

Abstract
The fields of C–H functionalization and photoredox catalysis have garnered enormous interest and utility in the past several decades. Many different scientific disciplines have relied on C–H functionalization and photoredox strategies including natural product synthesis, drug discovery, radiolabeling, bioconjugation, materials, and fine chemical synthesis. In this Review, we highlight the use of photoredox catalysis in C–H functionalization reactions. We separate the review into inorganic/organometallic photoredox catalysts and organic-based photoredox catalytic systems. Further subdivision by reaction class—either sp2 or sp3 C–H functionalization—lends perspective and tactical strategies for use of these methods in synthetic applications.
Graphical Abstract
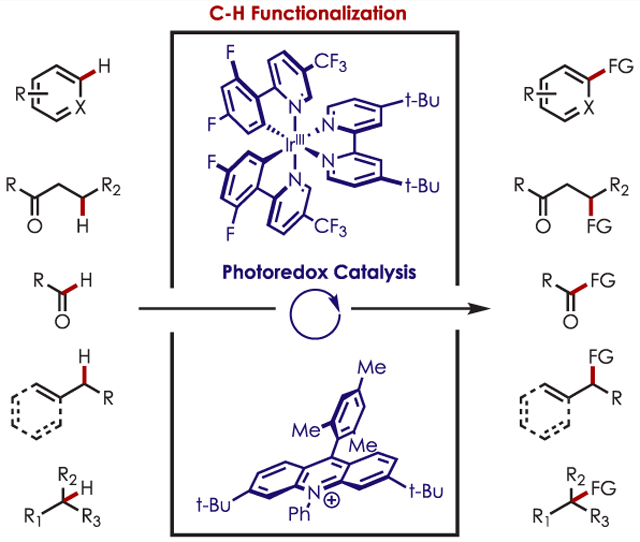
1. INTRODUCTION
1.1. Background and Importance
The field of catalytic C–H functionalization has continued to grow at a rapid pace over the past four decades both in terms of the scope of transformations that are possible as well as the types of catalytic manifolds that enable C–H functionalization. Catalytic strategies involving transition metals, enzymatic systems, photochemical, electrochemical and photoredox systems have all been utilized for the selective functionalization of C–H bonds.1–14 These strategies have been deployed in a number of settings including the selective functionalization of natural products,7,15 late-stage functionalization of pharmaceutical derivatives,16 petroleum feedstocks and polymers.17,18 Adoption of catalytic C–H functionalization tactics has begun in industry, particularly in pharmaceutical and agrochemical discovery settings.19
Photoredox catalysis continues to expand at a rapid pace and touches many different fields of scientific inquiry including applications to renewable energy and chemical feedstocks, new reaction development, natural product synthesis, materials and biological applications. While there have been numerous reviews and perspective articles on the subject of photoredox catalysis in general,20–24,24–49 this review will be a comprehensive overview of the use of photoredox catalysis specifically in transformations that involve C–H functionalization. We have elected to bifurcate this review into C–H functionalization reactions that employ either inorganic/organometallic or organic photoredox catalysts in order to compare and contrast the reactions possible with each class of catalyst and also to highlight the cases where overlap exists. We have further divided each of these sections to cover both aromatic and aliphatic C–H functionalization reactions and, subsequently, the types of intermediate radical or charged open-shell species that participate in these transformations.
1.2. Inorganic/Organometallic Photoredox Catalysts
The photochemistry and photophysics of inorganic and organometallic photoredox catalysts have been described in detail in a number of articles and reviews.23,28,36 In general, inorganic/organometallic photoredox catalysts offer advantages due to their high molar absorptivities, often efficient intersystem crossing (ISC), and triplet (T1) lifetimes (~100 ns to 1 ms). Many derivatives of Ru(II) and Ir(III) photocatalysts have been prepared as it is convenient to rapidly and systematically alter the ligand sets with either commercially available or easily prepared ligands to tune the absorption and excited state properties of these molecules. This class of photoredox catalysts generally absorbs in the ~390–480 nm window; however, development of near-IR-absorbing Os photoredox catalysts have also been recently described.50
The main drawback to the use of inorganic/organometallic photoredox catalyst systems is the higher cost involved (Ru ~ $15/g; Ir ~ $220/g), the inability to tune the redox potential at the metal center, and smaller excited state redox windows (ca. −2.2 to +1.8 V vs SCE)28 compared to organic photoredox catalysts (ca. −3.4 to +2.7 V vs SCE).35,51 Nonetheless, a plethora of transformations involving inorganic and organometallic photoredox catalysts have been reported to date.
1.3. Organic Photoredox Catalysts
We have covered the general photophysical and photochemical properties in detail for a range of organic photocatalysts in a prior review.35 As mentioned previously, excited-state organic photoredox catalysts have a larger redox window than their inorganic/organometallic counterparts, which may be key to achieve certain classes of transformations or to access substrates in particular transformations that are inaccessible using metal-based systems. The absorption profile of these catalysts is similar to that of inorganic/organometallic-based systems however often have lower extinction coefficients than their metal-based counterparts. Often organic photoredox catalysts operate via their S1 states rather than their T1 states, giving rise to the larger redox window available to these catalysts, however, at the expense of shorter lifetimes overall (10–50 ns).
Certainly, organic photoredox catalysts offer a more cost-effective option for achieving single electron redox reactions; however, their drawbacks include more complex syntheses for certain catalysts and shorter excited-state lifetimes which can require higher catalyst loadings and irradiation times. Overall, organic photoredox catalysts have been able to accomplish an impressive array of transformations including C–H functionalization reactions.
1.4. Definitions
In the case of this of review, C–H functionalization is defined as any organic transformation of a C–H into a C–X bond without a change in the oxidation state of the substrate. The scope of this review includes C–H functionalizations with a catalytic amount of a photooxidant or reductant. It does not cover noncatalytic transformations, those using semiconducting nanomaterials, or electrochemistry.52 This review includes transformations acting through a proton coupled electron transfer (PCET) in which electrons and protons are transferred together; a pertinent review on this topic appears in this issue of Chemical Reviews.53,54 Scheme 1 contains a summary of both the organic and inorganic catalysts described in this review, their redox properties, and their abbreviations.
Scheme 1.
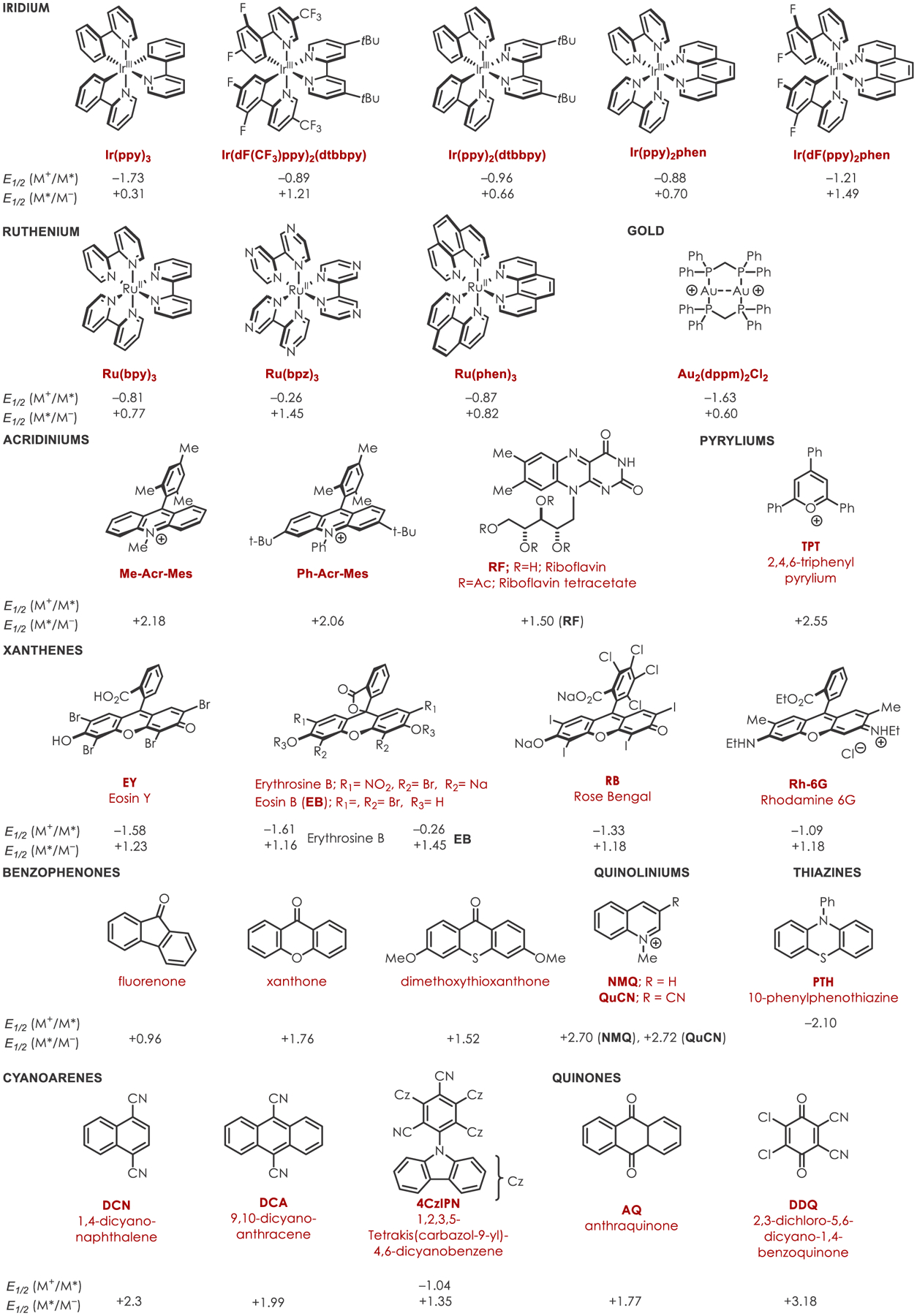
Table of Common Photoredox Catalysts, Abbreviations, and Excited-State Redox Values (vs SCE)28,35,55,56
2. INORGANIC/ORGANOMETALLIC PHOTOREDOX-CATALYZED C–H FUNCTIONALIZATION REACTIONS
2.1. (Hetero)aromatic C–H Functionalization
2.1.1. Transformations Involving Carbon sp3-Centered Radicals.
Direct C–H functionalization via radical intermediates is a valuable approach for the formation of C–C or C–X bonds in (hetero)arenes. Classically, the formation of carbon radicals required the use of toxic reagents, such as tributyltin hydride, thus limiting the utility of such reactions.57–59 More recently, photoredox catalysis has offered an alternative avenue for accessing reactive sp3 carbon-centered radicals through hydrogen atom transfer (HAT) sequences or single-electron transfer (SET) with alkyl halides, sulfonyl chlorides, acids or peroxides. Inorganic photoredox catalysts are particularly attractive for generating sp3-centered radicals through reductive quenching pathways due to the relative redox potentials between the carbon-centered radical precursor and the catalyst. In general, the resulting alkyl radical species undergo Minisci-type reactivity with heteroarenes (2.1), adding to the more electron-deficient carbon (Scheme 2). This has allowed for C–C, C–N, C–O, C–S, and C–P bond formation through C–H functionalization.
Scheme 2.
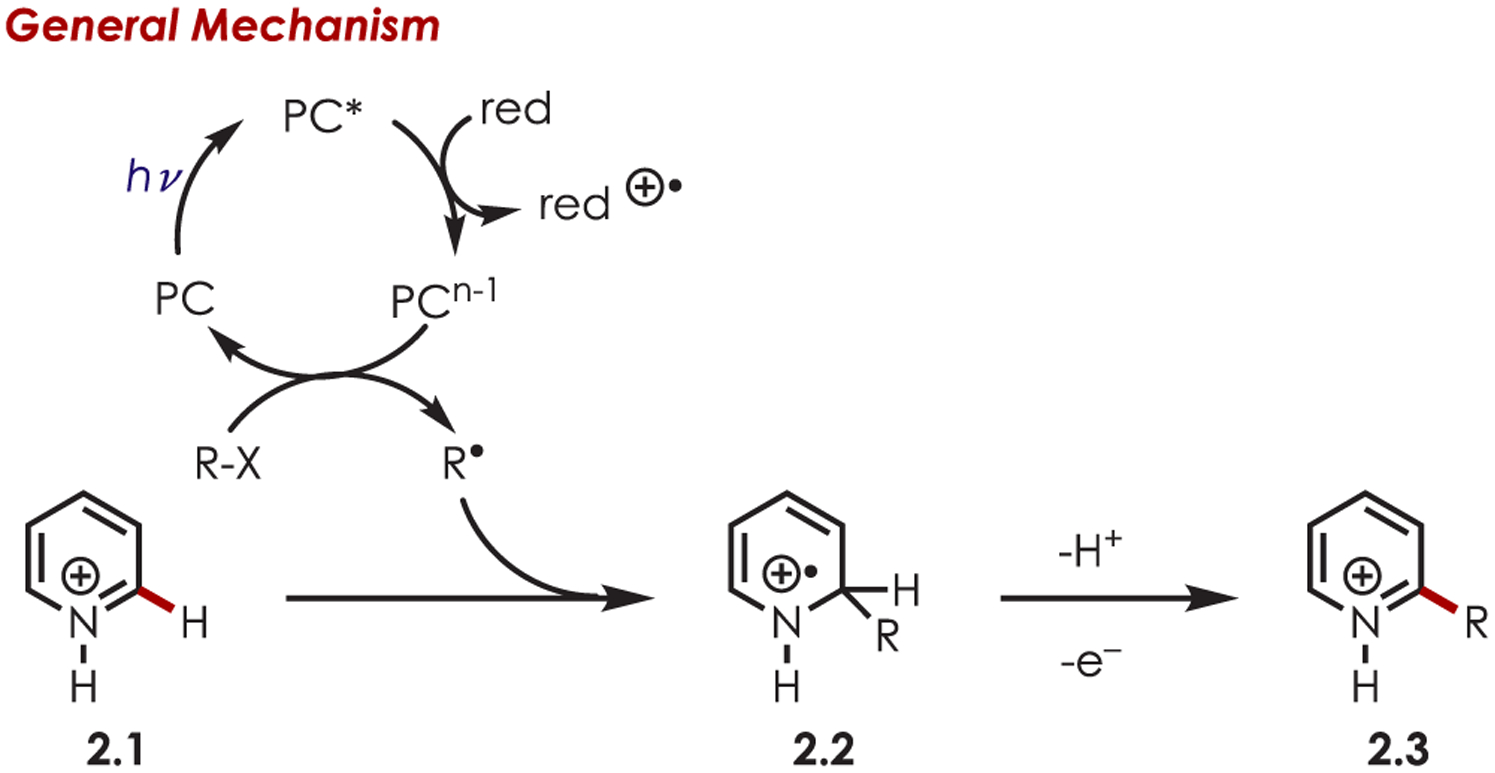
A General Mechanism of a Photoredox-Mediated Minisci-Type Radical Addition into Heteroarenes with a Sacrificial Reductant
Accessed via Alkyl Halides.
In 2009, Stephenson reported the seminal heteroaryl C–H functionalization using alkyl bromides in an intramolecular cyclization of indoles (Scheme 3).60 Irradiation of Ru(bpy)32+ results in a MLCT event forming excited state Ru(II)*. Reductive quenching of the excited state by triethylamine generates Ru(I), which then undergoes an electron transfer with the bromomalonate functionality. This selective reduction of the sp3 C–Br bond produces the carbon-centered radical (3.4) while regenerating the Ru(II) catalyst. The electron-rich heterocycle triggers an intramolecular cyclization with the electron-deficient alkyl radical, affording an intermediate benzylic radical, 3.5. Oxidation of the benzylic radical, from either Ru(II)* or bromomalonate, followed by elimination furnishes the desired product (3.6). Triethylamine was employed as the sacrificial reductant, which gave the greatest reaction efficiency for the selective formation of five- and six-membered rings. Functionalized indoles and pyrroles with electron-poor or electron-rich substitution were compatible, with the latter generally producing the desired product in higher yields.
Scheme 3.
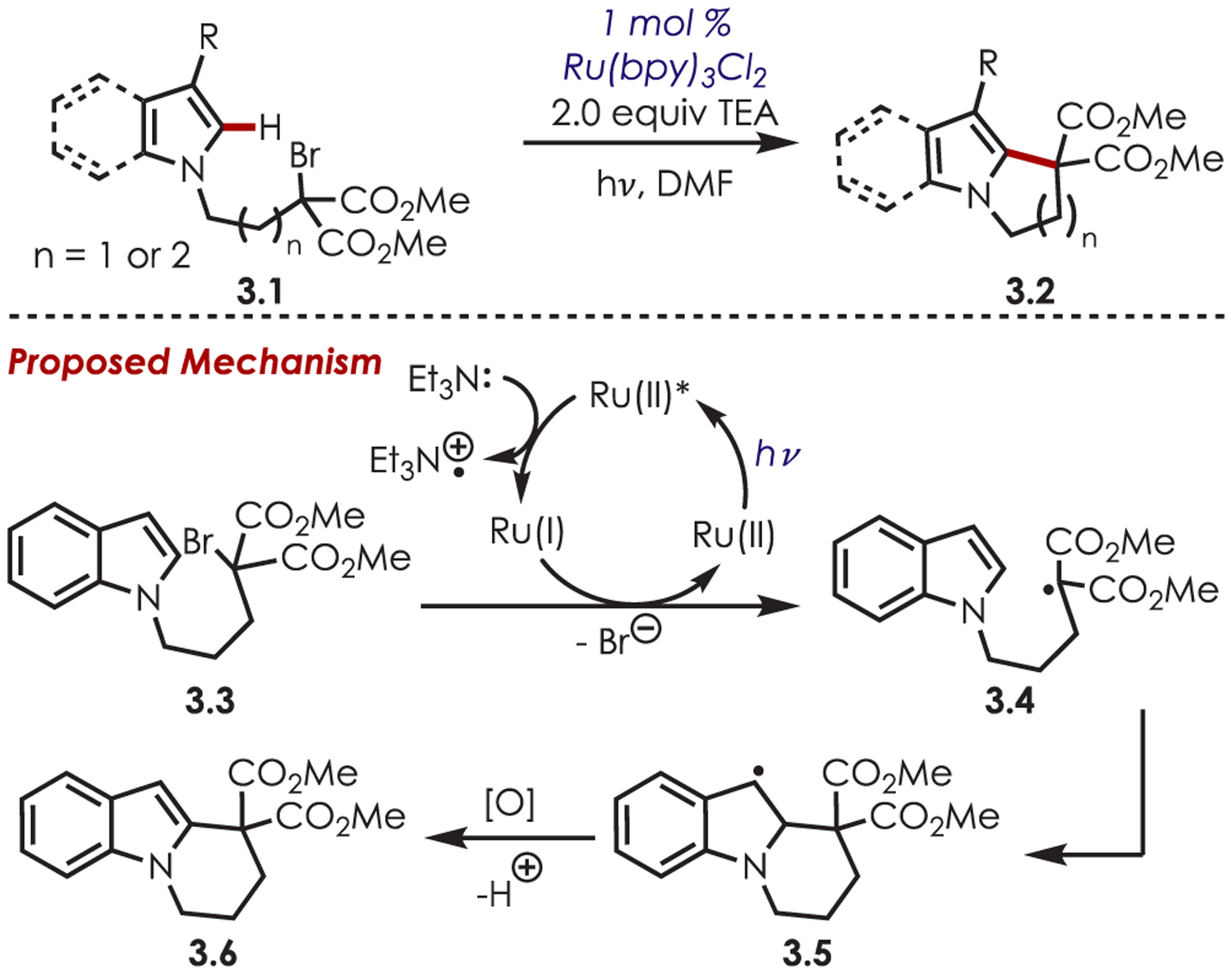
Intramolecular Heteroaryl C–H Alkylation
Shortly thereafter, Stephenson disclosed an expansion of this method for the intermolecular coupling of heteroaromatics (4.1) with bromomalonate (Scheme 4).61 This coupling relied on using an aromatic amine reductant, 4-methoxy-N,N-diphenylaniline, which prevented the undesired malonate and alkylated enamine side products from a reductive HAT of trialkylamines. C2 functionalization of indoles, azaindoles, pyrroles, and furans with bromomalonate was reported in 40–92% yield.
Scheme 4.
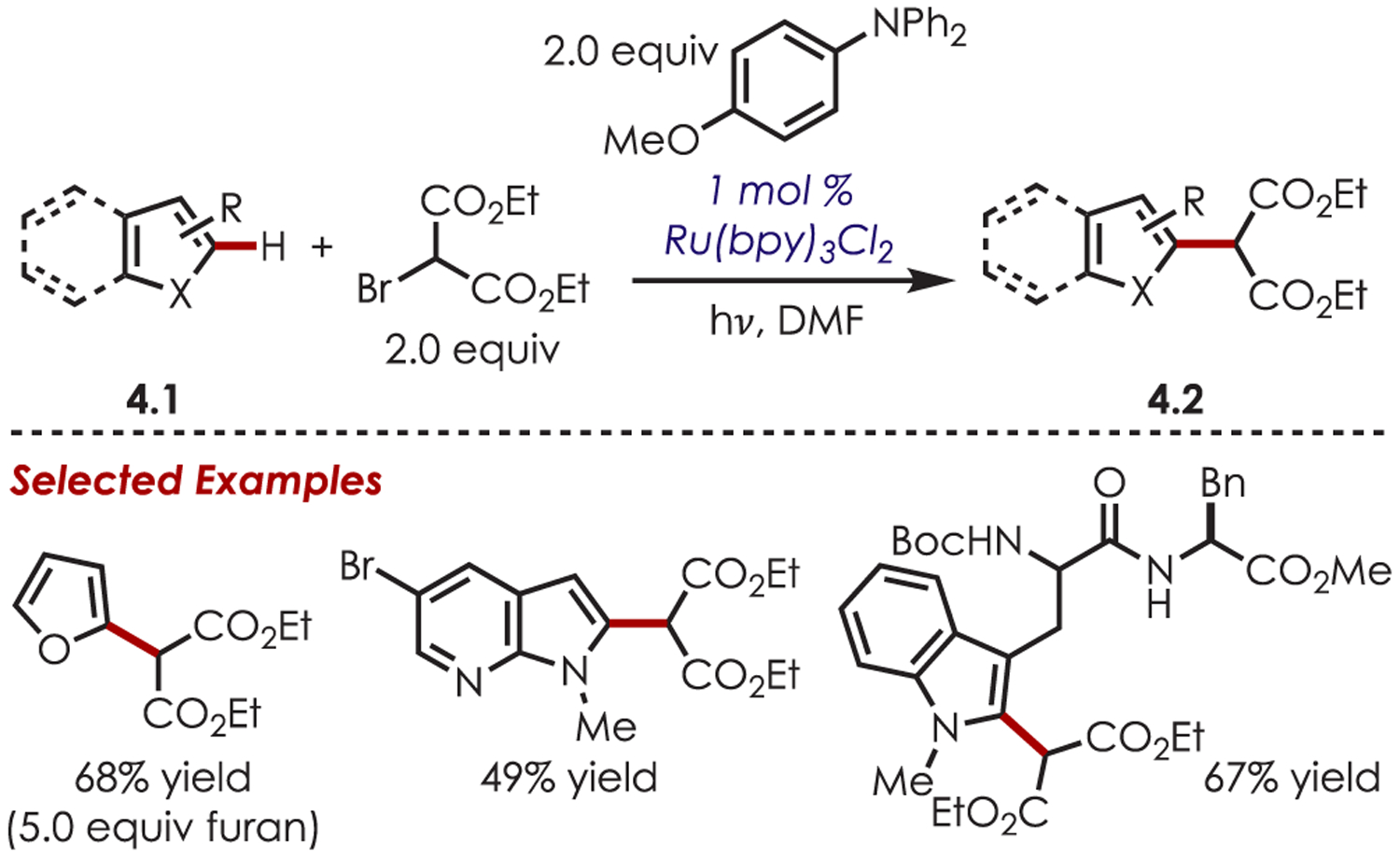
Scope of the Intermolecular Heteroaryl C–H Alkylation using Ir(ppy)3 with Respect to the Heteroarene
The scope of the bromoacetate partner (5.2) was also explored (Scheme 5).62 Ir(ppy)3 allowed for direct reduction of the tertiary bromide from the excited state, eliminating the requirement of an external reductant (E*ox = −1.73 V vs SCE). Tertiary bromoacetates containing an electron-deficient group with primary or secondary alkyl, allyl, or benzyl substitution were all found to be viable substrates. The mechanism is proposed to be similar to their other work, where the carbon-centered radical adds to the heteroarene in a Minisci-type fashion.
Scheme 5.
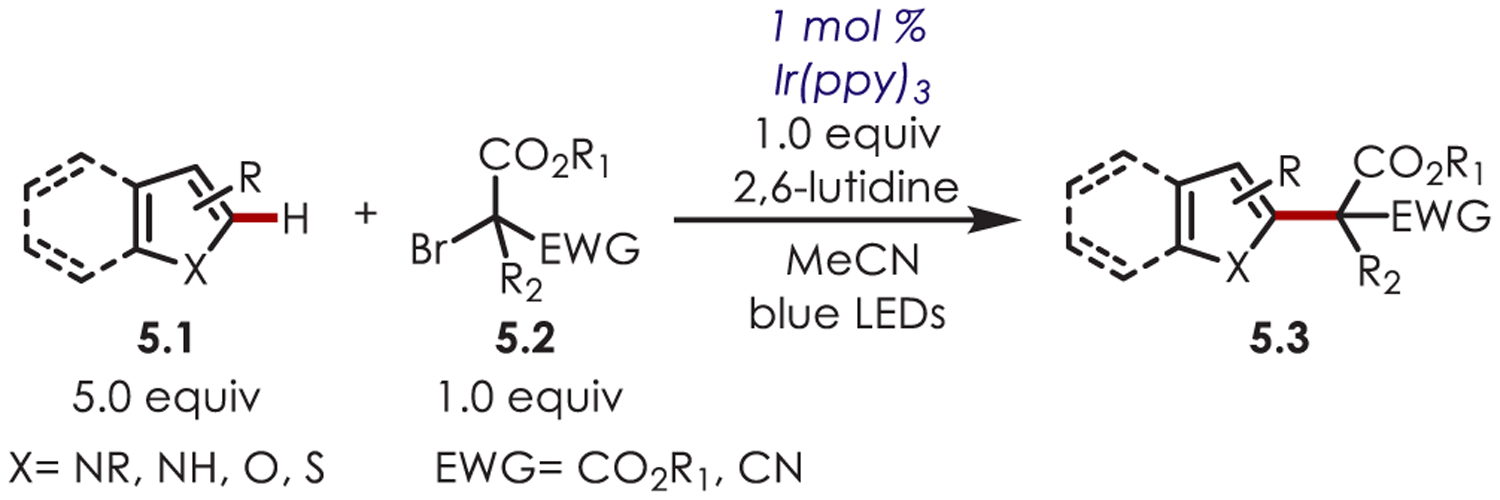
Scope of Alkyl Bromides for an Intermolecular Heteroaryl C–H Alkylation Using Ir(ppy)3
Alkylation of aniline derivatives (6.1) via sp3-centered radicals has also been reported employing tertiary alkyl bromides (6.2) as the radical surrogates (Scheme 6).63 Aryl and heteroaryl α-bromoketones were utilized as radical precursors and underwent selective ortho-functionalization with alkyl-, phenyl-, or methoxy-substituted anilines.
Scheme 6.
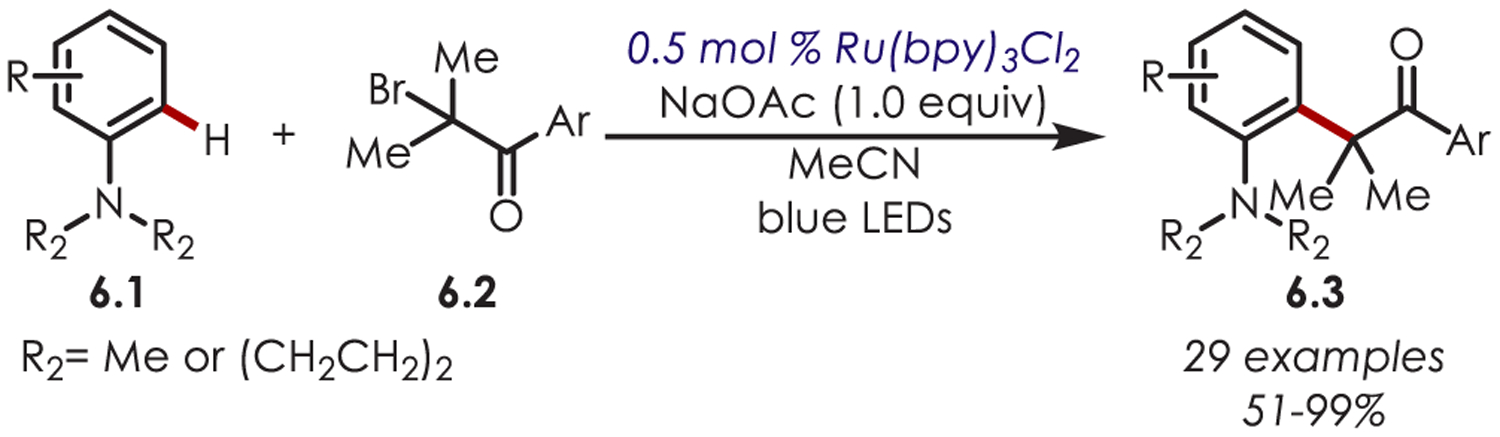
Alkylation of Aniline Derivatives through α-Bromoketones as sp3 Carbon-Centered Radical Precursors
Direct reduction of nonactivated bromoalkanes to their corresponding alkyl radicals is generally unfeasible for most inorganic photoredox catalysts on thermodynamic grounds because of the highly negative redox potentials of these substrates (Ered = −2.09 V vs SCE for 1-bromopentane).64 Despite this fact, a dimeric gold photoredox catalyst (Eox* = −1.63 V vs SCE) has been disclosed that generates nucleophilic alkyl radicals for a Minisci-type addition to heteroarenes (7.1) (Scheme 7).65 Binuclear Au(I) phosphine complexes were previously known to form dimeric Au–Au complexes upon excitation.56 The excited dimer opens a coordination site, which allows for the formation of an exciplex via association of a bromoalkane (7.2). Oxidative quenching of the dimeric gold complex (7.4) generates an alkyl carbon-centered radical. Ultimately, an inner sphere PET is invoked, rather than an outer sphere PET via an MLCT excited state typical of Ru or Ir polypyridyl photoredox catalysts, and allows for the formation of alkyl radicals from bromoalkanes with reduction potentials more negative than that of the dimeric catalyst while also preventing any stability issues that may be observed with free alkyl radicals. Primary, secondary, and tertiary bromoalkanes were acceptable radical precursors.
Scheme 7.
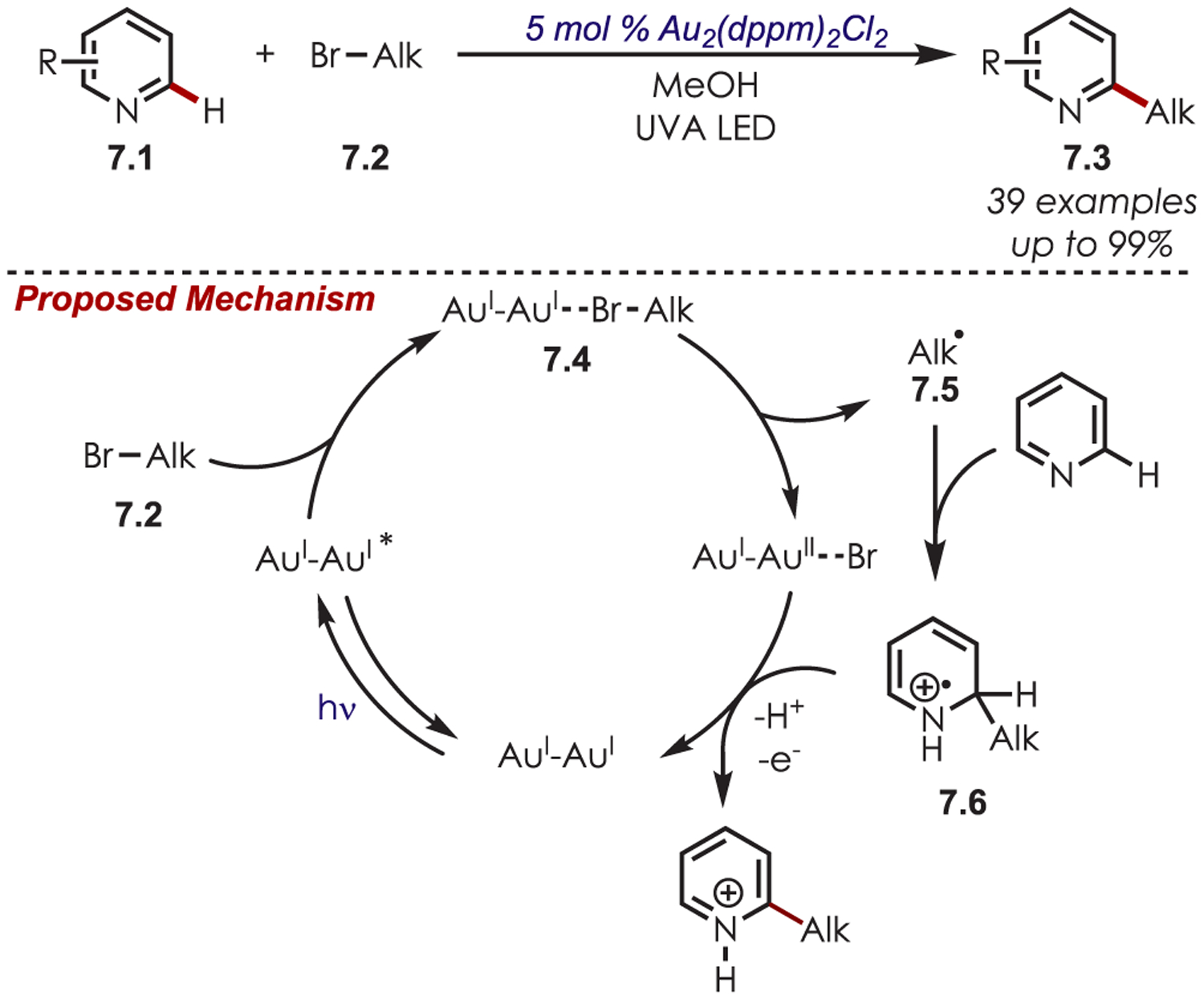
Dimeric Gold Complexes in Heteroaryl C–H Functionalizations with Alkyl Bromides
Trifluoroalkyl iodides (8.1)66 and bromodifluoroacetamides (8.3)67 have also been used successfully as precursors to fluoroalkyl sp3 carbon-centered radicals for the functionalization of heteroarenes and imidazo heterocycles (Scheme 8).68 A mixture of regioisomers was observed in the trifluoromethylation of indoles with a slight preference for substitution at the 2-position due to the generation of the more stable benzylic radical. A mixture of ortho-, meta-, and para-regioisomers were observed in the difluoroacetamidation of (hetero)arenes, which is consistent with a radical substitution process.
Scheme 8.
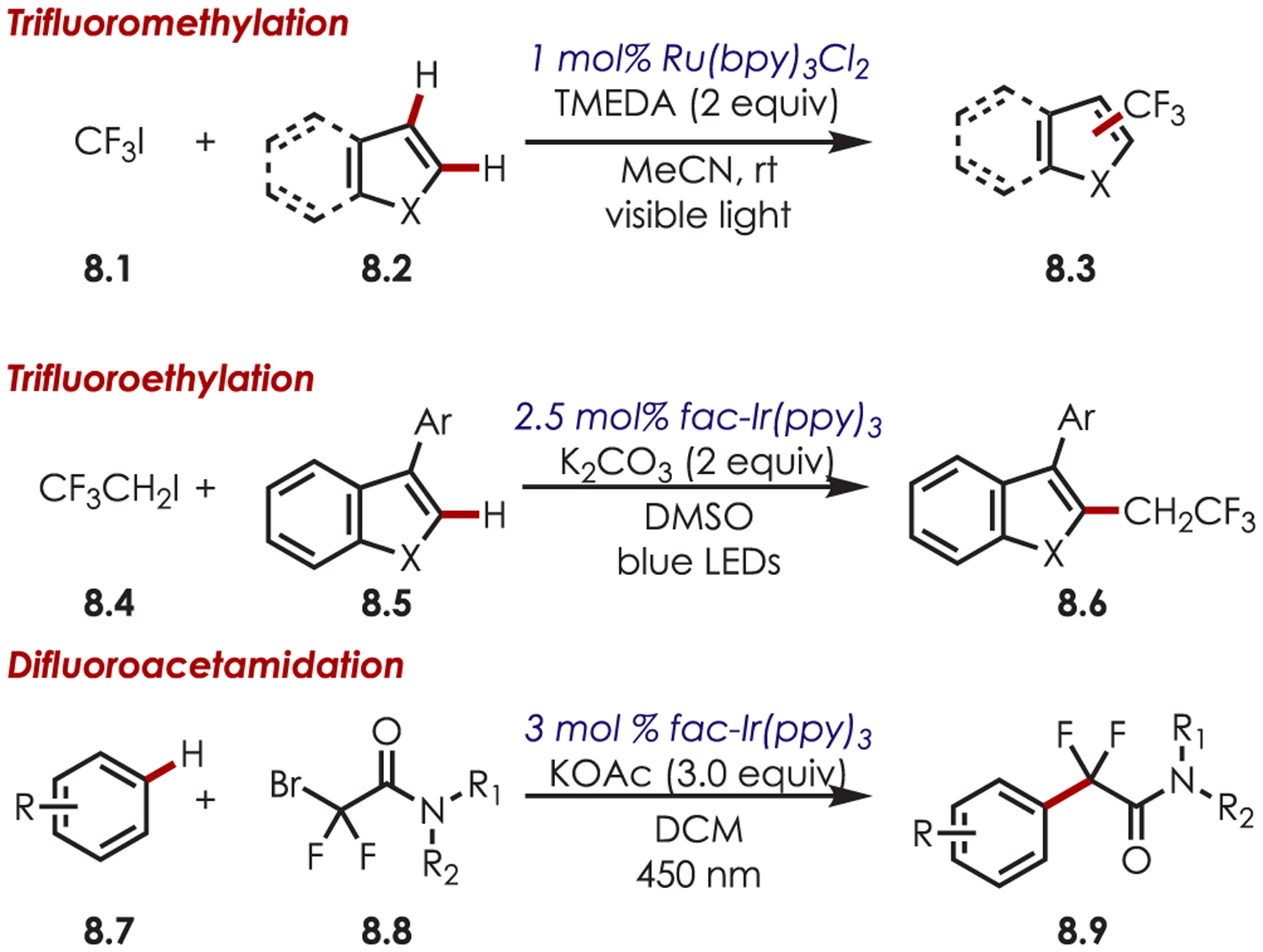
Heteroaryl C–H Fluoroalkylation
Accessed via Sulfonyl Chlorides.
Aryl C–H trifluoromethylation is an attractive transformation in medicinal chemistry, as the −CF3 group is known to have beneficial interactions with target proteins and enhanced metabolism.69,70 Nagib and MacMillan first reported a photoredox-catalyzed trifluoromethylation of alkyl, alkoxy or halogenated (hetero)arenes using trifluoromethanesulfonyl (triflyl) chloride (9.1) as the sp3-centered radical precursor.71 An SET from Ru(II)* (Eox* = −0.90 V vs SCE) to triflyl chloride (Ered = −0.18 V vs SCE) gives rise to a triflyl radical anion (9.2), which decomposes rapidly to generate a trifluoromethyl radical, SO2, and chloride anion (Scheme 9). The resulting electrophilic carbon-centered radical (9.3) can undergo addition to an electron-rich carbon of a (hetero)arene. The resultant cyclohexadienyl radical (9.4) is subsequently oxidized by Ru(III) and loss of a proton furnishes the trifluoromethylated arenes (9.5).
Scheme 9.
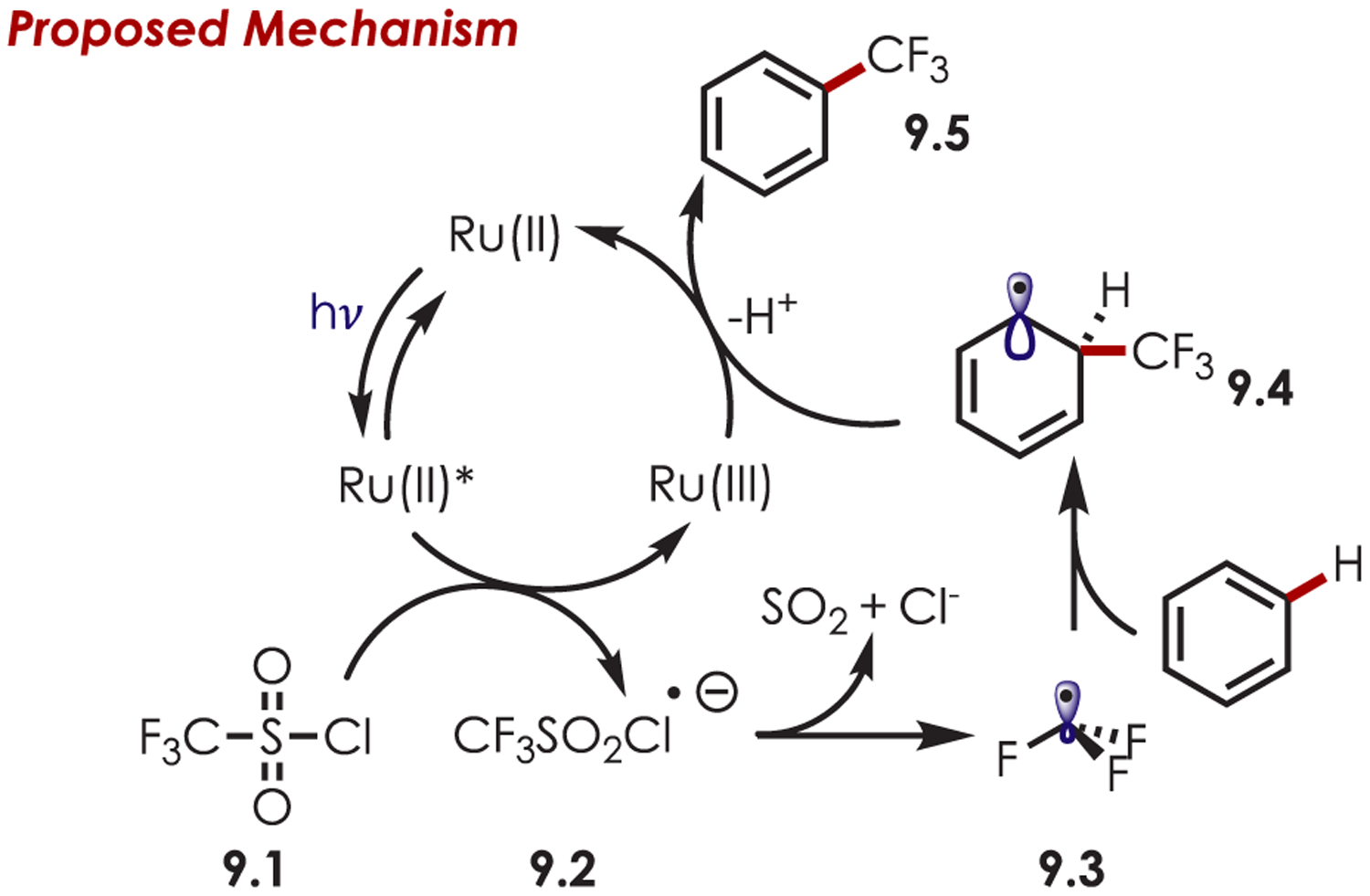
Mechanism of (Hetero)arene C–H Trifluoromethylation Using Sulfonyl Chlorides as Carbon-Centered Radical Precursors
Trifluoromethyl sulfonyl chlorides have also been demonstrated in the trifluoromethylation of five-membered heterocycles including pyrroles, furans, thiophenes, and thiazoles, which furnish the C2-trifluoromethylated products in excellent regioselectivities and yields (10.3–10.4) (Scheme 10). The high regioselectivity is rationalized by the stability of the radical and cationic intermediates, which are conjugated when functionalized at C2 but cross-conjugated when functionalized at C3. In addition to five-membered heterocycles, arenes such as anisoles, anilines, thioanisoles, and xylenes, as well as six-membered heterocycles (pyrazines, pyrimidines, pyridines, and pyrones), were efficient substrates. Six-membered rings yielded a mixture of regioisomers, with major substitution ortho- and minor para- to the electron-donating group on the arene.
Scheme 10.
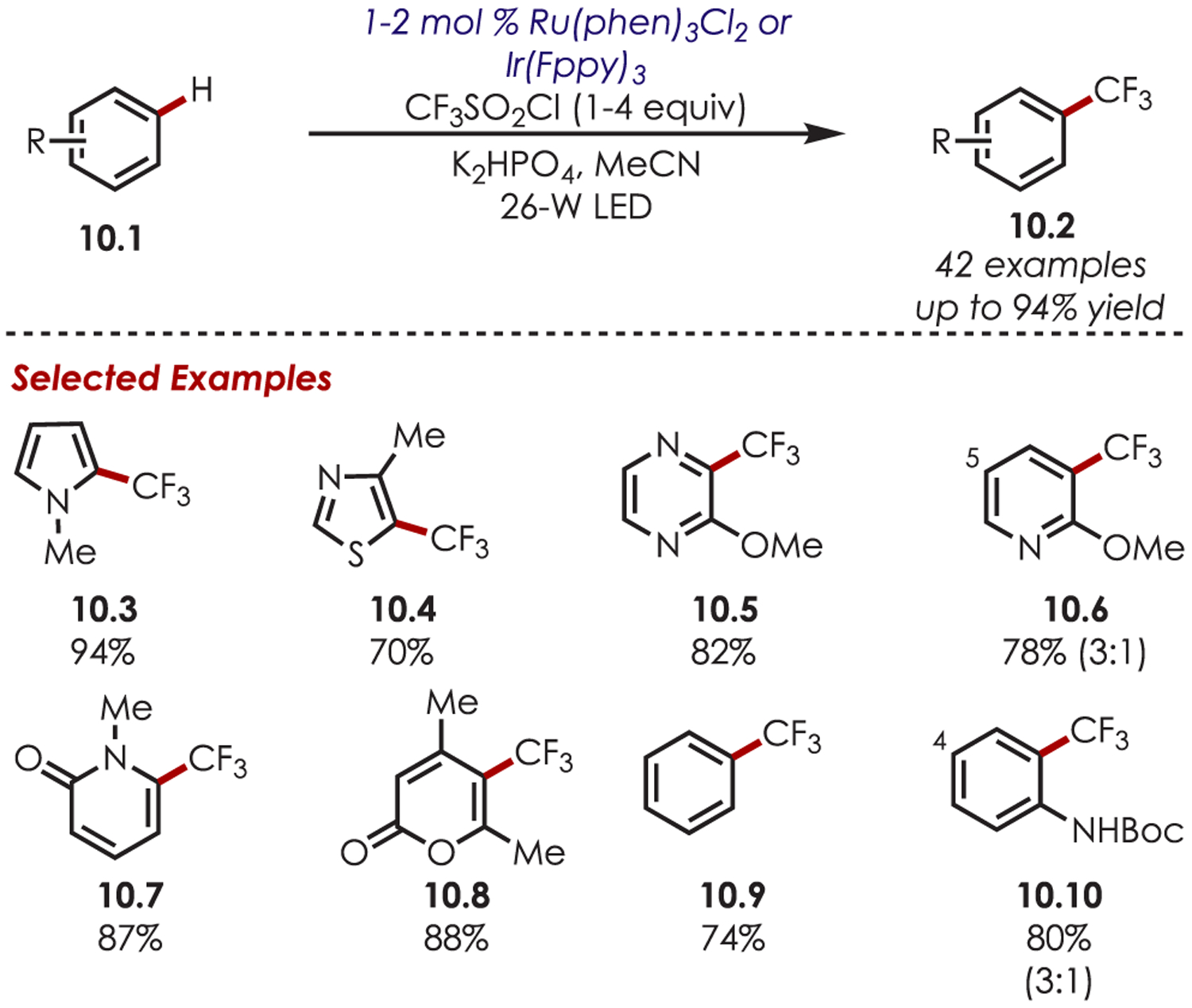
Scope of (Hetero)aryl C–H Trifluoromethylation Using Trifluoromethyl Sulfonyl Chloride
A radical cascade cyclization of ester arylpropiolates (11.1) initiated by a trifluoromethyl radical addition was later reported by Xiong and co-workers (Scheme 11).72 The proposed mechanism involved the addition of trifluoromethyl radical to the propiolate, furnishing an intermediate vinylic radical (11.3). A 5-exo cyclization generates cyclohexadienyl radical (11.4) which undergoes an ester migration, oxidation by Ru(III), and rearomatization by deprotonation to afford the 3-trifluoromethylcoumarin products (11.2).
Scheme 11.
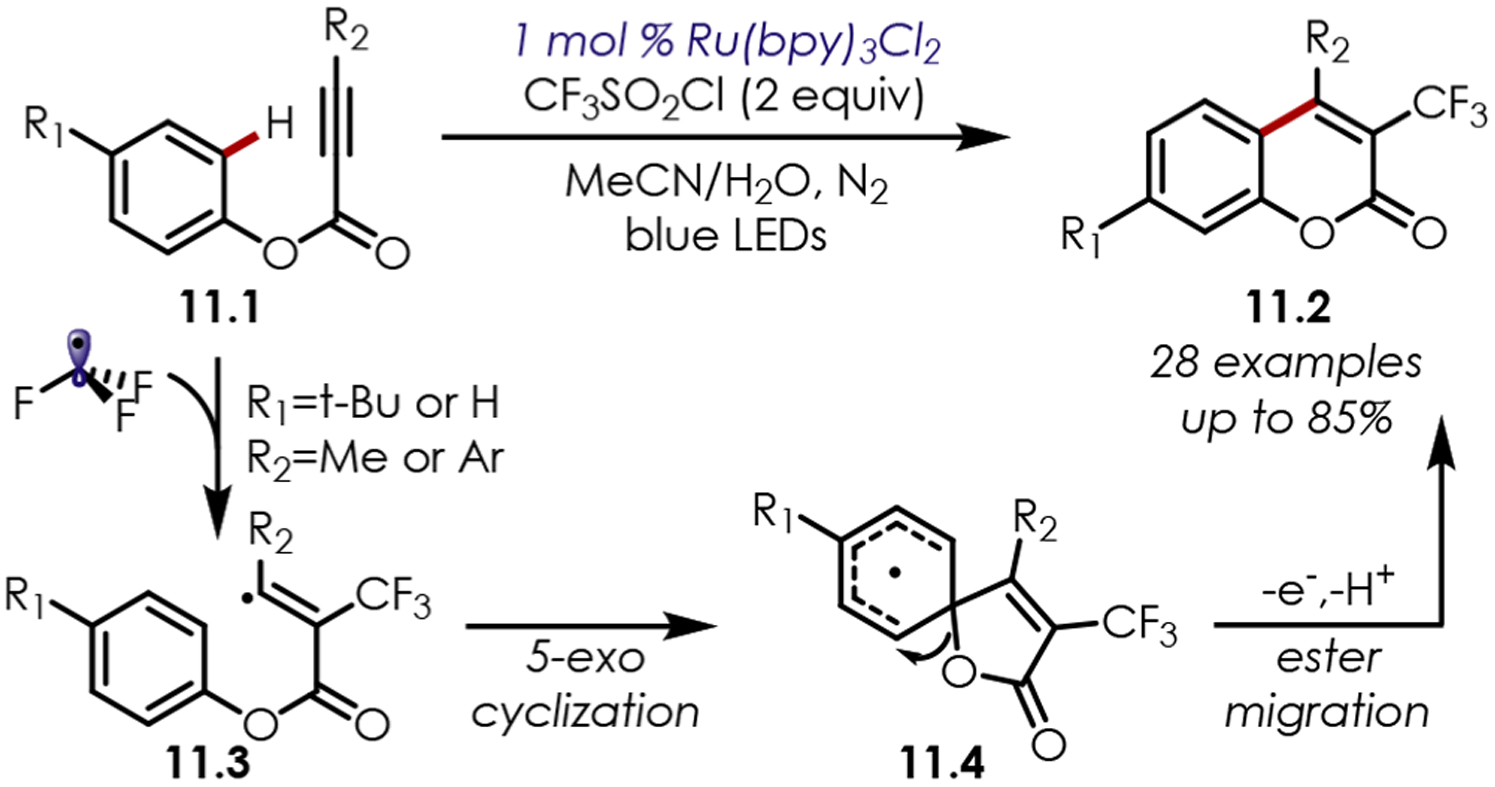
Trifluoromethyl Radical Addition/Cascade of Arylpropiolates for the Synthesis of Coumarin Derivatives
Accessed via Hydrogen Atom Transfer.
In 2014, MacMillan published a direct heteroarylation of ethers (12.2) through a photoredox-mediated Minisci-type C–H functionalization (Scheme 12).73 First, a PET between persulfate and Ir(III)* produces a persulfate radical anion (12.4). This proceeds to abstract the hydrogen atom adjacent to the oxygen atom, which generates the α-oxy radical (12.5). This key hydrogen atom transfer event grants access to ether-based α-oxy radicals, which are known to be relatively stable species.74 Such radicals are difficult to access through direct SET processes with photoredox catalysts due to their high oxidation potentials (e.g., THF, THP, and Et2O have Eox > +2.4 V vs SCE). A Minisci-type addition of the α-oxyalkyl radical (12.5) into the protonated heteroarene (12.6) generates an amine cation radical intermediate (12.7). After loss of a proton, the resulting α-amino radical (12.8) undergoes an oxidative SET with Ir(IV) to regenerate the catalyst and furnish the desired product. Electron-deficient heteroarenes, such as isoquinolines, quinoxaline, quinoline, pyridines, and pyrimidines, are compatible in this ether arylation. Both acyclic and cyclic ethers were successful radical precursors, including heterocyclic tetrahydropyran, tetrahydrofuran, oxetane, and dialkyl ethers. It is worth noting that Barriault later reported an identical Minisci-type addition with ethers that proceeds through the direct thermolysis of the persulfate to generate the HAT agent without requiring a transition-metal catalyst.75
Scheme 12.
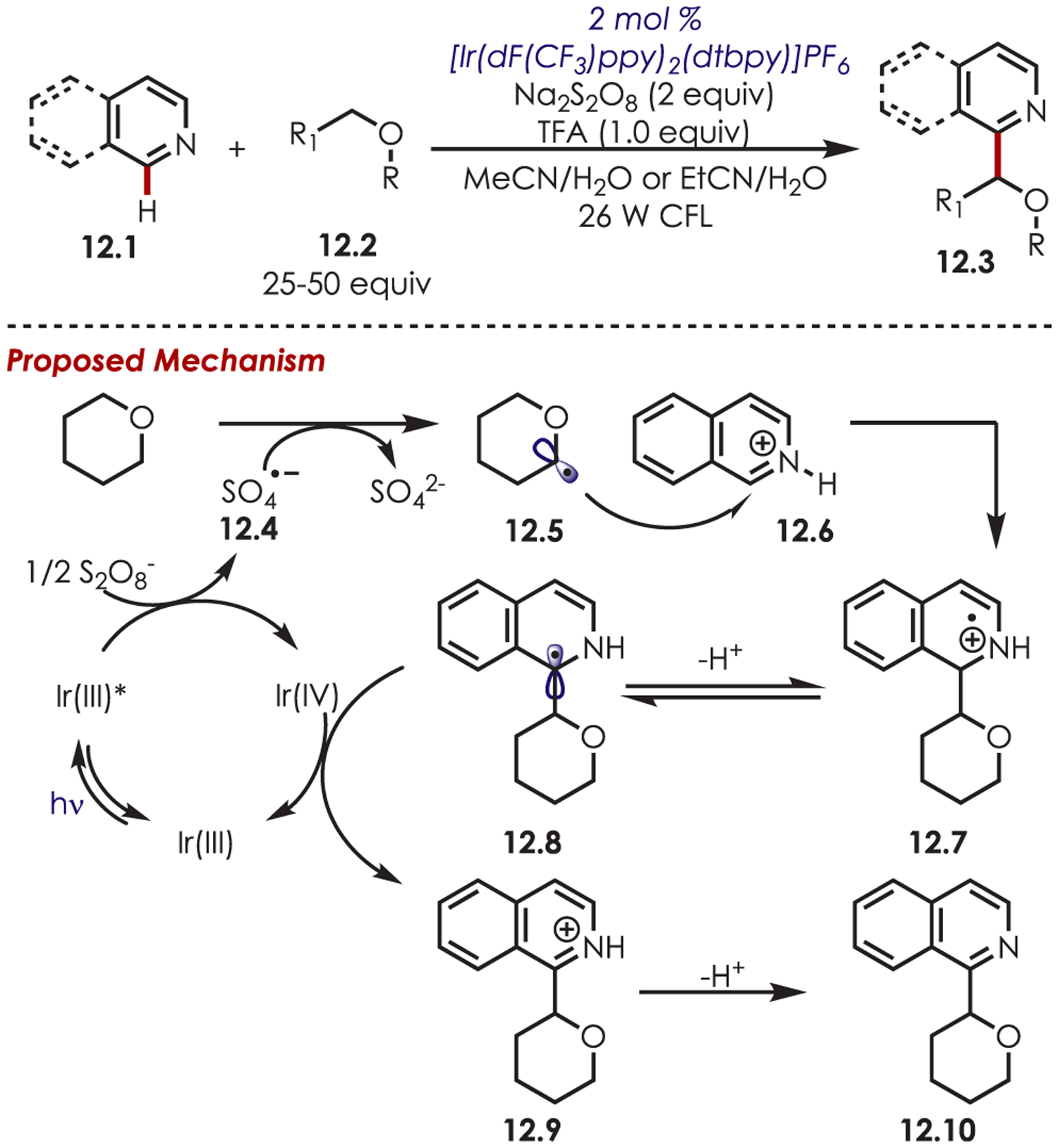
α-Heteroarylation of Ethers through the Generation of α-Oxy Radicals
The following year, MacMillan reported alcohols (13.2) as precursors to carbon-centered radicals for the C–H alkylation of heteroarenes (13.1) (Scheme 13).76 Under a Minisci-type pathway similar to the previous example, nucleophilic alkyl radicals add to electron-deficient heteroarenes to give a range of alkylated products (13.3). Simple alcohols (methanol and ethanol) as well as more complex alcohols were competent sp3 carbon-centered radical precursors. Cyclic ether derivatives could also be utilized; however, they undergo a radical ring opening to give the alkylated product with a pendent hydroxyl group.
Scheme 13.
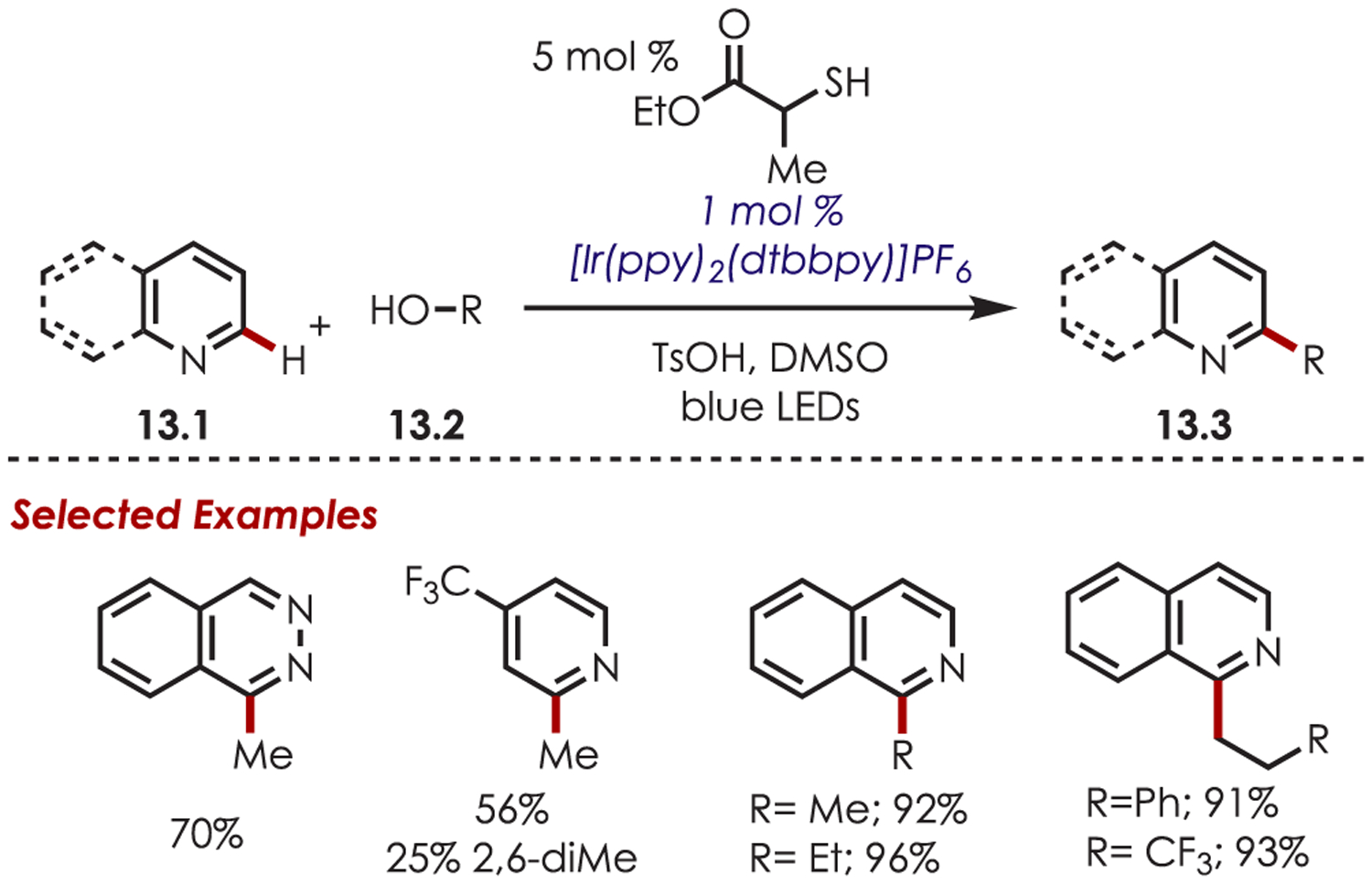
Minisci-Type C–H Functionalization Using Alcohols as Coupling Partners
This transformation varies mechanistically to that of the previous published ether arylation (Scheme 14). The authors performed Stern–Volmer analyses and found Ir(III)* is only quenched by the protonated heteroarene, which suggests an initial sacrificial oxidative quenching pathway to generate Ir(IV). The thiol 14.2 (Eox = +0.85 V vs SCE) is oxidized by Ir(IV) furnishing a thiyl radical (14.1) and regenerating Ir(III). A HAT event between the alcohol and thiyl radical produces an α-oxy carbon-centered radical (14.3) that undergoes nucleophilic addition into the protonated heteroarene. The resulting amine cation radical (14.4) is deprotonated and a spin-centered shift (SCS) elimination of water is proposed to occur to generate a benzylic radical (14.6). A final protonation and reduction by Ir(III)* furnishes the alkylated heteroarene.
Scheme 14.
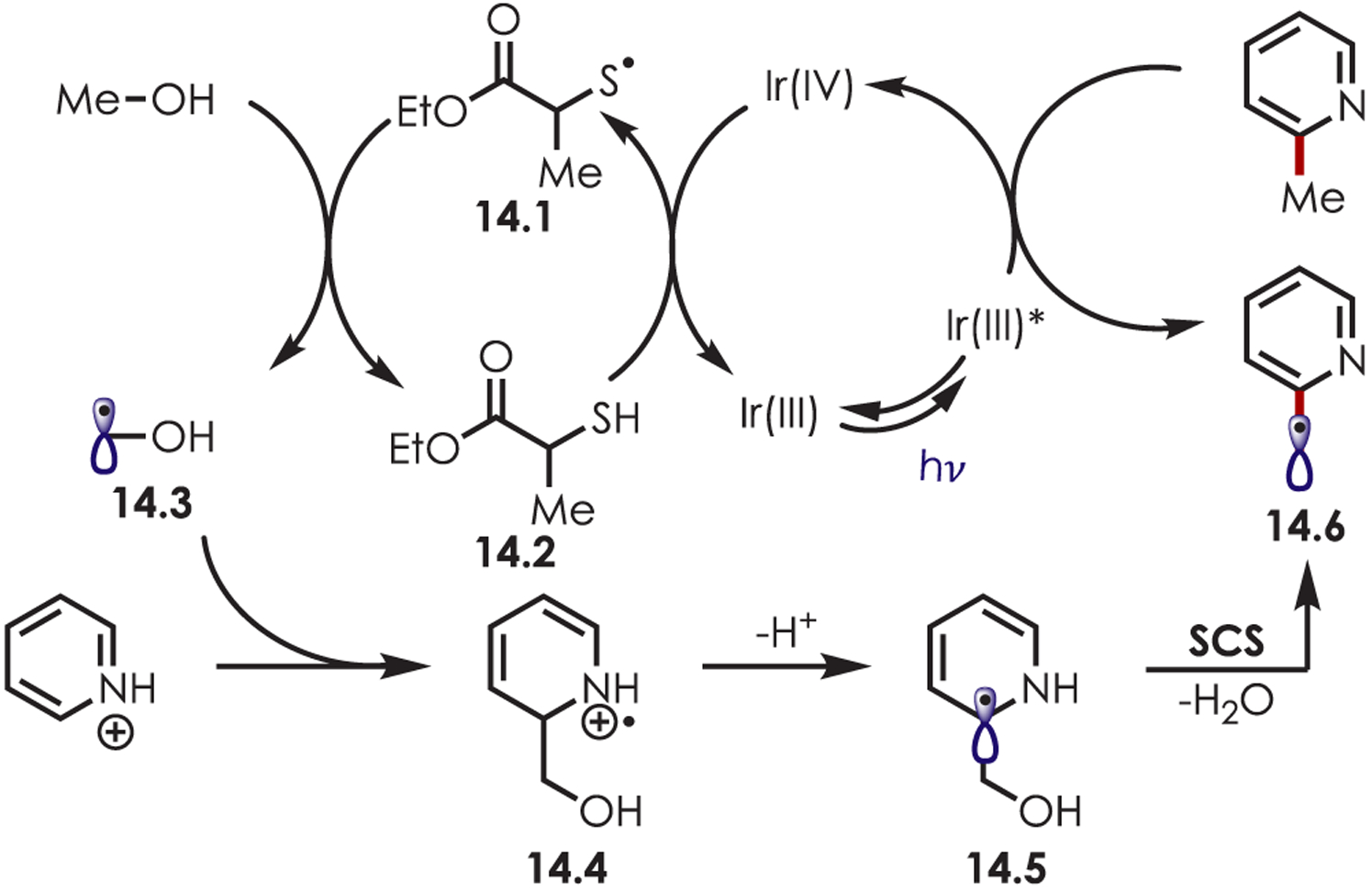
Mechanism of Minisci-Type C–H Functionalization Using Alcohols as Coupling Partners
The SCS is supported mechanistically through subjection of the hydroxymethyl intermediate (15.1) to net reductive conditions with the amine reductant and acid additives (Scheme 15). Parallel control experiments which included leaving out the photoredox catalyst, tributylamineformic acid, TsOH, or blue LEDs failed to produce significant product (<8%). With all additives present, the intermediate undergoes sufficient oxygen elimination furnishing the alkylated product in 60% yield. Additionally, a radical-trapping experiment with styrene gives the product (15.3) in 65% yield, which presumably arises from the formation of an intermediate β-amino radical. These results are suggestive of a SCS followed by reduction and protonation, but the authors note that a radical-chain pathway could not be ruled out.
Scheme 15.
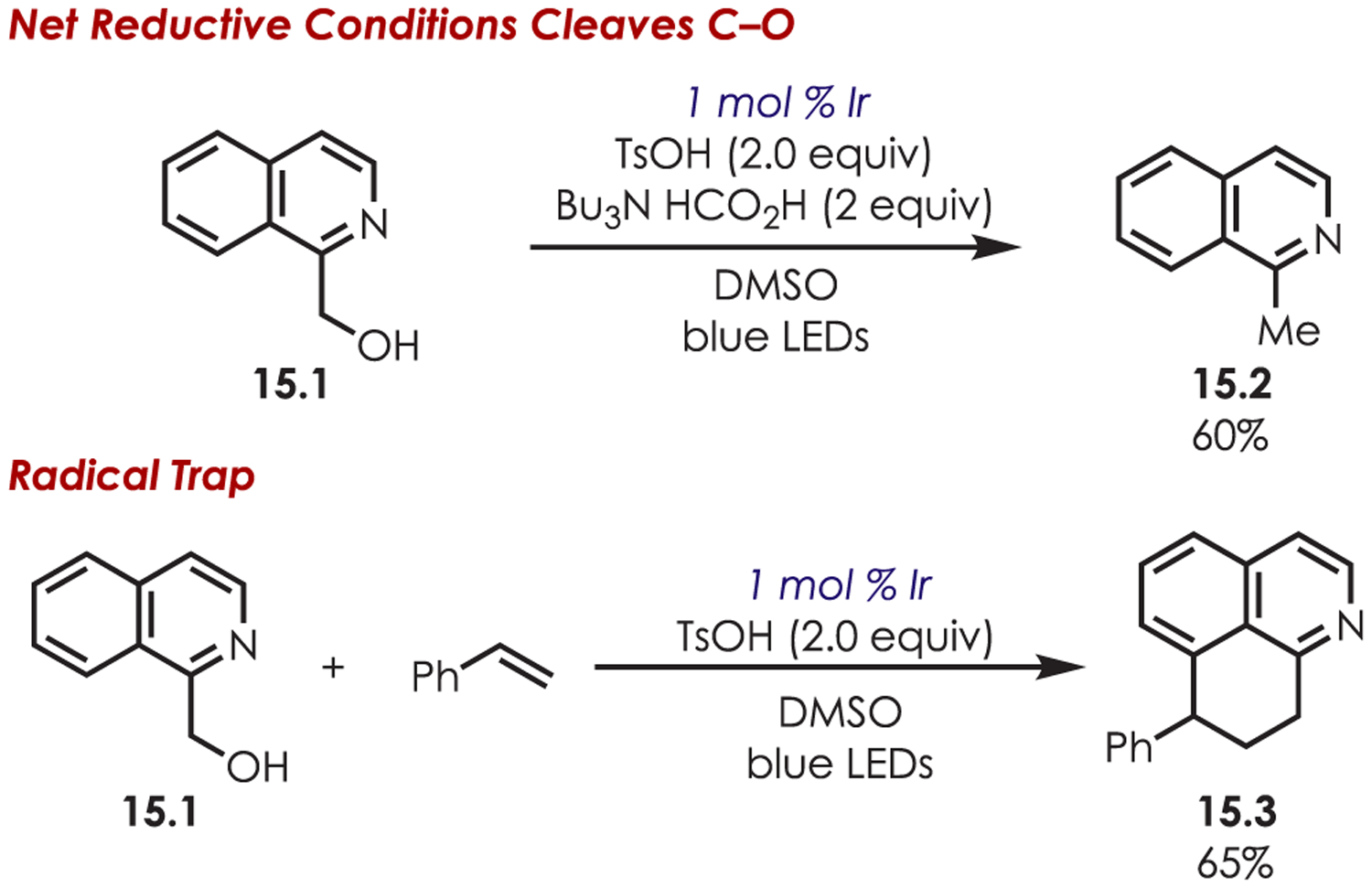
Mechanistic Support for a Spin-Centered Shift in the Minisci-Type C–H Functionalization Using Alcohols as Coupling Partners
In a similar transformation that builds on the MacMillan work, methanol has also been utilized as a hydroxymethylating reagent for a range of heteroarenes (16.1) in the absence of a thiol co-catalyst (Scheme 16).77 The hydroxymethyl radical species is generated from a hydrogen atom abstraction of methanol by a phenyl radical originating from benzoyl peroxide (BPO).
Scheme 16.
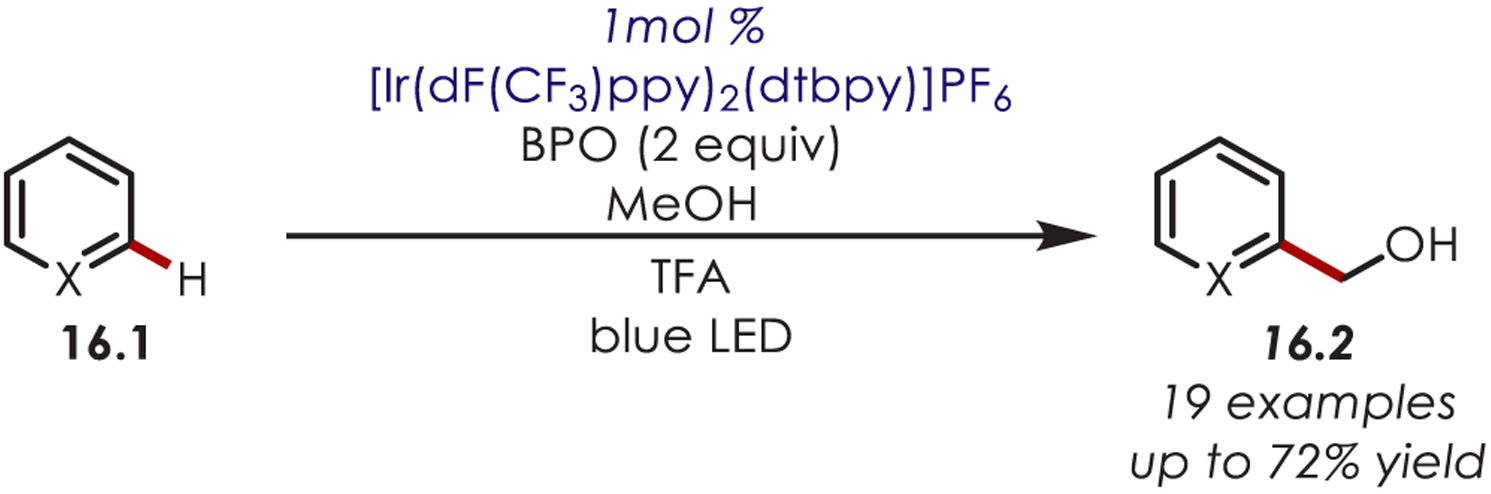
Hydroxymethylation of (Hetero)arenes
Accessed via Decarboxylation.
Carboxylic acids are a naturally abundant and commercially available alternative source of alkyl radicals. Glorius and co-workers have accessed sp3 carbon-centered radicals through a hydrogen atom transfer/decarboxylation sequence that was used in the functionalization of (hetero)arenes (Scheme 17).78 Reductive SET of persulfate with Ir(III)* generates a sulfate radical anion as the hydrogen atom transfer agent. A somewhat unusual mechanism involving the abstraction of the carboxylic acid hydrogen initiates a subsequent decarboxylation.79–81 Then the resulting carbon-centered radical undergoes Minsici reactivity with (hetero)arenes (17.1) to afford the alkylated products (17.3). Primary, secondary, and tertiary cyclic or acyclic carboxylic acids successfully underwent decarboxylation to generate the desired products. Amino and fatty acids were also found to be suitable radical precursors.
Scheme 17.
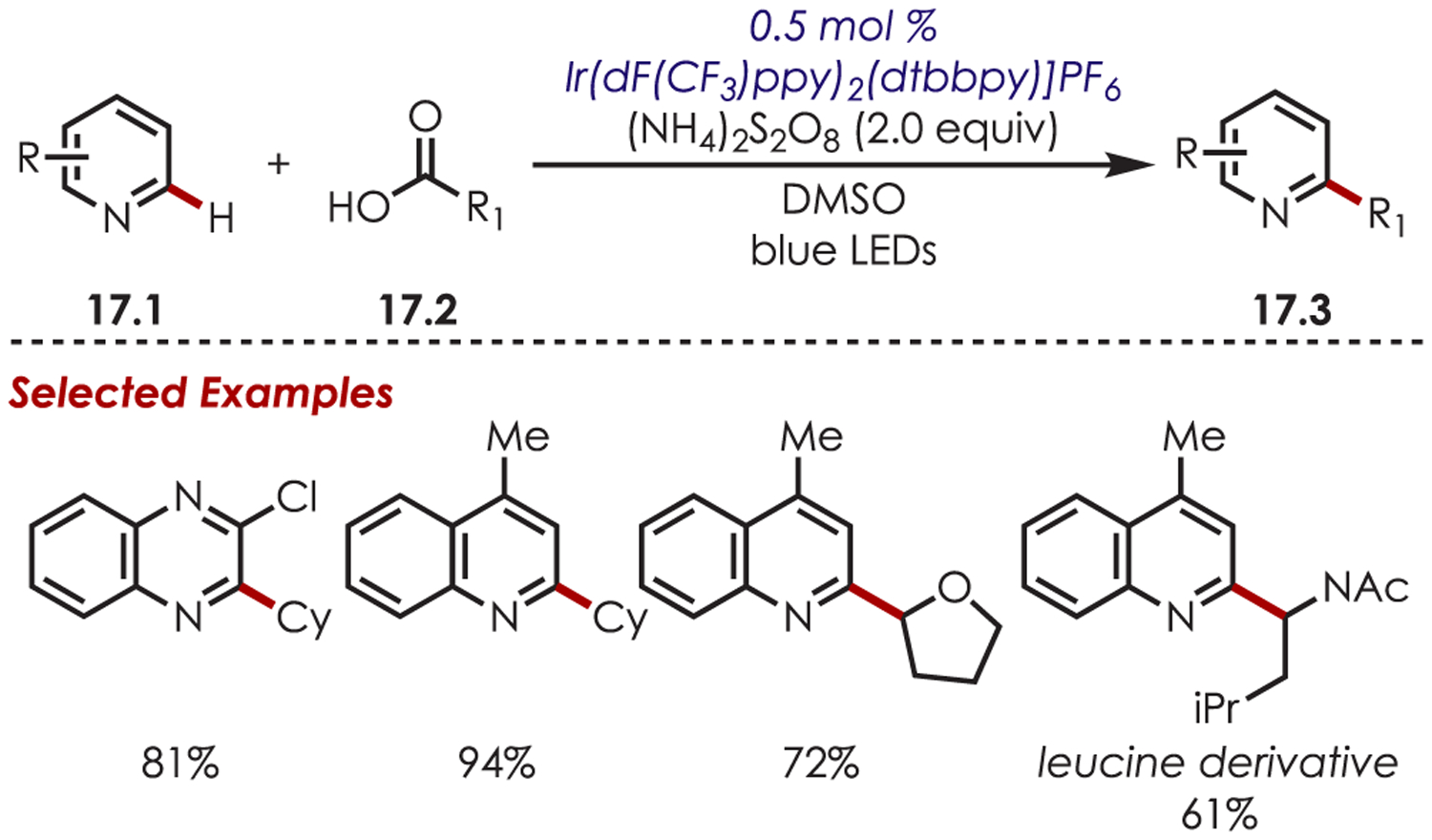
Heteroaryl C–H Functionalization Using Carboxylic Acids as Carbon-Centered Radical Precursors
A team at Merck led by DiRocco applied an analogous transformation for the late-stage functionalization of biologically active heterocycles (18.1) utilizing organic diacylperoxides (18.2) as alkylating reagents (Scheme 18).82 Pharmaceuticals and agrochemicals such as eszopiclone, diflufenican, fasudil, camptothecin, and fenarimol were alkylated in moderate to good yield as a mixture of the mono- and bis-alkylated products. Biscyclopropanecarbonyl peroxide was found to be another suitable carbon-centered radical source and afforded cyclo-propanated heteroarene products.
Scheme 18.
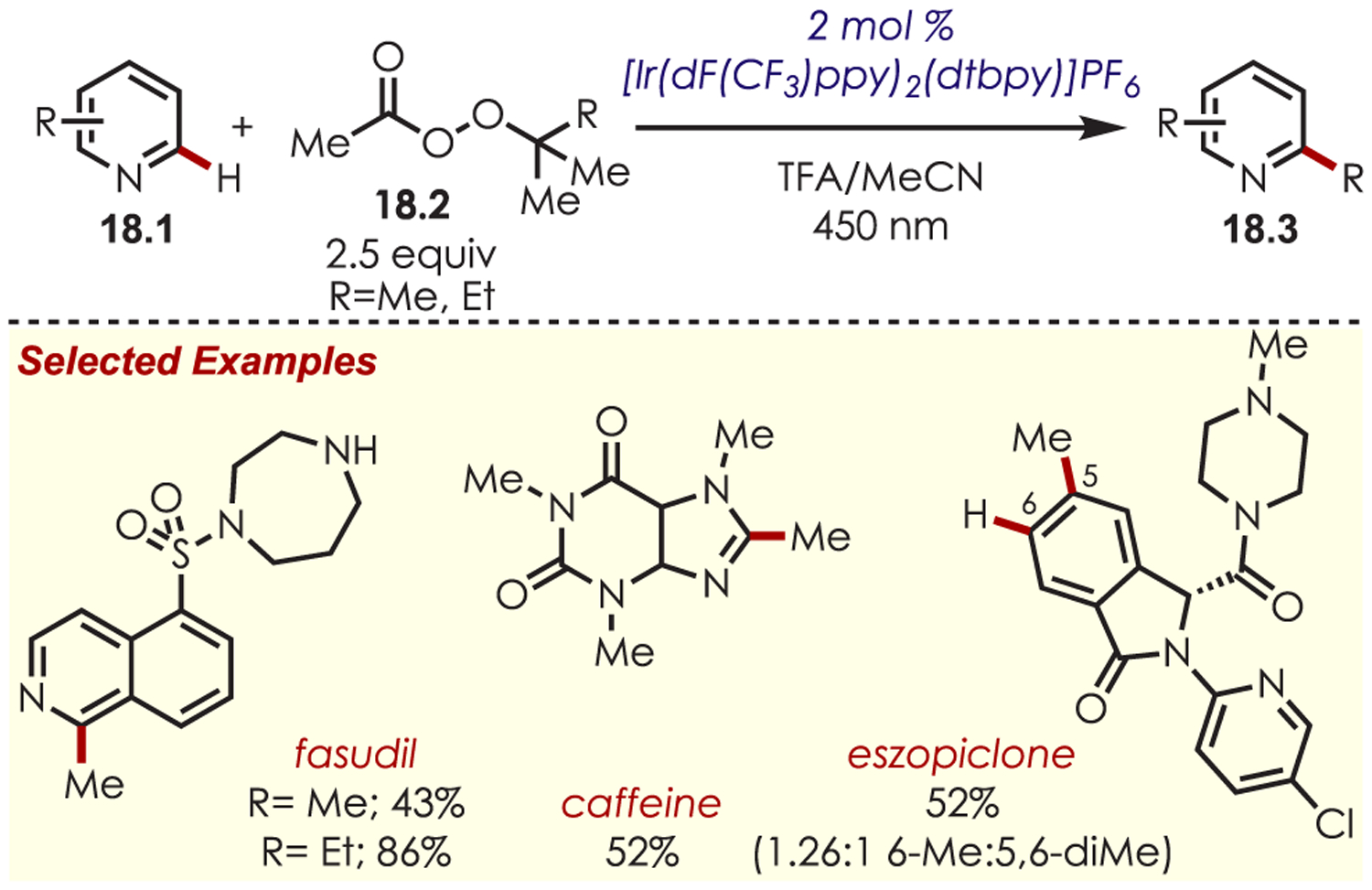
Peroxides Serving as Carbon-Centered Radical Precursors for a Minisci-Type C–H Functionalization
This transformation is proposed to proceed through a PCET of the peroxide (19.1) which overcomes the thermodynamic barriers of a direct reduction process (Ered = −1.95 V vs SCE; Eox*Ir(III) = −0.89 V vs SCE) (Scheme 19). After PCET, the unstable α-peroxy radical (19.2) decomposes to acetone and acetic acid while generating the key carbon-centered radical (19.3). A Minisci-type radical addition into the protonated heteroarene followed by oxidation with Ir(IV) regenerates the catalyst and affords the alkylated products.
Scheme 19.
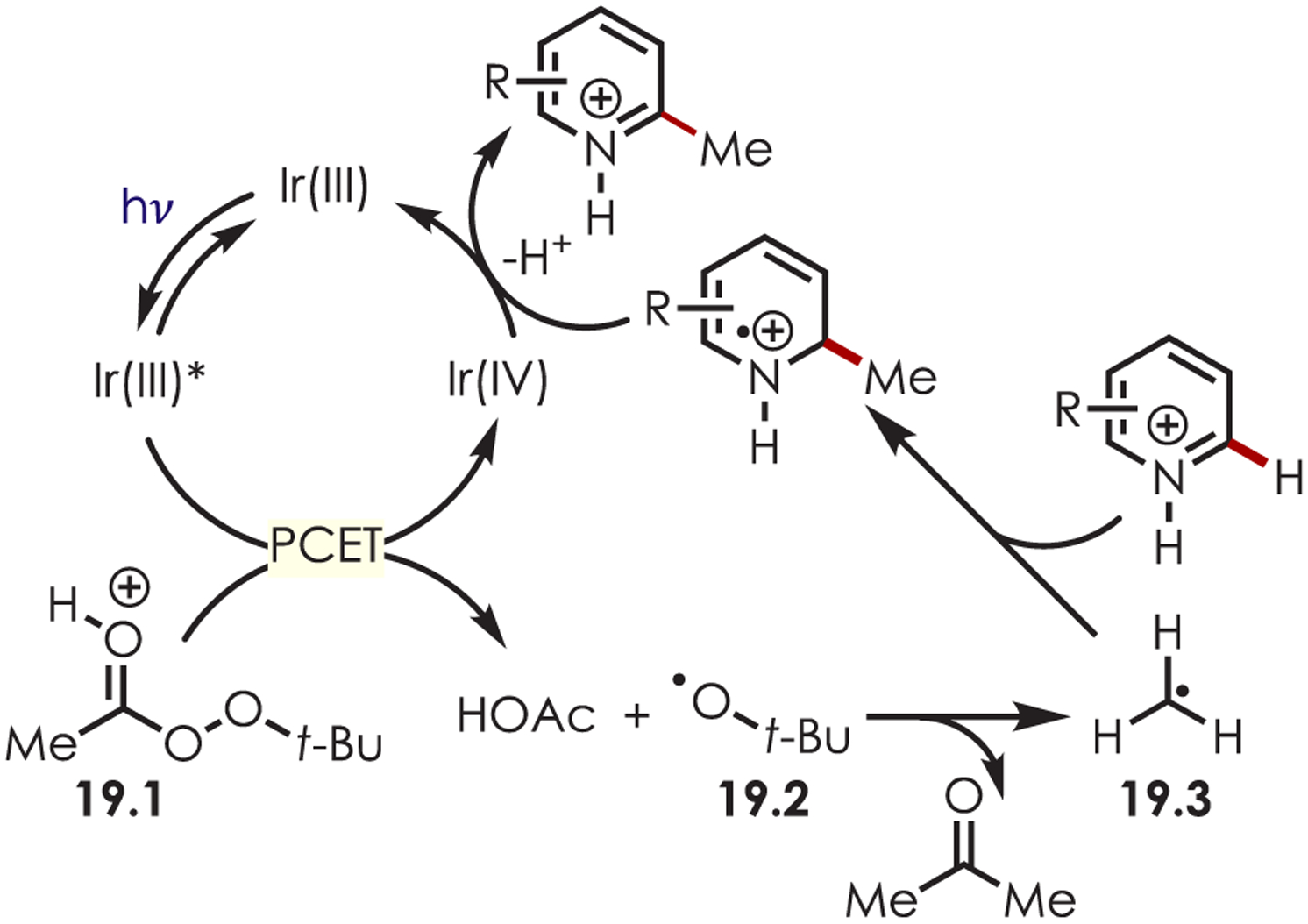
Mechanism for the Minisci-Type C–H Functionalization Using Acylperoxides as Radical Precursors
A decarboxylative C–H functionalization was reported by Zhu in 2013 for the synthesis of oxindoles, 20.3 (Scheme 20).83 Reductive SET between Ir(III)* and PhI(OAc)2 generates a phenyl iodine radical that triggers decarboxylation. The alkyl radical then adds to the olefin and initiates an intramolecular C–H cyclization.
Scheme 20.
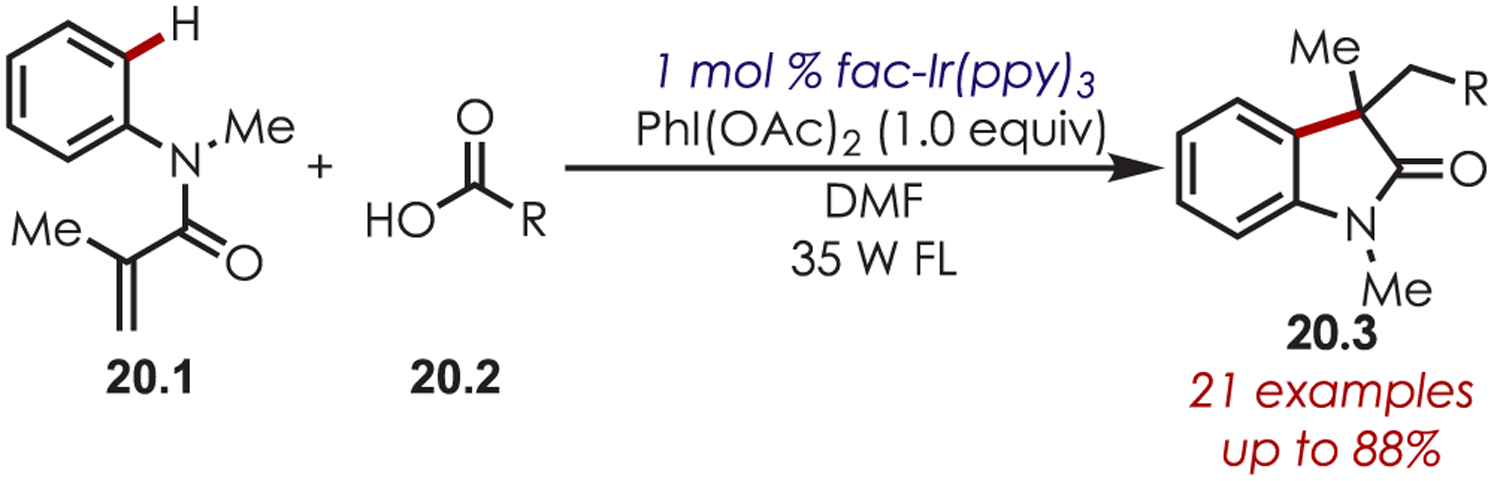
Decarboxylative C–H Functionalization for the Synthesis of Oxindoles
A decarboxylative trifluoromethylation of (hetero)arenes using iodine(III) trifluoroacetates (21.2) and C6F5I-(OCOCF3)2 as the trifluoromethyl radical source has been reported with a ruthenium photoredox catalyst (Scheme 21).84 The scope of the transformation is similar to that of the aforementioned (hetero)arene trifluoromethylation reactions and perfluoroalkylation is also possible with the requisite iodine(III) reagent.
Scheme 21.
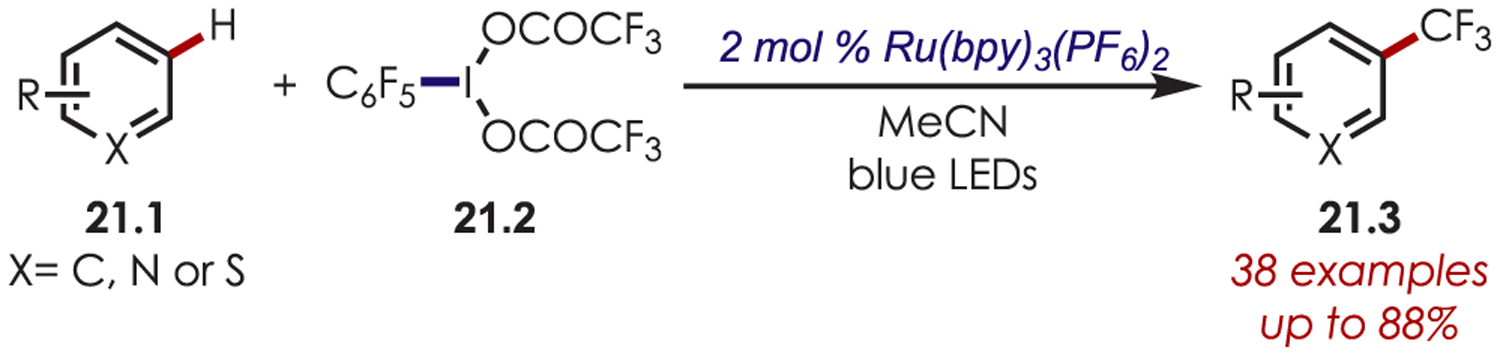
C–H Trifluoromethylation with Perfluoroarene Iodine(III) Trifluoroacetates as the Trifluoromethyl Radical Source
A Minisci radical alkylation with α-amino radicals was reported using dual Brønsted acid and photoredox catalysis (Scheme 22).85 The addition of α-amino radicals in this fashion has remained challenging due to overoxidation of the radical to the iminium ion (Eox = −1.03 V and +1.15 V vs SCE for the α-amino radical and starting amine, respectively).86,87 Other Minisci-type transformations typically proceed via Ir(III)/Ir(IV) cycles; however, an Ir(III)/Ir(II) cycle and the corresponding redox potentials were found to play a key role in regulating the oxidation of the intermediate radical, thus preventing the formation of the undesired iminium ion. Oxidation of Ir(II) (E1/2 [Ir(III/II)] = −1.37 V vs SCE) by N-acyloxy phthalimide, 22.2 (E1/2 = −1.26 to −1.37 V vs SCE), yields Ir(III). A long-lived triplet excited state of Ir(III) is proposed to selectively oxidize the intermediate cation radical, which subsequently rearomatizes via deprotonation. The scope of this chemistry includes (iso)quinolines, quinoxaline, phthalazine, phenanthridine, pyrimidine, and quinazoline with a broad scope of both natural and unnatural amino acids and peptides. This methodology was utilized in the late-stage functionalization of pharmaceutical derivatives including, caffeine, fasudil hydrochloride, and famciclovir.
Scheme 22.
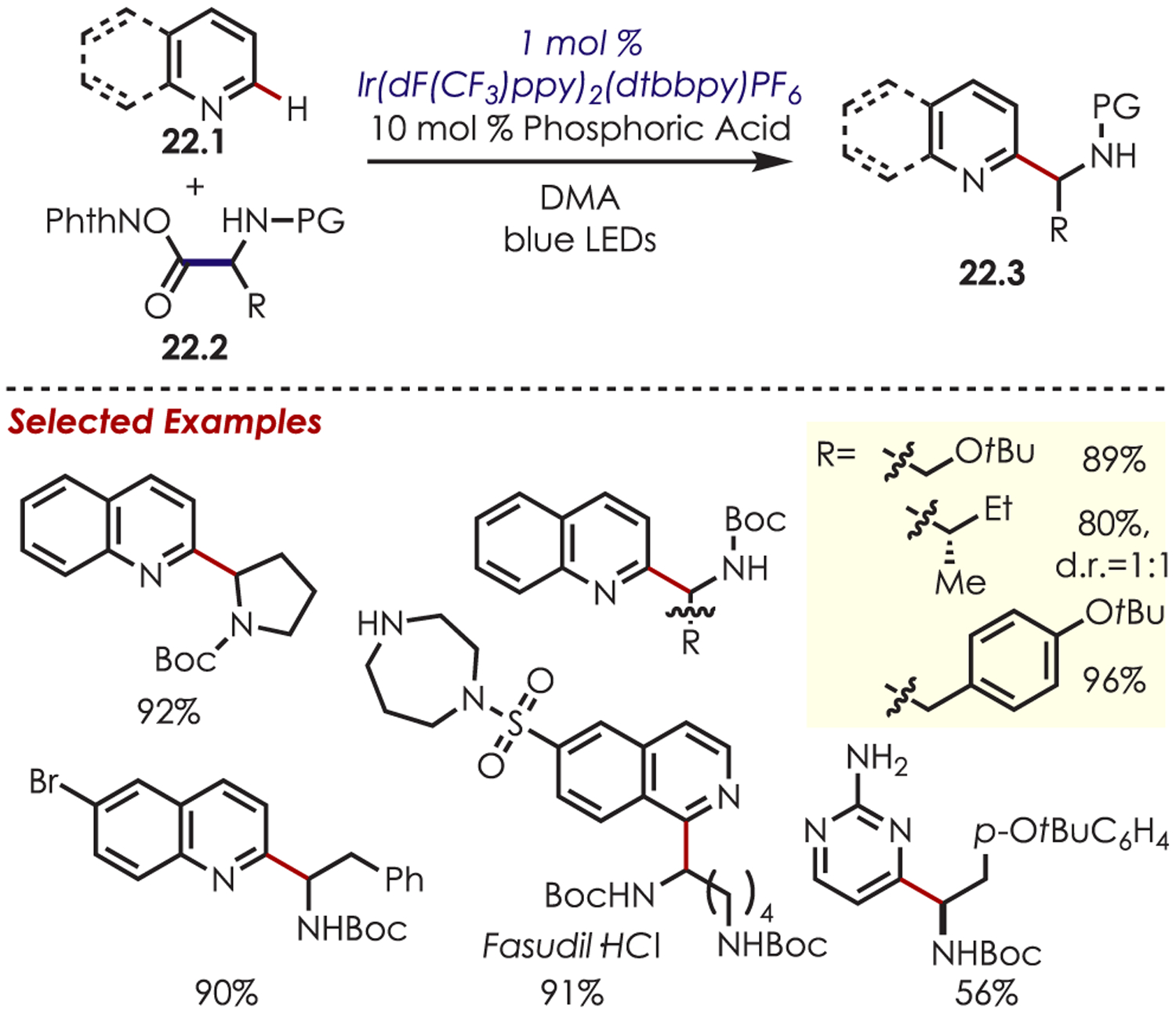
Addition of α-Amino Radicals in a Minisci-Type C–H Functionalization Reactions
One year later, Phipps et al. reported an enantioselective version of the Minisci-type radical addition of α-amino radicals via asymmetric Brønsted acid catalysis (Scheme 23).88 The selectivity of this alkylation relies on electrostatic and hydrogen-bonding interactions between the protonated heteroarene and the conjugate anion of a chiral phosphoric acid (23.1). This association creates a chiral environment while activating the pyridine for radical addition. A second hydrogen bonding interaction between the nucleophilic radical and the chiral phosphoric acid provides a defined ternary transition state, allowing for radical addition to occur with high enantioselectivity (23.2). A primary kinetic isotope effect of 3.6 was observed in a competition experiment between quinoline and quinolone-d7. This, along with other mechanistic experiments, suggests that radical addition may be reversible and deprotonation is product determining. Thus, the relative energies of the diastereomeric intermediates and barriers for the corresponding deprotonations are responsible for the observed enantioselectivity.
Scheme 23.
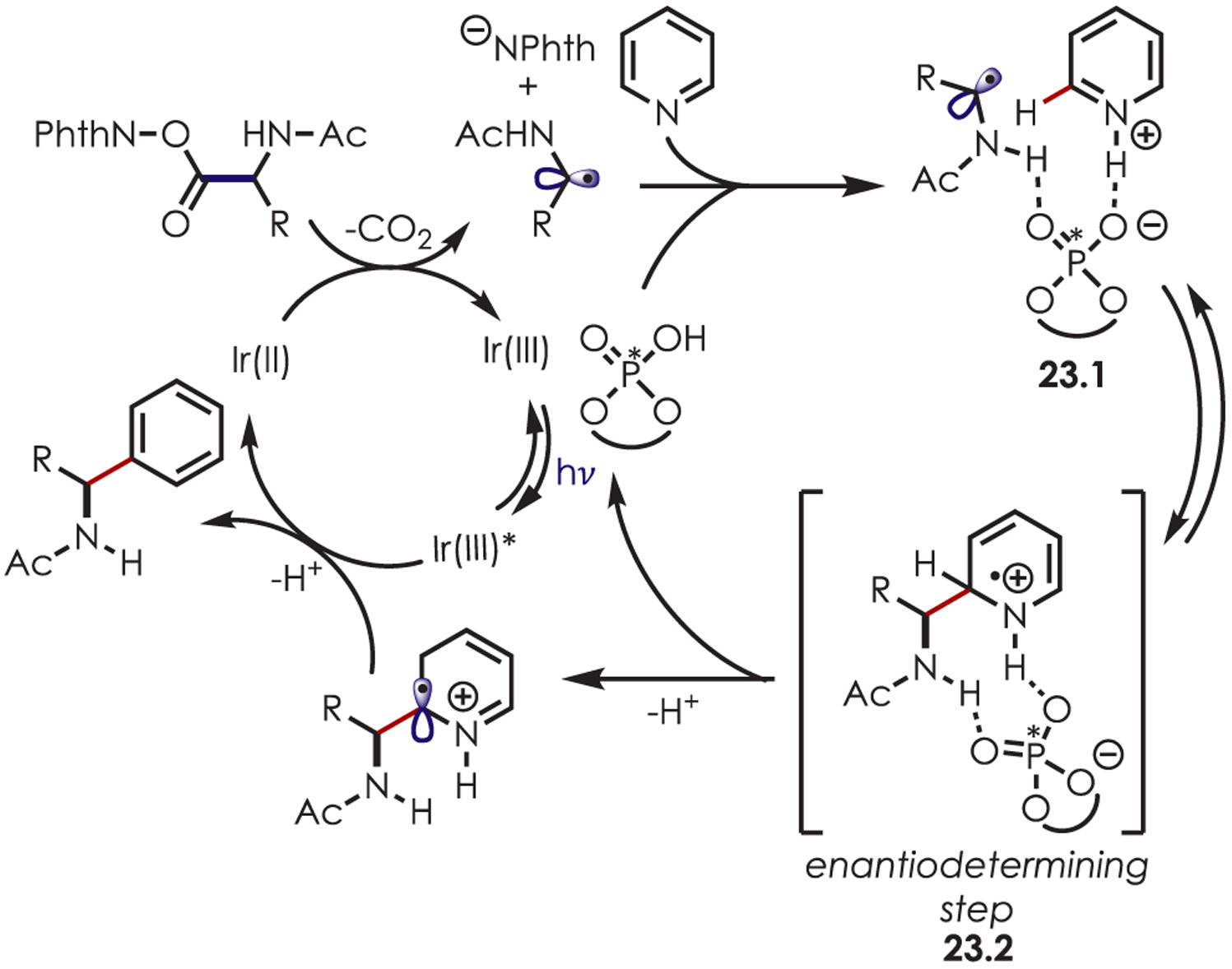
Mechanism for an Asymmetric Minisci-Type Radical Addition of α-Amino Radicals
A variety of redox-active esters (24.2) were suitable α-amino radical precursors (Scheme 24). Notably, the stereochemistry of the starting amino acid did not influence the enantiomer of the product. Quinolines demonstrated high C2 regioselectivity (>20:1) that was attributed to a combination of solvent effects, the N-Ac protecting group and the TRIP co-catalyst. Pyridines were also suitable coupling partners; however, they required electron-withdrawing substituents for efficient radical addition. This methodology was demonstrated in the late-stage functionalization of pharmaceutical derivatives: etofibrate, clofibrate, and niacin.
Scheme 24.
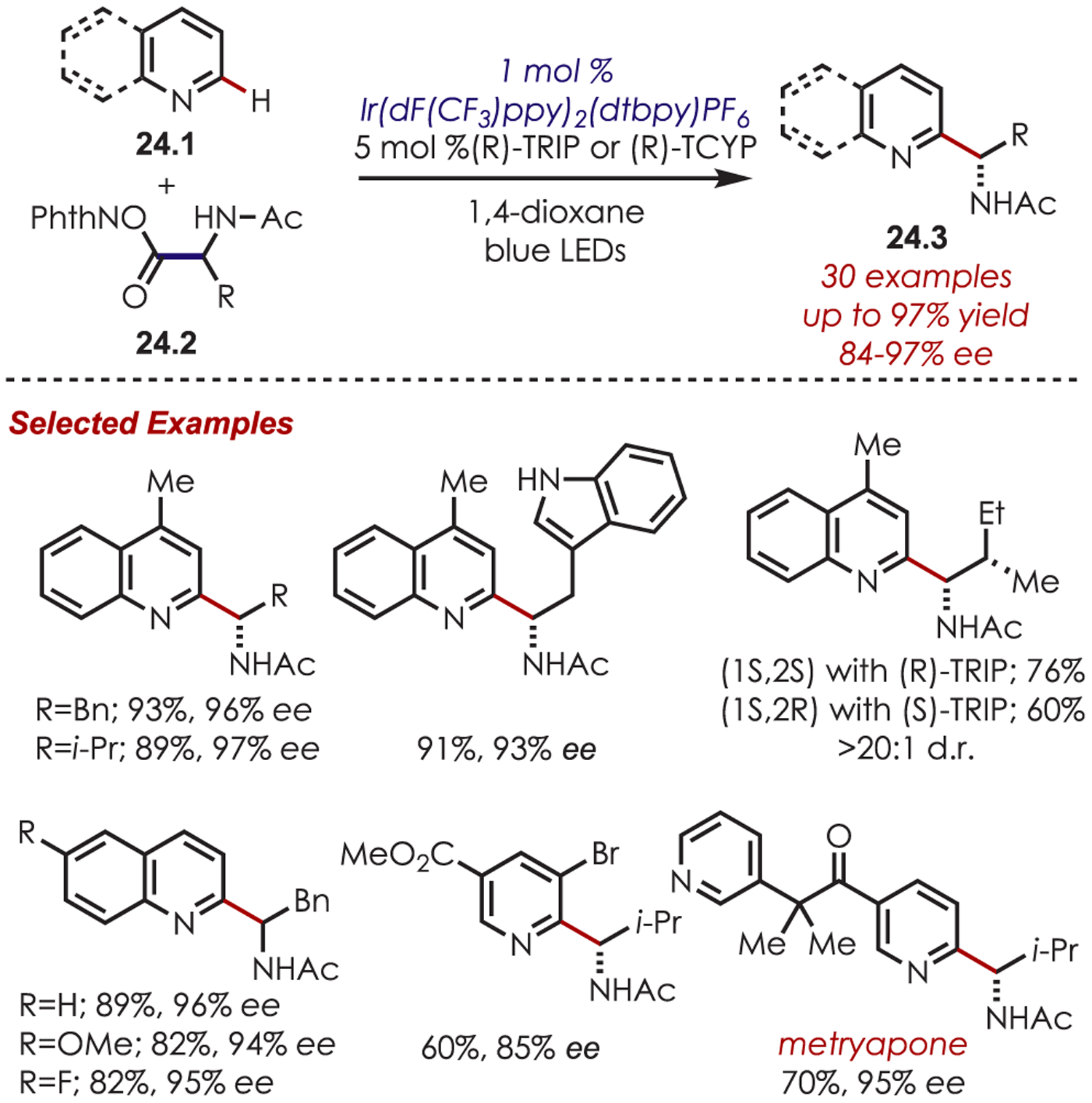
Scope of the Asymmetric Minisci-Type Radical Addition of α-Amino Radicals
2.1.2. Transformations Involving Carbon sp2-Centered Radicals.
Accessed via Diazoniums.
Photoredox catalysis has been a popular method for accessing aryl radicals from the corresponding diazonium salts to achieve Meerwein arylation products.89 PET between a catalyst and an aryldiazonium salt, 25.1 (Ered = −0.16 V vs MeCN),90 liberates nitrogen gas and produces a reactive aryl radical species, 25.2 (Scheme 25). This combines with an aromatic or heteroaromatic substrate and produces a cyclohexadienyl radical intermediate (25.4). A final oxidation via SET and deprotonation yields the desired C–H arylated products (25.5).
Scheme 25.
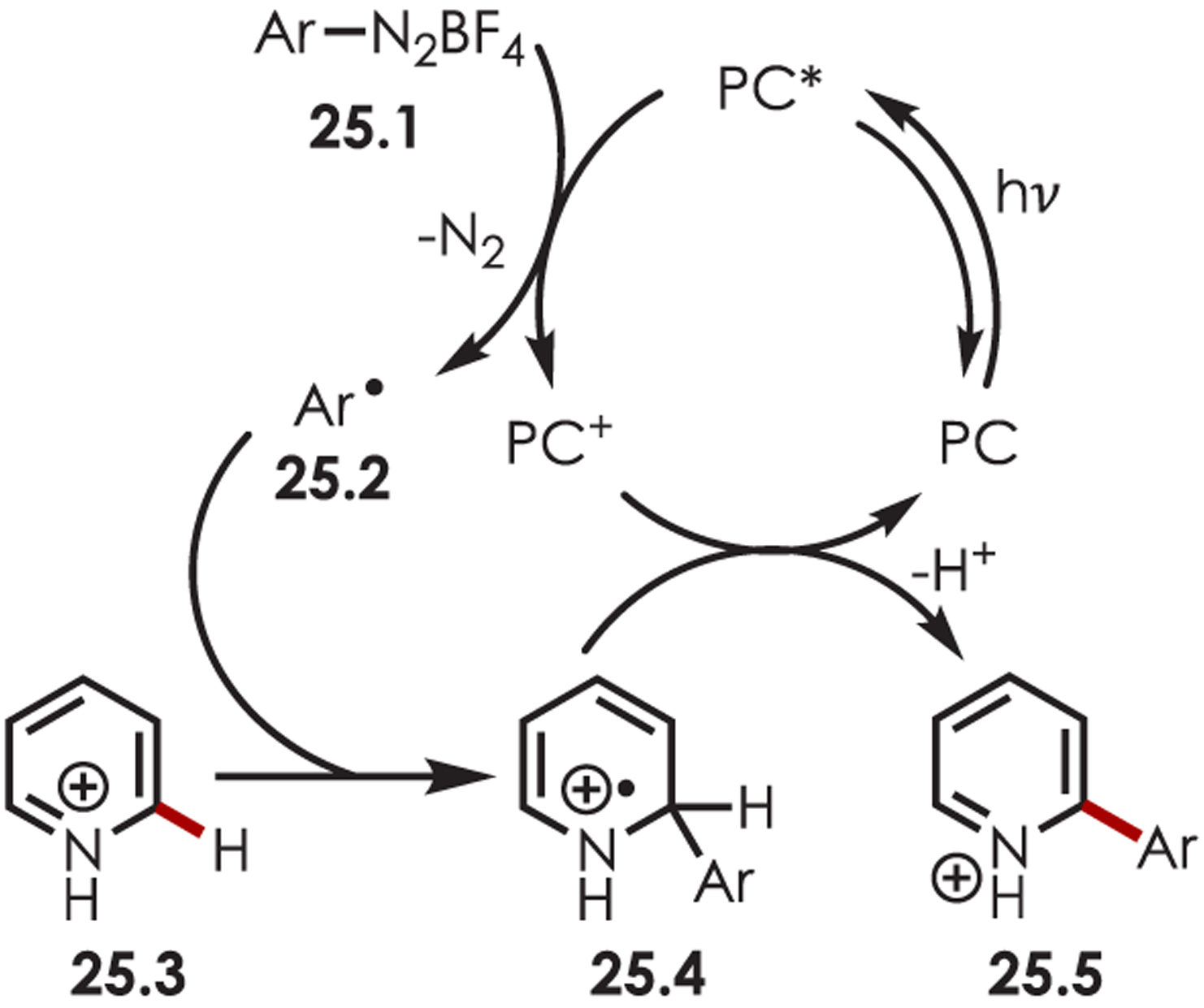
General Mechanism for the C–H Functionalization of Heteroarenes by Aryl Radicals Accessed via Aryldiazoniums
Cano-Yelo and Deronzier initially reported aryl radical generation from diazonium salts (26.1) with photoexcited Ru(bpy)32+.91 The aryl radical underwent an intramolecular Pschorr cyclization on a neighboring aryl C–H to form phenanthrene carboxylic acid derivatives in quantitative yields (Scheme 26). The primary mechanism was proposed as an electron transfer and was supported with quenching and flash photolysis experiments. The authors noted that direct photolysis in the absence of the catalyst gave poor yields of the cyclized product (<20%), suggesting a PET was involved. Later, the authors reported an intramolecular cyclization for the synthesis of fluorenone and fluorene (26.4) in poor yields. The rigidity of these systems was attributed to the low yields observed.92
Scheme 26.
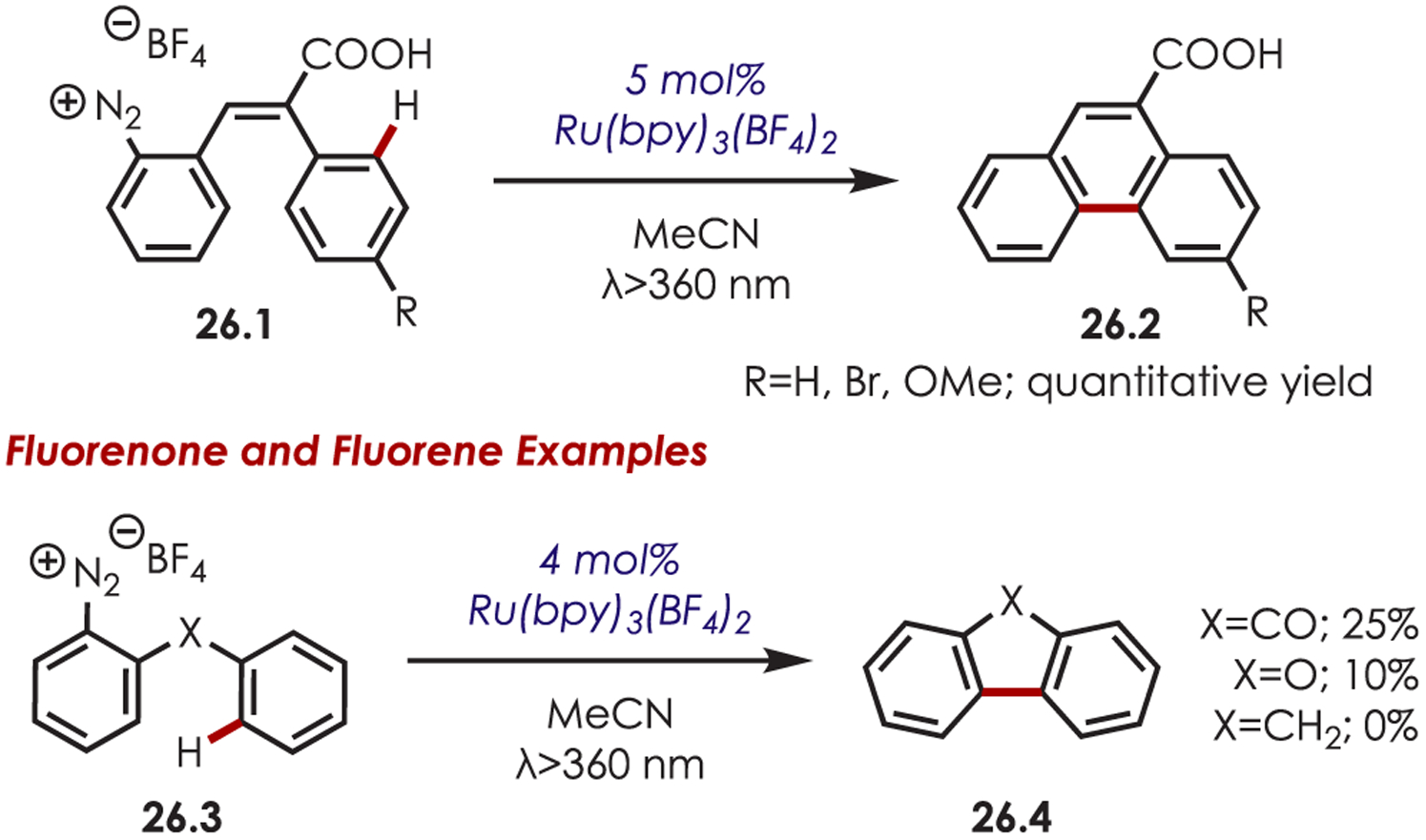
Early Reports of C–H Cyclization through Aryl Diazoniums
Deronzier’s seminal work using Ru(bpy)3Cl2 for the generation of aryl radicals from aryldiazoniums was not further explored in C–H functionalization until 2011. Sanford and co-workers employed this approach for generating aryl radicals in a dual palladium/photoredox-catalyzed intermolecular C–H functionalization reaction (Scheme 27).93 Methyl or alkoxy arenes (27.1) with a nitrogen-based directing group gave C–H arylation ortho to the directing group. A range of directing groups were compatible, including pyridines, amides, pyrazoles, pyrimidines and oxime ethers. Aryldiazonium salts with alkyl, chloro, fluoro, or trifluoromethyl substitution gave the products in 57–87% yield (27.2). Furthermore, an Ir(ppy)2(dtbbpy)PF6-catalyzed o-C–H arylation using pyrrolidinones as the directing group was reported by Sanford and Neufeldt.94
Scheme 27.
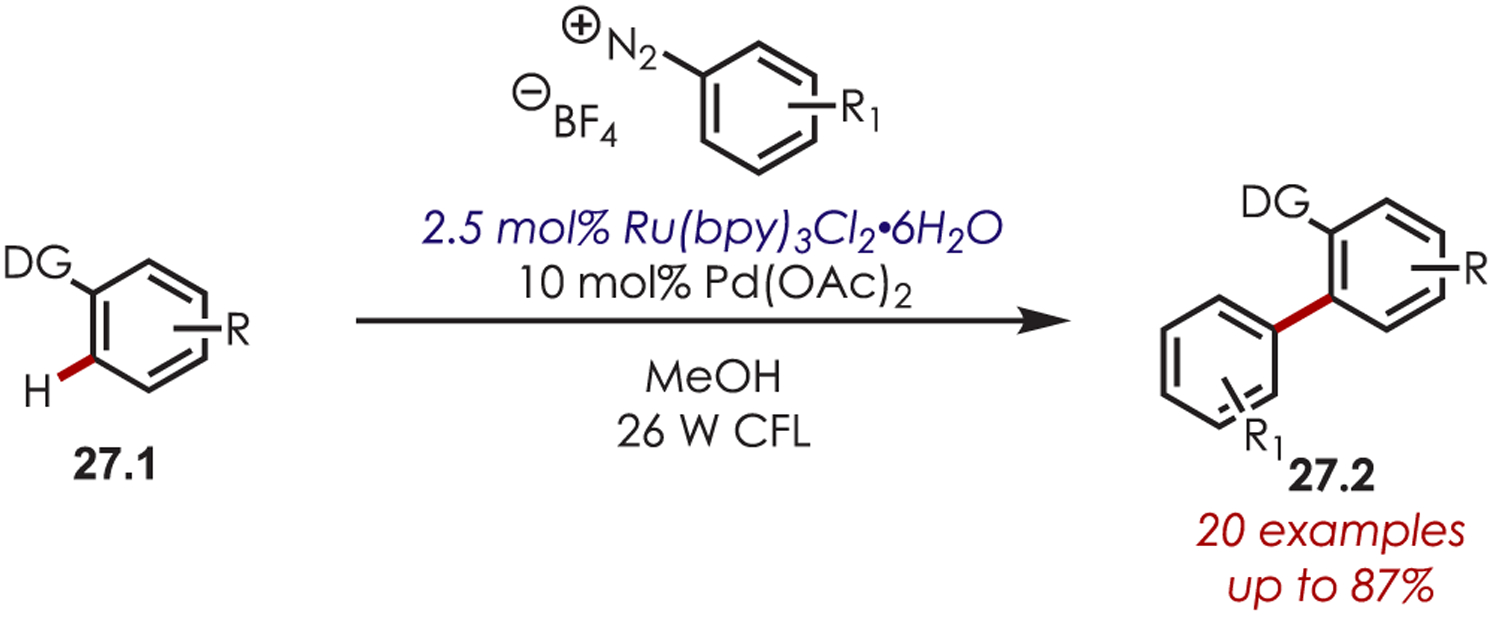
Dual Palladium-Photoredox-Catalyzed Intermolecular C–H Functionalization Using Aryl Diazoniums
An aryl radical (28.1) is generated by an ET from the photoexcited Ru(II)* to aryl diazonium (Scheme 28). A palladium(II) complex, which is preassociated with the aryl directing group, intercepts the phenyl radical giving rise to a putative Pd(III) intermediate (28.2). The resulting aryl Pd(III) species undergoes SET with Ru(III) to form a Pd(IV) intermediate (28.3) while simultaneously regenerating the photoredox catalyst. The product is formed via a reductive elimination and regenerates Pd(II).
Scheme 28.
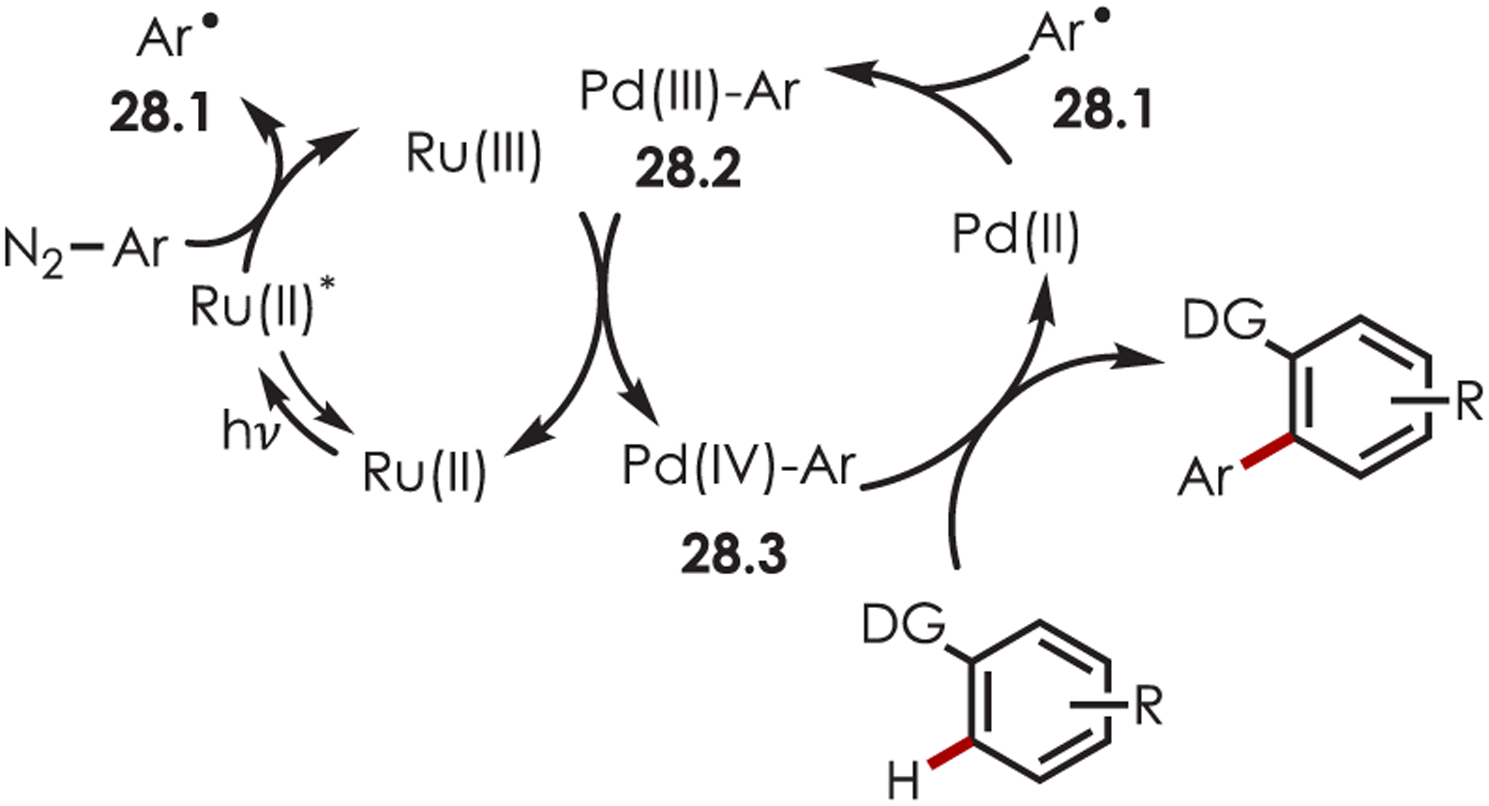
Mechanism of the Dual Palladium/Photoredox-Catalyzed Intermolecular C–H Functionalization Using Aryl Diazoniums
C–H arylation of heteroarenes (29.1) without a directing group was later reported by Xiao and occurs in water as the solvent (Scheme 29).95 This method yields C–H arylated products for electron deficient heteroarenes including pyridines, xanthenes, thiazole, pyrazine, and pyridazine. The regioselectivity varies for pyridine derivatives: C2 substitution was favored in substrates with substituents at the 4-position, while C3- or C2-substituted pyridines produced a mixture of 2-, 4-, and 6-substituted products. Additionally, under aqueous formic acid conditions, this method could be applied to C2 arylation of caffeine (29.3).
Scheme 29.
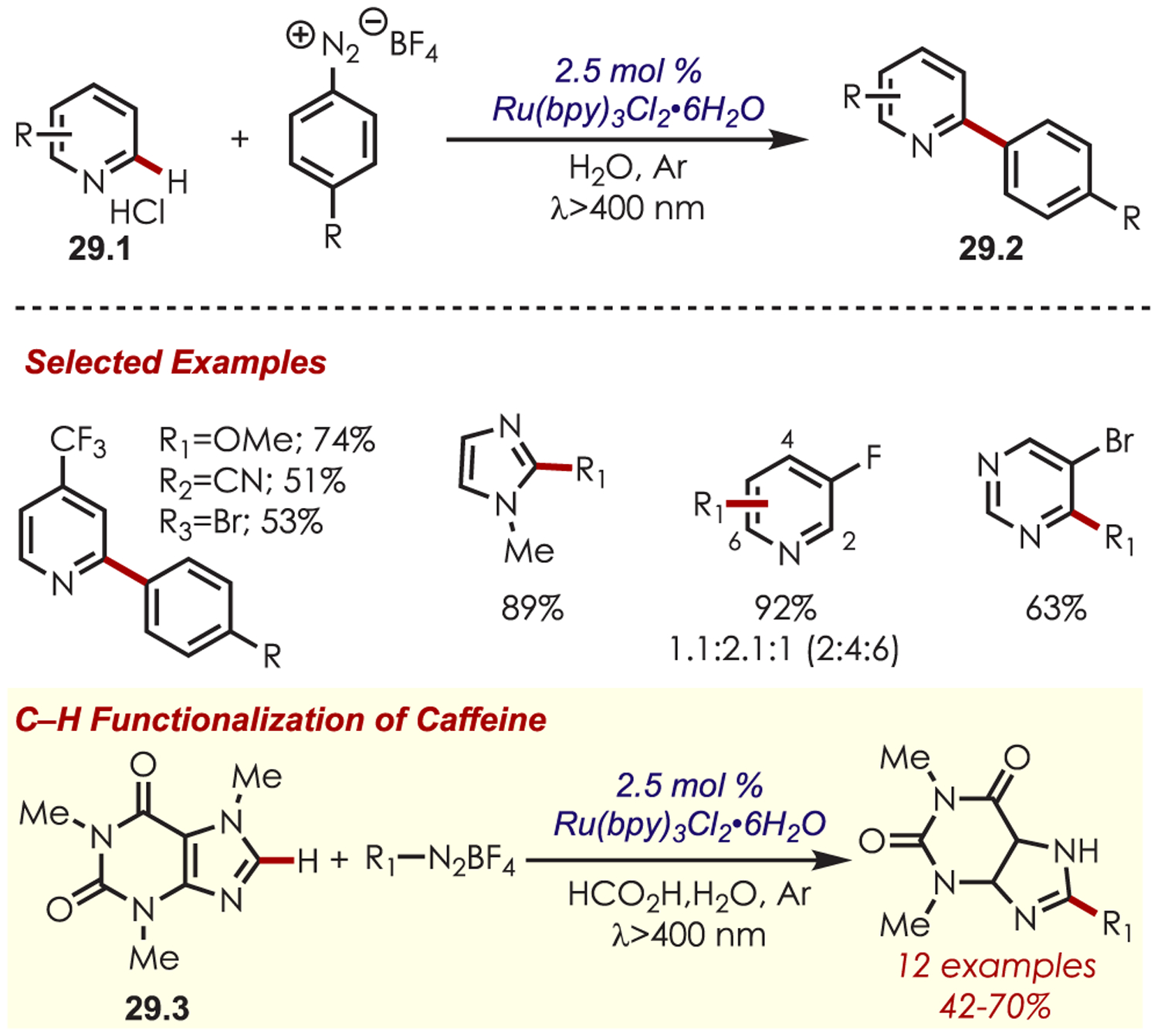
C–H Functionalization Using Aryl Diazoniums in Water
The same group also published a procedure for the arylation of arenes and heteroarenes using diaryliodonium salts (30.2) as aryl radical precursors (Scheme 30). A range of diaryliodonium salt coupling partners was compatible including electron-rich, electron-poor, and halogenated aryl salts. Additionally, unsymmetrical diaryliodonium salts were viable, with the less substituted arene typically being transferred.
Scheme 30.
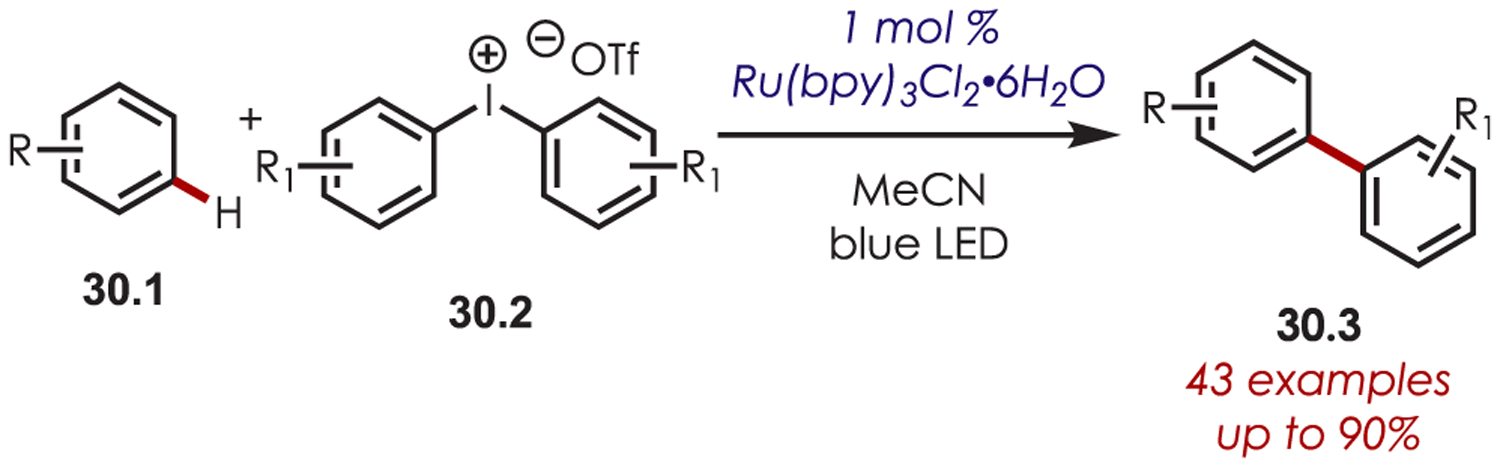
Diaryliodonium as Aryl Radical Precursors for (Hetero)aryl C–H Functionalization
Following the major contributions from Sanford, Konig, and Xiao, other photoredox catalysts have been demonstrated to be effective for C–H arylation via aryl radicals generated by their corresponding diazonium. These include rhodamine B,96 eosin Y,97 tetraphenylporphyrin (TPP),98 Ru(bpy)3,99–102 Cp-(MnCO)3,103 9,10-dihydroacridine (AcrH2),104 cercosporin,105 and iodo-BODIPYphotoredox catalysts.106
A three-component 1,2-diarylation of styrenes (31.2) using aryldiazoniums, 31.3, as carbon-centered radical precursors was reported by Li and co-workers (Scheme 31).107 Addition of the carbon-centered radical into an olefin generates a benzylic radical intermediate which is oxidized by Ir(IV) to the benzylic cation. This is trapped by a nucleophilic (hetero)arene to furnish Heck-type products. These authors later reported an analogous Heck-type coupling with sp3 carbon-centered radicals generated via Lewis acid promoted decarboxylation of N-hydroxyphthalimide esters.108
Scheme 31.
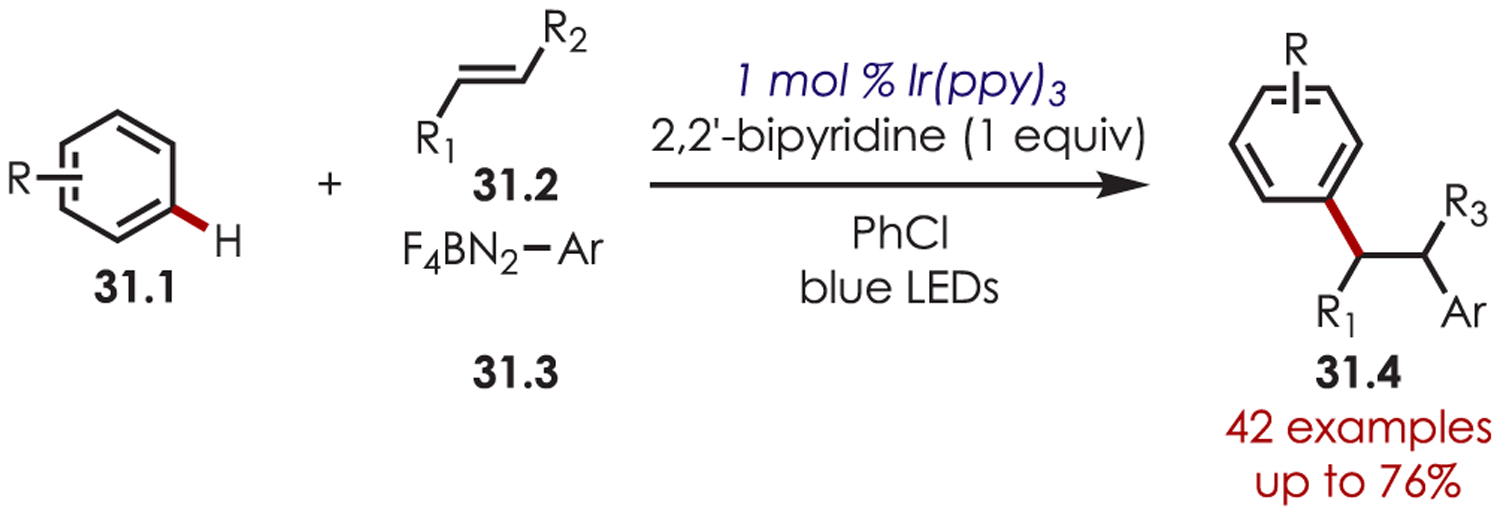
Three-Component 1,2-Diarylation of Styrenes with Aryl Diazoniums as Radical Precursors
The following year, a similar three-component dicarbofunctionalization of styrenes (32.2) was reported using pyridinium salts (32.1) as radical precursors (Scheme 32).109 The authors note that the use of a pyridinium salt as the radical precursor allows for the undirected dicarbofunctionalization of simple, unsubstituted benzyl radicals.
Scheme 32.
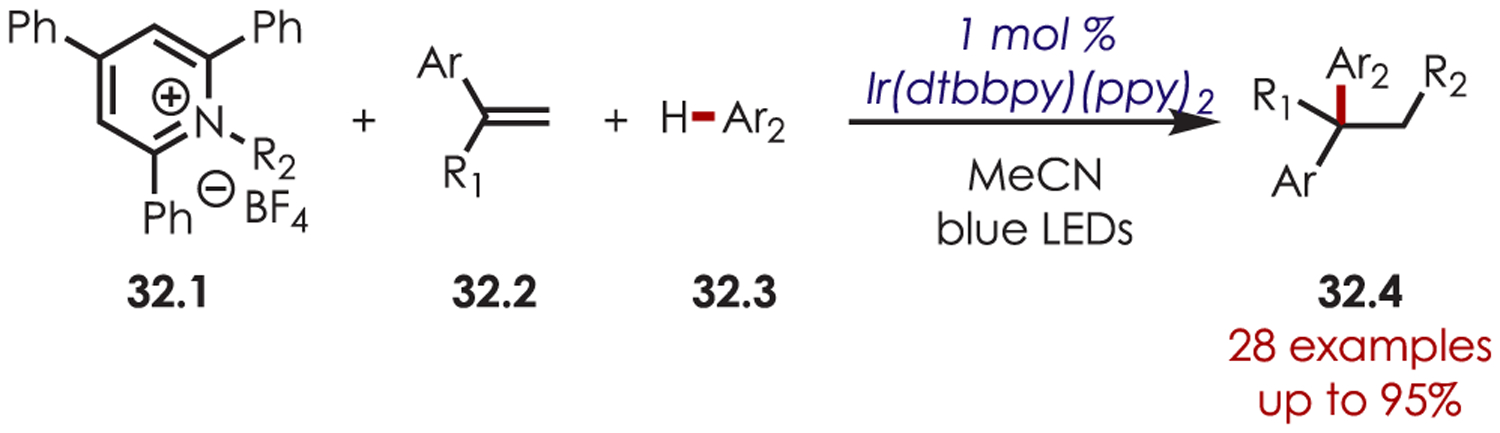
Pyridinium Salts as Radical Precursors for (Hetero)aryl C–H Functionalization
Accessed via Sulfonyl Chlorides.
Sulfonyl chlorides are an alternative source of aryl radicals and offer some advantages over their diazonium counterparts, including improved stability and commercial availability. Reductive PET to sulfonyl chlorides from an excited catalyst generates an aryl radical (vide infra), producing SO2 and hydrochloric acid as the sole byproducts. Addition of the aryl radical to the heteroarene, followed by single electron oxidation and deprotonation, furnishes the aryl-coupled (hetero)arenes.
In 2013, Li and co-workers reported a tandem cyclization of 1,6-enynes (33.1) with arylsulfonyl chlorides for the synthesis of 10a,11-dihydro-10H-benzo[b]fluorenes, 33.3 (Scheme 33).110 The aryl radical reacts with the alkyne and undergoes a subsequent carbocyclization on the pendant alkene to give rise to a primary alkyl radical. An intramolecular cyclization of this radical back onto the arene that bore to sulfonyl chloride, oxidation by Ru(III), and deprotonation furnished the tetracyclic adducts (33.3).
Scheme 33.
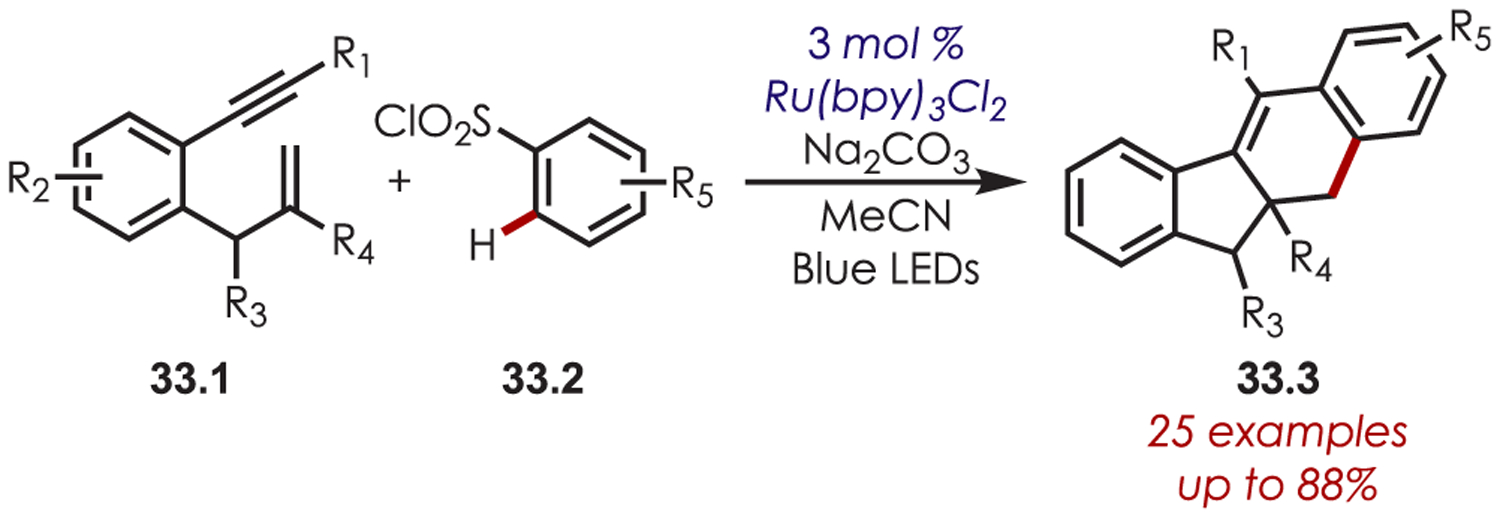
Sulfonyl Chlorides as Aryl Radical Precursors in a Cascade C–H Functionalization Reaction
Sulfonyl chlorides (34.1) have been used in the C2 arylation of heterocycles (34.2) including pyrroles, furans, and thiophenes under ruthenium catalysis (Scheme 34).111 An electronic and steric influence of the arylsulfonyl chlorides was noted to impact the efficiency of the reaction with more electron-deficient or ortho-substituted reagents resulting in a decreased yield. However, the coupled products were obtained for both electron-rich and -poor aryl coupling partners in moderate to excellent yields.
Scheme 34.
