

Abstract
Purpose.
Germline heterozygous mutations of GATA2 underlie a variety of hematological and clinical phenotypes. The genetic, immunological, and clinical features of GATA2-deficient patients with mycobacterial diseases in the familial context remain largely unknown.
Methods.
We enrolled 15 GATA2 index cases referred for mycobacterial disease. We describe their genetic and clinical features including their relatives.
Results.
We identified 12 heterozygous GATA2 mutations, two of which had not been reported. Eight of these mutations were loss-of-function and four were hypomorphic. None was dominant-negative in vitro and the GATA2 locus was found to be subject to purifying selection, strongly suggesting a mechanism of haploinsufficiency. Three relatives of index cases had mycobacterial disease and were also heterozygous, resulting in 18 patients in total. Mycobacterial infection was the first clinical manifestation in 11 patients, at a mean age of 22.5 years (range: 12 to 42 years). Most patients also suffered from other infections, monocytopenia or myelodysplasia. Strikingly, the clinical penetrance was incomplete (32.9% by age 40 years), as 16 heterozygous relatives aged between 6 and 78 years, including 4 older than 60 years, were completely asymptomatic.
Conclusion.
Clinical penetrance for mycobacterial disease was found to be similar to other GATA2 deficiency-related manifestations. These observations suggest that other mechanisms contribute to the phenotypic expression of GATA2 deficiency. A diagnosis of autosomal dominant GATA2 deficiency should be considered in patients with mycobacterial infections and/or other GATA2 deficiency-related phenotypes at any age in life. Moreover, all direct relatives should be genotyped at the GATA2 locus.
Keywords: Primary immunodeficiency, GATA2, mycobacteria, tuberculosis, haploinsufficiency
Introduction
GATA2 is a zinc-finger transcription factor expressed in hematopoietic stem cells and various hematopoietic progenitor cells in humans (1, 2). The first germline heterozygous mutations of human GATA2 were described in 2011 (3–7). The disease-causing mutations reported include missense, nonsense, and canonical essential splicing mutations, synonymous exonic mutations affecting RNA splicing, as well as small frameshift insertions/duplications, small frameshift deletions, small in-frame deletions, intronic mutations, synonymous and large deletions (Figure S1). One third of the mutations was inherited from a parent, and as many as two thirds occurred de novo. The probability of the mutation being damaging was estimated in silico in most cases, by determining its predicted effect on the protein. Only 12 germline mutations (p.A318T, p.R330*, p.T354M, p.T355del, p.T358N, p.R361L, p.C373R, p.R396Q, p.R396L, p.R398W, c.1017+572C>T and c.1017+512del28), have been characterized experimentally in vitro, in reporter assays, EMSA, or granulocyte colony-forming assays involving an overexpression system (6–18). Ten of these mutations were reported to be loss-of-function (LOF) (6–17) and two to be hypomorphic (p.R396Q and p.R396L) (10, 18). The mechanism of dominance has been described for only seven of these 12 mutant alleles (6, 17, 18), three of which have been reported to have a dominant negative (DN) effect in vitro (p.T354M, p.T355del and p.R330*) (6, 10, 15, 17), whereas the other four appear to cause disease by haploinsufficiency (10, 17, 18).
These mutations cause a broad spectrum of phenotypes, including monocytopenia with mycobacterial disease (monoMAC syndrome) (4, 19–21), familial myelodysplastic syndrome (MDS) (5, 6, 13, 22), chronic or acute myeloid leukemia (CML or AML) (5, 13, 22, 23), Emberger syndrome (primary lymphedema and MDS) (7, 11, 22), and, more rarely, isolated neutropenia (21), aplastic anemia (24), and isolated NK cell deficiency (3, 25, 26). The clinical features of the disease typically manifest in the second decade of life, but the age at onset varies widely, from early childhood to late adulthood. Clinical penetrance is variable, depending on the phenotype considered (1, 7, 27, 28). High levels of clinical penetrance have been reported for MDS and AML, at ages of 12 to 19 years, in the families of index cases carrying GATA2 mutations (1, 15, 21, 27, 29, 30), whereas lymphedema has a low penetrance up to the age of 40 years, in all published reports (7, 22, 28, 31). The published studies did not involve penetrance calculations per se and did not, therefore provide precise estimates of overall clinical penetrance, or of global or specific penetrance for any phenotype (1, 7, 27, 28). The reported estimates of penetrance were inferred from the number of probands and asymptomatic heterozygous relatives. Moreover, penetrance for infectious diseases, including mycobacterial diseases in particular, has not yet been estimated. The clinical phenotypes of autosomal dominant (AD) GATA2 deficiency differ considerably between patients, attesting to the variable expressivity of this deficiency and implying that the individual penetrance of these phenotypes at population level is incomplete, despite the absence of precise estimates.
Since 2011, 77 GATA2-deficient patients from 74 kindreds with a history of mycobacterial disease have been reported (3, 4, 8, 9, 20, 22, 25–28, 32–46). Mean age at first mycobacterial infection in these patients was 26 years (range: 8–51, including 57 patients with available information), and these patients had experienced a total of 92 mycobacterial episodes among them. Environmental mycobacteria (EM) were the most frequently documented infectious agents, in 78 episodes in 64 patients (M. avium complex, n=32 episodes; M. kansasii, n=25; M. fortuitum, n=8; M. abscessus, n=6; M. szulgai, n=1; M. malmoense, n=1; M. genavense, n=1; M. sherisii, n=1, M. fukienense, n=1; unidentified mycobacterial species n=2). Most EM infections were disseminated (n=58), but some were regional (n=7) or localized (pulmonary, n=3; extrapulmonary, skin n=1). The tissues sampled for EM isolation included lymph nodes, respiratory tissues, pleural fluid, blood, synovial fluid, bone marrow, spleen, and skin (4, 19, 34, 36–38, 43, 44, 47–50). Osteomyelitis was described in only one patient (36). Only one of the 64 patients who experienced EM infection had a history of Salmonella disease (prolonged Salmonella enterocolitis) (35). Six patients suffered from tuberculosis (8, 9, 27, 36, 46). Another patient had disseminated BCG disease after vaccination at 12 years of age (3, 26, 28); two other patients were vaccinated with BCG without complications (34, 35). The mycobacterial species isolated was not reported in seven episodes (36, 47). Monocytopenia (58 of the 58 patients tested), B lymphopenia (54/54), and NK lymphopenia (51/52) were the most frequently associated biological abnormalities; T lymphopenia (n=33/49) and neutropenia (22/39) were less common. In this study, we investigated a group of 18 patients with GATA2 mutations and mycobacterial diseases.
Materials and methods
Patients and kindreds
The patients were referred to the Laboratory of Human Genetics of Infectious Disease, France, for mycobacterial infectious and hematological abnormalities. Then, we excluded the diagnosis of Mendelian susceptibility to mycobacterial diseases (MSMD) in these patients. The parents and/or patients signed an informed consent form, in accordance with the requirements of the institutional review boards (IRB) of the various institutions involved. Approval for this study was obtained from the French Ethics Committee (CPP) and the French National Institute of Health and Medical Research (INSERM; 2010-A00650–39); and from the Rockefeller University (JCA-0699). The treating physicians of the patients with GATA2 deficiency completed a detailed questionnaire. Data were collected from 2011 to 2018 and sent to JB. The information recorded included date of birth, date of first clinical manifestations, BCG vaccination, infectious diseases (particularly those due to mycobacteria), hematological disorders, hematopoietic stem cell transplantation (HSCT), and vital status, including the cause of death for any patients who had died. Our analysis focused mainly on mycobacterial infectious diseases in GATA2-deficient patients. We did not consider clinical signs for other infectious diseases for the recruitment of these patients, and this is one of the limitations of this retrospective study. The experiments described here were performed in France, in accordance with local regulations, and with the approval of the IRB of Necker Hospital for Sick Children, France.
Diagnosis of mycobacterial infection
Infectious disease due to mycobacteria was diagnosed on the basis of clinical and radiologic features, staining for acid-fast bacillus (AFB), molecular PCR and/or microbiological culture results, when available. Recurrence was defined as a subsequent episode of mycobacterial disease caused by the same microbe after a period free from clinical disease and treatment. EM infections were classified as lung disease, according to the criteria of the American Thoracic Society (ATS)/Infectious Disease Society of America (IDSA), or extrapulmonary disease, in the form of skin and soft tissue, lymph node, bone and joint, disseminated or catheter-related infections (51, 52). Tuberculosis was diagnosed according to the criteria proposed by S. Graham et al. (53) and Lewinsohn et al (54). The diagnosis of tuberculosis was considered confirmed in the presence of at least one sign or symptom of tuberculosis with a positive culture for M. tuberculosis. Probable tuberculosis was defined as the presence of one sign or symptom suggestive of tuberculosis, a radiogical chest images (X-ray or CT-scan) consistent with intrathoracic tuberculosis and at least one of the following: i) positive clinical response to anti-tuberculosis treatment, ii) documented exposure to or close contact with a patient with tuberculosis, or iii) immunological evidence of M. tuberculosis in the form of a positive tuberculin skin test (TST) or tuberculosis interferon-γ release assay (IGRA). Possible tuberculosis was defined as signs or symptoms suggestive of tuberculosis infection, together with a chest X-ray not consistent with pulmonary tuberculosis but a positive clinical response to anti-tuberculosis treatment, documented exposure to or close contact with a patient with tuberculosis, or immunological evidence of M. tuberculosis on TST or IGRA.
Extraction of DNA, whole-exome sequencing (WES) and GATA2 sequencing
Genomic DNA was isolated from the whole blood of patients and healthy donors, by phenol/chloroform extraction or with the iPrep PureLink gDNA Blood kit, with the iPrep instruments from Thermo Fisher Scientific. For P9, genomic DNA was extracted from plasma with the QIAmp DNA Blood Mini Kit (Qiagen, 51104). WES was performed with 3 µg of DNA from P1, P7, P8 and P16. Exome capture was performed with the SureSelect Human All Exon 72 Mb kit (Agilent Technologies, Santa Clara, CA, USA). Paired-end sequencing was performed on a HiSeq 2000 machine (Illumina, San Diego, CA, USA) generating 100-base reads. The sequences were aligned with the GRCh37 reference build of the human genome, with BWA. For the other patients, GATA2 was sequenced with primers flanking each of the seven exons and part of their intronic borders, as well as for a region encompassing 1,500 base pairs (bp) upstream of exon 1 for the promoter region (PCR amplification conditions and primer sequences are available). In addition, we sequenced the intron 5 of GATA2 (containing the mutation at position c.1017+572, a common mutation in previous publications) in all patients (9). PCR products were analyzed by electrophoresis in 1% agarose gels, sequenced with the Big Dye Terminator cycle sequencing kit (Applied Biosystems, Foster City, CA), and analyzed on an ABI Prism 3700 machine (Applied Biosystems, Foster City, CA).
Public database
Two public and international database were used to check the frequency of GATA2 variants found in this study. It corresponds to Genome Aggregation Database (gnomAD, gnomad.broadinstitute.org) and Biomarker Recognition and Validation Online (BRAVO, bravo.coh.org). The database PubMed was used for the review of literature to find the information about mycobacterial infections in germline GATA2 deficiency. No publication data limit was applied; conference papers were excluded.
Plasmid cloning and mutagenesis
PCMV6 empty plasmid (EV), GATA2-wild type (WT) isoform 1 (Origene RC208554) and GATA2-WT isoform 2 (Origene RC227514) were used to transform competent TOP 10 cells (Invitrogen, C404006). The plasmids were then extracted with the HiSpeed Plasmid Maxi Kit (Qiagen, 12663). Mutated plasmids (p.R330*, p.M388T, p.R396Q, p.R396W, p.R398Q, p.R398W, c.599dup, c.915_916del, c.1023del, c.1035_1036insTCTGGCC and c.1099dup) were generated by site-directed mutagenesis (QuikChange II XL Site-Directed Mutagenesis Kit: Agilent 200521–5). Insertions (p.V382Gfs*23 and p.N381_V382ins41) were generated using In-Fusion® HD Cloning Plus (Takara) following the manufacturer’s instructions. Deletion was generated using Pfu Ultra Fusion II (Agilent) and Quick Blunting and Quick Ligation kits (NEB). Briefly, primers encompassing non-deleted parts of the plasmid were generated and the plasmid was amplified. PCR reaction was blunted and then ligated following manufacturer’s instructions. The ligation product was then transformed into competent bacteria.
