

Abstract
Metal-binding natural products contribute to metal acquisition and bacterial virulence, but their roles in metal stress response are underexplored. We show that a five-enzyme pathway in Pseudomonas aeruginosa synthesizes a small-molecule copper complex, fluopsin C, in response to elevated copper concentrations. Fluopsin C is a broad-spectrum antibiotic that contains a copper ion chelated by two minimal thiohydroxamates. Biosynthesis of the thiohydroxamate begins with cysteine and requires two lyases, two iron-dependent enzymes, and a methyltransferase. The iron-dependent enzymes remove the carboxyl group and the α carbon from cysteine through decarboxylation, N-hydroxylation, and methylene excision. Conservation of the pathway in P. aeruginosa and other bacteria suggests a common role for fluopsin C in the copper stress response, which involves fusing copper into an antibiotic against other microbes.
Metal-binding natural products allow microorganisms to acquire metal ions for survival and virulence (1, 2). Many microbes produce siderophores for iron acquisition (1, 3, 4), but few make natural products that bind copper under physiological conditions (5). These copper-binding compounds facilitate copper import when copper is limited or help detoxify copper when copper is in excess. Three examples are methanobactins, yersiniabactin, and xanthocillin. Methanobactins are produced by methane-oxidizing bacteria to scavenge copper for copper-dependent methane oxygenase (6, 7). Yersiniabactin, which is a virulence factor produced by uropathogenic Escherichia coli, serves a dual role in acquiring copper nutrients and protecting the bacterium from host-derived copper toxicity (8, 9). Xanthocillin and derivatives, which are fungal natural products, help regulate copper levels in the producing fungal strain and inhibit the growth of other fungal and bacterial species (10). In the immune system, copper is a bactericide that is marshalled by immune cells to poison invading microbes (11, 12). Thus, copper resistance and detoxification are essential for microbial virulence (13, 14). Although many proteins that help microbes traffic or efflux copper have been uncovered (15, 16), the potential for natural products to participate in copper homeostasis remains mostly unexplored.
We discovered a conserved biosynthetic pathway in Pseudomonas aeruginosa, which integrates copper into a broad-spectrum antibiotic in response to elevated copper concentrations. P. aeruginosa is a ubiquitous environmental bacterium found in soil, in water, and on human skin. It is responsible for many dangerous infections in hospitalized patients and people with compromised immune systems (17). P. aeruginosa tolerates high levels of copper (18), which is a common and persistent environmental pollutant; given that this bacterium is a prolific producer of natural products, we reasoned that it might synthesize copper-binding compounds to resist excess copper. Transcriptional analysis of P. aeruginosa strain PAO1 identified the expression of the operon PA3515–PA3519 as among the most highly (47- to 231-fold) up-regulated genes under high levels of copper stress (19). In response to an elevated but nontoxic level of copper, P. aeruginosa PAO1 induces expression of PA3515–PA3519 within 5 min, along with copZ1, which encodes a copper chaperone, and PA3521–PA3523, which encodes an efflux pump (20). This cluster of genes is controlled by the copper-response regulator CueR (Fig. 1A) (21), which senses cytoplasmic copper and activates copper efflux genes (22). The operon PA3515–PA3519 encodes five proteins of unknown functions, including a putative methyltransferase, two putative adenylosuccinate lyases, and two enzymes that contain a heme oxygenase domain (Fig. 1A). We proposed that these proteins synthesize a copper-binding natural product.
Fig. 1. Identification of the fluopsin C gene cluster (flc).
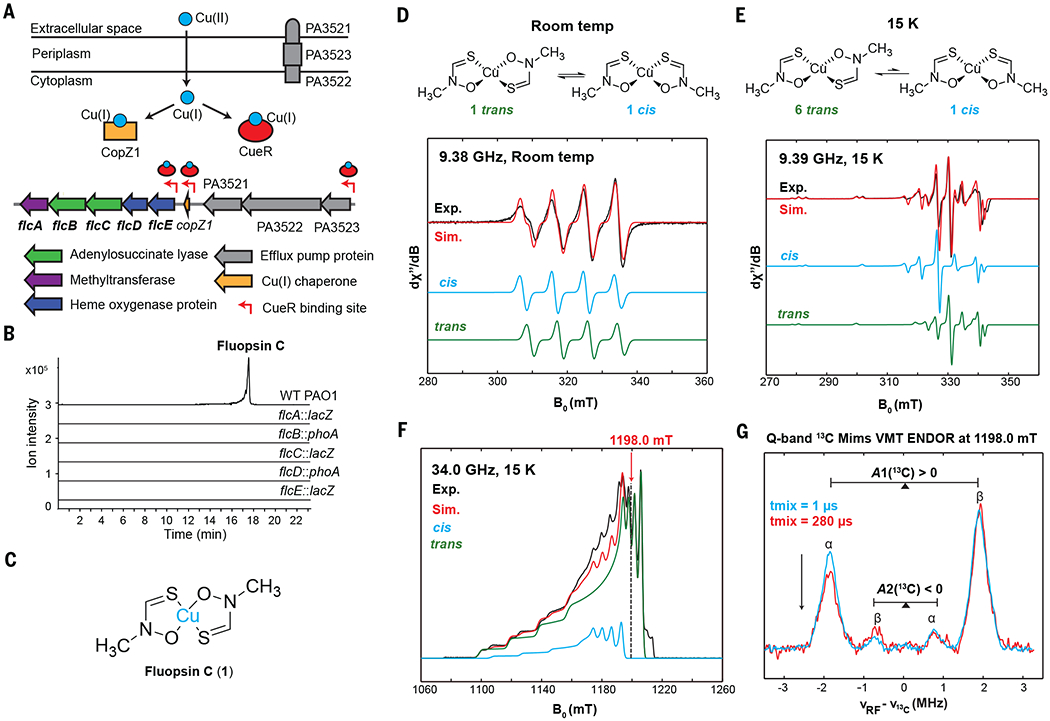