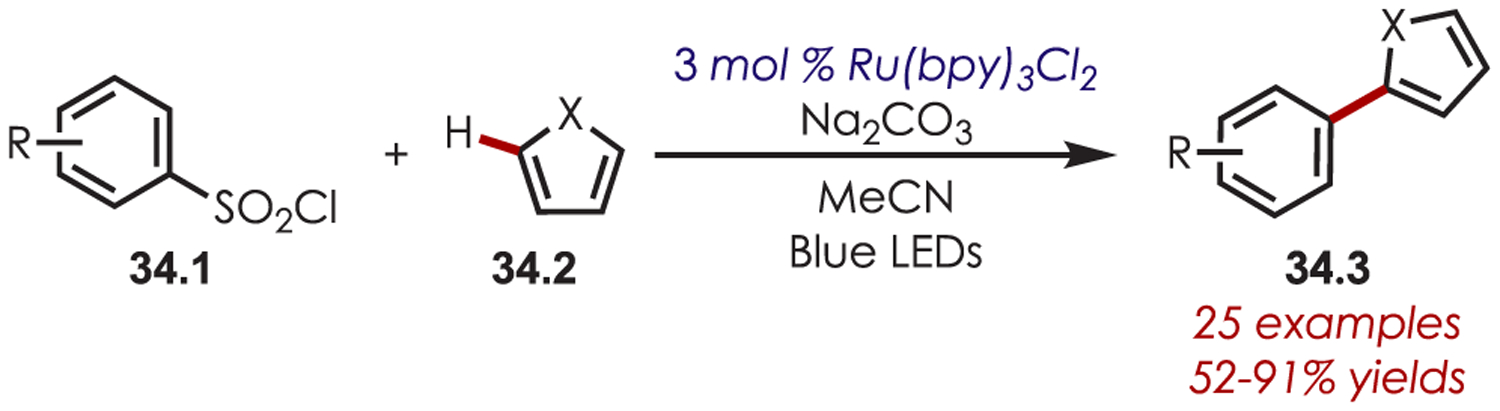
C2 Arylation of Heteroarenes using Sulfonyl Chlorides
Accessed via Aryl or Vinyl Halides.
The use of aryl halides (35.2) as aryl radical precursors was first developed by Stephenson and Lee, who reported the reduction of aryl iodides with fac-Ir(ppy)3 and Ir(ppy)2(dtbbpy)PF6 catalysts, respectively.112,113 Aryl and heteroaryl C–H functionalization using aryl iodide and bromide precursors was published shortly after this work.114 The authors propose similar mechanisms where a SET from the excited Ir(ppy)3 to the aryl halide generates an aryl halide radical anion. Dissociation of C–X releases a halogen anion and generates the sp2 aryl radical. Addition into a (hetero)arene affords biaryl products (35.3) as a mixture of ortho-, meta-, and para-isomers in moderate yield (Scheme 35).
Scheme 35.
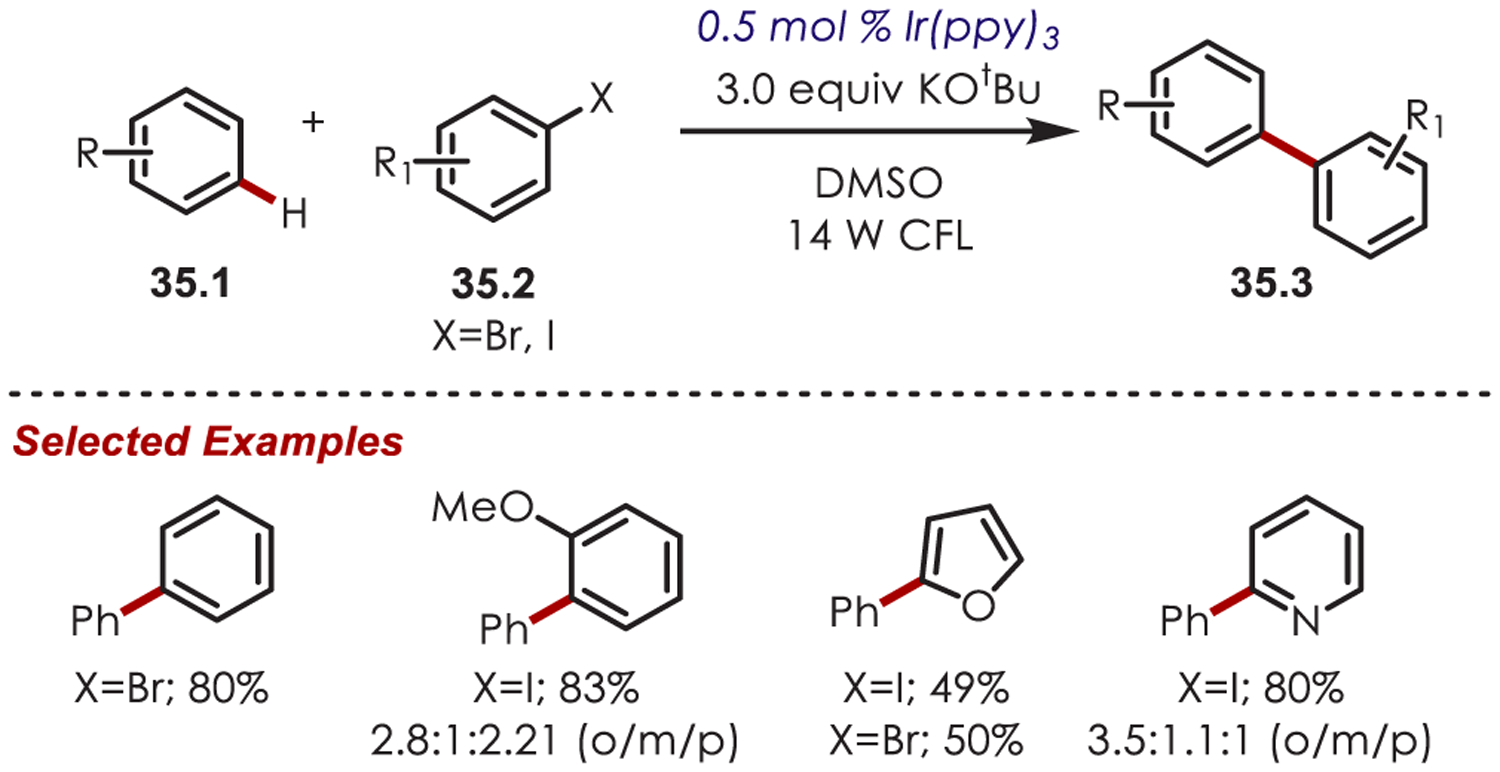
Aryl Halides as Aryl Radical Precursors for (Hetero)arene C–H Functionalizations
Reiser and co-workers generated vinylic sp2 carbon-centered radicals through the reduction of α-bromochalcones (36.2) (Scheme 36).115 A subsequent intermolecular and intramolecular cascade reaction with a heteroarene, such as furans, benzofurans, pyrroles, and indoles generates polycyclic heteroarenes (36.3) in high yields. The intramolecular reductive cyclization of α-bromochalcones has since been extended in the synthesis of urundeuvine116 and phenanthrenes derivatives117 and in the intermolecular coupling of alkenes.118
Scheme 36.
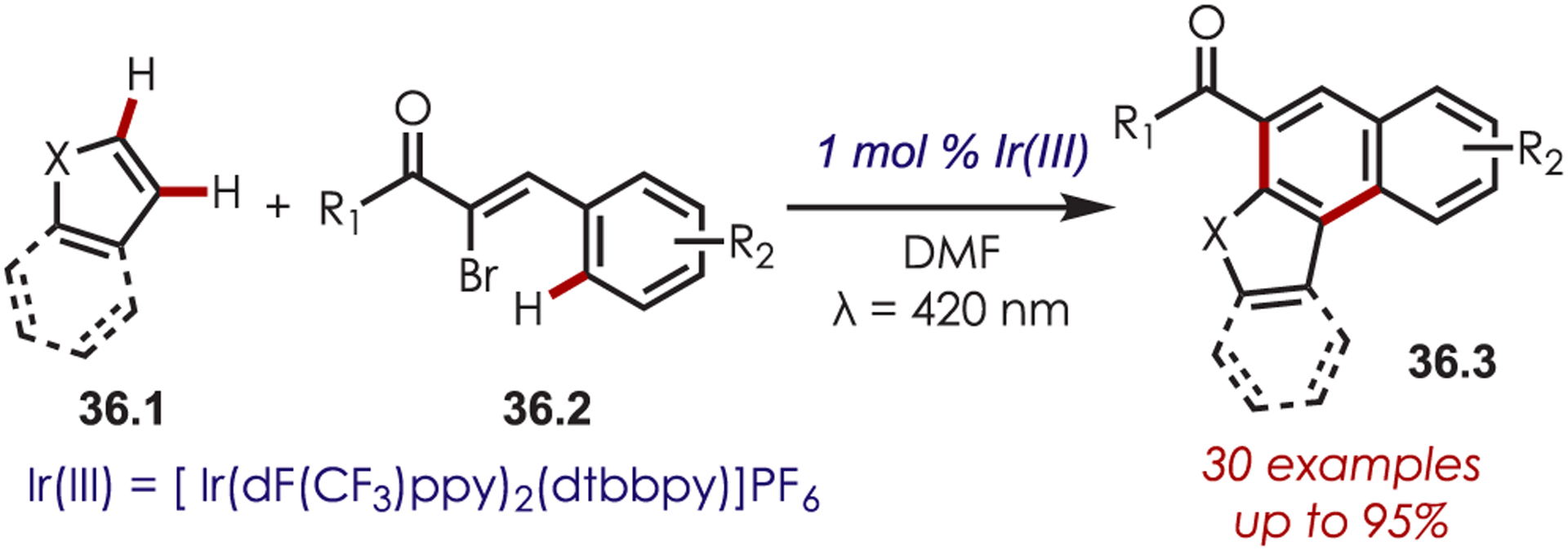
Generation of Vinylic Radicals in a Cascade C–H Functionalization Reaction for the Synthesis of Polycylic Heteroarenes
Accessed via Acyl Radical.
Photoredox catalysis has offered a mild approach for the formation of reactive acyl radical species compared to traditional methods requiring UV irradiation, radical initiators, or high temperatures.119 However, harnessing acyl radicals for aryl C–H functionalization is not well developed, and there are limited reports of Minisci-type C–H acylation of heteroarenes with relatively narrow scopes. The most common acyl radical precursors include aldehydes, α-oxy acids, and carboxylic acids.
In 2014, Zeng and co-workers published a Minisci-type C–H acylation of phenanthridine (37.2) with benzaldehydes using a sulfate radical anion as an H atom abstractor to produce the acyl radical (Scheme 37).120 Halogenated, alkyl-, methoxy-, and acetoxy-substituted benzaldehydes were successful partners with yields ranging from 27 to 73%. However, phenanthridine was the only reported radical acceptor. The persulfate anion (Ered[S2O82−/SO4•−] = +0.35 V vs SCE)121 is reduced by excited Ir(ppy)3 (E*ox[Ir(IV)/Ir(III)*] = −1.73 V vs SCE), generating a sulfate radical anion and Ir(ppy)3+. After H atom abstraction of the benzaldehyde, the resulting acyl radical (37.4) adds to the phenanthridine acceptor yielding an amidyl radical intermediate (37.5). This is subsequently deprotonated by the sulfate anion. A final ET between the radical anion and Ir(ppy)3+ produces the desired product (37.7) and regenerates Ir (ppy)3.
Scheme 37.
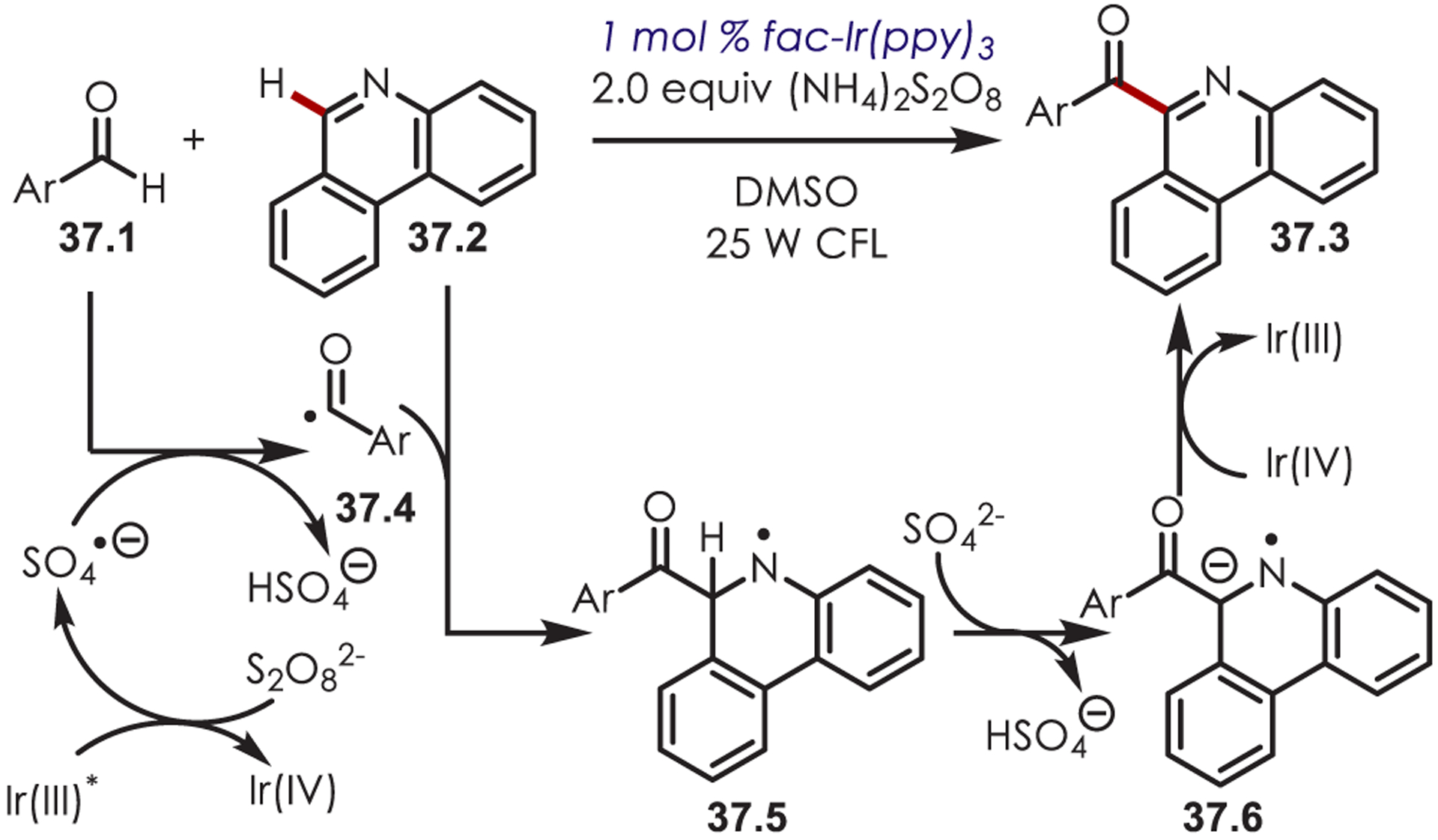
Minisci-Type Acylation Using Acyl Radicals
Benzaldehydes (38.2) have also been employed as acyl radicals precursors for the C–H acylation of indoles (38.1) using a dual palladium and iridium photoredox catalysis approach (Scheme 38).122 This method tolerated aliphatic and (hetero)aromatic aldehydes with alkylated or fluorinated indole derivatives and was demonstrated in batch and in flow. The mechanism proceeds through a C–H activation of the indole by palladium while forming a five-membered palladacycle (38.4). A tert-butyl alkoxy radical is generated by PET with Ir(III)* and proceeds to abstract the aldehyde hydrogen atom. An acylation of the palladacycle (38.5) followed by oxidation and reductive elimination produced the C–H acylated indoles (38.3).
Scheme 38.
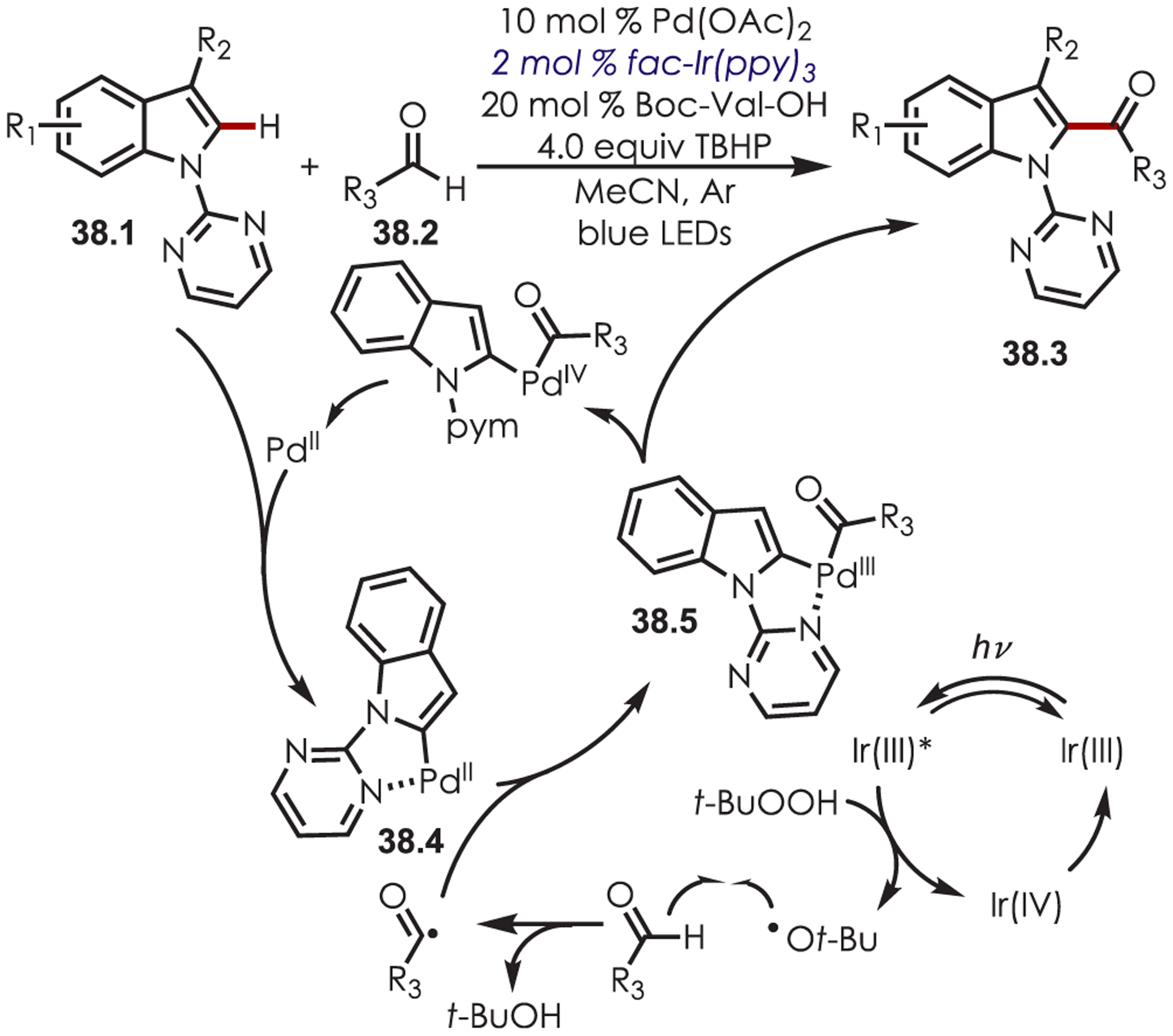
Dual Palladium and Photoredox Catalysis for C–H Acylation of Indoles
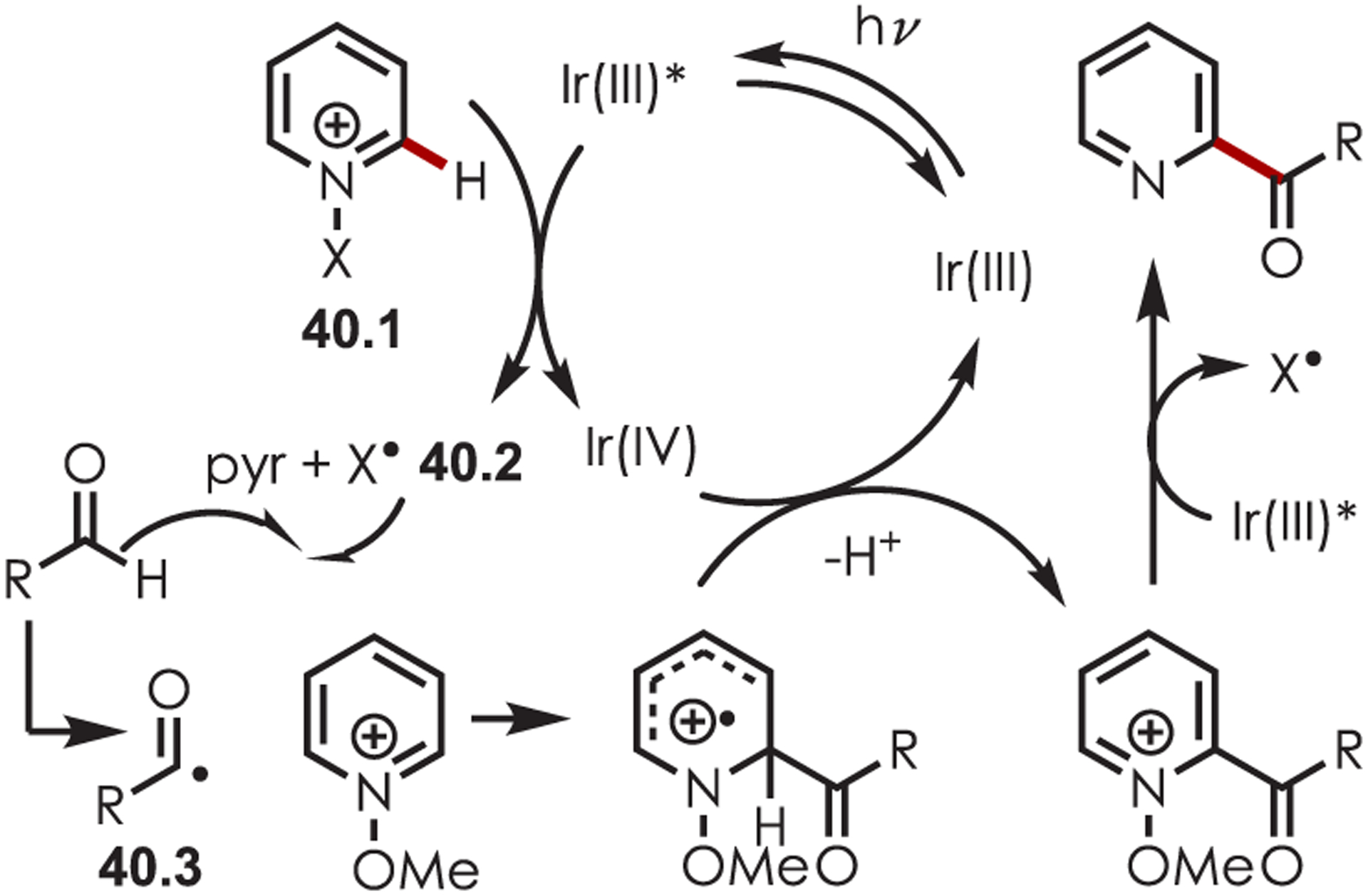
A method for site-selective acylation of pyridiniums was developed with an iridium photoredox catalyst and aldehydes as acyl radical precursors.123 N-Amino pyridinium salts (39.1) give excellent C4 selectivity while N-methoxy pyridinium salts (39.2) prefer C2 selectivity (Scheme 39).
Scheme 39. Site Selectivity for C–H Acylation of Pyridinium Saltsa.
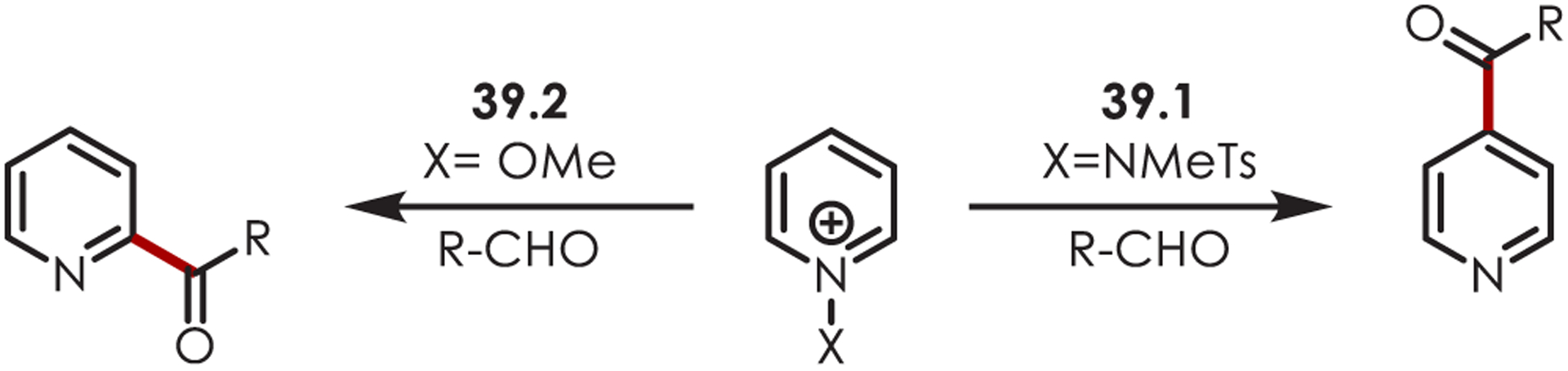
aConditions: 1.0 mol % [Ir(dF(CF3)ppy)2bpy]PF6, 1.2 equiv NaOAc, DCE, bLED
N-Alkoxy- and N-aminopyridinium salts (40.1) undergo SET with excited Ir(III)* generating either an alkoxy or amidyl radical species (40.2). This subsequently abstracts the hydrogen atom of an aldehyde generating an acyl radical, 40.3 (Scheme 40). The BDE’s of MeO-H (~104 kcal/mol), Ts(Me)N-H (~101 kcal/mol) relative to the aldehyde (~88 kcal/mol) provide a driving force for this hydrogen atom transfer event. The nucleophilic acyl radical inserts into a pyridinium salt, where the substitution at nitrogen dictates the site of selectivity. The more sterically hindered N-amino-substituted pyridiniums give C4 selectivity while smaller groups on nitrogen favors C2 addition. Supporting DFT calculations indicate that the transition state is 1.4 kcal/mol lower for C2 functionalization over C4 functionalization for N-methoxypyridinium salts. This is attributed to an attractive electrostatic interaction between the positively charged nitrogen and the oxo-functionality of the radical. The N-amino tosylates are too sterically hindered to undergo this favorable electrostatic interaction and the transition state for C4 acylation is 2.4 kcal/mol more stable than the C2 acylation. After insertion of the acyl radical into a pyridinium salt, the product is formed after deprotonation and N-heteroatom bond cleavage which releases another alkoxy or amidyl radical, continuing with a chain mechanism.
Scheme 40.
Mechanism for C2-Selective C–H Acylation of Pyridinium Salts
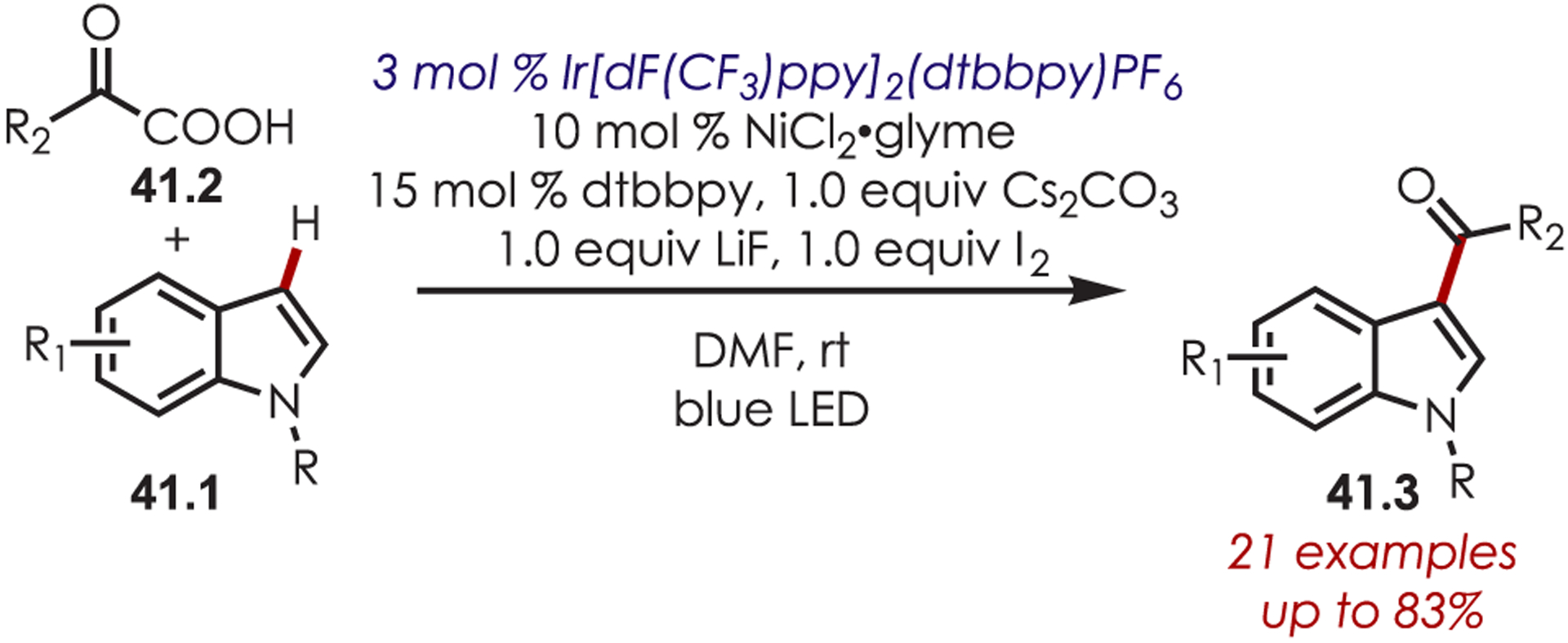
MacMillan’s seminal work merging nickel and photoredox catalysis for the functionalization of Csp2–X by acyl radicals generated from decarboxylation of α-oxo acids sparked an interest from several groups to employ similar strategies for C–H functionalization.124 Development of a C–H functionalization of indoles using α-oxo acids through a dual nickel/photoredox catalytic system was later published by Li and co-workers (Scheme 41).125 The proposed mechanism is similar to other metallophotoredox-mediated transformations. Single electron transfer between an α-oxo acid (41.2) and Ir(III)* triggers a decarboxylation and produces an acyl radical and Ir(II). C–H activation occurs through oxidative addition of Ni(0) into the indole derivative (41.1), which reacts rapidly with the acyl radical. The resulting intermediate undergoes reductive elimination to furnish the acylated products (41.3). Interestingly, this method produces C3-acylated indoles with high regioselectivity, while the other palladium-catalyzed C–H activation methods produced high C2 regioselectivity. No explanation for the contrasting C–H activation was provided by the authors.
Scheme 41.
Dual Nickel and Photoredox Catalysis for a C–H Acylation Using Carboxylic Acids
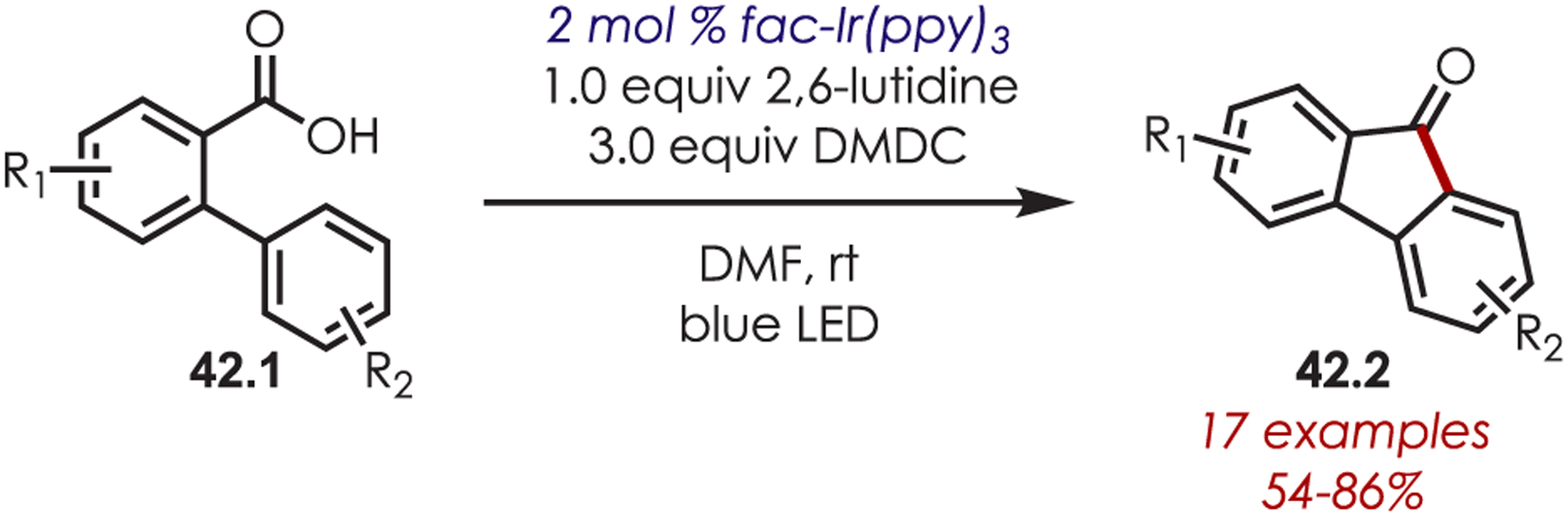
An intramolecular C–H functionalization via a deoxygenative Pschorr-like acyl radical cyclization was reported for the synthesis of fluorenones (42.2) with carboxylic acids (42.1) as acyl radical precursors (Scheme 42).126 Dimethyl dicarbonate (DMDC) as an additive generates an intermediate anhydride in situ. A subsequent decarboxylation furnishes the acyl radical that undergoes an intramolecular cyclization and a subsequent rearomative oxidation/deprotonation to afford the fluorenone. Both electron-poor and electron-rich substituted biaryls produced the desired fluorenone, including halogenated, alkylated, trifluoromethylated, and alkoxylated biaryls.
Scheme 42.
Synthesis of Fluorenones by a Pschorr-like Acyl Radical Cyclization
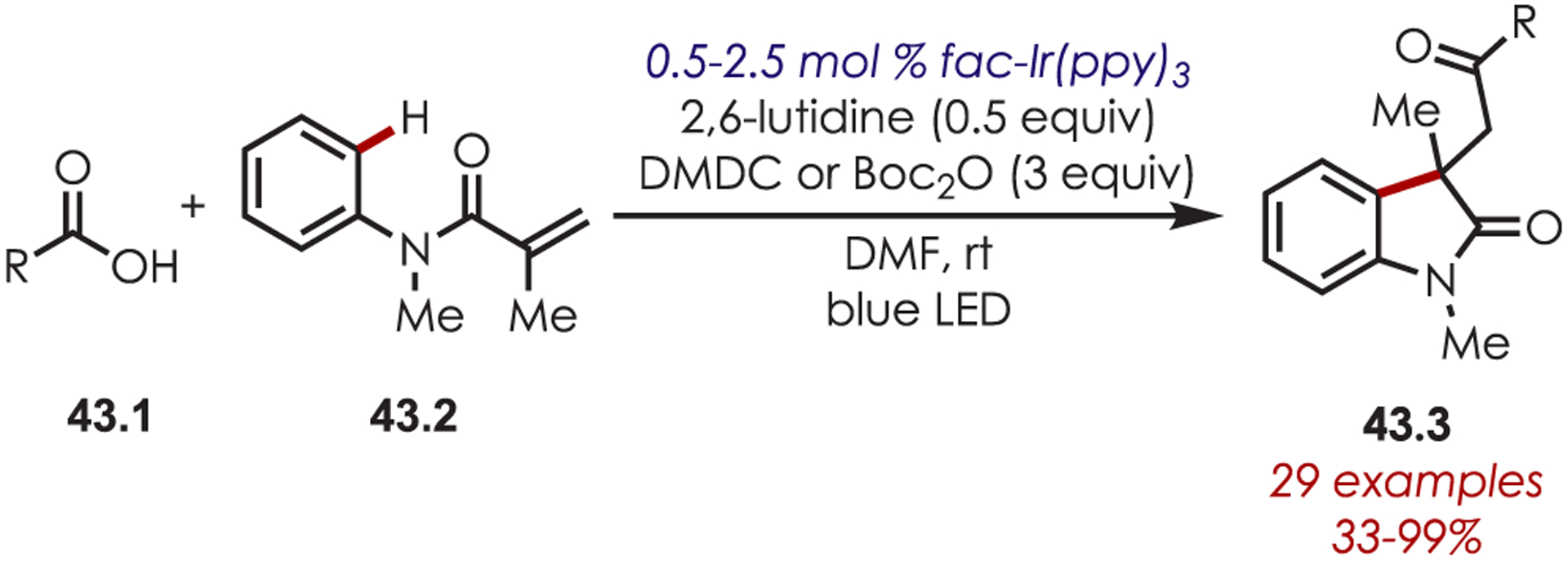
The Wallentin lab has recently reported that acyl radicals can be generated via in situ anhydride formation, reduction, and subsequent decarboxylation (Scheme 43).127 This catalytic acyl radical generation has been utilized in the synthesis of 2-oxoindole derivatives (43.3) via a radical cascade sequence. Aromatic carboxylic acids (43.1) were the acyl radical precursors formed via the aforementioned sequence, which underwent Giese addition to aryl methacrylamides and subsequent cyclization onto the pendant arenes to furnish the corresponding oxoindoles. This method was tolerant of both electron-rich and -poor substitution on the carboxylic acid reaction component. Additionally, a diverse range of methacrylamides (43.2) gave the product in high yields and was extended to other olefins, including α-alkyl styrene derivatives and a cinnamate amide substrate, giving access to more complex heterocycles.
Scheme 43.
Cascade Acyl Radical Addition to Amides for the Synthesis of 2-Oxoindoles
In 2018, terminal alkynes (44.2) were first reported as acyl radical precursors for C–H functionalization of electron-deficient heteroarenes (Scheme 44).128 Under oxidative conditions with persulfate as the terminal oxidant, phenylacetylene was shown to form phenylglyoxylic acid (44.4) in solution. This then proceeds via oxidative decarboxylation to produce the key acyl radical species (44.5). C2-selective acylation of quinoline and C1-selective acylation of isoquinolines were observed in moderate to good yields with a range of acetylene coupling partners. Outside of quinoline and isoquinoline derivatives, benzothiazole was the only other reported heterocycle successful in this C–H acylation. Although the acyl radicals reported in this method can be accessed through other precursors such as aldehydes, or α-oxy acids, the authors note an advantage of using terminal alkynes are shorter reaction times and only requiring stoichiometric amounts of the acyl radical precursor.
Scheme 44.
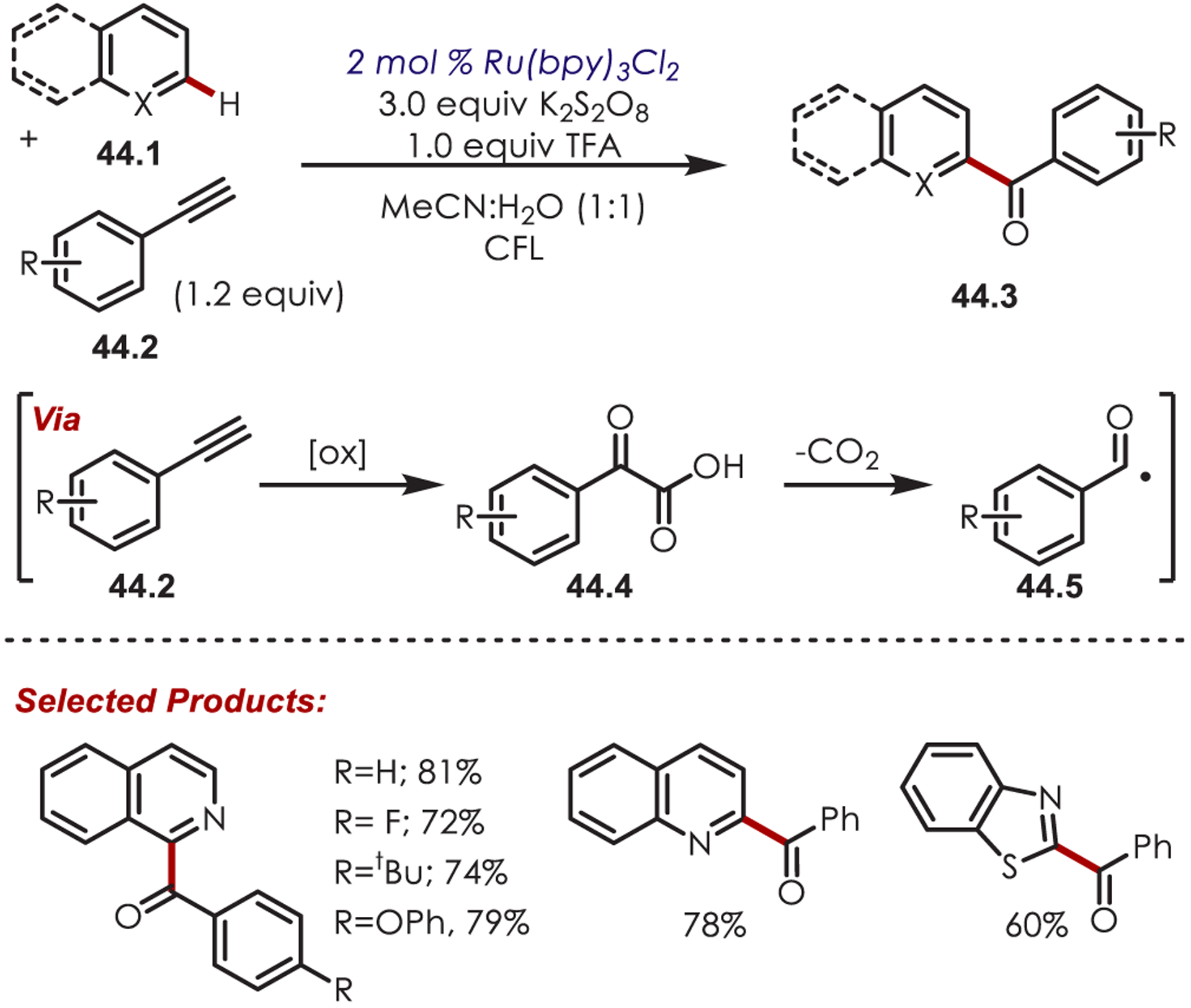
Alkynes as Acyl Radical Precursors in a C–H Acylation of Heteroarenes
2.1.3. Transformations Involving Nitrogen-Centered Radicals.
Nitrogen-centered radicals (NCR’s) which can be accessed through electron transfer with a photoredox catalyst, have been used in a range of aryl and heteroaryl C–H functionalizations.129 Amidyl radicals contain a high degree of electrophilic character on the nitrogen atom,130 which complements traditional two-electron reactivity. Generally, these reactive species are formed via reduction of N-halo amines (45.1), giving rise to the corresponding nitrogen-centered radical (45.2). Addition of 45.2 to an aromatic system leads to the formation of an intermediate cyclohexadienyl radical (45.3). Return ET to the oxidized catalyst then forms a cationic Wheland-type intermediate (45.4), which subsequently undergoes deprotonation to furnish the aminated (hetero)arenes (Scheme 45).
Scheme 45.
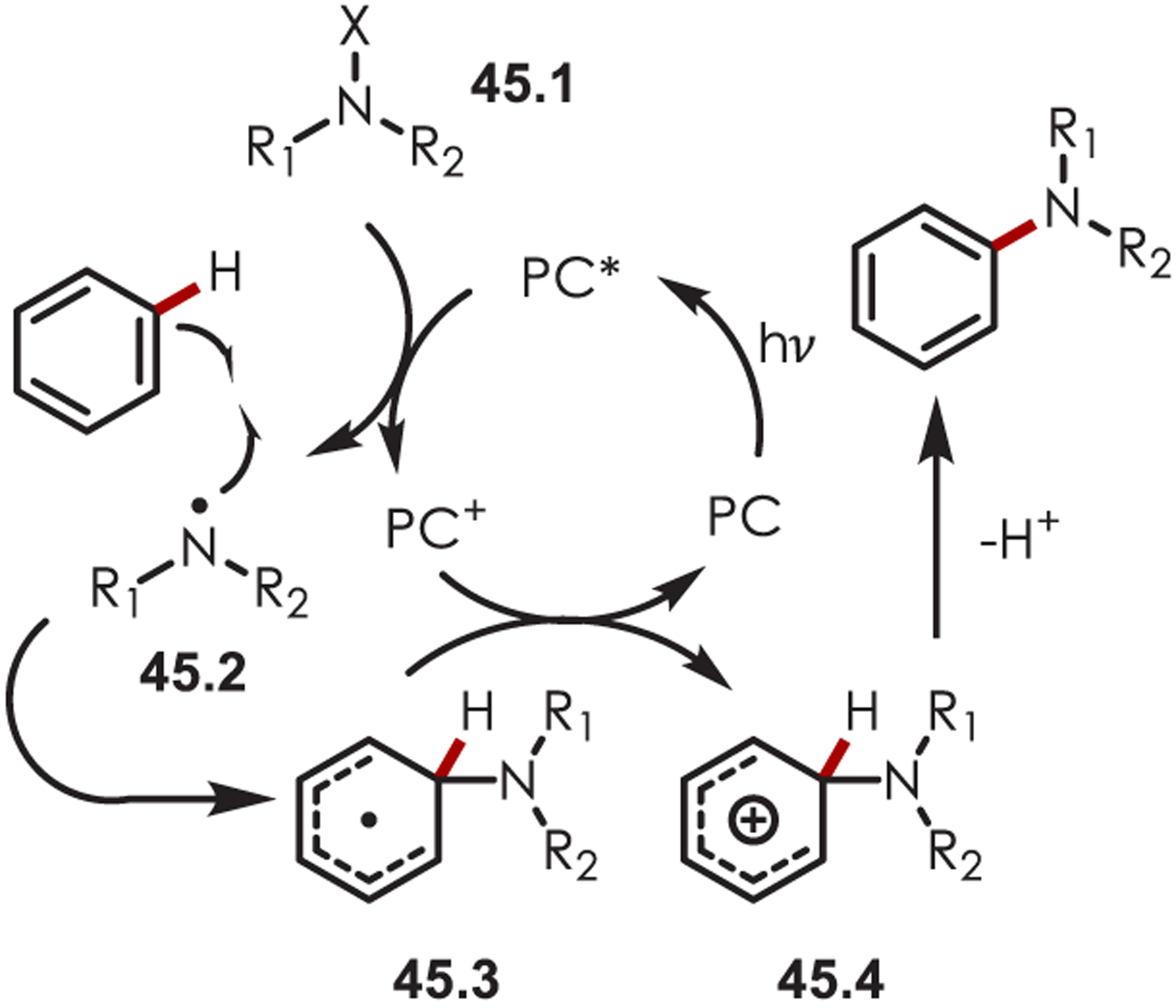
General Mechanism for the Addition of a Nitrogen-Centered Radical to Aromatics
Seminal work by Sanford and co-workers in 2014 demonstrated a photoredox-catalyzed aryl and heteroaryl C–H functionalization via NCR’s with an Ir(III) photoredox catalyst.131 It was found that N-acyloxyphthalimides (46.2) were efficient NCR radical precursors for the C–H amination of arenes and heteroarenes (Scheme 46). Both electron-rich and electron-poor arenes produced the desired aminated products as a mixture of ortho- and para-regioisomers, however, in lower yields for more electron-deficient arenes. The authors note that an advantage to this photoredox catalyzed approach over other C–H amination methods is the success of this method with a diverse range of heterocycles by avoiding excess oxidants and elevated temperatures.
Scheme 46.
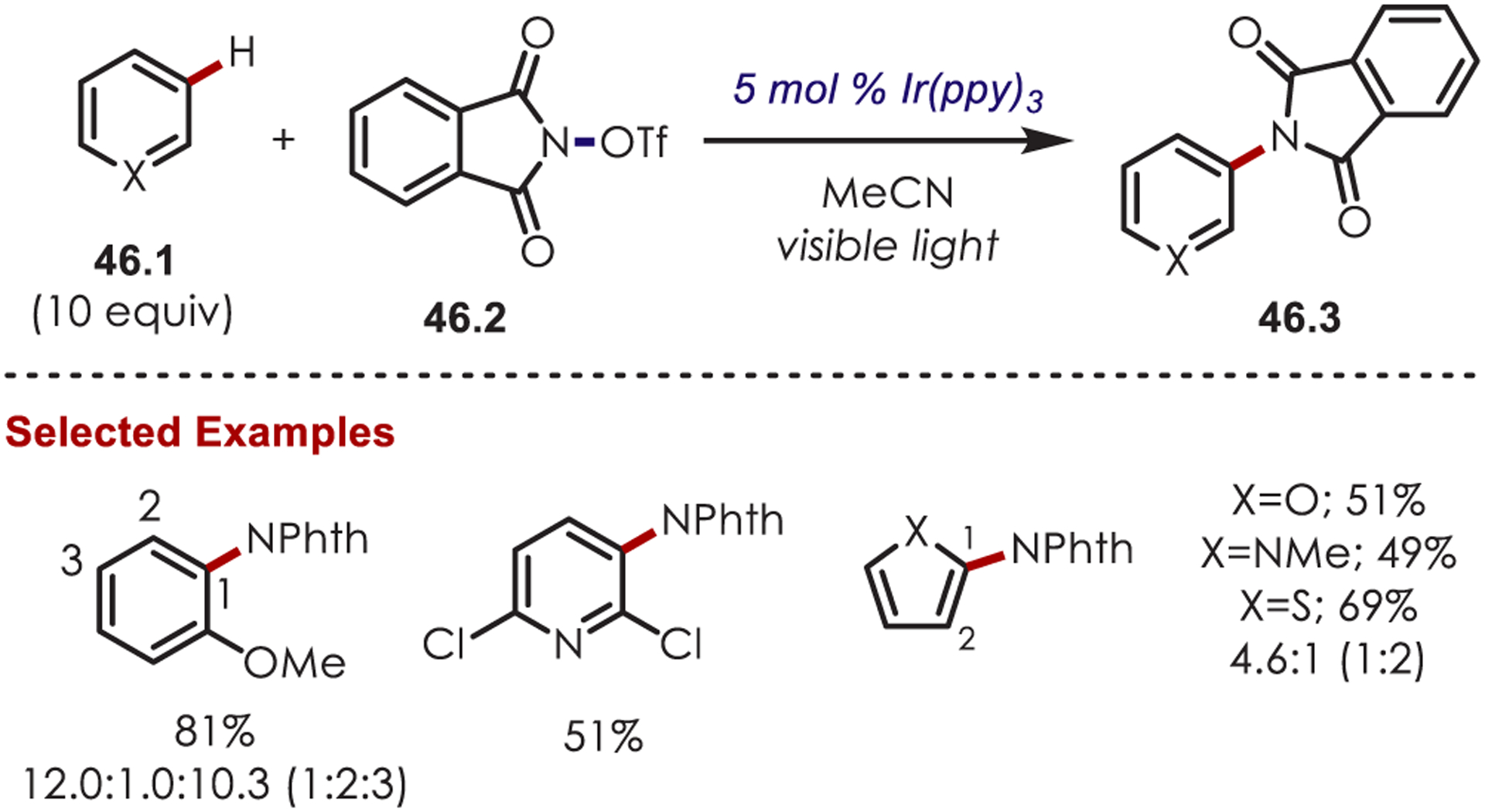
N-Acyloxyphthalimides as Nitrogen-Centered Radical Precursors for C–H Functionalization
Since the initial work reported by Sanford, a range of nitrogen-centered radical precursors (47.2) have been utilized to achieve (hetero)aryl C–H amination including N-acyloxypthalamides (47.4),131 N-chlorophthlamide (47.5),132 sulfonamides (47.6),133,134 benzenesulfonyl hydroxylamine (47.7),135 benzoyl azides (47.8),136 aryloxyamides (47.9),137,138 and N-aminopyridinium salts (47.10)139 (Scheme 47). Generally, these produce similar reactivity with arene and heteroarene derivatives. Functionalization of arenes gives a mixture of isomers, ortho-, para-, and meta-, with moderate selectivity influenced by the arene electronics. In contrast, substitution of heterocyclic derivatives gives high C2 selectivity for electron-rich heterocycles (i.e., indole, pyrrole, furan, and thiophene derivatives) and pyridine derivatives give high meta-selectivity.
Scheme 47.
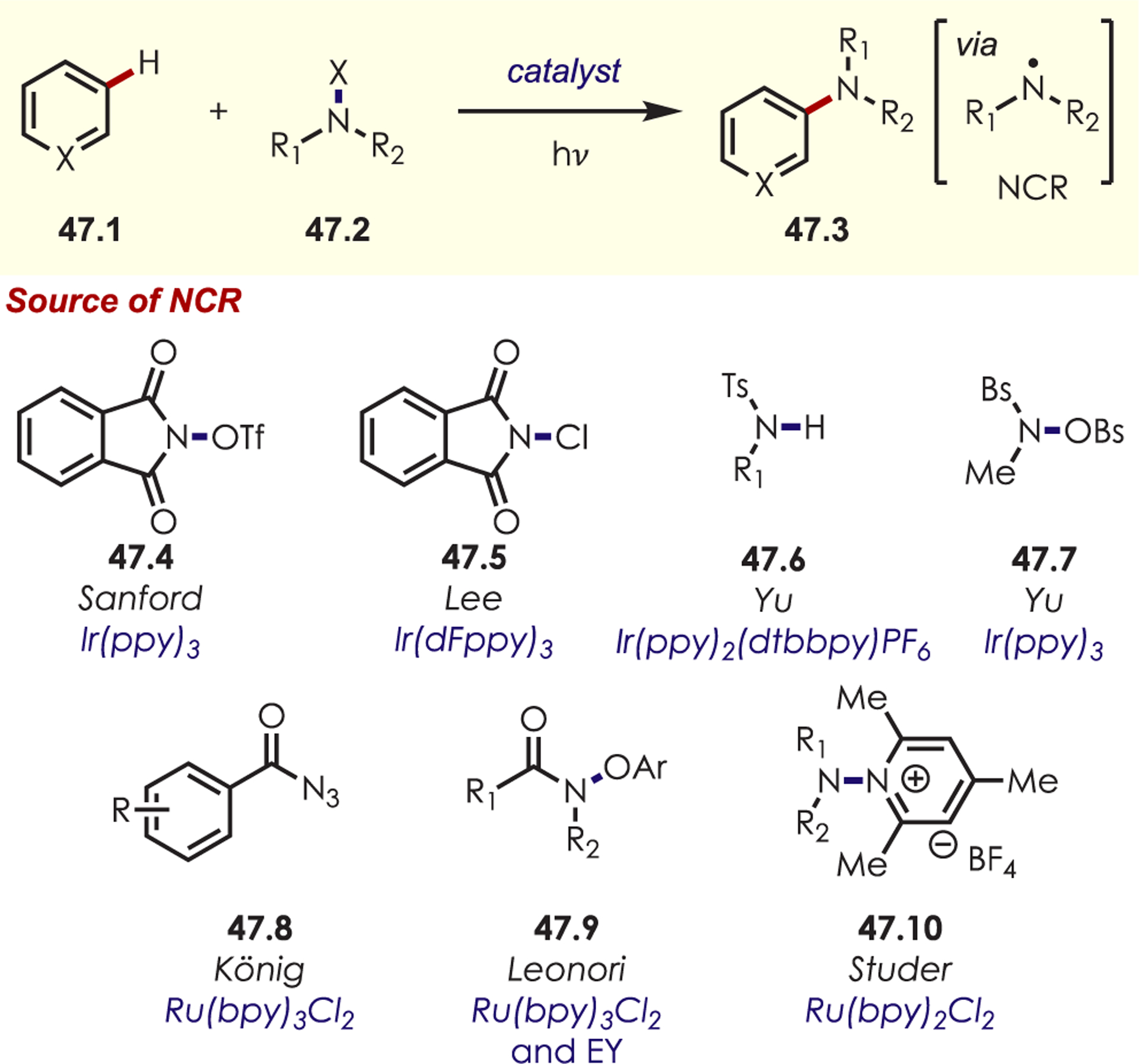
Other Nitrogen-Centered Radical Precursors for Photoredox Catalyzed (Hetero)aryl C–H Functionalization
Alkyl amines (48.2) serve as attractive NCR precursors that would eliminate the required synthesis of the activated amine. Leonori et al. reported a generalized method for arene C–H amination with alkyl amines with high site selectivity by generating N-haloamines in situ with N-chlorosuccinimide (NCS) (Scheme 48).140 A subsequent SET to the generated N-haloamine produces the key nitrogen-centered radical. Electron-rich and electron-poor arenes were tolerated in this method, including aryl halides and boranes. A range of functionalized piperidines were efficient coupling partners, in addition to pyrrolidine, azepanes, azetidines, secondary and primary alkylamines, and other pharmaceutically relevant amine derivatives. Xiao and co-workers reported an analogous C–H functionalization of benzoxazoles with amines through the in situ generation of N-chloro amines.141
Scheme 48.
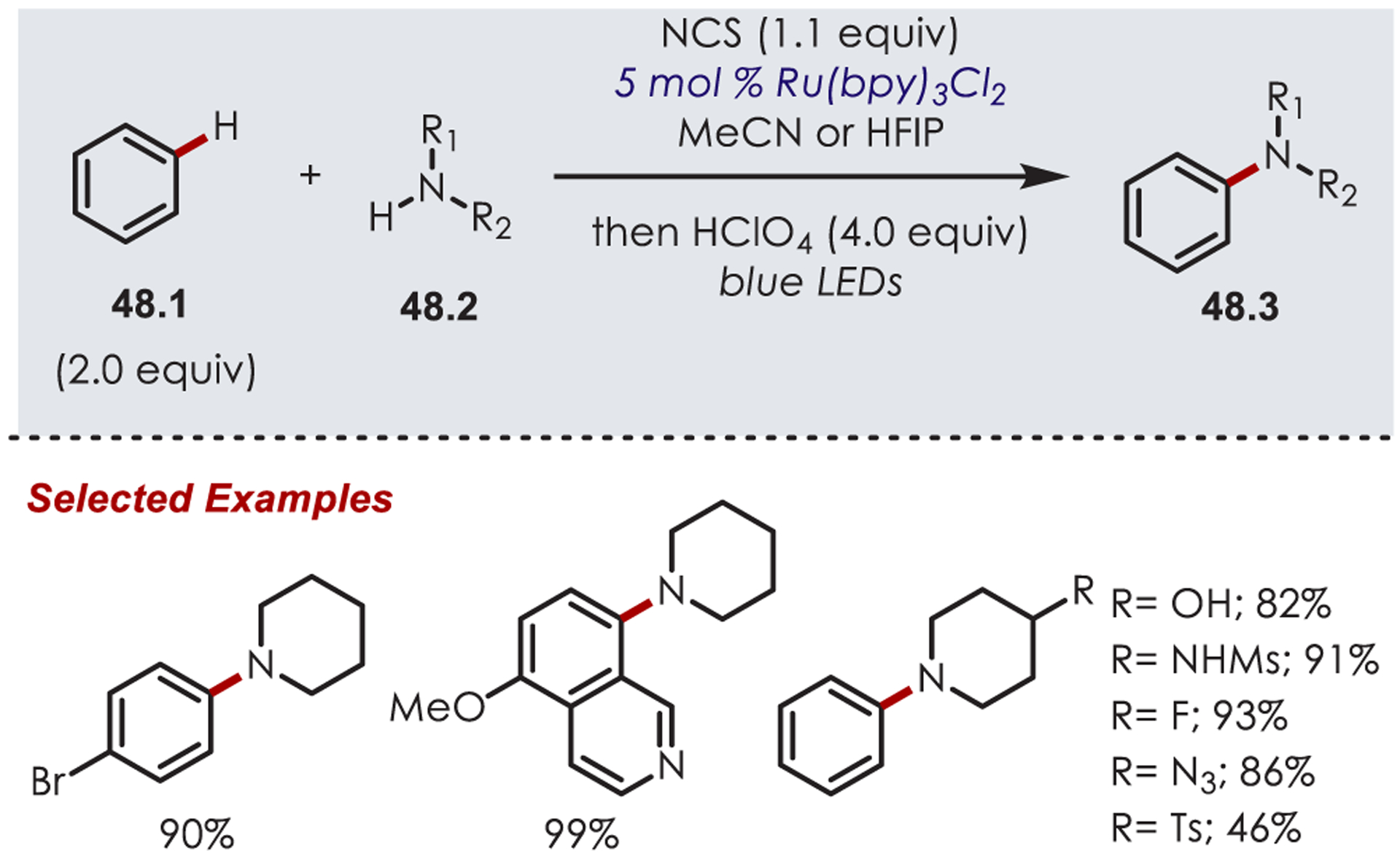
Alkyl Amines as Nitrogen-Centered Radical Precursors for C–H Functionalization
An intramolecular C–H amidation for the synthesis of phenanthridinones (49.2) and quinolinones (49.4) was reported via amidyl radicals generated by PCET of aryl amides (Scheme 49).142 The photoexcited Ir(III) and phosphate base are operative in a PCET mechanism that allows for sufficient generation of a nitrogen-centered radical from the strong amide N–H bond. In addition to forming the NCR, Ir(III)* is proposed to play a key role in initiating an E/Z isomerization of the olefin for the construction of quinolinones.
Scheme 49.
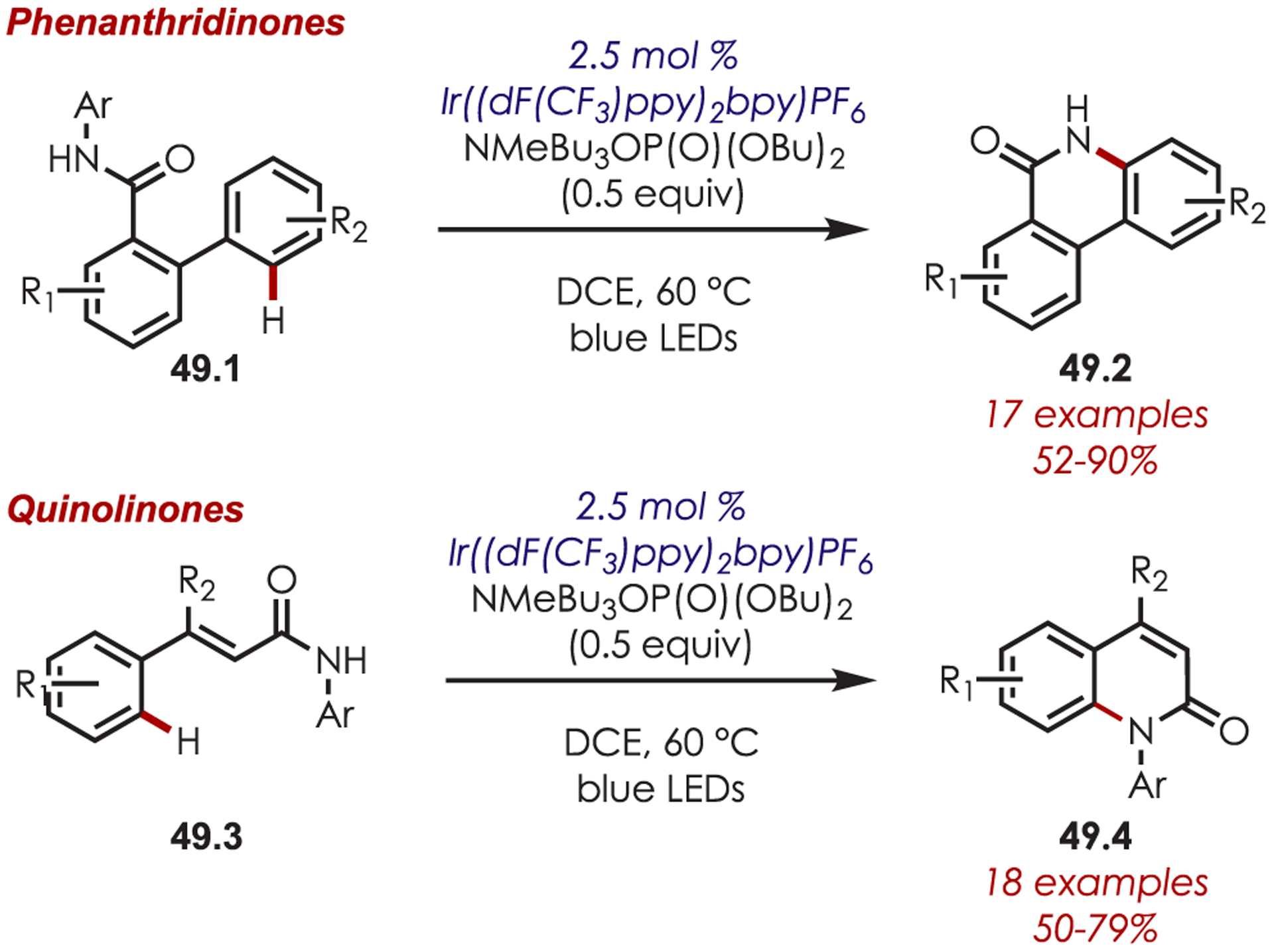
Synthesis of Phenanthridinones and Quinolinones through the Intramolecular Addition of a Nitrogen-Centered Radical
Zhang, Yu, and co-workers published an intramolecular amination through acyl oximes (50.1) as NCR precursors (Scheme 50).143 This method produces pyridine, quinolines, and phenanthridines using Ir(ppy)3 as the catalyst in good to excellent yields. Single-electron reduction of the acyl oximes generates the iminyl radical which undergoes an intramolecular homolytic aromatic substitution to afford the aza-arenes. The authors demonstrate this methodology with a five-step synthesis of benzo[c]phenanthridine alkaloids (50.3). The same year, the authors published a one-pot method that forms the acyl oximes in situ from the corresponding aldehyde, furnishing the phenanthridine or quinoline products.144
Scheme 50.
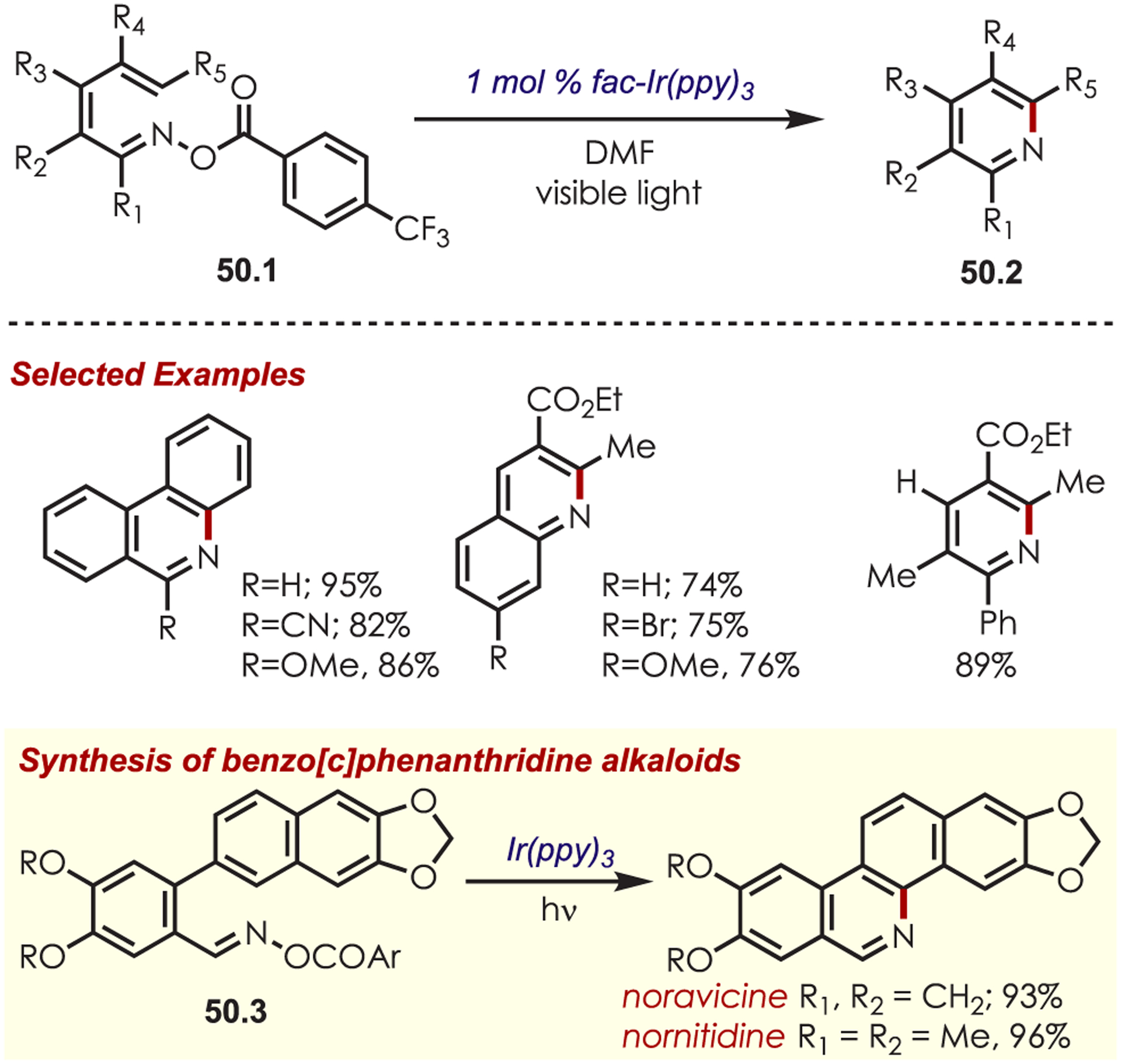
Synthesis of Substituted Pyridines through an Intramolecular Addition of a Nitrogen-Centered Radical
2.1.4. Transformations Involving Phosphorus, Oxygen, and Sulfur-Centered Radicals.
Oxygen-Centered Radicals via Carboxylates.
A direct benzoyloxylation of arenes (51.1) using benzoyl peroxide as an oxygen-centered radical precursor was reported by Li and co-workers (Scheme 51).145 Reductive SET of benzoyl peroxide from photoexcited Ru(II)* generates a benzoyl anion, a benzoyl radical (51.3), and Ru(III). The addition of the benzoyl radical to the (hetero)arene, followed by oxidation from Ru(III) and deprotonation affords the monobenzoylated products. The combination of acetonitrile as the solvent and sodium bicarbonate as an additive was noted to give the highest yield and ortho:para selectivity. Interestingly, no decarboxylation was detected, which was suggested to be a result of running the reaction at room temperature. Electron-rich arenes and heteroarenes were benzoyloxylated in poor to good yield.
Scheme 51.
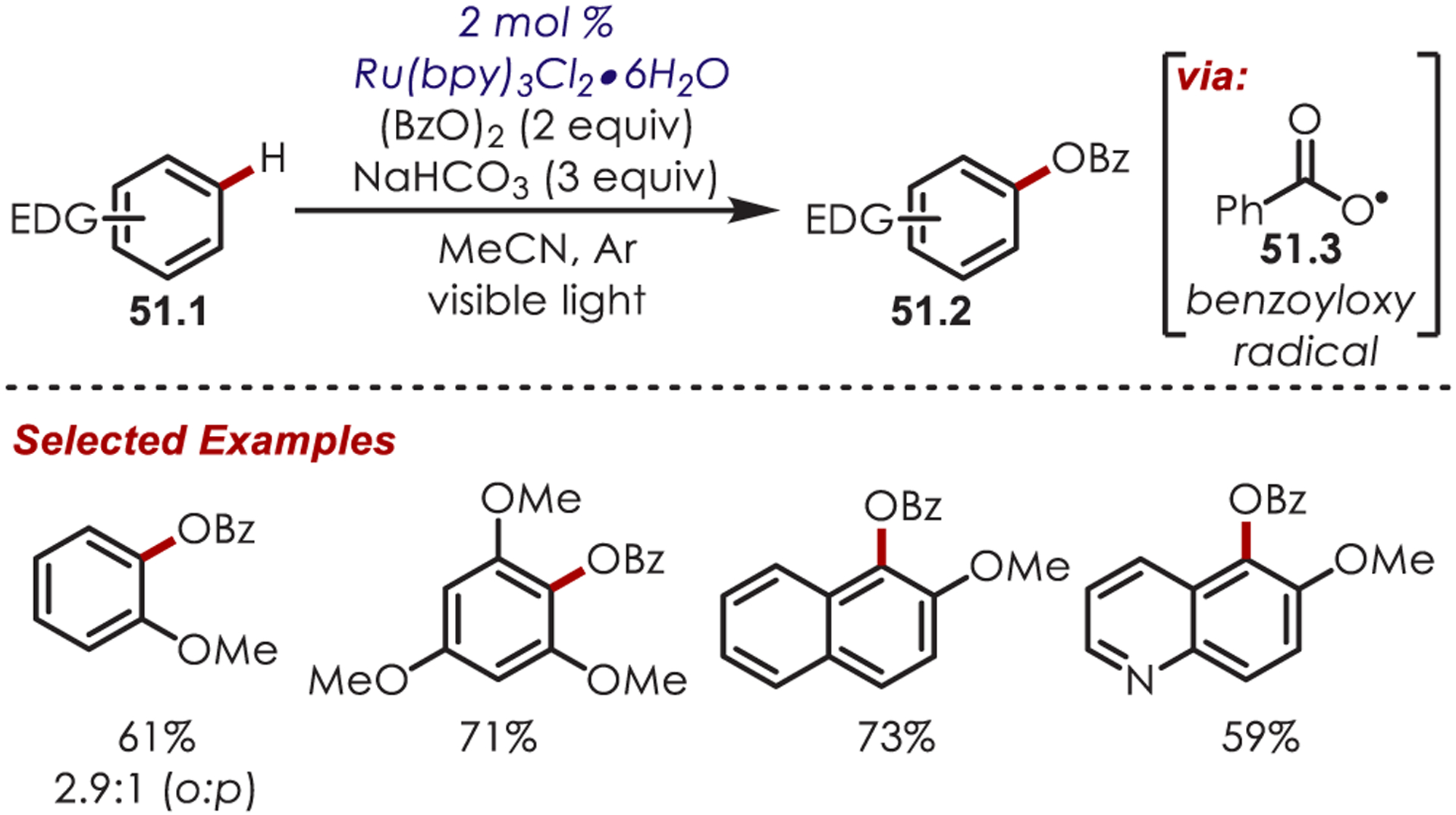
Benzoyloxylation through an Oxygen-Centered Radical
Sulfur-Centered Radicals Accessed via Thiols, Isothiacyanates, and Sulfonyl Chlorides.
Sulfur-centered radicals (SCRs) are commonly accessed by SET from a photoredox catalyst, but the addition of these reactive species to (hetero)arene C–H bonds remains limited.146 Sulfur-centered radical precursors include thioanilides (Eox = +0.90 V vs Ag/AgCl)147 and sulfonyl derivatives (Ered = −1.37 V vs SCE for 4-sulfonyltoluene).55 Both oxidative and reductive SET has been accomplished for the generation of SCR’s, which have been coupled with (hetero)arenes.
In 2012, Li and co-workers reported the synthesis of benzothiazoles (52.2) with thioanilides (52.1) as the SCR precursor in an intramolecular cyclization (Scheme 52).148 Excited Ru(bpy)32+* reduces molecular oxygen to produce O2•−. The thioanilide is deprotonated by DBU to form a sodium salt (52.3) with an Eox = +0.49 V vs Ag/AgCl which can then undergo a thermodynamically favorable oxidative PET by Ru(bpy)33+ to generate the SCR (52.4). A subsequent radical addition and hydrogen atom abstraction from O2•− yield the desired benzothiazole product. The authors note that an alternative mechanism involving oxidation of the sulfur anion directly from excited Ru(bpy)32+* could not be excluded.
Scheme 52.
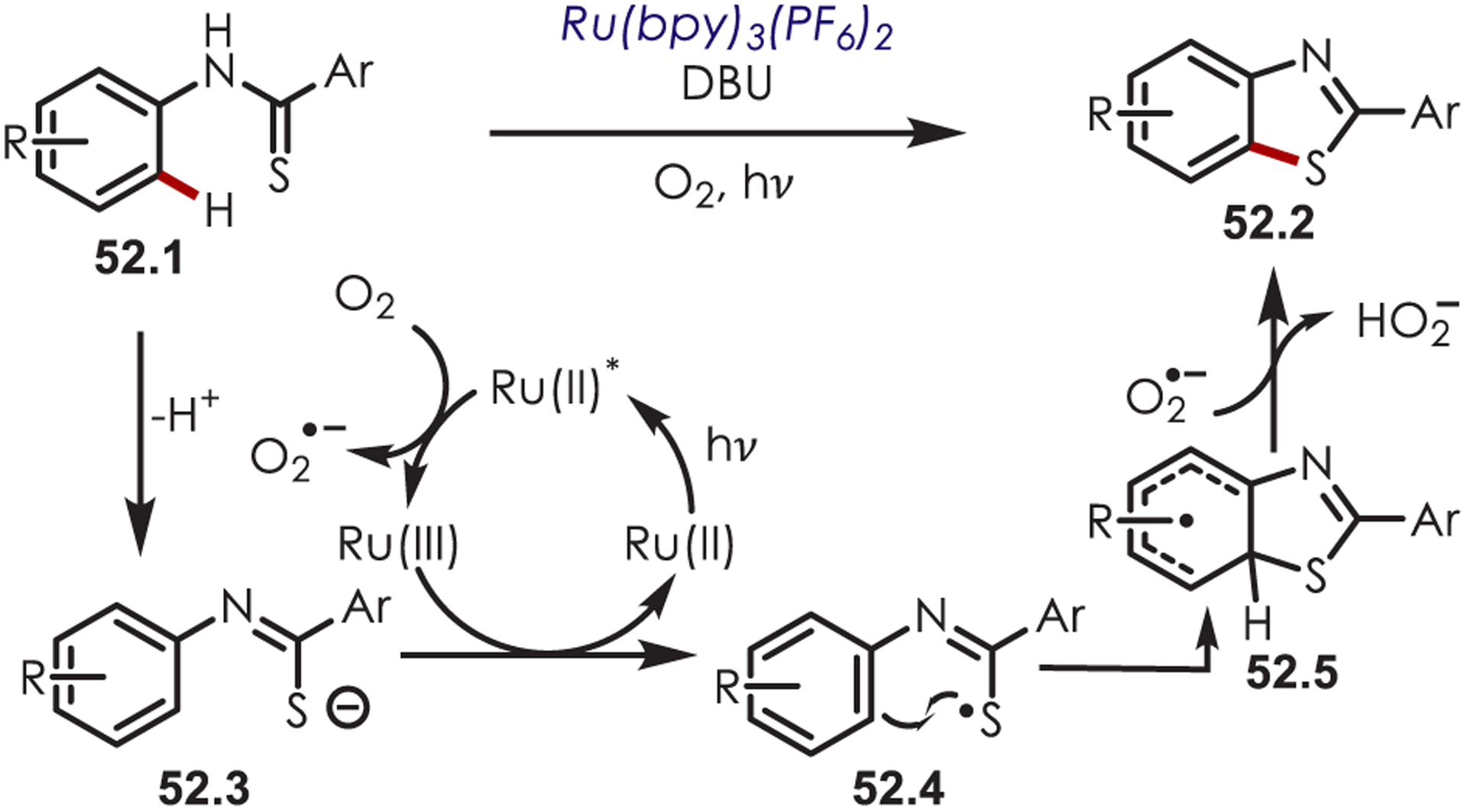
Synthesis of Benzothiazoles from the Addition of a Sulfur-Centered Radical to an Aryl C–H
Three years later, Lei reported an external oxidant-free variant of this work using dual photoredox-cobalt catalysis which gave H2 as the sole byproduct (Scheme 53).147 Various benzothiazoles (53.1), including halogenated, electron-rich, and electron-poor derivatives, were synthesized.
Scheme 53.
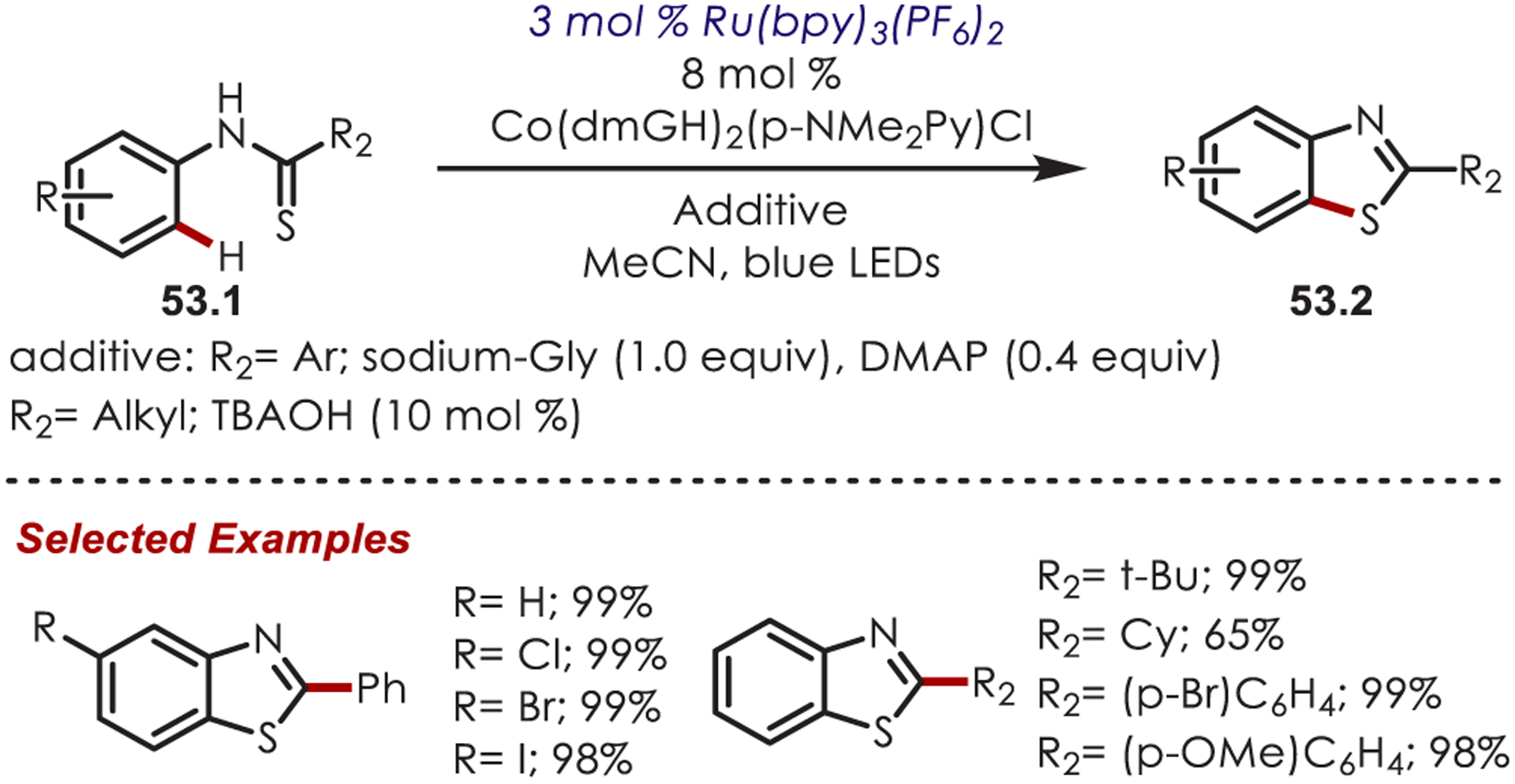
External Oxidant-Free Variant of the Sulfur-Centered Radical Cyclization for the Synthesis of Benzothiazoles
With several mechanistic studies in hand, the authors proposed the following mechanism (Scheme 54): photoexcited Ru(bpy)32+* oxidizes the thioamide anion (54.1) generating the SCR (54.2). Single-electron transfer from the resulting Ru(bpy)3+(E1/2red[Ru(bpy)3+/Ru(bpy)32+] = −1.33 V vs SCE) to Co(III) (E1/2red[CoIII/CoII] = −0.83 V vs SCE) turns over the ruthenium catalyst. Following cyclization of the SCR onto the arene, the resulting cyclohexadienyl radical (54.3) was oxidized by CoII and deprotonated by the base to produce the final benzothiazole (54.4). The resulting CoI species is protonated to CoIII–H and reacts with another proton to release H2, allowing this protocol to be dual catalytic in both the photooxidant and cobaloxime. This method was successful with electron-rich and poor aryl thioamides.
Scheme 54.
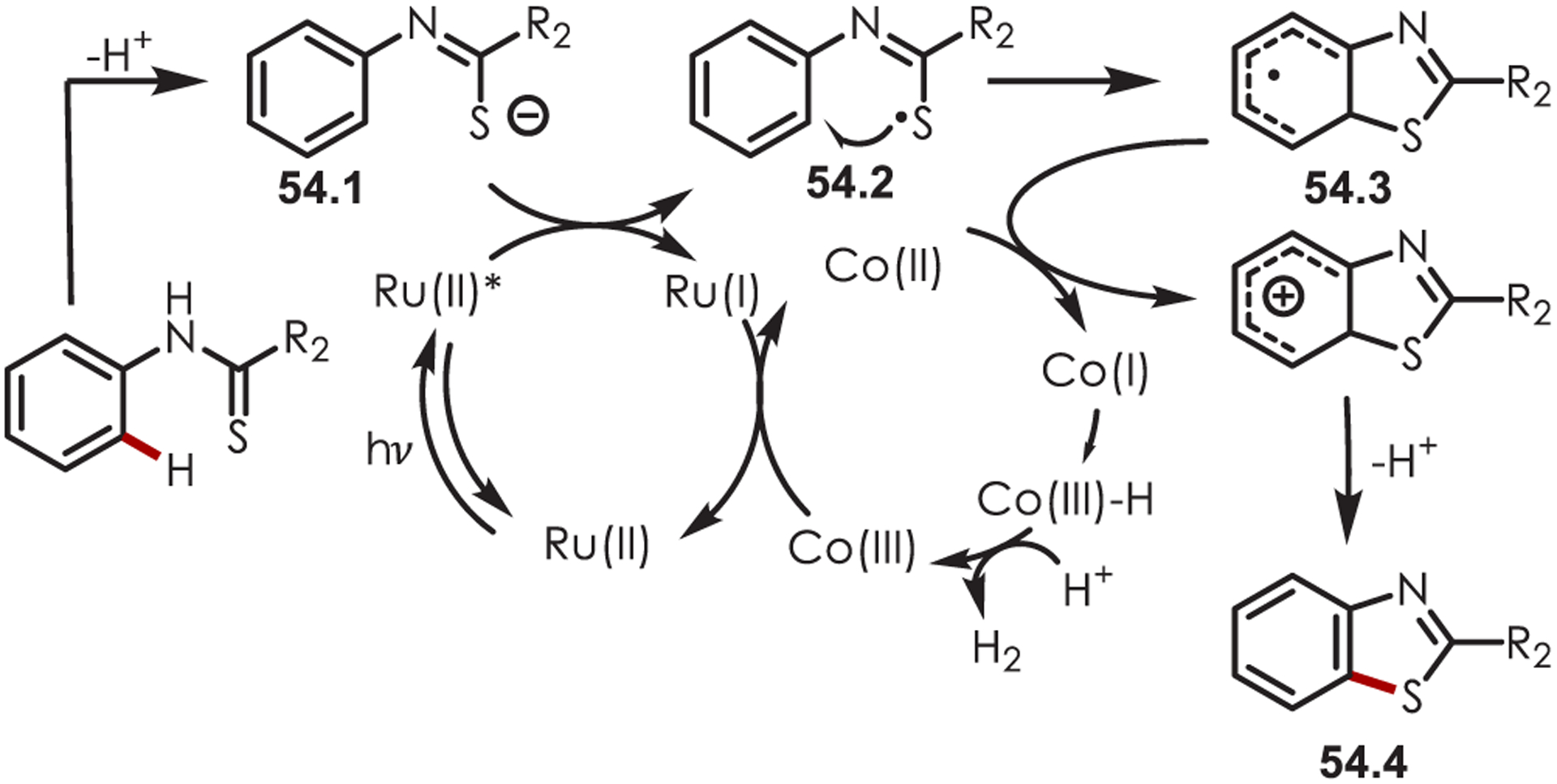
Mechanism for the Synthesis of Benzothiazoles through Sulfur-Centered Radicals
In 2012, the direct C3 sulfenylation of indoles (55.1) with arylsulfonyl chlorides (55.2) was reported by Zheng (Scheme 55).149 While the exact mechanism of this transformation is not known, the authors propose the sulfenylating agent is an aryl hypochlorothioite which is generated by multiple reductive SETs to an intermediate sulfonyl radical.
Scheme 55.
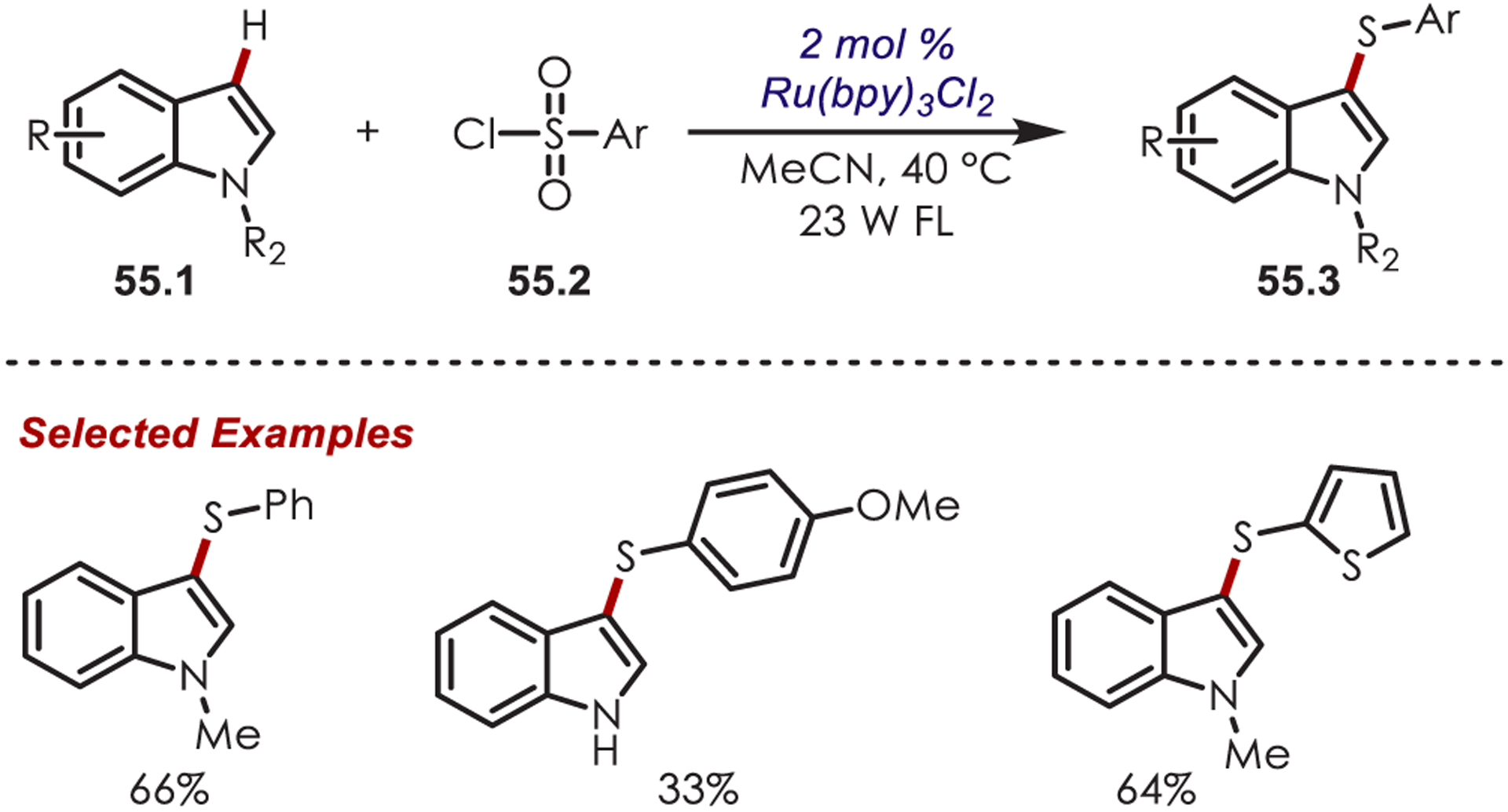
Sulfonyl Chlorides in a Heteroaryl C–H Sulfenylation
Similarly, sodium sulfinates (56.2) have served as SCR precursors in the C4 sulfonylation of naphthylamine derivatives (Scheme 56).150 The authors propose that an arylsulfonyl radical is generated from oxidation by photoexcited Ru(bpy)32+* or from K2S2O8. The copper co-catalyst plays an important role as it is proposed to ligate to the naphthylamide and oxidize the naphthalene to the corresponding cation radical. Combination of the sulfonyl radical and naphthylamide cation radical followed by rearomatization and deprotonation gives the product (56.3). The authors note the addition of the copper and persulfate additives were crucial for the success of the reaction. Substitution on the picolinamide directing group was tolerated and both alkyl and aryl sulfonates were suitable sulfonylating reagents. If the C4 position was unoccupied, sulfonylation was completely regioselective; otherwise, sulfonylation occurred ortho to the picolinamide group.
Scheme 56.
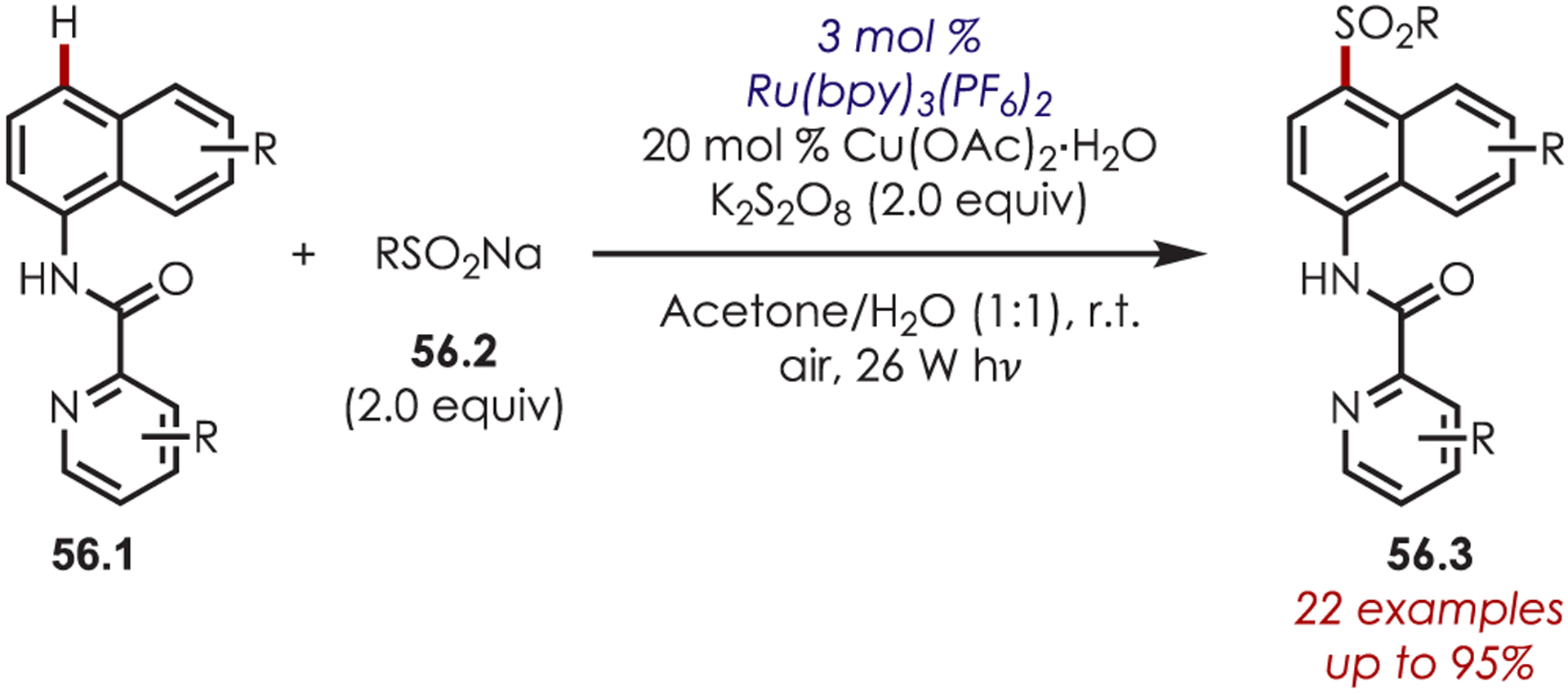
Aryl C–H Sulfonylation Using Sodium Sulfinates
Reiser subsequently reported the sulfonylation of heterocycles by sulfonyl chlorides (57.2) using an Ir(III) photoredox catalyst (Scheme 57).151 Oxidative quenching of IrIII* with the arylsulfonates generates the sufficiently stable sulfur-centered radical. At room temperature, this undergoes a radical addition and produces the sulfonylated heteroarenes. However, the authors found that at 45 °C the sulfonyl radical undergoes SO2 extrusion, producing an aryl radical species, which can subsequently undergo the radical addition to give the arylated products (57.3). Both alkyl and aryl sulfonates were suitable coupling partners in the sulfonylation of indoles, pyrroles, thiophenes, and thiazoles.
Scheme 57.
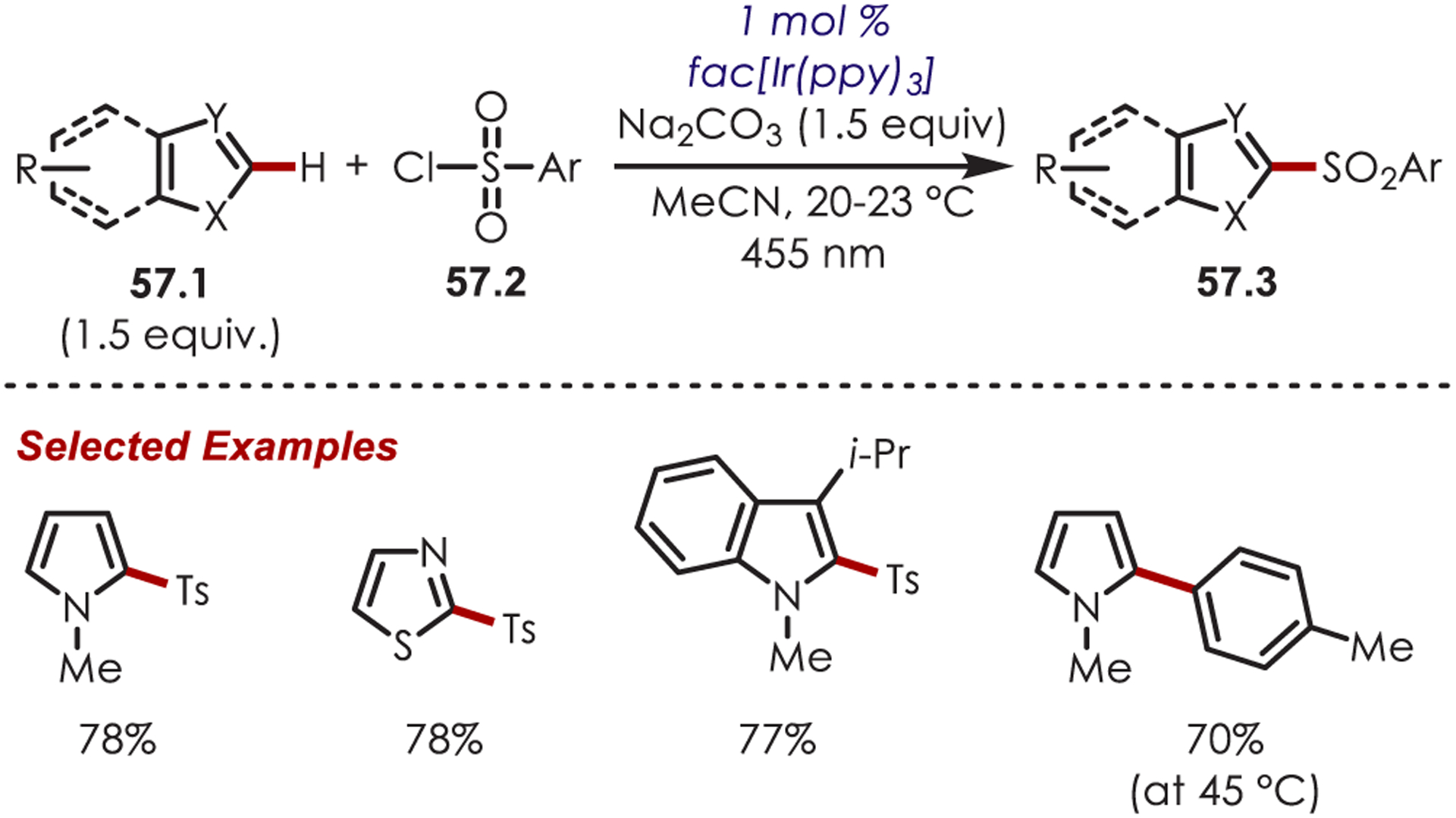
Aryl C–H Sulfonylation Using Sulfonyl Chlorides
2.1.5. Transformations Involving Charged Open-Shell Intermediates.
C–H functionalizations via intermediate arene cation radicals have been accomplished using Ru(II) photoredox catalysts; however, in general, inorganic catalysts are less frequently utilized for direct oxidation of arenes (generally Eox > + 1.0 V vs SCE)55 likely due to their low reduction potentials compared to organic photooxidants (E*red = +0.77 V for Ru(bpy)3).28 Therefore, direct oxidation of arenes by inorganic photoredox catalysts are only thermodynamically favorable for highly electron-rich arenes (i.e., dialkoxy or trialkoxy arenes). Consequently, any charged open-shell species are typically formed indirectly through redox processes that are thermodynamically feasible.
In 2016, Pandey and co-workers reported an arene amination of alkoxy arenes (58.1) with azoles (58.2) using Ru(bpy)3Cl2 (Scheme 58).152 The mechanism proceeds via a PET between excited Ru(II)* and Selectfluor, producing Ru(III) and Selectfluor cation radical (58.4). Ru(III) undergoes a SET with an electron-rich arene to generate an electrophilic arene cation radical (58.5). The aminated arenes are produced after nucleophilic addition of the azole, deprotonation, and subsequent hydrogen atom abstraction. The scope of this amination is limited to mono-, di-, and trialkoxy arenes but is tolerant of electronically diverse azoles including benzazoles, imidazoles, and tetrazole.
Scheme 58.
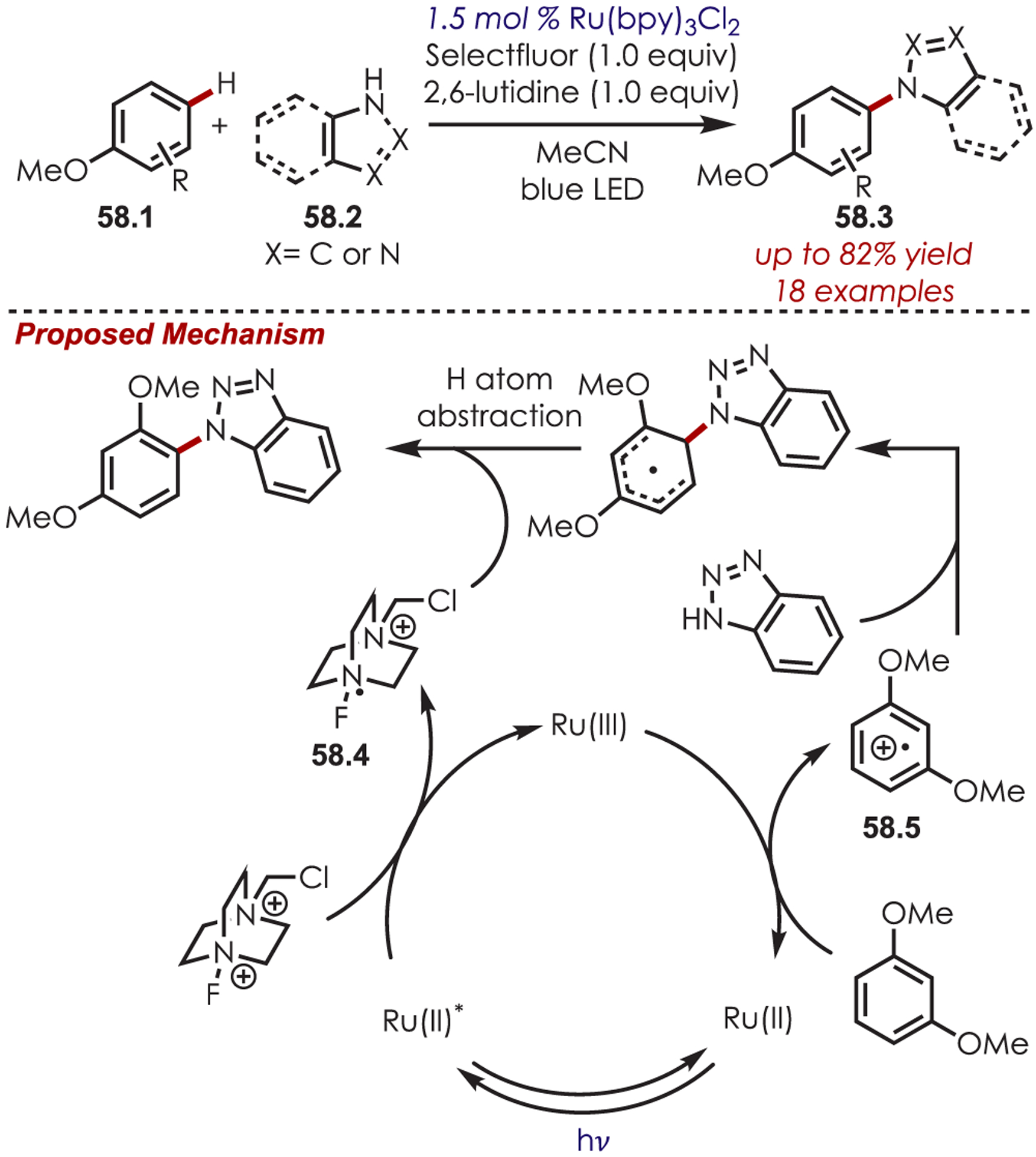
Alkoxy Aryl C–H Amination with Azole Nucleophiles via Aryl Cation Radical Intermediates
Extremely electron-rich arenes, such as tri- or dimethoxyarenes (Eox = +1.12 and +1.45 V vs SCE, respectively) can undergo a thermodynamically favored PET by photoexcited Ru(II) (Ered* = +1.45 V vs SCE). The resulting aryl cation radical (59.4) has been functionalized by König and co-workers in an arene C–H phosphonylation reaction (Scheme 59).153 Ammonium persulfate is required as both a sacrificial oxidant for the regeneration of Ru(II) from Ru(I) and for the generation of a sulfate anion in situ. The transient phosphonium intermediate (59.6) rearranges in the presence of the sulfate anion to the desired aryl phosphonate. Electron-rich heteroarenes, such as indoles, dimethoxypyridine, and dimethoxythiophene, were also successfully phosphonylated.
Scheme 59.
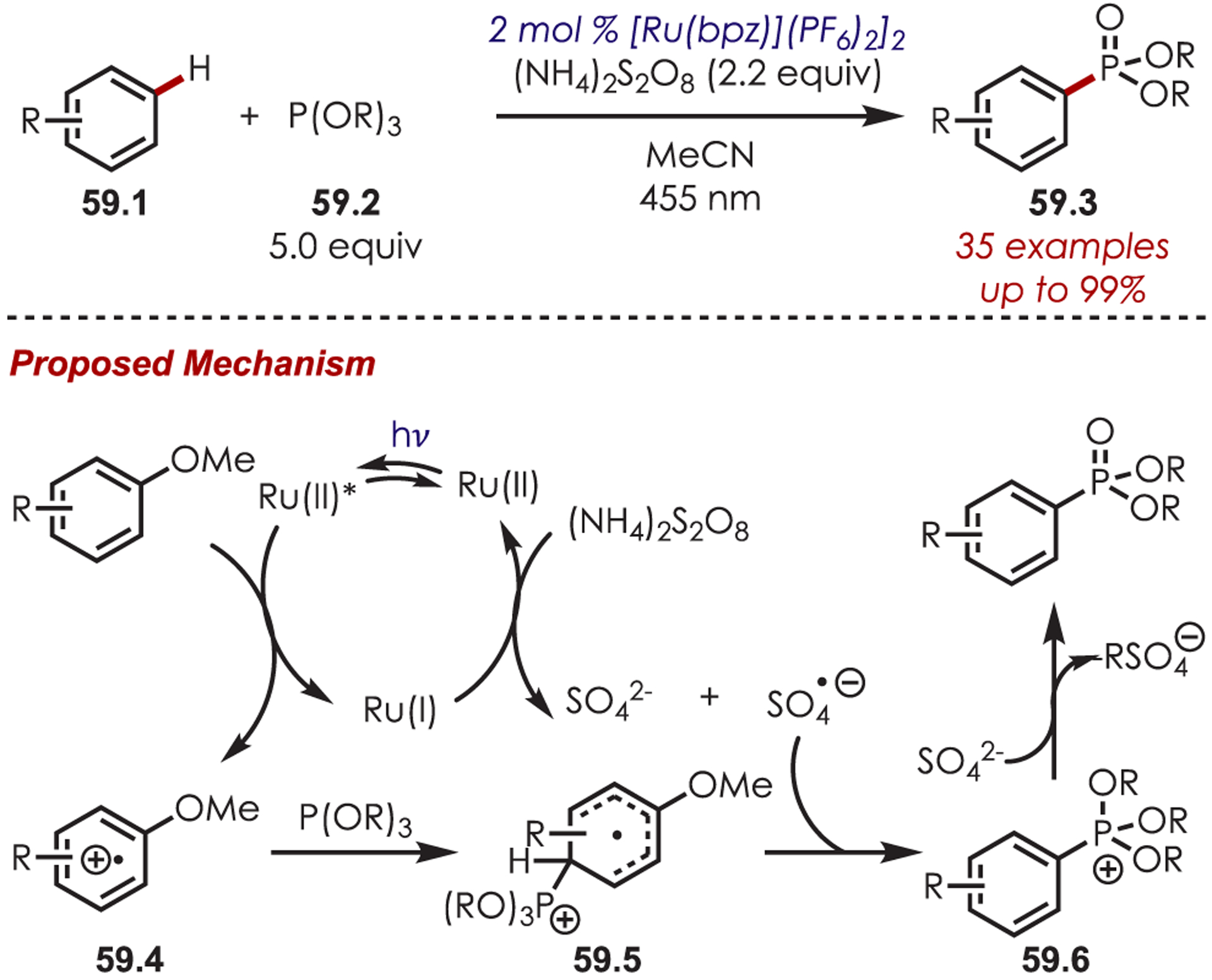
(Hetero)aryl C–H Phosphonylation through Aryl Cation Radicals
In 2019, Carreira and Ritter independently reported a C–H amination of arenes via pyridyl cation radicals.154,155 The mechanism of this amination proceeds through oxidative quenching of Ru(II)* by the O-triflylpyridine N-oxide, 60.4 (Scheme 60). The resulting pyridyl cation radical (60.2) couples with a (hetero)arene producing an intermediate cyclohexadienyl radical (60.3). A subsequent oxidation by Ru(III) and deprotonation furnishes the aminated products (60.4) while regenerating the photocatalyst.
Scheme 60.
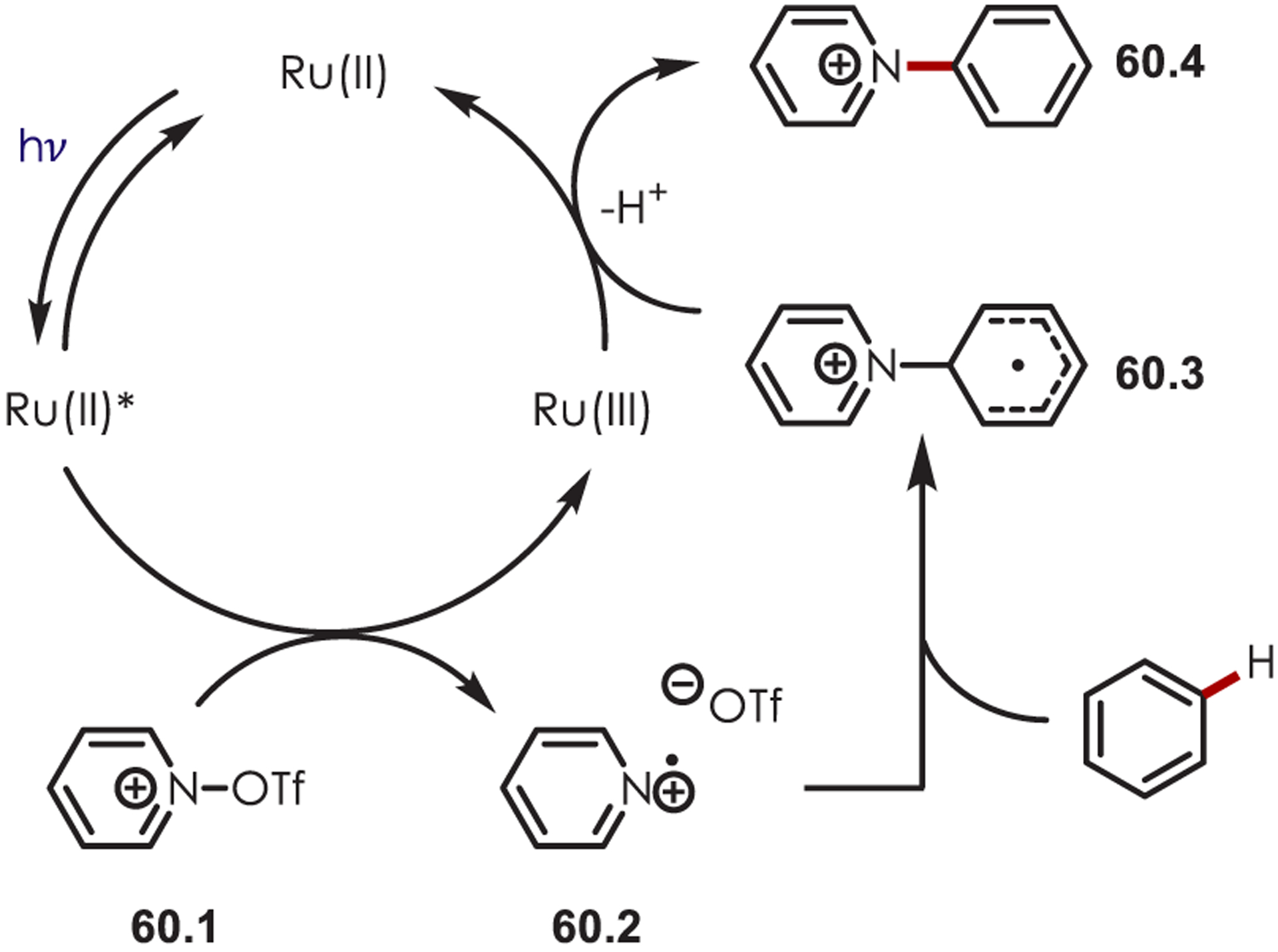
Mechanism of an Aryl C–H Amination via Pyridyl Cation Radicals υ
Carreira’s method was suitable with both electron-rich and electron-poor (hetero)arenes (61.1) with high functional group tolerance, including nitriles, nitro, ethers, amides, and esters (Scheme 61). The authors extended this methodology for an in situ Zincke aminolysis of the pyridinium by the addition of 10 equiv of piperidine, which generated the corresponding anilines. The N-arylpyridinium could also be hydrogenated by PtO2 to afford the N-arylated piperidines.
Scheme 61.
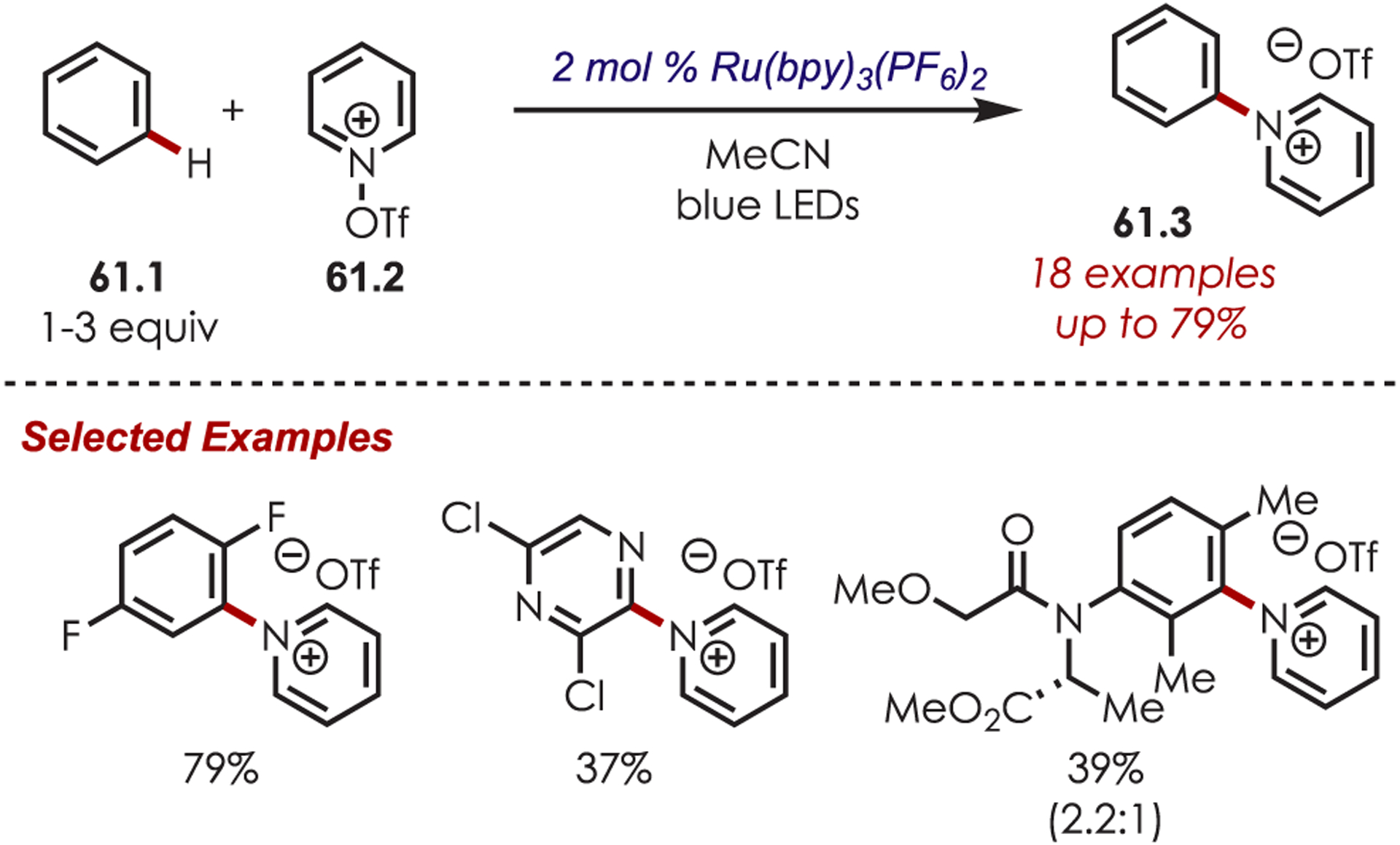
Scope of Aryl C–H Amination via Pyridyl Cation Radicals Developed by Carreira
Likewise, the method developed by Ritter and co-workers was applicable in the late-stage C–H aminations of more complex (hetero)arenes (Scheme 62). This highlights the utility of pyridinium cation radical species for accessing C–H functionalization of substrates that would typically be too electron-poor by other measures. Some noteworthy examples include sulfamethoxazole (62.4), an antibiotic, and vismodegib, an anticancer agent (62.5).
Scheme 62.
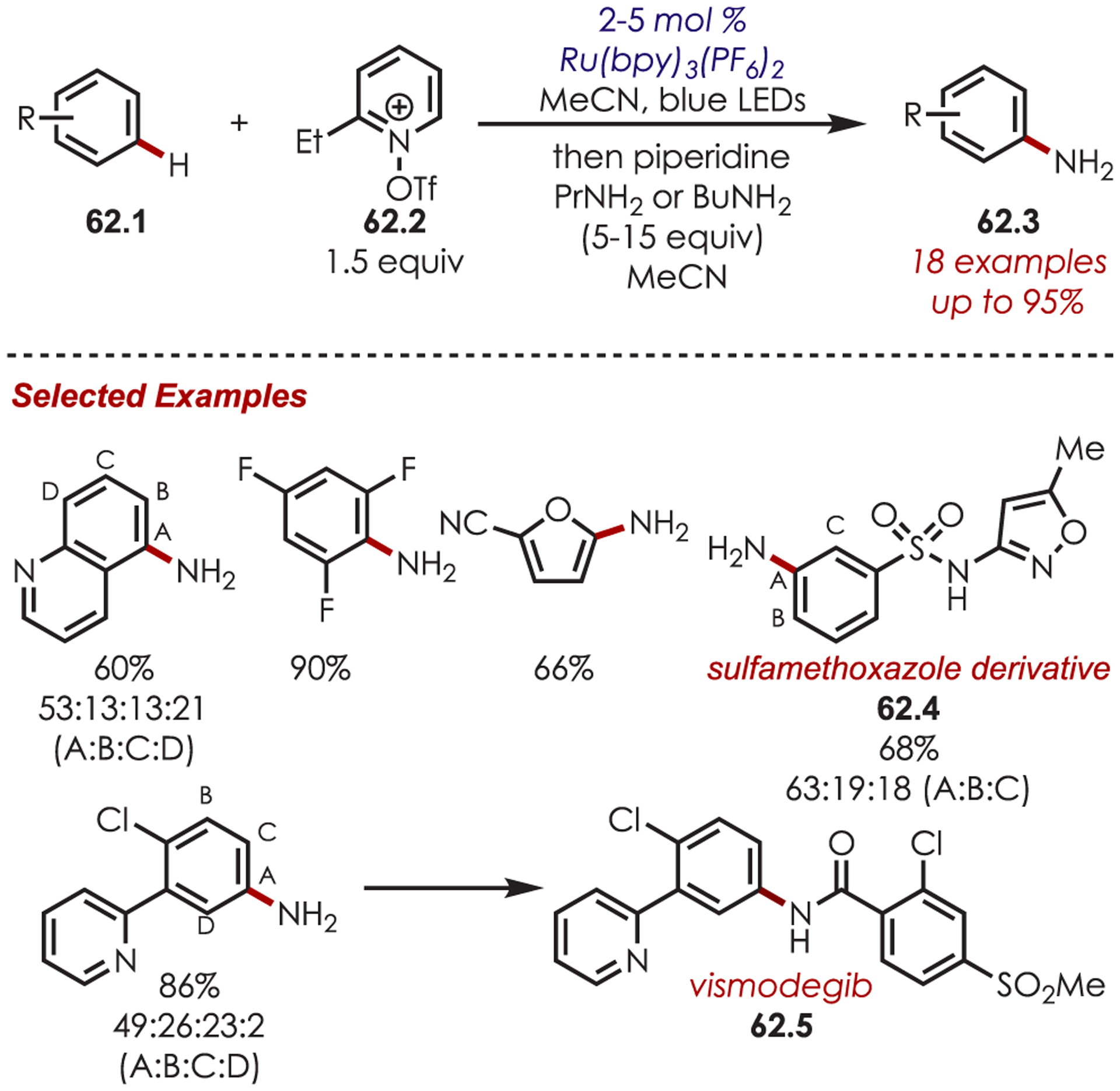
Scope of Aryl C–H Amination via Pyridyl Cation Radicals Developed by Ritter
Recently, Kano reported a Ru(III) catalytic system that overcomes the thermodynamic barrier for PET between (hetero)arenes and inorganic catalysts by forming intermediate N-arylpyridinium ions (63.3) in situ (Scheme 63).156 This system aminates more electron-poor arenes with oxidation potentials (Eox = +1.65 to +2.27 V vs SCE) outside the range of the catalyst (Ered[RuIII/RuII] = +1.37 V vs SCE). Ru(III) is generated by oxidative quenching of photoexcited Ru(II)* with ammonium persulfate. While electron transfer is unfavorable between Ru(III) and the arenes (up to 22.5 kcal/mol more endergonic), the authors take advantage of a known equilibrium system, similar to that reported by Kochi,157 that generates cation radicals through SET of the arene with Ru(III). The cation radical is then trapped by pyridine generating an N-arylpyridinium ion. The desired arylamine (63.2) is formed after treatment with pyrrolidine. Alternate mechanisms involving sulfate radical anions, formed photochemically or thermally, are excluded by Stern–Volmer quenching which showed only efficient quenching of Ru(II)* by ammonium persulfate. Conducting the reaction under thermal conditions capable of producing sulfate radical anions failed to give the desired aryl amine. While this offers an alternative avenue to amination of arenes with electron transfer disfavored up to 22.5 kcal/mol, it failed to aminate benzene, where ET is endergonic by 28.7 kcal/mol.
Scheme 63.
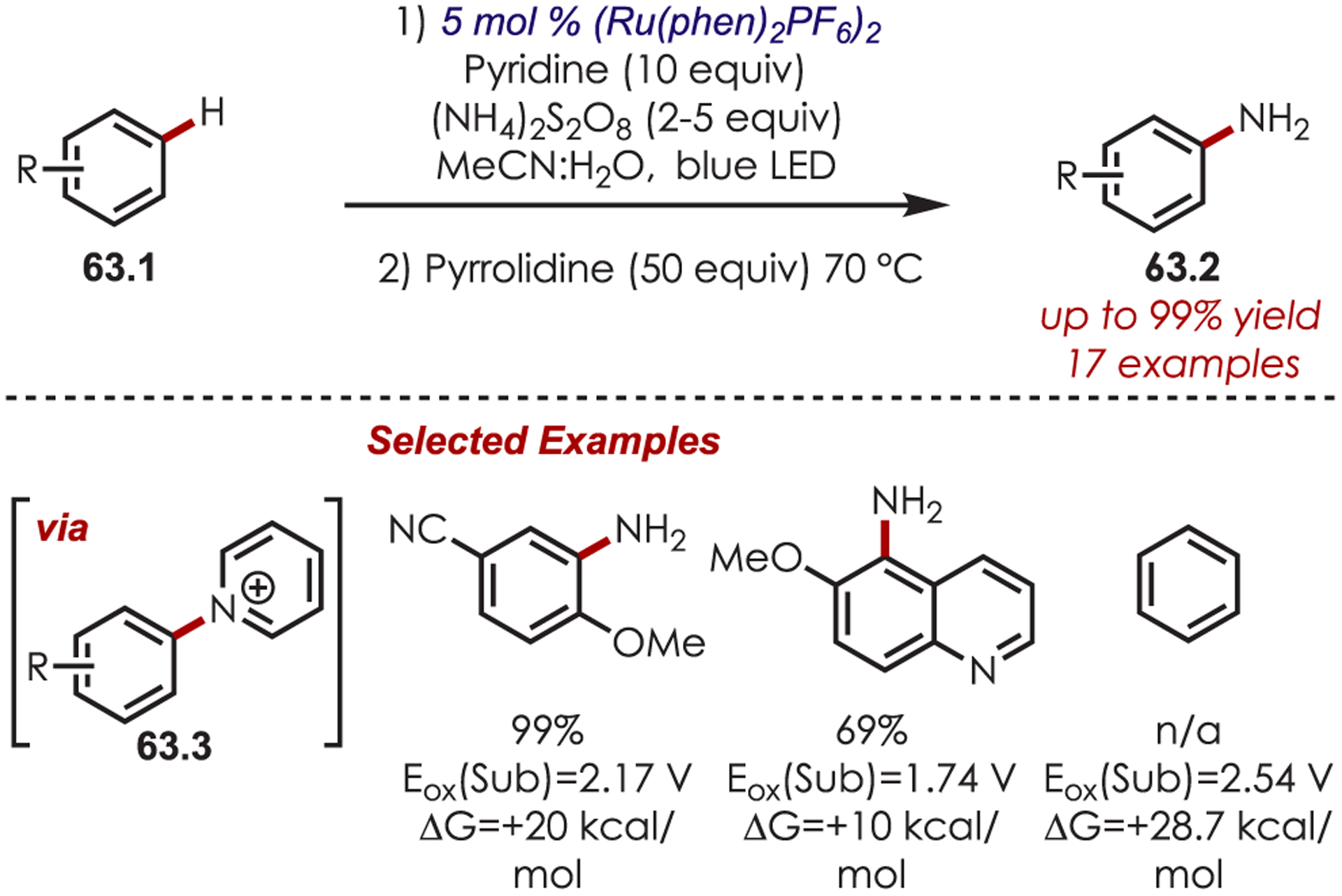
In Situ Generation of Pyridyl Cation Radicals for Aryl C–H Aminations
α-Amido sulfides (64.2) have been utilized as imine precursors in the coupling of nucleophilic (hetero)arenes (Scheme 64).158,159 SET from the amido sulfide to Ru(III) generates an intermediate sulfur cation radical (64.4) and Ru(II). A fragmentation of the C–S bond results in the intermediate iminium ion (64.5) and thiyl radical. Photoexcited Ru(II)* oxidizes molecular oxygen, regenerating the active Ru(III). Nucleophilic (hetero)arenes, cyanide, and 1,3-dicarbonyls were compatible with these methods, furnishing the desired products.
Scheme 64.
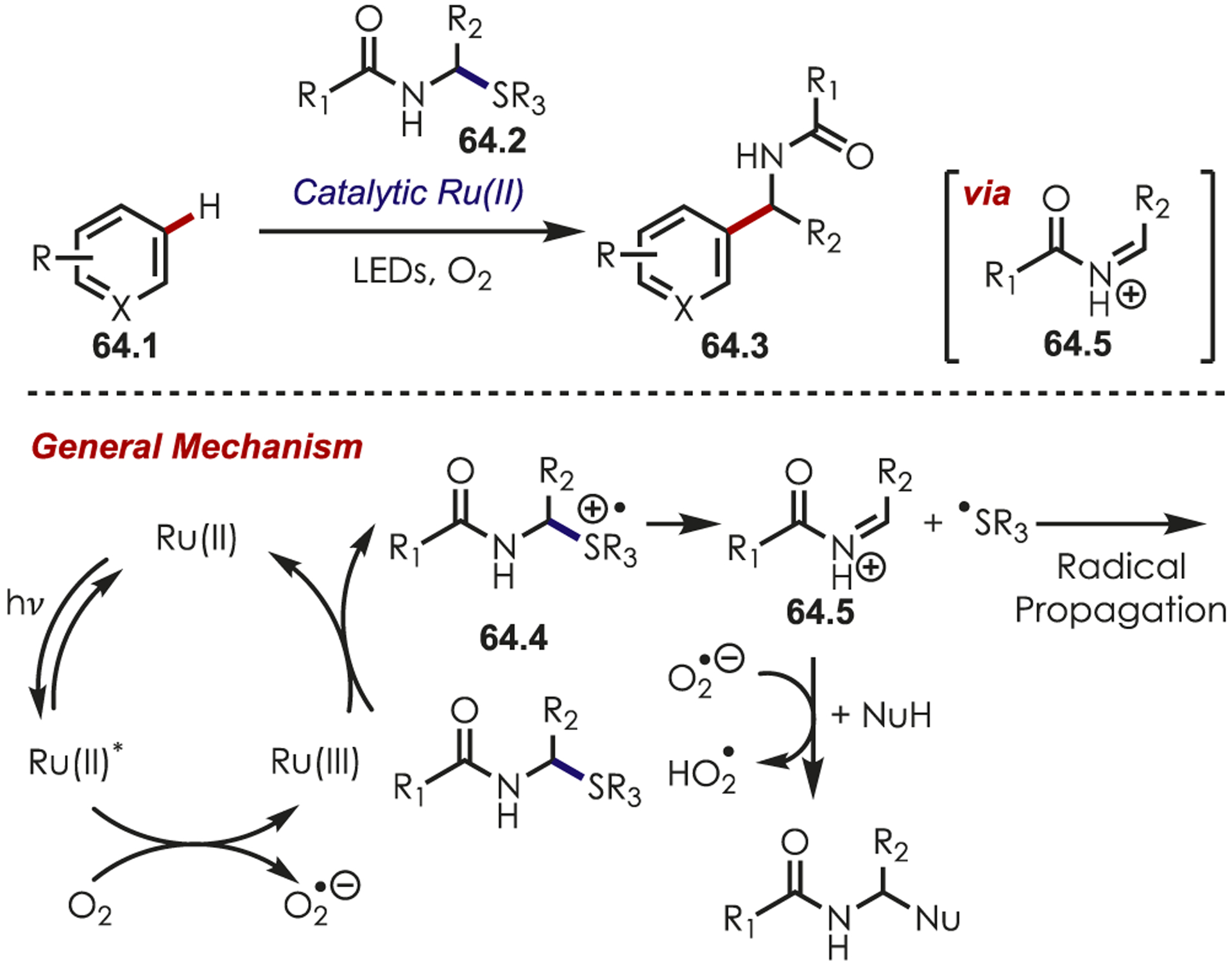
Coupling of Imines, Generated by α-Amido Sulfides, and Heteroarenes for C–H Functionalization
Generation of iminiums, via the corresponding nitrogen cation radical, has also been demonstrated for the functionalization of indoles (Scheme 65).160 Nucleophilic attack from the indole (65.1) to the iminium gives access to a range of 2-substituted indoles (65.3) with amino acid derivatives.
Scheme 65.
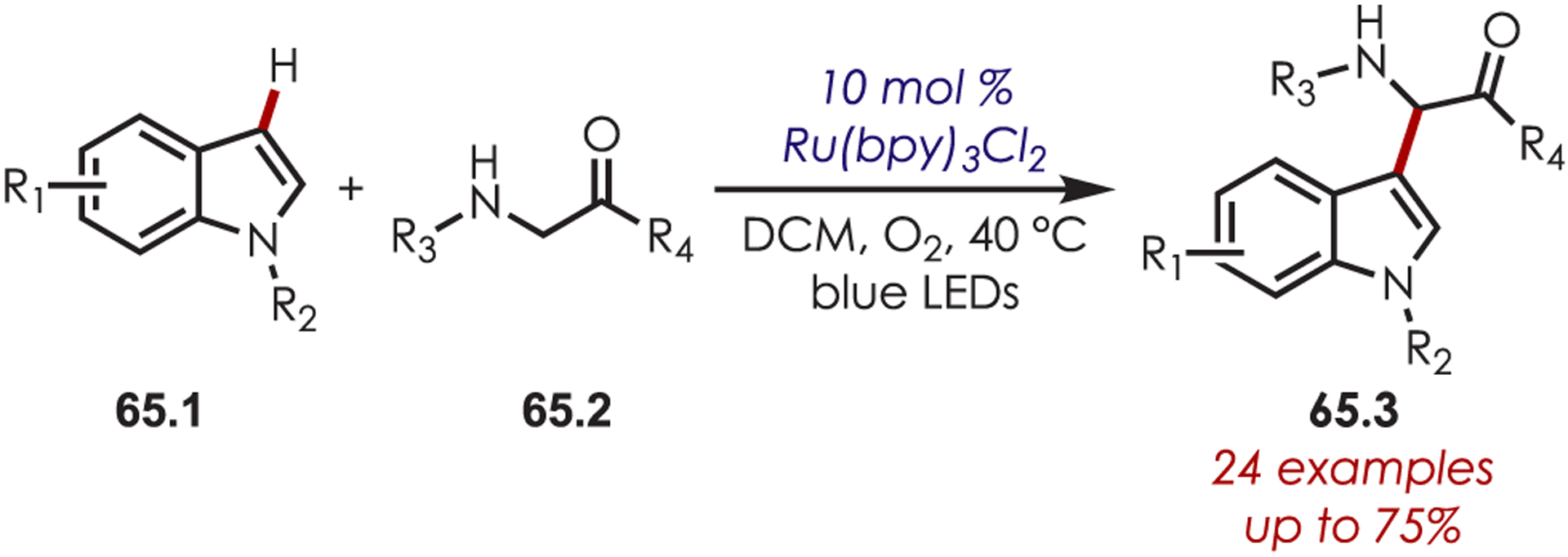
Indole C–H Functionalization with Photogenerated Iminium Ions
2.1.6. Miscellaneous.
Inorganic photoredox catalysts have been used in combination with transition-metal catalysts as an alternative to strong oxidants for the generation of reactive organometallic interemediates.37 In particular, a combination of rhodium and ruthenium catalysis has unlocked dual C–H activation and C–H functionalization of arenes. Rueping and co-workers were among the first to report a dual-catalyzed ruthenium/rhodium C–H olefination. The use of a Ru(II) photoredox catalyst is key for the reoxidation of a rhodium catalyst (Scheme 66).161 The Rh–hydride complex (66.3), is oxidized by photoexcited Ru(II)* to regenerate the active Rh catalyst (66.4). Molecular oxygen completes turnover of the photoredox catalytic cycle. This method provides an alternative to other metal-catalyzed oxidative Heck reactions, which typically require a directing group, prefunctionalization, stoichiometric external oxidant, and/or moderately harsh reaction conditions.
Scheme 66.
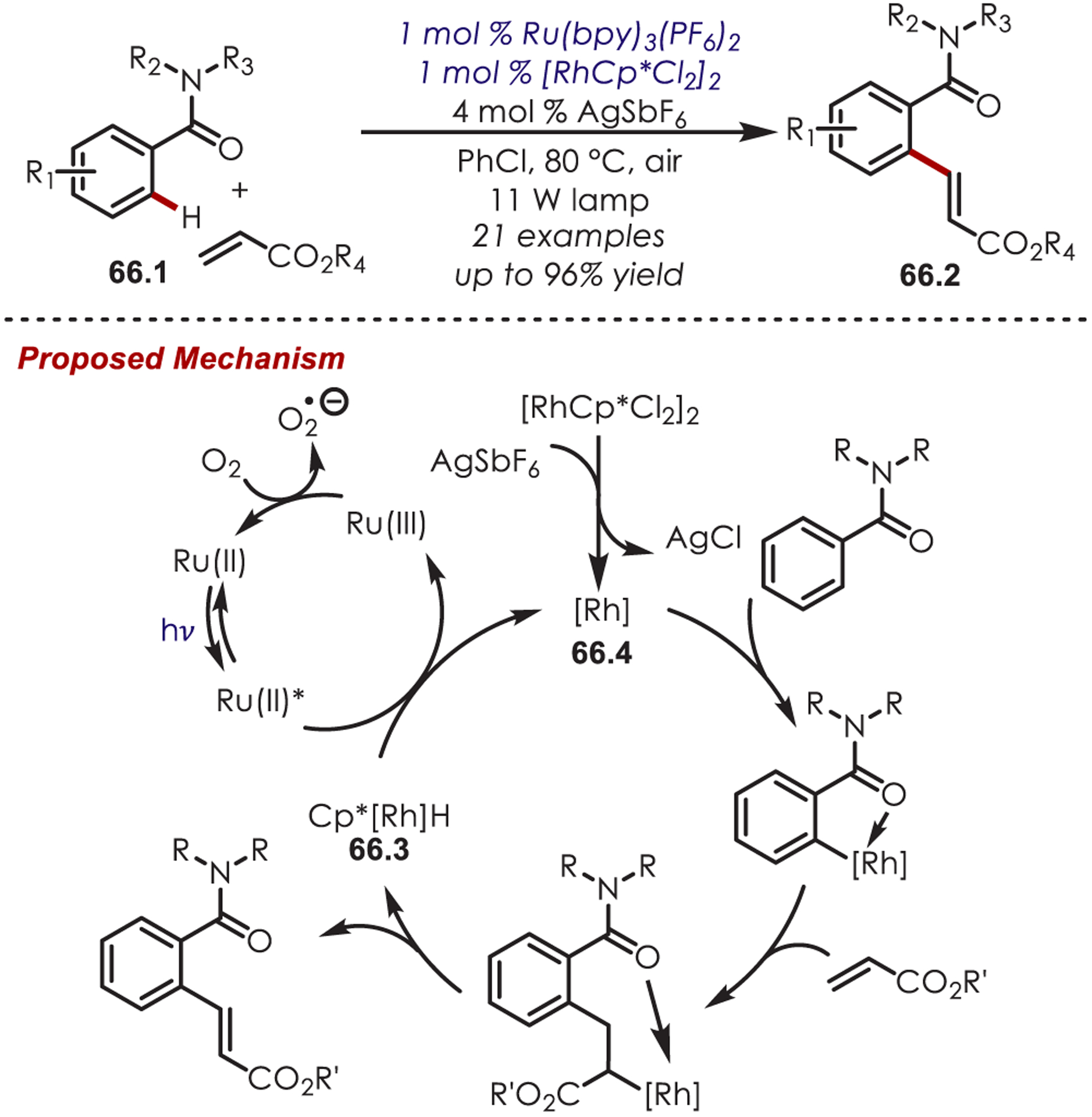
Heck-Type Reaction Using a Photoredox Catalyst as an Oxidant
A C–H ortho-olefination of o-(2-pyridyl)phenols (67.1) was later reported by Rueping using Ir(bpy)(ppy)2 as the catalyst (Scheme 67).162 This method was tolerant of various functional groups, including aldehydes, ketones, and esters; additionally, coupling with various acrylate esters was also demonstrated in good yields. The addition of the acetic acid as an additive was found to decrease the formation of an undesired byproduct that arose from hydrogenation of the olefin.
Scheme 67.
