

Abstract
Plant nucleotide-binding leucine-rich repeat receptors (NLRs) regulate immunity and cell death. In Arabidopsis, a subfamily of “helper” NLRs is required by many “sensor” NLRs. Active NRG1.1 oligomerized, was enriched in plasma membrane puncta, and conferred cytoplasmic calcium ion (Ca2+) influx in plant and human cells. NRG1.1-dependent Ca2+ influx and cell death were sensitive to Ca2+ channel blockers and were suppressed by mutations affecting oligomerization or plasma membrane enrichment. Ca2+ influx and cell death mediated by NRG1.1 and ACTIVATED DISEASE RESISTANCE 1 (ADR1), another helper NLR, required conserved negatively charged N-terminal residues. Whole-cell voltage-clamp recordings demonstrated that Arabidopsis helper NLRs form Ca2+-permeable cation channels to directly regulate cytoplasmic Ca2+ levels and consequent cell death. Thus, helper NLRs transduce cell death signals directly.
In plants, successful pathogens inject effectors into the host cell to dampen the immune response. Plants evolved a surveillance system consisting of intracellular nucleotide–binding leucine-rich repeat receptors (NLRs) capable of triggering immunity in response to effector activity or by direct effector recognition (1). Effector-triggered immunity leads to activation of pathogen defense and culminates in the death of the host cell, the “hypersensitive response,” which can further limit pathogen growth (2). NLR activation is sufficient to determine the outcome of a plant-pathogen interaction (1). In plants, NLRs are divided into two major classes based on their N-terminal domains: Toll/interleukin-1 receptor/Resistance (TIR)-NLRs, (hereafter, TNL) and the coiled-coil (CC)-NLRs (hereafter, CNL) (3, 4). All tested TNLs require ENHANCED DISEASE SUSCEPTIBILITY 1 (EDS1), as well as a subfamily of five redundant “helper” NLRs also known as RNLs because of their CC-R [RPW8 (Resistance to Powdery Mildew 8)-like CC] domain (5–7). There are two subfamilies of RNLs, NRG1 (N REQUIREMENT GENE 1) and ADR1 (ACTIVATED DISEASE RESISTANCE 1), in nearly all flowering plants (8).
Activation of the Arabidopsis RNL NRG1.1 can be mimicked using an autoactive allele (D485V; hereafter DV), mutated in the conserved methionine-histidine-aspartate motif. This is an accepted proxy for pathogen effector–mediated NLR activation (9–12). NRG1.1 DV–induced cell death is independent of the native RNLs NRG1 and ADR1 and of the EDS1-signaling module in the heterologous host, Nicotiana benthamiana [(4, 7, 13, 14); fig. S1].
Results
Structure of NRG1.1-signaling domain
We obtained x-ray crystal structures of two mutant NRG1.1 CC-R domains (residues 1 to 124), K94E/K96E/R99E/K100E/R103E/K106E/K110E [7K(R)/E] and K94E/K96E (2K/E), which diffracted to 3.0 and 2.5 Å, respectively (table S1). These putative surface mutations were required to achieve monodispersion of the protein. Structural homology modeling suggested that the CC-R domains share an N-terminal four-helical bundle with cell death pore forming mammalian mixed-linage kinase domain-like (MLKL) proteins and fungal HET-s/HELO domain proteins (6, 15, 16). Similar to predictions, the two mutant structures superimposed well with the four-helical bundles of the resting-state CC domain of ZAR1 (17) and the cation channel-forming domain of MLKL (18) (Fig. 1, A to D, and table S2).
Fig. 1. NRG1.1 CC-R resembles the ZAR1 and MLKL four-helical bundle.
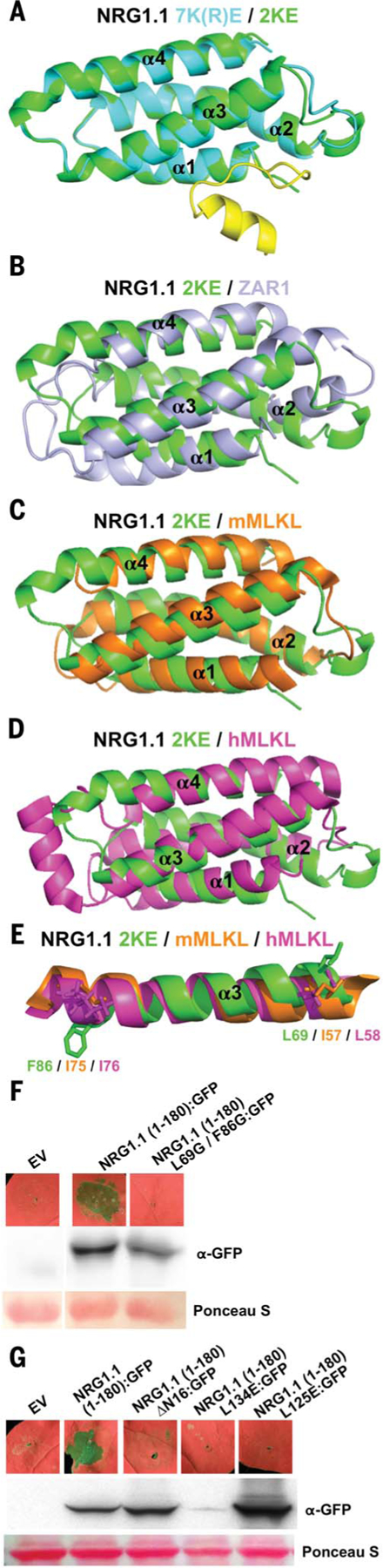
(A) Structural overlay of 7K(R)E (PBD: 7L7V) and 2KE (7L7W) with 2KE in green, 7K(R)E four-helical bundle (4HB) in cyan, and 7K(R)E N-terminal region in yellow. (B) Structural overlay of 2KE in green and the ZAR1 4HB (6J5V) structure in light purple. (C) Structural overlay of 2KE in green and mMLKL 4HB (4BTF) in orange. (D) Structural overlay of 2KE in green and hMLKL 4HB (2MSV) in magenta. (E) Conserved F86/I75/I76 and L69/I57/L58 shown in sticks in the α3 helix of superimposed 2KE, mMLKL and hMLKL 4HB structures, respectively. (F and G) In planta (N. benthamiana) phenotypes at 2 days after induction and protein accumulation of WT NRG1.1 CC-R and structure-derived mutants. EV, empty vector.
