

Abstract
The therapeutic use of small interfering RNAs (siRNAs) as gene regulation agents has been limited by their poor stability and delivery. Although arranging siRNAs into a spherical nucleic acid (SNA) architecture to form siRNA-SNAs increases their stability and uptake, prototypical siRNA-SNAs consist of a hybridized architecture that causes guide strand dissociation from passenger strands, which limits the delivery of active siRNA duplexes. In this study, a new SNA design that directly attaches both siRNA strands to the SNA core through a single hairpin-shaped molecule to prevent guide strand dissociation is introduced and investigated. This hairpin-like architecture increases the number of siRNA duplexes that can be loaded onto an SNA by 4-fold compared to the original hybridized siRNA-SNA architecture. As a result, the hairpin-like siRNA-SNAs exhibit a 6-fold longer half-life in serum and decreased cytotoxicity. In addition, the hairpin-like siRNA-SNA produces more durable gene knockdown than the hybridized siRNA-SNA. This study shows how the chemistry used to immobilize siRNA on nanoparticles can markedly enhance biological function, and it establishes the hairpin-like architecture as a next-generation SNA construct that will be useful in life science and medical research.
Graphical Abstract
INTRODUCTION
Small interfering RNAs (siRNAs) are short, double-stranded RNA molecules that silence genes via RNA interference (RNAi).1 Due to the efficiency and specificity of RNAi, in principle, siRNAs can be synthetically designed to silence any target sequence of interest, potentially serving as a gene regulation therapy for a broad range of diseases.2-4 Unfortunately, clinical use of siRNA has been restricted because siRNAs have limited biodistribution, are rapidly degraded in biological fluids, and cannot enter cells as single entities.5,6
Spherical nucleic acids (SNAs) consist of oligonucleotides radially arranged around a nanoparticle core in high density, and they exhibit properties that distinguish them from their linear counterparts. In particular, their biodistribution is markedly different, allowing them to enter tissues and cells that linear structures do not access, and they are more resistant to nuclease degradation with longer circulating half-lives.7-11 Thus, siRNA-SNAs have been shown to be promising as therapeutics in animal models of impaired wound healing and psoriasis as well as a first-in-human clinical trial for glioblastoma.12-16
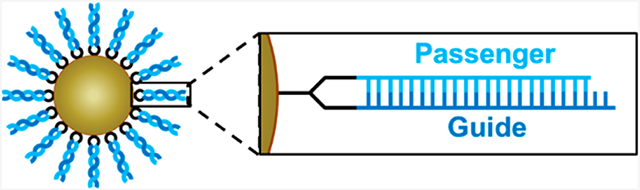
Prototypical siRNA-SNA designs exhibit a hybridized (hyb.) architecture, in which only the passenger strand of the siRNA is directly attached to the nanoparticle core, and the complementary guide strand is attached to the SNA conjugate via hybridization to the passenger strand (Figure 1A).9 With this SNA design, the hybridization of the guide strands to densely packed passenger strands on the nanoparticle core occurs with negative cooperativity; each binding event increases the charge density near the surface of the core, ultimately limiting the number of guide strands that can be loaded on the SNA due to electrostatic repulsion.17 According to previous studies, these siRNA-SNAs have only as many as 40 guide strands hybridized to the >80 passenger strands on the surface of the particle, a duplex efficiency of <50%.13,14 It is also possible that the guide strand can dehybridize before reaching the target cell with this design.18 Because intact siRNA duplexes are necessary for efficient gene silencing, the low functional siRNA concentrations delivered using this SNA design ultimately lead to limited target site accumulation, and a large amount of SNA material is required to induce silencing. Thus, methods for preparing siRNA-SNAs that bear a greater amount of active, stable siRNA are needed so that these conjugates can be delivered to target cells and potent gene silencing can be realized.19
Figure 1.

Architectures of siRNA-SNAs. (A) Hybridized architecture, in which the passenger strand is attached to the core and the guide strand is hybridized to it with low duplexing efficiency, and (B) hairpin-like architecture, in which a hairpin-like siRNA, a single RNA strand composed of a duplex and a hairpin-like region of PEG spacers, is attached to the core with high duplexing efficiency. Detailed chemical structures are shown in Figure S1.
Researchers have shown that the enzyme Dicer-2 cleaves siRNA duplexes from the SNA for RNA-induced silencing complex (RISC) processing, opening up the possibility to explore new, more stable chemistries (Figure S2).20 A straightforward approach involves attaching a thiol group to the end of both the passenger and guide strands so that both strands can be functionalized to the gold nanoparticle core (two points of chemical attachment). However, while such a strategy is suitable for gold nanoparticle cores, it is not extendable to many biocompatible cores, such as liposomes and poly(lactic-co-glycolic acid) (PLGA) nanoparticles.21,22 Hydrophobic anchor-conjugated RNA strands can separate and laterally diffuse on liposomal cores, and the chemistry of PLGA-SNAs features separate, single points of covalent attachment that are not conducive to the hybridization of adjacent complementary RNA strands. Lateral diffusion is also possible with gold nanoparticle cores, so there is no guarantee that duplex formation is maximized.23 Thus, it is ideal that both the passenger and guide strand on an siRNA-SNA are attached to each other and to the nanoparticle core through a single functional group.
Herein, we describe the design and synthesis of a hairpin-like (HP) siRNA-SNA architecture that fulfills this key criterion. In this novel design, both strands of the siRNA duplex are components of a single hairpin-shaped molecule, allowing for their direct attachment to the nanoparticle core via a single functional group (Figures 1B and S1). This design prevents guide strand dehybridization and permits increased duplex loading compared to the previously utilized hybridized siRNA-SNA design. As a result, improved serum nuclease resistance, decreased cytotoxicity, and increased gene silencing duration are observed. This hairpin-like siRNA-SNA design can be translated for use on a variety of nanoparticle cores due to its single functional group, making it a very attractive synthon for the development of next-generation siRNA-SNAs.
