

Abstract
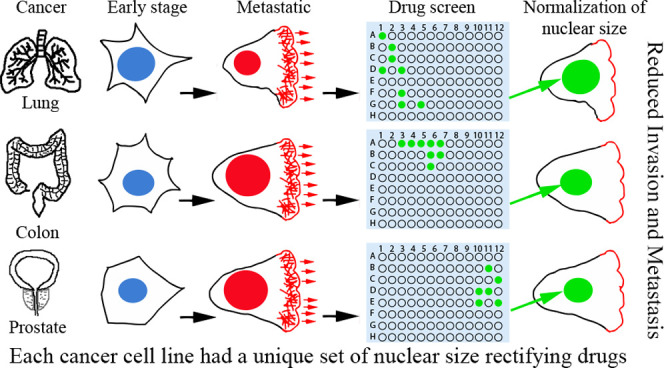
Background: Lower survival rates for many cancer types correlate with changes in nuclear size/scaling in a tumor-type/tissue-specific manner. Hypothesizing that such changes might confer an advantage to tumor cells, we aimed at the identification of commercially available compounds to guide further mechanistic studies. We therefore screened for Food and Drug Administration (FDA)/European Medicines Agency (EMA)-approved compounds that reverse the direction of characteristic tumor nuclear size changes in PC3, HCT116, and H1299 cell lines reflecting, respectively, prostate adenocarcinoma, colonic adenocarcinoma, and small-cell squamous lung cancer. Results: We found distinct, largely nonoverlapping sets of compounds that rectify nuclear size changes for each tumor cell line. Several classes of compounds including, e.g., serotonin uptake inhibitors, cyclo-oxygenase inhibitors, β-adrenergic receptor agonists, and Na+/K+ ATPase inhibitors, displayed coherent nuclear size phenotypes focused on a particular cell line or across cell lines and treatment conditions. Several compounds from classes far afield from current chemotherapy regimens were also identified. Seven nuclear size-rectifying compounds selected for further investigation all inhibited cell migration and/or invasion. Conclusions: Our study provides (a) proof of concept that nuclear size might be a valuable target to reduce cell migration/invasion in cancer treatment and (b) the most thorough collection of tool compounds to date reversing nuclear size changes specific to individual cancer-type cell lines. Although these compounds still need to be tested in primary cancer cells, the cell line-specific nuclear size and migration/invasion responses to particular drug classes suggest that cancer type-specific nuclear size rectifiers may help reduce metastatic spread.
Background
Nuclear size changes have been used in cancer diagnosis since the 1860s1 and remain an important parameter for diagnosis and prognostic grading of later-stage, higher-grade tumors.2 Characteristic nuclear size changes are independent of ploidy for at least 19 cancer types, suggesting a primary dysfunction in the regulation of size/scaling in the cell. The directionality and degree of size changes are tumor/tissue type-specific.3,4 For example, in small-cell squamous lung cancer and osteosarcoma, smaller nuclear size correlates with increased metastasis5,6 while for breast, prostate, colon, and several other cancer types, increased nuclear size correlates with increased metastasis (see Table 1 in ref (3)).
While absolute nuclear size changes are used in current diagnostics, in some tumor types, the size change is associated with the disruption of cell scaling.7 In normal cells and tissues, the nuclear-to-cytoplasm (N/C) ratio is maintained during the cell cycle,8,9 throughout which the nucleus doubles in volume.10−12 In contrast, the N/C ratio is altered in cancer cells by noncorrelated changes in nuclear size and/or cell size. A number of proteins have been implicated in regulating the absolute nuclear size and N/C scaling. Nuclear size is influenced by nuclear envelope proteins, such as the lamins that form the nucleoskeleton13 and the outer nuclear membrane nesprins that connect to the cytoskeleton.14 The inner nuclear membrane protein LAP2-Emerin-Man1 domain-containing protein 2 (LEM2) also regulates nuclear size by controlling membrane flow,15 possibly through interactions with lamins, chromatin,16,17 and protein kinases.18 Recent genetic screens in yeast and mammalian cells have implicated nucleocytoplasmic transport, linker of nucleoskeleton and cytoskeleton (LINC) complexes, and RNA processing/splicing in nuclear size and N/C ratio control.19−21
As nuclear size or scaling is disrupted in cancer cells with increased metastatic potential, we reasoned that such changes might facilitate cell migration/invasion. As the nuclear envelope connects to both the genome and the cytoskeleton, nuclear size dysregulation could achieve this by altering gene regulation, signaling, and/or nucleocytoplasmic transport or by mechanical effects enabling faster migration and/or easier invasion of cancer cells through epithelial barriers. Indeed, disruption of lamin A—known to be limiting for nuclear size changes13—reduces cell migration.22 Moreover, mechanical forces expected to be altered by nuclear size changes are important for the progress of metastasis23 and cellular traction stresses are linked to metastatic potential.24 Thus, we speculated that restoring nuclear size to a more normal range might reduce migration/invasion and thus metastatic potential, irrespective of the biological mechanism(s) that altered nuclear size control during carcinogenesis. We designed a proof-of-concept study to test this hypothesis and probe the chemical space in search of tool compounds that rectify cancer-associated nuclear size changes.
Here, we screened an Food and Drug Administration (FDA)/European Medicines Agency (EMA)-approved drug library for compounds that rectify nuclear size in three distinct cancer cell lines derived from different tissues: two in which nuclear size increases correlate with worse-grade tumors (prostate, colon adenocarcinoma) and one in which nuclear size decrease correlates with worse-grade tumors (lung). Most compounds identified had cell line-specific effects on nuclear size or scaling, possibly via indirect and pleiotropic mechanisms. Clustering compounds according to their therapeutic classes/mechanism of action identified many compound classes with characteristic nuclear size phenotypic signatures across cell lines and conditions. Detailed investigation of seven compounds revealed cell line-specific effects on cell migration and invasion that mirrored to a large extent nuclear size-rectifying effects, suggesting that their addition to treatment regimens might aid in combating metastatic cancer once animal testing has been completed.
Results
Nuclear Size/Scaling Chemical Screen
We screened for compounds that alter nuclear size and/or N/C ratio in cancer cell lines representing three tumor types (Figure 1). PC3 and HCT116 cells, respectively, represented late-stage prostate cancer and colonic adenocarcinoma where nuclear size increases compared to the healthy tissues/early-stage tumors reflect worse cancer grades. H1299 cells represented small-cell squamous lung carcinoma, where decreased nuclear size correlates with a worse grade. The cells were treated with the Prestwick library of 1120 previously approved drugs (PADs) at 10 μM for 6 h to reveal compounds that did not require postmitotic nuclear envelope reassembly to elicit size changes and for 36 h to identify compounds with low toxicity. To align with standard cancer diagnostic procedures, we used nuclear/cell area from imaging (focal) cross sections as our core size metric. The nuclear area was monitored with stably expressed H2B-mRFP, and cell area from CellMask DeepRed cytosol staining (Figure 1A). A total of 350–1000 cells/conditions were imaged for each screen using a PerkinElmer OPERA confocal instrument. Two full replicate screens were undertaken for each cell line and treatment duration, based on reproducibility in a triplicate pilot screen (Methods section; Figure S1 and Table S1).
Figure 1.

High-throughput screen for compounds affecting nuclear size. (A) Schematics of cell growth, treatment, and imaging methodology. (B) Schematics of outlier-based analysis methodology. Within each well, individual cells (left, bottom image) and their nuclei (left, top image) were masked (Methods section). For nuclear and N/C ratio metrics, well-based distributions of individual cell metric values were computed for dimethyl sulfoxide (DMSO) (blue) and compound-treated wells (red, middle plot). The former was used to define outlier thresholds (blue arrows; see the Methods section), from which the fractions of outlier cells (red-shaded regions) were computed for compound-treated wells (shown regions) and DMSO-treated cells (not shown). Blue and red asterisks show the mean value of the metric for DMSO- and compound-treated wells, respectively, defining the average-based analysis (Methods section). For each cell line/time point, data were collected from all wells across the library/replicates (right plot, example shown is plate 14 for PC3, 6 h). Standard statistical analyses of each dataset were used to identify compounds (e.g., oxyphenbutazone) that increased the fractions of outlier cells with either small/large nucleus and/or N/C ratio beyond nonspecific effects of DMSO and inactive compounds (referred to as “hits”).
For each cell line/compound/treatment duration condition, we calculated both absolute nuclear size and N/C ratio metrics for individual cells from high-content OPERA microscopy images (Figure 1, Methods section). The former is traditionally used by cytologists to grade tumors,25,26 while the latter identifies disruption of nuclear size scaling relative to cell size, a hallmark in certain cancer types.7 We next averaged these individual cell values, and also estimated the nuclear size variability (average- and outlier-based analysis strategies, respectively) across the cell population in each condition (see Figure 1B, Methods section, and the Supporting Information (SI) for details). We finally analyzed those datasets statistically to identify hit compounds that perturb absolute nuclear size and/or N/C ratio metrics beyond the typical metric variability across the library (Tables S2–S5, Methods section), thereby reducing nonspecific compound effects on nuclear size. Hit compounds were confirmed using Wilcoxon rank tests versus in-plate DMSO controls. Comparing absolute and relative nuclear size (N/C ratio) metrics allowed us to identify compounds that affect the nuclear size in different ways, while the two analysis strategies enabled us to distinguish compounds with large effects on a fraction of the cell population (outlier-based) from those that uniformly affect most cells in the population (average-based), as expanded in the Supporting Information (Complementary Analyses of Nuclear Size Chemical Screen Data section).
Compounds that induced changes in average absolute nuclear size or N/C ratio in the direction opposite to that associated with increased metastasis in the corresponding tumor type are referred to as nuclear size rectifiers (NSRs) throughout the manuscript.
After 6 and 36 h of treatment, respectively, 88 and 124 compounds were NSR for the absolute nuclear size (Figure 2A, left), while 151 and 178 compounds were NSRs for the N/C ratio (Figure 2A, right). NSR compound sets for the absolute and relative nuclear size only partially overlapped, likely owing to compound-associated cell size renormalization (Figure S2B). Likewise, compound sets aggravating cancer-associated nuclear size changes showed little overlap across cell lines (Figure 2B,D; Table S2) and compound sets with effects after 6 and 36 h of treatment were also distinct (Figure 2C,E). Compounds with the strongest effects are listed in Table 1. Most NSRs were cell line-specific and only a few affected nuclear size in all lines. Strikingly, a handful of compounds were NSRs for both PC3 and H1299 cell lines, or HCT116 and H1299 cell lines, despite rectifying nuclear size in both lines meant shifting the nuclear size in opposite directions. Notably, most NSRs had homogeneous effects on the cell population as opposed to large effects in a fraction of the population.
Figure 2.

Tumor/tissue-type-specific effect of NSR compounds. Colored regions on Venn diagrams show the number of hit compounds for PC3 (red), HCT116 (blue), and H1299 (yellow) tumor cell types. (A) Overlap in absolute nuclear size rectifiers (left) and N/C ratio rectifiers (right). (B) Overlap in compounds increasing/decreasing absolute nuclear size after 6 h (left) and 36 h (right). (C) Overlap in compounds increasing (left) or decreasing (right) mean nuclear size upon 6 h (light colors) and 36 h (dark colors) treatment. (D) Overlap in compounds increasing/decreasing N/C ratio after 6 h (left) and 36 h (right). (E) Overlap in compounds increasing (left) or decreasing (right) N/C ratio upon 6 h (light colors) and 36 h (dark colors) treatment.