The four-helical bundle of MLKL is sufficient to cause cell death, and this requires two hydrophobic residues, L58 and I76, which maintain the four-helical bundle hydrophobic core (19). Structural overlay predicted that the two hydrophobic residues are conserved in NRG1.1 as L69 and F86 (Fig. 1E); mutating them abolished cell death activity of an NRG1.1 fragment from residues 1 to 180 (hereafter NRG1.1 1–180) (Fig. 1F). In ZAR1, a hydrophobic groove made by α2 and α4B is important for oligomerization and function (17). We found two hydrophobic residues, L134 and L125, in the homologous region in NRG1.1; mutating them to glutamic acid abolished cell death in NRG1.1 1–180 (Fig. 1G). These data validate the NRG1.1 four-helical bundle structure.
Active ZAR1 oligomerizes into a pentamer in which the α1 helix of the four-helical bundle rearranges, flips out, and forms a plasma membrane (PM)–penetrating, funnel-like structure (17, 20, 21), consistent with models proposed for fungal Het-s/HELO activation (22). This first helix was essential for ZAR1 function at the PM (17). One of the NRG1.1 CC-R structures [7(K)R/E] revealed a potentially flexible N terminus (residues 1 to 16; absent in ZAR1; Fig. 1A) that could extend the four-helical bundle α1 helix of a putative funnel in the active NRG1.1 protein. This was disordered in the 2K/E structure. This region was required for NRG1.1 1–180 cell death induction (ΔN16; Fig. 1G).
Active NRG1.1 oligomerizes and forms puncta
We introduced several of the structure-derived mutations into the full-length NRG1.1 in cis with the autoactive DV allele because the wild-type (WT) resting NRG1.1 is inactive in the absence of sensor NLR activation (Fig. 2A). We found that the ΔN16, L134E, and L125E cis mutations each abolished the cell death function of NRG1.1 DV (Fig. 2A). Blue native-PAGE analyses revealed that active NRG1.1 DV formed high-molecular-weight complexes, whereas the inactive WT, catalytic p-loop (G199A/K200A), and DV p-loop cis double mutants did not (Fig. 2B). Mutation of L134E, but not of L125E, in cis also abolished NRG1.1 DV oligomer formation (Fig. 2B). Unlike the WT or inactive p-loop mutants, NRG1.1 DV was enriched in the PM fraction, whereas DV L125E showed substantially reduced PM enrichment (Fig. 2C). Although it oligomerized and was enriched at the PM (figs. S2 to S4), NRG1.1 DV ΔN16 failed to induce cell death (Fig. 2, A to C).
Fig. 2. Active NRG1.1 (D485V) oligomerizes on the PM and triggers cell death.
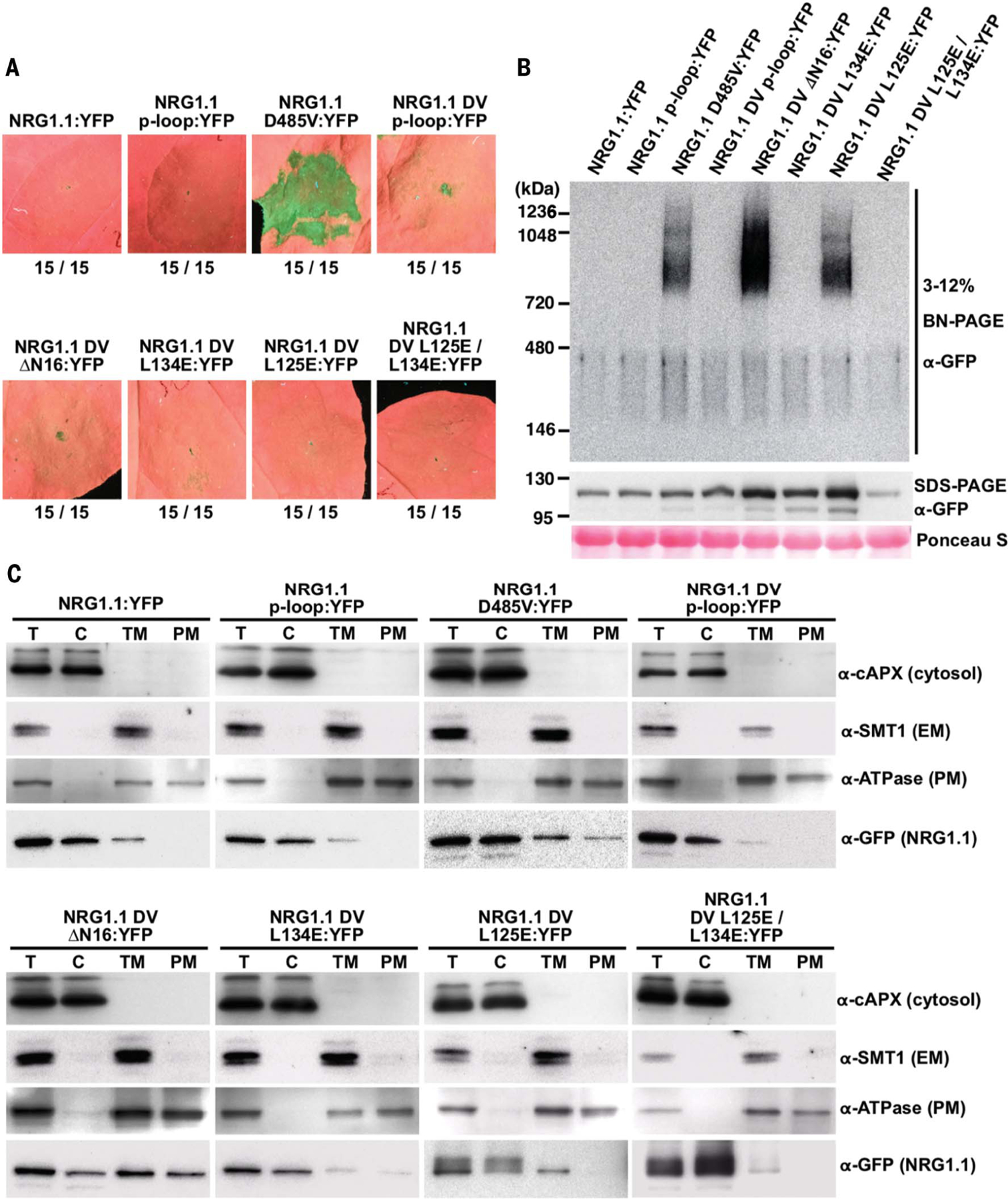
(A) In planta phenotypes of NRG1.1 variants in N. benthamiana (Nb) at 2 dpi. YFP, yellow fluorescent protein. (B) Oligomerization of NRG1.1 DV variants. Accumulation of YFP-tagged NRG1.1 variants was verified by SDS–polyacrylamide gel electrophoresis (PAGE) immunoblotting, and their oligomeric states were checked by blue native-PAGE immunoblotting in Nb adr1 nrg1. (C) Activation promotes PM enrichment. Total proteins (T) extracted from Nb-expressing NRG1.1:YFP variants were fractionated into cytosolic (C), total membrane (TM), and PM fractions and verified by marker proteins: Cytosol, cytosolic ascorbic peroxidase; endoplasmic reticulum membrane (EM), sterol methyltransferase 1; PM, H+ATPase.