RESULTS AND DISCUSSION
Hairpin-like siRNA Efficiently Self-Hybridizes to Form a Functional siRNA Duplex.
The hairpin-like siRNA molecule consists of a complementary passenger and guide strand joined by polyethylene glycol (PEG) spacers and a dithiol serinol group (Figure 2A). These constructs were synthesized as a single 44-mer RNA oligonucleotide containing the PEG and dithiol serinol groups in the middle using solid phase synthesis via standard protocols. Synthesis of the RNA oligo was confirmed using matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry (Table S1). The average synthesis yield was 7%, and the hairpin-like RNAs were stored at −20 °C and remained functional after 6 months.
Figure 2.

Hairpin-like RNAs self-hybridize to function as active siRNA duplexes. (A) Chemical structure of hairpin-like siRNA with passenger strand (light blue) and guide strand (dark blue). The sequence shown is used for targeting HER2 mRNA. (B) Scheme showing hairpin-like RNA conformations and predicted native PAGE band locations. (C) Native PAGE gel of hairpin-like siRNA, noncomplementary RNA, and cocomplementary RNA. (D) Gene silencing activity of hybridized and hairpin-like siRNA. SK-OV-3 cells were transfected with 100 nM siRNA. Relative (rel.) gene expression (exp.) was measured 48 h after siRNA administration by RT-qPCR, normalized to transfection agent-only treatment. Error bars are standard deviation (SD) of 3 experimental replicates (***P ≤ 0.001).
Next, it was confirmed that the passenger and guide strands within the hairpin-like siRNA hybridize at a high efficiency to form the intact siRNA duplex required for gene silencing. In native polyacrylamide gel electrophoresis (PAGE) analysis, the hairpin-like siRNA strands appear as three bands, one with a much higher intensity than the other two, indicating that the hairpin-like RNA likely exists in three conformations: closed, where the RNA self-hybridized; open, where the RNA did not self-hybridize; and sandwich, where two of the RNA molecules dimerized due to their complementarity (Figure 2B). To determine which band corresponded to which conformation, a similar RNA with noncomplementary passenger and guide strands was used; this strand cannot self-hybridize and only exists in the open conformation. This noncomplementary RNA was run through the gel, revealing the location of the band for the open conformation. Then, a second RNA molecule that could not self-hybridize but that was entirely complementary to the previous noncomplementary RNA was prepared. These two cocomplementary strands form a sandwich conformation and reveal the location of that band. Based on these results, we conclude that the majority (89% intensity according to ImageJ analysis) of the hairpin-like siRNA molecules exists in the closed conformation (Figure 2C). Thus, the hairpin-like siRNA molecule self-hybridizes at a high efficiency. The hairpin-like RNA was heated to 95 °C and then slowly cooled in duplex buffer to investigate if the percentage of strands in the closed conformation can be increased, but this process did not significantly affect the ratios of the observed conformations (Figure S3).
To investigate if the hairpin-like siRNA functions as an active siRNA duplex, its gene silencing activity was compared to that of standard siRNA that consists of two hybridized strands. As a model system, the human epidermal growth factor receptor 2 (HER2) gene was targeted for silencing in SK-OV-3 human ovarian cancer cells since it is a well-established oncogene and the protein it expresses is a common therapeutic target for breast cancer.24 HER2-targeting hairpin-like siRNAs were transfected with Lipofectamine RNAiMAX into SK-OV-3 cells, and gene knockdown was measured by reverse transcription quantitative polymerase chain reaction (RT-qPCR). HER2-targeting hairpin-like siRNAs significantly knocked down the HER2 gene as compared to a nontargeting control hairpin-like siRNA and at a similar level to a standard hybridized siRNA of the same sequence (Figure 2D). Thus, hairpin-like siRNAs self-hybridize and function as active siRNA duplexes.
Hairpin-like Design Enables Higher siRNA Duplex Loading on SNAs than Hybridized Design.
HER2-targeting hairpin-like siRNA-SNAs were synthesized by adding hairpin-like siRNA to gold nanoparticles and salt aging them and then washing away unbound oligonucleotides.7 Dynamic light scattering (DLS) shows an increase in hydrodynamic diameter when hairpin-like siRNAs are conjugated to the gold nanoparticles, indicating the successful formation of SNAs (bare gold nanoparticle: 10 nm, hyb. siRNA-SNA: 17 nm, HP siRNA-SNA: 23 nm) (Figures 3A and S4). Although the hairpin-like siRNA-SNA is larger than the hybridized siRNA-SNA, the difference in size is minor in the context of these experiments and may be due to a higher density of siRNA duplexes. OliGreen fluorescence quantification revealed 25 siRNA duplexes per hybridized siRNA-SNA particle (14% duplex efficiency) (Figure S5). Significantly, the hairpin-like SNA architecture has a 4-fold higher duplex loading, bearing 103 siRNA duplexes per particle (Figure 3B). This effect is sequence-independent, as siRNA-SNAs with other sequences exhibit a similar increase in duplex loading between the hybridized and hairpin-like architectures (Figure S6). Thus, the hairpin-like architecture allows for a significantly greater loading of siRNA on its surface than the hybridized architecture.
Figure 3.

Hairpin-like design increases duplex loading of siRNA-SNAs. (A) DLS of bare gold nanoparticles and hybridized and hairpin-like siRNA-SNAs. Diameter shown is number mean. Error bars are SD of 10 measurements (****P ≤ 0.0001). (B) siRNA duplex loading for each SNA architecture. Error bars are SD of 3 batches of SNAs (****P ≤ 0.0001).