(A) The flc cluster (PA3515–PA3519) in P. aeruginosa PAO1 is located in a copper island regulated by CueR. PA3521–PA3523 encodes an efflux pump (MexPQ-OpmE). (B) Genes flcA to flcE in PAO1 are essential for fluopsin C production in CuSO4-containing media. Extracted ion chromatograms (EICs) of fluopsin C (1) (m/z 152.9304 [M–C2H4NOS]+) are shown. (C) Structure of fluopsin C. (D) X-band room temperature EPR spectra of fluopsin C in toluene show the cis isomer (giso = 2.0900, Aiso(63Cu) = 259 MHz) and trans isomer (giso = 2.0808, Aiso(63Cu) = 247 MHz) with approximately equal concentrations. giso, isotropic g value; Aiso, isotropic A value; Exp., experimental; Sim., simulated. (E) X-band frozen-solution EPR spectra of fluopsin C in toluene at 15 K show different electronic structures for the cis isomer (g = [2.151, 2.064, 2.064], A(63Cu) = [580, 173, 173] MHz) and trans isomer (g = [2.166, 2.036, 2.036], A(63Cu) = [583, 108, 108] MHz) in ~1:6 ratio. (F) Q-band electron spin-echo–detected field-swept EPR spectrum confirms 1:6 ratio of cis:trans. (G) Q-band variable mixing time (VMT) Mims-ENDOR spectra of [15N,13C2]-fluopsin C in toluene at the magnetic field position (1198.0 mT) with contribution from only the trans isomer. tmix, mixing time.
To test this hypothesis and identify the natural product made by the PA3515–PA3519 operon, we obtained mutants in which each gene in the operon is disrupted by a lacZ or phoA insertion (23). We cultured P. aeruginosa PAO1 in the presence of 250 μM of CuSO4, a concentration that is well tolerated by P. aeruginosa, and compared the metabolomes of wild-type PAO1 with those of individual insertional mutants by means of liquid chromatography coupled–high resolution mass spectrometry (LC-HRMS). We identified two coeluting ions produced only by the wild type with mass-to-charge (m/z) ratios of 152.929 and 243.938 (Fig. 1B and fig. S1A). Both ions exhibit an isotopic distribution pattern indicative of a copper ion with molecular formulae of CuC2H4NOS+ and CuC4H9N2O2S2+, respectively (fig. S1A), which suggests that the 243.938 ion is a copper(II) complex that contains two identical ligands. We identified this complex as fluopsin C (Fig. 1C). The complex exhibits a signature d-d transition ultraviolet–visible light (UV-vis) absorption band for copper(II) at 550 to 600 nm (fig. S1B). Fluopsin C is a potent copper-containing antibiotic and cytotoxic agent that was first isolated >50 years ago from environmental Pseudomonas and Streptomyces strains (24–26). These early studies revealed that fluopsin C contains two identical N-methylthiohydroxamate ligands (Fig. 1C) by means of nuclear magnetic resonance (NMR) and infrared analysis after removal of copper from fluopsin C (27, 28) because the paramagnetic effect of copper(II) prevents direct NMR analysis of the copper complex. Fluopsin C was also identified from the P. aeruginosa LV strain that was isolated from a plant lesion, and the compound inhibits the growth of multidrug-resistant bacterial pathogens (29). We confirmed that fluopsin C inhibits the growth of a broad spectrum of Gram-positive and Gram-negative bacteria and yeast (table S1). We also found that fluopsin C is effective against multidrug-resistant strains of Acinetobacter baumannii and Staphylococcus aureus (table S1). By contrast, P. aeruginosa strains are much more resistant to fluopsin C than any other bacterial or fungal strains that were tested (24, 26, 28). For example, P. aeruginosa PAO1, the fluopsin C producer, can resist >70 μg/ml of fluopsin C (table S1). Because the structure of fluopsin C is reminiscent of copper pyrithione (fig. S1C), a synthetic copper-thiohydroxamate complex and an important antimicrobial and antifouling agent (30), we synthesized copper pyrithione (fig. S2) and measured its efficacy against the same panel of microbes that we also used to test the efficacy of fluopsin C. Except for P. aeruginosa PAO1, all microbes that we tested are susceptible to copper pyrithione (table S1 and fig. S2). Thus, PAO1 is resistant to both the fluopsin C it produces and copper pyrithione.