Confocal microscopy demonstrated that NRG1.1 DV exhibits increased PM localization compared with inactive alleles (figs. S2 to S4). Active NRG1.1 DV exhibited numerous puncta on the PM, whereas the NRG1.1 DV p-loop double mutant exhibited many fewer (figs. S2 to S4). These puncta were also observed for the NRG1.1 DV ΔN16 double mutant but were less common for the missense loss-of-function alleles, which colocalized more with the endoplasmic reticulum marker than the PM marker (fig. S3). These results show that the NRG1.1 N-terminal 16 residues may extend a funnel-like structure similar to that of ZAR1.
NRG1.1 is functional in human HeLa cells
We investigated the possibility that NRG1.1 forms PM pores and functions as a channel. We expressed NRG1.1 variants in HeLa cells. If NRG1.1 caused cell death, formed pores, and exhibited channel activity in this evolutionarily distant cellular background, then the most parsimonious conclusion would be that it did so autonomously. The alternative hypothesis would require a signaling partner conserved between plants and humans. NRG1.1 DV induced significant cell death at 6 hours after induction (fig. S5). This did not occur when WT NRG1.1 was expressed or when NRG1.1 DV activity was suppressed by p-loop, ΔN16, L125E, or L134E mutations in cis (fig. S5). Oligomerization and PM localization of the NRG1.1 variants in HeLa cells were similar to the in planta results (fig. S6). Thus, the genetic requirements for NRG1.1 DV cell death initiation, (p-loop, N16, L125, and L134) are similar in HeLa cells and plant cells.
We observed the morphology of the dying HeLa cells using scanning electron microscopy. We found that the number of PM pores, appearing as dark spots (23), correlated with the cell death activity of NRG1.1 variants (fig. S7 and Fig. 3, A to C). NRG1.1 DV–expressing cells exhibited 8-nm (average) pores in these processed samples (Fig. 3, E and F), although this may not represent the actual pore size. This apparent pore size was significantly different from the size of larger pores formed by GASDERMIN-D 1–275 L192D, a partial loss-of-function mutant of the pore-forming protein GASDERMIN-D that induces cell death and allows high protein accumulation [(17, 24); Fig. 3, D to F, and fig. S5B]. Cell fractionation experiments indicated that NRG1.1 DV was enriched in the PM fraction in HeLa cells, whereas the NRG1.1 DV p-loop cis double mutant was not (fig. S6B). Overall, active NRG1.1 DV localized to the PM and its cell death activity was associated with the occurrence of PM pores in HeLa cells.
Fig. 3. NRG1.1 forms ion channels permeable to Ca2+ and not Cl−.
