19 first-line treatment were retrieved from the drugvirus.info server, and their action mechanisms were studied using the Inxight: Drugs database (https://drugs.ncats.io/) (Table S1).
Based on the action mechanism of the standard drugs, HIV-1 protease I50V isolate, influenza virus hemagglutinin, SARS-CoV NSP12 polymerase, and HIV-1 protease A02 isolate were selected for the docking studies. The protein structures were retrieved from protein databank (https://www.rcsb.org/) and were prepared for docking studies. For each target, residues forming the binding site were retrieved using the PDBsum server. The antiviral drug; Lopinavir and its related natural products were docked against anti-retroviral protease inhibitor (I50V isolate) (PDB ID 3OXV), Ritonavir and its related natural products were docked against anti-retroviral protease inhibitor (A02 isolate) (PDB ID 4NJV), Remdesivir, and its related natural products were docked against anti-retroviral protease inhibitor (PDB ID 7BV2), and Arbidol and its related natural products were docked against anti-retroviral protease inhibitor (PDB ID 5T6S). During molecular docking, the energy range was set to 3, exhaustiveness to 8, and the number of modes to 9. For the ligand molecules, all the torsions were allowed to rotate during docking. The in-silico studies were performed on a local machine equipped with AMD Ryzen 5 six-core 3.4 GHz processor, 8 GB graphics, and 16 GB RAM with Microsoft Windows 10 and Ubuntu 16.04 LTS dual boot operating systems.
2.5. Molecular dynamic simulations to predict the protein structural stability
The structural stability of the free and bound targets was assessed using MD simulations run for a time scale of 20 ns [43,44] by employing the GROMOS96 54a7 [45] force field implemented in the GROMACS-2018 package [46]. A periodic cubic solvated box was created around the target proteins with at least 10 Å distance from the edge of the box and solvated using the simple point charge (SPC) model [47] and neutralized using sodium or chloride ions. The energy minimization was done using the steepest descent method. The system was equilibrated with a temperature coupling at 300K using V-rescale thermostat [48] in NVT ensemble and pressure coupling at 105 Pa using Parrinello-Rahman barostat [49] in NPT ensemble for a period of 500 ps. Bond parameters were adjusted using the LINCS algorithm [50], and the particle mesh Ewald method (PME) [51] was used to evaluate electrostatic interactions. A cut-off at 1.0 nm was set for the long-distance interactions as MD simulation accurately predicts properties of the system for a larger cut-off distance [52]. The final MD trajectories were prepared for a time scale of 20ns at a time step of 2fs with trajectory coordinates updated at 10ps intervals. The final trajectories were analyzed using gmx energy, gmx rms, gmx rmsf, gmx gyrate, gmx do_dssp, and gmx sasa modules of GROMACS along with interaction energies in terms of electrostatic and van der Waals energy between the ligand and the macromolecule.
2.6. Biding free energy calculations using g_mmpbsa
For Molecular mechanics/Poisson-Boltzmann surface area (MMPBSA) calculations, trajectory files were created from the final production MD trajectory with coordinates updated every 200ps. The g_mmpbsa package was used for binding energy calculations [53]. The g_mmpbsa package uses the following equation to calculate the binding energy of the protein-ligand complex;
| ΔG Binding = G Complex − (G Protein + G Ligand) | (I) |
The ‘G' term can be further decomposed into the following components
| ΔG = ΔE MM + ΔG Solvation - TΔS = ΔE (Bonded+Non-bonded) + ΔG (Polar+Non-polar) - TΔS | (II) |
Where,
G Complex = total free energy of the binding complex,
G Protein and G Ligand = total free energies of protein and ligand, respectively.
EMM = vacuum potential energy; G Solvation = free energy of solvation
3. Results
3.1. Structure-based screening of natural products
The natural product library consisting of 26,311 structures was screened against the local Pubchem COVID-19 library of COVID-19 prescribed drugs and COVID-19 clinical trials drug library. Among the total number of molecules screened, 17,798 natural product structures were found to have more than 60% structural similarities against the Pubchem COVID-19 library entries, of which 41 molecules were flavans, 41 were flavones, and 272 were isoflavonoids. The comparison against clinical trials drug library yielded 14,689 natural products with more than 60% structure similarity consisting of 30 flavans, 18 flavones, and 78 isoflavonoids. The study was extended to compare the complete natural product library against the most promising investigational drugs, viz. Remdesivir, Arbidol, Lopinavir, and Ritonavir molecules. This comparison yielded a total of 35 natural product structures with more than 60% structural similarity. The natural products with structural similarities to respective investigational drugs are detailed in Table 1 .
Table 1.
Natural products (with NPASS accession) that are structurally similar to prescribed COVID-19 drugs and their similarity score.
| Synthetic drug and the identified NPs | Molecular structure | Similarity score |
|---|---|---|
| Remdesivir | 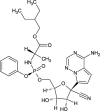 |
|
| 12,28-Oxa-8-Hydroxy-Manzamine A (NPC471891) | 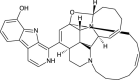 |
0.7676 |
| Marineosin A (NPC141377) | 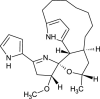 |
0.7558 |
| Bis(Gorgiacerol)Amine (NPC128683) |
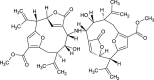 |
0.8107 |
| Methylstemofoline (NPC477986) | 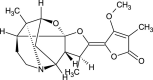 |
0.7645 |
| Chetracin B (NPC470488) | 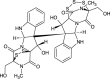 |
0.7877 |
| Oxyprotostemonine (NPC173173) | 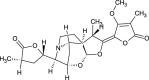 |
0.7644 |
| Stemocurtisine (NPC43648) | 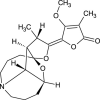 |
0.7569 |
| Munroniamide (NPC176245) | 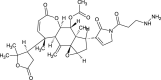 |
0.7705 |
| Alstolobine A (NPC277350) | 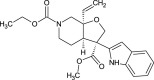 |
0.7598 |
| Discorhabdin H (NPC477417) | 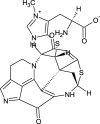 |
0.7665 |
| Arbidol | 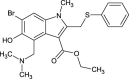 |
|
| Phellibaumin A (NPC29411) | 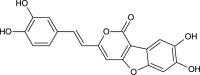 |
0.6838 |
| Difloxacin (NPC318183) | 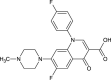 |
0.7363 |
| Lamellarin D (NPC129897) | 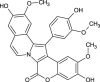 |
0.7175 |