Immunoblots and luciferase reporter assay
Human embryonic kidney (HEK)293T cells were plated at a density of 0.6×106 in 2 mL of Dulbecco’s modified Eagle medium (DMEM) (Gibco) supplemented with 10% fetal bovine serum (FBS) (referred as complete medium) in 60 mm culture dishes, which were incubated at 37°C under an atmosphere containing 5% CO2. After 24 hours of culture, the cells were transfected with a Lipofectamine transfection kit (LTX and PLUS reagent, Invitrogen 15338100). For each plasmid, 5 µg of plasmid DNA was used to transfect the cells of the corresponding plate. After 48 hours, the cells were collected and proteins were extracted in modified RIPA buffer (Sigma). Protein concentration was determined by the Bradford method. The proteins extracted were subjected to polyacrylamide gel electrophoresis (Criterion XT precast gel 10% Bis-Tris, 1.0 mm, BIO-RAD). The resulting protein bands were transferred onto a membrane with the iBlot system (Invitrogen), and analyzed with an anti-GATA2 antibody (Santa Cruz, sc-267) diluted 1:500 in 1x PBS supplemented with 3% skim milk powder or BSA, and anti-DDK antibody (Origene, TA5001) at a dilution of 1:2,000. An anti-GAPDH antibody (Santa Cruz, sc-25778) was used as the internal expression control for immunoblotting.
Luciferase reporter assays were performed in HEK293T cells. We plated 3×106 cells in 100 µL of DMEM in each well of a 96-well culture plate, and incubated the plates at 37°C, under an atmosphere containing 5% CO2. Lipofectamine was used to transfect the cells with various plasmids: EV, GATA2-WT and mutated isoform 1. After 24 hours, the supernatant was discarded and luciferase activity was measured with the Dual-Luciferase Reporter Assay kit (Promega), according to the manufacturer’s instructions. The other plasmids used in the experiment were promyelocytic myeloid leukemia (PML; Myc-DDK-tagged, Origene RC220236), the GATA2 luciferase reporter plasmid (Panomics, LR0030) and Renilla pRL-SV40-d238, created by Ho and Strauss (55). Samples were analyzed in a luminometer (Victor X4 model 2030, Perkin Elmer). Experiments were performed in triplicates.
Enzyme-linked immunosorbent assay (ELISA)
ELISA was performed on plasma from patients, family members (when samples were available) and healthy donors, for human Flt-3 ligand (FLT3L) (R&D Systems, DFK00) and human M-CSF [R&D Systems, DMC00B], in accordance with the kit manufacturer’s instructions.
Peripheral blood mononuclear cells (PBMCs), RNA analysis and TOPO cloning
PBMCs were obtained from heparin-treated whole blood from patients and healthy controls, by centrifugal separation on a Ficoll gradient and washing with RPMI 1640. Total RNA was extracted from transfected cells or frozen PBMCs from patients and healthy donors, with the RNeasy Micro Kit (Qiagen, 74004). Reverse transcription was performed with SuperScript II Reverse Transcriptase (Invitrogen, 1804). Quantitative real-time PCR (qPCR) was performed in an ABI Prism 7500 machine (Applied Biosystems), with SYBR Green master mix (Biorad, 1725124) and specific primers for GATA2. GAPDH expression was used as an internal control. Data were analyzed by the comparative cycle threshold (ΔCt) method. TOPO TA cloning was performed using PCR products from cDNA of GATA2 patients following the manufacturer’s instructions (K450001, Thermo Fisher Scientific).
Statistical analysis
Statistical analysis for the luciferase assay experiments was performed with GraphPad Prism version 5.03. We checked for normal distribution, and then performed an unpaired t-test (Wilcoxon matched-pairs signed-rank test) for statistical analysis, as two groups were compared in each case: WT versus mutants, combinations of WT and EV versus combinations of WT and mutant, or WT versus combinations of WT and mutant. Data were considered significant if p<0.01.
Penetrance calculation
We calculated the penetrance using the strategy that we previously described (56, 57). Briefly, the index patient of each family was excluded from this calculation, considering only other symptomatic and asymptomatic family members. We estimated penetrance as a function of age for all types of GATA2-related disease and by disease type (focused in mycobacterial infection), in GATA2-deficient first-degree relatives of index cases. We did not assess individually the penetrance for all phenotypes identified in the spectrum of GATA2 deficiency. For the calculation, we excluded the proband or index case of each kindred because per se they were sick and they suffered all parameters of GATA2-related disease. The first strategy was based on the use of data for all relatives with identified GATA2 mutation only (n=18). The second strategy was based on the assumption that non-genotyped relatives suffering from GATA2-related disease, including those with relevant information in the estimation of penetrance (n=22). We used the non-parametric cumulative incidence function taking competing risks into account, as implemented in the R package “cmprsk” (http://cran.r-project.org). Confidence intervals (CIs) were calculated by log-log transformation, to ensure that the lower limits of the 95% CIs were positive.
Selective pressure measures
GATA2 negative selection was analyzed using previously described models. SnIPRE (58) makes use of a generalized linear mixed model to model genome-wide variability for categories of mutations. It estimates two key population genetic parameters for each gene, one of which, f, quantifies the strength of purifying selection acting on human genes, with 0 corresponding to strong negative selection and 1 to neutral selection. We used the alignment of the hg19 human genome and the PanTro3 chimp genome from UCSC Genome Browser. All differences between the two species were annotated functionally by snpEff 50, using the GRCh37.65 build. We retrieved all human coding sequences (CDS) of more than 20 bp in length, and we considered the longest available transcript for each gene. We then retrieved all polymorphisms identified in the WES data of phase 1 of the 1,000 Genomes Project. Accordingly, we analyzed single-nucleotide polymorphisms (SNPs) annotated as non-synonymous or synonymous, located outside of gaps in the human-chimp alignment and shown to be polymorphic in at least one human population. We also used the dbSNP136 chimp database to remove positions that were polymorphic in humans or chimps from the list of positions divergent between humans and chimps. We finally determined the number of divergent and polymorphic synonymous and nonsynonymous mutations, and the proportion of synonymous and nonsynonymous sites, for a total of 18,969 genes. SnIPRE was then used to estimate the f parameter.
RVIS (59) uses allele frequency data to rank genes according to whether functional genetic variation for the gene concerned is more or less frequent than expected based on the amount of apparently neutral variation observed for the gene. A RVIS close to 0 indicates low levels of functional variation, whereas genes with RVIS values close to 1 are subject to high levels of variation. We downloaded the RVIS values calculated with the ExAC dataset from the RVIS website.
The intolerance of each gene to LOF variants was assessed by calculating the pLI score (60). Values of pLI close to 1 indicate a high degree of intolerance to LOF for the gene concerned. We considered pLI values of 0.9 and above to define a set of genes with extremely low tolerance to LOF. We downloaded the pLI scores from the ExAC website (ftp://ftp.broadinstitute.org/pub/ExAC_release/release0.3.1/functional_gene_constraint/fordist_cleaned_exac_r03_march16_z_pli_rec_null_data.txt).
Results
Demographics and diagnosis of GATA2 deficiency in 15 kindreds
We studied 15 index cases and three symptomatic relatives from 14 non-consanguineous families and one consanguineous family presenting mycobacterial diseases and hematological abnormalities. In total, 18 patients (Figure 1, Table 1 and Table S1) were referred to our laboratory between 2011 and 2018. These patients were from Brazil (kindreds A and B), Colombia (C), France (D, E and O), Germany (F), Mexico (G), Portugal (H), Spain (I, J and K), Tunisia (L), Turkey (M), and the USA (N). Kindreds B, D and E have been reported elsewhere (36, 61). We performed Sanger sequencing for the seven exons of GATA2, their flanking intronic regions and the intron 5 in 11 of the 15 index cases (P2-P5, P9, P11-P13, P15, P17 and P18). WES was performed for four of the 15 index cases (P1, P7, P8 and P16). We identified five missense mutations [c.1163T>C (p.M388T) in kindreds A and J, c.1193G>A (p.R398Q) in kindred D, c. 1192C>T (p.R398W) in kindreds B and F, c.1187G>A (p.R396Q) in kindred G and c.1186C>T (p.R396W) in kindred M]; one nonsense mutation [c.988C>T (p.R330*) in kindred I]; one small insertion [c.1035_1036insTCTGGCC (predicted p.G346Sfs*40) in kindred K]; two small duplications [c.599dup (predicted p.S201*) in kindreds H and L and c.1099dup (predicted p.D367Gfs*17) in kindred N]; two small deletions [c.915_916del (predicted p.W306Afs*77) in kindred C and c.1023del (predicted p.A342Pfs*45) in kindred E]; and one essential splicing site substitution [c.1143+2T>A (predicted to cause the loss of the donor site of exon 5), Kindred O], all in the heterozygous state (Figure 1). By TOPO TA cloning we evaluated the cDNA consequences of the essential splice site mutation c.1143+2T>A and found: a deletion of exon 5 (predicted p. S340_N381del), an insertion of 65 intronic base pairs (predicted p.V382Gfs*23) or an insertion of 123 intronic base pairs (predicted p.N381_V382ins41, Figure S2). Ten of these mutations have already been reported in other patients (9, 10, 21, 25–27, 36, 62), whereas two are novel (c.1035_1036insTCTGGCC and c.1143+2T>A, Figure S1). Intron five was sequenced by Sanger in all the patients to investigate the potential presence of intronic mutations (9). These novel variants were not found in public databases (GnomAD and BRAVO database) or in our own WES database of about 8,000 exomes from patients with various infectious diseases. We analyzed the familial segregation of the mutant alleles in the 15 kindreds. Three family members with clinical features suggestive of GATA2 deficiency (mycobacterial disease and cutaneous warts for P6; pulmonary tuberculosis for P14; mycobacterial disease and MDS for P10) (Table 1 and table S1) were found to be heterozygous for GATA2 mutations. Nineteen individuals were wild-type (WT). Surprisingly, 16 individuals were carriers of a heterozygous GATA2 mutation, but were not clinically affected (kindreds A, B, D, J, L, M and N; Figure 1). Biological material was not available for 18 other individuals (Figure 1). Together, these data suggest that 34 individuals (18 patients and 16 carriers) had AD GATA2 deficiency.
Figure 1. GATA2 deficiency.