Table 1. Nuclear Size Rectifier (NSR) Compounds That Correct Cancer-Related Absolute Nuclear Size (N) or Nuclear-to-Cytoplasm (N/C) Ratio Changes in One or Several of the Three Tumor Types beyond Detection Thresholdsa.
| compound—therapeutic group | nuclear change correction | strength, replicates | nuclear change aggravation |
|---|---|---|---|
| disulfiram—dopamine β hydrolase inhibitor | PC3, N, 6 h | mild, 2/3 | |
| PC3, N/C, 36 h | strong, 2/2 | ||
| H1299, N/C, 36 h | strong, 2/2 | ||
| tomatine—cholinergic inhibition | PC3, N&N/C, 6 h | strong, 3/3 | |
| PC3, N&N/C, 36 h | strong, 1/2 | ||
| HCT116, N, 6 h | strong, 1/2 | ||
| HCT116, N, 36 h | strong, 1/2 | ||
| astemizole—histamine antagonist | PC3, N&N/C, 6 h | mild, 3/3 | H1299, N/C, 6–36 h |
| PC3, N&N/C, 36 h | strong, 2/2 | ||
| perhexiline maleate—Ca2+ channel blocker | PC3, N, 6 h | strong, 2/3 | |
| PC3, N, 36 h | strong, 2/2 | ||
| HCT116, N, 36 h | mild, 1/2 | ||
| ebselen—Cox inhibitor | PC3, N, 6 h | mild, 3/3 | |
| PC3, N/C, 6 h | strong, 3/3 | ||
| alexidine dihydrochloride—detergent | PC3, N&N/C, 6 h | mild, 2/3 | H1299, N/C, 36 h, repl |
| PC3, N&N/C, 36 h | strong, 2/2 | ||
| HCT116, N&N/C, 36 h | mild to strong, 2/2 | ||
| HCT116, N/C, 6 h | |||
| H1299, N/C, 6 h | strong, 1/2 | ||
| H1299, N/C, 36 h, rep2 | strong 2/2 | ||
| cantharidin—serine protease releaser | PC3, N, 6 h | mild, 3/3 | HCT116, N, 6 h |
| PC3, N, 36 h | mild, 2/2 | H1299, N, 6 h | |
| HCT116, N, 36 h | mild, 2/2 | PC3, N/C, 6–36 h | |
| oxyphenbutazone—Cox inhibitor | PC3, N&N/C, 6 h | strong, 2/3 | H1299, N, 6 h |
| PC3, N&N/C, 36 h | strong, 2/2 | H1299, N, 36 h | |
| HCT116, N, 6 h | strong, 1/2 | ||
| HCT116, N, 36 h | strong, 2/2 | ||
| H1299, N/C, 6 h | strong, 2/2 | ||
| H1299, N/C, 36 h | strong, 1/2 | ||
| anisomycin—acetylcholine estherase inhibitor | PC3, N, 36 h | strong, 2/2 | Possibly HCT116, N/C |
| H1299, N&N/C, 36 h | strong, 2/2 | ||
| roxatidine acetate—histamine antagonist | PC3, N, 36 h | strong, 2/2 | PC3, N, 6 h |
| H1299, N, 6 h | mild, 2/2 | ||
| H1299, N/C, 36 h | strong, 2/2 | ||
| metixene HCl—cholinergic inhibition | PC3, N, 36 h | mild, 2/2 | H1299, N, 6 h |
| parthenolide—MAO inhibitor | PC3, N, 36 h | mild, 2/2 | HCT116, N/C, 36 h |
| H1299, N, 36 h | mild, 2/2 | ||
| prenylamine lactate—Ca2+ channel stimulation | PC3, N&N/C, 36 h | strong, 2/2 | |
| emetine dihydrochloride—protein synthesis inhibitor | PC3, N, 36 h | strong, 2/2 | H1299, N, 36 h |
| H1299, N, 6 h | mild, 2/2 | ||
| piperlongumine—alkaloid | PC3, N, 36 h | strong, 2/2 | PC3, N/C, 6 h |
| H1299, N/C, 36 h | strong, 2/2 | HCT116, N/C, 6 h | |
| lycorine HCl—protein translation inhibitor | PC3, N, 36 h | mild, 2/2 | PC3, N/C, 6 h |
| HCT116, N, 36 h | mild, 2/2 | ||
| sertraline—5HT uptake inhibitor | PC3, N, 6 h | mild, 3/3 | PC3, N/C, 6 h |
| PC3, N, 36 h | strong, 2/2 | H1299, N, 6 h | |
| HCT116, N, 36 h | strong, 1/2 | ||
| H1299, N/C, 6 h | mild, 2/2 | ||
| danazol—hormone estrogen antagonist | HCT116, N, 36 h | mild, 2/2 | PC3, N/C, 6 h |
| griseofulvin—antifungal drug | HCT116, N, 36 h | mild to strong, 2/2 | |
| puromycin dihydrochloride—antimetabolite | HCT116, N, 36 h | strong, 2/2 | H1299, N, 36 h |
| H1299, N, 6 h | strong, 2/2 | ||
| resveratrol—Cox inhibitor | PC3, N, 36 h | mild, 2/2 | H1299, N, 36 h |
| HCT116, N, 36 h | strong, 2/2 | PC3, N/C, 6 h | |
| cilostazol—phosphodiesterase inhibitor | HCT116, N, 36 h | strong, 2/2 | PC3, N/C, 6 h |
| eburnamonine—alkaloid | HCT116, N, 36 h | mild to strong, 2/2 | |
| monensin sodium salt—membrane ionophores producer | HCT116, N, 36 h | strong, 2/2 | H1299, N, 36 h |
| dienestrol—nonsteroidal estrogen | HCT116, N, 36 h | mild, 2/2 | |
| azacytidine—5-antimetabolite | HCT116, N, 36 h | mild, 2/2 | H1299, N, 36 h |
| hydroflumethiazide—Na+ Cl-transport inhibitor | H1299, N, 6 h | strong, 2/2 | |
| metaraminol bitartrate—α adrenergic agonist | H1299, N&N/C, 6 h | strong, 2/2 | |
| flufenamic acid—Cox inhibitor | H1299, N, 6 h | strong, 2/2 | |
| trimethoprim—folic acid antagonist | H1299, N, 6 h | mild, 2/2 | |
| fenspiride HCl—bradykinin antagonist | H1299, N, 6 h | strong, 2/2 | |
| methylprednisolone 6-α—glucocorticoid | H1299, N, 6 h | strong, 2/2 | HCT116, N, 36 h |
| HCT116, N/C, 36 h | mild, 2/2 | ||
| Debrisoquin sulfate—catecholamine depletor | H1299, N, 6 h | strong, 2/2 | |
| H1299, N, 36 h | mild, 2/2 | ||
| harmol HCl monohydrate—anxiolytic | H1299, N, 6 h | strong, 2/2 | |
| methoxy-6-harmalan—benzodiazepine receptor ligand | H1299, N, 6 h | mild, 2/2 | |
| HCT116, N/C, 36 h | strong, 1/2 | ||
| levonordefrin—α adrenergic agonist | H1299, N, 6 h | strong, 2/2 | HCT116, N, 36 h |
| HCT116, N/C, 36 h | strong, 2/2 | ||
| N-acetyl-l-leucine—anti-vertigo drug | H1299, N, 6 h | mild, 2/2 | |
| (+)-isoproterenol (+)-bitartrate salt—bronchodilator | H1299, N, 6 h | strong, 2/2 | |
| HCT116, N/C, 6 h | strong, 2/2 | ||
| proscillaridin A—cardiac glycoside | H1299, N, 6 h | strong, 2/2 | |
| H1299, N, 36 h | strong, 1/2 | ||
| etilefrine HCl—α adrenergic agonist | H1299, N, 6 h | strong, 2/2 | |
| HCT116, N/C, 6 h | strong, 2/2 | ||
| letrozole—P450 inhibitor | H1299, N, 6 h | strong, 2/2 | |
| HCT116, N/C, 6 h | strong, 2/2 | ||
| (+,−)-synephrine—α adrenergic agonist | H1299, N, 6 h | mild, 2/2 | |
| HCT116, N/C, 6 h | mild, 2/2 | ||
| verteporfin—treatment of ARMD | H1299, N, 6 h | mild, 2/2 | |
| monobenzone—melanin inhibition | H1299, N, 36 h | strong, 2/2 | HCT116, N, 36 h |
| trifluridine—DNA replication inhibitor | H1299, N, 36 h | strong, 2/2 | PC3, N, 6 h |
| PC3, N/C, 36 h | strong, 2/2 | HCT116, N, 36 h | |
| antimycin A—mitochondrial ETC inhibitor | PC3, N/C, 6 h | mild, 2/3 | |
| mitoxantrone dihydrochloride—topoisomerase II inhibitor | H1299, N/C | strong, 1/2 | HCT116, N/C, 36 h |
| PC3, N/C, 36 h | mild, 2/2 | ||
| paroxetine HCl—5HT uptake inhibitor | PC3, N, 6 h | mild, 2/3 | PC3, N/C, 6 h |
| parbendazole—antihelmintinc | HCT116, small N outliers, 36 h | mild, 2/2 | PC3, large N outliers, 6 h |
| H1299, large and small N outliers, 36 h | mild, 2/2 | ||
| camptothecine (S,+)—topoisomerase I inhibitor | all lines, large and small N outliers, 36 h | mild to strong, 2/2 |
Compound classes currently used in chemotherapy (e.g., microtubule inhibitors, DNA intercalating agents) and classes with consistent phenotypes across the class (e.g., β adrenergic receptor agonists (BAAs), Na+/K+ ATPase inhibitors) have been removed from this table and are listed in Figures S3, 4, and S4, respectively.
Generically Active vs Cell Line-Specific Compound Classes
All compounds were sorted according to pharmacological class/mechanism of action. The entire datasets of increased/decreased absolute nuclear size phenotypes across the sorted compound collection were plotted against conditions (cell line, replicate, time point) as red and blue heat maps, respectively, both for average- and outliers-based analyses separately (Figure 3). Strikingly, certain compound classes showed coherent nuclear size patterns within the class, clustering phenotypes across cell lines or for a particular cell line. For instance, DNA intercalating agents showed little effects on the N/C ratio but increased both the absolute nuclear size set point (average) and its variability (outliers), in all three cell lines with the strongest effects after 36 h (Figure S3A). Microtubule poisons showed more drastic effects on nuclear size variability than on the average, with, e.g., colchicine, paclitaxel, podophyllotoxin, nocodazole, and albendazole strongly increasing the fractions of both small and large nuclei in all cell lines, especially after 36 h treatment (Figures 3 and S3B). Statistical analysis of microtubule poisons compound class enrichment for such phenotypes strongly indicated that the latter are not due to off-target effects (Figures 3 and S3B, * and **). Phenotypic signatures were similar for microtubule poisons with distinct molecular mechanisms of action. Indeed, the microtubule stabilizer paclitaxel and the polymerization inhibitor colchicine (but not its UV-inactivated isomer lumicolchicine) showed overlapping phenotypic signatures on nuclear size.
Figure 3.

NSR compounds that increase (red) or decrease (blue) absolute nuclear size. Heat maps showing the mean nuclear size (first and third columns) and the fraction of large (second column) and small (fourth column) outliers across the compounds collection (vertical) and across tumor type/replicates (horizontal). Mean nuclear size and fraction of outlier cells were normalized to the detection thresholds (see the Methods section), and color-coded as indicated (darker red/blue reflecting a stronger phenotype). Compounds were ranked by the mechanism of action/therapeutic class, in alphabetic order. Drug categories with interesting phenotypic clustering are highlighted to the right: (i) β-Adrenergic receptor agonists; (ii) DNA intercalants and replication inhibitors; (iii) glucocorticoids; (iv) mitogen-activated protein (MAP) kinase inhibitors; (v, x) microtubule poisons; (vi) Na+/K+ ATPase inhibitors; (vii) 5-hydroxytryptamine (5-HT) (serotonin) reuptake inhibitors; (viii) cyclo-oxygenase (Cox) inhibitors; (ix) detergents-surfactants. Enrichment of compound classes for particular phenotypes was statistically significant, with class enrichment scores larger than 95%, and often 99% (see the Methods section).
In addition to DNA intercalants and microtubule poisons, which are mainstays of chemotherapy regimens, our screens recovered most known antineoplastic compounds present in the Prestwick library, buttressing the potential of nuclear size screens to identify anticancer compounds. However, antineoplastic compounds had extremely variable effects on nuclear size (Figure S3D) and are generally toxic. Hence, we focused on identifying compounds not commonly used in cancer therapies and with cell-type-specific effects on nuclear size, under the assumption that this cell-type specificity might reduce systemic toxicity.
Several compound classes were NSRs in only one or two of the three cancer cell lines (Figure 4). β-Adrenergic receptor agonists (BAAs, Figure 4A) such as, e.g., salbutamol, fenoterol, and clenbuterol, but not antagonists such as propafenone, levobunolol, and betaxolol (Figure S4A), reduced the relative nuclear size of HCT116 cells and increased the absolute nuclear size of H1299 cells (NSR for those lines, compare Figure 4A with Figure S4A). This example illustrates that the same compound class can elicit distinct nuclear size changes in different cell lines and thus potentially in different cancer types. In contrast, Na+/K+ ATPase inhibitors, such as digitoxigenin and lanatoside, selectively increased absolute nuclear size only in H1299 cells (Figure 4A) and thus may exhibit selectivity toward lung cancer. Glucocorticoids27 increased the absolute nuclear size in HCT116 cells specifically, exacerbating the cancer-associated nuclear size defect as opposed to functioning as an NSR (Figure S4B).
Figure 4.

Functional groupings for NSR compounds segregated to particular tumor types. Class enrichment scores larger than 95% (resp. 99%) are indicated as * (resp. **) for particular phenotypes. Absolute nuclear size and N/C ratio were normalized to the detection thresholds (Methods section), and color-coded as indicated. Compounds are numbered and listed in order of appearance at the bottom of each panel. (A) Heat maps showing the mean nuclear size (left) and mean N/C ratio (right) across tumor type/replicates (horizontal) treated with β-adrenergic receptor agonists (top heat maps) or Na+/K+ ATPase inhibitors (bottom heat maps). (B) Heat maps showing the mean nuclear size (left) and mean N/C ratio (right) across tumor type/replicates (horizontal) treated with cyclo-oxygenase (Cox) inhibitors (top heat maps) or 5HT-reuptake inhibitors (bottom heat maps). (C) Absolute vs relative nuclear size change profiles similarly extracted for eight other notable compounds.
