

Abstract

In this study, we successfully synthesized boranophosphate (PB), phosphorothioate (PS), and phosphate (PO) chimeric oligonucleotides (ODNs) as a candidate for the antisense oligonucleotides (ASOs). The PB/PS/PO-ODNs were synthesized utilizing H-boranophosphonate, H-phosphonothioate, and H-phosphonate monomers. Each monomer was condensed with a hydroxy group to create H-boranophosphonate, H-phosphonothioate, and H-phosphonate diester linkages, which were oxidized into PB, PS, and PO linkages in the final stage of the synthesis, respectively. As for condensation of an H-phosphonothioate monomer, regulating chemoselectivity was necessary since the monomer has two nucleophilic centers: S and O atoms. To deal with this problem, we used phosphonium-type condensing reagents, which could control the chemoselectivity. In this strategy, we could synthesize PB/PS/PO oligomers, including a 2′-OMe gapmer-type dodecamer. The physiological and biological properties of the synthesized chimeric ODNs were also evaluated. Insights from the evaluation of physiological and biological properties suggested that the introduction of suitable P-modification and sugar modification at proper sites of ODNs would control the duplex stability, nuclease resistance, RNase H-inducing ability, and one base mismatch discrimination ability, which are critical properties as potent ASOs.
Introduction
Many efforts have been devoted to the development of antisense oligonucleotides (ASOs) since it is demonstrated that an oligonucleotide that is complementary to a target mRNA could control the translation of mRNA into a protein.1,2 There are two kinds of ASOs according to a mechanism of translation regulation, namely, a steric blocking type and an RNase H-dependent type. RNase H is an endonuclease located mainly in the nucleus, recognizes DNA/RNA duplexes, and selectively cleaves the RNA strand.3,4 Required properties for potent RNase H-dependent ASO include high nuclease resistance, duplex stability, RNase H-inducing ability, high retention in tissues, and low cytotoxicity. Since the properties of ASOs can be modulated by introducing chemical modifications to phosphate (PO) moieties, sugar, and the nucleobase of nucleotides,5 many groups have investigated a wide variety of modifications. A phosphorothioate (PS) backbone, in which one of the nonbridging oxygen atoms is replaced with a sulfur atom, is the most used chemical modification applied to ASOs. PS is still widely used as a chemically modified analogue owing to its high nuclease resistance and the facility of its synthesis. In addition to this, it has been demonstrated that PS linkages are crucial for improving the pharmacokinetics of ASOs due to their high affinity with certain kinds of protein such as serum albumin.6,7 For example, it has been found that PS linkages prevent ASOs from glomerular filtration clearance, which led to the extension of blood retention. In addition to this, ASOs containing PS linkages bind specific proteins outside the cell, which leads to the uptake of ASOs into cells,8 and intracellular proteins determining the intracellular distribution of ASOs.9 However, it has been reported that PS linkages reduce duplex stabilities of ASOs with target mRNAs. Moreover, some PS oligonucleotides (ODNs) are cytotoxic and trigger undesired immunoresponse events, which are major obstacles for clinical trials.10,11
To improve duplex stability, nucleotides with sugar modifications have been introduced, such as 2′-O-modifications including 2′-OMe and 2′-O-MOE,12 and locked nucleic acids (LNAs).13−15 These modifications are typically used for gapmer-type ASOs, namely, the nucleotides with a sugar modification are placed in the 5′- and 3′-end (wing) regions of an ASO to acquire a duplex stability with a complementary RNA while deoxyribonucleotides are located in the central (gap) region to maintain an RNase H-inducing ability16 Some ASOs that were approved by the Food and Drug Administration (FDA) contain both PS linkages and sugar modifications.17 This suggests that a combination of P-modifications and sugar modifications dramatically improves their properties as ASOs. Alternatively, although some reports suggested that 2′-O-MOE gapmers containing PS linkages lower their cytotoxicity,18 further suppression of cytotoxicity would be needed for the more secured ASOs.
Under these circumstances, boranophosphate (PB), in which a nonbridging oxygen atom of a PO linkage is replaced with a borano group, has received a lot of attention as another promising ASO candidate since PB modification offers higher nuclease resistance than the PS counterpart19 and exhibits low cytotoxicity.20,21 However, a full PB modification reduces the duplex stability of ASOs with target mRNAs and RNase H-inducing activities.19,22−24
To overcome these problems, Caruthers et al.24−26 and our group27 introduced both PB and PO linkages in ODNs (PB/PO chimeric ODNs). PB/PO chimeric ODNs have improved duplex stability and some of them show higher RNase H-inducing activity compared to the fully PB-modified counterparts. Notably, PB/PO chimeric ODNs have substantial nuclease resistance.25 These indicate that P-modified chimeric ODNs can take advantage of each other’s strength. This strategy utilizing chimeric ODNs also works for ODNs containing PS linkages. The research from IONIS Pharmaceuticals, Inc. revealed that replacing a few PS linkages with alkylphosphonate linkages significantly reduces toxicity while maintaining the antisense activity of ASOs.28 Furthermore, replacing a few PS linkage with mesylphosphoramidate29 linkages can also suppress toxicity and in certain cases improve the antisense activity of ASOs.30 Taking that into consideration, we expected that introducing PB and PO linkages to PS-ODNs (PB/PS/PO and/or PB/PS chimeric ODNs) would be a promising way to modulate cytotoxicity along with maintaining the favorable pharmacokinetics of ODNs.
Although PB derivatives have been viewed as promising ASO candidates, examples for their synthesis are still limited. The biggest hurdle for the synthesis of PB derivatives is the fact that acyl-type amino protecting groups on nucleobases are not compatible with the synthesis of boranophosphate by the general phosphoramidite method since N-acyl groups were easily reduced with a boronating reagent to N-alkyl groups31 which cannot be removed. To deal with this problem, Caruthers et al. and our group obtained PB derivatives in different synthetic strategies. Caruthers et al. have synthesized PB-ODNs via the phosphoramidite method using N-di-tert-butylisobutylsilyl (N-BIBS)-protected phosphoramidite monomers. This protecting group is stable under the general reaction conditions of the phosphoramidite method26 and tolerant toward boronation. In contrast, we have developed the H-boranophosphonate method using an H-boranophosphonate monoester, which contains characteristic H–P → BH3 groups as monomer units.32,33 In this method, an H-boranophosphonate monomer unit is condensed with a 5′-hydroxy group using a condensing reagent to form an H-boranophosphonate diester linkage followed by detritylation step without a transformation of the resultant internucleotidic linkages. These two steps are repeated and after the designed length is achieved, all internucleotidic H-boranophosphonate diesters are oxidized to PB linkages by treatment with CCl4 and water in the presence of a base, followed by the removal of amino protecting groups and release from a solid support. Caruthers’s and our synthetic strategies were also applicable to the synthesis of PB/PO chimeric ODNs. We have synthesized PB/PO chimeric ODNs using H-boranophosphonate monomers and H-phosphonate monomers.27 Briefly, we utilized H-boranophosphonate monomers and H-phosphonate monomers to the chain elongation reactions with a condensing reagent to give H-boranophosphonate and H-phosphonate diester linkages, and these synthetic intermediates were simultaneously oxidized by treatment with CCl4 in the presence of water to PB and PO linkages, respectively. Alternatively, to the best of our knowledge, the synthesis of PB/PS/PO chimeric ODNs has never been reported.
In this research, we introduced H-phosphonothioate monomers into our previous synthetic strategy for PB/PO chimeric ODNs to synthesize PB/PS/PO chimeric ODNs (Scheme 1). This method also allows the synthesis of PB/PS chimeric ODNs. H-Phosphonothioate intermediates were synthesized by Stawinski et al. for the first time34,35 as the thio analogues of H-phosphonate. They condensed the H-phosphonothioate monoesters with 5′-hydroxy groups of thymidine derivatives and then oxidized the resultant H-phosphonothioate internucleotidic linkages to synthesize sulfur-containing nucleotide analogues such as a PS and a phosphorodithioate.34,36 From these insights, we expected that an introduction of PS linkages would be possible by utilizing H-phosphonothioate monoesters and the proper oxidation conditions. The major problem associated with using H-phosphonothioate monoesters is the chemoselectivity of a condensation reaction since H-phosphonothioate monoesters have two different nucleophilic centers, namely, sulfur and oxygen atoms. Desired PS derivatives are obtained by the O-activation followed by oxidation while the S-activation results in the formation of PO derivatives as byproducts. Stawinski et al. solved the problem using a chlorophosphate derivative, as a condensing reagent since soft S-nucleophiles have low reactivity toward the hard phosphorus centers.36 However, there are few reports using an H-phosphonothioate monomer for solid-phase synthesis.37−39 Hence, we tried to synthesize H-PS internucleotidic linkages on a solid support using phosphonium-type condensing reagents that is expected to avoid the S-activation since these have a hard character. In this research, we demonstrated that this synthetic strategy was applicable to the synthesis of PB/PS/PO chimeric ODNs including gapmer-type ODNs, which have 2′-OMe-modified nucleotides in the wing region.
Scheme 1. Strategy for the Synthesis of PB/PS/PO Chimeric ODNs in This Study.
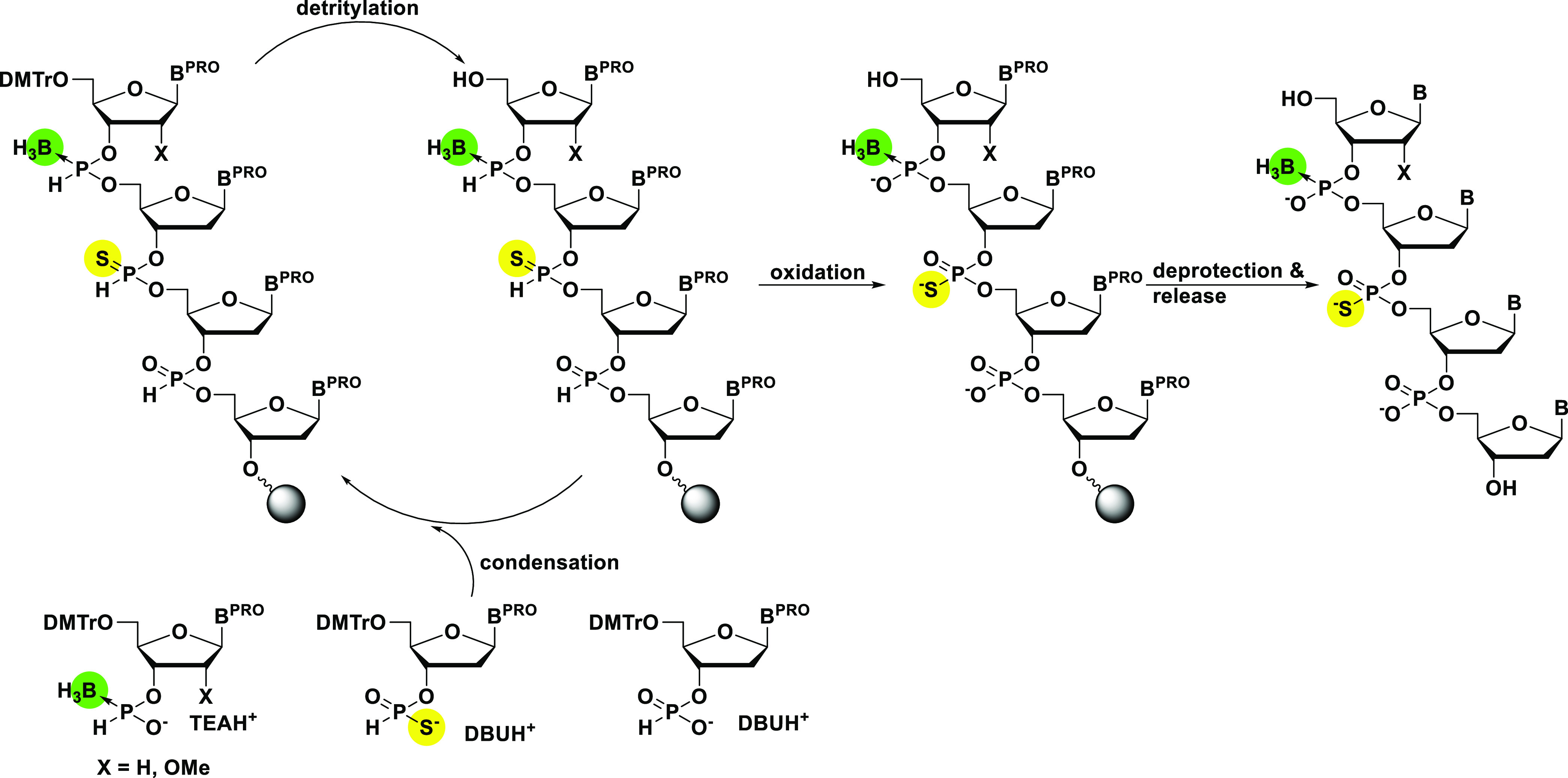
Results and Discussion
Synthesis of Monomer Units
Deoxyribonucleoside 3′-H-boranophosphonate monomers 3a, 3c, 3g, and 3t were synthesized by following our previous report from the nucleosides 1a, 1c, 1g, and 1t.27,33 An O6-unprotected deoxyguanosine derivative 3g was chosen as a monomer unit since it has been shown that 3g did not cause detectable side reactions27 (Scheme 2, method A). The 3′-H-phosphonate monomers 4a, 4c, 4g, and 4t were also synthesized according to a procedure in our preceding publication from the nucleosides 1a, 1c, 1g, and 1t.27,40 The monomers 4a, 4c, 4g, and 4t were isolated as 1,8-diazabicyclo undecanium salts (DBU salts) because DBU salts were reported to be more stable than the commonly used triethylammonium counterparts (TEA salts)41 (Scheme 2, method B). The 3′-H-phosphonothioate monomers 5a, 5c, 5g, and 5t were synthesized by following the literature from the nucleosides 1a, 1c, 1g, and 1t.35 Briefly, the nucleosides 1a, 1c, 1g, and 1t were condensed with triethylammonium phosphinic acid using PivCl as a condensing reagent, followed by oxidation with elemental sulfur. The 3′-H-phosphonothioate monomers 5a, 5c, 5g, and 5t were also isolated as DBU salts because of the above-mentioned reason. (Scheme 2, method C) (Yields; 5a 95%, 5c 99%, 5g 64%, and 5t 95%). The 2′-O-methyl-ribonucleoside 3′-H-boranophosphonate monomers 6a, 6c, 6g, and 6u were synthesized in a similar way to the synthesis of deoxyribonucleoside 3′-H-boranophosphonate monomers27,33 with good yields (Scheme 2, method D) (Yields: 6a 95%, 6c 90%, 6g 92%, and 6u 74%).