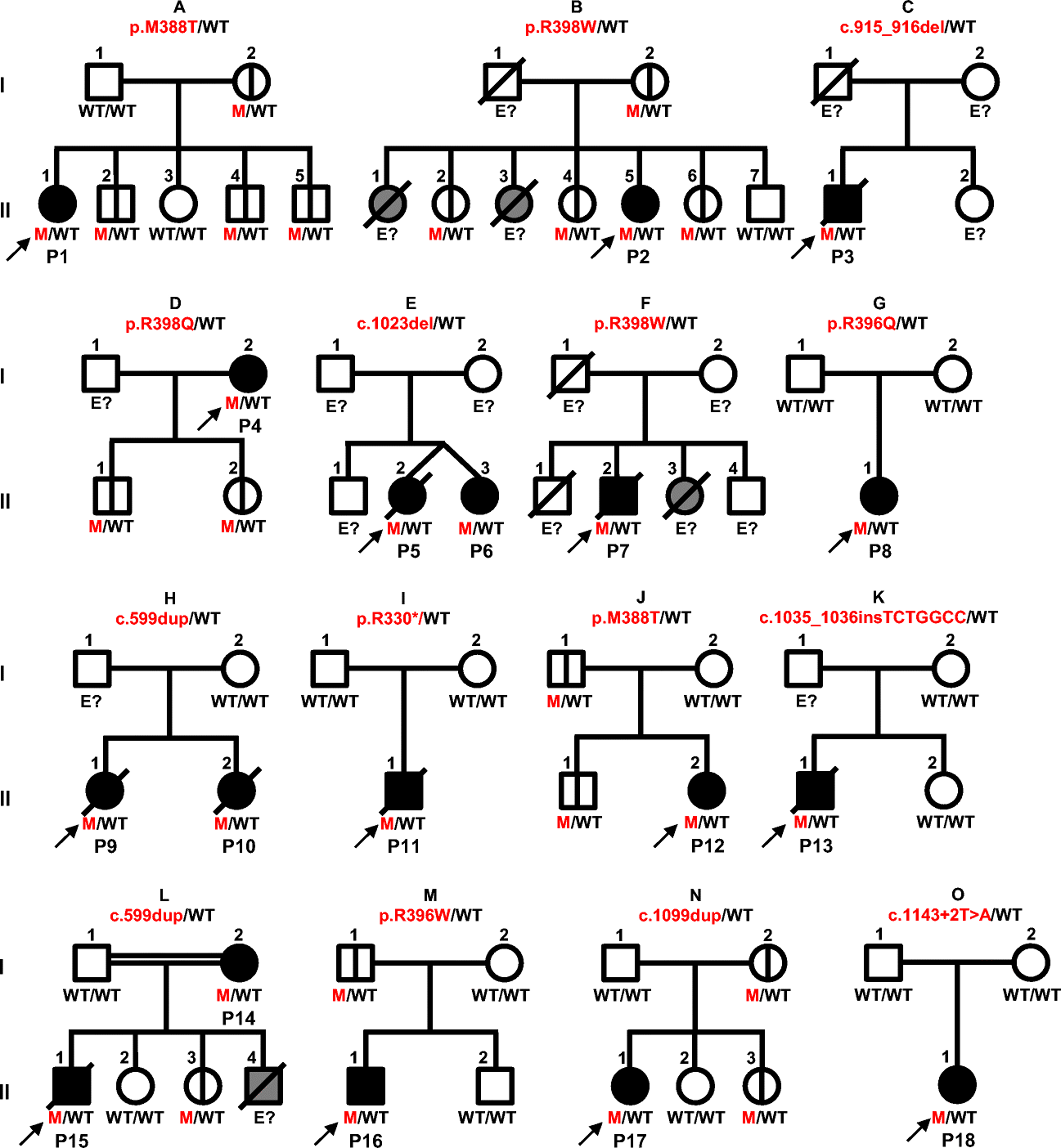
Schematic representation of the 15 kindreds with germline GATA2 mutations. Each kindred is designated by a capital letter (A-O), each generation by a Roman numeral (I-II), and each individual by an Arabic numeral. The proband is indicated by arrows. Solid black shapes indicate GATA2-deficient patients and clinical disease. Solid gray shapes indicate individuals with GATA2 deficiency-related clinical features (lymphedema or acute myeloid leukemia [AML]) who have not been genotyped. Mutations are indicated in red and wild type (WT) allele in black. Individuals whose genetic status could not be determined are denoted “E?” Individuals with symbols crossed by a black vertical line have asymptomatic GATA2 deficiency. Deceased individuals are represented by symbols crossed by a diagonal black line. Healthy individuals are shown in white.
Table 1.
Clinical features of GATA2-deficient patients
| Kindred | Code | No. | Mutation | Sex | Origin | Vital status | Mycobacterial species | Other microorganisms | Other clinical features | Transplantation |
|---|---|---|---|---|---|---|---|---|---|---|
| A | II. 1 | P1 | p.M388T/WT | Female | Brazil | Alive | M. tuberculosis, M. kansasii | S. aureus, HPV | Hypothyroidism, basal cell carcinoma, intermittent anemia, chronic sinusitis | - |
| B | II. 4 | P2A | p.R398W/WT | Female | Brazil | Alive | M. kansasii | K. pneumoniae, HPV | Lymphedema | - |
| C | II. 1 | P3 | c.915_916del/WT | Male | Colombia | Dead | M. tuberculosis | P. aeruginosa, HPV, Candida spp. | Deafness, cellulitis, spinocellular carcinoma | - |
| D | I.1 | P4B | p.R398Q/WT | Female | France | Alive | Mycobacterium spp. | H. influenzae, S. pneumoniae | Mouth ulcers, pneumonia, cellulitis, hemophagocytic syndrome | HSCT |
| E | II. 2 | P5B | c.1023del/WT | Female | France | Dead | M. avium, M. kansasii | HPV, Candida spp., Aspergillus spp. | Lymphedema, psoriasis, sinusitis | - |
| E | II. 3 | P6B | c.1023del/WT | Female | France | Alive | M. kansasii | HPV | Lymphedema | - |
| F | II. 1 | P7 | p.R398W/WT | Male | Germany | Dead | M. kansasii | - | - | - |
| G | II. 1 | P8 | p.R396Q/WT | Female | Mexico | Alive | M. kansasii | C. difficile | Obesity, subclavian thrombosis, hepatomegaly | - |
| H | II. 1 | P9 | c.599dup/WT | Female | Portugal | Dead | M. avium complex | EBV, Candida spp. | Granulomatous hepatitis | - |
| H | II. 2 | P1 0 | c.599dup/WT | Female | Portugal | Dead | Mycobacterium spp. | HPV, HHSV-2 | Mouth ulcers, hemochromatosis | Liver |
| I | II. 1 | P11 | p.R330*/WT | Mal e | Spain | Dead | Mycobacterium spp. | Candida spp. | Lymphedema, monoclonal gammapathy, sarcoidosis | - |
| J | II. 2 | P12 | p.M388T/WT | Female | Spain | Alive | M. avium intracellulare | S. salivarius, A. fumigatus | Deafness | HSCT |
| K | II. 1 | P13 | c.1035_1036insTC TGGCC/WT | Male | Spain | Dead | M. kansasii | C. freundii, Staphylococcus spp., Candida spp. | Inferior vena cava thrombosis, deafness | - |
| L | I.1 | P14 | c.599dup/WT | Female | Tunisia | Alive | M. tuberculosis | HPV | Lymphedema, thrombosis | - |
| L | II. 1 | P15 | c.599dup/WT | Male | Tunisia | Dead | M. tuberculosis | HPV | Lymphedema | - |
| M | II. 1 | P16 | p.R396W/WT | Male | Turkey | Alive | M. tuberculosis | HSV 1, HPV, Molluscum contagiosum | Hepatosplenomegaly, psoriasis, sarcoidosis | HSCT |
| N | II. 1 | P17 | c.1099dup/WT | Female | USA | Alive | M. avium intracellulare | C. difficile, S. pneumoniae, H. capsulatum, Brucella spp. | Mouth ulcers | - |
| O | II. 1 | P18 | c.1143+2T>A/WT | Female | France | Alive | M. avium | Undetermined | Erythema nodosum | - |
HSCT, Hematopoietic stem cell transplantation; HPV, Human papilloma virus; HSV, Herpes simplex virus; EBV, Epstein-Barr virus; -, Not present; patients previously reported by Jobim et al.60 (P2A) or Donadieu et al.36 (P4-P6B).
Expression of the mutant GATA2 alleles in HEK293T cells
Human GATA2 encodes two isoforms: isoform 1, which has 480 amino acids (aa, 50.5 kDa) and isoform 2, which has 466 aa (49.1 kDa) (1). We evaluated GATA2 expression, by transiently transfecting HEK293T cells with constructs encoding C-terminally DDK-tagged WT or mutant GATA2 cDNAs. Similar levels of GATA2 mRNA were detected in HEK293T cells with the WT allele and in cells with mutant alleles (data not shown). The WT GATA2 proteins had the expected molecular weight (MW) on western blots. The missense mutations (p.M388T, p.R396Q, p.R396W, p.R398Q and p.R398W) were detected at the expected MW. Protein levels for all missense mutations were normal for both isoforms, as shown by detection with a DDK-specific Ab (Figure 2 and Figure S3). The c.915_916del, c.1023del, p.R330*, p.V382Gfs*23, p.N381_V382ins41 and p. S340_N381del mutations produced a truncated protein for both isoforms, as shown by detection with an Ab against GATA2. The c c.1035_1036insTCTGGCC mutation produced a truncated protein in isoform 1 (Figure 2). The c.1099dup and c.599dup produced low levels of GATA2 protein of a lower MW for isoform 1 (Figure 2) and no detectable GATA2 protein for isoform 2 (Figure S3).
Figure 2. Protein expression of GATA2 alleles in an overexpression system.
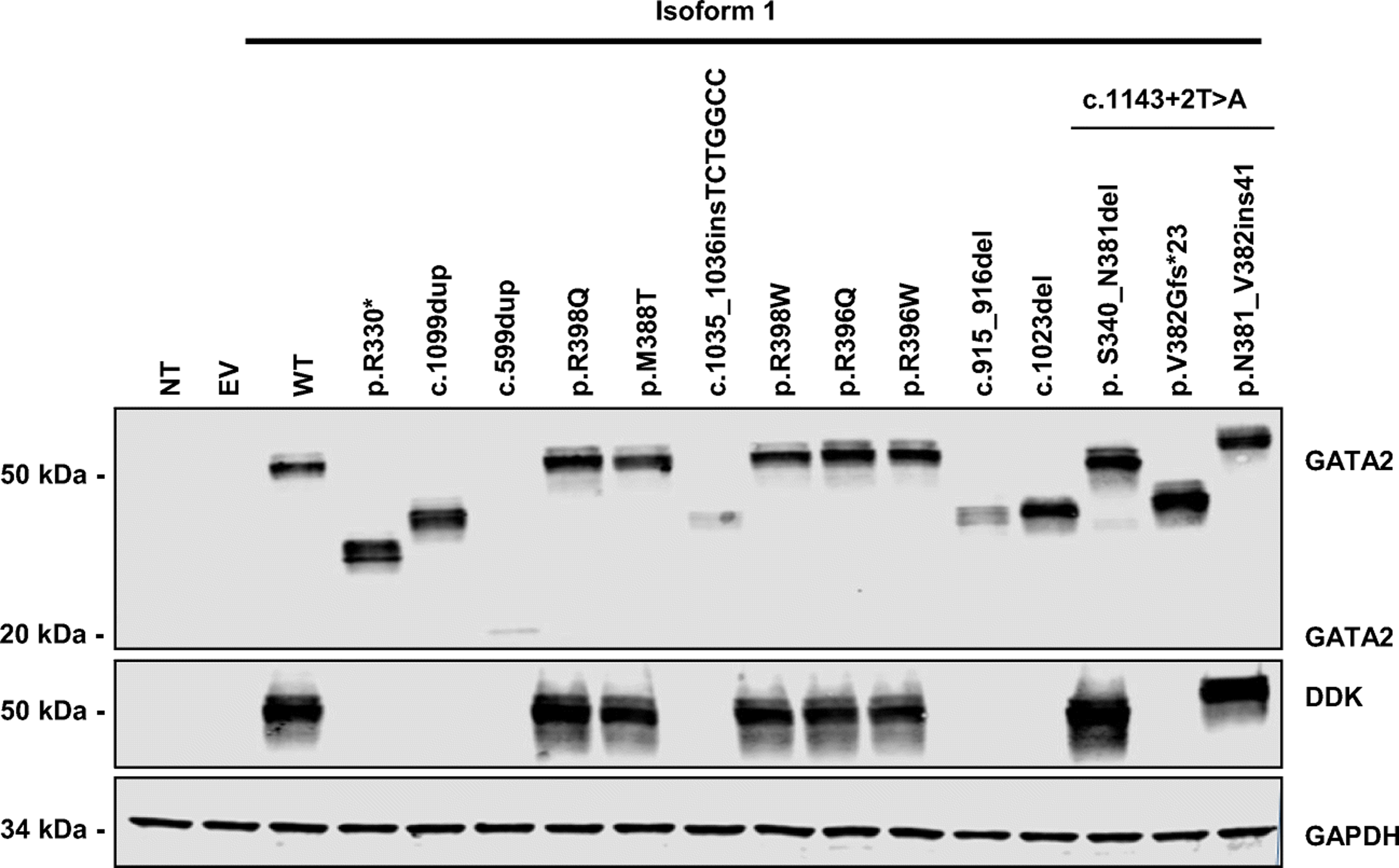
HEK293T cells were transfected with an empty plasmid (EV), a construct encoding GATA2 wild-type (WT)-DDK-tagged or mutated alleles corresponding to isoform 1 of GATA2 (p.R330*, p.M388T, p.R396W, p.R396Q, p.R398Q, p.R398W, c.599dup, c.915_916del, c.1023del, c.1035_1036insTCTGGCC, c.1099dup, p.V382Gfs*23, p.N381_V382ins41 and p. S340_N381del) for 48 hours. GATA2 protein levels were evaluated by western blotting with antibodies against GATA2 and against the DDK tag. GAPDH was used as a loading control. NT indicates non-transfected cells. The results shown are representative of three independent assays.