PC3 NSRs included many cyclo-oxygenase inhibitors and 5-HT reuptake inhibitors (e.g., sertraline, paroxetine, Figure 4B), where the decrease in absolute nuclear size was sometimes accompanied by an increased N/C ratio on the short term specifically. Likewise, γ-aminobutyric acid (GABA) receptor ligands/agonists were NSRs on HCT116 cells as they decreased absolute nuclear size; yet, their effect on cell size was even stronger such that the N/C ratio was increased (Figure S4B). The terpenoid natural product cantharidin, which has antiviral and antiproliferative effects, was a strong PC3 NSR both after 6 and 36 h treatment (Figure 4C). In addition to the above illustrative examples, our nuclear size screens identified ∼50 other NSR compounds for the different cancer types (Table 1; Figure S5). Thus, just as nuclear size changes are characteristic for each tumor type, many compounds that rectify these defects are often specific to each cell line representing these tumor types and exert distinctive effects on relative and absolute nuclear size metrics.
Cell Line-Specific NSRs Affect Coherent Protein Subnetworks
We next performed a cheminformatics-driven protein target analysis of NSR compounds to gain mechanistic insight into the cancer cell line specificity of nuclear size rectification. Known human protein targets of NSRs expressed in our cell lines (Cancer Cell Line Encyclopedia (CCLE) database, Methods section) were extracted from the ChEMBL database. In agreement with the key role of Lamin A in regulating nuclear size and stiffness,22,28,29 Prelamin A/C appeared as the most often-hit target of NSRs for all cell lines (Table S6). Hence, our screen and analysis clearly identified direct regulators of nuclear mechanical properties. Other often-hit targets across cell lines included the cytochrome P450 family, microtubules and associated proteins, Histone-lysine N-methyltransferase MLL, hypoxia-inducible factor 1 α, and huntingtin. These generic targets were also targets of compounds accentuating the cancer-associated nuclear size changes and showed no cell line specificity (see Table S6). In contrast, proteins targeted by cell line-specific NSR compounds were largely nonoverlapping across the three cancer cell lines and represented unique cellular pathways/functions as revealed by gene ontology (GO)-term enrichment analysis with p-values <10–4 (Methods section, Figures 5 and S6).
Figure 5.

PC3-specific NSRs target specific highly-connected protein networks. The default node color is pink. Edge thickness is proportional to the number of interactions. Blue nodes represent phosphorylation GO terms (kinases/phosphatases, e.g., 6468; 16310); nodes circled in blue indicate cAMP metabolism/catabolism and downstream response (GO 51591; 46058; 6198); nodes circled in green indicate cell surface receptor-linked signaling (GO 7166) or cell communication (10646). ABL2, further studied in Figure 8, is highlighted with a yellow circle.
PC3-only NSRs formed a well-connected protein-interaction network containing many kinases including SRC, ERBB, and many mitogen-activated protein (MAP) kinases (Figure 5). Moreover, PC3-only NSRs targeted cell–cell communication (via, e.g., receptors for histamine, ephrin), many of which were G-protein-coupled receptors (GPCR: e.g., α-1B adrenergic-, prostaglandin E2-, HT-receptors). Interestingly, H1299-specific NSRs also targeted GPCR/cAMP/adenylyl cyclase regulation, but via different protein targets from PC3-specific NSRs (e.g., melanocortin-, prostacyclin-, or κ-type opioid-receptor, Figure S6A). HCT116-specific NSRs mostly targeted lipid metabolism and catabolism pathways and, to a lesser extent, ion homeostasis and vitamin D pathways (Figure S6B). Overall, nearly all of the targets of the cancer cell line-specific NSRs were distinct to a particular cell line.
NSRs Have Heterogeneous Effects on Cell Viability
It is unclear whether nuclear size changes directly contribute to aggressive metastatic behavior or if the alterations in nuclear size are a byproduct of other aspects of tumor progression. In the former hypothesis, NSR treatment would be expected to reduce aspects of metastasis (viability, migration, invasion).
To test this hypothesis, we selected seven NSR compounds covering a range of target specificities (Table S7) and tested properties relating to tumor growth and spread: oxyphenbutazone, a nonsteroid anti-inflammatory drug which also depolymerizes microtubules, decreased absolute nuclear size in all cell lines and treatment durations (NSR for PC3 and HCT116); paroxetine and sertraline (Figure 4B), two antidepressants that block serotonin reuptake, and astemizole (Figure 4C), a histamine H1-receptor antagonist, as PC3-specific NSRs; digitoxigenin, a steroid lactone used to strengthen heartbeat, increased absolute nuclear size dramatically in H1299 (H1299 NSR) and slightly in HCT116; piperlongumine, an alkaloid natural product with claimed anticancer properties,30 increased the N/C ratio in PC3 at 6 h but decreased absolute nuclear size at 36 h and increased nuclear size in HCT116 and H1299 at 36 h; and parbendazole, an antihelminthic that increased the N/C ratio and the fractions of both large and small nuclear size outliers in all lines after 36 h.
Out of these seven compounds, oxyphenbutazone, parbendazole, digitoxigenin, piperlongumine, and paroxetine were tested in two viability assays. All compounds reduced the viability of each cancer cell line to different degrees, as quantified by the fraction of metabolically active cells (Figure 6A). Parbendazole showed its strongest viability effect on H1299 cells, where it corrects the N/C ratio. Oxyphenbutazone decreased cell viability similarly for PC3 and HCT116 cells where it is NSR, but viability decreased even more rapidly in H1299 cells where oxyphenbutazone is not NSR. All compounds were also tested in apoptosis assays, where PC3 cells were co-stained with Annexin V and propidium iodide (PI) to label early-stage apoptotic cells (positive only for Annexin V), late-stage apoptotic cells (positive for Annexin V and PI), necrotic cells (positive only for PI), in contrast to healthy cells negative for both stains. The fraction of cells in each population was quantified by fluorescence-activated cell sorting (FACS) using the gating strategy depicted in Figure 6B to detect only singlets and intact cells. All compounds but paroxetine induced apoptosis in PC3 cells to a variable extent, with digitoxigenin showing the strongest effect in comparison with PC3 NSRs parbendazole, oxyphenbutazone, and piperlongumine. From these viability assays, we concluded that there is no obvious correlation between nuclear size correction and cell viability.
Figure 6.

Proliferative and apoptotic effects induced by selected NSR compounds. (A) Fraction of viable cells (vertical axis) as a function of drug concentration (horizontal) for paroxetine-, oxyphenbutazone-, parbendazole-, digitoxigenin-, and piperlongumine-treated PC3, HCT116, and H1299 cells as indicated, as determined by resazurine fluorescence normalized to fluorescence in DMSO-treated control wells. Dose–response curves were obtained using least-square fitting of the data points in the Prism software. (B) Gating strategy for Annexin V/propidium iodide (PI) FACS analysis. Forward scattering (FSC) vs side scattering (SSC) plots (middle and bottom rows, left plots) were used to eliminate cell debris from the population, while distributions of Annexin V and PI signals (top row plots) and FSC-area vs FSC-width plots (middle and bottom rows, central plots) were used to remove cell doublets/triplets/groups. As a result, plotting individual cells according to their Annexin V vs PI signals (middle and bottom rows, right plots) accurately identified populations of healthy (low Annexin V, low PI), early apoptotic (finite Annexin V, low PI), late apoptotic (finite Annexin V, finite PI), and necrotic (low Annexin V, finite PI) cells. (C) FACS results plotted as the percentage of the total cell population under each condition representing healthy, early apoptotic, late apoptotic, and necrotic PC3 cells after 36 h incubation with the indicated drugs.
NSRs Inhibit Cell Migration and Invasion
To investigate potential cell line-specific effects of NSRs on migration, we next tested the seven selected compounds at concentrations from 1 nM to 10 μM in a scratch-wound healing assay (Figures 7A,B, S7A, and Methods section). PC3 and H1299 cells were analyzed as several compounds were unique NSRs for each.
Figure 7.

NSR compounds affecting nuclear size also affect cell migration and invasion. (A) Wound healing assay. Wound confluence (top row) and average nuclear size (bottom row) for PC3 cells treated with varying concentrations of oxyphenbutazone, piperlongumine, digitoxigenin, and sertraline (from left to right) as indicated, in % of DMSO-treated control cells. Red dashed lines represent cutoff values for strong reduction of migration (top row) and strong NSR phenotype (bottom row); see Figure S8A. (B) Wound healing assay. Wound confluence (top row) and average nuclear size (bottom row) for H1299 cells, under the same conditions as (A). (C) Orientation assay. Percentage of PC3 cells from panel (A) showing correctly oriented (left bars) or mis-oriented (right bars) centrioles, after 12 h of wound closure in the presence of 10 μM of indicated compounds, as assessed using γ tubulin staining. (D) Microfluidics migration assay. (Top) Representative images of cells migrating in the microfluidics chamber through confined (≤2 μm × 5 μm, top) and unconfined (15 μm × 5 μm, bottom) constrictions. Arrow shows migration direction. Scale bar, 50 μm. (Bottom) Mean nuclear transit time (time required to move through an individual constriction) for DMSO-, piperlongumine-, and digitoxigenin-treated PC3 cells. Error bars show SEM. (E) Invasion assay. Optical density (OD, arbitrary unit) characterizing the fraction of PC3 cells that successfully invaded a Boyden chamber within 24 h, in the presence of 10 μM of the indicated compounds.
Analysis of PC3 wound closure after 24 h incubation revealed that PC3 NSRs oxyphenbutazone, piperlongumine (Figure 7A), and astemizole (Figure S7A) reduced migration in a dose-dependent manner at concentrations starting at 100 nM or 1 μM, concentrations at which those compounds were NSR for this cell line. Parabendazole, which increased the fraction of large and small nuclear size outliers in PC3 (Table S4), behaving thus as an NSR for a fraction of cells, similarly inhibited migration but at higher concentrations of 1 μM or greater (Figure S7A). PC3-specific NSR sertraline reduced both migration and nuclear size as expected, but only at the maximal concentration of 10 μM (Figure 7A). Out of the five PC3 NSRs tested, only paroxetine failed to slow migration in wound healing assays (Figure S7A). Strikingly, digitoxigenin (an H1299-specific NSR that does not affect nuclear size in PC3), did not affect migration at any concentration (Figure 7A), in full agreement with our working hypothesis.
This pattern of migration phenotypes was in large part inverted in H1299 cells (Figures 7B and S7A): the H1299-specific NSR digitoxigenin increased nuclear size already only slightly at 100 nM, and strongly at 1 and 10 μM, and reduced cell migration at these three concentrations. In contrast, PC3 NSRs oxyphenbutazone, piperlongumine, parbendazole, astemizole, sertraline, and paroxetin showed no effects on H1299 cell migration at intermediate concentrations 100 nM or 1 μM, although some reduced H1299 cell migration at the highest 10 μM concentration. In summary, dose-dependent effects of compounds on nuclear size were mirrored to a large extent by dose-dependent effects on wound closure, in a cell line-specific manner.
Since the overlap between NSR and cell migration phenotypes was not perfect for some compounds at 10 μM, we next sought to assess how likely an NSR is to decrease cell migration, in probabilistic terms. We grouped together the PC3 and H1299 data across all tested compounds at concentrations where NSR or migration effects were detectable under any condition (i.e., ≥100 nM). From these 39 cell line/compound/concentration conditions, we estimated the conditional probability that a given compound at a given concentration shows a migration effect given it is NSR (see the Methods section for details). This approach estimated that an NSR compound has an 89% chance to show a migration defect in wound healing assays, substantially larger than random expectation (33% in our dataset, see the Methods section).
Effects in the wound healing assays could reflect reduced mobility or loss of directionality or both. After a scratch wound, normal cells orient with the centriole facing the wound to direct inward migration and wound closure. To test for loss of cytoskeletal polarization, the cells were stained for γ tubulin to determine centriole orientation. Piperlongumine, digitoxigenin, and oxyphenbutazone were tested in PC3 cells and each yielded defective centriole orientation in 65–85% of cells scored (Figure 7C). Hence, the 3 compounds tested altered the directionality of cell motion, making this parameter unlikely to explain why piperlongumine and oxyphenbutazone slow down wound closure while digitoxigenin did not.