Hairpin-like Design Increases Serum Nuclease Resistance and Decreases SNA Cytotoxicity.
In general, siRNAs bound to a particle surface in SNA form exhibit higher serum nuclease resistance than free siRNAs due to the negatively charged surface of the SNA, steric congestion, and high local salt concentrations.9,10,25 siRNA-SNAs have a higher half-life in serum than linear siRNAs, making them better suited for use as therapeutics. The hairpin-like siRNA-SNA, with its higher duplex density, may provide even greater serum nuclease resistance than the prototypical hybridized siRNA-SNA. The serum stability of hairpin-like and hybridized siRNA-SNAs was compared by incubating them in 10% fetal bovine serum (FBS) and measuring the amount of siRNA duplex remaining on the SNA over time (Figure 4A,B). Hairpin-like siRNA-SNAs were found to have a 6-fold longer half-life in serum compared to hybridized siRNA-SNAs (132 min vs 22 min), indicating that the hairpin-like architecture improves siRNA-SNA resistance to serum nucleases. This enhancement could enable SNAs with the hairpin-like design to deliver a greater amount of intact siRNA to target sites. Interestingly, hairpin-like siRNA-SNAs with a much lower duplex loading (39 duplexes/SNA, comparable to that of the hybridized siRNA-SNAs) had a half-life in serum (17 min) that was closer to that of the hybridized siRNA-SNAs (22 min) (Figure S7), indicating that the increased serum nuclease resistance of hairpin-like siRNA-SNAs likely stems from their high siRNA duplex density.
Figure 4.

Hairpin-like design increases serum stability and decreases cytotoxicity of siRNA-SNAs. (A) Degradation of siRNA-SNAs in serum. Error bars are SD of 3 experimental replicates. (B) Half-lives of siRNA-SNAs in serum, derived from curves in (A). Error bars are SD of 3 experimental replicates (***P ≤ 0.001). (C) Cytotoxicity of siRNA-SNAs. Dotted lines are 95% confidence interval (CI). Error bars are SD of 3 biological replicates. (D) LD50 of siRNA-SNAs, derived from curves in (C). Error bars are SD of 3 biological replicates (**P ≤ 0.01).
siRNA-SNAs are cytotoxic at high concentrations because the gold accumulates in cells, limiting the maximum dose of siRNA that can be delivered. Since hairpin-like siRNA-SNAs contain more active siRNA duplexes per gold nanoparticle than hybridized siRNA-SNAs, the hairpin-like siRNA-SNAs should be able to deliver greater amounts of siRNA before cytotoxic concentrations of gold are reached. SK-OV-3 cells were treated with increasing concentrations of hybridized and hairpin-like siRNA-SNAs, and cell viability was measured (Figure 4C,D). Indeed, the median lethal dose (LD50) occurs at a higher siRNA concentration with hairpin-like siRNA-SNAs than with hybridized siRNA-SNAs, indicating that the hairpin-like architecture is less cytotoxic. The increased serum nuclease resistance and decreased cytotoxicity resulting from the hairpin-like design will allow for higher amounts of active siRNA to be delivered by SNAs for therapeutic applications.
Hairpin-like siRNA-SNAs Produce a More Durable Gene Silencing Effect than Hybridized siRNA-SNAs.
The gene silencing activity of hybridized and hairpin-like siRNA-SNAs was compared to determine if the hairpin-like architecture affects interactions with RNAi proteins. The hairpin-like siRNA-SNAs were observed to enter cells as single-entity agents like their hybridized siRNA-SNA counterparts but delivered higher quantities of siRNA (hyb. siRNA-SNAs: 1.3 × 106 siRNA/cell, HP siRNA-SNAs: 2.5 × 106 siRNA/cell), in agreement with previous results that cellular uptake of SNAs is affected by oligonucleotide density (Figure S8).26 To remove the difference in uptake and compare the interactions of siRNA-SNAs with RNAi proteins, HER2-targeting siRNA-SNAs were transfected into SK-OV-3 cells such that the same amount of siRNA was delivered for both architectures.20 Both siRNA-SNAs achieved equal gene knockdown at a 100 nM siRNA concentration, demonstrating that the hairpin-like architecture does not prevent the surface-bound strands from interacting with RNAi proteins and allows for the same gene silencing functionality as the hybridized siRNA-SNA (Figure 5A).
Figure 5.

Hairpin-like design increases gene silencing durability of siRNA-SNAs. (A) Full gene silencing activity of HER2-targeting siRNA-SNAs. SK-OV-3 cells were transfected with 100 nM siRNA equivalent SNAs. Relative gene expression was measured 48 h after SNA administration by RT-qPCR, normalized to transfection agent-only treatment. Error bars are SD of 3 experimental replicates. (B) Knockdown potency of HER2-targeting siRNA-SNAs 2 days after SNA administration. SK-OV-3 cells were transfected with a range of SNA concentrations. Protein expression was measured using an in-cell Western assay, normalized to transfection agent-only treatment. Dotted lines are 95% CI. Error bars are SD of 3 biological replicates. (C) Knockdown potency of HER2-targeting siRNA-SNAs 4 weeks after SNA administration. Dotted lines are 95% CI. Error bars are SD of 3 biological replicates. (D) IC50 of HER2-targeting siRNA-SNAs at 2 days and 4 weeks after SNA administration, based on protein expression curves in (B) and (C). Error bars are SD of 3 biological replicates (ns, P > 0.05, *P ≤ 0.05).