Functional activity of the mutant GATA2 alleles in HEK293T cells
We then assessed the functional activity of the GATA2 mutants in a luciferase assay using HEK293T cells transfected with the different cDNAs and a renilla to assess transfection efficiency. The renilla used in this assay was modified to delete GATA2 binding sites (55). Only isoform 1 of GATA2 displayed transcriptional activity. The PML gene is a member of the tripartite motif (TRIM) family, the members of which have been reported to potentiate the transactivational activity of GATA2 (63). We therefore evaluated the impact of the various mutant alleles relative to GATA2-WT, with or without PML in overexpression. Similar levels of GATA2 mRNA were detected in HEK293T cells with the WT allele and in cells with mutant alleles (data not shown). HEK293T cells transfected with GATA2-WT displayed functional activity that was enhanced when the cells were cotransfected with PML (Figure 3A). However, the function of the various GATA2 alleles was severely impaired or abolished in both sets of conditions, depending on the allele tested, as shown by the significant difference relative to the WT (Figure 3A). Luciferase activity was impaired but not completely abolished for the missense alleles and the inframe deletion and insertion (p.M388T, p.R396Q, p.R396W, p.R398Q, p. S340_N381del and p.N381_V382ins41), suggesting that these alleles are functionally hypomorphic, whereas one missense variant (p.R398W), and the nonsense (p.R330*) and all frameshift alleles (c.599dup, c.915_916del, c.1023del, c.1035_1036insTCTGGCC, c.1099dup and p.V382Gfs*23) resulted in a complete LOF, at least in these overexpression conditions (Figure 3A).
Figure 3. Functional characterization of GATA2 alleles.
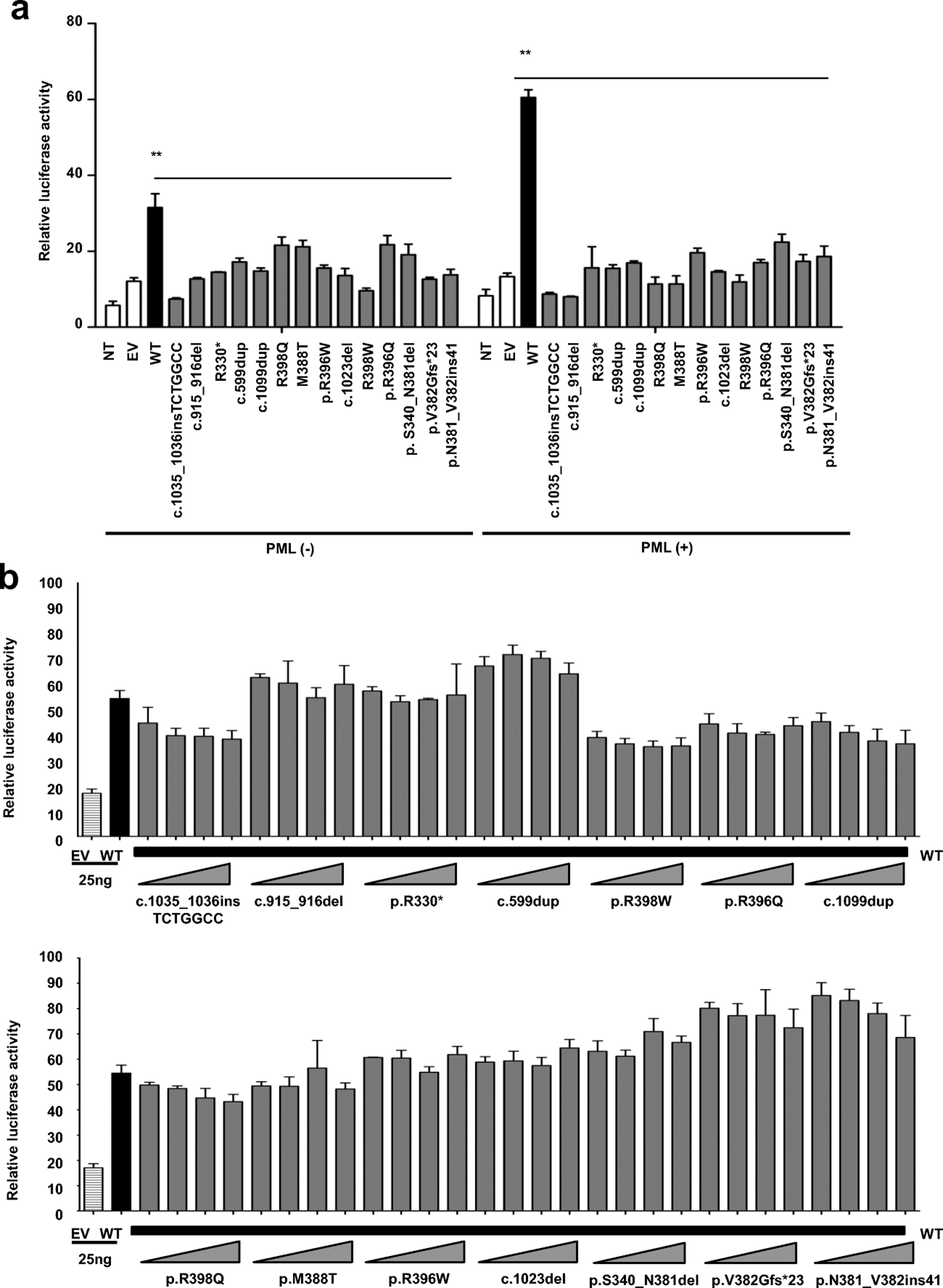
A. GATA2-luciferase activity in HEK293T cells. GATA2-dependent transactivation potential was evaluated with a GATA2-luciferase reporter plasmid. Cells were transiently transfected with (+) or without (–) PML, empty plasmid (EV) and various GATA2-plasmids (WT, p.R330*, p.M388T, p.R396W, p.R396Q, p.R398Q, p.R398W, c.599dup, c.915_916del, c.1023del, c.1035_1036insTCTGGCC, c.1099dup, p.V382Gfs*23, p.N381_V382ins41 and p. S340_N381del). B. Transient cotransfection of cells with PML and equivalent amounts of GATA2-WT plasmid (25 ng) but with various amounts of mutant plasmids (p.R330*, p.M388T, p.R396W, p.R396Q, p.R398Q, p.R398W, c.599dup, c.915_916del, c.1023del, c.1035_1036insTCTGGCC, c.1099dup, p.V382Gfs*23, p.N381_V382ins41 and p. S340_N381del), with different amounts of EV used to keep the final plasmid concentration constant. Error bars indicate the standard error of the mean. The results shown are representative of two independent assays with three replicates per experiment.
Study of the mechanism of dominance in HEK293T cells
We then investigated the mechanism underlying dominance (haploinsufficiency or negative dominance) in heterozygous cells. We performed luciferase assays in cells transiently co-transfected with PML and combinations of GATA2-WT and GATA2-mutant plasmids in different amounts, mimicking heterozygosity (Figure S4). Higher levels of luciferase activity were observed for combinations of WT-GATA2 with the various mutant GATA2 plasmids than for combinations of the mutant GATA2 plasmids with EV. The luciferase activities of the WT-mutant plasmids combination were in the range of the WT-EV combination (Figure S4), suggesting that the mechanism of dominance does not involve negative dominance. We also performed a different kind of assay in which the amount of WT plasmid was kept constant whilst that of the mutant plasmid was variant (Figure 3B). Similar levels of luciferase activity were recorded for the different amounts of mutant plasmid used, all these levels being higher than that for EV, but not significantly different from the corresponding WT with EV point. This finding provides support for the haploinsufficiency hypothesis because luciferase activity would be predicted to decrease in the presence of larger amounts of mutant plasmid in a negative dominance model.
Selective pressure selection of GATA2 gene
Purifying selection or negative selection is the selective removal of deleterious alleles from the population. We say that a gene is under purifying selection if the rate of its non-synonymous to synonymous variants (dN/dS) in a given population is significantly more reduced compared that expected by random chance (dN/dS=1) (64). We analyzed several selection parameters, to estimate the evolutionary pressure on the GATA2 locus (see methods). GATA2 is predicted to be intolerant to LOF variants (pLI score of 0.97, with 0.9 taken as the threshold beyond which genes are considered to be extremely intolerant of LOF variants) (Figure 4A). Moreover, GATA2 was found to be under negative selection according to the f parameter from Selection inference using Poisson random effects (SnIPRE), which is 0.388 (Figure 4B) (95% confidence interval [CI]: 0.223 – 0.675), placing GATA2 in the 26.4% of genes under the strongest evolutionary pressure. The residual variation intolerance score (RVIS) of 11.839 (Figure 4C) places GATA2 in the 11.8% of genes under the strongest negative pressure. Moreover, consensus negative selection (CoNeS) score (65) of GATA2 was low, consistent with a negative selection of the gene (Figure 4D). These results strongly suggest that the GATA2 locus has evolved under purifying selection or negative selection, providing additional support for the hypothesis that GATA2 deficiency results from haploinsufficiency (9, 66–68).
Figure 4. List of variants and strength of the purifying selection acting on GATA2.
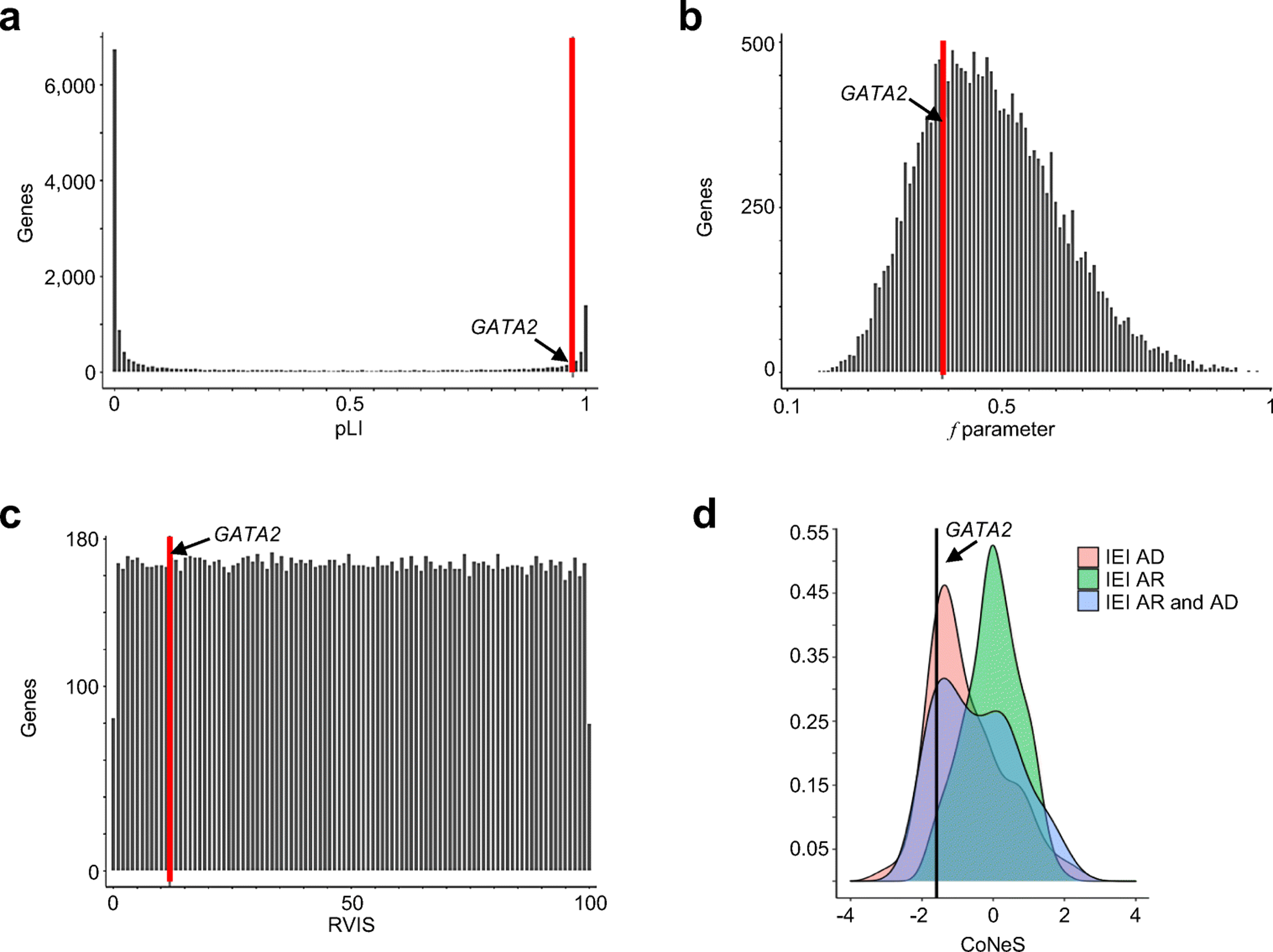
Genome-wide distribution of the strength of purifying selection, estimated by A. Probability of loss of function intolerance (pLI) score, B. the f parameter, C. residual variation intolerance (RVIS) score on bar plots, as described in the methods and D. consensus negative selection (CoNeS) score. The position of GATA2 is indicated by a black arrow and a red vertical bar.