Scheme 2. Synthesis of the Monomers Used in This Study.
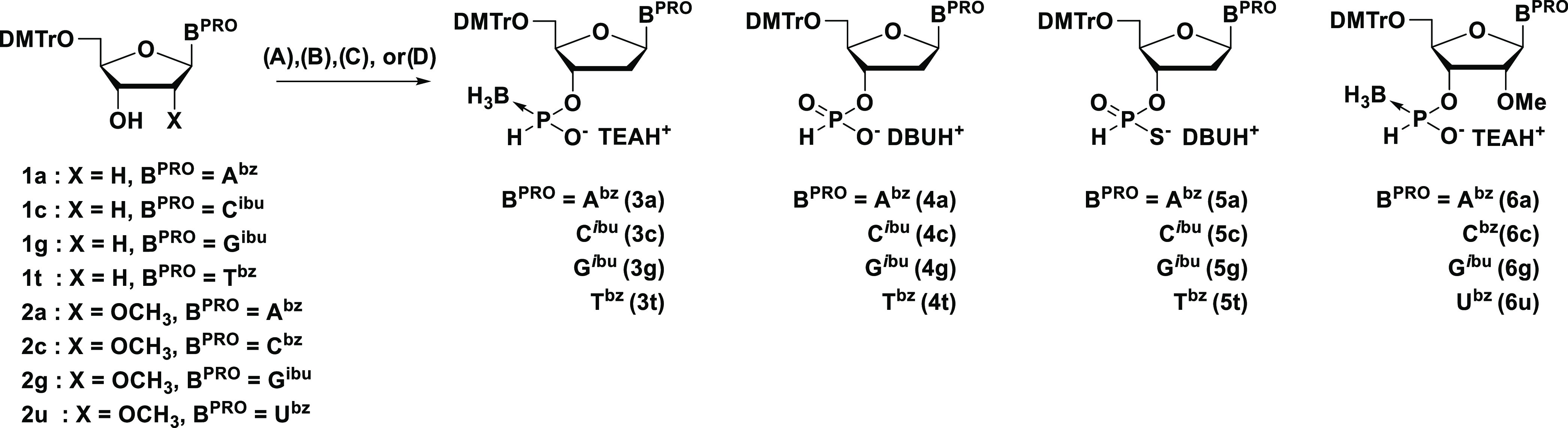
Method A (i) 1a, 1c, 1g, or 1t (1.0 equiv), pyridinium H-boranophosphonate (2.0 equiv), bis(2-oxo-3-oxazolidinyl) phosphinic chloride (2.0 equiv), pyridine, 0 °C to room temperature (rt), 1 h (ii) 1.0 M triethylammonium bicarbonate (TEAB) buffer; Method B (i) 1a, 1c, 1g, or 1t (1.0 equiv), diphenyl H-phosphonate (7.0 equiv), pyridine, 0 °C to rt, (ii) H2O–TEA (1:1, v/v), rt, (iii) 0.2 M DBU hydrogen carbonate aqueous solution; Method C (i) 1a, 1c, 1g, or 1t (1.5 equiv), triethylammonium phosphinate (1.0 equiv), pivaloyl chloride (1.5 equiv), 0 °C to rt, 15 min, (ii) elemental sulfur (1.5 equiv), rt, 1 h, (iii) 0.2 M DBU hydrogen carbonate aqueous solution; Method D (i) 2a, 2c, 2g, or 2u (1.0 equiv), pyridinium H-boranophosphonate (2.0 equiv), bis(2-oxo-3-oxazolidinyl) phosphinic chloride (2.0 equiv), pyridine, 0 °C to rt, 1 h, (ii) 1.0 M TEAB buffer.
Solid-Phase Synthesis of Phosphorothioate Dimers
Next, we investigated the reaction conditions for solid-phase synthesis using the H-phosphonothioate monomer 5t (Scheme 3). The reaction conditions for the solid-phase synthesis using H-boranophosphonate and H-phosphonate monomers had already been optimized in our previous report.27 First, the H-phosphonothioate monomer 5t (0.1 M) was condensed with the 5′-hydroxy group of thymidine on a highly cross-linked polystyrene (HCP) support42 via a succinyl linker using CH3CN as a solvent and a phosphonium-type condensing reagent (0.25 M) in the presence of a base, then detritylation was conducted by treatment with 3% dichloroacetic acid (DCA) in CH2Cl2 in the presence of Et3SiH as a trityl cation scavenger.43 Afterward, oxidation of the resultant H-phosphonothioate linkage was carried out using a mixture of CCl4 and H2O in the presence of triethylamine as a base and 2,6-lutidine as a cosolvent. Finally, deprotection of the N3-benzoyl group and cleavage of the linker by treatment with concentrated aqueous NH3 and EtOH afforded the PS dimer. The crude mixture was analyzed by reversed-phase high-performance liquid chromatography (RP-HPLC). The chemoselectivity was estimated by the area ratios of the PS diester and the PO diester which were derived from the S-activation. In addition to this, the HPLC yield was estimated by the area ratios of the PS diester (8) to unreacted thymidine and the PO diester (7).
Scheme 3. Solid-Phase Synthesis of TPST Dimera.
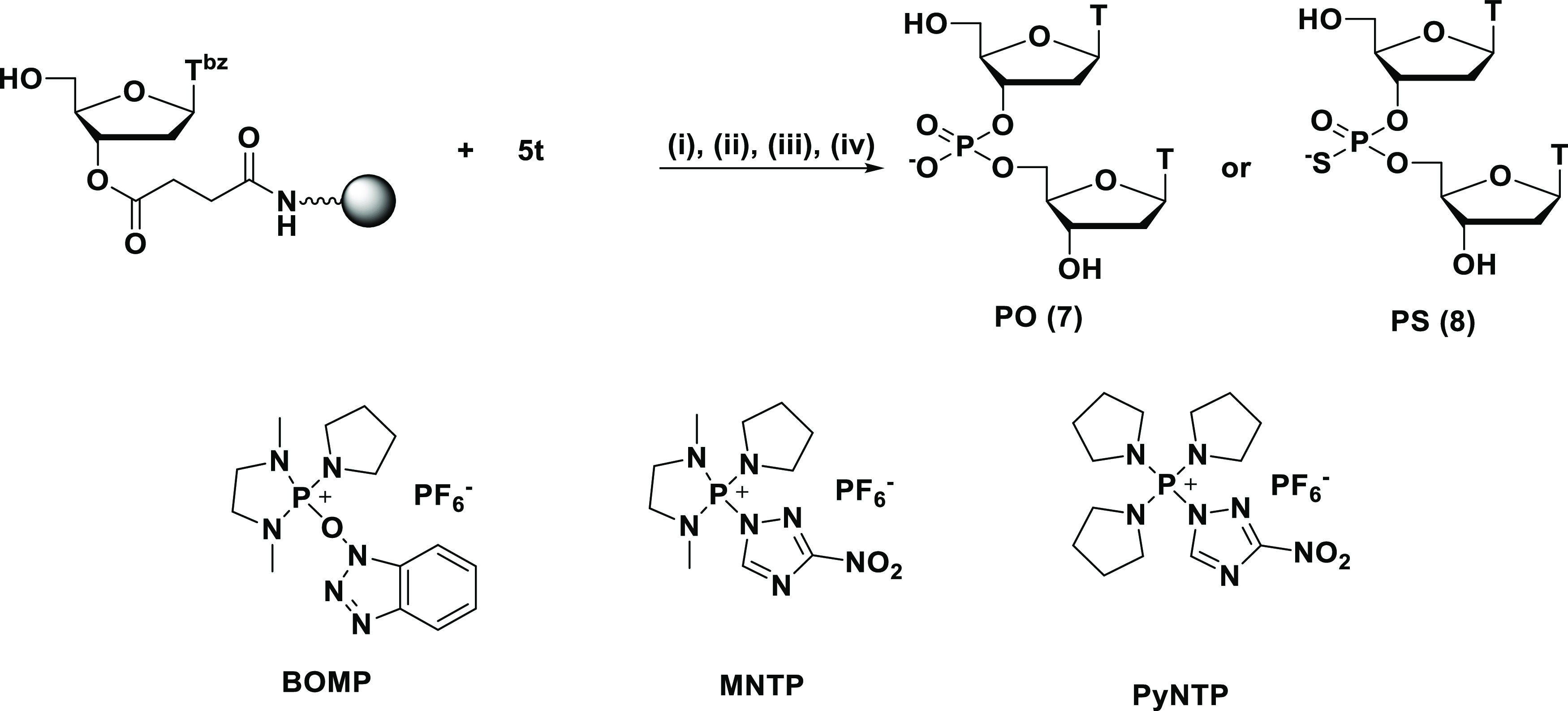
Reagents and conditions: (i) 0.1 M monomer, 0.25 M condensing reagent, 0.6 M base, CH3CN, rt, 3 min; (ii) 3% DCA, CH2Cl2–Et3SiH (1:1, v/v), rt, 4 × 15 s; (iii) 0.1 M Et3N in CCl4–2,6-lutidine–H2O (5:12.5:1, v/v/v), rt, 90 min; (iv) concentrated aqueous NH3–EtOH (3:1, v/v), 30 °C, 3 h.
First, the effect of condensing reagents on the reaction was investigated (Table 1, entries 1–3). In these experiments, 2,6-lutidine was used as a base in the condensation reaction. When the H-phosphonothioate monomer was condensed by 2-(benzotriazol-1-yloxy)-1,3-dimethyl-2-pyrrolidin-1-yl-1,3,2-diazaphospholidinium hexafluorophosphate (BOMP)44 which has HOBt as a leaving group, only a trace amount of the PS and the PO diester was obtained (entry 1). On the other hand, the use of condensing reagents such as 1,3-dimethyl-2-(3-nitro-1,2,4-triazol-1-yl)-2-pyrrolidin-1-yl-1,3,2-diazaphospholidinium hexafluorophosphate (MNTP)45 (entry 2) and 3-nitro1,2,4-triazol-1-yl-tris(pyrrolidin-1-yl) phosphonium hexafluorophosphate (PyNTP)45 (entry 3) which have 3-nitro 1,2,4-triazole (NT) as a leaving group afforded PS diester with over 90% HPLC yields. These results suggested that the presence of NT was critical for the condensation of the H-phosphonothioate monomer and the 5′-hydroxy group. Compared with MNTP, PyNTP gave better chemoselectivity and condensation efficiency. Therefore, PyNTP was chosen as a condensing reagent for H-phosphonothioate monomers in the following investigations.
Table 1. Solid-Phase Synthesis of TPST Dimer.
| condensation
conditions |
||||
|---|---|---|---|---|
| entry | condensing reagents | base (0.6 M) | chemoselectivity (PO:PS)a | HPLC yield (%)b |
| 1 | BOMP | 2,6-lutidine | <1 | |
| 2 | MNTP | 2,6-lutidine | 6:94 | 91 |
| 3 | PyNTP | 2,6-lutidine | 4:96 | 94 |
| 4 | PyNTP | pyridine | 3:97 | 94 |
| 5 | PyNTP | quinoline | 2:98 | 94 |
| 6 | PyNTP | quinolinec | 3:97 | 95 |
| 7 | PyNTP | 1:99 | 97 | |
Determined by RP-HPLC: area ratio of TPST and TPOT.
Determined by RP-HPLC: area ratio of TPST/(TPST + TPOT + T).
1.8 M quinoline was used.
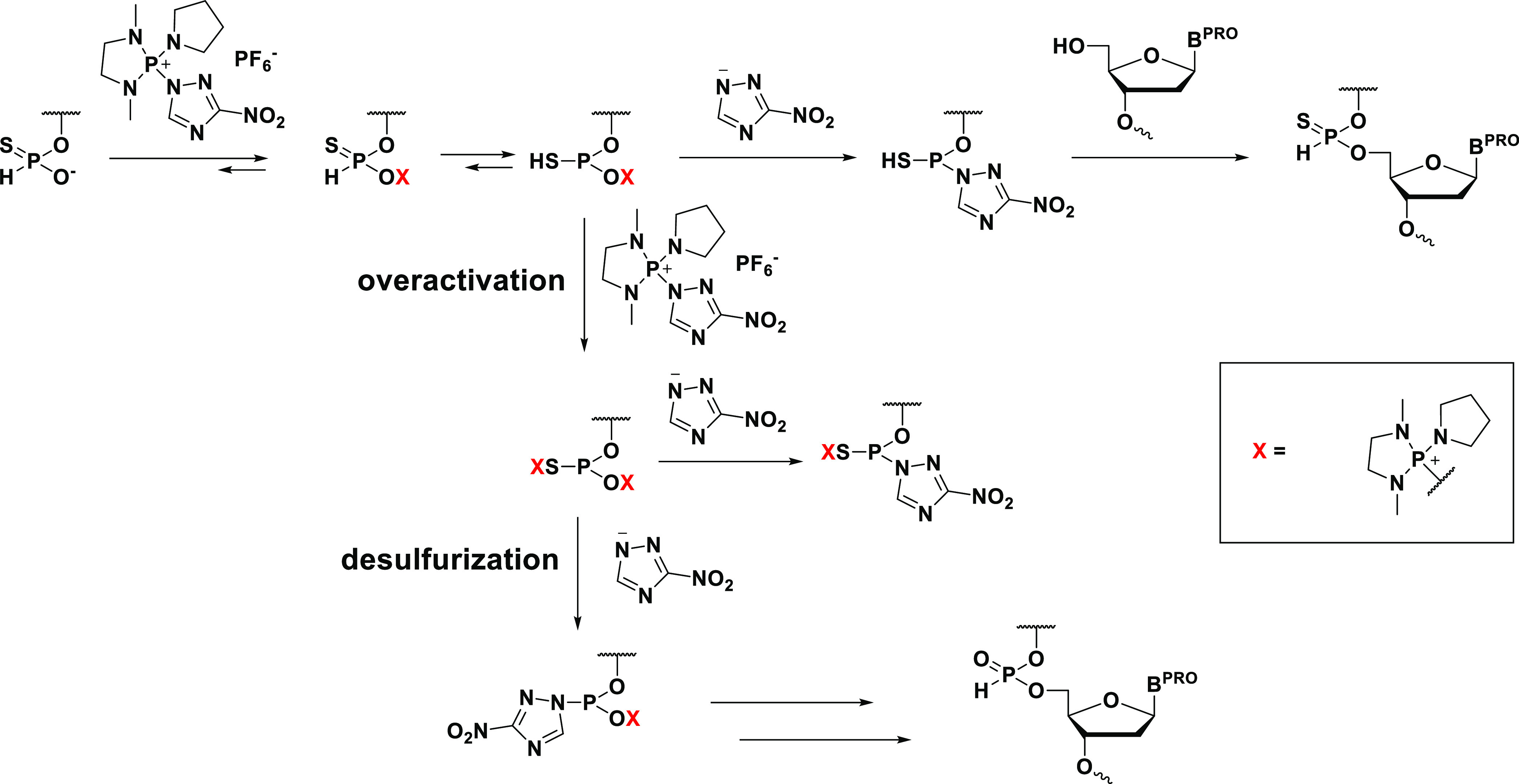
Next, bases for the condensation reaction were examined (entries 3–7). It was found that the use of the weaker base (pyridine, entry 4, quinoline, entry 5) slightly improved chemoselectivities while maintaining high condensation efficiency. Raising the ratio of quinoline in the reaction solvent had a marginal effect on the reaction outcome (entry 6). Surprisingly, the condensation reaction in the absence of base improved both coupling yield and chemoselectivity (entry 7). Therefore, we concluded that the utilization of PyNTP in the presence of 1.8 M quinoline or without a base was the optimum conditions for the condensation reaction. It has been shown that MNTP had a higher activity as a condensing reagent for phosphorylation and phosphonylation reactions than PyNTP,45 likely due to less steric hindrance and the strained cyclic structure of the phosphonium center. However, for the condensation reaction using the H-phosphonothioate 5t, PyNTP gave better chemoselectivity and condensation efficiency. In addition, the use of a stronger base for the condensation reaction was found to cause lower chemoselectivity and condensation efficiency. From these observations, an overactivation of the monomer 5t seemed to affect the reaction outcome. The activation of 5t by MNTP might be prone to lead the overactivation as shown in Scheme 4 and resulted in inferior chemoselectivity and condensation efficiency. High basicity of the reaction medium may also cause the overactivation of the monomer.