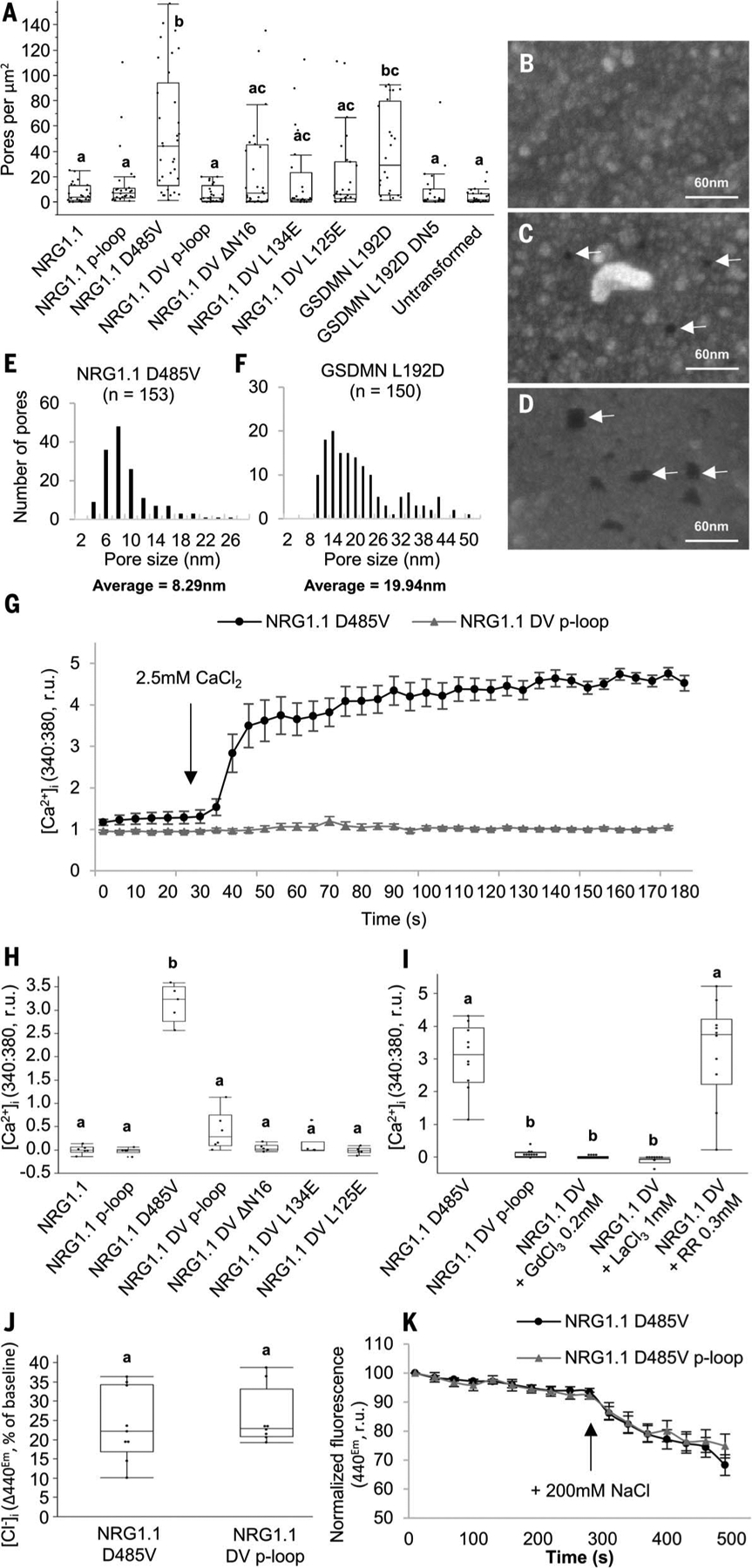
(A) Quantification of apparent PM pores in HeLa cells expressing NRG1.1 variants. (B to D) Representative scanning electron micro-graphs of cells exhibiting apparent PM pores in cell lines expressing the NRG1.1 DV p-loop (B), NRG1.1 D485V (C) or the pore-forming protein GSDMN 1–275 L192D (D). White arrows indicate the visible PM pores. (E and F) Distribution of the diameter of PM pores visible after NRG1.1 D485V (E) or GSDMN L192D (F) expression. The average diameters are significantly different (t test, P < 0.0001). (G) [Ca2+]i in NRG1.1 D485V or NRG1.1 D485V p-loop–expressing HeLa cells. Black arrow indicates CaCl2 addition. (H) Ca2+ influx in HeLa cells expressing NRG1.1 variants. (I) Effect of the Ca2+ channel blockers LaCl3, GdCl3, and ruthenium red (RR) on NRG1.1 D485V–induced Ca2+ influx. (J) Intracellular [Cl−] accumulation in cells expressing the NRG1.1 D485V or NRG1.1 DV p-loop. (K) Representative time course experiment showing variation of [Cl−]. Black arrow indicates 200 mM NaCl addition. Letters indicate statistical significance (ANOVA with post hoc Tukey, P < 0.01).
Helper NLRs form Ca2+-permeable channels
Ca2+ influx is a hallmark of programmed cell death in both the animal and plant kingdoms (25, 26) and is a requirement for NLR signaling (25, 27). Using Fura-2–based Ca2+ imaging (28, 29), we measured the cytosolic free Ca2+ concentration ([Ca2+]i) in HeLa cells expressing NRG1.1 DV or the DV p-loop double mutant to determine whether NRG1.1 DV affected cytoplasmic Ca2+ homeostasis. We observed sustained [Ca2+]i increases specifically in the NRG1.1 DV–expressing cells seconds after CaCl2 addition (Fig. 3G and fig. S8). Loss of the cell death cis mutations ΔN16, L125E, or L134E all suppressed NRG1.1 DV–driven Ca2+ influx (Fig. 3H and fig. S8). The general Ca2+ influx channel blockers LaCl3 and GdCl3, but not the Ca2+ release channel blocker ruthenium red (30–32), blocked NRG1.1 DV–driven Ca2+ influx (Fig. 3I and fig. S9). These observations are consistent with NRG1.1 DV directly or indirectly facilitating Ca2+ influx. We investigated the specificity of NRG1.1 DV–driven ion flux by measuring cytosolic [Cl−] using 6-methoxy-N-ethylquinolinium iodide (MEQ)/dihydroMEQ (33). There was no difference between the NRG1.1 DV and NRG1.1 DV p-loop–expressing cells (Fig. 3, J and K, and fig. S10), indicating that NRG1.1 DV channels do not drive Cl−influx. Thus, we conclude that NRG1.1 might form Ca2+-permeable channels, or facilitate their formation, in the PM of HeLa cells.