Environmental mycobacterial diseases in GATA2-deficient patients
The mycobacterial diseases in the patients were caused by EM (n=10), M. tuberculosis (n=4), both EM and M. tuberculosis (n=1), and unidentified Mycobacterium spp. (n=3) (Figure 5A, Table 1). Eleven patients developed EM disease due to M. avium (n=5) and/or M. kansasii (n=7). One of these patients (P5) had infectious diseases caused by two different EM: M. avium and M. kansasii. P1 presented a combination of EM disease and tuberculosis. EM disease recurred in P13, who developed a M. kansasii infection of the lymph nodes at the age of 24 years, and a disseminated infection three years later. No other EM was identified in this cohort. Mean age at the diagnosis of EM infection was 20 years (range: 12 years to 29 years). EM infection was diagnosed on the basis of a positive culture (n=10), PCR (n=4), the direct detection of acid-fast bacilli (AFB; n=2), or on clinical grounds (n=2). Positive cultures were obtained from two sites in four patients: the lungs and blood (n=3), or the liver and blood (n=1). EM disease was detected in the lungs (n=7), lymph nodes (n=6), liver (n=3), spleen (n=2), and bone marrow (n=3) (Table 2 and Figure 5B). Up to four organs were affected in patients with EM infections. None of the patients of this cohort presented EM disease of the soft tissues or skin. Most episodes involved simultaneous infections of the lungs and lymph nodes. Disseminated EM disease was documented in seven patients (P5, P6, P7, P8, P9, P13 and P17). Three patients with pulmonary EM disease had fever and coughing (P2, P6 and P17) and three patients had chest pain, fever and coughing (P5, P7 and P12).
Figure 5. Mycobacterial infections and distribution in GATA2-deficient patients.
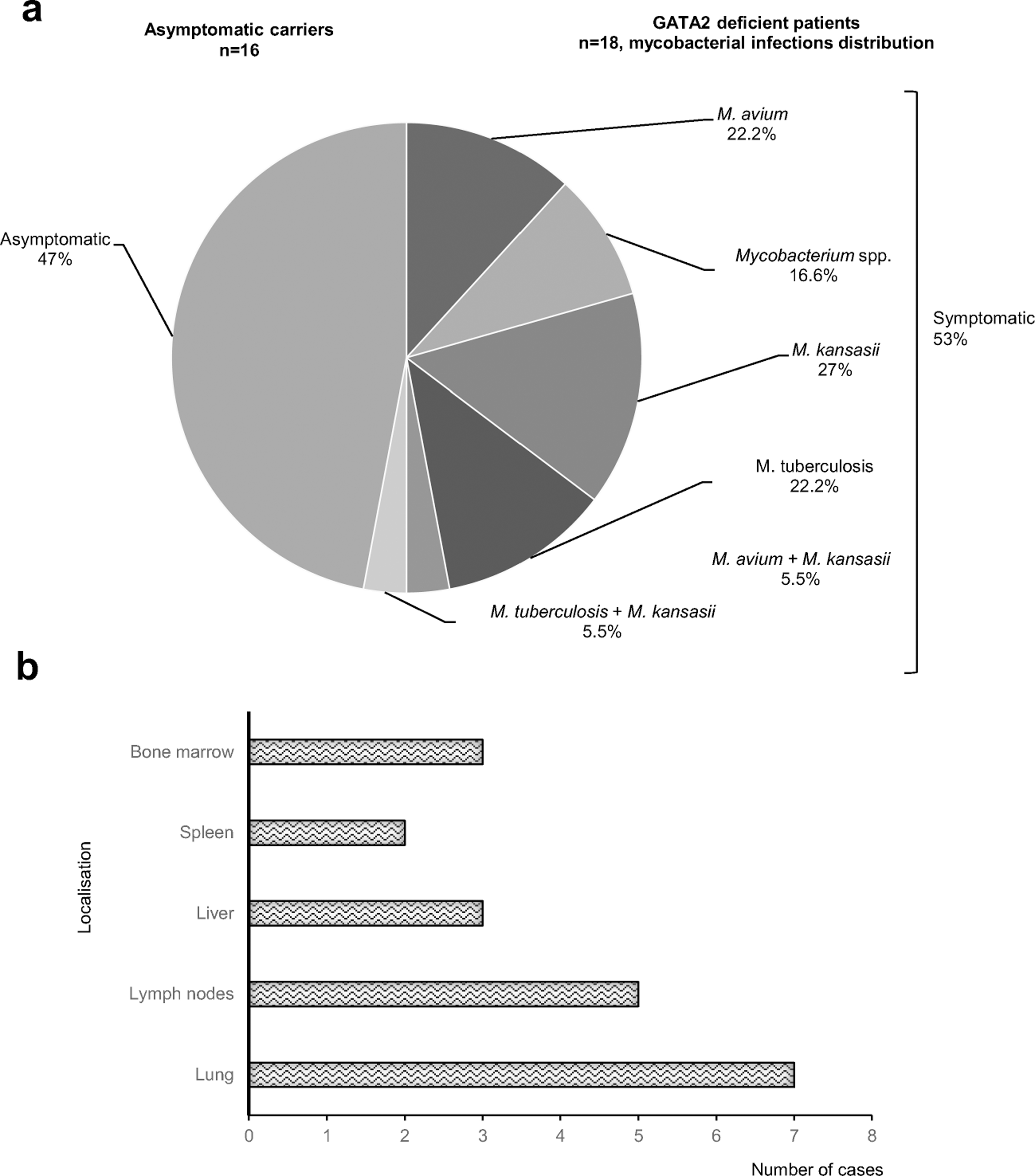
A. Spectrum of mycobacterial species causing disease in GATA2-deficient patients. The patients infected with a particular “type” of mycobacterial species are represented as percentages, with infections with two different species considered as a type of infection. Asymptomatic carriers are individuals carrying the mutation with no clinical phenotype. B. Mycobacterial infection sites in patients infected with EM infection.
Table 2.
Summary table of the GATA2 deficient-patients
| Total number of GATA2-deficient patientsA | n= 18 |
| Sex | |
| -Male | n = 6 |
| -Female | n = 12 |
| Age at diagnosis of GATA2 deficiency (years), mean (range) | 26.9 years (13–47) |
| Age at diagnosis of mycobacterial infection (years), mean (range) | 22.5 years (12–42) |
| Patients vaccinated with BCG | n = 13 |
| BCG-associated manifestations | n = 0 |
| Tuberculosis-associated manifestations | n = 7 |
| Localized | |
| -Lungs | n = 4 |
| -Lymph nodes | n = 1 |
| Disseminated | n = 2 |
| EM-associated manifestations | n = 16 |
| Localized | |
| -Lungs | n = 3 |
| -Lymph nodes | n = 3 |
| Disseminated | n = 10 |
| Unidentified mycobacterium-associated manifestations | n = 3 |
| Localized | |
| -Lungs | n = 2 |
| -Lymph nodes | n = 0 |
| Disseminated | n = 1 |
| Vital status | |
| Alive | n = 10 |
| Dead | n = 8 |
16 healthy GATA2 mutation carriers are not included in this table; BCG, Bacillus Calmette-Guérin; EM, environmental mycobacteria
Tuberculosis in GATA2-deficient patients
Thirteen of the 18 patients studied here were vaccinated with BCG at birth (Table 2). No complications of vaccination [local-regional BCG disease (BCG-itis) or disseminated BCG (BCG-osis)] were reported in this cohort. Tuberculosis was diagnosed in five patients (P1, P3, P14, P15 and P16, Table 1), all of whom had been vaccinated with BCG. Mean age at tuberculosis diagnosis was 29 years (range: 17 years to 42 years). Tuberculosis was thoracic in four patients (P1, P3, P14 and P15) and disseminated in one patient (P16). The diagnosis of tuberculosis was confirmed in two patients (P1 and P16) by cultures positive for M. tuberculosis, whereas tuberculosis was considered probable in the other three cases (P3, P14 and P15). In this cohort, tuberculosis was mostly found in the lungs (n=4) (Table 2). Recurrences of tuberculosis were documented in P3, who had two pulmonary episodes (at 28 years and 32 years), and P16 (29 years and 31 years), who suffered two episodes of disseminated tuberculosis affecting the lungs, lymph nodes, spleen and bone marrow. Patients with infections caused by unidentified Mycobacterium spp. displayed disseminated (P4) or localized (P10 and P11) forms of infectious disease.
Other clinical manifestations in GATA2-deficient patients
All patients suffered from non-mycobacterial infections, caused by viruses, bacteria, or fungi (Table 1, Table S2). The microorganisms isolated were: Streptococcus pneumoniae (n=2), Klebsiella pneumoniae (n=1), Citrobacter freundii (n=1), Clostridium difficile (n=2), Haemophilus influenzae (n=1), Pseudomona aeruginosa (n=1), Brucella spp. (n=1), Staphylococcus aureus (n=1) and Streptococcus salivarius (n=1). Eight patients suffered from bacterial infections other than mycobacterial infections (Table S2). In addition, five patients displayed mucocutaneous fungal infections caused by Candida spp.: mouth infections in three of these cases (P3, P9 and P11), cutaneous in another (P5) and esophageal in two cases (P11 and P13). Candida infections may have been favored by cytopenia (neutropenia in particular, documented in P3 and P13) and certain drug treatments (such as long-term antibiotic therapy with or without immunosuppressants, P13). One patient had disseminated histoplasmosis at the age of 23 years (P17). P5 and P12 suffered from disseminated aspergillosis, resulting in the death of P5. One patient suffered from Epstein-Barr virus (EBV) infection (P9). One patient suffered fulminant hepatitis due to Herpes simplex virus type 2 (HSV)-2 (P10). Eight patients had recurrent warts caused by human papillomavirus (P1, P2, P3, P5, P6, P10, P14 and P15). P1 had to be hospitalized for 10 days due to adverse reactions to yellow fever vaccine. In addition to infectious diseases, some patients had lymphedema (P1, P2, P5, P6, P14 and P15), deafness (P3 and P13), sarcoidosis (P11 and P16), psoriasis (P5 and P16), monoclonal gammopathy (P11), obesity (P8), thrombosis (P8, P13 and P14), autoimmune hemolytic anemia (P12), or non-infectious mouth ulcers (P4 and P17).
Hematological and immunological abnormalities in patients with GATA2 mutations
GATA2 mRNA is present in total peripheral mononuclear blood cells (PBMCs), in various lymphoid (T cells, B cells, NK cells) and myeloid cell (monocytes, total dendritic cells, myeloid dendritic cells, plasmacytoid dendritic cells) subsets and in CD34+ cells (1, 69–71). An evaluation of circulating blood cell counts for the patients for whom such data were available showed that some patients had low neutrophil counts (n=8), with few or no B cells (n=16), low T cell counts (n=11), few or no NK cells (n=15), and low dendritic cell counts (n=6) (Table S1). Monocytopenia was the most common feature among these patients, being found in 17 of 18 patients (Table S1). Bone marrow analysis was performed for 13 patients. Hypoplasia was observed in eight patients (P3, P5, P9, P10, P11, P13, P15 and P17), MDS was observed in seven patients (P4, P5, P9, P10, P11, P13 and P15), AML was observed in P7, and hemophagocytic syndrome secondary to disseminated mycobacterial infection was observed in P13 (Table S1). Cytogenetic analysis was performed for nine patients, six of whom were considered cytogenetically “normal” by metaphase cytogenetics. However, cytogenetic analysis results were aberrant for three patients (P10, P11 and P18; Table S1), with deletions and aneuploidy detected in P10; and duplication (15; 1qter->1q12:15p11->15qter) in P11. FLT3L levels in plasma were high in all the patients tested (n=9) and in one carrier, but was undetectable in four carriers (Figure S5A). No plasma samples were available for the other patients. Only two patients had high plasma M-CSF concentrations. Therefore, FLT3L is more sensitive than M-CSF in symptomatic patients and it is useful as parameter of GATA2 deficiency. This protein remained low in the other patients (n=7; Figure S5B).