Scheme 4. Plausible Mechanism for Overactivation and Formation of TPOT.
The optimized reaction conditions were then applied to monomers containing other nucleobases (Scheme 5 and Table 2). The deoxyadenosine 5a, deoxycytidine 5c, and deoxyguanosine 5gH-phosphonothioate monomers were used to synthesize the dimers 10, 12, and 14 following the procedures in entries 6 and 7 of Table 1. In contrast to the thymidine H-phosphonothioate monomer, the presence of quinoline as a base was critical for efficient condensation reactions of deoxyadenosine and deoxycytosine counterparts (Table 2, entries 1 vs 2, entries 3 vs 4, respectively). As for the deoxyguanosine H-phosphonothioate monomer 5g, the coupling yield and chemoselectivity were better in the absence of a base, but still not satisfactory (Table 2, entries 5 vs 6). Doubling the equivalents of the deoxyguanosine H-phosphonothioate monomer and PyNTP afforded the product in 93% HPLC yield (entry 7). Thus, conditions in entries 1, 3, and 7 were chosen as the optimum conditions for each H-phosphonothioate monomer.
Scheme 5. Solid-Phase Synthesis of NPST Dimers.
Reagents and conditions: (i) 0.1 M monomer, 0.25 M condensing reagent in the presence of 1.8 M base or in the absence of base, CH3CN, rt, 3 min; (ii) 3% DCA, CH2Cl2–Et3SiH (1:1, v/v), rt, 4 × 15 s; (iii) 0.1 M Et3N in CCl4–2,6-lutidine–H2O (5:12.5:1, v/v/v), rt, 90 min; (iv) concentrated aqueous NH3–EtOH (3:1, v/v), 30 °C, 12 h for entries 1–4, 50 °C, 12 h for entries 5–7.
Table 2. Solid-Phase Synthesis of NPST Dimers.
| condensation
reaction conditions |
|||||
|---|---|---|---|---|---|
| entry | monomer | base | product | chemoselectivity (PO:PS)a | HPLC yield (%)b |
| 1 | 5a | quinoline | dAPST (10) | 2:98 | 95 |
| 2 | 5a | dAPST (10) | 3:97 | 70 | |
| 3 | 5c | quinoline | dCPST (12) | 2:98 | 96 |
| 4 | 5c | dCPST (12) | 2:98 | 94 | |
| 5 | 5g | quinoline | dGPST (14) | 1:>99 | 88 |
| 6 | 5g | dGPST (14) | 1:>99 | 91 | |
| 7c | 5g | dGPST (14) | 1:>99 | 93 | |
Determined by RP-HPLC: area ratio of NPST and NPOT.
Determined by RP-HPLC: area ratio of NPST/(NPST + NPOT + T).
0.2 M monomer and 0.5 M condensing reagent were used.
In addition to this, we carried out the solid-phase synthesis of PB dimers using 2′-O-methyl-3′-H-boranophosphonate monomers (6a, 6c, 6g, and 6u) for the synthesis of gapmer-type PB/PS/PO chimeric oligonucleotides. 2′-O-Methyl-3′-H-boranophosphonate monomers were condensed with the 5′-hydroxy group of thymidine on an HCP support via a succinyl linker under the same conditions with 2′-deoxynucleoside counterparts using MNTP as a condensing reagent in the presence of 2,6-lutidine (Scheme 6). The following procedures were the same as in the synthesis of TPST. Dimers APBT, CPBT, GPBT, and UPBT (underline indicates 2′-OMe nucleoside) were obtained in 95–98% HPLC yields (Table 3), indicating that the condensation reaction proceeded efficiently regardless of the presence of 2′-O-modification.
Scheme 6. Solid-Phase Synthesis of NPBT Dimers Using 2′-OMe-3′-H-Boranophosphonate Monomers.

Reagents and conditions: (i) 0.1 M monomer, 0.25 M MNTP, 0.6 M 2,6-lutidine, CH3CN, rt, 3 min; (ii) 3% DCA, CH2Cl2–Et3SiH (1:1, v/v), rt, 4 × 15 s; (iii) 0.1 M Et3N in CCl4–2,6-lutidine–H2O (5:12.5:1, v/v/v), rt, 90 min; (iv) concentrated aqueous NH3–EtOH (3:1, v/v), 30 °C, 12 h for entries 1–2, 50 °C, 12 h for entry 3, 30 °C, 3 h for entry 4.
Table 3. Solid-Phase Synthesis of NPBT Dimers Using 2′-OMe-3′-H-Boranophosphonate Monomers.
| entry | monomer | producta | HPLC yield (%)b |
|---|---|---|---|
| 1 | 6a | APBT (15) | 95 |
| 2 | 6c | CPBT (16) | 96 |
| 3 | 6g | GPBT (17) | 98 |
| 4 | 6u | UPBT (18) | 97 |
Subscript PB = boranophosphate, underline = 2′-OMe modification.
Determined by RP-HPLC: area ratio of NPBT/(NPBT + T).
Solid-Phase Synthesis of PB/PS/PO Chimeric ODNs
Next, to elucidate whether the synthetic strategy is applicable to the synthesis of PB/PS/PO chimeric ODNs, the synthesis of tetramers (d(CPOAPBGPST) (19) and d(CPSAPBGPOT) (20)) containing PB, PS, and PO linkages was conducted using the H-phosphonothioate, H-boranophosphonate, and H-phosphonate monomers. The cycle consisting of the condensation and detritylation was repeated, and oxidation of internucleotidic linkages followed by removal of the amino protecting groups and cleavage of the linker afforded the tetramers. It was confirmed that the tetramers were formed as main products by the RP-HPLC analysis of the reaction mixtures (Figure S4). These results indicated that these different internucleotidic linkages were simultaneously oxidized without side reactions, and thus this strategy enables the synthesis of PB/PS/PO chimeric ODNs.
These results prompted us to synthesize a PB/PS/PO chimeric DNA dodecamer, and d(CPSAPSGPSTPSCPBAPBGPBTPBCPOAPOGPOT) (21) was chosen as a synthetic target to demonstrate the potential of the strategy, since 21 contains almost all of the potential combination of internucleotidic linkages and nucleobases. The dodecamer 21 was synthesized by repeating the condensation and detritylation cycles, oxidation, and the deprotection and the release step and was isolated in 6% yield (Table 4, entry 3). Encouraged by the success, we designed and synthesized types of chimeric ODNs containing PB modifications (Table 4, entries 4–6). These sequences were antisense sequences to apoB protein mRNA46 and the site of chemical modifications of the PB/PS/PO-ODN 22 was designed as follows: (1) PB or PS linkages were placed at the 3′ and 5′-ends to gain resistance toward exonucleases; (2) four consecutive PO linkages were introduced at the central position of ODNs to improve duplex forming and RNase H-inducing abilities. As for the PB/PS-ODN 23, PB or PS modifications were introduced at the central position of the sequence instead of PO linkages. Also, the PB/PO-ODN 24 was designed as shown in entry 6. These ODNs were successfully synthesized and isolated in 5–19% yields (Table 4, entries 4–6). Subsequently, we attempted to apply this synthetic strategy to a 2′-OMe gapmer and synthesized GPBCPBAPB d(TPSTPOGPOGPOTPSAPB)UPBUPBC (25). The RP-HPLC analysis of crude mixture indicated that the desired oligonucleotide was formed as the main product and 25 was isolated in 13% yield (Table 4, entry 7). This result indicated that some 2′-O-modified gapmers would be synthesized by this synthetic strategy. It is worth noting that although we had purified PB containing ODNs by anion-exchange HPLC, it was found that RP-HPLC purification using a mixture of methanol and a buffer containing hexafluoroisopropanol and triethylamine as an eluent was effective for isolation of these ODNs.47 Thus, all of the ODNs but 22 were purified by RP-HPLC. Although there is room for further improvement of yields, some kinds of chimeric ODNs were successfully synthesized in the strategy.
Table 4. Results of the Solid-Phase Synthesis of PB/PS/PO Chimeric ODNs.
| isolated yieldb |
m/z |
||||
|---|---|---|---|---|---|
| entry | abbreviation of ODNs | producta | (%) | calcd | found |
| 1 | d(CPSAPBGPOT) (19) | 592.6236 | 592.6209 | ||
| 2 | d(CPOAPBGPST) (20) | 592.6236 | 592.6217 | ||
| 3 | d(CPSAPSGPSTPSCPBAPBGPBTPBCPOAPOGPOT) (21) | 6 | 615.7786 | 615.7767 | |
| 4 | PB/PS/PO | d(GPBCPSAPBTPOTPOGPOGPOTPSAPBTPSTPBC) (22) | 19 | 614.1138 | 614.1136 |
| 5 | PB/PS | d(GPBCPSAPBTPSTPSGPBGPBTPSAPBTPSTPBC) (23) | 5 | 618.6194 | 618.6175 |
| 6 | PB/PO | d(GPBCPBAPBTPOTPOGPOGPOTPBAPBTPBTPBC) (24) | 19 | 604.9781 | 604.9774 |
| 7 | PB/PS/PO-gapmer | GPBCPBAPB d(TPSTPOGPOGPOTPSAPB)UPBUPBC (25) | 13 | 635.9695 | 635.9670 |
Subscript PB = boranophosphate, PS = phosphorothioate, PO = phosphate, underline = 2′-OMe modification.
Determined by UV absorbance 260 nm.
Hybridization Properties
Next, we moved on to the evaluation of properties that are crucial for ASOs. First, hybridization properties of the obtained ODNs were investigated by thermal denaturation tests. The UV melting curves and Tm values of the ODNs 22–29 with the complementary RNA (cRNA) are shown in Figure 1 and Table 5, respectively. As for PO, PS, and PB-ODNs, the order of hybridization ability was PO-ODN (Tm value of the duplex with cRNA: 45.0 °C) > PS-ODN (34.0 °C) > PB-ODN (32.2 °C), which was in good agreement with the previous reports.19,27 Introduction of PO linkages to PB or PS-ODNs (PB/PO or PS/PO-ODN) improved the duplex stability (PB (Tm value of the duplex with cRNA: 32.2 °C) vs PB/PO-ODNs (35.0 °C) and PS (34.0 °C) vs PS/PO-ODNs (37.3 °C)). Although PB/PS chimeric ODN reduced its duplex stability compared to the other ODNs, replacing PB or PS linkages with PO linkages (PB/PS/PO chimeric ODN) also improved the duplex stability and the Tm value was as almost the same as the PS-ODN (PB/PS (Tm value of the duplex with cRNA: 28.8 °C) vs PB/PS/PO-ODNs (34.0 °C)). In addition, PB/PS/PO chimeric gapmers having 2′-OMe modifications on the wing region showed improved duplex stability (42.3 °C). From these results, it was verified incorporation of sugar modifications and introducing suitable P-modifications into proper positions of ASOs could regulate the duplex stability.
Figure 1.

UV Melting Curves of ODNs (22–29) and cRNA (30) Duplexes.
Table 5. Tm Values of The ODN/cRNA Duplexesa.
| entry | abbreviation of ODNs | sequenceb | Tm (°C) | ΔTm (°C)c |
|---|---|---|---|---|
| 1 | PO | d(GPOCPOAPOTPOTPOGPOGPOTPOAPOTPOTPOC) (26) | 45.0 | |
| 2 | PS | d(GPSCPSAPSTPSTPSGPSGPSTPSAPSTPSTPSC) (27) | 34.0 | –11.0 |
| 3 | PB | d(GPBCPBAPBTPBTPBGPBGPBTPBAPBTPBTPBC) (28) | 32.2 | –12.8 |
| 4 | PB/PS/PO | d(GPBCPSAPBTPOTPOGPOGPOTPSAPBTPSTPBC) (22) | 34.0 | –11.0 |
| 5 | PB/PS | d(GPBCPSAPBTPSTPSGPBGPBTPSAPBTPSTPBC) (23) | 28.8 | –16.2 |
| 6 | PB/PO | d(GPBCPBAPBTPOTPOGPOGPOTPBAPBTPBTPBC) (24) | 35.0 | –10.0 |
| 7 | PS/PO | d(GPSCPSAPSTPOTPOGPOGPOTPSAPSTPSTPSC) (29) | 37.3 | –7.7 |
| 8 | PB/PS/PO-gapmer | GPBCPBAPB d(TPSTPOGPOGPOTPSAPB)UPBUPBC (25) | 42.3 | –2.7 |
cRNA (30): r(GPOAPOAPOUPOAPOCPOCPOAPOAPOUPOGPOC).