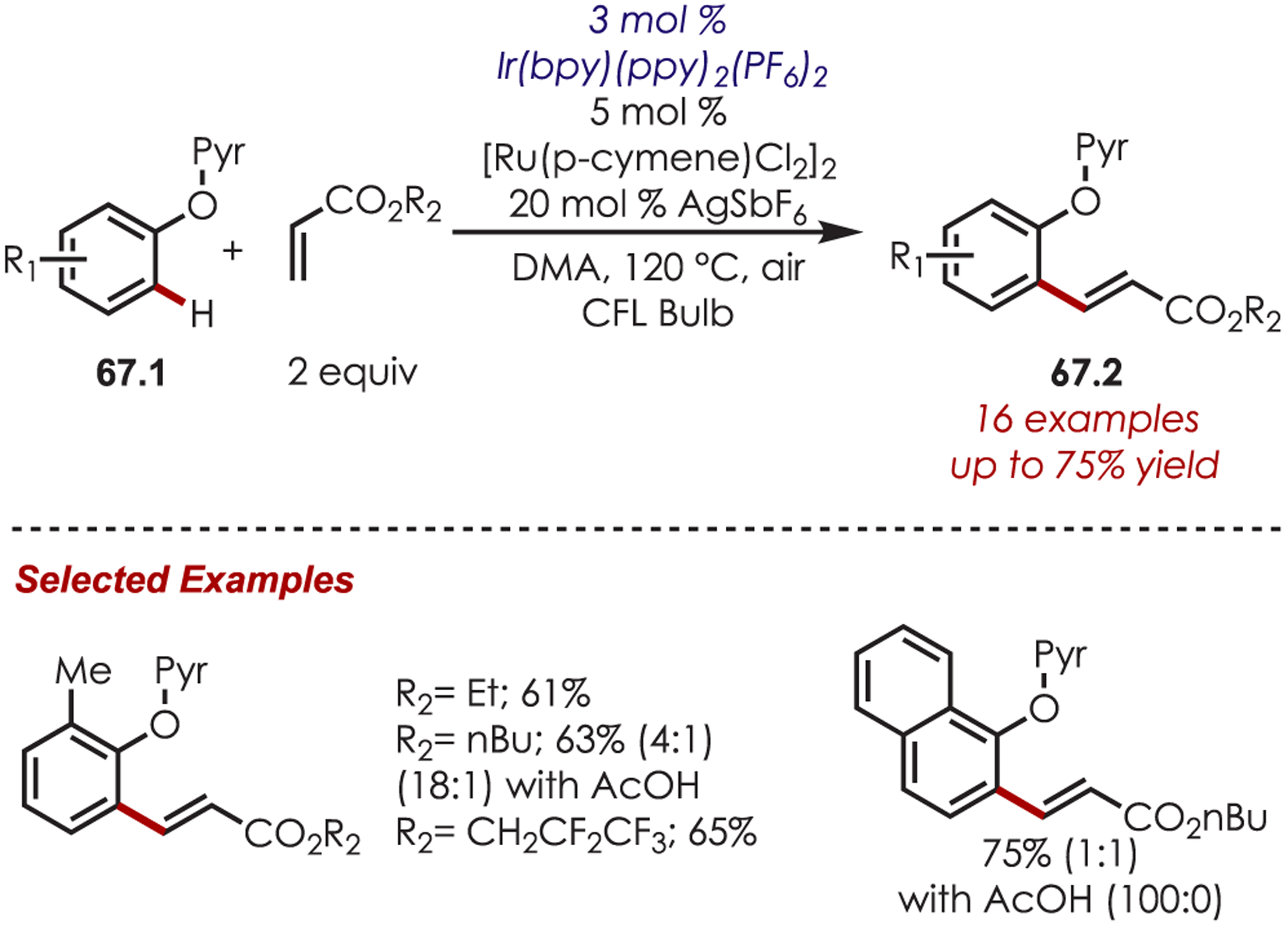
Ortho-Selective C–H Olefination of Phenol Derivatives
An intramolecular derivative of the ortho-C–H activation and functionalization was extended to the synthesis of indole derivatives 68.2 (Scheme 68).163 In this case, a Pd(II) catalyst leads to the intermediate Pd–H species, and Ir(III)*, along with O2•−, oxidizes Pd(0) to Pd(II).
Scheme 68.
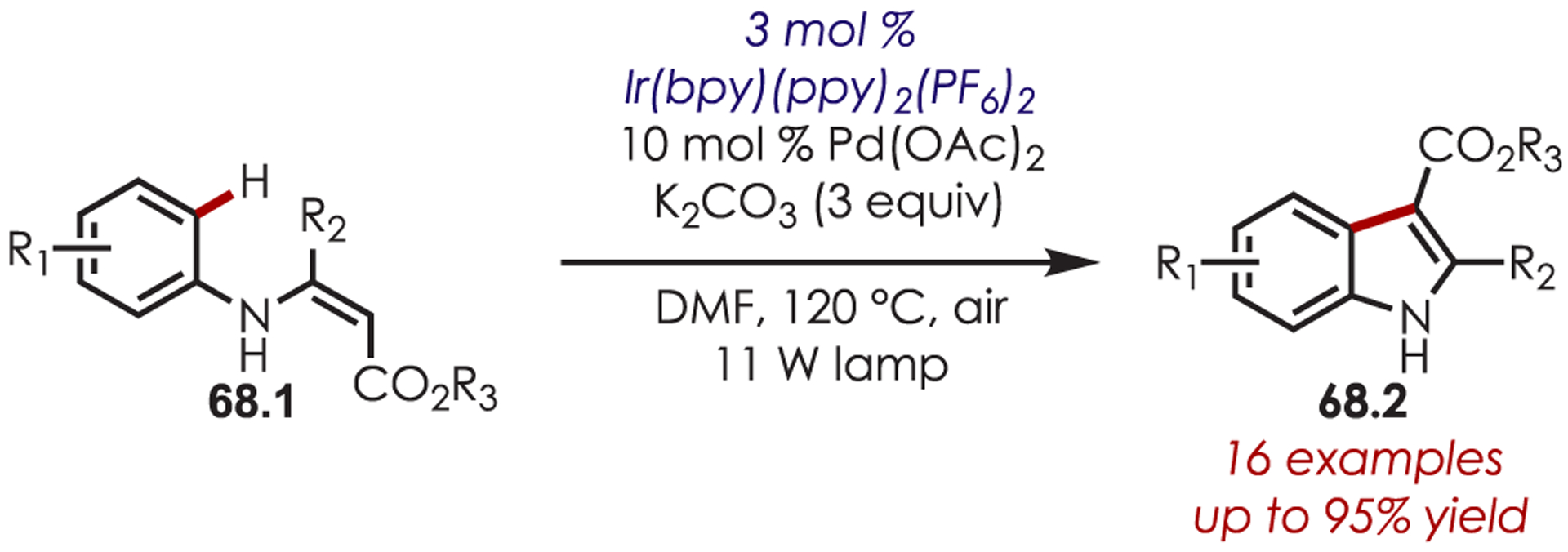
Intramolecular C–H Ortho-Olefination for the Synthesis of Indole Derivatives
Cho and co-workers reported a dual palladium- and visible-light-catalyzed intramolecular C–H amination of N-substituted 2-amidobiaryls (69.1). Again, photoexcited Ir(III) is used to regenerate the active Pd catalyst (Scheme 69).164 Electrochemical and transient photoluminescence spectroscopy were utilized to study the reaction mechanism and suggest that SET from a palladacyclic intermediate (69.2) to the photoexcited Ir(IV)* is at play. Molecular oxygen is responsible for regenerating both the photoredox catalyst and Pd(II) from Pd(I).
Scheme 69.
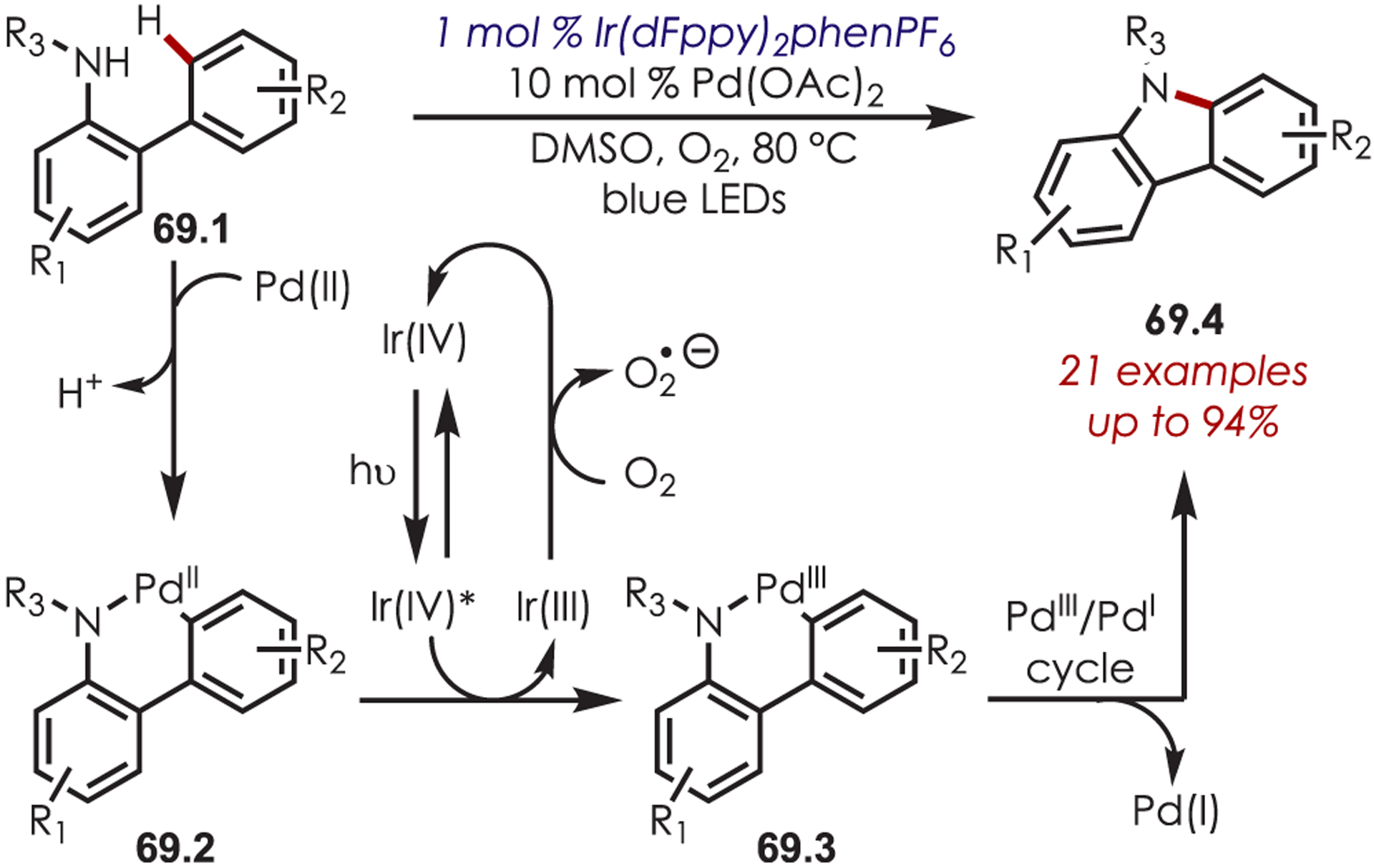
Dual Palladium and Photoredox Catalysis for an Intramolecular C–H Amination
A photoexcited Ru(II) complex has been utilized for a dual meta-C–H activation and alkylation of arenes (Scheme 70).165 A cyclometalated Ru(II) species (70.4) was found to be the active photoredox catalyst, and after excitation, it undergoes a SET with an alkyl halide (70.2). This generates a carbon-centered radical and a Ru(III) cyclometalated complex (70.5). Nucleophilic attack of the radical occurs at the para- position of the arene, relative to Ru(III). The high selectivity for para-functionalization is attributed to the active Ru(II) species rather than Ru(III) undergoing SET, which is noted by Ackermann and co-workers to be more selective.166 A LMCT then regenerates Ru(II), and a deprotonation/rearomatization furnishes the meta-functionalized arene (70.3). The arene coupling partner was tolerant of electron-donating or -withdrawing substituents, as well as halogenated arenes. Other directing groups including isoquinolines, pyrimidine, pyrazole, and purine were successful. Both cyclic and acyclic alkyl bromides and iodides were competent carbon radical precursors. Piperidinyl and azetidine derivatives gave the products in 62% and 58% yield, respectively.
Scheme 70.
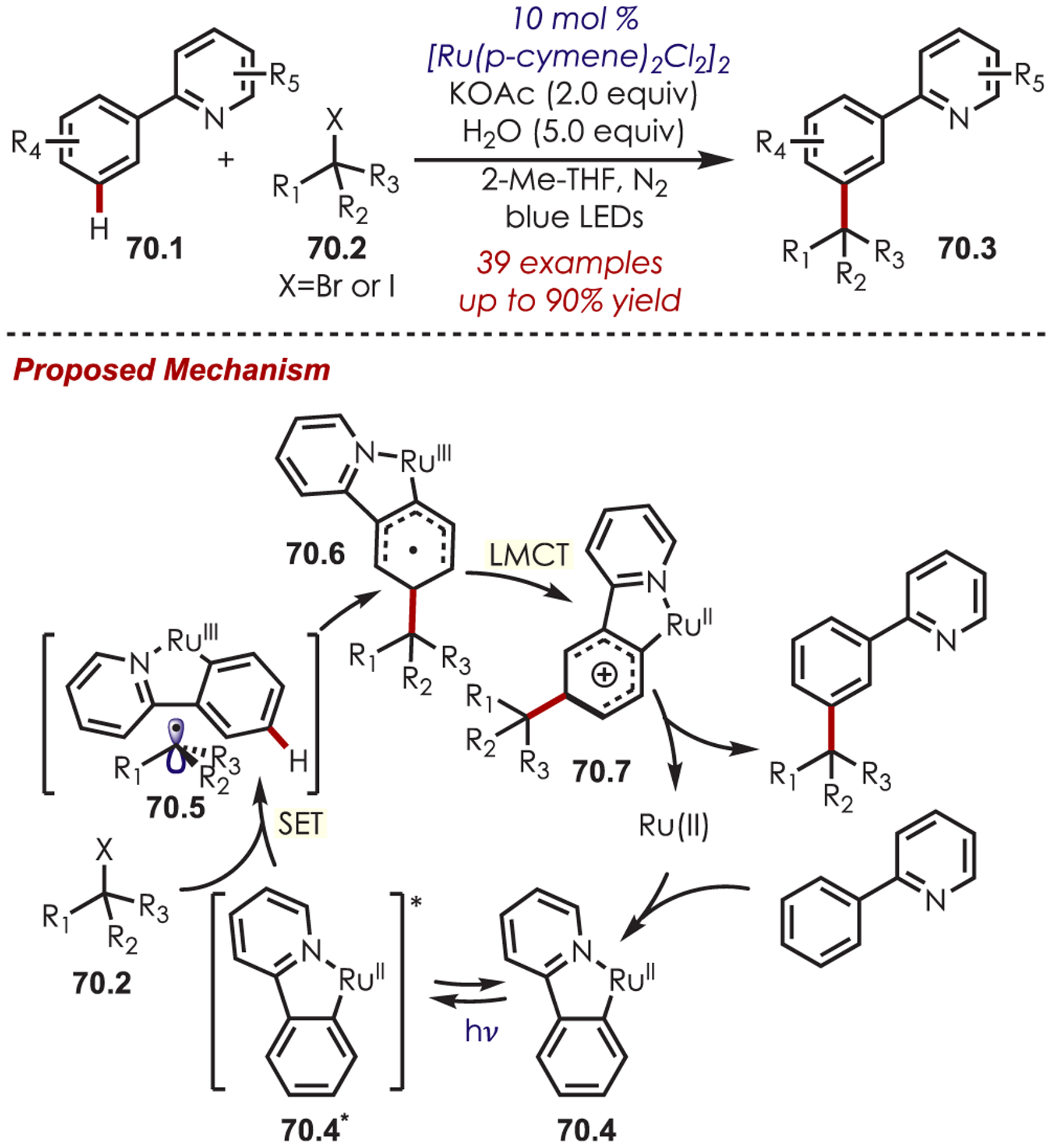
Directed Meta-C–H Alkylation of Pyridyl-Substituted Arenes
2.2. Aliphatic C–H Functionalization
C–H functionalization of activated C–H bonds, typically those with a BDE < 92 kcal/mol, is generally accomplished through hydrogen atom abstraction pathways. These include C–H bonds which are α to a heteroatom (BDE = 90.1 kcal/mol for pyrrolidine167 or 92.0 kcal/mol for THF),168 α to a carbonyl (BDE = 88.0 kcal/mol for cyclohexanone),169 or at benzylic (BDE = 89.9 kcal/mol for toluene),170 allylic (BDE = 88.8 kcal/mol for propylene),170 or aldehyde (BDE = 88.9 kcal/mol for acetylaldehyde)171,172 positions are commonly accessed through one of these weaker C–H bonds. However, the functionalization at more remote and unactivated aliphatic C–H bonds with BDEs > 92 kcal/mol has remained a significant challenge, but C–H abstraction strategies have also been utilized to accomplish a range of C–X bond-forming reactions.
2.2.1. α-Heteroatom C–H Functionalization.
Functionalization of amines has been a longstanding synthetic interest due to their prevalence in naturally occurring molecules, agrochemicals, materials, and pharmaceuticals.16 As a result, there has been interest in accomplishing transformations involving amine-containing substrates through C–H functionalization pathways and particularly through photoredox catalysis.
In general, photoredox activation of amines (71.1) occurs through two major pathways that both proceed first through an oxidation event, generating an intermediate amine cation radical (71.2). Then the amine cation radical can form either an electrophilic iminium ion (71.3) or a nucleophilic α-amino radical (71.4) (Scheme 71).27,173–175 Functionalization α- to oxygen and sulfur has also been explored mainly through the photochemical generation of radicals at that position. The contrasting electronic nature of these intermediates has been exploited in several synthetic strategies and many methods have been reported for either nucleophilic or electrophilic coupling α- to heteroatoms.
Scheme 71.
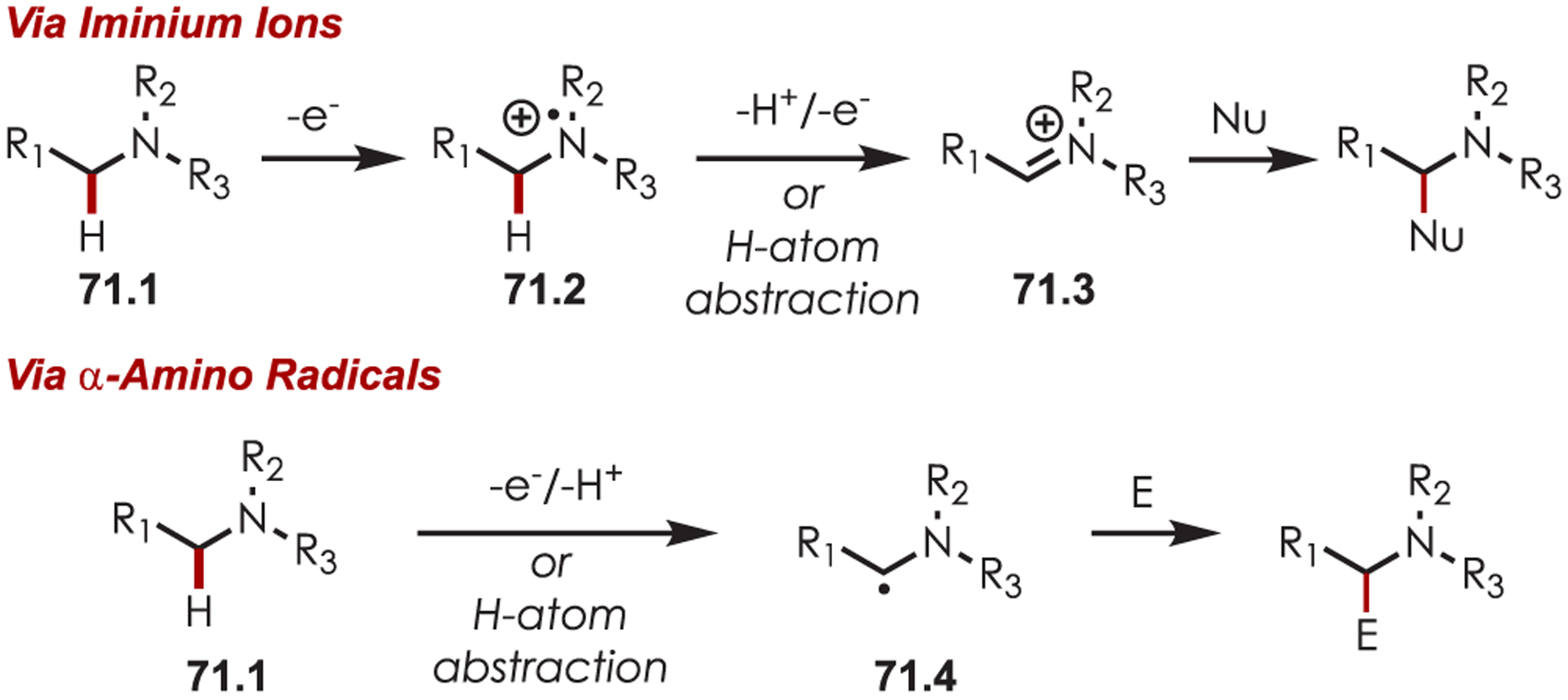
General Pathways for α-Amino C–H Functionalization via the Generation of Electrophilic Iminium Ions or Nucleophilic α-Amino Radicals
Via Iminium Ions.
Generation of amine cation radicals has been reported through metal or electrochemical pathways.176–179 In 2010, Stephenson first reported the development of α-amino C–H bond functionalization utilizing inorganic photoredox catalysis in an aza-Henry reaction (Scheme 72).180 The authors found that Ir(ppy)2(dtbbpy)PF6 was an optimal catalyst, undergoing reductive quenching of the excited state with an amine (72.1) to generate amine cation radicals (72.3). A subsequent hydrogen atom abstraction generated an electrophilic iminium ion (72.4) that can be trapped with a nucleophile. Reduction of nitromethane, or molecular oxygen, is required to regenerate Ir(III) from Ir(II) (Ir(III)/Ir(II) = −1.51 V vs SCE). The resulting radical anion is proposed to be responsible for the generation of the iminium ion. This photoredox catalyst allowed for α-alkylation of N-aryl tetrahydroisoquinolines in excellent yields using nitromethane or nitroethane as the nucleophile. A nonbenzylic amine, N-phenylpyrrolidine, also gave the alkylated product, however, in 27% yield. The authors noted that while the corresponding Cu(I) facilitated aza-Henry reactions with TBHP were faster,181 this method furnished the desired C–H-functionalized products in higher yields.
Scheme 72.
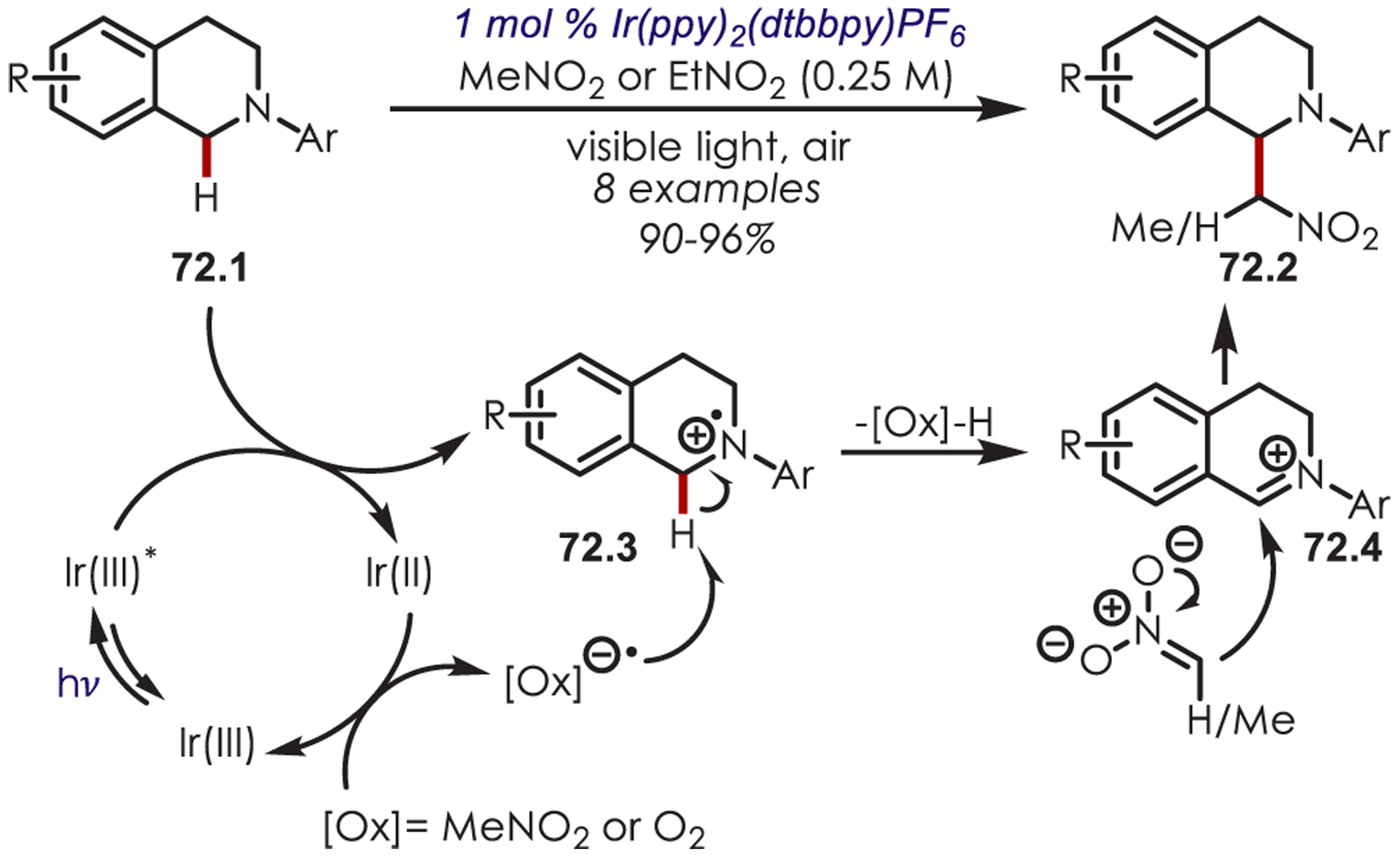
Mechanism of the Aza-Henry Reaction for the C–H Alkylation of Tetrahydroisoquinolines
Following these initial reports of α-amino functionalization, Stephenson and co-workers disclosed improved conditions for the aza-Henry reaction (Scheme 73).182 Using BrCCl3 as the stoichiometric oxidant in DMF allowed for efficient generation of the iminium ion with full conversion in 3 h.183 The resulting iminiums were trapped with a range of diverse nucleophiles including cyanide, copper acetylides, enolates, electron-rich aromatics and malonates to give the functionalized products (73.2) in 43–95% yields. These transformations were also reported in flow for an increased reaction scale (>1 g), which also showed improved reaction times.184
Scheme 73.
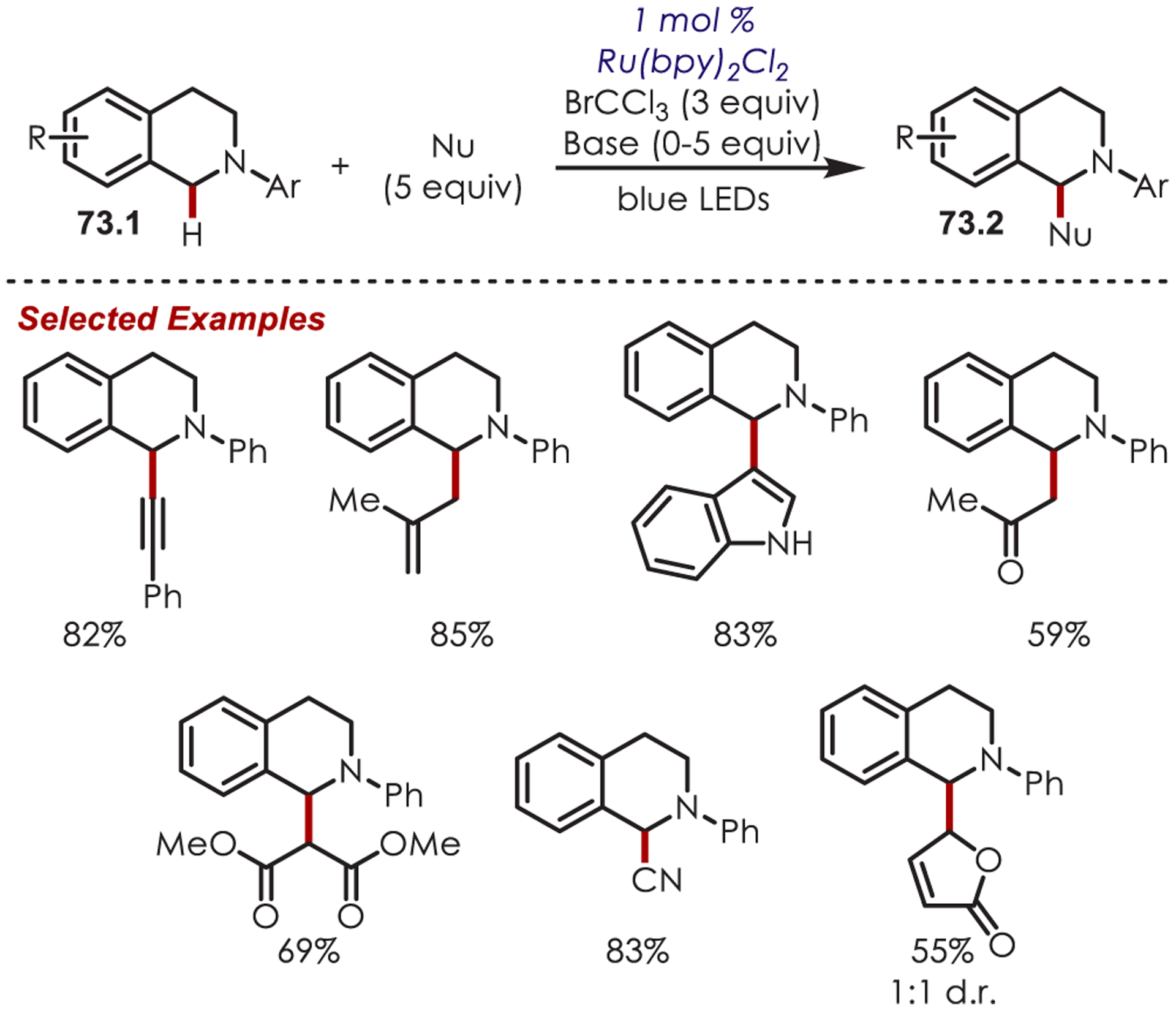
Scope of Elaborated Aza-Henry Reaction for the C–H Alkylation of Tetrahydroisoquinolines
In 2010, Fabry and co-workers published a direct Mannich reaction by combining photoredox and Lewis base catalysis (Scheme 74).185 A carefully executed interplay of the two catalytic cycles allowed for the catalyst to be turned over without oxygen, which was found to be critical to obtain high yields by preventing the iminium from rapidly undergoing undesired side reactions (i.e., oxidation to an amide). Fabry employed Ru(bpy)3PF6 and proline catalysis to afford the Mannich-type products with tetrahydroisoquinolines (74.1) and acyclic ketones (74.2). The authors noted that more sterically hindered ortho-substituted N-aryl substituents gave decreased yields.
Scheme 74.
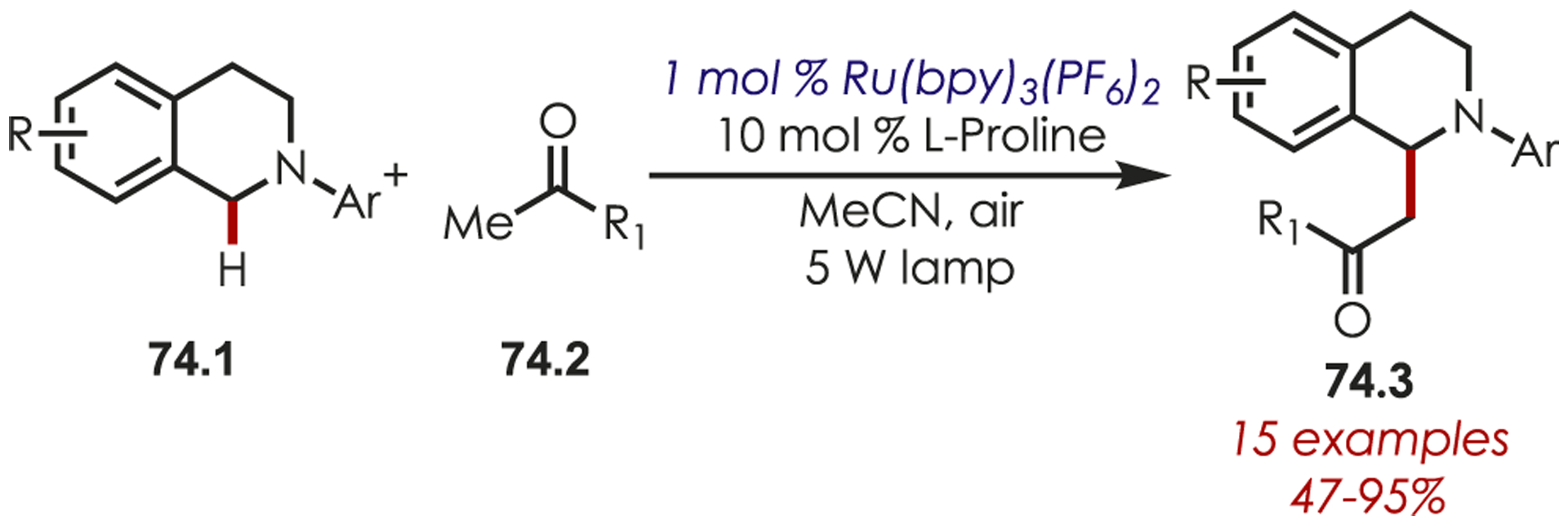
Mannich Reaction for the C–H Alkylation of Tetrahydroisoquinolines
Similarly, a Mannich-type reaction of tertiary amines (75.1) using silyl enol ethers (75.2) catalyzed by Ru(bpy)3Cl2 was reported by Xia (Scheme 75).186 The generated iminium ions were trapped with nucleophilic enolsilanes, which then eliminated trimethylsilyl cation to furnish the desired products (75.3).
Scheme 75.
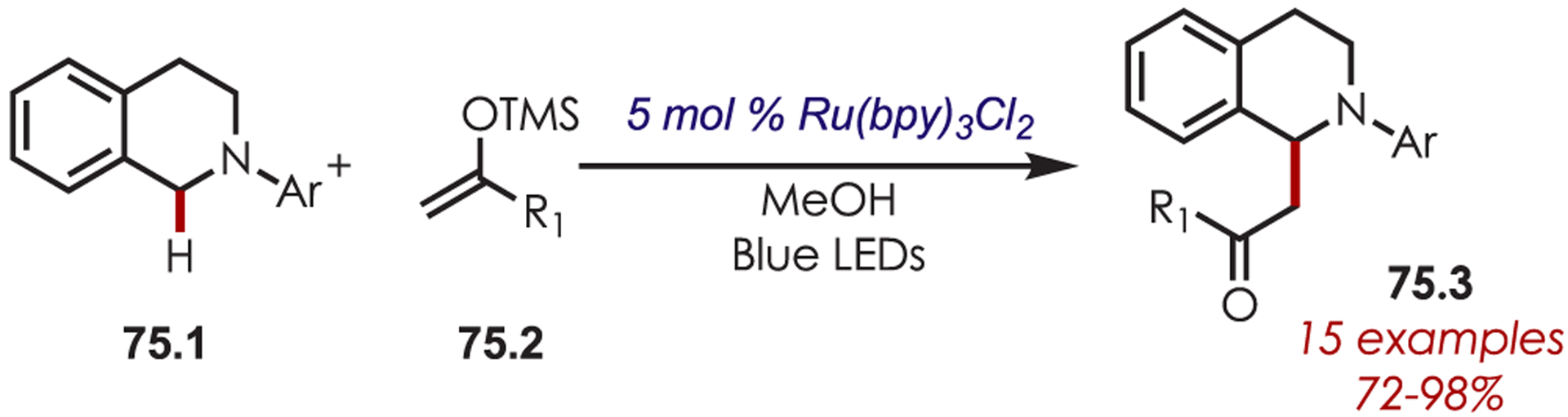
Mannich Reaction for the C–H Alkylation of Tetrahydroisoquinolines Using Silyl Enol Ethers as Nucleophiles
Other variants of α-functionalization for the cyanation (76.1),187 phosphonylation (76.2),188 copper-catalyzed alkynylation (76.3),189 alkyl- or arylation via organometallics (76.4),190 DABCO-mediated acroleination though nucleophilic catalysis (76.5),191 alkylation via decarboxylative addition of β-keto acids (76.6),192 or diastereoselective fluororoxoindolation193 (76.7) of tetrahydroisoquinolines have also been reported (Scheme 76).
Scheme 76.
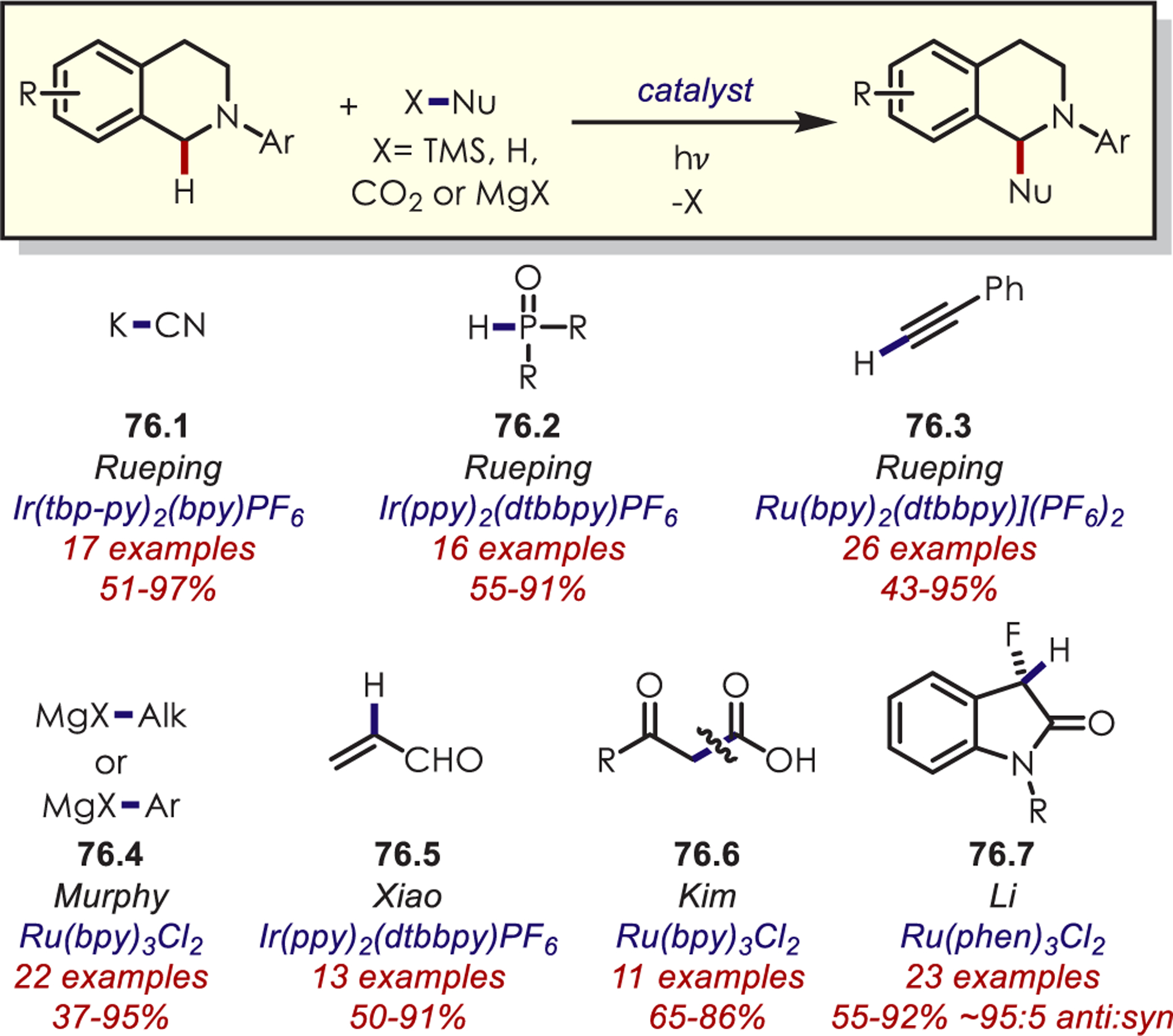
Other Nucleophiles for the Addition into Photoredox-Generated Iminiums for the C–H Functionalization of Tetrahydroisoquinolines
Additionally, photoexcited gold complexes have been utilized for generating tetrahydroisoquinioline iminiums with varying nucleophilic partners including cyanide (77.3),194 enolates (77.4), and phosphonate (77.5) nucleophiles (Scheme 77).195
Scheme 77.
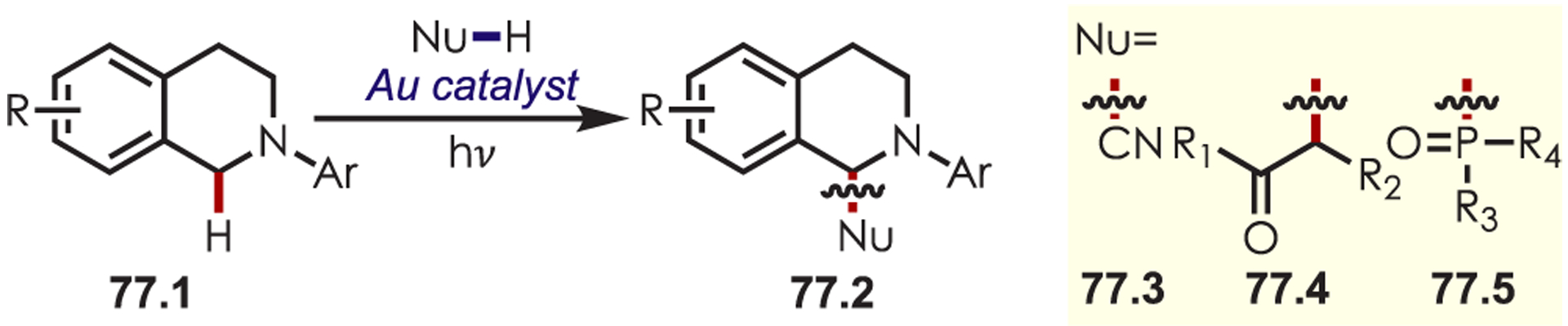
Dimeric Gold Complexes as Photoredox Catalysts in C–H Alkylation and Phosphonylation of Tetrahydroisoquinolines
N-Alkylation of tetrahydroisoquinoline derivatives (78.1 and 78.4) followed by an intramolecular α-amino cyclization can give access to isoquino[3,4-a][1,2,4]-triazines196 (78.3) and pyrazole (78.6) fused heterocycles, (Scheme 78).197 Both manifolds utilize Ru(bpy)32+ to generate an iminium intermediate.
Scheme 78.
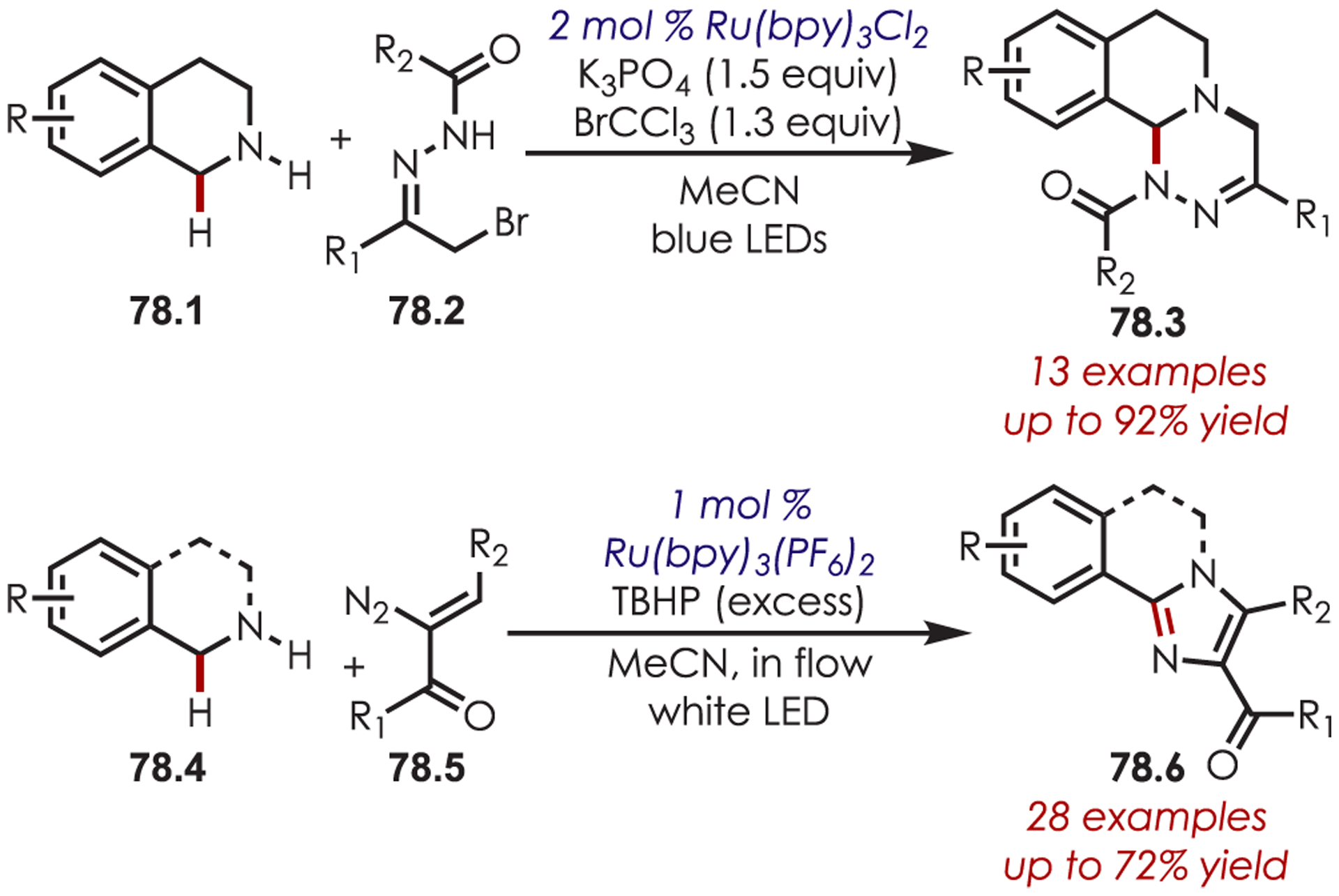
C–H Alkylation and Subsequent Cyclization of Tetrahydroisoquinolines
Similarly, the synthesis of pyrrole-fused isoquinolines (79.2) was accomplished by Xiao through deprotonation of the photoredox-generated iminium to afford a 1,3-azomethine ylide (79.3) (Scheme 79).198 A subsequent [3 + 2] cycloaddition and oxidative aromatization afforded the cyclized pyrrole products. Rueping independently published a [3 + 2] cycloaddition of photogenerated azomethine ylides for the synthesis of pyrrolo[2,1-a]isoquinolines the same year.199
Scheme 79.
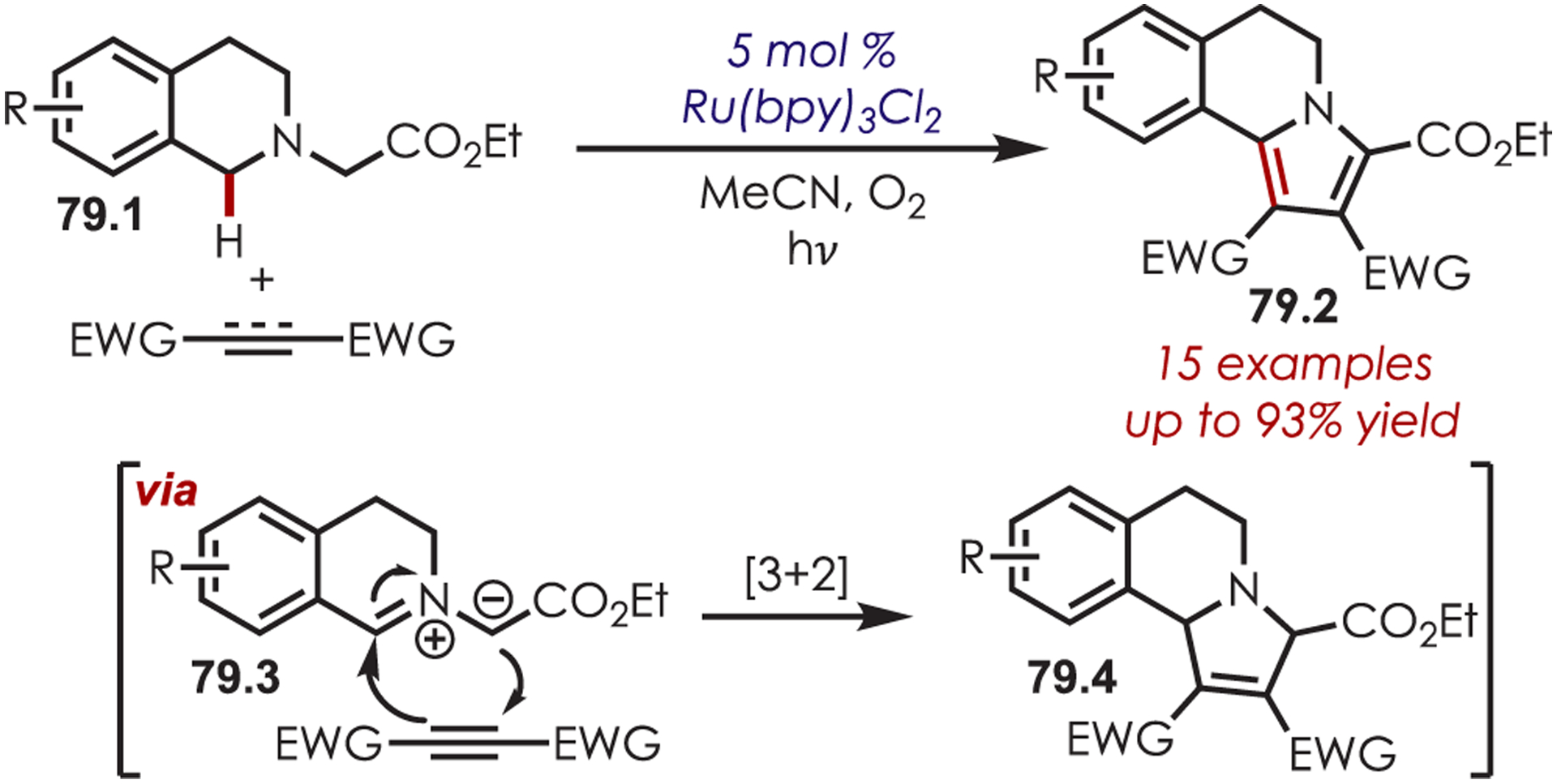
[3 + 2] Cyclization of Photogenerated Iminium Ions with Alkynes
Intramolecular cyclizations of diamines (80.1) and amino alcohol (80.2) derivatives were reported in the synthesis of isoquinopyrimidines (80.3) and isoquinooxazines (80.4) (Scheme 80).200 Ir(III)* serves as the photooxidant to generate an electrophilic iminium ion which can undergo subsequent addition from a tethered amine or alcohol nucleophile. Marvin later reported an analogous transformation for the synthesis of 1,3-oxazines using Ru(bpy)3Cl2.201
Scheme 80.
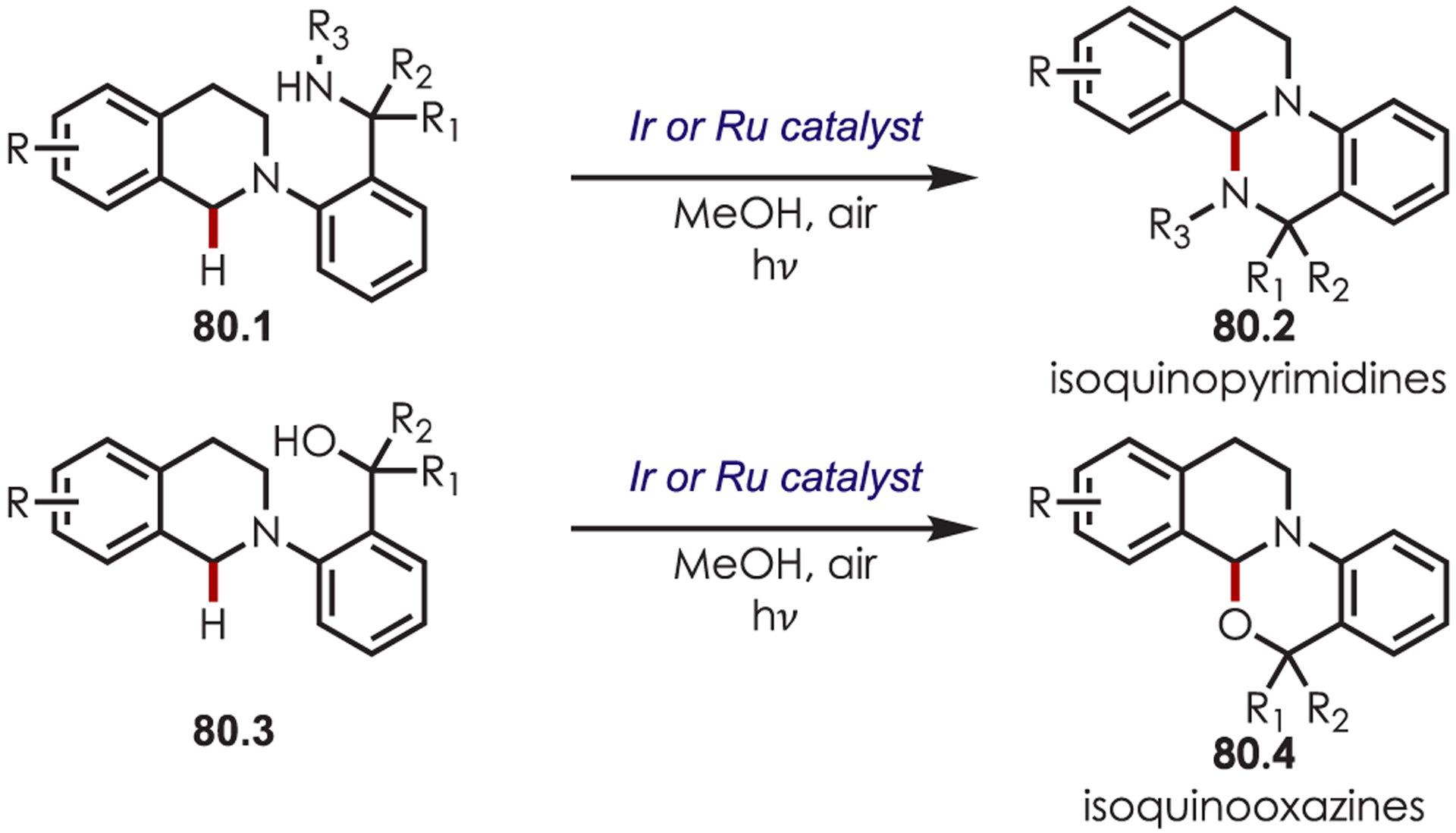
Intramolecular C–H Amination or Alkoxylation through Iminium Ions
Rovis and DiRocco developed the first asymmetric α-acylation of tetrahydroquinolines (81.1) by using dual NHC and photoredox catalysis (Scheme 81).202 NHC catalysis has previously been shown to convert aldehydes to acyl anion or homoenolate equivalents under mild conditions.203,204 Formation of a Breslow intermediate (81.4) from the NHC and the aldehyde results in a stereoselective addition into iminium ions generated via photoredox catalysis. The inclusion of m-dinitrobenzene (m-DNB) as an additive was found to be required for maximum reactivity which was suggested to oxidatively quench Ru(II)* with adventitious oxygen serving as the terminal oxidant.
Scheme 81.
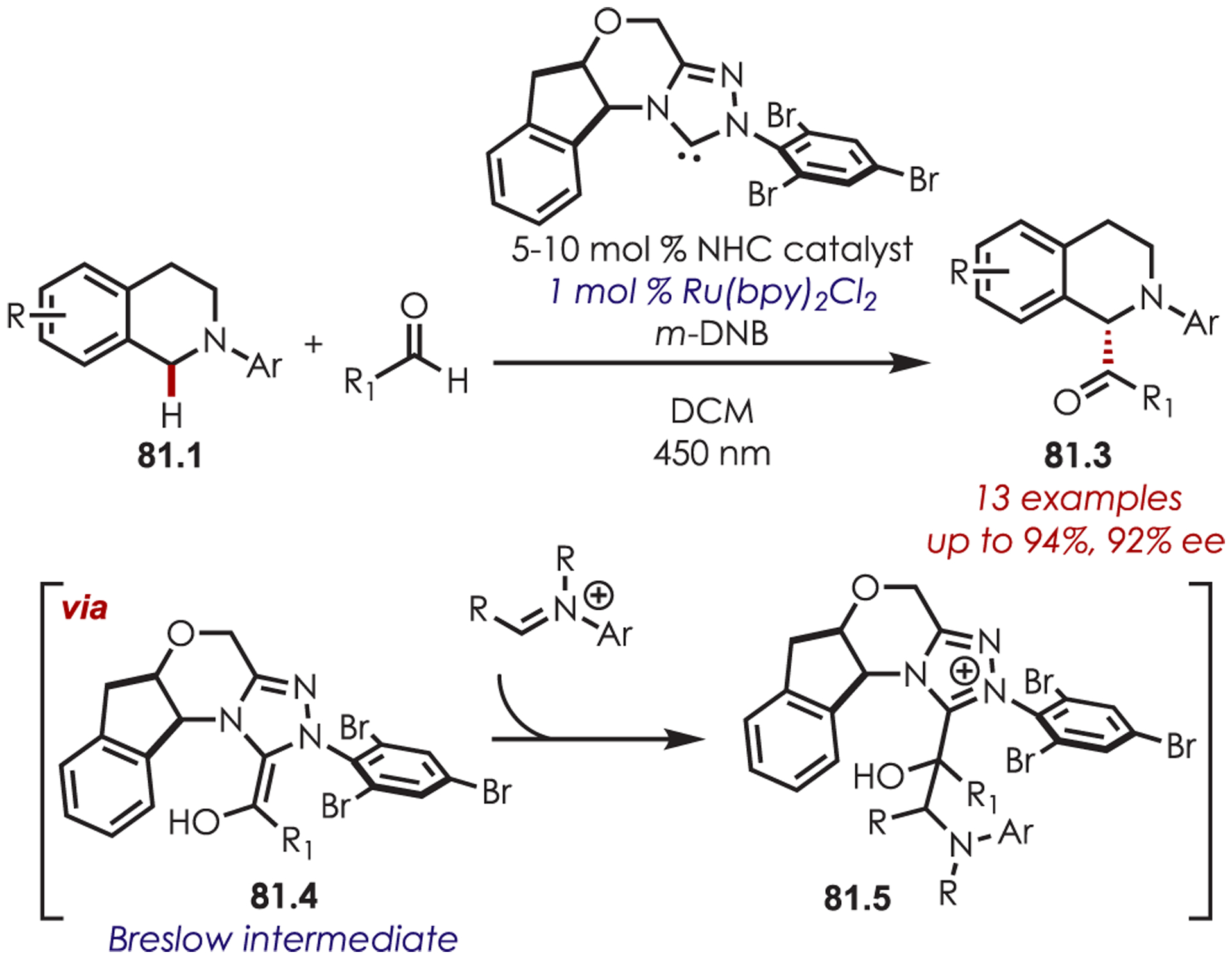
NHC Catalysis for Stereoselective Addition into Iminium Ions
In 2013, Stephenson accomplished an enantioselective oxidative C–H alkylation of tetrahydroisoquinolines (82.1) through dual photoredox and asymmetric anion-binding catalysis (Scheme 82).205 The photogenerated iminium forms a chiral ion pair with a chiral thiourea catalyst that directs stereoselective nucleophilic addition.
Scheme 82.
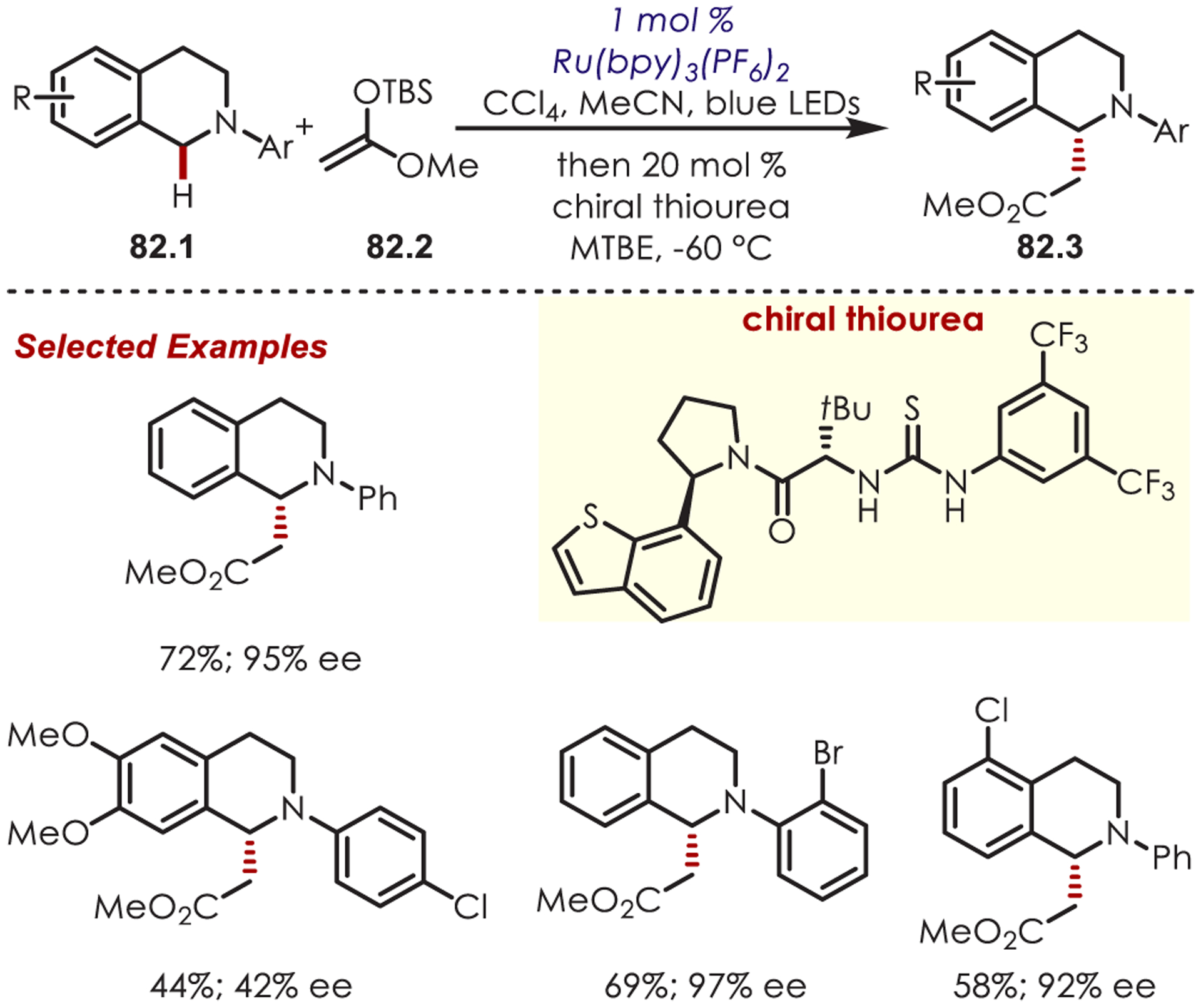
Chiral Ion Pairing for Stereoselective Addition into Iminium Ions
Likewise, chiral PyBox ligands have been used in copper and Ir(ppy)2(dtbbpy)PF6-catalyzed asymmetric tetrahydroisoquinoline arylation reactions with aryl boronic acids (83.2) (Scheme 83).206 However, the levels of enantioselectivity were somewhat low for the majority of the scope. Additionally, the dehydrogenative asymmetric addition of enols into photoredox-generated iminiums by combined Co and chiral enamine catalysis has also been reported.207
Scheme 83.
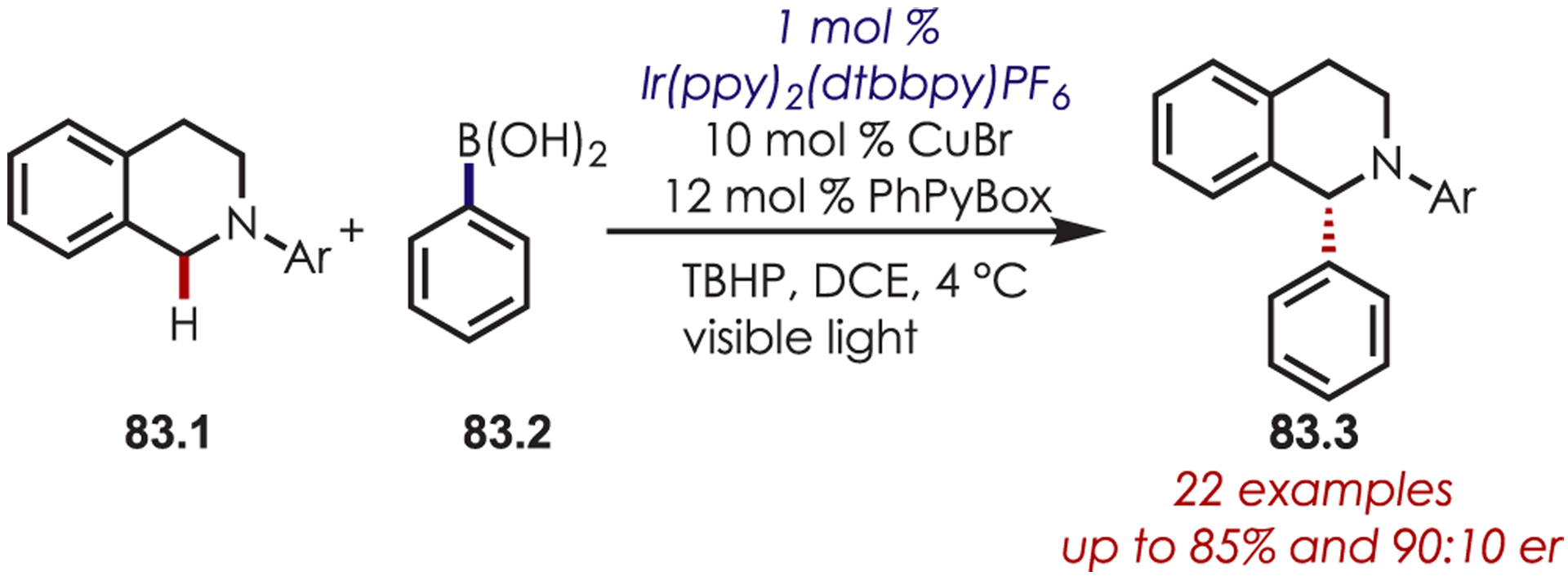
Chiral Copper Catalysis for Stereoselective Coupling with Iminium Ions
Up to this point, the discussion of iminium generation via photoredox catalysis has focused on mechanisms that proceed through single electron oxidation via reductive quenching cycles. However, Stephenson and co-workers reported the formation of iminiums by an H atom abstraction then oxidation through an oxidative quenching cycle with Ru(bpy)3Cl2 and a persulfate terminal oxidant (Scheme 84).208 Persulfate, a known oxidative quencher of Ru(II)*, generates a sulfate radical anion which serves as a strong H atom abstracting agent. Abstraction of the α-amino C–H, followed by an oxidation or a radical chain process with persulfate, generates the intermediate acyliminium ion (84.4). This was coupled with electron-rich (hetero)arenes or alcohol nucleophilic partners. Amides are generally more difficult to directly oxidize, so this oxidative quenching pathway allows for the generation of N-acyliminium ions that would otherwise be difficult to access.
Scheme 84.
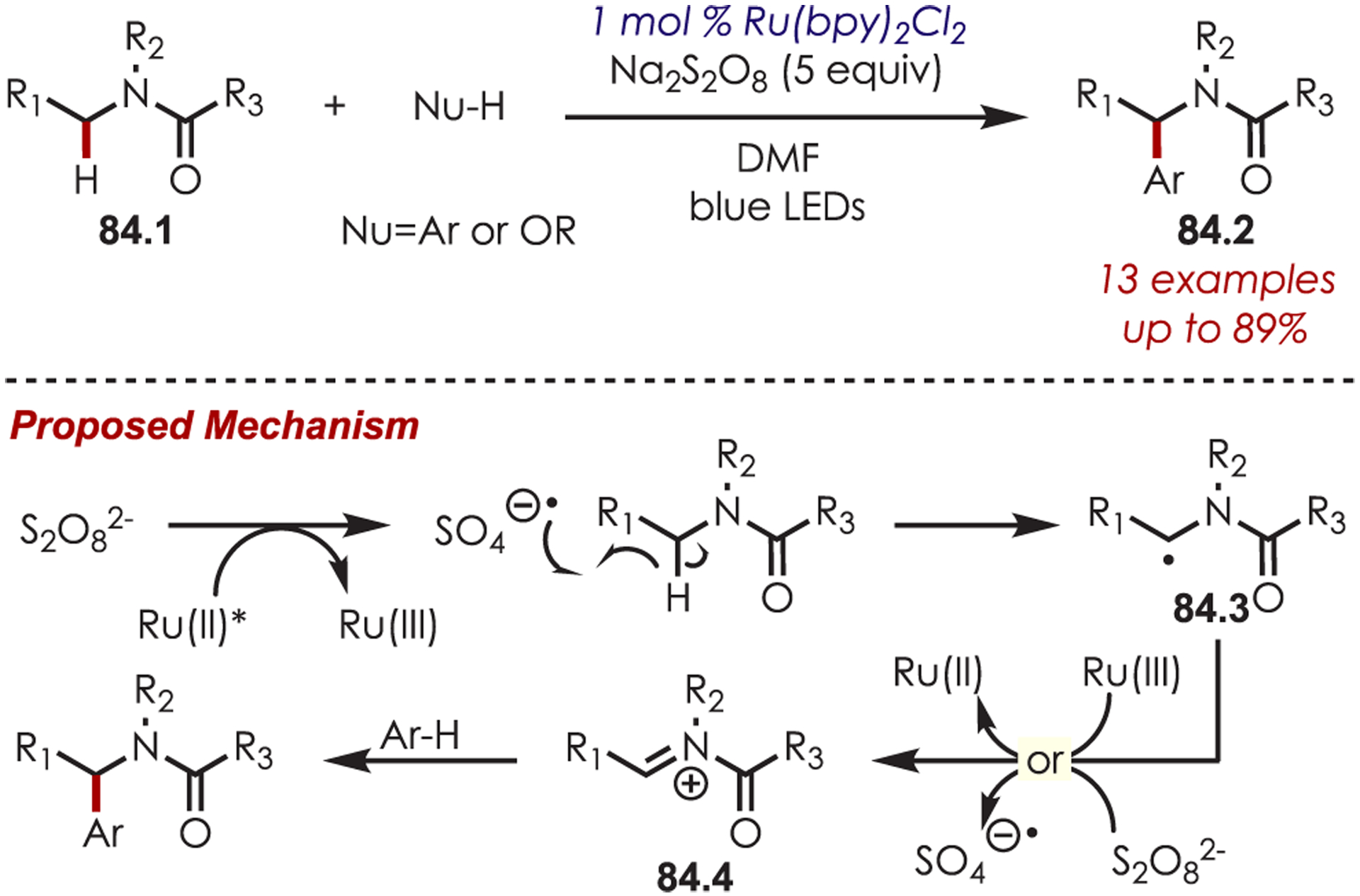
Amide-Derived Iminium Ions Couple with Electron-Rich Aromatics or Alcohols
Zhu and Rueping published an arylation reaction of glycine derivatives and peptides (85.1) with nucleophilic indole partners, 85.2 (Scheme 85).209 Heteroarylation of nonpeptidic amines using indole as a nucleophile was also reported with Ru(bpy)3Cl2.160
Scheme 85.
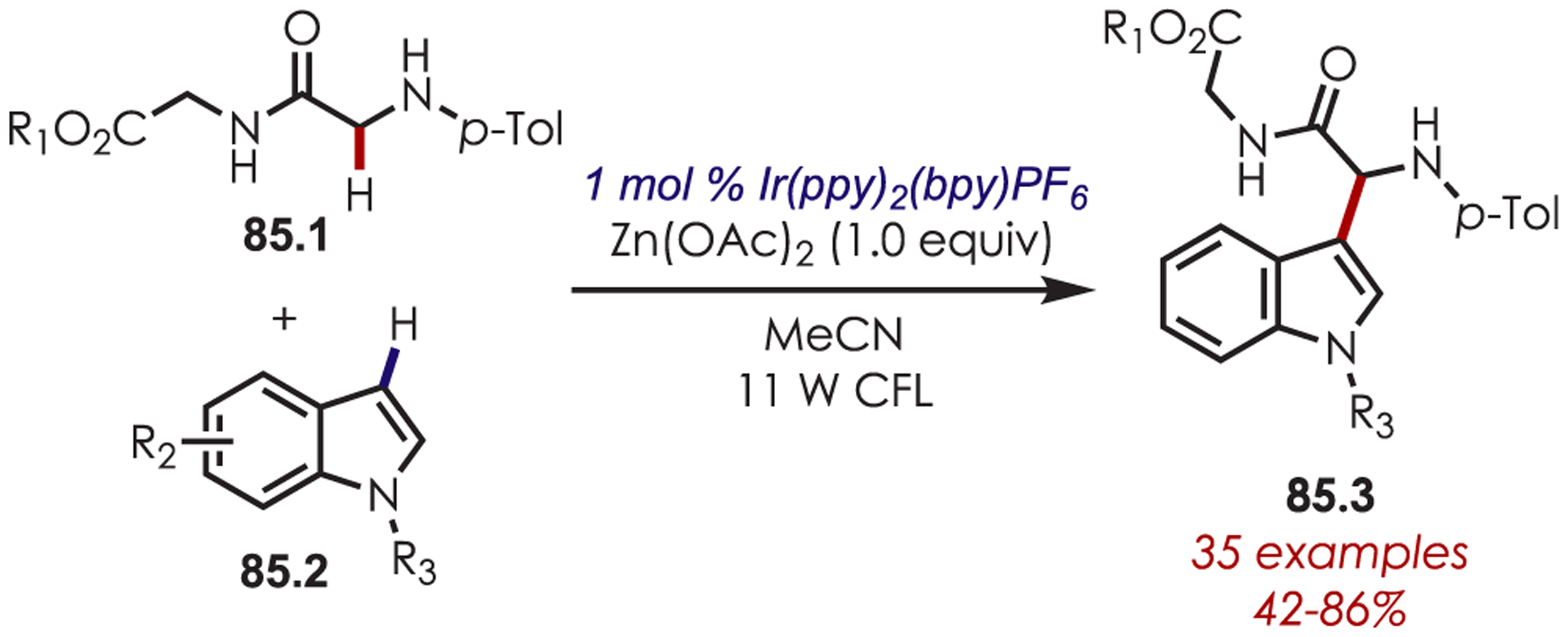
α-Arylation of Glycine Derivatives via Photogenerated Iminium Ions
Xiao and co-workers developed a highly diastereoselective method for the formation of tetrahydroimidazoles (86.3) through an intramolecular sulfonamide addition to iminiums (Scheme 86).210 Longer reaction times were found to increase the diastereoselectivity by allowing for epimerization of the product under the reaction conditions, which afforded the 1,3-syn cyclization products.
Scheme 86.
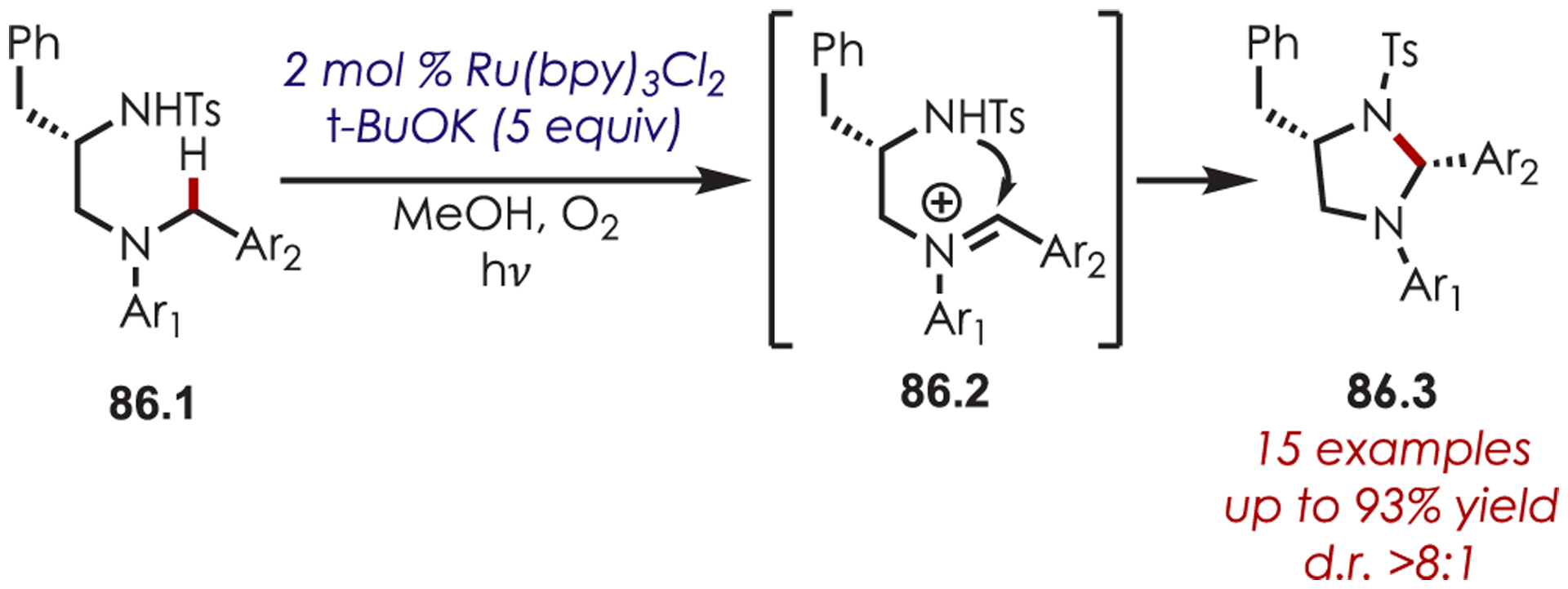
Intramolecular Sulfonamide Cyclization to Photogenerated Iminiums for the Synthesis of Tetrahydroimidazoles
The addition of cyanide into iminiums generated through Ir(III) photoredox catalysis has also been demonstrated for the functionalization of tertiary aliphatic amines (87.1), including complex amines and pharmaceuticals (Scheme 87).211
Scheme 87.
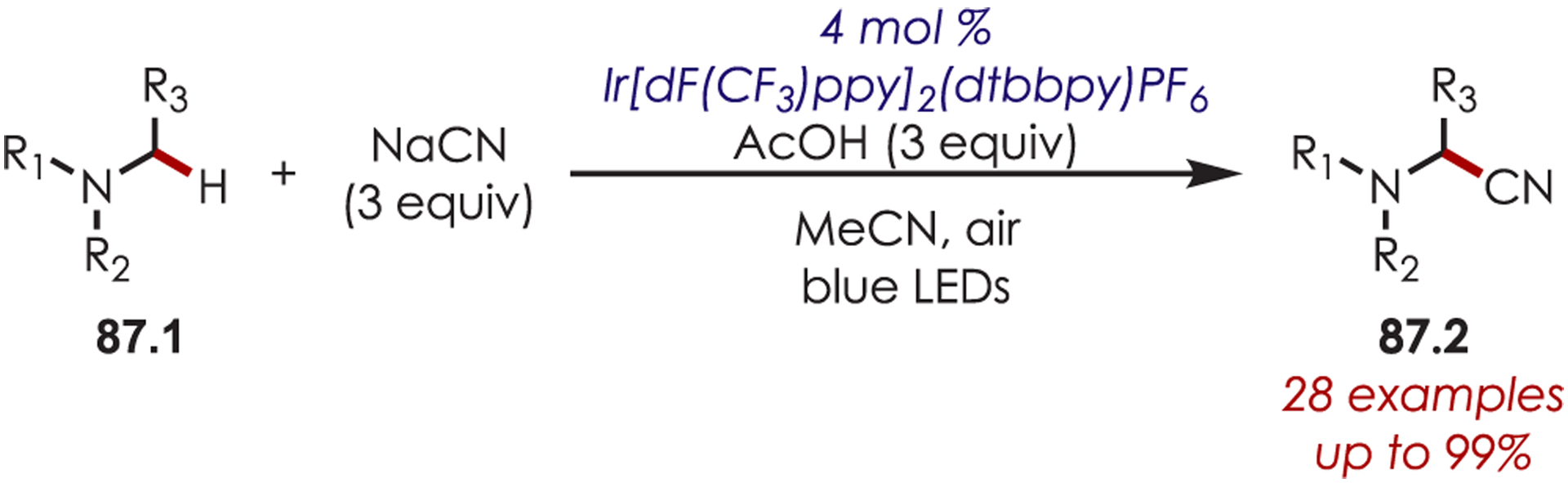
C–H Cyanation of Tertiary Aliphatic Amine Derived Iminium Ions
Zhang and co-workers reported the synthesis of substituted benzimidazoles (88.1) using Ru(bpy)3Cl2 as the photoredox catalyst (Scheme 88).212 Intramolecular cyclization of a sulfonamide onto the intermediate iminium followed by an oxidation–elimination and detosylation sequence affords the benzimidazole product (88.1).
Scheme 88.
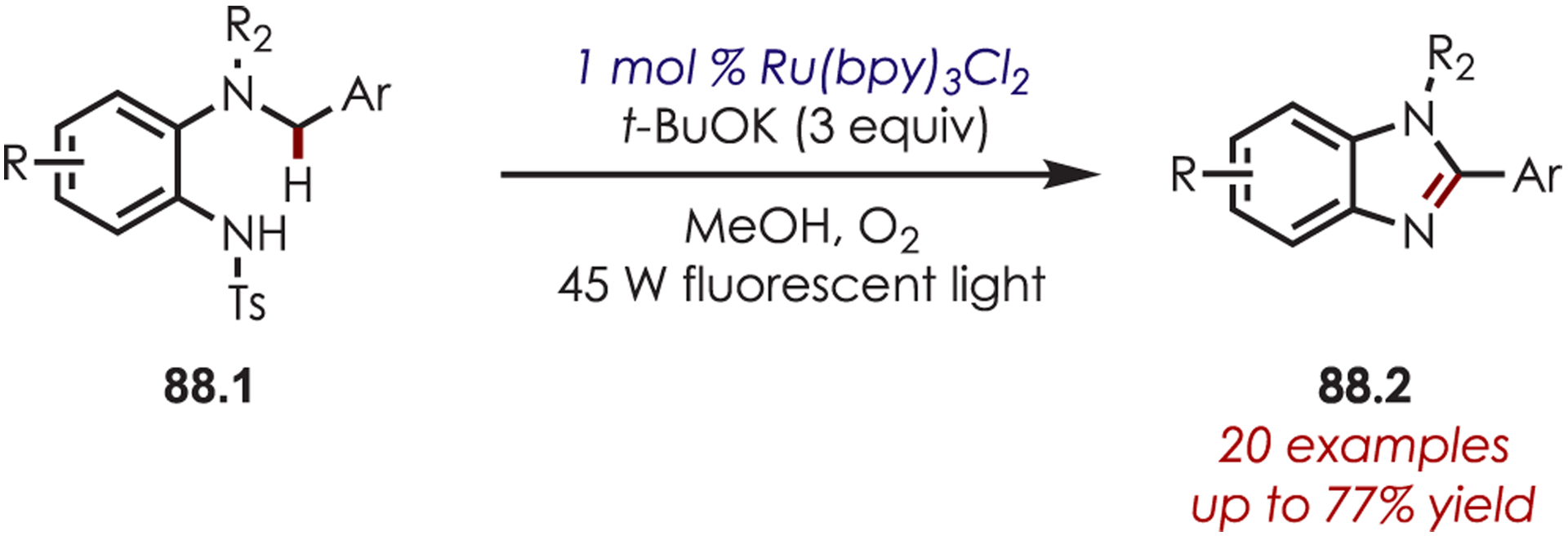
Benzimidazole Synthesis through an Intramolecular C–H Amination of Iminium Ions
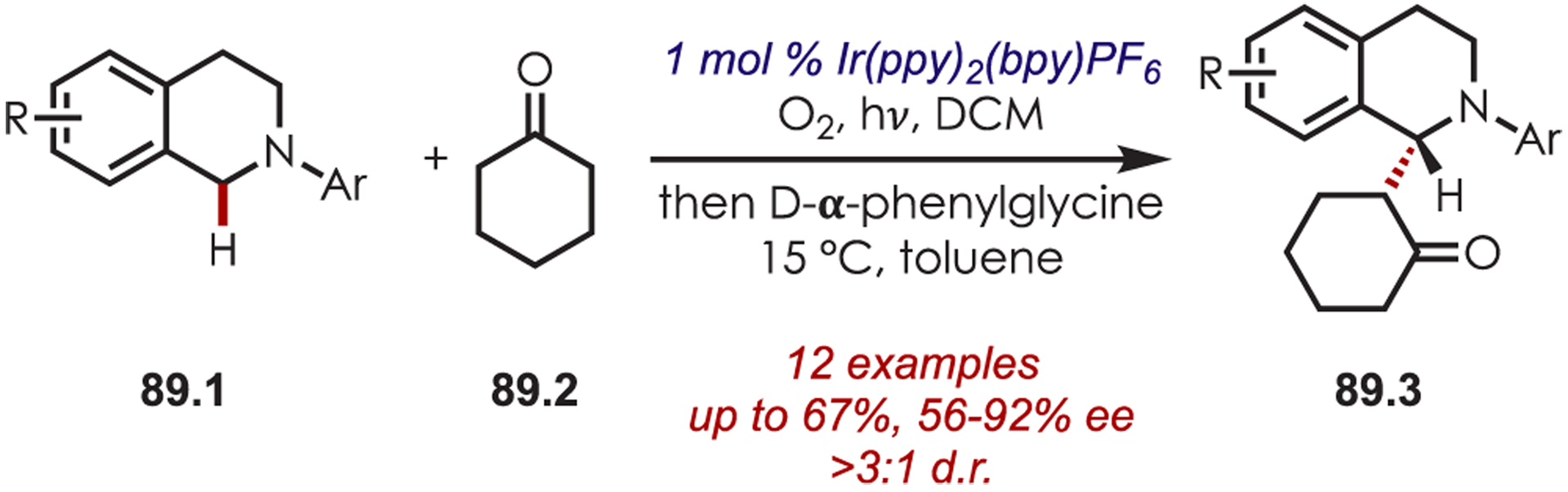
An enantioselective α-alkylation of tetrahydroisoquinolines (89.1) was reported via the addition of chiral enamines generated in situ from cyclic ketones (89.2) to a photoredox-generated iminium ion (Scheme 89).213 The amino acid organocatalyst afforded moderate to good levels of diastereo- and enantiocontrol.
Scheme 89.
Stereoselective Alkylation of Tetrahydroisoquinolines Using Dual Organo- and Photoredox Catalysis
Recently, Zhang accomplished an enantioselective coupling of glycine derivatives (90.2) with ketones and aldehydes (90.1) through an asymmetric Mannich-type reaction (Scheme 90).214 Catalytically formed chiral enamines (90.4) intercept imines (90.5) that are generated from deprotonation of the photoredox-generated aminium cation radicals derived from the starting amines (90.2) as a key step in the transformation. This afforded the unnatural α-alkylated amino acid products in good yield and with excellent stereoselectivity. Various glycine esters, including glycine amides or dipeptides, furnished the desired coupling products. Cyclic or acyclic ketones including substituted cyclohexanones, tetrahydrothiapyrone, tetrahydropyranone, or cycloheptanone gave the products in high enantioselectivity, however acylic ketones gave the product in lower yield. A range of aliphatic aldehydes with alkyl-, allyl-, and benzyl-protected esters were also suitable nucleophilic partners.
Scheme 90.
Asymmetric Mannich-Type Reaction for the C–H Alkylation of Glycine-Derived Iminium Ions
In 2015, Marvin reported the synthesis of (±)-tetrabenazine (91.2) via visible-light-photoredox catalysis (Scheme 91).215 Oxidation of the amine by photoexcited Ru(II) generates the intermediate iminium, which undergoes cyclization with the pendant silyl enol ether to afford the product in 57% yield and 5:1 diastereoselectivity.
Scheme 91.
Synthesis of (±)-Tetrabenazine via a Photogenerated Tetrahydroisoquinoline Iminium Ion
In a related synthesis, Marvin and co-workers utilized a photoredox-mediated cyclization as a key step in the synthesis of (±)-5-epi-cermizine C (92.3) and (±)-epimyrtine (92.4) (Scheme 92).216 The iminium ion undergoes cyclization in the presence of a tethered allylsilane, affording the fused ring product in 50% yield and 4.7:1 dr. This intermediate was subjected to two subsequent parallel transformations to furnish the two desired natural products.
Scheme 92.
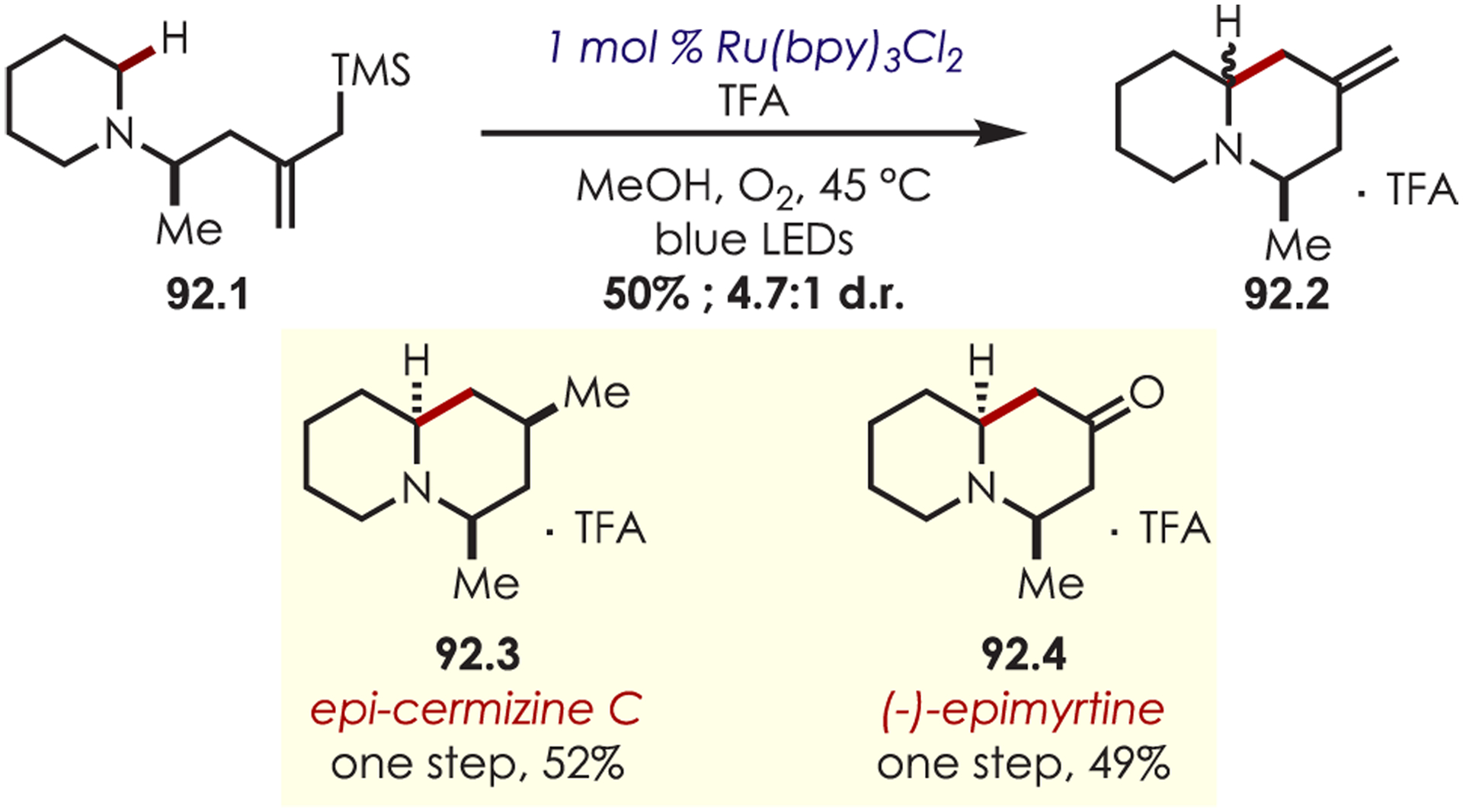
Synthesis (±)-5-epi-Cermizine C and (±)-Epimyrtine via Iminium Ions
Via α-Amino and α-Heteroatom Radicals.
Functionalization of amines via the intermediacy of an iminium ion relies on a single-electron oxidation of amines to the corresponding nitrogen-centered cation radical, which is further oxidized to the iminium in the presence of a stoichiometric oxidant. These processes are typically limited to tetrahydroisoquinolines that generate benzylic iminium ions, with few exceptions. Alternatively, in the absence of a terminal oxidant, the intermediate α-amino radical (93.2) formed in this process may be intercepted by a range of radical acceptor groups, affording formal C–H functionalized products (93.3) (Scheme 93). The generation of other heteroatom nucleophilic radicals have also been explored including α-oxy and α-thiyl radicals.
Scheme 93.
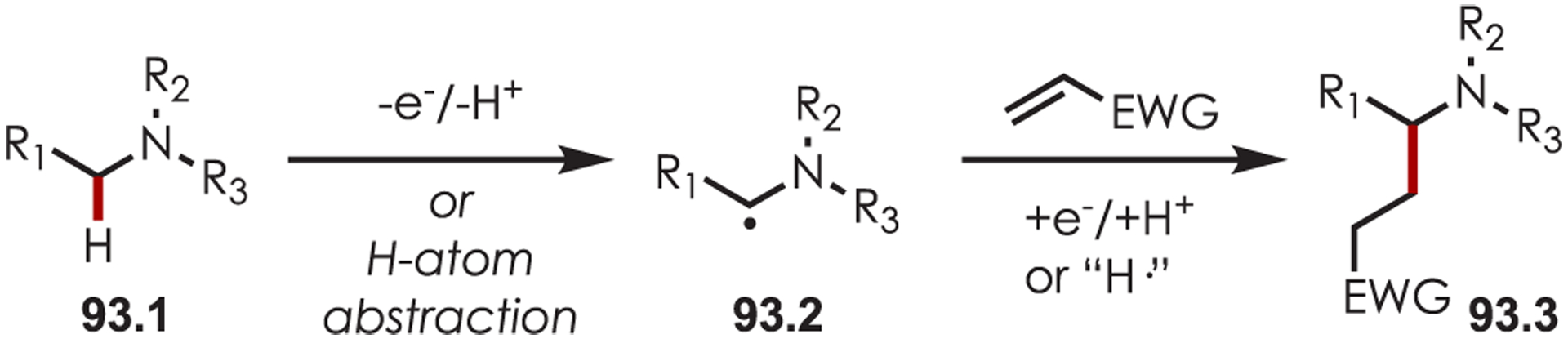
α-Amine Functionalization via Radical Trapping
In 2011, McNally and MacMillan discovered an α-arylation of amines (94.1) by an accelerated serendipity strategy (Scheme 94).217 This reaction couples cyanobenzenes (94.2) with α-amino radicals to generate valuable benzylic amine products (94.3). Cyclic amines ranging from 5 to 7 membered rings all gave the desired product in excellent yields as did acyclic amines. Pharmaceutically relevant amines, including indolines and tetrahydroquinolines, were compatible giving the product in excellent yield and good regioselectivity. Notably, functionalization is not observed at the acyclic benzylic carbon if present and favors functionalization at the cyclic carbon adjacent to nitrogen. Other electron-deficient (hetero)arenes were suitable as coupling partners. Esters, amides, phosphonate esters, tetrazoles, and pyridine derivatives gave the desired products in 26–92% yields. Furthermore, linezolid underwent heteroarylation in 58% yield.
Scheme 94.
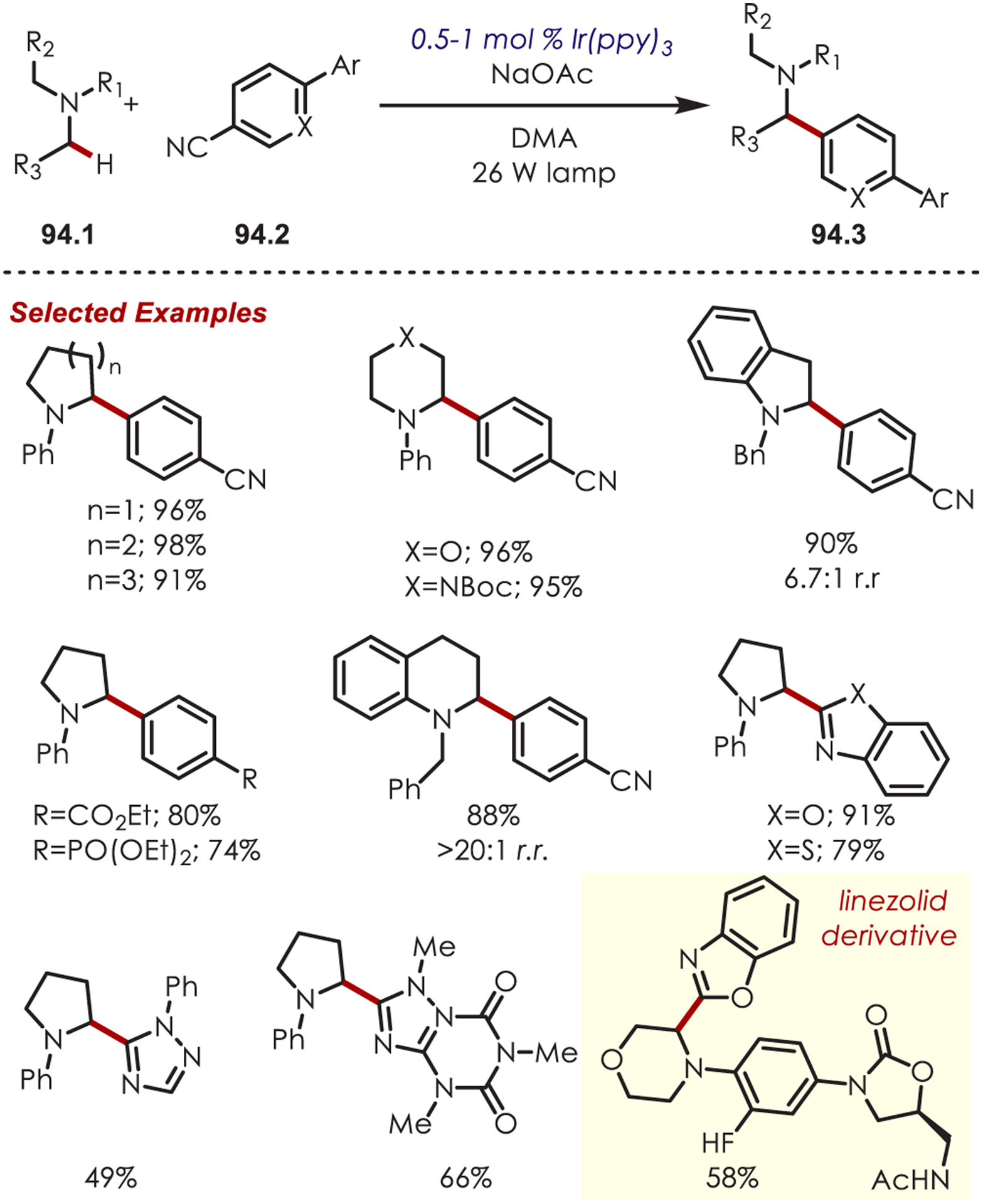
α-Amino C–H Arylation with Cyanobenzenes
The proposed mechanism proceeds through an Ir(III/IV) cycle where first an Ir(III)* reduces 1,4-dicyanobenzene to the corresponding anion radical 95.1 (E*ox = −1.73 V and Ered = −1.61 V vs SCE, respectively), yielding an Ir(IV) oxidant (Scheme 95). A SET between the amine and Ir(IV) then generates an amine cation radical (95.2) and regenerates Ir(III). The α-C–H bonds of the amine cation radical are significantly weakened, by ~40 kcal/mol, resulting in a more facile deprotonation which affords the reactive α-amino radical (95.3). Radical–radical coupling of the aryl anion radical and the α-amino radical followed by loss of cyanide anion furnished the final α-arylated product.
Scheme 95.
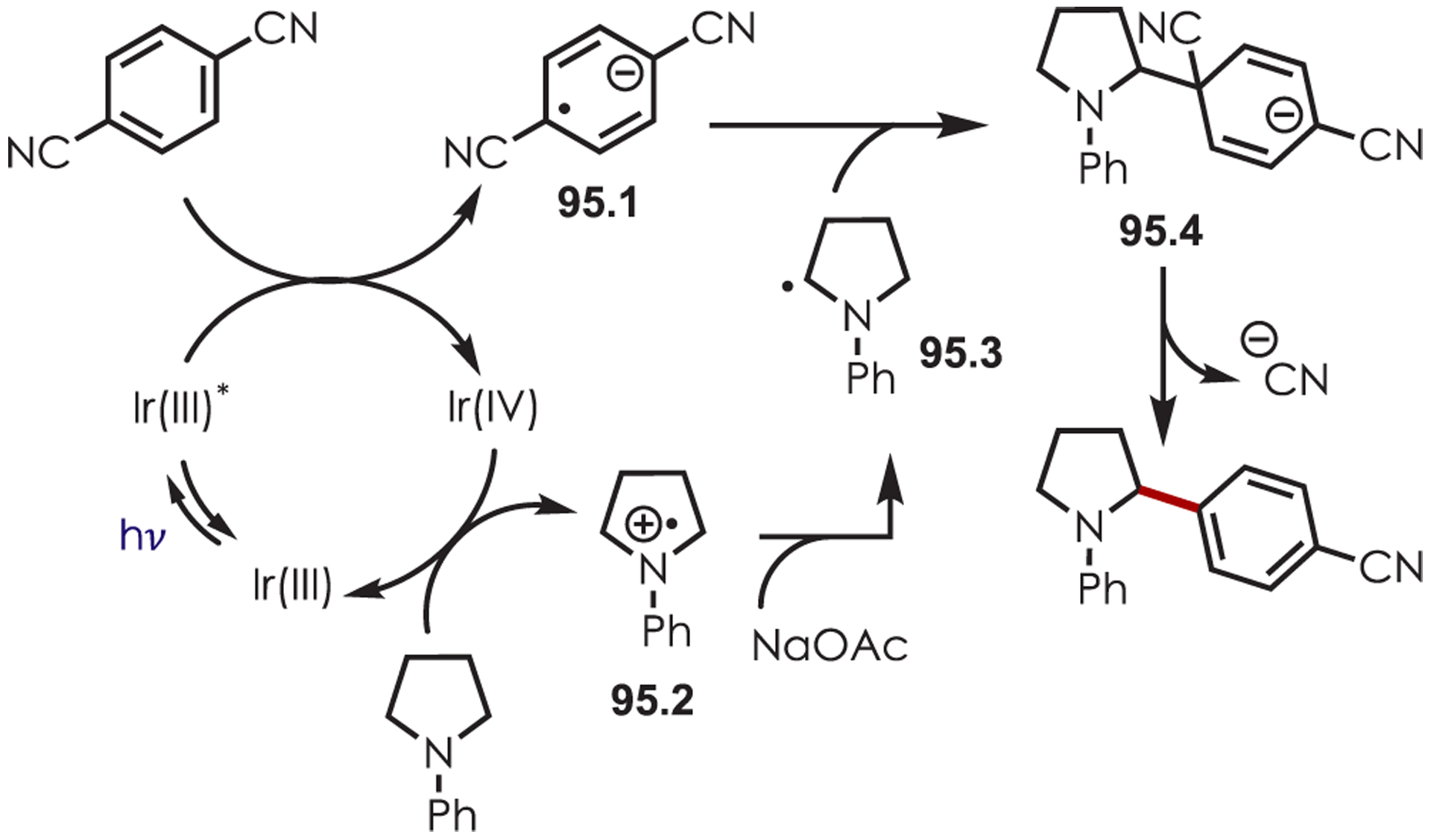
Mechanism of the α-Amino C–H Arylation with Cyanobenzenes
Recently, a diastereoselective α-amino C–H arylation of piperidines (96.1) has been reported by Houk, Mayer, and Ellman that operates via a similar mechanism to the McNally and MacMillan work (Scheme 96).218 The high diastereoselectivity of this transformation was determined to be a result of an in situ light-driven epimerization process from an unselective C–H arylation step via the intermediacy of the amine cation radical. This mecahnism generally converted the mixture of epimers at C1 when R1 = H (e.g., 96.4 and 96.5) to the syn diasteromer and when R1 = alkyl (e.g., 96.6), the anti-adduct predominated.
Scheme 96.
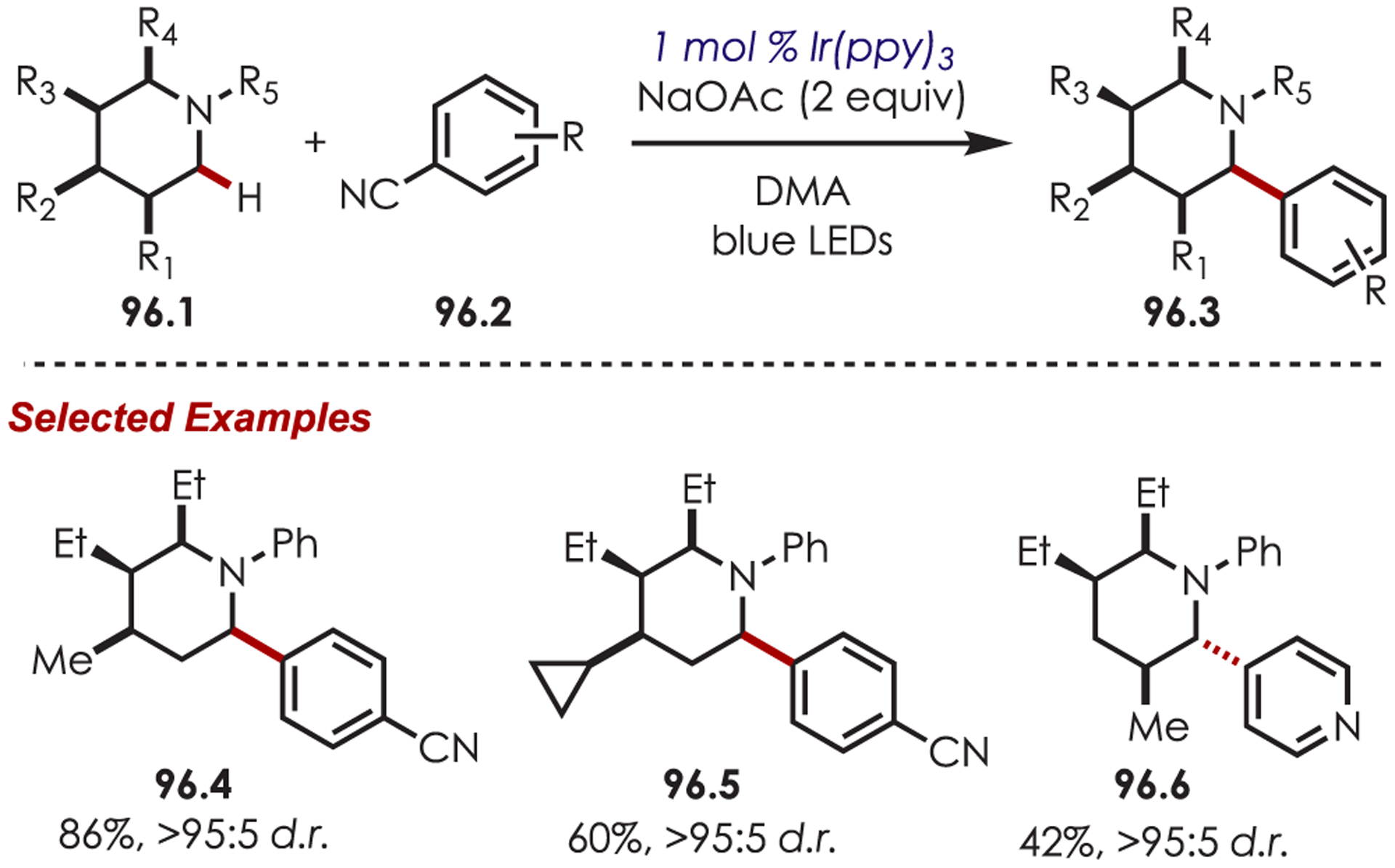
Diastereoselective α-Amino C–H Arylation with Cyanobenzenes
Developments have also been made for the addition of nucleophilic α-amino radicals into other radical acceptors in both an inter- and intramolecular fashion. Reiser and co-workers disclosed an α-amino radical addition of tetrahydroisoquinolines (97.1) with Michael acceptors (Scheme 97).219 They noted that a delicate electronic balance was required between the coupling partners and the catalyst. In particular, optimal results with an Ir(III) photoredox catalyst were obtained when a N-PMP substituted tetrahydroisoquinoline was reacted with the more electron-rich p-methoxyphenyl vinyl ketone. Furthermore, an intriguing intramolecular Michael addition followed by a subsequent spontaneous dehydrogenation afforded 5,6-dihydroindolo[2,1-a]tetrahydroisoquinoline (97.5).
Scheme 97.
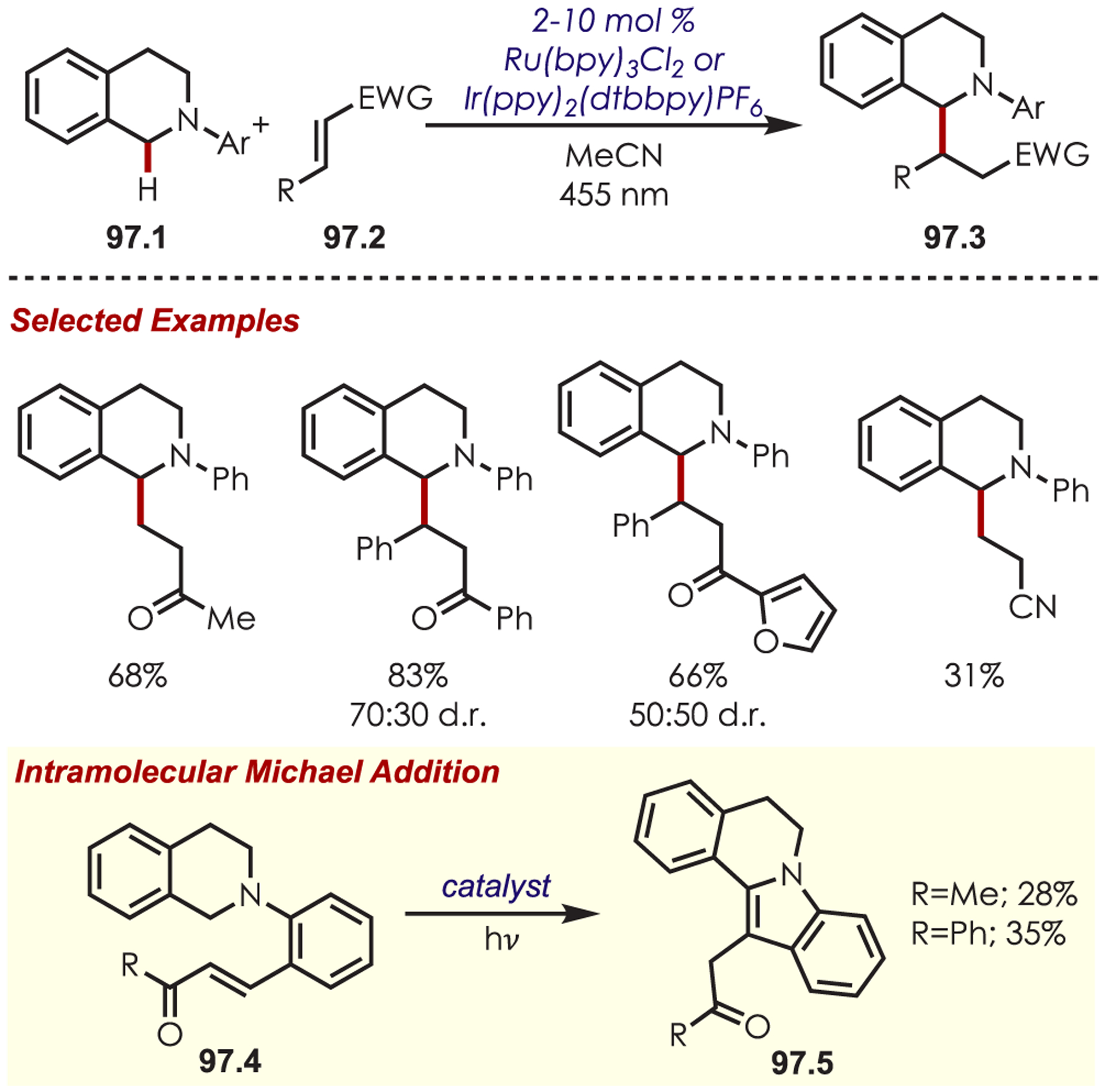
α-Amino C–H Alkylation by Trapping with Michael Acceptors
Nishibayashi published an analogous addition of α-amino radicals to electron-deficient alkenes (98.2) (Scheme 98).220 The resulting alkyl radical intermediate is reduced by Ir(II) and protonated to afford the desired products while simultaneously regenerating the starting Ir(III) species. A screen of the alkene acceptors found that two electron-withdrawing groups were required for high yields. Tertiary dialkyl aryl and diaryl alkyl amines gave the coupled product in 73–97% yield. Diisopropylmethylamine was also a successful substrate, producing the product in 83% yield with substitution occurring through the generation of a primary radical. When N-(methyl-d3)-N-phenylaniline (98.4) was subjected to the reaction conditions, 51% D-enrichment (98.5) was observed at the methine, suggesting the proton mainly derives from the α-amino C–H. However, other hydrogen abstraction pathways cannot be excluded. Furthermore, a radical-clock experiment with a cyclopropyl radical acceptor (98.7) results in the ring-opened product (98.8) in 64% yield, supporting the formation of the alkyl radical intermediate under the reaction conditions. The quantum yield was determined to be 0.32, which suggests a radical-chain process would be inefficient. Ultimately, the authors conclude that the mechanism supported by their investigation is comparable to those previously discussed, proceeding via generation of the α-amino radical, addition into the alkene acceptor, and reduction/protonation to afford the product.
Scheme 98.
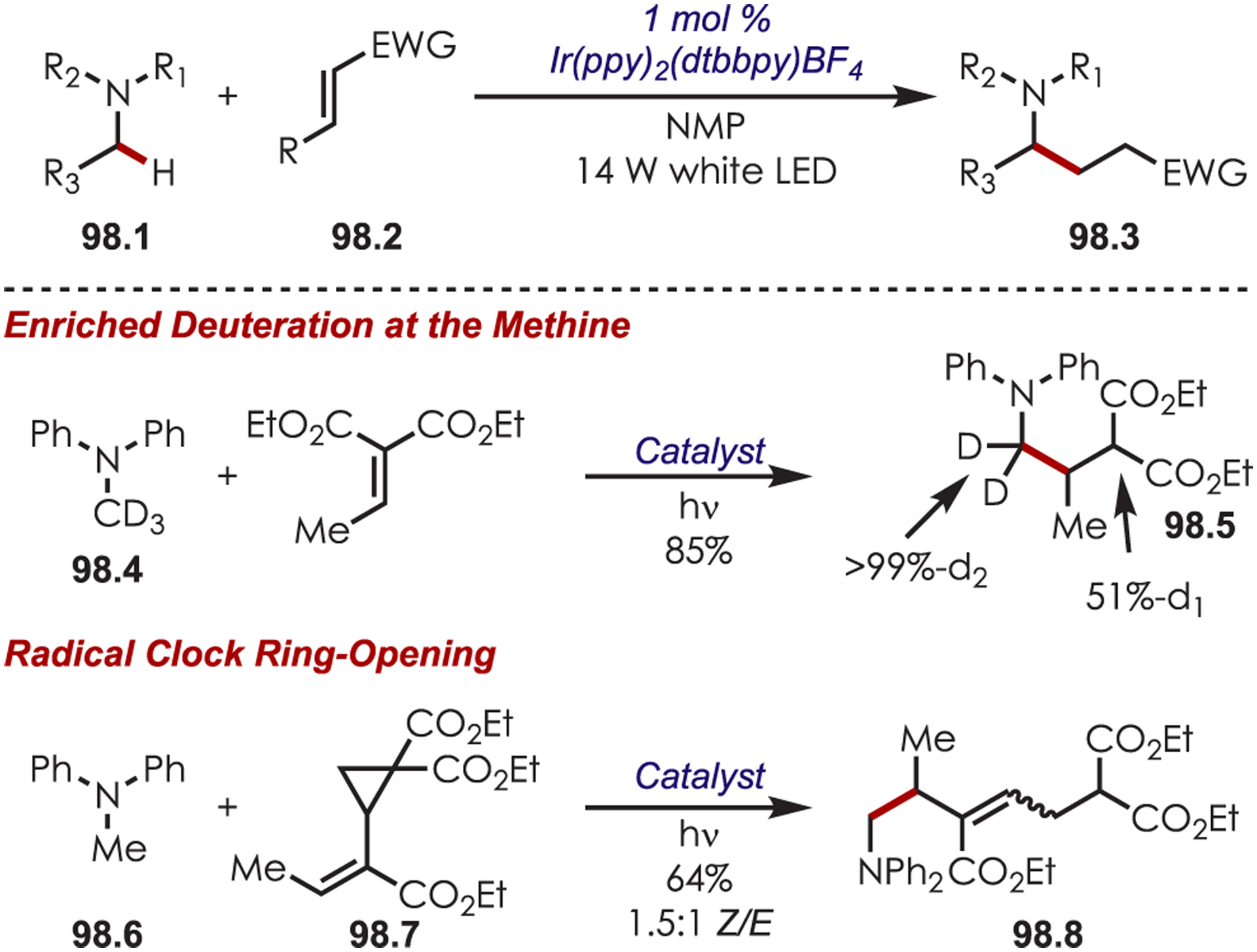
Mechanistic Support of the α-Amino C–H Alkylation with Electron-Deficient Alkenes
Nishibayashi later reported a radical–radical amination of benzocylic α-amino radicals (99.5) and di-tert-butyl azodicarboxylate radical anion (99.4) (Scheme 99).221 Both cyclic and acyclic amines were compatible and furnished the aminated products (99.3) in 25–90% yields.
Scheme 99.
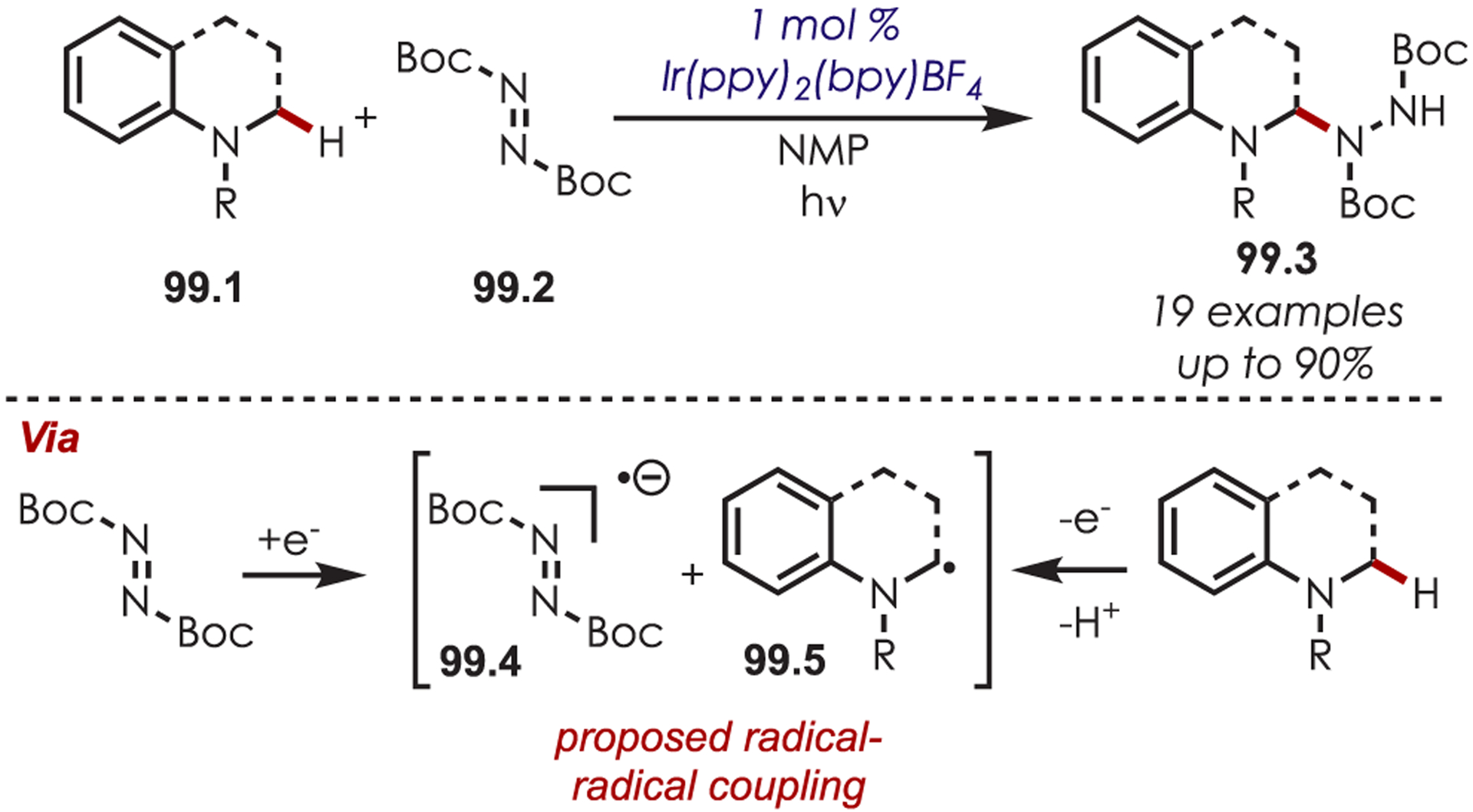
C–H Amination via an α-Amino Radical-Azodicarboxylate Radical Anion Coupling Reaction
Yoon reported a Michael-type alkylation of tetrahydroisoquinoline derivatives (100.1). The intermediacy of nucleophilic α-amino radicals in this process offers reactivity complementary to that of previously discussed nucleophilic addition into iminiums (Scheme 100).222 Ru(bpy)3Cl2, in combination with TFA as a Brønsted acid, was found to greatly improve reaction yields and reaction rates. In addition, the introduction of TFA reduced the yield of undesired byproducts and avoided a catalyst deactivation pathway which was observed in the initial reaction kinetics. KIE experiments determined that in the absence of TFA a negligible KIE was determined for the amine and an inverse second-order KIE for the enone. The addition of TFA gave a first-order KIE with the amine and a normal second-order KIE with the enone. These results suggests that the addition of the acid co-catalyst changed the rate-determining step. Without added acid, the RDS was consistent with addition of the α-amino radical into the Michael acceptor. However, with the added acid, the rate limiting step was found to involve chain-propagating hydrogen atom abstraction of the amine from the alkyl radical intermediate. This observation accounts for an acceleration of the slow C–C bond forming step and prevents catalyst deactivating dimerization. Ultimately, the Brønsted acid additive results in a change in the reaction mechanism to a chain propagating hydrogen abstraction which was found to overall increase the reaction efficiency. Isoquinolines gave the highest yield, however N-arylpyrrolidine gave the product in a lower yield of 30%. Suitable radical acceptors included aliphatic and aromatic enones, as well as acrylates.
Scheme 100.
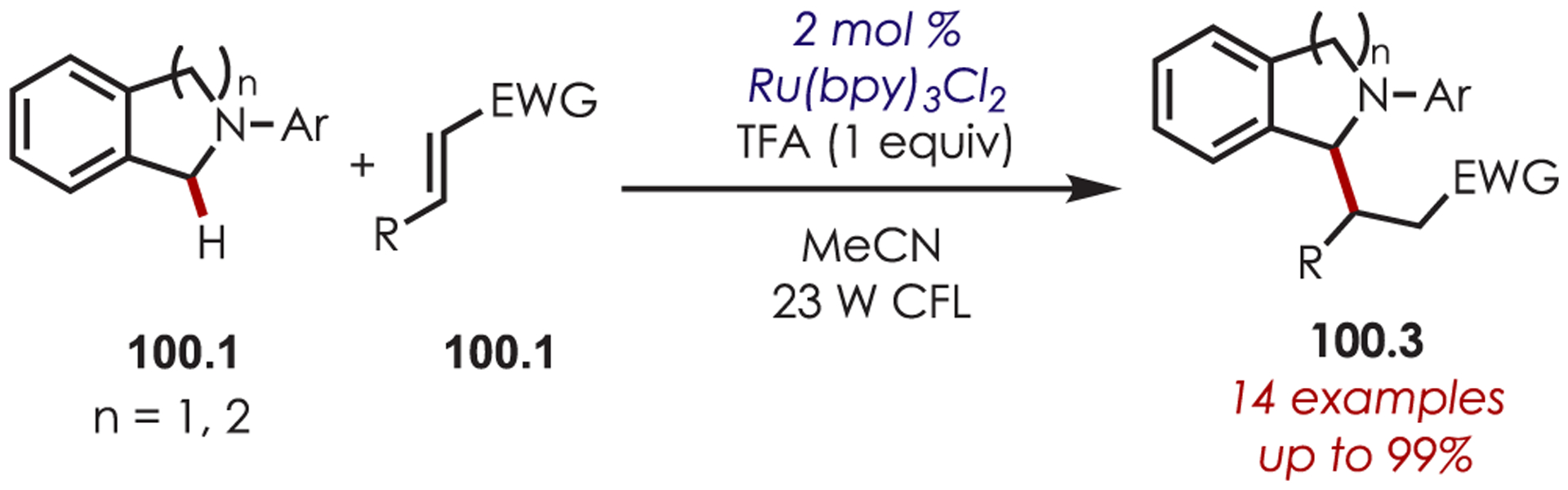
α-Amino C–H Alkylation Reported by Yoon
Addition of α-amino radicals into acrylate derivatives for the synthesis of various γ-aminobutyric esters (101.2) was reported by Li in 2012 (Scheme 101).223 Acetoxyacrylates (101.3) also served as suitable radical acceptors, resulting in a subsequent elimination of an acetate anion to generate an unsaturated product following a single electron reduction of the corresponding α-keto radical.
Scheme 101.
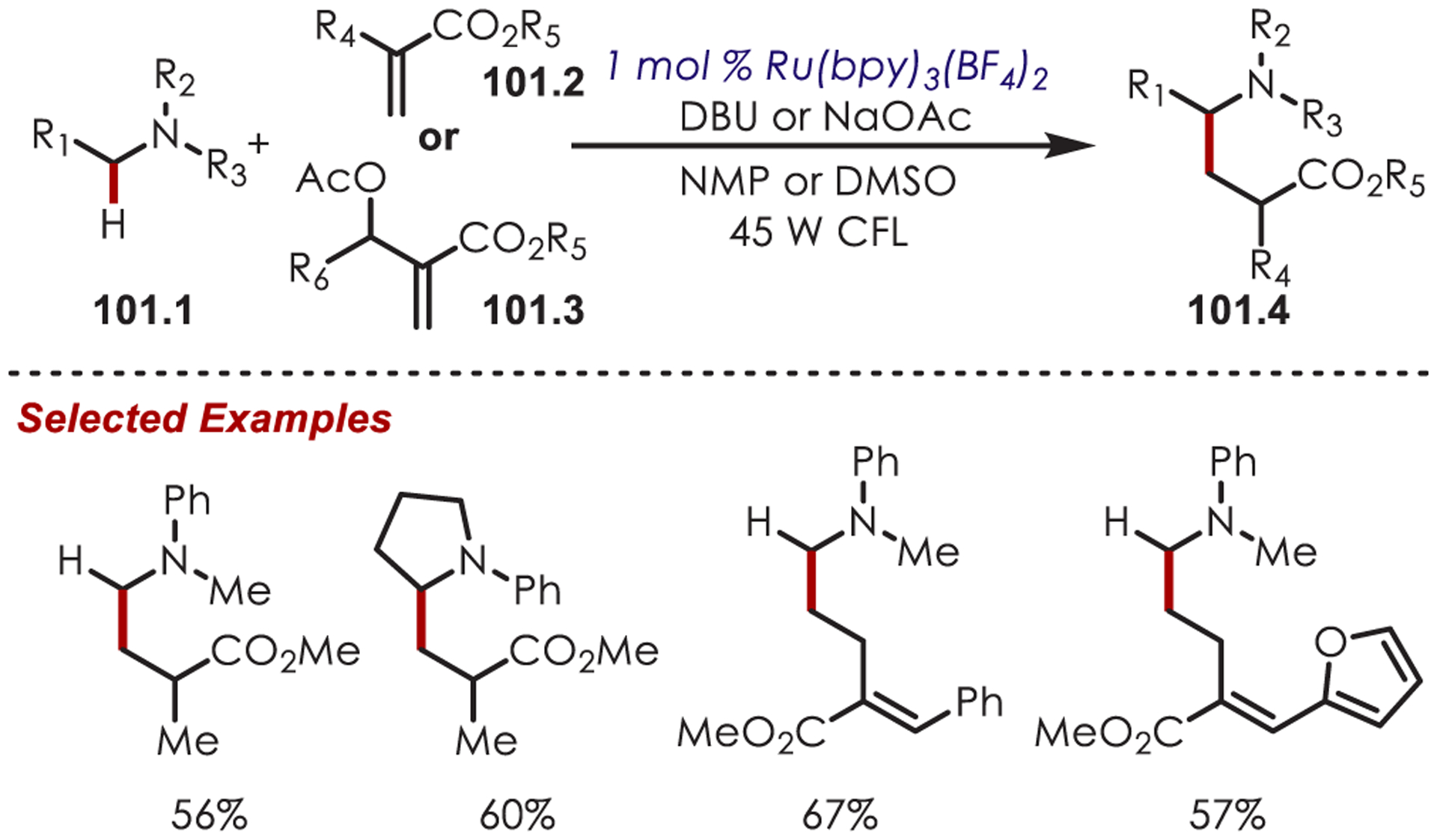
Acrylates as α-Amino Radical Acceptors for C–H Alkylations
Moreover, Li reported that 2,3-allenoates (102.2) are suitable α-amino radical acceptors that yield the corresponding unsaturated γ-aminobutyric esters (102.3) (Scheme 102).224
Scheme 102.
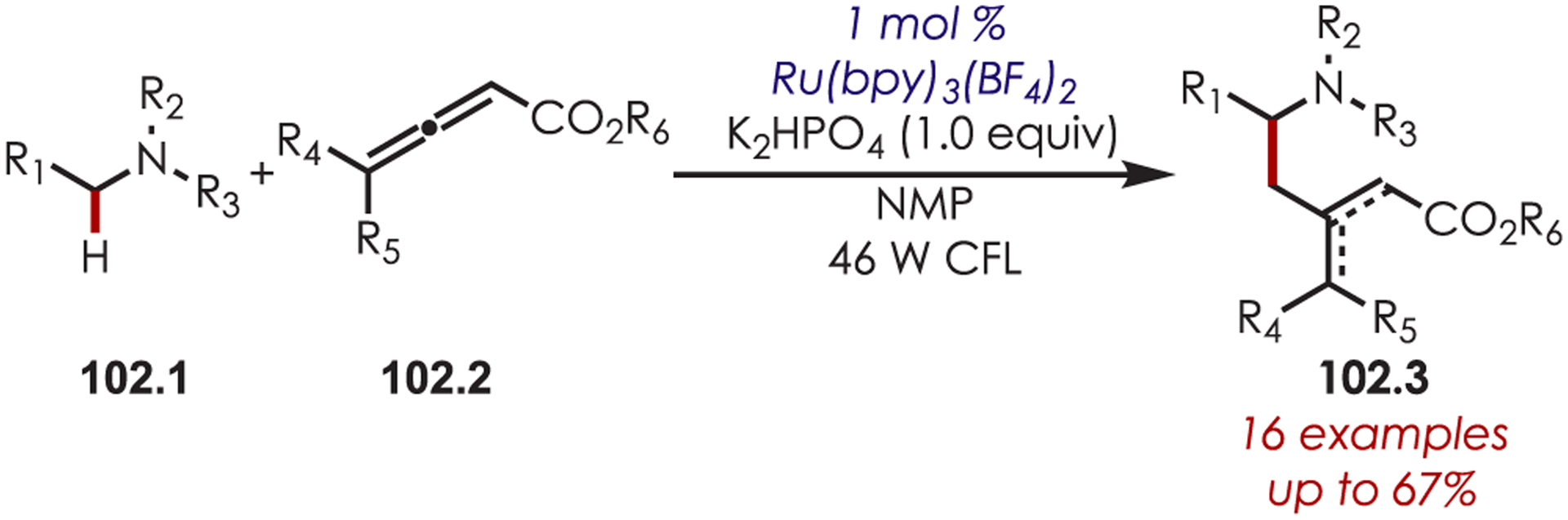
2,3-Allenoates as α-Amino Radical Acceptors for C–H Alkylations
Hashmi later reported a gold-catalyzed alkynylation of aliphatic amines (Scheme 103).225 Reduction of the alkyne (103.2) from [Au2(μ-dppm)2]2+* generates an alkynyl radical (103.4) and the gold oxidant. Subsequent generation of the α-amino radical (103.5) and then a radical–radical coupling furnishes the α-alkynylated amines.
Scheme 103.
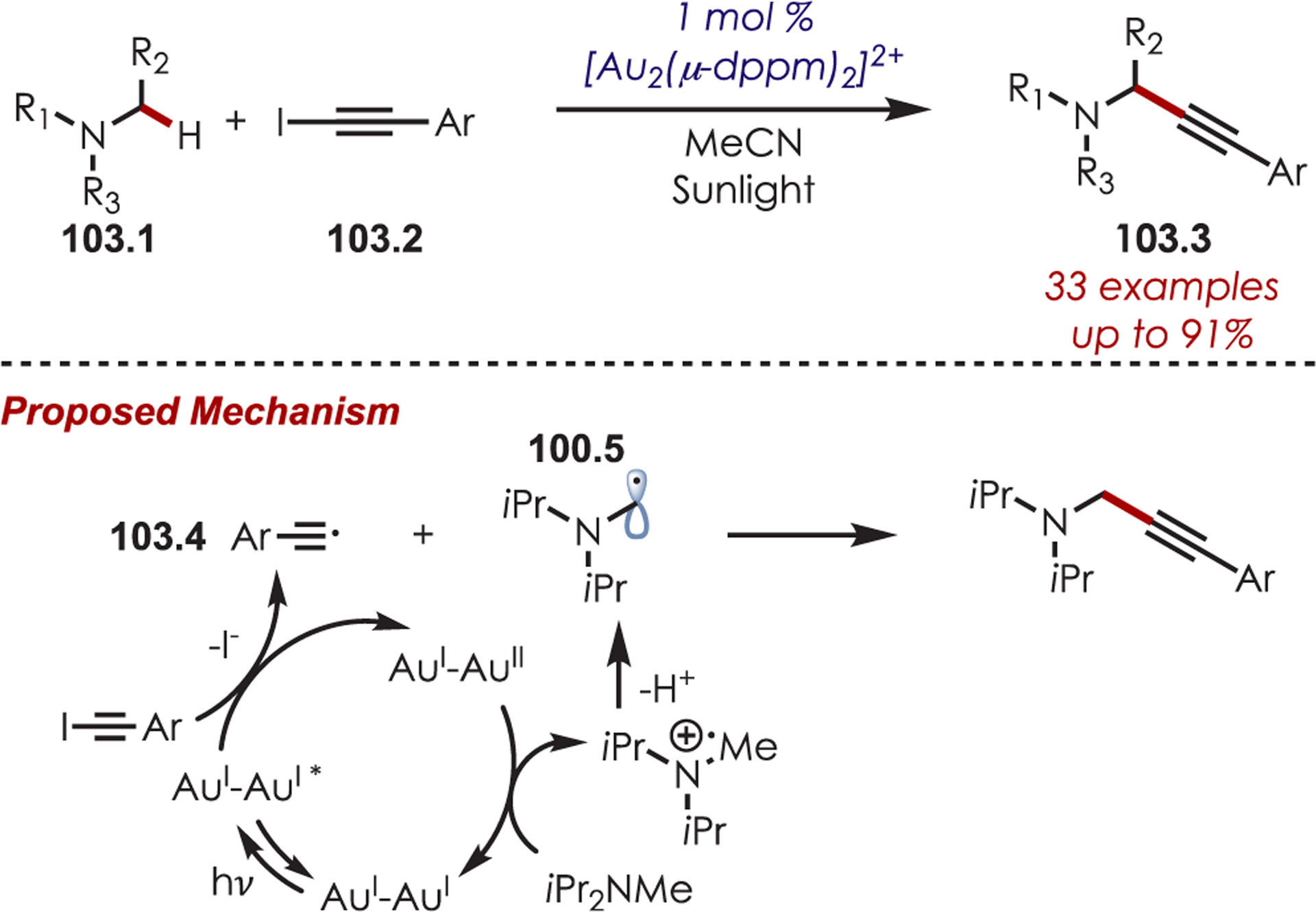
Photoredox Gold Catalysis of the Alkynylation of Amines through an α-Amino Radical
Vinyl azides (104.2) have also been successful radical traps for α-amino radicals (Scheme 104).226 The resulting intermediate iminyl radical is reduced and subsequently hydrolyzed to furnish β-amino ketones (104.3).
Scheme 104.
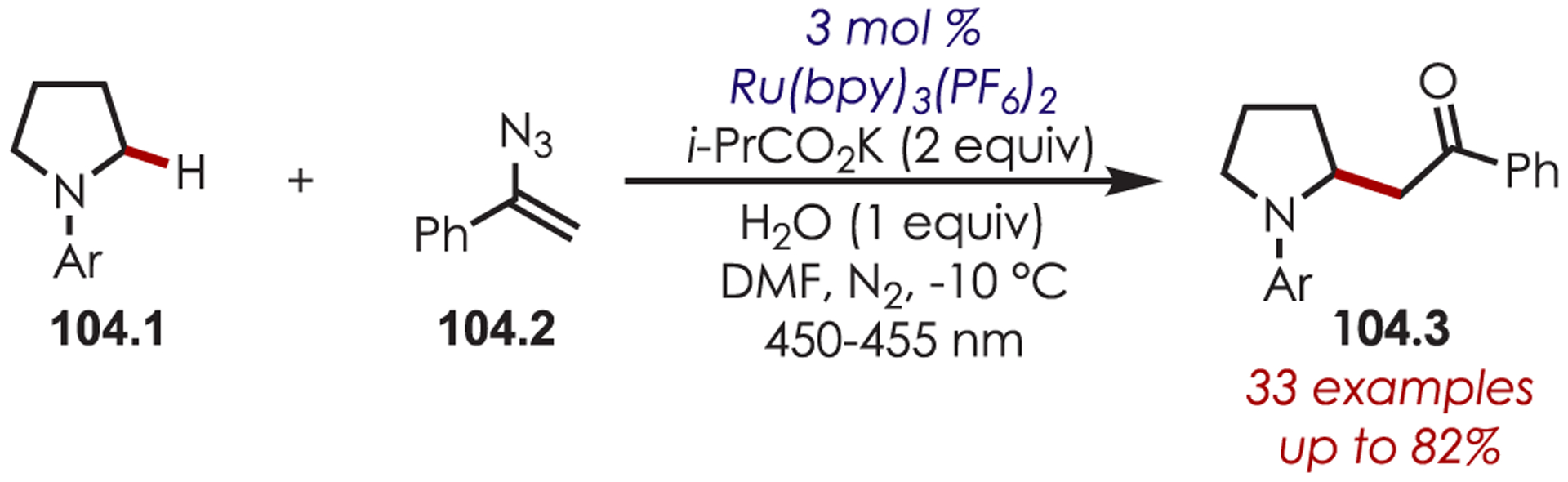
Vinyl Azides as α-Amino Radical Acceptors for C–H Alkylations
Inspired by their earlier results utilizing α-amino radicals generated via decarboxylation in combination with nickel catalysis,227 MacMillan accomplished a direct C–H α-amino and α-oxy radical cross coupling with aryl halides (105.2) through a triple catalytic system (Scheme 105).228 Halogenated arenes with both electron-withdrawing and rich functional groups were tolerated. Heteroaromatic coupling partners including pyridines, pyrimidines, and pyrrolidines as well as various N-protecting groups were tolerated under the optimized reaction conditions. The method was additionally tolerant of potentially sensitive functionalities, including lactams, ureas, alcohols, and amines. Unsymmetrical amines were arylated with moderate to excellent regioselectivity depending on the substitution patterns, generally at the less hindered position. Furthermore, THF and oxetane were found to undergo α-oxy arylation in good yields, 76% and 53%, respectively.
Scheme 105.