We characterized the structure of the copper-containing fluopsin C by electron paramagnetic resonance (EPR) spectroscopy. EPR confirms the presence of copper(II) (3d9, electron spin S = 1/2) in adx2−y2 ground state. Toluene was chosen as a solvent because it allows fluopsin C to produce well-defined EPR signals at cryogenic temperatures. When fluopsin C is dissolved in toluene, two components are observed in ~1:1 ratio in the solution state at room temperature (Fig. 1D), whereas one component dominates (85%) at cryogenic temperature (15 K) in the frozen-solution state (Fig. 1, E and F). Although the EPR signals of these components overlap through the entire X-band envelope at the microwave frequency of 9.39 GHz (Fig. 1E), the signals are well separated at the Q-band (34.0 GHz), at which only the major component contributes to the EPR envelope at magnetic field positions higher than 1198.0 mT (Fig. 1F). The electron-nuclear double resonance (ENDOR) spectra of [15N,13C2]-fluopsin C at 1198.0 mT (Fig. 1G) exhibit two 13C hyperfine coupling signals with different magnitudes and opposite signs (A1 ≈ +4 MHz, A2 ≈ −2 MHz). Combining the ENDOR data with theoretical calculations (fig. S3), we identify the major component as the trans isomer and assign the hyperfine coupling values A1 and A2 to the methyl and thiohydroxamate 13C, respectively. Thus, the trans isomer of fluopsin C is favored (cis:trans = 1:6) in the frozen-solution state (Fig. 1, E and F).
We also assessed the stability of fluopsin C in the presence of Fe(III) and reductants in aqueous solutions. Fluopsin C remains intact in the presence of Fe(III) at pH 7.4, as evidenced by minimal changes to the UV-vis spectrum of fluopsin C (fig. S1D). Titrating 1 to 5 equiv of dithiothreitol or glutathione into fluopsin C resulted in a notable shift in the UV-vis spectrum, including a reduction of the d-d absorption band for Cu(II) at 550 to 600 nm (fig. S1, E and F), which is evidence of the reduction of Cu(II) to Cu(I) in fluopsin C. These results suggest that fluopsin C exists as a Cu(I) complex in the reducing cytoplasm.
The structure of fluopsin C bears little resemblance to that of other metal-binding natural products. Specifically, the thiohydroxamate differs from other copper ligands, including the paired thioamides and oxazolone heterocycles in methanobactins (6), the thiazoli(di)nes in yersiniabactin (31), and the diisonitriles in xanthocillin (10), as well as the diisonitriles in other compounds known to mediate copper or zinc acquisition (32, 33). A natural thiohydroximate with S-glucosyl-O-sulfonyl moieties is found in the plant defense compounds glucosinolates (fig. S1C) (34). In comparison, the thiohydroxamate in fluopsin C is minimal, containing only five nonhydrogen atoms. Although the structure and antimicrobial activity of fluopsin C have been known since 1970, its biosynthesis and biological function were unknown.
To confirm the role of PA3515–PA3519 in fluopsin C biosynthesis, we expressed this operon under a lacUV5 promoter in a heterologous host, Pseudomonas fluorescens SBW25, which lacks homologous genes. P. fluorescens that expresses the operon produces fluopsin C in the presence of CuSO4 (fig. S4), which indicates that PA3515–PA3519 is sufficient for fluopsin C production and confirms our assignment of the biosynthetic gene cluster (flc, PA3515–PA3519 as flcA to flcE). Having identified the flc cluster, we investigated the biosynthetic pathway that generates the unusual structure of fluopsin C. We cultured P. aeruginosa PAO1 in M9 minimal media that contained CuSO4 (fig. S5) and conducted feeding studies by using either [15N,13C5] l-methionine or [15N,13C3] l-cysteine as potential precursors. Analysis by LC-HRMS revealed that one carbon from [15N,13C5] l-methionine and one nitrogen and one carbon from [15N,13C3] l-cysteine were incorporated in each ligand of fluopsin C (Fig. 2A). We reasoned that the N-methyl comes from methionine, likely through an S-adenosyl-l-methionine (SAM)–dependent methylation, and the thiohydroxamate backbone comes from l-cysteine.
Fig. 2. FlcB catalyzes S-succinylation of cysteine, and FlcE performs oxidative decarboxylation.