Subscript PB = boranophosphate, PS = phosphorothioate, PO = phosphate, underline = 2′-OMe modification.
The difference in Tm value relative to that of the PO-ODN/cRNA duplex.
Nuclease Resistance
Second, nuclease digestion experiments were conducted using snake venom phosphodiesterase (SVPDE) from Crotalus adamanteus venom, a representative of 3′-exonuclease. Aqueous solutions of each ODN (22–29) were treated with SVPDE solution (0.4 U/mL) at 37 °C for 12 h. After SVPDE was denatured at 95 °C, the mixtures were analyzed by RP-HPLC. The results are shown in Figures 2 and S21. The PO-ODN was completely digested while the PS-ODN was partially degraded in the same conditions, respectively (Figure S21). On the other hand, the PB-ODN remained almost intact (Figure S21). Therefore, it was suggested that the order of nuclease resistance toward SVPDE was PB-ODN > PS-ODN > PO-ODN, which was in good agreement with the previous report.19 On the other hand, the PB/PO-ODN, PS/PO-ODN, PB/PS/PO-ODN, and PB/PS/PO-gapmer, which have three or four consecutive PO linkages at the central positions, were completely digested and their full-length ODNs were not detected by RP-HPLC (Figures 2 and S21). Although SVPDE is known as a 3′-exonuclease, there are reports that SVPDE also has endonuclease activity.48−50 Hence, SVPDE may recognize consecutive PO linkages at the central position as endonuclease. In sharp contrast, there was a slight sign of decomposition of the PB/PS-ODN indicating that combination of PB and PS modifications confers substantial nuclease resistance to ODNs (Figure 2). In the nuclease digestion experiments, it was suggested that the ODNs containing a higher ratio of PO linkages would significantly reduce nuclease resistance.
Figure 2.

RP-HPLC profiles of ODN before (top) and after (bottom) the treatment with snake venom phosphodiesterase (SVPDE) for 12 h at 37 °C. (* indicates an artifact peak).
RNase H Activity
Finally, we studied the effect of ODNs on RNase H activity, which is crucial for the efficacy of RNase H-dependent antisense therapeutics. Escherichia coli RNase H was used for the experiments. Aqueous solutions of each ODN (22–29) and 10 equiv of cRNA were treated with 25 or 50 U/mL RNase H at 37 °C for 30 min. After RNase H was denatured at 95 °C, the mixtures were analyzed by RP-HPLC. In the assay using 25 U/mL RNase H, although almost all of the ODNs did not show clear effects on RNase H activities, the PO-ODN (26) and the PB/PS/PO-gapmer (25) were able to induce cleavage of the cRNA strand by RNase H to some extent since small fragments of cRNA were detected by RP-HPLC (Figure S22). Compared with the PO-ODN, the PB/PS/PO-gapmer showed slightly better RNase H activity. In the assay using 50 U/mL RNase H, almost all cRNA was digested by RNase H with all ODNs (Figures 4 and S23). Hence, we calculated the amount of the intact cRNA compared with benzamide as an internal standard based on the area ratio of each peak in HPLC profiles. In the presence of PB/PS/PO-gapmer, PB/PS/PO, PS/PO, PS, and PO-ODNs, over 95% of cRNA was cleaved by RNase H. In contrast, when using PB/PS, PB/PO, and PB-ODN, approximately 90% of cRNA was cleared by RNase H. From these results, it was suggested that increasing the ratio of PB linkages suppressed RNase H activity.
Figure 4.

RP-HPLC profiles of ODN/cRNA (30) before (top) and after (bottom) the treatment with RNase H for 30 min at 37 °C.
On the other hand, in the RNase H assay, two pairs of cleaved fragments, namely, p4mer and 8mer (cleavage site a in Figure 3) and p5mer and 7mer (cleavage site b in Figure 3) (p indicates phosphate group at the 5′ end of fragments) were mainly detected by RP-HPLC and electrospray ionization mass spectrometry (ESI-MS) analysis. The peaks derived from the fragments were shown in Figure 3 (fragment A was from cleavage of RNA (30) at cleavage site a; fragment B was from cleavage of RNA (30) at cleavage site b). The area ratios of the peaks A and B in HPLC profiles were shown in Table 6 and the representative profiles were shown in Figure 4. In the presence of ODNs without PB modification, the area ratios of peaks A and B were approximately 1:2 (Table 6, entries 1, 2, 7), whereas ODNs containing PB linkages offered varied cleavage site preferences, except for the PB/PS/PO-ODN (22). Therefore, it was suggested that ODNs containing PB linkages would change the cleavage site preference. Hence, the effect of PB linkages on the recognition of one base mismatch was studied as follows.
Figure 3.

Plausible Cleavage Site of the DNA/RNA Duplex.
Table 6. Ratio of Digested Fragmentsa.
| area
ratiob |
||||
|---|---|---|---|---|
| entry | abbreviation of ODNs | sequenceb | A | B |
| 1 | PO | d(GPOCPOAPOTPOTPOGPOGPOTPOAPOTPOTPOC) (26) | 1 | 2.3 |
| 2 | PS | d(GPSCPSAPSTPSTPSGPSGPSTPSAPSTPSTPSC) (27) | 1 | 2.2 |
| 3 | PB | d(GPBCPBAPBTPBTPBGPBGPBTPBAPBTPBTPBC) (28) | 1 | 3.5 |
| 4 | PB/PS/PO | d(GPBCPSAPBTPOTPOGPOGPOTPSAPBTPSTPBC) (22) | 1 | 2.5 |
| 5 | PB/PS | d(GPBCPSAPBTPSTPSGPBGPBTPSAPBTPSTPBC) (23) | 1 | >4.5 |
| 6 | PB/PO | d(GPBCPBAPBTPOTPOGPOGPOTPBAPBTPBTPBC) (24) | 1 | 1.3 |
| 7 | PS/PO | d(GPSCPSAPSTPOTPOGPOGPOTPSAPSTPSTPSC) (29) | 1 | 2.0 |
| 8 | PB/PS/PO-gapmer | GPBCPBAPB d(TPSTPOGPOGPOTPSAPB)UPBUPBC (25) | 1 | 4.0 |
Fragment A: r(pAUGC); fragment B: r(pAAUGC).
Determined by the area ratio of A and B in RP-HPLC profile.
Similar RNase H cleavage experiments were conducted using PB/PS/PO-gapmer (25) or PO-ODN (26) and the RNA (31, r(GAACACCAAUGC), boldface indicates a mismatch to the ODNs) having one base mismatch to the ODNs. In the presence of PO-ODN, less than 5% of the RNA (31) remained, whereas with PB/PS/PO-gapmer, approximately 20% of the RNA (31) was intact (Figure 5). To evaluate the duplex stability of PB/PS/PO-gapmer (25) and PO-ODN (26) with the RNA (31), thermal denaturation tests were conducted in a similar way. The UV melting curves of PB/PS/PO-gapmer (25) and PO-ODN (26) with the RNA (31) are shown in Figure S20. The calculated Tm value of PO-ODN (26) and RNA (31) duplex was 33.4 °C, 11.6 °C lower than that of the matched duplex, whereas the melting curve of the mixture of the PB/PS/PO-gapmer (25) and the RNA (31) had no apparent inflection point, indicating the low duplex stability of PB/PS/PO-gapmer (25) with the RNA (31). Taking these results into consideration, the reduced cleavage of the mismatched RNA strand by RNase H in the presence of the PB/PS/PO-gapmer would be attributed to both the cleavage preference induced by the PB/PS/PO-gapmer and the inferior duplex stability of the PB/PS/PO-gapmer with the complementary RNA with a mismatched base. Thus, although the experiment was a preliminary study, it was demonstrated that proper chemical modification would improve the ability of one base mismatch recognition.
Figure 5.

RP-HPLC profiles of ODN/cRNA (31) before (top) and after (bottom) the treatment with RNase H for 30 min at 37 °C.
Conclusions
We developed an efficient synthetic strategy for PB/PS/PO chimeric ODNs without a limitation of the nucleobase and position of P-modification utilizing H-boranophosphonate, H-phosphonothioate, and H-phosphonate monomers. In addition to this, we showed that the strategy was applicable to the synthesis of a PB/PS/PO chimeric 2′-OMe gapmer. One of the key points pertaining to the synthesis of these chimeric ODNs was regulating the chemoselectivity of the condensation reaction using an H-phosphonothioate monomer. It was found that phosphonium-type condensing reagents having NT as a leaving group, especially PyNTP, provided excellent chemoselectivity. The properties of obtained ODNs were also evaluated. As for a thermal denaturation study, introducing PO linkages to PS-ODN, PB-ODN, and PB/PS-ODN (PS/PO, PB/PO, and PB/PS/PO chimeric ODNs) improved their duplex stability. An SVPDE assay revealed that the introduction of three or four consecutive PO linkages reduced their nuclease resistance significantly. In the RNase H activity assay, it was suggested that ODNs containing a higher ratio of the PB linkages reduced their RNase H activity to some extent, and altered the cleavage site preference of RNase H. From these results, it was expected that introducing proper P-modifications at appropriate sites of ASOs would regulate the duplex stability, nuclease resistance, RNase H activity, and one base mismatch discrimination. In addition, this synthetic strategy would also work for the synthesis of any other gapmers containing LNA or 2′-O-MOE which are widely used sugar modifications of ASOs. Thus, we expect that a PB/PS/PO chimeric ODN is one of the promising candidates of potent ASOs. Hence, the details of the biological and physiological properties of PB/PS/PO chimeric ODNs are under investigation.
Experimental Section
General Information
All reactions were conducted under an Ar atmosphere. Dry organic solvents were prepared by appropriate procedures. A thermostatic chamber was used for the reaction run at 50 °C. 1H NMR spectra were recorded at 400 MHz with tetramethylsilane (δ 0.0) as an internal standard in CDCl3. 13C NMR spectra were recorded at 100 MHz with CDCl3 (δ 77.0) as an internal standard in CDCl3. 31P NMR spectra were recorded at 162 MHz with H3PO4 (δ 0.0) as an external standard in CDCl3. Analytical thin-layer chromatography was performed on commercial glass plates with a 0.25 mm thickness silica gel layer. Silica gel column chromatography was performed using spherical, neutral, 63–210 μm silica gel. Manual solid-phase synthesis was carried out using a glass filter (10 mm × 50 mm) with a stopper at the top and a stopcock at the bottom as a reaction vessel. Synthesized dimers were analyzed by reversed-phase HPLC. Synthesized oligomers (tetramer and dodecamers) were analyzed by reversed-phase HPLC and/or anion-exchange HPLC, purified by reverse-phase HPLC or anion-exchange HPLC, and identified by electrospray ionization (ESI) mass spectroscopy. Isolated yields of oligomers were estimated by measuring UV–vis spectra. PO-ODN (26), PS-ODN (27), PS/PO-ODN (29), and cRNA (30) were purchased and used for denaturation, nuclease resistance, and RNase H activity tests without further purification.
General Procedure for the Synthesis of 5′-O-Dimethoxytrityl-nucleoside 3′-H-phosphonothioates (5a, 5c, 5g, and 5t)
Nucleoside 1a (0.90 g, 1.5 mmol, 1.5 equiv), 1c (0.98 g, 1.5 mmol, 1.5 equiv), 1g (0.92 g, 1.5 mmol, 1.5 equiv), or 1t (0.96 g, 1.5 mmol, 1.5 equiv) was dried by repeated coevaporation with dry pyridine. On the other hand, dry triethylamine was added to phosphinic acid (0.19 mL, 1 mmol, 1.0 equiv) and the solution was dried by coevaporation with dry pyridine. Thereafter, nucleoside and triethylammonium phosphinate were dissolved in the same solvent (5 mL). Pivaloyl chloride (0.17 mL, 1.5 mmol, 1.5 equiv) was added to the solution at 0 °C while stirring. After 15 min, the solution was warmed to rt and after further 5 min, elemental sulfur (48 mg, 1.5 mmol, 1.5 equiv) was added. Stirring was continued for 1 h, and then the mixture was diluted with CHCl3 (30 mL) and washed with 1.0 M TEAB buffers (3 × 30 mL). The aqueous layers were combined and back-extracted with CHCl3 (3 × 30 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (20 g of neutral silica gel, 2 × 10 cm) using AcOEt–MeOH–Et3N and CH2Cl2-MeOH-Et3N as the eluent. The fractions containing 5a, 5c, 5g, or 5t were collected and concentrated under reduced pressure. CHCl3 (30 mL) was added to the residue and washed with 0.2 M DBU hydrogen carbonate aqueous solutions. The aqueous layers were combined and back-extracted with CHCl3. The organic layers were combined, dried over Na2SO4, filtered, and concentrated under reduced pressure to yield 5a, 5c, 5g, or 5t.
1,8-Diazabicyclo [5.4.0] undec-7-enium 5′-O-dimethoxytrityl-N6-benzoyladenosine 3′-H-phosphonothioate as a Mixture of (Sp)- and (Rp)-Diastereomers (5a)
The crude mixture containing 5a was purified by silica gel column chromatography using AcOEt–MeOH–Et3N (100:0:1–100:4:1, v/v/v) and then CHCl3–MeOH–Et3N (100:2:1, v/v/v) as the eluent. Then, the fractions containing 5a were collected and concentrated under reduced pressure. CHCl3 (30 mL) was added to the residue and washed with 0.2 M DBU hydrogen carbonate aqueous solutions (3 × 30 mL). The aqueous layers were combined and back-extracted with CHCl3 (3 × 30 mL). 5a was obtained as colorless foam (0.88 g, 0.95 mmol, 95% yield). 1H NMR (400 MHz, CDCl3) δ 11.9–11.7 (br, 1H), 9.2–8.9 (br, 1H), 8.71 (s, 1H), 8.20 (s, 1H), 8.17 (d, J = 574 Hz, 0.5H), 8.13 (d, J = 579 Hz, 0.5H), 8.01 (d, J = 7.3 Hz, 2H), 7.62–7.57 (m, 1H), 7.54–7.48 (m, 2H), 7.46–7.38 (m, 3H), 7.34–7.10 (m, 6H), 6.79 (d, J = 8.7 Hz, 4H), 6.65–6.57 (m, 1H), 5.43–5.35 (m, 1H), 4.57–4.54 (m, 0.5H), 4.48–4.44 (m, 0.5H), 3.76 (s, 6H), 3.52–3.36 (m, 8H), 2.98–2.84 (m, 4H), 1.99 (quintet, J = 5.8 Hz, 2H), 1.80–1.60 (m, 6H); 13C{1H} NMR (100 MHz, CDCl3) δ 166.0, 164.5, 158.3, 152.4, 151.5, 149.2, 144.5, 141.4, 135.6, 135.6, 133.6, 132.6, 130.0, 128.8, 128.1, 127.8, 127.7, 126.7, 123.2, 113.1, 86.4, 86.4, 86.2 (d, 3JC–P = 5.8 Hz), 85.5 (d, 3JC–P = 6.7 Hz), 84.6, 84.5, 75.4 (d, 2JC–P = 4.8 Hz), 74.8 (d, 2JC–P = 4.8 Hz), 63.6, 63.5, 55.1, 54.2, 48.6, 39.8, 38.1, 32.2, 29.7, 28.9, 26.8, 24.0, 19.5; 31P{1H} NMR (162 MHz, CDCl3) δ 53.0, 52.9. HRMS (ESI/Q-TOF) m/z calcd for C38H35N5O7PS– [M–DBU–H]−, 736.2000; found 736.1982.