| Lamellarin Gamma Acetate (NPC476995) | 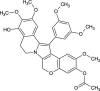 |
0.6682 |
| Hydroxy-6-Methylpyran-2-One Derivative (NPC470989) | 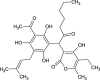 |
0.6822 |
| Cyathuscavin C (NPC474779) | 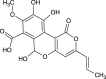 |
0.7363 |
| Cyathusal B (NPC317781) | 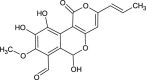 |
0.7135 |
| Clausarin (NPC83535) | 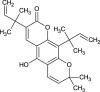 |
0.6770 |
| Cyathuscavin B (NPC474763) | 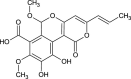 |
0.7135 |
| Pulvinatal (NPC327652) | 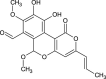 |
0.7010 |
| Lopinavir | 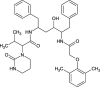 |
|
| Hexahydrodipyrrolo trione derivative (NPC122886) | 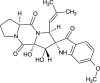 |
0.7458 |
| Beauvericin (NPC89923) | 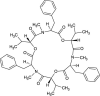 |
0.7478 |
| Chaetocin (NPC128582) | 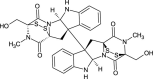 |
0.7615 |
| Mollenine A (NPC285622) | 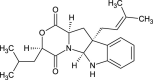 |
0.7462 |
| Chetracin B (NPC470488) | 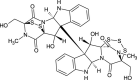 |
0.7877 |
| Beauvericin H1 (NPC157311) | 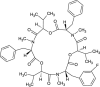 |
0.7549 |
| Dragonamide A (NPC222466) | 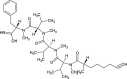 |
0.7703 |
| Chetracin D (NPC470491) | 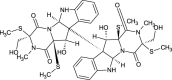 |
0.7892 |
| Dimethyl-3-Oxodecanamide derivative (NPC324081) | 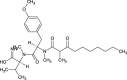 |
0.7569 |
| Symplocamide A (NPC473450) | 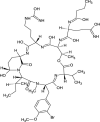 |
0.7481 |
| Ritonavir | 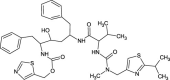 |
|
| Bionectin B (NPC475859) | 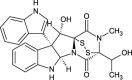 |
0.7146 |
| Luteoalbusin A (NPC470731) | 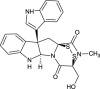 |
0.6906 |
| Bionectin A (NPC165743) | 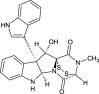 |
0.7122 |
| Oidioperazine A (NPC297642) | 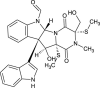 |
0.7153 |
| Holstiine (NPC11149) | 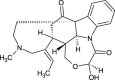 |
0.6876 |
| Chetracin B (NPC470488) | 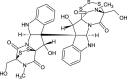 |
0.7142 |
| Mollenine A (NPC285622) | 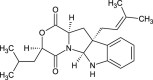 |
0.6796 |
| Methaniminium derivative (NPC194699) | 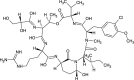 |
0.7274 |
| Verticillin E (NPC191817) | 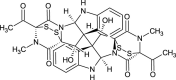 |
0.7181 |
| Chaetocin (NPC128582) | 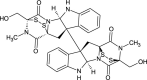 |
0.6888 |
3.2. Molecular properties based PK/PD analysis
Molecular properties and Pharmacokinetics prediction of natural products were predicted using Osiris data warrior software and the admetSAR server. The drug-likeness estimated based on the molecular properties of the selected structures indicated that out of 35 molecules, 23 molecules with positive scores indicated their potential drug-like effects (Table 2 A). Gastrointestinal (GI) absorption is an important parameter to screen orally administered drugs. A positive value shown in Table 2A for gastrointestinal (GI) absorption suggests a high probability of success for absorption into the intestinal tract [54]. While the blood-brain barrier (BBB) penetration indicates the potential of a drug to cross into the brain, it can bind to specific receptors and activate specific signaling pathways. Therefore, the prediction of BBB penetration is crucial in the drug development pipeline [55]. In the present study, 33 molecules were found to penetrate the human intestine barrier, 17 molecules penetrating the blood-brain barrier. None of them was the substrate for Cytochromes P450 group of isozymes that regulates drug metabolism, indicating a high possibility of their bioavailability (Table 2A). Further, out of 35 molecules, 34 were predicted to be non-mutagenic, non-tumorigenic, and non-irritant, with 10 molecules predicted to have reproductive effects (Table 2 B). Among the 35 structures, 29 compounds were non-AMES toxic, 34 non-carcinogens, and 34 were not readily biodegradable.
Table 2A.
) Molecular properties and Pharmacokinetics prediction of natural products filtered in for screening against COVID-19 condition.
| Identified NPs | Bioavailability and Druglikeness |
In silico Pharmacokinetics |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cLogP | Mol. wt | H-Acceptors | H-Donors | Rotatable Bonds | Total Surface Area | Polar Surface Area | Druglikeness | Human intestinal absorption | Caco-2 permeability | Blood-brain barrier | CYP2D6 substrate | |
| 12,28-Oxa-8-Hydroxy-Manzamine A | 5.2547 | 562.755 | 6 | 2 | 1 | 414.34 | 60.33 | −2.227 | 0.696+ | 0.541- | 0.800+ | 0.671- |
| Alstolobine A | 2.4707 | 398.457 | 7 | 1 | 6 | 297.41 | 80.86 | −8.1671 | 0.988+ | 0.566- | 0.896+ | 0.816- |
| Beauvericin | 5.2239 | 783.96 | 12 | 0 | 9 | 610.8 | 139.83 | 4.3764 | 0.991+ | 0.661+ | 0.678+ | 0.825- |
| Beauvericin H1 | 5.3247 | 801.95 | 12 | 0 | 9 | 617.15 | 139.83 | 3.0364 | 0.990+ | 0.599+ | 0.786+ | 0.829- |
| Bionectin A | 2.9631 | 450.542 | 7 | 3 | 1 | 282.66 | 139.27 | 5.5488 | 0.889+ | 0.508+ | 0.608+ | 0.831- |
| Bionectin B | 2.6488 | 494.595 | 8 | 4 | 2 | 311.68 | 159.5 | 5.0182 | 0.900- | 0.525- | 0.832- | 0.838- |
| Bis(Gorgiacerol)Amine | 5.4399 | 757.83 | 13 | 3 | 10 | 560.06 | 183.97 | −19.005 | 0.965- | 0.616- | 0.932- | 0.848- |
| Chaetocin | 2.7962 | 696.852 | 12 | 4 | 3 | 409.82 | 246.96 | 5.8356 | 0.900+ | 0.626- | 0.661- | 0.801- |
| Chetracin B | 1.092 | 760.916 | 14 | 6 | 3 | 437.49 | 312.72 | 5.4873 | 0.885+ | 0.574- | 0.816- | 0.805- |
| Chetracin D | 0.3976 | 788.99 | 14 | 6 | 7 | 496.04 | 287.42 | 5.6124 | 0.922+ | 0.589- | 0.869- | 0.805- |
| Clausarin | 5.9768 | 380.482 | 4 | 1 | 4 | 295.44 | 55.76 | −5.9217 | 0.975+ | 0.840- | 0.825+ | 0.867- |
| Cyathusal B | 0.5256 | 346.29 | 8 | 3 | 3 | 243.75 | 122.52 | −4.326 | 0.915+ | 0.592+ | 0.767- | 0.909- |
| Cyathuscavin B | 0.5053 | 376.316 | 9 | 3 | 4 | 264.22 | 131.75 | −4.7328 | 0.878+ | 0.627+ | 0.775- | 0.894- |
| Cyathuscavin C | 0.0774 | 362.289 | 9 | 4 | 3 | 248.31 | 142.75 | −2.2479 | 0.868+ | 0.592+ | 0.767- | 0.909- |
| Difloxacin | 1.251 | 399.396 | 6 | 1 | 3 | 283.48 | 64.09 | 5.1997 | 0.985+ | 0.879+ | 0.968- | 0.911- |
| Discorhabdin H | −10.123 | 762.664 | 10 | 3 | 5 | 337.61 | 198.19 | 2.7192 | 0.734+ | 0.603- | 0.903- | 0.795- |
| Dragonamide A | 3.7111 | 653.905 | 10 | 2 | 18 | 539.59 | 125.32 | −3.0172 | 0.969+ | 0.543- | 0.628- | 0.783- |
| Hexahydrodipyrrolo trione derivative | 0.2123 | 427.456 | 9 | 3 | 2 | 288.36 | 119.41 | 6.7335 | 0.946+ | 0.615- | 0.978- | 0.802- |