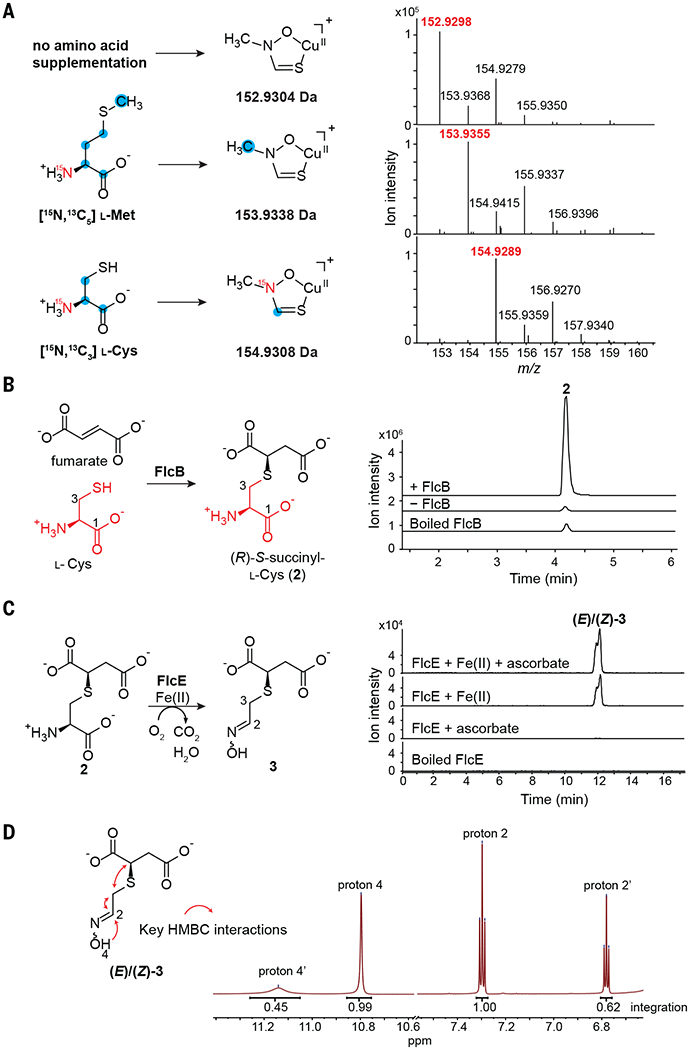
(A) Feeding studies in P. aeruginosa PAO1 using [15N,13C5] l-methionine or [15N,13C3] l-cysteine. Mass spectra of fluopsin C with a single ligand are shown. Positions of isotopic labeling are inferred. (B) EICs of 2 (m/z 238.0380 [M+H]+) from the FlcB reaction and controls. A small amount of racemic 2 forms nonenzymatically. (C) FlcE converts 2 to 3 in an Fe(II)-dependent manner. EICs of 3 (m/z 230.0094 [M+Na]+) are shown. (D) NMR spectrum of 3 in deuterated dimethylsulfoxide supports an oxime structure that exists in two diastereomers. HMBC, heteronuclear multiple bond correlation; ppm, parts per million.
To identify the first biosynthetic step for fluopsin C, we overexpressed each Flc protein in E. coli and analyzed the metabolomes of the corresponding strains by LC-HRMS. The FlcB-overexpressing E. coli produced a species with a m/z of 238.040 (2, C7H12NO6S+) at a substantially higher level than the other strains (fig. S6). This mass is consistent with S-succinyl cysteine. FlcB belongs to the adenylosuccinate lyase superfamily that catalyzes the cleavage of adenylosuccinate into adenine monophosphate and fumarate (35). Thus, we hypothesized that FlcB catalyzes a reverse reaction: conjugate addition of l-cysteine to fumarate to form 2. Indeed, purified FlcB catalyzed the in vitro conversion of l-cysteine and fumarate to a single isomer of 2 with an apparent turnover number (kcat) of 0.74/s, whereas a small amount of racemic 2 slowly formed in the absence of FlcB (Fig. 2B and fig. S7), indicating that FlcB is required for efficient and stereoselective synthesis of 2. NMR characterization reveals identical chemical shifts between 2 and the synthetic R isomer but not the S isomer (figs. S8 to S13 and table S3). Thus, FlcB catalyzes the first step in fluopsin C biosynthesis by conjugating l-cysteine with fumarate (Fig. 2B).
We investigated the remaining Flc enzymes for activity toward 2. Each purified enzyme (FlcA, FlcC, FlcD, or FlcE) was incubated with 2 in the presence of E. coli cell-free lysate (figs. S14 and S15). The incubation of FlcE with 2 produced a distinct species, 3 (m/z 208.028, C6H10NO5S+) (Fig. 2C and fig. S15), which corresponds to the net loss of C, O, and two H from 2. FlcE can modify (R)-2 but not (S)-2, which confirms that the first intermediate is (R)-2 (fig. S16). Sequence alignment suggests that both FlcD and FlcE contain a heme oxygenase domain. Heme oxygenase domains also exist in the Chlamydia protein associating with death domains (CADD) and the enzyme SznF, and the crystal structures of these proteins show that this domain contains a diiron center (36, 37). Therefore, we incubated FlcE with 2 in the presence of Fe(II) without cell lysate and observed the formation of 3 (Fig. 2C). The activity of FlcE requires Fe(II) and is only modestly increased by addition of ascorbate as a reductant; no other common metal cofactors reconstitute FlcE activity (Fig. 2C and fig. S17). These results suggest that FlcE is an iron-dependent enzyme that does not require an external reductant. NMR characterization of 3 indicates that it lacks the cysteine amine or carboxylate but contains an oxime instead (Fig. 2D, tables S4 and S5, and figs. S18 to S22), which suggests that FlcE catalyzes both an oxidative decarboxylation and N-hydroxylation.