Next, the effect of SNA design on gene silencing potency was compared. Specifically, it was investigated whether RNAi machinery recognized both SNAs identically regardless of differences in architecture and loading, or if the higher loading of hairpin-like siRNA-SNAs enabled each SNA to be more potent, or if the denser loading or attachment architecture of hairpin-like siRNA-SNAs inhibited interactions with RNAi machinery. To study differences due to these interactions, a range of SNA concentrations was transfected into SK-OV-3 cells for 24 h so that at each concentration, the same amount of siRNA of either design was delivered regardless of differences in uptake.20 HER2 protein expression was measured 2 days after SNA administration using an in-cell Western assay, since it is higher-throughput than RT-qPCR and correlated the previously measured gene expression with protein expression. The potency of both SNAs was found to be similar in terms of SNA concentration (hybridized siRNA-SNA half maximal inhibitory concentration (IC50): 0.020 nM, hairpin-like siRNA-SNA IC50: 0.017 nM) (Figure 5B,D). The dependency of knockdown on SNA concentration, rather than siRNA concentration, is in agreement with previous observations that the SNA is the active entity in gene silencing.9,20 RNAi proteins recognize both architectures of SNA similarly regardless of attachment architecture or loading, and the hairpin-like attachment strategy does not impair gene silencing activity.
Gene silencing duration is a critical property of siRNA that affects the ability to drive a persistent phenotypic change, the portion of treatment time during which the target protein is below the therapeutic threshold, and the required dosing schedule.27 The kinetics of siRNA-SNA gene silencing is slower than that of linear siRNA; with siRNA-SNAs, it takes longer to reach maximum knockdown as well as return to pretreatment gene expression levels.9,12 Studies have suggested that Dicer-2 cleavage is the rate-limiting step in the SNA gene silencing pathway, resulting in a sustained rate of siRNA removal from SNAs and slower subsequent processing by RNAi machinery.20 Thus, at early time points, both SNA architectures have a similar gene silencing effect regardless of loading since siRNA release occurs at a similar rate (Figures 5B and S9A). However, because the hairpin-like siRNA-SNAs have a higher siRNA loading than the hybridized siRNA-SNAs, it will take longer for intracellular hairpin-like siRNA-SNAs to be depleted of siRNA so their knockdown should be more persistent than that of hybridized siRNA-SNAs at later time points (Figure S9B). Indeed, 4 weeks after a 24-h SNA treatment, SK-OV-3 cells treated with the hairpin-like siRNA-SNAs had persistent HER2 gene silencing at lower SNA concentrations, reducing the hairpin-like siRNA-SNA IC50 to 0.00038 nM; conversely, cells treated with hybridized siRNA-SNAs had a weakening of the gene silencing effect, with a rise in IC50 to 0.070 nM (Figure 5C,D). This result suggests that the hairpin-like siRNA-SNA allows for the release of siRNA for a longer period than the hybridized siRNA-SNA, lengthening the duration of knockdown. Therefore, not only is the hairpin-like architecture able to deliver more siRNA at low SNA concentrations than the hybridized architecture, it also provides a more persistent gene silencing effect.
CONCLUSION
From these data, one can conclude that the incorporation of a hairpin structure into SNA design can lead to marked improvements in SNA performance. Indeed, the novel hairpin-like architecture improves siRNA-SNA duplex loading, serum nuclease resistance, and gene silencing durability, while reducing cytotoxicity. These factors should work synergistically to enhance the delivery of active siRNA duplexes to target tissues and increase the window for gene silencing. Due to these advantages and the universality of the design, hairpin-like siRNA-SNAs are an important synthetic advance in the development of next-generation siRNA-SNA constructs, potentially driving the development of siRNA therapeutics and significantly expanding their scope of utility.
EXPERIMENTAL SECTION
Oligonucleotide Synthesis.
RNA oligonucleotides were synthesized on a MerMade 12 system (BioAutomation) using 2′-O-triisopropylsilyloxymethyl-protected phosphoramidites (Chem-Genes). In the hybridized siRNA system, the passenger and guide RNA strands were synthesized separately. The passenger strand consisted of RNA base cyanoethyl phosphoramidites (Glen Research), two spacer-18 (18-O-dimethyoxytritylhexaethylene glycol,1-[(2-cyanoethyl)-(N,N-diisopropyl)] phosphoramidites, and a thiol modification added by using 1-O-dimethoxytrityl-propyl-disulfide,1′-succinyl-lcaa-controlled pore glass beads. The guide strand consisted of RNA base cyanoethyl phosphoramidites. For the hairpin-like siRNA system, the single strand consisted of RNA base cyanoethyl phosphoramidites (the guide portion of the strand), two spacer-18 phosphoramidites, a dithiol serinol phosphoramidite (the hairpin turn and nanoparticle attachment moiety), two spacer-18 phosphoramidites, and RNA base cyanoethyl phosphoramidites (the passenger portion of the strand). After synthesis, RNA oligonucleotides were deprotected following the manufacturer’s protocol (Bioautomation). Deprotected RNA oligonucleotides were purified using high-performance liquid chromatography on a C18 column using 0.1 M triethylammonium acetate and acetonitrile as the solvents. The 5′-DMT group was removed from the purified RNA oligonucleotides via treatment with 20% acetic acid at room temperature for 1 h and extracting 3 times with ethyl acetate. The oligonucleotide solution was lyophilized and suspended in DNase/RNase-free water. The oligonucleotide sequences used in this work are listed in Table S1.
Hairpin-like siRNA Characterization.