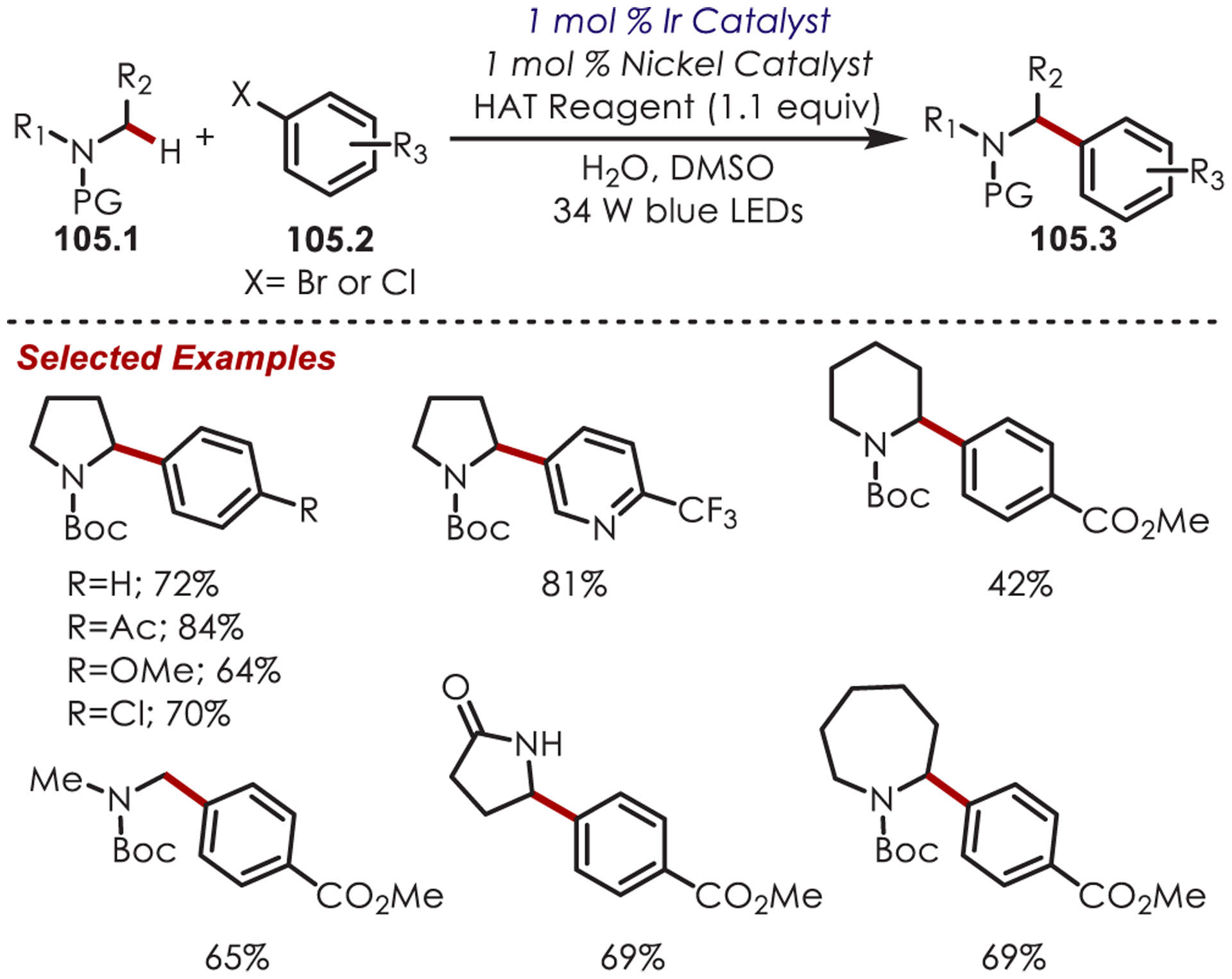
α-Amino C–H Arylation through a Triple Catalytic System
The mechanism for this transformation is proposed to proceed through a three-component photoredox-HAT-nickel tricatalytic cycle (Scheme 106). The photoexcited Ir(III)* oxidizes a tertiary amine HAT catalyst to generate the reduced Ir(II) catalyst and an amine cation radical (106.1) (E*red = +1.21 V vs SCE and Eox = +1.22 V vs SCE, for Ir(III)* and the amine, respectively). The generated electrophilic amine cation radical can undergo a kinetically selective HAT with the most electron-rich C–H site of the substrate. The resulting α-amino radical (106.2) is intercepted by a nickel catalytic cycle to furnish the arylated products.
Scheme 106.
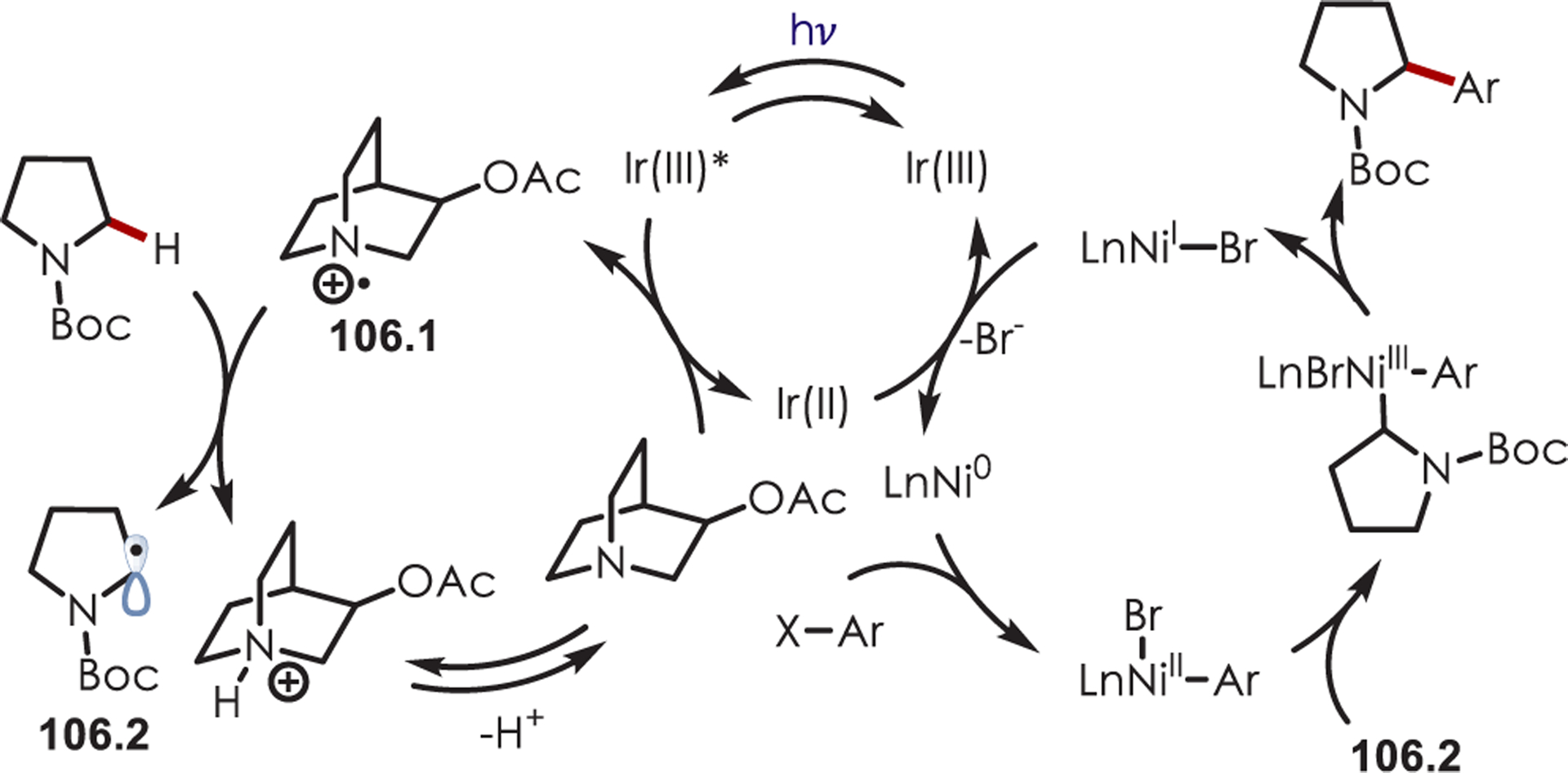
Mechanism of the α-Amino C–H Arylation through a Triple Catalytic System
This methodology was later extended to an α-hydroxy (hetero)arylation (Scheme 107).229 This reaction relied on a zinc-mediated deprotonation to activate the α-hydroxy C–H (107.1) for hydrogen atom abstraction from the amine cation radical. The authors highlighted this methodology in a three-step synthesis of Prozac (107.4).
Scheme 107.
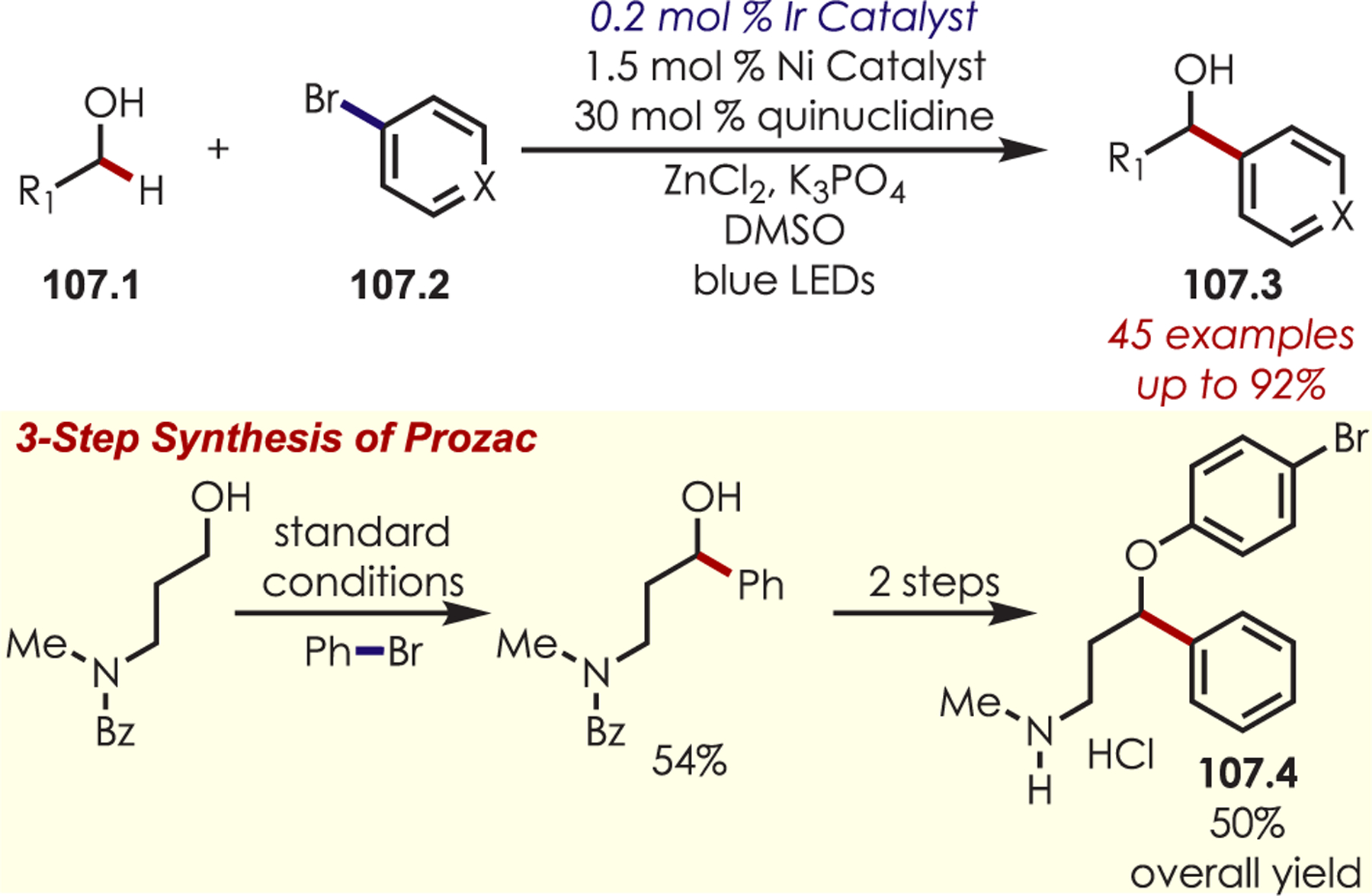
α-Hydroxy C–H Arylation through A Triple Catalytic System
Later that year, Molander independently reported a nickel- and photoredox-catalyzed α-oxy arylation of ethereal solvents, 108.1 (Scheme 108).230 Investigation of the reaction revealed an unexpected Ir(III)* to Ni(II) energy transfer that may trigger a Ni–Br homolysis event from the photoexcited nickel species. The resulting bromine radical is proposed to serve as the hydrogen atom abstractor to generate the α-hetero radical species. Additionally, analogous uranyl photoredox-catalyzed alkylations of tetrahydrofurans via direct hydrogen atom transfer have been reported.231
Scheme 108.
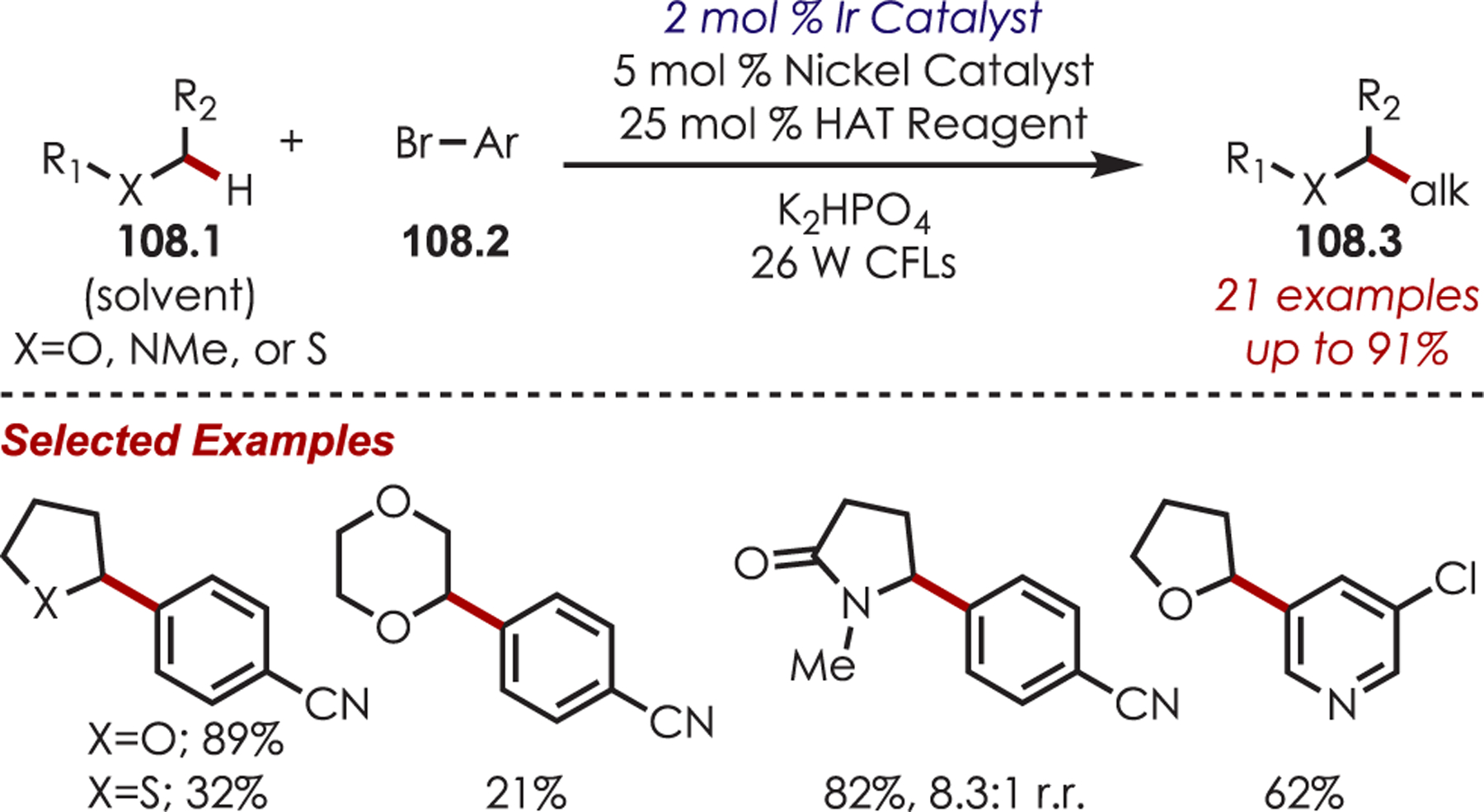
α-Oxy C–H Arylation through a Triple Catalytic System Reported by Molander
The following year, MacMillan extended this triple catalytic cross coupling to include alkyl bromides, 109.2 (Scheme 109).232 Aliphatic and cyclic amines, including amino acids and peptide derivatives, ethers, and thioethers, were successfully alkylated with alkyl bromides as the coupling partner. Notably, the α-methyl products could be accessed in 41–61% yields with methyl p-toluenesulfonate. This alkylation was highlighted in the late-stage functionalization of Prozac, which gave the product in 45–52% yields and good to excellent regioselectivity with three different alkyl bromides.
Scheme 109.
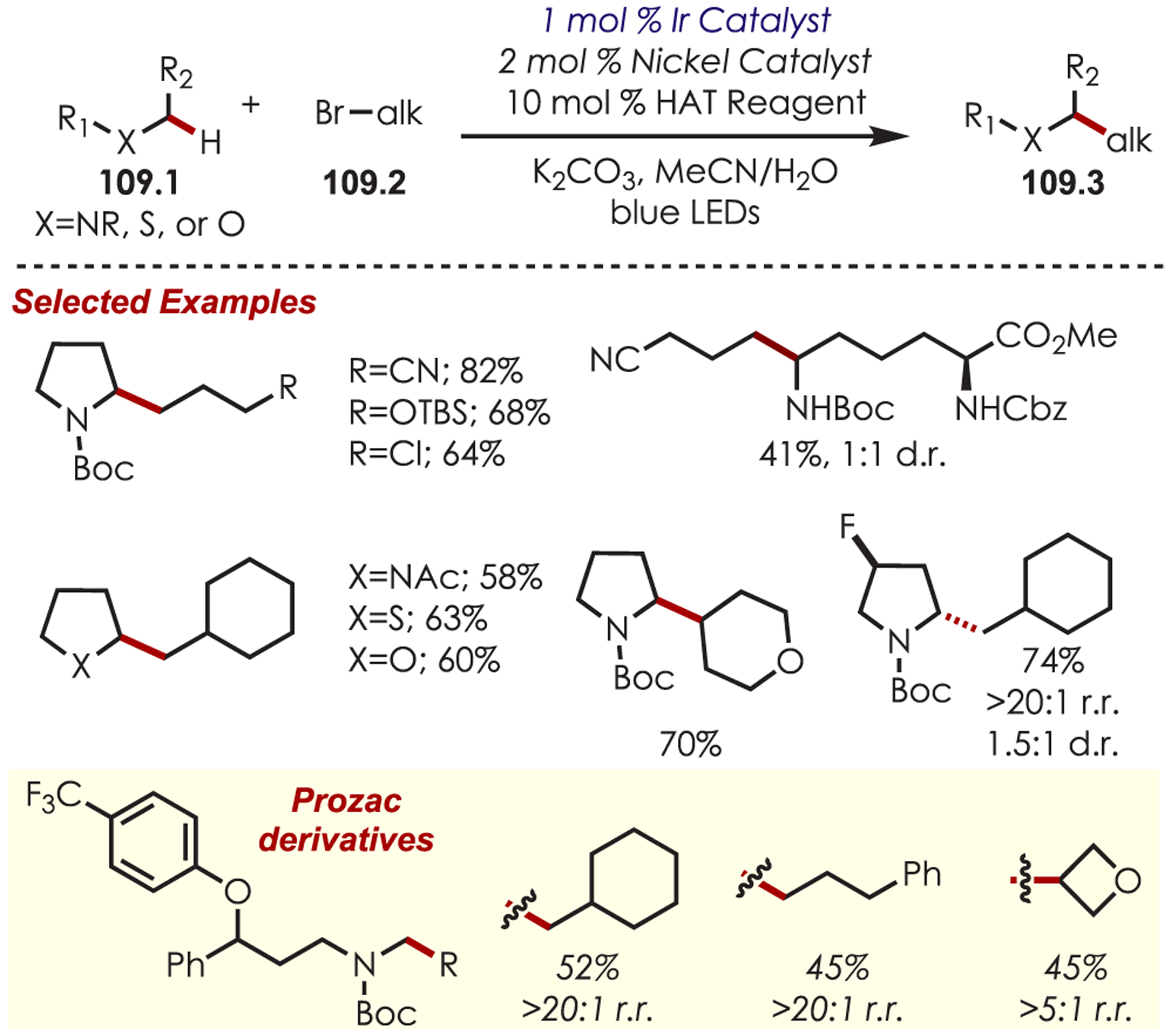
α-Hetero C–H Alkylation
In the same year, Doyle independently reported a related cross coupling of aryl iodides (110.2) via α-amino radicals (Scheme 110).233 However, this method does not rely on a hydrogen atom abstraction to generate the radical, which is alternatively generated by oxidation and subsequent deprotonation. Bis(oxazoline), or BiOX, ligands were noted to be crucial for high reaction efficiency which limited the undesired β-hydride elimination product observed with more sterically bulky ligands. This generates a range of benzylic amines from electronically diverse (hetero)aryl iodides and the corresponding cyclic or acyclic amine.
Scheme 110.
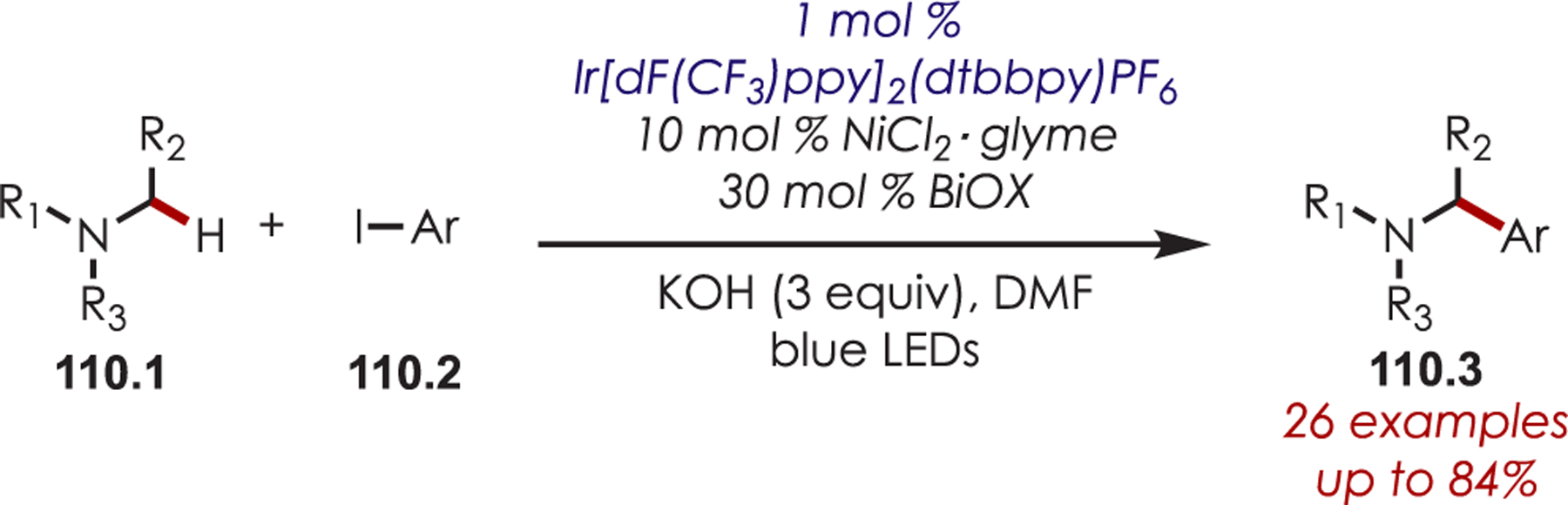
α-Amino C–H Arylation through Dual Nickel and Photoredox Catalysis Reported by Doyle
Doyle and co-workers reported the coupling of aryl chlorides (111.2) and α-oxy radicals generated through a hydrogen atom abstraction event via chlorine radicals around the same time (Scheme 111).234 Photolysis of an intermediate Ni(III) aryl chloride complex generates a chlorine radical through an MLCT process. Next, a thermodynamically favorable α-oxy C–H abstraction by the chlorine atom (BDEs of HCl and THF are 102 and 92 kcal/mol, respectively) afforded an alkyl radical intermediate. Interception of this alkyl radical by Ni(II) followed by reductive elimination afforded the coupled benzylic ether products.
Scheme 111.
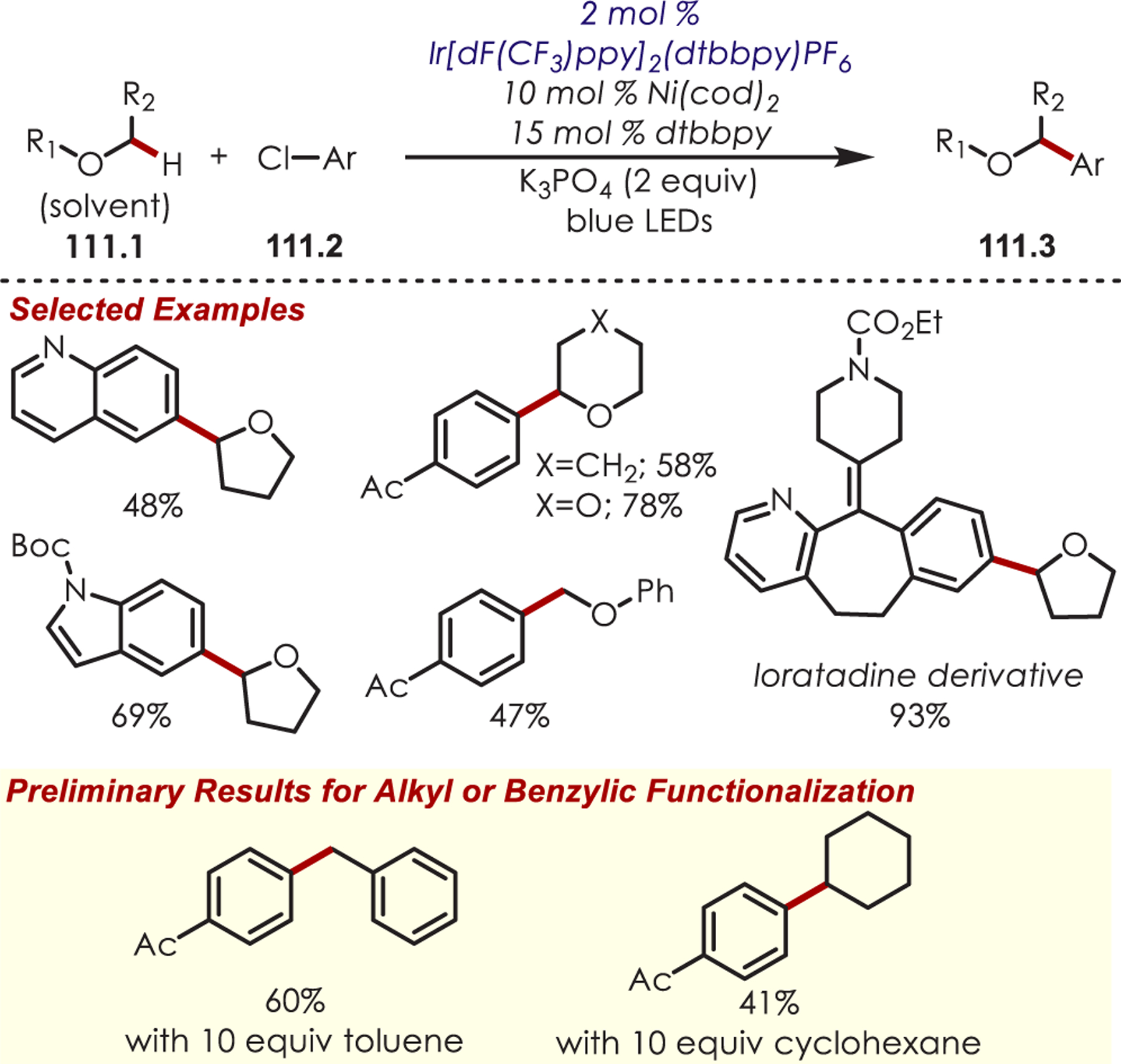
α-Oxy C–H Arylation Using Aryl Chlorides via Dual Nickel and Photoredox Catalysis
Notably, the coupling of aryl chlorides (112.1) with 1,3-dioxolane (112.2) can be subjected to a hydrolytic workup which furnishes the formylated arenes, 109.3 (Scheme 112).235
Scheme 112.
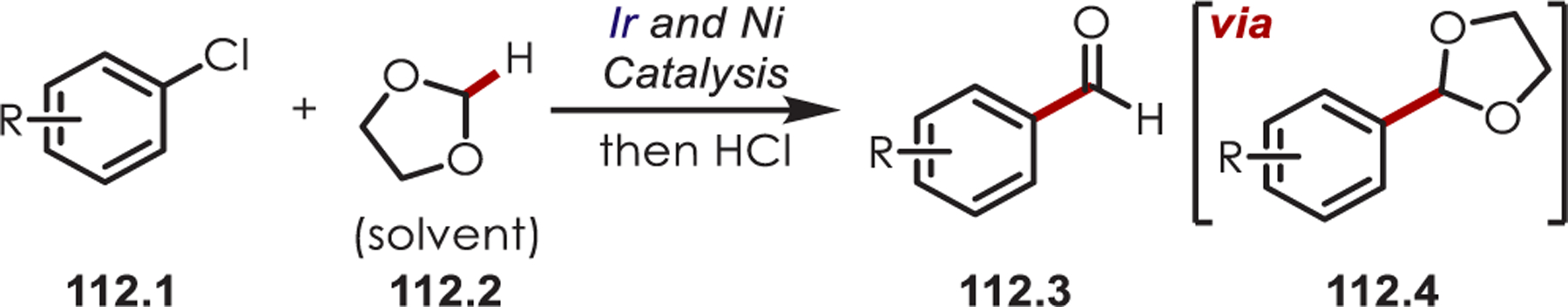
Coupling of 1,3-Dioxolane and Aryl Chlorides and Subsequent Hydrolytic Workup to Access the Formylated Arenes
Chlorine radicals as hydrogen-abstracting reagents have also been reported with maleate and fumarate radical acceptors (113.2) for α-hetero alkylation (Scheme 113).236 Aliphatic alkanes, silanes, and aldehyde C–H bonds were alkylated under these conditions; however, the latter likely proceeds through a radical-chain mechanism which is known to occur in the literature.237
Scheme 113.
α-Hetero C–H Alkylation with Maleate and Fumarate Radical Acceptors
The formation of α-amino radicals through thiyl radical HAT has been utilized for the deuteration (114.2) or tritiation (114.3) of pharmaceutical derivatives (Scheme 114).238 The driving force for the H–D/T exchange is the relative BDE of the α-amino C–H and thiol S–H bonds, with values of 93 and 87 kcal/mol, respectively. Introduction of either D2O or T2O enables in situ formation of the active deuterating or tritiatng agent by rapid D/H or T/H exchange with the thiol co-catalyst, which intercepts the α-amino radical. This method was used for the selective labeling of 18 drugs and drug candidates.
Scheme 114.
α-Amino C–H Deuteration and Tritiation of Pharmaceutical Derivatives
Recently, a photoredox and nickel-mediated methylation was reported using alkyl peroxides (115.2) as the HAT and methylating agent (Scheme 115).239 Visible-light-promoted homolysis of the peroxyl O–O bond generates an alkoxy radical that serves as the hydrogen atom transfer agent. A nickel-mediated coupling of the alkyl radical and a methyl radical, from β-methyl scission of the alkoxy radical, furnishes the methylated products. This was demonstrated in both α-amino and benzylic C–H methylation, including in the late-stage functionalization of pharmaceutical and natural product derivatives.
Scheme 115.
C–H Methylation via Peroxide Sensitization and Nickel Radical Coupling
In 2019, a zwitterionic 1,2,3-triazolium amidate (116.4) was utilized, alongside an Ir(III) catalyst, as an HAT catalyst for the alkylation of α-amino and α-oxy C–H bonds (Scheme 116).240 The goal was to develop a more robust intermolecular HAT reagent that would allow for sufficient tunability of the N–H BDE, which was previously not easily accessible using quinuclidinium-, benzoyloxy-, thiyl-, or phosphoryloxy-based HAT catalysts. However, the authors found that single-electron oxidation of triazolium amidates suffers from considerable back-electron transfer, particularly for other amidate or Ir(III) derivatives. Still, one amidate (BDE = 100.2 kcal/mol) was successful in the alkylation of α-amino or α-oxy C(sp3)–H bonds with vinyl ketones/esters, bis(phenylsulfonyl)ethylene, and benzylidenemalononitrile acceptors (116.2) and, furthermore, was used for the generation of nucleophilic acyl radicals. Single-electron oxidation of the zwitterionic triazolium amidate by Ir(III)* generates the amidyl radical which serves as the active hydrogen atom abstracting agent.
Scheme 116.
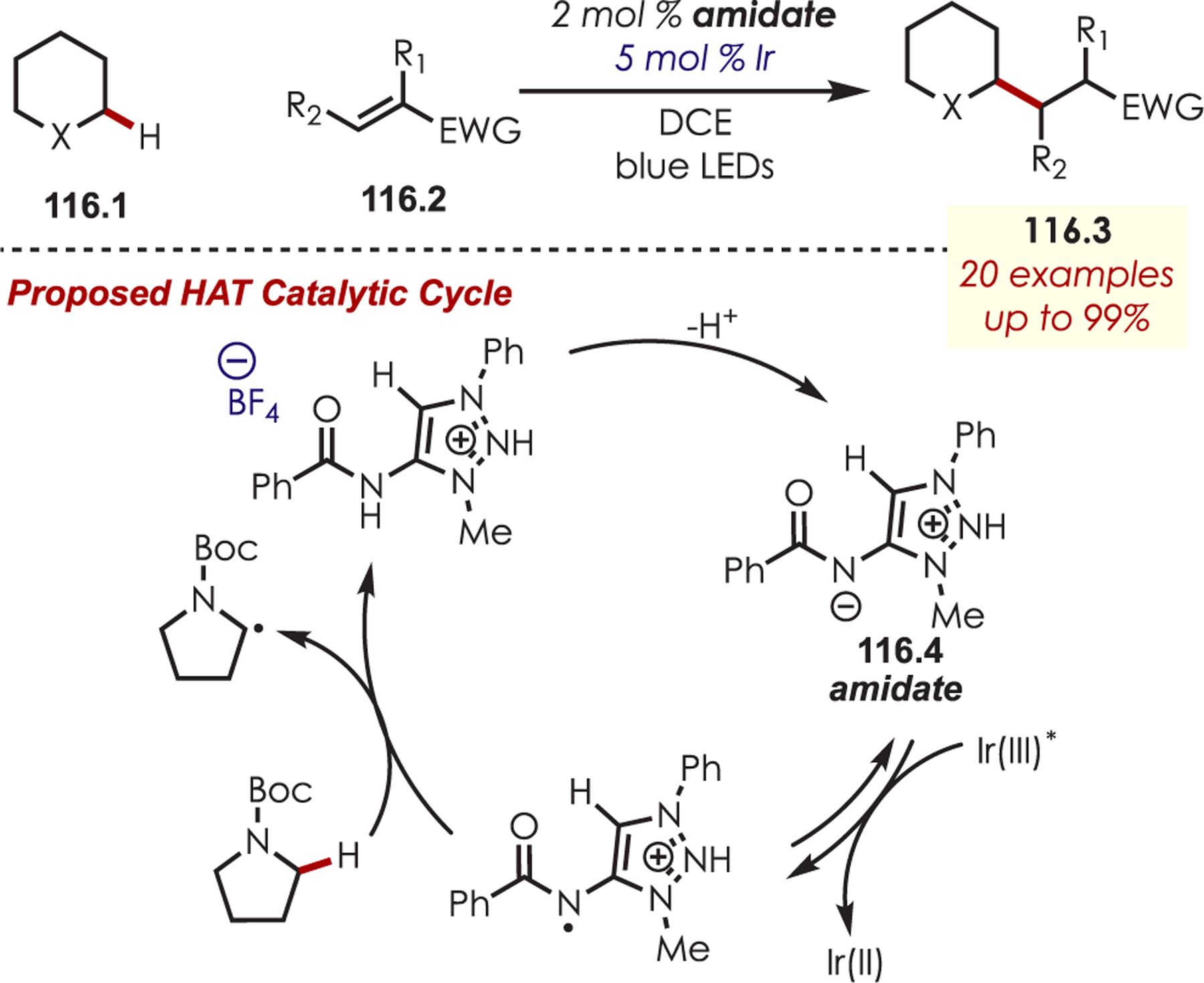
α-Hetero C–H Alkylation Using an Amidate HAT Reagent
The merging of nickel and photoredox catalysis has been successful in the α-acylation of amines241 (117.1) and ethers242 (117.3) with anhydrides or acyl chlorides (Scheme 117). In this case, oxidative addition of the Ni(0) into the acyl species leads to the Ni(II) acyl complex. Interception of the α-hetero species by Ni(II) and reductive elimination furnishes the acylated amines (117.2) or ethers (117.4).
Scheme 117.
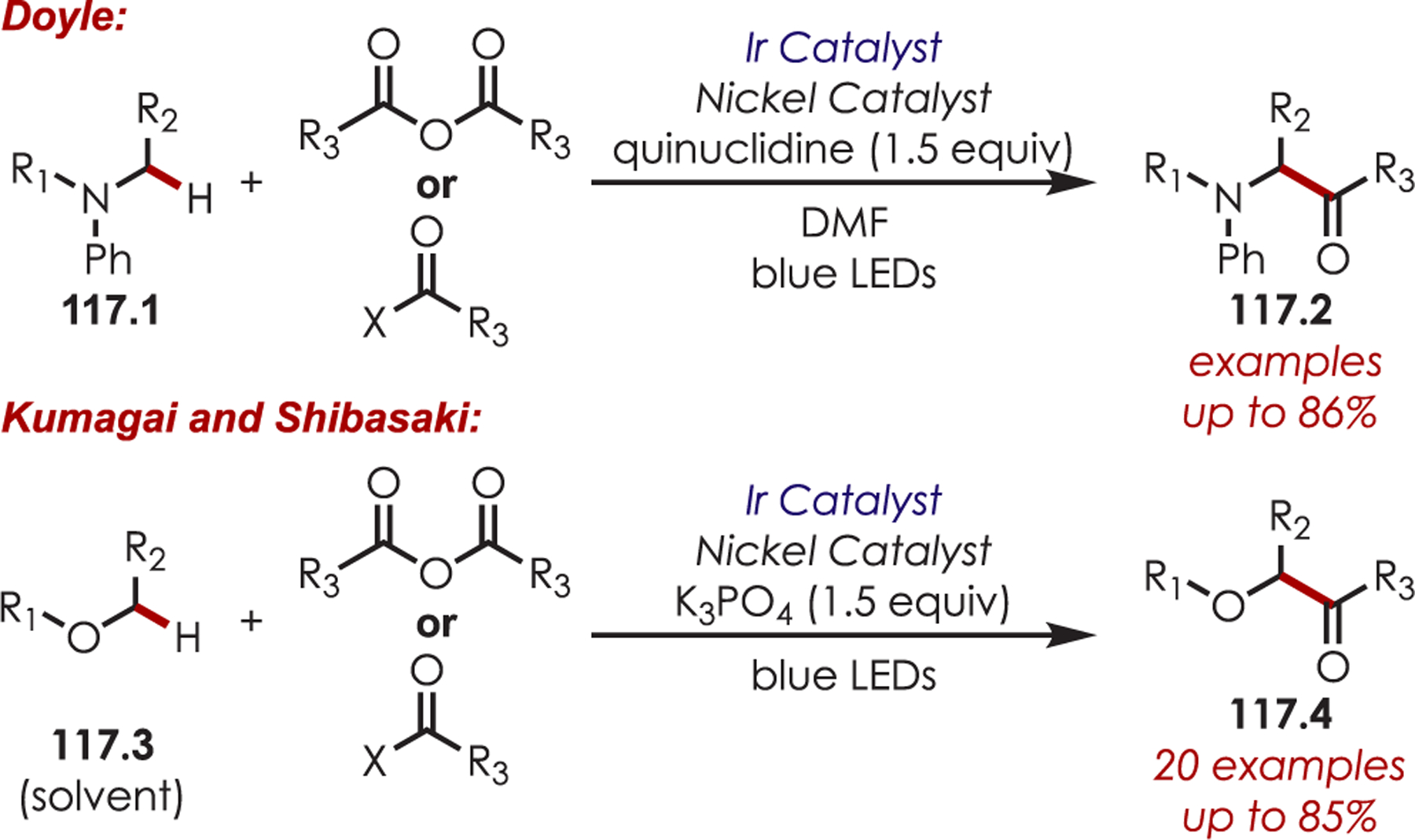
α-Hetero C–H Acylation through Dual Nickel and Photoredox Catalysis
Recently, an enantioselective α-acylation of amines (118.1) was reported by Huo and co-workers utilizing photoredox catalysis in combination with a chiral bis-oxazoline nickel complex (Scheme 118).243 The in situ generation of acyl halides from carboxylic acids via activation with dimethyl dicarbonate (DMDC) and nucleophilic displacement by chloride allowed for the use of readily available acids as acyl radical precursors. A broad scope was examined giving the acylated products (118.3) in good yields and enantioselectivities. The application of this method to late-stage functionalization of several more complex amines was demonstrated in good stereoselectivities and moderate yields.
Scheme 118.
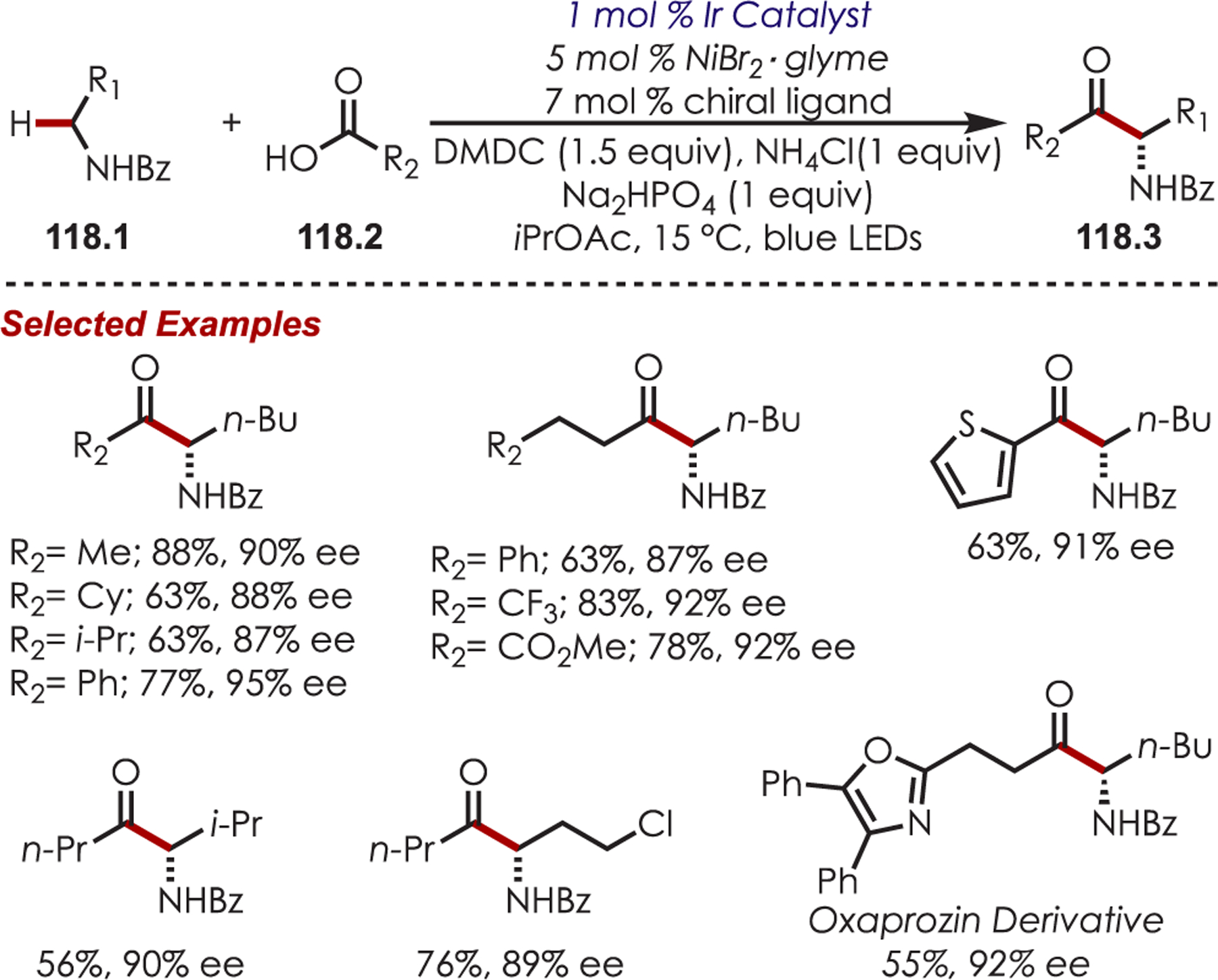
Enantioselective α-Amino C–H Acylation
In general, HAT pathways for C–H functionalizations generate a carbon-centered radical from abstraction of the most hydridic C–H or the C–H bond with the lowest BDE.244 A remaining challenge is the selective abstraction of other less activated C–H bonds in the presence of other potentially weak C–H bonds. One strategy that has been presented by MacMillan to overcome this is to use bond-weakening co-catalysts to activate the desired C–H bond. Using either a monobasic phosphate or trifluoroacetate salt as a hydrogen-bonding catalyst, the C–H bonds adjacent to alcohols (119.1) were activated toward HAT and underwent a subsequent alkylation/lactonization reaction (Scheme 119).245 Remarkably, this method selectively alkylated α-hydroxyl C–H bonds in the presence of weaker ether or allylic or benzylic C–H bonds with excellent regioselectivity. DFT calculations revealed that the hydrogen-bonding interaction weakened the α-hydroxy BDE by approximately 3 kcal/mol (e.g., hexanol = 94.1 kcal/mol vs 91.0 kcal/mol with the catalyst). In addition to the change in BDE, polarization is proposed to play a significant role in the selectivity, resulting in decreased s-character of the hybridized carbon orbitals when hydrogen-bonding occurs.
Scheme 119.
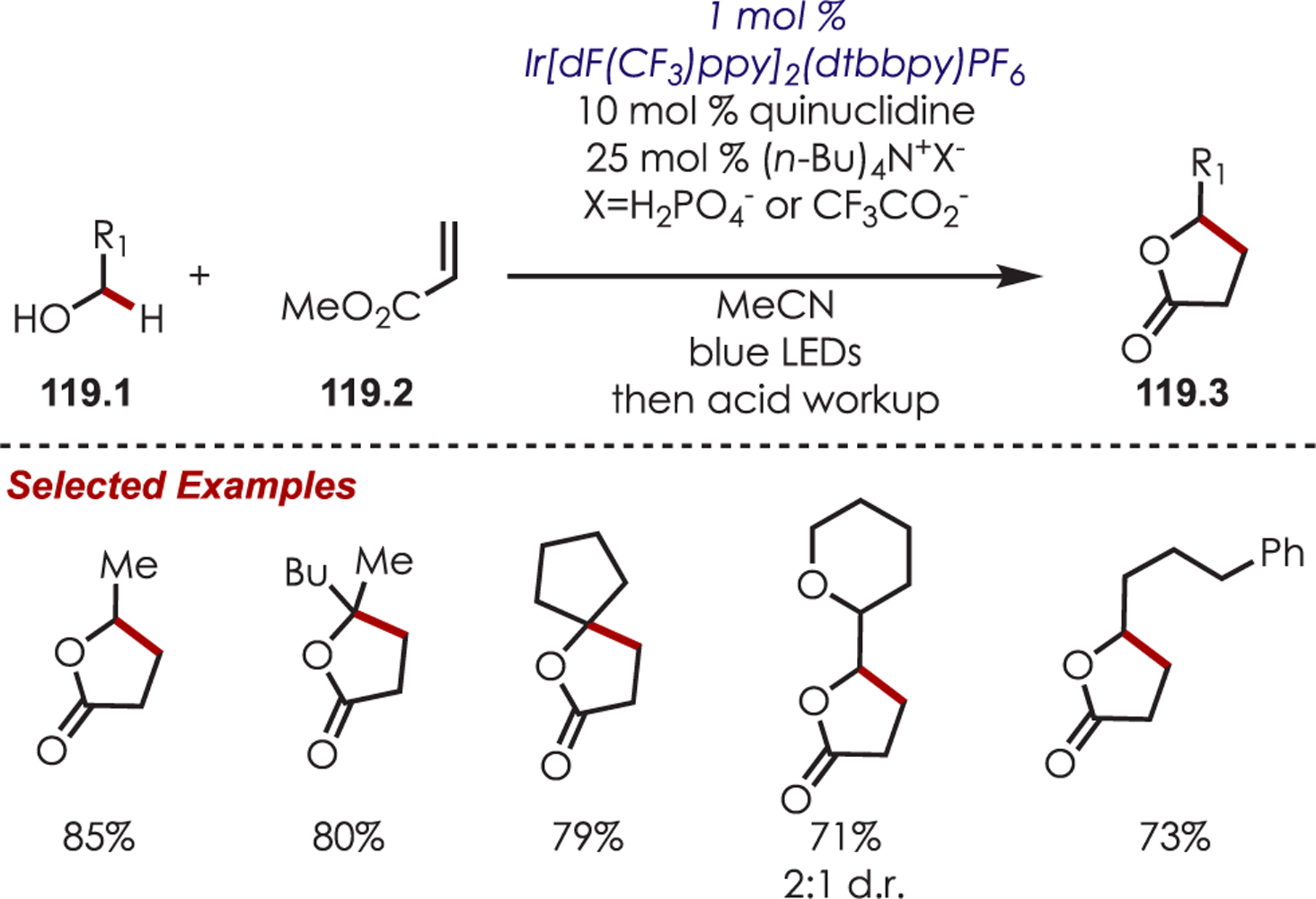
Direct α-Hydroxy C–H Alkylation Using a Hydrogen-Bonding Co-catalyst to Activate α-C–H Bonds on Alcohols
In an analogous manner, the α-alkylation of ethanol (120.1) has been accomplished by utilizing a pentavalent silicate species in order to weaken the α-C–H bond, thus allowing for subsequent HAT and radical addition (Scheme 120).246
Scheme 120.
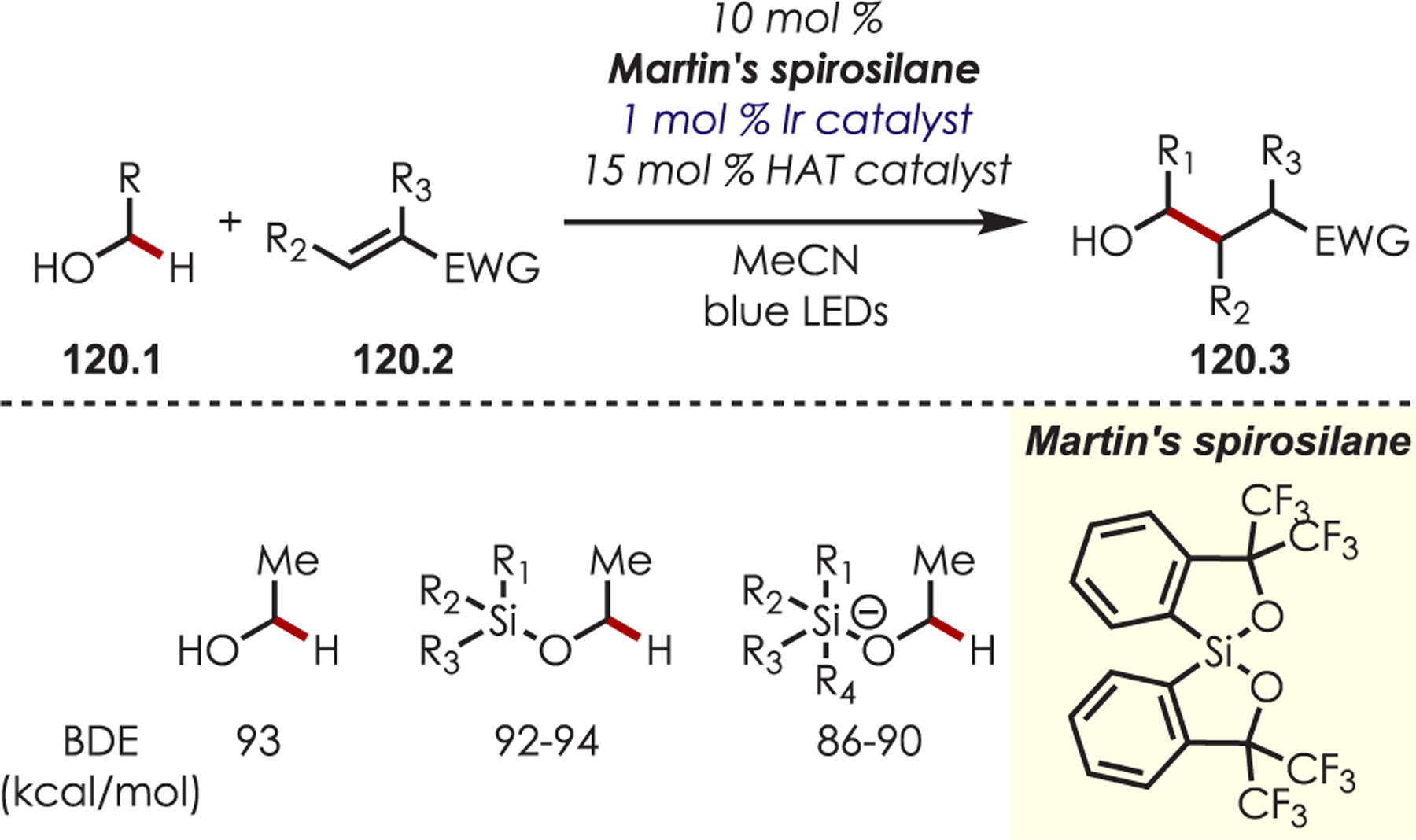
Effect on the BDEs of the α-Hydroxy C–H with the Coordination of a Pentavalent Silicate Species as a Bond-Weakening Catalyst
The scope of this alkylation was demonstrated with a variety of radical acceptors (121.2) and showed high selectivity for alkylation of the α-oxy C–H bond in the presence of benzylic C–H bonds, cyclic ethers, and amines (Scheme 121).
Scheme 121.
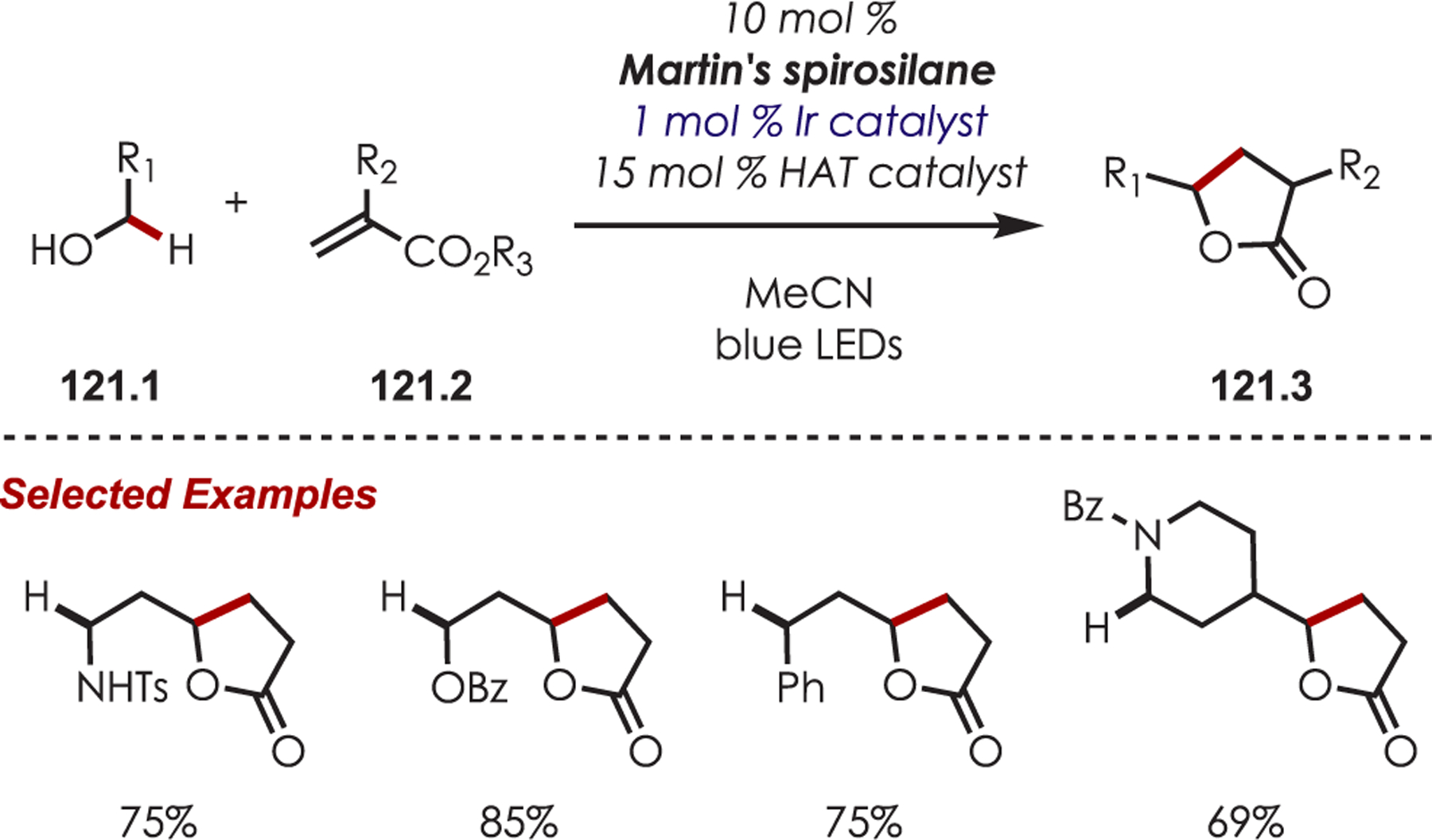
Scope of the Direct α-Hydroxy C–H Alkylation using a Pentavalent Silicate Species as a Bond Weakening Catalyst
Diarylborinic acids247 (122.4) and dihydrogen phosphate248 (122.5) have also been utilized as C–H bond-weakening co-catalysts for the α-alkylation of sugar derivatives (Scheme 122).
Scheme 122.
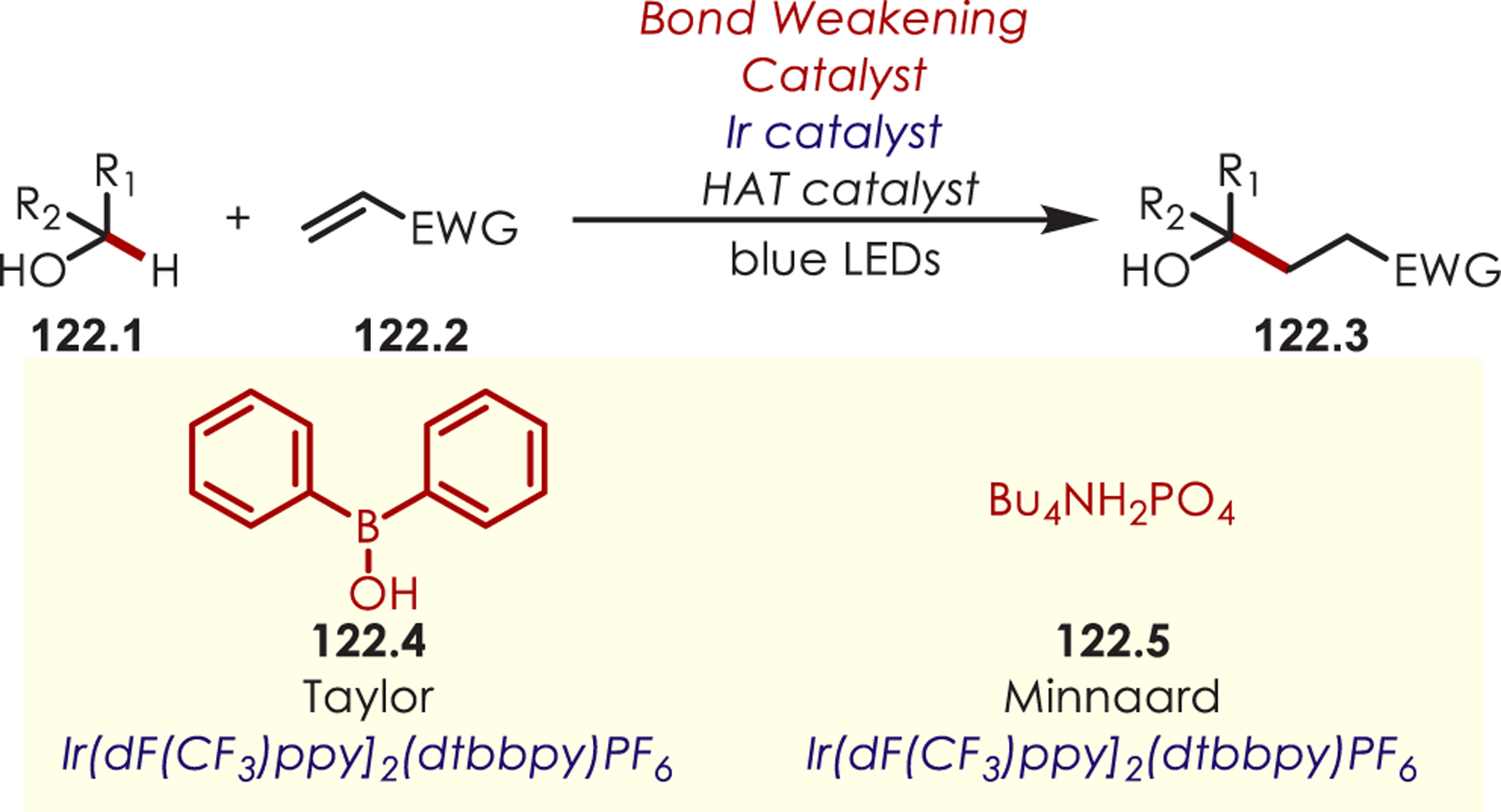
Borinic Acids and Tetrabutyl Ammonium Phosphate as Bond Weakening Catalysts for α-Hydroxy C–H Alkylations
Moreover, alkynes (123.2) were found to be amenable to cross coupling with ethers and amides (123.1) using a combination of nickel and photoredox catalysis (Scheme 123).249 A mixture of regioisomeric allylic ethers is observed with alkyl-substituted alkynes, but increasing steric hindrance resulted in increased selectivity, favoring alkylation to afford the corresponding cis-olefin product. Ultimately, trimethylsilyl-substituted alkynes were found to give >20:1 regioselectivity. The alkenylated products were proposed to be generated from reductive elimination of a key nickel hydride species. Mechanistic evidence suggests that an oxidative addition to HCl, from the hydrogen atom abstraction event between a chlorine atom generated by photolysis of NiCl2 and the ether, produces the nickel hydride.
Scheme 123.
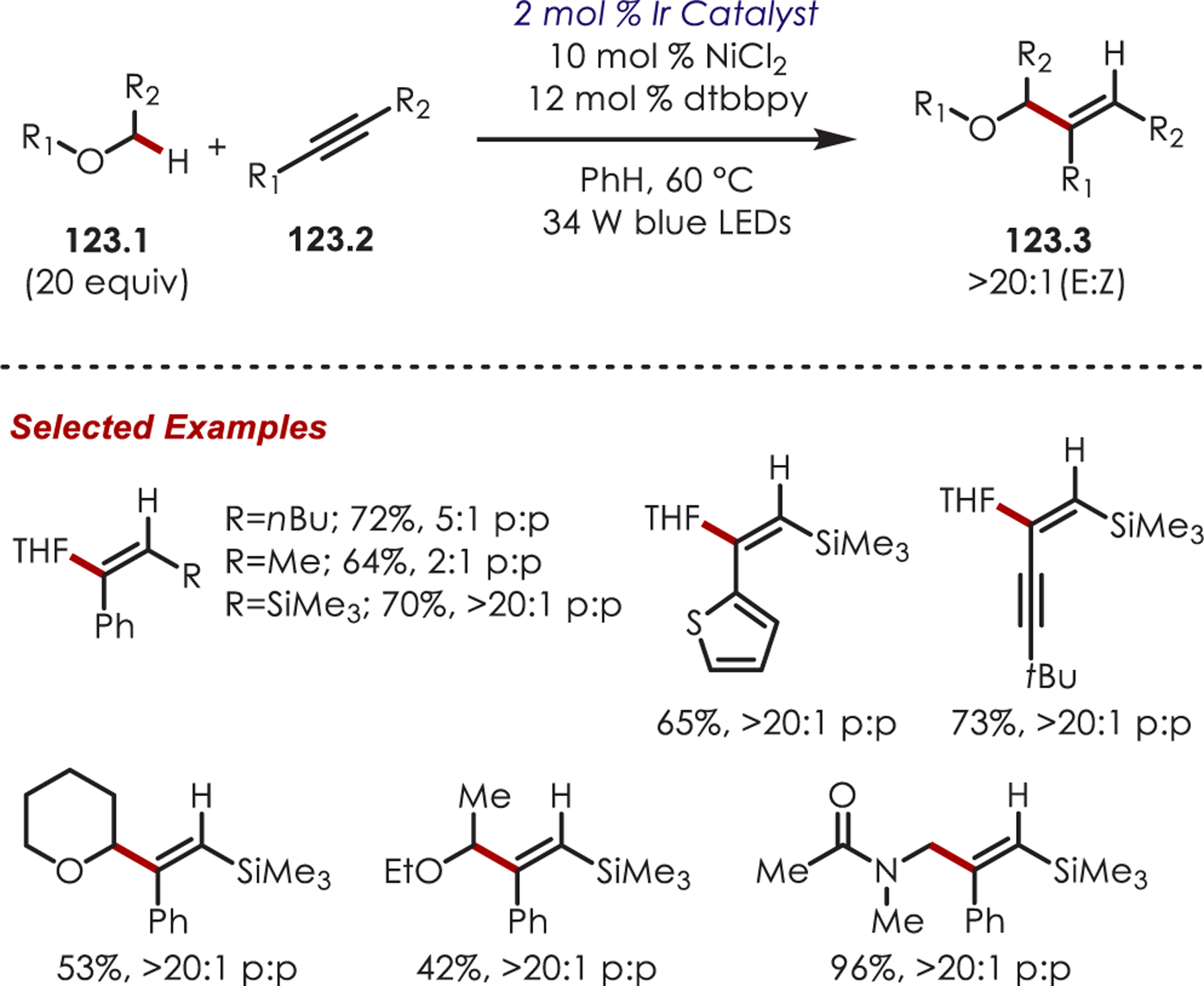
Coupling of Alkynes with α-Oxo Radicals via Dual Nickel and Photoredox Catalysis
Recently, a comprehensive investigation of the reaction mechanism for arylation and alkylation of amines through photoredox–nickel–HAT triple catalytic systems, such as those used by MacMillan, was studied by Rueping and Cavallo through computational analysis (Scheme 124).250 They found both reductive and oxidative quenching cycles are possible, but the reductive quenching cycle is thermodynamically favored with quenching of Ir(III)* by quinuclidine at the M06(SMD-MeCN)/BS2/PBE/BS1 level of theory. A reductive quenching event generates the amine cation radical as the hydrogen abstracting reagent along with the reduced Ir(II) species. Site-selective hydrogen atom abstraction generates a carbon-centered α-hetero radical (124.1). Two potential catalytic nickel cycles were determined feasible, but the energetically favored pathway is triggered by radical addition into Ni(I). The resulting Ni(II) species, 124.2, is reduced by Ir(II), losing bromide anion and regenerating the quinuclidine HAT reagent and Ni(I) (124.3). An oxidative addition of the aryl or alkyl halide coupling partner occurs and a subsequent reductive elimination from the Ni(III) complex furnishes the coupled products (124.5) while regenerating the active Ni(I) catalyst. The alternative, less energetically favored catalytic nickel cycle goes through a sequence of SET, radical addition, oxidative addition and reductive elimination involving Ni(I)–Ni(0)–Ni(I)–Ni(III) oxidation states.
Scheme 124.
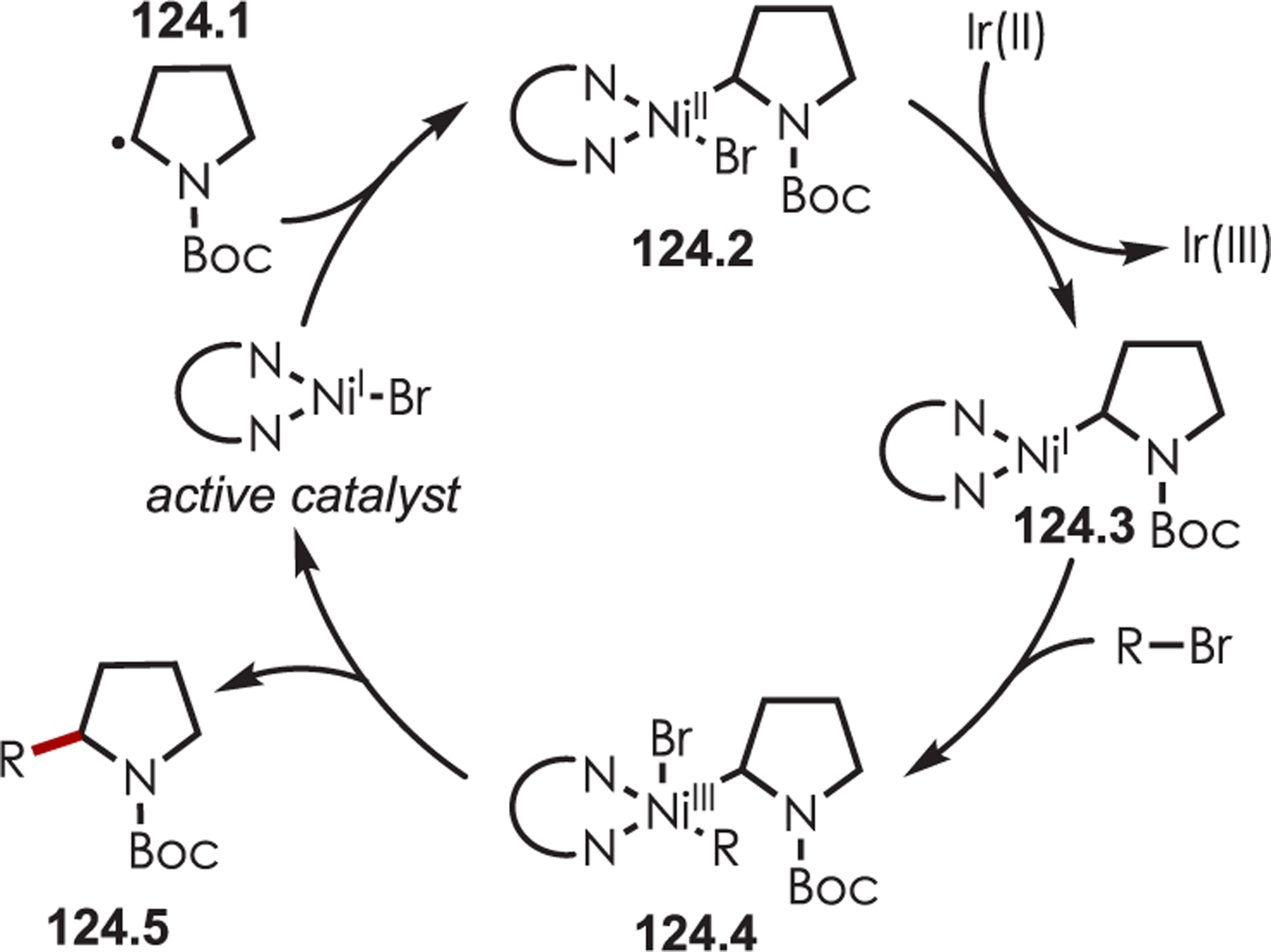
Mechanism of Nickel-Mediated α-Amino Cross-Couplings with Aryl Halides
An α-amino radical–radical coupling has been utilized in the monofluoroalkenylation of dialkylamines (Scheme 125).251 Oxidation of the amine to an amine cation radical by photoexcited Ir(III) forms Ir(II). Then, a reductive SET of gem-difluoroalkenes (125.2) by the Ir(II) generates monofluoroalkenyl radicals that were coupled with the generated α-amino radicals. A related quinuclidine HAT and photoredox-catalyzed transformation was reported for the multifluoroarylation of ethers which couples α-amino radicals and aryl fluoride anion radicals.252
Scheme 125.

C–H Monofluoroalkenylation of Dialkylamines
In 2018, Rovis and co-workers reported an α-alkylation of primary amines by HAT and photoredox catalysis (Scheme 126).253 A key component of the success of this chemistry is the modulation of the high nucleophilic reactivity on the free amine (126.2) nitrogen by using CO2 to form carbamates in situ (126.4). This ultimately decreases the nucleophilicity of the amine and enables the α-amino hydrogen abstraction event to occur. In particular, the quinuclidinium cation radical was chosen as an ideal hydrogen atom transfer reagent in order to facilitate a favorable electrostatic interaction between the electrophilic amine cation radical and the carbamate anion, thus overcoming the lower acidity of the electron-withdrawing carbamyl α-C–H bond. After radical addition into an acceptor, the α-acyl radical intermediate (126.5) is reduced, forming the corresponding enolate. Upon proton transfer, decarboxylation of the in situ generated carbamate to form the free amine and subsequent lactamization affords the γ-lactam product.
Scheme 126.
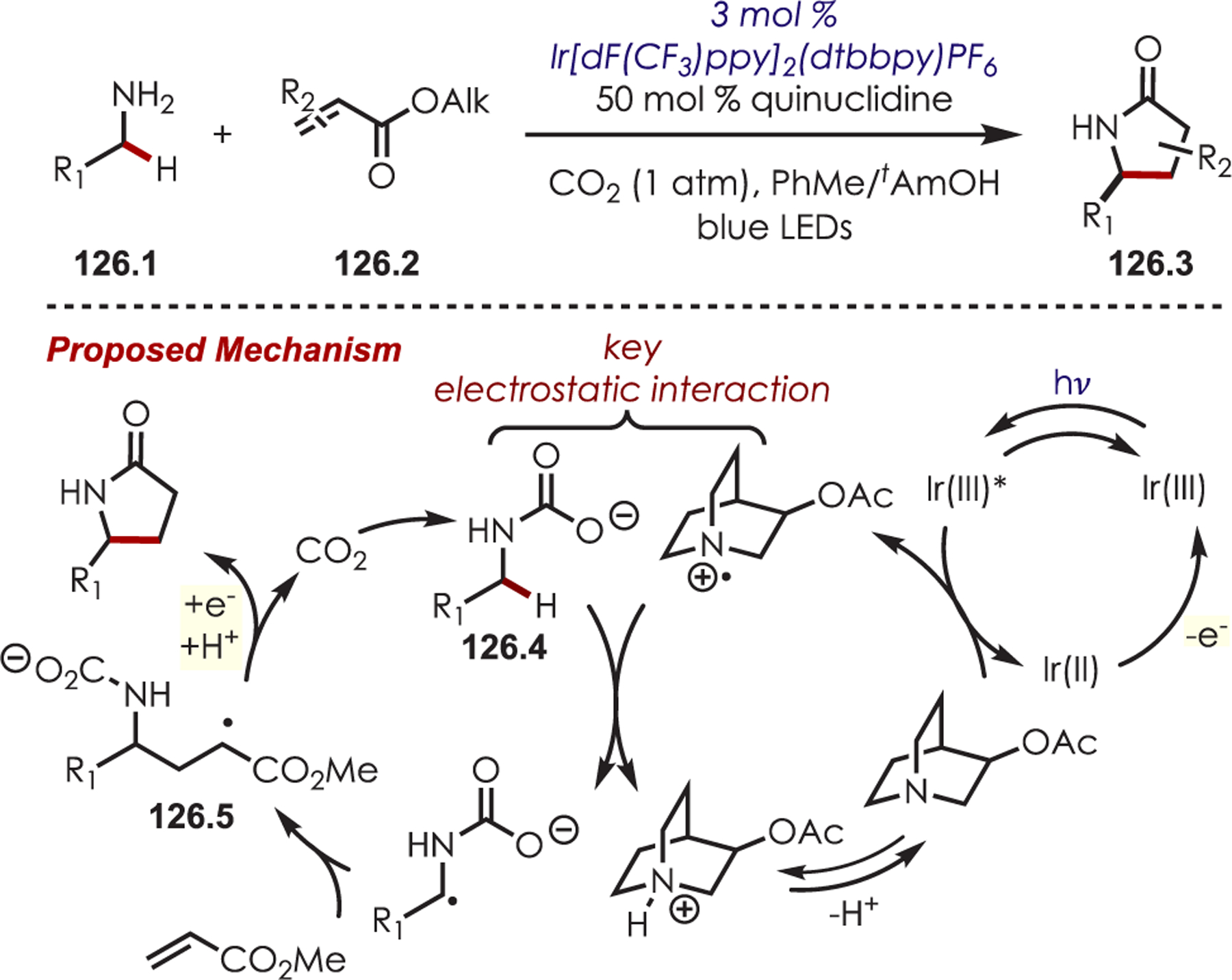
Mechanism of a Primary Amine C–H Alkylation and Lactamization through In Situ Generated Carbamates
A range of primary amines and acrylates were suitable in this lactam synthesis and afforded a mixture of diastereomers (127.1–127.7) between 1:1 and 2.3:1 dr when applicable (Scheme 127). Simple aliphatic amines and cyclic amines (127.3) were tolerated, furnishing the product with an α-quaternary center. Functional groups that were tolerated include sterically hindered, fluorinated, silyl-protected alcohols and benzylic, acetal-, alkenyl-, alkynyl-, or heteroaryl-containing amines. Acrylates substituted with α- or β-alkyl or phenyl groups and methylacrylonitrile were found to be compatible radical acceptors, giving substitution in the lactam ring.
Scheme 127.
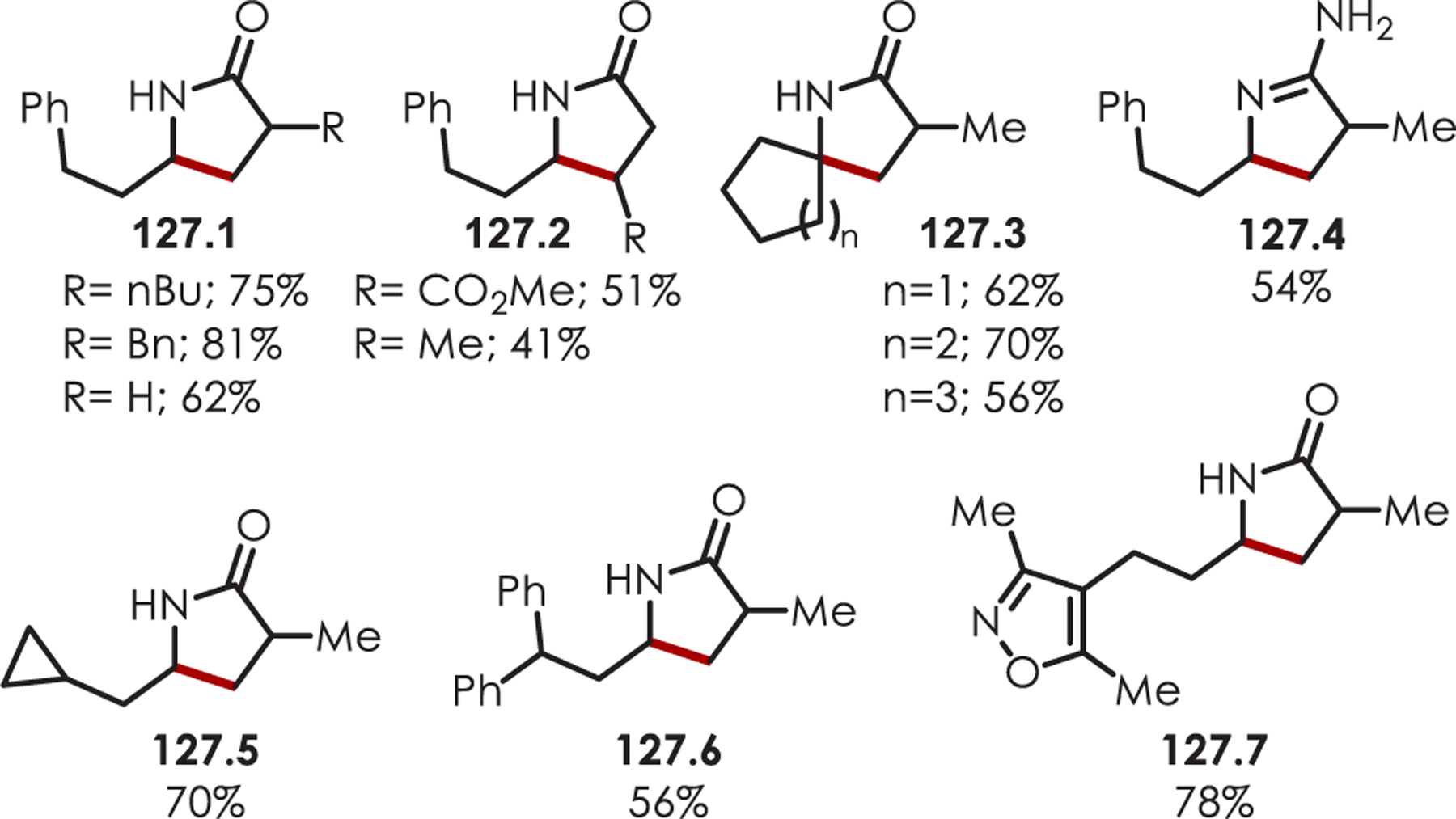
Scope of Primary Amine C–H Alkylation and Lactamization through In Situ Generated Carbamates
In 2017, Reiser reported the synthesis of 2,3-disubstituted indolines (128.2) through the generation of α-amino radical species (Scheme 128).254 A vinylic carbon-centered radical is generated through reduction of the α-bromochalcone by Ir(II), and a subsequent 1,6-hydrogen atom transfer generates the α-amino radical. A 5-exo-trig cyclization followed by hydrogen atom abstraction furnishes the indoline products.
Scheme 128.
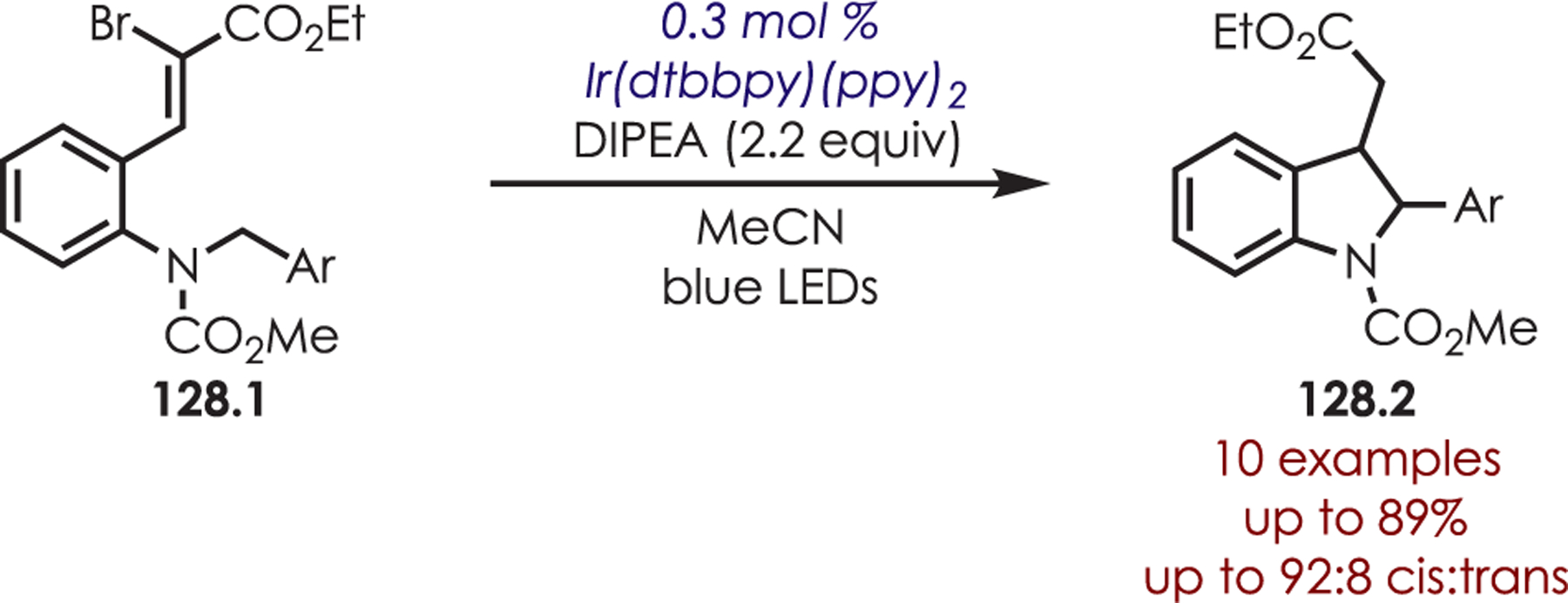
Generation of α-Amino Radicals through 1,6-HAT: Synthesis of Indolines
C–H functionalization reactions adjacent to aliphatic secondary amides are generally less explored due to competitive amidyl radical formation and subsequent 1,5-hydrogen atom transfer. However, Rovis and Yorimitsu developed a divergent α-alkylation of aliphatic primary amines (129.1) that prevents the intramolecular ring cyclization by judicious selection of a protecting group (Scheme 129).255 Trifluoromethanesulfonamide protecting groups on the amines render the α-amino hydrogen atoms of conjugate base (129.5) more hydridic, thus providing a driving force for HAT by an electrophilic quinuclidinium cation radical (129.4) to furnish an α-amino anion radical (129.6). Subsequent alkylation occurred adjacent to the amine followed by a reduction and protonation events adjacent to the electron-withdrawing group on the alkene to give the final adducts. None of the potentially competing 1,5-HAT events were observed under these reactions.
Scheme 129.
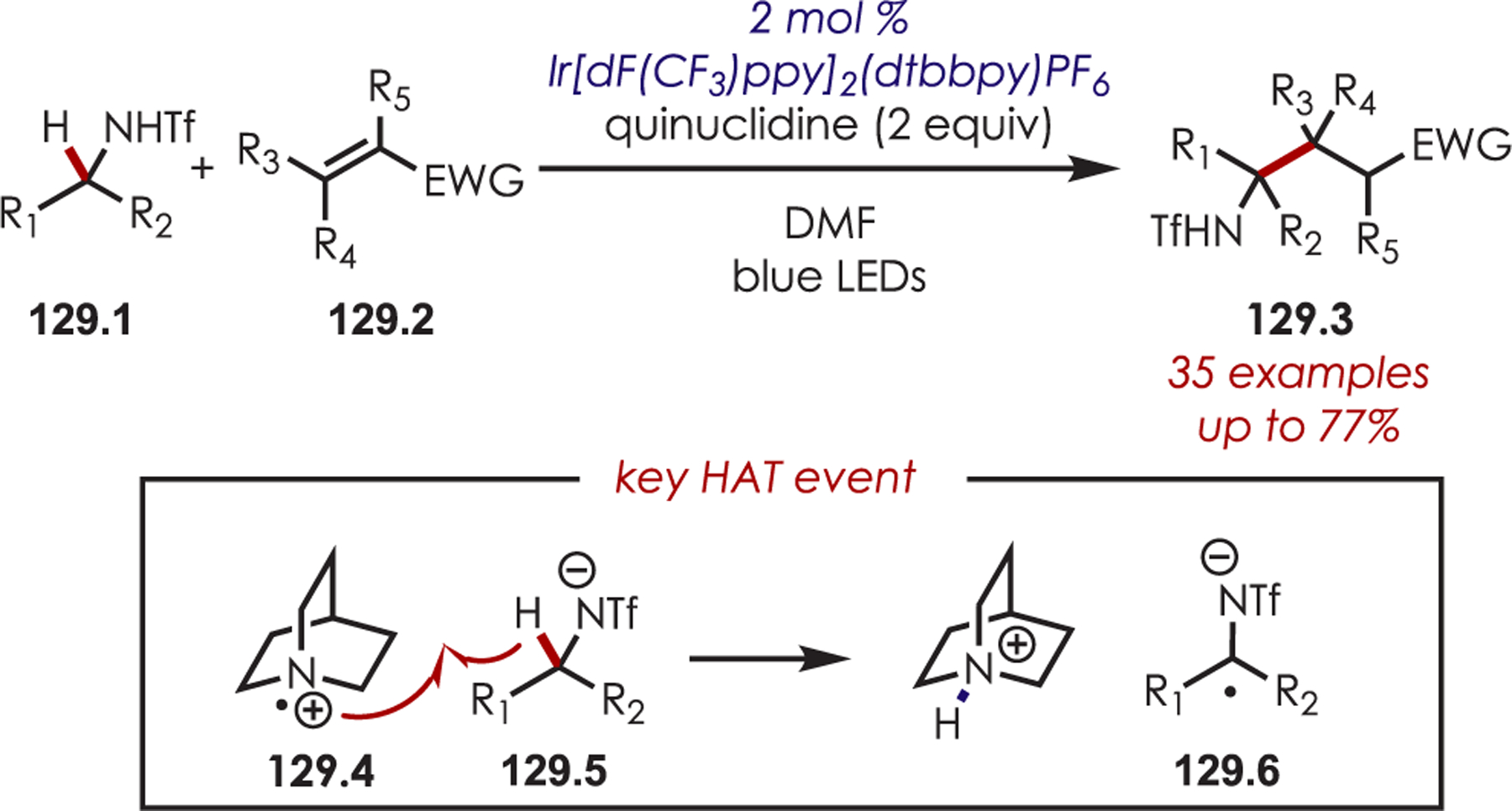
C–H Alkylation of Aliphatic Secondary Amides
In addition to quinuclidinium cation radicals (130.1), other hydrogen atom abstracting agents have been utilized in functionalization reactions adjacent to heteroatoms, including sulfonamides (130.2), phosphates (130.3) for α-cyanation,256 and thiocarboxylates (130.4) for α-arylation (Scheme 130).257,258
Scheme 130.
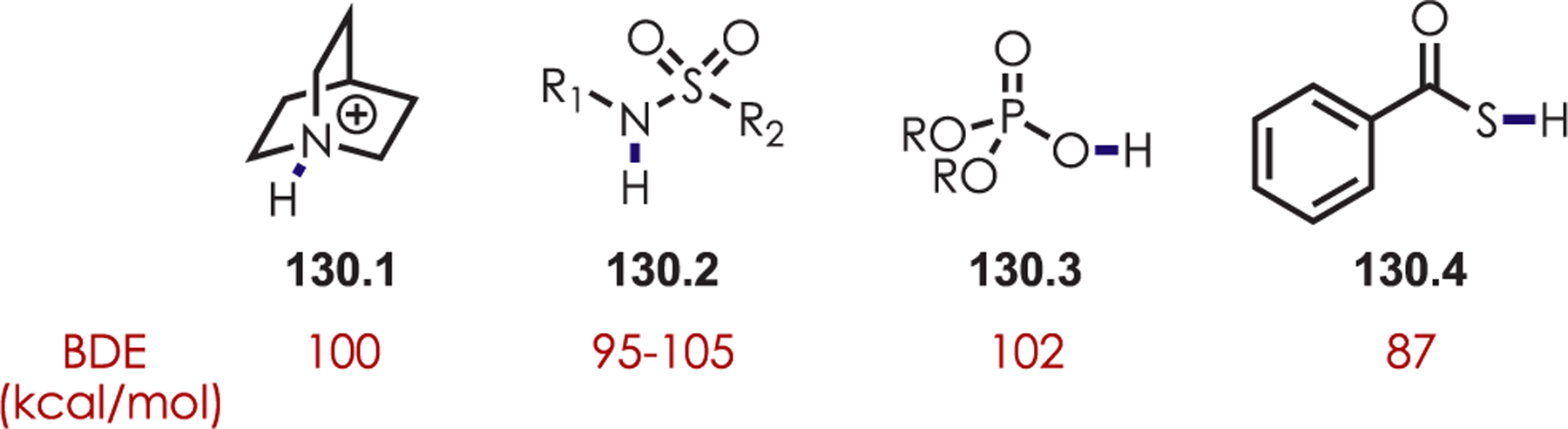
Conjugate Acids of Common Hydrogen Atom Abstracting Reagents and Corresponding BDEs
More recently, Montgomery and Martin reported a dual catalytic strategy for α-arylation and alkylation of amides (131.1) with unactivated organic halides (131.2) that exhibit complementary reactivity to the previously reported α-hetero alkylations (Scheme 131).259 This method is not limited to electron-deficient radical acceptors and does not require stoichiometric HAT mediators. The orthogonality of this transformation for both α- and δ-functionalization was demonstrated by alternate reaction conditions, where in the absence of the nickel catalyst a 1,5-HAT leads to the δ-substituted product. Additionally, asymmetric catalysis was achieved by using a chiral BiOx ligand. Investigation of the mechanism for this transformation was reportedly ongoing.
Scheme 131.
C–H Alkylation and Arylation of Amides
Meggers accomplished an asymmetric radical–radical cross coupling for the synthesis of enantioenriched 1,2-amino alcohols (132.4) though a chiral iridium photoredox catalyst (132.3; Scheme 132).260 This proceeds through coordination of the catalyst to the ketone (132.1), which then undergoes excitation to form the active photoexcited species (132.4). Irradiation triggers single-electron transfer between the electron donor amine and the ligated ketone resulting in the generation of a chiral radical pair. A stereocontrolled radical–radical recombination (132.6) yields the products in high enantio- and diastereoselectivity. An asymmetric model was proposed that prevented addition at the more sterically hindered Si face, thus being the source of stereoinduction. Heteroaryl trifluoromethyl ketones were found as the only competent substrates and were suggested to arise from a key persistent radical effect of the intermediate ketyl radical that prevented homodimerization. Diaryl amines and tetrahydroisoquinolines were found to be suitable amine-coupling partners.
Scheme 132.
Chiral Ir-Mediated C–H Alkylation with Trifluoromethyl Aryl Ketones
While Meggers’ catalyst functions as both the Lewis acid and photoredox catalyst, chiral Brønsted catalysis has also been reported in combination with photoredox catalysis for the asymmetric coupling of α-amino radicals with aldimines (Scheme 133).261 The Ooi group reported a range of (hetero)aromatic imines (133.1) that were suitable substrates, but aliphatic imines were not compatible. Both N,N-diarylaminomethanes and N-alkyl derivatives gave the product in moderate yields and high enantioselectivities. The enantiodetermining step is proposed as a radical–radical coupling between the α-amino radical and a photogenerated iminyl radical anion that is positioned in a chiral environment through hydrogen bonding with the phosphonium ion.
Scheme 133.
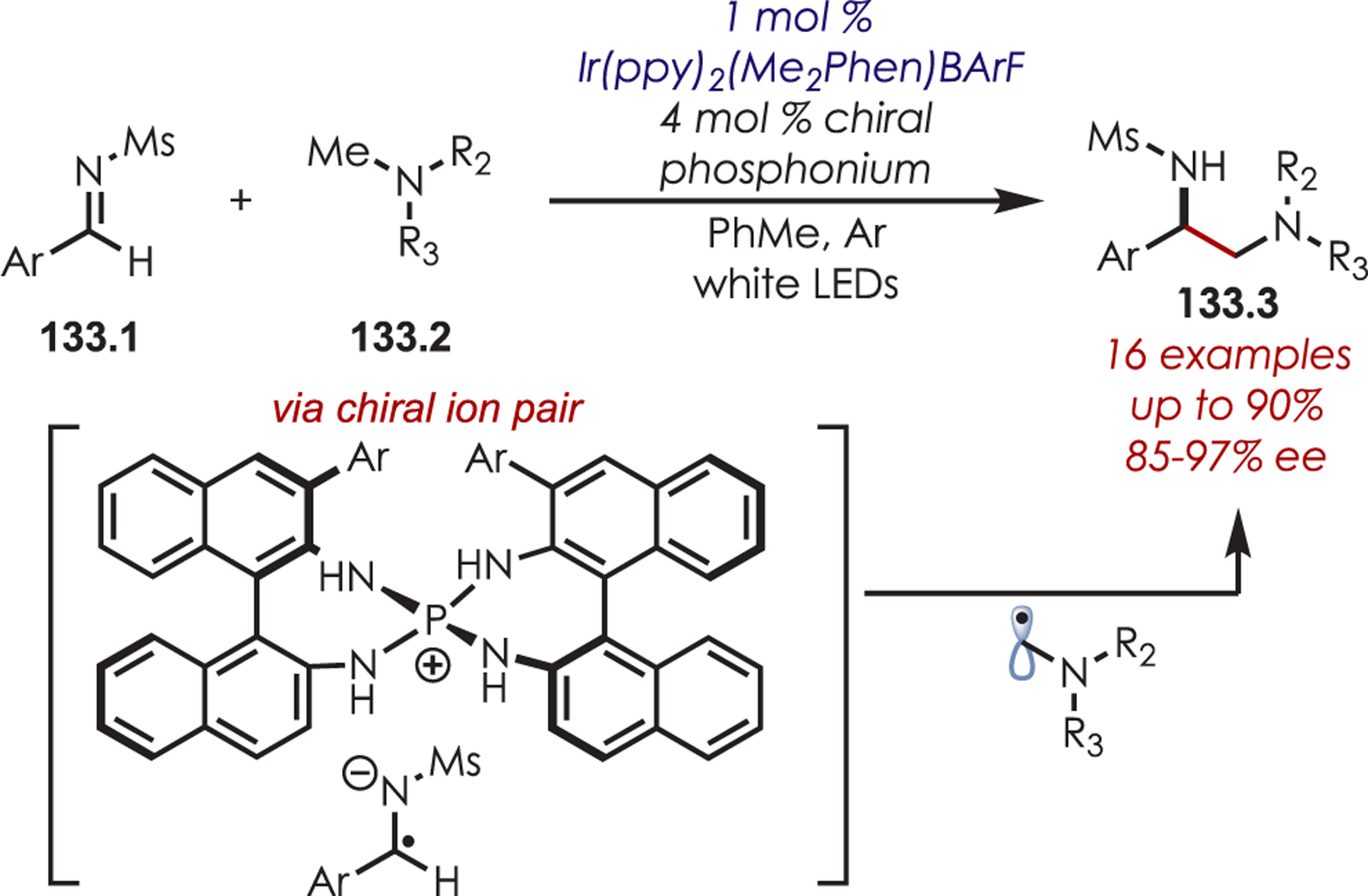
Asymmetric Coupling of α-Amino Radicals with Aldimines
Hydroaminoalkylations have been reported by the groups of Rovis and Breit using dual transition-metal and photoredox catalysis for the coupling of α-amino and allyl radicals (Scheme 134).262,263 These reactions proceed through the generation of a metal π-allyl species that is intercepted by the photogenerated α-amino radicals.
Scheme 134.
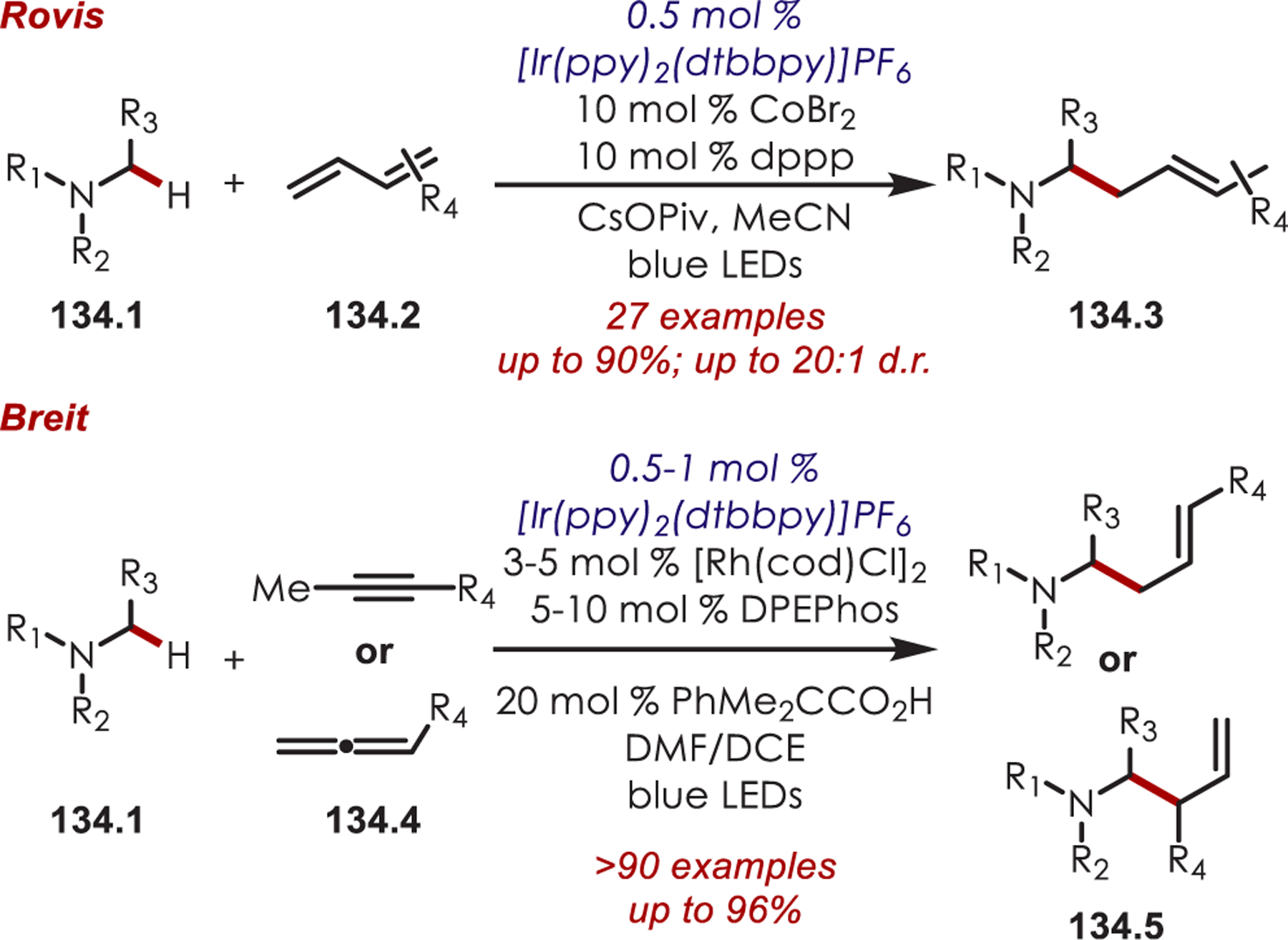
Hydroaminoalkylations through Dual Transition-Metal and Photoredox Catalysis
An α-allylation of amines via a proposed radical–radical coupling was reported by Xiao and Alper using dual palladium and photoredox catalysis (Scheme 135).264 The proposed catalytic cycle involves formation of an allyl radical from the reduction of a π-allylpalladium complex by the iridium photocatalyst. The allyl radical combines with an α-amino radical, formed in a similarly discussed single-electron oxidation and deprotonation event, to furnish the allylated amines (135.3) in moderate to good yields. This allylation was utilized in the formal synthesis of oxoprotoberberine precursors (135.4). A related C–H allylation of N-aryl tetrahydroisoquinolines via dual iodine and photoredox catalysis has also been reported through a radical–radical coupling mechanism.265
Scheme 135.
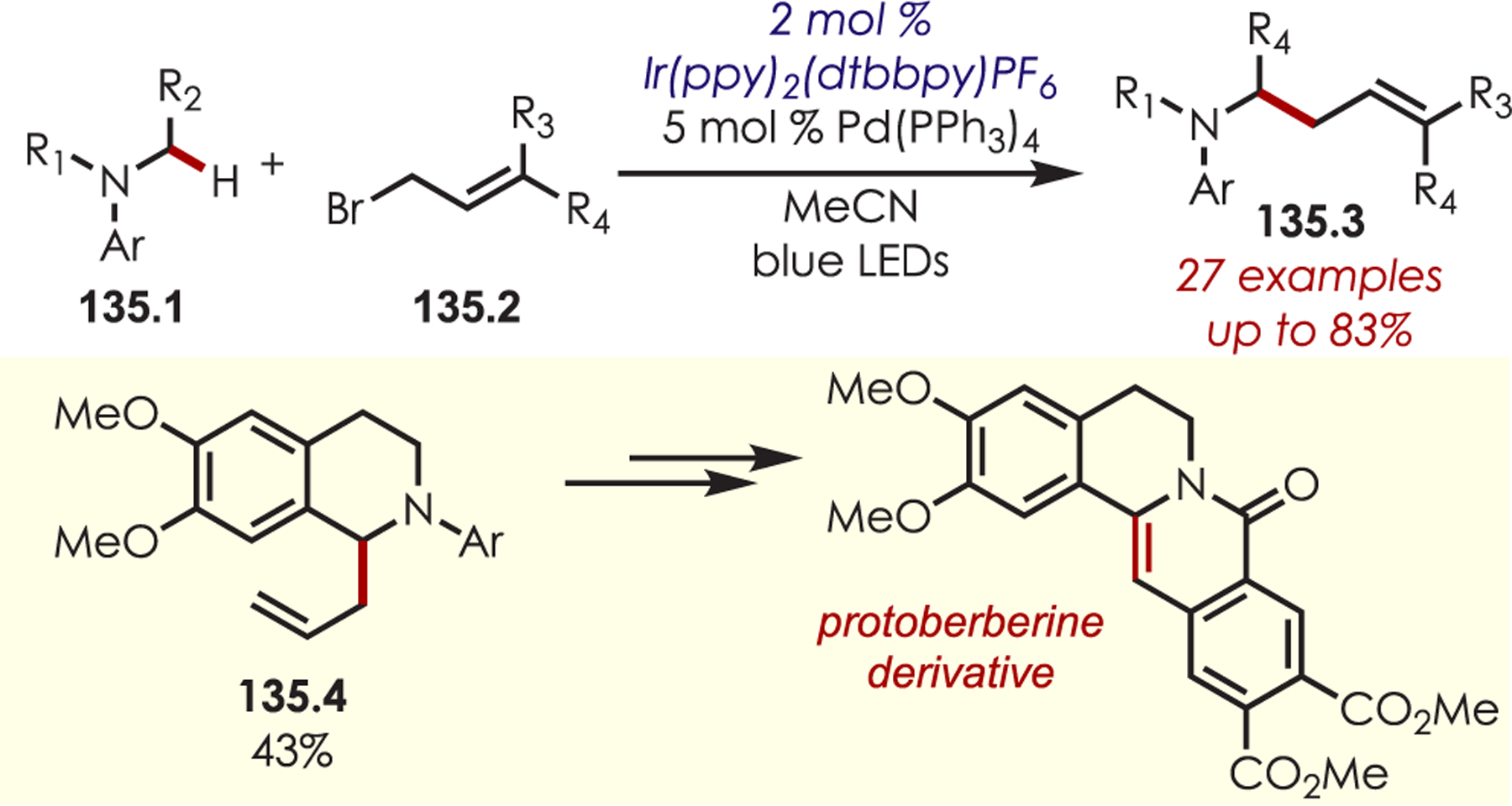
C–H α-Allylation of Amines Using Allylic Bromides
Recently, an enantioselective α-allylation of anilines (136.1) through synergistic combination of palladium and photoredox catalysis was reported (Scheme 136).266 A chiral Pd(II) allyl complex couples with photogenerated α-amino radicals and a subsequent reductive elimination affords the allylated anilines (136.3) in high enantioselectivity.
Scheme 136.
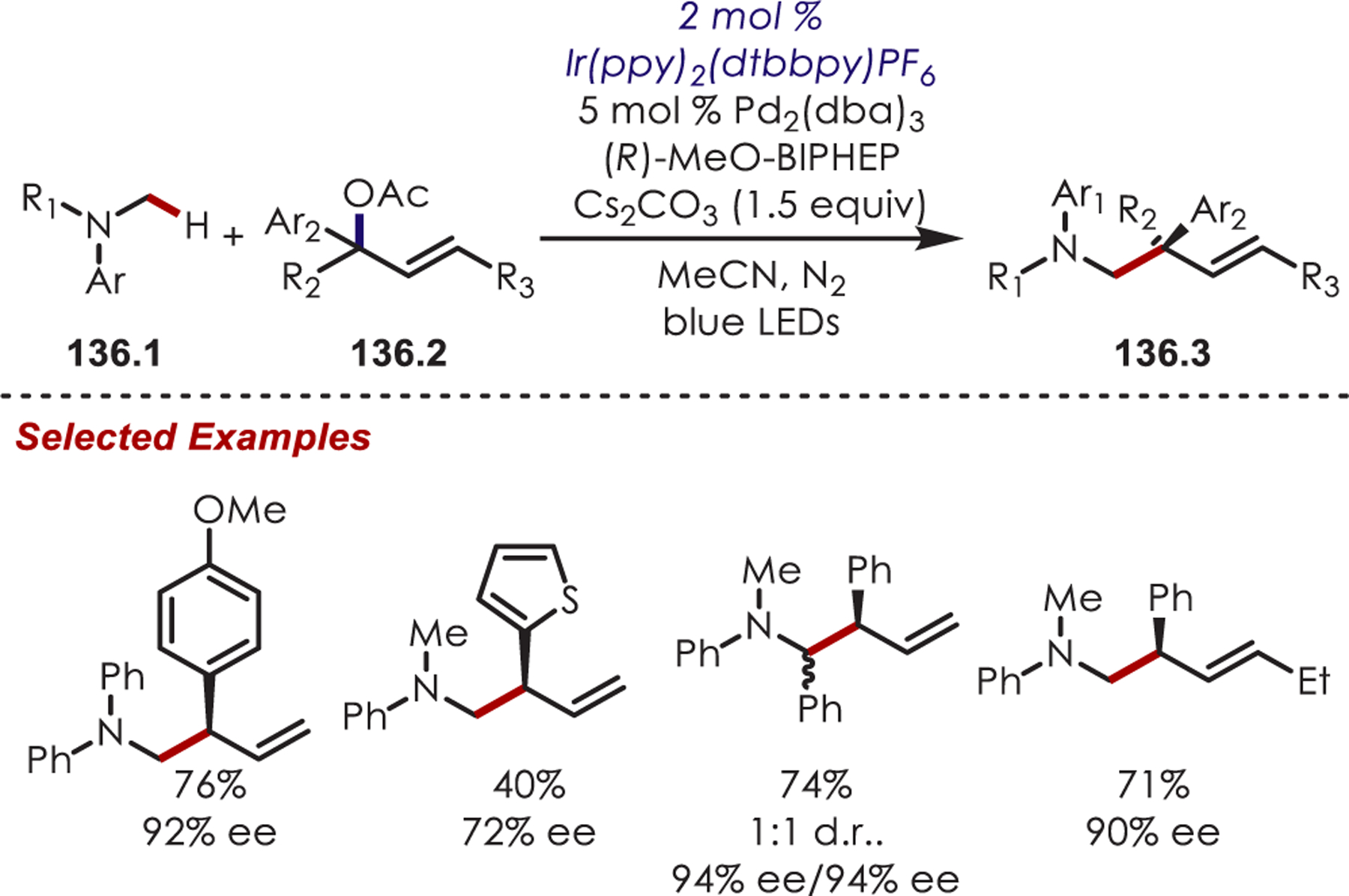
Asymmetric C–H α-allylation of Amines via Dual Palladium and Photoredox Catalysis
It is worth noting that α-amino radicals can be directly accessed through imine reduction and have been successfully trapped with an array of radical acceptors; however, this falls outside of the scope of this review.267
2.2.2. Carbonyl α- and β-C–H Functionalization.
The α- and β-functionalization of aldehyde and ketone derivatives has been explored through photoredox-catalyzed single-electron pathways. The most common strategy for α-carbonyl derivatives relies on either catalytic generation of enamine or enolate intermediates or enol silanes in reactions with electron-deficient radicals, typically formed from alkyl halides via single-electron reduction by the photoredox catalyst. Employing electron-rich enamines, enolates, or enol silanes is critical for the success of these reactions, as the reaction partners are better electronically matched with electron-deficient alkyl radical species.
Strategies for direct β-functionalization of carbonyls largely rely on catalytic enamine or enolate generation, followed by single-electron oxidation and depronation β-to the starting carbonyl. The subsequent allylic radicals can engage a range of electrophiles in net carbonyl β-functionalization reactions.
α-Functionalization of Carbonyl Derivatives.
MacMillan pioneered the field of enantioselective enamine cation radical (SOMO) catalysis for α-aldehyde and ketone functionalization using ceric ammonium nitrate (CAN) as a stoichiometric oxidant.268–270 Chiral enamines, formed by condensation of chiral amines onto ketones, are oxidized by CAN to an enamine cation radical or SOMO-actived species. It was demonstrated that a range of electron-rich alkenes, arenes, and halides intercepted these catalytically generated SOMO intermediates to afford enantioenriched α-functionalized products. The drawbacks to this strategy are that it relies upon super stoichiometric quantities of an oxidant (CAN) and were limited to more nucleophilic reaction partners.
In an effort to address these limitations, Nicewicz and MacMillan introduced the merger of photoredox catalysis with chiral enamine catalysis in seminal work from 2008 that accomplished enantioselective aldehyde α-alkylation by alkyl bromides (137.2) (Scheme 137).271 This report takes advantage of PET to affect reductive cleavage of alkyl halides to generate electron-deficient carbon-centered radicals. The aforementioned electron-deficient radicals reacted readily with the catalytically generated chiral enamine species to afford the α-alkylated aldehydes in good to excellent enantioselectivities (137.1). α-Bromoketones and esters were found to be suitable alkyl radical sources, and racemic tertiary substituted bromides could be used to generate an α-quaternary carbon stereocenter with good diastereocontrol. Mechanistically, this addresses the shortcomings of SOMO catalysis as the electron-rich alkene (enamine) is electronically matched with the electron-deficient radical and expands the chemical reactivity space in enamine-catalyzed alkylation reactions and only catalytic quantities of electron-transfer reagent are required.
Scheme 137.
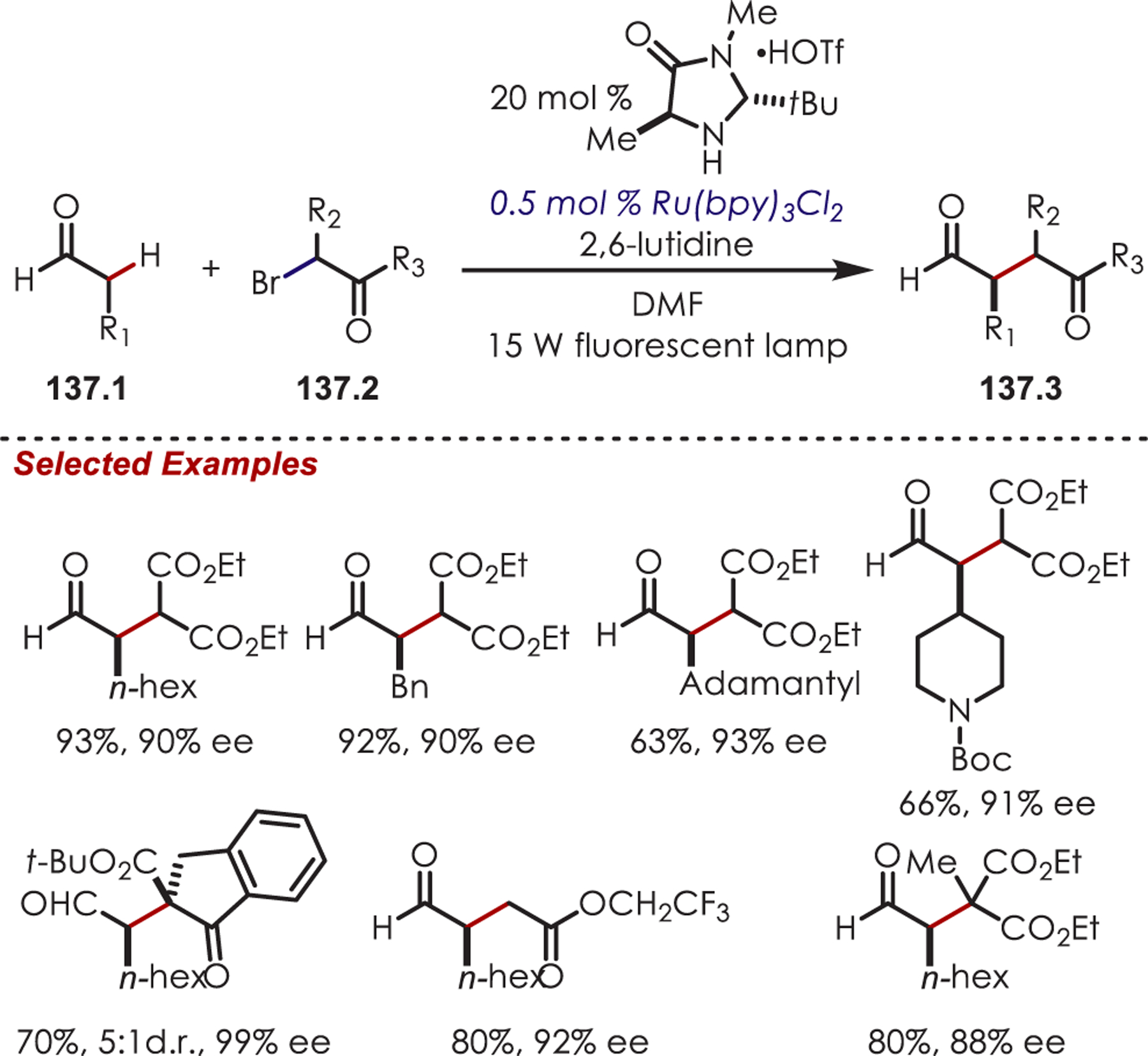
Asymmetric α-C–H Alkylation of Aldehydes Using Dual Photoredox and Organocatalysis
The proposed mechanism begins with reductive quenching of Ru(II)* by an enamine to generate Ru(I) (Scheme 138). Ru(I) is a known reductant (Eox = −1.33 V vs SCE) and undergoes SET with alkyl bromides to generate carbon-centered radicals (138.1). In another catalytic cycle, a chiral enamine (138.2) is formed through condensation of the chiral imidazolidinone and the aldehyde. Interception of the electron-deficient radical by the electron-rich enamine generates an α-amino radical intermediate (138.3). This species is readily oxidized by Ru(II)*, forming an iminium ion (138.4) which is hydrolyzed to the final product, while closing both catalytic cycles. In a related transformation, Luo reported the photoalkylation of β-ketocarbonyls by the generation of α-keto radicals using chiral enamine and ruthenium photoredox catalysis.272
Scheme 138.
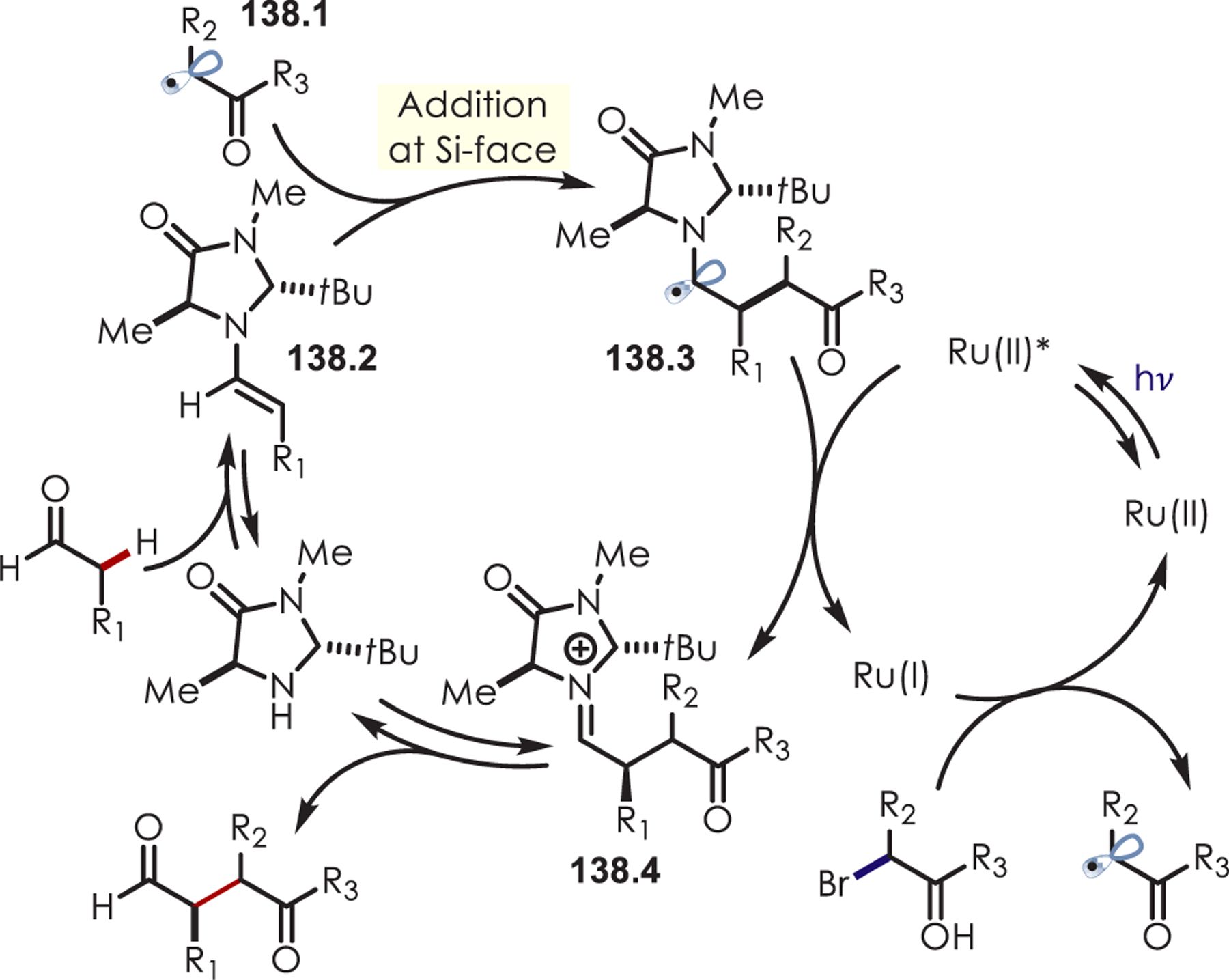
Mechanism of Asymmetric α-C–H Alkylation of Aldehydes Using Dual Photoredox and Organocatalysis
In the following years, MacMillan reported extensions of the Nicewicz methodology for enantioselective α-trifluoroalkylation,273 and α-benzylation of aldehydes (Scheme 139).274 Trifluoroalkyl iodides (139.2) or benzylic bromides (139.3) were used as sp3 carbon-centered radical precursors. The addition of these radicals to the enamine from the more sterically accessible Si face furnishes the α-functionalized products in excellent yields and enantioselectivities.
Scheme 139.
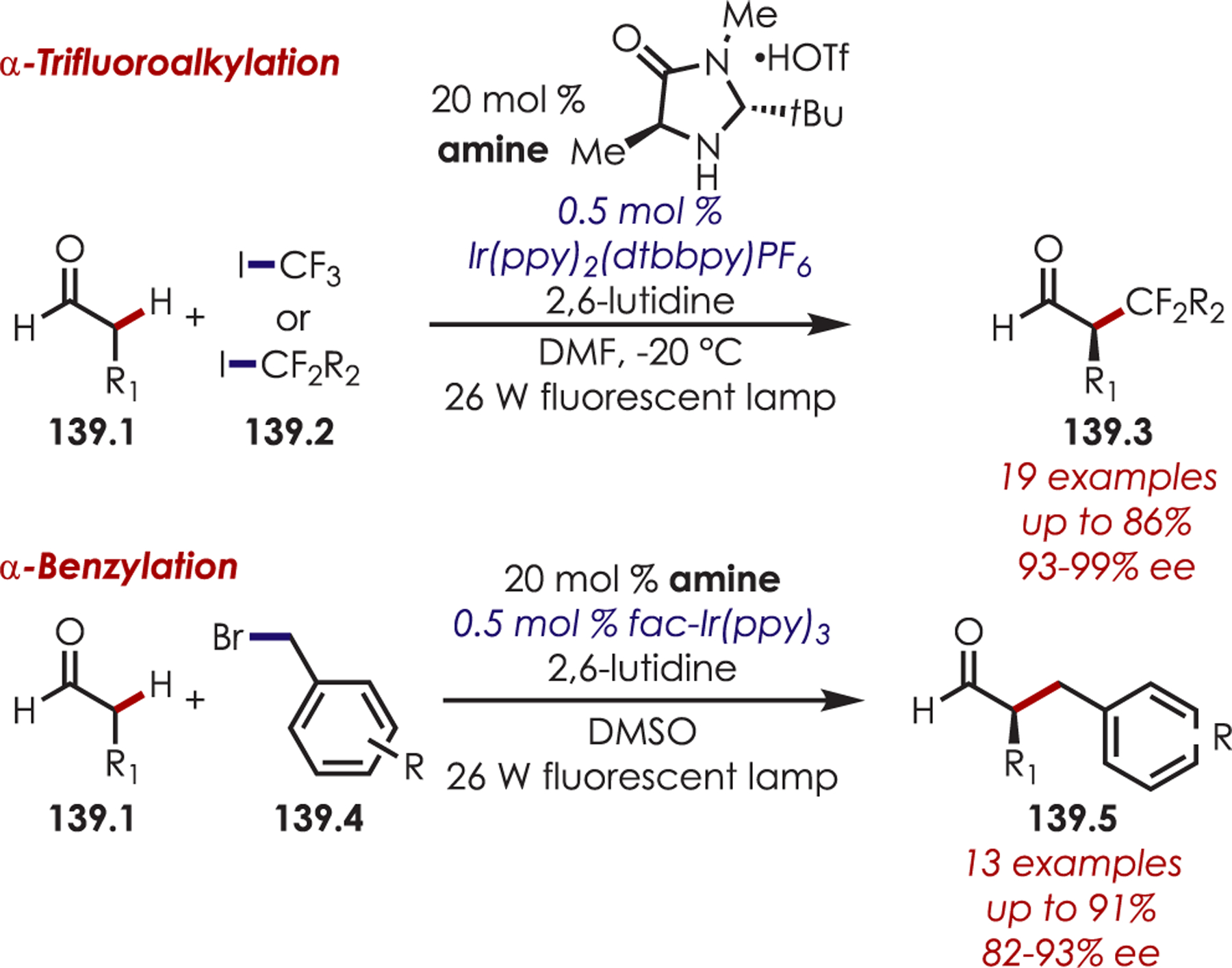
Asymmetric α-C–H Fluoroalkylation of Aldehydes using Dual Photoredox and Organocatalysis
An enantioselective α-alkylation of aldehydes (140.1) with α-bromonitriles (140.2) was also reported by MacMillan in 2015 (Scheme 140).275 The enantioselective synthesis of oxonitriles is particularly useful for accessing pharmacophore fragments which was highlighted in a four-step synthesis of the lignan natural product, (−)-bursehernin (140.4).
Scheme 140.
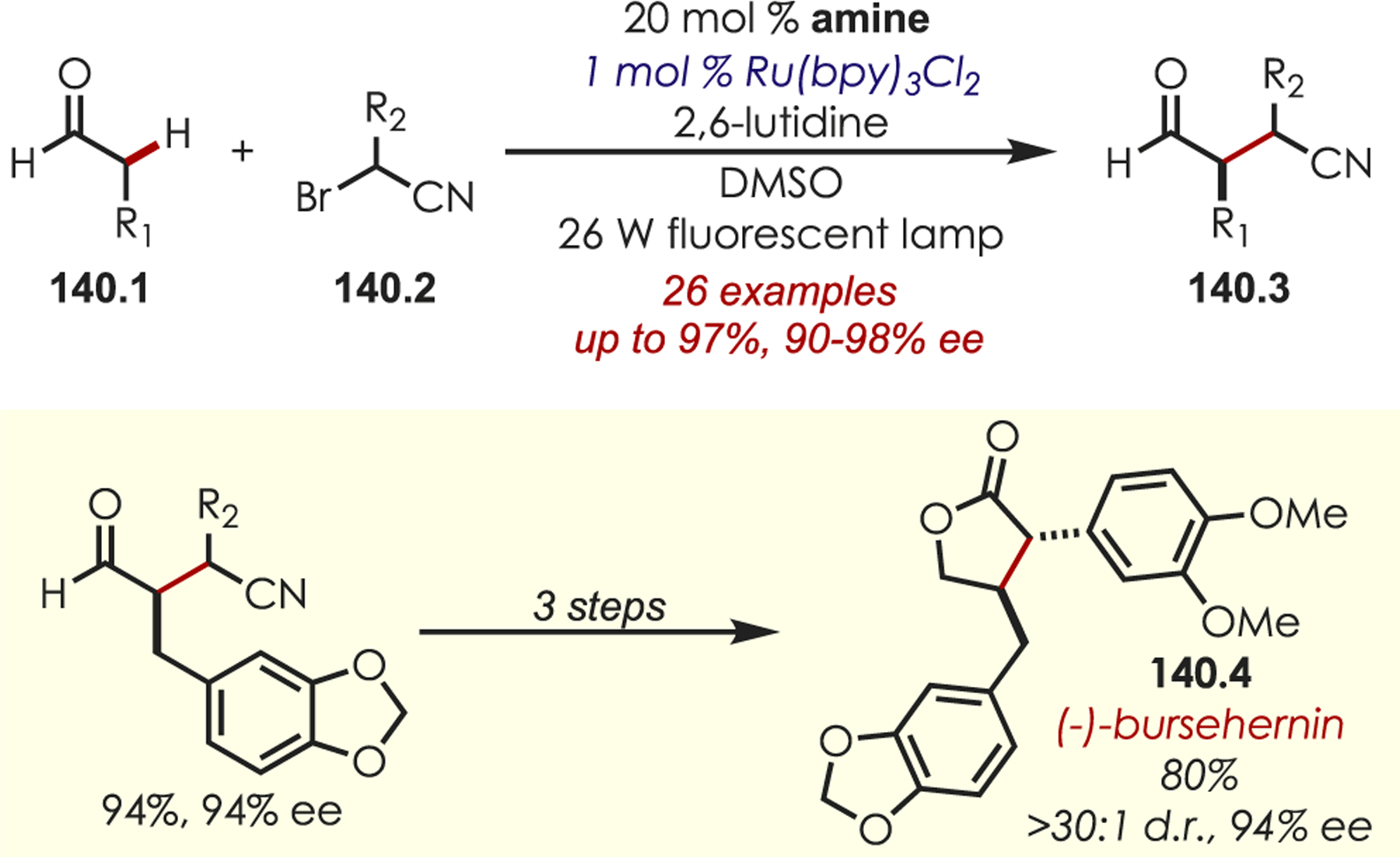
Asymmetric Synthesis of Oxonitriles via α-C–H Functionalization of Aldehydes
Cheng later investigated the origin of stereocontrol for these asymmetric α-functionalizations via enamine and photoredox catalysis and found that the enantioselectivity is largely controlled by steric effects and pseudo-C2 symmetry of the imidazolidinone catalyst.276 Two transition states were found to influence the stereoselectivity where the bulky tert-butyl group of the imidazolidinone shielded the bottom face, thus leading to selective radical attack at the Si face of E-cis enamine 141.1 or at the Si face of E-trans enamine 141.2 (Scheme 141).
Scheme 141.
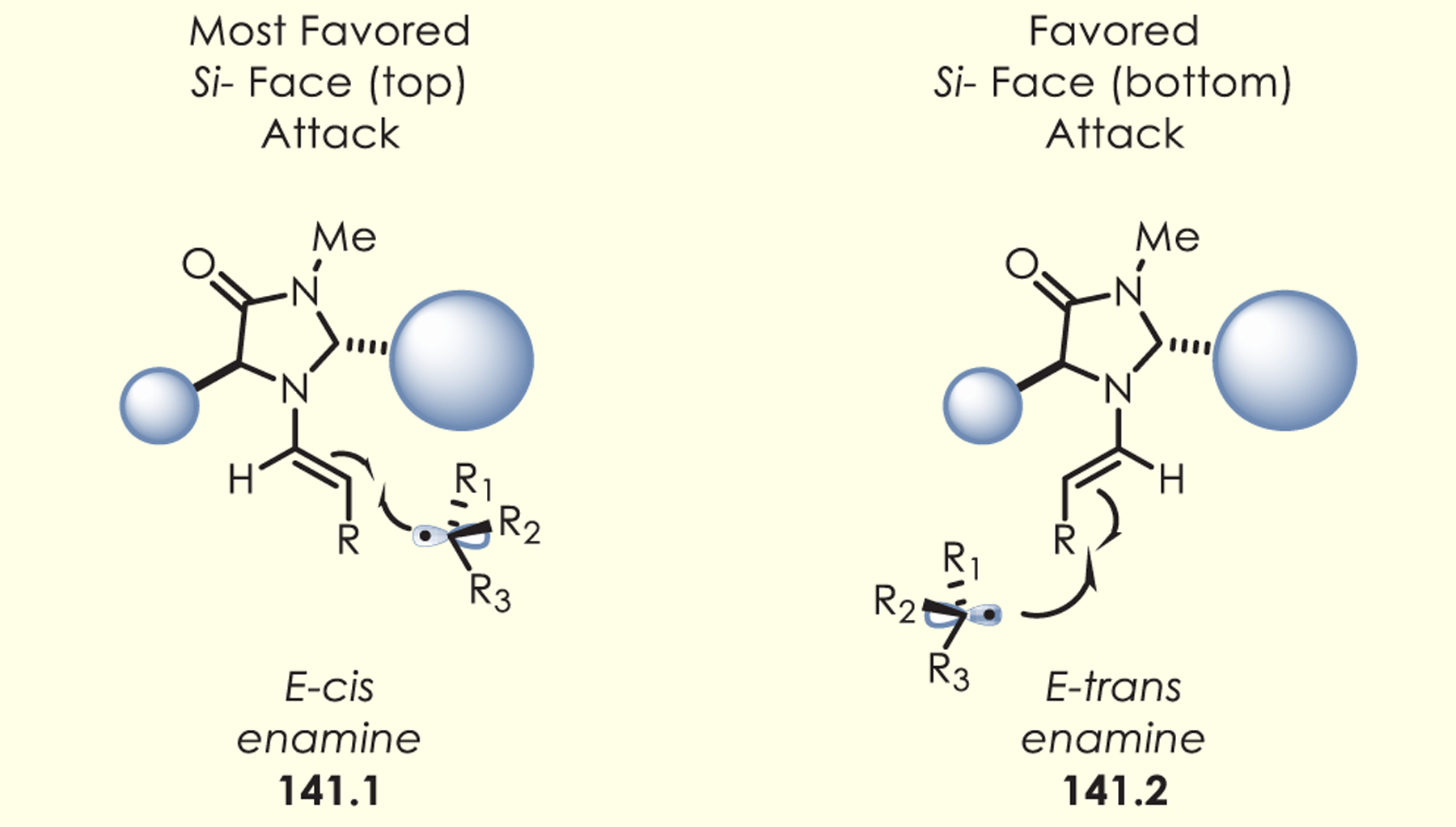
Two Energetically Favored Transition States for Addition of Radicals to Chiral Imidazolidinone Derived Enamines
Following these seminal studies, MacMillan developed a related enantioselective α-alkylation of aldehydes (142.2) using alcohols (142.1) as radical surrogates (Scheme 142).277 A key spin-centered shift enabled the generation of the reactive benzylic radical intermediate (142.4), which added to the Si-face of the organocatalytically generated enamine in analogous fashion to Scheme 138. Aliphatic and aryl-substituted aldehydes were coupled to benzylpyridines in excellent yields and enantioselectivities. Nucleofuges other than alcohols, including acetates and ethers, were also suitable in this catalytic alkylation strategy.
Scheme 142.
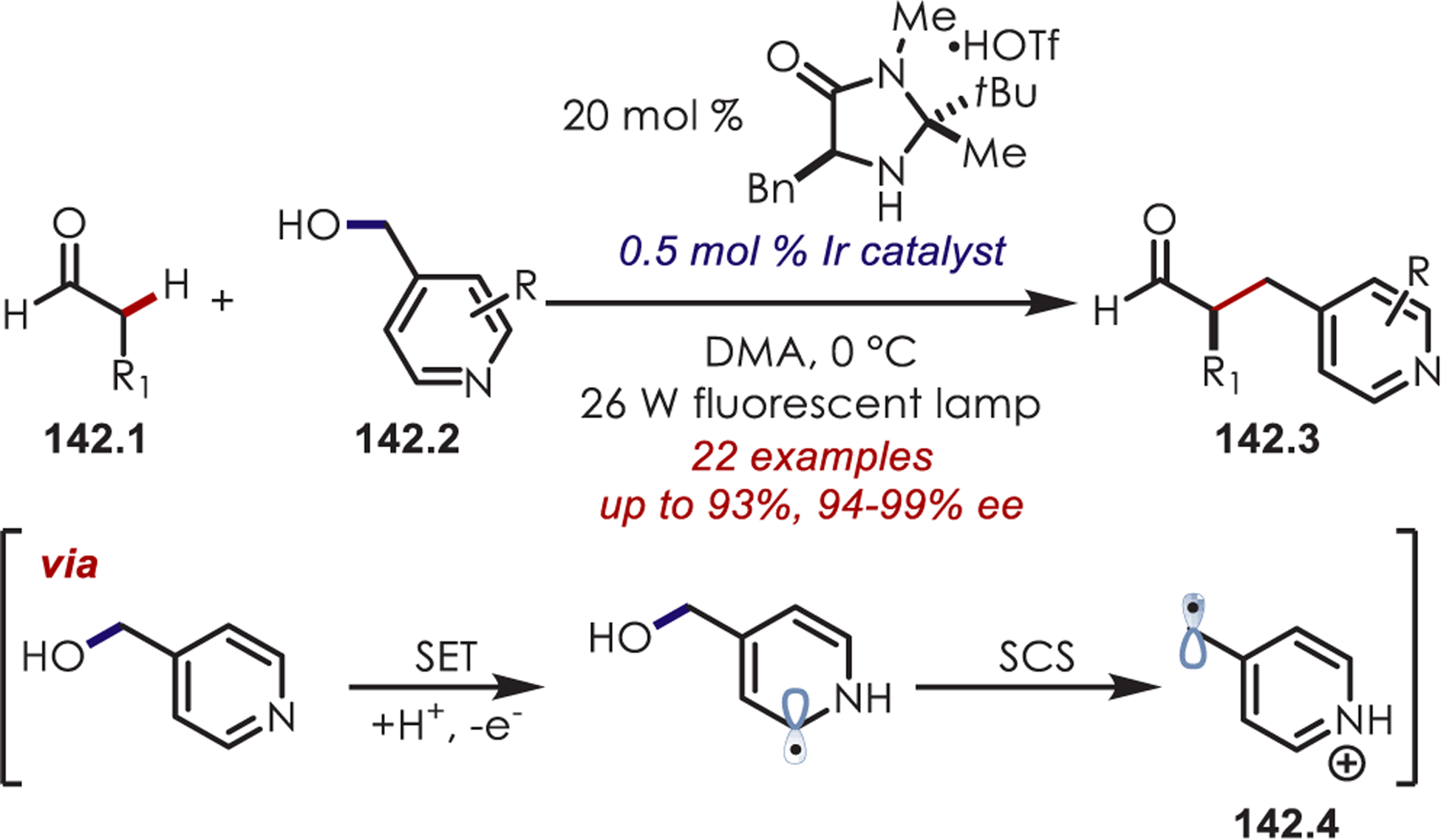
Asymmetric α-C–H Benzylation of Aldehydes Using Dual Photoredox and Organocatalysis
It is worth noting that Melchiorre has developed an asymmetric α-alkylation of aldehydes through direct excitation of the catalytically generated chiral enamine intermediates,278–281 which was also noted in the seminal work of Nicewicz and MacMillan.271 Investigation of the mechanism revealed a radical-chain pathway is at play, is initiated by light, and generates either photoexcited enamine or an electron-donor–acceptor complex with the alkyl bromide, ultimately resulting in the generation of the sp3 carbon-centered radical and radical–radical coupling to give the alkylated adducts.
An alternative to dual enamine/photoredox catalysis is using chiral photoredox catalysts that become the active photochemical species after association of a substrate. However, it is worth noting that this association causes changes in the photophysical properties of the catalyst that may be difficult to predict. Despite this, Meggers reported a chiral iridium-catalyzed α-alkylation of 2-acyl imidazoles (143.1) that develops the active photoredox catalyst after coordination to the substrate, thus allowing for efficient stereocontrol (Scheme 143).282 This method unites 2-acyl imidazoles with benzyl or phenacyl bromides (143.2) serving as carbon-centered radical precursors. First, coordination of the acyl imidazole and deprotonation generates the active photoredox catalyst. A photoreductively generated alkyl radical combines with the nucleophilic enolate species, generating an iridium ketyl radical. Subsequent oxidation furnished the α-alkylated 2-acyl imidazoles in high yields and enantioselectivities.
Scheme 143.