Conserved negatively charged RNL residues are required for Ca2+ influx
We focused on the first 16 amino acids of NRG1.1 across 334 plant RNL sequences because NRG1.1 DV ΔN16 retained oligomerization and PM enrichment but lost Ca2+ influx. We observed a pattern of glycine or negatively charged or polar residues separated by two to three hydrophobic residues (table S3 and fig. S11). This motif was conserved in the ADR1 clade of RNLs, partially degenerated in the NRG1 clade, and further degenerated in CNLs (fig. S12). Although different from an N-terminal motif recently implicated in CNL function (34), these two domains share regularly spaced, negatively charged residues. Such residues are critical for ion selectivity and permeability in Ca2+ channels (35, 36). We targeted the negatively charged residues within the first 16 amino acids of NRG1.1 DV and the autoactive WT ADR1 (37) for structurally conservative but uncharged cis mutations (NRG1.1 DV: D3N, E14Q, or D3N/E14Q; ADR1: D6N, D11N, or D6N/D11N).
We assayed Ca2+ influx triggered by these alleles in planta using the intracellular Ca2+ reporter GCaMP3 in transgenic N. benthamiana (38). We confirmed that either NRG1.1 DV or ADR1 expression triggered [Ca2+]i increases in planta 2 to 3 hours after estradiol treatment (a typical time required for estradiol-induced protein accumulation), which was accompanied by cell death 16 hours after [Ca2+]i increase (Fig. 4, A and B). LaCl3 and GdCl3 abolished cell death (Fig. 4A) and reduced Ca2+ influx (Fig. 4B). In both plant (Fig. 4, C to F) and HeLa cells (Fig. 4, G to J), mutations in the RNL-conserved N-terminal motif, especially E14Q (in NRG1.1 DV) and D11N (in ADR1), significantly reduced the rate of Ca2+ influx (Fig. 4, C, E, and G to J, and fig. S13). These mutations also reduced cell death induction (Fig. 4, D and F, and figs. S14 and S15).
Fig. 4. Negatively charged residues in the conserved RNL motif are required for Ca2+ influx and cell death.
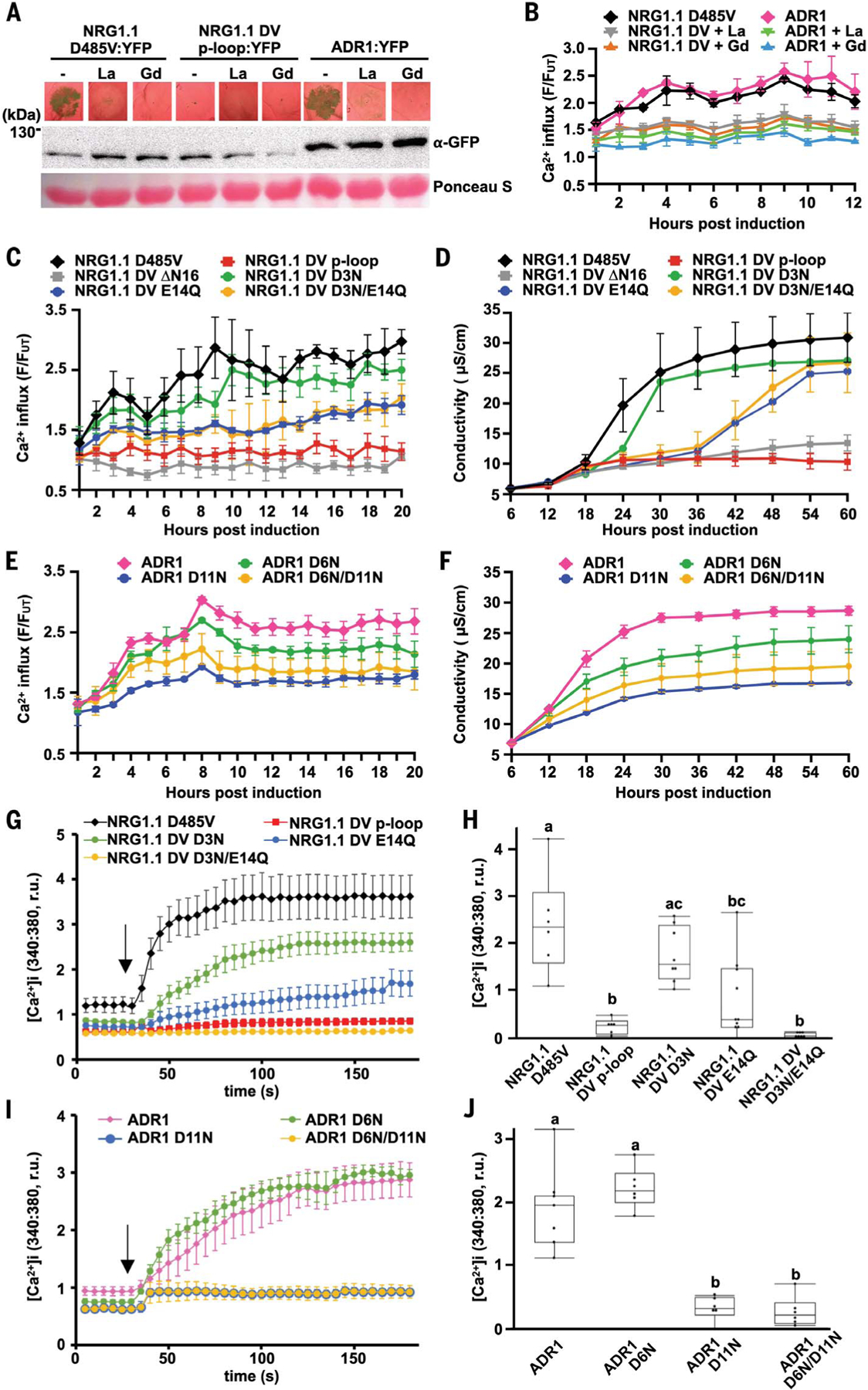
(A) In planta phenotypes (2 dpi) of plant RNLs in the presence of the Ca2+ channel blockers LaCl3 (La) and GdCl3 (Gd). Accumulation of YFP-tagged RNL variants was verified by SDS-PAGE and-immunoblotting, in Nb adr1 nrg1. (B, C, and E) Time course RNL-induced Ca2+ influx measurements with GCaMP3-transgenic Nb. (D and F) Time course conductivity measurement depicting RNL-triggered cell death in Nb. (G to J) [Ca2+]i in HeLa cells expressing NRG1.1 DV and variants (G and H) or ADR1 and variants (I and J). Black arrows indicate the addition of CaCl2. Letters indicate statistical significance (ANOVA with post hoc Tukey, P < 0.05). YFP-tagged RNL protein expression in Nb and HeLa cells was verified (fig. S16).