The masses of the hairpin-like siRNAs were measured using MALDI-TOF mass spectrometry. Hairpin-like siRNAs were mixed with 2′,4′-dihydroxyacetophenone (DHAP) and dried on a MALDI plate. MALDI-TOF was performed using an Autoflex III Smartbeam MALDI-TOF mass spectrometer. Conformation analysis of the hairpin-like siRNAs was performed using native PAGE. The two cocomplementary hairpin-like siRNAs were mixed at an equal molar ratio in duplex buffer (30 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (pH 7.3), 100 mM potassium acetate, 2 mM magnesium acetate). The solution was then heated to 95 °C for 2 min and slowly cooled to room temperature to allow for hybridization to occur. RNA samples were run through a 10% native PAGE gel at 150 V for 45 min. SYBR Gold was used to stain the RNA. The gel was imaged using an Amersham Typhoon biomolecular imager.
SNA Salt-Aging Synthesis.
Citrate-capped AuNPs 13 nm in diameter were synthesized by reducing chloroauric acid with sodium citrate in water following the Turkevich-Frens method.28 To prepare hybridized siRNA-SNAs, 100 μM passenger RNA and 100 μM guide RNA were added to duplex buffer, heated to 95 °C for 2 min, and slowly cooled to room temperature to allow for hybridization. The AuNPs were functionalized with these hybridized siRNAs to form SNAs via a previously described salt-aging procedure.20 To prepare the hairpin-like siRNA-SNAs, 100 μM hairpin-like siRNA was added to duplex buffer, heated to 95 °C for 2 min, and slowly cooled to room temperature. Hairpin-like siRNA (final concentration = 4 μM) was then added to a solution of 10 nM 13 nm AuNPs, 0.2% Tween-20, and 150 mM NaCl. The solution was incubated overnight. The next day, the salt concentration was gradually increased to 1 M NaCl by adding a concentrated solution of NaCl every 2 h while shaking. The solution was allowed to equilibrize overnight. Unattached oligonucleotides were removed by washing with 1× Dulbecco’s phosphate-buffered saline (DPBS) 3 times in Amicon Ultra 100 K molecular weight cutoff spin filters (MilliporeSigma). The SNAs were stored at 4 °C for up to 3 months.
SNA Characterization.
SNAs and AuNPs were diluted to 1 nM in 1× DPBS and water, respectively, and size was analyzed with dynamic light scattering using a Malvern Zetasizer. The SNA concentration was determined by measuring the absorbance spectra of the SNAs using a Cary-5000 spectrophotometer (Agilent). The AuNP’s extinction coefficient is not affected by functionalization with oligonucleotides, so AuNP (and, in effect, SNA) concentration was calculated using the absorbance at 520 nm and an extinction coefficient of 2.27 × 108 M−1 cm−1.29
To measure the siRNA duplex loading of hybridized siRNA-SNAs, 3 μL of 100 nM SNAs was incubated with 100 μL of 9 M urea at 45 °C for 20 min, mixed with 100 μL of 0.01% Tween-20, and centrifuged at maximum speed for 25 min to dissociate the guide strands from the SNAs. The passenger strand-conjugated SNAs formed a pellet, while dissociated guide strands were in the supernatant. A portion of the supernatant (25 μL) was added to 75 μL of water and 100 μL of 0.5% Quant-iT OliGreen reagent (Invitrogen) in 1X TE buffer. The samples were analyzed in a 96-well plate by measuring OliGreen fluorescence (λex = 480 nm, λem = 520 nm) with a BioTek Cytation 5 imaging reader and comparing to a standard curve of guide RNA. siRNA duplex loading of hairpin-like siRNA-SNAs was then calculated by taking the ratio of guide RNA concentration to AuNP concentration. To measure the number of passenger strands on each SNA, the remaining passenger strand-conjugated SNAs were added to 9 M urea to dissolve the gold. The OliGreen assay was repeated with these SNAs to measure passenger RNA concentration. Passenger strand loading on the SNA was then calculated by taking the ratio of passenger RNA concentration to AuNP concentration.
To measure the siRNA duplex loading of hairpin-like siRNA-SNAs, 7 μL of 100 nM SNAs was mixed with 7 μL of 40 mM potassium cyanide (KCN) and heated at 55 °C until the gold dissolved. The solution was diluted in 112 μL of 1× DPBS. Then, 25 μL of this solution was mixed with 175 μL of 0.28% Quant-iT OliGreen reagent (Invitrogen) in 1× DPBS. OliGreen fluorescence was measured and compared to a standard curve of hairpin-like siRNA. siRNA duplex loading of hairpin-like siRNA-SNAs was then calculated by taking the ratio of hairpin-like siRNA concentration to AuNP concentration.
Serum Nuclease Resistance of SNAs.
The serum nuclease resistance of siRNA-SNAs was measured as previously described.25 To degrade the siRNA on the SNAs with nucleases, 20 nM SNAs were incubated in 10% fetal bovine serum (FBS) in 1× DPBS at 37 °C. As a negative control, 20 nM SNAs were incubated in 1× DPBS at 37 °C. At each time point, an 18-μL aliquot was removed and mixed with 30 mM sodium dodecyl sulfate (SDS) to inactivate the nucleases and stop the reaction. The degraded SNAs were then washed 3 times by suspending in 0.01% Tween-20 in 1× DPBS, centrifuging at 21,000 × g for 25 min, and removing the supernatant to remove serum and degraded RNA fragments. The amount of siRNA remaining on the SNA at each time point was measured following the previously described SNA characterization methods. The AuNP concentration of the SNA samples was determined by measuring absorbance at 520 nm using a BioTek Cytation 5 imaging reader.
Cytotoxicity of SNAs.