We next considered the possibility that compound effects on nuclear size might reverse an advantage of the tumor cells to migrate through tight junctions. We tested PC3 migration through 5 × 15 and 5 × 2 μm2 constrictions, the latter requiring substantial nuclear deformation, while the former exceeding the size of the nucleus and thus partly serving as controls for migratory ability in a three-dimensional (3D) environment (Figure 7D). The mean migration speed through the 5 × 15 μm2 constrictions was slightly slower for piperlongumine-treated cells (79 min) versus DMSO solvent control (91 min). Similarly, digitoxigenin-treated cells exhibited a slightly slower mean migration through the smaller 5 × 2 μm2 constrictions (73 versus 82 min for controls). However, in both cases, the differences were not statistically significant (two-tailed t-test p > 0.05). In regard to compound-induced centriole orientation defects but minimal 3D migration defects, the compound-induced slower wound closure suggests that tested NSRs affect specifically two-dimensional (2D) cell polarization/mobility rather than mechanical stiffness as cells preferentially use these two distinct mechanisms, respectively, for 2D vs 3D migration.31−33
A transwell migration assay was also performed (Figure 7E) to investigate compound effects on PC3 invasion (Methods section). Cells treated with piperlongumine and parbendazole had ∼50% fewer cells migrate through the Boyden chamber membrane compared to DMSO-treated controls, where oxyphenbutazone and digitoxigenin reduced the invasion capability by 15 and 30% respectively. Thus, NSR compounds reduced both cell migration and invasion in tissue culture assays, although the cell line specificity of migration phenotypes mirrored the cell line specificity of NSR phenotypes to a larger extent in wound healing assays.
Network Analysis of Cell Line-Specific NSRs Identifies Candidate Protein Targets Mediating Cell Migration Effects
Finally, we selected a functional protein target indicated by the cell line-specific NSR targets network analysis (Figure 5) to test if knocking it down could affect nuclear size and migration in a similar cell line-specific manner as the NSR compounds. A major node of the PC3-specific network of NSR targets is the tyrosine-protein kinase ABL2, targeted by the (−)-epicatechin PC3-specific NSR (Table S6), a natural flavanoid with antioxidant and anticancer properties in vivo34,35 (Tables S2–S5). ABL2 is expressed in both PC3 and H1299; so we siRNA inhibited ABL2 in these lines and monitored changes in nuclear size and cell migration.
ABL2 siRNA treatment significantly decreased ABL2 expression levels to the limit of detection (Figure 8A) and decreased the absolute nuclear size compared to siRNA control, acting as a PC3-specific NSR (Figure 8B). Consistent with our working hypothesis, ABL2 siRNA treatment also decreased cell migration as siABL2-treated cells closed the wound slower, as reflected by both a significantly lower confluence and larger wound width than siCTRL-treated cells (Figure 8C, left). ABL2 siRNA treatment had no effect on H1299 migration (Figure 8C, right), and slightly increased migration in HCT116 (Figure S8E), in agreement with ABL2 being a protein specific to the network of PC3-only NSRs.
Figure 8.

Network analysis of PC3-specific NSR identifies protein targets to reduce migration. (A) Quantification of ABL2 mRNA level in the presence of the indicated siRNAs in PC3 cells. (b) (Left) Whisker and Box plots showing the distribution of absolute nuclear size in the migrating PC3 cells (from (C)), treated with control siRNA and siABL2 as indicated. Three replicates (R1–R3) are shown (N = 110, 111, and 100 for siCTRL-treated cells; N = 109, 113, and 104 siABL2-treated cells). The distribution of nuclear sizes in cells treated with siCTRL and siABL2 was compared using Wilcoxon rank tests for each replicate (**: p < 10–4, data from all replicates aggregated: p < 10–9). (Right) Example images of siCTRL (top)- and siABL2 (bottom)-treated cells from the edges of the wound. (C) ABL2 silencing reduces wound closure. Wound confluence (top row) and wound width in % of its value at the first time point (bottom row) as a function of time for PC3 (left) and H1299 (right) cells, treated with 20 nM siCTRL (blue) and 20 nM siABL2 (orange). Data points show confluence and width values averaged over eight replicates, and error bars represent the standard error on the mean (SEM).
Discussion
We hypothesized that for the 19 cancer types for which nuclear size changes have clearly been shown to correlate with increased metastasis (reviewed in ref (3)), the size change might contribute at least in part to metastasis through changes in cellular mechanics and/or migration. This idea is based on the well-established fact that disruption of the nucleoskeleton and/or connections between the nucleoskeleton and the cytoskeleon invariably had corresponding effects on nuclear size and/or cell migration where tested.36−38 The direct relationship between nuclear size changes and migration is more speculative, although nuclear size regulation and migration share a set of effector proteins.13,14 As the nucleus is the largest and sturdiest organelle in the cell, it can be the greatest impediment to cell migration through tight constrictions between tissues; however, nuclear size changes could increase nuclear plasticity to facilitate squeezing through constrictions (Figure S9A) or increase tumor cell motility as experimentally validated in the references above, possibly through effects on actin23,28,39 (Figure S9B). Hence, migrating cancer cells could in principle benefit from a broad range of nuclear size/stiffness alterations.
Notably, the directionality of nuclear size changes correlating with increased metastasis was cell type-specific. Therefore, we hypothesized that using nuclear size as a readout in high-throughput compounds screening across cell types could help identify compounds able to mitigate a migratory advantage that the initial size change contributed to the tumor cells, in a selective, cell-type-specific manner. To test this idea, we screened an FDA/EMA-approved drug library for compounds that rectify cancer-associated nuclear size defects in three different cancer cell lines where nuclear size increase or decrease correlates with worse-grade tumors. With our choice of library, we anticipate that the mechanistic understanding of approved drugs and their easy commercial accessibility will facilitate rapid follow-up research based on our discoveries.
While we have not yet proven a causal relationship between the nuclear size change and altered migration, we identified 50–100 NSRs for each cancer cell line, many of which showed cell line-specific NSR activity and have not been previously tested for cancer therapies for the tumor types represented in our screens. We selected seven of these NSRs for detailed investigation, all of which reduced cell migration under most conditions (cell line, concentration), where they also rectified nuclear size. Hence, the simple nuclear size readout appears to be a reasonable predictor of the effect of drugs on cell migration, irrespective of whether these drugs have pleiotropic effects or impinge on nuclear size and/or migration via many indirect mechanistic routes.
Notably, the cell invasion assay did not correlate as strongly with nuclear size correction as the cell migration assays. While this discrepancy could be attributable to technical aspects (differences in treatment duration, effective compound doses on cells in the two very different assay systems, etc.), it could also stem from different compounds targeting different aspects of cell migratory behavior. More detailed testing of NSR compounds in a variety of assays, in primary tumor cells, and in xenograft models is required before considering these compounds further for clinical use on cancer patients.
An important finding from this study is that many compounds identified in our screen showed cell-type-specific effects on nuclear size and on cell migration in overall agreement with their NSR status. These compounds represent groups not traditionally used to treat these cancer types (Table 1), and so may indicate new compound classes with potential use for specific cancer types. Moreover, their cell-type specificity would likely limit their systemic toxicity. We note, though, that several compounds were NSR only on the shorter (6 h) term but not on the longer (36 h) term, possibly indicating cellular adaptation to perturbation that might reduce compound efficiency in a therapeutical context. Moreover, some PC3-specific NSRs (e.g., sertraline, astemizole, oxyphenbutazone) showed anti-migratory effects in H1299 when applied at high concentrations with minimal effects on nuclear size (Figures 7, S7A, and S8B, bottom left corner), indicating that additional effects of compounds at high concentrations could change the character of the cellular response, further indicating the need to investigate individual hits before translational application.
The cell-type specificity might also extend to NSR protein targets: central to the protein network of PC3-specific NSR targets was the nonreceptor tyrosine kinase ABL2 that is frequently overexpressed in Hepatocellular carcinoma40 and whose silencing significantly attenuates migration and invasion in HepG2 cells.41 Likewise, ABL2 knockdown in breast cancer cells reduces migration.42 In prostate cancer cells, knockdown of ABL2 has contrasted effects on invasion and migration,43,44 indicating that (1) ABL2 might be the target of cell line-specific signals and (2) ABL2 levels need to be finely tuned for optimal repression of migration. The only NSR which directly targets ABL2 in our screen, epicatechin, might modulate AMPK/Akt/mTOR pathway.34 In agreement with our hypothesis, the related compound epicatechin gallate significantly restrains the migration of prostate cancer cells, possibly via Akt/mTOR.45 Other catechins have been associated with reduced invasion of melanoma cells.46
The potential value of the new compound types identified here is underscored by the fact that our screen uncovered most known antineoplastic agents present in the compound library, among which were DNA intercalants and microtubule poisons commonly used in anticancer treatments. However, such commonly used compounds showed neither directional nor tissue specificity. Microtubule poisons affected the N/C ratio in the short term but the nuclear size only in the longer term, in agreement with short-term reduction of cell size,47 possibly owing to increased cell compressibility upon microtubule depolymerization.48 They also strongly affected nuclear size variability, possibly arising from asymmetric nuclear division. We postulate that the nonspecificity of antineoplastic agents may play a role in their systemic side effects and propose in contrast that the cell line-specific effects of NSR compounds identified in our study, provided they can be replicated in primary tumors in vivo, may improve tissue specificity of chemotherapy regimens, by targeting tumor spread while reducing systemic toxicity.
Although our screen identified NSRs directly targeting nuclear mechanics (e.g., Prelamin A/C), which in turn affect migration via partially elucidated mechanisms,22 how other NSRs affect nuclear size may be indirect and diverge from their established mechanisms of action.49 Digitoxigenin induces cytotoxic effects on non-small-cell lung cancer cells through a second function inhibiting Na+/K+ ATPase activity.50 Digitoxigenin is also an effective chemotherapeutic against metastatic uveal melanoma,51 and its derivative, amantadig, is effective in inhibiting hormone-refractory prostate cancer cells,52 but the mechanism by which it acts is not clear. Piperlongumine has been reported to inhibit glycolysis and FOXO3A and to have senolytic and antimicrobial activity,53−56 yet its effects on breast cancer in xenograft models57,58 are thought to function through reactive oxygen species (ROS)-mediated apoptosis.30,58−60 Interestingly, our screen also identified other therapeutically promising ROS-mediated apoptosis inducers such as the sodium ionophore monensin, or compounds that inhibit the oxidative stress-protective aldehyde dehydrogenase enzyme ALDH2, such as disulfiram (Figure S5). Both compounds are PC3-specific NSRs and have been separately shown to differentially affect growth and/or migration of cancer and healthy prostate epithelium cells.61,62
The other compounds we tested for migration effects have not been used to our knowledge as anticancer agents, nor can their known targets readily explain their effects on nuclear size regulation. Paroxetine and sertraline are antidepressants of the selective serotonin reuptake inhibitor (SSRI) class used to treat depressive and obsessive–compulsive disorders. Paroxetine interacts with cytochrome P450 complex enzymes64 and promotes mitochondrial-induced apoptosis in astrocytes.65 Astemizole and other cationic amphiphilic antihistamines are currently being assessed as anticancer agents.66−68 In the context of prostate cancer, astemizole has been recently used to downregulate androgen receptor pathway via the inhibition of the polycomb repressive complex 2 (PRC2), which otherwise promotes proliferation, migration, invasion, and metastasis.69 Parbendazole is a benzimidazole derivative showing broad-spectrum anthelminthic activity and is also a potent microtubule poison.70,71 Parbendazole induced apoptosis in nearly 40% of PC3 cells and inhibited cell migration by nearly 75% in wound healing assays and by 50% in invasion assays. Thus, even though this compound is from a group of drugs with antihelminthic activity, it may hold promise as a therapeutic against prostate cancer.
Other antihelminthics affected nuclear size in our study as well as compounds from other therapeutic groups not previously used in cancer treatments that might be repurposed to treat particular cancer types if these effects replicate in cells from primary tumors. This is underscored by such repurposing already underway for BAAs that were NSRs here for HCT116 cells representing colonic adenocarcinoma and for H1299 cells representing small-cell lung cancer. Interestingly, BAAs suppress the epithelial-to-mesenchymal transition of bronchial epithelial cells and have been used to treat lung cancer.72,73 BAAs are promising candidates also for triple-negative breast cancer and gliomas.74,75
Conclusions
This study provides a proof of principle that high-throughput screening for compounds that rectify nuclear size changes can identify drugs that might be able to reduce metastasis in a cancer type-specific manner. It also provides support to the hypothesis that targeting tissue/cancer type-specific nuclear size changes can affect cell migration with the potential to reduce metastasis. However, there is still much more work to be done to determine causality and to ensure that the possible pleiotropic effects of identified compounds, of which compounds with the strongest NSR effects are listed in Table 1, do not confound the potential benefits with unintended negative effects. Although drug-associated nuclear size corrections might often be indirect and we did not infer mechanistic causes of drug-associated nuclear size phenotypes, we found that the nuclear size readout was a significantly good predictor of cell migration. Engaging similar screens for other cancer/tissue types, testing the effects of compounds on multiple primary tumor cells and controls for each cancer, and testing actual effects on metastatic spread in animal models will all be needed before the full value of this approach can be assessed. While this will require many additional studies, our study is a necessary first step that introduces a potential new paradigm in both nuclear size scaling and cancer research.