| Holstiine | 1.5964 | 382.458 | 6 | 1 | 0 | 270.15 | 70.08 | 5.6428 | 0.972+ | 0.631+ | 0.567+ | 0.784- |
| Hydroxy-6-Methylpyran-2-One Derivative | 5.228 | 500.586 | 8 | 4 | 11 | 387.58 | 141.36 | −13.889 | 0.984+ | 0.563+ | 0.660- | 0.866- |
| Lamellarin D | 4.3105 | 499.474 | 9 | 3 | 4 | 352.71 | 119.09 | 1.8379 | 0.983+ | 0.604+ | 0.606+ | 0.448- |
| Lamellarin Gamma Acetate | 5.3 | 573.596 | 10 | 1 | 8 | 423.68 | 106.84 | 2.3739 | 0.987+ | 0.683+ | 0.747+ | 0.628- |
| Luteoalbusin A | 3.1416 | 464.569 | 7 | 3 | 2 | 295.59 | 139.27 | 5.9847 | 0.890+ | 0.581- | 0.575+ | 0.812- |
| Marineosin A | 4.6896 | 409.572 | 5 | 2 | 2 | 323.4 | 62.4 | −2.232 | 0.986+ | 0.638- | 0.824+ | 0.764- |
| Methaniminium derivative | −0.4649 | 910.463 | 21 | 10 | 14 | 679.31 | 329.1 | 6.1103 | 0.795+ | 0.647- | 0.976- | 0.831- |
| Methylstemofoline | 0.9975 | 345.394 | 6 | 0 | 1 | 220.03 | 57.23 | 4.0559 | 0.922+ | 0.670+ | 0.679+ | 0.741- |
| Mollenine A | 3.3511 | 368.475 | 5 | 1 | 4 | 275.46 | 58.64 | 3.6919 | 0.980+ | 0.516- | 0.608+ | 0.824- |
| Munroniamide | −0.4894 | 597.663 | 12 | 2 | 7 | 419.88 | 166.86 | −8.9989 | 0.940+ | 0.628- | 0.500+ | 0.808- |
| Oidioperazine A | 1.9865 | 538.647 | 9 | 3 | 4 | 357.08 | 167.78 | 6.2082 | 0.843+ | 0.510- | 0.807- | 0.506- |
| Oxyprotostemonine | 1.004 | 431.483 | 8 | 0 | 2 | 289.89 | 83.53 | 2.3627 | 0.890+ | 0.648+ | 0.549+ | 0.803- |
| Phellibaumin A | 2.8483 | 352.297 | 7 | 4 | 2 | 248.17 | 120.36 | 0.0022 | 0.952+ | 0.828- | 0.725+ | 0.905- |
| Pulvinatal | 0.9535 | 360.317 | 8 | 2 | 4 | 259.66 | 111.52 | −6.9354 | 0.919+ | 0.627+ | 0.775- | 0.894- |
| Stemocurtisine | 1.4247 | 347.41 | 6 | 0 | 1 | 239.64 | 57.23 | 2.9196 | 0.922+ | 0.668+ | 0.758+ | 0.744- |
| Symplocamide A | 0.6976 | 1052.03 | 23 | 11 | 18 | 763.41 | 359.52 | 1.3524 | 0.915+ | 0.634- | 0.959- | 0.830- |
| Verticillin E | 1.7482 | 752.872 | 14 | 4 | 3 | 439.8 | 281.1 | 4.5639 | 0.895+ | 0.536- | 0.836- | 0.825- |
Table 2B.
) In-silico Pharmacodynamics prediction of natural products selected for screening against COVID-19 condition.
| Identified NPs | Mutagenic | Tumorigenic | Reproductive effective | Ocular irritancy | Aerobic biodegradibility | Ames tooxicity score | Carcinogen |
|---|---|---|---|---|---|---|---|
| 12,28-Oxa-8-Hydroxy-Manzamine A | NONE | NONE | NONE | 0.946- | 1.00- | 0.707- | 0.607- |
| Alstolobine A | NONE | HIGH | NONE | 0.979- | 1.00- | 0.714- | 0.573- |
| Beauvericin | NONE | NONE | NONE | 0.925- | 0.912- | 0.772- | 0.622- |
| Beauvericin H1 | NONE | NONE | NONE | 0.922- | 0.996- | 0.776- | 0.536- |
| Bionectin A | NONE | NONE | NONE | 0.972- | 0.986- | 0.733- | 0.609- |
| Bionectin B | NONE | NONE | NONE | 0.965- | 0.988- | 0.870- | 0.611- |
| Bis(Gorgiacerol)Amine | NONE | NONE | HIGH | 0.901- | 0.623- | 0.573- | 0.487- |
| Chaetocin | NONE | NONE | NONE | 0.918- | 0.994- | 0.645- | 0.623- |
| Chetracin B | NONE | NONE | NONE | 0.911- | 0.973- | 0.679- | 0.644- |
| Chetracin D | NONE | NONE | NONE | 0.904- | 0.996- | 0.678- | 0.627- |
| Clausarin | NONE | NONE | HIGH | 0.607+ | 0.993- | 0.506- | 0.472- |
| Cyathusal B | NONE | NONE | HIGH | 0.561- | 0.937- | 0.707+ | 0.465- |
| Cyathuscavin B | NONE | NONE | HIGH | 0.590- | 0.966- | 0.712+ | 0.515+ |
| Cyathuscavin C | NONE | NONE | HIGH | 0.574- | 0.937- | 0.707+ | 0.465- |
| Difloxacin | NONE | NONE | NONE | 0.949- | 1.00- | 0.885+ | 0.610- |
| Discorhabdin H | NONE | NONE | NONE | 0.960- | 1.00- | 0.593- | 0.532- |
| Dragonamide A | NONE | NONE | NONE | 0.922- | 1.00- | 0.812- | 0.678- |
| Hexahydrodipyrrolo trione derivative | NONE | NONE | NONE | 0.927- | 1.00- | 0.658- | 0.597- |
| Holstiine | NONE | NONE | NONE | 0.986- | 0.951- | 0.572- | 0.501- |
| Hydroxy-6-Methylpyran-2-One Derivative | NONE | NONE | NONE | 0.732- | 0.500+ | 0.815- | 0.723- |
| Lamellarin D | NONE | NONE | HIGH | 0.833- | 0.993- | 0.586- | 0.389- |
| Lamellarin Gamma Acetate | NONE | NONE | HIGH | 0.989- | 0.995- | 0.880- | 0.599- |
| Luteoalbusin A | NONE | NONE | NONE | 0.986- | 0.987- | 0.670- | 0.630- |
| Marineosin A | NONE | NONE | NONE | 0.972- | 1.00- | 0.655- | 0.651- |
| Methaniminium derivative | NONE | NONE | NONE | 0.905- | 0.962- | 0.615- | 0.570- |
| Methylstemofoline | NONE | NONE | NONE | 0.891- | 1.00- | 0.755- | 0.470- |
| Mollenine A | NONE | NONE | NONE | 0.986- | 0.997- | 0.572- | 0.528- |
| Munroniamide | LOW | HIGH | LOW | 0.978- | 1.00- | 0.512- | 0.562- |
| Oidioperazine A | NONE | NONE | NONE | 0.987- | 0.997- | 0.670- | 0.606- |
| Oxyprotostemonine | NONE | NONE | NONE | 0.943- | 0.994- | 0.681- | 0.440- |
| Phellibaumin A | HIGH | NONE | HIGH | 0.528- | 0.911- | 0.550+ | 0.419- |
| Pulvinatal | NONE | NONE | HIGH | 0.547- | 0.966- | 0.712+ | 0.515+ |
| Stemocurtisine | NONE | NONE | NONE | 0.914- | 0.995- | 0.781- | 0.420- |
| Symplocamide A | NONE | NONE | NONE | 0.901- | 0.945- | 0.644- | 0.594- |
| Verticillin E | NONE | NONE | HIGH | 0.900- | 0.986- | 0.763- | 0.610- |
3.3. Molecular interactions studies using automated docking
The in-silico molecular interaction studies were used to predict the most effective natural product drug to bind to the appropriate target involved in the regulation of virus entry, replication, assembly, and release, as well as host-specific interactions. In the present study, the docking studies were carried out for synthetic antiviral agents as well as their structurally similar natural products against different targets proteins of SARS-CoV-2 to deduce the structural insight of molecular interactions. The study yielded natural products being effectively bound to their respective targets (Table 3 ). The results were expressed in terms of docking energy (kcal/mol). Many selected natural products have displayed docking energies lower than their structurally similar standard drug counterparts. The natural products similar to Remdesivir interact with SARS-CoV NSP12 polymerase with docking energies comparably lower than the standard drug. The natural products tested as influenza virus hemagglutinin inhibitors are also bound to the target with docking energies lower than the standard drug arbidol. The binding interactions of natural products tested as viral protease inhibitors were compared with standard drugs Lopinavir and Ritonavir. The effectiveness of these binding of natural products with the highest interaction energy in each group was selected for protein stability assessment using molecular dynamics simulations (Fig. 4 ).