In 96-well cell culture plates with 2,500 SK-OV-3 cells per well, siRNA-SNAs in Opti-MEM were incubated with SK-OV-3 cells in triplicate for 48 h. The wells were washed with 1× DPBS 3 times to remove dead cells and SNAs that remained outside the cells. A mixture of 50 μL of 1× DPBS and 50 μL of CellTiter-Glo 2.0 reagent (Promega) was added to the wells, and the luminescence was measured using a BioTek Cytation 5 imaging reader. Luminescent readout from adenosine triphosphate (ATP) concentration was used to indicate live cell count. Cell viability was normalized to cells treated with Opti-MEM only.
Cellular Uptake of SNAs.
In 24-well cell culture plates with 50,000 SK-OV-3 cells per well, SK-OV-3 cells were treated with 1 nM SNAs in Opti-MEM for 24 h. At the end of each time point, the cells were washed with 1× DPBS 3 times to remove SNAs that remained outside the cells. The cells were then trypsinized using 150 μL of TrypLE Express (Thermo Fisher). The number of cells in each sample was measured by staining 10 μL of cells with 10 μL of Trypan Blue (Thermo Fisher) and measuring their concentration using an Invitrogen Countess II automated cell counter. Next, 120 μL of cells was dissolved in 50 mL of 2% hydrochloric acid (HCl) and 2% nitric acid (HNO3). The gold concentration in this solution was measured by performing inductively coupled plasma mass spectrometry (ICP-MS) on the acidified samples. ICP-MS was performed on a computer-controlled (QTEGRA software) Thermo iCapQ ICP-MS (Thermo Fisher Scientific, Waltham, MA, USA) operating in STD mode and equipped with an ESI SC-2DX PrepFAST autosampler (Omaha, NE, USA). Internal standard was added inline using the prepFAST system and consisted of 1 ng/mL of a mixed element solution containing Bi, In, 6Li, Sc, Tb, and Y (IV-ICPMS-71D from Inorganic Ventures). Online dilution was also carried out by the prepFAST system and used to generate a calibration curve consisting of 2, 4, 20, 100, and 200 ppb Au. Each sample was acquired using 1 survey run (10 sweeps) and 3 main (peak jumping) runs (40 sweeps). The isotopes selected for analysis were 197Au (for SNA quantification) and 89Y, 115In, 159Tb, and 209Bi (chosen as internal standards for data interpolation and machine stability). Instrument performance was optimized daily through autotuning followed by verification via a performance report (passing manufacturer specifications). The number of SNAs taken up into each cell was calculated by dividing the number of gold nanoparticles per sample by the number of cells per sample. The amount of siRNA taken up into each cell was calculated by multiplying the SNA uptake by the siRNA duplex loading on the SNA.
Quantification of Gene Knockdown by RT-qPCR.
In 12-well cell culture plates with 25,000 SK-OV-3 cells per well, HER2-targeting or nontargeting siRNA or siRNA-SNAs were transfected at a 100 nM siRNA concentration with Lipofectamine RNAiMAX (Thermo Fisher Scientific) using the manufacturer’s instructions. After transfection for 24 h, the treatment solution was replaced with McCoy’s 5A (modified) medium (Gibco) supplemented with 10% FBS and 1% penicillin-streptomycin, and the cells were incubated for another 24 h. RNA was isolated from the cells using a PureLink RNA Mini Kit (Thermo Fisher Scientific). mRNA levels were measured in triplicate by RT-qPCR using qScript XLT One-Step RT-qPCR ToughMix (Quanta Biosciences), TaqMan Gene Expression Assays (HER2: Hs01001580_m1, GAPDH: Hs03929097_g1; Thermo Fisher Scientific), a Bio-Rad C1000 Touch Thermal Cycler, and a Bio-Rad CFX384 Real-time System. CT values were normalized to the housekeeping gene and untreated cells using the Pfaffl method.30
Quantification of Protein Knockdown by in-Cell Western.
In 96-well cell culture plates with 20,000 SK-OV-3 cells per well, siRNA-SNAs targeting HER2 were transfected with Lipofectamine RNAiMAX using the manufacturer’s instructions. After transfection for 24 h, the SNA treatment solution was replaced with McCoy’s 5A (modified) medium (Gibco) supplemented with 10% FBS and 1% penicillin-streptomycin, and the cells were incubated for the remainder of the treatment period. The medium was replaced twice per week to continuously provide the cells with nutrients. At the end of the treatment period, the wells were washed with 1× DPBS 3 times and fixed in methanol chilled at −20 °C for 15 min. The wells were washed with 0.05% Tween-20 in 1× DPBS 2 times and then 1× DPBS and then incubated with Intercept blocking buffer (LI-COR) for 90 min with shaking. The cells were then incubated with HER2 antibody (29D8) diluted 1:200 in Intercept blocking buffer (Cell Signaling) for 2 h. The wells were washed with 0.1% Tween-20 in 1× DPBS 3 times and incubated with 2 μg/mL IRDye 800CW goat anti-rabbit secondary antibody (LI-COR) and 500 nM CellTag 700 (LI-COR) diluted 1:500 in Intercept blocking buffer for 1 h protected from light, with shaking. The wells were washed with 0.1% Tween-20 in 1× DPBS 3 times and imaged on an Odyssey CLx system (LI-COR). HER2 protein expression was normalized to cell count by normalizing fluorescence at 800 nm to fluorescence at 700 nm.
Statistics.
All statistical analyses were performed using GraphPad Prism. For comparisons between two groups, means were compared using an unpaired, two-tailed t test. For comparisons between more than two groups, means were compared using an ordinary one-way analysis of variance (ANOVA) with Tukey’s multiple comparison test. P values were multiplicity adjusted to account for multiple comparisons. Serum nuclease resistance results were fit with an exponential one-phase decay using a least-squares fit. Cytotoxicity results and in-cell Western protein expression results were fit with a 3-parameter logistic curve using a least-squares fit.