Incomplete clinical penetrance for GATA2 deficiency
We then estimated clinical penetrance as a function of age at onset of the first GATA2 deficiency-related symptom in 18 relatives of the index cases confirmed to carry GATA2 mutations (P14 was not considered due to a lack of information about age). Only two of these 18 individuals presented GATA2 deficiency-related symptoms (Table 1) (P6, suffered from M. kansasii infection at 17 years of age; and P10 suffered from MDS at 12 years of age). The other 16 individuals remained healthy at their most recent follow-up visit (mean follow-up: 39.33 years; range: 6 to 78 years). Estimated clinical penetrance in these 18 individuals reached a plateau at 14%, at the age of 20 years (95% CI: 0.02–0.36) (Figure 6A). However, only living relatives of the index case and one dead relative (P10) were tested for genetic etiologies of GATA2 deficiency, which may have led to an ascertainment bias and underestimation of penetrance. We therefore performed a second calculation including four non-genotyped family members who died from diseases very likely to be related to GATA2 deficiency: AML or lymphedema (kindred B, II.1 and II.3, both of whom died from AML at ages of 36 and 33 years, respectively; kindred F, II.3 who died from AML at the age of 26 years; and kindred L, II.4, who died from lymphedema at birth, taken an age of 0). Penetrance in these 22 individuals was estimated at 15.5% by the age of 25 years (95% CI: 0.034–0.35), and increased to 32.9% by the age of 40 years (95% CI: 0.12–0.55) (Figure 6A). This result suggests that penetrance for GATA2 deficiency increases with age, but remains incomplete at the age of 40 years. Mycobacterial infections were the main clinical manifestation in the 14 index cases. We therefore analyzed the penetrance of mycobacterial diseases relative to the other clinical phenotypes associated with GATA2 deficiency (AML, viral infections or lymphedema) in the 22 aforementioned individuals. Penetrance for mycobacterial disease was estimated at 10% by the age of 20 years (95% CI: 0.017–0.3), increasing to 22.6% by the age of 40 years (95% CI: 0.06–0.44) (Figure 6B), whereas the penetrance for other clinical phenotypes was estimated at 10% at the age of 20 years (95% CI: 0.15–0.28), increasing to 28% by the age of 40 years (95% CI: 0.095–0.5) (Figure 6B). We observed no significant differences in penetrance between mycobacterial disease and other clinical phenotypes, probably due to the small number of individuals available for this analysis. Estimates of clinical penetrance may be biased by incomplete information about the relatives of the index case (as illustrated by the observed difference between the two strategies of analysis). The observation of four GATA2-mutation carriers who remain physically healthy at more than 60 years of age (kindred A, I.2, 78 years; kindred B, I.2, 63 years; kindred J, I.1, 68 years; and kindred M, I.1, 66 years) confirms the incomplete penetrance of these mutations. These results suggest that other elements, such as genetic modifiers or pathogen exposure, may play a role in the development of GATA2 deficiency-related diseases (31, 72).
Figure 6. Penetrance of clinical diseases in patients with GATA2 deficiency.
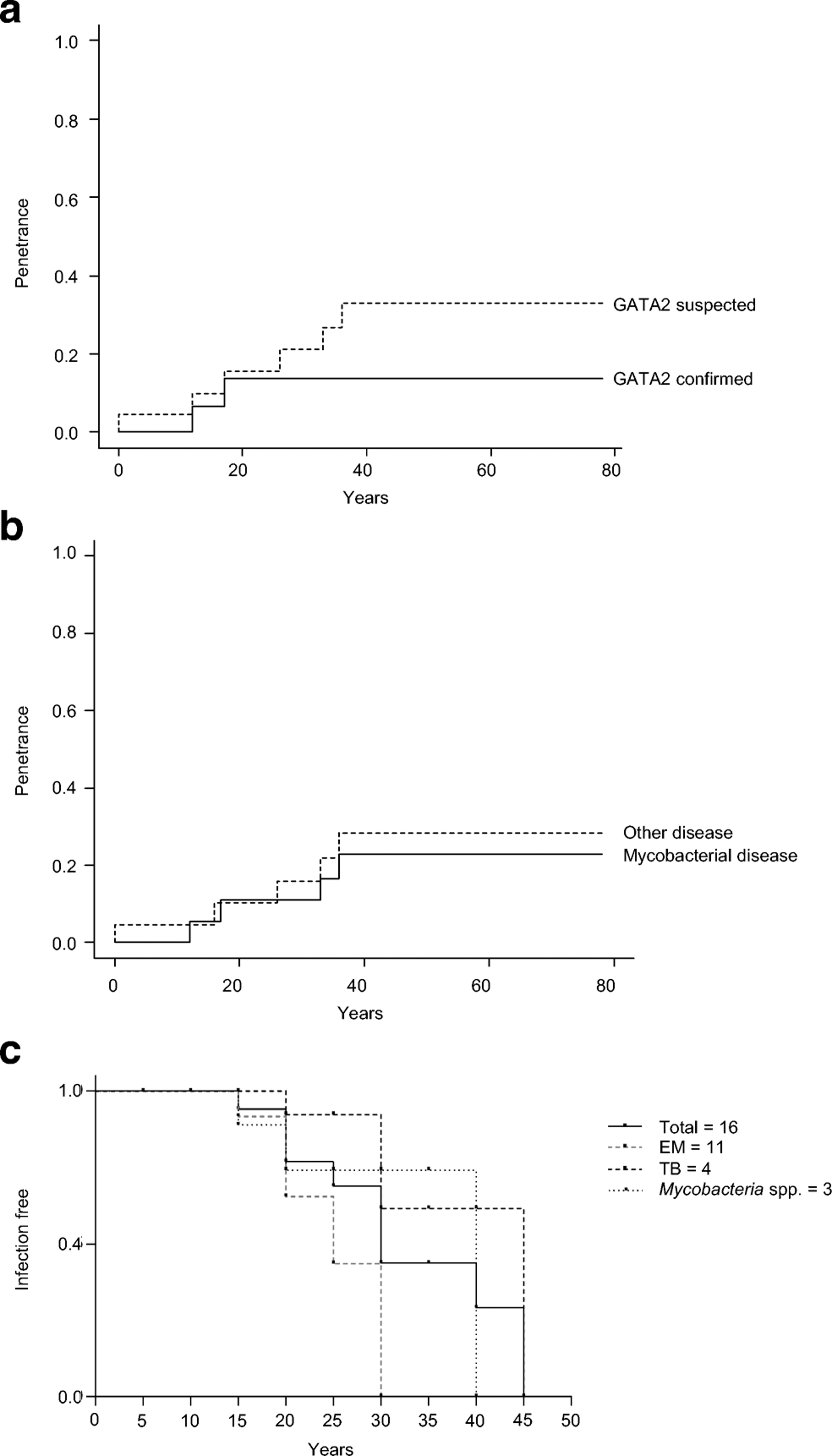
A. Kaplan-Meier curves showing the penetrance of GATA2 in confirmed carriers (GATA2 confirmed, continuous line) and family members with suspected GATA2 deficiency (GATA2 suspected, discontinuous line) with clinical manifestations at 20 and 40 years. B. Penetrance of mycobacterial infection (mycobacterial disease, continuous line) and other clinical manifestations of GATA2 deficiency (other disease, discontinuous line). C. Kaplan-Meier curve showing the percentage of mycobacterial infection-free patients from 5 to 45 years. The total number of patients (n=16) is indicated by a continuous black line, patients infected with environmental mycobacteria (EM, n=11) are indicated by a discontinuous gray line, patients infected with Mycobacterium tuberculosis (TB, n=4) are indicated by a discontinuous black line and patients infected with unidentified mycobacteria (Mycobacterium spp., n=3) are indicated in a black dotted line.
Clinical outcome
We focused on the onset of mycobacterial infection and survival in the 18 patients. Age at onset of infection was known for 16 of these patients, with infection typically beginning in early adulthood. Mean age at onset of the first mycobacterial infection was 22.5 years (range: 12 to 42 years) (Table 2 and Figure 6C). The first mycobacterial infection was caused by EM in 11 of these cases (mean age: 20 years; range: 12 to 29 years), by TB in four cases (mean age: 29 years; range: 17 to 42 years) and unidentified mycobacteria in three cases (mean age: 22.6 years; range: 12 to 38 years) (Figure 6C). Ten of the 18 patients and all the asymptomatic individuals are still alive (Table 2). The most recent follow-up visit occurred at a mean age of 31.3 years (range: 13 to 51 years) for the patients and 41.18 years (range: 11 to 78 years) for the asymptomatic individuals. Three patients (P4, P12 and P16) underwent HSCT, who are alive and well, and one patient underwent liver transplantation (P10). Mortality was 44.4% for the patients with symptoms (P3, P5, P7, P9, P10, P11, P13 and P15). The cause of death was infectious disease in three patients (disseminated Aspergillus infection for P5, disseminated M. avium infection for P9 and disseminated M. kansasii infection for P13), hematological disease in two patients (myelodysplasia for P3 and AML for P7), and unknown in two patients (P11 and P15). Fulminant hepatitis associated with encephalitis and multiorgan failure caused the death of P10.
Discussion
We describe here an international cohort of 18 patients with AD GATA2 deficiency and mycobacterial disease from 15 kindreds in 10 countries. Various patients with this disorder have already been reported, but information about their ethnicity is often missing. The patients reported here were from Brazil, Colombia, France, Germany, Mexico, Portugal, Spain, Tunisia, Turkey, and the USA. The key clinical presentation in these GATA2-deficient patients is mycobacterial disease, with a high proportion of infections caused by EM, such as M. kansasii and M. avium, in particular. M. tuberculosis was the second most frequently identified mycobacterium, identified in five of these patients. No significant differences in terms of mycobacterial species and geographic or ethnic origin were found between this cohort and other reported patients (3, 8, 9, 26–28, 34–36). No complications of BCG vaccination were reported in this cohort, consistent with the low susceptibility to BCG documented in other published studies of GATA2-deficient patients (3, 26, 28). This probably reflects the age-dependent decline of immunity in these patients. Interestingly, one patient had an adverse reaction to the yellow fever vaccine, requiring hospitalization. The loss of peripheral dendritic cells, including plasmacytoid dendritic cells in particular, may have contributed to the adverse effects of this vaccine, as reported for GATA2-deficient patients with severe influenza (18, 73). Patients with GATA2 deficiency are also susceptible to viral, pyogenic, and fungal infections.
Clinical penetrance for any individual GATA2 deficiency-related disease phenotype was incomplete at the age of 40 years (32.9%), as 16 relatives of the index cases carried GATA2 mutations but remained asymptomatic, up to the age of 78 years for one of these individuals. Penetrance for mycobacterial infection was incomplete at the age of 40 years (22.6%), and there were no significant differences in penetrance between mycobacterial disease and other clinical phenotypes. The penetrance of GATA2 deficiency was not sufficiently well characterized in previous published, in which a high but incomplete penetrance was assumed for MDS and AML (1, 15, 21, 29) and a low penetrance was assumed for lymphedema (7, 22, 28, 31, 72, 74–77) based on the number of patients affected, in the absence of a detailed penetrance analysis. We show here, by calculating the clinical penetrance of any GATA2 deficiency-associated disease phenotype or for mycobacterial disease in particular, excluding the index cases, that the penetrance of GATA2 deficiency increases with age but remains incomplete at the age of 40 years. Various factors may underlie this clinical heterogeneity, including gene modifiers in humans, promoter methylation, allele-specific expression, somatic rescue environmental exposure and the type of microbe (31, 72, 78). Our findings therefore formally demonstrate the incomplete penetrance of GATA2 deficiency, which has been assumed for many years, but never analyzed directly (36, 72, 74, 75).