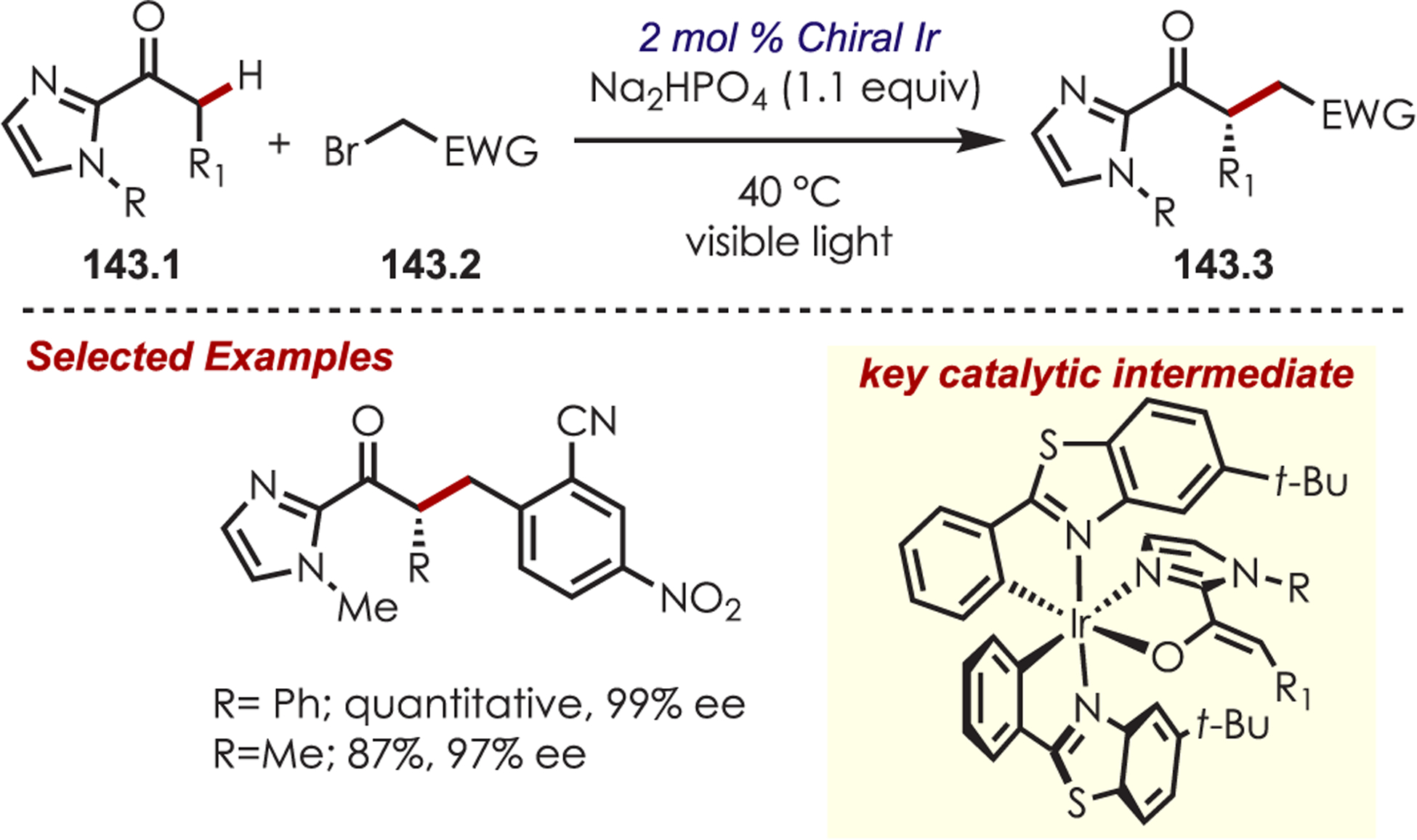
Asymmetric α-C–H Alkylation through a Chiral Ir Photoredox Catalyst
Shortly thereafter, the authors demonstrated an asymmetric trichloromethylation (144.4),283 alkylation (144.5), and amidation (144.6) with organic diazo and azide compounds, respectively.284 These diazo species generate the reactive alkyl radical through reduction from a chiral Rh–enolate complex and loss of nitrogen gas (Scheme 144). Difluoroalkyl bromides were later shown to be suitable carbon radical precursors for the asymmetric synthesis of chiral γ-ketoamides (144.7) using a chiral Rh complex as the Lewis acid but required an additional Ru(bpy)3(PF6)2 co-catalyst to generate the carbon-centered radical through PET.285 The authors noted that no desired product was observed when employing the chiral iridium as both the photoredox and Lewis acid catalyst.
Scheme 144.
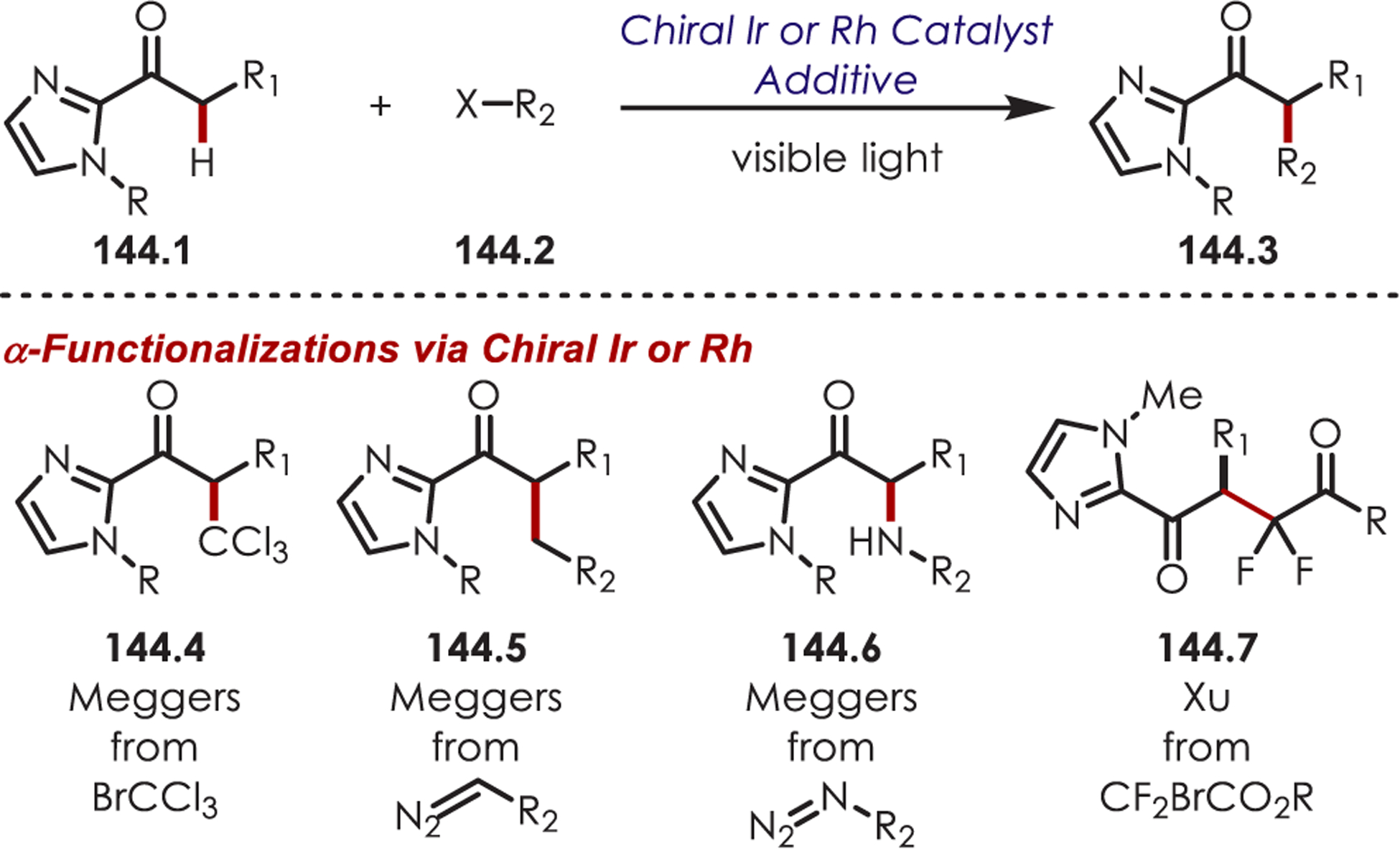
Compatible Nucleophiles for the Asymmetric α-C–H Functionalization of Aldehydes Using Chiral Ir or Rh Photoredox Catalysis
In 2015, Meggers reported an enantioselective α-alkylation of imidazole-derived ketones (145.1) using a chiral Rh catalyst which served as a dual photoredox/chiral Lewis acid catalyst (Scheme 145).286 Coordination of the carbonyl to the rhodium and a subsequent deprotonation generates a nucleophilic enolate complex. A photoinduced electron-transfer event between the excited rhodium(III) species and the tertiary amine generates the corresponding Rh(II) complex and a nitrogen-centered cation radical. Deprotonation of this amine cation radical and secondary oxidation by molecular oxygen generates the iminium species, which couples with the nucleophilic enolate through the less hindered Re-face to afford the products (145.3) in high enantioselectivity.
Scheme 145.
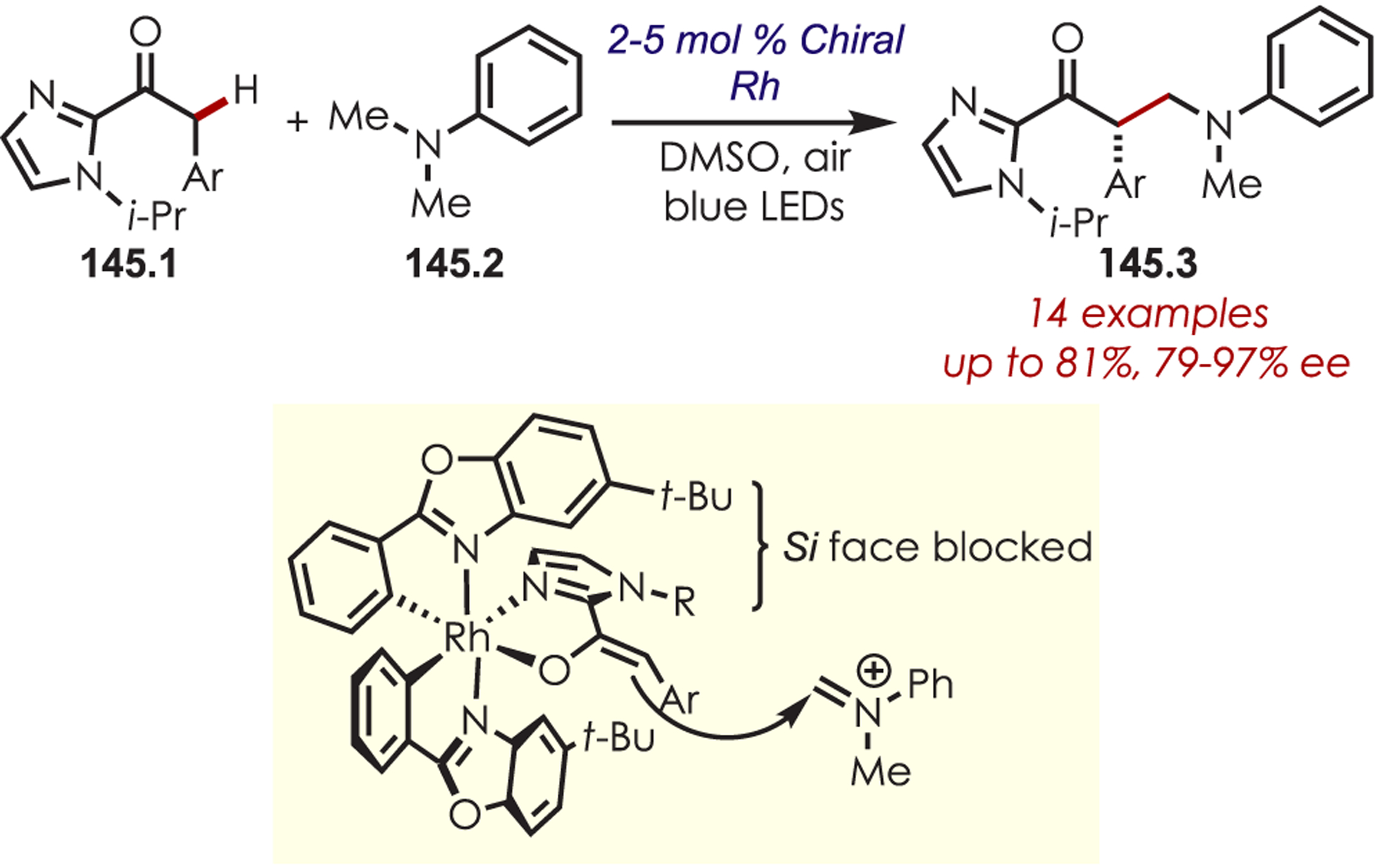
Asymmetric Coupling of Aldehydes and α-Amino Radicals Using a Chiral Rh Photoredox Catalyst
In contrast to the previous examples of asymmetric α-functionalization of aldehydes and ketones which rely on the addition of a photogenerated radical to an enamine, MacMillan reported that the enamines can be oxidized via SET to generate a 3πe− enaminyl radical (146.1) in an analogy to their prior SOMO work (Scheme 146).287 These electrophilic radicals can be trapped with olefins to access the enantioenriched α-alkylated products with complementary reactivity in comparison to the prior art. This was applied in both an intramolecular alkylation for the generation of 5–7 membered rings (146.4) as well as intermolecular alkylation (146.8) with styrenyl acceptors. A thiophenol co-catalyst was necessary to capture the radical formed on the alkene following addition to 146.1 in order to complete the transformations. The subsequent thiyl radical is responsible for reoxidation of the photoredox catalyst and turnover of the catalyst system.
Scheme 146.
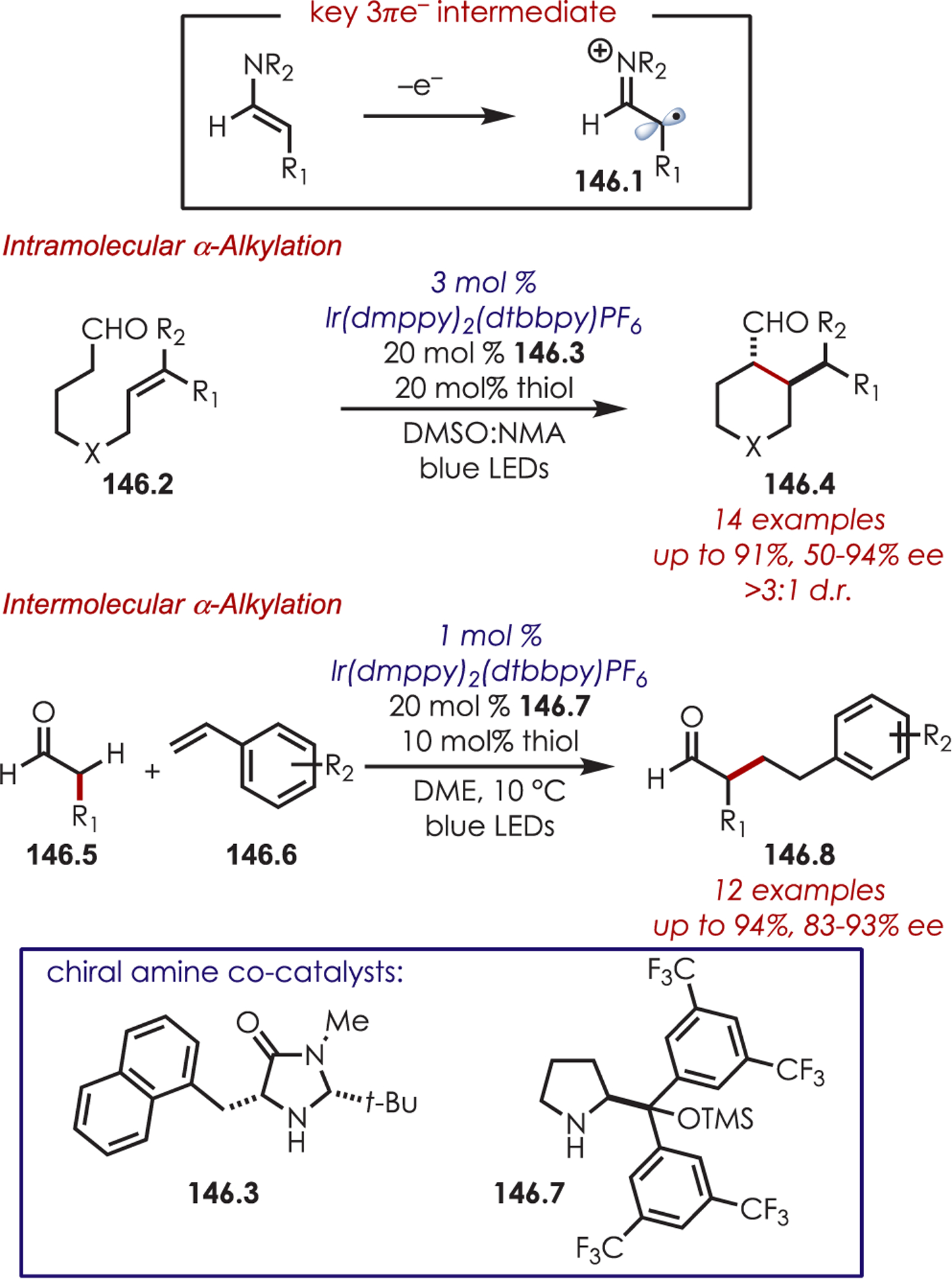
3πe− Enaminyl Radical for Asymmetric Aldehyde C–H Functionalization
An asymmetric C–C coupling of aldehydes (147.1) with xanthenes (147.2) was reported using a dual photoredox and enamine organocatalysis system (Scheme 147).288 Oxidative quenching of Ru(II)* by BrCCl3 generates a CCl3 radical that subsequently abstracts a hydrogen atom from xanthene to form a benzylic radical. A computational analysis of three potential mechanisms, where a benzylic radical adds to an enamine or enamine cation radical, or a benzylic cation is attacked by the enamine were compared. The authors found the latter was the most exergonic pathway with a ΔG⧧ = 4.3 kcal/mol in contrast to addition of the benzylic radical to the catalytically generated enamine or enamine cation radical, which was 20.6 or 30.9 kcal/mol, respectively. The benzylic cation is generated via oxidation of the benzylic radical by Ru(III) (Eox = +0.12 V and Ered = +1.29 V vs SCE, respectively). The addition of the enamine to the benzylic cation was found to be the key stereodefining step with ΔΔG⧧ = 1.7 kcal/mol for the (R) and (S) diastereomeric transition states which matched well with the experimentally determined value for 147.4 (ΔΔG⧧ = 2.0 kcal/mol).
Scheme 147.
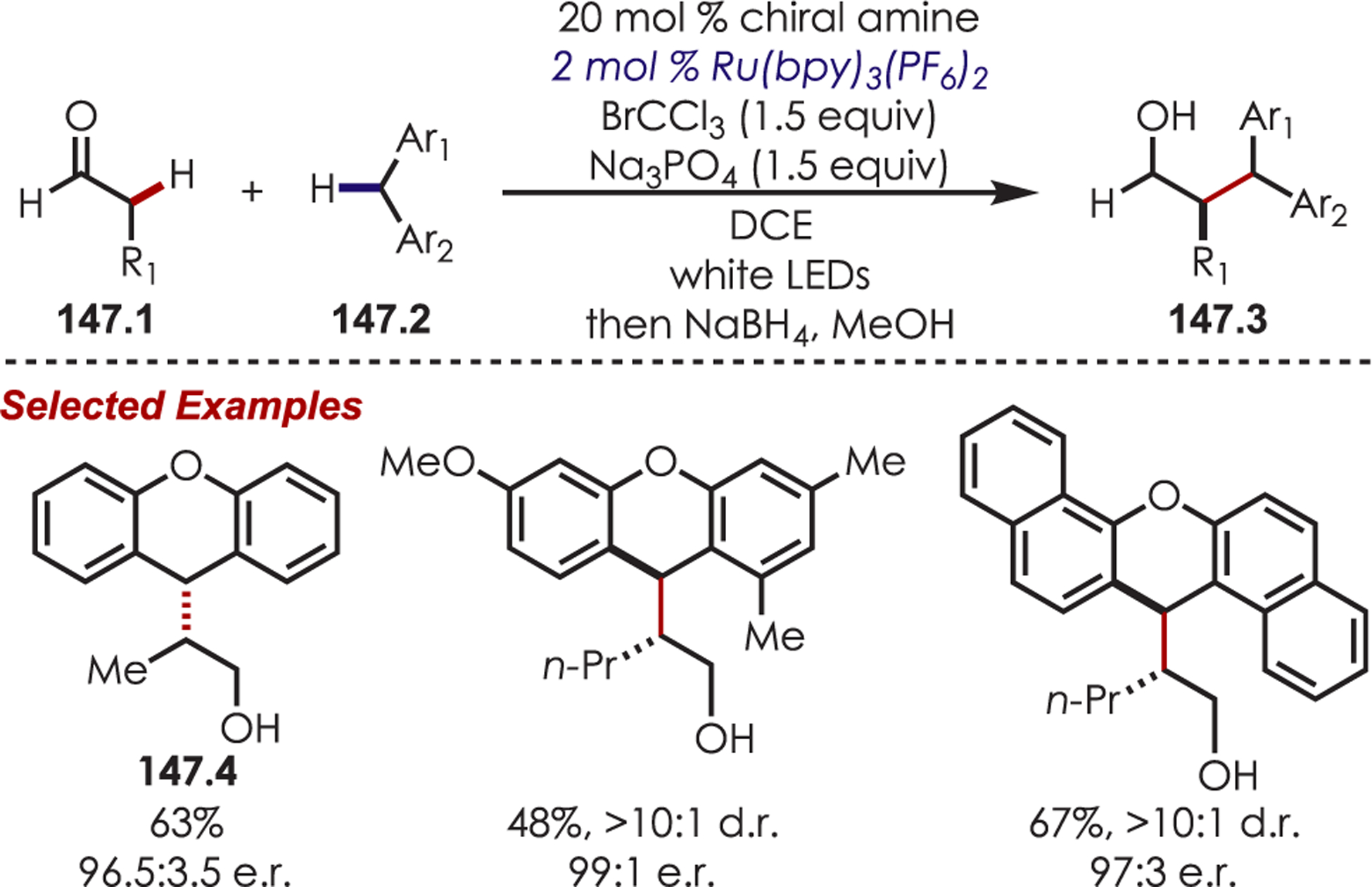
Photoredox-Mediated Asymmetric Coupling of Aldehydes and Xanthenes and Subsequent Reduction
MacMillan also reported an α-trifluoromethylation of ketones using enolsilanes (148.1), either pregenerated or formed in situ with trifluoromethyl iodide (Scheme 148).289 SET generates a trifluoromethyl radical, which adds to the silyl enol ether to generate an α-silyloxy radical. Oxidation by Ru(II)* generates a silyloxycarbenium ion that is rapidly hydrolyzed to the desired product (148.3). This was extended to the α-functionalization of amide and ester derivatives and was also successful at forming α-quaternary centers.
Scheme 148.
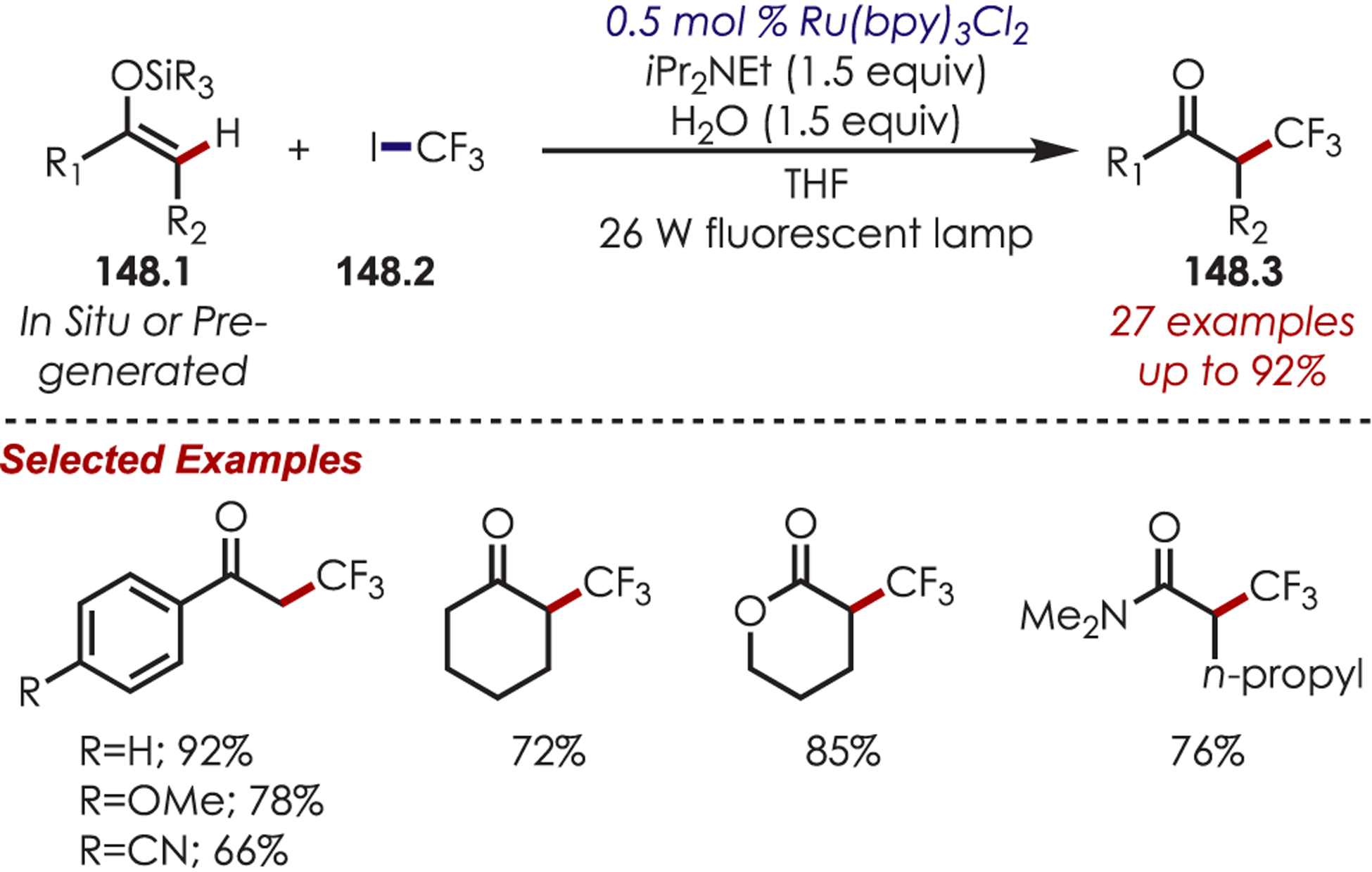
α-Trifluoromethylation of Ketones
An oxyamination of aldehydes (149.1) was reported in 2009 by Akita and co-workers via oxidation of enamines and trapping with TEMPO (Scheme 149).290 This work is analogous that of Sibi, who employed an Fe(III) oxidant to generate the key enamine cation radical intermediate.291 In this work, the Ru(bpy)32+ photooxidant replaces the inorganic oxidant.
Scheme 149.
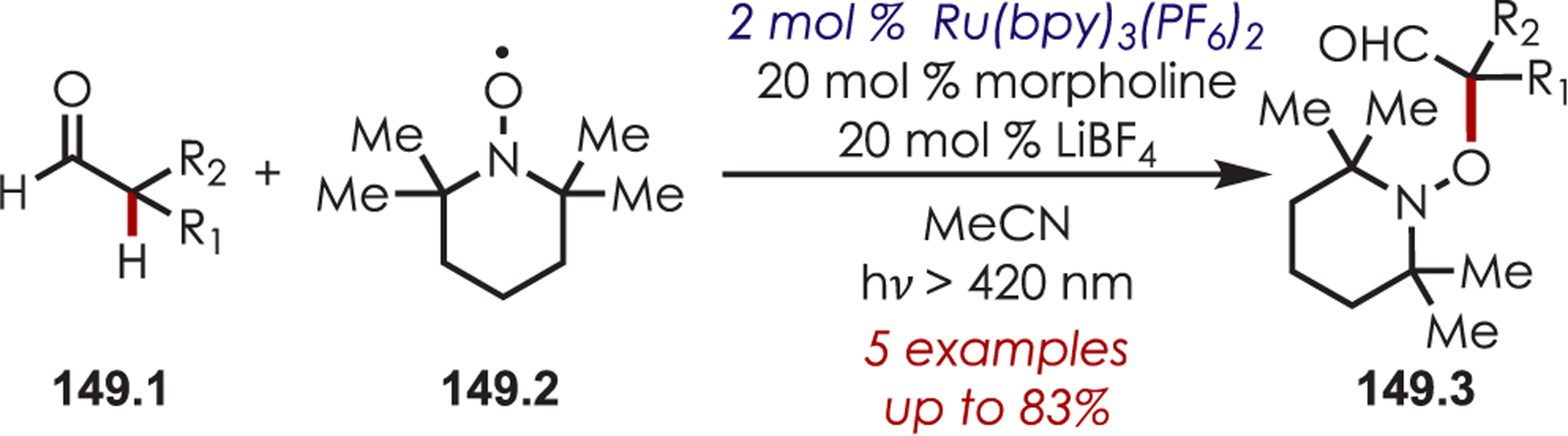
Aldehyde C–H Oxyamination with TEMPO
Gryko later reported an α-alkylation of aldehydes (150.1) via enamine and iminium catalysis with diazoacetates (150.2) as the radical coupling partner (Scheme 150).292 The enamine is oxidized by Ru(II) to an enamine cation radical that couples with the diazoacetate. A concurrent loss of N2 generates an α-ester radical intermediate that is reduced by Ru(I) and protonated to furnish the desired alkylated aldehydes.
Scheme 150.
Aldehyde C–H Alkylation with Diazoacetate Coupling Partners
Ketimines (151.1) have also been reported by Dixon as competent substrates in α-alkylation reactions through carbon-centered radical addition to their enamine tautomers (Scheme 151).293 A NiCl2(PPh3)2 additive was found to increase the reaction efficiency, and while the exact role was unclear, it was proposed that it may stabilize the radical species, either the alkyl radical or amine cation radical.
Scheme 151.
Ketimine C–H Alkylation Using Dual Photoredox and Nickel Catalysis
An α-allylation of 1,3-dicarbonyls has been reported in the presence of air and triphenylcarbenium tetrafluoroborate as a stoichiometric oxidative quencher (Scheme 152).294 Allyl stannanes and allyl sulfones were found to be suitable acceptors with ethyl benzoyl acetate as the α-keto radical precursor. Two intramolecular variants were also demonstrated, however, in lower yields (29–31%).
Scheme 152.
α-Allylation of 1,3-Dicarbonyls with Allyl Stannanes and Allyl Sulfones
β-C–H Functionalization of Carbonyl Compounds.
Following MacMillan’s α-arylation of amines through the coupling of α-amino radicals with benzonitrile derivatives,217 they recognized the potential to extend this strategy for the β-arylation of aldehydes and ketones.295 Prior art for direct β-functionalization of these ketone and aldehyde derivatives was limited to Saegusa–Ito-like mechanisms via the intervention of palladium catalysis. This achievement opens a new avenue for β-C–C bond functionalization of carbonyl-containing compounds.
This transformation relies on both the generation of an enamine and a dicyanobenzene radical anion coupling partner (Scheme 153). First, dicyanobenzene is reduced by Ir(III)*, generating the corresponding dicyanobenzene anion radical (153.5) and Ir(IV) oxidant. The enamine, generated in situ, is subsequently oxidized by Ir(IV), giving rise to an enaminyl cation radical (153.3) and regenerating the ground state Ir(III) species. Deprotonation at the now significantly weakened allylic C–H bond forms the key β-enamine radical (153.4). This intermediate undergoes a radical–radical coupling with the dicyanobenzene anion radical, which rapidly eliminates a cyanide anion and is hydrolyzed to furnish the β-arylated aldehydes.
Scheme 153.
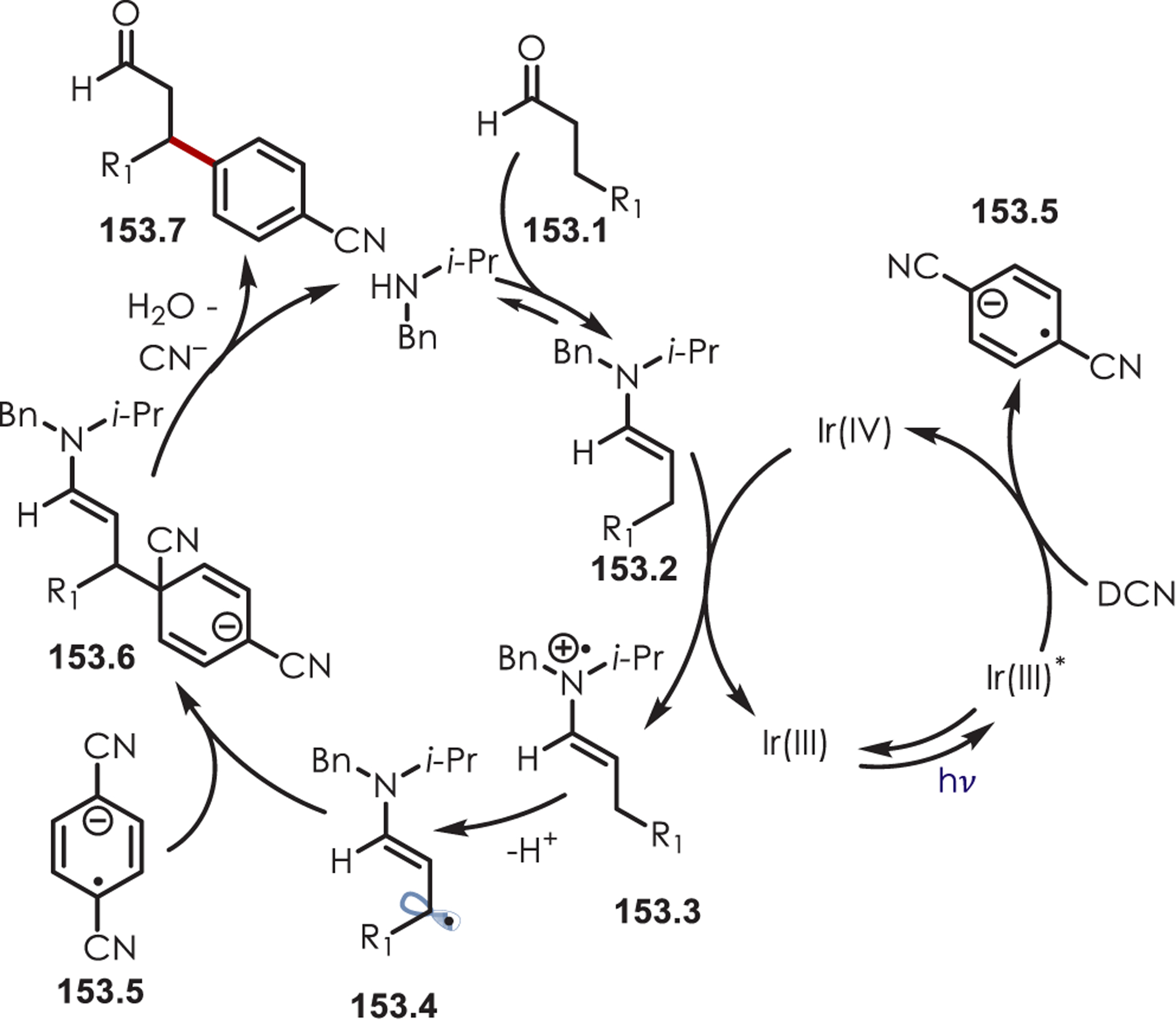
Mechanism of β-Aldehyde C–H Arylation with Cyanobenzenes
This β-functionalization has high functional group tolerance including aldehydes (154.1) substituted with (hetero)arenes, alkenes, alkynes, and amines (Scheme 154). The cyanobenzene coupling partner (154.2) was extended to a range of electron-poor (hetero)arenes including benzonitriles, aryl sulfones or esters, pyridines, and 7-azaindole.
Scheme 154.
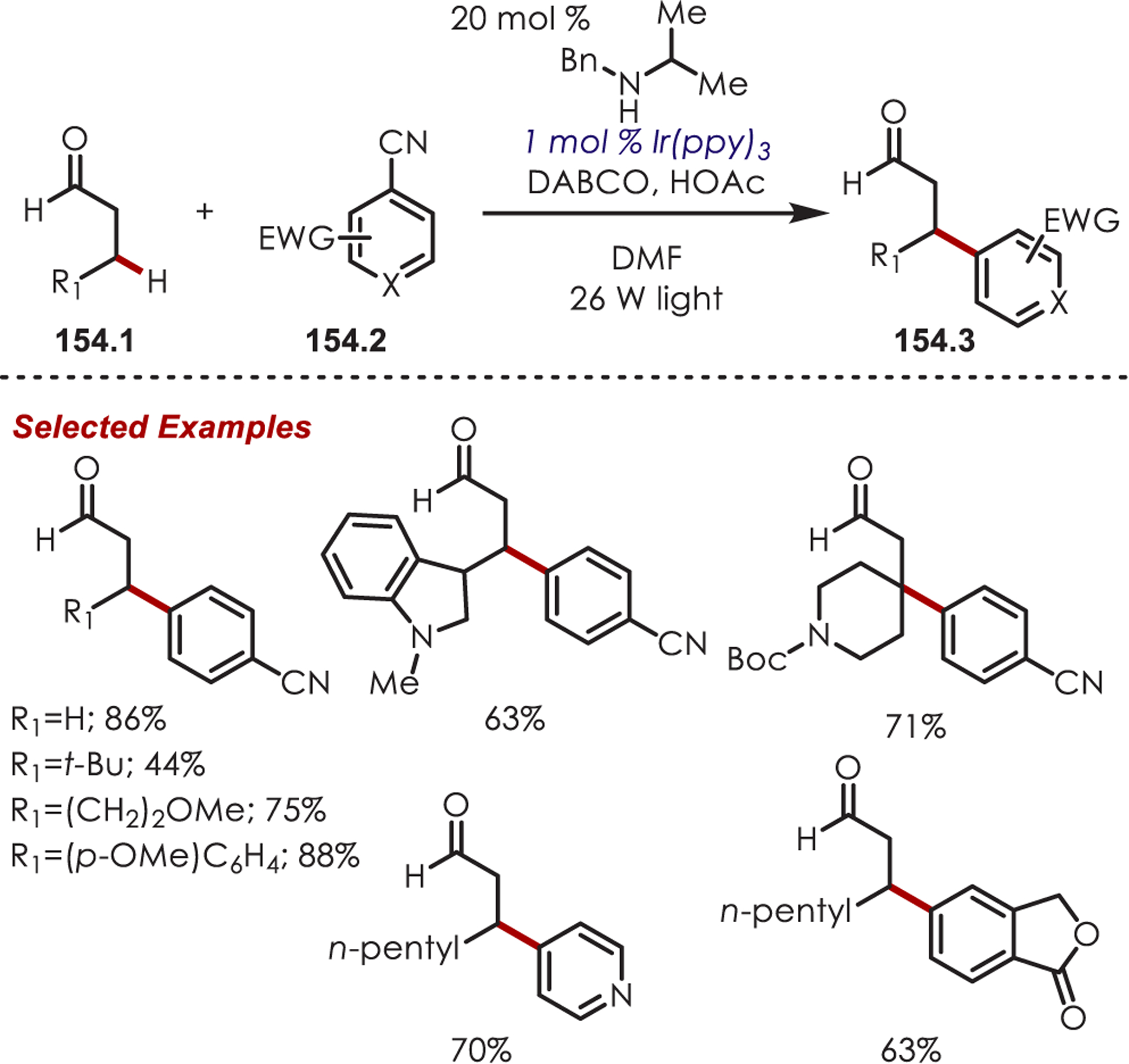
Scope of β-Arylation of Aldehydes with Cyanobenzenes
Cyclohexanone (155.1) was also identified as a suitable substrate for Ir(III)-catalyzed β-functionalization with 1,4-dicyanobenzene (155.2) as the coupling partner and shows the potential to extend this methodology to ketone substrates (Scheme 155). A chiral cinchona derived catalyst in combination with cyclohexanone as a substrate led to the formation of the desired product in 82% yield and 50% ee.
Scheme 155.
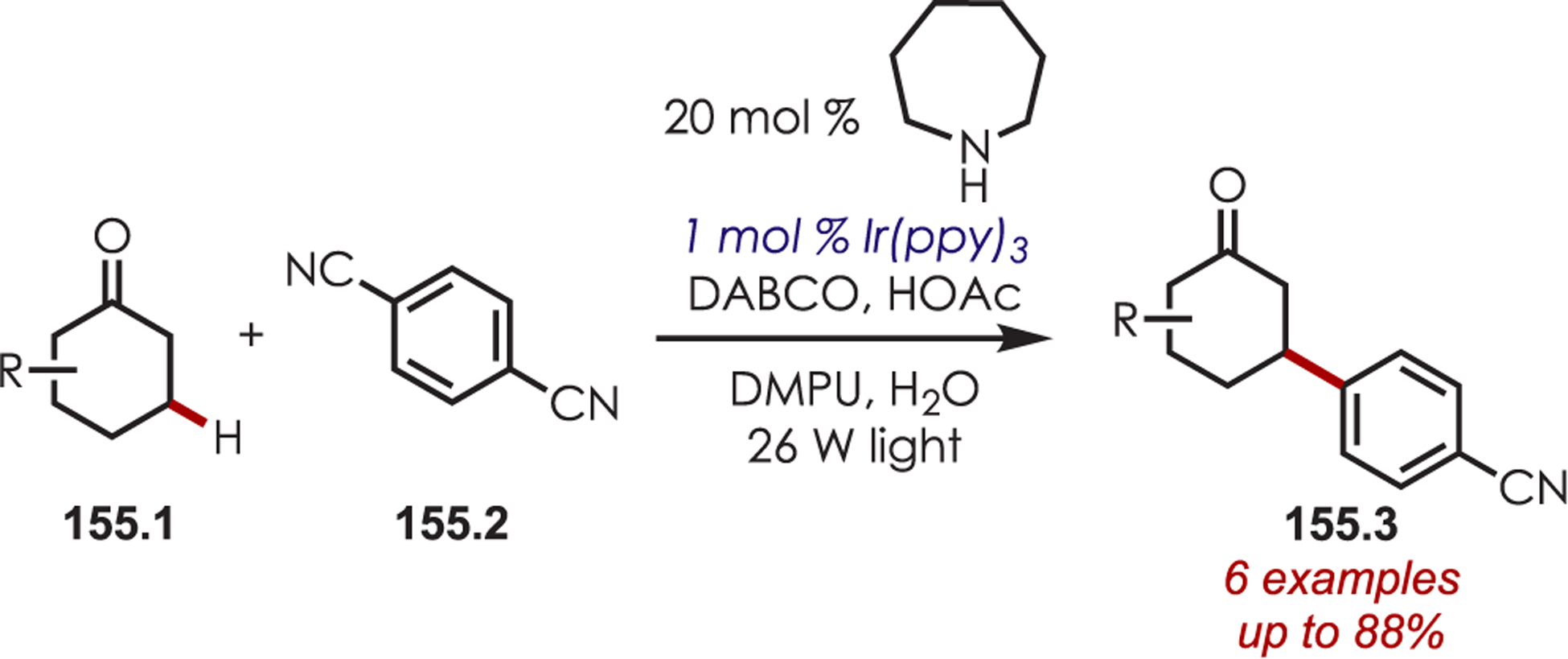
β-C–H Arylation of Cyclohexanone with Cyanobenzenes
Following this report, the MacMillan lab disclosed that the β-functionalization of aldehydes (156.2) could be extended to include a range of Michael acceptors (Scheme 156).296 Additionally, this system is compatible with intramolecular variants, affording the corresponding 6-exo (156.4) and 5-exo (156.5) product fused ring systems in 54% and 47% yields with 4:1 and 9:1 dr, respectively.
Scheme 156.
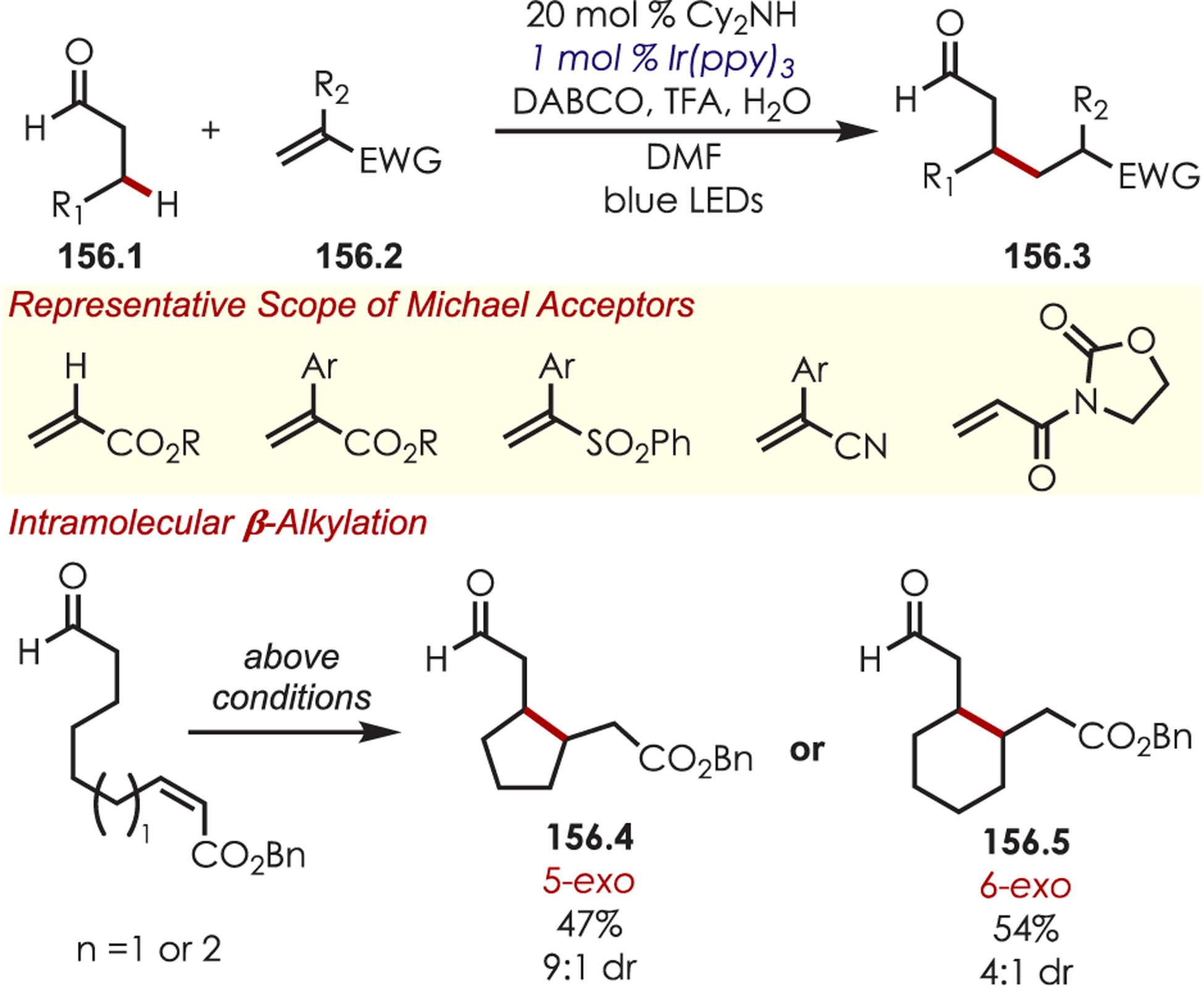
β-C–H Alkylation of Aldehydes with Electron-Deficient Alkene Radical Traps
Additionally, the β-hydroxyalkylation of cyclic ketones (157.1) was accomplished through an enaminyl and ketyl radical–radical coupling (Scheme 157).297
Scheme 157.
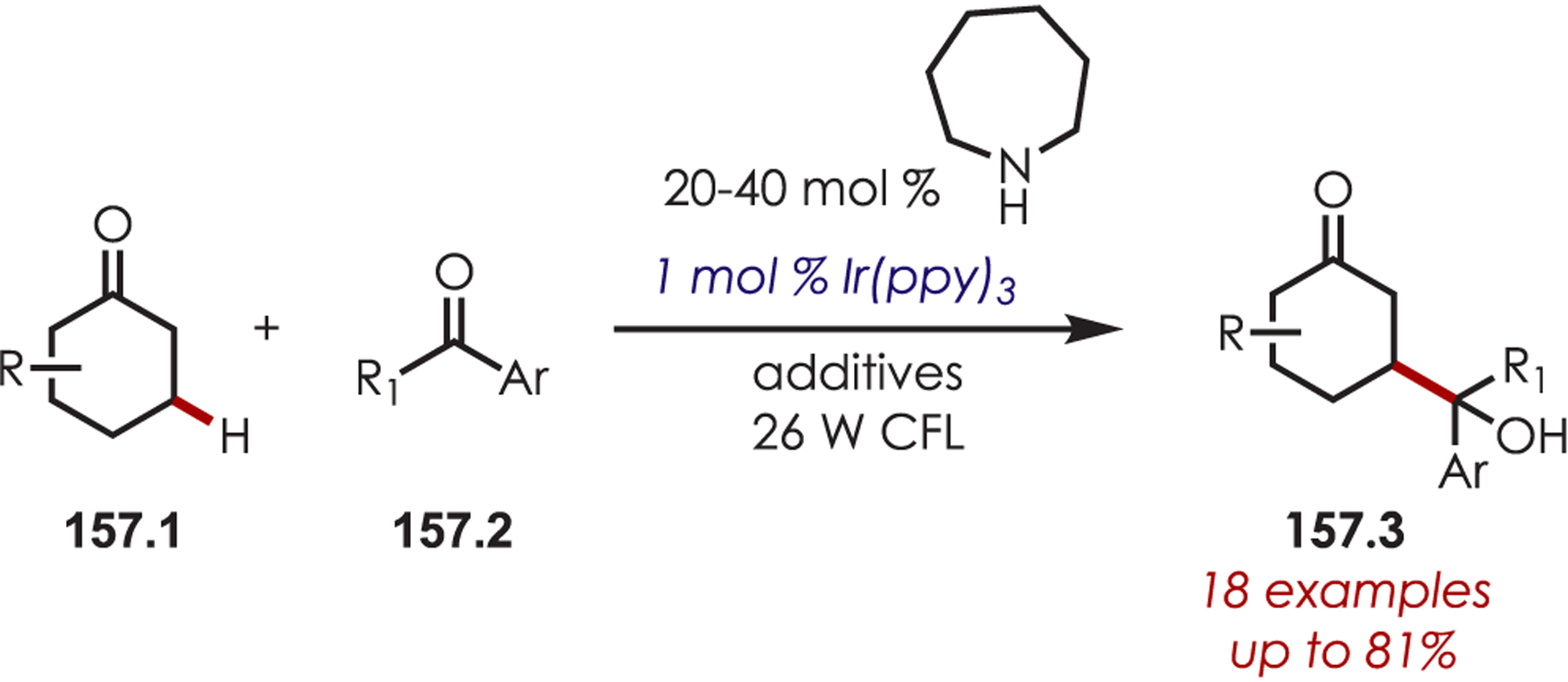
Scope of β-Aldehyde C–H Arylation with Cyanobenzenes
After their development of chiral Rh catalysis in α-functionalization,286 Meggers and co-workers reported a similar strategy for the asymmetric β-functionalization of 2-acyl imidazoles and 2-acyl pyridines (158.1) using 1,2-diketones (158.2) as electrophiles (Scheme 158).298 Following bidentate binding of the amide carbonyl to the Rh center, deprotonation forms an enolate, which undergoes excitation to 158.4 and undergoes PET with the 1,2-diketone, affording a ketal anion radical. Proton transfer from the acidified β-position of the Rh-bound enol cation radical affords an α-keto radical. Upon radical–radical recombination, the desired β-functionalized product (158.3) is formed. The optimized conditions were compatible with substrates containing, ether, thioether, free alcohol, bromide, alkene, and indole motifs. More electron-poor groups on the arene lead to lower yields, which were improved upon heating. Notably, substrates lacking an aryl substituent at the β-position failed to give any desired product, suggesting the aryl group is required for efficient reactivity, likely due to increased radical stability. The scope of 1,2-dicarbonyl compounds was limited to aryl α-ketoesters but also included 3,4-hexanedione.
Scheme 158.
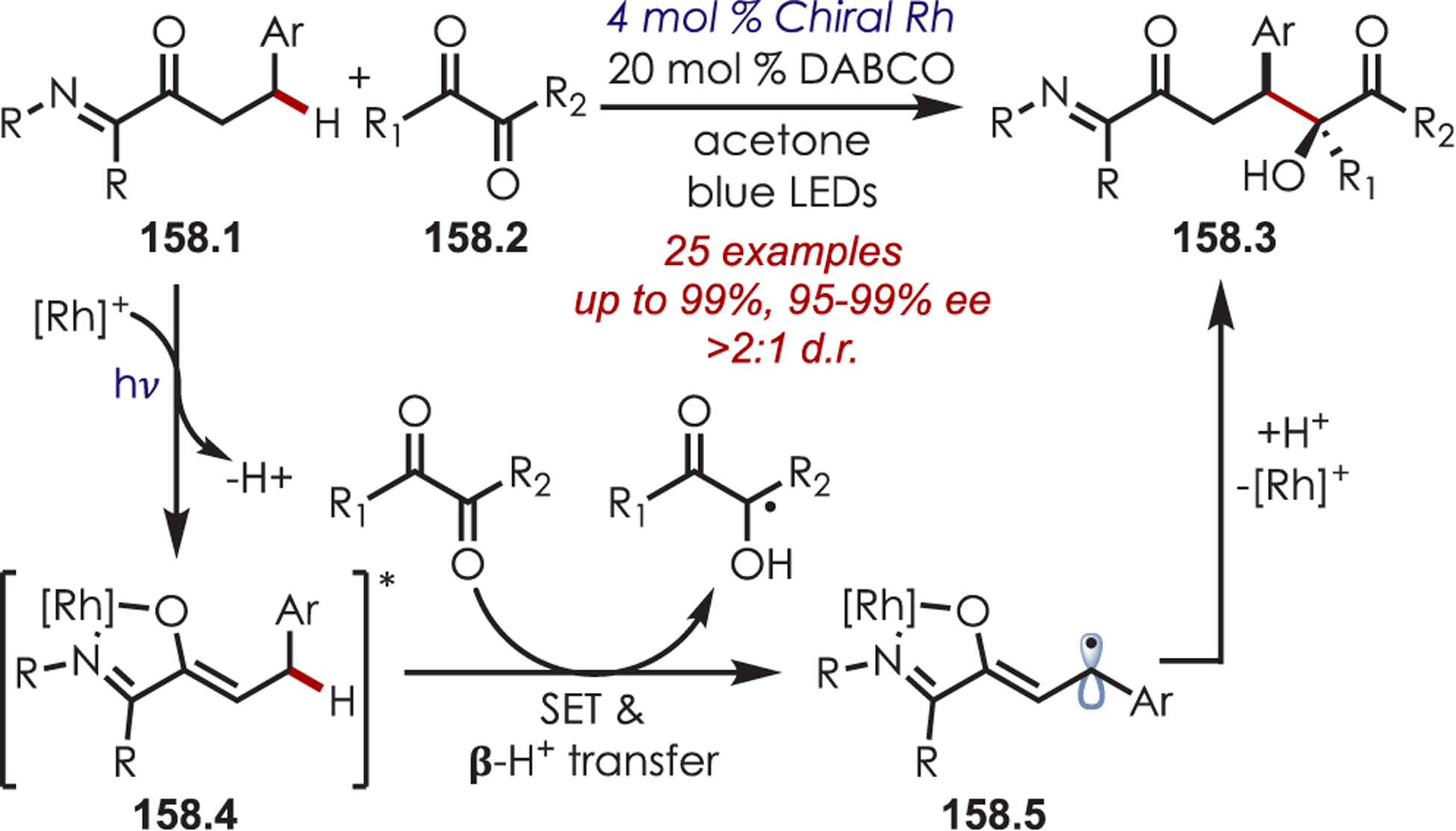
Chiral Rh Photoredox Catalyst for β-Functionalization of Ketones
Xiao reported an enantioselective di- and perfluoroalkylation of β-ketoesters (Scheme 159).299 This transformation utilized an Ir(III) photoredox catalyst to generate the reactive alkyl sp3 radical which undergoes a radical addition into enolates bound to a chiral nickel co-catalyst.
Scheme 159.
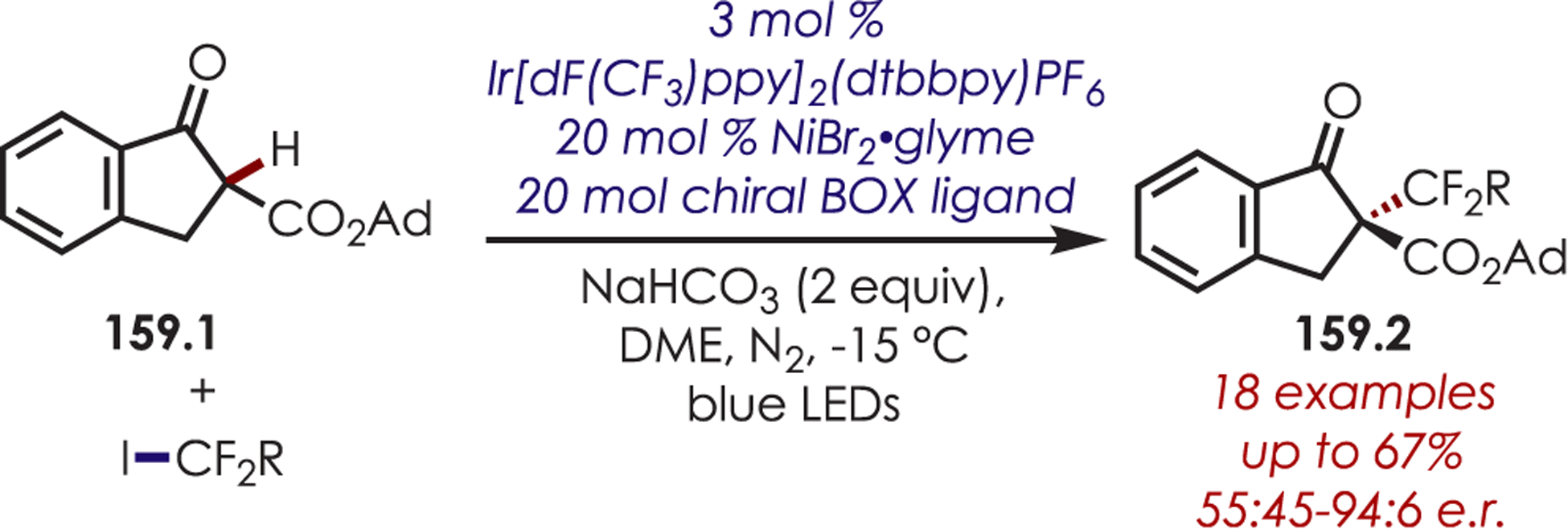
Asymmetric Difluoroalkylation of Aldehydes
An alternative approach for β-selective functionalization of amides was reported by Polyzos in 2017 using dual palladium and photoredox catalysis (Scheme 160).300 This transformation is reminiscent of the synergistic palladium and photoredox catalysis C–H arylation using diazonium as an sp2 aryl radical precursor.94 Following C–H activation of the amide substrate, aided by the quinolone directing group, a palladacyclic intermediate (160.4) is formed. In tandem, single-electron transfer from the excited state Ru(II) complex to the aryl diazonium results in the expulsion of nitrogen and formation of an aryl radical. Addition of the radical to the palladacycle (160.4) forms a Pd(III)–aryl complex (160.5). Oxidation of the palladium center by Ru(I) generates a Pd(IV) intermediate while regenerating the ground state Ru(II) photoredox catalyst. Finally, a reductive elimination from the Pd (IV) species furnishes the β-arylated amide (160.2) and Pd(II). This highly selective β-arylation was compatible with aliphatic amides with α-quaternary centers.
Scheme 160.
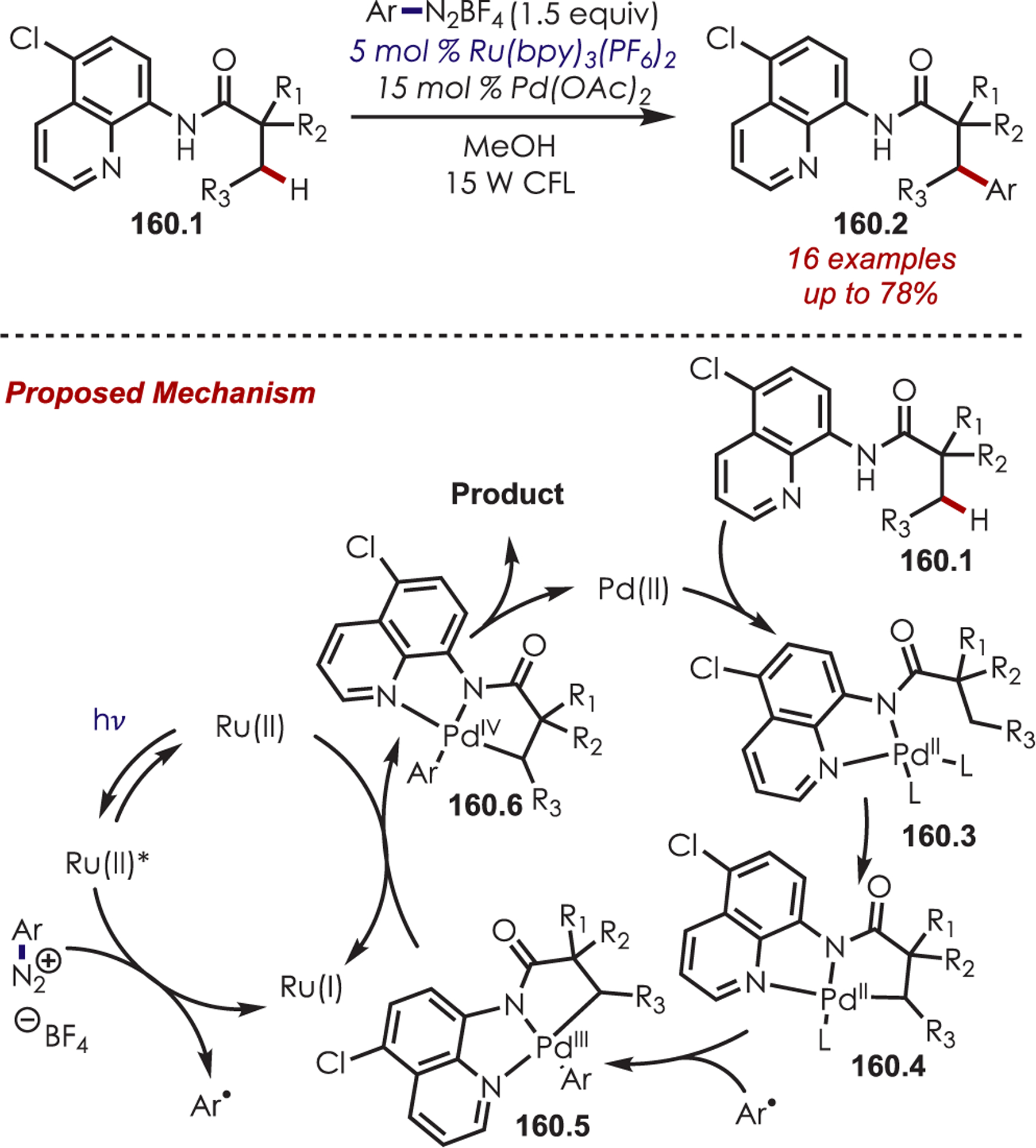
β-Arylation of Ketones by Dual Palladium and Photoredox Catalysis with Aryl Diazoniums
Direct Aldehyde C–H Functionalization.
In 2013, an aerobic oxidation of aldehydes (161.1) to the corresponding carboxylic acids via singlet oxygen was reported using an Ir(III) photoredox catalyst (Scheme 161).301 Photoexcited Ir(III) is known to generate singlet oxygen through an energy transfer process. The generated singlet oxygen is capable of abstracting an aldehyde C–H to produce anacyl radical. Radical–radical coupling with hydroperoxyl radical formed following HAT affords a peroxyacid. Reaction between this peracid (161.3) and a second equivalent of aldehyde affords 2 equiv of the desired carboxylic acid product through a Bayer–Villiger-type rearrangement (161.4). Aliphatic and aromatic aldehydes were competent substrates giving the corresponding acids in excellent yields.
Scheme 161.
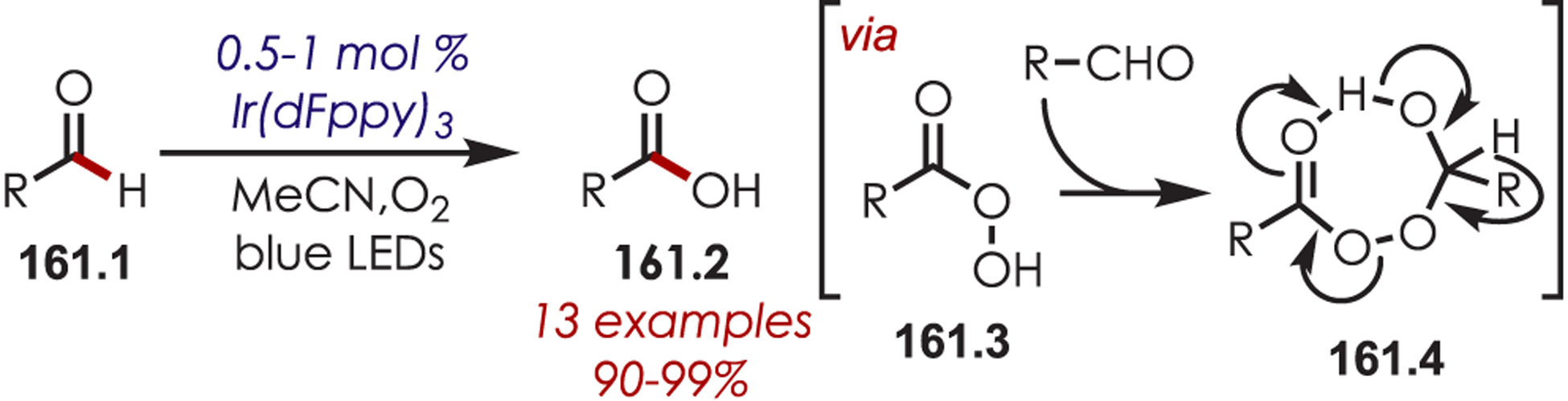
Direct Aldehyde C–H Hydroxylation via Acyl Radicals
Aldehydes (162.1) may also be converted to the corresponding N-hydroxyphthalimide esters (162.3) under photoredox conditions. (Scheme 162).302 Following absorption of visible light, Ru(II)* undergoes oxidative quenching in the presence of oxygen, affording a Ru(I) complex and superoxide radical anion. Hydrogen atom abstraction between this radical and a N-hydroxypthalimide derivative (162.2) affords a hydroxyphthalimide radical and the conjugate base of hydrogen peroxide. This couples with aldehyde derivative to afford the N-hydroxyester products in yields ranging from 21%–92%.
Scheme 162.
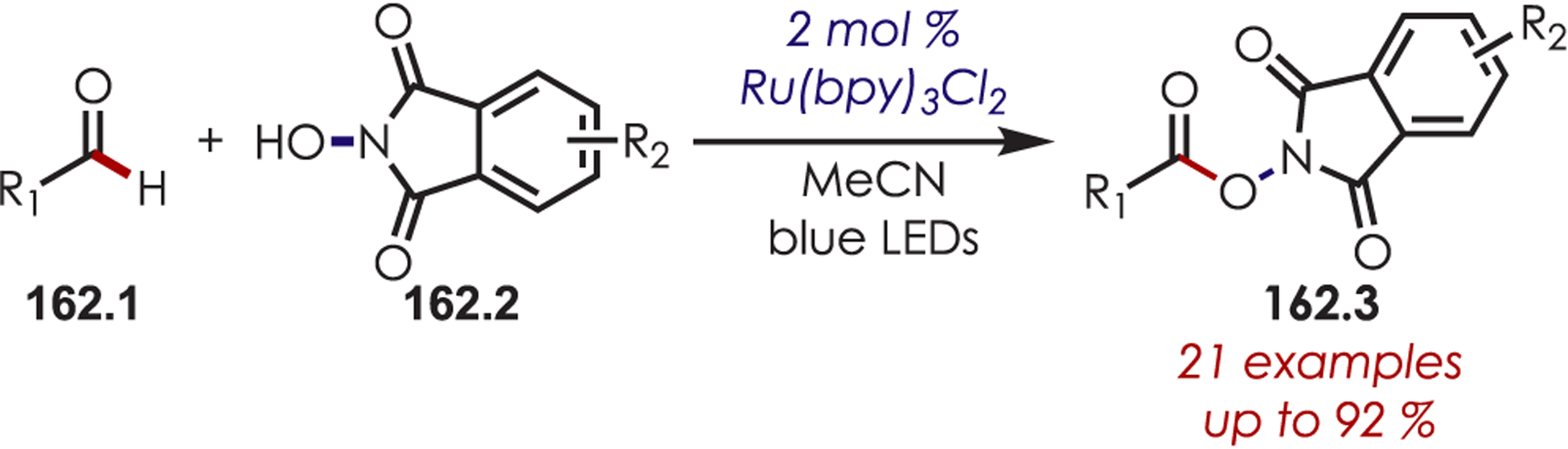
Synthesis of N-Hydroxyphthalimide Esters through Direct Aldehyde C–H Functionalization
An in situ generation of acid chlorides from aldehydes (163.1) was accomplished through the photogeneration of acyl radicals followed by trapping with N-chlorosuccinimide(163.2) (Scheme 163).303 A subsequent addition of aliphatic or aromatic secondary amines afforded the desired amides in a one-pot process.
Scheme 163.
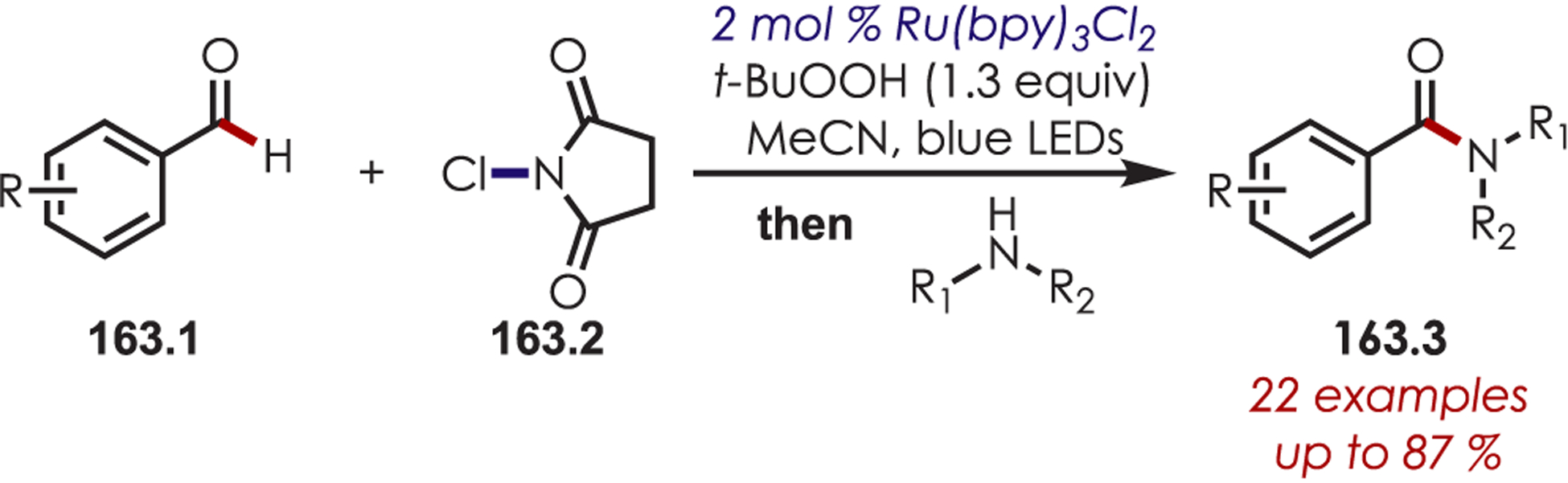
Acyl Chlorides Generated from Acyl Radicals for the Synthesis of Amides
Glorius and co-workers reported a cooperative dual catalytic system for the synthesis of trifluoromethyl thioesters (164.3) via acyl radicals (Scheme 164).304 Following deprotonation and single electron oxidation of the organic co-catalyst sodium benzoate, the resulting benzoate acyloxy radical engages the aldehyde substrate in HAT, forming an acyl radical. In a second step, radical group transfer between the N-trifluoromethylth-iophthalimide and the acyl radical affords the desired thiotrifluoromethyl ester. This system was successful at generating a range of trifluoromethyl esters, including with more complex cholesterol, lithocholic, and trans-andosterone derivatives.
Scheme 164.
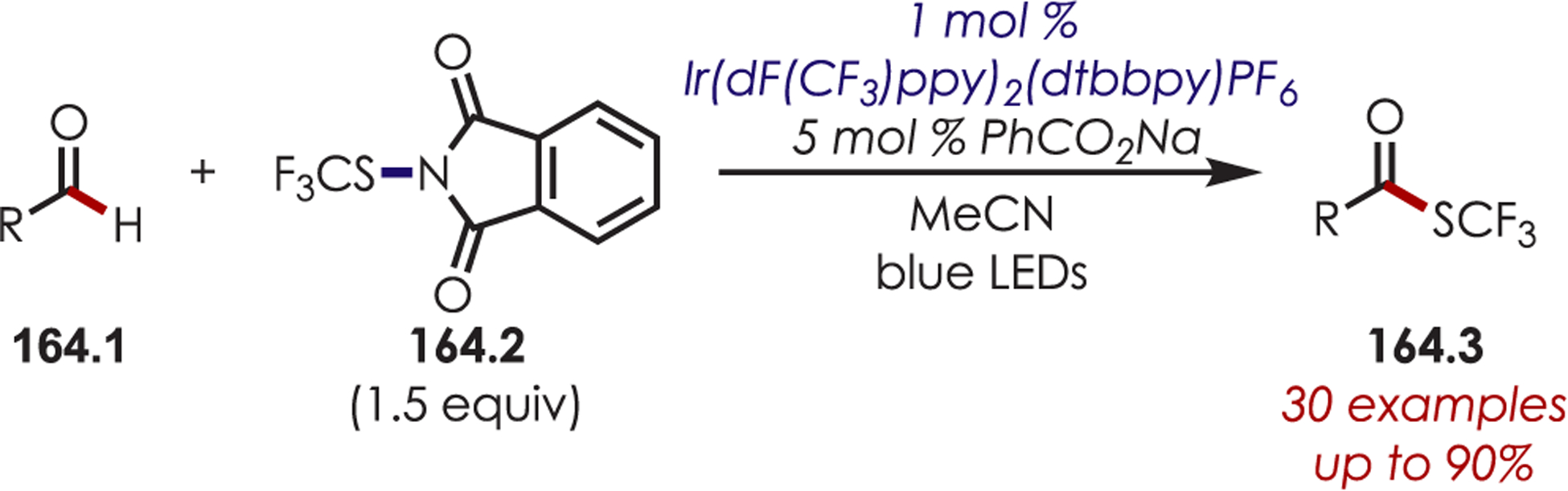
Synthesis of Trifluoromethyl Thioesters through Acyl Radicals
Acyl radicals (165.1) generated via HAT can also be utilized as cross-coupling partners in combination with a Ni/Ir dual catalytic system (Scheme 165).305 Aryl and alkyl bromides were found to successfully couple with acyl radicals when using 3-acetoxyquinuclidine (165.2) as the hydrogen atom transfer catalyst, as discussed previously for α-heteroatom hydrogen atom abstraction.228 The acyl radical is intercepted by a nickel intermediate, which subsequently undergoes reductive elimination to afford the ketone products.
Scheme 165.
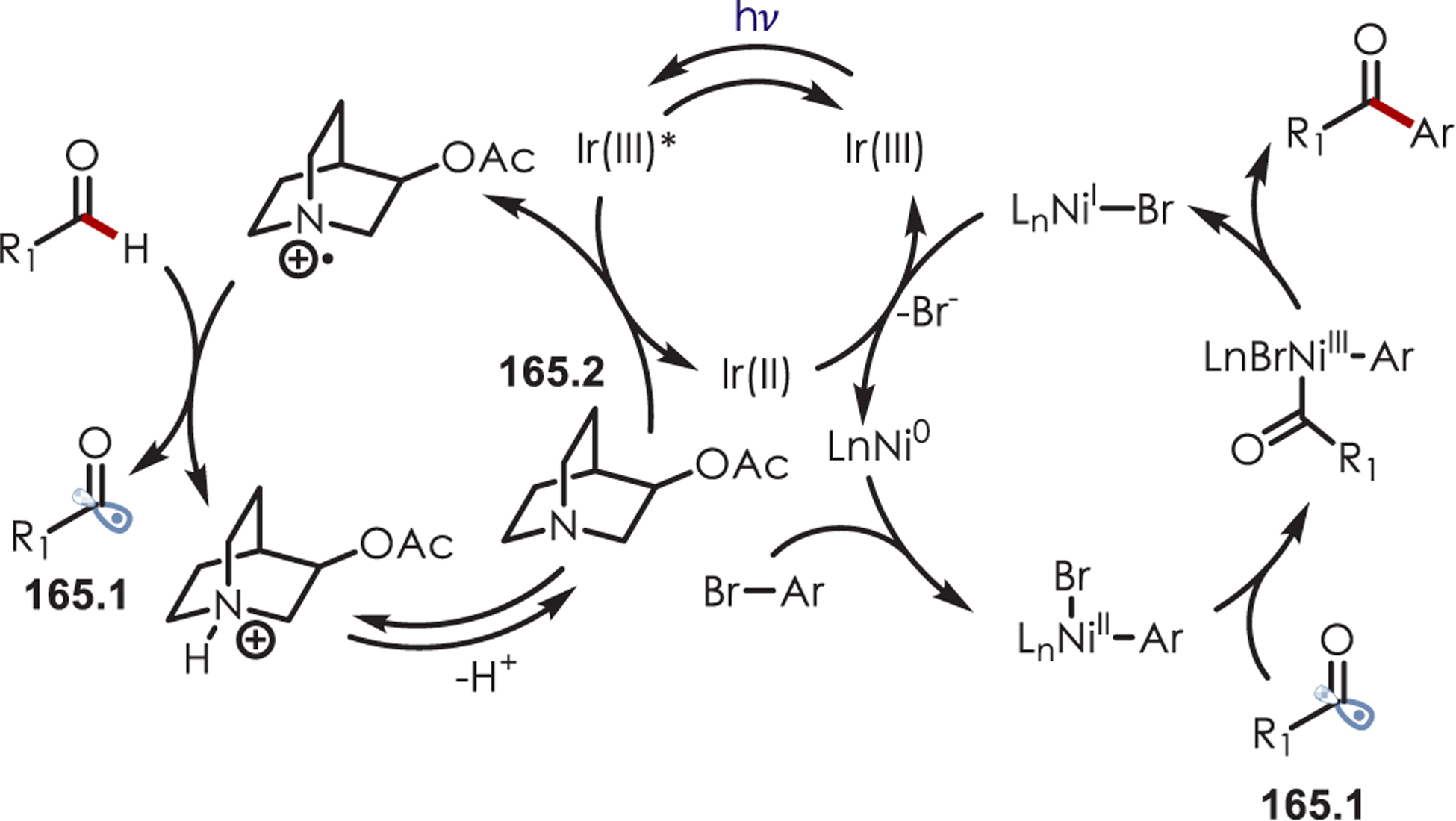
Mechanism of Direct Aldehyde C–H Arylation through a Triple Catalytic System
The aryl bromide (166.2) scope included electron-deficient or electron-rich arenes and heterocycles, generating the products in excellent yields (Scheme 166). Vinyl and alkyl bromides were also compatible; however, lower yields were observed (55–74% yield). Cyclic or acylic aliphatic aldehydes were competent substrates, and this methodology was tolerant of carbamates, alcohols, and tert-butyl functional groups. Interestingly, benzaldehydes were successful substrates, despite having a less hydridic C–H bond for abstraction. Recently, acyl radicals, generated through dual HAT and decatungstate or anthraquinone photoredox catalysts were successfully intercepted by a palladium catalytic cycle to access direct arylation and alkenylation of aldehydes.306
Scheme 166.
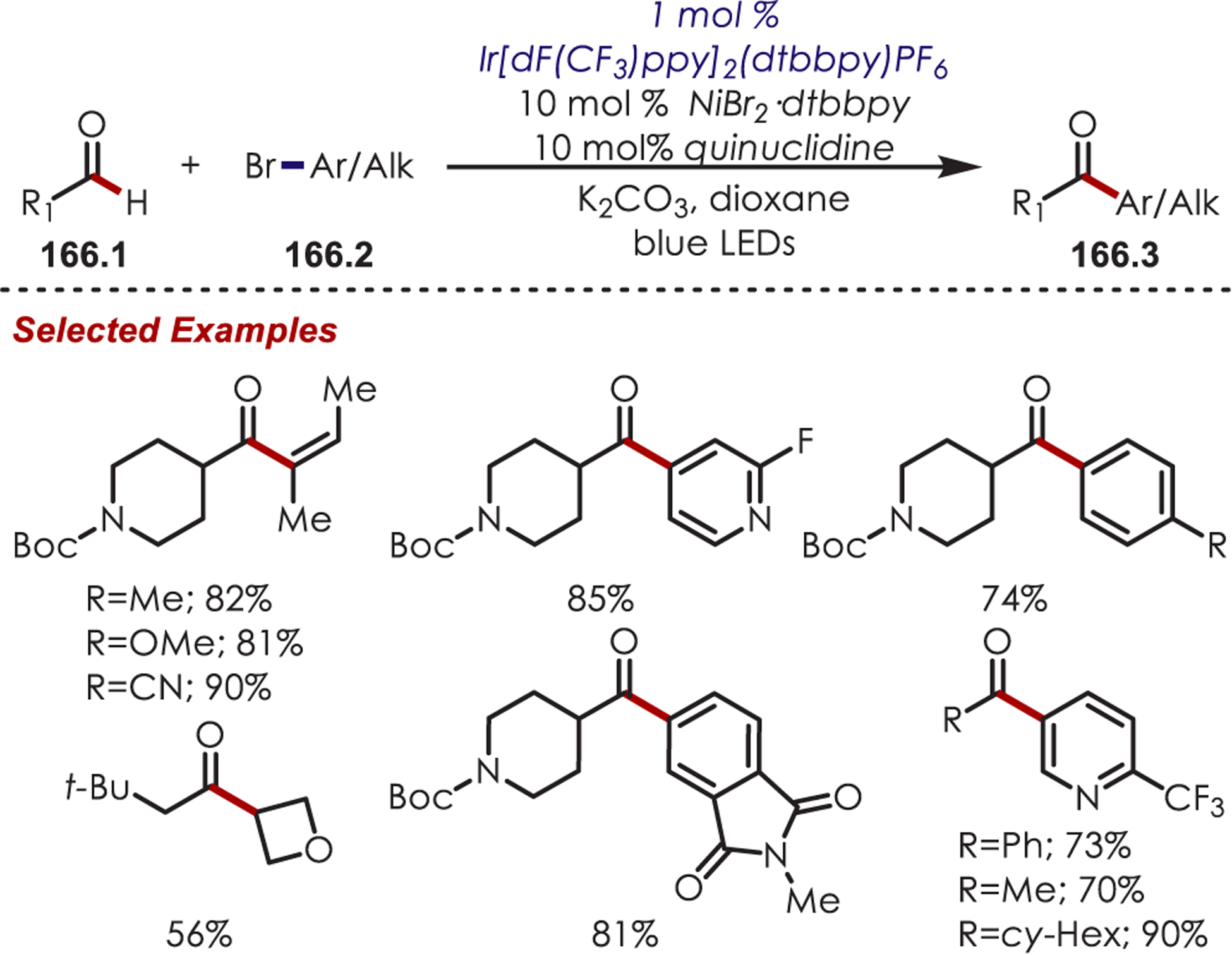
Scope of Direct Aldehyde C–H Arylation through a Triple Catalytic System
Liu reported the trapping of photogenerated nucleophilic acyl radicals with electron-poor alkenes (167.2; Scheme 167).307 Additionally, these authors independently reported the interception of aryl bromides (167.4) through synergistic nickel catalysis for the synthesis of aryl ketones by direct aldehyde C–H functionalization.
Scheme 167.
Direct Aldehyde C–H Alkylation with Electron-Deficient Alkenes and Arylation with Nickel Catalysis
Glorius and co-workers had envisioned that direct C–H abstraction of aldehydes (168.1) could be accomplished through an electrophilic benzoyloxy radical, thus providing a high degree of selectivity for the synthesis of ynones, ynamides, and ynoates (Scheme 168).308 This system uses ethynylbenziodoxolone (EBX) as the alkynylating reagent (168.2) of the acyl radical while simultaneously producing a benzoiodoxolonyl radical. Ultimately, this radical can undergo two potential pathways: either turning over the photocatalytic cycle by a reductive SET from Ir(II) or entering the HAT cycle and serving as the hydrogen atom abstracting agent for generation of the acyl radical. This direct alkynylation was compatible with benzaldehydes and aliphatic aldehydes, as well as a variety of aryl or TIPS EBX reagents. Additionally, the authors demonstrated the selective alkynylation of complex derivatives including cholesterol, lithocholic, and adapalene derivatives.
Scheme 168.
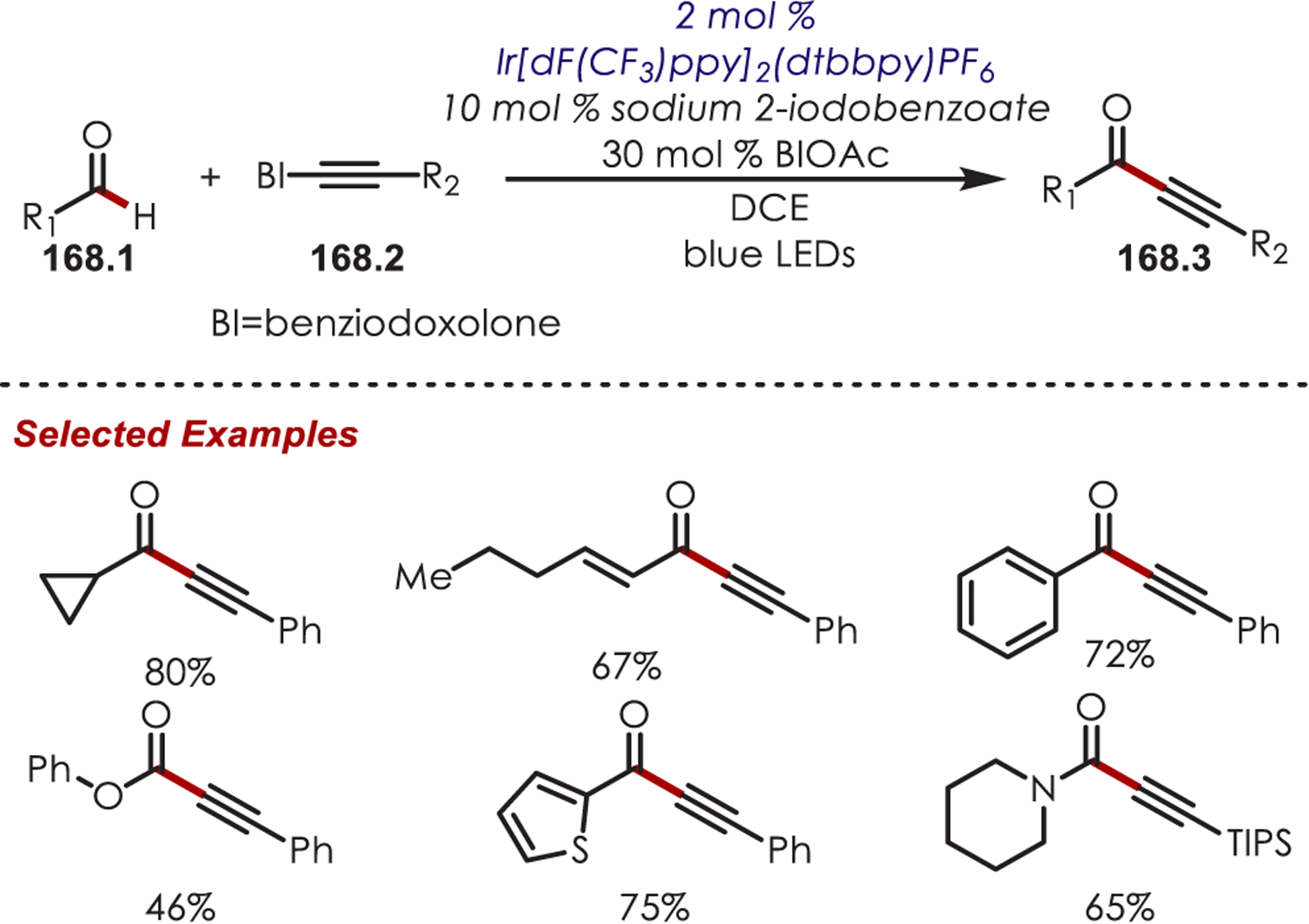
Alkynylation of Aldehydes Using a Benziodoxolonyl Radical as a Hydrogen Abstractor
The direct C–H functionalization of hydrazones (169.1 and 169.4) by photoredox catalysis has been demonstrated by Zhu and co-workers for difluoroalkylation309 and dihydropyrazole synthesis (Scheme 169).310 Oxidative quenching of Ir(III)* by alkyl bromides generates acarbon-centered radical. Radical addition into electrophilic imines results in the formation of a nitrogen center radical. Subsequent oxidation by Ir(IV) and tautomerization affords the difluoroalkylated hydrazones products. Fused dihydropyrazoles (169.6) are formed when using geminal dibromomalonate derivatives. Following formation of the α-bromohydrazone product from the first catalytic cycle, a second PET reduction event generates a carbon-centered radical, which undergoes 1,5-HAT, generating an α-amino radical species. Oxidation of this intermediate radical by Ir(IV) affords an iminium species which is trapped by the pendant malonate in a polar process, resulting in the formation of the fused ring product. Difluoroalkylated hydrazones have also been synthesized through a gold-catalyzed photoredox method via a similar mechanism.311
Scheme 169.
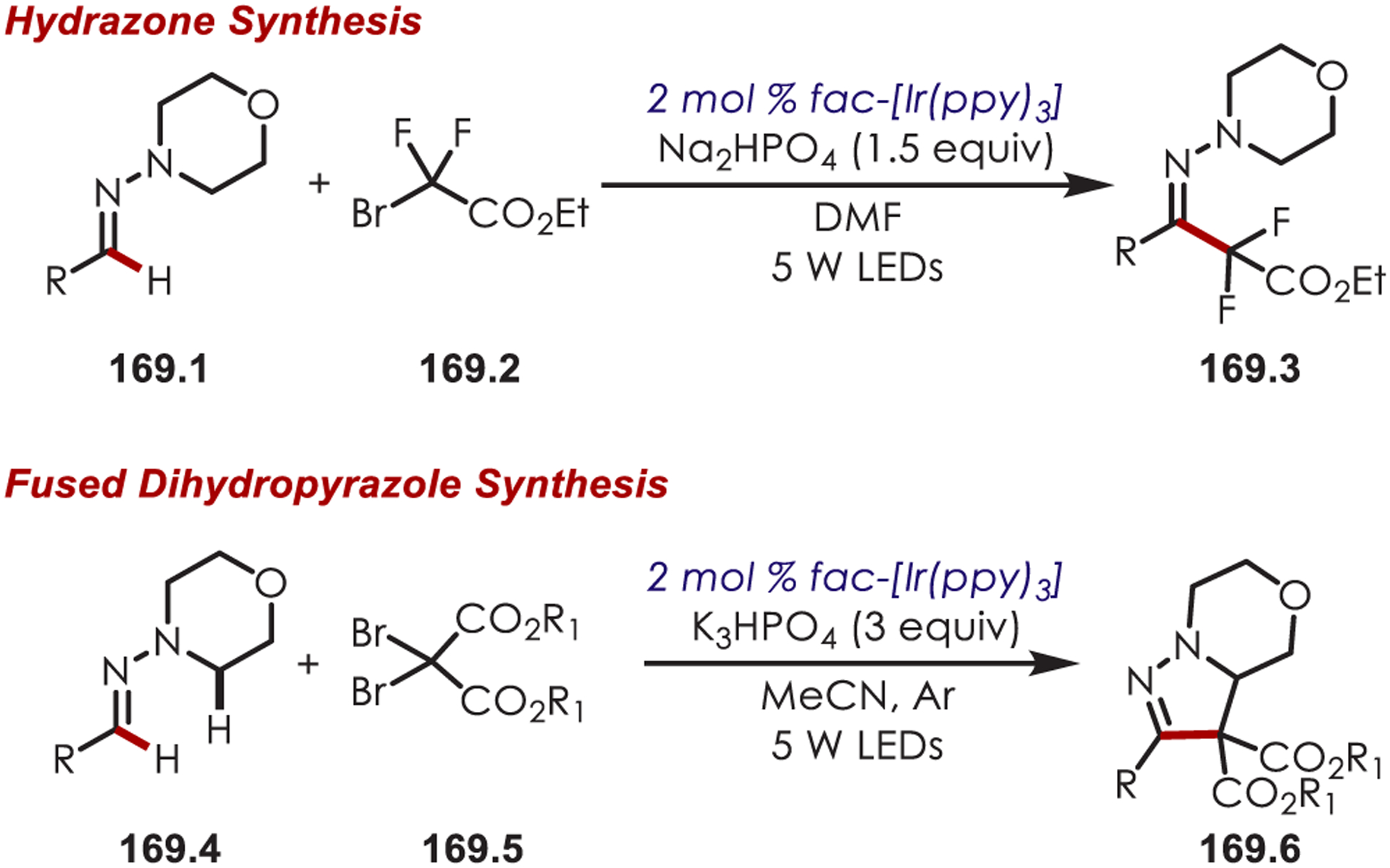
Hydrazone C–H Alkylations
2.2.3. Benzylic and Allylic C–H Functionalization.
Early reports of benzylic C–H functionalization enabled by photoredox catalysis are Mannich-type reactions of tetrahydroisoquinolines; however, these are covered under α-heterofunctionalization (section 2.2.1). The relatively weaker BDE of benzylic and allylic C–H bonds has been exploited through hydrogen atom abstractions (BDE ~85 and ~80 kcal/mol for benzylic or allylic C–H bonds, respectively) for the generation of reactive sp3 carbon-centered radicals.312 These radicals have been trapped with electrophilic reagents or coupled to alkyl and aryl halides through metallophotoredox catalysis. In general, direct allylic C–H functionalization is more elusive than benzylic functionalization.
Via Hydrogen Atom Abstractions.
The MacMillan lab reported an arylation of benzylic ethers via the synergistic combination of Ir(III) photoredox catalyst and thiol HAT co-catalyst (Scheme 170).313 The photoexcited Ir(III) catalyst undergoes a reductive SET with the cyanobenzene partner, generating a benzene anion radical. Simultaneously, an oxidative PCET generates a thiyl radical that serves as the hydrogen atom abstracting agent for the generation of the benzylic radical. A radical–radical coupling of the benzylic radical and the benzene anion radical, followed by elimination of cyanide affords the coupled products. This method was tolerant of a range of functionality on both arene coupling partners in good to excellent yield.
Scheme 170.
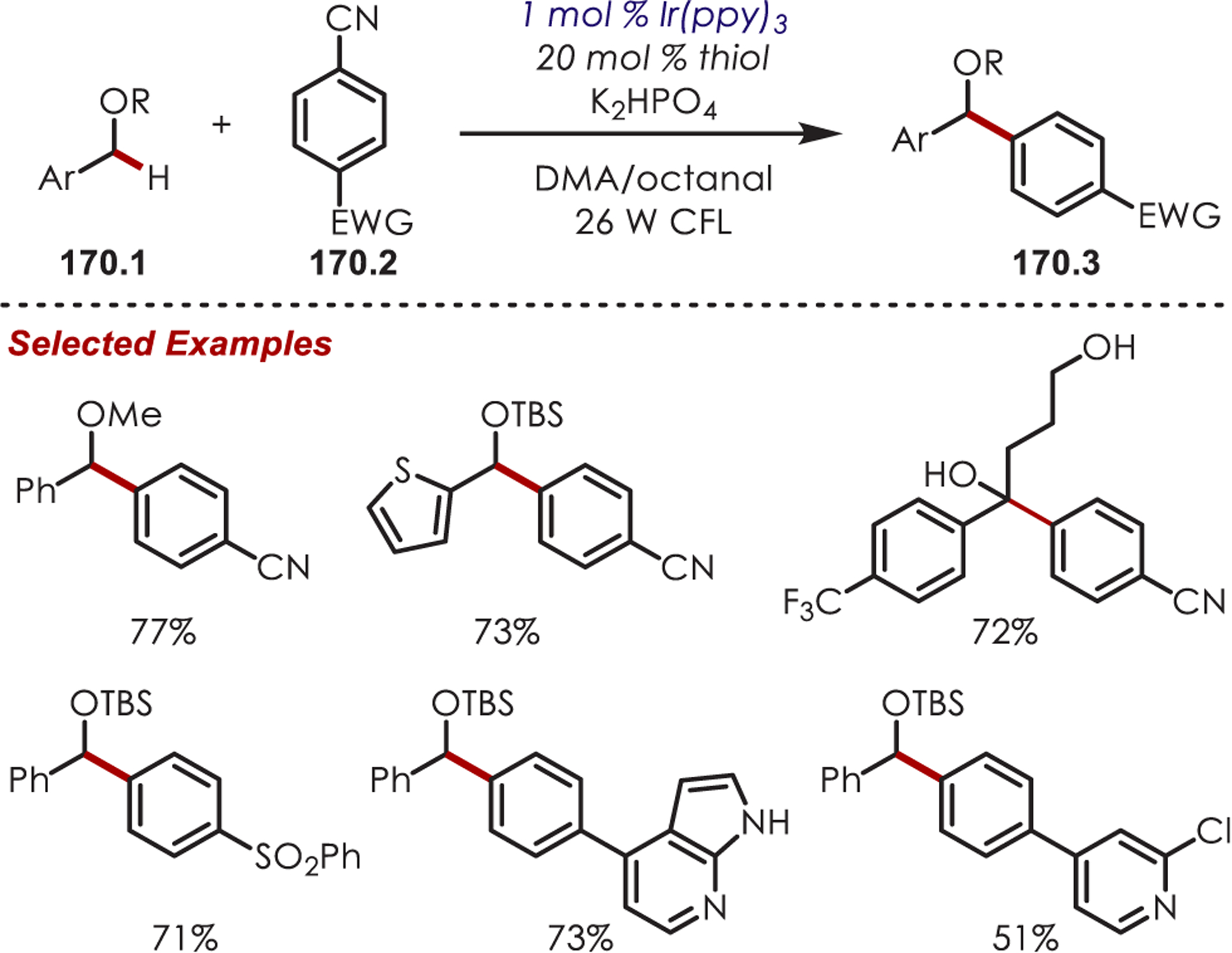
Benzylic C–H Arylation with Cyanobenzenes and a Thiol Hydrogen-Atom Abstracting Reagent
Soon thereafter, the same group reported the coupling of aryl ethers with N-aryl imines through a similar PCET-mediated transformation with Ir(ppy)3 as the photoredox catalyst in conjuction with a thiol HAT co-catalyst.314 In this case, a reduction of the imine by Ir(II) regenerates the ground state catalyst while providing an iminyl anion radical that couples with the benzylic radical.
Under an analogous mechanism, the direct allylic arylation via a dual photoredox and organic catalyst system was accomplished (Scheme 171).315 Allylic C–H bonds (171.1) are relatively weak (82.3 kcal/mol for cyclohexene) and can undergo hydrogen atom abstraction by the thiyl radical. A radical–radical coupling with a cyanobenzene anion radical, generated via reductive SET from Ir(III)* and subsequent elimination of cyanide furnished the arylated products.
Scheme 171.
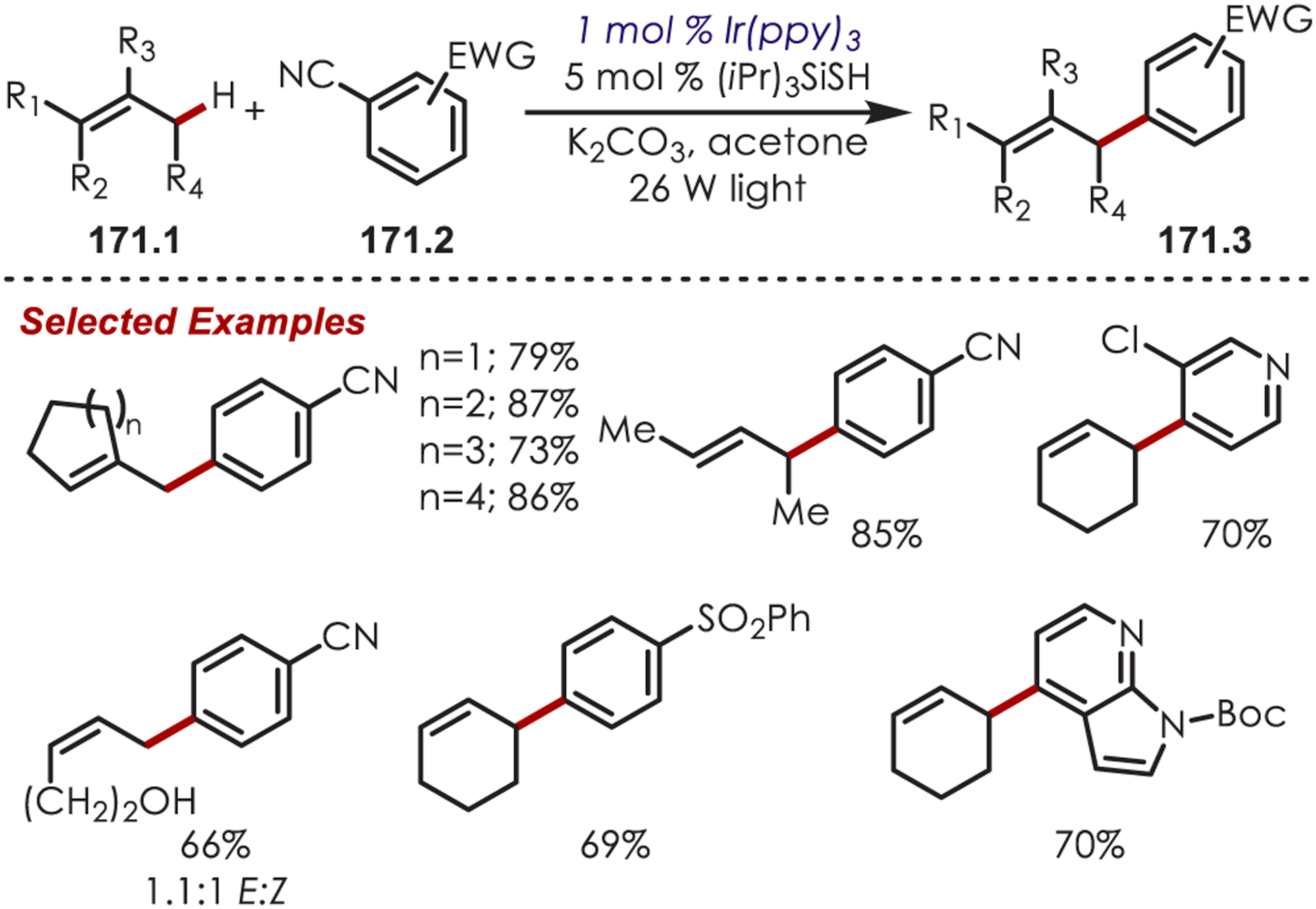
Allylic C–H Arylation with Cyanobenzenes and a Thiol Hydrogen Atom Abstracting Reagent
Thiols have also been reported as suitable hydrogen atom abstracting agents in an aryl ketone (172.2) alkylation reaction (Scheme 172).316 The photogenerated allylic radical was successfully trapped with a range of electronically diverse aryl ketones, including more complex derivatives such as tamoxifen. Additionally, complex allylic substrates including citronellyl, α-pinene, and γ-terpinene were functionalized with this methodology.
Scheme 172.
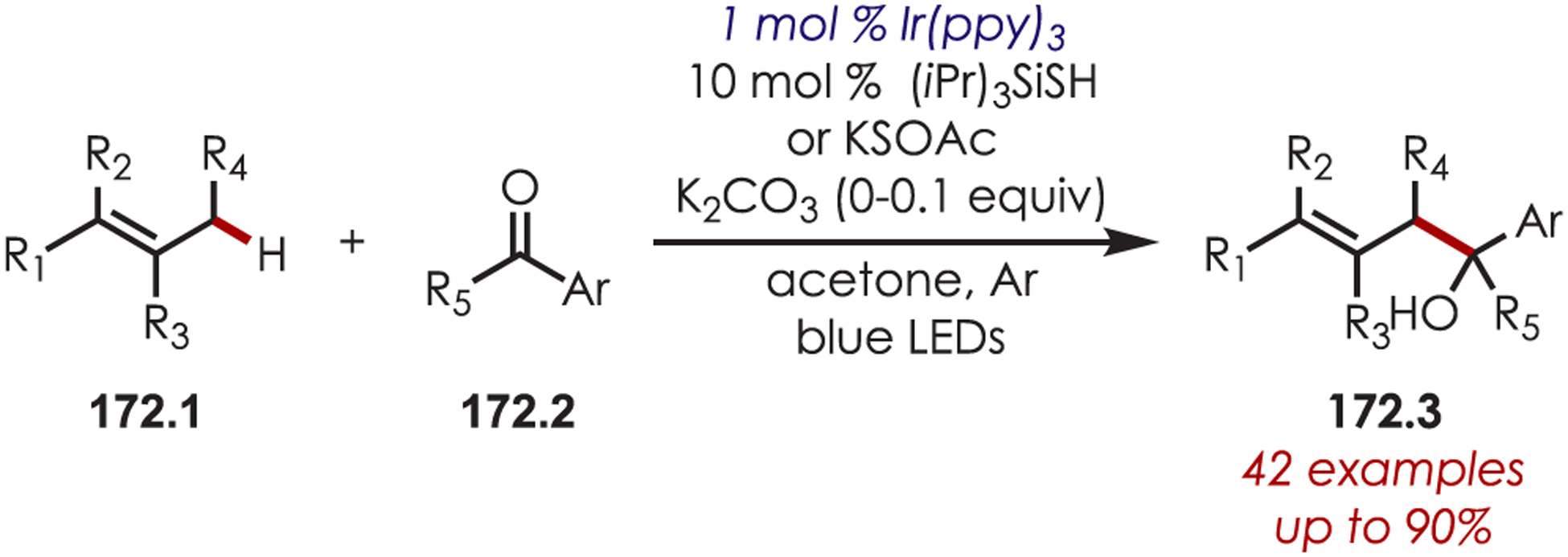
Allylic Radicals Trapped with Aryl Ketones
Further expanding on this HAT theme, triisopropylsilylthiol has been utilized to form nucleophilic allylic radicals from enol silanes (173.1) that can be trapped with imines (173.2) in a Mannich-type reaction (Scheme 173).317 The thiyl radical is generated through a PCET from the photoexcited iridium(III) catalyst. Huang and co-workers reported a comparable allyl radical addition to imines using dual iridium and thiol catalysis and provided mechanistic support for allyl radical addition into the imine.318
Scheme 173.
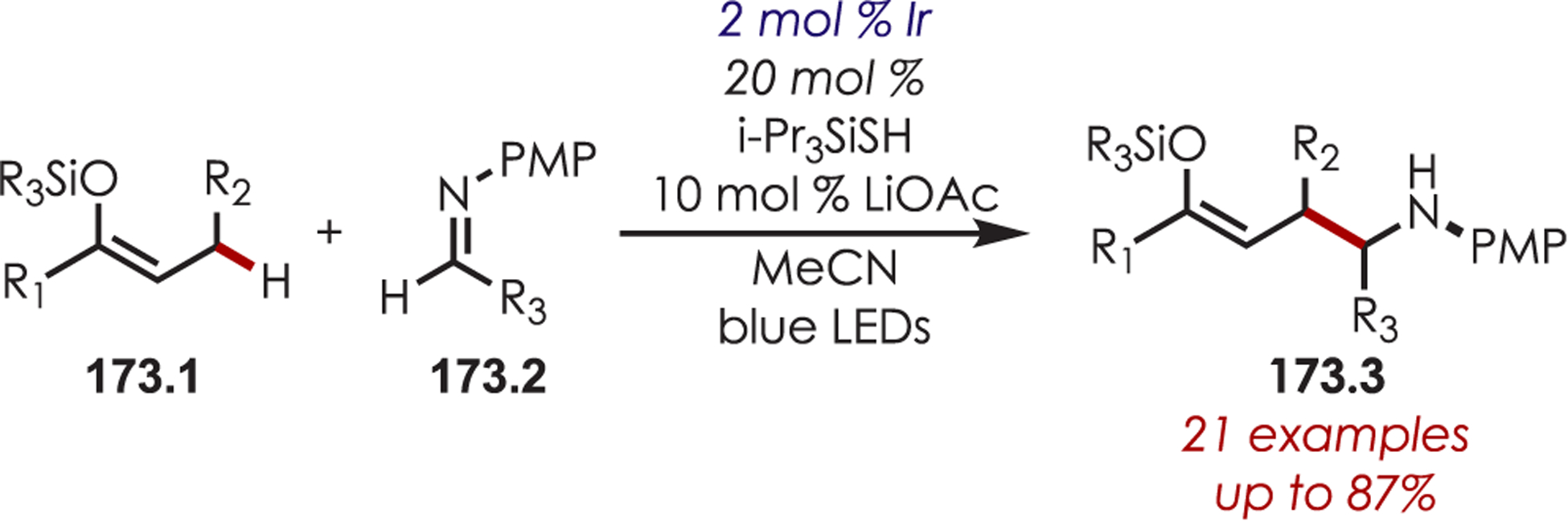
Allylic Radicals Trapped with Imines
Kanai and Oisaki reported that diarylsulfonamides could serve as an HAT catalyst for the abstraction of allylic and benzylic C–H bonds (174.1) with dicyanobenzene (174.2) coupling partners (Scheme 174).319 The sulfonamide HAT catalyst has a N–H BDE of 95 kcal/mol, which allows for the selective abstraction of C–H bonds, and helps prevent undesired reactivity that may be observed with quinuclidinium cation radical (100 kcal/mol for N–H) or benzoate radical (110 kcal/mol for O–H) hydrogen atom abstracting agents. Electron-rich and halogenated arenes were successful in the benzylic functionalization, including those that resulted in quaternary centers. The scope of allylic functionalization was limited to five cyclic derivatives ranging from 54 to 95% yield, but included a late-stage functionalization of prasterone.
Scheme 174.
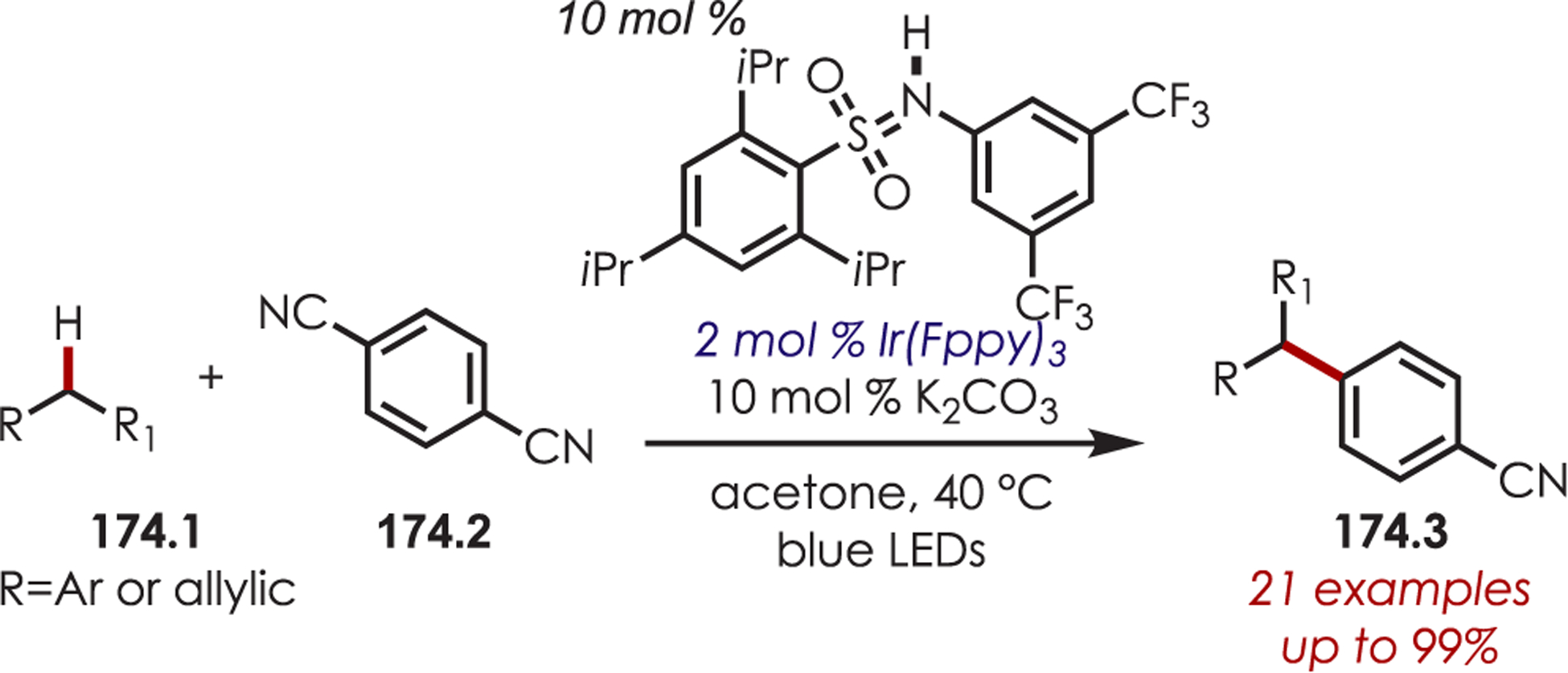
Benzylic or Allylic Radical Coupling with Cyanobenzenes using Sulfonamide as the HAT Catalyst
Recently, a benzylic heteroarylation was accomplished using (benzyloxy)phthalimides (175.1) as benzyloxy radical precursors (Scheme 175).320 Reduction of Ir(III)* from DIPEA generates Ir(II). The reduced form of the iridium catalyst undergoes a reductive SET with the (benzyloxy)phthalimides and a subsequent hydrogen atom transfer with an amine cation radical to generate an benzyloxy radical. A subsequent intramolecular 1,2-HAT and radical arylation cascade furnished the (hetero)arylated products (175.3). Electron-rich and electron-poor benzyloxy and substituted pyridines were tolerated in this reaction and furnished the products in good yields.
Scheme 175.
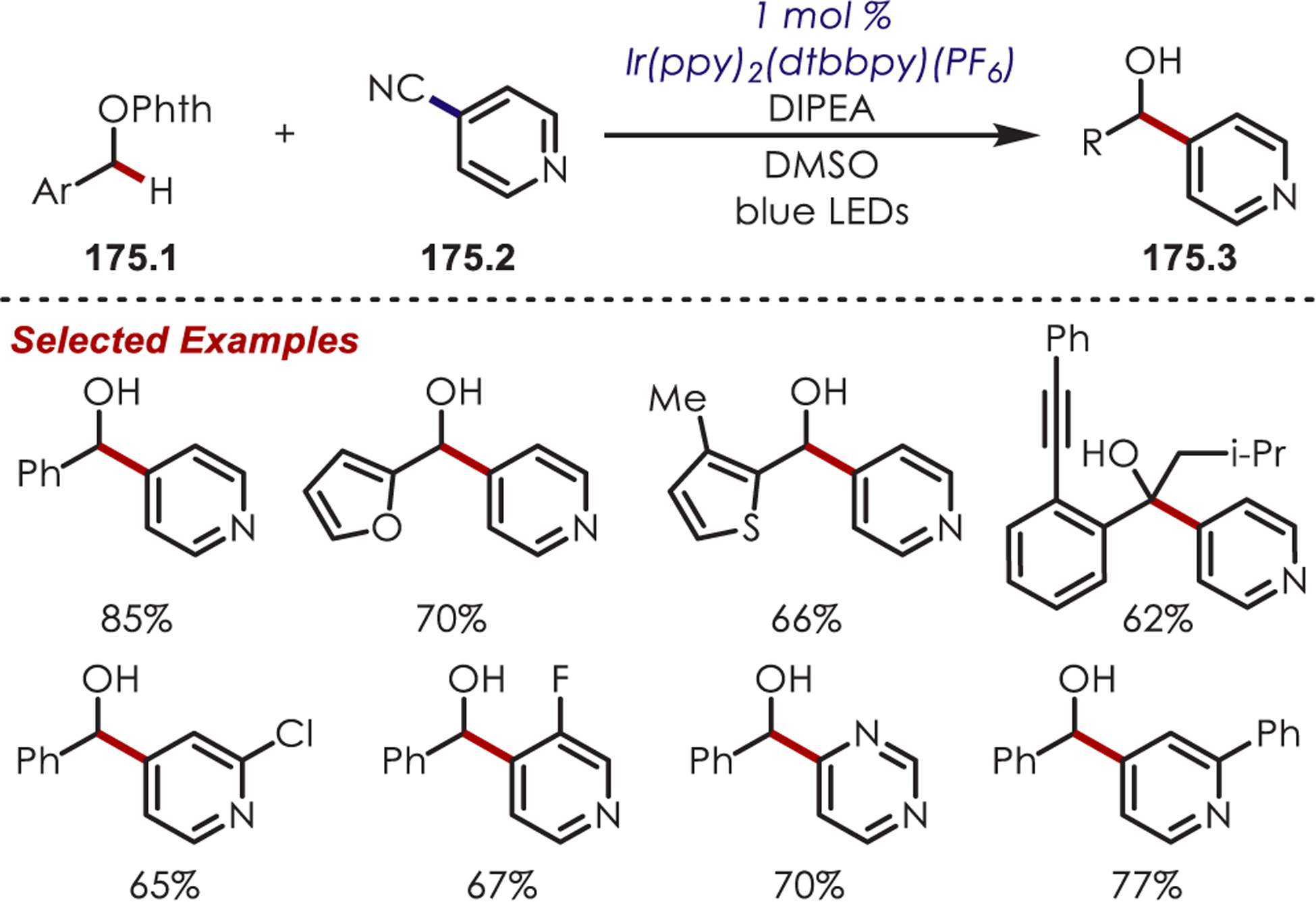
Benzylic Radical Coupling with Cyanopyridines by Photogenerated Benzyloxy Radicals
Following this work, Bolm reported a sulfoximidation of benzylic C–H bonds with a sulfoximidoyl hypervalent iodine(III) reagent 176.2 as a nitrogen-centered radical (NCR) precursor (Scheme 176).321 The NCR radical abstracts a benzylic C–H and a subsequent oxidation by Ru(III) furnished the reactive benzylic cation. The arene scope was extended to anisoles, halogenated arenes, naphthylenes, and allyl-substituted arenes. A variety of substitutions were also tolerated at the benzylic position, including alkyl or aryl functional groups.
Scheme 176.
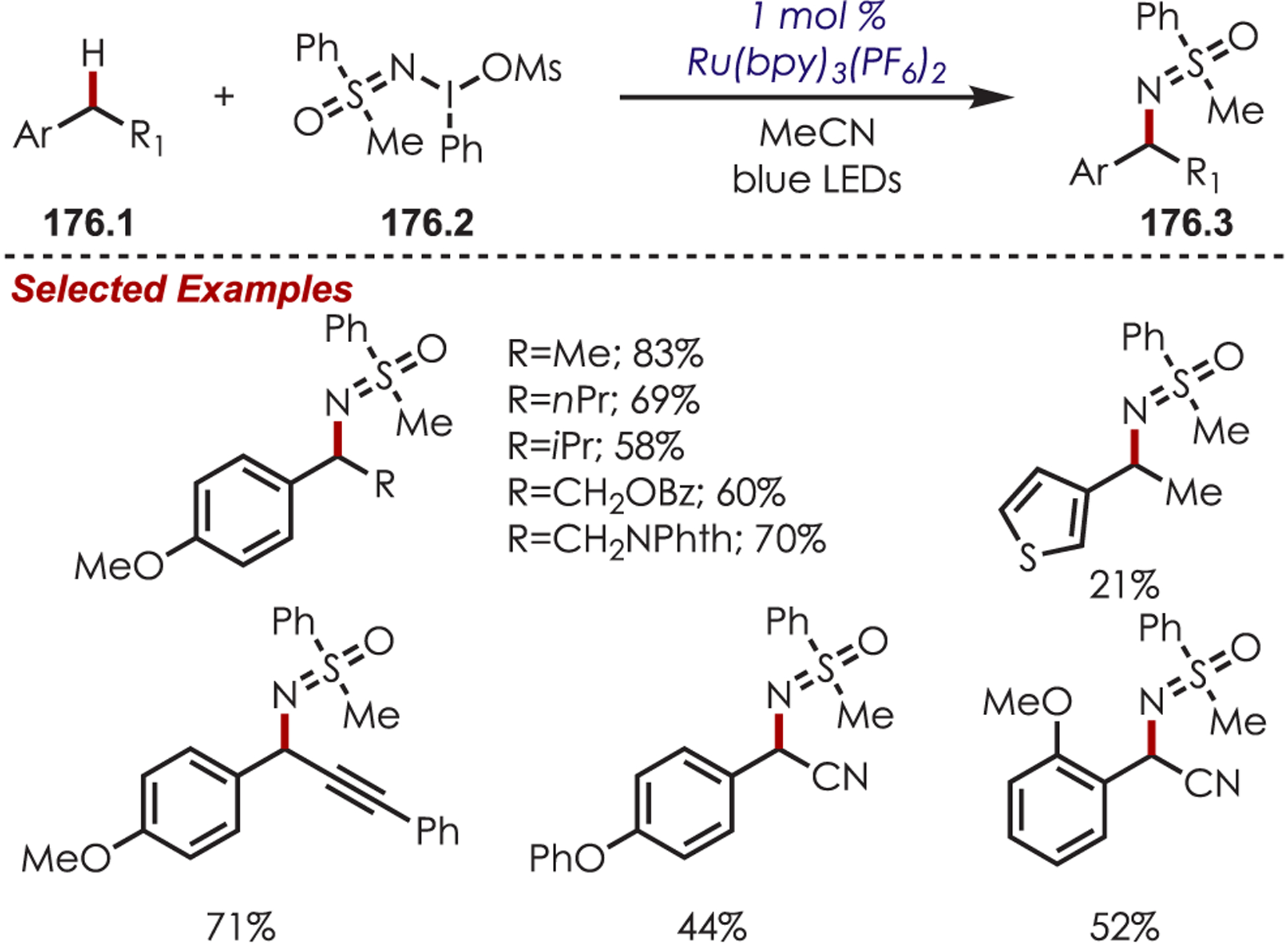
Benzylic Sulfoximidation Using a Nitrogen-Centered Radical as the HAT Reagent
A photoredox-catalyzed benzylic sulfonylation was accomplished by a three-component coupling with a sulfur dioxide surrogate, an aryl diazonium salt as an aryl radical precursor, and 4-methylphenols (177.1), enabled by an Ir(III) catalyst (Scheme 177).322 Photoexcited Ir(III)* participates in a PET with an aryldiazonium, furnishing a reactive aryl radical. Radical trapping with SO2 generates the key arylsulfonyl radical (177.3). Concurrently, a phenoxy radical (177.4) is generated through oxidation of the arene and subsequent deprotonation. An intermolecular HAT with another molecule of 177.1 and gives rise to a benzylic radical (177.5) and returned starting arene. A radical–radical recombination of the benzylic and arylsulfonyl radical furnished the desired products. This methodology was limited to alkyl-substituted 4-phenols. The aryldiazonium partner was tolerant of electron-rich and electron-poor substitution.
Scheme 177.
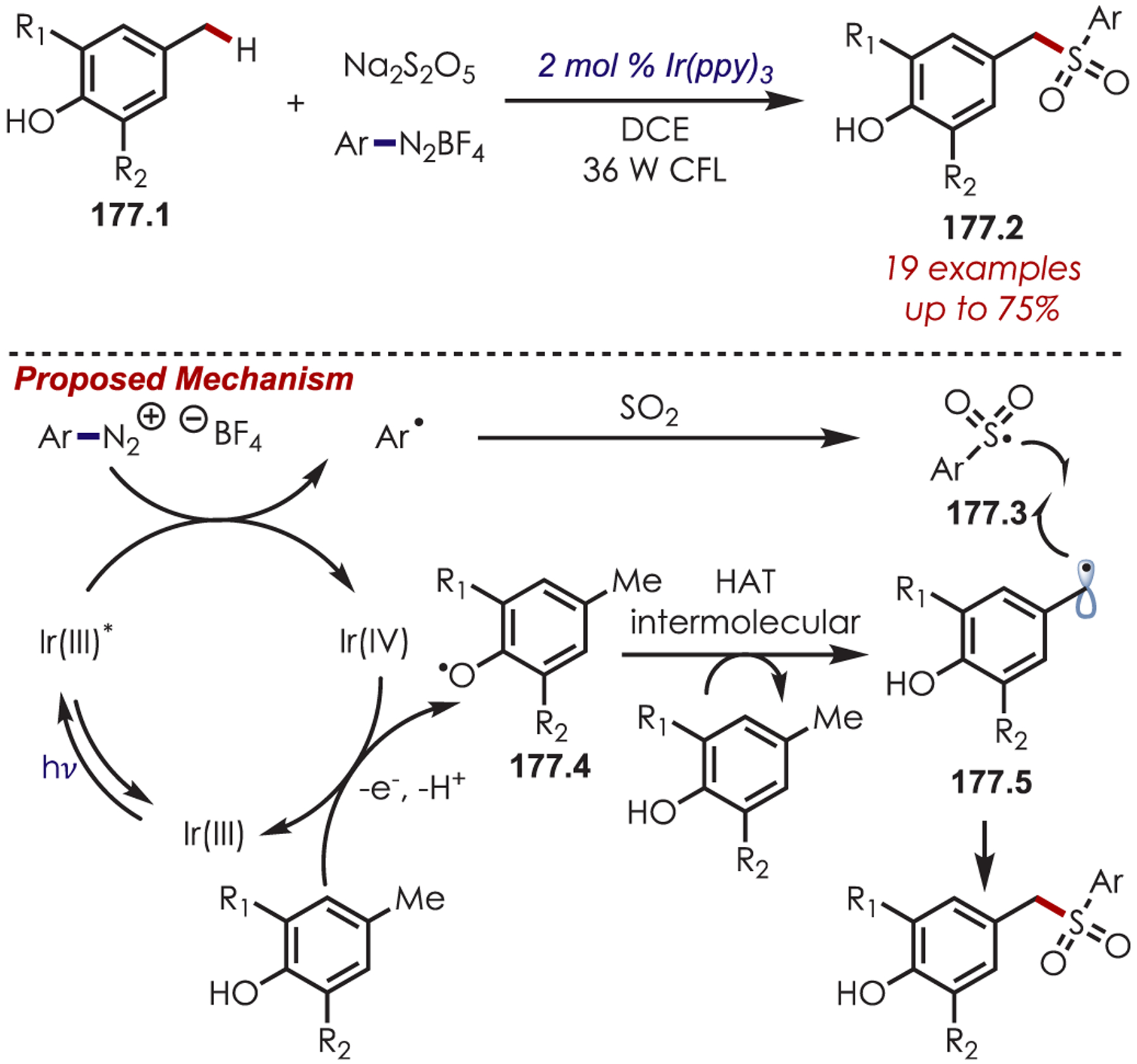
Benzylic Sulfonylation through a Three-Component Coupling
Tetrabutylammonium decatungstate (TBADT) is a widely used dual photoredox catalyst and hydrogen atom abstractor and has been utilized in a benzylic fluorination reaction (Scheme 178).323 Alkyl and halogenated ethyl benzene derivatives (178.1) were competent coupling partners, giving the desired product in moderate to poor yield. Interestingly, p-ethyl toluene was selectively functionalized at the ethyl benzylic position, and no fluorination was observed at the methyl.
Scheme 178.
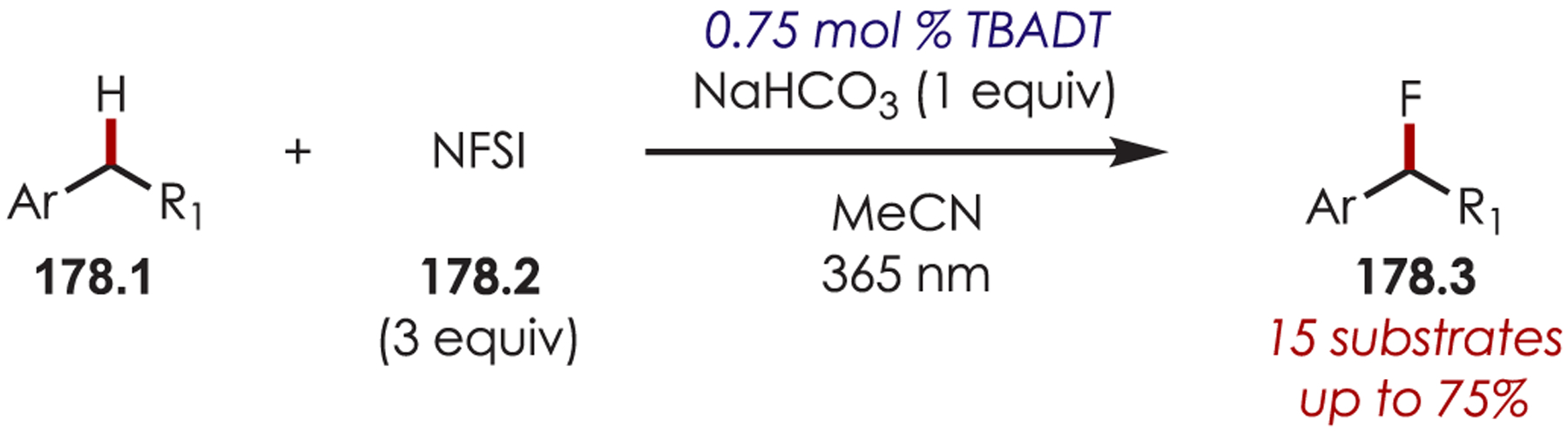
Benzylic Fluorination Using TBADT as a Photoredox and HAT Catalyst
Pandey and co-workers developed a benzylic amination with azole nucleophiles (179.2) enabled by an Ir(III) photoredox catalyst (Scheme 179).324 Bromotrichloromethane oxidatively quenches the photoexcited Ir(III)* and generates the active hydrogen atom abstractor, trichloromethyl radical. Ir(IV) subsequently oxidizes the arene (179.1) to the aryl cation radical, which undergoes a hydrogen atom abstraction to generate a benzylic cation. The addition of a variety of azole nucleophiles furnished the desired products. The aryl scope was limited to anisole derivatives, but gave excellent para-selectivity for derivatives containing multiple benzylic sites. The authors also reported a method for benzylic oxidation to afford the benzophenones by adding water as the nucleophile. A benzylic C–H azidation using the Zhadankin reagent and Cu(dap)2+ as the photoredox catalyst has been reported but proceeds through a radical-chain process initiated by generation of an azide radical from the photoexcited catalyst.325
Scheme 179.
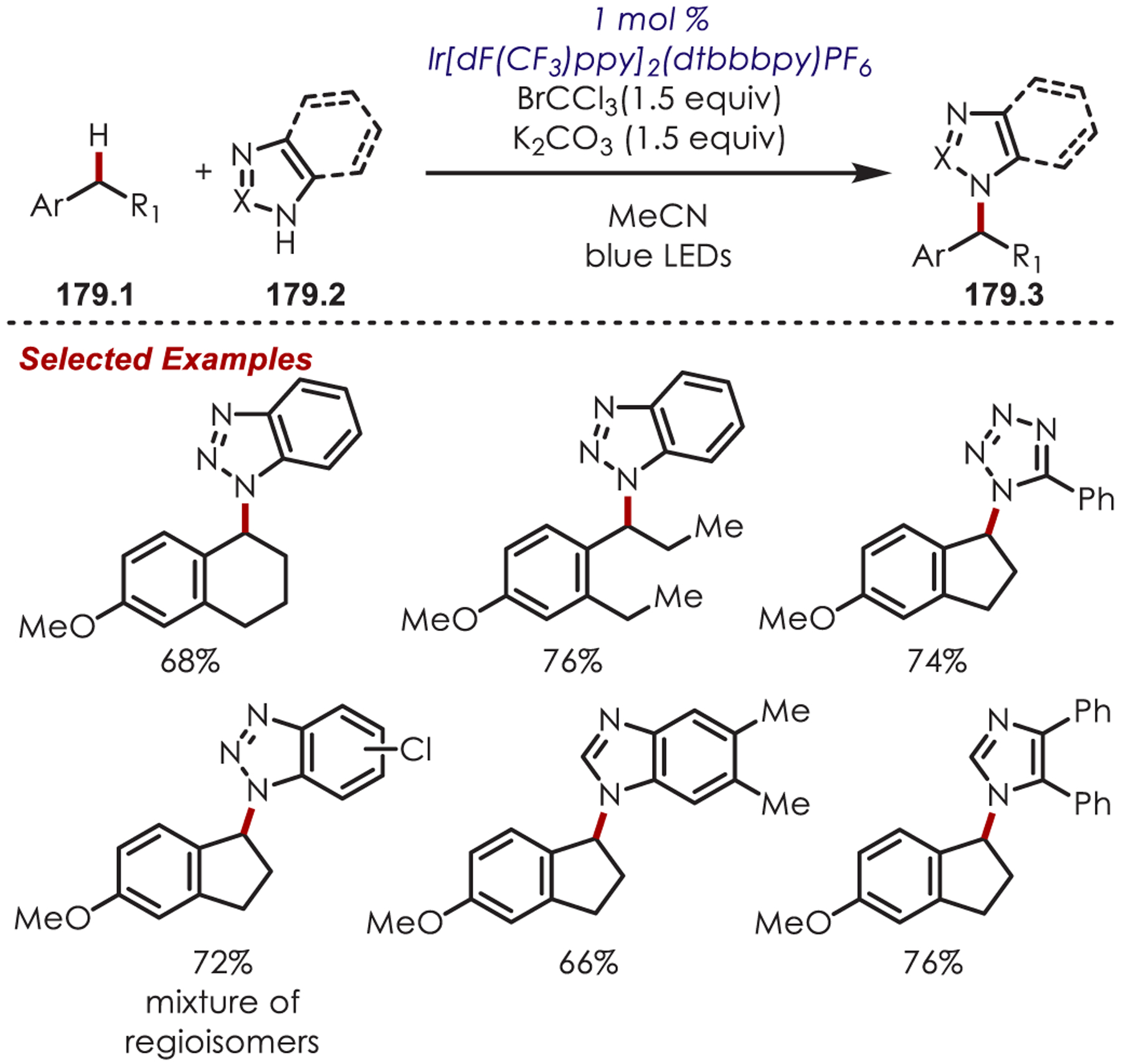
Benzylic Amination through the Nucleophilic Addition of Azoles to Benzylic Cations
These authors later reported the intramolecular cyclization of tethered amine or alcohol nucleophiles (180.1) onto photogenerated benzylic cations for the synthesis of heterocycles (Scheme 180).326 Selectfluor is utilized as an oxidative quencher and hydrogen atom abstractor. This methodology was highlighted in the total synthesis of (−)-codonopsinine (180.3) and (+)-centrolobine (180.4).
Scheme 180.
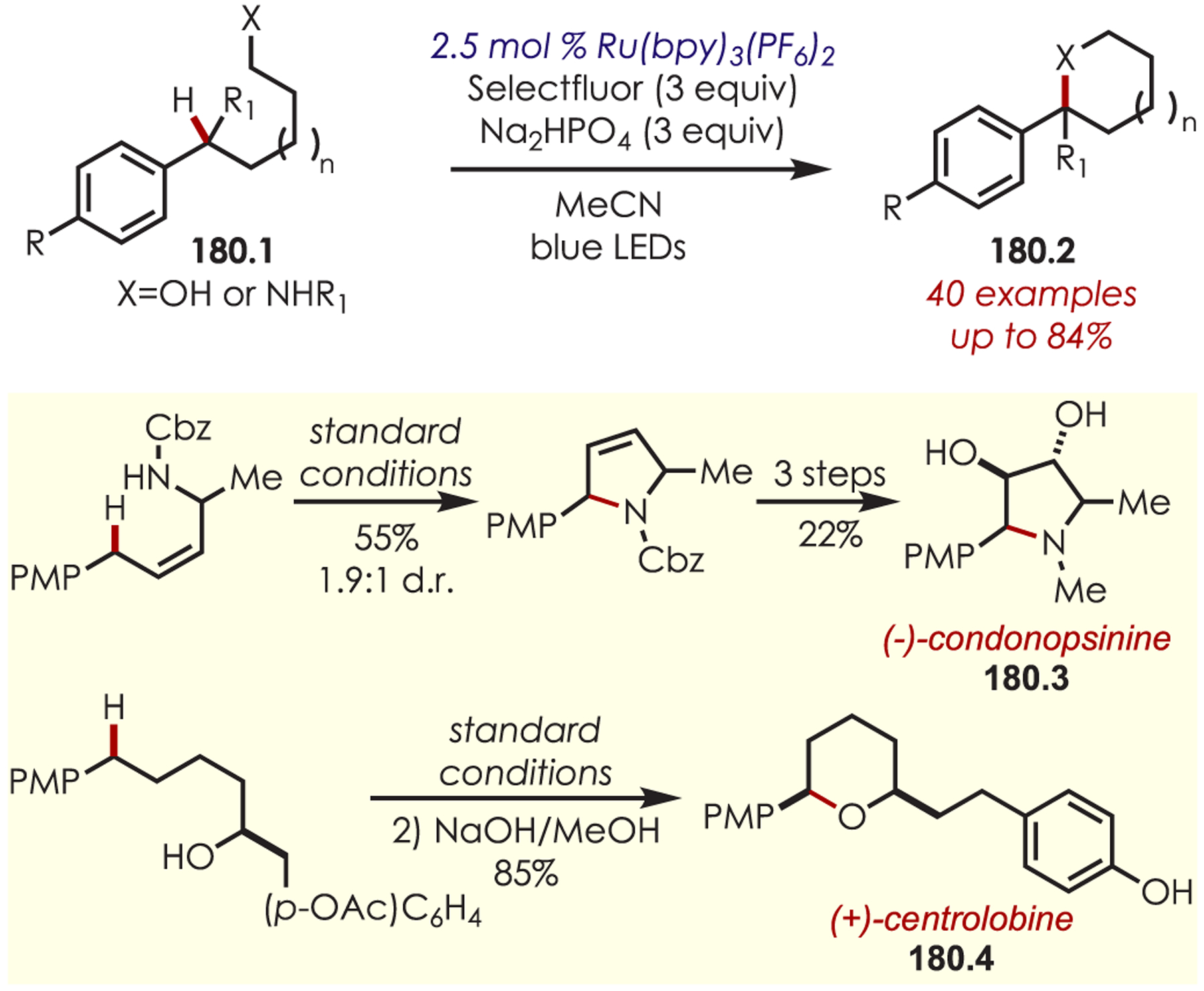
Intramolecular Benzylic Amination and Alkoxylation
In 2019, Yoon published a benzylic alkoxylation mediated by Cu(II) and an Ir(III) photoredox catalyst (Scheme 181).327 Cu(II) was used in both the oxidation of the benzylic radical to the cation and for Ir(III)* to Ir(IV). A wide range of alcohol nucleophiles (181.2) was compatible and gave the products in high chemoselectivity. Notably, this method is compatible for the late-stage functionalization of more complex molecules, including tocopherol, catechin, and methyl podocarpate, and with more complex alcohol nucleophiles such as amino acids, carbohydrates, and terpenes.
Scheme 181.
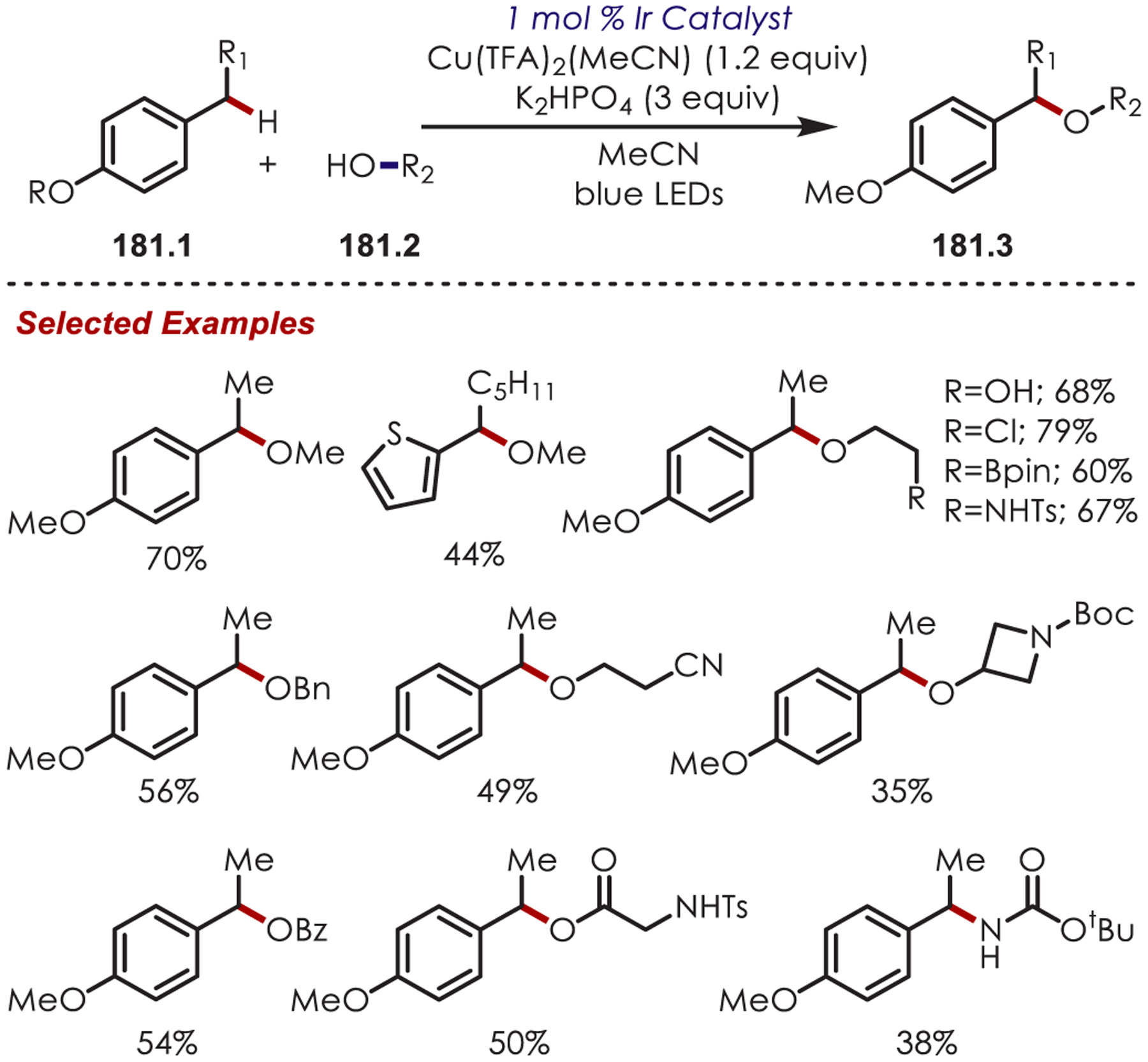
Benzylic Alkoxylation through Phosphate-Mediated Hydrogen Atom Abstraction
In 2020, a coupling of benzylic radicals with aldehydes (182.2) was reported for the synthesis of α-arylketones through a dual nickel and iridium photoredox catalytic cycle (Scheme 182).328 SET from a bromide anion to Ir(III) generates a bromine atom, which abstracts both benzylic and aldehydic C–H bonds. The acyl radical, 182.4, and benzyl radical are consecutively trapped by Ni(I)Br to give an intermediate Ni(III) species, 182.6, which undergoes reductive elimination to afford the final products, 182.3.