Methods
Cell Lines
PC3, HCT116, and H1299 cells were stably transfected to express H2B-mRFP in the pEGFP-N2 clontech vector and maintained under G418 selection (500 μg/mL) for general culture. Selection was removed for one passage prior to plating for experiments to avoid drug interference effects.
Cell Culture and Manipulation
The Prestwick compound library (PAD, 2015 version) was contained in fourteen 96-wells plates, each of which consists of 80 drugs mostly approved by the FDA and EMA agencies (1 mM stock concentration in DMSO) and 16 in-plate DMSO controls, for a total of 1120 compounds. For each screen, ∼5000 H2B-mRFP tagged cells were plated on each well of 14 Greiner Screenstar 96-wells glass-bottom imaging plates and left overnight in 99 μL of Roswell Park Memorial Institute (RPMI) medium for adhesion and growth prior to compound treatment (addition of 1 μL of stock to a final well concentration of 10 μM). The cells were incubated for 6 or 36 h before preparation for imaging. Cell densities prior to compound treatment were adjusted to account for cell doubling during the 36 h long-term treatment to minimize differences in cell densities at the time of imaging. The cells were fixed in 3.7% EMD FX0410-5 formaldehyde solution for 15 min at room temperature, washed with phosphate-buffered saline (PBS), and then incubated for 30 min in 50 μL of HCS CellMask DeepRed 0.5X (Molecular Probes) for cytosol staining. Stained fixed cells were washed again and stored in PBS at 4 °C. Plates were wrapped in parafilm and aluminum foil and imaged within 1 week.
Imaging Data Acquisition
H2B-mRFP tagged cells were imaged using an Opera High-Content Screening instrument (PerkinElmer), equipped with a 20× air objective (LUCPLFLN, NA = 0.45) and Peltier-cooled charge-coupled device (CCD) cameras, or with the latest Opera Phenix for the 6 h time point in H1299, for technical reasons. H2B-mRFP (nucleus) and CellMask DeepRed (cytosol) fluorochromes were exposed sequentially at 561 and 640 nm for 320 and 200 ms, respectively, and fluorescence was measured on distinct channels with 600/40 and 690/70 bandpass detection filters. Bleed-through of the cytosolic channel into the nuclear channel was negligible. For each well, 20 fields of view (FOVs) were acquired, avoiding the edge of the well where cells accumulate due to capillary action. A similar imaging protocol was used to determine the compound dose–response curve of nuclear size (Figures 7 and S7). The drug incubation time was set to 24 h. Only six cells were scored with 10 μM astemizole treatment in this replicate experiment, making an accurate estimation of average nuclear size impossible under this condition. For this compound, these missing data have been replaced by the data from the original screen, averaging the data from 6 and 36 h to interpolate treatment duration (although the nuclear size was reduced very similarly after 6 and 36 h astemizole treatment).
Data Processing and Quantitative Analysis
An adapted Acapella (PerkinElmer) software script was used to automatically mask the cytoplasm and nucleus of individual cells within each FOV. Cell and nuclear sizes were, respectively, defined as the areas (in pixels) of the masked regions in the focal plane, based on, respectively, the CellMask DeepRed and H2B-mRFP signals. Intensity threshold filters, size filters, and morphological filters (which threshold on cell/nuclei size, nuclear roundness, width-to-length ratio, distance between nuclei) were used to filter out detection artifacts, multiple detections of single cell/nuclei, or unique detection of cell clusters. Identical processing parameters were used in Acapella for all conditions to prevent postacquisition processing biases. Single-cell data were exported to a .txt file for subsequent analysis in MATLAB (The Mathworks; see below and Figure 1).
Data Filtering and Computation of Nuclear Size Metrics
For conditions that yielded higher cell densities at the time of imaging, the above filtering step was sometimes insufficient to remove clusters of cells so cell size outliers were eliminated from all datasets. For each condition separately, the entire cell size distribution was analyzed, and we extracted the 25, 50, and 75 size quartiles (Q1, Q2, and Q3, respectively). Next, we filtered out detection areas with a size larger than the standard outlier threshold Q2 + 6 × (Q3 – Q2) (i.e., likely cell clusters), and the detections smaller than Q2 – 6 × (Q2 – Q1) (i.e., imaging artifacts or cell debris). All DMSO control wells for each plate were processed together in this step. For each cell that passed filtering, we computed two metrics: the absolute nuclear size N, and the relative nuclear size, or N/C ratio, defined as the ratio of the absolute nuclear size to the absolute cell size.
Statistical Analysis of Distributions—Average-Based and Outlier-Based Strategies
Compounds that altered absolute or relative (N/C) nuclear size metrics were identified using two complementary strategies for each metric.
Average-Based Strategy
The average value of the metric across all scored cells was determined for each compound- and DMSO-treated well alike. Hits were selected as compounds with an average metric value that stands out from the distribution of average metric values across the library. Specifically, we computed the 25, 50, and 75% percentiles of the distribution of well-averaged metric values across the entire Prestwick library for each cell line/replicate separately (respectively, denoted Q1, Q2, and Q3). We computed low and high outlier thresholds Q2 – s × (Q2 – Q1) and Q2 + s × (Q3 – Q2) with the standards choices of s = 6 for the nuclear size metric (hard selection) and s = 3 for the N/C ratio (softer selection), to account for the larger cell-to-cell variability in N/C ratio compared to absolute nuclear size (N/C ratio variability also reflects cell size variability). These pairs of thresholds are represented as dashed red lines on Figure S1B, top row and bottom row for absolute nuclear size and N/C metrics, respectively. Finally, compounds that reduced (respectively, increased) the mean value of the metric below (respectively, above) those thresholds were identified as hits for the corresponding metric and the average-based analysis strategy. The slightly less stringent criterion for hit selection based on the N/C ratio may explain, in part, the increased sensitivity of the N/C ratio relative to nuclear size in identifying active compounds.
Outlier-Based Strategy
For each plate, we computed the 25, 50, and 75% percentiles characterizing the distribution of metric values across the population of individual DMSO-treated control cells only (i.e., 16 wells per plate). We used these percentiles (Q1, Q2, Q3) to compute low and high (soft) outlier thresholds as defined above DMSOlowThres = Q2 – 3 × (Q2 – Q1) and DMSOhighThres = Q2 + 3 × (Q3 – Q2). Individual cells with values below (resp. above) the low (resp. high) threshold were designated as having an abnormally small (resp. large) metric. We then scored the fractions of those cells with abnormally small and large metric values for all DMSO control and compound wells (Figure 1B, central plot, red-shaded areas). These fractions were typically 0–10% for DMSO controls and cells treated with compounds showing no specific effect on nuclear size. However, these fractions could reach 20–30% of the cells or even more for the strongest compounds (e.g., oxyphenbutazone, Figure 1B, right plot). Thus, compounds that deregulate the metric beyond the expected variability in our entire dataset are compounds that enrich one or both of those fractions of outlier cells above this typical level defined by DMSO controls and other compounds. To detect these compounds, we proceeded as for the average-based metric: we computed the distributions of each of these fractions of outlier cells across the library, computed the 25, 50, and 75% percentiles that characterize the distributions of those fractions of outlier cells (Q1small/large, Q2small/large, and Q3small/large) and defined hit detection thresholds as Fracsmall > Q2small + 6 × (Q3small – Q2small) and Fraclarge > Q2large + 6 × (Q3large – Q2large).
Hit Confirmation
Hits were confirmed by computing direct compound-vs-DMSO Wilcoxon rank tests, where the multiple well-averaged values of the metric across all DMSO and compound replicates were used to perform the tests. Reported p-values account for variability between plates in well-averaged metric values from both DMSO and compound treatments.
Statistical Analysis of Compound Class Enrichment for Phenotypes of Interest—Class Enrichment Scores
The analysis above revealed clustering of particular compound classes with consistent absolute nuclear size and/or N/C ratio phenotypes across the set of cell line/treatment duration/replicate conditions (i.e., clusters in Figure 3). To estimate the statistical significance of compound class enrichment for any given phenotype, we performed individual compound-vs-entire library Wilcoxon rank tests using data from all replicates. The Wilcoxon p-values obtained from these tests indicate whether a given compound produces the phenotype significantly above the typical level in the library. Compounds showing p-values lower than 0.05 were counted for the entire library (1120 compounds in total) and for each of the particular drug classes identified by the heat maps, i.e., β adrenergic receptor agonists (15 compounds), DNA intercalants (5), replication inhibitors (5), glucocorticoids (26), MAP kinase inhibitors (2), microtubule poisons (9), Na+/K+ ATPase inhibitors (5), 5-HT (serotonin) uptake inhibitors (8), cyclo-oxygenase inhibitors (48), detergents-surfactants (11), antineoplastic agents (25), β adrenergic antagonists (14), and GABA receptor ligand and stimulants (8 compounds). For each class of compounds (encompassing n compounds), the probability that the number k of compounds scored with p < 0.05 is not related to their therapeutic class equals the probability to randomly pick k active compound within a group of n and is given by the hypergeometric law H(N,m,n,k) with parameters N = 1120 and m = total number of active (p < 0.05) compounds for this particular phenotype. Thus, the probability that the compound class is statistically relevant to the observed pattern is 1 – H(N,m,n,k) and defines the class enrichment scores (given in %) reported in the legends of Figures 3, 4, S3, and S4. All compound classes showing characteristic patterns on the heatmap were statistically significant, with class enrichment scores larger than 95%, and often 99%. For antineoplastic agents, class enrichment scores were computed the same way, with the exception that active compounds for all phenotypes were gathered. Using a similar approach for Cox inhibitors yielded a class enrichment score of 93–94% for both the average absolute nuclear size and N/C ratio.
Cheminformatics Analysis
The ChEMBL81 version 27 database was queried using the webresource82 client for Python and dictionaries built mapping Prestwick compound IDs to a SMILES representation of the active compound. This was then in turn used to extract ChEMBL compound IDs for entries matching each of the unique SMILES codes of the Prestwick compounds. These IDs were then used to identify recorded activities to human protein targets within ChEMBL, which had an IC50, KD, MIC, MIC90, EC50, GI50, or potency recorded as less than or equal to 10 μM. Subsequent Python programs were used to query this data in the form of dictionaries stored in the JSON format and interrogate the commonly hit targets. The Python code used to achieve this data curation is available at https://github.com/stevenshave/NuclearSizeCheminformatics. We note that polypharmacological profiles curated and reported in Table S6 are incomplete and skewed toward historically well-explored protein targets and established drugs, as there is not a public record of all human proteins assayed against these compounds. From the incomplete data, we can however reveal proteins meriting further investigation. Gene nodes identified as being targets of cell line-specific NSR with any metric/methodology were first filtered against genome-wide transcriptomics data for our three cell lines (from the Cancer Cell Line Encyclopedia, CCLE, CCLE_RNAseq_rsem_genes_tpm_20180929 dataset) to ensure that networks presented include only genes expressed in the relevant cell line. Proteins targeted by cell line-specific NSR compounds pulled by any of our metrics/analysis methodologies were represented on interaction maps using Cytoscape (Figures 5 and S6). Targets of NSRs shared by at least two cell lines were de facto excluded. Documented proteins interactions were loaded from the iRefIndex human interactome database. GO enrichment analysis was performed using a hypergeometric function-based statistical test in the BINGO Cytoscape plugin. Key enriched GO terms were manually selected on each network, typically eliminating nonspecific GO terms (e.g., “molecular processes”) and rather selecting GO terms represented by 1–500 genes typically in the entire genome. Enrichment p-values were generally less than 10–5, often less than 10–10, possibly emphasized by certain GO terms being enriched in targets of the Prestwick compounds. To quantify chemical scaffolds, ScaffoldHunter83 version 2.6.3 was used on SMILES representations of active molecules and default settings for ring sizes used to profile molecular scaffold counts.
Pilot Screen
To estimate the accuracy of these analysis strategies and assess screen reproducibility, we ran a pilot screen on PC3 cells at the 6 h time point in triplicate (Table S1 and Figure S1). For each hit, obtained using absolute or relative (N/C ratio) nuclear size metrics (rows 1–2 and 3–4, respectively), the distributions of mean metric values (rows 1–3) and the fraction of outlier cells (rows 2–4) across the three replicates (thus, three values per metric–analysis strategy pair) were compared to the similar distributions from all DMSO controls of the collection using a Wilcoxon rank test. These tests yielded for every hit a p-value score that is represented as scatter plots in Figure S1A. Among hits replicated exactly once, twice, or three times, the fraction of hits showing significant (p < 0.05) p-values is shown in Table S1. As a result, ca. 85–100% of the hits that replicated twice and three times had low p-values irrespective of the metric/analysis strategy. A large fraction of hits that scored only once out of three replicates still showed statistical significance upon direct comparison with DMSO controls, in particular for the absolute nuclear size metric analyzed with the average-based strategy. This reflected scores slightly below hit detection thresholds, as can be viewed on scatter plots showing the raw data across replicates for three plates (240 compounds, Figure S1B). The rather low false discovery rate <20% for hits determined using average-based analysis of absolute nuclear size prompted us to downsample larger screens to duplicate screens, and to use this metric and analysis method as the default hit discovery methodology, unless otherwise specified.