After identifying the reaction catalyzed by FlcE, we found that the other heme oxygenase domain–containing enzyme, FlcD, catalyzes the conversion of 3 to a distinct species, 4 (m/z 194.012, C5H8NO5S+) in the presence of Fe(II) (Fig. 3A and fig. S23). The mass difference between 3 and 4 corresponds to a loss of CH2. Only Fe(II), among the common metal co-factors, reconstitutes the activity of apo-FlcD, and the reductant ascorbate modestly increases FlcD activity (Fig. 3A and fig. S24). We isolated 4 from a one-pot reaction containing 2, FlcD, and FlcE for structural characterization. NMR analysis revealed that 4 also contains an oxime and is one methylene shorter than 3 (table S6 and figs. S25 to S29), which indicates that FlcD catalyzes a methylene excision of 3. To identify which carbon atom is excised by FlcD, we conducted a one-pot reaction containing FlcB, FlcE, and FlcD using [3-13C] l-cysteine as a precursor to 4. The 3-13C atom in [3-13C] l-cysteine is retained in 4, which suggests excision of 2-C by FlcD (Fig. 3B). To determine the fate of the excised 2-C, we examined formaldehyde and formate as potential oxidation products formed by FlcD. No formaldehyde was detected (fig. S30), whereas formate and 4 were produced in 1:1 stoichiometry (Fig. 3C and figs. S31 and S32). Therefore, FlcD catalyzes excision of 2-C from 3 as formate in a net four-electron oxidation. The four-electron oxidation and the lack of need for external reductants suggest that FlcD incorporates oxygens from O2 into formate while regenerating the ferrous center (fig. S33).
Fig. 3. FlcD excises a methylene carbon from 3 as formate.
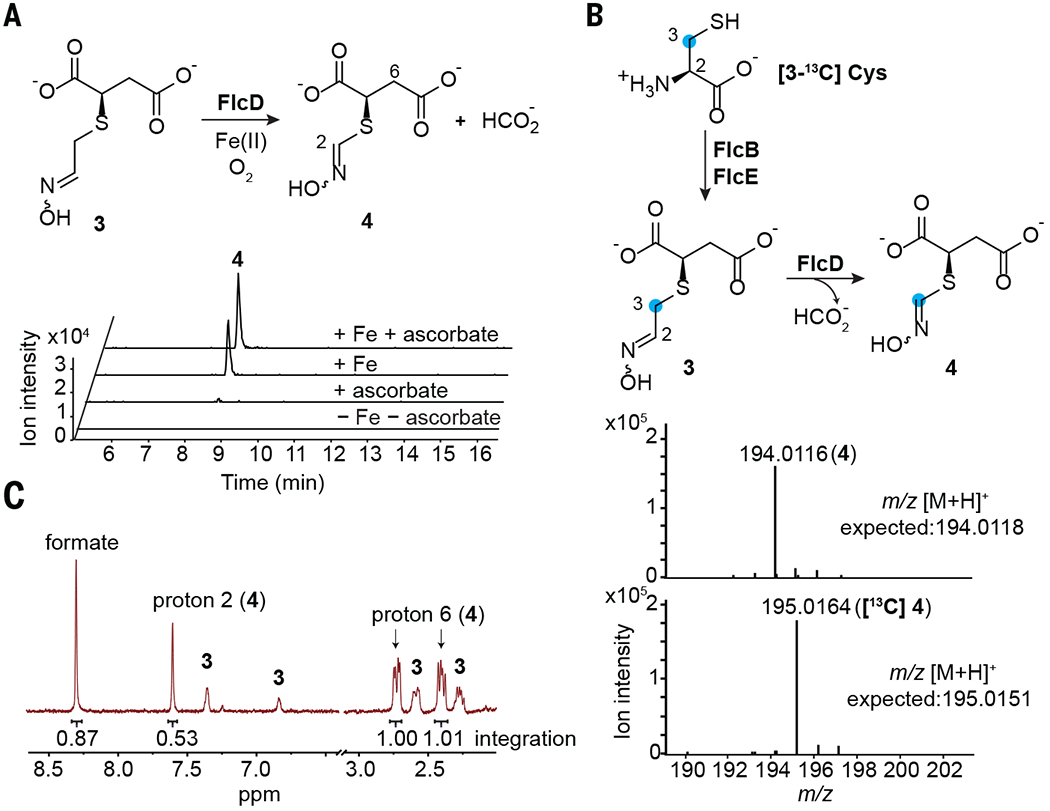
(A) FlcD-catalyzed formation of 4 (m/z 194.0118 [M+H]+) depends on Fe(II). (B) Mass spectra show that the 3-13C atom of [3-13C] l-cysteine is retained in 4 in a one-pot reaction containing FlcB, FlcE, and FlcD. (C) NMR analysis of the FlcD reaction in D2O reveals the production of formate and 4 in a 1:1 stoichiometry.