Table 3.
Molecular interactions between the selected natural products with targets of their structurally similar chemical drugs expressed as docking energies along with their structure similarity score. The table also details the number of hydrogen bonds formed between the Natural product and the amino acid residues from the target molecule.
| Target protein | Synthetic drug and the identified NPs | Docking Energya | H-bonds | Receptor residues involved in H-bond formation |
|---|---|---|---|---|
| SARS-CoV NSP12 POLYMERASE | Remdesivir | −7.2 | 03 | ILE23, LEU126, GLY48 |
| 12,28-Oxa-8-Hydroxy-Manzamine A | −10.4 | 02 | GLY130, ALA38 | |
| Marineosin A | −7.9 | 00 | – | |
| Bis(Gorgiacerol)Amine | −7.8 | 02 | ILE23, GLY130 | |
| Methylstemofoline | −7.7 | 02 | SER128, ALA129 | |
| Chetracin B | −7.5 | 01 | PHE156 | |
| Oxyprotostemonine | −7.5 | 02 | SER128, ALA129 | |
| Stemocurtisine | −7.5 | 01 | GLY48 | |
| Munroniamide | −6.9 | 05 | VAL49, ILE131, GLY48, GLY130, LEU126 | |
| Alstolobine A | −6.8 | 03 | PHE156, ASP157, ALA154 | |
| Discorhabdin H | −6.7 | 02 | GLY48, ASP22 | |
| INFLUENZA VIRUS HEMAGGLUTININ | Arbidol | −7.1 | 01 | GLU64 |
| Phellibaumin A | −9.4 | 04 | ASP280, SER290, LYS58, ILE288 | |
| Difloxacin | −8.4 | 03 | LYS58, LEU292, PRO293 | |
| Lamellarin D | −8.4 | 02 | LYS58, CYS305 | |
| Lamellarin Gamma Acetate | −7.8 | 01 | GLU57 | |
| Hydroxy-6-Methylpyran-2-One Derivative | −7.6 | 03 | THR59, GLU57, THR59 | |
| Cyathuscavin C | −7.5 | 02 | GLU57, PRO306 | |
| Cyathusal B | −7.4 | 02 | GLU57, PRO306 | |
| Clausarin | −7.3 | 02 | GLU64, ARG85 | |
| Cyathuscavin B | −7.3 | 00 | – | |
| Pulvinatal | −7.3 | 01 | THR59 | |
| HIV-1 PROTEASE I50V ISOLATE | Lopinavir | −6.5 | 03 | GLY49, GLY51, GLY52 |
| Hexahydrodipyrrolo trione derivative | −8.4 | 03 | PRO81, ASP25, GLY48 | |
| Beauvericin | −7.2 | 01 | GLY49 | |
| Chaetocin | −7.1 | 06 | THR74, ASN88, GLN92, ASP30, ILE72, GLY73 | |
| Mollenine A | −7.1 | 00 | – | |
| Chetracin B | −6.9 | 00 | – | |
| Beauvericin H1 | −6.6 | VAL50, GLY51, THR80 | ||
| Dragonamide A | −6.3 | 02 | ASP30, VAL50 | |
| Chetracin D | −6.2 | 04 | THR74, ARG87, ASP29, GLY73 | |
| Dimethyl-3-Oxodecanamide derivative | −5.6 | 03 | VAL50, GLY51, PHE53 | |
| Symplocamide A | −4.6 | 00 | – | |
| HIV-1 PROTEASE A02 ISOLATE | Ritonavir | −7.7 | 04 | ASP29, ASP30, GLY48, GLY49 |
| Bionectin B | −8.1 | 03 | ILE50, THR82, GLY51 | |
| Luteoalbusin A | −8.0 | 04 | GLY51, GLY52, PRO81, PRO79 | |
| Bionectin A | −7.7 | 02 | THR96, ASN98 | |
| Oidioperazine A | −7.7 | 02 | ILE50, ASP25 | |
| Holstiine | −7.1 | 00 | – | |
| Chetracin B | −7.0 | 02 | ARG87, LUE97 | |
| Mollenine A | −6.9 | 00 | – | |
| Methaniminium derivative | −6.6 | 01 | PRO81 | |
| Verticillin E | −6.6 | 02 | THR74, ASN88 | |
| Chaetocin | −6.4 | 02 | ARG08, THR26 |
kcal/mol.
Fig. 4.

Docking interaction between Remdesivir (a), and 12,28-Oxa-8-Hydroxy-Manzamine A (b) with SARS-CoV NSP12 polymerase, Arbidol (c), and Phellibaumin A (d) with influenza virus hemagglutinin, Lopinavir (e), and Hexahydrodipyrrolo trione derivative (f) with HIV-1 protease I50V isolate and Ritonavir (g), and Bionectin B (h) with HIV-1 protease A02 isolate.
3.4. Molecular dynamic simulations to predict the protein structural stability
In the present study, united-atom MD simulations were performed to confirm the accuracy of binding resulting from docking studies [56]. The result of the MD simulation displayed the conformational changes acquired by different target proteins of SARS-CoV-2 upon binding and inferred the structural insight on molecular stability (Fig. 5 ).
Fig. 5.

RMSD (a–d), RMSF (e–h), Rg (i–l) and SASA (m–p) plots obtained from MD trajectories analysis of native, Natural product bound, and chemical drug bound structure of SARS-CoV NSP12 polymerase, influenza virus hemagglutinin, and HIV-1 protease of I50V isolate and A02 isolate.