Apoptosis Assay
Cells were incubated with each compound at a concentration of 10 μM in standard six-well tissue culture plates for 36 h, refreshing the medium every day. Approximately 106 cells were counted, washed with ice-cold PBS, and stained with 5 μL of the Annexin V apoptosis marker conjugate with the 647-Alexafluor chromophore (Thermo Fisher Scientific) and 5 μL of 50 μg/mL propidium iodide (Biotium) for cell death detection, in 10 mM N-(2-hydroxyethyl)piperazine-N′-ethanesulfonic acid (HEPES), 140 mM NaCl, and 2.5 mM CaCl2, pH 7.4 for 15 min before FACS analysis. The 647 nm Annexin V signal was used for the detection of early stages of apoptosis as cells are not permeable to propidium iodide at this stage. Late-stage apoptosis was detected by the presence of both of the markers and necrotic cells by the presence of propidium iodide signal only.
Viability Assay
For viability analysis, a resazurine-based assay was performed. Metabolically active cells reduce resazurine (a.k.a. alamarBlue) to resorufine, a red and highly fluorescent compound. The cells were seeded onto PE96 plates (PerkinElmer) at a concentration of 5000 cells/well in a final volume of 100 μL and allowed to recover/grow for 24 h before compound addition. The media was then replaced with media containing six serial dilutions of each compound, and the cells were allowed to grow for either 6 or 36 h. alamarBlue (10 μL, Thermo Fisher, 10× stock) was added on each single well and allowed to react for 3 h prior to taking an absorbance reading at 530 nm and fluorescence reading with excitation at 560 nm (substrate excitation 530–570 nm) and emission at 590 nm (substrate emission 580–590 nm) on a microplate reader (JASCO V-550). The fluorescence signal in each well normalized to DMSO control well was used as a readout for cell viability.
Wound Healing Assay
Approximately 25 000 cells per well were seeded on Sartorius 96-well LockView or ImageLock microplates (Sartorius AG) the night prior to wound formation. The cell monolayer was scratched with the IncuCyte WoundMaker that simultaneously makes equivalently sized scratch wounds in the monolayer in all wells, and the medium was replaced with a compound-containing medium supplemented with 1% fetal bovine serum (FBS) to induce cell migration and reduce cell proliferation. Serial dilutions (1 μL) of the compounds in DMSO were added to obtain the 1 nM to 10 μM final concentrations, and the plate was mixed gently. The plates were placed in the IncuCyte incubator and imaged in a bright field with a 10× objective every 3 h for 48 h. Analysis of the wound confluence and width was performed with an automated script in IncuCyte 2021C software provided by Sartorius.
Probabilistic Analysis of the Correlations between Migration Phenotypes (Wound Healing Assays) and Nuclear Size Phenotypes (NSR Status)
To estimate the (frequentist) conditional probability that a given compound at a given concentration shows a migration effect given it is NSR (P(migration|NSR), see below), we grouped the PC3 and H1299 data across all seven tested compounds and concentrations, excluding concentrations where no NSR effects were detectable under any condition (≤10 nM). The nuclear size versus parbendazole concentration profile in H1299 cells was noisy/irregular and henceforth no wound healing assay has been performed under this condition, yielding a total of 39 cell line/compound/concentration conditions (Figure S8A). For each condition, two numbers were computed: the migration index, defined as the % of wound confluence, normalized to the % of wound confluence in the DMSO control (both after 24 h incubation), and the NSR index, defined as the % of average nuclear size decrease compared to DMSO for PC3 cells, and % of nuclear size increase compared to DMSO for H1299 cells. Conditions showing a nuclear size larger (resp. smaller) than the corresponding DMSO control in PC3 (resp. H1299) cells yielded therefore a negative NSR index. Each cell line/compound/concentration yielded a dot in (migration index–NSR index) coordinates (total 39 data points, Figure S8A). The data distribution segregated conditions with migration impairment (migration <80% of DMSO control), nuclear size rectification (>20% downregulation for PC3; upregulation for H1299, compared to DMSO), both phenotypes or no phenotype (Figure S8A). With these definitions, out of 39 conditions, 5 had migration defects but no NSR phenotype, 8 had migration defects and an NSR phenotype, 1 had no migration defect although it had an NSR phenotype, and 25 showed no phenotype. From this, we estimated the following (frequentist) marginal probabilities
and used them to compute the conditional probabilities reported in the Results section
P(migration|NSR) = 0.89 ≫ P(migration|not NSR) = 0.17, P(migration) = 0.33 strongly deviates from the expectation if the migration phenotype was statistically independent of the NSR status of a compound, P(migration|NSR) = P(migration|not NSR) = P(migration). We note that the estimation of the marginal probabilities above was performed on 39 compound/concentration/cell line conditions, which represents only a small fraction of the compound/concentration/cell line sample space.
ABL2 Silencing Assay
PC3, H1299-H2BRFP, and HCT116-H2BRFP cells were reverse-transfected with 20 nM siRNA using INTERFERin transfection reagent (Polyplus Transfection SA, Illkirch, France) under the conditions recommended by the manufacturer. siRNAs, obtained from Thermo Fisher Scientific (Waltham, MA), targeting ABL2 (Silencer Select Pre-designed siRNA, Cat# 4427037, ID: s872/s229383, Ambion). ABL2 silencing upon s872 treatment (ca. 90–100% in 2/3 replicates) was clearly stronger than upon s229383 treatment (∼75%) in PC3, as quantified by qPCR (Figure S8C). Therefore, s872 (referred to as siABL2 in the manuscript) was selected for wound healing and nuclear size assays in PC3 and H1299. Silencer Select Negative Control 1 (Cat# 4390843, Ambion) was used as a control for off-target effects. Wound healing assays were performed and analyzed as described above, with cells transfected with siRNAs 48 h prior to wound healing and Incucyte imaging. Wound confluence at the first time point (3 h) was subtracted to the confluence at all other time points to obtain the data represented in Figures 8C and S8E. The wound width was normalized to the width at the first time point and expressed in %. The nuclear size of the migrating PC3 cells was analyzed manually from Incucyte images in ImageJ, to make sure that the same cell cultures were assessed for nuclear size and migration. As an additional control, nuclear size of PC3-H2BRFP cells plated on coverslips and treated with siCtrl and siABL2 for 48 h prior to fixation and staining with CellMask DeepRed was measured using fluorescent imaging on a Leica Thunder Imager 3D tissue slide scanner at the Cell and Tissue Imaging Unit, Institute of Biomedicine, University of Eastern Finland (Figure S8D). Nuclear size analysis was performed automatically using custom Matlab scripts (Mathworks), and statistics were performed in Microsoft Excel.
Microfluidics Migration Assay
PC3 cells were seeded into microfluidic chambers consisting of migration channels with a fixed height of 5 μm and constrictions of 1–2 or 15 μm in width, coated with a solution of 5 μg/mL of fibronectin 24 h prior to experiments as described previously.84,85 A total of 30 000 cells were seeded into each device 6 h prior to imaging and were treated with 10 μM piperlonguine, digitoxigenin, or equivalent DMSO. Imaging was performed on a Zeiss LSM700 laser scanning confocal microscope with a 20× air objective. The cells were imaged at 10 min intervals in a temperature-controlled stage (37 °C) for 14 h. The time required for cells to migrate through an individual constriction was quantified using a previously described MATLAB script for measuring nuclear transit through these microfluidic devices.86
Invasion Assay
The QEM endothelial cell invasion assay was carried out in accordance with manufacturer’s recommendations (ECM210, Millipore). The cells were seeded at a concentration of 105 on the inside Boyden chamber in the absence of FBS on a 3 μm pore membrane coated with ECM proteins and submerged on the outside Boyden chamber in the presence of conditioned media and 10% FBS as chemoattractant. The cells were allowed to migrate through the membrane for 24 h in the presence of compounds at the 10 μM concentration used in the size screen, prior to fixation and staining of the lower part of the membrane. Noninvading cells were removed with a cotton stab from the top of the membrane, and the cells were lysed. Absorbance values of the cell lysate were analyzed on a microplate reader (JASCO V-550) with a 540 nm wavelength, to quantify the relative number of migrating cells by colorimetry.
Acknowledgments
The authors thank E. Jarman and B. Hormann for initiating pilot studies as part of their undergraduate Honours and M.Sc. diplomas, respectively. They also thank the McGill University core facility for allowing them to perform some imaging on their Opera Phenix system and the Institute for Research in Immunology and Cancer, University of Montreal, High Throughput Core Facility for general support. They thank the Cell and Tissue Imaging Unit at the Institute of Biomedicine, University of Eastern Finland, for assistance with imaging of siRNA-treated cells.
Glossary
List of Abbreviations
- FDA
Food and Drug Administration
- EMA
European Medicines Agency
- N/C ratio
nuclear-to-cytoplasm ratio
- LEM2
LAP2-Emerin-Man1 domain-containing protein 2
- LINC
linker of nucleoskeleton and cytoskeleton
- PAD
previously approved drugs
- mRFP
monomeric red fluorescent protein
- DMSO
dimethyl sulfoxide
- NSR
nuclear size rectifier
- BAA
β-adrenergic receptor agonist
- 5-HT
5-hydroxytryptamine (serotonin)
- GABA
γ-aminobutyric acid
- FACS
fluorescence-activated cell sorting
- FBS
fetal bovine serum
- ROS
reactive oxygen species
- SSRI
selective serotonin reuptake inhibitor
- SEM
standard error on the mean
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.2c00004.
Additional methods; statistics and detailed listings of all compounds identified by average-based and outlier-based approaches; screen data reproducibility (Figure S1); analysis methodology, but not the choice of metric, yielded overlapping results (Figure S2); comparison of absolute and relative nuclear size responses to different treatment duration informs on compound mechanism of action (Figure S3); on the importance of knowing which of absolute or relative nuclear size actively contributes to metastasis (Figure S4); individual compounds spanning many pharmacological classes can correct cancer-associated nuclear size changes (Figure S5); cell line-specific NSRs target specific protein networks, additional data (Figure S6); NSR compounds affecting nuclear size also affect cell migration, additional data (Figure S7); nuclear size rectification is a good predictor of cell migration impairment (Figure S8); model for how nuclear size changes may affect cancer cell invasiveness (Figure S9); summary table showing the fraction of hits replicated once, twice or three times, using the two metrics and two analysis strategies introduced in the main text (Table S1); compounds tested in detail with chemical structures (Table S7) (PDF)
Effects across cell lines and duration of treatment of compounds identified as affecting the average absolute nuclear size in at least one condition (Table S2) (XLSX)
Effects across cell lines and duration of treatment of compounds identified as affecting the average N/C ratio in at least one condition (Table S3) (XLSX)
Effects across cell lines and duration of treatment of compounds identified as affecting the fractions of outlier cells with abnormally large or small nucleus in at least one condition (Table S4) (XLSX)
Effects across cell lines and duration of treatment of compounds identified as affecting the fractions of outlier cells with abnormally large or small N/C ratio in at least one condition (Table S5) (XLSX)
Targets of NSR compounds (Table S6) (XLSX)
Author Present Address
∇ University of Manchester, Manchester M13 9PL, U.K
Author Contributions
○ S.T. and A.R. contributed equally to this work. S.T., A.R., M.T., E.C.S. conceived/designed experiments. A.R., S.T., S.K., A.S., N.T.P., N.Z., J.T.K., J.C., and Y.Z. performed experiments. S.T., S.S., J.W., N.T.P., J.L., M.A., N.O.C., V.G.B., L.L., and M.T. provided analysis tools and scientific input. S.T., A.R., L.L., M.T., and E.C.S. wrote the manuscript. All authors contributed to editing the manuscript.
This work was principally supported by Wellcome grants 095209 (E.C.S.) and 085178 (M.T.), the European Research Council SCG-233457 (M.T.), the Canadian Institutes of Health Research FDN-167277 (M.T.), a Canada Research Chair in Systems and Synthetic Biology (M.T.), and Medical Research Council MR/R018073/1 to E.C.S. This work was also supported by the Sigrid Jusélius Foundation (S.T., L.L.), the Cancer Society of Finland (L.L.), the Foundation of the Finnish Cancer Institute (L.L.), and the Academy of Finland 317871 (L.L.). It was also supported by Wellcome Centre for Cell Biology 092076, the National Institutes of Health R01GM137605, R01HL082792, and U54CA210184 (J.L.), the Department of Defense Breast Cancer Research Program Breakthrough Award BC150580 (J.L.), and the National Science Foundation CAREER Award CBET-1254846 (J.L.). A.R. was supported by a Darwin Trust Studentship and J.T.K. from the Knight@KIC 2018–2019 Graduate Fellowship. This work was performed in part at the Cornell NanoScale Science & Technology Facility (CNF), a member of the National Nanotechnology Coordinated Infrastructure (NNCI), which is supported by the National Science Foundation (Grant NNCI-1542081).