Comparing the structure of 4 to that of fluopsin C, we hypothesized that the remaining two enzymes, the putative methyltransferase FlcA and lyase FlcC, catalyze N-methylation and cleavage of thiohydroxamate from succinate, respectively. Purified 4 was incubated with either FlcA or FlcC. FlcA does not catalyze methylation of 4 in the presence of SAM, whereas FlcC rapidly catalyzes consumption of 4 and production of fumarate (figs. S34 and S35). Monitoring the FlcC reaction by NMR reveals a time-dependent consumption of 4 (apparent kcat = 0.46/s) and concomitant production of fumarate and a distinct species, 5 (Fig. 4A and fig. S35). Compound 5 contains a sole slowly exchanged singlet proton at 8.0 parts per million in D2O (Fig. 4A and fig. S36) and does not react with Ellman’s reagent that modifies free thiols (fig. S37), which suggests that 5 is a thiohydroxamate rather than an oxime thiol. Therefore, FlcC catalyzes the C–S bond cleavage of 4 to yield the thiohydroxamate 5 and fumarate.
Fig. 4. FlcC and FlcA catalyze the final steps of fluopsin C biosynthesis.
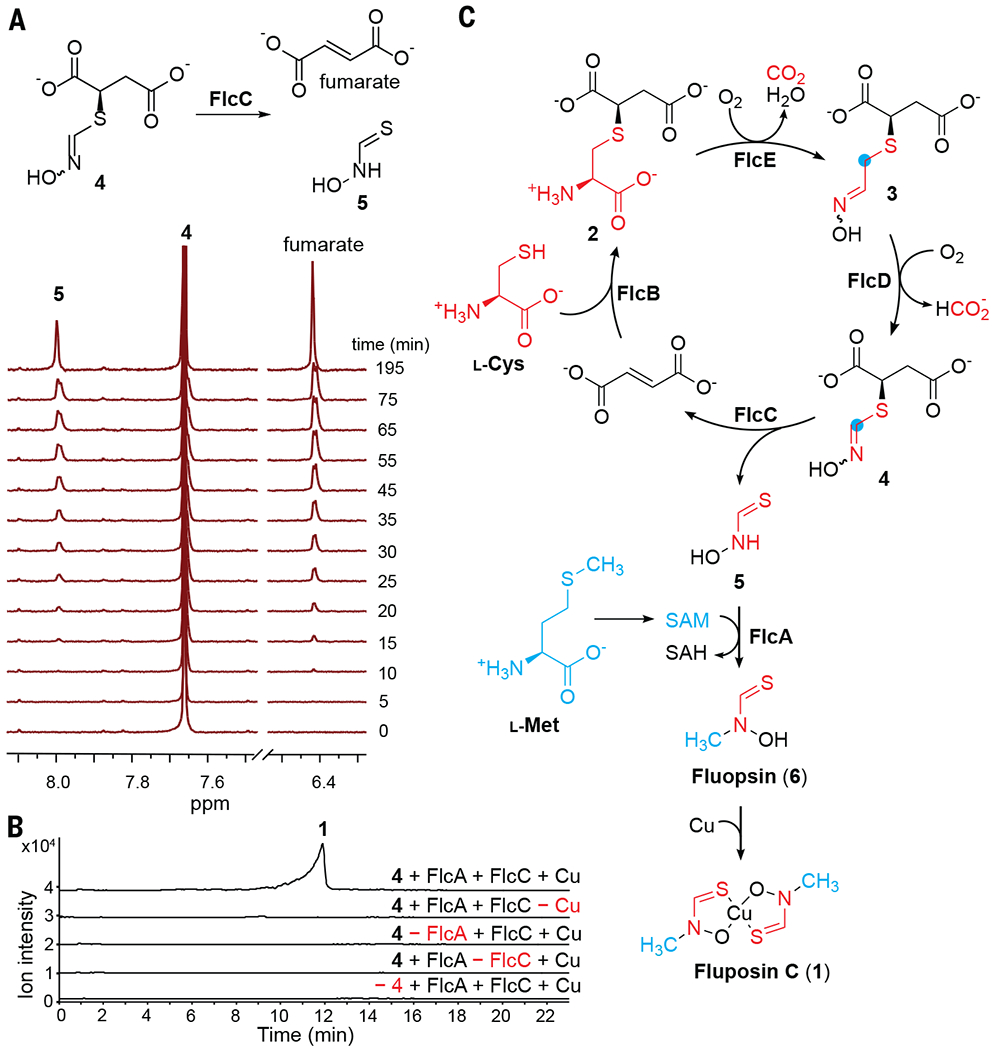
(A) NMR time-course study of the lyase FlcC. (B) FlcA and FlcC catalyze tandem transformations of 4 to fluopsin C, with addition of CuSO4 at the end of the reaction. Combined EICs of 1 (m/z 152.9304 [M–C2H4NOS]+ and 265.9215 [M+Na]+) are shown. (C) Complete fluopsin C biosynthetic pathway. SAH, S-adenosyl-l-homocysteine.