Only 12 of the germline GATA2 mutations reported in previous studies have been properly characterized in vitro, limiting our understanding of the mechanism underlying GATA2 deficiency (6–18). We report here an in vitro evaluation of all GATA2 variants found, two of which have never before been described. Protein levels were affected, with all variants generating lower levels of protein than the WT, a truncated protein or no protein at all. We also showed, in luciferase assays, that WT GATA2 was able to bind and activate its own promoter, consistent with the findings of Cortes-Lavaud et al (10). The mutant alleles tested displayed a complete LOF or were hypomorphic in terms of this binding. However, functional differences were not correlated with clinical outcome. Clinical phenotypes did not differ markedly between patients with hypomorphic alleles and patients carrying LOF alleles. A comparison of these patients with other reported patients carrying the same mutations revealed their clinical phenotypes to be similar (in terms of infections and hematological manifestations), but not identical (21, 24, 27, 28). This observation has been made before in other studies in which clinical outcome was found to differ between individuals carrying the same mutation, and even between family members (27, 75).
Our findings suggest that haploinsufficiency is the mechanism underlying dominance. This conclusion is based on (i) the truncating nature of most mutations, (ii) the lack of negative dominance in vitro, and (iii) the negative selection acting on GATA2. Our results are at odds with a previous report on p.R330* (17), which suggested that this variant was dominant-negative. The differences in the experimental conditions used probably account for this discrepancy. The use of different promoters, normalization strategies, and transfection reagents, may account for the differences in the results obtained. A previous report demonstrated that the same allele could be complete LOF or hypomorphic, depending on the promoter used for the luciferase assay (15), highlighting the different roles of GATA2 as a transcription factor. GATA2 deficiency is a complex inborn error of hematopoiesis requiring further study and characterization, given the observed differences between patients and the incomplete penetrance for any GATA2 deficiency-related disease considered. It has already been shown that epigenetic factors may play a role in the severity of GATA2 deficiency (72). Therefore, the search of modifier genes and somatic variations might help to characterize the broad spectrum within this deficiency (annulling partial or totally the effect of the germline causing mutation) (72, 74, 78). A diagnosis of AD GATA2 deficiency should be considered in adults of any age with mycobacterial infections, with hematological disorders. FL3TL on plasma constitute a good monitoring in asymptomatic individuals to see the evolution of the disease. Moreover, all direct relatives of these patients should undergo genotyping for the GATA2 locus. Complete familial segregation is strongly considered in the selection of donor for HSCT in GATA2 deficiency.
Supplementary Material
Acknowledgments
We would like to thank the patients and their families, whose cooperation was essential for the collection of the data used in this study. We thank all members of the Laboratory of Human Genetics of Infectious Diseases for helpful discussions and Christine Rivalain, Cécile Patissier, Dominick Papandrea, Dana Liu and Yelena Nemirovskaya for their assistance. We would also like to thank Annarita Miccio for providing us with the sequences for the GATA2 and GAPDH qPCR primers, and Clement KM Ho and Jerome F Strauss for providing us with the modified pRL-SV40-d238 Renilla.
The Laboratory of Human Genetics of Infectious Diseases is supported in part by institutional grants from INSERM, Paris Descartes University, The Rockefeller University and the St. Giles Foundation, the National Institute of Allergy and Infectious Diseases (NIAID) of the National Institutes of Health (NIH) (R37AI095983), and grants from the French National Research Agency (ANR) under the “Investments for the future” program (ANR-10-IAHU-01) and IFNGPHOX (ANR13-ISV3–0001-01 for JB and ACN), GENMSMD (ANR-16-CE17–0005-01 for JB) grants, ECOS-NORD (C19S01–63407 for JB and JFR) and SRC2017 (for JB). CO-Q is supported by ANR-HGDIFD (ANR-14-CE15–006-01). AG was supported by the ANR-IFNGPHOX (ANR13-ISV3–0001-01), GENMSMD (ANR-16-CE17–0005-01) and the Imagine Institute. AC-N and EBO-J are supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP grants 2012/11757–2, 2010/51814–0, and 2012/51094–2) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPQ grant 303809/2010–8). MMG and RC are supported by Instituto de Salud Carlos III, grants PI11/01086 and PI14/00405, co-financed by the European Regional Development Fund (ERDF). JFR and AAA are supported by Colombia-France (ECOS-NORD/COLCIE NCIAS/MEN/ICETEX; 619–2013, Diana García de Olarte foundation PID and Colciencias grant 713–2016 #111574455633).
Footnotes
Compliance with ethical standards
Research involving human participants. Informed consent for participation in this study was obtained in accordance with local regulations, with approval from the IRB. The experiments described here were performed in Mexico and France, in accordance with local regulations.
Informed consent. Written informed consent was obtained from the guardians of the pediatric patients or directly from adult patients.
Conflict of interest: The authors have no conflict of interest to declare
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
References
- 1.Collin M, Dickinson R, Bigley V. Haematopoietic and immune defects associated with GATA2 mutation. Br J Haematol. 2015;169(2):173–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wlodarski MW, Collin M, Horwitz MS. GATA2 deficiency and related myeloid neoplasms. Semin Hematol. 2017;54(2):81–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bigley V, Haniffa M, Doulatov S, Wang XN, Dickinson R, McGovern N, et al. The human syndrome of dendritic cell, monocyte, B and NK lymphoid deficiency. J Exp Med. 2011;208(2):227–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hsu AP, Sampaio EP, Khan J, Calvo KR, Lemieux JE, Patel SY, et al. Mutations in GATA2 are associated with the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome. Blood. 2011;118(10):2653–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hyde RK, Liu PP. GATA2 mutations lead to MDS and AML. Nat Genet. 2011;43(10):926–7. [DOI] [PubMed] [Google Scholar]
- 6.Hahn CN, Chong CE, Carmichael CL, Wilkins EJ, Brautigan PJ, Li XC, et al. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat Genet. 2011;43(10):1012–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ostergaard P, Simpson MA, Connell FC, Steward CG, Brice G, Woollard WJ, et al. Mutations in GATA2 cause primary lymphedema associated with a predisposition to acute myeloid leukemia (Emberger syndrome). Nat Genet. 2011;43(10):929–31. [DOI] [PubMed] [Google Scholar]
- 8.Johnson KD, Hsu AP, Ryu MJ, Wang J, Gao X, Boyer ME, et al. Cis-element mutated in GATA2-dependent immunodeficiency governs hematopoiesis and vascular integrity. J Clin Invest. 2012;122(10):3692–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hsu AP, Johnson KD, Falcone EL, Sanalkumar R, Sanchez L, Hickstein DD, et al. GATA2 haploinsufficiency caused by mutations in a conserved intronic element leads to MonoMAC syndrome. Blood. 2013;121(19):3830–7, S1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cortes-Lavaud X, Landecho MF, Maicas M, Urquiza L, Merino J, Moreno-Miralles I, et al. GATA2 germline mutations impair GATA2 transcription, causing haploinsufficiency: functional analysis of the p.Arg396Gln mutation. J Immunol. 2015;194(5):2190–8. [DOI] [PubMed] [Google Scholar]
- 11.Kazenwadel J, Betterman KL, Chong CE, Stokes PH, Lee YK, Secker GA, et al. GATA2 is required for lymphatic vessel valve development and maintenance. J Clin Invest. 2015;125(8):2979–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fujiwara T, Fukuhara N, Funayama R, Nariai N, Kamata M, Nagashima T, et al. Identification of acquired mutations by whole-genome sequencing in GATA-2 deficiency evolving into myelodysplasia and acute leukemia. Ann Hematol. 2014;93(9):1515–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hahn CN, Brautigan PJ, Chong CE, Janssan A, Venugopal P, Lee Y, et al. Characterisation of a compound in-cis GATA2 germline mutation in a pedigree presenting with myelodysplastic syndrome/acute myeloid leukemia with concurrent thrombocytopenia. Leukemia. 2015;29(8):1795–7. [DOI] [PubMed] [Google Scholar]
- 14.Greif PA, Dufour A, Konstandin NP, Ksienzyk B, Zellmeier E, Tizazu B, et al. GATA2 zinc finger 1 mutations associated with biallelic CEBPA mutations define a unique genetic entity of acute myeloid leukemia. Blood. 2012;120(2):395–403. [DOI] [PubMed] [Google Scholar]
- 15.Chong CE, Venugopal P, Stokes PH, Lee YK, Brautigan PJ, Yeung DTO, et al. Differential effects on gene transcription and hematopoietic differentiation correlate with GATA2 mutant disease phenotypes. Leukemia. 2017;32(1):194–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Niimi K, Kiyoi H, Ishikawa Y, Hayakawa F, Kurahashi S, Kihara R, et al. GATA2 zinc finger 2 mutation found in acute myeloid leukemia impairs myeloid differentiation. Leuk Res Rep. 2013;2(1):21–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ping N, Sun A, Song Y, Wang Q, Yin J, Cheng W, et al. Exome sequencing identifies highly recurrent somatic GATA2 and CEBPA mutations in acute erythroid leukemia. Leukemia. 2017;31(1):195–202. [DOI] [PubMed] [Google Scholar]
- 18.Sologuren I, Martinez-Saavedra MT, Sole-Violan J, de Borges de Oliveira E Jr., Betancor E, Casas I, et al. Lethal Influenza in Two Related Adults with Inherited GATA2 Deficiency. J Clin Immunol. 2018;38(4):513–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Camargo JF, Lobo SA, Hsu AP, Zerbe CS, Wormser GP, Holland SM. MonoMAC Syndrome in a Patient With a GATA2 Mutation: Case Report and Review of the Literature. Clin Infect Dis. 2013;57(5):697–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koegel AK, Hofmann I, Moffitt K, Degar B, Duncan C, Tubman VN. Acute lymphoblastic leukemia in a patient with MonoMAC syndrome/GATA2 haploinsufficiency. Pediatr Blood Cancer. 2016;63(10):1844–7. [DOI] [PubMed] [Google Scholar]
- 21.Pasquet M, Bellanne-Chantelot C, Tavitian S, Prade N, Beaupain B, Larochelle O, et al. High frequency of GATA2 mutations in patients with mild chronic neutropenia evolving to MonoMac syndrome, myelodysplasia, and acute myeloid leukemia. Blood. 2012;121(5):822–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kazenwadel J, Secker GA, Liu YJ, Rosenfeld JA, Wildin RS, Cuellar-Rodriguez J, et al. Loss-of-function germline GATA2 mutations in patients with MDS/AML or MonoMAC syndrome and primary lymphedema reveal a key role for GATA2 in the lymphatic vasculature. Blood. 2012;119(5):1283–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fasan A, Eder C, Haferlach C, Grossmann V, Kohlmann A, Dicker F, et al. GATA2 mutations are frequent in intermediate-risk karyotype AML with biallelic CEBPA mutations and are associated with favorable prognosis. Leukemia. 2013;27(2):482–5. [DOI] [PubMed] [Google Scholar]
- 24.Ganapathi KA, Townsley DM, Hsu AP, Arthur DC, Zerbe CS, Cuellar-Rodriguez J, et al. GATA2 deficiency-associated bone marrow disorder differs from idiopathic aplastic anemia. Blood. 2015;125(1):56–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mace EM, Hsu AP, Monaco-Shawver L, Makedonas G, Rosen JB, Dropulic L, et al. Mutations in GATA2 cause human NK cell deficiency with specific loss of the CD56(bright) subset. Blood. 2013;121(14):2669–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dickinson RE, Griffin H, Bigley V, Reynard LN, Hussain R, Haniffa M, et al. Exome sequencing identifies GATA-2 mutation as the cause of dendritic cell, monocyte, B and NK lymphoid deficiency. Blood. 2011;118(10):2656–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spinner MA, Sanchez LA, Hsu AP, Shaw PA, Zerbe CS, Calvo KR, et al. GATA2 deficiency: a protean disorder of hematopoiesis, lymphatics, and immunity. Blood. 2014;123(6):809–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dickinson RE, Milne P, Jardine L, Zandi S, Swierczek SI, McGovern N, et al. The evolution of cellular deficiency in GATA2 mutation. Blood. 2014;123(6):863–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wlodarski MW, Hirabayashi S, Pastor V, Stary J, Hasle H, Masetti R, et al. Prevalence, clinical characteristics, and prognosis of GATA2-related myelodysplastic syndromes in children and adolescents. Blood. 2016;127(11):1387–97; quiz 518. [DOI] [PubMed] [Google Scholar]