The RMSD analysis was done to understand the deviation of Cα atoms of the protein from its backbone, and RMSF analysis was done to study the fluctuations associated with the amino acid residues of the protein during the simulation. The average RMS deviations and RMS fluctuations were calculated from the MD trajectories of natural product, and synthetic drug bound HIV-1 protease (I50V isolate), Influenza virus hemagglutinin, SARS-CoV NSP 12 polymerase, and HIV-1 protease (A02 isolate) and were compared with their respective unbound structures (Table 4 ). The RMSD plots indicate that Remdesivir bound SARS CoV NSP 12 polymerase (Fig. 4a) and Arbidol bound Influenza virus hemagglutinin (Fig. 4b) displayed lesser deviations than their structurally similar natural products, Hydroxy Manzamin A and Phellibaurin_A bound structures. However, the macromolecular structures HIV-1 protease (I50V isolate) (Fig. 4c) and HIV-1 protease (A02 isolate) (Fig. 4d) displayed lesser RMS deviations upon binding with respective natural products compared to their synthetic drug counterparts. It can also be seen from these plots that the deviations in the macromolecular structures are relatively low after 5ns in both conditions. From the RMSF plots (Fig. 4e–h), it can be inferred that, though the residues displayed minor fluctuations at certain positions, all proteins were able to retain their secondary structure's packability. This was inferred based on the Rg plots (Fig. 5i-l), where the structures were found to be very tightly packed, as the secondary structure elements like α-helix, β-sheet, and turn were remodeled at each time step of the MD simulation. The SASA plots (Fig. 5m–p) also supported these findings.
Table 4.
Calculated MD parameters for native and ligand-bound SARS CoV2 drug targets obtained from the MD simulation along with binding energies and the contributing energy terms of the prescribed drugs and their most similar natural product calculated using g_mmpbsa module.
| SARS-CoV NSP12 POLYMERASE | INFLUENZA VIRUS HEMAGGLUTININ | HIV-1 PROTEASE I50V ISOLATE | HIV-1 PROTEASE A02 ISOLATE | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gromacs Modules | Native Protein | Remdesivir | 12,28-Oxa-8-Hydroxy-Manzamine A | Native Protein | Arbidol | Phellibaumin A | Native Protein | Lopinavir | Hexahydrodipyrrolo trione derivative | Native Protein | Ritonavir | Bionectin B | |
| Potential Energy (x 10−6) | −0.638 | −0.638 | −0.637 | −4.605 | −4.604 | −4.604 | −0.436 | −0.434 | −0.436 | −0.519 | −0.518 | −0.518 | |
| RMSD (nm) | 0.213 | 0.195 | 0.186 | 0.481 | 0.429 | 0.549 | 0.247 | 0.270 | 0.254 | 0.265 | 0.447 | 0.287 | |
| RMSF (nm) | 0.105 | 0.055 | 0.099 | 0.176 | 0.216 | 0.231 | 0.130 | 0.141 | 0.130 | 0.140 | 0.144 | 0.139 | |
| Rg (nm) | 1.558 | 1.523 | 1.524 | 2.802 | 2.835 | 2.763 | 1.316 | 1.307 | 1.342 | 1.343 | 1.488 | 1.352 | |
| SASA (nm2) | 92.95 | 85.00 | 87.13 | 175.24 | 176.47 | 177.43 | 59.57 | 60.05 | 60.92 | 64.83 | 71.84 | 64.44 | |
| Secondary Structure | 210.49 | 221.97 | 219.29 | 283.92 | 295.63 | 285.29 | 119.47 | 112.07 | 118.78 | 117.81 | 117.54 | 120.51 | |
| Coul-SRa | – | −47.22 | −3.84 | – | −9.45 | −65.80 | – | −40.29 | −30.24 | – | −83.47 | −66.90 | |
| LJ-SRa | – | −92.72 | −64.15 | – | −109.61 | −114.98 | – | −113.62 | −109.54 | – | −326.78 | −160.13 | |
| MMPBSA Module | Binding Energya | – | −63.68 | −57.17 | – | −104.39 | −60.86 | – | −77.59 | −93.53 | – | −180.45 | −161.08 |
| SASA Energya | – | −19.36 | −8.68 | – | −14.03 | −13.62 | – | −14.11 | −13.84 | – | −34.91 | −16.52 | |
| Polar Solvation Energya | – | 146.39 | 35.27 | – | 43.27 | 132.86 | – | 97.20 | 67.11 | – | 179.49 | 73.50 | |
| Electrostatic Energya | – | −36.09 | −3.27 | – | −6.71 | −51.28 | – | −29.45 | −17.77 | – | −49.11 | −32.73 | |
| van der Waals Energya | – | −154.62 | −80.48 | – | −126.92 | −128.81 | – | −131.22 | −129.03 | – | −375.91 | −185.33 | |
kJ/mol.
The binding free energy calculations performed using the g_mmpba module displayed better binding of natural products with their respective target proteins. The binding free energies of natural products, 12,28-Oxa-8-Hydroxy-Manzamine A (−57.17 kJ/mol), Phellibaumin A (−60.86 kJ/mol), and Bionectin B (−161.08 kJ/mol) were found to be encouraging in deducing their interactions with their respective targets. However, their structurally similar synthetic drug counterparts viz. Remdesivir (−63.68 kJ/mol), Arbidol (−104.39 kJ/mol), and Ritonavir (−180.45 kJ/mol) displayed higher binding free energies towards their respective targets. Nevertheless, the binding free energy of the natural product Hexahydrodipyrrolo trione derivative (−93.53 kJ/mol) was found to be higher than its structurally similar standard drug counterparts; Lopinavir (−77.59 kJ/mol). The associated terms for binding free energy calculations along with the calculated MD parameters for unbound and ligand-bound targets detailing RMSD, RMSF, Rg, SASA, Secondary structure, Coul-SR energy, and LJ-SR energy are detailed in Table 4.
4. Discussion
Viral infections have always created challenges in human healthcare research. Identifying efficacious and cost-effective antiviral drugs in the absence of potential vaccines or standard therapies thus holds utmost importance. Due to the advancements in virology, molecular biology, and computational biology, we can now decipher the pathophysiology of many viral diseases, including COVID-19 infection [57]. Natural products have been playing an important role as complementary medicine to combat viral infections, and herbal medicines have been an excellent source for modern drug research programs for a long time [58]. Their origin, availability, safety, and cost-effectiveness make them a reliable choice alongside modern medicine [59].
With the advancements in computational techniques, new strategies have been designed to predict the interactions of potential human target proteins with specific viral strains [60]. These models rely on the available interaction information to predict the novel host-virus interactions. These predictions have been reliable in the past in understanding the infection mechanism of SARS-CoV [61], MERS-CoV [61], Ebola virus [62], and Zika virus [63]. However, these computational methods play a significant role in modern drug research; the experimental verifications of virus-host interactions are needed to substantiate the potential interactions [[64], [65], [66]]. Along with this, the availability of verified interactions and relevant information is a prerequisite for computational drug discovery methods. Most of the chemical structure search and identification methods today rely on the conventional string search to modern data structure module-based approaches [[67], [68], [69], [70]]. The structural comparison and screening based on physiochemical properties, topological indices, molecular graphs, pharmacophore features, molecular shapes, molecular fields, and quantitative measures are expected to reduce false-positive results and yield more effective structures [13,14]. In the current investigation, we were able to shortlist some of the natural products as potential drug-like molecules based on their structural similarities with synthetic chemical drugs. The screening is based on the central foundation of medicinal chemistry that the structurally similar molecules will have similar biological effects [12,14]. The yielded structures were docked to the binding site of the target protein of their respective structurally similar synthetic drugs. To confirm the structural stability, molecular dynamic simulation studies were performed, and the results were compared with unbound and synthetic drug-bound macromolecular structures. The results identified the selected natural products as potential drug leads, and with critical screening, could serve as promising molecules in the first-line treatment for COVID-19 infections. Several studies have also shown the therapeutic potentials of natural products and their interactions with the key viral proteins associated with virulence [9,[71], [72], [73], [74]]. Nevertheless, the future studies comprising in-vitro studies targeting specific proteins followed by their in-silico structure stability assessment with nano and microsecond simulations will validate their use as potential therapeutic molecules.