To reconstitute the activity of the final methyltransferase FlcA, we incubated 4 with FlcC, FlcA, and SAM, adding CuSO4 at the end of the reaction. Fluopsin C is produced only when both Flc enzymes are present (Fig. 4B), supporting the role of FlcA in methylating 5 and generating the Cu-free fluopsin (6). Having reconstituted each biosynthetic step, we performed a one-pot reaction containing l-cysteine, fumarate, and all five Flc enzymes and their cofactors, and we added 2 mM CuSO4 at the end. A 24-hour one-pot reaction containing 0.25 to 0.75 mol % of each Flc enzyme resulted in an 87% yield across five steps. Omission of any Flc enzyme abolished the production of fluopsin C; in particular, omission of FlcE, FlcD, or FlcC resulted in the accumulation of 2, 3, and 4, respectively, which further supports the biosynthetic pathway (fig. S38 and Fig. 4C).
We uncovered the biosynthetic origin of fluopsin C, a metal complex produced by bacteria, and characterized its electronic structure. Its biosynthesis entails a series of unusual enzymatic transformations that convert cysteine to a minimal thiohydroxamate (Fig. 4C). The lyases FlcB and FlcC add and subsequently remove an S-succinyl group while recycling fumarate. The heme oxygenase enzymes, FlcE and FlcD, remove both 1-C and 2-C from cysteine and oxidize the amine to an oxime, generating the thiohydroximate form that is protected by the S-succinyl group. Only a few heme oxygenase enzymes have been characterized, such as SznF in the biosynthesis of the anticancer drug streptozotocin (38), UndA in the biosynthesis of the biofuel undecene (39, 40), and BesC in the biosynthesis of a terminal alkyne-containing amino acid (41). FlcD and FlcE share low sequence identity with these enzymes and contain divergent sequences at conserved positions (figs. S39 and S40), which may hint at different mechanisms used by FlcD and FlcE. In particular, the carbon excision reaction catalyzed by FlcD has limited precedence in enzyme chemistry (42, 43).
Nonclassical roles of siderophores are emerging, such as the transport of noniron metals (44). Our discovery of the fluopsin C biosynthetic pathway further expands the functions of metal-binding natural products beyond metal acquisition. As we and others have shown, fluopsin C exhibits broad-spectrum antibiotic activity against microbes and potent cytotoxicity against mammalian cells and whole animals, and it is much less active against various P. aeruginosa strains (24, 26, 29). Because fluopsin C contains copper, its antimicrobial mechanism likely differs from existing antibiotic classes or the copper-binding compound xanthocillin, which exhibits diminished antimicrobial effect in the presence of copper (10). We found that the flc gene cluster is preserved in 4937 of the 4955 (99.6%) P. aeruginosa genomes, which include both clinical and environmental isolates (45). The preservation of the flc cluster suggests that almost all P. aeruginosa strains can produce and resist fluopsin C. Given that elevated levels of copper occur in water, soil, and activated macrophages (2, 46), P. aeruginosa may integrate copper into fluopsin C to inhibit the growth of other microbes or the activities of host cells during infection as part of the copper stress response. The flc cluster is also distributed in the genomes of many other bacteria (figs. S41 and S42 and table S7), including A. baumannii, Enterobacter cloacae, and Streptococcus dysgalactiae, particularly isolates from infected human patients and in hospital settings. Additionally, we identified the flc cluster in the genomes of P. fluorescens, Lysobacter enzymogenes, and Lysobacter capsica strains (fig. S41 and S42 and table S7), which colonize the roots and leaves of plants and exert biocontrol activity against phytopathogenic organisms. In particular, the flc-containing L. capsica AZ78 strain is resistant to copper and more effectively protects plants against oomycetes in combination with a copper fungicide (47); the production of fluopsin C may contribute to the copper resistance and copper-augmented fungicidal activity of this strain. Overall, the wide distribution of flc in both pathogenic and environmental bacteria suggests that fluopsin C serves a conserved role in helping the bacteria that produce it to interact with other microbes in the environment and their eukaryotic hosts in response to elevated copper levels.
Supplementary Material
ACKNOWLEDGMENTS
We thank W. van der Donk, G. Pielak, A. Bowers, E. Patteson, and members of the Li lab for helpful discussions and K. Trippe (USDA-ARS National Forage Seed Production Research Center) for P. fluorescens SBW25.
Funding:
This work was supported by National Institutes of Health grant DP2HD094657 (B.L.), the Packard Fellowship for Science and Engineering (B.L.), The University of North Carolina at Chapel Hill (B.L.), and National Institutes of Health grant R35GM126961 (R.D.B.). NMR experiments were performed in the University of North Carolina at Chapel Hill Department of Chemistry NMR Core Laboratory supported in part by National Science Foundation grants CHE-0922858 and CHE-1828183.
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data and materials availability:
All data are available in the main text or the supplementary materials. The sequence similarity network is available on Zenodo (48). Materials are available by request from the corresponding author.