Scheme 182.
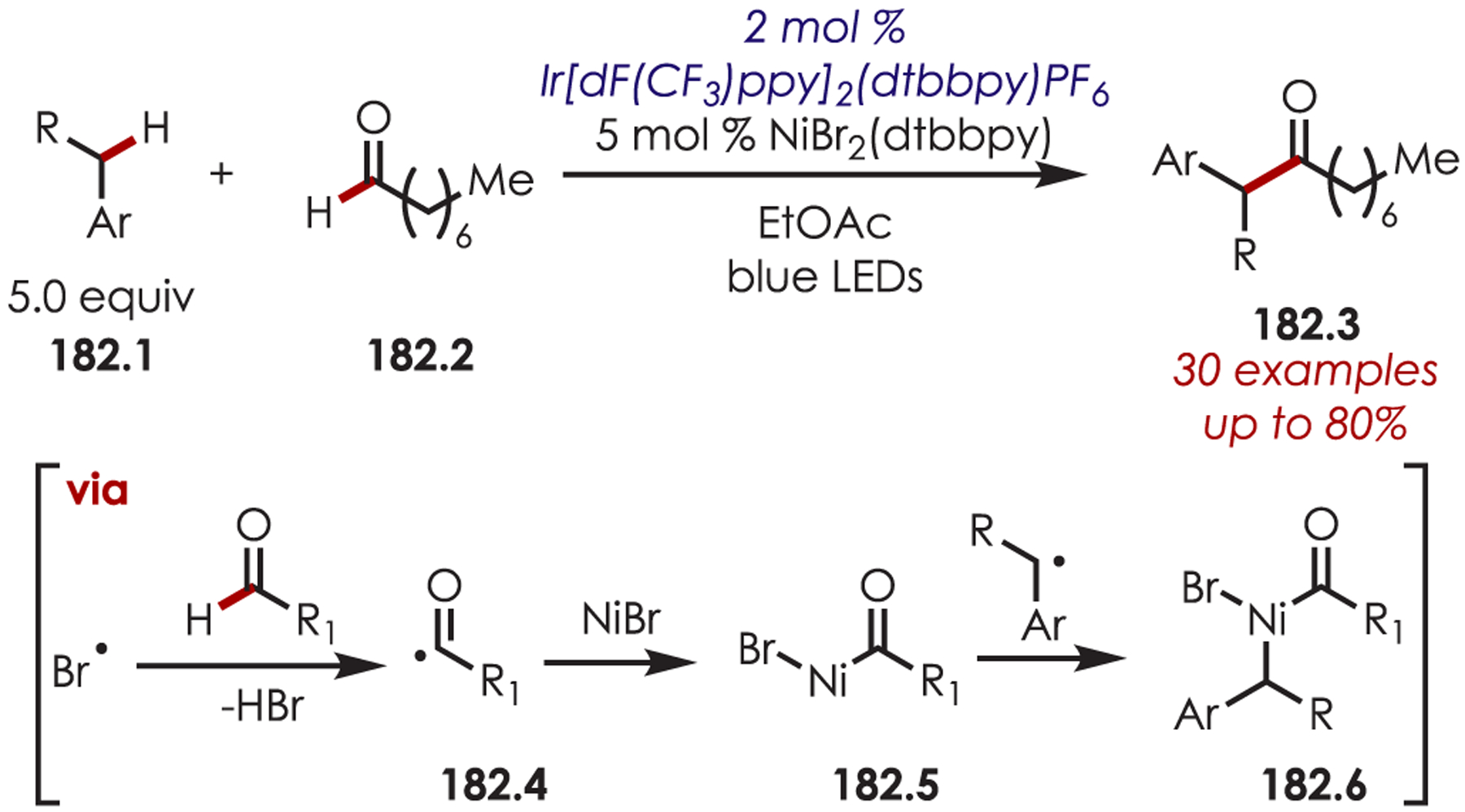
Benzylic and Aldehydic C–H Bond Coupling
A vinylic C–H functionalization of olefins, 183.2, with acyl chlorides, (183.1) was accomplished by Ngai and co-workers (Scheme 183).329 Experimental and computational studies suggest a PCET generates the benzylic radical 183.4, which undergoes addition into an olefin and a subsequent 1,3-chlorine atom shift producing intermediate 183.6. Oxidation by Ir(IV), deprotonation, and hydrolysis upon workup afforded the enone products.
Scheme 183.
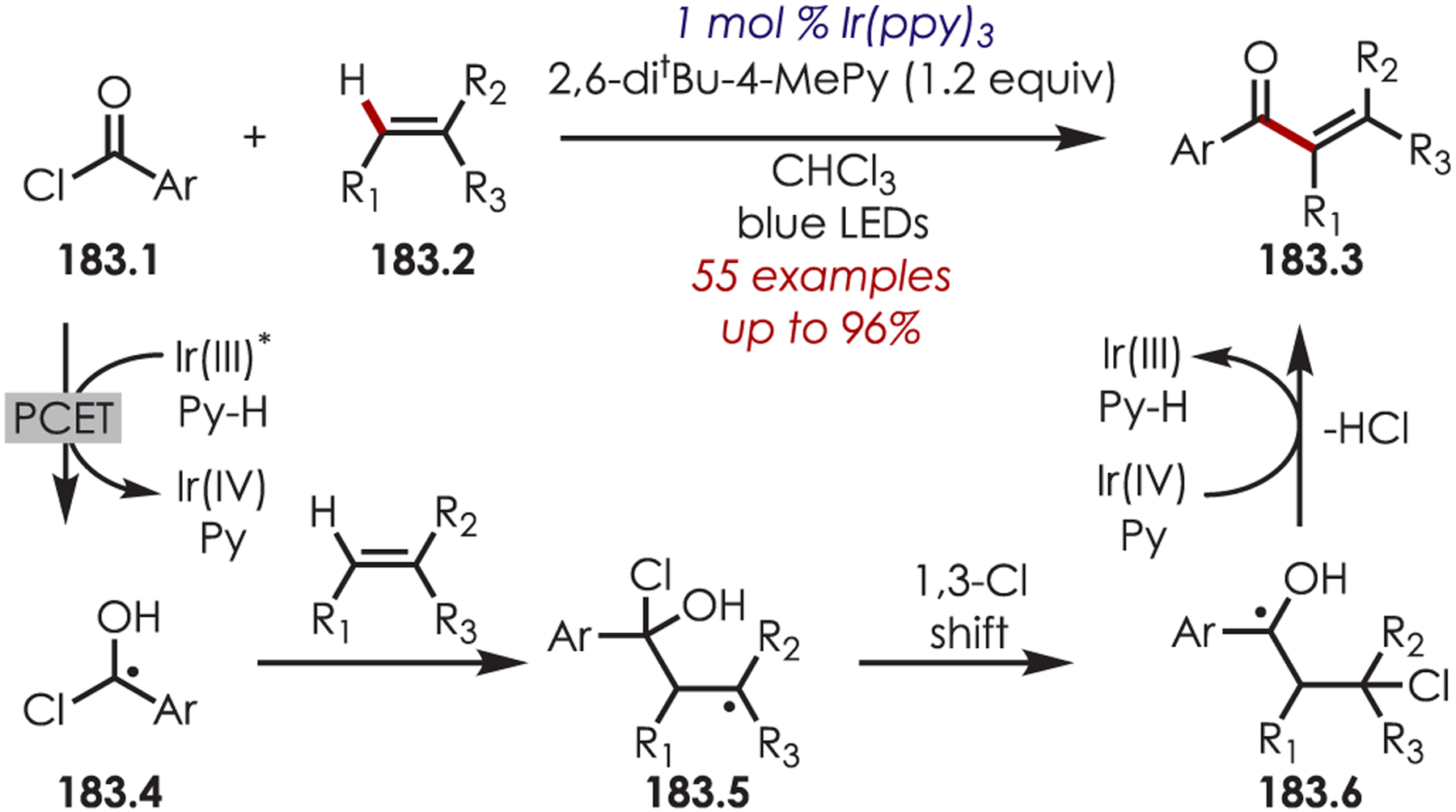
Vinylic C–H Functionalization with Acyl Chlorides
The prior examples of allylic C–H functionalization have generally relied on the union of nucleophilic allylic radicals with an electrophilic partners. However, the generation of an allylic cation through SET of the allylic radical would provide complementary reactivity for coupling with a nucleophilic partner. Recently, Hong and co-workers reported the thiolation of allylic C–H bonds via a HAT then oxidation to an allylic cation (184.3) and subsequent addition of a thiol nucleophile (Scheme 184).330 This produces allyl thioethers and alkyl allyl sulfides in moderate yields. The authors noted that an added base was required to prevent undesired hydrothiolation by immediately deprotonating the thiol.
Scheme 184.
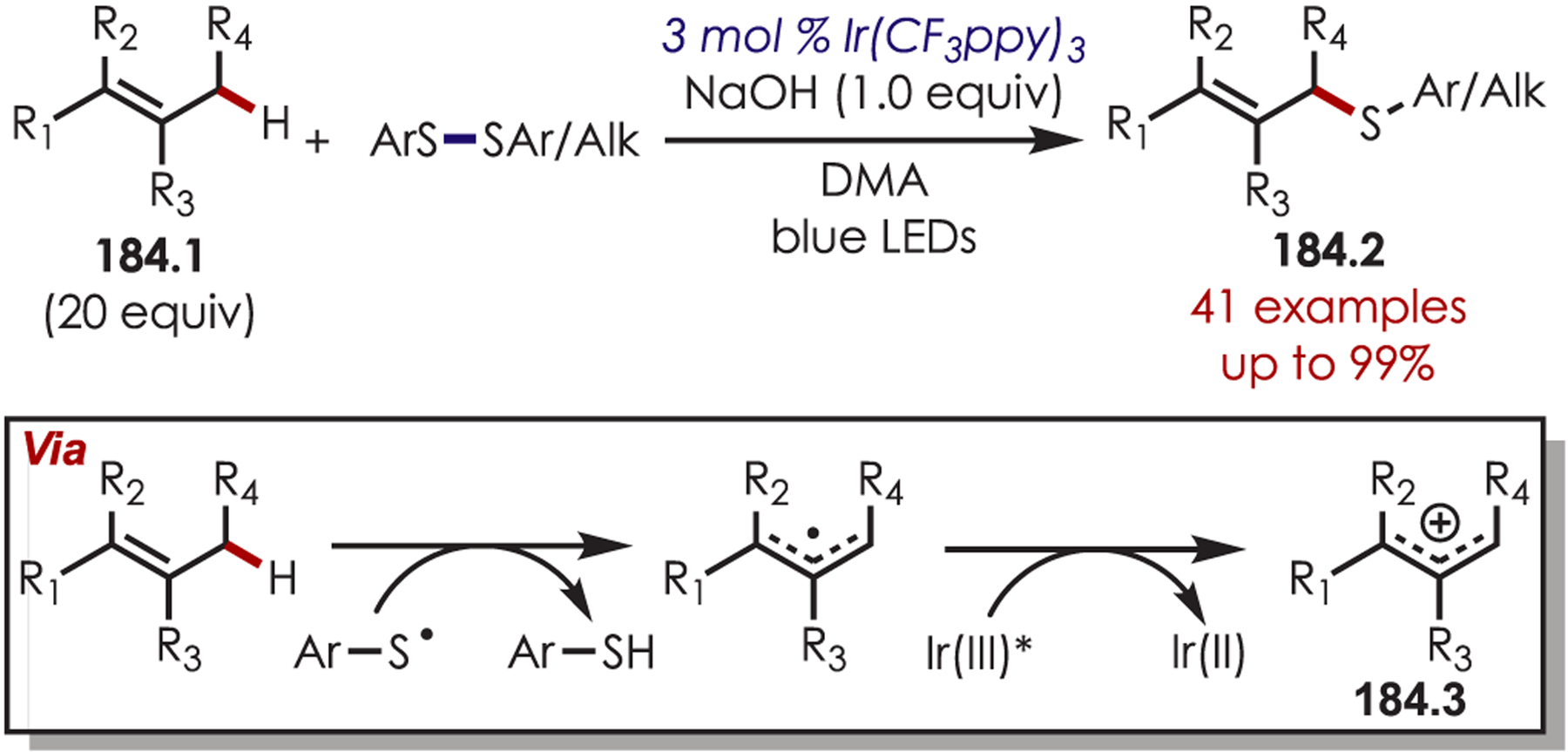
Allylic Thiolation via Allylic Cations
Via Radical Addition.
In 2012, a benzylic C–H carbocyclization was reported through an oxidative Ru(II) quenching mechanism to generate an aryl radical from the sulfonyl chloride (Scheme 185).331 Both electron-rich and electron-poor aryl sulfonyl chloride (185.1) coupling partners yielded the desired products with a diverse scope of the o-alkyl arylalkyne partner (185.2).
Scheme 185.
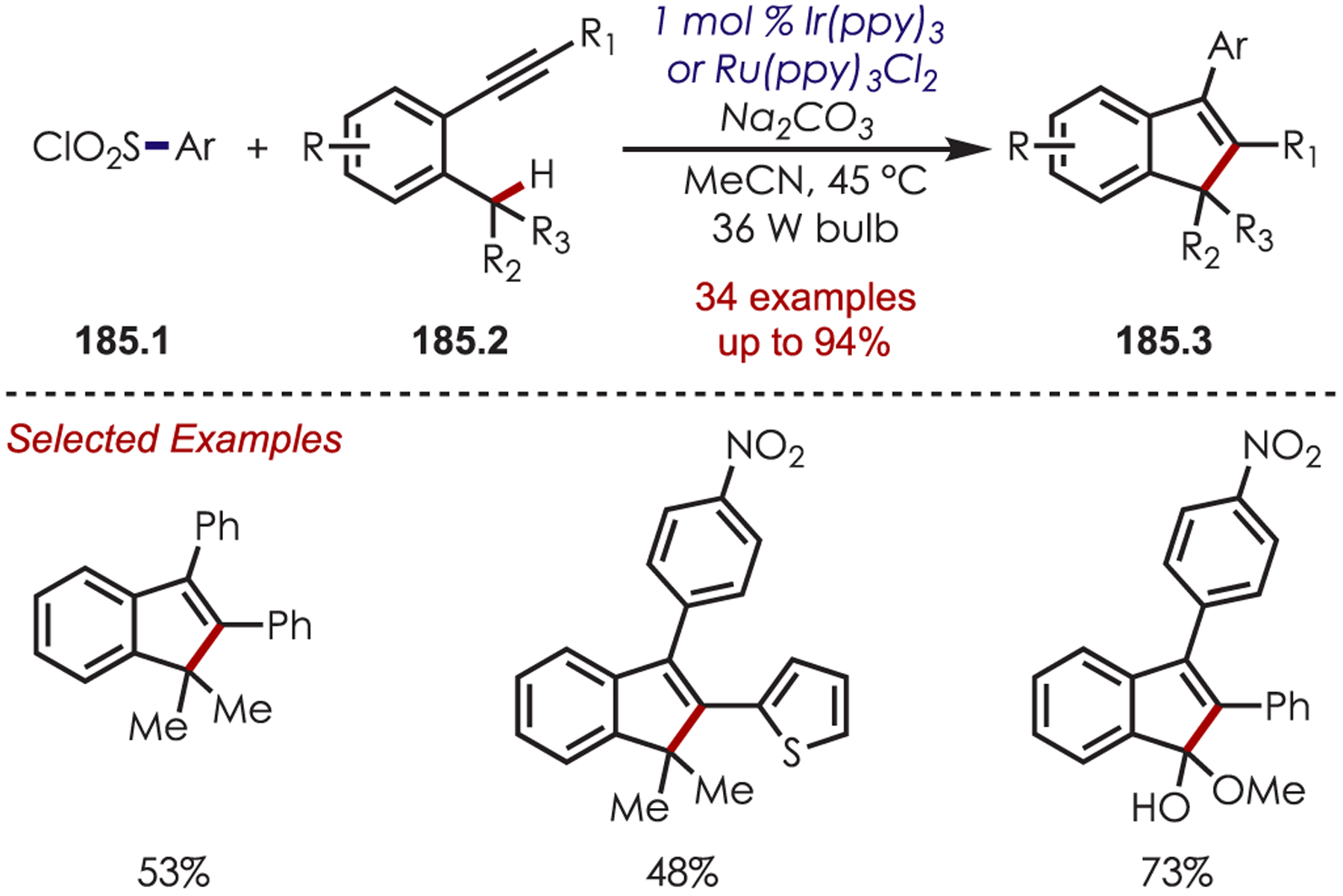
Benzylic C–H Carbocylization for the Synthesis of Indene Derivatives
Via Electron Transfer.
Electron-rich olefins can undergo photoinduced electron-transfer processes to generate the corresponding cation radical and subsequent deprotonation of the now acidic allylic C–H affords an allylic radical intermediate which can be intercepted by electrophiles. Glorius and co-workers utilized this general strategy in a reported diastereoselective allylation of aldehydes a by dual photoredox and chromium catalyst system to furnish homoallylic alcohols (Scheme 186).332 Reductive quenching of photoexcited Ir(III) generates an aryl radical cation radical (186.2) which is deprotonated to generate the allyl radical (186.3). A key allyl chromium intermediate (186.4) is formed from Cr(II) and adds to the aldehyde substrates via a Zimmerman–Traxler transition state that selectively affords the anti- allylation product (186.5).
Scheme 186.
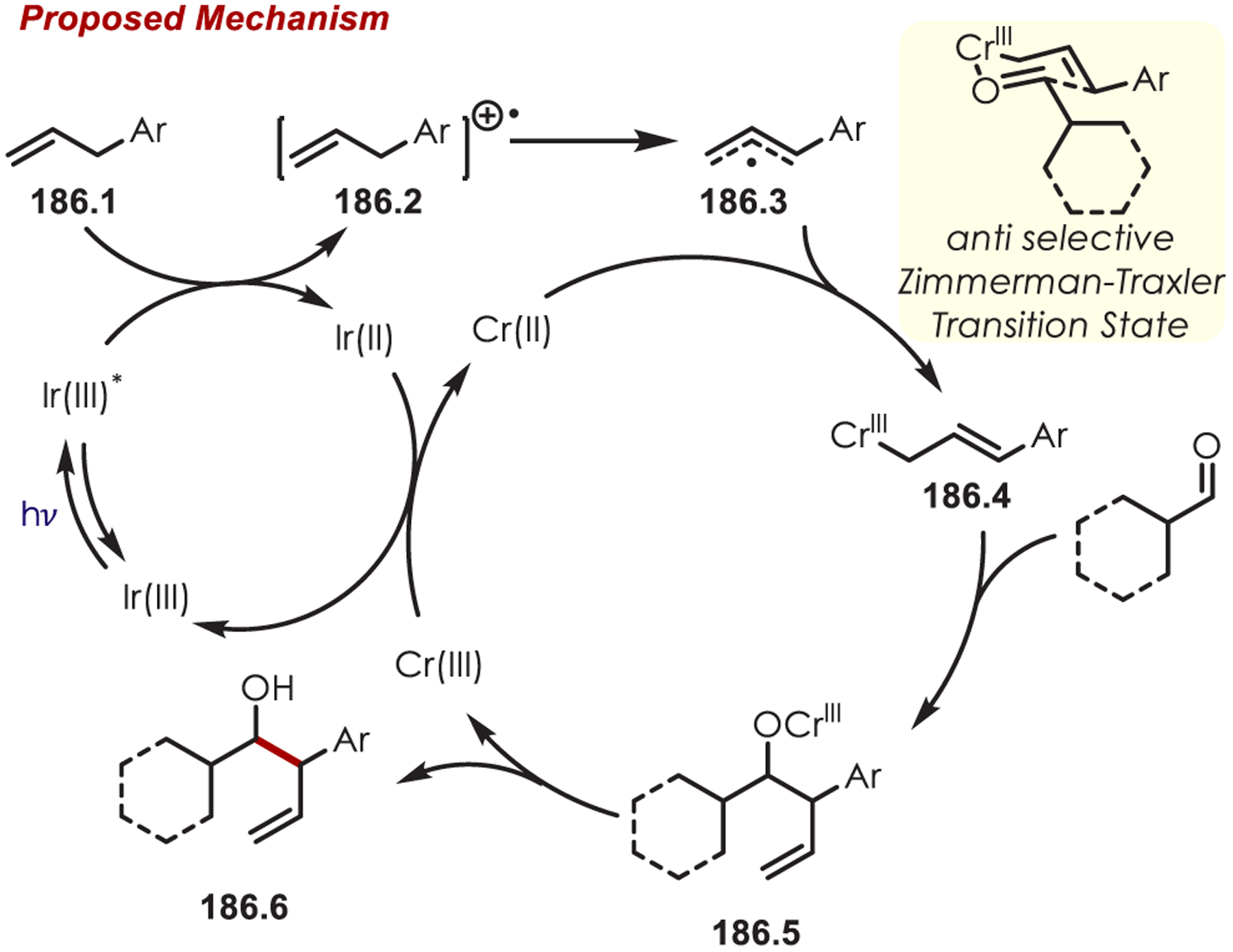
Mechanism of Diastereoselective Allylation of Aldehydes via Dual Photoredox and Chromium Catalysis
The scope of this asymmetric allylation utilizes allyl (hetero)arenes (187.2) including anisoles, indoles, carbazoles, and diarylamines with a diverse range of aliphatic or aldehyde (187.1) coupling partners (Scheme 187). This method was compatible in the late-stage functionalization of an epiandrosterone derivative which afforded the product in 68% yield and >19:1 dr.
Scheme 187.
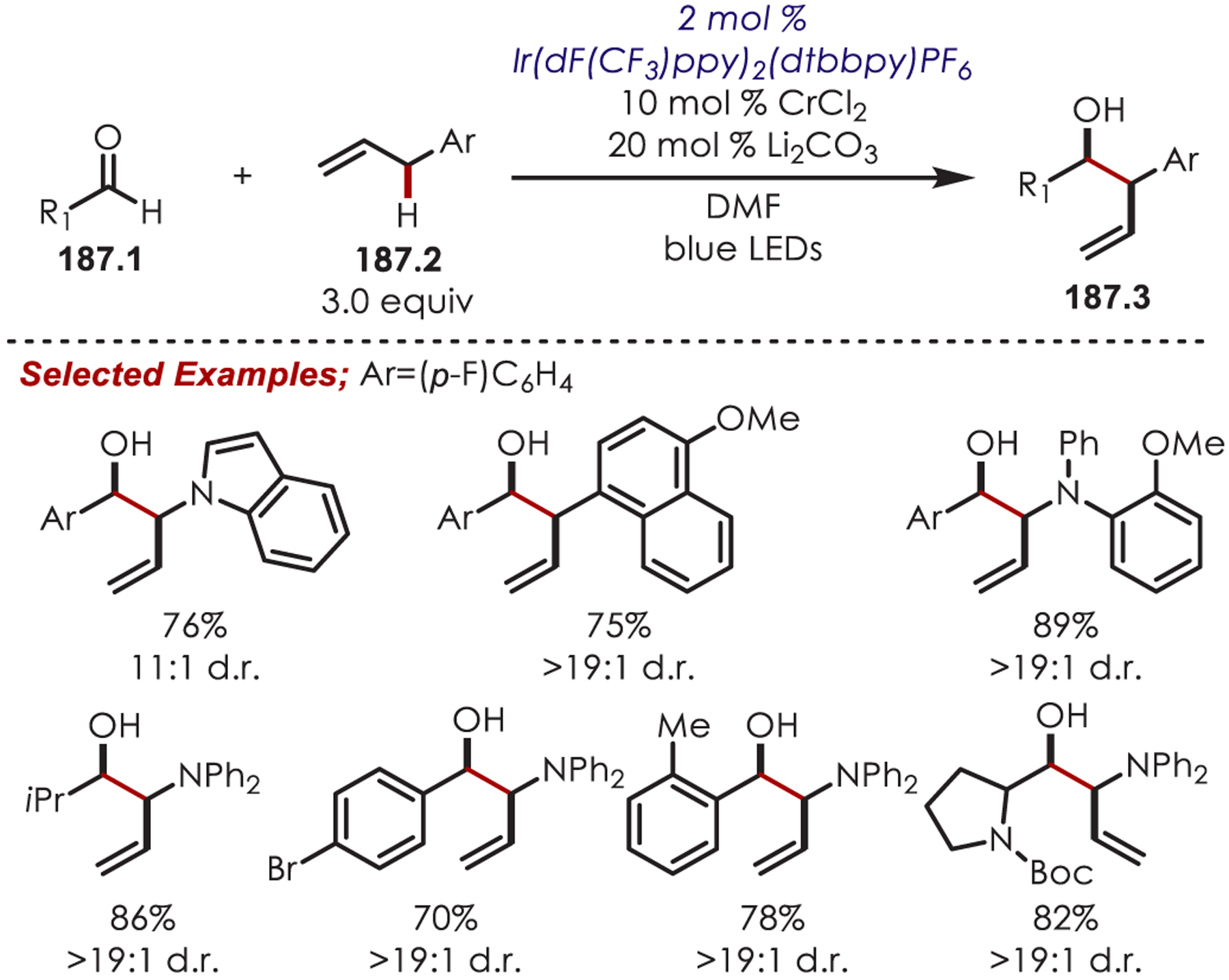
Scope of Diastereoselective Allylation of Aldehydes via Dual Photoredox and Chromium Catalysis
Recently, Glorius reported an extension of this methodology using enol ethers (188.2) as the allylic radical precursor for direct access to monoprotected homoallylic 1,2-diols (188.3;Scheme 188).333 The selectivity of this methodology gave a moderate preference for the anti-diastereomer, which is attributed to a conformational equilibrium of the γ-silyloxyallylchromium species from an intramolecular coordination between oxygen and chromium, which forms a five-membered intermediate.
Scheme 188.
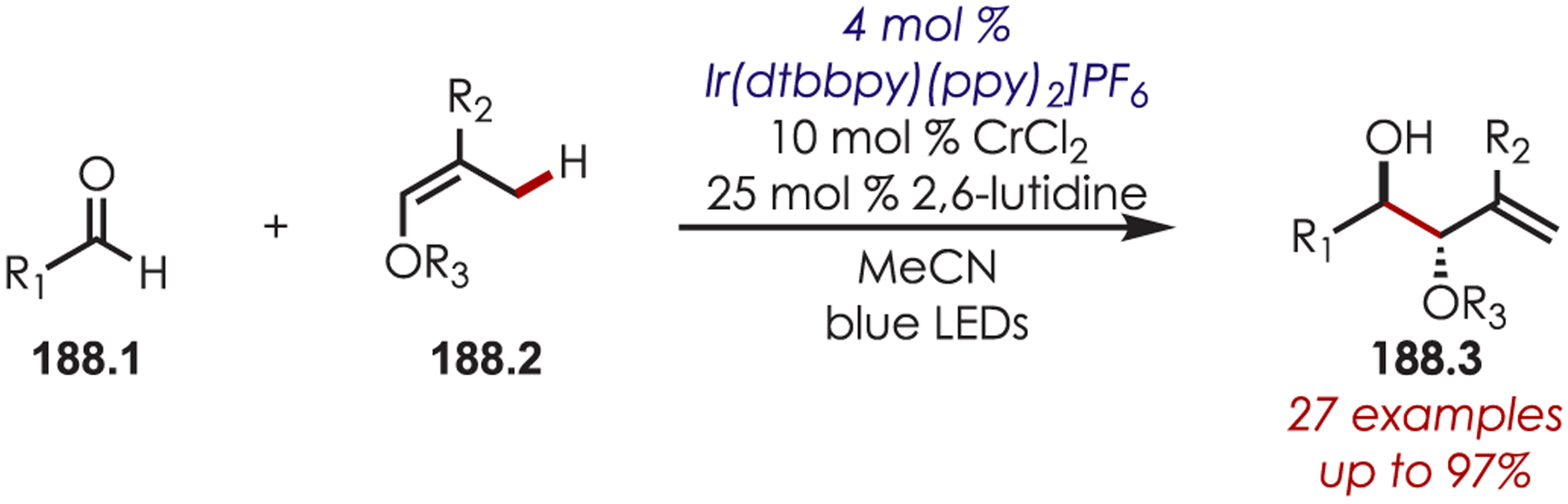
Diastereoselective Allylation of Aldehydes via Dual Photoredox and Chromium Catalysis Using Enol Ethers
In 2019, Ooi reported a direct C–H allylation of silyl enol ethers (189.1) by generation of an allylic radical via oxidative SET and deprotonation (Scheme 189).334 Subsequent trapping of the nucleophilic radical with a range of electron-deficient alkenes (189.2) was demonstrated. A broad scope of cyclic and acyclic ketones afforded the β-alkylated ketone derivatives.
Scheme 189.
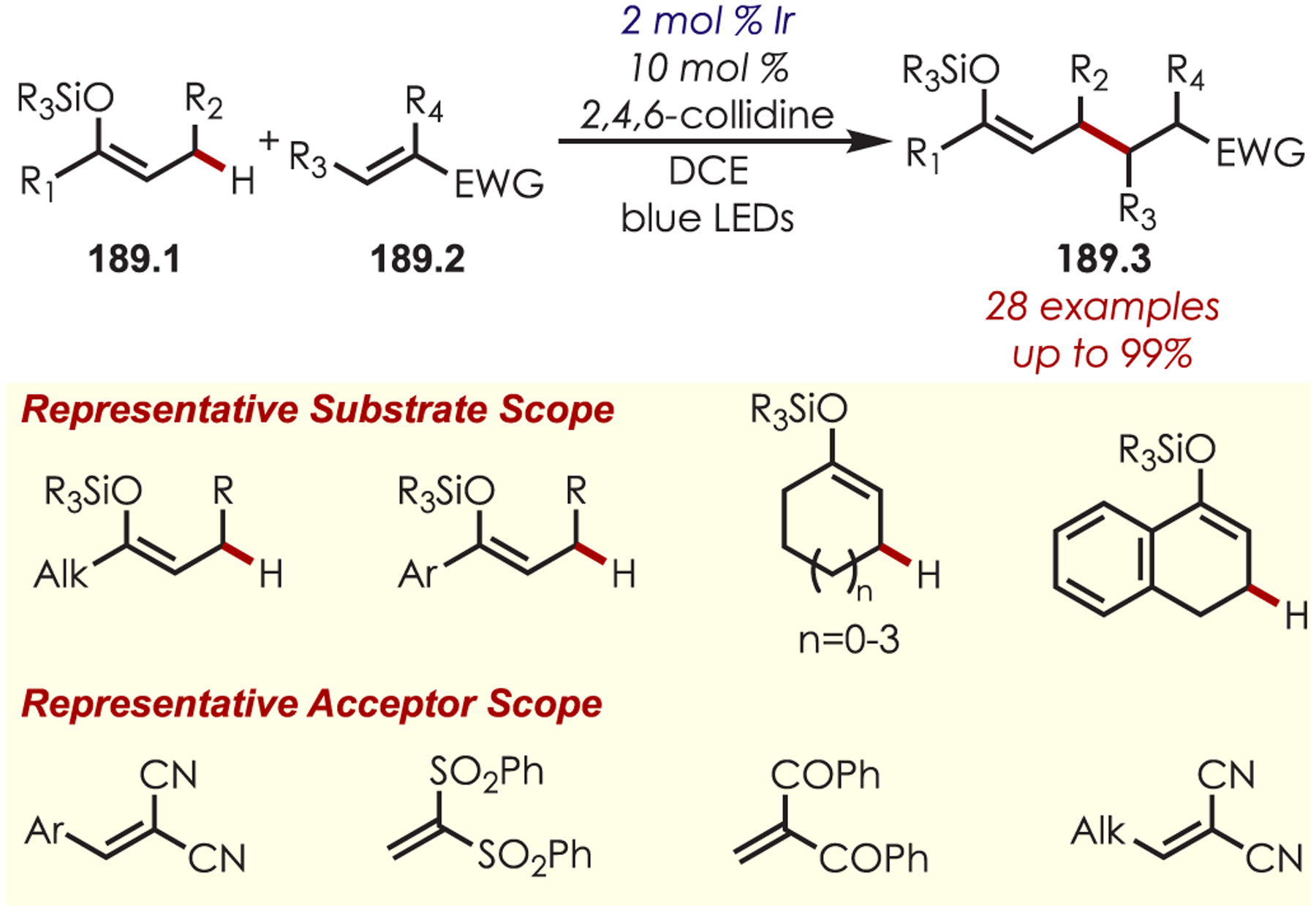
Allylic Alkylation of Silyl Enol Ethers
2.2.4. Unactivated sp3 C–H Functionalization.
The activation of typically unreactive C–H bonds by photoredox catalysis manifolds is generally accomplished through either a hydrogen atom abstraction event, which is difficult to accomplish with high selectivity and generality, or through Hofmann–Löffler–Freytag-type 1,5-hydrogen atom transfers of amide derivatives.
Via Hydrogen Atom Abstraction.
Hydrogen atom transfer catalysts are utilized for the abstraction of unactivated sp3 C–H bonds distal to functional group handles (BDE ~ 96–98 kcal mol).335 The challenge is to select these generally unreactive C–H bonds over allylic, benzylic, or C–H bonds α-to heteroatoms (BDE ~ 88.8, 89.7, and ~92 kcal/mol, respectively).
Early reports of remote sp3 C–H functionalization were accomplished through hydrogen atom transfer events for aliphatic fluorination. Inspired by prior non-photoredox oxidative fluorination of alkanes,336–338 Britton developed a method to generate and trap alkyl radicals using a decatungstate photocatalyst and NFSI (Scheme 190).339,340 This proceeds through a hydrogen abstraction from the excited polyoxometalate catalyst to generate the alkyl radical that is subsequently trapped with fluorine. The steric bulk and redox potential of the catalyst is proposed to functionalize C–H bonds that mimic in vivo oxidative metabolism by abstracting less congested C–H bonds while avoiding destabilizing interactions. While this provides a more direct route to functionalizing remote C–H bonds, the scope of this chemistry was limited. Later, Britton extended this to18F-fluorination using [18F]NFSI341,342 and found that a cationic ammonium forms a precursor complex with the decatungstate catalyst which significantly increases the rate of C–H abstraction.268 Uranyl photoredox catalysts have also been utilized in aliphatic C–H fluorinations with NFSI.343
Scheme 190.
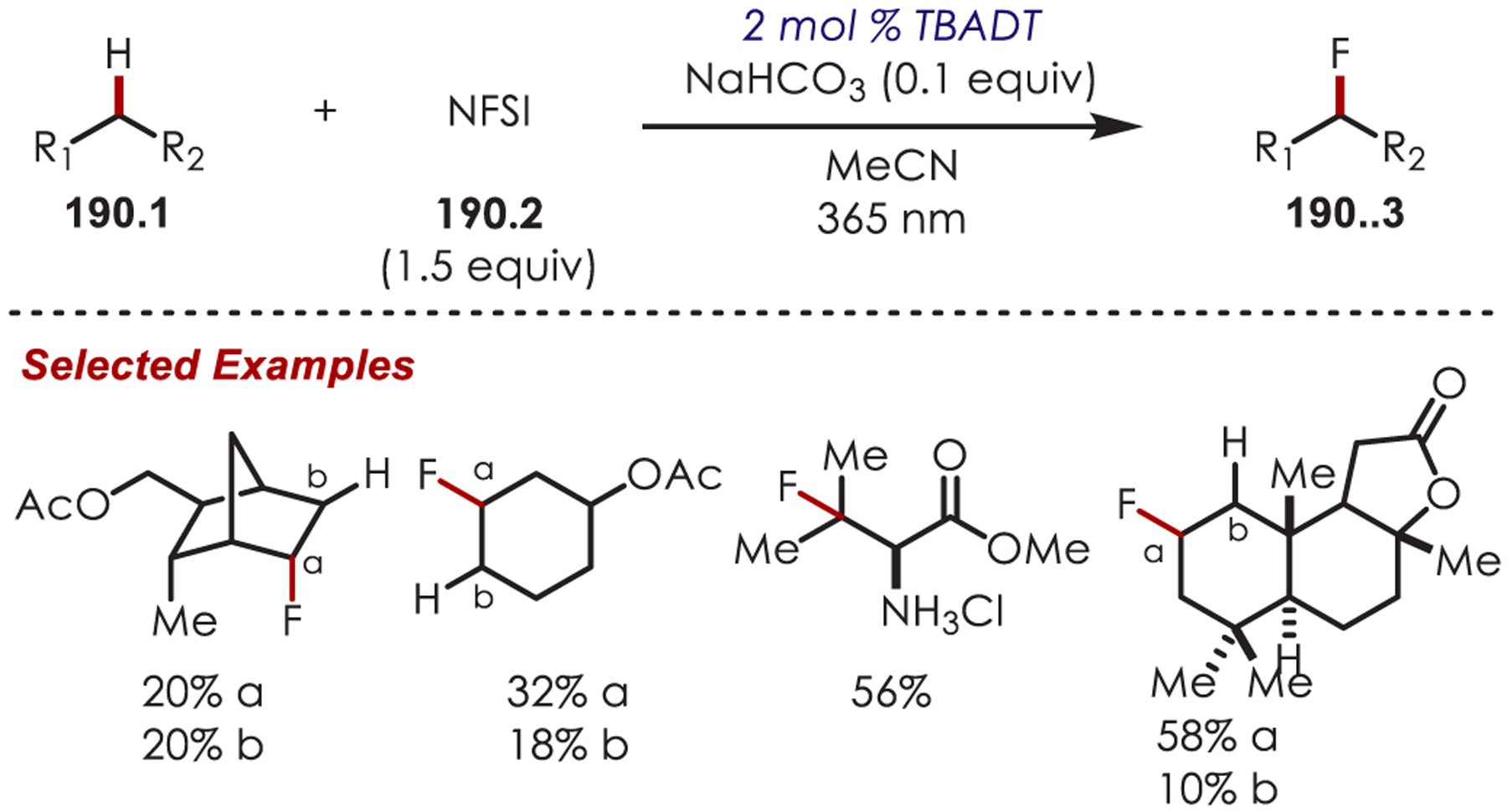
C–H Fluorination Remote from Functional Groups Using TBADT as a Photoredox and HAT Catalyst
TBADT-catalyzed C–H functionalizations have also been utilized for the coupling of olefins (191.2) by Ryu (Scheme 191).344–346 Additionally, uranyl-catalyzed direct C–H abstraction of cycloalkanes, ethers, acetals, and amides for generation of alkyl radicals and subsequent trapping with electron deficient alkenes was reported by Ravelli.347
Scheme 191.
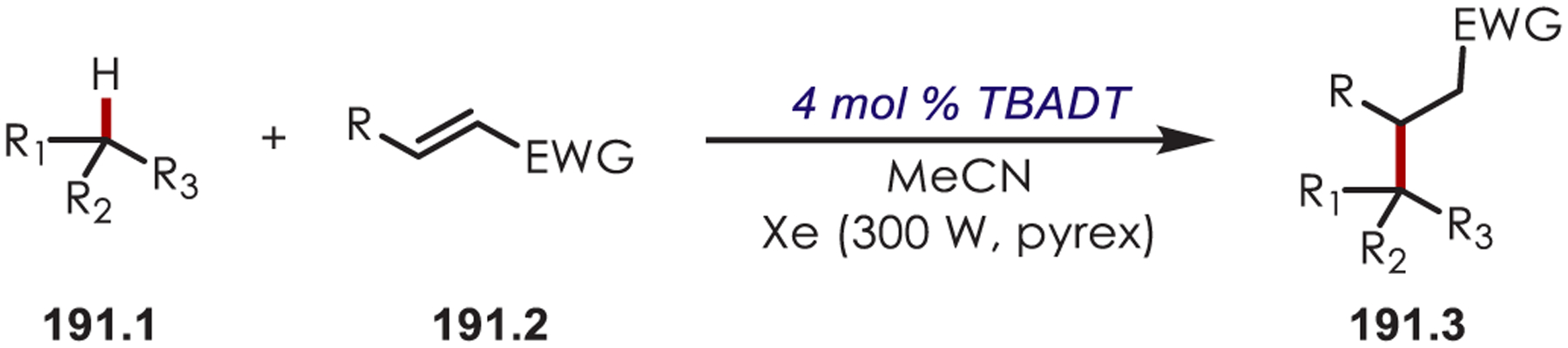
C–H Alkylation Remote from Functional Groups Using TBADT as a Photoredox and HAT Catalyst
In 2018, a seminal communication by Zuo and co-workers demonstrated the use of hydrocarbon gas feedstocks (192.1) such as methane, ethane, propane, and butane in alkylation, arylation, and amination reactions using cerium salts as catalysts (Scheme 192).348 Abstracting alkoxy radicals were generated in situ from a photoinduced LMCT of Ce(IV) alkoxides generated from Ce salts and simple alcohols. The alkoxy radical served as a hydrogen atom transfer catalyst to produce an alkyl radical that was trapped with electron-deficient alkenes, arenes and di-tert-butyl azodicarboxylate. While hydrocarbons with 2° C–H bonds (propane and butane) gave rise to an approximate statistical mixture of regioisomers using 2,2,2-trichloroethanol as the co-catalyst, switching to methanol as the alcohol co-catalyst gave up to 8:1 regioselectivity for 2° C–H amination when employing an azodicarboxylate electrophile (192.7).
Scheme 192.
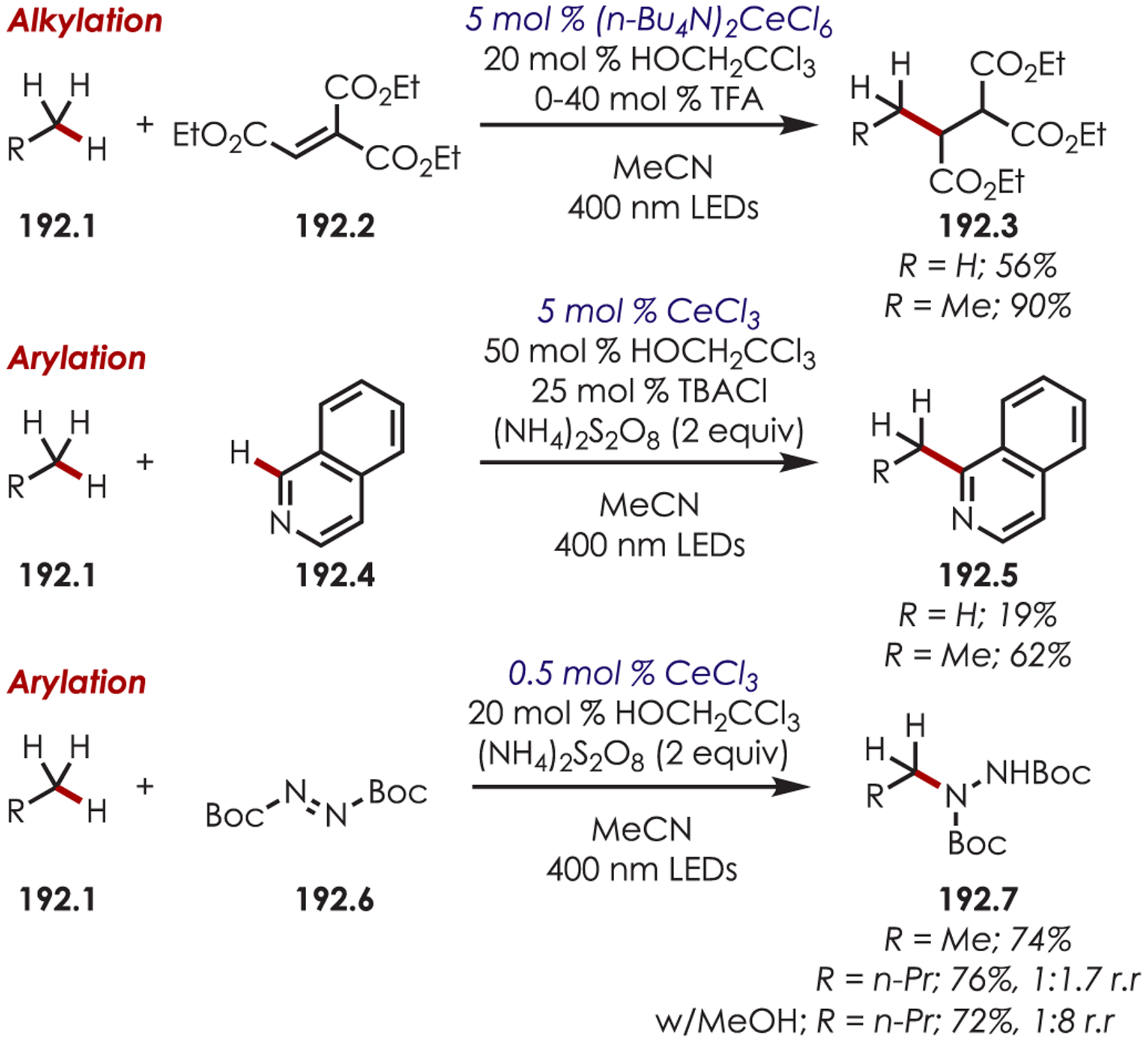
Hydrocarbon C–H Functionalization Using Cerium Salts
The following year, Zuo reported an extension to the cerium-catalyzed C–H functionalization of alkanes with alcohols serving as the HAT reagent (Scheme 193).349 Mechanistic studies confirmed that an LMCT excitation, followed by a rapid bond homolysis generates the alkoxy radical. Diphenylantracene was used as an additive to improve turnover of the cerium catalytic cycle. This was demonstrated in an amination using di-tert-butyl azodicarboxylate with cyclic alkanes and an alkylation of cyclohexane with electron-deficient alkenes.
Scheme 193.
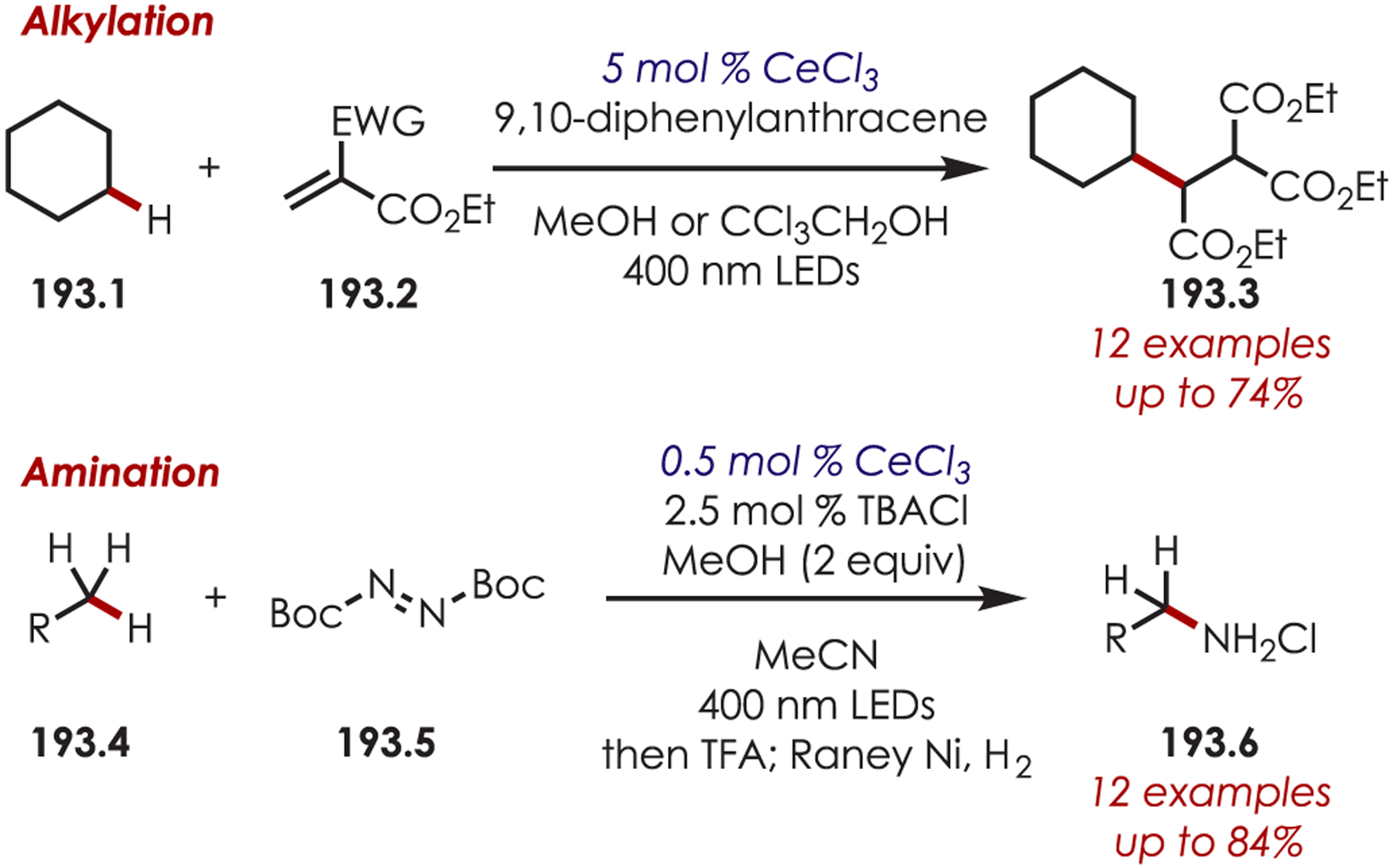
C–H Alkylation and Amination of Alkanes Using Cerium Salts
Noël accomplished the TBADT-catalyzed alkylation of gaseous hydrocarbons (methane, ethane, propane, and isobutane, 194.1) with electron-deficient alkenes in good yields and high selectivity in continuous flow (Scheme 194).350 A commercially available flow reactor, Vaportec UV-150 photochemical flow reactor, was used. Malononitriles, α-cyanocinnamate, triethyl ethylenetricarboxylate, N-phenylmaleimide, and 3-methylene-2-norbornanes were suitable radical acceptors.
Scheme 194.
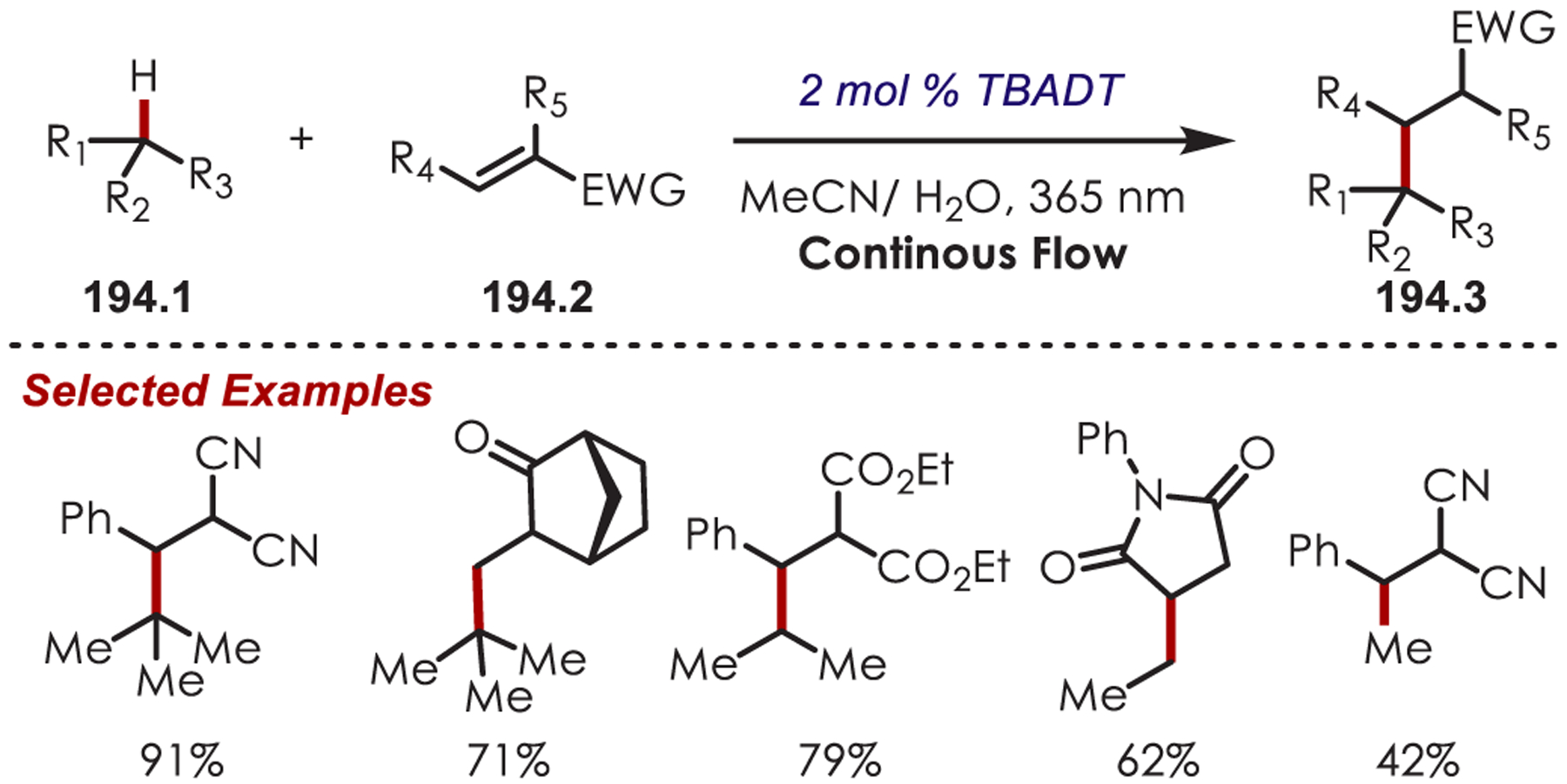
Hydrocarbon C–H Alkylation Using TBADT in Flow
In 2018, MacMillan reported a C–H arylation via dual decatungstate and nickel catalysis (Scheme 195).351 The alkyl radical, formed from hydrogen atom abstraction, was intercepted by nickel and ultimately generates a range of (hetero)arylated products (195.3). Cyclic and acyclic alkanes were compatible, however, in some cases giving a mixture of regioisomers. A diverse range of aryl and heteroaryl bromide coupling partners were explored including pyridyl, pyrimidyl, and thiazole derivatives. Additionally, the authors demonstrated this method for the natural product epipatidine, which was synthesized in two steps with a 28% yield.
Scheme 195.
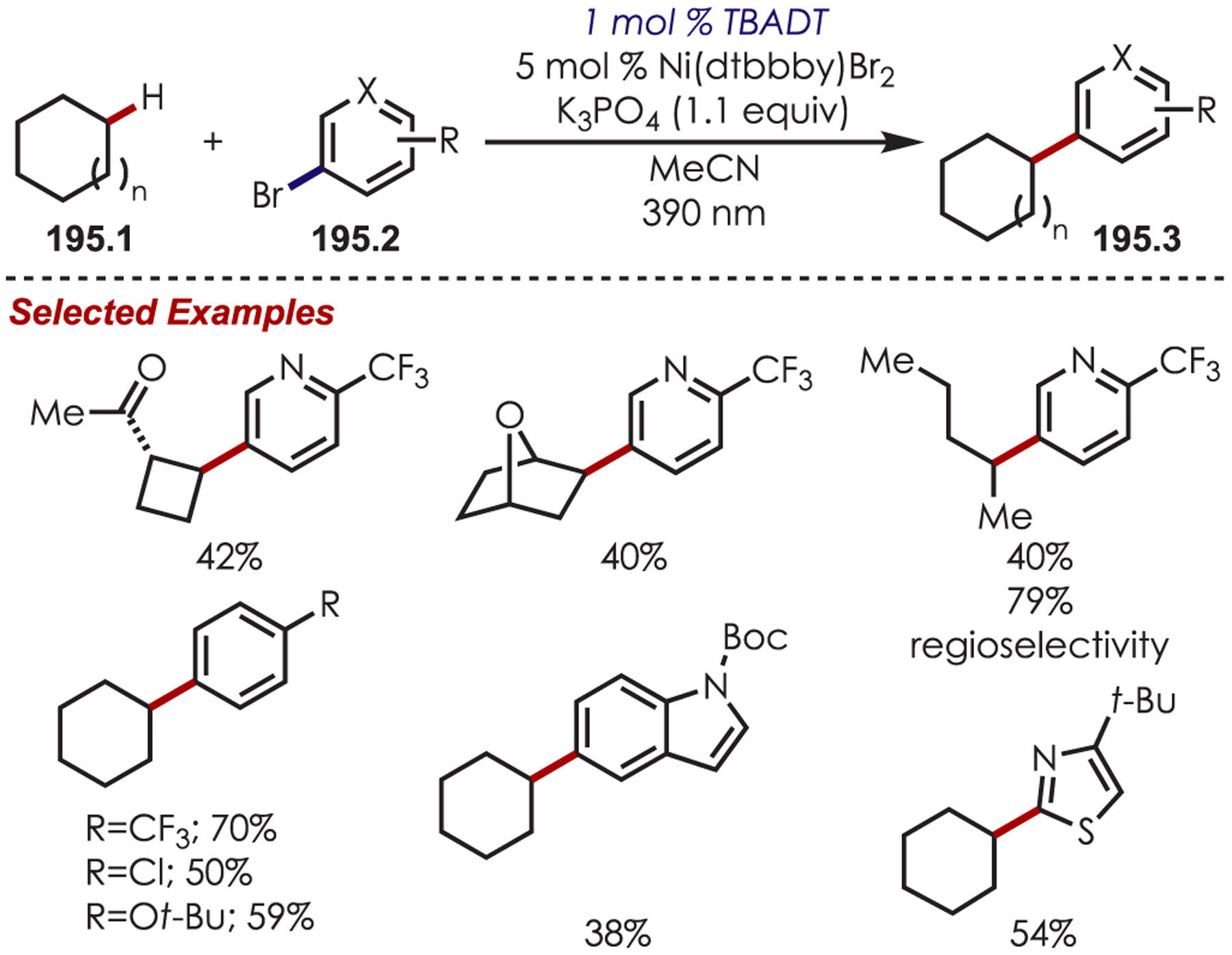
C–H Coupling with (Hetero)aryl Bromides Remote from Functional Groups Using Dual TBADT and Nickel Catalysis
Doyle has also reported a C–H acylation with acid chlorides via nickel catalysis that proceeds via the intermediacy of chlorine radicals as hydrogen atom abstractors for the synthesis of ketones (196.3; Scheme 196).352 This strategy abstracts unactivated C–H bonds (196.1) as well as benzylic and α-heteroatom C–H bonds. While this affords useful ketone adducts, site selectivity remains a challenge as mixtures of regioisomers were often observed.
Scheme 196.
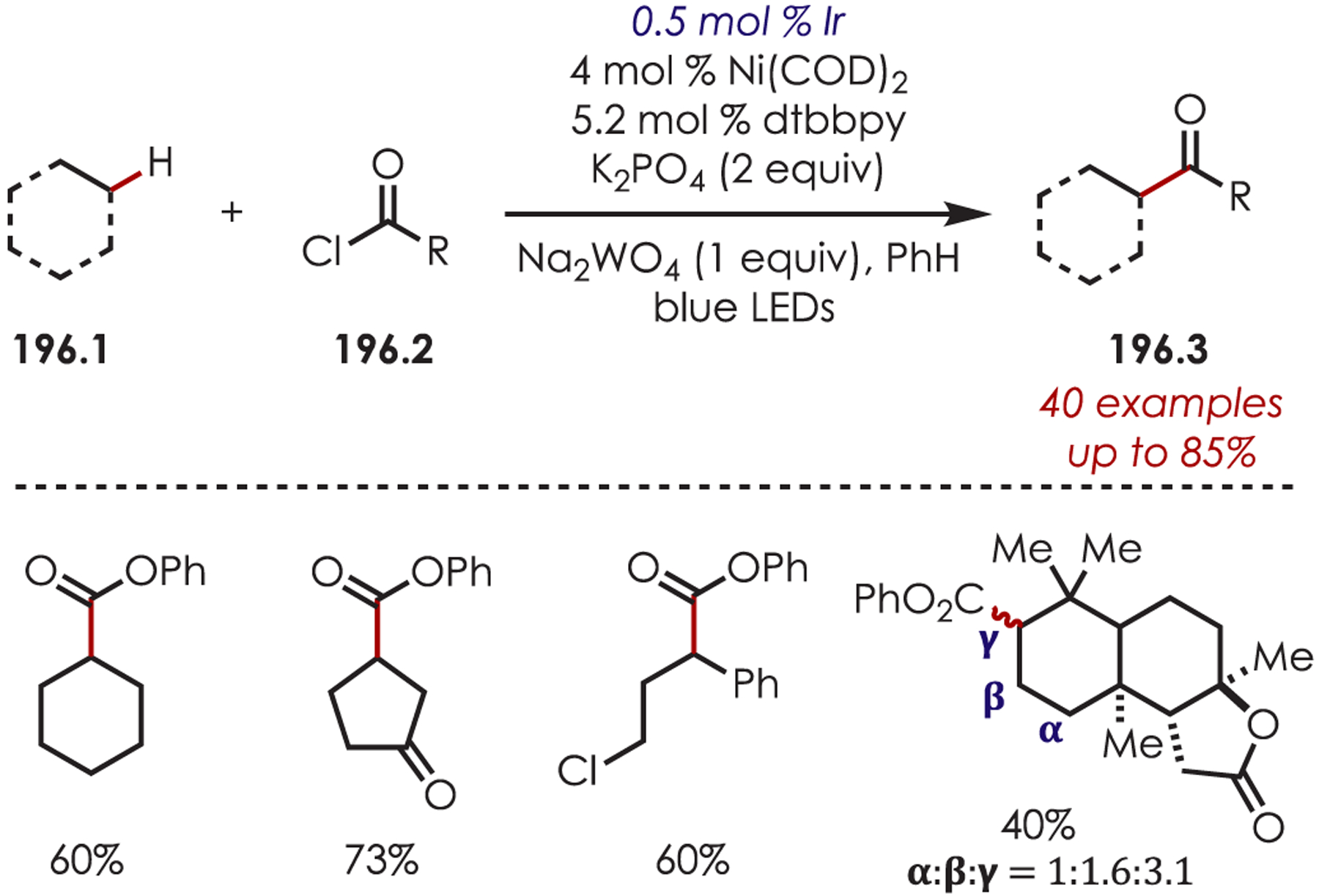
Remote C–C Bond Formation Using Dual Nickel and Iridium Photoredox through Chloride Radical HAT
Additionally, MacMillan has demonstrated the use of decatungstate and copper catalysis for the trifluoromethylation of unactivated alkyl C–H bonds (Scheme 197).353 Decatungstate undergoes predictable selective hydrogen atom abstraction at the most hydridic and sterically accessible C–H bond,354 giving substitution at the site most distal to an electron-withdrawing group. In the case of amine substrates, protonation renders the C–H bonds adjacent to the ammonium less hydridic and therefore less susceptible to abstraction. Copper is proposed to generate a Cu(II)-CF3 intermediate that is catalytically active and involved in the key bond-forming step. A broad range of substrates (197.1) was alkylated under these conditions, including amines and alkanes, which were applicable to benzylic functionalization.
Scheme 197.
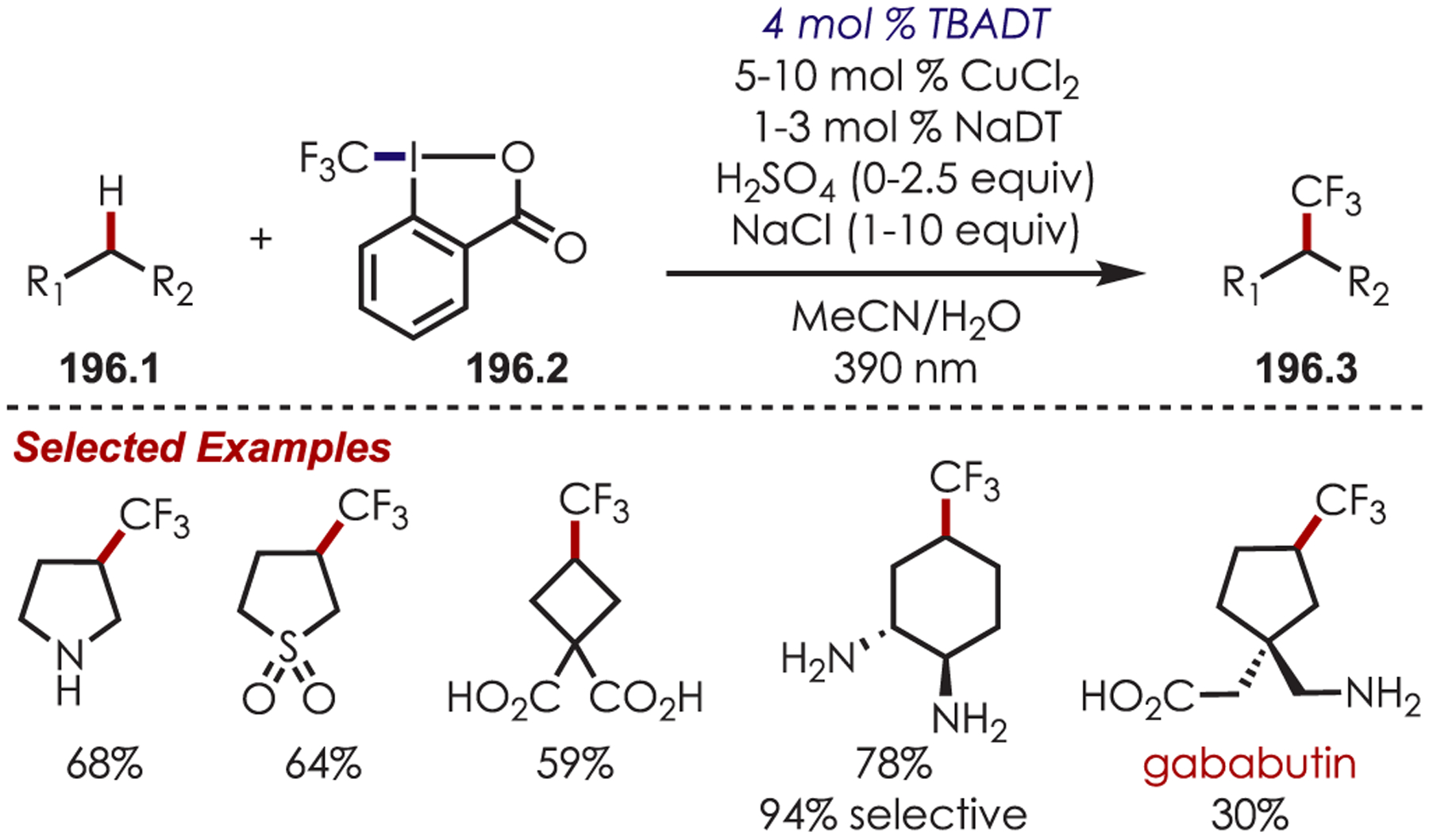
Remote C–H Trifluoromethylation through Dual TBADT and Copper Catalysis
Recently, the coupling of aldehydes and alkyl radicals via a reductive radical-polar crossover pathway was reported by utilizing CrCl3 to generate nucleophilic organochromium carbanions in situ (Scheme 198).355 Cyclic alkanes were functionalized with aryl aldehyde partners. Additionally, an aminoalkylation and oxoalkylation through the generation of α-hetero radicals was demonstrated.
Scheme 198.
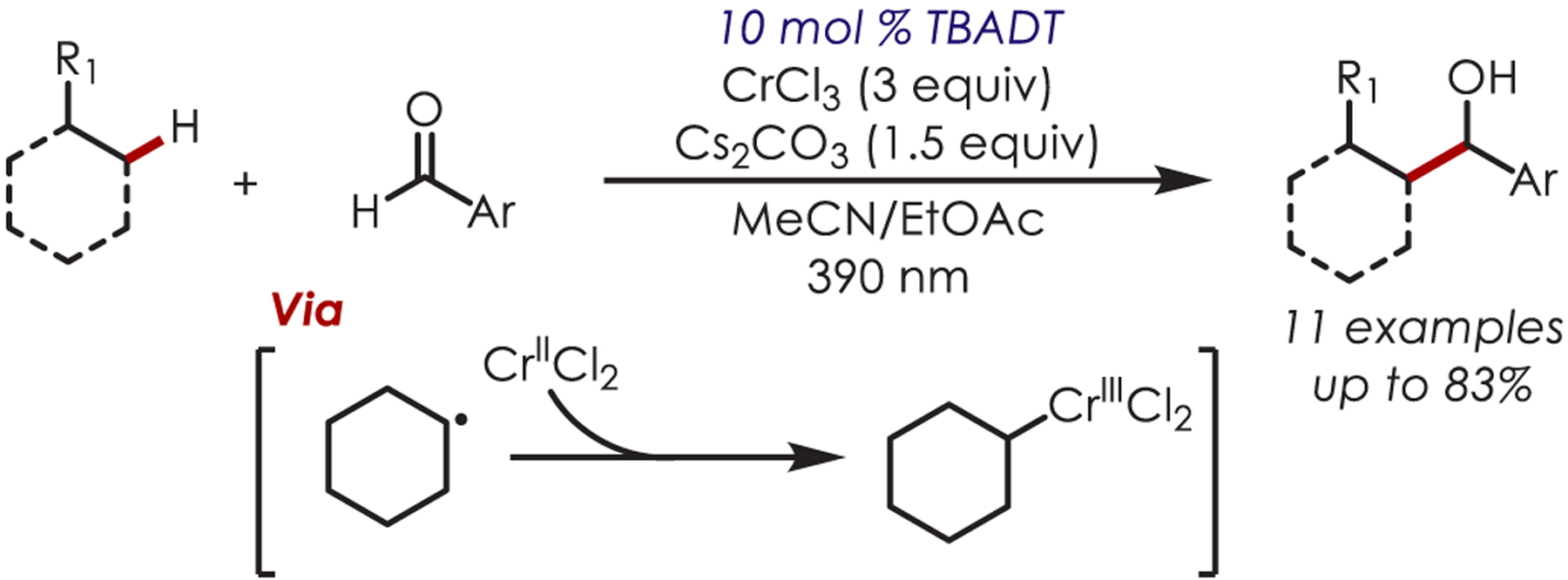
Remote Radical C–H Coupling with Aldehydes through the Generation of Nucleophilic Organochromium Carbanions
Wang and co-workers utilized decatungstate with a chiral phosphoric acid to achieve an asymmetric C–H alkylation (Scheme 199).356 The chiral phosphoric acid plays a critical role as a proton transfer shuttle during tautomerization of the enol to the ketone and provides a method for asymmetric protonation of the enol tautomer.
Scheme 199.
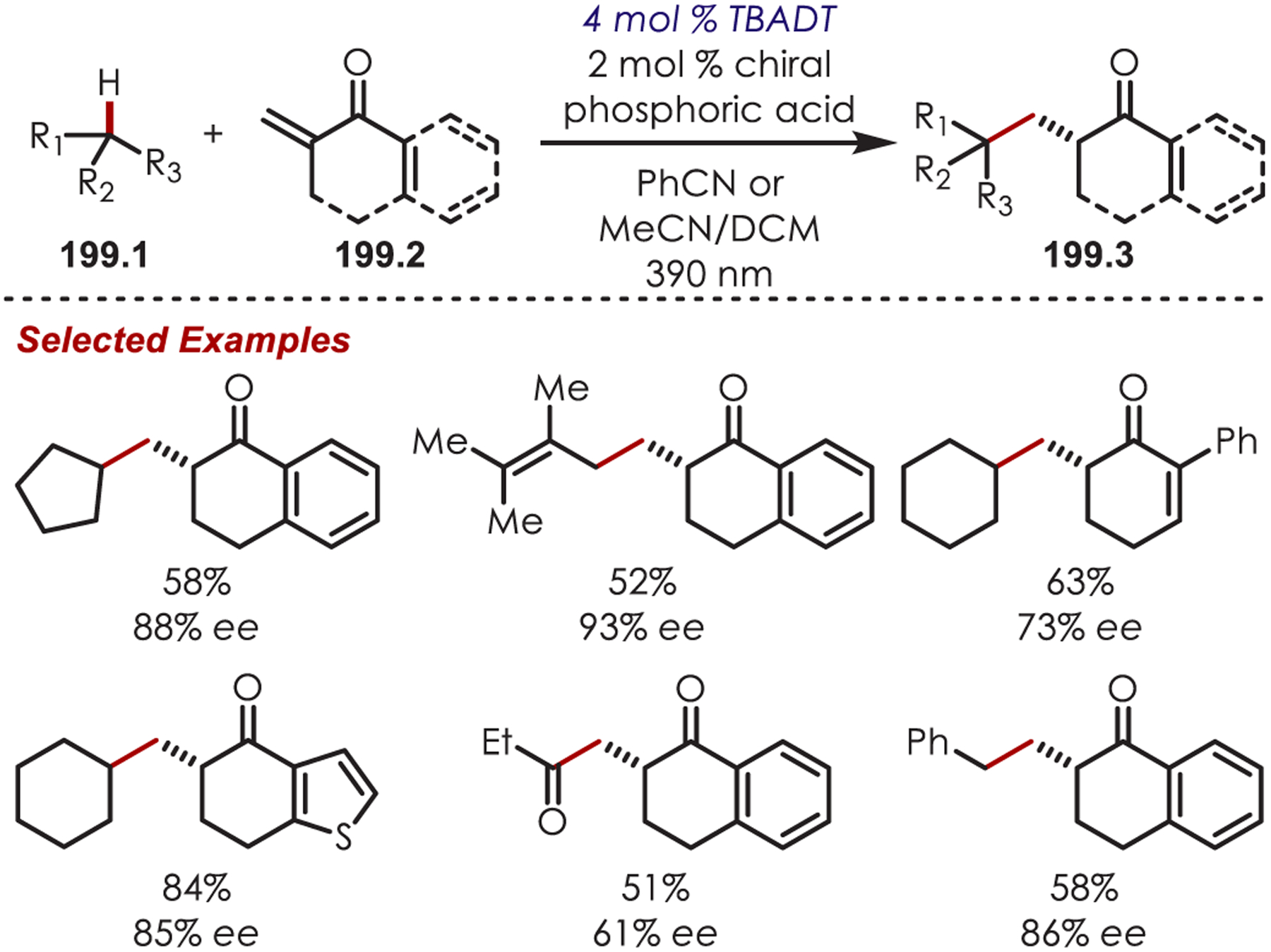
Asymmetric C–H Alkylation with TBADT and a Chiral Phosphoric Acid
Persulfate anion radicals as hydrogen atom abstractors have been utilized to generate alkyl radicals for coupling with heteroarenes (200.2) for Minisci-type products (Scheme 200).357 Cycloalkanes and nonfunctionalized aliphatic alkanes were tolerated with a diverse range of heteroarenes. It should be noted that the alkane coupling partner (200.1) was required in large excess (50 equiv).
Scheme 200.
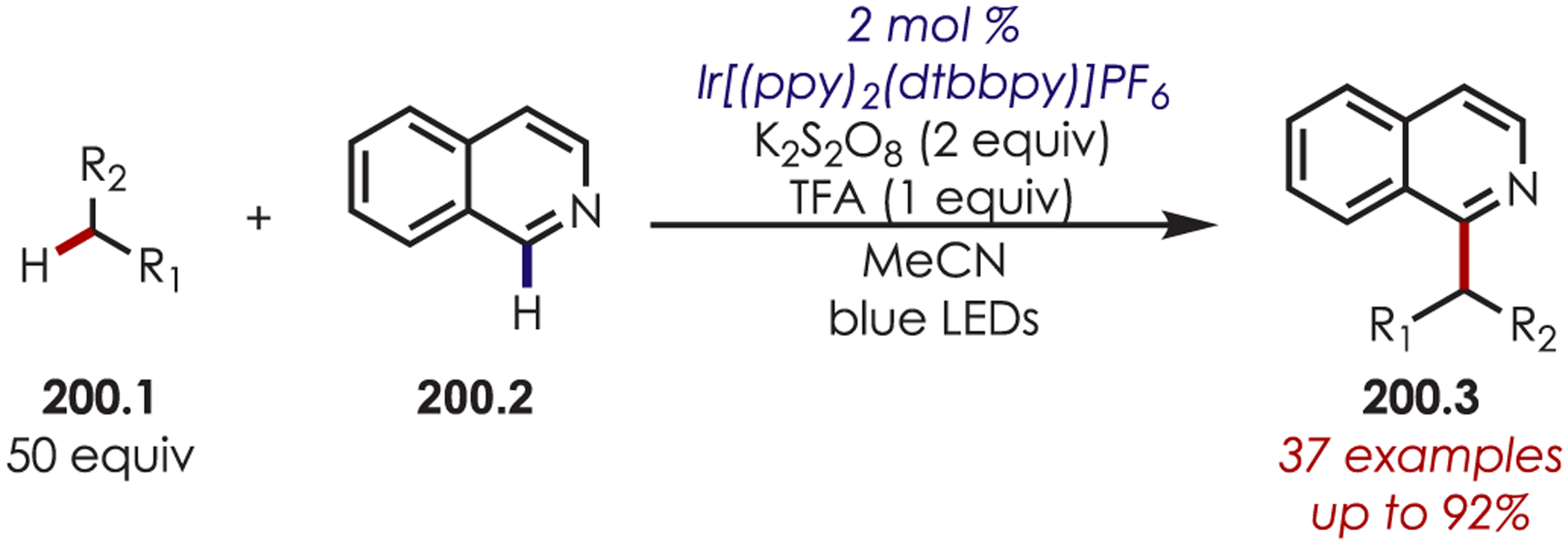
Remote C–H Heteroarylation via a Minisci-Type Reaction
Martin reported a new highly selective quinuclidine HAT catalyst derivative for the C–H abstraction of adamantanes (201.1) and subsequent trapping with radical acceptors (201.2; Scheme 201).358 This catalyst selectively abstracts the tertiary C–H in the adamantane core in the presence of other weaker tertiary or α-hetero C–H bonds. This catalyst provides high chemoselectivity by leveraging the charge transfer character in the C–H functionalization step which is enhanced by the electron-withdrawing substituent on the quinuclidine. Increased positive charge formation in the transition state of the adamantane is favored due to the higher stability of the tertiary adamantane carbocation relative to other potential carbocations.359 Adamantyl derivatives with alkyl, aryl, hydroxy, halide, and nitrile functional groups were well tolerated. The following year, the authors extended this methodology for the coupling of adamantyl radicals with imines and hydrazones to access the aminoalkylated products.360
Scheme 201.
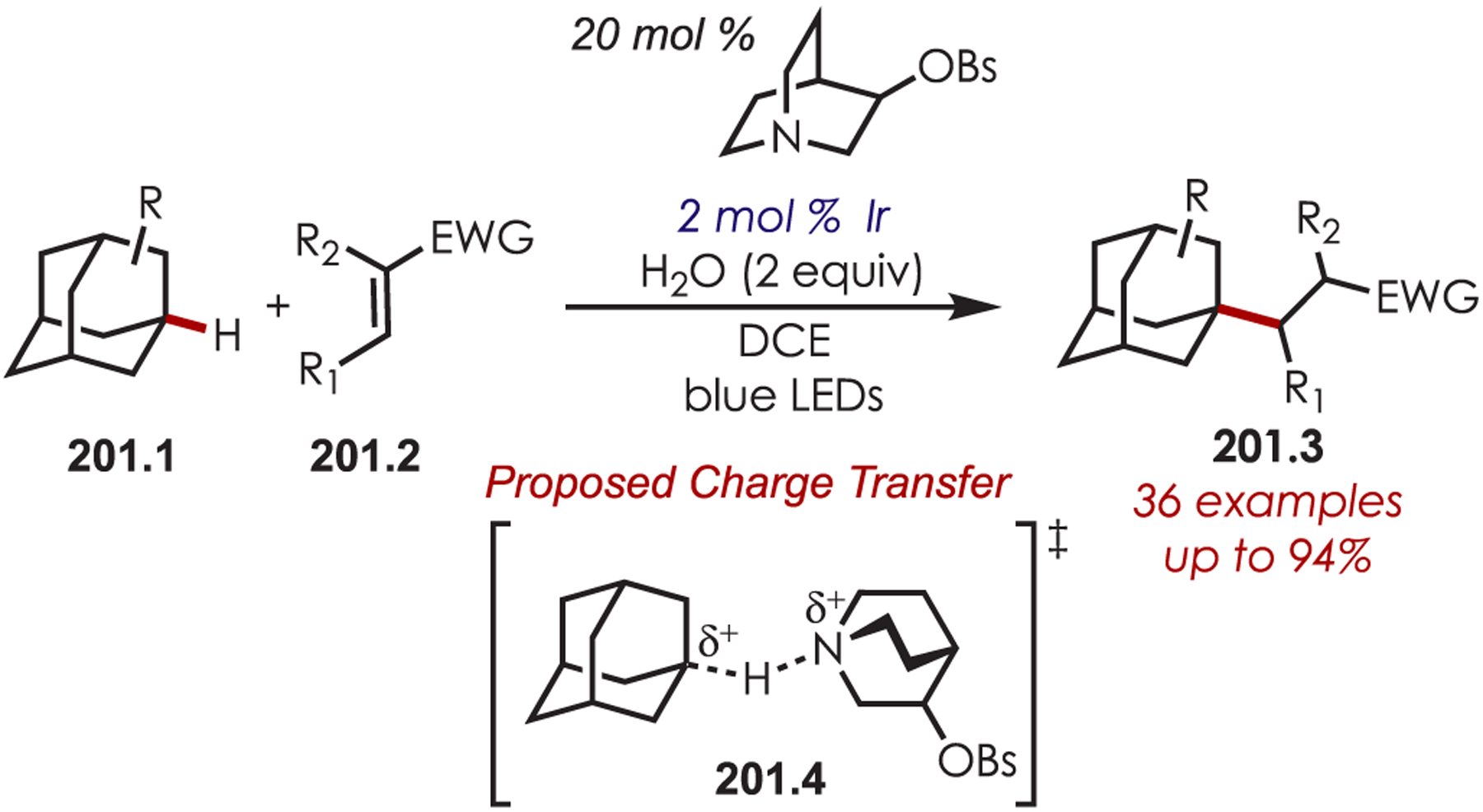
Adamantyl C–H Alkylation with Activation through a Charge-Transfer Event
Glorius reported an unactivated sp3 C–H trifluoromethylthiolation via Ir(III) photoredox catalysis (Scheme 202).361 A selective C–H activation via a hydrogen transfer from a benzoyloxy radical is proposed to abstract alkyl C–H bonds. The radical formed through this HAT is then trapped with a trifluoromethylthiol phthalimide, furnishing the desired trifluoromethylthiolated products. Tertiary C–H bonds were favored over secondary and primary, and the reaction was applicable to a range of hydrocarbons including cyclopropyl, adamantyl, and alcohols. Amine, ester, and amide functional groups were tolerated, and this methodology was also demonstrated on a range of natural products and heterocycles including ambroxide, androsterone, and pregabalin derivatives.
Scheme 202.

Remote C–H Trifluoromethylthiolation Using a Benzoyloxy Radical Hydrogen Atom Abstractor
Via 1,5-Hydrogen Atom Transfer.
A popular strategy for a Hofmann–Löffler–Freytag 1,5-hydrogen atom transfer is homolysis of the N–X bond through photoredox-mediated pathways (Scheme 203).362 The resulting nitrogen-centered radical (203.1) undergoes a δ-C–H abstraction to afford the remote carbon-centered radical (203.2). Either radical trapping or oxidation of the radical and subsequent nucleophilic addition furnishes the substituted amine products. In order for an efficient 1,5-HAT to occur, the nitrogen-centered radical must be sufficiently electrophilic through either protonation or substitution with an electron-withdrawing group.
Scheme 203.
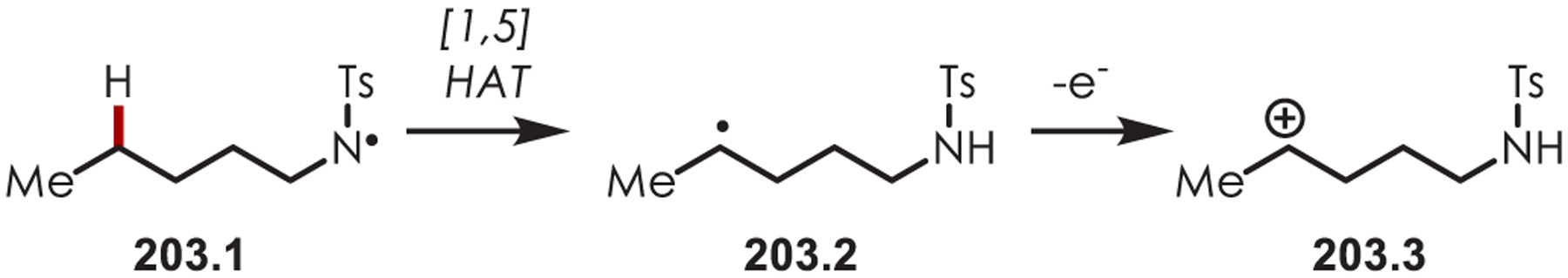
General Scheme of a 1,5-HAT
In 2015, Yu reported a remote amidation and chlorination by harnessing the innate reactivity of aliphatic α-amino radicals to undergo Hofmann–Löffler–Freytag-type 1,5-HAT (Scheme 204).363 First, a nitrogen-centered radical is generated through oxidative quenching of Ir(III)* by N-chlorosulfonamides. The resulting radical undergoes a 1,5-HAT to generate a carbon-centered radical. A subsequent oxidation by Ir(IV) and nucleophilic addition furnishes the functionalized products (204.2 and 204.4). This method was utilized in the late-stage functionalization of (−)-cis-myrtanylamine and (+)-dehydroabietylamine.
Scheme 204.
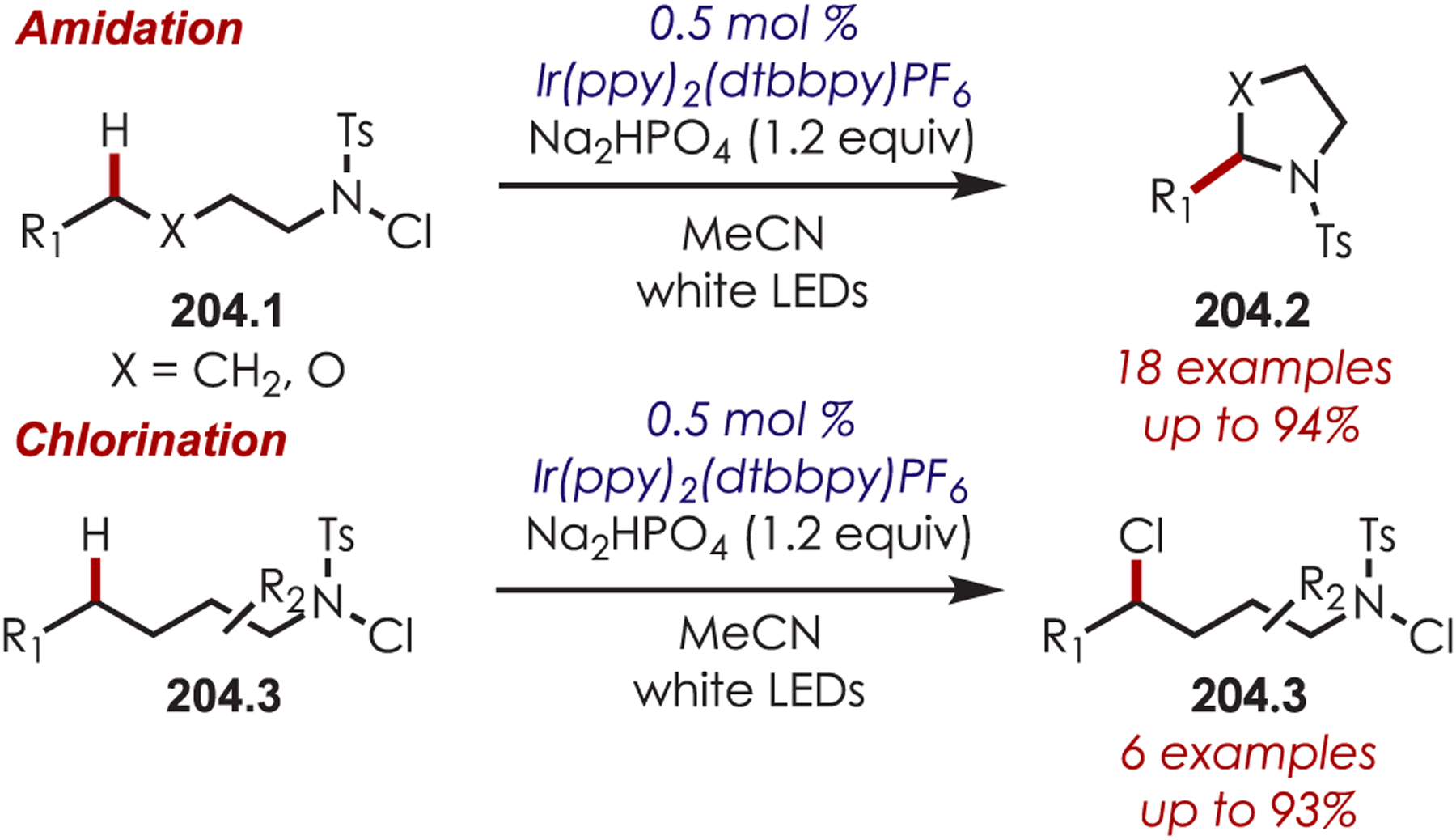
Remote C–H Amidation and Chlorination via a 1,5 HAT of Chlorosulfonamides
Oxygen-centered radicals have also been employed in 1,5-HAT processes; however, intermolecular C–C bond formation following HAT has been challenging, often resulting in unproductive chemistry. Despite this, a remote allylation and alkenylation was reported using N-alkoxy phthalimides (205.1) as alkoxy radical precursors (Scheme 205).364 Selective δ-functionalization was observed, and a variety of allyl acceptors were appropriate including aryl substituted allyl sulfones and vinyl sulfones, which gave the corresponding alkenes (205.3). The Hantzsch ester played a key role by first quenching Ir(III)* to form an amine cation radical. The cation radical then protonates an N-alkoxyphthalimide radical anion, facilitating the formation of the alkoxyl radical and subsequently, the 1,5-HAT.
Scheme 205.
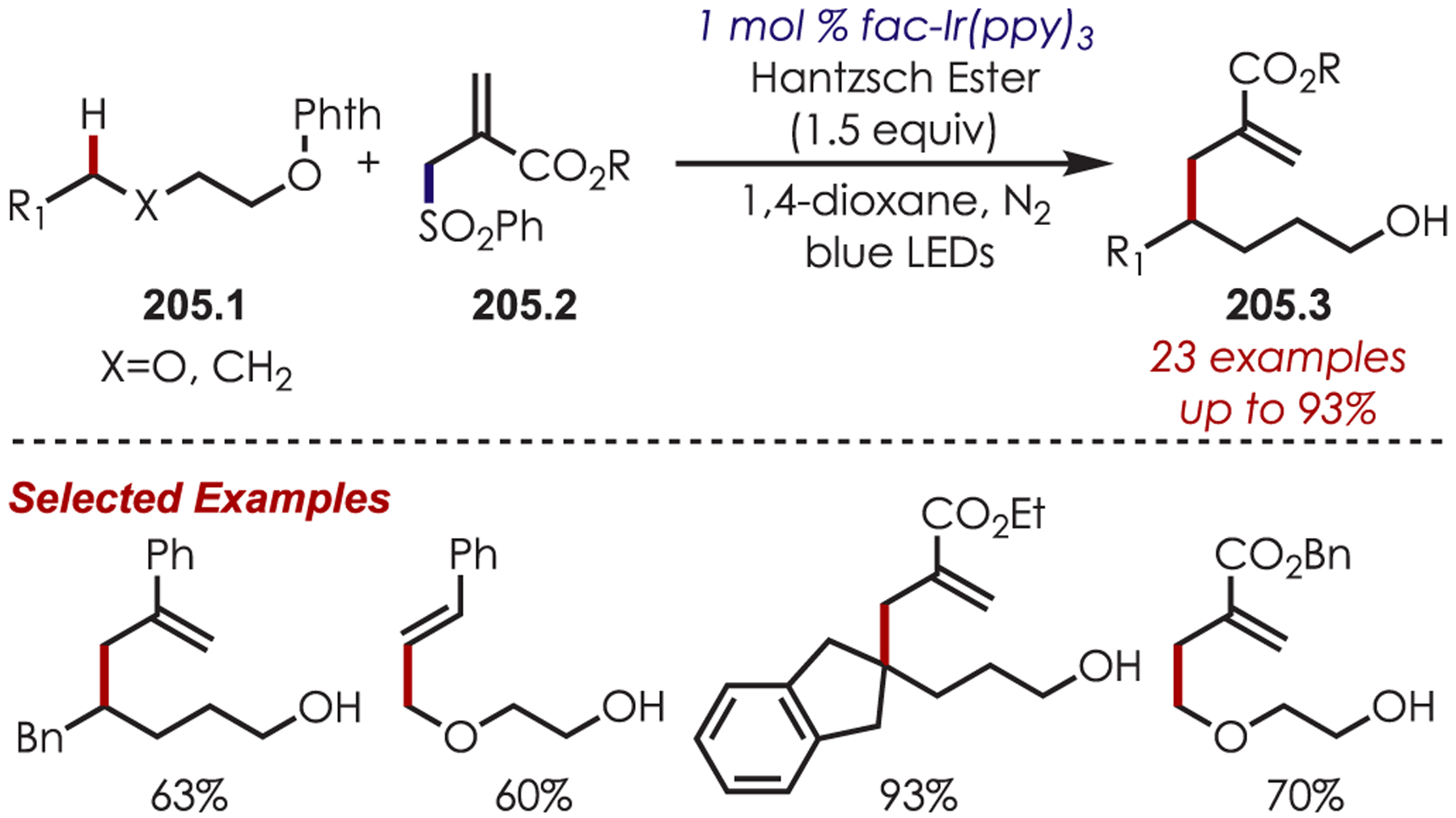
Remote C–H Allylation and Alkenylation via a 1–5 HAT of N-Alkoxyphthalimides
In 2016, Knowles269 and Rovis270 independently reported a strategy for remote functionalization of N-alkyl amides (206.1) by PCET for the homolysis of strong N–H bonds, BDFE = ~107 kcal/mol (Scheme 206).365,366 Both methods proceed via oxidation of the amine by photoexcited Ir(III)* in a concerted PCET to generate an amidyl radical (206.2). A 1,5-HAT results in the formation of a remote carbon-centered radical (206.3) which is trapped with a radical acceptor, reduced by Ir(II), and protonated to furnish the alkylated product (206.5) while regenerating the catalyst.
Scheme 206.
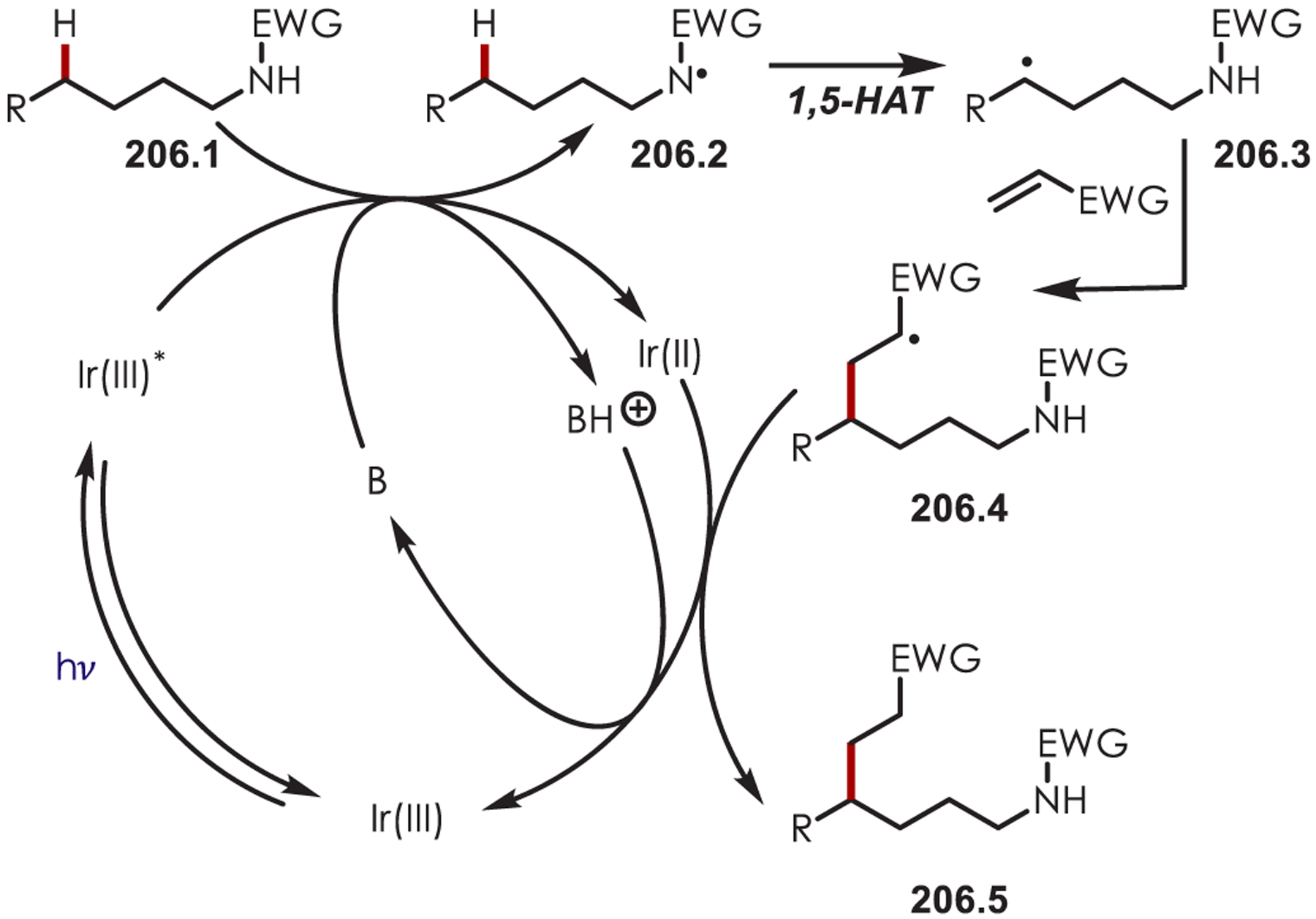
Mechanism for the Remote C–H Functionalization of N-Alkyl Amides through a PCET-Initiated 1,5-HAT
The scope of both methods is comparable; a variety of functionalized amide derivatives (207.1) were suitable, including cyclic and acyclic systems and substrates containing heteroatoms or functionalized with electron-donating groups (Scheme 207). Knowles demonstrated that a variety of radical acceptors are appropriate including alkyl and enone derivatives, dicarbonyls, fumarates, arylsulfonate, methacrolein, α-phenyl acrylonitrile and methylacrylate; the Rovis work reported acrylate derivatives as acceptors.
Scheme 207.
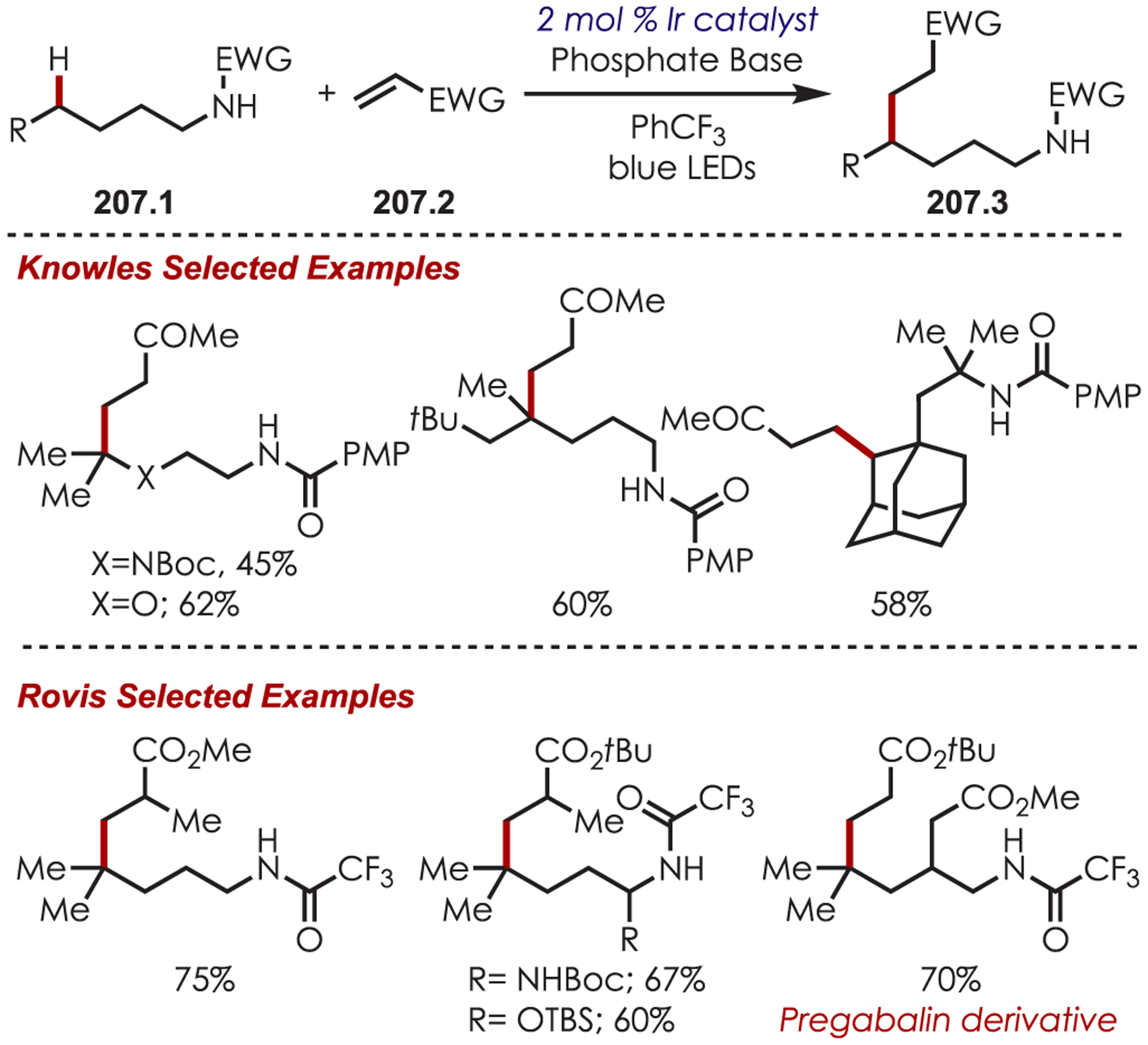
Scope of the Remote C–H Functionalization of N-Alkyl Amides through a PCET-Initiated 1,5-HAT by Knowles and Rovis
Rovis later reported the γ-alkylation of carboxylic acid derivatives (208.1) by utilizing a key alkoxycarbonyl protecting group (Scheme 208).367 The role of the protecting group was critical in acidifying the N–H for deprotonation, modifying the resultant amidyl anions’ oxidation potential for SET, and providing a higher N–H BDE relative to C–H bond. The mechanism occurs first by deprotonation of the amide (pka ~11.0) by K3PO4. The resulting potassium amide salt (Eox = +1.04 V vs SCE) undergoes SET with Ir(III)* generating the nitrogen-centered radical. A range of electron deficient alkenes (208.2) were employed as coupling partners and various amides were also found to be suitable including those containing tertiary or secondary C–H bonds.
Scheme 208.
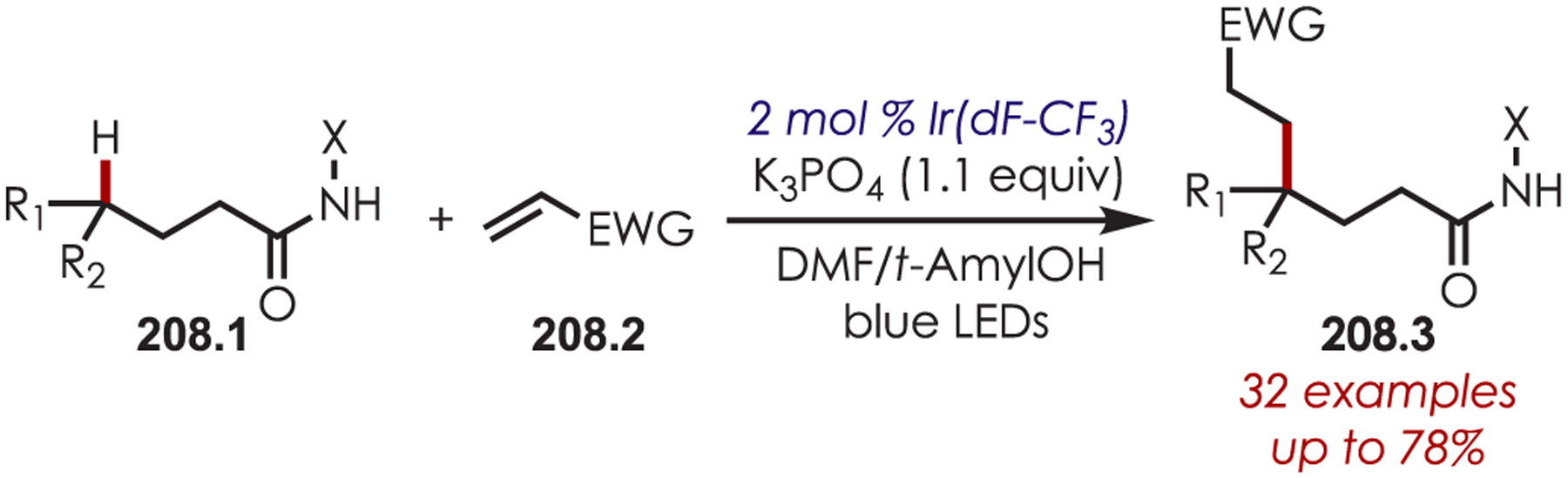
γ-Alkylation of Carboxylic Acids through a 1,5-HAT
The synthesis of γ-alkylated ketones (209.3) was reported by Studer using α-aminooxy acid auxiliaries (209.1; Scheme 209).368 The carbon-centered radicals formed via 1,5-HAT from the photoredox-generated iminyl radical were trapped with various Michael acceptors and a subsequent hydrolysis of the imine afforded the γ-functionalized ketones in moderate to good yields (31–84%).
Scheme 209.
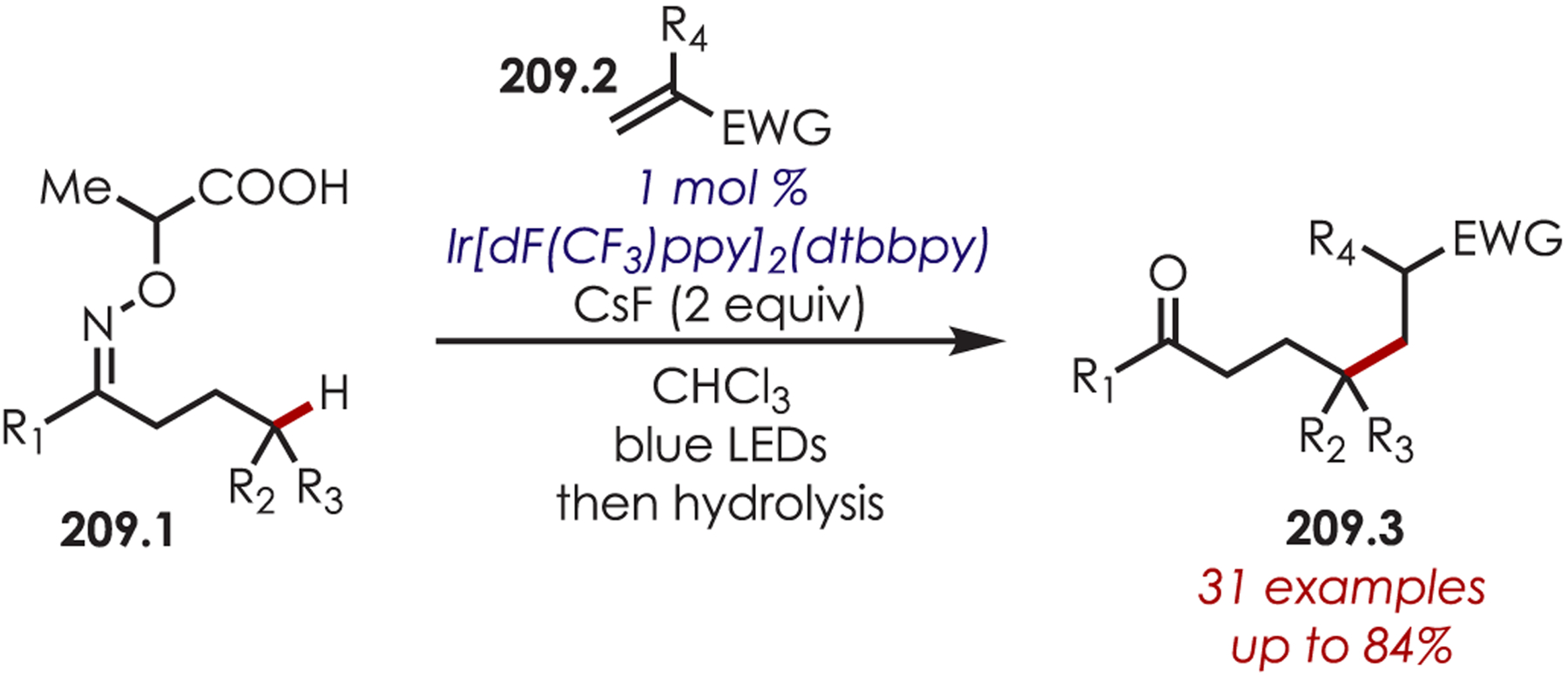
Allylic Chlorides as Radical Traps for the Allylation of Remote C–H Bonds
Allyl chlorides (210.2) have been shown utilized as successful carbon-centered radical acceptors from radicals formed via a 1,5-HAT from pendant amidyl radicals in work from the Tambar lab. Following initial radical addition to the allyl chloride, β-scission leads to elimination of the chloride for the allylation of remote C–H bonds (Scheme 210).369 The exact role of the nickel co-catalyst is unclear but is proposed to stabilize the allyl chloride. This method has high functional group tolerance and is compatible with cyclic and acyclic substrates that contain heteroatoms or functionalized with electron-donating groups. The scope of acceptors includes aliphatic, ester, or aryl-substituted allyl chlorides.
Scheme 210.
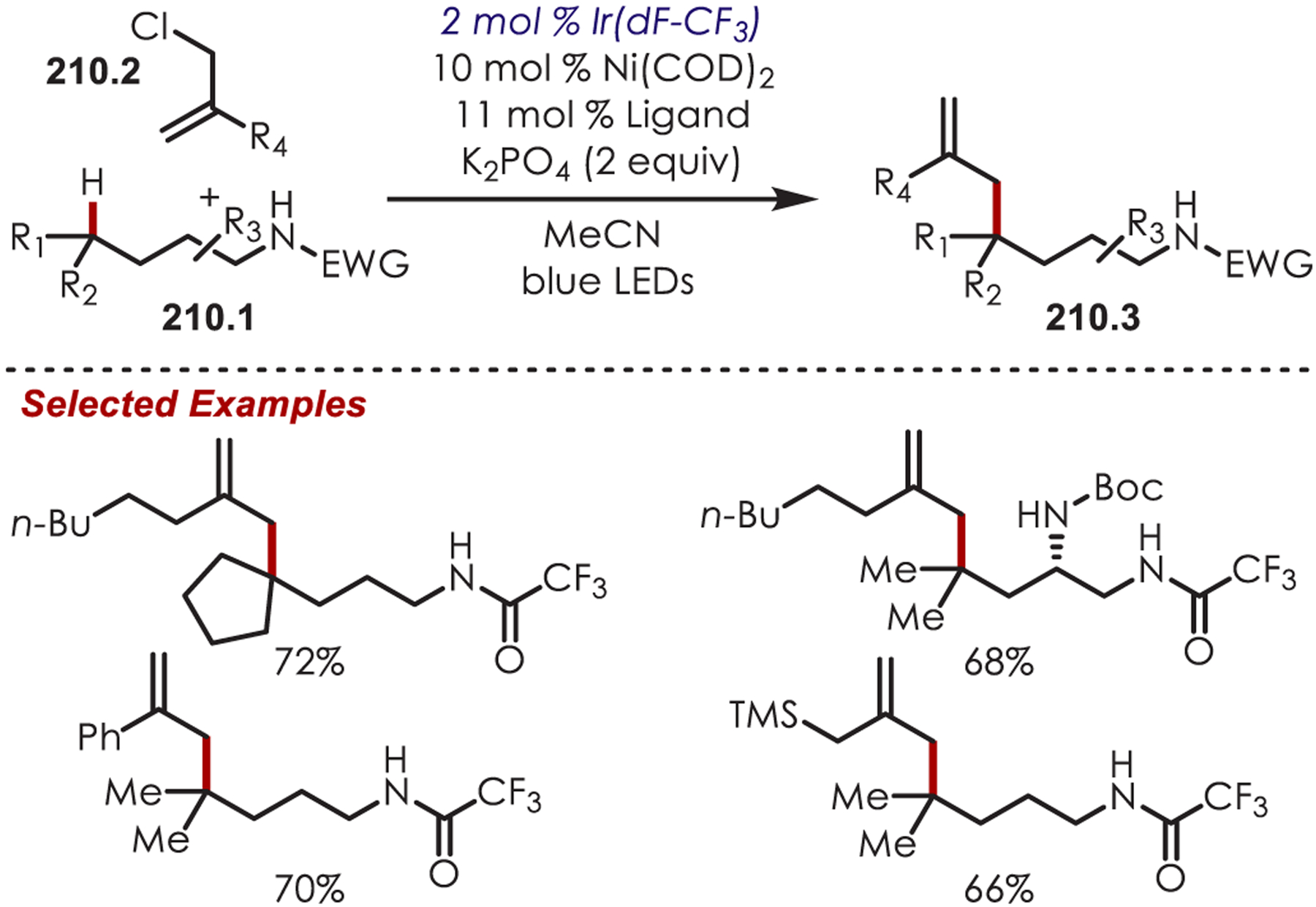
Allylic Chlorides as Radical Traps for the Allylation of Remote C–H Bonds
A remote vinylation through iminyl radicals has been reported with aryl vinyl boronic acid coupling partners (211.2) and O-acyloximes (Scheme 211).370 Primary, secondary, and tertiary γ-functionalized products were tolerated with alkyl-, halogenated, alkoxy-, or amine-substituted aryl boronic acids.
Scheme 211.
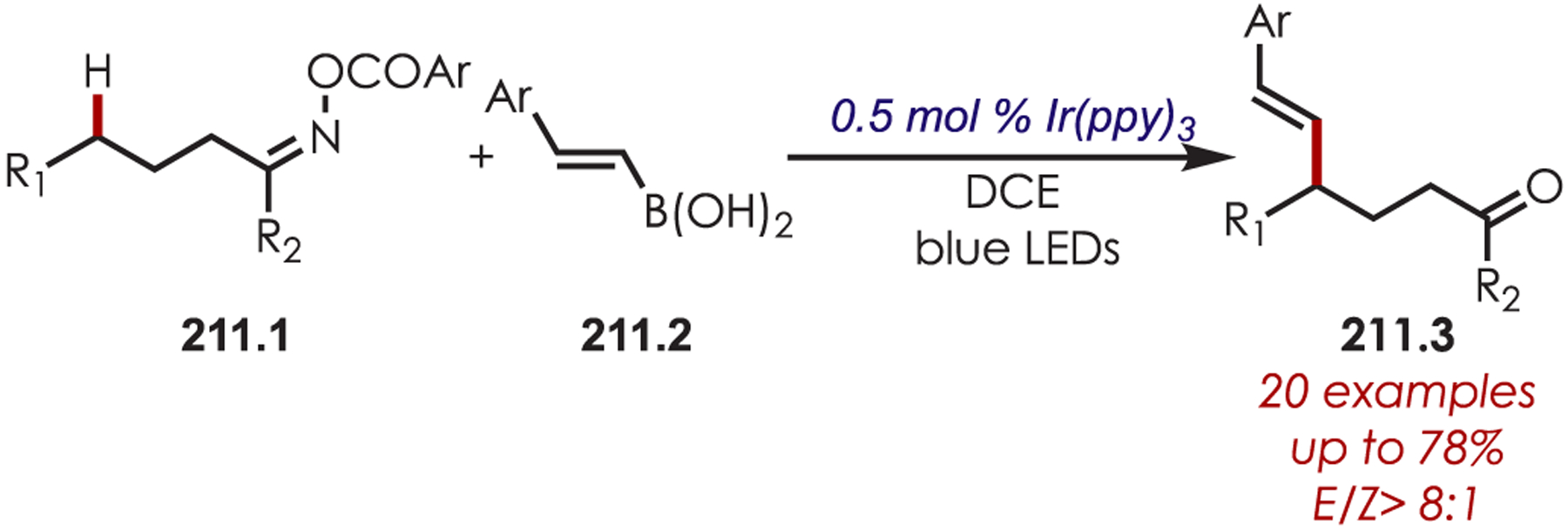
Remote C–H Vinylation of O-Acyloximes
The merging of PCET, 1,5-HAT, and chiral Lewis acid catalysis for a remote asymmetric alkylation of alcohols (212.1) and amides (212.3) was reported by Meggers (Scheme 212).371,372 This methodology utilized a chiral Rh catalyst that precomplexes with the amide or ketone acceptors. The scope of the hydroxy or amide derivatives included mostly symmetrical aliphatic substituted substrates. Various alkyl or aryl pyrazole derivatives were suitable acceptors of the NCR 1,5-HAT event. However, the scope of the amide functionalization was quite limited by the requirement of an imidazole-derived acceptor and did not tolerate any modifications. The mechanism proceeds through first a single electron reduction of the chiral Rh-coordinated imidazole, which diverges from their previous work which forms a chiral environment for the addition of the radical to the alkene.371,373 The authors note that the reduction must outcompete direct conjugate addition of the radical intermediate to prevent racemic background reactivity, thus limiting the scope of acceptors.
Scheme 212.
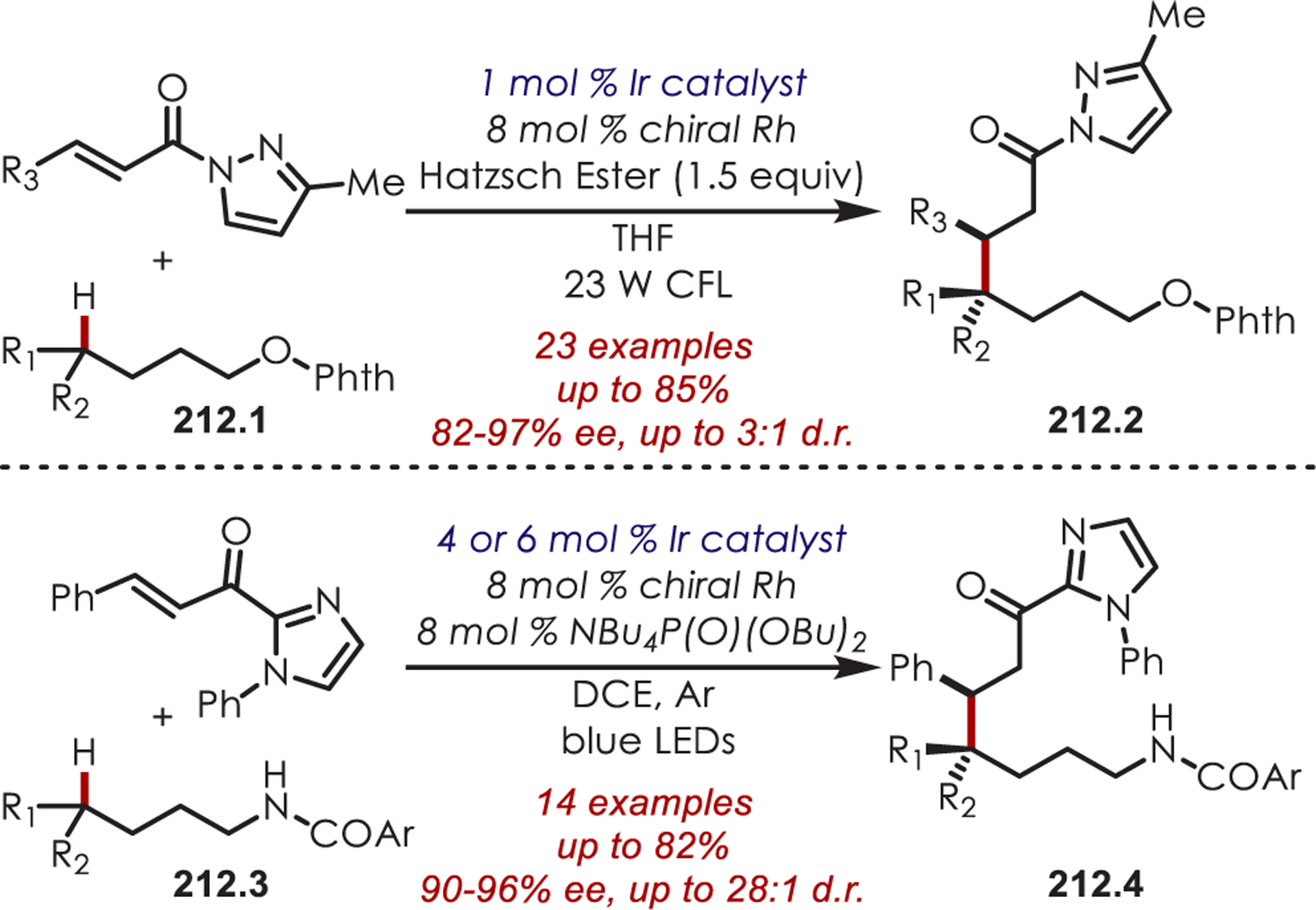
Chiral Lewis Acid Catalysis for the Asymmetric Remote Alkylation of Imidazole Amides
Dual photoredox and Cu catalysis has become a strategy for intercepting carbon-centered radicals through a chiral Cu complex for the enantioselective remote C–H bond functionalization. In 2020, Nagib reported an enantioselective radical C–H amination for the synthesis of β-amino alcohols (213.3) through the interception of an alkyl radical by a chiral copper catalyst (Scheme 213).374 This selectively generates chiral β-amino alcohol precursors in the presence of other alkyl, allyl, benzyl, and propargyl C–H bonds. The proposed mechanism proceeds through an energy transfer between the photoexcited iridium and Cu bound oxime, generating a nitrogen-centered radical. A subsequent 1,5-HAT and intramolecular amination affords the oxazoline products.
Scheme 213.
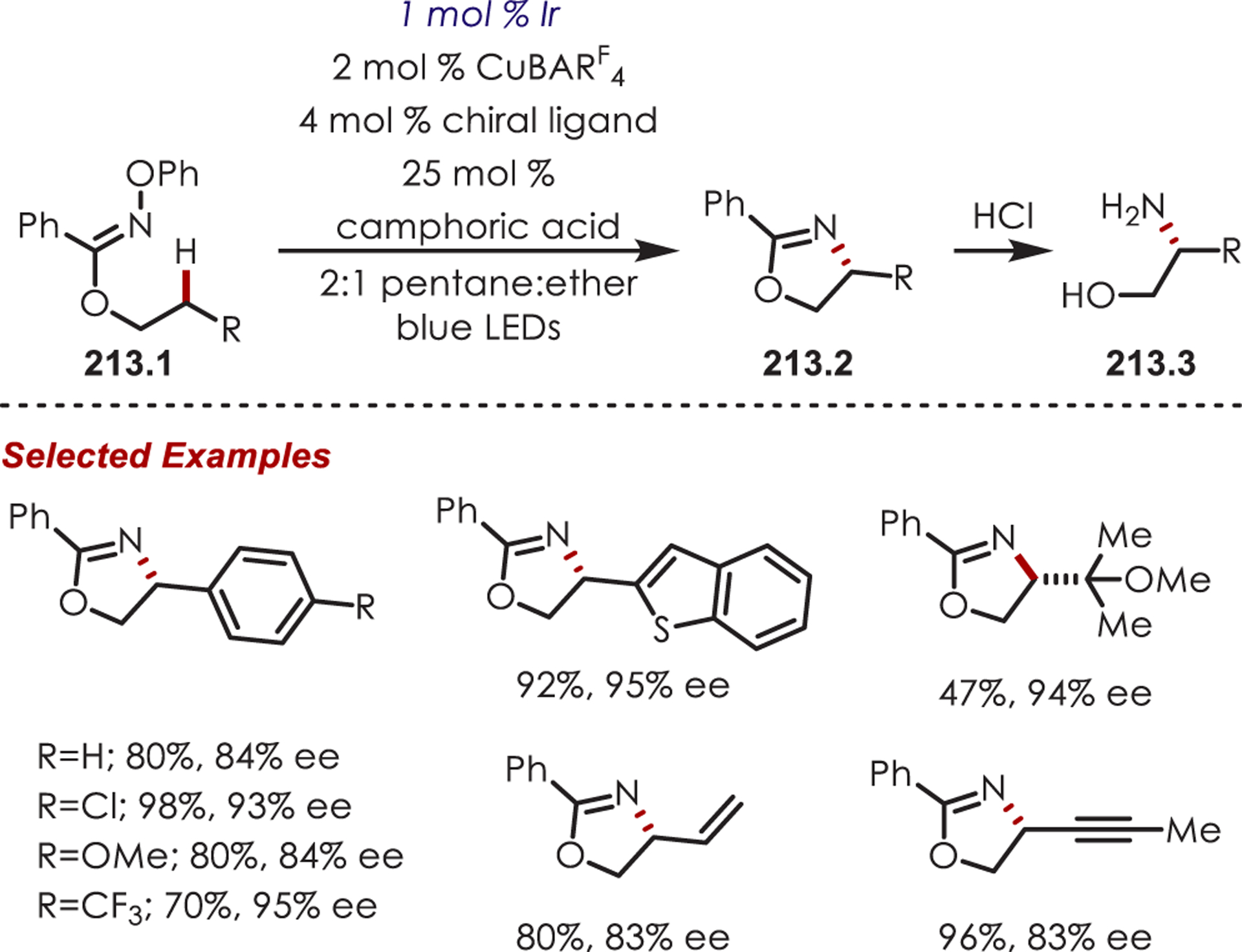
Enantioselective C–H Bond Functionalization with Imidate Chiral Copper Catalysis
The same year, Yu reported an enantioselective C–H cyanation via dual photoredox and copper catalysis (Scheme 214).375 The carbon-centered radical, generated by 1,5-HAT by an amidyl radical, was intercepted by a chiral BOX-bound Cu(II) cyanide intermediate. Electron-withdrawing or donating substituents alongside halogenated arenes all afforded the products (214.2) in good yields and enantioselectivities. While functionalization of nonbenzylic C–H bonds was explored, the enantioselectivies of these derivatives dropped significantly to 12–17% ee. Goosheg also reported an analogous copper and Ir(III)-catalyzed asymmetric cyanation.376
Scheme 214.
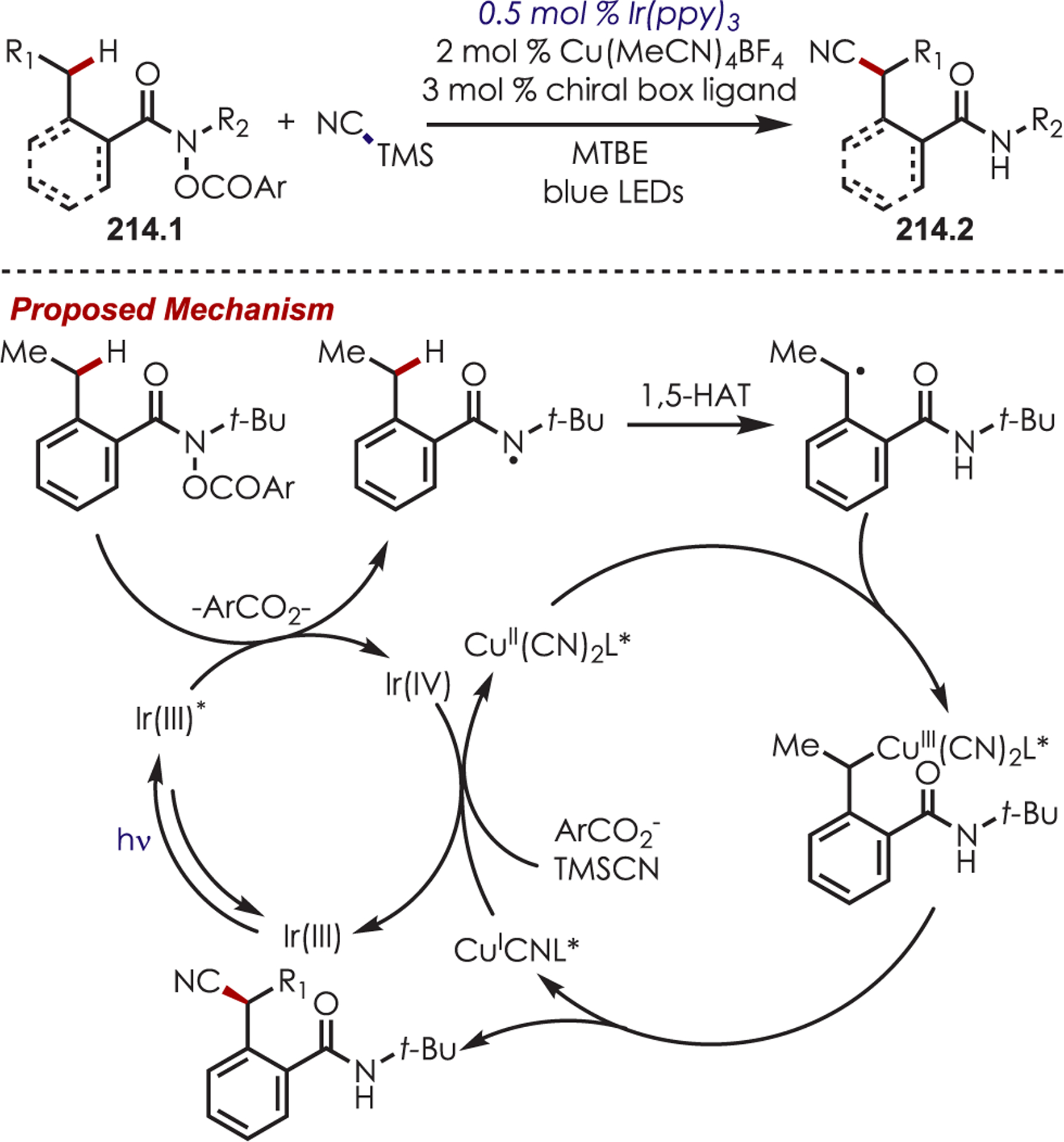
Enantioselective C–H Cyanation via Photoredox and Chiral Copper Catalysis
Lastly, a dual copper and photoredox strategy has been utilized for the functionalization of alcohol and alkyl halide derivatives (Scheme 215).377 In this instance, N-alkoxypyridinium salts (215.1) were employed as oxygen-centered radical surrogates, which formed following single-electron reduction by the photocatalyst. As with many other mechanisms discussed in this section, a 1,5 HAT ensues and the resultant radical was intercepted by copper in the presence of TMSN3, TMSCN, or TMSSCN nucleophiles. Aliphatic or aryl γ-substituted alkanes were compatible and yielded the γ-functionalized products in good yields.
Scheme 215.
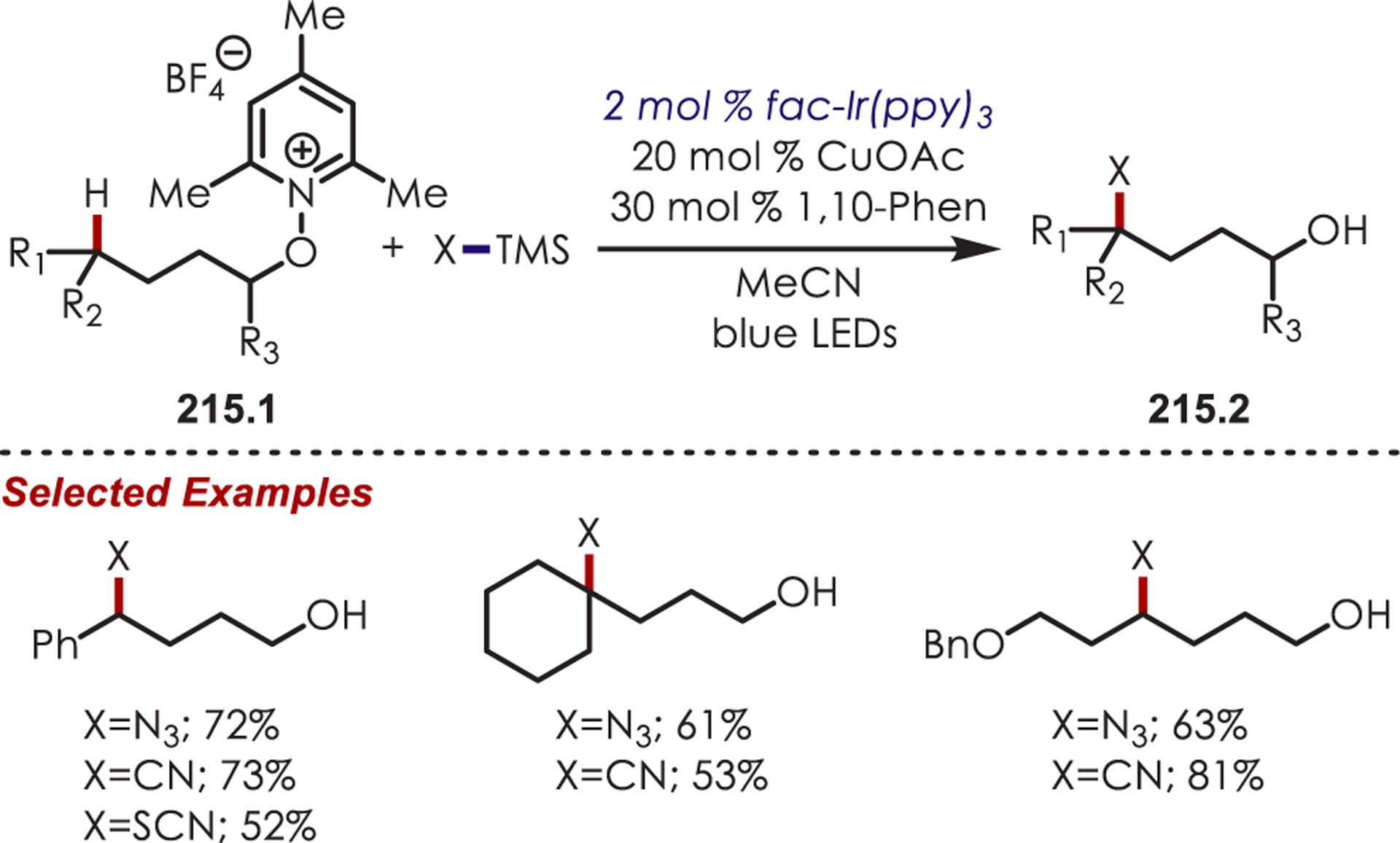
C–H Azidation, Thiocyanation, and Isothiocyanation for the Synthesis of γ-Substituted Alcohols
3. ORGANIC PHOTOREDOX-CATALYZED C–H FUNCTIONALIZATION REACTIONS
3.1. (Hetero)aromatic C–H Functionalization
(Hetero)aryl C–H functionalization has been accomplished using organic photoredox catalysts for the generation of reactive open-shell intermediates including carbo-, nitrogen-, oxygen-, phosphorus-, and sulfur-centered radicals. Additionally, organic photooxidants have enabled the direct oxidation of arenes, which can be coupled with nucleophilic partners. The generation of these open-shell species has provided a valuable synthetic tool for the functionalization of C(sp2)–H bonds.
3.1.1. Transformations Involving Carbon sp2- and sp3-Centered Radicals.
The functionalization of (hetero)arenes using organic photoredox catalysts is accomplished through reactive sp2- and sp3-centered radicals similar to the inorganic catalyzed transformations in 2.1. Carbon-centered sp2-hybridized radicals have been generated through aryldiazoniums, aryl halides, and carboxylic acids by using organic photoreductants and oxidants. Though sp3 carbon-centered radicals are commonly generated through reduction of alkyl bromides, this reactivity is less developed with organic catalysts likely due to their weaker reducing ability relative to their inorganic counterparts. Alternative methods for sp3 radical generation via oxidative decarboxylative and desulfonylative pathways are more common for organic photoredox catalyst systems.
Accessed via Diazoniums.
Diazoniums are frequently utilized precursors to aryl radicals, which have been typically activated through transition-metal photoredox catalysis. However, König reported a metal-free arylation of heteroarenes (216.1) via aryldiazoniums (216.2) in 2012 (Scheme 216).378 An aryl radical, formed via single electron reduction with photoexcited Eosin Y (EY)* undergoes an addition to a heteroarene in a Minisci-type fashion. The resulting cyclohexadienyl radical intermediate is oxidized and deprotonated to furnish the final product. This method gives products with high C2-selectivity for furan, thiophene, and pyrrole derivatives with a broad scope of diazonium salts in moderate to good yields.
Scheme 216.
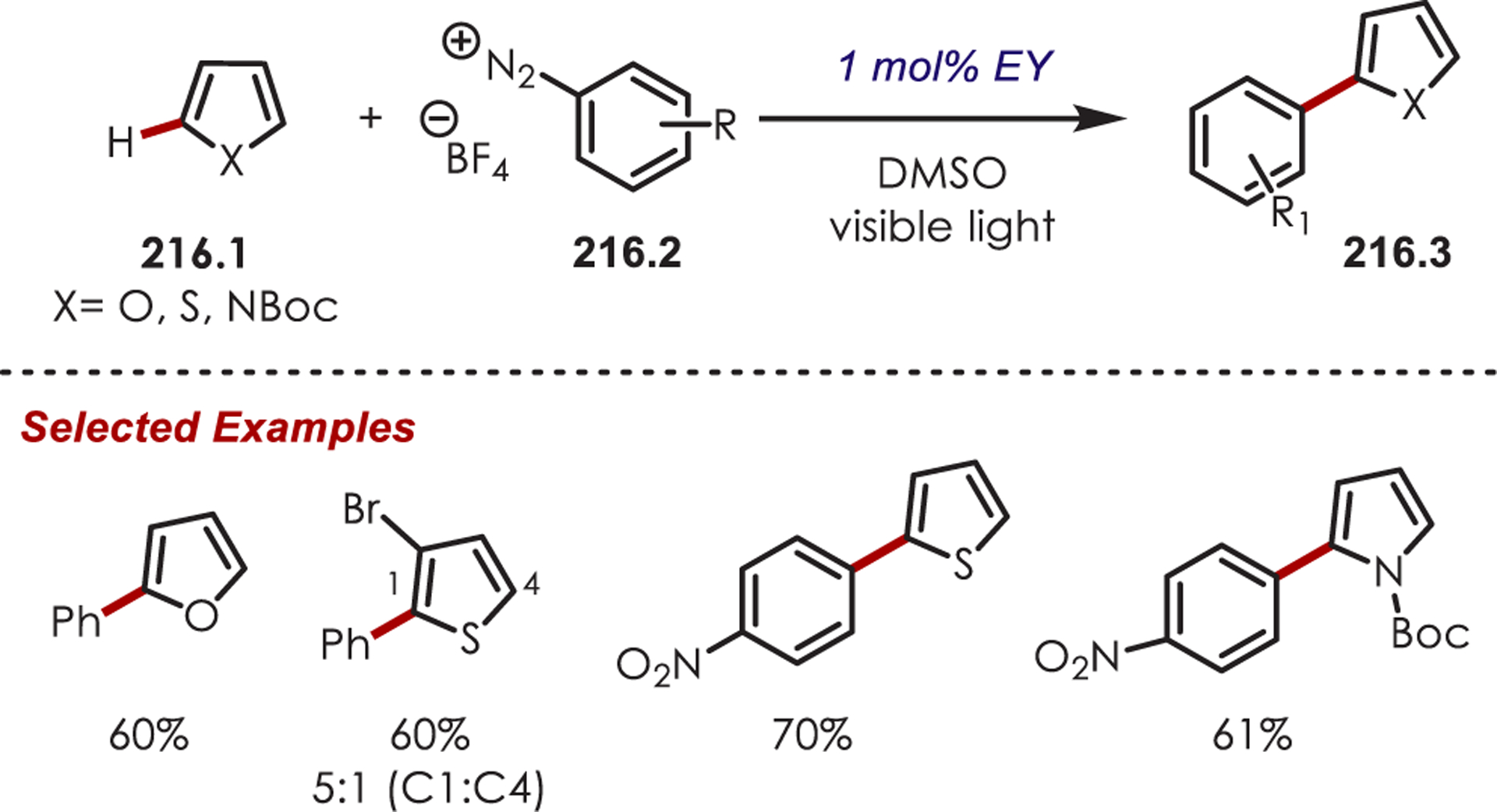
Generation of Aryl Radicals from Aryldiazoniums Using an Eosin Y Photoredox Catalyst for C–H Heteroarylation
Likewise, a [4 + 2] benzannulation of biaryldiazonium salts (217.1) with alkynes was reported in the synthesis of phenanthrenes (Scheme 217).379 PET to a diazonium from eosin Y generates the aryl radical. A subsequent addition to the alkyne generates a vinyl radical which then undergoes an intramolecular addition into the arene.
Scheme 217.