Ethics approval and consent to participate
Not applicable.
Funding
This work is an extension of a research project supported by the Department of Science and Technology (DST)- Science and Engineering Research Board (SERB), Govt. of India (Grant number: PDF/2018/00237).
Consent for publication
Not applicable.
Availability of data and materials
All the data used during the current study are available from the corresponding author on reasonable request.
CRediT authorship contribution statement
S.J. Aditya Rao: Conceptualization, Investigation, Methodology, Software, Validation, Writing – original draft, Writing – review & editing. Nandini P. Shetty: Formal analysis, Supervision, Writing – review & editing.
Declaration of competing interest
The authors declare that, the above mentioned manuscript has not been published and is not under consideration for publication elsewhere. The authors are aware of the submission and have no conflicts of interest to disclose.
Acknowledgment
The authors thank the Department of Science and Technology (DST) - Science and Engineering Research Board (SERB), Govt. of India for their financial support.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.micpath.2022.105497.
Abbreviations
- ADME
Absorption, Distribution, Metabolism, and Excretion
- APBS
Adaptive Poisson-Boltzmann Solver
- HTVS
High Throughput Virtual Screening
- MD
Molecular Dynamics
- MM
Molecular mechanical
- MMPBSA
Molecular mechanics/Poisson-Boltzmann surface area
- NCATS
National Center for Advancing Translational Sciences
- NPASS
Natural product activity and species source database
- NPs
Natural products
- PD
Pharmacodynamics
- PDB
Protein Data Bank
- PK
Pharmacokinetics
- PME
Particle Mesh Ewald method
- Rg
Radius of Gyration
- RMSD
Root Mean Square Deviation
- RMSF
Root Mean Square Fluctuation
- RO5
Rule-of-Five
- SASA
Solvent Accessible Surface Area
- SMILES
Simplified Molecular Input Line Entry System
- SPC
Simple Point Charge
- TPSA
Topological polar surface area
Appendix A. Supplementary data
The following is the Supplementary data to this article:
Data availability
Data will be made available on request.
References
- 1.Lin L.T., Hsu W.C., Lin C.C. Antiviral natural products and herbal medicines. J. Tradit. Complement. Med. 2014;4:24–35. doi: 10.4103/2225-4110.124335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li X., Wang W., Zhao X., Zai J., Zhao Q., Li Y., Chaillon A. Transmission dynamics and evolutionary history of 2019-nCoV. J. Med. Virol. 2020;92:501–511. doi: 10.1002/jmv.25701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.WHO Global research on coronavirus disease (COVID-19) 2021. https://www.who.int/emergencies/diseases/novel-coronavirus-2019/global-research-on-novel-coronavirus-2019-ncov
- 4.Rehman S.U., Shafique L., Ihsan A., Liu Q. Evolutionary trajectory for the emergence of novel coronavirus SARS-CoV-2. Pathogens. 2020;9 doi: 10.3390/pathogens9030240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harcourt J., Tamin A., Lu X., Kamili S., Sakthivel S.K., Murray J., Queen K., Tao Y., Paden C.R., Zhang J., Li Y., Uehara A., Wang H., Goldsmith C., Bullock H.A., Wang L., Whitaker B., Lynch B., Gautam R., Schindewolf C., Lokugamage K.G., Scharton D., Plante J.A., Mirchandani D., Widen S.G., Narayanan K., Makino S., Ksiazek T.G., Plante K.S., Weaver S.C., Lindstrom S., Tong S., Menachery V.D., Thornburg N.J. Severe acute respiratory syndrome coronavirus 2 from patient with coronavirus disease, United States, Emerg. Inf. Disp. 2020;26:1266–1273. doi: 10.3201/EID2606.200516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang W., Tang J., Wei F. Updated understanding of the outbreak of 2019 novel coronavirus (2019-nCoV) in Wuhan, China. J. Med. Virol. 2020;92:441–447. doi: 10.1002/jmv.25689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chan J.F.W., Yuan S., Kok K.H., To K.K.W., Chu H., Yang J., Xing F., Liu J., Yip C.C.Y., Poon R.W.S., Tsoi H.W., Lo S.K.F., Chan K.H., Poon V.K.M., Chan W.M., Ip J.D., Cai J.P., Cheng V.C.C., Chen H., Hui C.K.M., Yuen K.Y. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet. 2020;395:514–523. doi: 10.1016/S0140-6736(20)30154-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li H., Liu Z., Ge J. Scientific research progress of COVID-19/SARS-CoV-2 in the first five months. J. Cell Mol. Med. 2020;24:6558–6570. doi: 10.1111/jcmm.15364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chilamakuri R., Agarwal S. COVID-19: characteristics and therapeutics. Cells. 2021;10 doi: 10.3390/cells10020206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yuan H., Ma Q., Ye L., Piao G. The traditional medicine and modern medicine from natural products. Molecules. 2016;21 doi: 10.3390/molecules21050559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walsh C.T., Tang Y. The Royal Society of Chemistry; 2017. Natural Product Biosynthesis. [Google Scholar]
- 12.Martin Y.C., Kofron J.L., Traphagen L.M. Do structurally similar molecules have similar biological activity? J. Med. Chem. 2002;45:4350–4358. doi: 10.1021/jm020155c. [DOI] [PubMed] [Google Scholar]
- 13.Gfeller D., Grosdidier A., Wirth M., Daina A., Michielin O., Zoete V. SwissTargetPrediction: a web server for target prediction of bioactive small molecules. Nucleic Acids Res. 2014;42 doi: 10.1093/nar/gku293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kumar A., Zhang K.Y.J. Advances in the development of shape similarity methods and their application in drug discovery. Front. Chem. 2018;6 doi: 10.3389/fchem.2018.00315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nobile M.S., Cazzaniga P., Tangherloni A., Besozzi D. Graphics processing units in bioinformatics, computational biology and systems biology. Briefings Bioinf. 2017;18:870–885. doi: 10.1093/bib/bbw058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aditya Rao S.J., Ramesh C.K., Raghavendra S., Paramesha M. Dehydroabietylamine, A diterpene from carthamus tinctorious L. Showing antibacterial and anthelmintic effects with computational evidence. Curr. Comput. Aided Drug Des. 2020;16:231–237. doi: 10.2174/1573409915666190301142811. [DOI] [PubMed] [Google Scholar]
- 17.Gange S.J., Golub E.T. From smallpox to big data: the next 100 years of epidemiologic methods. Am. J. Epidemiol. 2016;183:423–426. doi: 10.1093/aje/kwv150. [DOI] [PubMed] [Google Scholar]
- 18.Docherty A.B., Lone N.I. Exploiting big data for critical care research. Curr. Opin. Crit. Care. 2015;21:467–472. doi: 10.1097/MCC.0000000000000228. [DOI] [PubMed] [Google Scholar]
- 19.Greene C.S., Tan J., Ung M., Moore J.H., Cheng C. Big data bioinformatics. J. Cell. Physiol. 2014;229:1896–1900. doi: 10.1002/jcp.24662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wasser T., Haynes K., Barron J., Cziraky M. Using “big data” to validate claims made in the pharmaceutical approval process. J. Med. Econ. 2015;18:1013–1019. doi: 10.3111/13696998.2015.1108919. [DOI] [PubMed] [Google Scholar]