The authors declare no competing financial interest.
Notes
All authors are consenting to publication of these results in the current form.
Notes
Data generated or analyzed during the current study are included in the published manuscript and its Supporting Information files.
Supplementary Material
References
- Beale L. S. Examination of sputum from a case of cancer of the pharynx and the adjacent parts. Arch. Med. 1860, 2, 1860. [Google Scholar]
- de Las Heras J. I.; Batrakou D. G.; Schirmer E. C. Cancer biology and the nuclear envelope: A convoluted relationship. Semin. Cancer Biol. 2013, 23, 125–137. 10.1016/j.semcancer.2012.01.008. [DOI] [PubMed] [Google Scholar]
- de Las Heras J. I.; Schirmer E. C.. The nuclear envelope and cancer: a diagnostic perspective and historical overview. Cancer Biology and the Nuclear Envelope; Advances in Experimental Medicine and Biology; Springer, 2014; Vol. 773, pp 5–26. [DOI] [PubMed] [Google Scholar]
- Zink D.; Fischer A. H.; Nickerson J. A. Nuclear structure in cancer cells. Nat. Rev. Cancer 2004, 4, 677–687. 10.1038/nrc1430. [DOI] [PubMed] [Google Scholar]
- de Andrea C. E.; Petrilli A. S.; Jesus-Garcia R.; Bleggi-Torres L. F.; Alves M. T. Large and round tumor nuclei in osteosarcoma: good clinical outcome. Int. J. Clin. Exp. Pathol. 2011, 4, 169–174. [PMC free article] [PubMed] [Google Scholar]
- Ladekarl M.; Boek-Hansen T.; Henrik-Nielsen R.; Mouritzen C.; Henriques U.; Sorensen F. B. Objective malignancy grading of squamous cell carcinoma of the lung. Stereologic estimates of mean nuclear size are of prognostic value, independent of clinical stage of disease. Cancer 1995, 76, 797–802. . [DOI] [PubMed] [Google Scholar]
- Chen L. Cytology: diagnostic principles and clinical correlates. Am. J. Surg. Pathol. 2010, 34, 286. 10.1097/PAS.0b013e3181cc7ca3. [DOI] [Google Scholar]
- Cavalier-Smith T. Economy, speed and size matter: evolutionary forces driving nuclear genome miniaturization and expansion. Ann. Bot. 2005, 95, 147–175. 10.1093/aob/mci010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edens L. J.; White K. H.; Jevtic P.; Li X.; Levy D. L. Nuclear size regulation: from single cells to development and disease. Trends Cell Biol. 2013, 23, 151–159. 10.1016/j.tcb.2012.11.004. [DOI] [PubMed] [Google Scholar]
- Fidorra J.; Mielke T.; Booz J.; Feinendegen L. E. Cellular and nuclear volume of human cells during the cell cycle. Radiat. Environ. Biophys. 1981, 19, 205–214. 10.1007/BF01324188. [DOI] [PubMed] [Google Scholar]
- Jorgensen P.; Edgington N. P.; Schneider B. L.; Rupes I.; Tyers M.; Futcher B. The size of the nucleus increases as yeast cells grow. Mol. Biol. Cell 2007, 18, 3523–3532. 10.1091/mbc.e06-10-0973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann F. R.; Nurse P. Nuclear size control in fission yeast. J. Cell Biol. 2007, 179, 593–600. 10.1083/jcb.200708054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jevtić P.; Edens L. J.; Li X.; Nguyen T.; Chen P.; Levy D. L. Concentration-dependent Effects of Nuclear Lamins on Nuclear Size in Xenopus and Mammalian Cells. J. Biol. Chem. 2015, 290, 27557–27571. 10.1074/jbc.M115.673798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W.; Schneider M.; Neumann S.; Jaeger V. M.; Taranum S.; Munck M.; Cartwright S.; Richardson C.; Carthew J.; Noh K.; et al. Nesprin interchain associations control nuclear size. Cell. Mol. Life Sci. 2012, 69, 3493–3509. 10.1007/s00018-012-1034-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kume K.; Cantwell H.; Burrell A.; Nurse P. Nuclear membrane protein Lem2 regulates nuclear size through membrane flow. Nat. Commun. 2019, 10, 1871 10.1038/s41467-019-09623-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachner A.; Reipert S.; Foisner R.; Gotzmann J. LEM2 is a novel MAN1-related inner nuclear membrane protein associated with A-type lamins. J. Cell Sci. 2005, 118, 5797–5810. 10.1242/jcs.02701. [DOI] [PubMed] [Google Scholar]
- Hirano Y.; Kinugasa Y.; Asakawa H.; Chikashige Y.; Obuse C.; Haraguchi T.; Hiraoka Y. Lem2 is retained at the nuclear envelope through its interaction with Bqt4 in fission yeast. Genes Cells 2018, 23, 122–135. 10.1111/gtc.12557. [DOI] [PubMed] [Google Scholar]
- Huber M. D.; Guan T.; Gerace L. Overlapping functions of nuclear envelope proteins NET25 (Lem2) and emerin in regulation of extracellular signal-regulated kinase signaling in myoblast differentiation. Mol. Cell. Biol. 2009, 29, 5718–5728. 10.1128/MCB.00270-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantwell H.; Nurse P. A systematic genetic screen identifies essential factors involved in nuclear size control. PLoS Genet. 2019, 15, e1007929 10.1371/journal.pgen.1007929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantwell H.; Nurse P. Unravelling nuclear size control. Curr. Genet. 2019, 1281–1285. 10.1007/s00294-019-00999-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jevtić P.; Schibler A. C.; Wesley C. C.; Pegoraro G.; Misteli T.; Levy D. L. The nucleoporin ELYS regulates nuclear size by controlling NPC number and nuclear import capacity. EMBO Rep. 2019, 20, e47283 10.15252/embr.201847283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. S.; Hale C. M.; Panorchan P.; Khatau S. B.; George J. P.; Tseng Y.; Stewart C. L.; Hodzic D.; Wirtz D. Nuclear lamin A/C deficiency induces defects in cell mechanics, polarization, and migration. Biophys. J. 2007, 93, 2542–2552. 10.1529/biophysj.106.102426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirtz D.; Konstantopoulos K.; Searson P. C. The physics of cancer: the role of physical interactions and mechanical forces in metastasis. Nat. Rev. Cancer 2011, 11, 512–522. 10.1038/nrc3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraning-Rush C. M.; Califano J. P.; Reinhart-King C. A. Cellular traction stresses increase with increasing metastatic potential. PLoS One 2012, 7, e32572 10.1371/journal.pone.0032572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussolati G.; Maletta F.; Asioli S.; Annaratone L.; Sapino A.; Marchio C.. “To Be or Not to Be in a Good Shape”: Diagnostic and Clinical Value of Nuclear Shape Irregularities in Thyroid and Breast Cancer. Cancer Biology and the Nuclear Envelope; Advances in Experimental Medicine and Biology; Springer, 2014; Vol. 773, pp 101–121. [DOI] [PubMed] [Google Scholar]
- Veltri R. W.; Christudass C. S.. Nuclear Morphometry, Epigenetic Changes, and Clinical Relevance In Prostate Cancer. Cancer Biology and the Nuclear Envelope; Advances in Experimental Medicine and Biology; Springer, 2014; Vol. 773, pp 77–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pufall M. A.Glucocorticoids and Cancer. Glucocorticoid Signaling; Advances in Experimental Medicine and Biology; Springer, 2015; Vol. 872, pp 315–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. K.; Louhghalam A.; Lee G.; Schafer B. W.; Wirtz D.; Kim D. H. Nuclear lamin A/C harnesses the perinuclear apical actin cables to protect nuclear morphology. Nat. Commun. 2017, 8, 2123 10.1038/s41467-017-02217-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bausch A. R.; Ziemann F.; Boulbitch A. A.; Jacobson K.; Sackmann E. Local measurements of viscoelastic parameters of adherent cell surfaces by magnetic bead microrheometry. Biophys. J. 1998, 75, 2038–2049. 10.1016/S0006-3495(98)77646-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harshbarger W.; Gondi S.; Ficarro S. B.; Hunter J.; Udayakumar D.; Gurbani D.; Singer W. D.; Liu Y.; Li L.; Marto J. A.; Westover K. D. Structural and Biochemical Analyses Reveal the Mechanism of Glutathione S-Transferase Pi 1 Inhibition by the Anti-cancer Compound Piperlongumine. J. Biol. Chem. 2017, 292, 112–120. 10.1074/jbc.M116.750299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lautscham L. A.; Kammerer C.; Lange J. R.; Kolb T.; Mark C.; Schilling A.; Strissel P. L.; Strick R.; Gluth C.; Rowat A. C.; et al. Migration in Confined 3D Environments Is Determined by a Combination of Adhesiveness, Nuclear Volume, Contractility, and Cell Stiffness. Biophys. J. 2015, 109, 900–913. 10.1016/j.bpj.2015.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson P. M.; Denais C.; Bakshi M. C.; Lammerding J. Nuclear deformability constitutes a rate-limiting step during cell migration in 3-D environments. Cell. Mol. Bioeng. 2014, 7, 293–306. 10.1007/s12195-014-0342-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner C. A.; Ehrlicher A.; Kohlstrunk B.; Knebel D.; Kas J. A.; Goegler M. Cell migration through small gaps. Eur. Biophys. J. 2006, 35, 713–719. 10.1007/s00249-006-0079-1. [DOI] [PubMed] [Google Scholar]
- Almaguer G.; Ortiz-Vilchis P.; Cordero P.; Martinez-Vega R.; Perez-Duran J.; Meaney E.; Villarreal F.; Ceballos G.; Najera N. Anticancer potential of (-)-epicatechin in a triple-negative mammary gland model. J. Pharm. Pharmacol. 2021, 73, 1675–1682. 10.1093/jpp/rgab133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdulkhaleq L. A.; Assi M. A.; Noor M. H. M.; Abdullah R.; Saad M. Z.; Taufiq-Yap Y. H. Therapeutic uses of epicatechin in diabetes and cancer. Vet. World 2017, 10, 869–872. 10.14202/vetworld.2017.869-872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell E. S.; Lammerding J. Causes and consequences of nuclear envelope alterations in tumour progression. Eur. J. Cell Biol. 2016, 95, 449–464. 10.1016/j.ejcb.2016.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crisp M.; Liu Q.; Roux K.; Rattner J. B.; Shanahan C.; Burke B.; Stahl P. D.; Hodzic D. Coupling of the nucleus and cytoplasm: role of the LINC complex. J. Cell Biol. 2006, 172, 41–53. 10.1083/jcb.200509124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause M.; Yang F. W.; Te Lindert M.; Isermann P.; Schepens J.; Maas R. J. A.; Venkataraman C.; Lammerding J.; Madzvamuse A.; Hendriks W.; et al. Cell migration through three-dimensional confining pores: speed accelerations by deformation and recoil of the nucleus. Philos. Trans. R. Soc., B 2019, 374, 20180225 10.1098/rstb.2018.0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ananthakrishnan R.; Ehrlicher A. The forces behind cell movement. Int. J. Biol. Sci. 2007, 3, 303–317. 10.7150/ijbs.3.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K.; Lim H. Y.; Shi S.; Lee J.; Deng S.; Xie T.; Zhu Z.; Wang Y.; Pocalyko D.; Yang W. J.; et al. Genomic landscape of copy number aberrations enables the identification of oncogenic drivers in hepatocellular carcinoma. Hepatology 2013, 58, 706–717. 10.1002/hep.26402. [DOI] [PubMed] [Google Scholar]
- Xing Q. T.; Qu C. M.; Wang G. Overexpression of Abl2 predicts poor prognosis in hepatocellular carcinomas and is associated with cancer cell migration and invasion. OncoTargets Ther. 2014, 7, 881–885. 10.2147/OTT.S62348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil-Henn H.; Patsialou A.; Wang Y.; Warren M. S.; Condeelis J. S.; Koleske A. J. Arg/Abl2 promotes invasion and attenuates proliferation of breast cancer in vivo. Oncogene 2013, 32, 2622–2630. 10.1038/onc.2012.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang X. F.; Zhang Z. W.; Liu Q.; Sun N.; Pan L. L.; Shen J.; Li T.; Yun C.; Li H.; Shi L. H. miR-20a promotes prostate cancer invasion and migration through targeting ABL2. J. Cell. Biochem. 2014, 115, 1269–1276. 10.1002/jcb.24778. [DOI] [PubMed] [Google Scholar]