1,8-Diazabicyclo [5.4.0] undec-7-enium 5′-O-dimethoxytrityl-N4-isobutyrylcytidine 3′-H-phosphonothioate as a Mixture of (Sp)- and (Rp)-diastereomers (5c)
The crude mixture containing 5c was purified by silica gel column chromatography using AcOEt–MeOH–Et3N (100:0:1–100:4:1, v/v/v) and then CHCl3–MeOH–Et3N (100:4:1, v/v/v) as the eluent. Then, the fractions containing 5c were collected and concentrated under reduced pressure. CHCl3 (20 mL) was added to the residue and washed with 0.2 M DBU hydrogen carbonate aqueous solutions (3 × 20 mL). The aqueous layers were combined and back-extracted with CHCl3 (3 × 20 mL). 5c was obtained as colorless foam (0.89 g, 0.99 mmol, 99% yield). 1H NMR (400 MHz, CDCl3) δ 11.7–11.4 (br, 1H), 8.8–8.5 (br, 1H), 8.19 (d, J = 5.0 Hz, 0.5H), 8.17 (d, J = 5.0 Hz, 0.5H), 8.13 (d, J = 574 Hz, 0.5H), 8.10 (d, J = 581 Hz, 0.5H), 7.46–7.39 (m, 2H), 7.37–7.20 (m, 7H), 7.13 (d, J = 6.0 Hz, 0.5H), 7.11 (d, J = 6.0 Hz, 0.5H), 6.90–6.81 (m, 4H), 6.30 (t, J = 6.0 Hz, 0.5H), 6.29 (t, J = 6.0 Hz, 0.5H), 5.37–5.29 (m, 0.5 H), 5.25–5.17 (m, 0.5 H), 4.48 (q, J = 3.0 Hz, 0.5 H), 4.36 (q, J = 3.3 Hz, 0.5 H), 3.80 (s, 6H), 3.54–3.39 (m, 8H), 2.92–2.82 (m, 2H), 2.69–2.61 (m, 1H), 2.38–2.26 (m, 1H), 2.00 (quintet, J = 5.8 Hz, 2H), 1.82–1.60 (m, 7H), 1.25–1.17 (m, 6H); 13C{1H} NMR (100 MHz, CDCl3) δ 176.7, 166.0, 162.1, 158.4, 155.0, 144.6, 144.1, 135.4, 135.2, 130.0, 129.9, 128.1, 127.9, 126.9, 113.2, 96.0, 87.1, 87.0, 86.7, 86.0 (d, 3JC–P = 6.7 Hz), 85.3 (d, 3JC–P = 5.8 Hz), 74.2 (d, 2JC–P = 4.8 Hz), 73.1 (d, 2JC–P = 5.8 Hz), 62.6, 62.5, 55.1, 54.2, 48.5, 41.2, 40.8, 38.1, 36.5, 32.3, 28.9, 26.7, 24.0, 19.4, 19.0, 18.9; 31P{1H} NMR (162 MHz, CDCl3) δ 53.6, 52.6. HRMS (ESI/Q-TOF) m/z calcd for C34H37N3O8PS– [M–DBU–H]−, 678.2044; found 678.2032.
1,8-Diazabicyclo [5.4.0] undec-7-enium 5′-O-dimethoxytrityl-N2-isobutyrylguanosine 3′-H-phosphonothioate as a Mixture of (Sp)- and (Rp)-diastereomers (5g)
The crude mixture containing 5g was purified by silica gel column chromatography twice using CHCl3–MeOH–Et3N (100:2:1–100:4:1, v/v/v) as the eluent for the first time, and AcOEt–MeOH–Et3N (100:2:1, v/v/v) and then CHCl3–MeOH–Et3N (100:3:1–100:5:1, v/v/v) as the eluent for the second time. Then, the fractions containing 5g were collected and concentrated under reduced pressure. CHCl3 (20 mL) was added to the residue and washed with 0.2 M DBU hydrogen carbonate aqueous solutions (3 × 20 mL). The aqueous layers were combined and back-extracted with CHCl3 (3 × 20 mL). 5g was obtained as colorless foam (0.58 g, 0.64 mmol, 64% yield). 1H NMR (400 MHz, CDCl3) δ 8.12 (d, J = 581 Hz, 0.5H), 8.05 (d, J = 574 Hz, 0.5H), 7.82 (s, 0.5H), 7.80 (s, 0.5H), 7.44–7.31 (m, 3H), 7.31–7.22 (m, 4H), 7.22–7.13 (m, 2H), 6.79–6.67 (m, 4H), 6.18 (t, J = 6.0 Hz, 0.5H), 6.18 (t, J = 6.4 Hz, 0.5H), 5.75–5.62 (m, 0.5H), 5.62–5.51 (m, 0.5H), 4.37 (q, J = 3.7 Hz, 0.5H), 4.26 (q, J = 3.8 Hz, 0.5H), 3.75 (s, 6H), 3.50–3.25 (m, 8H), 3.02–2.83 (m, 1H), 2.83–2.74 (m, 2H), 2.74–2.58 (m, 2H), 2.03–1.87 (m, 2H), 1.83–1.53 (m, 6H), 1.20–1.07 (m, 6H); 13C{1H} NMR (100 MHz, CDCl3) δ 179.8, 166.0, 158.3, 155.9, 148.1, 148.0, 147.7, 147.6, 144.6, 144.6, 138.2, 137.9, 135.6, 130.0, 130.0, 128.1, 128.1, 127.7, 126.7, 121.3, 121.2, 113.0, 86.2, 86.1, 85.4 (d, 3JC–P = 5.8 Hz), 84.6 (d, 3JC–P = 5.8 Hz), 84.1, 84.1, 74.4, 73.5, 70.5, 63.4, 63.1, 55.1, 54.3, 48.6, 46.0, 39.0, 38.1, 35.8, 35.8, 32.4, 28.9, 26.7, 23.9; 31P{1H} NMR (162 MHz, CDCl3) δ 52.8, 52.5. HRMS (ESI/Q-TOF) m/z calcd for C35H37N5O8PS– [M–DBU–H]−, 718.2106; found 718.2087.
1,8-Diazabicyclo [5.4.0] undec-7-enium 5′-O- dimethoxytrityl-N3-benzoylthymidine 3′-H-phosphonothioate as a Mixture of (Sp)- and (Rp)-diastereomers (5t)
The crude mixture containing 5t was purified by silica gel column chromatography using AcOEt–MeOH–Et3N (100:0:1–100:1:1, v/v/v) and then CHCl3–MeOH–Et3N (100:0:1–100:3:1, v/v/v) as the eluent. Then, the fractions containing 5t were collected and concentrated under reduced pressure. CHCl3 (20 mL) was added to the residue and washed with 0.2 M DBU hydrogen carbonate aqueous solutions (3 × 20 mL). The aqueous layers were combined and back-extracted with CHCl3 (2 × 20 mL). 5t was obtained as yellow foam (0.85 g, 0.95 mmol, 95% yield). 1H NMR (400 MHz, CDCl3) δ 11.63–11.49 (br, 1H), 8.13 (d, J = 572 Hz, 0.5H), 8.08 (d, J = 578 Hz, 0.5H), 7.92 (d, J = 8.2 Hz, 2H), 7.78 (s, 0.5H), 7.77 (s, 0.5H), 7.64 (t, J = 7.3 Hz, 1H), 7.53–7.39 (m, 5H), 7.38–7.27 (m, 5H), 7.26–7.18 (m, 1H), 6.91–6.82 (m, 4H), 6.44 (t, J = 8.7 Hz, 0.5H), 6.43 (t, J = 8.9 Hz, 0.5H), 5.51–5.41 (m, 0.5H), 5.41–5.32 (m, 0.5H), 4.41 (d, J = 1.4 Hz, 0.5H), 4.30 (d, J = 2.3 Hz, 0.5H), 3.79 (s, 6H), 3.63–3.46 (m, 1H), 3.45–3.31 (m, 7H), 2.87–2.62 (m, 3H), 2.54–2.38 (m, 1H), 1.93 (quintet, J = 5.8 Hz, 2H), 1.78–1.54 (m, 6H), 1.37 (d, J = 6.9 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 169.1, 165.9, 162.8, 158.5, 158.5, 149.2, 144.3, 144.2, 135.7, 135.4, 135.3, 135.2, 135.1, 134.9, 131.5, 130.3, 130.0, 129.0, 128.2, 127.9, 126.9, 113.2, 111.0, 111.0, 86.9, 86.0 (d, 3JC–P = 5.8 Hz), 85.2 (d, 3JC–P = 6.7 Hz), 84.8, 75.5 (d, 2JC–P = 4.8 Hz), 74.6 (d, 3JC–P = 4.8 Hz), 63.5, 63.4, 55.1, 54.1, 48.4, 40.2 (d, 3JC–P = 2.9 Hz), 39.7 (d, 3JC–P = 2.9 Hz), 38.0, 32.1, 28.8, 26.6, 23.9, 19.3, 11.4; 31P{1H} NMR (162 MHz, CDCl3) δ 54.5, 53.4. HRMS (ESI/Q-TOF) m/z calcd for C38H36N2O9PS– [M–DBU–H]−, 727.1885; found 727.1884.
General Procedure for the Synthesis of 5′-O-Dimethoxytrityl-2′-O-methyl-nucleoside 3′-H-boranophosphonate
Nucleoside 2a (0.69 g, 1.0 mmol, 1.0 equiv), 2c (0.68 g, 1 mmol, 1.0 equiv), 2g (0.70 g, 1 mmol, 1.0 equiv), or 2u (0.67 g, 1 mmol, 1.0 equiv) and pyridinium H-boranophosphonate (0.28 g, 2.0 mmol, 2.0 equiv) were dried individually by repeated coevaporation with dry pyridine and dissolved together in dry pyridine (25 mL) at 0 °C under argon. Bis(2-oxo-3-oxazolidinyl) phosphinic chloride (Bop-Cl) (0.51 g, 2.0 mmol, 2.0 equiv) was added at 0 °C, and the mixture was stirred for 1 h at 0 °C. The mixture was warmed to rt and diluted with CH2Cl2 (30 mL) and washed with 1 M triethylammonium bicarbonate (TEAB) buffers (pH 7) (3 × 30 mL). The aqueous layers were combined and back-extracted with CH2Cl2 (3 × 30 mL). The organic layers were combined, dried over Na2SO4, filtered, and concentrated under reduced pressure. The residue was then purified by silica gel column chromatography (20 g of neutral silica gel) using AcOEt–MeOH–Et3N and CHCl3–MeOH–Et3N as the eluent. The residue was dissolved in CHCl3 (30 mL for 6a, 6c, and 6g) or CH2Cl2 (30 mL for 6u) and washed with 1 M TEAB buffers (3 × 30 mL). The aqueous layers were combined and back-extracted with CHCl3 (3 × 30 mL for 6a, 6c, and 6g) or CH2Cl2 (30 mL for 6u). The organic layers were combined, dried over Na2SO4, filtered, and concentrated under reduced pressure to yield 6a, 6c, 6g, or 6u
Triethylammonium 5′-O-Dimethoxytrityl-N6-benzoyl-2′-O-methyladenosine 3′-H-boranophosphonate as a Mixture of (Sp)- and (Rp)-diastereomers (6a)
The crude mixture containing 6a was purified by silica gel column chromatography using AcOEt–MeOH–Et3N (100:0:1–100:4:1, v/v/v) and then CHCl3–MeOH–Et3N (100:4:1, v/v/v) as the eluent. 6a was obtained as colorless foam (0.81 g, 0.95 mmol, 95%). 1H NMR (400 MHz, CDCl3) δ 9.09 (s, 1H), 8.70 (s, 0.5H), 8.69 (s, 0.5H), 8.24 (s, 0.5H), 8.20 (s, 0.5H), 8.02 (d, J = 7.8 Hz, 2H), 7.9–7.8 (br, 0.5H), 7.60 (t, J = 7.3 Hz, 1H), 7.51 (t, J = 7.8 Hz, 2H), 7.48–7.42 (m, 2H), 7.36–7.31 (m, 4H), 7.30–7.23 (m, 2H), 7.23–7.17 (m, 1H), 6.84–6.78 (m, 4H), 6.27 (t, J = 5.0 Hz, 0.5H), 6.27 (t, J = 5.0 Hz, 0.5H), 5.09–5.02 (m, 1H), 4.74–4.68 (m, 1H), 4.55–4.50 (m, 1H), 3.77 (s, 6H), 3.55–3.48 (m, 5H), 2.99 (q, J = 7.3 Hz, 6H), 1.27 (t, J = 7.3 Hz, 9H), 0.9–0.1 (br, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 164.5, 158.4, 152.5, 151.8, 151.7, 149.3, 144.4, 141.7, 135.6, 135.6, 135.5, 135.5, 133.6, 132.7, 130.0, 128.8, 128.1, 128.1, 127.8, 127.8, 127.7, 126.8, 123.4, 113.1, 113.1, 86.6, 86.5, 86.4, 86.4, 84.1 (d, 3JC–P = 2.9 Hz), 84.0 (d, 3JC–P = 2.9 Hz), 82.3 (d, 3JC–P = 2.9 Hz), 81.9 (d, 3JC–P = 2.9 Hz), 73.1 (d, 2JC–P = 5.8 Hz), 72.2 (d, 2JC–P = 5.8 Hz), 63.0, 62.8, 58.5, 58.4, 55.1, 45.2, 8.4; 31P{1H} NMR (162 MHz, CDCl3) δ 108.8–103.9 (br). HRMS (ESI/Q-TOF) m/z calcd for C39H40BN5O8P– [M–Et3N–H]−, 748.2713; found 748.2691.