We analyzed the electrophysiological properties of NRG1.1 DV and NRG1.1 DV D3N E14Q in human embryonic kidney (HEK) 293 cells using the patch-clamp technique (28, 29). Having confirmed that NRG1.1 DV drives Ca2+ influx in transfected HEK293 cells similar to HeLa cells using Fura-2 imaging (fig. S17), we proceeded to record whole-cell voltage-clamp currents with voltage ramp (from +50 mV to −200 mV). We recorded robust currents at both positive and negative potentials only in cells expressing NRG1.1 DV with CsCl and CaCl2 in the pipette and bath solution, respectively (Fig. 5, A and C). Both inward and outward currents were abolished when Cs+ and Ca2+ were substituted for the channel-impermeable cation TEA+ (Fig. 5, B and C), indicating that NRG1.1 DV formed nonselective cation channels mediating Ca2+ influx rather than Cl− efflux. The NRG1.1 D3N E14Q double mutant eliminated Ca2+ currents (Fig. 5, A to C), consistent with D3 and/or E14 acting as a selectivity filter. Substitution of Mg2+ for Ca2+ in the bath resulted in a similar permeability for Mg2+ and Ca2+ (Fig. 5D), confirming that NRG1.1 is a nonselective, cation-permeable channel.
Fig. 5. NRG1.1 D485V forms nonselective cation channel.
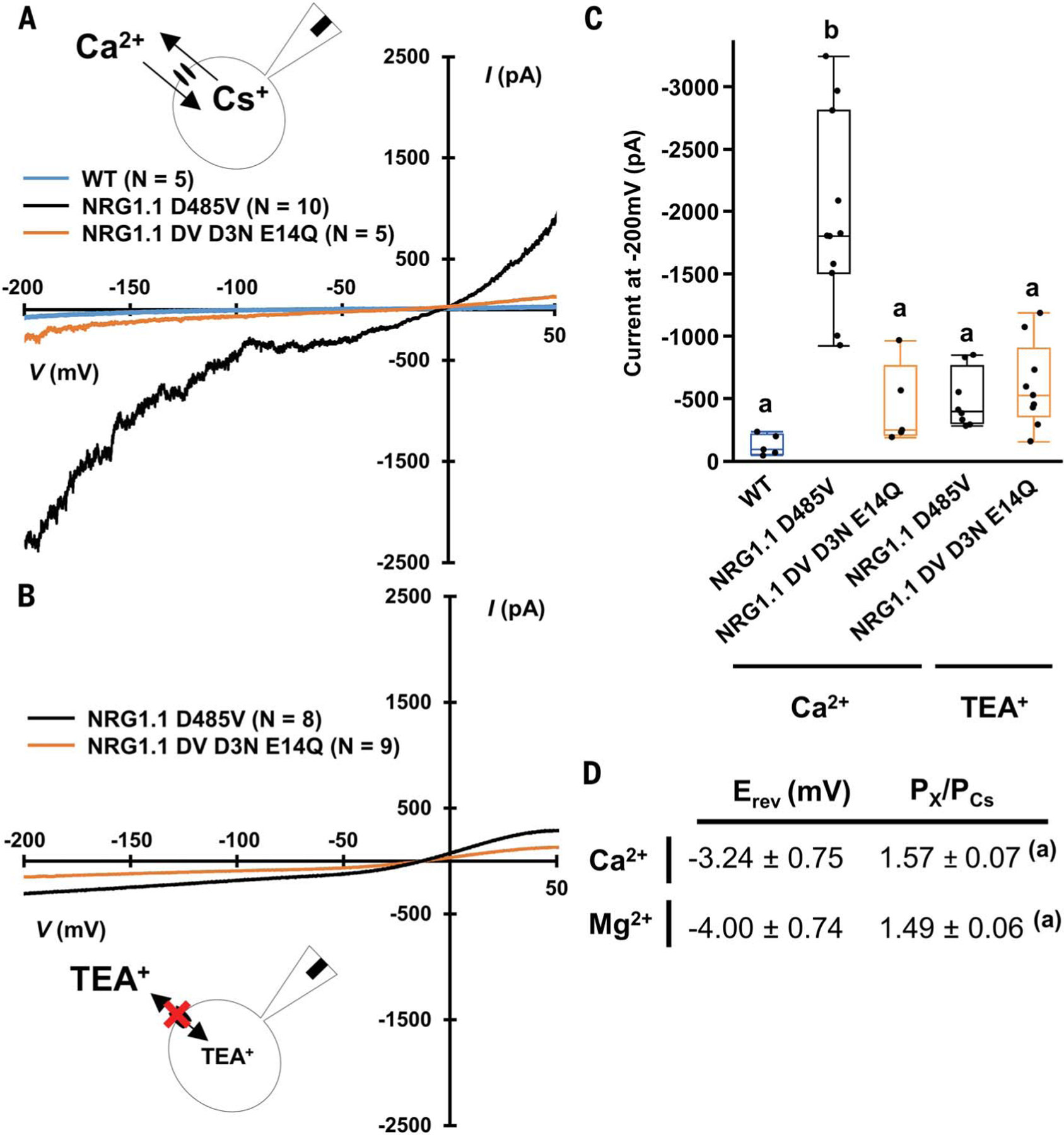
(A) Whole-cell current recorded in voltage ramp (+50 to ‒200 mV) in a CaCl2 bath solution. (B) Whole-cell current recorded in voltage ramp (+50 to ‒200 mV) in a TEA-Cl bath solution. (C) Mean current recorded at ‒200 mV during experiments in (A) and (B). (D) Reversal potential (Erev) and relative ion permeability (PX/PCs) measured in voltage ramp experiments in CaCl2 or MgCl2 solutions in HEK293 cells expressing NRG1.1 D485V (N = 10 Ca2+ and N = 8 Mg2+). Letters indicate statistical difference (ANOVA with post hoc Tukey, P < 0.05).
Discussion
Ca2+ signaling is known to regulate plant immunity (26, 39). Constitutive cytoplasmic Ca2+ influx induces constitutive defense activation and cell death (40, 41). We found that the NRG1.1-signaling domain structure resembles the pore-forming domain of the cation channel MLKL (Fig. 1), oligomerizes in puncta on the PM (Fig. 2), and is sufficient to drive cytoplasmic Ca2+ influx in plants and human cells (Figs. 3 to 5 and fig. S2). Our data are consistent with NRG1.1 acting as a nonselective, Ca2+-permeable cation channel.
All TNL immune receptors tested so far required the redundant RNLs of the ADR1 and NRG1 subfamilies (5). Thus, we propose that TNL activation induces RNL-dependent Ca2+ influx to initiate cell death and, likely, immune responses. Supporting this view, we observed that TNL RPS4 activation is associated with Ca2+ influx (fig. S18). Cyclic nucleotide–gated channels may also contribute to sustain the RNL-initiated Ca2+ influx because