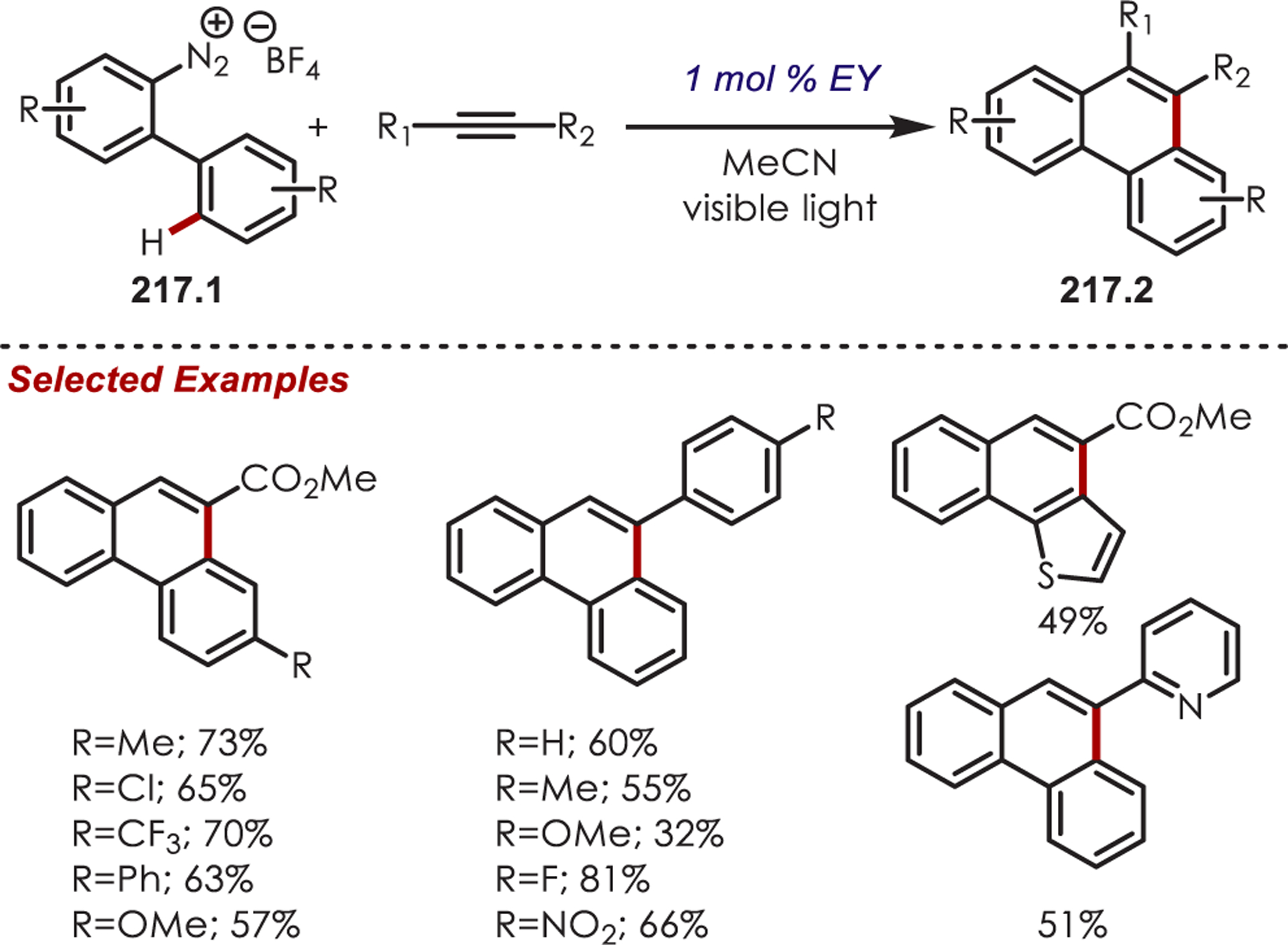
[4 + 2] Benzannulation of Biaryldiazonium Salts with Alkynes through Aryl Radicals Generated by Aryldiazoniums
Accessed via Aryl Halides.
Aryl chlorides are significantly harder to reduce (Ered[PhCl/PhCl*−] = −2.88 V vs SCE) than the corresponding bromides (Ered[PhBr/PhBr*−] = −1.56 vs SCE)380 and thus are not as commonly utilized as a precursor for sp2 carbon-centered radicals. Nonetheless, a highly reducing carbazole-derived catalyst (CAR1; E*ox = −2.75 V vs SCE) was reported that generates sp2 aryl radicals through reductive PET (Scheme 218).381 This carbon-centered radical was found to couple (hetero)aryl chlorides (218.2) with donating groups, thus allowing for a thermodynamically favored PET to occur. Successful C2-selective arylation of pyrrole and simple arenes was possible with this method; however, the scope and yield of these reactions are less efficient compared to their aryl bromide or diazonium counterparts.
Scheme 218.
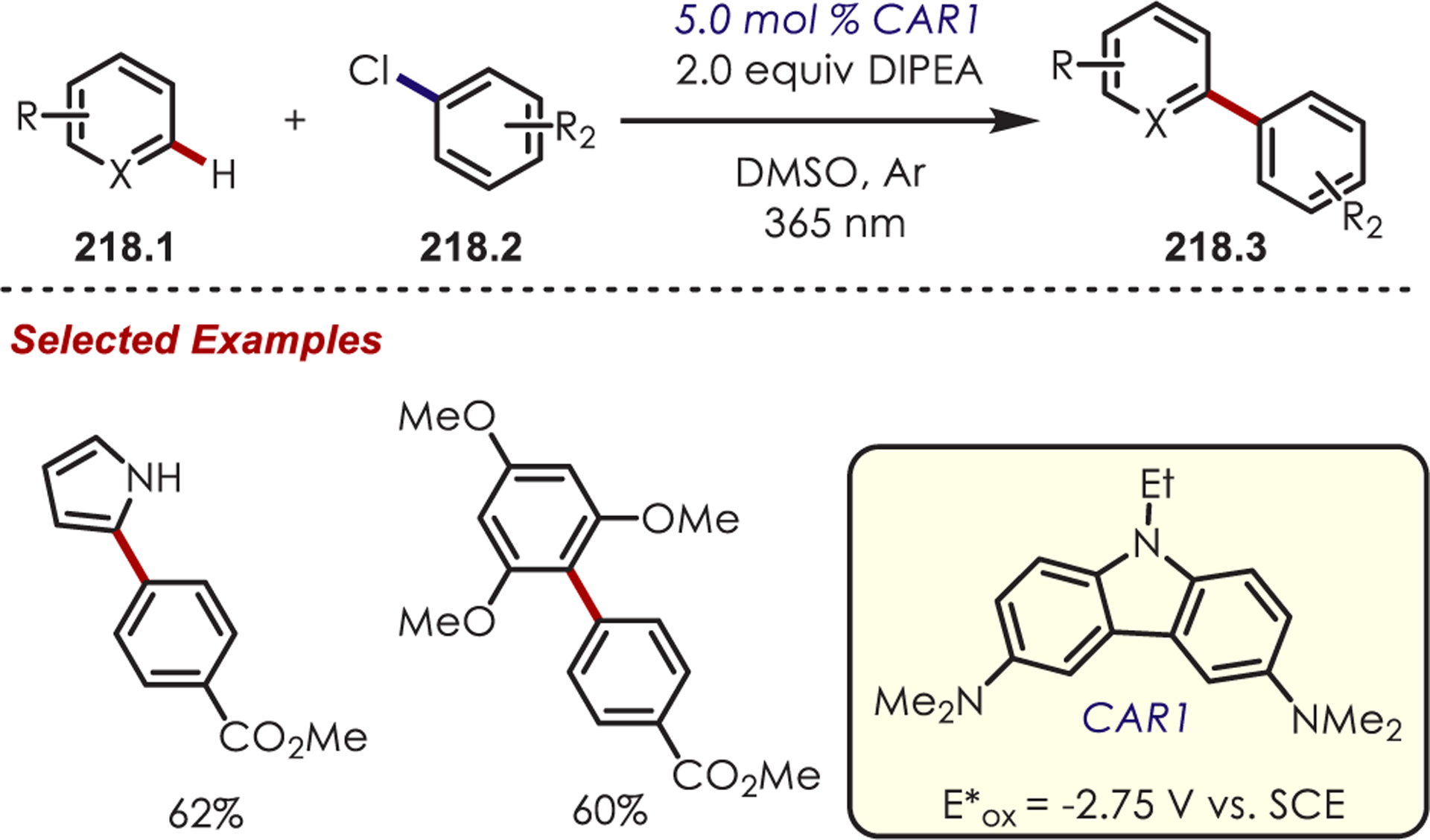
(Hetero)aryl Coupling through C–H Functionalization with Aryl Radicals Generated from Aryl Chlorides
Accessed via Aryl and Alkyl Sulfonyl Chlorides.
Aryl sulfonyl chlorides (219.1) have been employed as aryl radical surrogates with eosin Y* as the photoreductant in the cyclization of 2-isocyanobiphenyls (Scheme 219).382 Addition of the resultant aryl radical to isocyanide forms an intermediate imidoyl radical. An intramolecular cyclization onto the pendant aryl C–H, followed by oxidation and deprotonation afforded the desired phenanthridines (219.3). Electron-rich and electron-poor aryl sulfonyl chlorides were compatible in the intramolecular cyclization of isocyanobiphenyls.
Scheme 219.
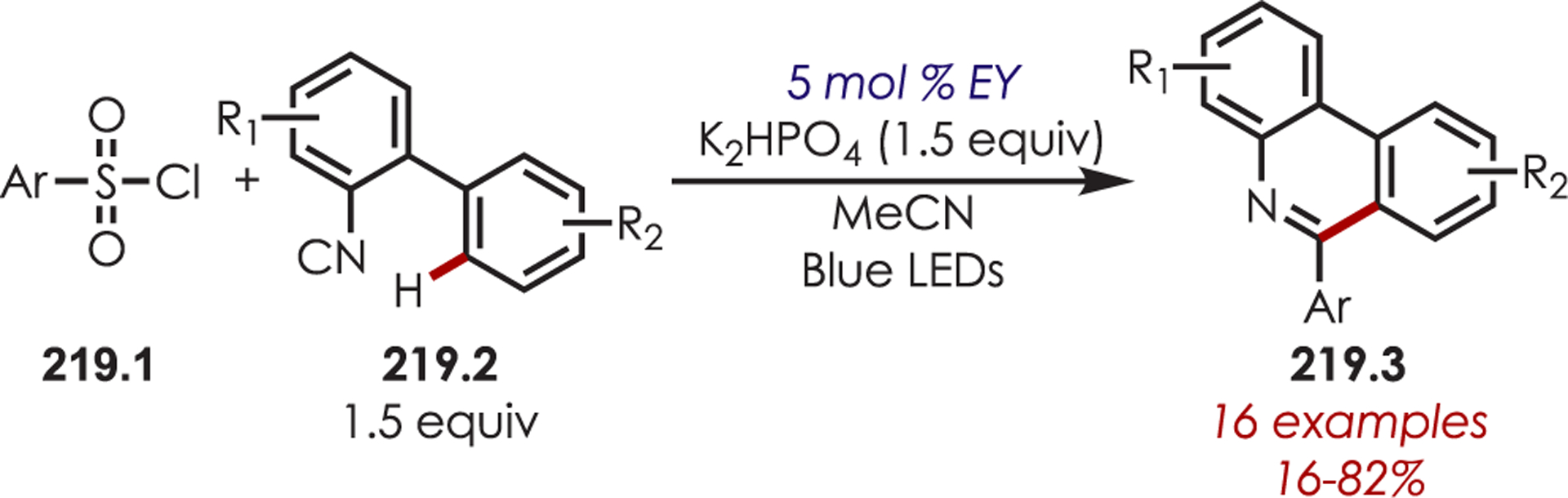
Generation of Aryl Radicals from Aryl Sulfonyl Chlorides for Arene C–H Functionalization
An anthraquinone-mediated oxidative photoredox process was demonstrated in the perfluoroalkylation of arenes by Itoh and co-workers in 2013 via sp3 carbon-centered radicals (Scheme 220).383 The mechanism is analogous to transition metal counterparts; oxidation of sulfinate 220.2 by AQN* (Eox = +0.6 V and Ered* > +1.5 V vs SCE, respectively) produces a sulfonyl radical. Rapid loss of SO2 generates the perfluoroalkyl radical. Then, an addition to the arene and an oxidation/deprotonation sequence rearomatizes the substrate to afford the perfluoroalkyl arene (220.3). Electron-rich and electron-deficient arenes generated the desired products, however, in lower yields for the latter.
Scheme 220.
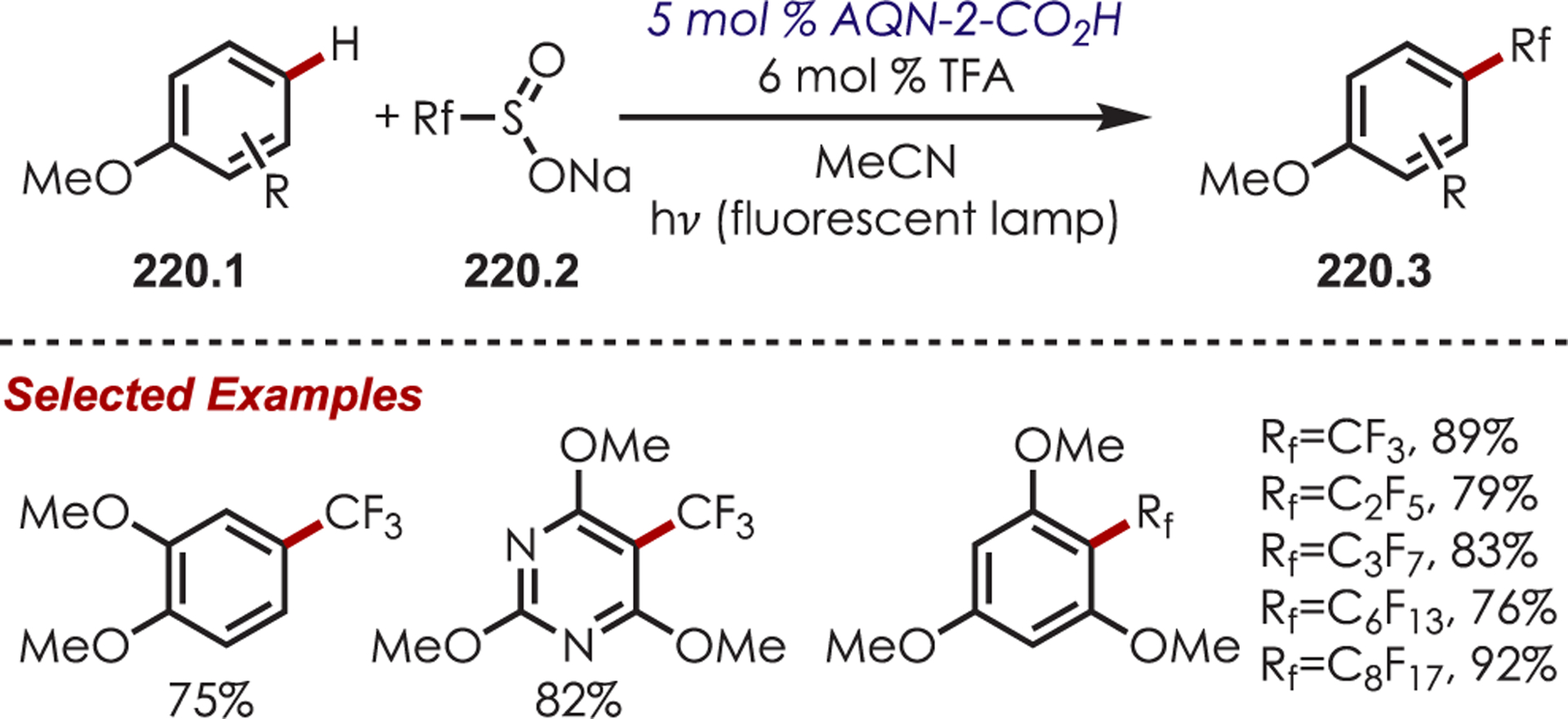
Perfluoroalkylation of Arenes Using Sulfinates as Fluoroalkyl Radical Precursors
Accessed by Carboxylic Acids.
An ortho-acylation of acetanilides (221.1) was accomplished via a dual eosin Y and palladium catalytic system (Scheme 221).384 Oxidation of phenylglyoxylic acid (221.2) by photoexcited eosin Y* generates a benzoyl radical (221.4). Molecular oxygen regenerates the catalyst and produces a superoxide radical anion. The palladium catalytic cycle is initiated by an ortho-C–H activation of the acetanilide (221.5). The benzoyl radical reacts with the intermediate palladacycle, generating a Pd(III) intermediate (221.6). Oxidation of the palladium complex by the superoxide anion radical, followed by reductive elimination, regenerates Pd(II) and yields the ortho-acylated acetanilides. Both electron-withdrawing and donating groups on the arene were compatible with alkyl, alkoxy, halo, and trifluoromethyl phenylglyoxylic acids.
Scheme 221.
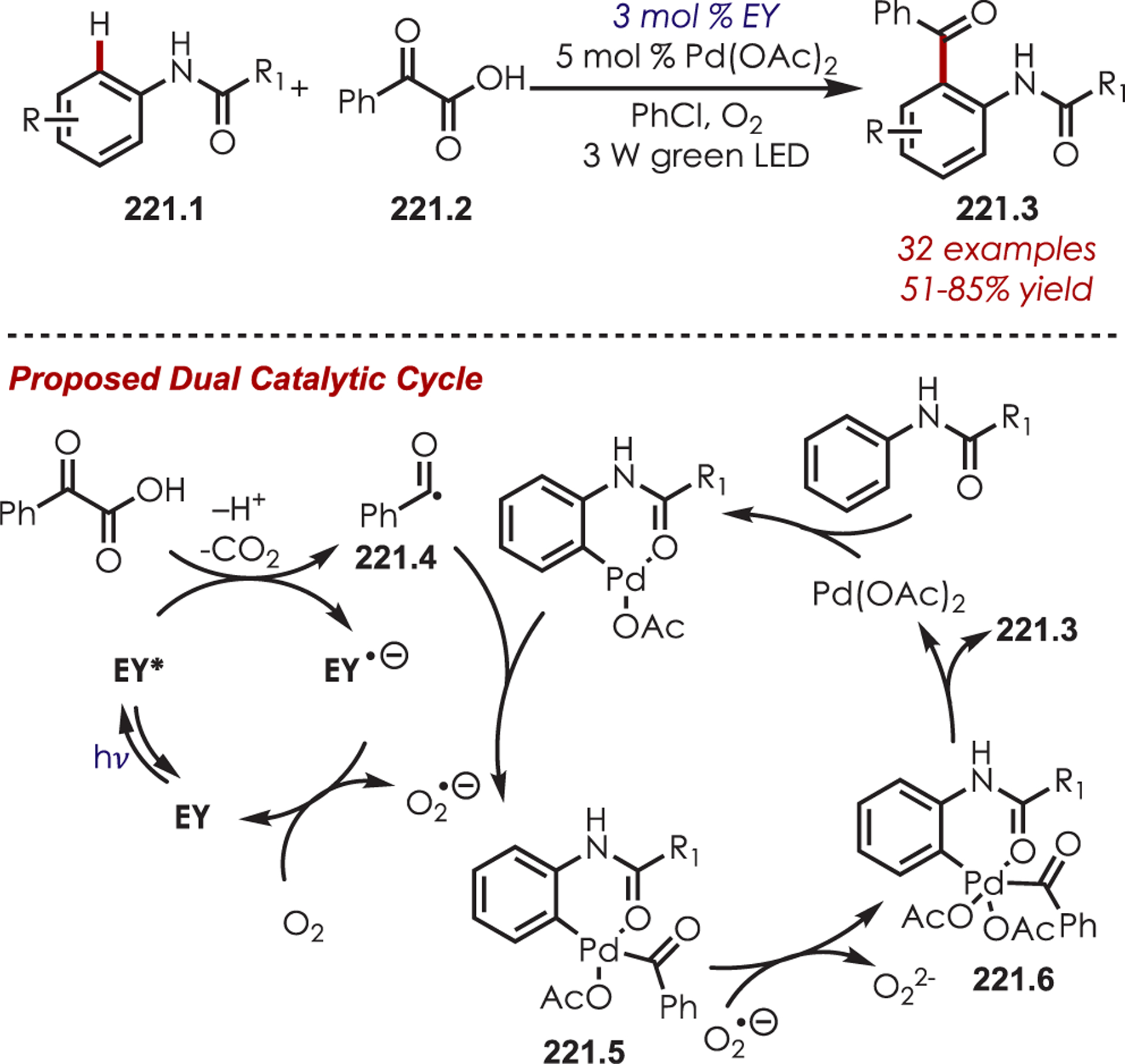
C–H Acylation of Acetanilides through Dual Photoredox and Palladium Catalysis
α-Oxo acids have further been demonstrated for C–H acylation of quinoxalin-2(1H)-ones (222.1) using acridine red as a photosensitizer (Scheme 222).385 The production of singlet oxygen enables the formation of an acyl radical species, which adds into a quinoxaline-2(1H)-one. A subsequent 1,2-hydrogen atom shift, oxidation, and deprotonation sequence affords the acylated products.
Scheme 222.
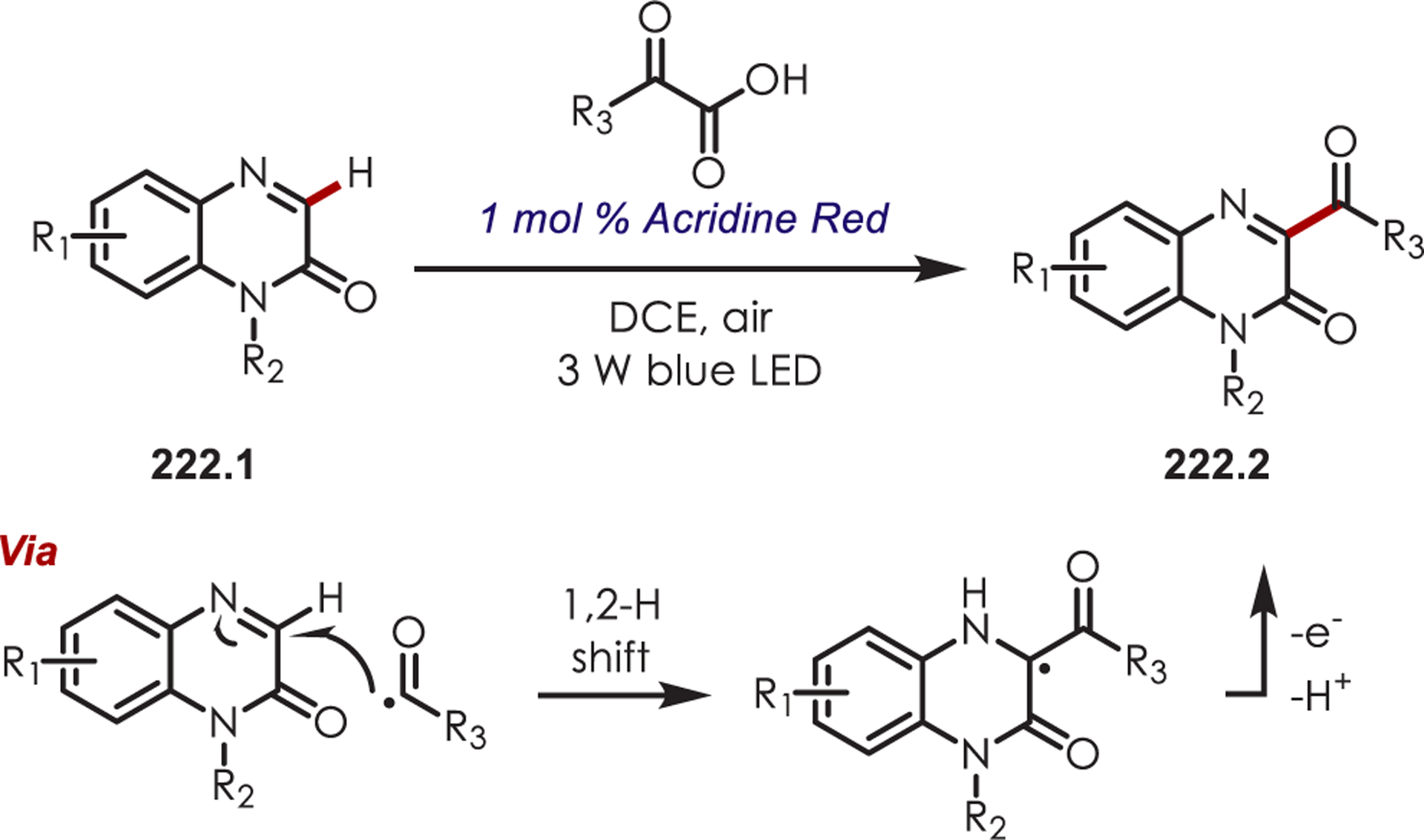
α-Oxo Acids as Acyl Radial Precursors for C–H Acylation
A decarboxylative Heck-type coupling of aliphatic carboxylic acids and olefins was reported using dual acridinium and cobaloxime catalysis (Scheme 223).386 A diverse scope of olefins including vinyl (hetero)arenes, vinyl silanes and vinyl boronates underwent C–H functionalization to afford the alkylated products. The alkyl radicals generated from SET of the carboxylate to the excited catalyst added into the styreneyl derivatives to produce an intermediate benzylic radical. The radical is trapped with Co(II) and undergoes a subsequent homolytic Co–C bond cleavage and β-hydrogen abstraction to afford the Heck-type products.
Scheme 223.
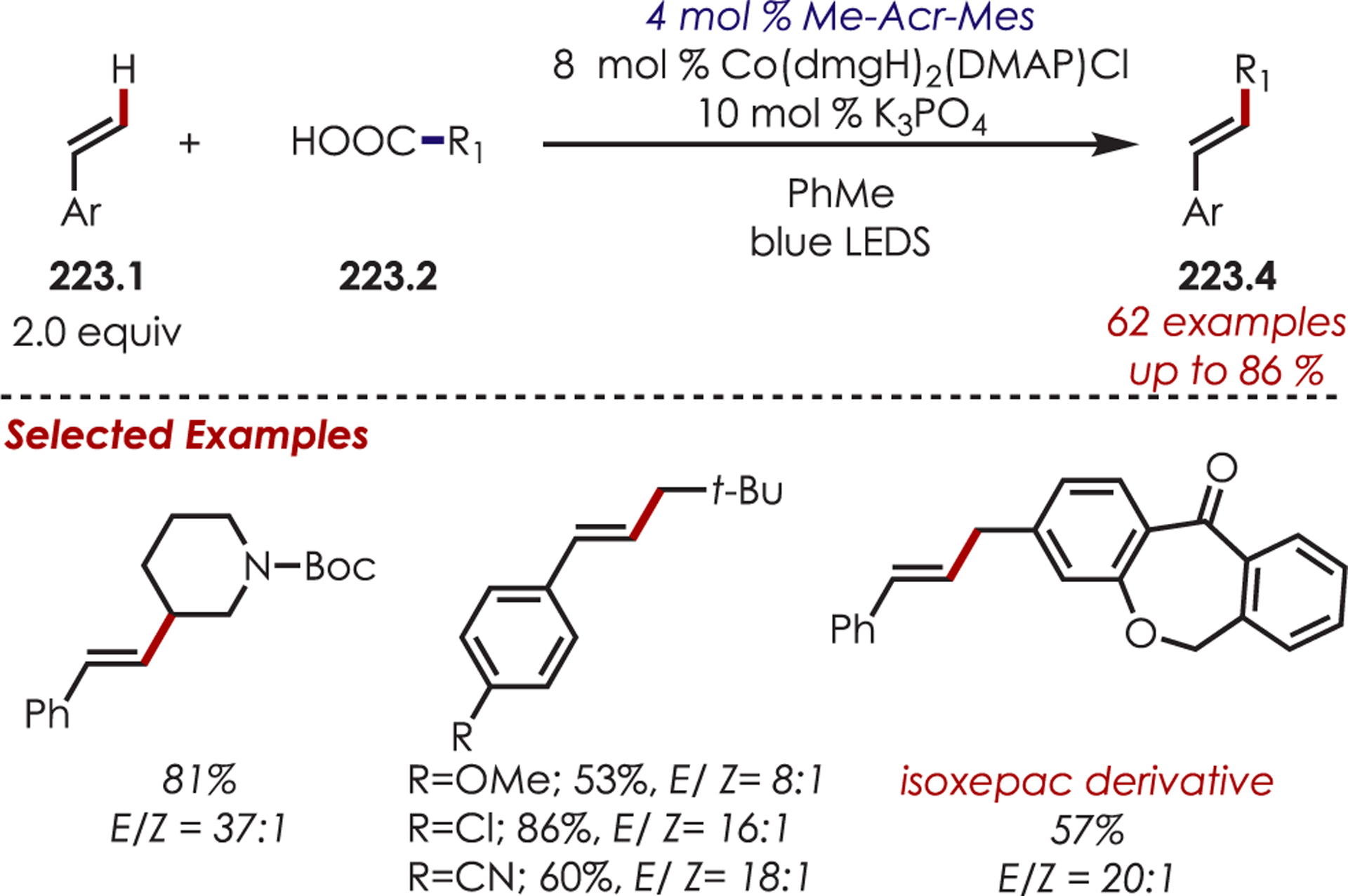
Decarboxylative Heck-Type Coupling of Aliphatic Acids and Vinyl (Hetero)arenes
Accessed via Sulfonium Salts.
Procter and co-workers have reported a unique strategy for the formation of aryl radicals from sulfonium salts. The sulfonium salts (224.3) were accessed in situ via an interrupted Pummerer reaction by treatment of benzothiophene S-oxide with triflic anhydride and the desired arene (224.1; Scheme 224).387 Alkyl arenes, anisoles and anilines were compatible and the arene scope had high functional group tolerance including halogens, triflates, mesylates, ketone, amido, ester, trifluoromethyl, and cyano groups. In the subsequent step, the intermediate aryldibenzo-thiophenium salt (224.3) then underwent a single electron reduction from PTH* to result in the expulsion of dibenzothiophene and afforded the reactive aryl radical species. An assortment of heterocycles was employed as coupling partners with the generated aryl radicals including pyrroles, thiophenes, furans, and indoles. Interestingly, complete regioselectivity was observed for 3-substituted heterocycles, giving selective substitution at C1. The authors highlighted this methodology in the synthesis of both natural and unnatural pseudilins (224.4), marine natural products.
Scheme 224.
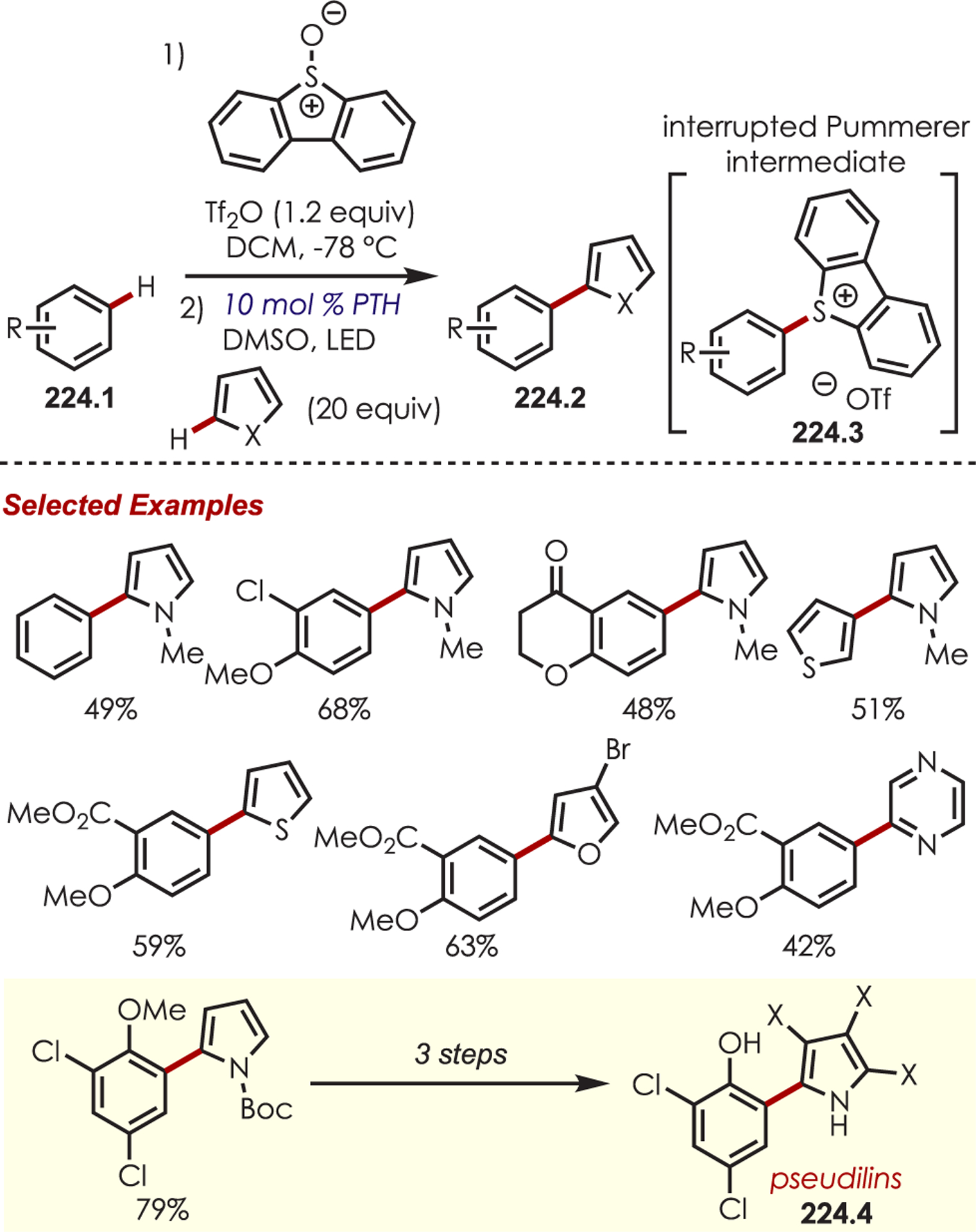
Generation of Aryl Radicals through C–H Activation by an Interrupted Pummerer Activation Allows for a Direct C–H/C–H Coupling of (Hetero)arenes
3.1.2. Transformations involving Nitrogen-Centered Radicals.
The addition of nitrogen-centered radicals into arenes via organic photoredox catalysis is far less explored than transition-metal alternatives.388 Nevertheless, there are reports of arene functionalizations through NCRs formed via organic photoredox catalysis.
A C–H imidation of (hetero)arenes (225.1) was reported using 3,6-dimethoxy-9H-thioxanthen-9-one as the photoreductant with an N-acyloxyphthalimide NCR precursor (Scheme 225).389 This proceeds through an analogous mechanism as discussed for NCR addition into arenes through inorganic mediated pathways (section 2.1.3). Experimental investigations of the mechanism suggest a PET between the N-acyloxyphthalimide and triplet photoexcited state of the catalyst to generate the NCR. A mixture of o/m/p isomers is observed for electron-deficient arenes. Simultaneously, Itoh and co-workers published an analogous transformation starting with phthalimide as the NCR precursor for indole functionalization using 2-tert-butylanthraquinone as the catalyst.390
Scheme 225.
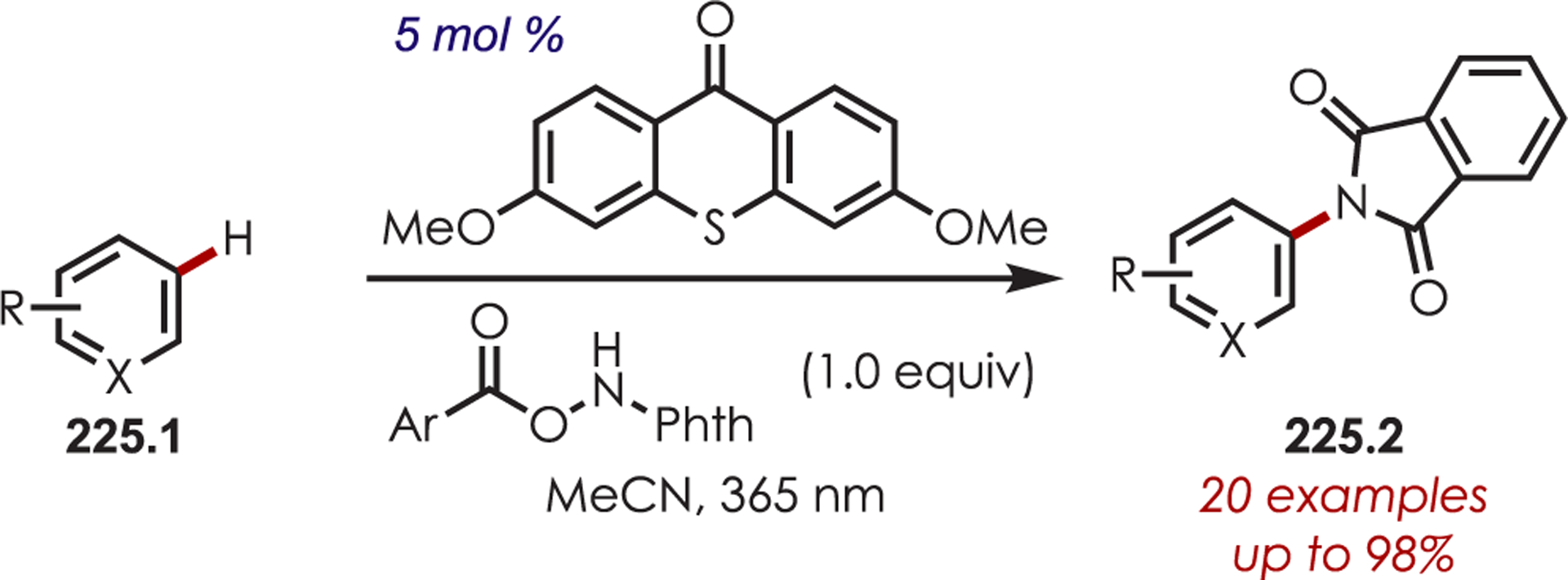
N-Acyloxyphthalimides for the Generation of Nitrogen-Centered Radical in Heteroaryl C–H Functionalization
o-2,4-Dinitrophenyloximes have been reported as nitrogen-centered radical precursors for the synthesis of phenanthridines with eosin Y (Scheme 226).391 Photoexcited eosin Y is reduced by DIPEA to furnish eosin Y radical anion that reduces the dinitrophenyloximes to the reactive NCR, which cyclizes onto the pendant arene.
Scheme 226.
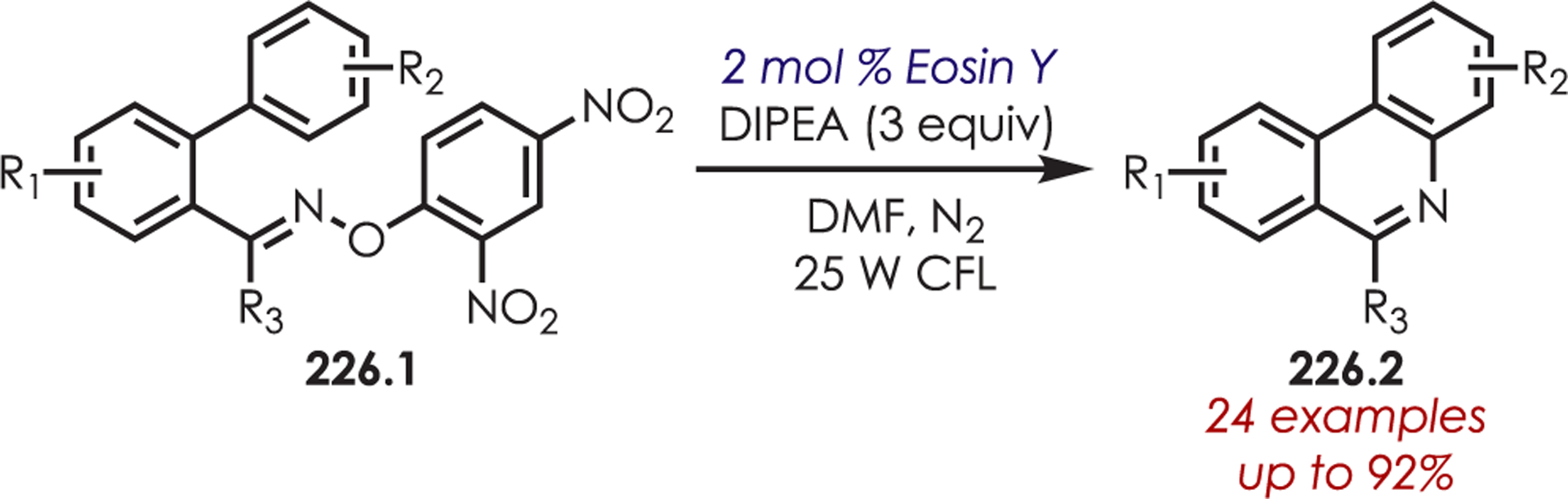
Synthesis of Phenanthridine Derivatives using o-2,4-Dinitrophenyloximes as NCR Precursors
A dehydrogenative coupling reaction of phenols (227.1) with acyclic diarylamines was accomplished using triphenylpyrylium (TPT) as the photooxidant (Scheme 227).392 The scope of this methodology is limited to coupling electron-rich phenol derivatives with electron-rich or neutral anilines in poor to good yields.
Scheme 227.
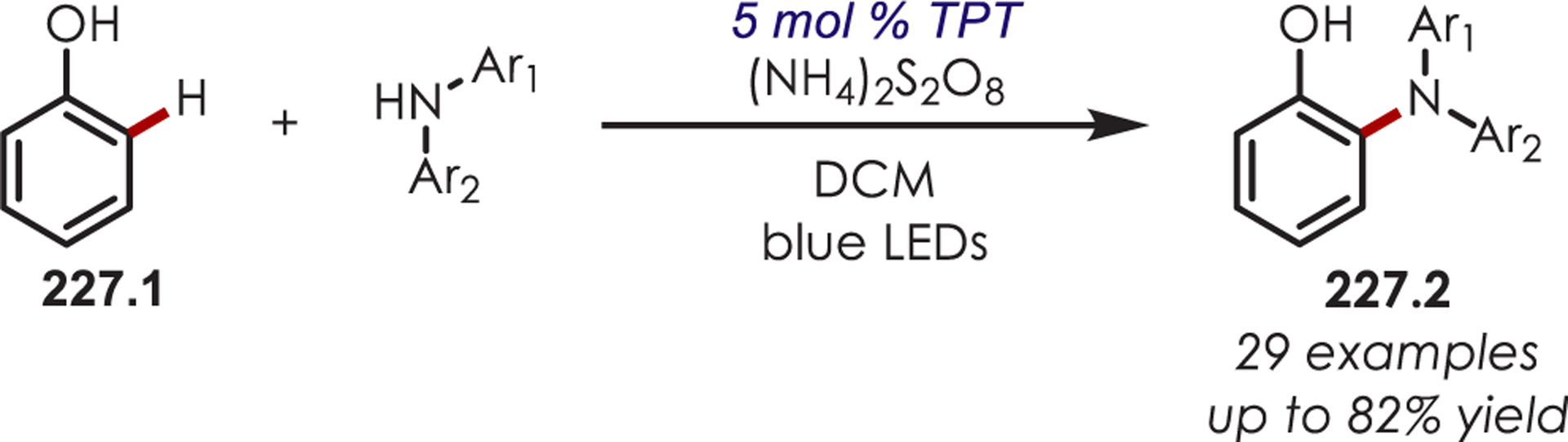
Phenol C–H Dehydrogenative Coupling with Diarylamines
An analogous transformation was reported one year later by Zhao and co-workers for the coupling of quinoxalinones (228.1) with aliphatic amines (Scheme 228).393 Eosin Y oxidizes the amine to the amine cation radical, which is then deprotonated by superoxide to the NCR. A Minisci-type addition into the quinoxalinone, followed by SET and deprotonation affords the aminated products with cyclic or acyclic aliphatic amines as suitable NCR precursors. The NCR was observed via ESR and control experiments confirmed the catalyst, light and oxygen were all required for efficient conversion to product.
Scheme 228.
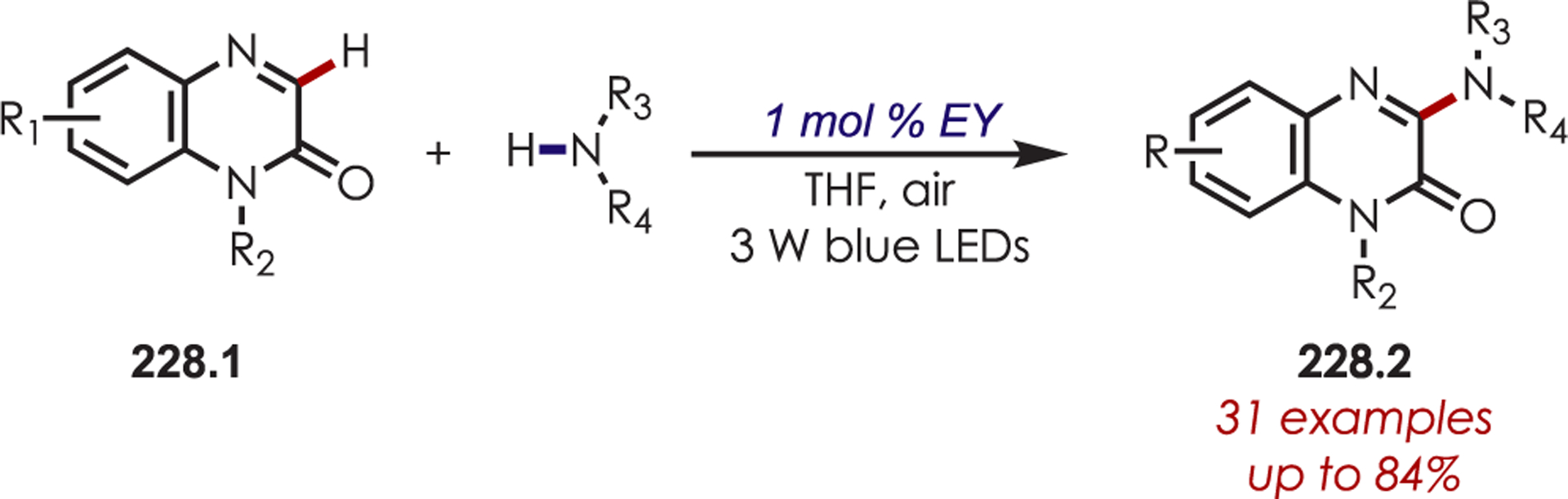
Quinoxalinone C–H Functionalization via Minisci-Type Addition of a Nitrogen-Centered Radical
3.1.3. Transformations involving Phosphorus-, Oxygen-, and Sulfur-Centered Radicals.
Accessed via Diaryl Phospine Oxide Arylphosphonates.
Single electron oxidation of phosphorus and sulfur derivatives produces reactive P- or S-centered radicals, PCR or SCR, respectively, which have been used for (hetero)aryl C–H functionalization. Inspired by the phosphorylation of aryl halides or pseudohalides via PCRs generated by photoredox catalysis,394,395 Wu and co-workers developed a method for C–H functionalization of heteroarenes via these intermediates (Scheme 229).396 The phosphorus-centered radical (229.4) is generated via a reductive quenching of photoexcited eosin B cation radical with diarylphosphine oxides. Addition of the PCR to thiazole and a final deprotonation produces C–H phosphorylated heteroarenes. The authors do not comment on regeneration of the catalyst. Around the same time, Lei reported a similar C–H phosphonylation of benzothiazoles using arylphosphonates with Na2eosin Y as the photooxidant.397
Scheme 229.
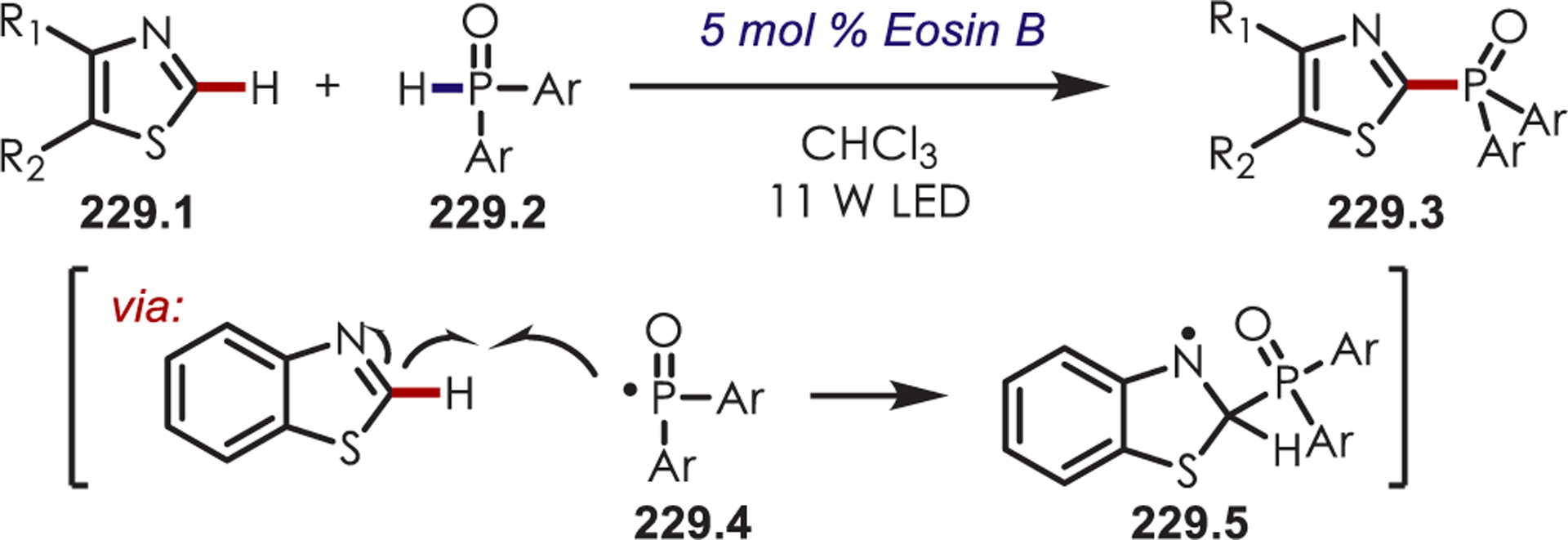
Addition of Phosphate-Centered Radicals for Heteroaryl C–H Functionalization
Arylphosphonates as PCR precursors have also been utilized in the phosphonylation of aminoquinolines (230.1) via dual transition-metal and photoredox catalysis (Scheme 230).398 High regioselectivity for C4 or C5 functionalization was controlled by the coordination of Ag or Na to the quinoline or amide nitrogen, respectively. Later, the authors expanded this work to C4-selective phosphonation of 8-hydroxyquinoline esters through a dual silver and eosin Y photoredox-mediated process.399
Scheme 230.
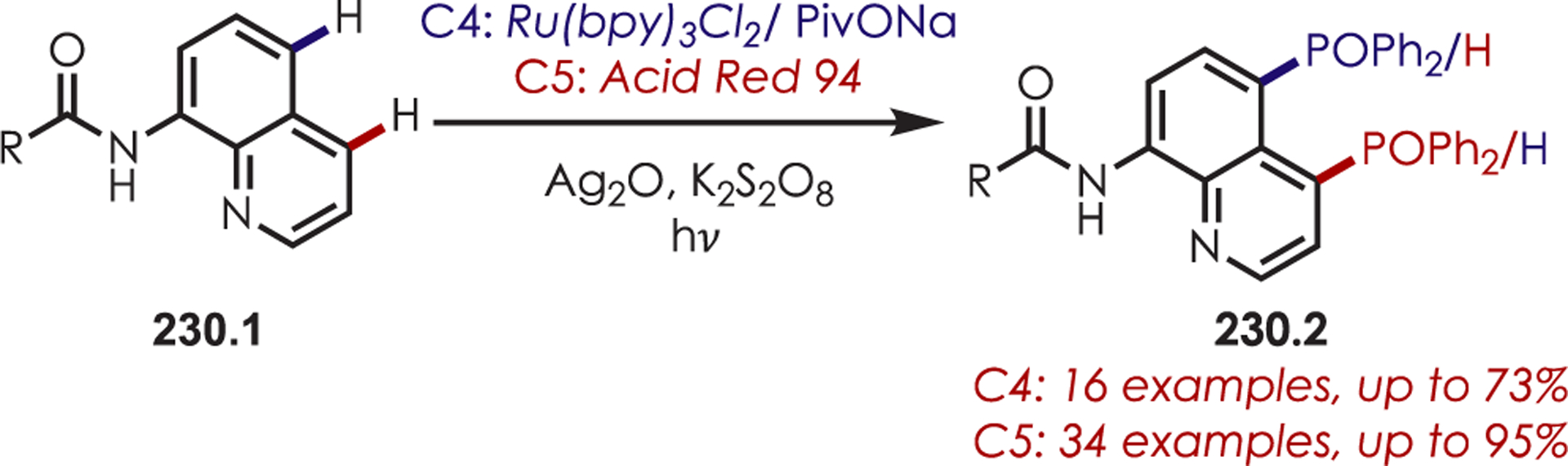
Heteroaryl C–H Phosphonylation of Aminoquinolines
Accessed via Carboxylates.
Gonzalez-Gomez and co-workers accomplished a metal-free dehydrogenative intramolecular lactonization of 2-arylbenzoic acids (231.1) using an acridinium salt as the photoredox catalyst (Scheme 231).400 The benzoyloxy radical (231.3), formed by PET with the photoexcited acridinium, undergoes a 6-endo-trig cyclization to give an intermediate cyclohexadienyl radical (231.4). A subsequent oxidative HAT or SET/deprotonation sequence with the sulfate radical anion generates the desired substituted benzocoumarins. The catalyst is regenerated by oxidation from the persulfate anion. The resulting sulfate radical anion may act as the oxidant in the final step. The authors propose that for more electron-rich biphenyls an oxidation of the arene to form an aryl cation radical may be possible; however, the regioselectivity observed is not in accordance with an aryl cation radical mechanism. This lactonization was later reported under oxidant and acceptor-free conditions by using a hydrogen-evolving cobalt catalyst in conjuction with an acridinium photooxidant.401
Scheme 231.
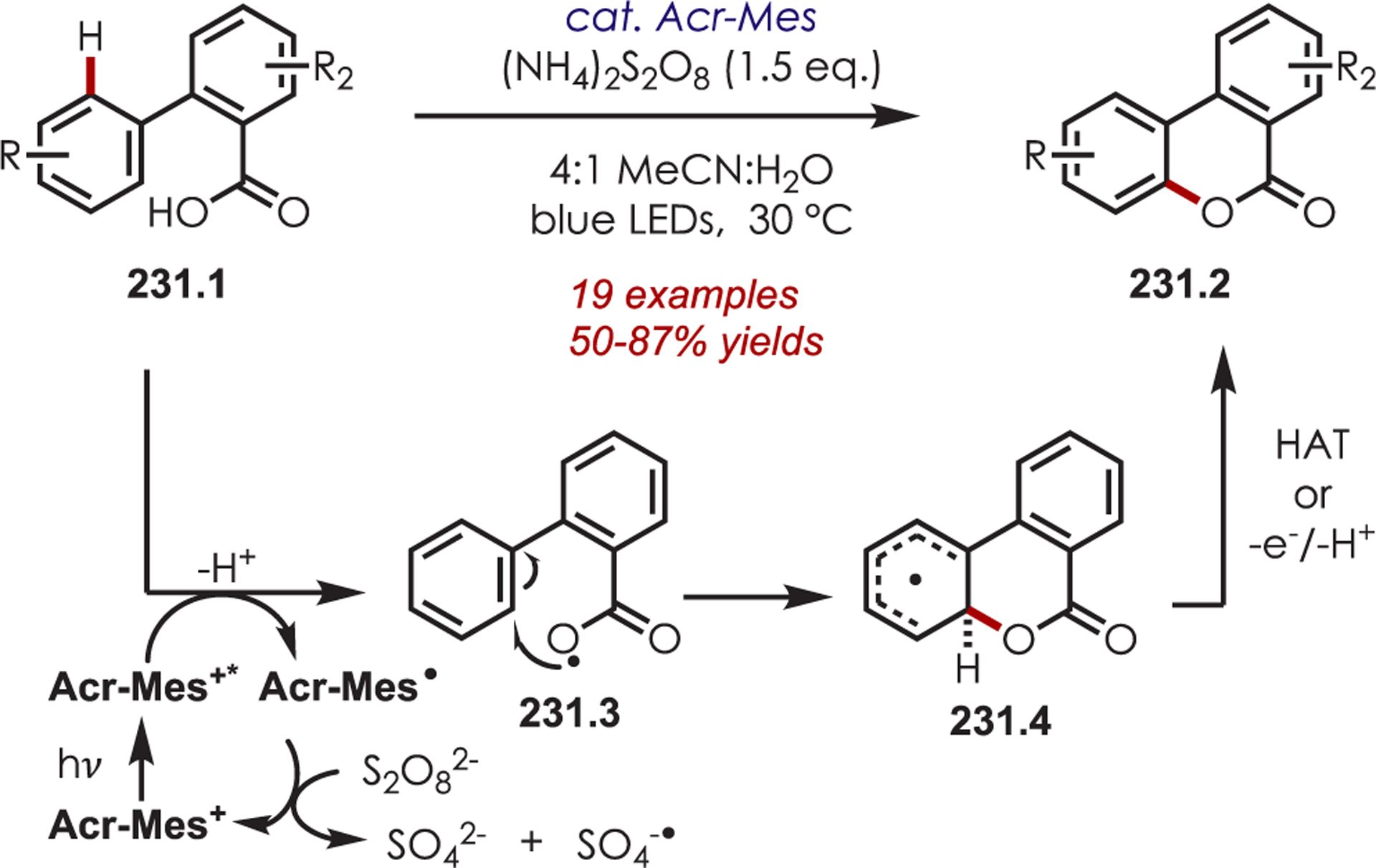
Intramolecular Lactonization of 2-Arylbenzoic Acids through a Benzyoyloxy Radical Cyclization
SCR Accessed via Thioanlilides, Thiols, Isothiacyanates, and Sulfonyl Chlorides.
A metal-free synthesis of benzothiazoles and thiadiazoles has been reported using thioanilides as sulfur-centered radical precursors (Scheme 232).402,403 Oxidation of the thiolamide anion by photoexcited Eosin Y generated the reactive SCR, which proceeded to cyclize onto the pendant arene or imine to furnish a benzothiazole or thiadiazole, respectively.
Scheme 232.
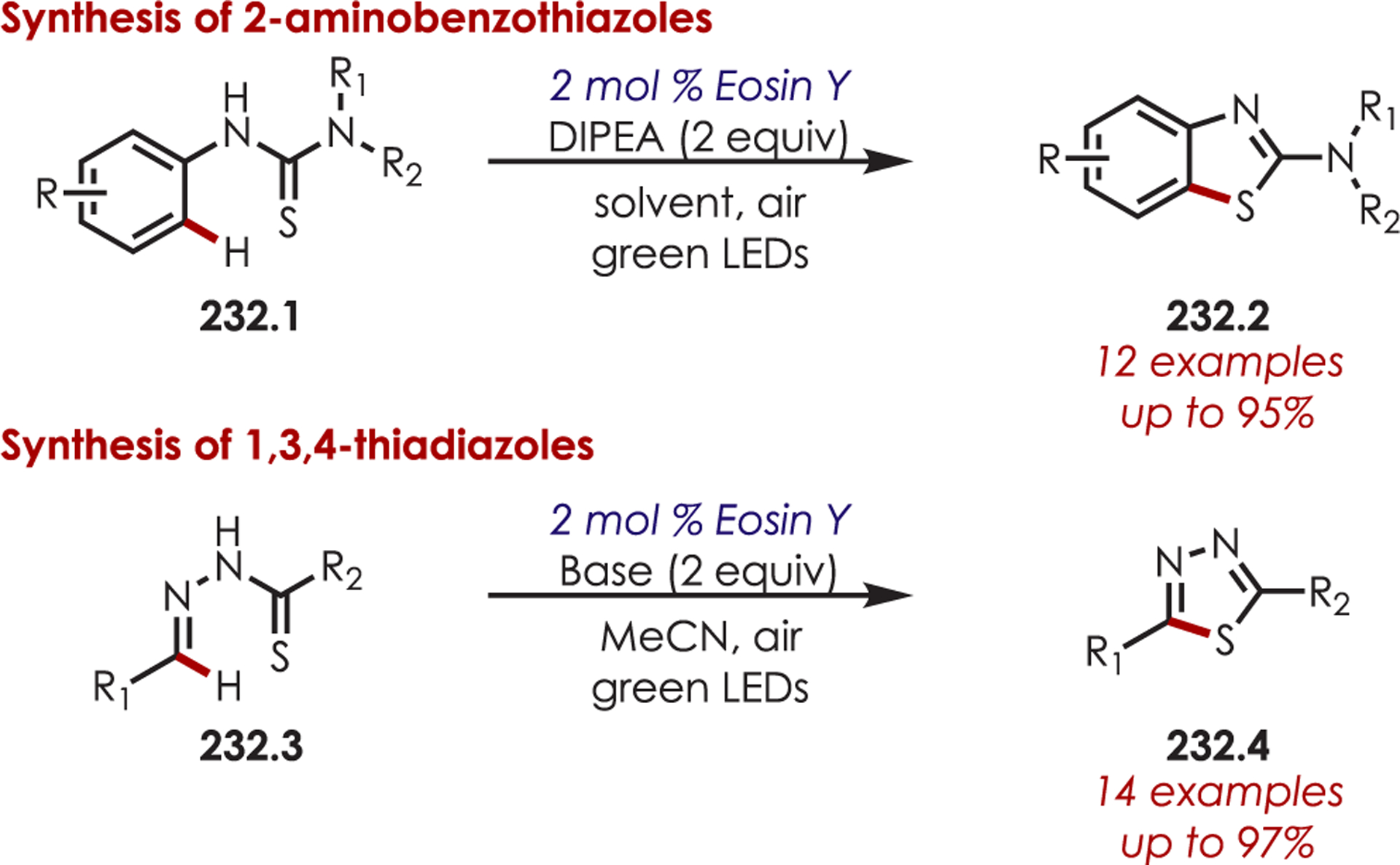
Sulfur-Centered Radicals for the Synthesis of Thiazoles
The direct C3 sulfenylation of indoles (233.1) was accomplished with thiophenols as SCR precursors and rose bengal (RB) serving as the photooxidant (Scheme 233).404 The authors propose the SET of molecular oxygen by excited RB* generating singlet oxygen which abstracts a hydrogen atom from the arylthiol. This undergoes a radical addition to indole, which is then rearomatized by an oxidation/deprotonation sequence to give the sulfenylated indole product.
Scheme 233.

C3 Selective C–H Sulfenylation of Indoles through a Minisci-Type Reaction with Thiyl Radicals
Following these reports, methods for sulfenylation of imidazo[1,2-a]pyridines and indoles (234.1) have been developed with thiophenol SCR precursors (Scheme 234).405
Scheme 234.
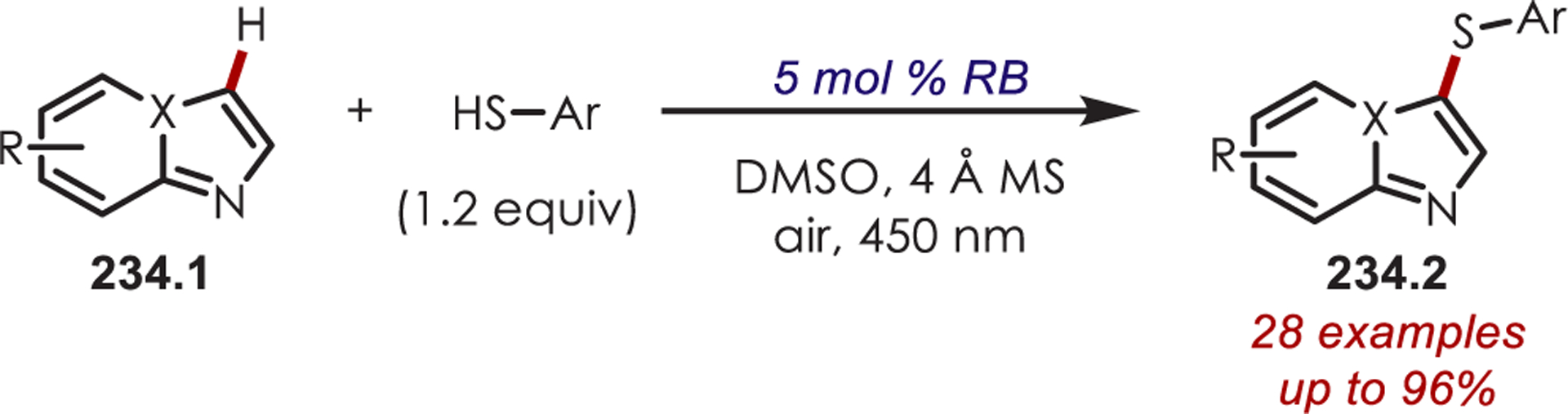
C–H Sulfenylation of Heteroarenes through a Minisci-Type Reaction with Arylthiyl Radicals
In addition to thiols, thiocyanates have been proposed as SCR precursors and have been successful in the C3 thiocyanation of indoles. An operationally simple photoredox catalyzed thiocyanation was first reported by Li in 2014 (Scheme 235).406 Oxidative SET between ammonium thiocyanate and RB* produces the reactive thiocyanate radical. Subsequent radical addition to indole, oxidation and deprotonation yields the C3 thiocyanated indoles (235.1). Molecular oxygen is utilized as the terminal oxidant to regenerate the photocatalyst. Electron-rich and weakly electron-withdrawing indoles worked well in this procedure, giving the product in high yields. However, strongly electron-deficient indoles failed to give the desired product. Some N-substitution was tolerated including N-Me-, N-Ph-, and N-Bn-substituted indoles.
Scheme 235.
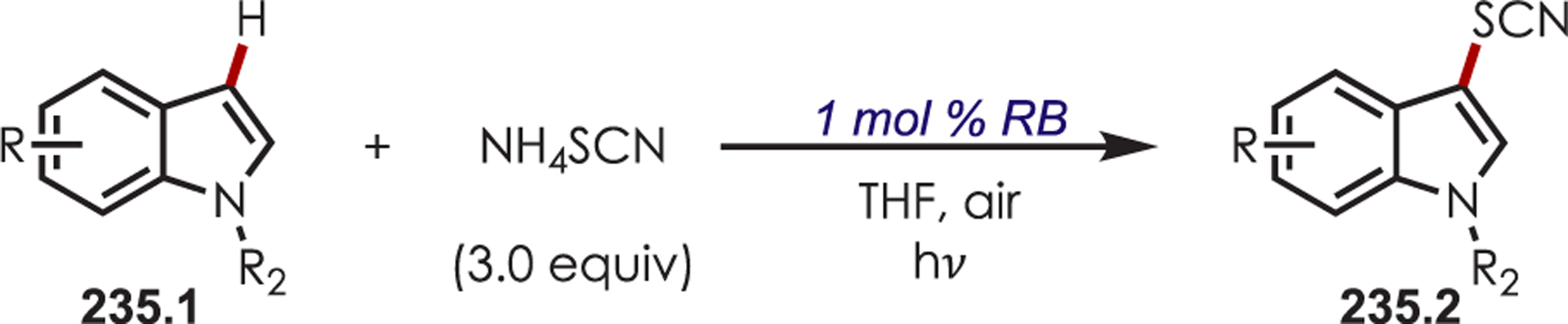
C3 Thiocyanation of Indoles through a Minisci-Type Reaction with Thiocyanate Radical
Hajra reported a thiocyanation of imidazoheterocycles (236.1) using eosin Y as the photooxidant (Scheme 236).407 This was also successful in the selenocyanation of imidazopyridines when using KSeCN. The proposed mechanism follows the previous examples of SCR addition to indole derivatives.
Scheme 236.
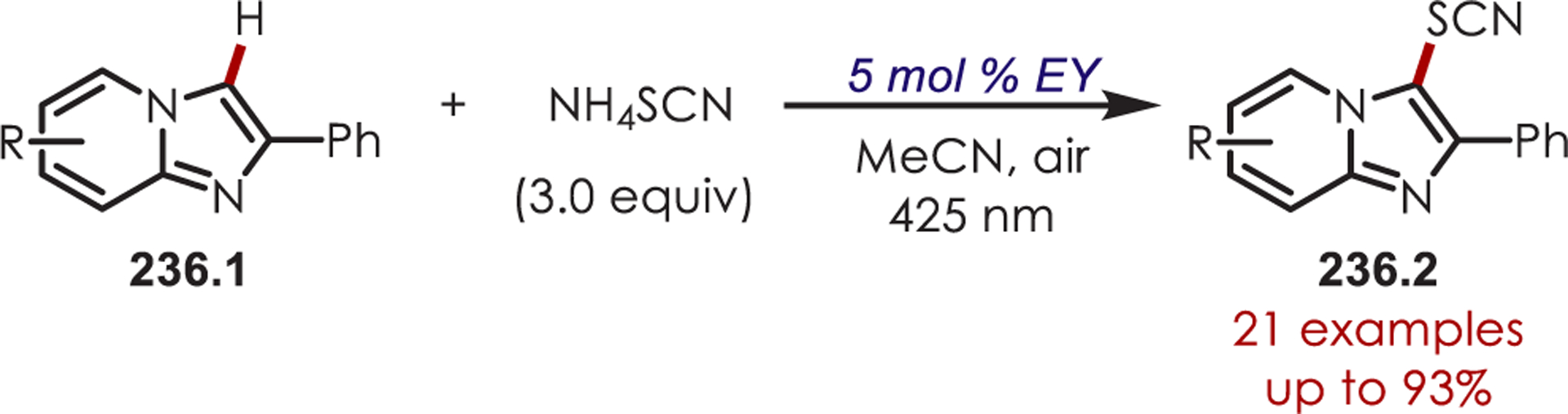
C3 Thiocyanation of Imidazoles through a Minisci-Type Reaction with Thiocyanate Radical
Additionally, sulfinic acids (237.2) have been reported as SCR precursors for the sulfenylation of heteroarenes.408 Wang and co-workers developed a method with eosin B as the photoredox catalyst (Scheme 237). Oxidation of the sulfinic acid generates a sulfonyl radical which is converted into a thiol sulfur-centered radical through a series of reductions. A Minisci radical addition into the heteroarene, oxidation, and deprotonation produces the final sulfenylated product.
Scheme 237.
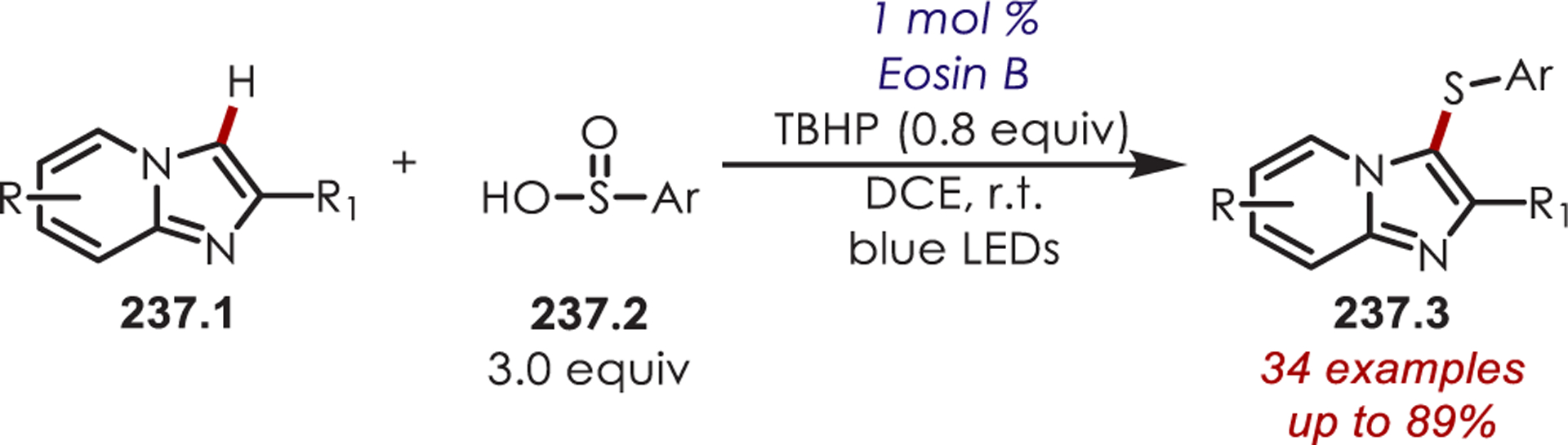
Heteroaryl C–H Sulfenylation Using Sulfinic Acids
3.1.4. Transformations involving Charged Open-Shell Intermediates.
Arene Cation Radicals.
(Hetero)arene cation radicals, formed via single-electron oxidation of (hetero)-aromatics, have been utilized as reactive species for a range of C–C, C–N, C–O, and C–X bond formations. Organic photoredox catalysts are particularly attractive for accomplishing the direct oxidations of arenes due to their high excited state reduction potentials, up to +2.72 V vs SCE for QuCN+.409
Earlier efforts involving the photosensitization of organic compounds with dicyanobenzenes410–414 inspired Pandey and co-workers’ seminal work on the functionalization of arene cation radicals. Following oxidation by dicyanobenzene (DCN) photooxidants, Pandey reported the intermediate arene cation radical would undergo intramolecular cyclizations with pendant nucleophiles to produce coumarins (238.1),415 chromenes (238.2),416 benzofurans (238.3),417,418 and carbonannulated products (238.4) (Scheme 238).419,420 The authors note the regioselectivity of these transformations were consistent with the calculated electron densities (Huckel or MNDO) of the HOMO of the arene cation radical species.
Scheme 238.
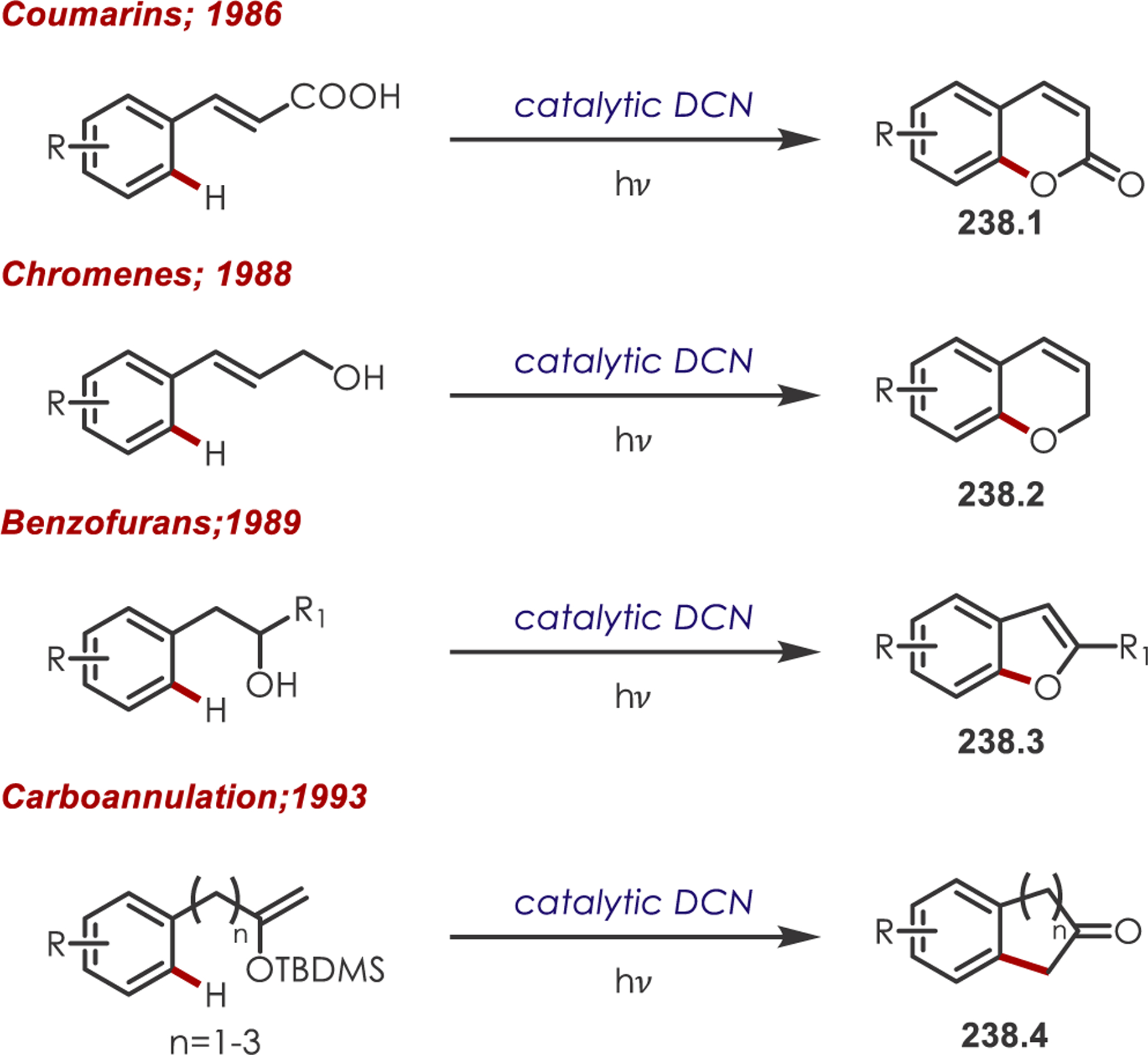
DCN-Catalyzed C–H Cyclizations through Arene Cation Radicals
In 1990, Pandey reported an extension of this method in an intramolecular amine cyclization (Scheme 239).421 Following oxidation by DCN*, nucleophilic attack of the ethylamine to the cation radical produces the desired substituted dihydroindole derivatives. Further oxidation from air was noted to give the oxidized aromatic indole products. This method was utilized in the formation of benzopyrrolizidines in one pot, which have potential as mitomycin precursors. Using DCA as the photoredox catalyst, oxidation of veratrole (239.3) gave the desired dihydroindole intermediate (239.4). Switching the wavelength 8 h later generates singlet photoexcited DCA, which enables oxidation of the iminium cation intermediate and in the presence of MeOH yields the mitomycin precursor (239.5) in 62% in one pot.
Scheme 239.

Intramolecular C–H Amination and Application to the Synthesis of Mitomycin Precursors
One year later, Qian reported the addition of benzotriazoles (240.2) to benzenoids via aryl cation radical intermediates employing 9,10-dicyanthracene (DCA) as the photooxidant (Scheme 240).422 The authors propose that the mechanism proceeds through an electron transfer between a cation radical and benzotriazole, which after deprotonation generates the azole radical. The addition of the radical to another equivalent of the neutral arene, followed by aerobic oxidation, furnishes the final product. The authors support this mechanism based on the observation of an ESR signal they assign to the benzotriazole radical. Additionally, this intermediate radical was successfully trapped with phenyl tert-butyl nitrone. The scope of this chemistry is limited to biphenyls, naphthalenes, and anisoles and, unfortunately, the selectivity of the transformation is not discussed.
Scheme 240.

Arene C–H Amination with Benzotriazole Nucleophiles
While the work in the 1990s by Pandey, Bhalerao, Qian, and Karthikeyan gave some insight into the reactivity of aryl cation radicals, it went largely unnoticed, conceivably due to the limited substrate scopes and inadequate mechanistic understanding of arene cation radicals at this time. Several decades later, the interest in C–H functionalization via reactive arene cation radicals was reawakened by efforts from Fukuzumi.
Fukuzumi first reported the photocatalytic oxygenation of benzene to phenol (241.2) through arene cation radicals formed by 3-cyano-1-methylquinolinium (QuCN+)423 or DDQ,424 serving as the photoredox catalyst and O2 serving as the stoichiometric oxidant (Scheme 241). Single-electron oxidation of benzene (Eox = +2.48 V vs SCE in MeCN) was accomplished using QuCN+, which is capable of oxidizing benzene in its singlet photoexcited state (Ered* = +2.72 V vs SCE) or via an electron transfer from benzene to the triplet excited DDQ (E*red = +3.18 V vs SCE).
Scheme 241.
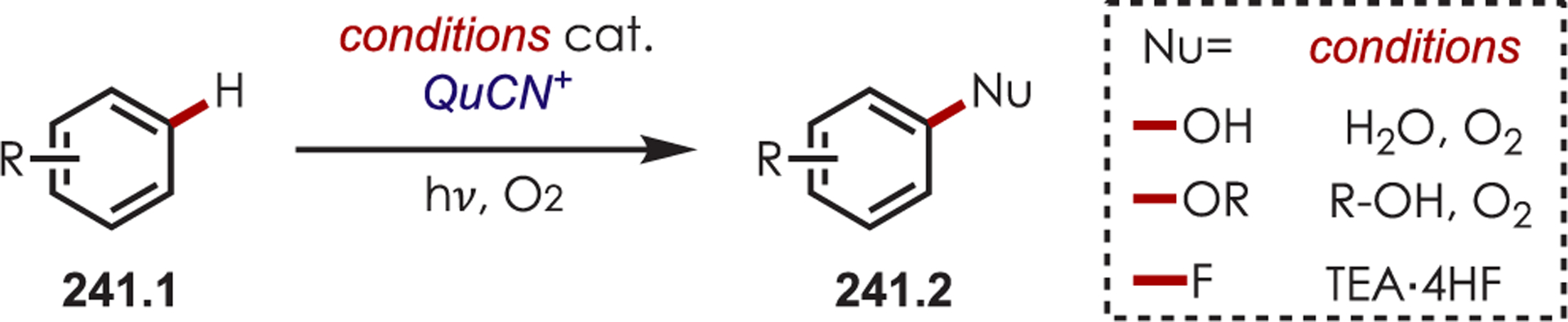
Arene C–H Functionalization with Alcohol, Fluoride, and Bromide as a Nucleophile
Under the same putative mechanism for the hydroxylation of benzene, Fukuzumi later accomplished the C–H functionalization of benzene with alcohols,425 fluoride,426 and bromide.427 These methods produced a range of halogenated or alkoxylated arenes; however the scope of arenes investigated was limited by its oxidation potential, where E*red[Cat] > Eox[substrate]. In the case of bromination, monoselective halogenation is observed, but the regioselectivity of the transformation was not well understood for nonsymmetrical arenes.
The mechanism of these transformations is proposed to proceed via excitation of the catalyst by visible light to generate the photoexcited quinolinium ion (Scheme 242). Single-electron transfer from the arene (242.1) to the photoexcited catalyst gives an arene cation radical and reduced quinolinium.A benzene cation radical dimer (242.3) was observed via IR spectroscopy, which is in equilibrium with the nondimeric cation radical.425 Molecular oxygen is reduced by the quinolinium radical, regenerating the catalyst and producing HO2• after protonation. Addition of the nucleophile to the cation radical and formal hydrogen atom abstraction by HO2• afforded the substituted arene (242.5). The resulting hydrogen peroxide reacts with hydrogen bromide and the starting arene to generate another equivalent of product and water. The same mechanism was proposed for the addition of fluoride and alcohols to aryl cation radicals.
Scheme 242.
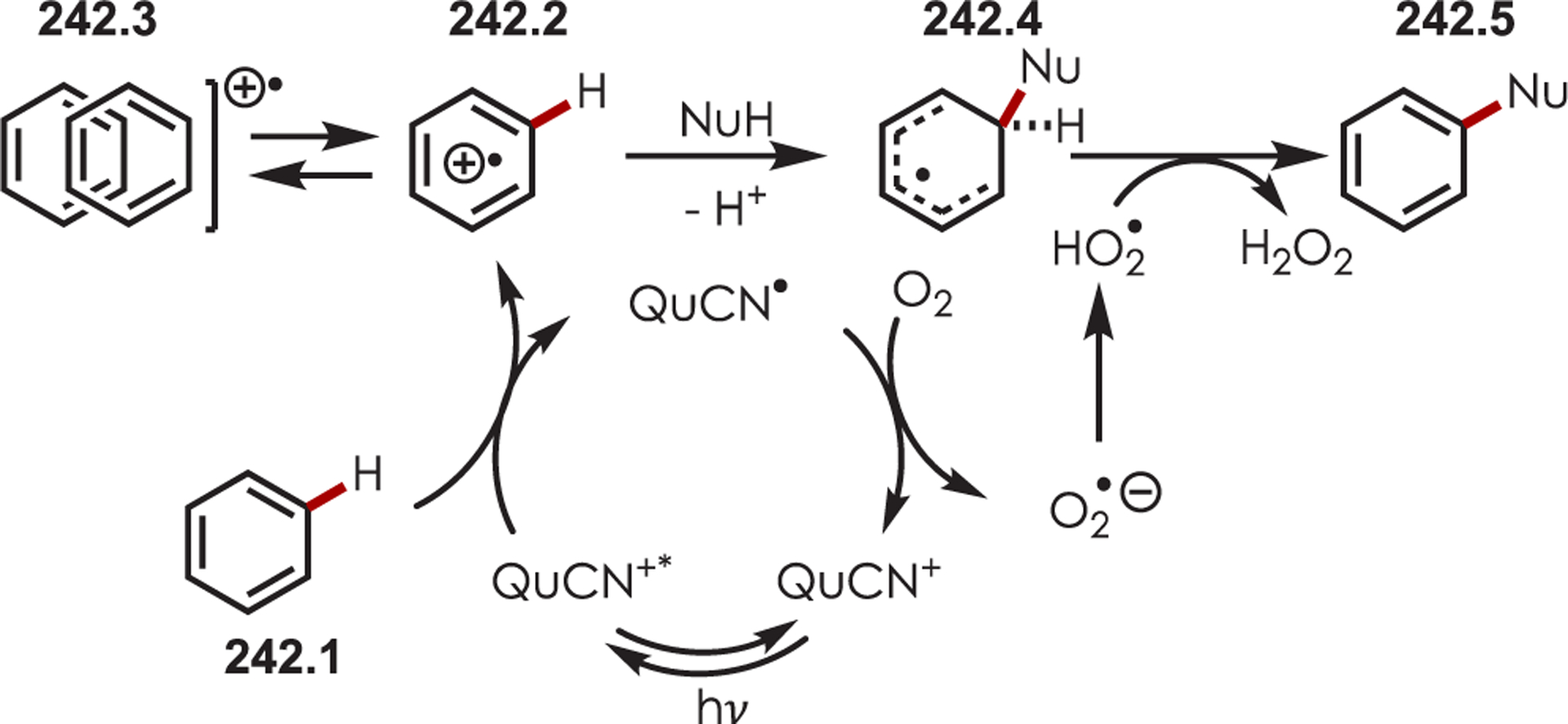
Proposed Mechanism of Aryl C–H Functionalization with QuCN+ as the Photooxidant
In 2001, Fukuzumi first reported the discovery of a 10-alkylacridinium ion which enabled a PET with various electron donors (Scheme 243).428 The high oxidizing strength of the photoexcited state of acridinium ions have been utilized in the oxidation of a large collection of substrates in order to access their cation radicals for a range of transformations.429
Scheme 243.
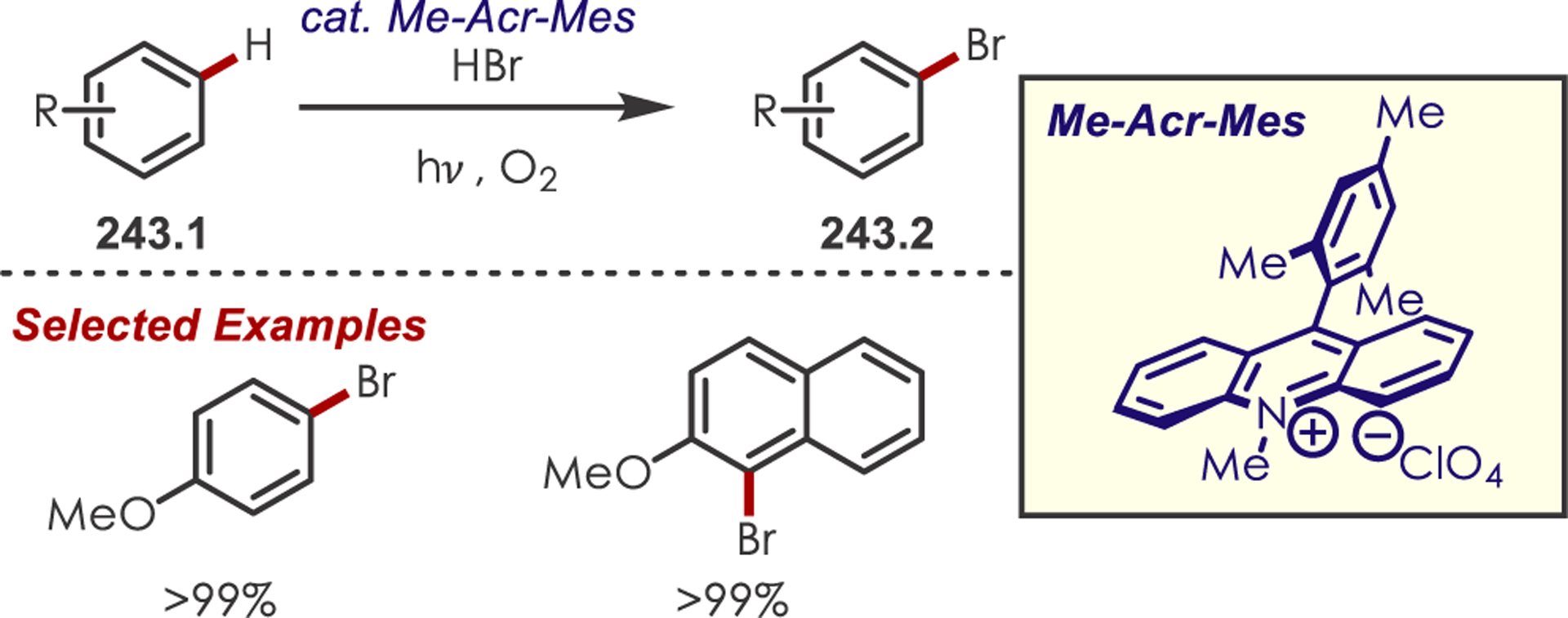
Aryl C–H Bromination with an Acridinium Salt as the Photooxidant
In their efforts toward developing an oxidation of toluenes to benzoic acids, Fukuzumi noted that trimethoxybenzenes (244.1) underwent a chlorination with Acr-Mes and hydrogen chloride.430 However, other anisole derivatives only produced trace product (Scheme 244).
Scheme 244.
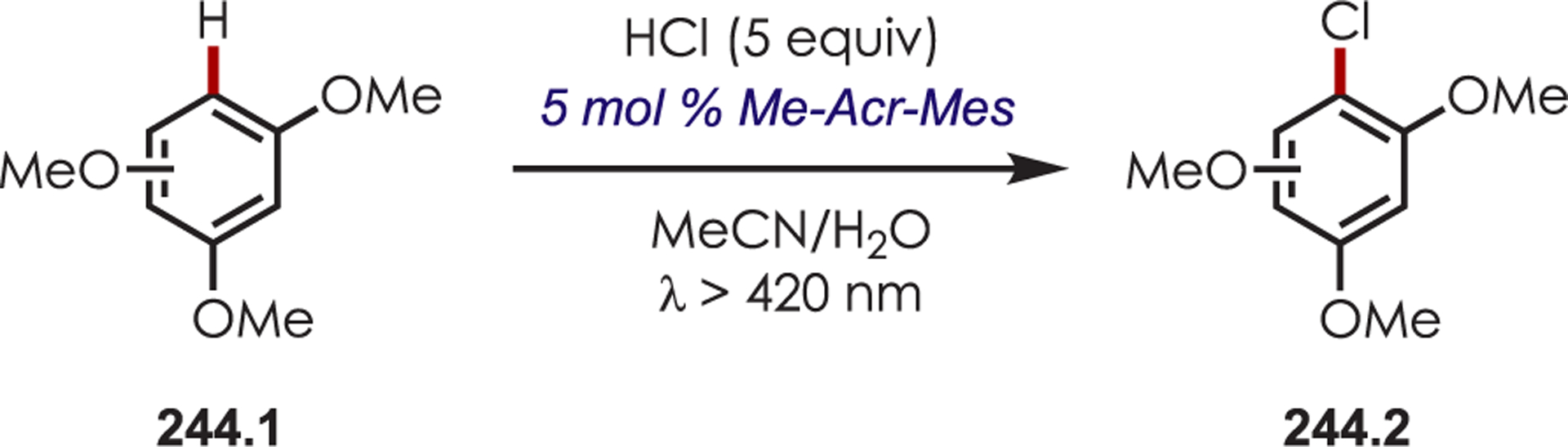
Chlorination of Trialkoxyarenes with an Acridinium Salt as the Photooxidant
Recently, Nicewicz and co-workers utilized a derivative of Fukuzumi’s highly oxidizing acridinium, Ph-Acr-Mes, catalyst to accomplish site-selective arene C–H aminations (Scheme 245).431 The scope of this methodology extends to a variety of electron-rich arenes (245.1), including phenol derivatives, halogenated arenes, biphenyls, dimethoxypyridine, and more complex pharmaceutical derivatives, such as naproxen methyl ester. The nucleophilic coupling partner includes pyrazoles, triazoles, tetrazole, and di-N-Boc adenine. High para-regioselectivity was observed, with minor formation of the ortho-regioisomer.
Scheme 245.
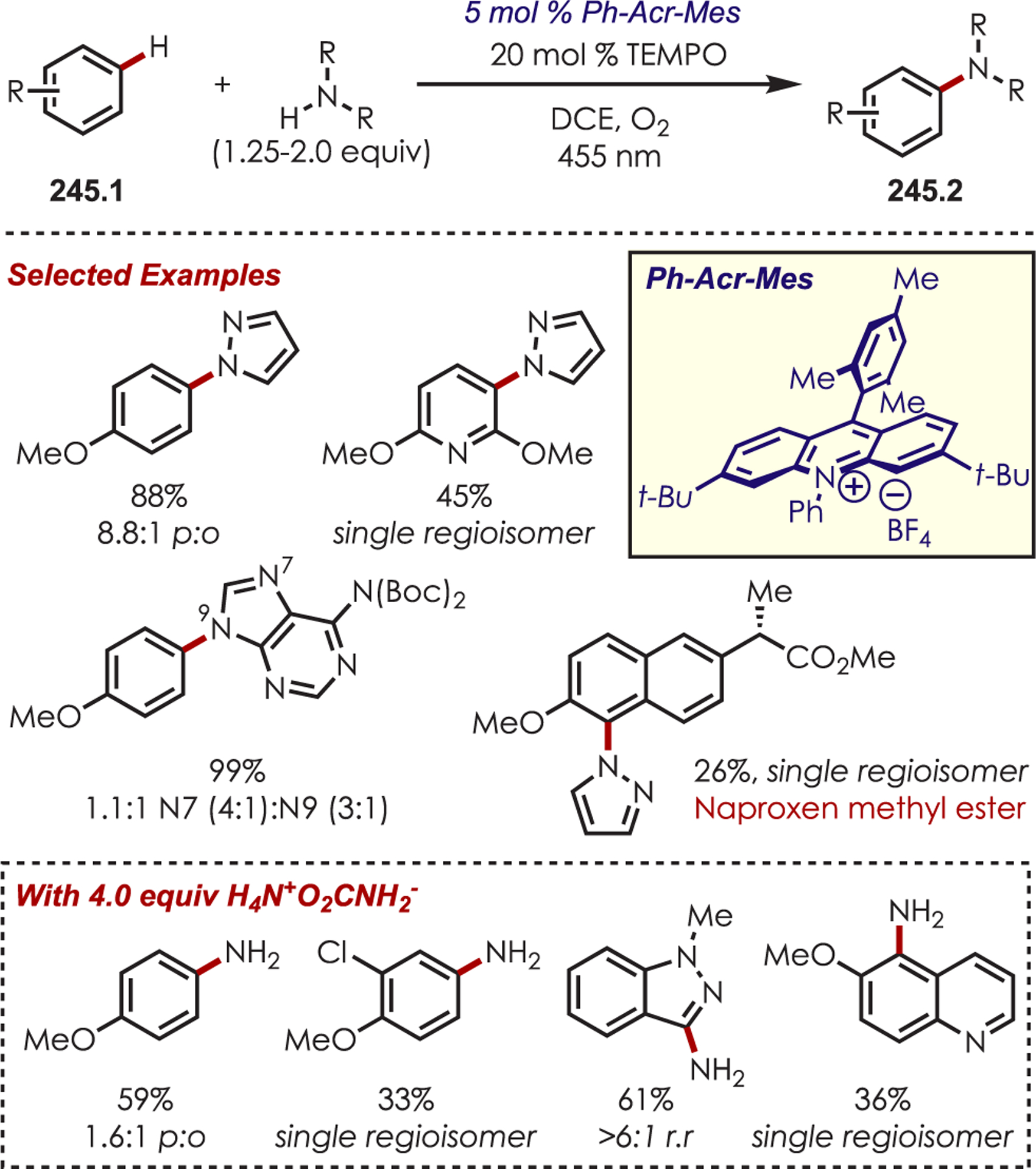
Arene C–H Amination with Azole Nucleophiles
The arene cation radical (246.1) was generated by oxidation from photoexcited Acr-Mes+*. Addition of a nitrogen nucleophile, including azoles and ammonium carbamate, to an aryl cation radical generates an intermediate cyclohexadienyl radical 246.2 (Scheme 246). A proposed subsequent deprotonation and rearomatization from catalytic TEMPO furnished the aminated arenes. Oxidation of Mes-Acr• by dioxygen turns over the catalytic cycle and generates O2•−. TEMPO is regenerated by a hydrogen atom transfer event from the O2•− or other superoxide intermediates (Scheme 246). The authors also proposed an alternative pathway, supported from prior art from Sonnatag,432–434 which involves the trapping of O2 by the cyclohexadienyl radical, followed by an intramolecular hydrogen atom transfer and elimination of peroxyl radical.
Scheme 246.
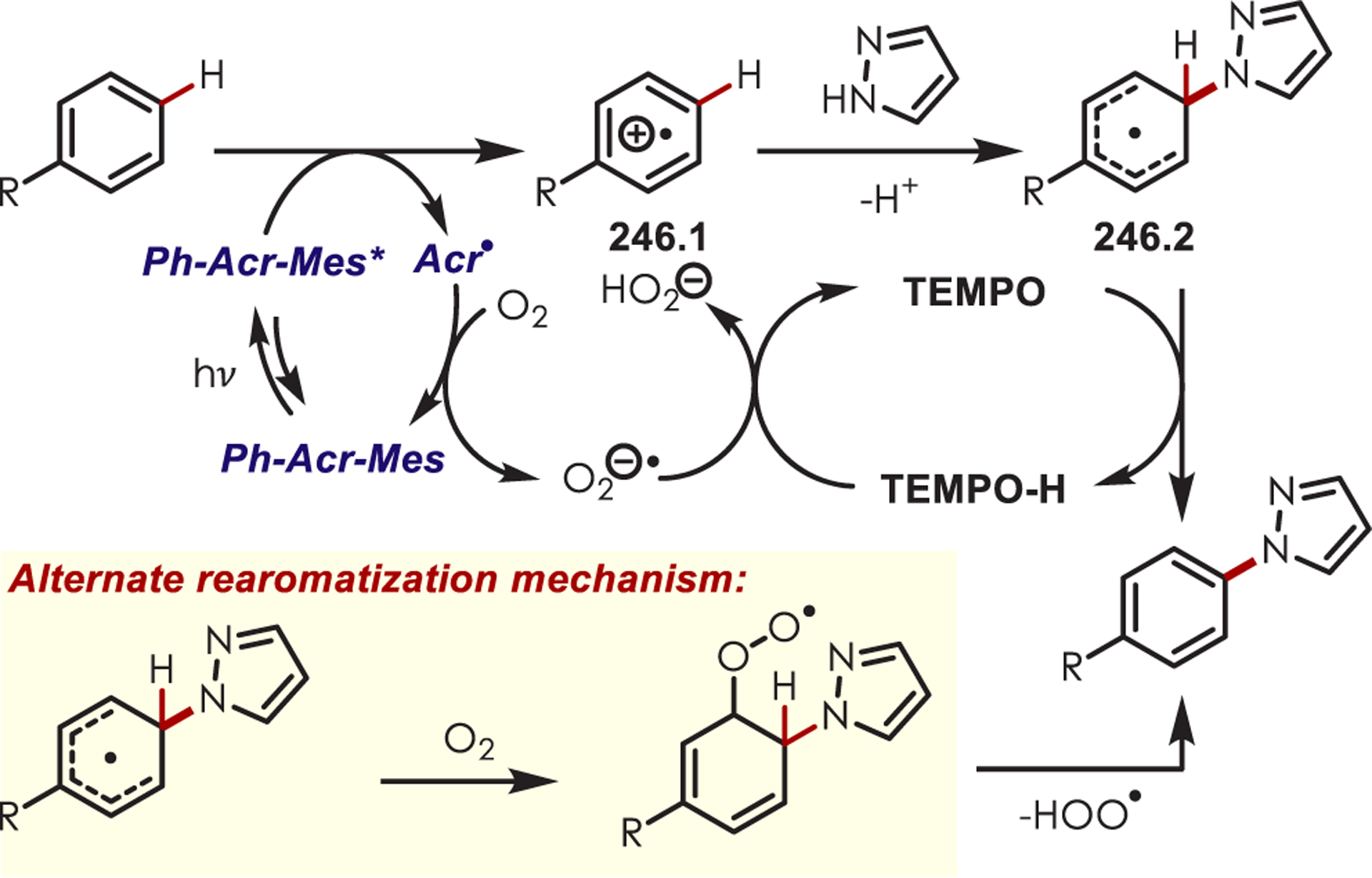
Proposed Mechanism of Arene C–H Amination with Azole Nucleophiles
The site-selectivity of these transformations was later found to be correlated with the electron density of the arene and cation radical intermediates.435 Nicewicz and co-workers developed a computational model to predict the site-selectivity of C–H functionalization and identified trends in the classes of (hetero)arenes. This was accomplished by using a natural population analysis (NPA) electron density calculations (B3LYP/6–31G+(d,p)) of the arene and the respective arene cation radical. The difference in the NPA values between the arene and aryl cation radical correlated to the major site of functionalization. In general, they found substituted benzenoids, pyridines, and quinolines undergo reactivity that is directed by the electron-donating group, with most models predicting the major product as the para-regioisomer (Scheme 247). The predicted selectivity matched well with the experimentally observed results, successfully predicting the site selectivity of the product as either a single regioisomer or a mixture of ortho-/para-regioisomers.
Scheme 247.
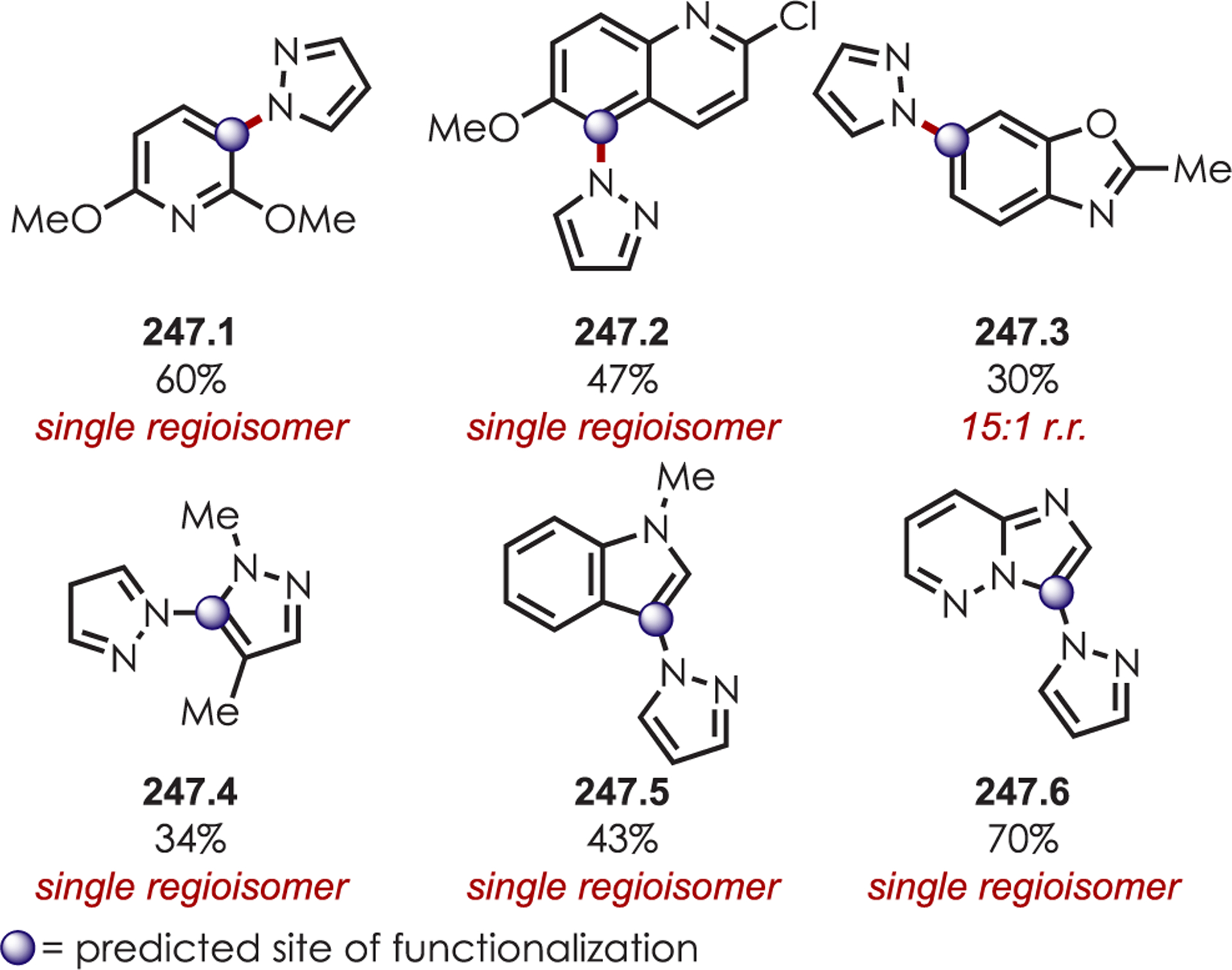
Predicted Site-Selectivity of Arene C–H Amination with Azole Nucleophiles
In 2016, an aryl C–H amination and hydroxylation via dual photoredox catalysis and hydrogen evolution was published with QuCN+ as the photooxidant (Scheme 248).436 The scope of the hydroxylation was extended to a variety of electron-deficient and halogenated arenes, producing a mixture of ortho- and para-isomers as the major products with meta-functionalization observed as a minor product. A Co(III) cobaloxime co-catalyst regenerates QuCN+ by oxidizing the quinolinium radical. The resulting Co(II) oxidizes the intermediate cyclohexadienyl radical, formed from addition of water or amine into the arene cation radical, and a final deprotonation-rearomatization affords the desired phenols (248.2).
Scheme 248.
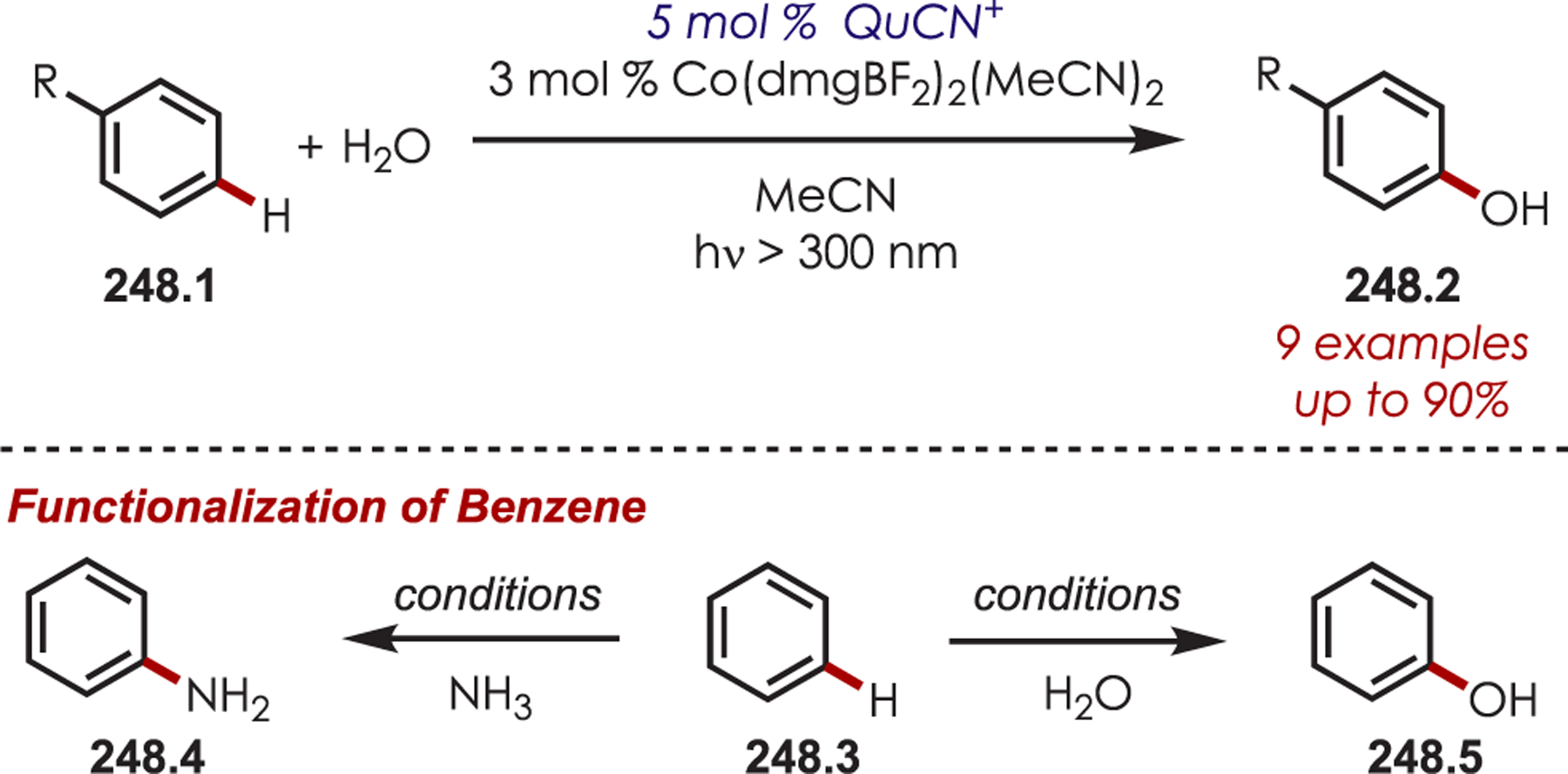
Aryl C–H Amination and Hydroxylation with QuCN+ as the Photooxidant
Since the development of acridinium-catalyzed arene amination with azoles, C–H functionalization of arenes using cyanide (249.1),437 primary amines (249.2),438 and radiofluoride439,440 (249.3) as nucleophiles were reported (Scheme 249). These proceed under a similar reaction mechanism: arene oxidation to generate the aryl cation radical, addition of a nucleophile, and rearomatization via an oxidation/deprotonation sequence.
Scheme 249.
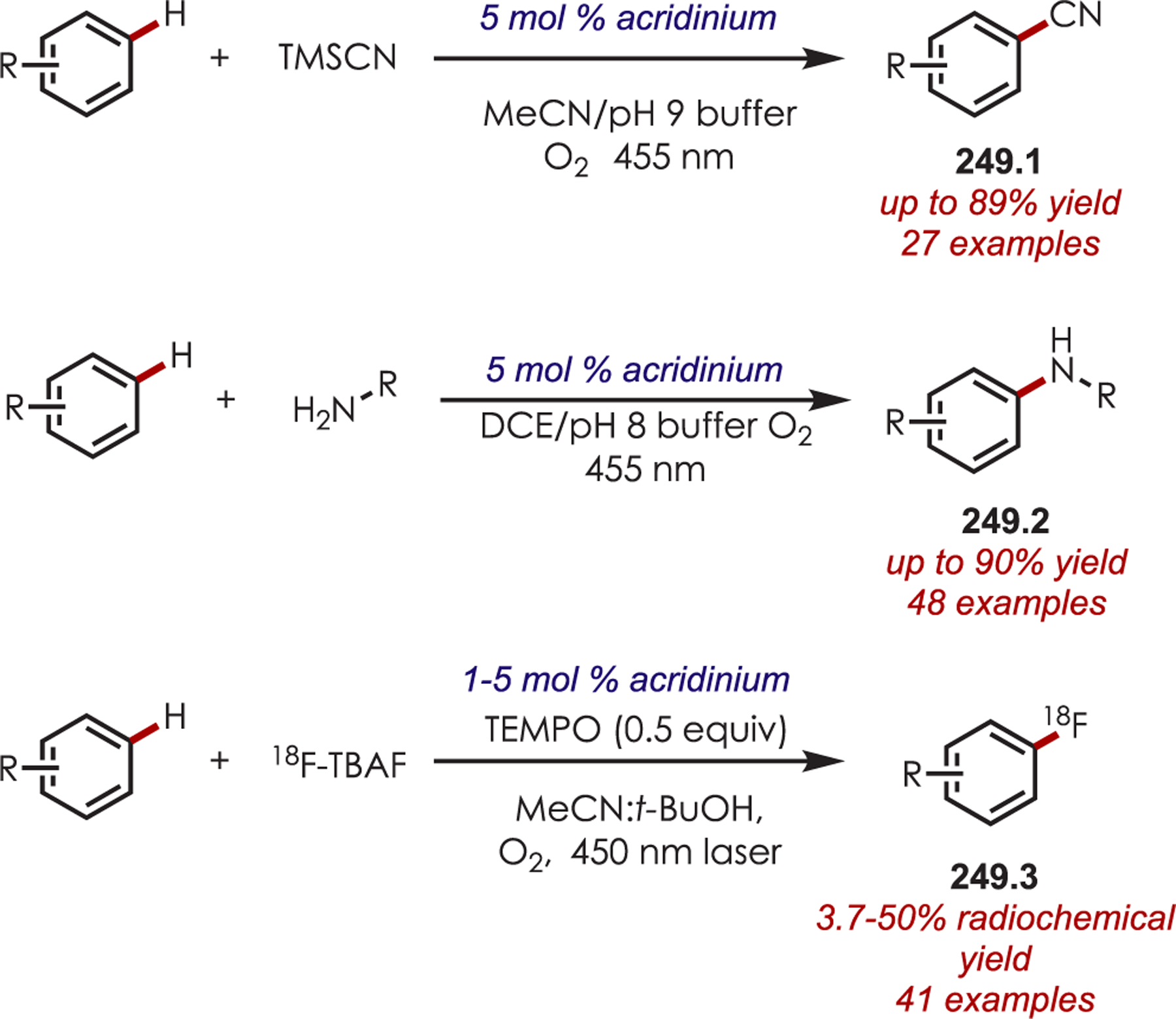
Arene C–H Functionalizations with Radiofluoride, Cyanide, and Primary Amines as the Nucleophiles
Other acridinium-catalyzed arene C–H amination reactions that proceed via the intermediacy of arene cation radical have been reported with azole nucleophiles and persulfate as the terminal oxidant,441 in water,442 or in coupling reactions with quinoxaline-2(1H)-ones.443 Lei has reported the use of a cobaloxime co-catalyst in conjunction with an acridinium photooxidant that allows for a dehydrogenative coupling between arenes and azoles that obviates the need for a terminal oxidant due to the evolution of dihydrogen facilitated by the cobalt co-catalyst.444,445 Recently, a direct C–H amination of 2H-indazoles, a previously unexplored class of substrate in these transformations, was accomplished with primary and secondary amines, azoles, and sulfoximines using an acridinium photooxidant under aerobic conditions.446
A dehydrogenative coupling of arenes and styrenes (250.2) was disclosed using a dual acridinium and cobaloxime system (Scheme 250).447 Radical addition into the aryl cation radical (250.4) from the styrene generates a distonic cation radical intermediate that is rearomatized through deprotonation. The resulting benzylic radical (250.5) undergoes a subsequent oxidation and elimination to afford the product. The authors note that another potential mechanism is radical addition of the alkene cation radical to the arene. The cobaloxime is responsible for regeneration of the photoredox catalyst by SET to Co(II), affording a basic Co(I) species. The Co(II) is regenerated via protonation and hydrogen evolution to generate Co(III) and a final reductive SET from the benzylic radical to Co(II). Electron-rich (hetero)arenes were compatible, including thiophenes, furans, and pyrroles. Styrene derivatives with electron-donating and -withdrawing groups gave the product in moderate to excellent yield.
Scheme 250.
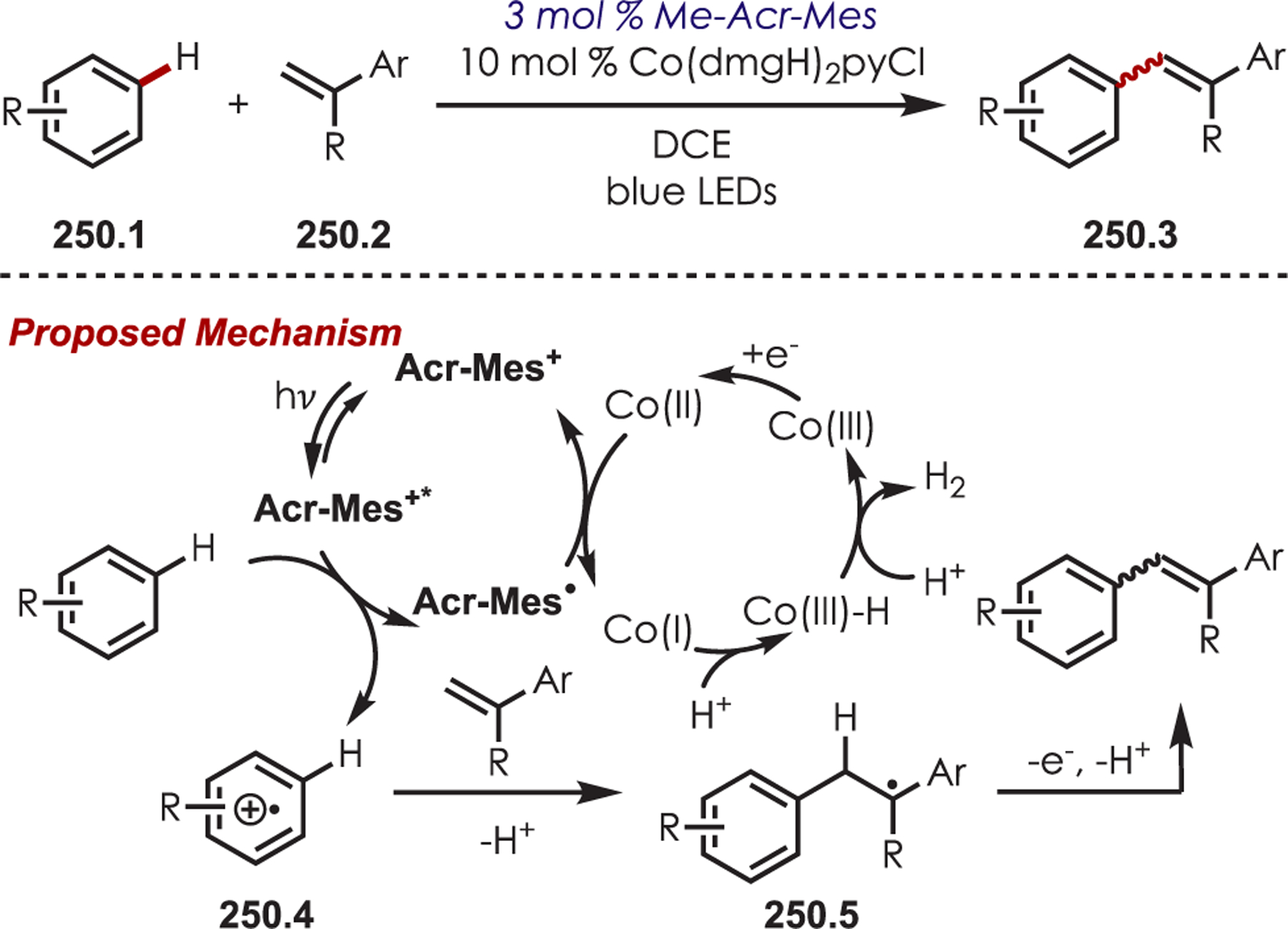
Synthesis of Styrene Derivatives through a Dehydrogenative Coupling of Arenes and Styrenes through Dual Photoredox and Cobaloxime Catalysis
Diazoacetates (251.2) have also been demonstrated as competent nucleophilic partners for C–H functionalization of arenes via arene cation radicals by Nicewicz and co-workers (Scheme 251).448 However, the proposed mechanism involves a unique cyclopropanation of the diazoacetate, releasing nitrogen gas. The generated arene cation radical undergoes nucleophilic addition with diazoacetates, producing the reduced catalyst and a distonic cation radical (251.4). This intermediate is reduced by the acridine radical, generating an equivalent of nitrogen gas, intermediate norcaradiene (251.5), and regenerates the ground state acridinium. A second photoredox cycle then begins with oxidation of the norcaradiene/cycloheptatriene. Ring opening of the norcaradiene forms a distonic, benzylic cation radical (251.6). A final reduction, from acridine radical, and deprotonation affords the desired alkylated products. The authors support the mechanism with both experimental studies (deuterium labeling, subjection of reaction intermediates) and computational studies. The scope of this alkylation includes a range of electron-rich (hetero)arenes and pharmaceutical derivatives. Furthermore, a formal C–H methylation was demonstrated in two steps from the alkylated biphenyl ester through an organophotoredox catalyzed hydrodecarboxylation.449
Scheme 251.
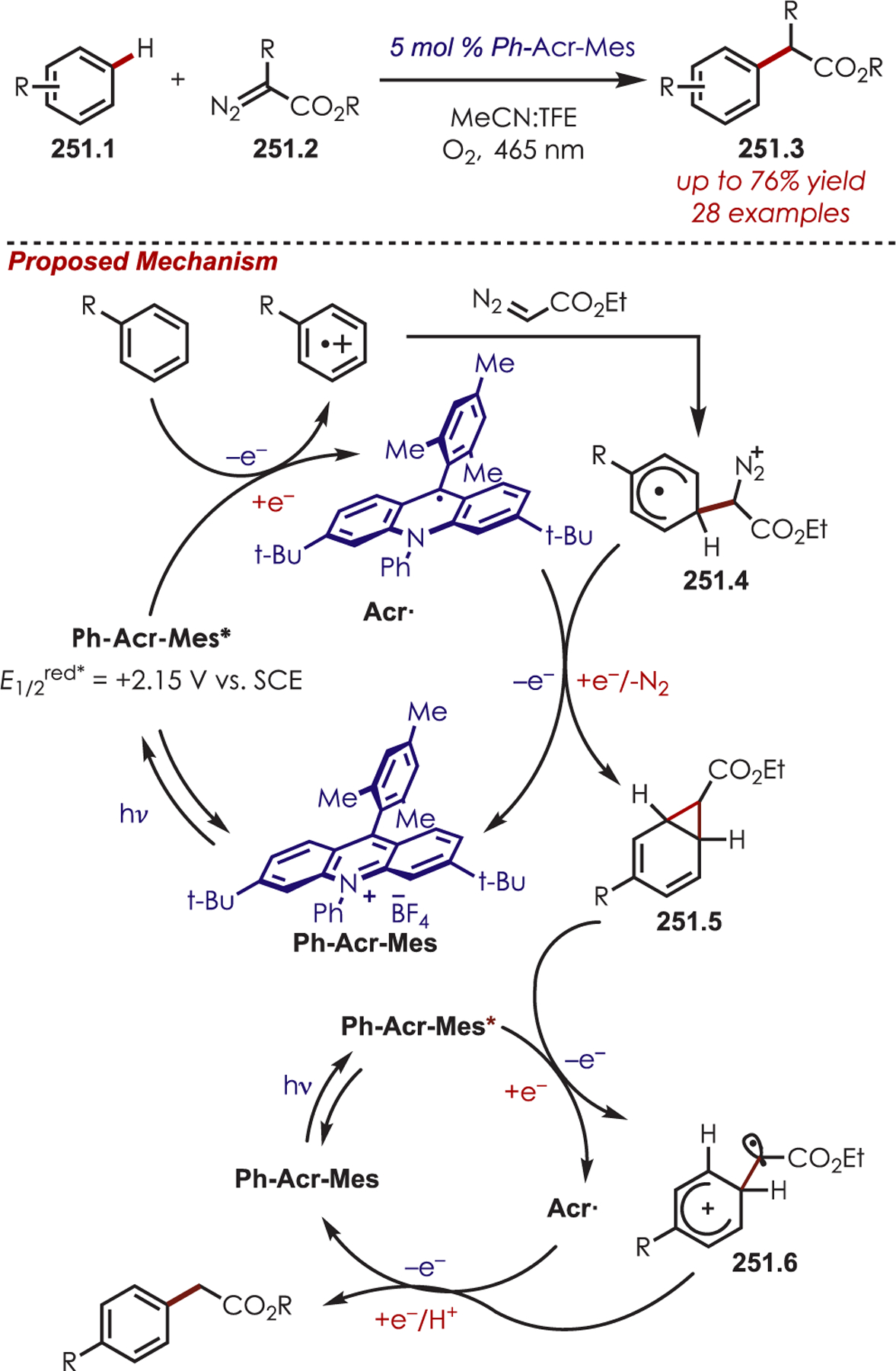
Mechanism of an Arene C–H Alkylation Using Diazoacetates as Polar Nucleophiles Catalyzed by an Acridinium Salt
An oxidative C–H amination of thiophenes (252.1) has been achieved by utilizing DDQ as a photooxidant in combination with tert-butyl nitrite (TBN) as an electron mediator for selective C2 amination (Scheme 252).450 It is proposed that the reaction proceeds via oxidation of thiophene to the cation radical, which undergoes selective C2 substitution with a range of azole nucleophiles. The resulting alkyl radical is further oxidized by DDQ•− to form the desired product and DDQH2. tert-Butyl nitrite is proposed to regenerate DDQ through oxidation by NO2, and TBN is regenerated with molecular oxygen.
Scheme 252.
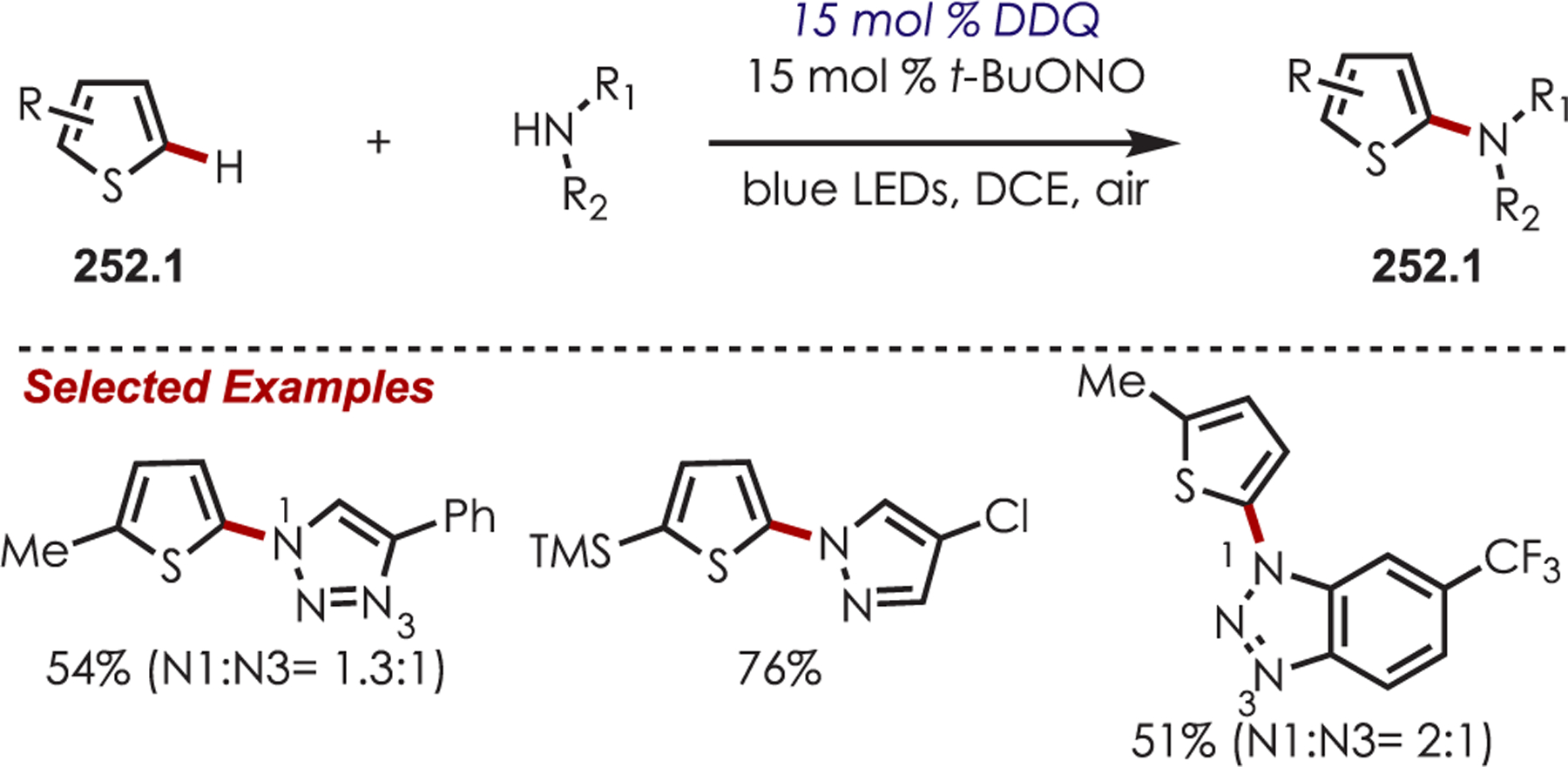
Thiophene C2-Selective C–H Amination with DDQ
Recently, a sulfoximidation of arenes using an acridinium salt with molecular oxygen or persulfates as terminal oxidants has been reported (Scheme 253).451 Both simple alkyl or aryl sulfoximidates (253.1) were competent nucleophiles in the functionalization of electron-rich arenes.
Scheme 253.
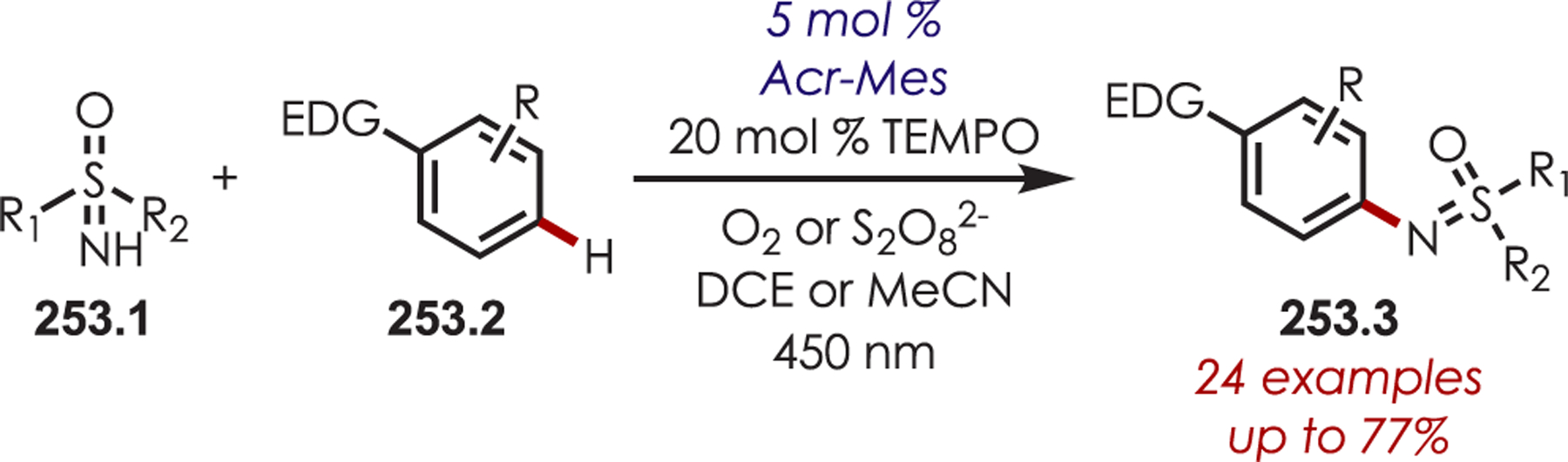
Arene C–H Sulfoximidation Catalyzed by an Acridinium Salt
Similarly, a C–H sulfonamidation of (hetero)arenes using an acridinium photooxidant was reported in 2016 by König (Scheme 254).452 N-Substituted pyrroles with electron-poor or rich sulfonamides were generally compatible, giving the C2 sulfonamidated products (254.3) in moderate to excellent yields.
Scheme 254.
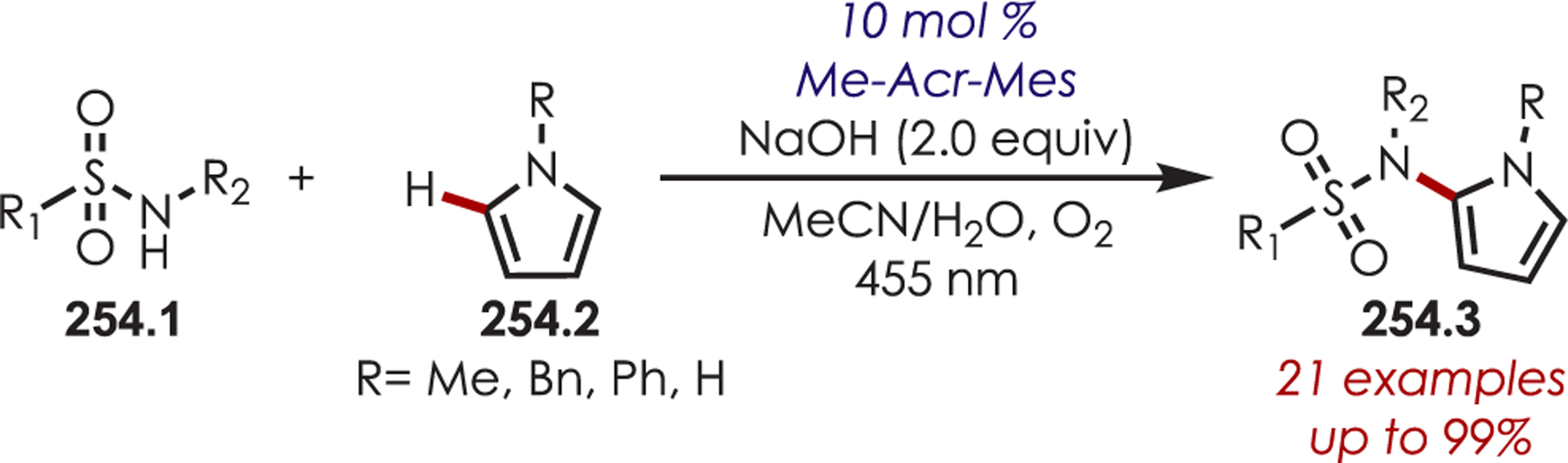
Selective C2 Sulfonamidation of Pyrroles Catalyzed by an Acridinium Salt
König and co-workers later extended this work in the sulfonamidation of electron-rich arenes (255.2) by utilizing a cobaloxime co-catalyst (Scheme 255).453 They propose a radical–radical coupling with a nitrogen-centered radical (255.4) formed via SET of sulfoximine from the charge-transfer state of Me-Acr-Mes*. The resulting amine cation radical undergoes fast deprotonation, generating a NCR that adds to an aryl cation radical, also generated via one electron oxidation by Me-Acr-Mes*. Co(III) regenerates the ground-state catalyst and produces Co(II), which is further reduced to Co(I) by the acridine radical. Hydrogen evolution through a series of protonations regenerates the Co(III) species, and a final deprotonation/rearomatization affords the product. Electron-rich and -poor aromatic or aliphatic NH-sulfoximines were successful NCR precursors, furnishing the desired substituted pyrroles.
Scheme 255.
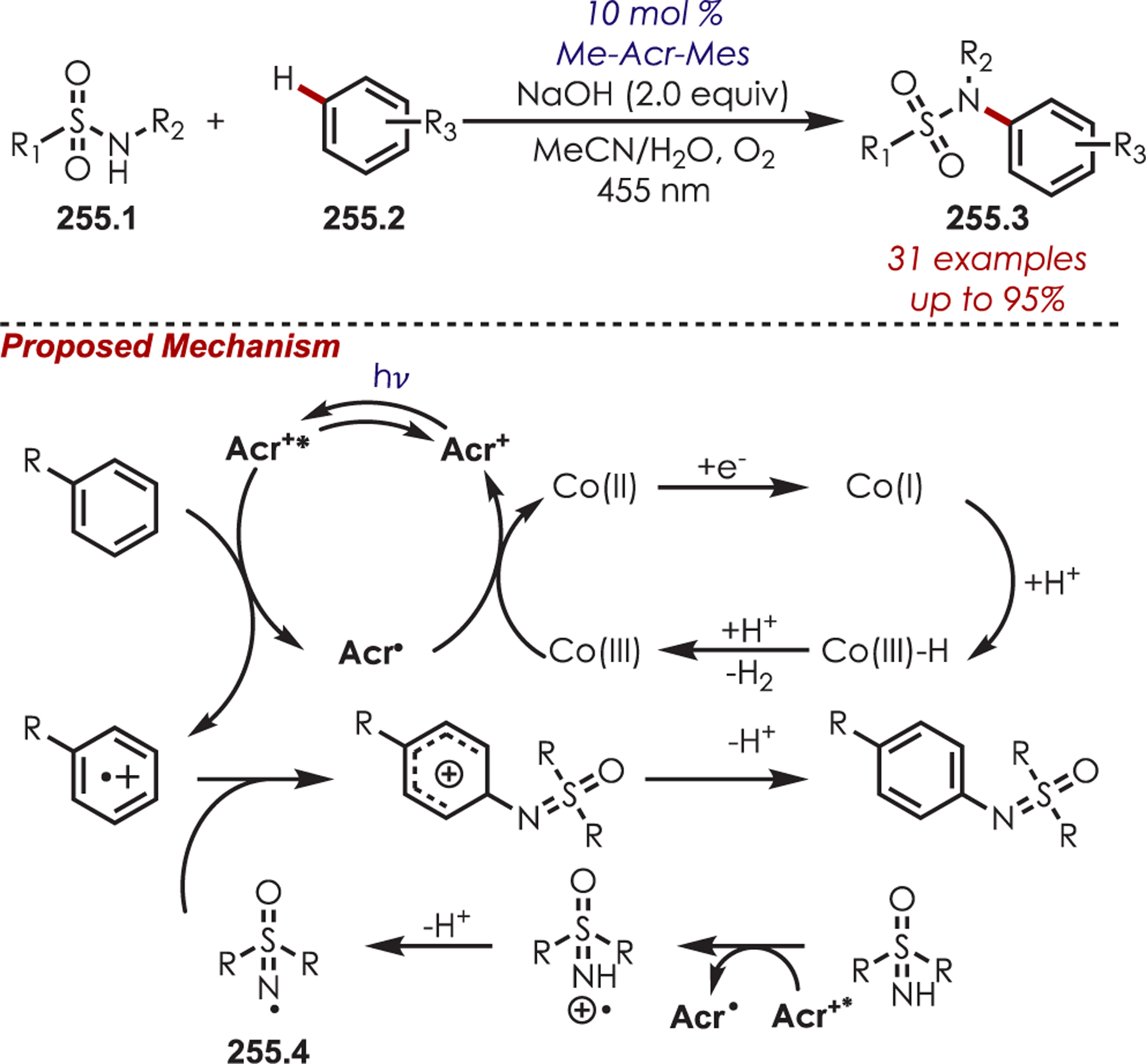
Mechanism of Arene C–H Sulfonamidation by Dual Photoredox and Cobaloxime Catalysis
In addition to the aforementioned examples, König and co-workers have developed C–H functionalizations utilizing other organic photooxidants for the bromination (256.2),454 amination (256.4),455 and nitration456 (256.6) of arenes (Scheme 256). In each case, arene cation radicals are proposed as key intermediates, and the desired products are observed as a mixture of ortho- and para-isomers.
Scheme 256.
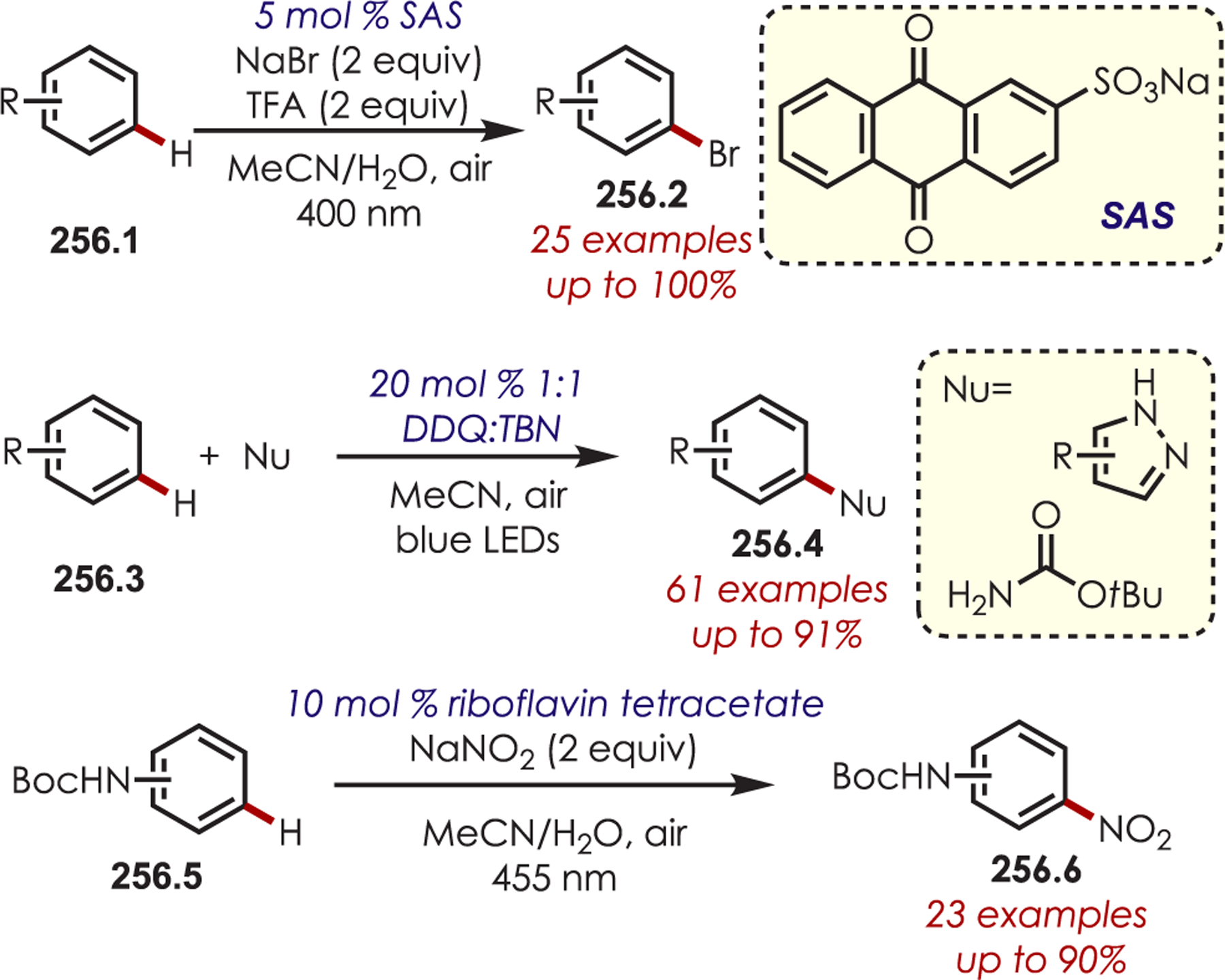
Arene C–H Bromination, Amination, Amidation, and Nitration via Arene Cation Radicals
Organophosphites (257.2) have also been successful nucleophiles for addition into electrophilic arene cation radicals (Scheme 257).457 The intermediate cyclohexadienyl radical undergoes an oxidation, deprotonation, and rearomatization to generate a phosphonium salt. Ammonium acetate is used as an additive to convert the arylated phosphonium cation to the final phosphonylated product. Catalytic Co(II) regenerates the catalyst through SET and hydrogen evolution. Electron-rich arenes, such as simple alkyl (hetero)arenes, anisoles, anilines, and polycyclic arenes, were demonstrated in this phosponylation.