- 21.Raghavendra S., Aditya Rao S.J., Kumar V., Ramesh C.K. Multiple ligand simultaneous docking (MLSD): a novel approach to study the effect of inhibitors on substrate binding to PPO. Comput. Biol. Chem. 2015;59:81–86. doi: 10.1016/j.compbiolchem.2015.09.008. [DOI] [PubMed] [Google Scholar]
- 22.Janakirama A.R.S., Shivayogi S.M., Satyanarayana J.K., Kumaran R.C. Characterization of isolated compounds from Morus spp. and their biological activity as anticancer molecules. Bioimpacts. 2021;11:187–197. doi: 10.34172/bi.2021.09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arantes P.R., Saha A., Palermo G. Fighting covid-19 using molecular dynamics simulations. ACS Cent. Sci. 2020;6:1654–1656. doi: 10.1021/acscentsci.0c01236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dr. Duke's phytochemical and ethnobotanical databases, U S Dep. Agric. Agric. Res. Serv. (REP). 1992-2016. (n.d.). 10.15482/USDA.ADC/1239279.. [DOI]
- 25.Zeng X., Zhang P., He W., Qin C., Chen S., Tao L., Wang Y., Tan Y., Gao D., Wang B., Chen Z., Chen W., Jiang Y.Y., Chen Y.Z. NPASS: natural product activity and species source database for natural product research, discovery and tool development. Nucleic Acids Res. 2018;46:D1217–D1222. doi: 10.1093/nar/gkx1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Das K., Gezici S. Review article Plant secondary metabolites, their separation, identification and role in human disease prevention. Ann. Phytomedicine An Int. J. 2018;7:13–24. doi: 10.21276/ap.2018.7.2.3. [DOI] [Google Scholar]
- 27.Hussein R.A., El-Anssary A.A. Herb. Med.; 2019. Plants Secondary Metabolites: the Key Drivers of the Pharmacological Actions of Medicinal Plants. [DOI] [Google Scholar]
- 28.Singh B., Bhat T.K., Singh B. Potential therapeutic applications of some antinutritional plant secondary metabolites. J. Agric. Food Chem. 2003;51:5579–5597. doi: 10.1021/jf021150r. [DOI] [PubMed] [Google Scholar]
- 29.Kim S., Chen J., Cheng T., Gindulyte A., He J., He S., Li Q., Shoemaker B.A., Thiessen P.A., Yu B., Zaslavsky L., Zhang J., Bolton E.E. PubChem 2019 update: improved access to chemical data. Nucleic Acids Res. 2019;47 doi: 10.1093/nar/gky1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.U.S. National Institute of Health (NIH) U.S. Natl. Libr. Med.; 2008. U.S. National Library of Medicine (NLM), ClinicalTrials.Gov Database.https://clinicaltrials.gov/ct2/home [Google Scholar]
- 31.Sander T., Freyss J., Von Korff M., Rufener C. DataWarrior: an open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015;55:460–473. doi: 10.1021/ci500588j. [DOI] [PubMed] [Google Scholar]
- 32.Divyashri G., Murthy T.P.K., Sundareshan S., Kamath P., Murahari M., Saraswathy G.R., Sadanandan B. In silico approach towards the identification of potential inhibitors from Curcuma amada Roxb against H. pylori: ADMET screening and molecular docking studies. Bioimpacts. 2020;11:119–127. doi: 10.34172/BI.2021.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kikiowo B., Ogunleye A.J., Inyang O.K., Adelakun N.S., Omotuyi O.I., Metibemu D.S., David T.I., Oludoyi O.O., Ijatuyi T.T. Flavones scaffold of chromolaena odorata as a potential xanthine oxidase inhibitor: induced fit docking and ADME studies. Bioimpacts. 2020;10:227–234. doi: 10.34172/bi.2020.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aditya Rao S.J., Shetty N. Structure-based Assessment of Homologous Analogues of Natural products: a computational approach to predict the therapeutic effects of natural products. Res. Sq. 2021 doi: 10.21203/rs.3.rs-620117/v1. [DOI] [Google Scholar]
- 35.Liu J.-H., Yu B.-Y. Biotransformation of bioactive natural products for pharmaceutical lead compounds. Curr. Org. Chem. 2010;14:1400–1406. doi: 10.2174/138527210791616786. [DOI] [Google Scholar]
- 36.Lipinski C.A., Lombardo F., Dominy B.W., Feeney P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997;23:3–25. doi: 10.1016/S0169-409X(96)00423-1. [DOI] [PubMed] [Google Scholar]
- 37.Doak B.C., Over B., Giordanetto F., Kihlberg J. Oral druggable space beyond the rule of 5: insights from drugs and clinical candidates. Chem. Biol. 2014;21:1115–1142. doi: 10.1016/j.chembiol.2014.08.013. [DOI] [PubMed] [Google Scholar]
- 38.Aditya Rao S.J., Venugopal T., Jayanna N., Paramesha M., Ramesh C. Bioactive isolates of Morus species as antibacterial agents and their insilico profiling. Lett. Drug Des. Discov. 2021;18:445–453. doi: 10.2174/1570180817999201104120815. [DOI] [Google Scholar]
- 39.Jarrahpour A., Motamedifar M., Zarei M., Youssoufi M.H., Mimouni M., Chohan Z.H., Ben Hadda T. Petra, Osiris, and molinspiration together as a guide in drug design: predictions and correlation structure/antibacterial activity relationships of new N-sulfonyl monocyclic β-lactams, phosphorus. Sulfur. Silicon Relat. Elem. 2010;185:491–497. doi: 10.1080/10426500902953953. [DOI] [Google Scholar]
- 40.Cheng F., Li W., Zhou Y., Shen J., Wu Z., Liu G., Lee P.W., Tang Y. AdmetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J. Chem. Inf. Model. 2012;52:3099–3105. doi: 10.1021/ci300367a. [DOI] [PubMed] [Google Scholar]
- 42.Trott O., Olson A.J. Software news and update AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin J.-H. Accommodating protein flexibility for structure-based drug design. Curr. Top. Med. Chem. 2012;11:171–178. doi: 10.2174/156802611794863580. [DOI] [PubMed] [Google Scholar]
- 44.Salsbury F.R. Molecular dynamics simulations of protein dynamics and their relevance to drug discovery. Curr. Opin. Pharmacol. 2010;10:738–744. doi: 10.1016/j.coph.2010.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schmid N., Eichenberger A.P., Choutko A., Riniker S., Winger M., Mark A.E., Van Gunsteren W.F. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. 2011;40:843–856. doi: 10.1007/s00249-011-0700-9. [DOI] [PubMed] [Google Scholar]
- 46.Abraham M.J., Murtola T., Schulz R., Páll S., Smith J.C., Hess B., Lindah E. Gromacs: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. Software. 2015;1–2:19–25. doi: 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- 47.Berendsen H.J.C., Postma J.P.M., van Gunsteren W.F., Hermans J. 1981. Interaction models for water in relation to protein hydration; pp. 331–342. [DOI] [Google Scholar]
- 48.Bussi G., Donadio D., Parrinello M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007;126 doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- 49.Parrinello M., Rahman A. Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 1981;52:7182–7190. doi: 10.1063/1.328693. [DOI] [Google Scholar]
- 50.Hess B., Bekker H., Berendsen H.J.C., Fraaije J.G.E.M. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 1997;18:1463–1472. doi: 10.1002/(SICI)1096-987X(199709)18:12<1463::AID-JCC4>3.0.CO;2-H. [DOI] [Google Scholar]