Triethylammonium 5′-O-Dimethoxytrityl-N4-benzoyl-2′-O-methylcytidine 3′-H-boranophosphonate as a Mixture of (Sp)- and (Rp)-diastereomers (6c)
The crude mixture containing 6c was purified by silica gel column chromatography using AcOEt–MeOH–Et3N (100:0:1–100:4:1, v/v/v) and then CHCl3–MeOH–Et3N (100:4:1, v/v/v) as the eluent. 6c was obtained as colorless foam (0.75 g, 0.90 mmol, 90%). 1H NMR (400 MHz, CDCl3) δ 8.65 (d, J = 7.8 Hz, 0.5H), 8.59 (d, J = 7.3 Hz, 0.5H), 7.95–7.86 (m, 2H), 7.86–7.77 (br, 0.5H), 7.51 (t, J = 7.8 Hz, 2H), 7.44 (dd, J = 7.1, 5.7 Hz, 2H), 7.39–7.24 (m, 7H), 7.20–7.02 (br, 0.5H), 6.92–6.85 (m, 4H), 6.05 (s, 0.5H), 6.02 (s, 0.5H), 5.05–4.92 (m, 0.5H), 4.82–4.75 (m, 0.5H), 4.40 (dd, J = 2.1, 8.9 Hz, 1H), 4.12–4.03 (m, 1H), 3.83 (s, 6H), 3.67 (s, 3H), 3.63–3.53 (m, 2H), 3.01 (q, J = 7.2 Hz, 6H), 1.27 (t, J = 7.3 Hz, 9H), 0.90–0.10 (br, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 166.1, 162.1, 158.5, 145.0, 144.0, 135.6, 135.5, 135.2, 135.1,133.1, 133.0, 130.2, 130.0, 130.0, 129.9, 128.9, 128.3, 128.1, 128.0, 128.0, 127.4, 127.0, 113.3, 113.2, 96.5, 96.4, 88.9, 88.7, 86.9, 86.9, 83.3, 82.7, 81.4 (d, 3JC–P = 5.8 Hz), 81.3 (d, 3JC–P = 5.2 Hz), 72.9 (d, 2JC–P = 9.6 Hz), 69.2 (d, 2JC–P = 4.8 Hz), 60.5, 58.5, 58.4, 55.1, 45.3, 8.4; 31P{1H} NMR (162 MHz, CDCl3) δ 111.8–102.0. HRMS (ESI/Q-TOF) m/z calcd for C38H40BN3O9P– [M–Et3N–H]−, 724.2601; found 724.2591.
Triethylammonium 5′-O-Dimethoxytrityl-N2-isobutyryl-2′-O-methylguanosine 3′-H-boranophosphonate as a Mixture of (Sp)- and (Rp)-diastereomers (6g)
The crude mixture containing 6g was purified by silica gel column chromatography using AcOEt–MeOH–Et3N (100:2:1, v/v/v) and then CHCl3–MeOH–Et3N (100:3:1–100:5:1, v/v/v) as the eluent. 6g was obtained as colorless foam (0.78 g, 0.92 mmol, 92%). 1H NMR (400 MHz, CDCl3) δ 12.54–11.63 (br, 1H), 10.47–9.66 (br, 1H), 7.88 (s, 0.5H), 7.87 (s, 0.5H), 7.81–7.67 (br, 0.5 H), 7.53–7.36 (m, 2H), 7.34–7.28 (m, 4H), 7.26–7.13 (m, 3H), 6.93–6.83 (br, 0.5H), 6.83–6.66 (m, 4H), 5.93 (d, J = 4.6 Hz, 0.5H), 5.90 (d, J = 4.6 Hz, 0.5H), 5.37–5.29 (m, 0.5H), 5.29–5.20 (m, 0.5H), 4.58 (t, J = 4.8 Hz, 0.5H), 4.56 (t, J = 4.8 Hz, 0.5H), 4.44–4.36 (m, 0.5H), 4.36–4.28 (m, 0.5H), 3.75, 3.75, 3.74 (s, s, s, 6H), 3.54–3.37 (m, 4H), 3.37–3.22 (m, 1H), 3.00 (q, J = 7.2 Hz, 6H), 2.62–2.40 (m, 1H), 1.25 (t, J = 7.3 Hz, 9H), 1.11 (d, J = 6.9 Hz, 3H), 1.04 (d, J = 5.5 Hz, 3H), 0.95–0.10 (br, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ179.6, 179.5, 158.3, 158.3, 155.7, 148.2, 147.6, 147.6, 144.5, 138.1, 137.9, 138.1, 137.9, 135.6, 135.5, 135.4 129.8, 127.9, 127.6, 127.6 126.6, 126.6, 121.3, 121.2, 112.9, 112.8, 86.5, 86.3, 86.2, 86.1, 83.2, 82.9, 82.3, 82.2, 72.5 (d, 2JC–P = 4.8 Hz), 72.0 (d, 2JC–P = 3.9 Hz), 62.7, 62.4, 58.4, 55.0, 45.4, 35.6, 35.6, 18.6, 8.5; 31P{1H} NMR (162 MHz, CDCl3) δ 108.0–102.0. HRMS (ESI/Q-TOF) m/z calcd for C36H42BN5O9P– [M–Et3N–H]−, 730.2819; found 730.2791.
Triethylammonium 5′-O-Dimethoxytrityl-N3-benzoyl-2′-O-methyluridine 3′-H-boranophosphonate as a Mixture of (Sp)- and (Rp)-diastereomers (6u)
The crude mixture containing 6u was purified by silica gel column chromatography using AcOEt–MeOH–Et3N (100:0:1, v/v/v) and then CHCl3–MeOH–Et3N (100:0:1–100:1:1, v/v/v) as the eluent. 6u was obtained as colorless foam (0.62 g, 0.74 mmol, 74%). 1H NMR (400 MHz, CDCl3) δ 8.23 (d, J = 8.2 Hz, 0.5H), 8.17 (d, J = 8.2 Hz, 0.5H), 8.01–7.90 (m, 2H), 7.65 (t, J = 7.3 Hz, 1H), 7.51 (t, J = 7.8 Hz, 2H), 7.47–7.38 (m, 2H), 7.38–7.21 (m, 7H), 6.87 (m, 4H), 5.97 (d, J = 2.3 Hz, 0.5 H) 5.97 (d, J = 2.1 Hz, 0.5 H), 5.30 (d, J = 2.7 Hz, 0.5H), 5.27 (d, J = 2.7 Hz, 0.5H), 5.10–5.03 (m, 0.5 H), 4.95–4.86 (m, 0.5 H), 4.36 (m, 1H), 4.15–3.91 (m, 1H), 3.80 (s, 6H), 3.73–3.58 (m, 1H), 3.55 (d, J = 4.6 Hz, 4H), 3.14–2.91 (m, 6H), 1.44–1.18 (m, 9H), 1.03–0.16 (m, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 168.9, 168.8, 162.0, 158.6, 158.6, 149.1, 144.2, 144.2, 140.1, 135.2, 135.1, 135.1, 134.9, 134.8, 131.3, 130.5, 130.2, 130.1, 130.0, 129.1, 128.2, 128.0, 128.0, 127.1, 113.3, 101.8, 101.8, 87.7, 87.1, 83.4, 82.9, 82.1, 73.1 (d, 2JC–P = 8.7 Hz), 69.9 (d, 2JC–P = 4.8 Hz), 61.0, 60.5, 58.4, 58.3, 55.2, 45.3, 8.5; 31P{1H} NMR (162 MHz, CDCl3) δ 111.0–103.0. HRMS (ESI/Q-TOF) m/z calcd for C38H39BN2O10P– [M–Et3N–H]−, 725.2441; found 725.2427.
General Procedure for the Manual Solid-Phase Synthesis of TPST Dimer (Table 1)
HCP-loaded 5′-O-DMTr-N3-benzoylthymidine, via a succinyl linker (0.5 μmol), in a reaction vessel was treated with 3% DCA in dry CH2Cl2 (5 × 12 s, 1 mL each) and washed with dry CH2Cl2 (4 × 1 mL) and CH3CN (4 × 1 mL). Thereafter, it was dried in vacuo for 10 min. To the reaction vessel, the 3′-H-phosphonothioate thymidine monomer 5t (19.7 mg, 20 μmol, 40 equiv, 0.1 M) and condensing reagent (50 μmol, 100 equiv, 0.25 M) (BOMP, 23.3 mg, for entry 1; MNTP, 22.3 mg for entry 2, PyNTP, 25.0 mg for entries 3–7) were added. Then, a solution of 2,6-lutidine (0.6 M, 120 μmol for entry 1–3), pyridine (0.6 M, 120 μmol for entry 4), quinoline (0.6 M, 120 μmol for entry 5, 1.8 M, 355 μmol for entry 6), or without base (entry 7) in dry CH3CN (200 μL) was added under Ar, and the reaction vessel was stirred slowly with hands for 3 min. The HCP was washed with dry CH3CN (4 × 1 mL) and CH2Cl2 (4 × 1 mL), and the detritylation reaction was carried out by treatment with 3% DCA in dry CH2Cl2–Et3SiH (1:1, v/v) (5 × 12 s, 1 mL each). Thereafter, the HCP was washed with dry CH2Cl2 (4 × 1 mL) and CH3CN (4 × 1 mL), and dried in vacuo for 10 min. The H-phosphonothioate internucleotide linkage was oxidized by treatment with a solution (500 μL) of 0.1 M TEA in CCl4–2,6-lutidine–H2O (5:12.5:1, v/v/v) for 90 min. The HCP was washed with dry CH3CN (4 × 1 mL) and CH2Cl2 (4 × 1 mL), and dried in vacuo. The HCP was treated with concentrated aqueous NH3–EtOH (3:1, v/v, 5 mL) at rt for 3 h, filtered, and washed with EtOH (3 × 1 mL). The filtrate and washings were combined and concentrated under reduced pressure. The residue was analyzed by RP-HPLC. RP-HPLC was performed with a linear gradient of 0–30% CH3CN for 60 min in 0.1 M triethylammonium acetate (TEAA) buffer (pH 7.0) at 30 °C with a flow rate of 0.5 mL/min using a C18 column (100 Å, 3.9 mm × 150 mm).
General Procedure for the Manual Solid-Phase Synthesis of NPST Dimers (Table 2)
HCP-loaded 5′-O-DMTr-N3-benzoylthymidine, via a succinyl linker (0.5 μmol, 30.65 μmol/g), in a reaction vessel was treated 3% DCA in dry CH2Cl2 (5 × 12 s, 1 mL each) and washed with dry CH2Cl2 (4 × 1 mL) and CH3CN (4 × 1 mL). Thereafter, it was dried in vacuo for 10 min. To the reaction vessel, the 3′-H-phosphonothioate monomer unit (5a (19.0 mg, 20 μmol, 40 equiv, 0.1 M), 5c (17.8 mg, 20 μmol, 40 equiv, 0.1 M), or 5g (18.1 mg, 20 μmol, 40 equiv, 0.1 M for entries 5 and 6, 40 μmol, 80 equiv, 0.2 M for entry 7)) and PyNTP (25 mg, 50 μmol, 100 equiv, 0.25 M for entries 1–6; 50 mg, 100 μmol, 200 equiv, 0.5 M for entry 7) as condensing reagent were added. Then, a solution (200 μL) of quinoline (1.8 M, 355 μmol for entries 1, 3, and 5) or without base (for entries 2, 4, 6, and 7) in dry CH3CN was added under Ar, and the reaction vessel was stirred slowly with hands for 3 min. The HCP was washed with dry CH3CN (4 × 1 mL) and CH2Cl2 (4 × 1 mL), and the detritylation reaction was carried out by treatment with 3% DCA in dry CH2Cl2–Et3SiH (1:1, v/v) (4 × 15 s, 1 mL each). Therefore, the HCP was washed with dry CH2Cl2 (4 × 1 mL) and CH3CN (4 × 1 mL), and dried in vacuo for 10 min. The H-phosphonothioate internucleotide linkage was oxidized by treatment with a solution (500 μL) of 0.1 M TEA in CCl4–2,6-lutidine–H2O (5:12.5:1, v/v/v) for 90 min. The HCP was washed with dry CH3CN (4 × 1 mL) and CH2Cl2 (4 × 1 mL), and dried in vacuo. The HCP was treated with concentrated aqueous NH3–EtOH (3:1, v/v, 5 mL) at rt for 12 h (entries 1–4) or at 50 °C in a thermostatic chamber for 12 h (entries 5–7), filtered, and washed with EtOH (3 × 1 mL). The filtrate and washings were combined and concentrated under reduced pressure. The residue was analyzed by RP-HPLC. RP-HPLC was performed with a linear gradient of 0–30% CH3CN for 60 min in 0.1 M triethylammonium acetate (TEAA) buffer (pH 7.0) at 30 °C with a flow rate of 0.5 mL/min using a C18 column (100 Å, 3.9 mm × 150 mm).