- 30.Hirabayashi S, Wlodarski MW, Kozyra E, Niemeyer CM. Heterogeneity of GATA2-related myeloid neoplasms. Int J Hematol. 2017;106(2):175–82. [DOI] [PubMed] [Google Scholar]
- 31.Brambila-Tapia AJL, Garcia-Ortiz JE, Brouillard P, Nguyen HL, Vikkula M, Rios-Gonzalez BE, et al. GATA2 null mutation associated with incomplete penetrance in a family with Emberger syndrome. Hematology. 2017;22(8):467–71. [DOI] [PubMed] [Google Scholar]
- 32.Holland SM, Eisenstein EM, Kuhns DB, Turner ML, Fleisher TA, Strober W, et al. Treatment of refractory disseminated nontuberculous mycobacterial infection with interferon gamma. A preliminary report. N Engl J Med. 1994;330(19):1348–55. [DOI] [PubMed] [Google Scholar]
- 33.Mutsaers PG, van de Loosdrecht AA, Tawana K, Bodor C, Fitzgibbon J, Menko FH. Highly variable clinical manifestations in a large family with a novel GATA2 mutation. Leukemia. 2013;27(11):2247–8. [DOI] [PubMed] [Google Scholar]
- 34.Vila A, Dapas JI, Rivero CV, Bocanegra F, Furnari RF, Hsu AP, et al. Multiple Opportunistic Infections in a Woman with GATA2 Mutation. Int J Infect Dis. 2017;54:89–91. [DOI] [PubMed] [Google Scholar]
- 35.Ishida H, Imai K, Honma K, Tamura S, Imamura T, Ito M, et al. GATA-2 anomaly and clinical phenotype of a sporadic case of lymphedema, dendritic cell, monocyte, B- and NK-cell (DCML) deficiency, and myelodysplasia. Eur J Pediatr. 2012;171(8):1273–6. [DOI] [PubMed] [Google Scholar]
- 36.Donadieu J, Lamant M, Fieschi C, Sicre de Fontbrune F, Caye A, Ouachee M, et al. Natural history of GATA2 deficiency in a survey of 79 French and Belgian patients. Haematologica. 2018;103(8):1278–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mishra SS, Williams JF, B GP, George A. GATA2 deficiency in a young man with lymphoedema. Br J Haematol. 2020. Jul 9. doi: 10.1111/bjh.16941. [DOI] [PubMed]
- 38.Eguchi K, Ishimura M, Sonoda M, Ono H, Shiraishi A, Kanno S, et al. Nontuberculous mycobacteria-associated hemophagocytic lymphohistiocytosis in MonoMAC syndrome. Pediatr Blood Cancer. 2018;65(7):e27017. [DOI] [PubMed] [Google Scholar]
- 39.Egenlauf B, Schuhmann M, Giese T, Junghanss T, Stojkovic M, Tintelnot K, et al. Disseminated Mycosis by Arthrocladium fulminans Jeopardizing a Patient with GATA2 Deficiency. Respiration. 2019;97(5):472–5. [DOI] [PubMed] [Google Scholar]
- 40.Bogaert DJ, Laureys G, Naesens L, Mazure D, De Bruyne M, Hsu AP, et al. GATA2 deficiency and haematopoietic stem cell transplantation: challenges for the clinical practitioner. Br J Haematol. 2020;188(5):768–73. [DOI] [PubMed] [Google Scholar]
- 41.Abou Dalle I, Bannon SA, Patel KP, Routbort MJ, Cortes JE, Ferrajoli A, et al. Germline Genetic Predisposition to Myeloid Neoplasia From GATA2 Gene Mutations: Lessons Learned From Two Cases. JCO Precis Oncol. 2019;3. [DOI] [PMC free article] [PubMed]
- 42.Simonis A, Fux M, Nair G, Mueller NJ, Haralambieva E, Pabst T, et al. Allogeneic hematopoietic cell transplantation in patients with GATA2 deficiency-a case report and comprehensive review of the literature. Ann Hematol. 2018;97(10):1961–73. [DOI] [PubMed] [Google Scholar]
- 43.Mendes-de-Almeida DP, Andrade FG, Borges G, Dos Santos-Bueno FV, Vieira IF, da Rocha L, et al. GATA2 mutation in long stand Mycobacterium kansasii infection, myelodysplasia and MonoMAC syndrome: a case-report. BMC Med Genet. 2019;20(1):64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mardahl M, Jorgensen SE, Schneider A, Raaschou-Jensen K, Holm M, Veirum J, et al. Impaired immune responses to herpesviruses and microbial ligands in patients with MonoMAC. Br J Haematol. 2019;186(3):471–6. [DOI] [PubMed] [Google Scholar]
- 45.Fakhri B, Cashen AF, Duncavage EJ, Watkins MP, Wartman LD, Bartlett NL. Fifty Shades of GATA2 Mutation: A Case of Plasmablastic Lymphoma, Nontuberculous Mycobacterial Infection, and Myelodysplastic Syndrome. Clin Lymphoma Myeloma Leuk 2019;19(9):e532–e5. [DOI] [PubMed] [Google Scholar]
- 46.Monif M, Huq A, Chee L, Kilpatrick T. MonoMac syndrome with associated neurological deficits and longitudinally extensive cord lesion. BMJ Case Rep. 2018;2018. [DOI] [PMC free article] [PubMed]
- 47.Lovell JP, Zerbe CS, Olivier KN, Claypool RJ, Frein C, Anderson VL, et al. Mediastinal and Disseminated Mycobacterium kansasii Disease in GATA2 Deficiency. Ann Am Thorac Soc. 2016;13(12):2169–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vinh DC, Patel SY, Uzel G, Anderson VL, Freeman AF, Olivier KN, et al. Autosomal dominant and sporadic monocytopenia with susceptibility to mycobacteria, fungi, papillomaviruses, and myelodysplasia. Blood. 2010;115(8):1519–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Griese M, Zarbock R, Costabel U, Hildebrandt J, Theegarten D, Albert M, et al. GATA2 deficiency in children and adults with severe pulmonary alveolar proteinosis and hematologic disorders. BMC Pulm Med. 2015;15:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.West RR, Hsu AP, Holland SM, Cuellar-Rodriguez J, Hickstein DD. Acquired ASXL1 mutations are common in patients with inherited GATA2 mutations and correlate with myeloid transformation. Haematologica. 2013;99(2):276–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Griffith DE, Aksamit T, Brown-Elliott BA, Catanzaro A, Daley C, Gordin F, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175(4):367–416. [DOI] [PubMed] [Google Scholar]
- 52.Blanc P, Dutronc H, Peuchant O, Dauchy FA, Cazanave C, Neau D, et al. Nontuberculous Mycobacterial Infections in a French Hospital: A 12-Year Retrospective Study. PLoS One. 2016;11(12):e0168290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Graham SM, Ahmed T, Amanullah F, Browning R, Cardenas V, Casenghi M, et al. Evaluation of tuberculosis diagnostics in children: 1. Proposed clinical case definitions for classification of intrathoracic tuberculosis disease. Consensus from an expert panel. J Infect Dis. 2012;205 Suppl 2:S199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lewinsohn DM, Leonard MK, LoBue PA, Cohn DL, Daley CL, Desmond E, et al. Official American Thoracic Society/Infectious Diseases Society of America/Centers for Disease Control and Prevention Clinical Practice Guidelines: Diagnosis of Tuberculosis in Adults and Children. Clin Infect Dis. 2017;64(2):111–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ho CK, Strauss JF 3rd. Activation of the control reporter plasmids pRL-TK and pRL-SV40 by multiple GATA transcription factors can lead to aberrant normalization of transfection efficiency. BMC Biotechnol. 2004;4:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.de Beaucoudrey L, Samarina A, Bustamante J, Cobat A, Boisson-Dupuis S, Feinberg J, et al. Revisiting human IL-12Rbeta1 deficiency: a survey of 141 patients from 30 countries. Medicine (Baltimore). 2010;89(6):381–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Prando C, Samarina A, Bustamante J, Boisson-Dupuis S, Cobat A, Picard C, et al. Inherited IL-12p40 deficiency: genetic, immunologic, and clinical features of 49 patients from 30 kindreds. Medicine (Baltimore). 2013;92(2):109–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Eilertson KE, Booth JG, Bustamante CD. SnIPRE: selection inference using a Poisson random effects model. PLoS Comput Biol. 2012;8(12):e1002806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 2013;9(8):e1003709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jobim M, Lima Abreu E, Bustamante J, Oleaga C, Garcia TS, Jobim L. Sindromes MonoMAC e Emberger em pacientes com mutaçao no gene GATA2. Arquivos de Asma, Alergia en Imunologia. 2019;3(1):89–93. [Google Scholar]
- 62.Overbeek MJ, van de Loosdrecht AA, Vonk-Noordegraaf A. Granulomatous lung disease in a patient with a family history of hematological disorders. Sarcoidosis Vasc Diffuse Lung Dis. 2015;31(4):350–3. [PubMed] [Google Scholar]
- 63.Tsuzuki S, Towatari M, Saito H, Enver T. Potentiation of GATA-2 activity through interactions with the promyelocytic leukemia protein (PML) and the t(15;17)-generated PML-retinoic acid receptor alpha oncoprotein. Mol Cell Biol. 2000;20(17):6276–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Quintana-Murci L, Clark AG. Population genetic tools for dissecting innate immunity in humans. Nat Rev Immunol. 2013;13(4):280–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rapaport F, Boisson B, Gregor A, Beziat V, Boisson-Dupuis S, Bustamante J, et al. Negative selection on human genes causing severe inborn errors depends on disease outcome and both the mode and mechanism of inheritance. bioRxiv. 2020;doi. 10.1101/2020.02.07.938894. [DOI] [PMC free article] [PubMed]
- 66.Bolze A, Mahlaoui N, Byun M, Turner B, Trede N, Ellis SR, et al. Ribosomal protein SA haploinsufficiency in humans with isolated congenital asplenia. Science. 2013;340(6135):976–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE, Avery DT, et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science. 2014;345(6204):1623–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rieux-Laucat F, Casanova JL. Immunology. Autoimmunity by haploinsufficiency. Science. 2014;345(6204):1560–1. [DOI] [PubMed] [Google Scholar]
- 69.Minegishi N, Suzuki N, Kawatani Y, Shimizu R, Yamamoto M. Rapid turnover of GATA-2 via ubiquitin-proteasome protein degradation pathway. Genes Cells. 2005;10(7):693–704. [DOI] [PubMed] [Google Scholar]
- 70.Pan X, Minegishi N, Harigae H, Yamagiwa H, Minegishi M, Akine Y, et al. Identification of human GATA-2 gene distal IS exon and its expression in hematopoietic stem cell fractions. J Biochem. 2000;127(1):105–12. [DOI] [PubMed] [Google Scholar]
- 71.Schlums H, Jung M, Han H, Theorell J, Bigley V, Chiang SC, et al. Adaptive NK cells can persist in patients with GATA2 mutation depleted of stem and progenitor cells. Blood. 2017;129(14):1927–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Al Seraihi AF, Rio-Machin A, Tawana K, Bodor C, Wang J, Nagano A, et al. GATA2 monoallelic expression underlies reduced penetrance in inherited GATA2-mutated MDS/AML. Leukemia. 2018; 32(11): 2502–2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Poyhonen L, Bustamante J, Casanova JL, Jouanguy E, Zhang Q. Life-Threatening Infections Due to Live-Attenuated Vaccines: Early Manifestations of Inborn Errors of Immunity. J Clin Immunol. 2019;39(4):376–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bodor C, Renneville A, Smith M, Charazac A, Iqbal S, Etancelin P, et al. Germline GATA2 p.THR354MET mutation in familial myelodysplastic syndrome with acquired monosomy 7 and ASXL1 mutation demonstrating rapid onset and poor survival. Haematologica. 2012;97(6):890–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Svobodova T, Mejstrikova E, Salzer U, Sukova M, Hubacek P, Matej R, et al. Diffuse parenchymal lung disease as first clinical manifestation of GATA-2 deficiency in childhood. BMC Pulm Med. 2015;15:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mojica AM, Elizalde A. GATA2 Deficiency in a Pediatric Patient. J Allergy Clin Immunol Pract. 2019;7(6):2021–2. [DOI] [PubMed] [Google Scholar]
- 77.Fertitta L, Fontbrune FS, Battistella M, De Masson A, Bergeron A, Ranta D, et al. Folliculotropic mycosis fungoides associated with GATA2 deficiency: a new skin manifestation. Br J Dermatol. 2018;179(6):1420–1. [DOI] [PubMed] [Google Scholar]
- 78.Catto LFB, Borges G, Pinto AL, Cle DV, Chahud F, Santana BA, et al. Somatic genetic rescue in hematopoietic cells in GATA2 deficiency. Blood. 2020;136(8):1002–5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.