- 51.Essmann U., Perera L., Berkowitz M.L., Darden T., Lee H., Pedersen L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577–8593. doi: 10.1063/1.470117. [DOI] [Google Scholar]
- 52.Huang C., Li C., Choi P.Y.K., Nandakumar K., Kostiuk L.W. Effect of cut-off distance used in molecular dynamics simulations on fluid properties. Mol. Simulat. 2010;36:856–864. doi: 10.1080/08927022.2010.489556. [DOI] [Google Scholar]
- 53.Kumari R., Kumar R., Consortium O.S.D.D., Lynn A. g _ mmpbsa - a GROMACS tool for MM-PBSA and its optimization for high-throughput binding energy calculations. J. Chem. Inf. Model. 2014;54:1951–1962. doi: 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- 54.Kwofie S.K., Broni E., Teye J., Quansah E., Issah I., Wilson M.D., Miller W.A., Tiburu E.K., Bonney J.H.K. Pharmacoinformatics-based identification of potential bioactive compounds against Ebola virus protein VP24. Comput. Biol. Med. 2019;113 doi: 10.1016/j.compbiomed.2019.103414. [DOI] [PubMed] [Google Scholar]
- 55.Suenderhauf C., Hammann F., Huwyler J. Computational prediction of blood-brain barrier permeability using decision tree induction. Molecules. 2012;17:10429–10445. doi: 10.3390/molecules170910429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aditya Rao S.J., Nandini Shetty P. Evolutionary selectivity of amino acid is inspired from the enhanced structural stability and flexibility of the folded protein. Life Sci. 2021;281 doi: 10.1016/j.lfs.2021.119774. [DOI] [PubMed] [Google Scholar]
- 57.Yuki K., Fujiogi M., Koutsogiannaki S. COVID-19 pathophysiology: a review. Clin. Immunol. 2020;215 doi: 10.1016/j.clim.2020.108427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Miryala S.K., Basu S., Naha A., Debroy R., Ramaiah S., Anbarasu A., Natarajan S. Identification of bioactive natural compounds as efficient inhibitors against Mycobacterium tuberculosis protein-targets: a molecular docking and molecular dynamics simulation study. J. Mol. Liq. 2021;341 doi: 10.1016/j.molliq.2021.117340. [DOI] [Google Scholar]
- 59.Islam M.T., Sarkar C., El-Kersh D.M., Jamaddar S., Uddin S.J., Shilpi J.A., Mubarak M.S. Natural products and their derivatives against coronavirus: a review of the non-clinical and pre-clinical data. Phyther. Res. 2020;34:2471–2492. doi: 10.1002/ptr.6700. [DOI] [PubMed] [Google Scholar]
- 60.Malathi K., Ramaiah S. Bioinformatics approaches for new drug discovery: a review. Biotechnol. Genet. Eng. Rev. 2018;34:243–260. doi: 10.1080/02648725.2018.1502984. [DOI] [PubMed] [Google Scholar]
- 61.Dyall J., Coleman C.M., Hart B.J., Venkataraman T., Holbrook M.R., Kindrachuk J., Johnson R.F., Olinger G.G., Jahrling P.B., Laidlaw M., Johansen L.M., Lear-Rooney C.M., Glass P.J., Hensley L.E., Frieman M.B. Repurposing of clinically developed drugs for treatment of Middle East respiratory syndrome coronavirus infection. Antimicrob. Agents Chemother. 2014;58:4885–4893. doi: 10.1128/AAC.03036-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cao H., Zhang Y., Zhao J., Zhu L., Wang Y., Li J., Feng Y.-M., Zhang N. Prediction of the Ebola virus infection related human genes using protein-protein interaction network. Comb. Chem. High Throughput Screen. 2017;20 doi: 10.2174/1386207320666170310114816. [DOI] [PubMed] [Google Scholar]
- 63.Barrows N.J., Campos R.K., Powell S.T., Prasanth K.R., Schott-Lerner G., Soto-Acosta R., Galarza-Muñoz G., McGrath E.L., Urrabaz-Garza R., Gao J., Wu P., Menon R., Saade G., Fernandez-Salas I., Rossi S.L., Vasilakis N., Routh A., Bradrick S.S., Garcia-Blanco M.A. A screen of FDA-approved drugs for inhibitors of Zika virus infection. Cell Host Microbe. 2016;20:259–270. doi: 10.1016/j.chom.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vasudevan K., Basu S., Arumugam A., Naha A., Ramaiah S., Anbarasu A., Veeraraghavan B. Identification of potential carboxylic acid-containing drug candidate to design novel competitive NDM inhibitors: an in-silico approach comprising combined virtual screening and molecular dynamics simulation. Res. Prepr. 2021 https://assets.researchsquare.com/files/rs-784343/v1/852884a0-692b-4972-8dc2-a63b773d8cc6.pdf?c=1637245347 [Google Scholar]
- 65.Thillainayagam M., Ramaiah S., Anbarasu A. Molecular docking and dynamics studies on novel benzene sulfonamide substituted pyrazole-pyrazoline analogues as potent inhibitors of Plasmodium falciparum Histo aspartic protease. J. Biomol. Struct. Dyn. 2020;38:3235–3245. doi: 10.1080/07391102.2019.1654923. [DOI] [PubMed] [Google Scholar]
- 66.Thillainayagam M., Malathi K., Anbarasu A., Singh H., Bahadur R., Ramaiah S. Insights on inhibition of Plasmodium falciparum plasmepsin I by novel epoxyazadiradione derivatives–molecular docking and comparative molecular field analysis. J. Biomol. Struct. Dyn. 2019;37:3168–3182. doi: 10.1080/07391102.2018.1510342. [DOI] [PubMed] [Google Scholar]
- 67.Gasteiger J., Engel T. Wiley‐VCH Verlag GmbH & Co. KGaA; 2003. Chemoinformatics: A Textbook. [DOI] [Google Scholar]
- 68.Volarath P., Wang H., Fu H., Harrison R. Annu. Int. Conf. IEEE Eng. Med. Biol. - Proc. 2004. Knowledge-based algorithms for chemical structure and property analysis; pp. 3011–3014. [DOI] [PubMed] [Google Scholar]
- 69.Ullmann J.R. An algorithm for subgraph isomorphism. J. ACM. 1976;23:31–42. doi: 10.1145/321921.321925. [DOI] [Google Scholar]
- 70.Cordella L.P., Foggia P., Sansone C., Vento M. vol. 1999. 1999. Performance evaluation of the VF graph matching algorithm; pp. 1172–1177. (Proc. - Int. Conf. Image Anal. Process. ICIAP). [DOI] [Google Scholar]
- 71.Prasansuklab A., Theerasri A., Rangsinth P., Sillapachaiyaporn C., Chuchawankul S., Tencomnao T. Anti-COVID-19 drug candidates: a review on potential biological activities of natural products in the management of new coronavirus infection. J. Tradit. Complement. Med. 2021;11:144–157. doi: 10.1016/j.jtcme.2020.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huang J., Tao G., Liu J., Cai J., Huang Z., Chen J.X. Current prevention of COVID-19: natural products and herbal medicine. Front. Pharmacol. 2020;11 doi: 10.3389/fphar.2020.588508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gasmi A., Chirumbolo S., Peana M., Noor S., Menzel A., Dadar M., Bjørklund G. The role of diet and supplementation of natural products in COVID-19 prevention. Biol. Trace Elem. Res. 2021 doi: 10.1007/s12011-021-02623-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chakravarti R., Singh R., Ghosh A., Dey D., Sharma P., Velayutham R., Roy S., Ghosh D. A review on potential of natural products in the management of COVID-19. RSC Adv. 2021;11:16711–16735. doi: 10.1039/d1ra00644d. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the data used during the current study are available from the corresponding author on reasonable request.
Data will be made available on request.