General Procedure for the Manual Solid-Phase Synthesis of NPBT Dimers Using 5′-O-Dimethoxytrityl-2′-O-methyl nucleoside 3′-H-boranophosphonate (Table 3)
HCP-loaded 5′-O-DMTr-N3-benzoylthymidine, via a succinyl linker (0.5 μmol, 30.65 μmol/g), in a reaction vessel was treated 3% DCA in dry CH2Cl2 (5 × 12 s, 1 mL each) and washed with dry CH2Cl2 (4 × 1 mL) and CH3CN (4 × 1 mL). Thereafter, it was dried in vacuo for 10 min. To the reaction vessel, the 2′-O-methyl-nucleoside 3′-H-boranophosphonate monomer unit (6a (17.1 mg, 20 μmol, 40 equiv, 0.1 M), 6c (16.7 mg, 20 μmol, 40 equiv, 0.1 M), 6g (16.9 mg, 20 μmol, 40 equiv, 0.1 M), or 6u (16.7 mg, 20 μmol, 40 equiv, 0.1 M)) and MNTP (22.3 mg, 50 μmol, 100 equiv) as a condensing reagent were added and 2,6-lutidine (0.6 M, 120 μmol) in dry CH3CN (200 μL) was added under Ar, and the reaction vessel was stirred slowly with hands for 3 min. The HCP was washed with dry CH3CN (4 × 1 mL) and CH2Cl2 (4 × 1 mL), and the detritylation reaction was carried out by treatment with 3% DCA in dry CH2Cl2–Et3SiH (1:1, v/v) (4 × 15 s, 1 mL each). Therefore, the HCP was washed with dry CH2Cl2 (4 × 1 mL) and CH3CN (4 × 1 mL), and dried in vacuo for 10 min. The H-boranophosphonate internucleotide linkage was oxidized by treatment with a solution (500 μL) of 0.1 M TEA in CCl4–2,6-lutidine–H2O (5:12.5:1, v/v/v) for 90 min. The HCP was washed with dry CH3CN (4 × 1 mL) and CH2Cl2 (4 × 1 mL), and dried in vacuo. The HCP was treated with concentrated aqueous NH3–EtOH (3:1, v/v, 5 mL) at rt for 12 h (entries 1 and 2), at 50 °C in a thermostatic chamber for 12 h (entry 3), or rt for 3 h (entry 4), filtered, and washed with EtOH (3 × 1 mL). The filtrate and washings were combined and concentrated under reduced pressure. The residue was analyzed by RP-HPLC. RP-HPLC was performed with a linear gradient of 0–30% CH3CN for 60 min in 0.1 M triethylammonium acetate (TEAA) buffer (pH 7.0) at 30 °C with a flow rate of 0.5 mL/min using a C18 column (100 Å, 3.9 mm × 150 mm).
HRMS (ESI/Q-TOF): m/z calcd for APBT (15) [M–H]−, 582.1890; found 582.1879. calcd for CPBT (16) [M–H]−, 558.1778; found 558.1765. calcd for GPBT (17) [M–H]−, 598.1839; found 598.1824. calcd for UPBT (18) [M–H]−, 559.1618; found 559.1607.
Synthesis of PB/PS/PO Chimeric Tetramer ODNs and Chimeric Dodecamer ODNs Containing PB Modifications (PB/PS/PO, PB/PO, PB/PS-ODNs, and PB/PS/PO-gapmer) (Table 4)
HCP-loaded 5′-O-DMTr-N3–benzoylthymidine (0.5 μmol, 30.65 μmol/g, entries 1–3), 5′-O-DMTr-N4-isobutyrylcytidine (0.5 μmol, 22.03 μmol/g, entries 4–6) or 5′-O-DMTr-2′-O-methyl N4-benzoylcytidine (0.5 μmol, 30.78 μmol/g, entry 7) via a succinyl linker, was treated with 3% DCA in dry CH2Cl2–Et3SiH (1:1, v/v, 4 × 15 s, 1 mL each) and washed with dry CH2Cl2 (4 × 1 mL) and CH3CN (4 × 1 mL), and then dried in vacuo for 10 min. Chain elongations were conducted by repeating steps (a) and (b) 3 times (for the synthesis of 19 and 20) or 11 times (for the synthesis of 21–25).
(a) Condensation step: to introduce a PB, PS, and PO linkages, the H-boranophosphonate, H-phosphonothioate, or H-phosphonate monomer and the condensing reagent were added to the reaction vessel and a condensation reaction was performed with optimized condensation conditions as shown in Table 7 in CH3CN for 3 min.
Table 7. Condensation Reaction Conditions of Each Monomer.
| monomer | quantity | condensing reagent | quantity | base | concentration |
|---|---|---|---|---|---|
| 3a, c, g, and t | 40 equiv, 20 μmol | MNTP | 100 equiv, 50 μmol | 2,6-lutidine | 0.6 M |
| 4a, c, g, and t | 40 equiv, 20 μmol | MNTP | 100 equiv, 50 μmol | 2,6-lutidine | 0.6 M |
| 5a, c | 40 equiv, 20 μmol | PyNTP | 100 equiv, 50 μmol | quinoline | 1.8 M |
| 5g | 80 equiv, 40 μmol | PyNTP | 200 equiv, 100 μmol | ||
| 5t | 40 equiv, 20 μmol | PyNTP | 100 equiv, 50 μmol | ||
| 6a, c, g, and u | 40 equiv, 20 μmol | MNTP | 100 equiv, 50 μmol | 2,6-lutidine | 0.6 M |
(b) Detritylation step: to the reaction vessel, 3% DCA in dry CH2Cl2–Et3SiH (1:1, v/v) (4 × 15 s, 1 mL) was added, and the HCP was washed with dry CH2Cl2 (4 × 1 mL) and CH3CN (4 × 1 mL), and then dried in vacuo for 10 min.
After the designed length was achieved, the resultant internucleotide linkages were oxidized with a solution (500 μL) of 0.1 M Et3N in CCl4–2,6-lutidine–H2O (5:12.5:1, v/v/v) for 90 min. The HCP was washed with dry CH2Cl2 (4 × 1 mL) and CH3CN (4 × 1 mL), and then dried in vacuo for 10 min. The HCP was treated with concentrated aqueous NH3–EtOH (3:1, v/v, 5 mL) at 50 °C in a thermostatic chamber for at least 20 h, filtered, and washed with EtOH. The filtrate and washings were combined and concentrated under reduced pressure. The residue was analyzed by RP-HPLC or anion-exchange HPLC, and then purified by anion-exchange HPLC (for 22) or reverse-phase HPLC (for 21, 23–25). RP-HPLC was performed with a linear gradient of 5–40% MeOH in a buffer containing 100 mM hexafluoroisopropanol (HFIP) and 8 mM triethylamine at 60 °C for 20 min with a flow rate of 0.5 mL/min. Anion-exchange HPLC was performed with a linear gradient of 0–0.5 M NaCl of 30% iPOH in a 10 mM Tris-HCl buffer (pH 7.5) at 30 °C for 40 min.
Isolated yields: 6% (d(CPSAPSGPSTPSCPBAPBGPBTPBCPOAPOGPOT) (21)); 19% (d(GPBCPSAPBTPOTPOGPOGPOTPSAPBTPSTPBC) (22)); 5% (d(GPBCPSAPBTPSTPSGPBGPBTPSAPBTPSTPBC) (23)); 19% (d(GPBCPBAPBTPOTPOGPOGPOTPBAPBTPBTPBC) (24)); 13% (GPBCPBAPB d(TPSTPOGPOGPOTPSAPB)UPBUPBC (25)); HRMS (ESI/Q-TOF): m/z calcd for d(CPSAPBGPOT) (19) [M–2H]2– 592.6236; found 592.6209. calcd for d(CPOAPBGPST) (20) [M–2H]2–, 592.6236; found 592.6217. calcd for d(CPSAPSGPSTPSCPBAPBGPBTPBCPOAPOGPOT) (21) [M–6H]6–, 615.7786; found 615.7767. calcd for d(GPBCPSAPBTPOTPOGPOGPOTPSAPBTPSTPBC) (22) [M–6H]6–, 614.1138; found 614.1136. calcd for d(GPBCPSAPBTPSTPSGPBGPBTPSAPBTPSTPBC) (23) [M–6H]6–, 618.6194; found 618.6175. calcd for d(GPBCPBAPBTPOTPOGPOGPOTPBAPBTPBTPBC) (24) [M–6H]6–, 604.9781; found 604.9774. calcd for GPBCPBAPB d(TPSTPOGPOGPOTPSAPB)UPBUPBC (25) [M–6H]6–, 635.9695; found. 635.9670.
Thermal Denaturation Study
A solution containing pairs of complementary strands (4.0 μM, 0.6 nmol each) and 100 mM NaCl in 10 mM NaH2PO4–Na2HPO4 buffer (pH 7.0) was heated for 10 min at 95 °C and cooled to 0 °C at a rate of 0.5 °C/min, and then left at 0 °C for 10 min. Denaturation tests were carried out in a 1 cm path length quartz cell. The UV absorbance values at 260 nm were recorded at a rate of 0.2 °C/min from 0 to 95 °C. The Tm value was determined from the peak value of the first derivative of the thermal melting curve. The sequence of cRNA was r(GPOAPOAPOUPOAPOCPOCPOAPOAPOUPOGPOC) or r(GPOAPOAPOCPOAPOCPOCPOAPOAPOUPOGPOC); subscript PO = phosphate.
Nuclease Resistance Assay
In the nuclease resistance assay, snake venom phosphodiesterase (SVPDE) from C. adamanteus was used. An aqueous solution of SVPDE solution (4 × 10–4 U in 45 μL) and a 200 mM Tris-HCl buffer (pH 8.5) at 37 °C containing 30 mM MgCl2 (50 μL) were successively added to 0.1 mM aqueous solution of each ODNs (5 μL, 0.5 nmol). After 12 h, the solution was heated to 95 °C for 1 min to denaturate SVPDE and then cooled to 4 °C. The mixture was diluted with 0.1 M TEAA buffer (80 μL) and CH3CN (20 μL) and then analyzed using RP-HPLC. RP-HPLC was performed with a linear gradient of 0–40% CH3CN for 60 min in 0.1 M TEAA buffer (pH 7.0) at 30 °C with a flow rate of 0.5 mL/min.
RNase H Activity Evaluation
Cleavage experiments of cRNA by RNase H were conducted using 0.5 μM ODN (22–29, 0.05 nmol) and 5 μM cRNA (30, 0.5 nmol) in a 10 mM Tris buffer containing 100 mM NaCl, 0.5 mM MgCl2, and 0.1 mM benzamide as an internal standard at pH 7.2, and the final volume of the solution was 100 μL. A solution of each one of ODNs (22–29) and 10 equiv of cRNA in the buffer was heated for 10 min at 95 °C, cooled to 0 °C at a rate of 0.5 °C/min, and then left at 37 °C for 10 min. A solution of 60 U/μL RNase H from E. coli in the buffer was added to the solution to afford a final concentration of 50 or 25 U/mL and left at 37 °C for 30 min. RNase H was denatured by heating at 95 °C for 1 min, and then the mixture was cooled and analyzed by RP-HPLC. RP-HPLC was performed with a linear gradient of 0–11% MeCN over 44 min followed by 11–40% over 16 min in 0.1 M TEAA buffer (pH 7.0) at 50 °C with a flow rate of 0.5 mL/min.
RNase H Activity Evaluation (One Base Mismatch)
Cleavage experiments of cRNA by RNase H were conducted using 0.5 μM ODN (25 or 26, 0.05 nmol) and 5 μM RNA (31, 0.5 nmol) in a 10 mM Tris buffer containing 100 mM NaCl, 0.5 mM MgCl2, and 0.1 mM benzamide as an internal standard at pH 7.2, and the final volume of the solution was 100 μL. A solution of each one of the ODNs (25, 26) and 10 equiv of RNA (31) in the buffer was heated for 10 min at 95 °C, cooled to 0 °C at a rate of 0.5 °C/min, and then left at 37 °C for 10 min. A solution of 60 U/μL RNase H from Escherichia coli in the buffer was added to the solution to afford a final concentration of 50 U/mL and left at 37 °C for 30 min. RNase H was denatured by heating at 95 °C for 1 min, and then the mixture was cooled and analyzed by RP-HPLC. RP-HPLC was performed with a linear gradient of 0–11% MeCN over 44 min followed by 11–40% over 16 min in 0.1 M TEAA buffer (pH 7.0) at 50 °C with a flow rate of 0.5 mL/min.
Acknowledgments
The authors thank Noriko Sawabe (Tokyo University of Science) for her technical assistance with the NMR measurements. They also thank Dr. Yayoi Yoshimura (Tokyo University of Science) for the mass spectra measurements. They thank Enago (www.enago.jp) for the English language review. This research was partially supported by AMED under Grant Number JP21ae0121026.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.1c01812.