Supplementary Material
ACKNOWLEDGMENTS
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under awards R01CA208783 and P50CA221747. This work was also supported by the Polsky Urologic Cancer Institute of the Robert H. Lurie Comprehensive Cancer Center of Northwestern University at Northwestern Memorial Hospital. M.K.V. and G.Y. thank Robert Stawicki, Matthew Capek, and Jennifer Delgado for synthesizing RNA. MALDI-MS analysis of RNA was performed at the Integrated Molecular Structure Education and Research Center (IMSERC) generously supported by the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource Grant NSF ECCS-2025633, the State of Illinois, and the International Institute for Nanotechnology (IIN). Gold analysis for cellular uptake quantification was performed at the Northwestern University Quantitative Bioelement Imaging Center (QBIC) generously supported by NASA Ames Research Center Grant NNA04CC36G.
ABBREVIATIONS
- DLS
dynamic light scattering
- exp
expression
- FBS
fetal bovine serum
- HER2
human epidermal growth factor receptor 2
- HP
hairpin-like
- hyb
hybridized
- IC50
half maximal inhibitory concentration
- LD50
median lethal dose
- MALDI-TOF
matrix-assisted laser desorption/ionization time-of-flight
- PAGE
polyacrylamide gel electrophoresis
- PBS
phosphate-buffered saline
- PEG
polyethylene glycol
- PLGA
poly(lactic-co-glycolic acid)
- rel
relative
- RISC
ribonucleic acid-induced silencing complex
- RNA
ribonucleic acid
- RNAi
ribonucleic acid interference
- RT-qPCR
reverse transcription quantitative polymerase chain reaction
- siRNA
small interfering ribonucleic acid
- SNA
spherical nucleic acid
- UV–vis
ultraviolet–visible
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c12750.
Schemes, RNA and SNA characterization data, additional experimental results, and RNA oligonucleotide sequences (PDF)
This content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The authors declare the following competing financial interest(s): C.A.M. has financial interests in Exicure, Inc., which could potentially benefit from the outcomes of this research.
Contributor Information
Matthew K. Vasher, Department of Biomedical Engineering, Northwestern University, Evanston, Illinois 60208, United States; International Institute for Nanotechnology, Northwestern University, Evanston, Illinois 60208, United States.
Gokay Yamankurt, International Institute for Nanotechnology, Northwestern University, Evanston, Illinois 60208, United States; Interdisciplinary Biological Sciences Graduate Program, Northwestern University, Evanston, Illinois 60208, United States.
Chad A. Mirkin, Department of Biomedical Engineering, Northwestern University, Evanston, Illinois 60208, United States; International Institute for Nanotechnology and Department of Chemistry, Northwestern University, Evanston, Illinois 60208, United States.
REFERENCES
- (1).Fire A; Xu S; Montgomery MK; Kostas SA; Driver SE; Mello CC Potent and Specific Genetic Interference by Double-Stranded RNA in Caenorhabditis Elegans. Nature 1998, 391 (6669), 806. [DOI] [PubMed] [Google Scholar]
- (2).Sørensen DR; Leirdal M; Sioud M. Gene Silencing by Systemic Delivery of Synthetic siRNAs in Adult Mice. J. Mol. Biol 2003, 327 (4), 761–766. [DOI] [PubMed] [Google Scholar]
- (3).Dorsett Y; Tuschl T siRNAs: Applications in Functional Genomics and Potential as Therapeutics. Nat. Rev. Drug Discovery 2004. 3 (4), 318–329. [DOI] [PubMed] [Google Scholar]
- (4).Setten RL; Rossi JJ; Han S The Current State and Future Directions of RNAi-Based Therapeutics. Nat. Rev. Drug Discovery 2019, 18 (6), 421. [DOI] [PubMed] [Google Scholar]
- (5).Petrocca F; Lieberman J Promise and Challenge of RNA Interference-Based Therapy for Cancer. JCO 2011, 29 (6), 747–754. [DOI] [PubMed] [Google Scholar]
- (6).Kim B; Park J-H; Sailor MJ Rekindling RNAi Therapy: Materials Design Requirements for In Vivo siRNA Delivery. Adv. Mater 2019, 31 (49), 1903637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Mirkin CA; Letsinger RL; Mucic RC; Storhoff JJ A DNA-Based Method for Rationally Assembling Nanoparticles into Macroscopic Materials. Nature 1996, 382 (6592), 607. [DOI] [PubMed] [Google Scholar]
- (8).Cutler JI; Auyeung E; Mirkin CA Spherical Nucleic Acids. J. Am. Chem. Soc 2012, 134 (3), 1376–1391. [DOI] [PubMed] [Google Scholar]