they have been shown to be activated upon immune response (42). It is plausible that cell death is a product of Ca2+-responsive factors that execute a cell death program rather than being a consequence of Ca2+ cytotoxicity. Cell death and defense activation can be uncoupled during effector-triggered immunity, suggesting that cell death has additional signaling requirements beyond elevated [Ca2+] (2, 43). Plants have MLKL homologs that are involved in immunity and can trigger cell death (44), possibly by regulating ion homeostasis [Mg2+ rather than Ca2+ (18, 45), by analogy with human MLKL].
CNLs likely also function like RNLs and trigger cytoplasmic Ca2+ influx. Recently, the CNL ZAR1 was shown to be a Ca2+-permeable channel that requires a negative charge ring for cation influx (21). It is thus likely that ZAR1 E11, NRG1.1 E14, and ADR1 D11 all form negative charge rings that act as cation selective filters to achieve fast Ca2+ influx and initiation of cell death and defense signaling. The combined data from our work and (21) provide a mechanistic explanation for how the two major classes of plant intracellular innate immune receptors, the TNLs and CNLs, control cell death triggered by pathogen recognition.
Supplementary Material
ACKNOWLEDGMENTS
We thank B. Staskawicz (UCB), R. Martin (UCB), J. Schroeder (UCSD), and S. Grant (UNC) for reading the manuscript; members of the Dangl laboratory for discussions about the work; B. Staskawicz and T. Qi for N. benthamiana nrg adr1 seeds; K. Yoshioka (Univ. of Toronto) for N. benthamiana GCaMP3 seeds; and V. Madden and K. White for electron microscopy sample preparation. UNC Microscopy Services are supported in part by Cancer Center Core Support Grant P30 CA016086 to the UNC Lineberger Comprehensive Cancer Center.
Funding:
This work was supported by the National Science Foundation (grant IOS-1758400 to J.L.D. and grant IOS-1457257 to Z.-M.P.), the National Institutes of Health (grants GM137286 and GM135218 to M.R.R.) and the Howard Hughes Medical Institute (HHMI). J.L.D. is an HHMI investigator. N.-H.K. was partially supported by the Basic Science Research Program through the National Research Foundation of Korea Fellowship funded by the Ministry of Education (grant 2014R1A6A3A03058629). F.E.-K. was supported by the University of Tübingen, the Deutsche Forschungsgemeinschaft (SFB/CRC1101 project D09), and the Reinhard Frank Stiftung (Project “helperless plant”) to F.E.-K. and J.L.D. L.W. was supported by National Key Laboratory of Plant Molecular Genetics, the Institute of Plant Physiology and Ecology/Center for Excellence in Molecular Plant Sciences, and the Chinese Academy of Sciences Strategic Priority Research Program (type B; project number XDB27040214).