Copies of 1H, 13C, 31P NMR spectra for new compounds, HPLC profiles, and results of thermal denaturation tests (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Kilanowska A.; Studzińska S. In Vivo and in Vitro studies of Antisense Oligonucleotides - a Review. RSC Adv. 2020, 10, 34501–34516. 10.1039/D0RA04978F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamecnik P. C.; Stephenson M. L. Inhibition of Rous Sarcoma Virus Replication and Cell Transformation by a Specific Oligodeoxynucleotide. Proc. Natl. Acad. Sci. U.S.A. 1978, 75, 280–284. 10.1073/pnas.75.1.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H.; Lima W. F.; Zhang H.; Fan A.; Sun H.; Crooke S. T. Determination of the Role of the Human RNase H1 in the Pharmacology of DNA-like Antisense Drugs. J. Biol. Chem. 2004, 279, 17181–17189. 10.1074/jbc.M311683200. [DOI] [PubMed] [Google Scholar]
- Nowotny M.; Gaidamakov S. A.; Ghirlando R.; Cerritelli S. M.; Crouch R. J.; Yang W. Structure of Human RNase H1 Complexed with an RNA/DNA Hybrid: Insight into HIV Reverse Transcription. Mol. Cell 2007, 28, 264–276. 10.1016/j.molcel.2007.08.015. [DOI] [PubMed] [Google Scholar]
- Sharma V. K.; Sharma R. K.; Singh S. K. Antisense Oligonucleotides: Modifications and Clinical Trials. Med. Chem. Commun. 2014, 5, 1454–1471. 10.1039/C4MD00184B. [DOI] [Google Scholar]
- Crooke S. T.; Vickers T. A.; Liang X. H. Phosphorothioate Modified Oligonucleotide-Protein Interactions. Nucleic Acids Res. 2020, 48, 5235–5253. 10.1093/nar/gkaa299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaus H. J.; Gupta R.; Chappell A. E.; Østergaard M. E.; Swayze E. E.; Seth P. P. Characterization of the Interactions of Chemically-Modified Therapeutic Nucleic Acids with Plasma Proteins Using a Fluorescence Polarization Assay. Nucleic Acids Res. 2019, 47, 1110–1122. 10.1093/nar/gky1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooke S. T.; Wang S.; Vickers T. A.; Shen W.; Liang X. H. Cellular Uptake and Trafficking of Antisense Oligonucleotides. Nat. Biotechnol. 2017, 35, 230–237. 10.1038/nbt.3779. [DOI] [PubMed] [Google Scholar]
- Liang X. H.; Sun H.; Hsu C. W.; Nichols J. G.; Vickers T. A.; De Hoyos C. L.; Crooke S. T. Golgi-Endosome Transport Mediated by M6PR Facilitates Release of Antisense Oligonucleotides from Endosomes. Nucleic Acids Res. 2020, 48, 1372–1391. 10.1093/nar/gkz1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler J.; Stessl M.; Amartey J.; Noe C. R. Off-Target Effects Related to the Phosphorothioate Modification of Nucleic Acids. ChemMedChem 2010, 5, 1344–1352. 10.1002/cmdc.201000156. [DOI] [PubMed] [Google Scholar]
- Levin A. A. A Review of Issues in the Pharmacokinetics and Toxicology of Phosphorothioate Antisense Oligonucleotides. Biochim. Biophys. Acta, Gene Struct. Expression 1999, 1489, 69–84. 10.1016/S0167-4781(99)00140-2. [DOI] [PubMed] [Google Scholar]
- Prakash T.; Bhat B. 2-Modified Oligonucleotides for Antisense Therapeutics. Curr. Top. Med. Chem. 2007, 7, 641–649. 10.2174/156802607780487713. [DOI] [PubMed] [Google Scholar]
- Singh S. K.; Wengel J. Universality of LNA-Mediated High-Affinity Nucleic Acid Recognition. Chem. Commun. 1998, 1247–1248. 10.1039/A801571F. [DOI] [Google Scholar]
- Singh S. K.; Koshkin A. A.; Wengel J.; Nielsen P. LNA (Locked Nucleic Acids): Synthesis and High-Affinity Nucleic Acid Recognition Nucleic Acids), Is Introduced. Following the Watson – Crick Base Pairing Rules, LNA Forms Duplexes with Complemen- Stabilities and Generally Improved Selectivities. Chem. Commun. 1998, 455–456. 10.1039/a708608c. [DOI] [Google Scholar]
- Wengel J.; Koshkin A.; Singh S. K.; Nielsen P.; Meldgaard M.; Rajwanshi V. K.; Kumar R.; Skouv J.; Nielsen C. B.; Jacobsen J. P.; Jacobsen N.; Olsen C. E. LNA (Locked Nucleic Acid). Nucleosides Nucleotides 1999, 18, 1365–1370. 10.1080/07328319908044718. [DOI] [Google Scholar]
- Crooke S. T.; Baker B. F.; Crooke R. M.; Liang X. H. Antisense Technology: An Overview and Prospectus. Nat. Rev. Drug Discovery 2021, 20, 427–453. 10.1038/s41573-021-00162-z. [DOI] [PubMed] [Google Scholar]
- Crooke S. T.; Seth P. P.; Vickers T. A.; Liang X. H. The Interaction of Phosphorothioate-Containing RNA Targeted Drugs with Proteins Is a Critical Determinant of the Therapeutic Effects of These Agents. J. Am. Chem. Soc. 2020, 142, 14754–14771. 10.1021/jacs.0c04928. [DOI] [PubMed] [Google Scholar]
- Bennett C. F.Antisense Drug Technology Principles, strategies, and Applications; Crooke S. T., Ed.; CRC Press: Florida, 2008; pp 276–277. [Google Scholar]
- Sergueev D. S.; Shaw B. R. H-Phosphonate Approach for Solid-Phase Synthesis of Oligodeoxyribonucleoside Boranophosphates and Their Characterization. J. Am. Chem. Soc. 1998, 120, 9417–9427. 10.1021/ja9814927. [DOI] [Google Scholar]
- Hall I. H.; Burnham B. S.; Rajendran K. G.; Chen S. Y.; Sood A.; Spielvogel B. F.; Shaw B. R. Hypolipidemic Activity of Boronated Nucleosides Nucleotides in Rodents. Biomed. Pharmacother. 1993, 47, 79–87. 10.1016/0753-3322(93)90295-V. [DOI] [PubMed] [Google Scholar]
- Hall A. H. S.; Wan J.; Shaughnessy E. E.; Shaw B. R.; Alexander K. A. RNA Interference Using Boranophosphate SiRNAs: Structure-Activity Relationships. Nucleic Acids Res. 2004, 32, 5991–6000. 10.1093/nar/gkh936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu M.; Saigo K.; Wada T. Solid-Phase Synthesis of Oligodeoxyribonucleoside Boranophosphates by the Boranophosphotriester Method. J. Org. Chem. 2006, 71, 4262–4269. 10.1021/jo0603779. [DOI] [PubMed] [Google Scholar]
- Higson A. P.; Sierzchala A.; Brummel H.; Zhao Z.; Caruthers M. H. Synthesis of an Oligothymidylate Containing Boranophosphate Linkages. Tetrahedron Lett. 1998, 39, 3899–3902. 10.1016/S0040-4039(98)00687-X. [DOI] [Google Scholar]
- McCuen H. B.; Noe M. S.; Olesiak M.; Sierzchala A. B.; Caruthers M. H.; Higson A. P. Synthesis and Biochemical Activity of New Oligonucleotide Analogs. Phosphorus, Sulfur Silicon Relat. Elem. 2008, 183, 349–363. 10.1080/10426500701734745. [DOI] [Google Scholar]
- McCuen H. B.; Noé M. S.; Sierzchala A. B.; Higson A. P.; Caruthers M. H. Synthesis of Mixed Sequence Borane Phosphonate DNA. J. Am. Chem. Soc. 2006, 128, 8138–8139. 10.1021/ja061757e. [DOI] [PubMed] [Google Scholar]
- Roy S.; Olesiak M.; Shang S.; Caruthers M. H. Silver Nanoassemblies Constructed from Boranephosphonate DNA. J. Am. Chem. Soc. 2013, 135, 6234–6241. 10.1021/ja400898s. [DOI] [PubMed] [Google Scholar]
- Sato K.; Imai H.; Shuto T.; Hara R. I.; Wada T. Solid-Phase Synthesis of Phosphate/Boranophosphate Chimeric DNAs Using the H-Phosphonate- H-Boranophosphonate Method. J. Org. Chem. 2019, 84, 15032–15041. 10.1021/acs.joc.9b01257. [DOI] [PubMed] [Google Scholar]
- Migawa M. T.; Shen W.; Brad Wan W.; Vasquez G.; Oestergaard M. E.; Low A.; De Hoyos C. L.; Gupta R.; Murray S.; Tanowitz M.; Bell M.; Nichols J. G.; Gaus H.; Xue-Hai L.; Swayze E. E.; Crooke S. T.; Seth P. P. Site-Specific Replacement of Phosphorothioate with Alkyl Phosphonate Linkages Enhances the Therapeutic Profile of Gapmer ASOs by Modulating Interactions with Cellular Proteins. Nucleic Acids Res. 2019, 47, 5465–5479. 10.1093/nar/gkz247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelobanov B. P.; Burakova E. A.; Prokhorova D. V.; Fokina A. A.; Stetsenko D. A. New Oligodeoxynucleotide Derivatives Containing N-(Methanesulfonyl)-Phosphoramidate (Mesyl Phosphoramidate) Internucleotide Group. Russ. J. Bioorg. Chem. 2017, 43, 664–668. 10.1134/S1068162017060024. [DOI] [Google Scholar]
- Anderson B. A.; Freestone G. C.; Low A.; De-Hoyos C. L.; Drury W. J. III; Østergaard M. E.; Migawa M. T.; Fazio M.; Wan W. B.; Berdeja A.; Scandalis E.; Burel S. A.; Vickers T. A.; Crooke S. T.; Swayze E. E.; Liang X.; Seth P. P. Towards next Generation Antisense Oligonucleotides: Mesylphosphoramidate Modification Improves Therapeutic Index and Duration of Effect of Gapmer Antisense Oligonucleotides. Nucleic Acids Res. 2021, 49, 9026–9041. 10.1093/nar/gkab718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergueeva Z. A.; Sergueev D. S.; Shaw B. R. Borane-Amine Complexes - Versatile Reagents in the Chemistry of Nucleic Acids and Their Analogs. Nucleosides, Nucleotides Nucleic Acids 2001, 20, 941–945. 10.1081/NCN-100002464. [DOI] [PubMed] [Google Scholar]
- Higashida R.; Oka N.; Kawanaka T.; Wada T. Nucleoside H-Boranophosphonates: A New Class of Boron-Containing Nucleotide Analogues. Chem. Commun. 2009, 10, 2466–2468. 10.1039/b901045a. [DOI] [PubMed] [Google Scholar]
- Uehara S.; Hiura S.; Higashida R.; Oka N.; Wada T. Solid-Phase Synthesis of P-Boronated Oligonucleotides by the H-Boranophosphonate Method. J. Org. Chem. 2014, 79, 3465–3472. 10.1021/jo500185b. [DOI] [PubMed] [Google Scholar]
- Stawinski J.; Thelin M.; Zain R. Nucleoside H-phosphonates. X. Studies On Nucleoside Hydrogenphosphonothioate Diester Synthesis. Tetrahedron Lett. 1989, 30, 2157–2160. 10.1016/s0040-4039(01)93737-2. [DOI] [Google Scholar]
- Stawinski J.; Thelin M.; Westman E.; Zain R. Nucleoside H-Phosphonates. 12. Synthesis of Nucleoside 3′-(Hydrogen-Phosphonothioate) Monoesters via Phosphinate Intermediates. J. Org. Chem. 1990, 55, 3503–3506. 10.1021/jo00298a023. [DOI] [Google Scholar]
- Zain R.; Stawiński J. Nucleoside H-Phosphonates. 17. Synthetic and 31P NMR Studies on the Preparation of Dinucleoside H-Phosphonothioates. J. Org. Chem. 1996, 61, 6617–6622. 10.1021/jo960810m. [DOI] [PubMed] [Google Scholar]
- Seeberger P. H.; Caruthers M. H.; et al. Synthesis Of Phosphorodithioate DNA by the H-Phosphonothioate Method. Tetrahedron 1999, 55, 5759–5772. 10.1016/s0040-4020(99)00241-0. [DOI] [Google Scholar]
- Kamaike K.; Hirose K.; Kayama Y.; Kawashima E. Synthesis of Oligonucleoside Phosphorodithioates by the H-Phosphonothioate Method. Tetrahedron Lett. 2004, 45, 5803–5806. 10.1016/j.tetlet.2004.06.025. [DOI] [PubMed] [Google Scholar]
- Kamaike K.; Hirose K.; Kayama Y.; Kawashima E. Synthesis of Oligonucleoside Phosphorodithioates on a Solid Support by the H-Phosphonothioate Method. Tetrahedron 2006, 62, 11814–11820. 10.1016/j.tet.2006.09.024. [DOI] [PubMed] [Google Scholar]
- Jankowska J.; Sobkowski M.; Stawiński J.; Kraszewski A. Studies on Aryl H-Phosphonates. I. An Efficient Method for the Preparation of Deoxyribo- and Ribonucleoside 3′-H-Phosphonate Monoesters by Transesterification of Diphenyl H-Phosphonate. Tetrahedron Lett. 1994, 35, 3355–3358. 10.1016/S0040-4039(00)76906-1. [DOI] [Google Scholar]
- Froehler B. C.; Matteucci M. D. The Use of Nucleoside H-Phosphonates in the Synthesis of Deoxyoligonucleotides. Nucleosides Nucleotides 1987, 6, 287–291. 10.1080/07328318708056206. [DOI] [Google Scholar]
- McCollum C.; Andrus A. An Optimized Polystyrene Support for Rapid, Efficient Oligonucleotide Synthesis. Tetrahedron Lett. 1991, 32, 4069–4072. 10.1016/S0040-4039(00)79865-0. [DOI] [Google Scholar]
- Shimizu M.; Wada T.; Oka N.; Saigo K. A Novel Method for the Synthesis of Dinucleoside Boranophosphates by a Boranophosphotriester Method. J. Org. Chem. 2004, 69, 5261–5268. 10.1021/jo0493875. [DOI] [PubMed] [Google Scholar]
- Wada T.; Sato Y.; Honda F.; Kawahara S. I.; Sekine M. Chemical Synthesis of Oligodeoxyribonucleotides Using N-Unprotected H- Phosphonate Monomers and Carbonium and Phosphonium Condensing Reagents: O- Selective Phosphonylation and Condensation. J. Am. Chem. Soc. 1997, 119, 12710–12721. 10.1021/ja9726015. [DOI] [Google Scholar]
- Oka N.; Shimizu M.; Saigo K.; Wada T. 1,3-Dimethyl-2-(3-Nitro-1,2,4-Triazol-1-Yl)-2-Pyrrolidin-1-Yl-1,3, 2-Diazaphospholidinium Hexafluorophosphate (MNTP): A Powerful Condensing Reagent for Phosphate and Phosphonate Esters. Tetrahedron 2006, 62, 3667–3673. 10.1016/j.tet.2006.01.084. [DOI] [Google Scholar]
- Straarup E. M.; Fisker N.; Hedtjärn M.; Lindholm M. W.; Rosenbohm C.; Aarup V.; Hansen H. F.; Ørum H.; Hansen J. B. R.; Koch T. Short Locked Nucleic Acid Antisense Oligonucleotides Potently Reduce Apolipoprotein B MRNA and Serum Cholesterol in Mice and Non-Human Primates. Nucleic Acids Res. 2010, 38, 7100–7111. 10.1093/nar/gkq457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton J. M.; Guimaraes G. J.; Annavarapu V.; van Dongen W. D.; Bartlett M. G. Current State of Oligonucleotide Characterization Using Liquid Chromatography-Mass Spectrometry: Insight into Critical Issues. J. Am. Soc. Mass Spectrom. 2020, 31, 1775–1782. 10.1021/jasms.0c00179. [DOI] [PubMed] [Google Scholar]
- Matsubara H.; Hasegawa S.; Fujimura S.; Shima T.; Sugimura T.; Futai M. Studies on Poly (Adenosine Diphosphate Ribose). J. Biol. Chem. 1970, 245, 3606–3611. 10.1016/S0021-9258(18)62969-2. [DOI] [PubMed] [Google Scholar]
- Pritchard A. E.; Kowalski D.; Laskowski M. An Endonuclease Activity of Venom Phosphodiesterase Specific for Single-Stranded and Superhelical DNA. J. Biol. Chem. 1977, 252, 8652–8659. 10.1016/S0021-9258(19)75271-5. [DOI] [PubMed] [Google Scholar]
- RAZZELL W. E.; KHORANA H. G. Studies on Polynucleotides. IV. Enzymic Degradation; the Stepwise Action of Venom Phosphodiesterase on Deoxyribo-Oligonucleotides. J. Biol. Chem. 1959, 234, 2114–2117. 10.1016/S0021-9258(18)69875-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.