- (9).Giljohann DA; Seferos DS; Prigodich AE; Patel PC; Mirkin CA Gene Regulation with Polyvalent siRNA-Nanoparticle Conjugates. J. Am. Chem. Soc 2009, 131 (6), 2072–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Seferos DS; Prigodich AE; Giljohann DA; Patel PC; Mirkin CA Polyvalent DNA Nanoparticle Conjugates Stabilize Nucleic Acids. Nano Lett. 2009, 9 (1), 308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Choi CHJ; Hao L; Narayan SP; Auyeung E; Mirkin CA Mechanism for the Endocytosis of Spherical Nucleic Acid Nanoparticle Conjugates. Proc. Natl. Acad. Sci. U. S. A 2013, 110 (19), 7625–7630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Zheng D; Giljohann DA; Chen DL; Massich MD; Wang X-Q; Iordanov H; Mirkin CA; Paller AS Topical Delivery of siRNA-Based Spherical Nucleic Acid Nanoparticle Conjugates for Gene Regulation. Proc. Natl. Acad. Sci. U.SA 2012, 109 (30), 11975–11980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Jensen SA; Day ES; Ko CH; Hurley LA; Luciano JP; Kouri FM; Merkel TJ; Luthi AJ; Patel PC; Cutler JI; Daniel WL; Scott AW; Rotz MW; Meade TJ; Giljohann DA; Mirkin CA; Stegh AH Spherical Nucleic Acid Nanoparticle Conjugates as an RNAi-Based Therapy for Glioblastoma. Science Translational Medicine 2013, 5 (209), 209ra152–209ra152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Randeria PS; Seeger MA; Wang X-Q; Wilson H; Shipp D; Mirkin CA; Paller AS siRNA-Based Spherical Nucleic Acids Reverse Impaired Wound Healing in Diabetic Mice by Ganglioside GM3 Synthase Knockdown. Proc. Natl. Acad. Sci. U.S.A 2015, 112 (18), 5573–5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Nemati H; Ghahramani M-H; Faridi-Majidi R; Izadi B; Bahrami G; Madani S-H; Tavoosidana G Using siRNA-Based Spherical Nucleic Acid Nanoparticle Conjugates for Gene Regulation in Psoriasis. J. Controlled Release 2017, 268, 259–268. [DOI] [PubMed] [Google Scholar]
- (16).Kumthekar P; Ko CH; Paunesku T; Dixit K; Sonabend AM; Bloch O; Tate M; Schwartz M; Zuckerman L; Lezon R; Lukas RV; Jovanovic B; McCortney K; Colman H; Chen S; Lai B; Antipova O; Deng J; Li L; Tommasini-Ghelfi S; Hurley LA; Unruh D; Sharma NV; Kandpal M; Kouri FM; Davuluri RV; Brat DJ; Muzzio M; Glass M; Vijayakumar V; Heidel J; Giles FJ; Adams AK; James CD; Woloschak GE; Horbinski C; Stegh AH A First-in-Human Phase 0 Clinical Study of RNA Interference-Based Spherical Nucleic Acids in Patients with Recurrent Glioblastoma. Sci. Transl. Med 2021, 13 (584), eabb3945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Randeria PS; Jones MR; Kohlstedt KL; Banga RJ; Olvera de la Cruz M; Schatz GC; Mirkin CA What Controls the Hybridization Thermodynamics of Spherical Nucleic Acids? J. Am. Chem. Soc 2015, 137 (10), 3486–3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Elbashir SM; Harborth J; Lendeckel W; Yalcin A; Weber K; Tuschl T Duplexes of 21-Nucleotide RNAs Mediate RNA Interference in Cultured Mammalian Cells. Nature 2001, 411 (6836), 494. [DOI] [PubMed] [Google Scholar]
- (19).Shim MS; Kwon YJ Efficient and Targeted Delivery of siRNA in Vivo. FEBS Journal 2010, 277 (23), 4814–4827. [DOI] [PubMed] [Google Scholar]
- (20).Yamankurt G; Stawicki RJ; Posadas DM; Nguyen JQ; Carthew RW; Mirkin CA The Effector Mechanism of siRNA Spherical Nucleic Acids. Proc. Natl. Acad. Sci. U.S.A 2020, 117 (3), 1312–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Banga RJ; Chernyak N; Narayan SP; Nguyen ST; Mirkin CA Liposomal Spherical Nucleic Acids. J. Am. Chem. Soc 2014, 136 (28), 9866–9869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Zhu S; Xing H; Gordiichuk P; Park J; Mirkin CA PLGA Spherical Nucleic Acids. Adv. Mater 2018, 30 (22), 1707113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Ionita P; Volkov A; Jeschke G; Chechik V Lateral Diffusion of Thiol Ligands on the Surface of Au Nanoparticles: An Electron Paramagnetic Resonance Study. Anal. Chem 2008, 80 (1), 95–106. [DOI] [PubMed] [Google Scholar]
- (24).Mitri Z; Constantine T; O’Regan R The HER2 Receptor in Breast Cancer: Pathophysiology, Clinical Use, and New Advances in Therapy. Chemother Res. Pract 2012, 2012, 743193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Barnaby SN; Lee A; Mirkin CA Probing the Inherent Stability of siRNA Immobilized on Nanoparticle Constructs. Proc. Natl. Acad. Sci. U.S.A 2014, 111 (27), 9739–9744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Giljohann DA; Seferos DS; Patel PC; Millstone JE; Rosi NL; Mirkin CA Oligonucleotide Loading Determines Cellular Uptake of DNA-Modified Gold Nanoparticles. Nano Lett. 2007, 7 (12), 3818–3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Bartlett DW; Davis ME Insights into the Kinetics of siRNA-Mediated Gene Silencing from Live-Cell and Live-Animal Bioluminescent Imaging. Nucleic Acids Res. 2006, 34 (1), 322–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Kimling J; Maier M; Okenve B; Kotaidis V; Ballot H; Plech A Turkevich Method for Gold Nanoparticle Synthesis Revisited. J. Phys. Chem. B 2006, 110 (32), 15700–15707. [DOI] [PubMed] [Google Scholar]
- (29).Demers LM; Mirkin CA; Mucic RC; Reynolds RA; Letsinger RL; Elghanian R; Viswanadham G A Fluorescence-Based Method for Determining the Surface Coverage and Hybridization Efficiency of Thiol-Capped Oligonucleotides Bound to Gold Thin Films and Nanoparticles. Anal. Chem 2000, 72 (22), 5535–5541. [DOI] [PubMed] [Google Scholar]
- (30).Pfaffl MW A New Mathematical Model for Relative Quantification in Real-Time RT-PCR. Nucleic Acids Res. 2001, 29 (9), No. e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.