REFERENCES AND NOTES
- 1.Sandy M, Butler A, Chem. Rev 109, 4580–4595 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Palmer LD, Skaar EP, Annu. Rev. Genet 50, 67–91 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Challis GL, ChemBioChem 6, 601–611 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Ghssein G et al. , Science 352, 1105–1109 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Kenney GE, Rosenzweig AC, Annu. Rev. Biochem 87, 645–676 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim HJ et al. , Science 305, 1612–1615 (2004). [DOI] [PubMed] [Google Scholar]
- 7.Ross MO et al. , Science 364, 566–570 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chaturvedi KS, Hung CS, Crowley JR, Stapleton AE, Henderson JP, Nat. Chem. Biol 8, 731–736 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koh E-I, Robinson AE, Bandara N, Rogers BE, Henderson JP, Nat. Chem. Biol 13, 1016–1021 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raffa N et al. , Proc. Natl. Acad Sci. U.S.A 118, e2015224118 (2021).33593906 [Google Scholar]
- 11.White C, Lee J, Kambe T, Fritsche K, Petris MJ, J. Biol. Chem 284, 33949–33956 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Djoko KY, Ong CL, Walker MJ, McEwan AG, J. Biol. Chem 290, 18954–18961 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wolschendorf F et al. , Proc. Natl. Acad. Sci. U.S.A 108, 1621–1626 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sheldon JR, Skaar EP, Curr. Opin. Immunol 60, 1–9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huffman DL, O’Halloran TV, Annu. Rev. Biochem 70, 677–701 (2001). [DOI] [PubMed] [Google Scholar]
- 16.Fu Y, Chang F-MJ, Giedroc DP, Acc. Chem. Res 47, 3605–3613 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lister PD, Wolter DJ, Hanson ND, Clin. Microbiol. Rev 22, 582–610 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Teitzel GM, Parsek MR, Appl. Environ. Microbiol 69, 2313–2320 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Teitzel GM et al. , J. Bacteriol 188, 7242–7256 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Quintana J, Novoa-Aponte L, Argüello JM, J. Biol. Chem 292, 15691–15704 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thaden JT, Lory S, Gardner TS, J. Bacteriol 192, 2557–2568 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Changela A et al. , Science 301, 1383–1387 (2003). [DOI] [PubMed] [Google Scholar]
- 23.Held K, Ramage E, Jacobs M, Gallagher L, Manoil C, J. Bacteriol 194, 6387–6389 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Egawa Y, Umino K, Awataguchi S, Kawano Y, Okuda T, J. Antibiot 23, 267–270 (1970). [DOI] [PubMed] [Google Scholar]
- 25.Otsuka H et al. , J. Antibiot 25, 369–370 (1972). [DOI] [PubMed] [Google Scholar]
- 26.Ito S, Inuzuka K, Suzuki T, J. Antibiot 23, 542–545 (1970). [DOI] [PubMed] [Google Scholar]
- 27.Egawa Y, Umino K, Ito Y, Okuda T, J. Antibiot 24, 124–130 (1971). [DOI] [PubMed] [Google Scholar]
- 28.Shirahata K, Deguchi T, Hayashi T, Matsubara I, Suzuki T, J. Antibiot 23, 546–550 (1970). [DOI] [PubMed] [Google Scholar]
- 29.Navarro MOP et al. , Front. Microbiol 10, 2431 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Helsel ME, White EJ, Razvi SZA, Alies B, Franz KJ, Metallomics 9, 69–81 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Drechsel H et al. , Liebigs Ann 1995, 1727–1733 (1995). [Google Scholar]
- 32.Wang L et al. , ACS Chem. Biol 12, 3067–3075 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Harris NC et al. , Proc. Natl. Acad. Sci. U.S.A 114, 7025–7030 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clay NK, Adio AM, Denoux C, Jander G, Ausubel FM, Science 323, 95–101 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Toth EA, Yeates TO, Structure 8, 163–174 (2000). [DOI] [PubMed] [Google Scholar]
- 36.Schwarzenbacher R et al. , J. Biol. Chem 279, 29320–29324 (2004). [DOI] [PubMed] [Google Scholar]
- 37.McBride MJ et al. , Proc. Natl. Acad. Sci. U.S.A 118, e2015931118 (2021).33468680 [Google Scholar]
- 38.Ng TL, Rohac R, Mitchell AJ, Boal AK, Balskus EP, Nature 566, 94–99 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang B et al. , J. Am. Chem. Soc 141, 14510–14514 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Manley OM, Fan R, Guo Y, Makris TM, J. Am. Chem. Soc 141, 8684–8688 (2019). [DOI] [PubMed] [Google Scholar]
- 41.Marchand JA et al. , Nature 567, 420–424 (2019). [DOI] [PubMed] [Google Scholar]
- 42.Cicchillo RM et al. , Nature 459, 871–874 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ting CP et al. , Science 365, 280–284 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Johnstone TC, Nolan EM, Dalton Trans 44, 6320–6339 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Winsor GL et al. , Nucleic Acids Res 44, D646–D653 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wagner D et al. , J. Immunol 174, 1491–1500 (2005). [DOI] [PubMed] [Google Scholar]
- 47.Puopolo G, Giovannini O, Pertot I, Microbiol. Res 169, 633–642 (2014). [DOI] [PubMed] [Google Scholar]
- 48.Patteson JB et al. , Heme oxygenase pfam14518 sequence similarity network, Zenodo; (2021); 10.5281/zenodo.5508248. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in the main text or the supplementary materials. The sequence similarity network is available on Zenodo (48). Materials are available by request from the corresponding author.