- Stoen M. J.; Andersen S.; Rakaee M.; Pedersen M. I.; Ingebriktsen L. M.; Donnem T.; Lombardi A. P. G.; Kilvaer T. K.; Busund L. R.; Richardsen E. Overexpression of miR-20a-5p in Tumor Epithelium Is an Independent Negative Prognostic Indicator in Prostate Cancer-A Multi-Institutional Study. Cancers 2021, 13, 4096 10.3390/cancers13164096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L.; Guo Y.; Wu Z.; Zhao S.; Zhang Z.; Zheng F.; Sun L.; Hao Z.; Xu C.; Wang T.; Peng Y. Epicatechin gallate prevents the de novo synthesis of fatty acid and the migration of prostate cancer cells. Acta Biochim. Biophys. Sin. 2021, 53, 1662–1669. 10.1093/abbs/gmab144. [DOI] [PubMed] [Google Scholar]
- Singh T.; Katiyar S. K. Green tea catechins reduce invasive potential of human melanoma cells by targeting COX-2, PGE2 receptors and epithelial-to-mesenchymal transition. PLoS One 2011, 6, e25224 10.1371/journal.pone.0025224. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Melmed R. N.; Karanian P. J.; Berlin R. D. Control of cell volume in the J774 macrophage by microtubule disassembly and cyclic AMP. J. Cell Biol. 1981, 90, 761–768. 10.1083/jcb.90.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y.; Stewart A. G.; Lee P. V. S. On-chip cell mechanophenotyping using phase modulated surface acoustic wave. Biomicrofluidics 2019, 13, 024107 10.1063/1.5084297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleem S.; Khan R.; Afzal M.; Kazmi I. Oxyphenbutazone promotes cytotoxicity in rats and Hep3B cellsvia suppression of PGE2 and deactivation of Wnt/beta-catenin signaling pathway. Mol. Cell. Biochem. 2018, 444, 187–196. 10.1007/s11010-017-3243-2. [DOI] [PubMed] [Google Scholar]
- Schneider N. F. Z.; Persich L.; Rocha S. C.; Ramos A. C. P.; Cortes V. F.; Silva I. T.; Munkert J.; Padua R. M.; Kreis W.; Taranto A. G.; et al. Cytotoxic and cytostatic effects of digitoxigenin monodigitoxoside (DGX) in human lung cancer cells and its link to Na,K-ATPase. Biomed. Pharmacother. 2018, 97, 684–696. 10.1016/j.biopha.2017.10.128. [DOI] [PubMed] [Google Scholar]
- Fagone P.; Caltabiano R.; Russo A.; Lupo G.; Anfuso C. D.; Basile M. S.; Longo A.; Nicoletti F.; De Pasquale R.; Libra M.; Reibaldi M. Identification of novel chemotherapeutic strategies for metastatic uveal melanoma. Sci. Rep. 2017, 7, 44564 10.1038/srep44564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva I. T.; Munkert J.; Nolte E.; Schneider N. F. Z.; Rocha S. C.; Ramos A. C. P.; Kreis W.; Braga F. C.; de Padua R. M.; Taranto A. G.; et al. Cytotoxicity of AMANTADIG - a semisynthetic digitoxigenin derivative - alone and in combination with docetaxel in human hormone-refractory prostate cancer cells and its effect on Na(+)/K(+)-ATPase inhibition. Biomed. Pharmacother. 2018, 107, 464–474. 10.1016/j.biopha.2018.08.028. [DOI] [PubMed] [Google Scholar]
- Zhou L.; Li M.; Yu X.; Gao F.; Li W. Repression of Hexokinases II-Mediated Glycolysis Contributes to Piperlongumine-Induced Tumor Suppression in Non-Small Cell Lung Cancer Cells. Int. J. Biol. Sci. 2019, 15, 826–837. 10.7150/ijbs.31749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z.; Shi Z.; Lin J.; Zhao S.; Hao M.; Xu J.; Li Y.; Zhao Q.; Tao L.; Diao A. Piperlongumine-induced nuclear translocation of the FOXO3A transcription factor triggers BIM-mediated apoptosis in cancer cells. Biochem. Pharmacol. 2019, 163, 101–110. 10.1016/j.bcp.2019.02.012. [DOI] [PubMed] [Google Scholar]
- Mgbeahuruike E. E.; Stalnacke M.; Vuorela H.; Holm Y. Antimicrobial and Synergistic Effects of Commercial Piperine and Piperlongumine in Combination with Conventional Antimicrobials. Antibiotics 2019, 8, 55 10.3390/antibiotics8020055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Chang J.; Liu X.; Zhang X.; Zhang S.; Zhang X.; Zhou D.; Zheng G. Discovery of piperlongumine as a potential novel lead for the development of senolytic agents. Aging 2016, 8, 2915–2926. 10.18632/aging.101100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharadwaj U.; Eckols T. K.; Kolosov M.; Kasembeli M. M.; Adam A.; Torres D.; Zhang X.; Dobrolecki L. E.; Wei W.; Lewis M. T.; et al. Drug-repositioning screening identified piperlongumine as a direct STAT3 inhibitor with potent activity against breast cancer. Oncogene 2015, 34, 1341–1353. 10.1038/onc.2014.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj L.; Ide T.; Gurkar A. U.; Foley M.; Schenone M.; Li X.; Tolliday N. J.; Golub T. R.; Carr S. A.; Shamji A. F.; et al. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature 2011, 475, 231–234. 10.1038/nature10167. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Jeong C. H.; Ryu H.; Kim D. H.; Cheng W. N.; Yoon J. E.; Kang S.; Han S. G. Piperlongumine Induces Cell Cycle Arrest via Reactive Oxygen Species Accumulation and IKKbeta Suppression in Human Breast Cancer Cells. Antioxidants 2019, 8, 553 10.3390/antiox8110553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q.; Chen W.; Lv X.; Weng Q.; Chen M.; Cui R.; Liang G.; Ji J. Piperlongumine, a Novel TrxR1 Inhibitor, Induces Apoptosis in Hepatocellular Carcinoma Cells by ROS-Mediated ER Stress. Front. Pharmacol. 2019, 10, 1180 10.3389/fphar.2019.01180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketola K.; Vainio P.; Fey V.; Kallioniemi O.; Iljin K. Monensin is a potent inducer of oxidative stress and inhibitor of androgen signaling leading to apoptosis in prostate cancer cells. Mol. Cancer Ther. 2010, 9, 3175–3185. 10.1158/1535-7163.MCT-10-0368. [DOI] [PubMed] [Google Scholar]
- Iljin K.; Ketola K.; Vainio P.; Halonen P.; Kohonen P.; Fey V.; Grafstrom R. C.; Perala M.; Kallioniemi O. High-throughput cell-based screening of 4910 known drugs and drug-like small molecules identifies disulfiram as an inhibitor of prostate cancer cell growth. Clin. Cancer Res. 2009, 15, 6070–6078. 10.1158/1078-0432.CCR-09-1035. [DOI] [PubMed] [Google Scholar]
- Sanchez C.; Reines E. H.; Montgomery S. A. A comparative review of escitalopram, paroxetine, and sertraline: Are they all alike?. Int. Clin. Psychopharmacol. 2014, 29, 185–196. 10.1097/YIC.0000000000000023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Then C. K.; Liu K. H.; Liao M. H.; Chung K. H.; Wang J. Y.; Shen S. C. Antidepressants, sertraline and paroxetine, increase calcium influx and induce mitochondrial damage-mediated apoptosis of astrocytes. Oncotarget 2017, 8, 115490–115502. 10.18632/oncotarget.23302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasanovic A.; Simsir M.; Choveau F. S.; Lalli E.; Mus-Veteau I. Astemizole Sensitizes Adrenocortical Carcinoma Cells to Doxorubicin by Inhibiting Patched Drug Efflux Activity. Biomedicines 2020, 8, 251 10.3390/biomedicines8080251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bens A.; Dehlendorff C.; Friis S.; Cronin-Fenton D.; Jensen M. B.; Ejlertsen B.; Lash T. L.; Kroman N.; Mellemkjaer L. The role of H1 antihistamines in contralateral breast cancer: a Danish nationwide cohort study. Br. J. Cancer 2020, 122, 1102–1108. 10.1038/s41416-020-0747-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegaard A. M.; Dehlendorff C.; Vind A. C.; Anand A.; Cederkvist L.; Petersen N. H. T.; Nylandsted J.; Stenvang J.; Mellemgaard A.; Osterlind K.; et al. Repurposing Cationic Amphiphilic Antihistamines for Cancer Treatment. EBioMedicine 2016, 9, 130–139. 10.1016/j.ebiom.2016.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q.; Wang G.; Li Q.; Jiang W.; Kim J. S.; Wang R.; Zhu S.; Wang X.; Yan L.; Yi Y.; et al. Polycomb group proteins EZH2 and EED directly regulate androgen receptor in advanced prostate cancer. Int. J. Cancer 2019, 145, 415–426. 10.1002/ijc.32118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havercroft J. C.; Quinlan R. A.; Gull K. Binding of parbendazole to tubulin and its influence on microtubules in tissue-culture cells as revealed by immunofluorescence microscopy. J. Cell Sci. 1981, 49, 195–204. 10.1242/jcs.49.1.195. [DOI] [PubMed] [Google Scholar]
- Quinlan R. A.; Roobol A.; Pogson C. I.; Gull K. A correlation between in vivo and in vitro effects of the microtubule inhibitors colchicine, parbendazole and nocodazole on myxamoebae of Physarum polycephalum. J. Gen. Microbiol. 1981, 122, 1–6. 10.1099/00221287-122-1-1. [DOI] [PubMed] [Google Scholar]
- Kainuma K.; Kobayashi T.; D’Alessandro-Gabazza C. N.; Toda M.; Yasuma T.; Nishihama K.; Fujimoto H.; Kuwabara Y.; Hosoki K.; Nagao M.; et al. beta2 adrenergic agonist suppresses eosinophil-induced epithelial-to-mesenchymal transition of bronchial epithelial cells. Respir. Res. 2017, 18, 79 10.1186/s12931-017-0563-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P.; Yan H.; Wang S.; Kai J.; Pi G.; Peng Y.; Liu X.; Sun J. Post-radiotherapy maintenance treatment with fluticasone propionate and salmeterol for lung cancer patients with grade III radiation pneumonitis: A case report. Medicine 2018, 97, e10681 10.1097/MD.0000000000010681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuglu M. M.; Bostanabad S. Y.; Ozyon G.; Dalkilic B.; Gurdal H. The role of dual specificity phosphatase 1 and protein phosphatase 1 in beta2adrenergic receptormediated inhibition of extracellular signal regulated kinase 1/2 in triple negative breast cancer cell lines. Mol. Med. Rep. 2018, 17, 2033–2043. 10.3892/mmr.2017.8092. [DOI] [PubMed] [Google Scholar]
- Wnorowski A.; Such J.; Paul R. K.; Wersto R. P.; Indig F. E.; Jozwiak K.; Bernier M.; Wainer I. W. Concurrent activation of beta2-adrenergic receptor and blockage of GPR55 disrupts pro-oncogenic signaling in glioma cells. Cell. Signalling 2017, 36, 176–188. 10.1016/j.cellsig.2017.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaulton A.; Bellis L. J.; Bento A. P.; Chambers J.; Davies M.; Hersey A.; Light Y.; McGlinchey S.; Michalovich D.; Al-Lazikani B.; Overington J. P. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies M.; Nowotka M.; Papadatos G.; Dedman N.; Gaulton A.; Atkinson F.; Bellis L.; Overington J. P. ChEMBL web services: streamlining access to drug discovery data and utilities. Nucleic Acids Res. 2015, 43, W612–W620. 10.1093/nar/gkv352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetzel S.; Klein K.; Renner S.; Rauh D.; Oprea T.; Mutzel P.; Waldemann H. Interactive exploration of chemical space with Scaffold Hunter. Nat. Chem. Biol. 2009, 5, 581–583. 10.1038/nchembio.187. [DOI] [PubMed] [Google Scholar]
- Davidson P. M.; Sliz J.; Isermann P.; Denais C.; Lammerding J. Design of a microfluidic device to quantify dynamic intra-nuclear deformation during cell migration through confining environments. Integr. Biol. 2015, 7, 1534–1546. 10.1039/C5IB00200A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keys J.; Windsor A.; Lammerding J.. Assembly and Use of a Microfluidic Device to Study Cell Migration in Confined Environments: Methods and Protocols. The LINC Complex; Methods in Molecular Biology; Humana Press, 2018; Vol. 1840, pp 101–118. [DOI] [PubMed] [Google Scholar]
- Elacqua J. J.; McGregor A. L.; Lammerding J. Automated analysis of cell migration and nuclear envelope rupture in confined environments. PLoS One 2018, 13, e0195664 10.1371/journal.pone.0195664. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.