Footnotes
Competing interests: The authors declare no competing interests.
SUPPLEMENTARY MATERIALS
science.sciencemag.org/content/373/6553/420/suppl/DC1
MDAR Reproducibility Checklist
Data and materials availability:
Data are available in the manuscript, the supplementary materials, or in the Protein Data Bank (PDB) files. NRG1.1 N-terminal domain atomic coordinates have been deposited in the PDB with accession codes 7L7W (2K/E native) and 7L7V (7K(R)/E native).
REFERENCES AND NOTES
- 1.Jones JD, Vance RE, Dangl JL, Science 354, aaf6395 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Laflamme B et al. , Science 367, 763–768 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Tamborski J, Krasileva KV, Annu. Rev. Plant Biol 71, 355–378 (2020). [DOI] [PubMed] [Google Scholar]
- 4.Saur IML, Panstruga R, Schulze-Lefert P, Nat. Rev. Immunol 21, 305–318 (2020). [DOI] [PubMed] [Google Scholar]
- 5.Saile SC et al. , PLOS Biol 18, e3000783 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jubic LM, Saile S, Furzer OJ, El Kasmi F, Dangl JL, Curr. Opin. Plant Biol 50, 82–94 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Lapin D, Bhandari DD, Parker JE, Annu. Rev. Phytopathol 58, 253–276 (2020). [DOI] [PubMed] [Google Scholar]
- 8.Van Ghelder C et al. , Sci. Rep 9, 11614 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tran DTN et al. , Curr. Biol 27, 1148–1160 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu Z et al. , New Phytol 222, 938–953 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Williams SJ et al. , Mol. Plant Microbe Interact 24, 897–906 (2011). [DOI] [PubMed] [Google Scholar]
- 12.Tameling WIL et al. , Plant Physiol 140, 1233–1245 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qi T et al. , Proc. Natl. Acad. Sci. U.S.A 115, E10979–E10987 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lapin D et al. , Plant Cell 31, 2430–2455 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bentham AR, Zdrzalek R, De la Concepcion JC, Banfield MJ, Plant Cell Physiol 59, 2398–2408 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Daskalov A et al. , Proc. Natl. Acad. Sci. U.S.A 113, 2720–2725 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang J et al. , Science 364, eaav5870 (2019).30948527 [Google Scholar]
- 18.Xia B et al. , Cell Res 26, 517–528 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cai Z et al. , Nat. Cell Biol 16, 55–65 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang J et al. , Science 364, eaav5868 (2019).30948526 [Google Scholar]
- 21.Bi G et al. , Cell 184, 3528–3541.e12 (2021). [DOI] [PubMed] [Google Scholar]
- 22.Seuring C et al. , PLOS Biol 10, e1001451 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herr DR et al. , Neuromolecular Med 22, 293–303 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ding J et al. , Nature 535, 111–116 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Huysmans M, Lema AS, Coll NS, Nowack MK, Curr. Opin. Plant Biol 35, 37–44 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Moeder W, Phan V, Yoshioka K, Plant Sci 279, 19–26 (2019). [DOI] [PubMed] [Google Scholar]
- 27.Grant M et al. , Plant J 23, 441–450 (2000). [DOI] [PubMed] [Google Scholar]
- 28.Caterina MJ et al. , Nature 389, 816–824 (1997). [DOI] [PubMed] [Google Scholar]
- 29.Yuan F et al. , Nature 514, 367–371 (2014). [DOI] [PubMed] [Google Scholar]
- 30.Gao X et al. , PLOS Pathog 9, e1003127 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Castro J, Aromataris EC, Rychkov GY, Barritt GJ, Biochem. J 418, 553–566 (2009). [DOI] [PubMed] [Google Scholar]
- 32.De Vriese K, Costa A, Beeckman T, Vanneste S, Int. J. Mol. Sci 19, 1506 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Inglefield JR, Schwartz-Bloom RD, Methods Enzymol 307, 469–481 (1999). [DOI] [PubMed] [Google Scholar]
- 34.Adachi H et al. , eLife 8, e49956 (2019).31774397 [Google Scholar]
- 35.Tang L et al. , Nature 505, 56–61 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fan M et al. , Nature 582, 129–133 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dong OX et al. , New Phytol 210, 960–973 (2016). [DOI] [PubMed] [Google Scholar]
- 38.DeFalco TA et al. , Plant Cell Physiol 58, 1173–1184 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Stael S et al. , Trends Plant Sci 20, 3–11 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yoshioka K et al. , Plant Cell 18, 747–763 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao C et al. , New Phytol 230, 1078–1094 (2021). [DOI] [PubMed] [Google Scholar]
- 42.Tian W et al. , Nature 572, 131–135 (2019). [DOI] [PubMed] [Google Scholar]
- 43.Gassmann W, Mol. Plant Microbe Interact 18, 1054–1060 (2005). [DOI] [PubMed] [Google Scholar]
- 44.Mahdi LK et al. , Cell Host Microbe 28, 813–824.e6 (2020). [DOI] [PubMed] [Google Scholar]
- 45.Ros U et al. , Cell Rep 19, 175–187 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available in the manuscript, the supplementary materials, or in the Protein Data Bank (PDB) files. NRG1.1 N-terminal domain atomic coordinates have been deposited in the PDB with accession codes 7L7W (2K/E native) and 7L7V (7K(R)/E native).