Scheme 257.
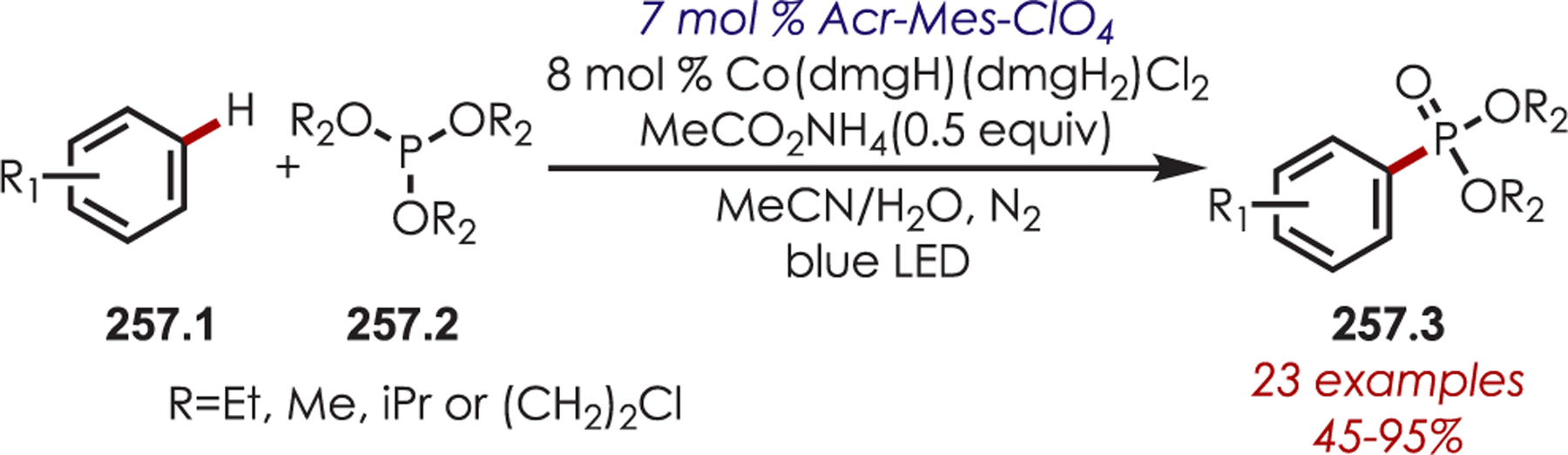
Arene C–H Phosphonylation through Addition to an Arene Cation Radical
Nitrogen Cation Radicals.
Indole C3 formylation has been accomplished by Li and co-workers by accessing reactive nitrogen cation radical intermediates (Scheme 258).458 Oxidation of TMEDA by RB* generates an amine cation radical (258.4) that subsequently forms an iminium from a hydrogen atom abstraction event. Electrophilic addition from the indole to the iminium (258.5) and a subsequent rearomatization by deprotonation furnishes an amino-methylated indole derivative. A second, more favorable SET from RB* generates an iminium ion which undergoes further hydrolysis to afford the formylated indole (258.3). The ground-state rose bengal is regenerated by molecular oxygen. The reaction scope included alkyl-, alkoxy-, or halo-substituted indoles with electron-rich N-protecting groups.
Scheme 258.
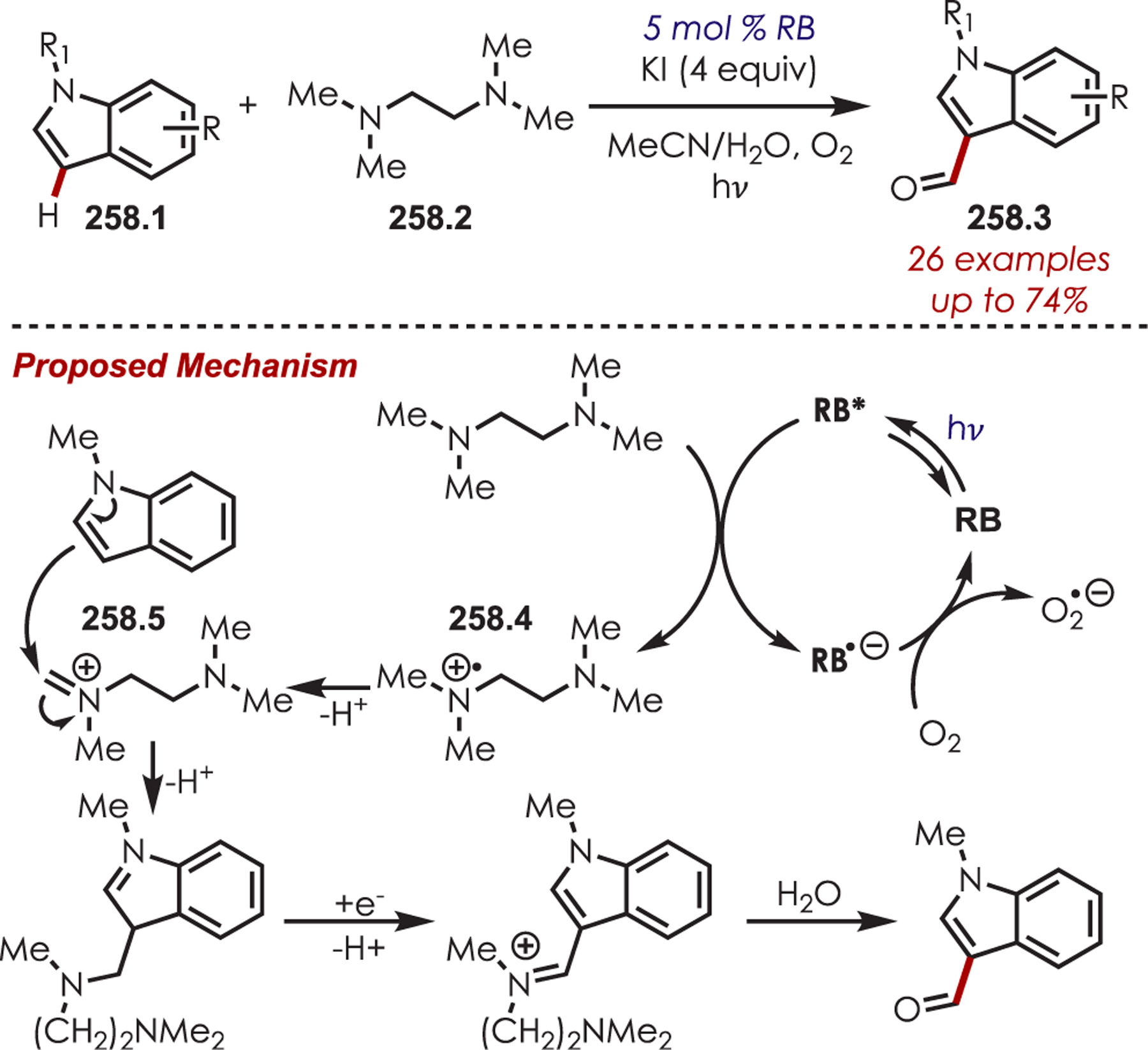
Indole C–H Formylation through Nitrogen Cation Radicals
Nitrogen cation radicals have also been utilized for (hetero)aryl bromination by activation of NBS, 259.2 (Scheme 259).459 NBS (Eox = +0.28 V vs Fc/Fc*) is oxidized by excited erythrosine B (Ered* = +0.76 V vs Fc/Fc*) to the nitrogen cation radical. This radical ion of NBS amplifies its electrophilicity and allows for an electrophilic aromatic bromination to occur without any negative side reactivity from bromination of benzylic C–H bonds. However, this method generally required electron-rich arenes, and arenes with electron-withdrawing groups failed to give any of the desired bromoarene.
Scheme 259.
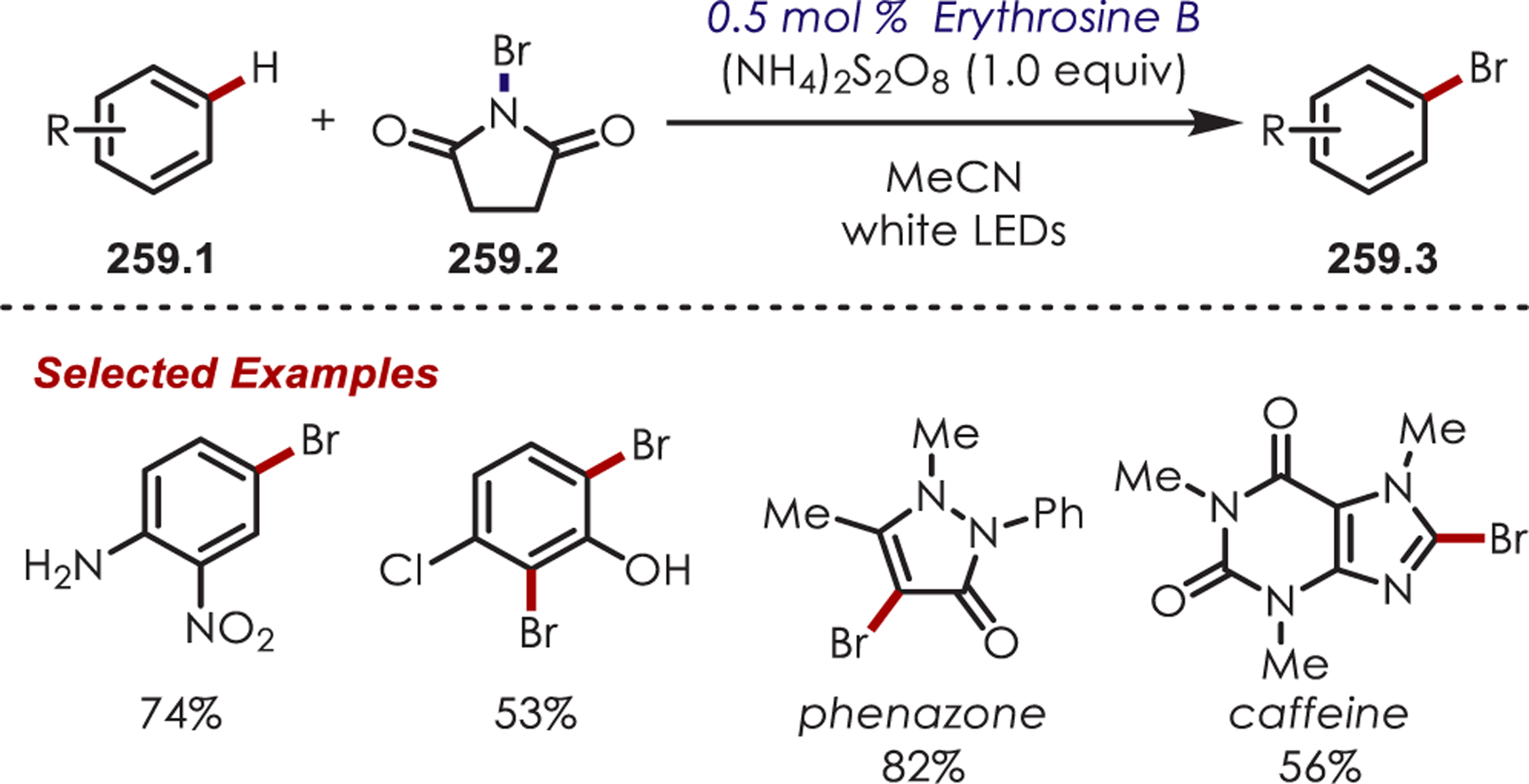
Arene C–H Bromination through Activation of NBS by Generation of an Amine Cation Radical
3.1.5. Miscellaneous.
Organic photoredox catalysts have been utilized as oxidants in a dual cobalt-mediated C–H and N–H bond annulation of aryl amides (260.1) with alkynes (Scheme 260).460 Substituted benzamides and heteroaromatics gave products in good to excellent yields. Meta-substituted benzamides were competent substrates but gave a mixture of regioisomers, with the major regioisomer at the less sterically hindered site. This method is compatible with other chelating groups on the substrates including thienyl, pyrazolyl, pyridyl, and azaindolyl. Both symmetrical and unsymmetrical alkynes with alkyl or aryl substitution were tolerated. In the case of unsymmetrical alkynes, high regioselectivity was observed for aryl substituients proximal to the nitrogen. Regioselective annulation was also observed with terminal alkynes, giving the product with substitution adjacent to nitrogen. Moreover, diynes were successful at generating the annulated products.
Scheme 260.
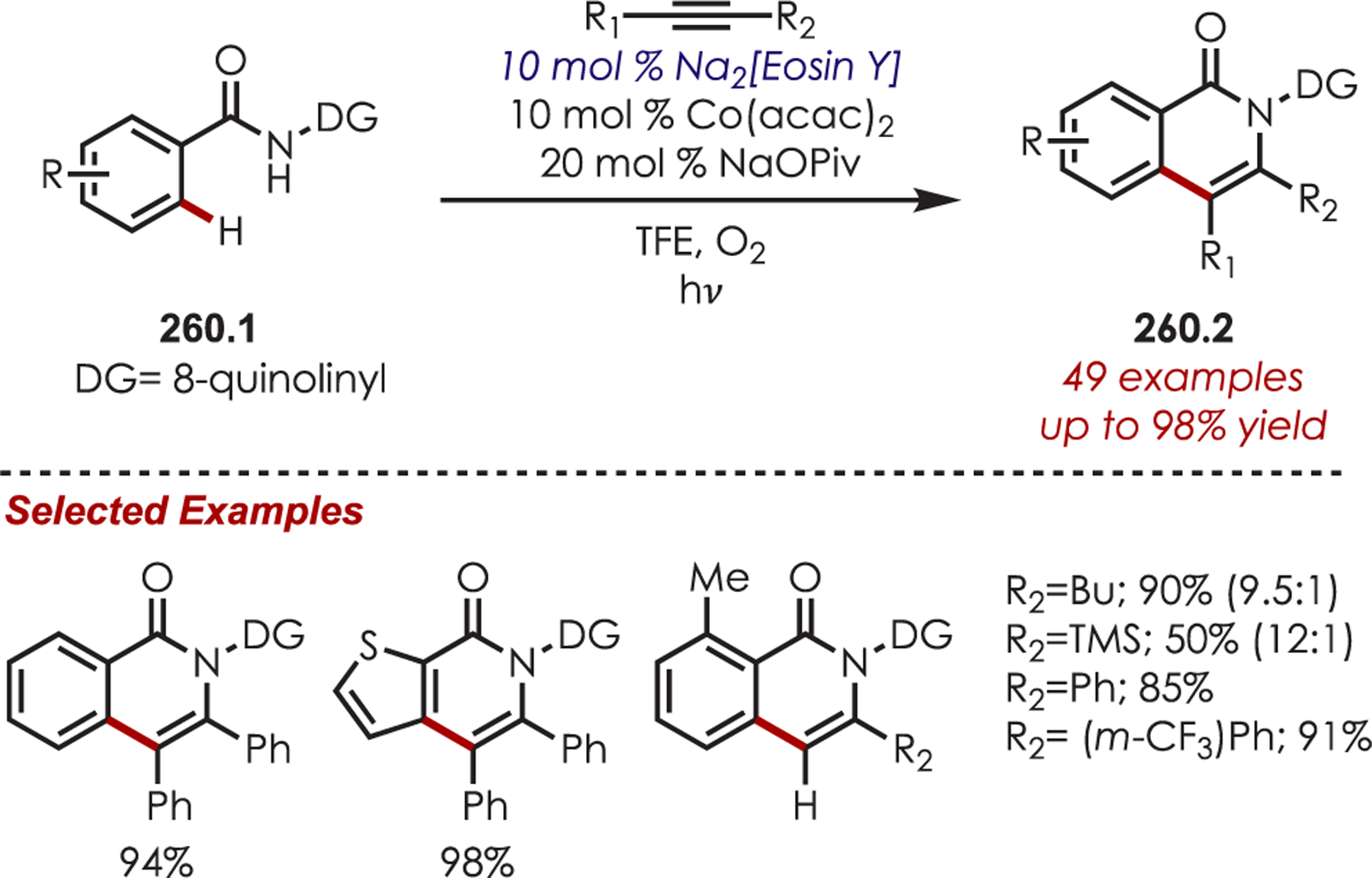
Directed Aryl C–H and N–H Bond Annulation of Aryl Amides with Alkynes
A series of mechanistic experiments led the authors to the following mechanism: after ligand exchange, an oxidative PET with eosin Y forms Co(III), which then undergoes cyclo-metalation via a concerted/metalation deprotonation pathway furnishing a Co(III)–amide complex, 261.1 (Scheme 261). The authors note this complex may also be formed by an aerobic oxidation of Co(II) followed by coordination and cyclo-metalation. Coordination of the alkyne to the carbocycle, followed by insertion and reductive elimination, furnishes the desired product (261.3) and Co(I). A second oxidation of Co(I) by eosin Y, regenerates the starting Co(II) species and eosin Y anion radical. Molecular oxygen is the terminal oxidant for the transformation and restores the eosin Y photoredox catalyst.
Scheme 261.
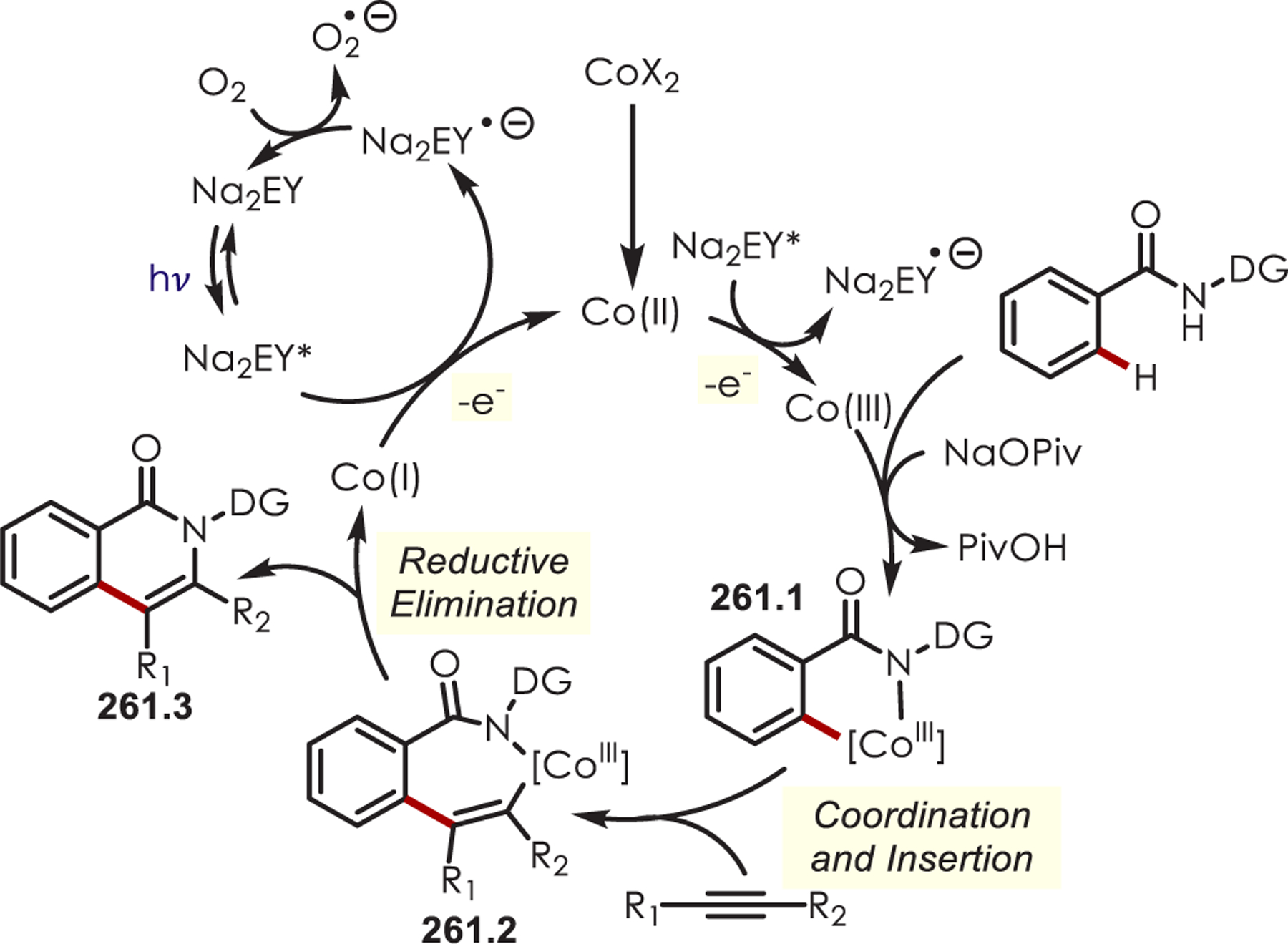
Mechanism of the Directed Aryl C–H and N–H Bond Annulation of Aryl Amides with Alkynes
Recently, a dual photoredox and cobalt cyclization has been reported for the synthesis of phenanthrenes (262.2) through α-keto radicals (Scheme 262).461 Oxidation of the enol tautomer of the 1,3-dicarbonyl and subsequent deprotonation generates an α-carbonyl radical. This undergoes an intramolecular cyclization on the pendent arene and is further oxidized and deprotonated to afford the phenanthrene products in good to excellent yields. Both electron-donating and electron-withdrawing functionality was tolerated on the biphenyls in this method.
Scheme 262.

Synthesis of Phenanthrenes through Aryl C–H by an α-Keto Radical
König et al. reported a chlorination of electron-rich arenes (263.1) by in situ bromination followed by an ipso-halogen exchange (Scheme 263).462 A dicyanobenzene derivative, 4CzIPN, was used as the photoredox catalyst (Ered* = +1.35 V vs SCE) as a direct oxidation of the arene (Eox > +1.7 V vs SCE for anisole derivatives) is thermodynamically unfavorable. Oxidation of the bromide anion is more exergonic than chloride (Eox = 0.74 and 1.05 V vs SCE, respectively) and is proposed to occur principally. This generates bromine as a brominating agent which regioselectivity brominates the arene para to the EDG. Ipso-substitution by a chlorine atom affords the chlorinated arenes in high regioselectivity. This method was successful with anisoles, halogenated arenes, thiazole, and pyrazole derivatives.
Scheme 263.
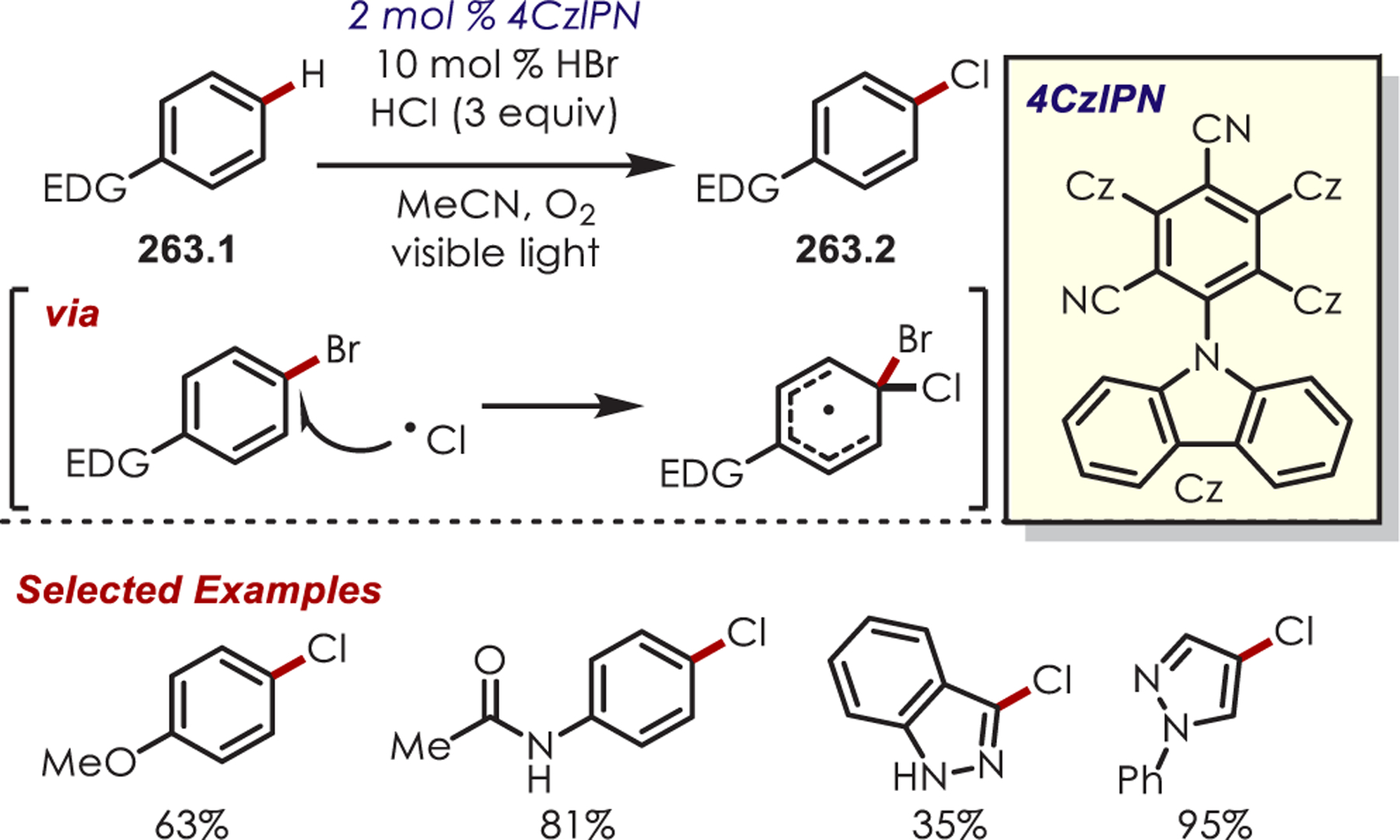
Arene C–H Chlorination via an In Situ Ipso-Halogen Exchange
3.2. Aliphatic C–H Functionalization
3.2.1. α-Heteroatom C–H Functionalization.
Via Iminium or Oxocarbenium Ions.
Following the work from Stephenson and co-workers180,182 for α-amino C–H functionalization via inorganic photoredox catalysis, several groups have explored the use of organic photoredox catalysts for related transformations. One of the early reports of an organic photoredox catalyzed amine functionalization was with rose bengal under air (Scheme 264).463 The proposed mechanism is identical to the previously discussed inorganic mediated pathways: generation of the iminium by amine oxidation and hydrogen atom transfer. Aza-Henry and Mannich type reactions were demonstrated with this metal-free alternative. Notably, a nonbenzylic substrate, 4-methyl-N,N-dimethylaniline, successfully gave the product in 48% yield. Eosin Y catalyzed Mannich-type reactions of tetrahydroisoquinolines were also reported in an alkylation using nitromethane464 and thiocyanation.465
Scheme 264.
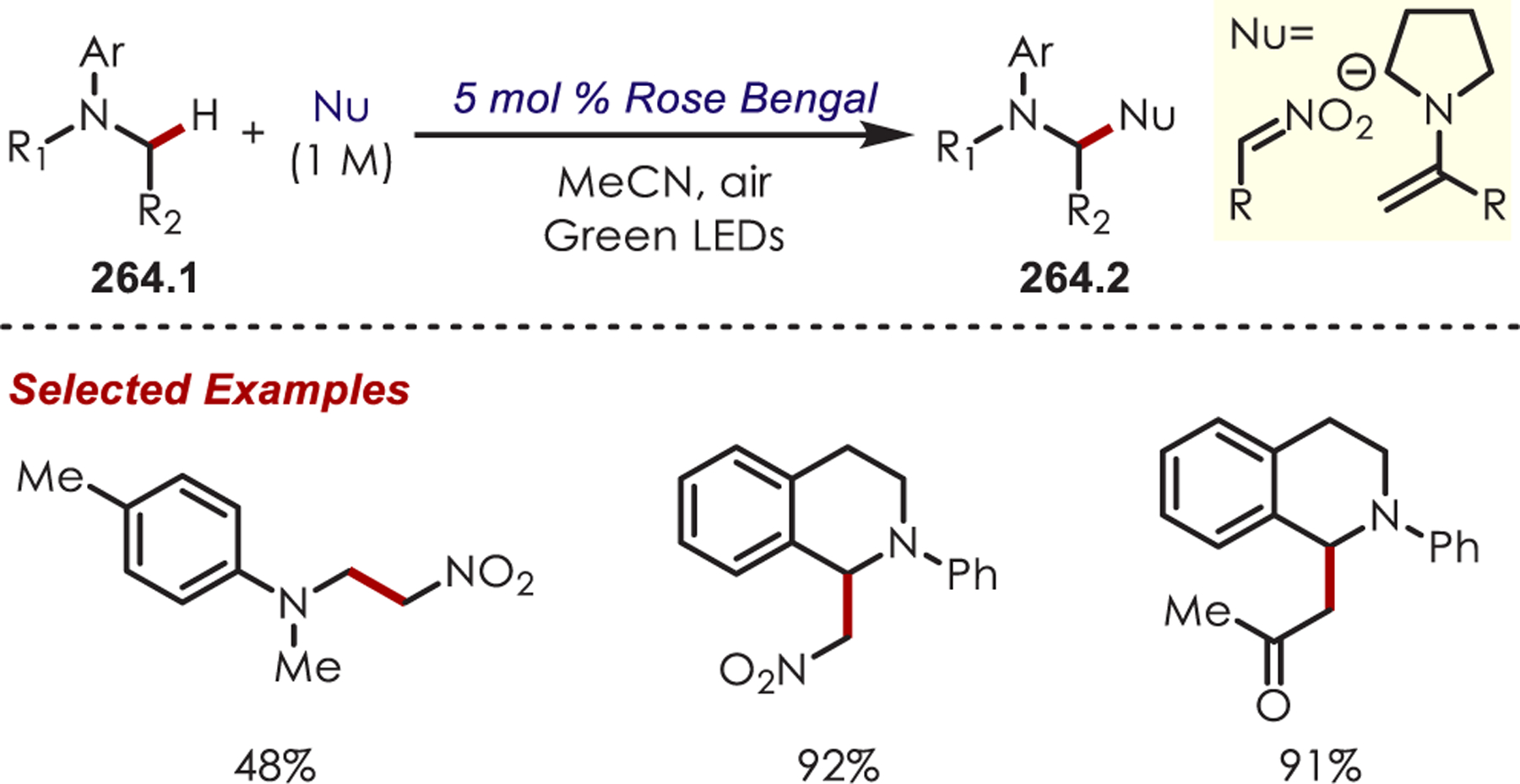
C–H Alkylation of Amines through Photogenerated Iminiums
Rose bengal was later shown to catalyze a cross-dehydrogenative coupling between diazo compounds (265.2) and tertiary amines via the intermediacy of iminium ions (Scheme 265).466 The products were subjected to subsequent ring expansion conditions for the synthesis of N-aryl-2,3-dihydrobenzo[d]-azepines. The mechanism proceeds through a nucleophilic addition of the diazoacetate to the photogenerated iminium ion.
Scheme 265.
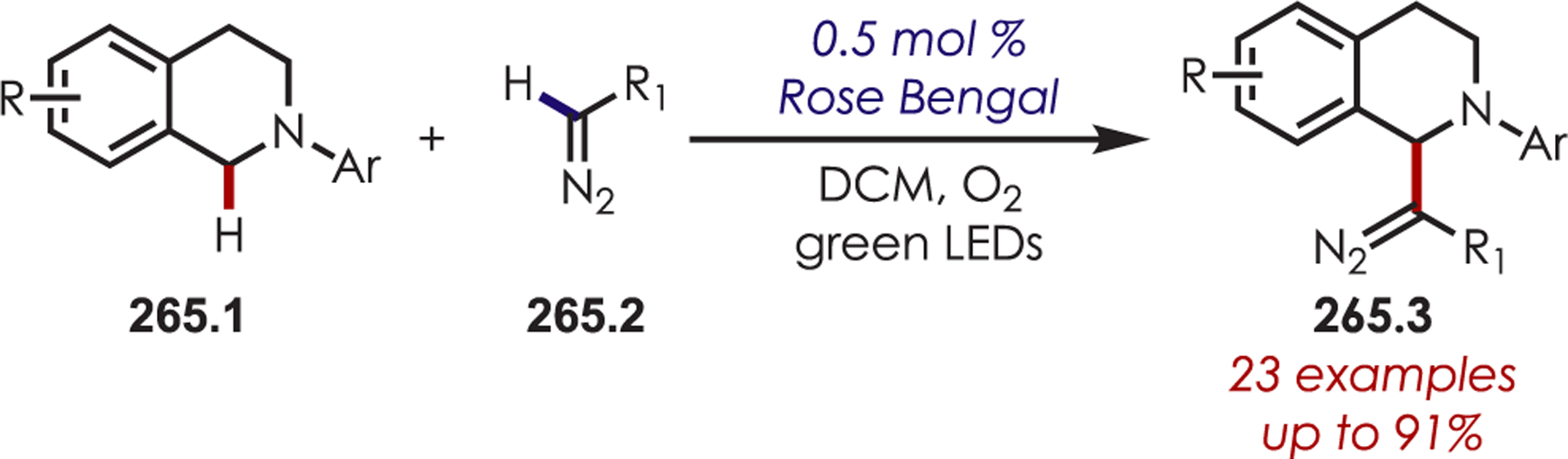
C–H Alkylation of Amines through Photogenerated Iminiums with Diazoacetates
Tan and co-workers later reported an α-cyanation and trifluoromethylation catalyzed by a cooperative Rose Bengal/graphene oxide (GO) system (Scheme 266.).467 The exact role of graphene oxide is not known, but it is suggested that the iminium intermediate may be stabilized by the slightly acidic GO, thus leading to rate enhancement. A selective C2 trifluoromethylation and alkynylation of tetrahydroisoquinolines catalyzed by rose bengal with oxygen as a terminal oxidant has also been reported.468
Scheme 266.
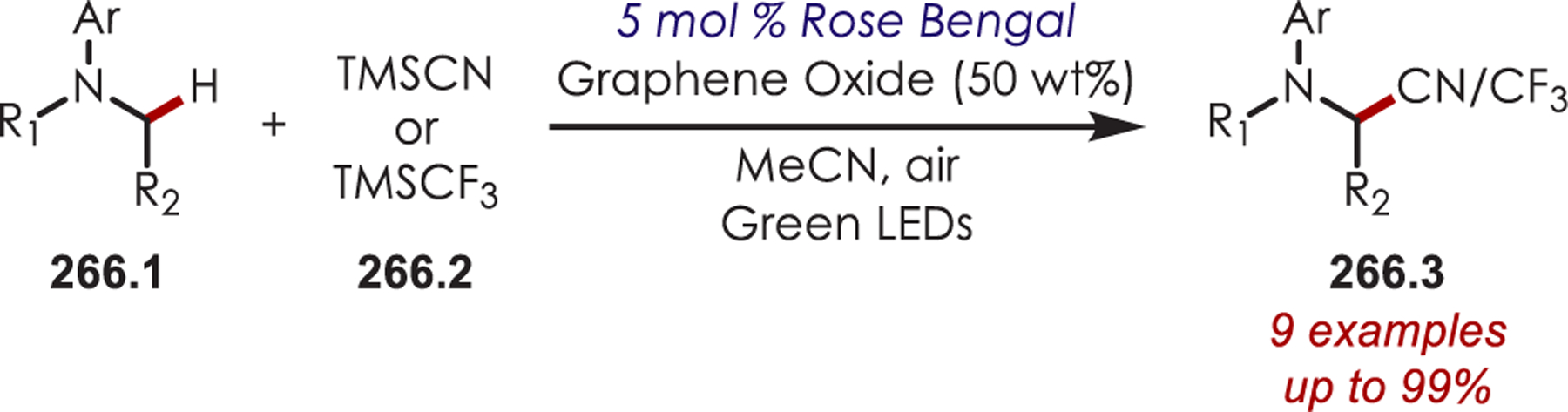
C–H Cyanation and Trifluoromethylation of Amines through Photogenerated Iminiums
Silyl enol ethers (267.2) have also been utilized as suitable nucleophilic partners for addition into photogenerated iminiums of glycine derivatives with 5-aminofluorescein and InBr3 as a Lewis acid additive (Scheme 267).469
Scheme 267.
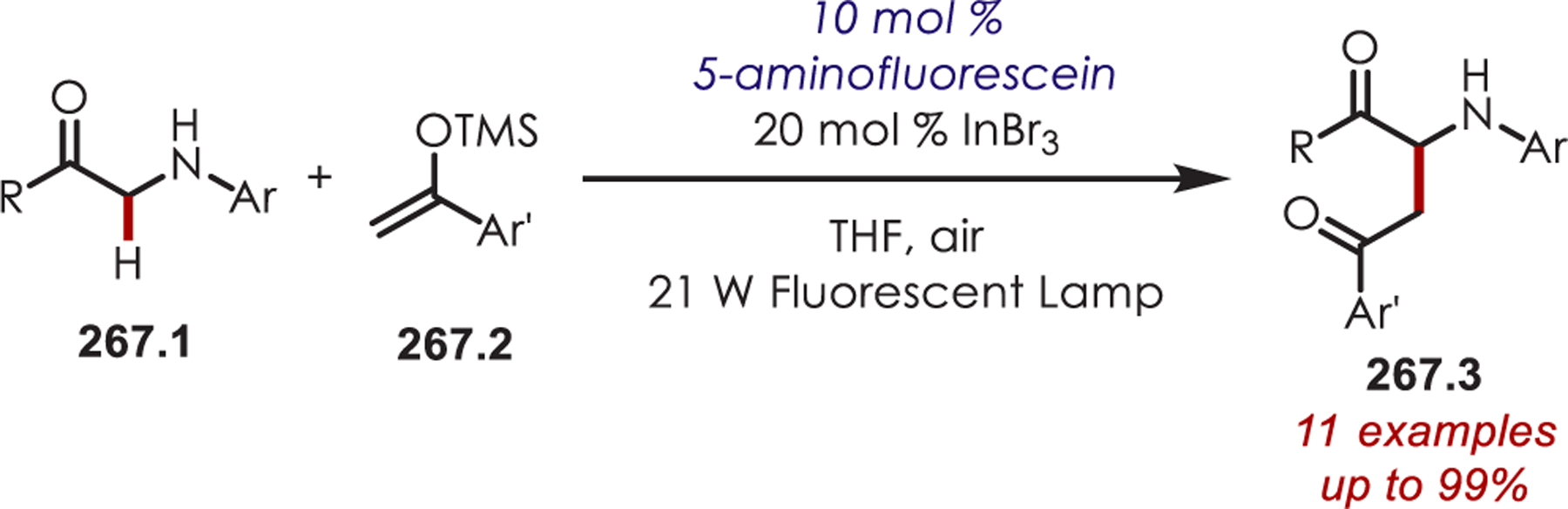
C–H α-Alkylation of Amines with Silyl Enol Ethers
A coupling of N-aryl glycine derivatives (268.1) and olefins via the intermediacy of photogenerated imines was accomplished for the synthesis of substituted dihydroquinolines (Scheme 268).470 The electrophilic iminium species formed in situ is trapped with an olefin through two consecutive C–C bond-forming events initiated by an EAS from the electron-rich arene. The intermediate tetrahydroquinoline is further oxidized to afford the final product. This transformation was later reported using Ru(bpy)3Cl2 as the photoredox catalyst.471
Scheme 268.
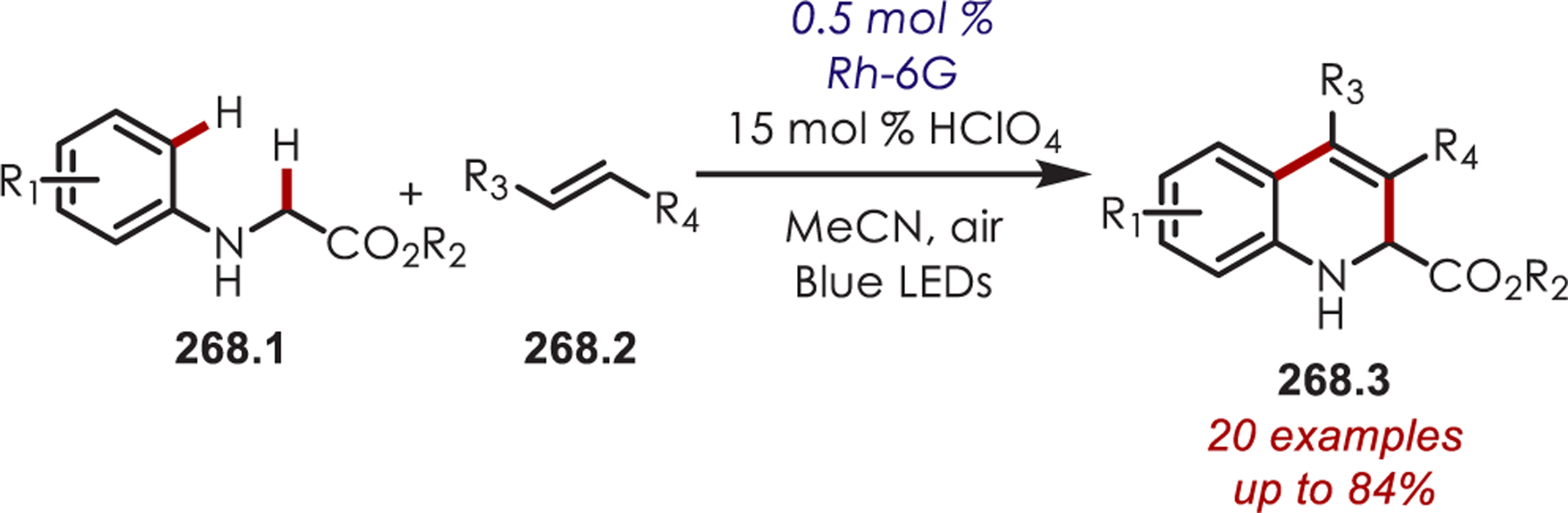
Synthesis of Quinolines through a Cascade EAS and Olefin Addition to the Photogenerated Iminium Ion
An in situ alkylation of tetrahydroisoquinolines (269.1) derivatives and subsequent 6π–electrocyclization has been reported for the synthesis of nitroindoloisoquinolines (Scheme 269).472 Nevertheless, the scope is limited, and low to moderate yields were observed.
Scheme 269.
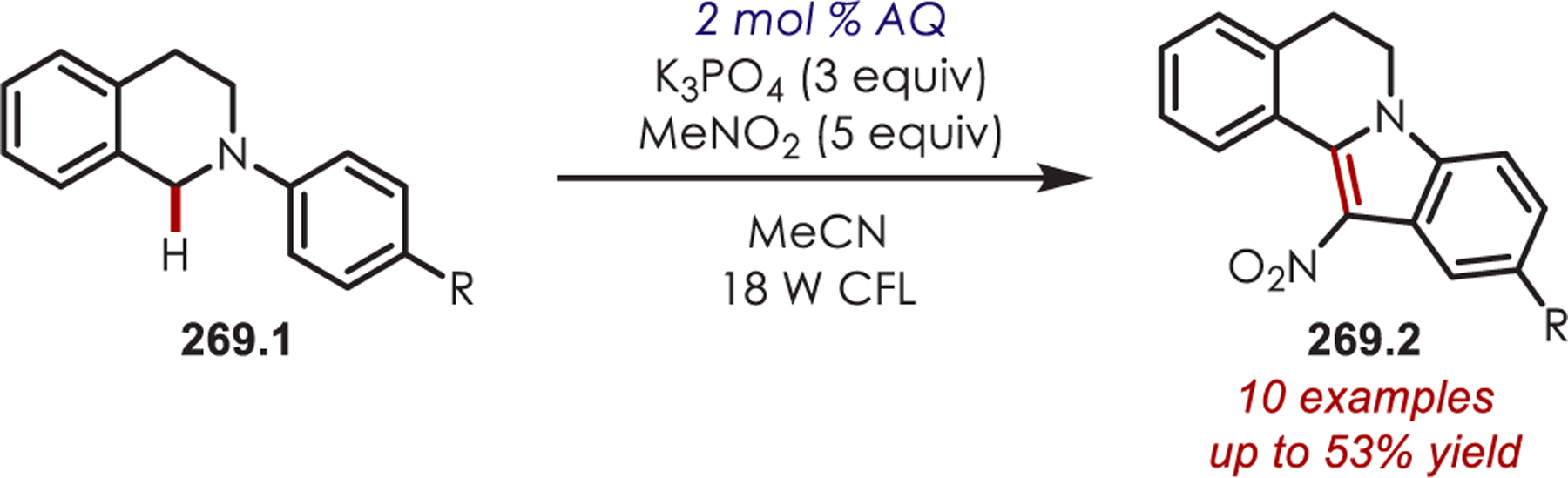
In Situ C–H Alkylation of Tetrahydroisoquinolines and Subsequent 6π Electrocyclization
An enantioselective α-functionalization of tetrahydroisoquinolines (270.1) and tetrahydro-β-carbolines (270.3) was reported the following year with a cooperative DPZ photoredox and chiral Lewis acid catalysis (Scheme 270).473 Both acrolein and acrylonitrile were suitable radical acceptors, which gave the products in good yields and enantioselectivities. The mechanism proceeds through an activation of the radical acceptor by β-isocupreidine (β-ICD) to generate a β-ICD-acrolein complex (270.5). This subsequently couples with a photogenerated iminium in a Mannich-type fashion to afford the α-olefinated amines. The tetrakis(3,5-bis(trifluoromethyl)phenylborate (BArF) counterion was proposed to have an important role in stabilizing the chiral intermediates, which in turn lowered the activation energy and increased the reaction rate, ultimately resulting in improved reaction efficiency.
Scheme 270.
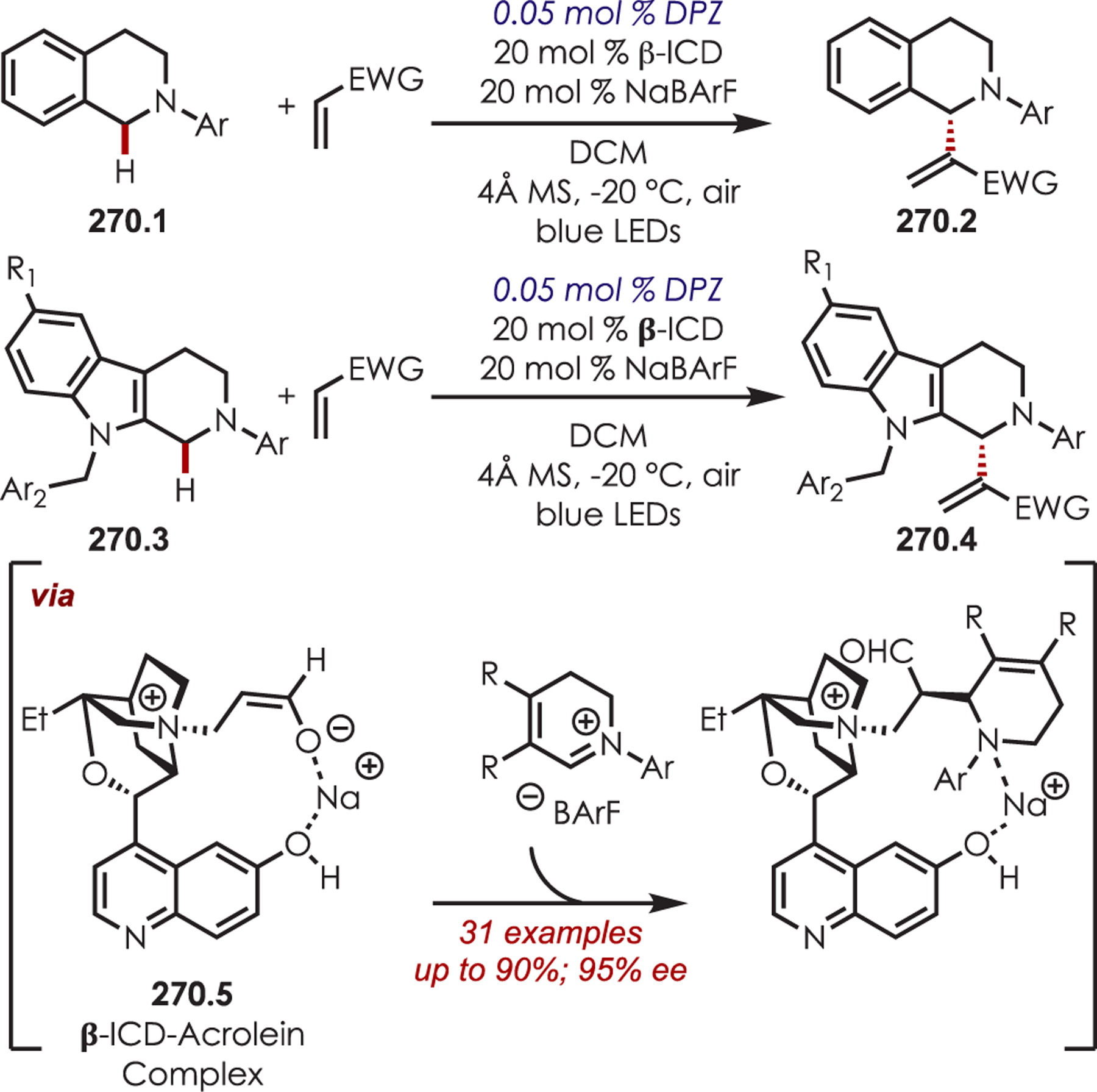
Asymmetric α-C–H Alkylation of Tetrahydroisoquinolines through a Chiral β-ICD Acrolein Complex
Wu and co-workers reported a related transformation for the generation of oxocarbenium ions (271.6) that couple with β-keto esters for the diversification of isochromans (Scheme 271).474 The proposed mechanism begins with oxidation of the isochroman to the aryl cation radical (271.4) by Acr-Mes*. Deprotonation and a second oxidation forms an oxocarbenium species. Addition of the Cu(II)-activated nucleophile followed by a deprotonation by Co(I) delivers the coupled product. While the scope of this chemistry is quite limited, the oxonium intermediate is quite unique and rarely generated through photoredox catalysis.
Scheme 271.
Oxocarbenium Ion Generation for the C–H Alkylation of Isochromans
Via α-Amino and α-Heteroatom Radicals.
In general, tetrahydroisoquinolines form reactive electrophilic iminium species that can undergo nucleophilic addition. However, Pandey and co-workers reported a radical–radical coupling of tetrahydroisoquinoline α-amino radicals with alkyl selenides and 9,10-dimethoxyanthracene (DMA) as the catalyst (Scheme 272).475 Reduction of the alkyl selenide by photoexcited DMA generates an alkyl radical, which is proposed to couple with the α-amino radical formed through an oxidation and deprotonation sequence from the DMA cation radical. This also could undergo an intramolecular variant (272.3) to afford indolines and tetrahydroquinoline derivatives.
Scheme 272.
C–H Alkylation of Tetrahydroisoquinolines through a Radical–Radical Coupling from Alkyl Selenides
A controllable chemodivergent functionalization of tetrahydroisoquinonlines (273.1) was reported through the α-amino radical addition to N-substituted itaconimide electrophiles using a dicyanopyrazine (DPZ) photooxidant (Scheme 273).476 The reaction proceeds through one of four different radical cascade pathways depending on the solvent, temperature, or additives. [4 + 2] adducts (273.4) were observed with catalytic LiPF6 as the Lewis acid and were suggested to occur through an EDA mechanism. Mannich-type products (273.3) from an addition and protonation were observed with shorter reaction times and 0.5 equiv of the Lewis acid. The use of base in the absence of a Lewis acid gives the conjugate adduct products (273.6). Finally, at low temperature and under aerobic conditions, the addition-coupling products (273.5) were observed.
Scheme 273.
Chemodivergent C–H Functionalization of Tetrahydroisoquinolines
Our laboratory has developed an α-alkylation of carbamate-protected amines (274.1) via carbamyl radicals generated by an acridinium photooxidant (Scheme 274).477 After direct oxidation of the carbamate, the protons adjacent to the resulting amine cation radical experience a dramatic decrease in pKa from 30 to 5. This facilitates a deprotonation and spin-centered shift event to produce the key intermediate α-carbamyl radical. A variety of radical acceptors were suitable, including aliphatic vinyl ketones with α- or β-substitution, methacrylate, dimethyl fumarate, nitrile, or sulfonyl derivatives. Additionally, this method was showcased in the synthesis of the indolizidine alkaloid, (+)-monomorine I.
Scheme 274.
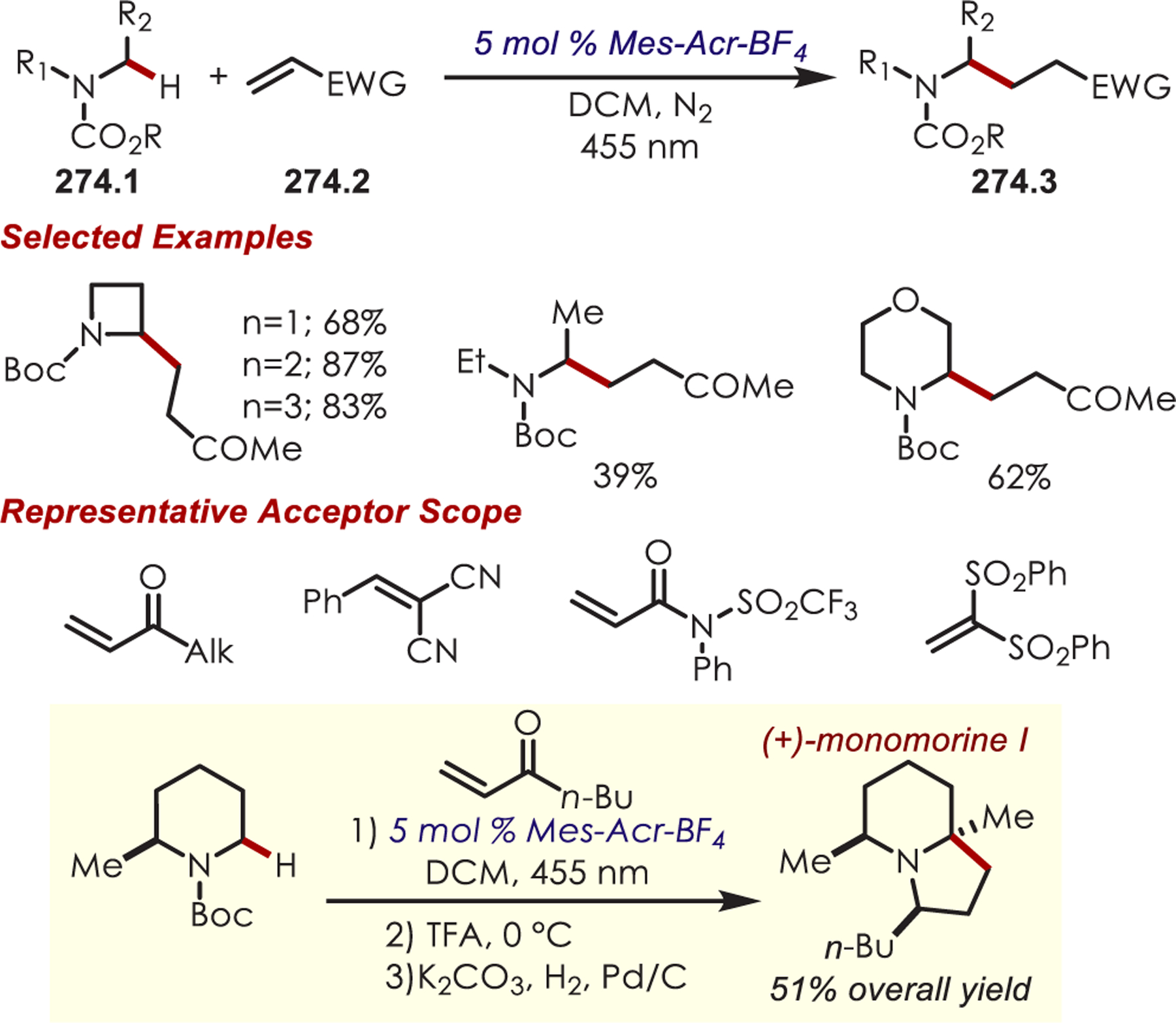
α-Carbamyl C–H Alkylation of Piperidines with an Acridinium Salt as the Photooxidant
Site-selective functionalization of piperazines was later accomplished by distinguishing the two possible C–H functionalization sites via differential protection of the respective nitrogen atoms.478 The mechanism is identical to the previously published α-amine functionalization wherein a key α-amino radical species (275.2) is generated and intercepted by a radical acceptor (Scheme 275).
Scheme 275.
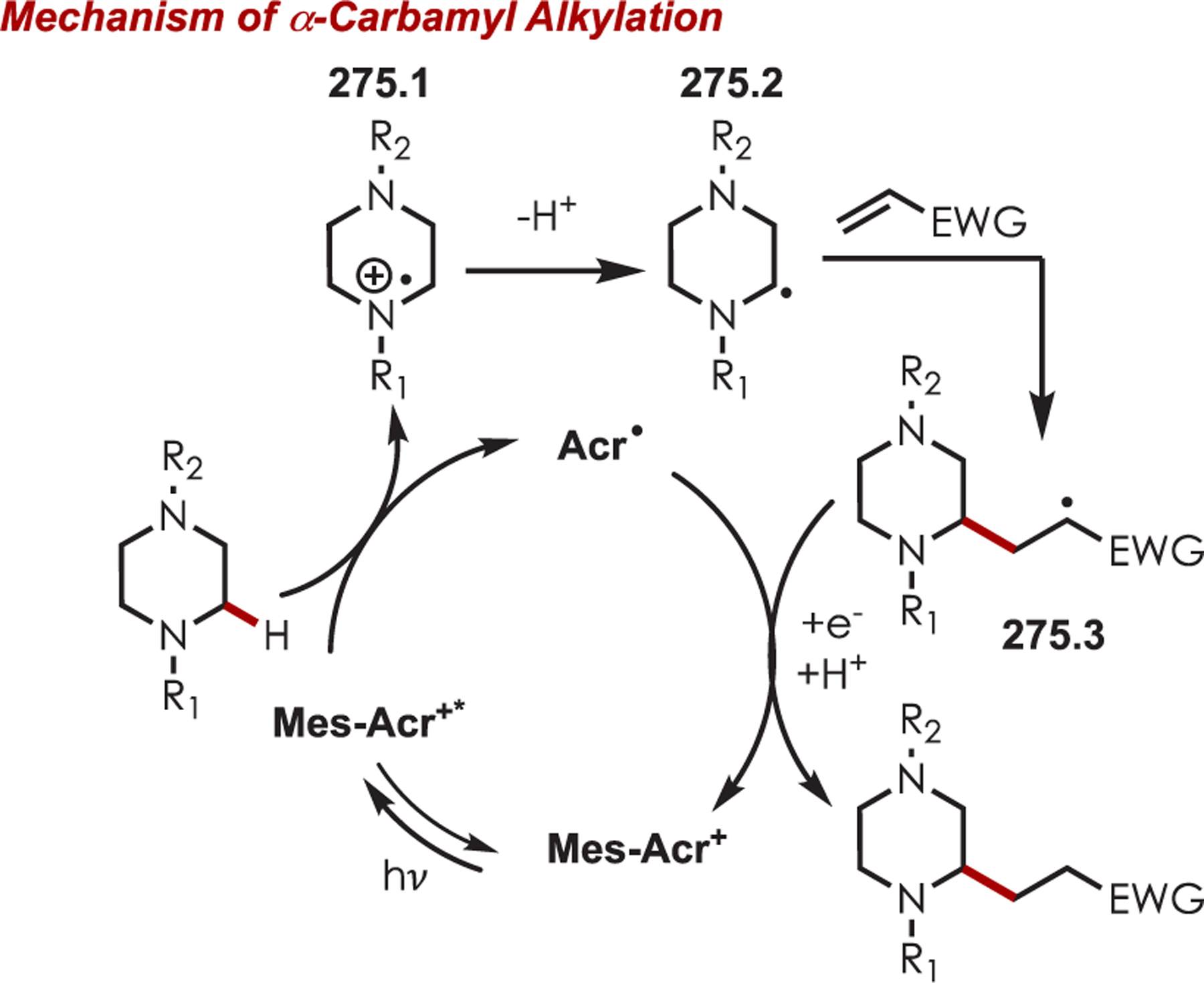
Mechanism of α-Carbamyl C–H Alkylation of Piperazines with an Acridinium Salt as the Photooxidant
Excellent selectivity was observed with benzamides and sulfonamides for functionalization adjacent to the Boc-protected nitrogen, however, in lower yields for the latter (Scheme 276). (Hetero)aryl amines resulted in a reversal of regioselectivity, giving high selectivity in proximity to the N-(hetero)aryl group. Other Michael acceptors including unsaturated sulfones, amides, and nitriles gave the alkylated piperazines with high to excellent regioselectivity. A DFT natural population analysis was performed and successfully predicted the product site-selectivity and outcome. The calculations found that the selectivity correlated to the charge density on the nitrogen atoms, wherein the nitrogen with the highest degree of positive charge correlated to the site of alkylation.
Scheme 276.
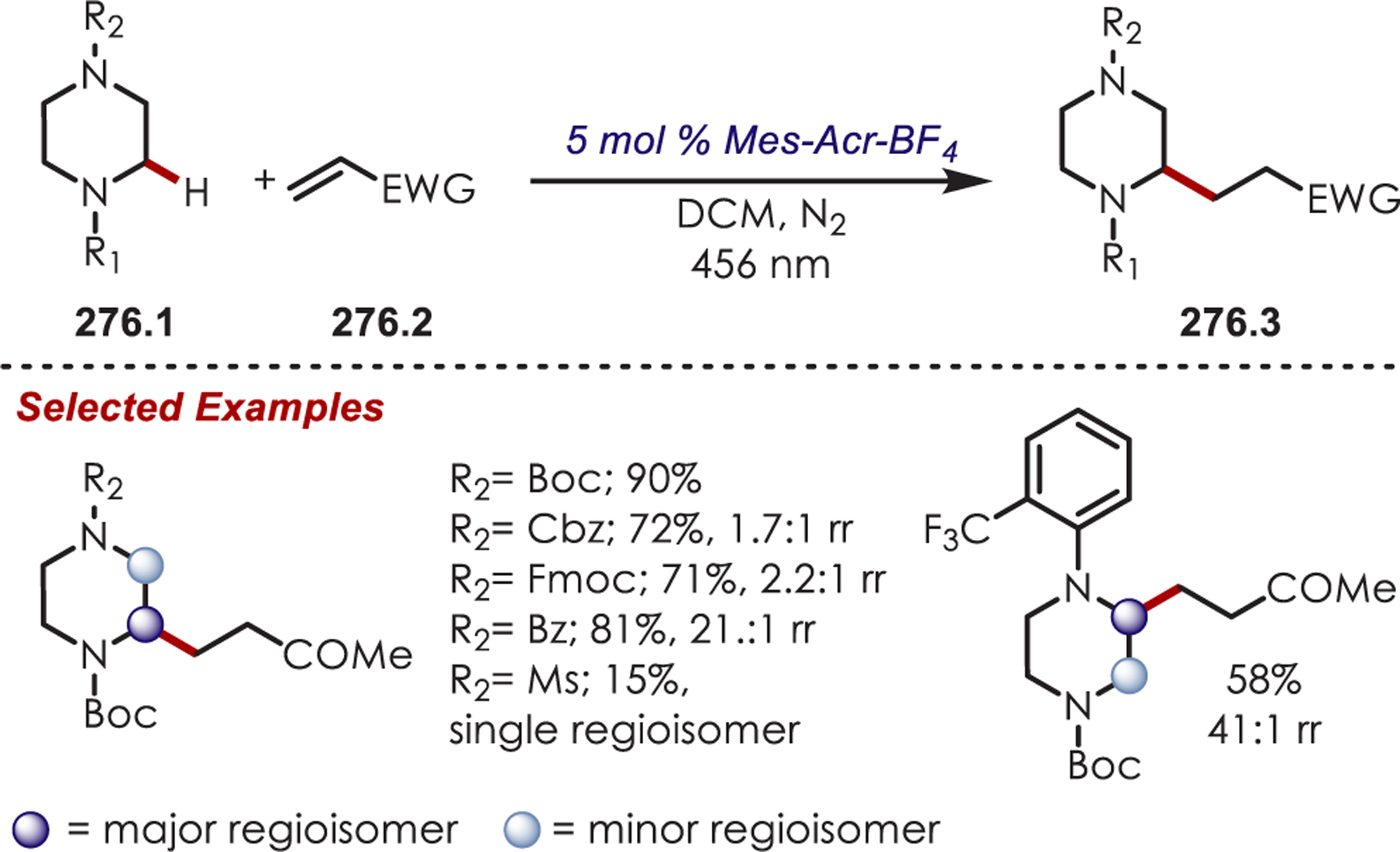
Site-Selective Piperazine C–H Alkylation with an Acridinium Salt as the Photooxidant
Many inorganic photoredox-catalyzed transformations have relied on HAT events to activate substrates rather than direct electron transfer between the catalyst and substrate. However, there remain limited examples of organic photocatalysts which function via excited-state hydrogen atom transfer, rather than photoinduced electron transfer. Nonetheless, Wu and co-workers developed an eosin Y-mediated alkylation by relying on a HAT to photoexcited EY (Scheme 277).479 Ethers, alcohols, amides, thioethers, aldehydes, benzyl, and cyclohexyl C–H bonds (277.1) were successfully alkylated with this method. Additionally, the scope of the olefin was explored and found to be successful with a diverse range of electron-withdrawing olefins.
Scheme 277.
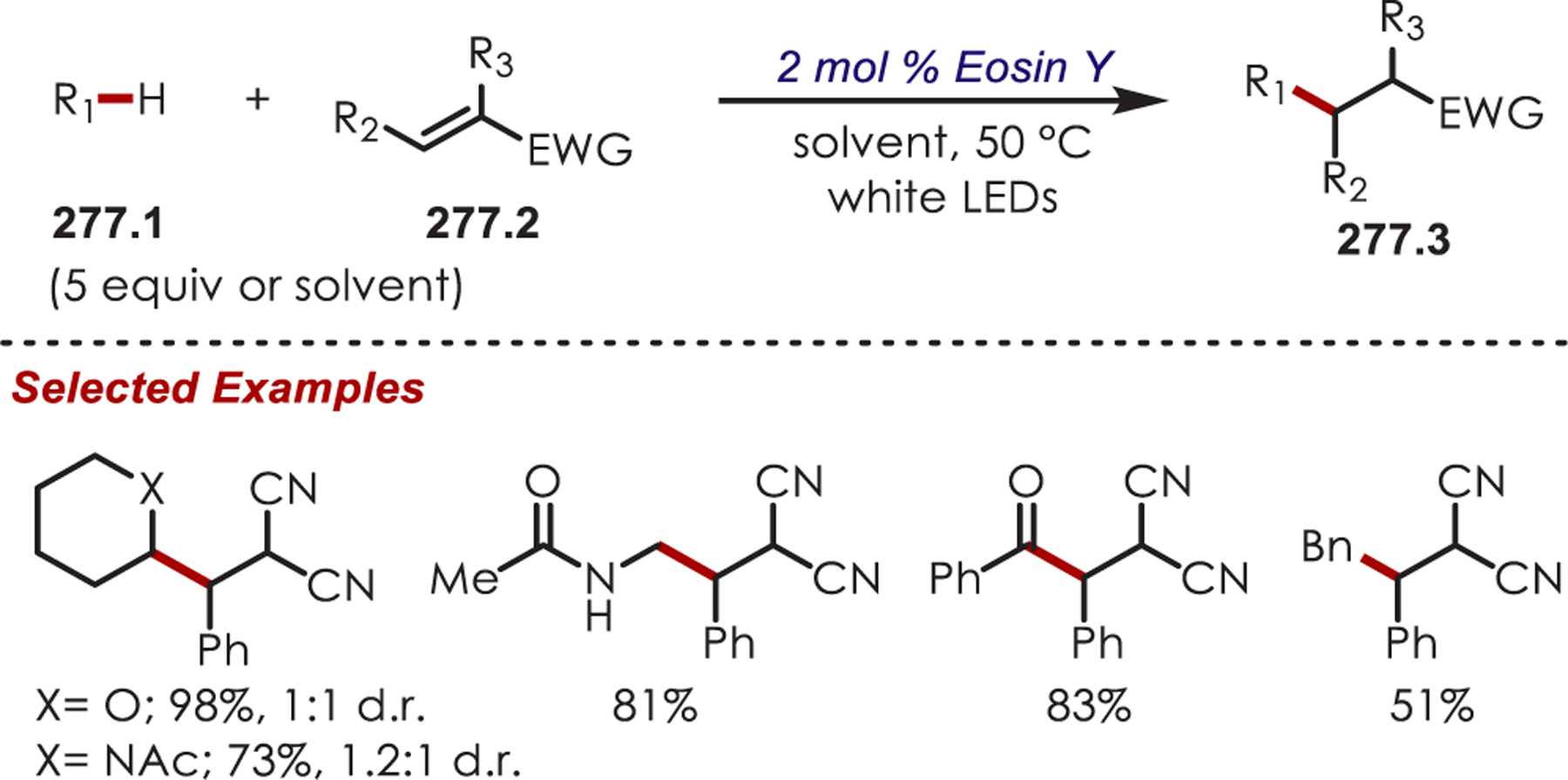
α-Hetero C–H Alkylation with Eosin Y as the Photooxidant
α-Heteroatom radicals generated through a quinuclidine-catalyzed hydrogen atom transfer have been trapped with alkenes in the presence of CO2 to afford the carbocarboxylated products 278.3 (Scheme 278).480 This C–H functionalization was demonstrated with amides, ethers, and thioethers coupled with electron-deficient alkenes.
Scheme 278.
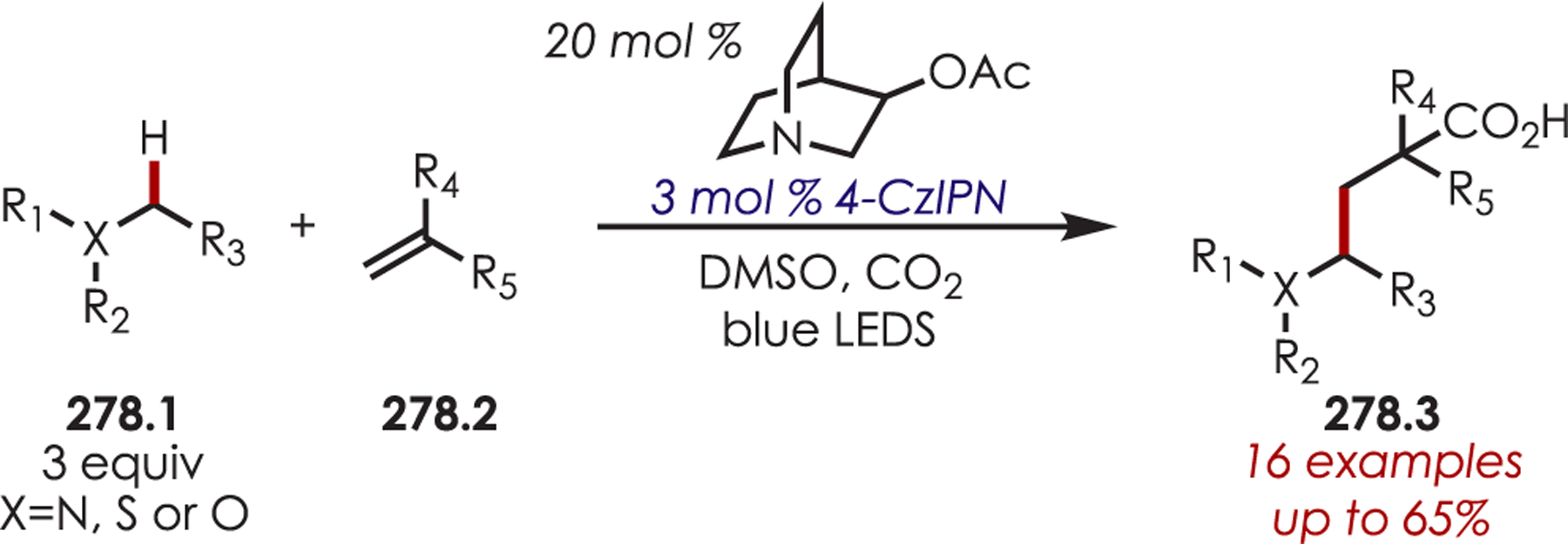
Carbocarboxylation of Alkenes via α-Hetero Radicals
An α-alkylation/arylation of saturated aza-heterocycles (279.1) was recently reported using dual nickel photoredox-catalyzed conditions (Scheme 279).481 Benzaldehyde was used as a photosensitizer HAT catalyst to generate the α-amino radical. A nickel-mediated coupling between aryl or alkyl bromides (279.2) afforded the α-arylated aza-heterocycles. The scope included pyrrolidine and piperidine aza-heterocycles, as well as tetrahydrothiophene. Electron-withdrawing or -donating aryl bromides and acyclic or cyclic aliphatic bromides with various functional groups were tolerated as coupling partners.
Scheme 279.
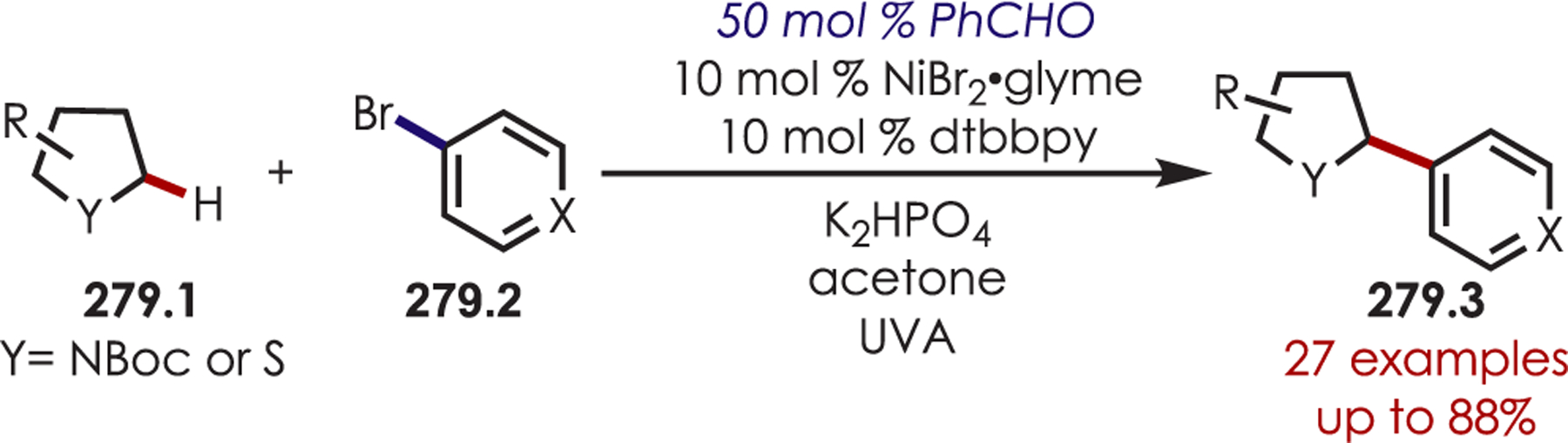
α-Heteroatom C–H Arylation with Aryl Bromides through a Benzaldehyde Photosensitization and HAT
Likewise, Martin reported a nickel photoredox-catalyzed α-arylation via triplet excited benzophenones as the key HAT catalysts (Scheme 280).482 The system is catalytic with respect to the HAT reagent by using nickel as both an electron donor and acceptor, thus obviating the need for sacrificial redox agents. A wide scope of aryl and alkyl bromide coupling partners (280.2) was compatible, including more complex cholesterol, menthol, allofuranose, or glycine derivatives. The scope of aryl bromide coupling partners displayed high functional group tolerance, allowing for the use of boronic esters, organotin reagents, and free alcohols or amines. Notably, electron-rich aryl bromides, benzothiophenes, and indoles were suitable substrates; often these classes of compounds are lower yielding with iridium photoredox-catalyzed pathways due to competitive catalyst quenching.228,230,234,483 Arylation and alkylation were demonstrated with both cyclic and acyclic ethers, oxetanes, pyrrolidines, and even cycloalkanes. Benzaldehydes were later reported as suitable photosensitizers/HAT catalysts in the cross coupling of ethers and alkyl/aryl bromides through combined nickel photoredox catalysis.484
Scheme 280.
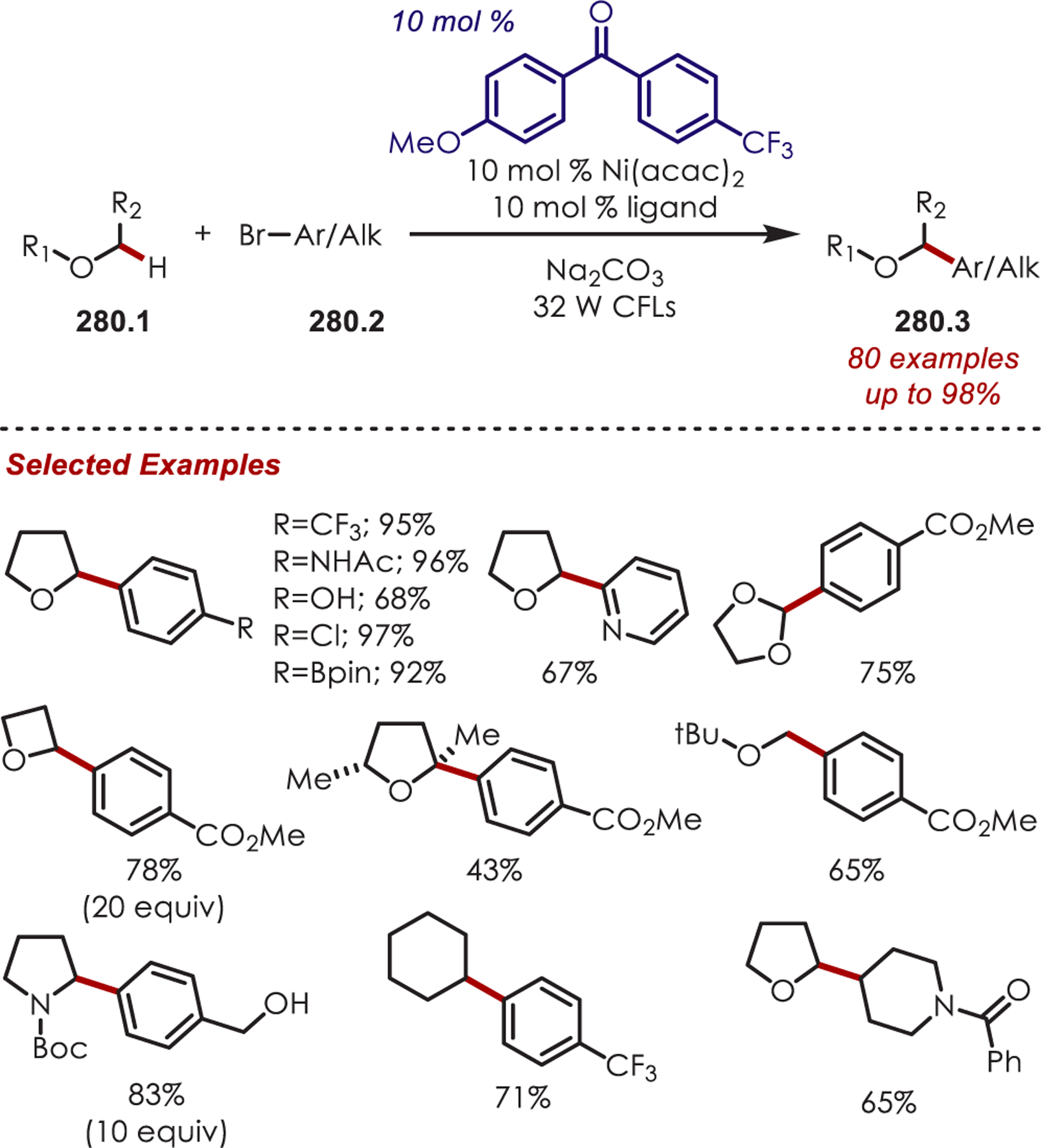
α-Oxo C–H Arylation and Alkylation Reactions with Benzophenones as a Photoredox and HAT Catalyst
In a similar vein, excited-state benzophenone derivatives were used as catalysts to generate α-heteroatom radicals via HAT in alkenyl-, alkynyl-, and allylation reactions (Scheme 281).485 A reductive PCET of sulfinyl radicals regenerates the diarylketone catalyst. Alkenyl-, alkynyl-, and allyl-substituted ethers and amides are furnished as the products in good yield with high E-selectivity.
Scheme 281.

α-Hetero C–H Alkynylation, Alkenylation, and Allylations Using a Diarylketone
Recently, a CzIPN-mediated α-amino radical addition into vinyl ketones was demonstrated for the synthesis of 1,4-dicarbonyls (Scheme 282).486 Further oxidation of the intermediate aminocarbonyl and HAT generated an iminium ion in situ, which was then hydrolyzed by water to the 1,4-dicarbonyl (282.2).
Scheme 282.
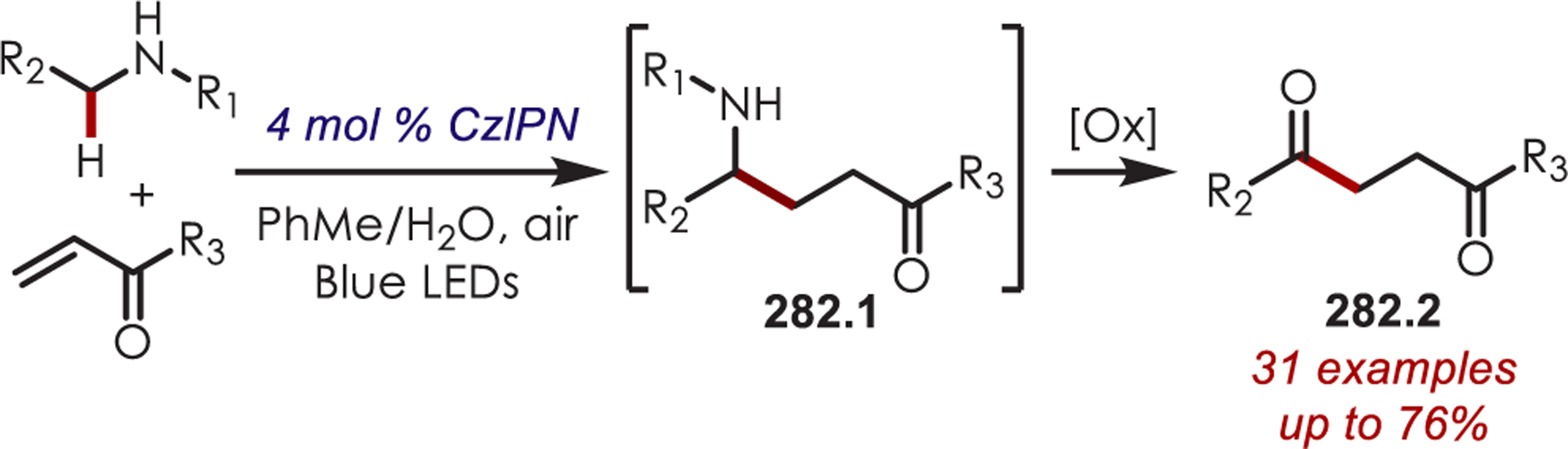
α-Amino Alkylation for the Synthesis of 1,4-Dicarbonyls
Cresswell has developed a CzIPN-catalyzed α-alkylation of unprotected amines, 283.1 (Scheme 283).487 Azide ion is used as the HAT precatalyst, which generates the strongly oxidizing azidyl radical, capable of abstracting unactivated alkyl hydrogen atoms through a reductive quenching of the catalyst. This method is particularly useful for the synthesis of α-tertiary primary amines and γ-lactams, which can be accessed through the condensation of the alkylated amines. A variety of spirocycles with varying ring size, including those containing heteroatoms were successful. Substituted Michael acceptors afforded the hydroxymethyl or amine lactam products. Other non-ester-based acceptors were tolerated, such as vinylpyridine, which afforded the uncyclized products.
Scheme 283.
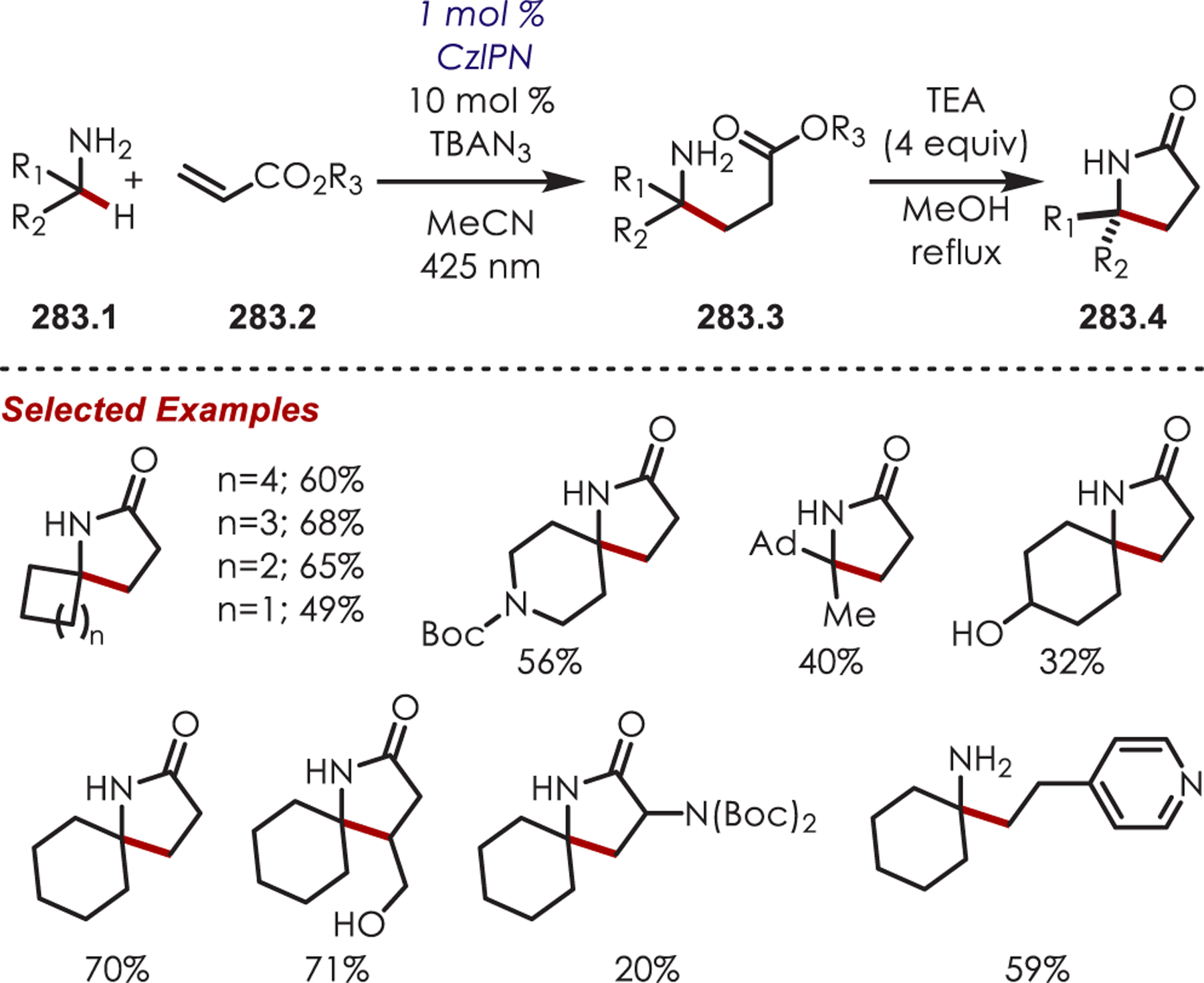
α-C–H Alkylation of Unprotected Amines through an Azidyl Radical HAT
Additionally, sulfonamides have been reported as suitable HAT agents for α-heteroatom alkylations (Scheme 284).488 Sulfonamides are attractive HAT catalysts due to the ability to modulate the N–H BDE from 95 up to 105 kcal/mol thus allowing for more selective hydrogen atom abstraction.
Scheme 284.
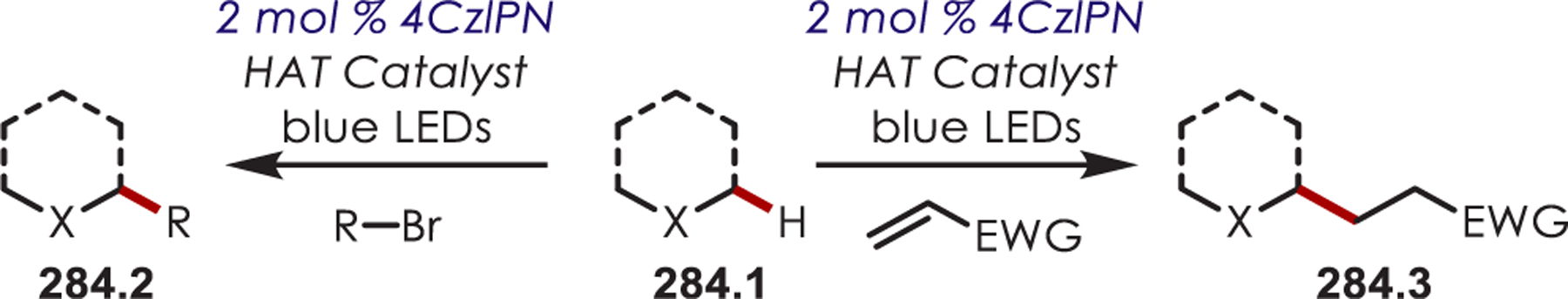
Sulfonamides as HAT Catalysts for α-Hetero C–H Alkylations
3.2.2. Carbonyl α-C–H Functionalization.
α-C–H functionalization reactions of carbonyl derivatives have been widely developed in the realm of inorganic photoredox catalysis, particularly for enantioselective transformations. Through an analogous mechanism, organic photoredox catalysts generate carbon- or oxygen-centered radicals, which selectively add to chiral enamines. However, there are no reports of organic photoredox-catalyzed β-C–H functionalization reactions of carbonyl derivatives.
Zeitler and co-workers developed an α-alkylation of aldehydes using Eosin Y as an organic photoredox catalyst that is nearly identical to the reaction developed by Nicewicz and MacMillan271 (Scheme 285).489 Eosin Y anion radical served as the reductant for the alkyl halide (285.2) to generate the sp3 carbon-centered radical. Addition of the α-keto radical to the enamine produced an intermediate α-amino radical. Oxidation from photoexcited EY and hydrolysis generated the desired product and EY anion radical, which serves as the reductant for another equivalent of alkyl bromide. A year later, this transformation was reported with Rose Bengal as the photoredox catalyst, but was also very limited in scope with seven examples in 51–94% yields and 80–85% ee’s.490
Scheme 285.
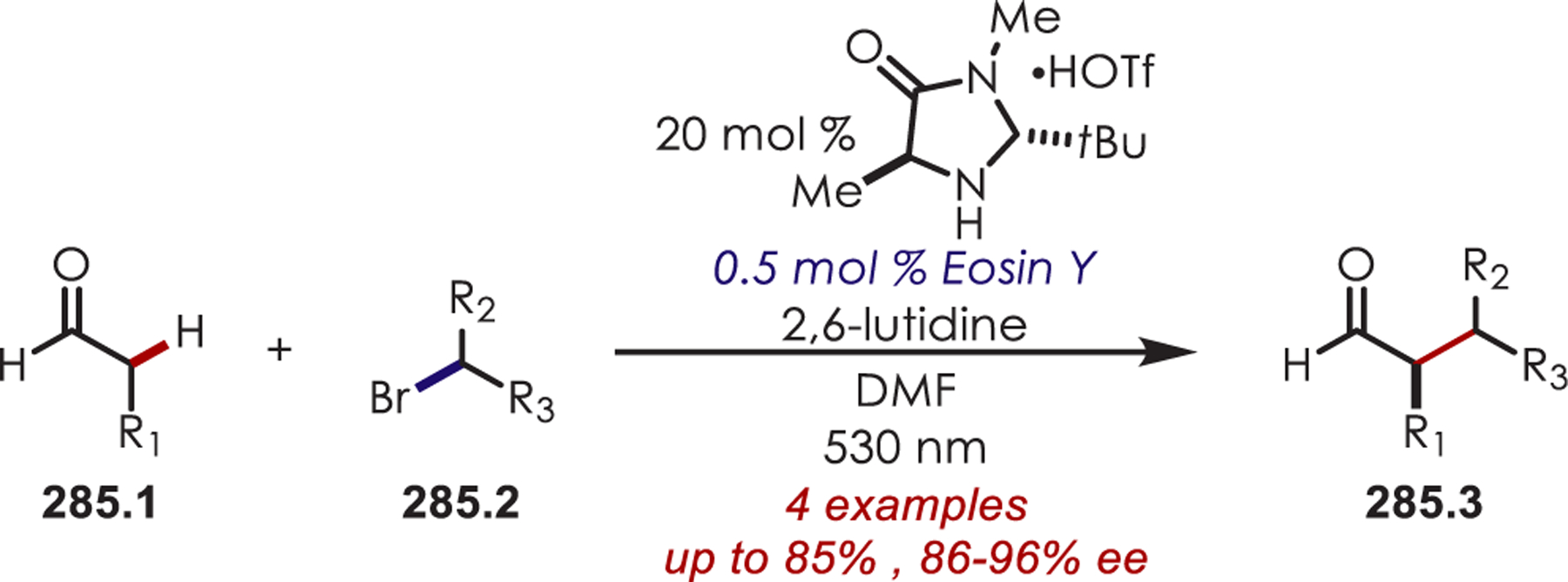
α-Alkylation of Aldehydes with Alkyl Bromides Using Eosin Y as Photocatalyst
Direct Aldehyde C–H Functionalization.
Acyl radicals, generated through a photomediated hydrogen atom abstraction event, have been utilized in the synthesis of aliphatic and aryl ketones.119 Itoh and co-workers intercepted acyl radicals for the synthesis of 3-acyl-4-arylcoumarin derivatives (Scheme 286).491 Hydrogen abstraction from photogenerated triplet AQN* generates an intermediate acyl radical which adds to alkynes (286.1), producing a vinyl radical. A subsequent cyclization through an aryl C–H or C–O ipso-functionalization and 1,2-ester migration gave access to coumarin derivatives (286.3).
Scheme 286.
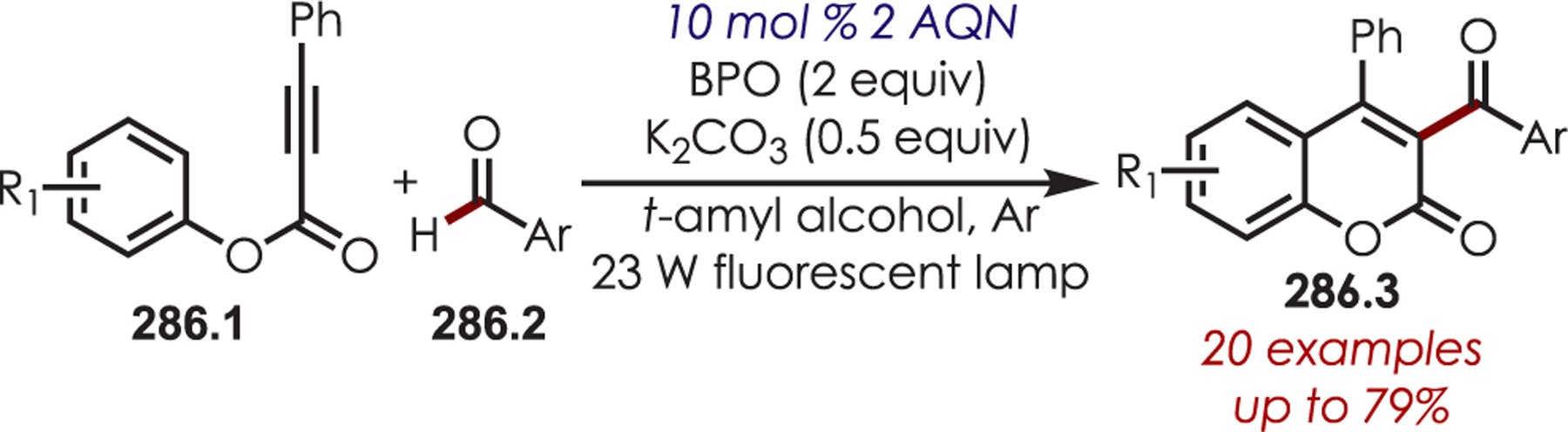
Generation of Acyl Radicals for a Radical Cascade with Alkynes and Arenes to Produce 3-Acyl-4-arylcoumarins
Salles and co-workers reported an alkene hydroacylation reaction with benzaldehyde derivatives catalyzed by a methylene blue and persulfate system (Scheme 287).492 The acyl radicals are proposed to be generated by C–H abstraction from a hydroperoxyl radical, formed from oxidation of hydroperoxide anion by photoexcited methylene blue. Additionally, acyl radicals generated with eosin Y have been reported by Wu and co-workers to couple in a similar manner with electron-deficient benzylidenemalononitrile.479
Scheme 287.
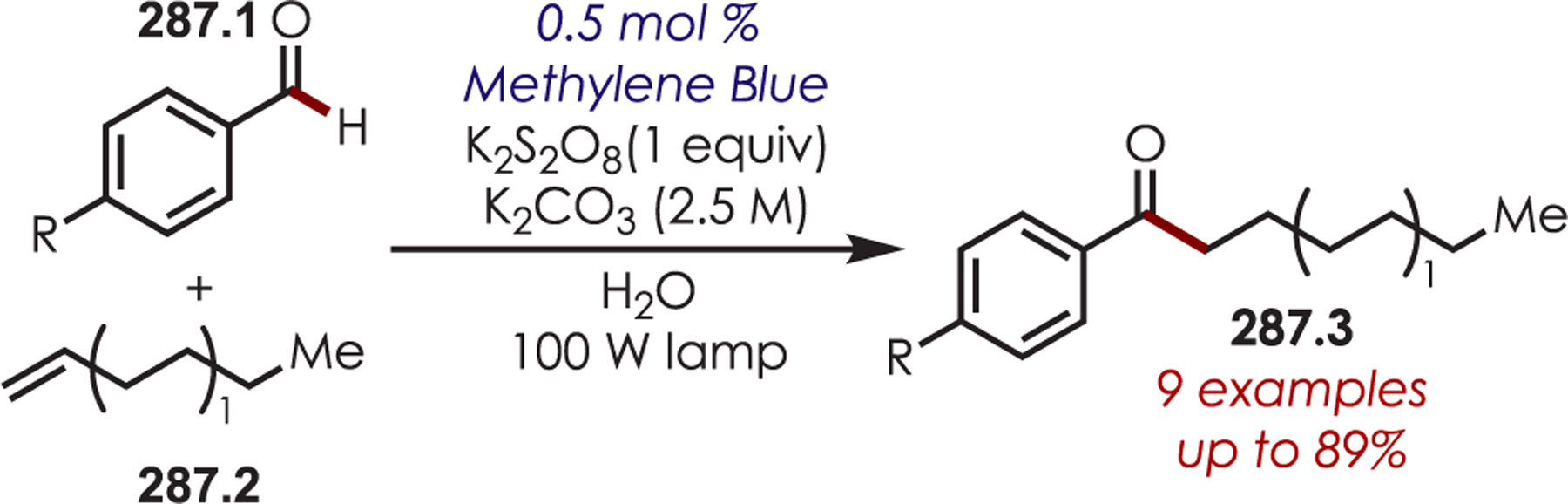
Direct Aldehyde C–H Alkylation through Acyl Radical Coupling to Olefins
The synthesis of amides (288.3) directly from aldehydes under catalytic conditions has been accomplished with a phenazine ethosulfate catalyst (Scheme 288).493 The photoredox catalytic cycle plays a key role in the generation of hydrogen peroxide, from superoxide, as an in situ oxidant for oxidation of the hemiaminal to the amide. Rose bengal was also reported as a catalyst for an analogous photooxidative amidation of aldehydes.494
Scheme 288.
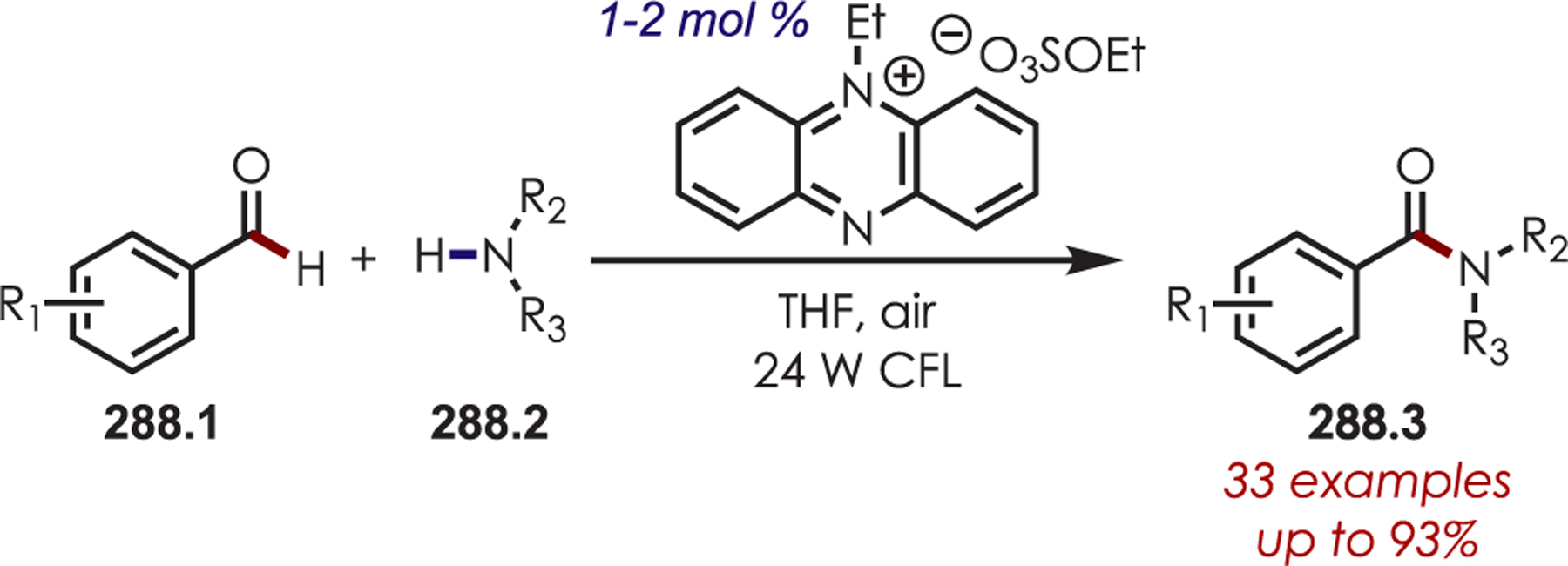
Amide Synthesis from Aldehydes with a Phenazine Ethosulfate
Kanai developed a diastereoselective allylation of aldehydes, 289.1, using dual chromium and acridinium catalytic system and a chiral thiophosphoric imide HAT catalyst (Scheme 289).495,496 Allylic C–H activation occurs through a HAT from a thiol radical and subsequent interception by Cr(II). The resulting intermediate, 289.4, reacts with an aldehyde through a six-membered chairlike transition state, which selectively yields the anti-products, 289.3, after protonation.
Scheme 289.
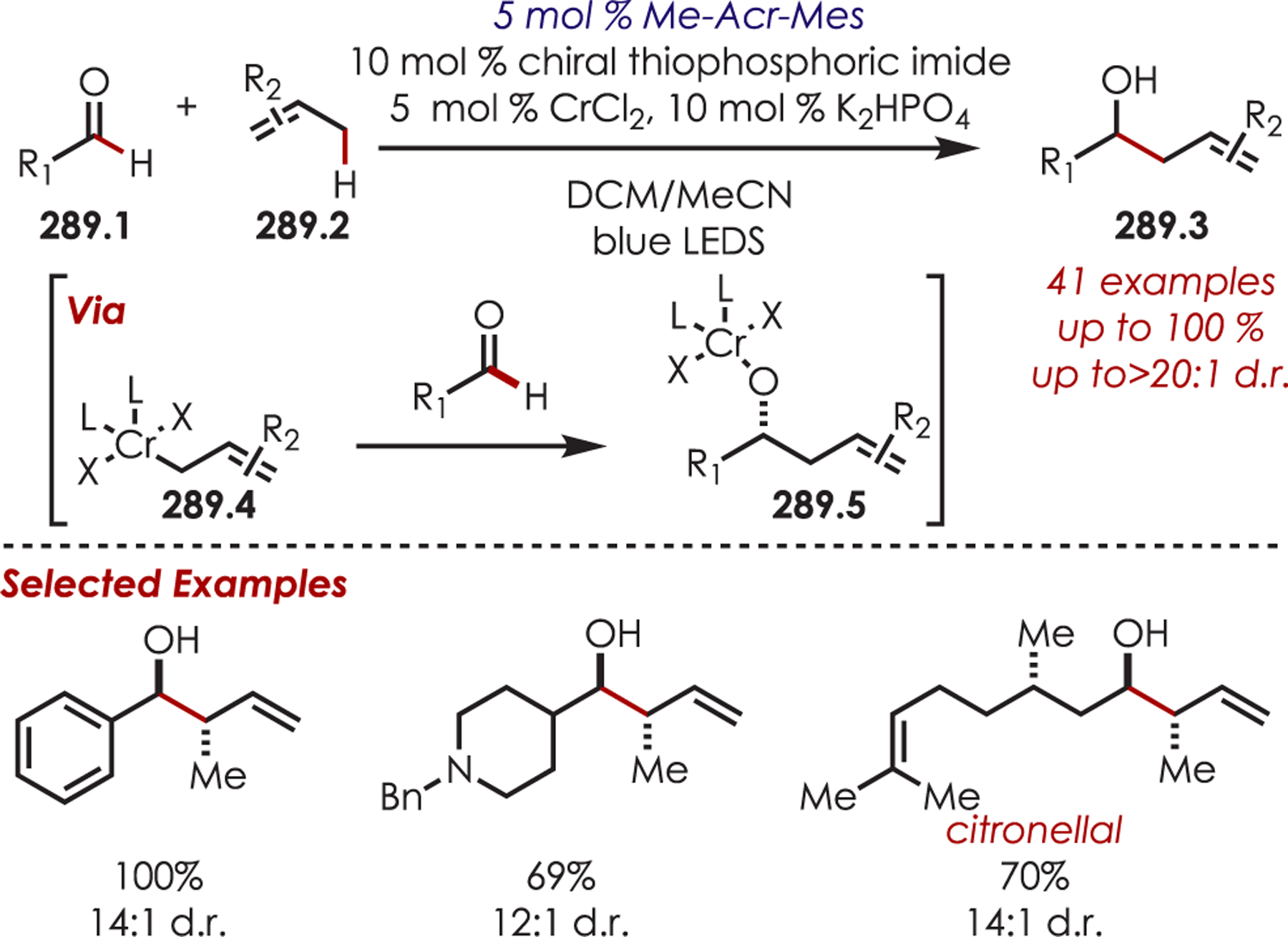
Dual Chromium and Photoredox Catalysis for the Synthesis of Homoallylic Alcohols
Likewise, α-keto amides (290.3) were synthesized through α-keto acyl radicals mediated through a rose bengal-generated hydroperoxyl radical HAT (Scheme 290).497 Following hydrogen atom abstraction from hydroperoxyl radical, the newly formed acyl radical is proposed to undergo a radical–radical coupling with an amine radical to afford the desired product.
Scheme 290.
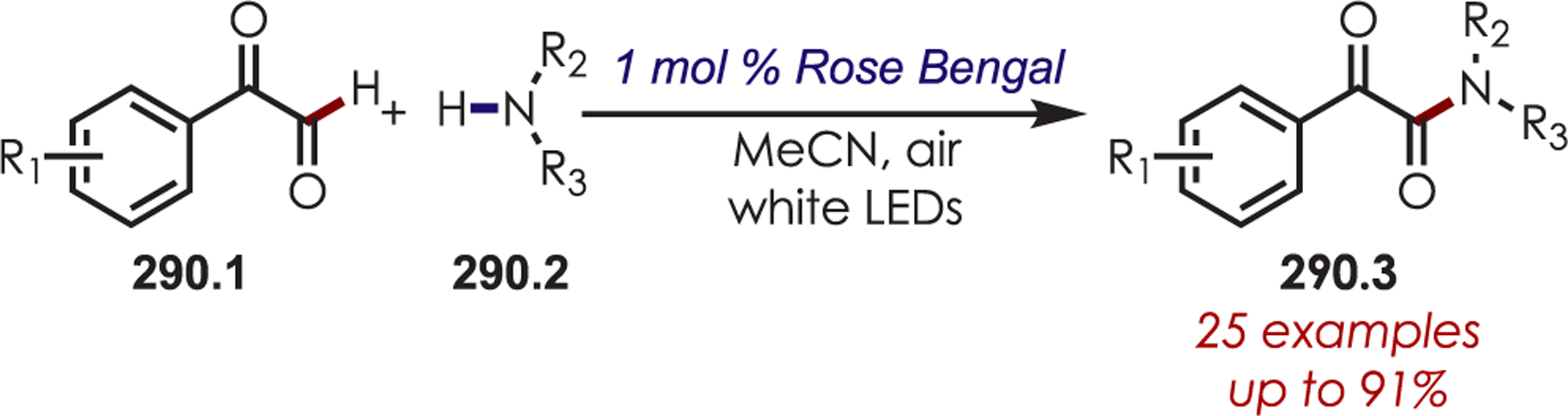
Synthesis of α-Keto Amides through an Acyl Radical
The asymmetric synthesis of 1,4-dicarbonyl compounds through a dual eosin Y and chiral Rh Lewis acid co-catalyst was achieved by Meggers and Wu (Scheme 291).498 Eosin Y serves as both the hydrogen atom transfer and photoredox catalyst while a chiral Rh catalyst generates a chiral enamine in situ (291.6), thus providing an asymmetric environment for the selective addition of the photogenerated acyl radical (291.3) to a rhodium-coordinated N-acyl pyrazole (291.6). Subsequent reverse hydrogen atom transfer regenerates both catalytic cycles while furnishing the product (291.2). The scope of this chemistry was extended to aliphatic or aryl aldehydes with aliphatic enamides in good to excellent enantioselectivity. This methodology was also extended to an asymmetric Giese-type addition with photogenerated α-sulfur and phosphorus radicals.
Scheme 291.
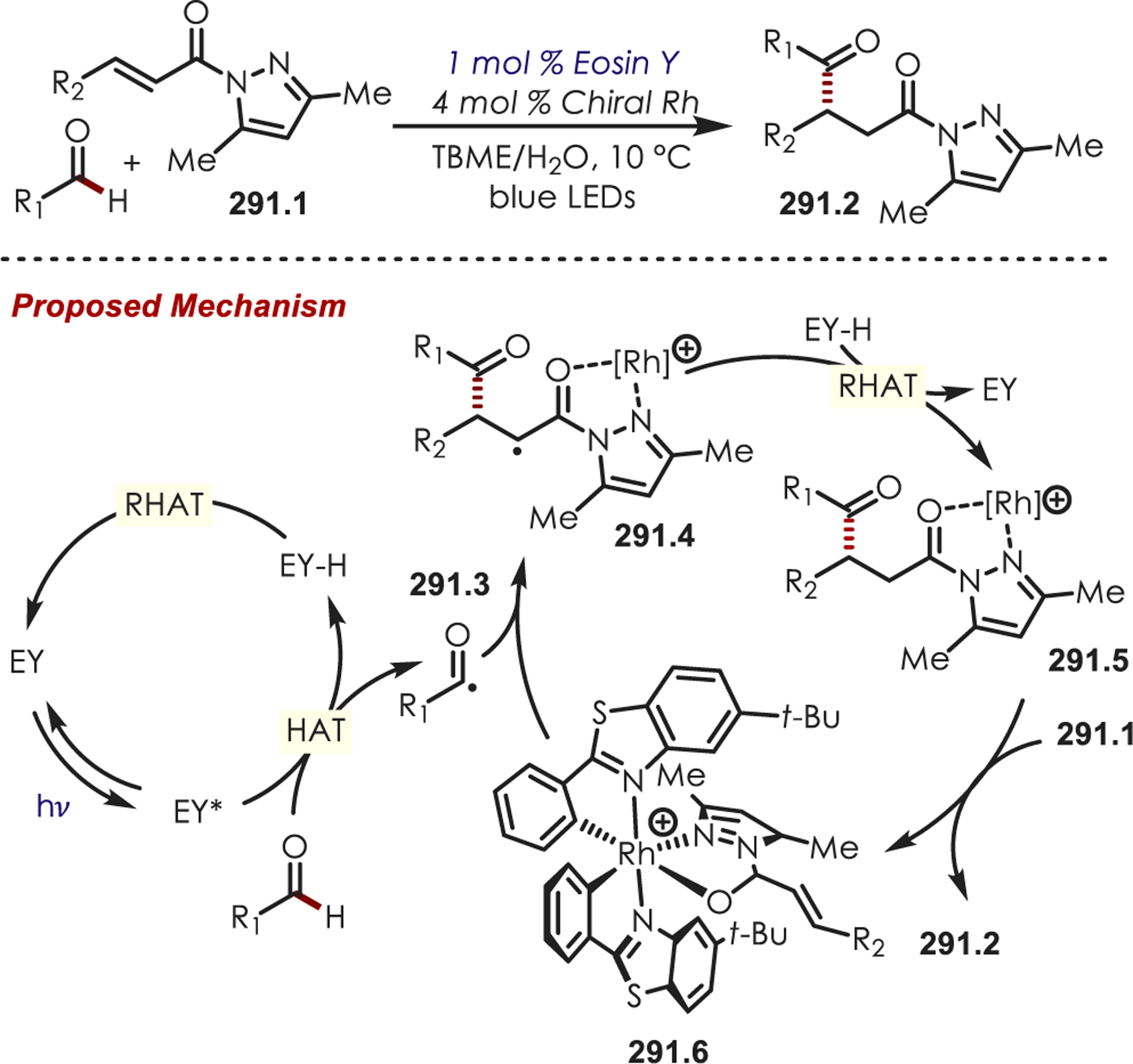
Dual Eosin Y Photoredox and Chiral Rh Lewis Acid Catalysis for the Asymmetric Synthesis of 1,4-Dicarbonyls
3.2.3. Benzylic and Allylic C–H Functionalization.
Via Hydrogen Atom Abstraction.
In 2015, Pandey developed a benzylic amination using DCA as the photoredox catalyst (Scheme 292).499 Interestingly, the employed amide coupling partner played a key role as the HAT catalyst in addition to acting as the nucleophile. Oxidative SET and subsequent deprotonation of the amine generates a NCR that serves as the HAT reagent. Hydrogen atom abstraction of the benzylic C–H, followed by oxidation by photoexcited DCA generates the benzylic cation. This transformation required N-OMe amides (292.2) as the coupling partner, which were proposed to form crucial captodative stabilized NCR’s.
Scheme 292.
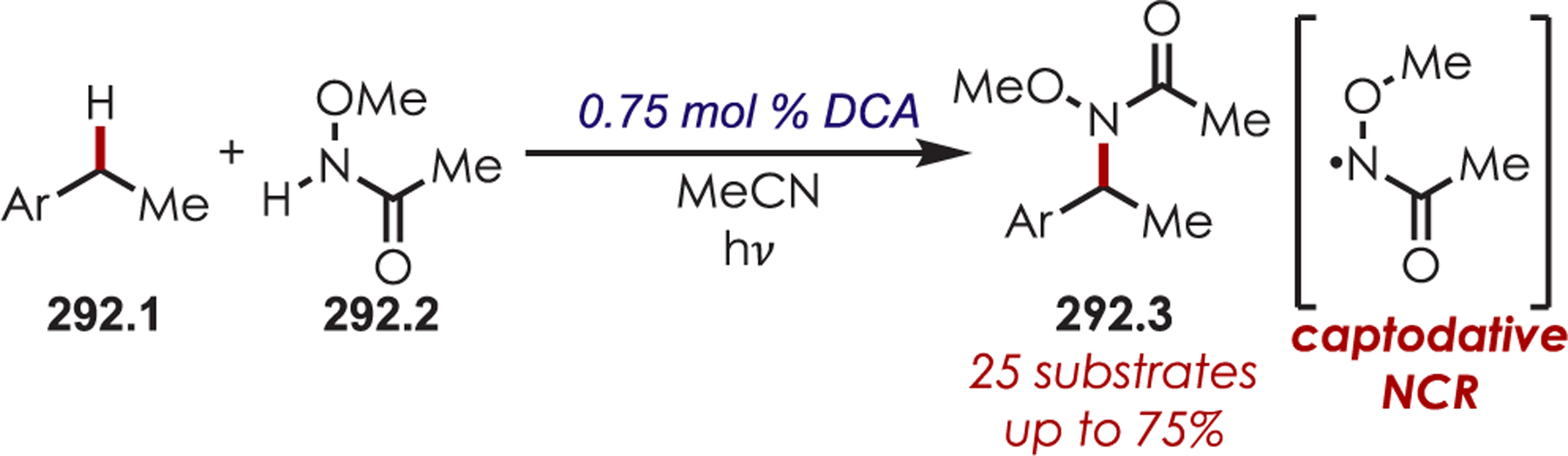
Benzylic C–H Amination via HAT by Stabilized Nitrogen-Centered Radicals
Selective mono- and difluorination of diarylketones was reported with 9-fluorenone or xanthone serving as the photoredox catalysts, respectively (Scheme 293).500 The benzylic radical was generated via an HAT event from the photoexcited catalyst. Subsequent trapping of the radical by Selectfluor affords the desired fluorinated product (293.2) and an amine cation radical. A HAT between the amine cation radical and the ketyl radical of the catalyst completes the catalytic cycle; the potential of a radical-chain mechanism was not discussed. The authors found that when xanthone was used as the catalyst with Selectfluor II, a sequential fluorination occurs to produce the gem-difluoro products (293.3); however, an exact rationale for the change in selectivity was not offered. The monofluorination method was compatible with a diverse range of electron-rich and poor arenes, and the difluorination was found suitable with electron-rich, neutral, or halogenated arene derivatives. Wu and co-workers later reported a benzylic fluorination with Selectfluor501 and chlorination502 using N-chlorosuccinamide with an acridinium ion as the photoredox catalyst.
Scheme 293.
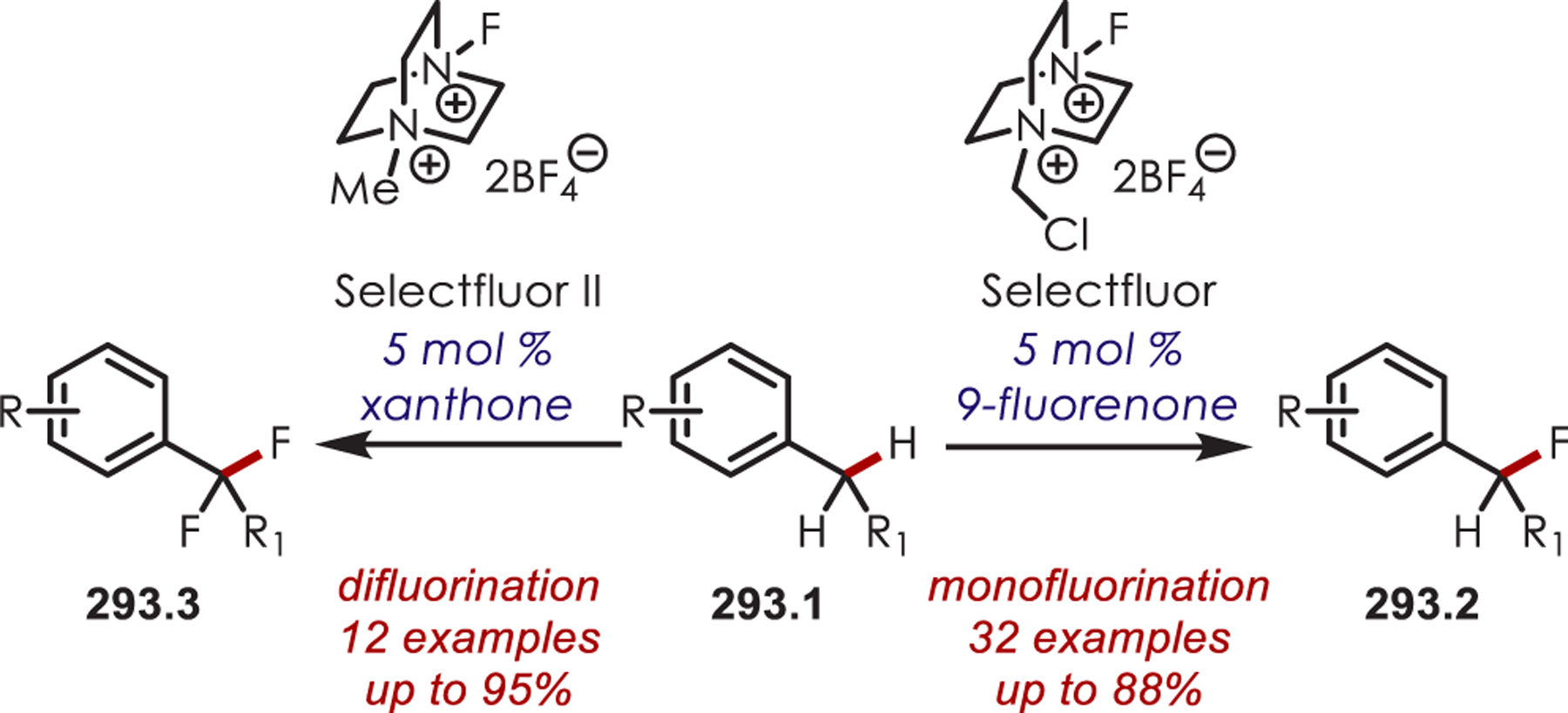
Benzylic C–H Mono- or Difluorination Using 9-Fluorenone and Xanthone as the Photoredox Catalysts
Xiao and co-workers utilized Selectfluor as a hydrogen atom transfer reagent for an intramolecular benzylic amination and oxidation (Scheme 294).503 A range of functionalized 3-hydroxyisoindolinones (294.2) were synthesized, and preliminary mechanistic work suggests that eosin Y may act as both a redox catalyst and photosensitizer.
Scheme 294.
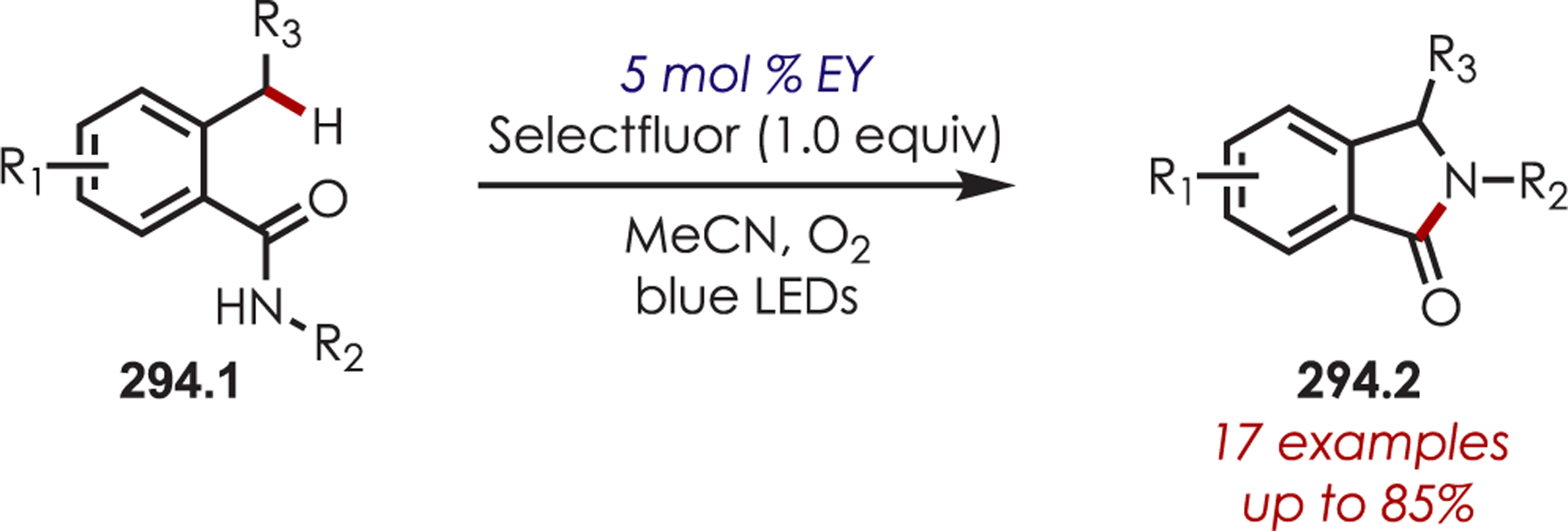
Intramolecular Benzylic C–H Amination and Oxidation for the Synthesis of 3-Hydroxyisoindolinones
König reported a carboxylation of benzylic C–H bonds with CO2 using triisopropylsilanethiol as the hydrogen atom transfer catalyst (Scheme 295).504 Single-electron reduction of the benzylic radical by the reduced form of the catalyst affords a benzylic anion that is readily trapped with CO2. This carboxylation tolerated a variety of functional groups including amides, esters and acetals for the benzylic C–H carboxylation of ortho-, meta-, and para-substituted arenes.
Scheme 295.
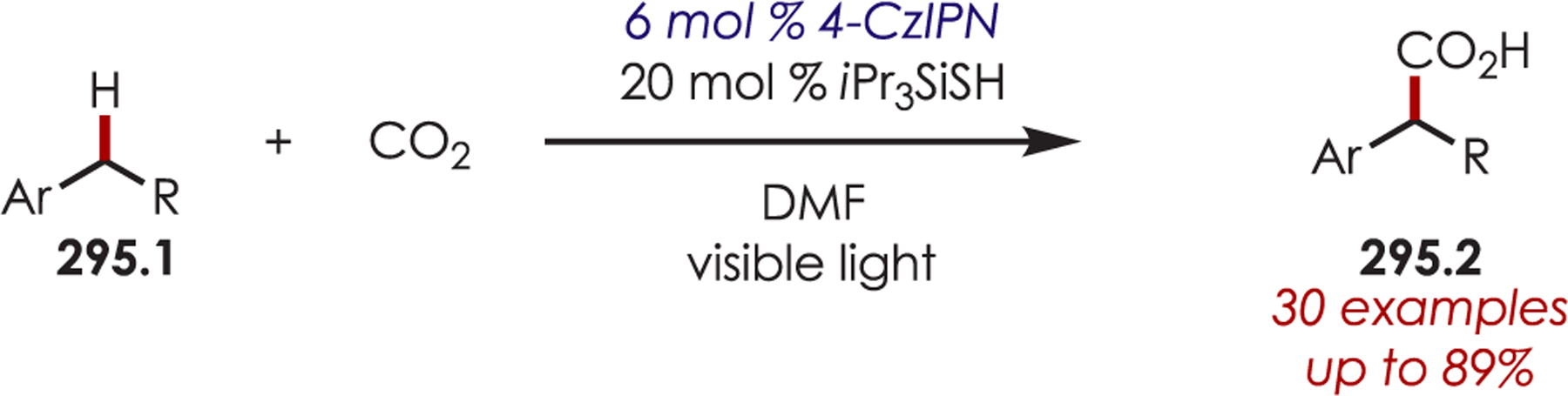
Benzylic C–H Carboxylation
Via SET Pathways.
In 2013, Pandey and co-workers reported a synthesis of C1-substituted isochromans (296.2) through the direct activation of benzylic C–H bonds and intramolecular coupling of tethered alcohols (Scheme 296).505 The proposed mechanism proceeds through oxidation of the arene to the aryl cation radical by photoexcited 1,4-dicyanonaphthalene (DCN), followed by a subsequent deprotonation. The resulting benzylic radical is further oxidized by a second equivalent of excited-state DCN to a benzylic cation, which then undergoes an intramolecular trapping by the pendant alcohol to afford the desired products. Molecular oxygen is responsible for catalyst turnover.
Scheme 296.
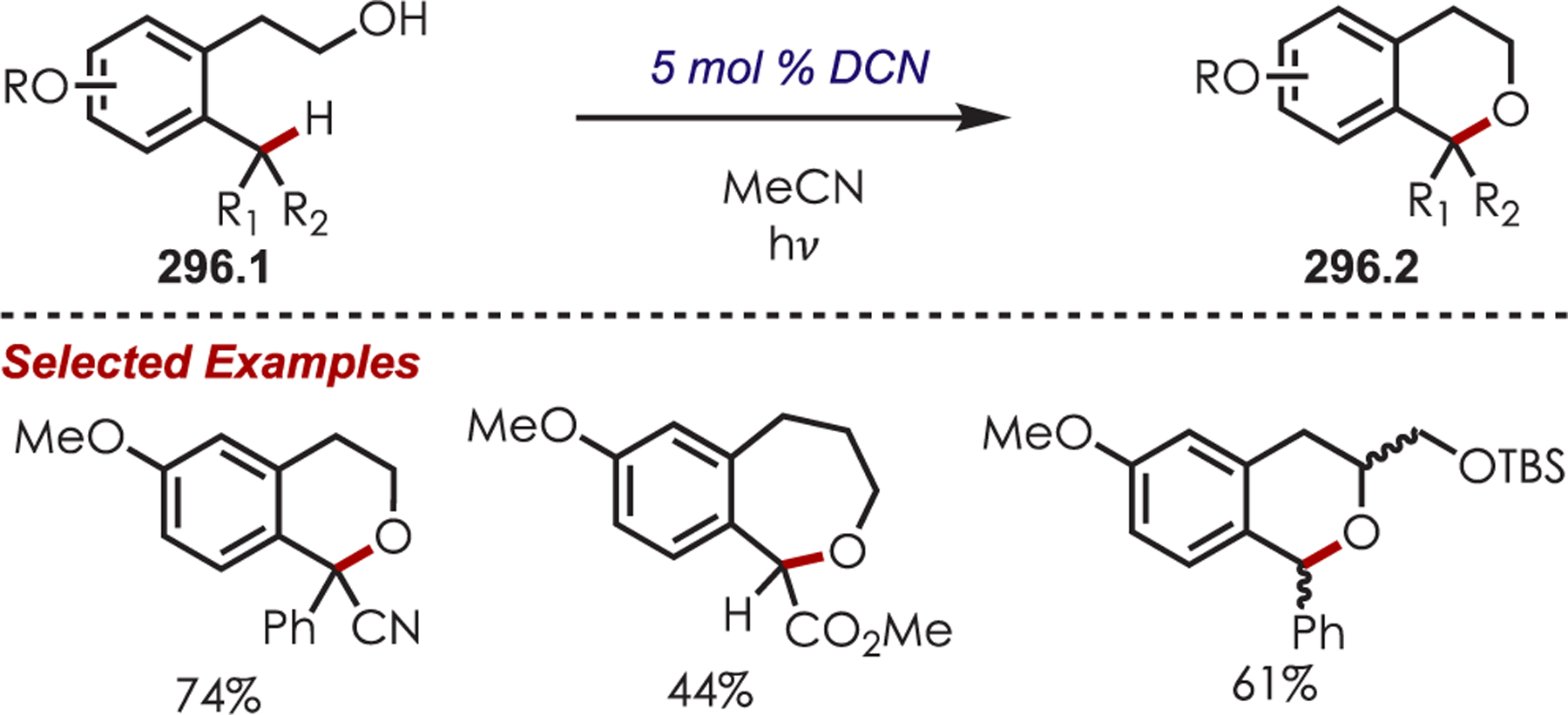
Intramolecular Benzylic C–H Alkoxylations with DCN
In a subsequent report, 2,4,6-triphenylpyrylium tetrafluoroborate (TPT) was employed as a catalyst in a similar intramolecular benzylic cyclization for the synthesis of ethers and lactones (Scheme 297).506 The mechanism is proposed to proceed through first oxidation of the arene (297.1), then deprotonation to form a benzylic radical. Trapping with molecular oxygen ultimately generates a benzylic alcohol that undergoes the intramolecular cyclization to afford the cyclized products. This protocol was successful in the synthesis of both five- and six-membered ethers.
Scheme 297.
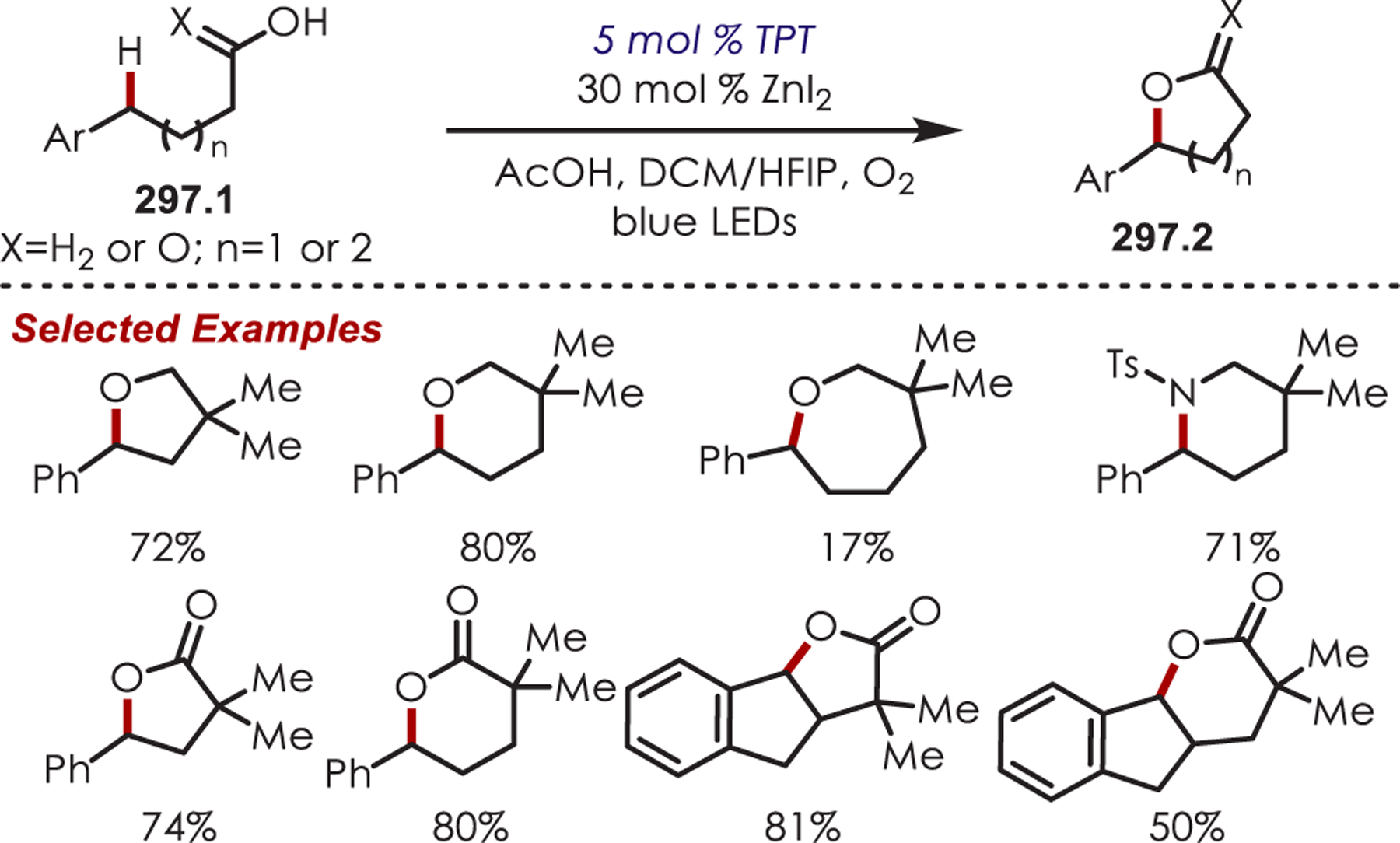
Intramolecular Benzylic C–H Alkoxylations with a Pyrylium Salt for the Synthesis of Ethers and Lactones
A benzylic trifluoromethylthiolation without an external hydrogen atom abstractor additive was reported through an inner-sphere hydrogen atom transfer mechanism (Scheme 298).507 The scope of this methodology was extended to heterocycles, thiophene, furan, and indoles as well as other alkyl- or halogen-substituted arenes. Additionally, good functional group tolerance was observed, including amines, halogens, and esters. Moreover, the authors demonstrated this method for the late-stage functionalization of complex derivatives, which demonstrated excellent site-selectivity.
Scheme 298.
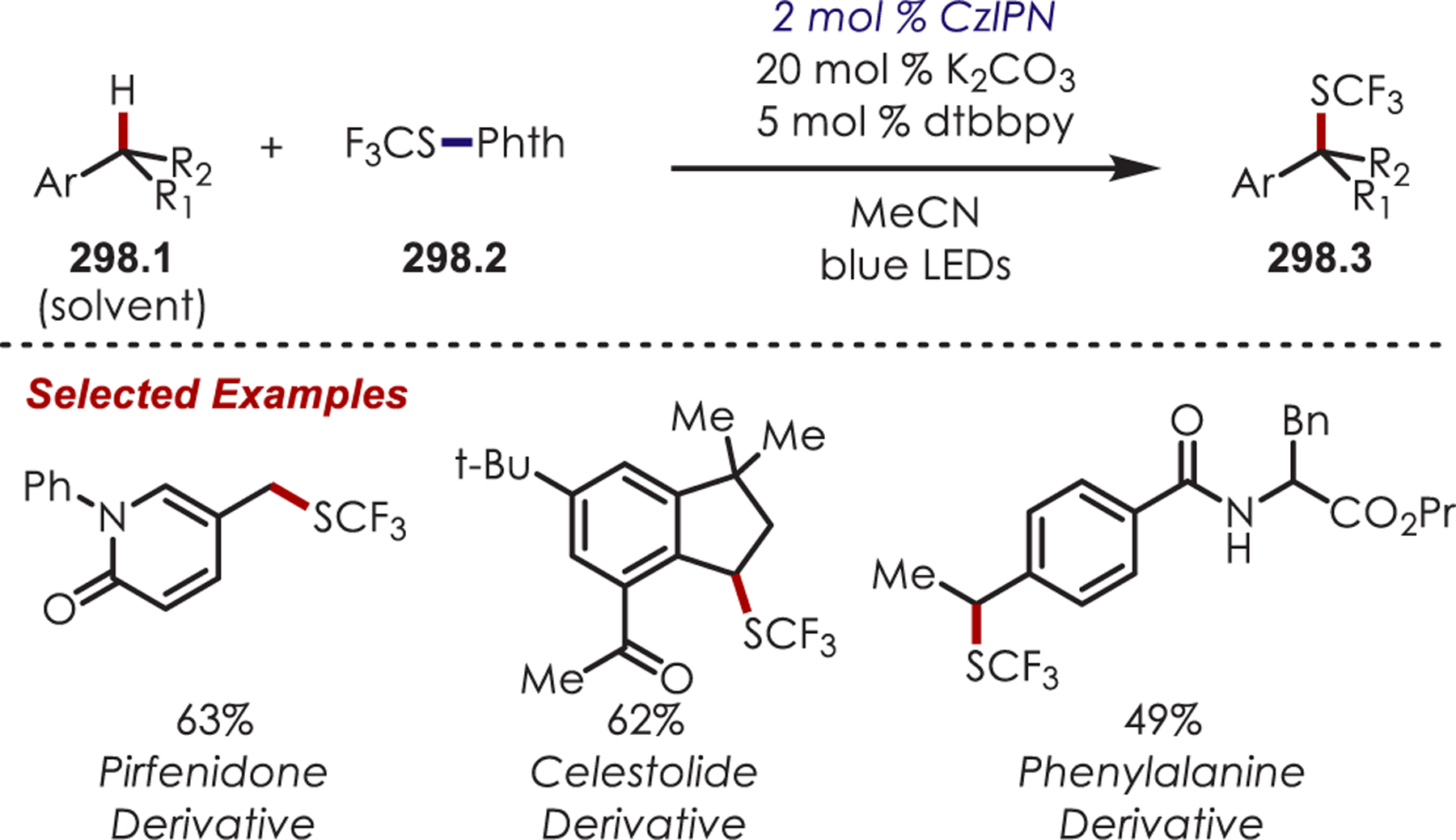
Benzylic C–H Trifluoromethylation through an Inner-Sphere Hydrogen-Atom-Transfer Mechanism
The high selectivity of this transformation, despite the presence of other benzylic, secondary, tertiary, or less acidic sp3 C–H bonds, was attributed to the mechanism involving an inner-sphere radical initiation (Scheme 299). They suggest the photogenerated aryl cation radical (299.4) undergoes an intramolecular 1,2-HAT with the benzylic C–H bond and subsequent deprotonation to generate the benzylic radical. The involvement of a benzylic radical was supported through EPR and radical clock experiments. The calculated quantum yield, φ = 0.33, suggests a radical chain pathway is less likely, but mechanistic support for an inner-sphere mechanism was not provided.
Scheme 299.
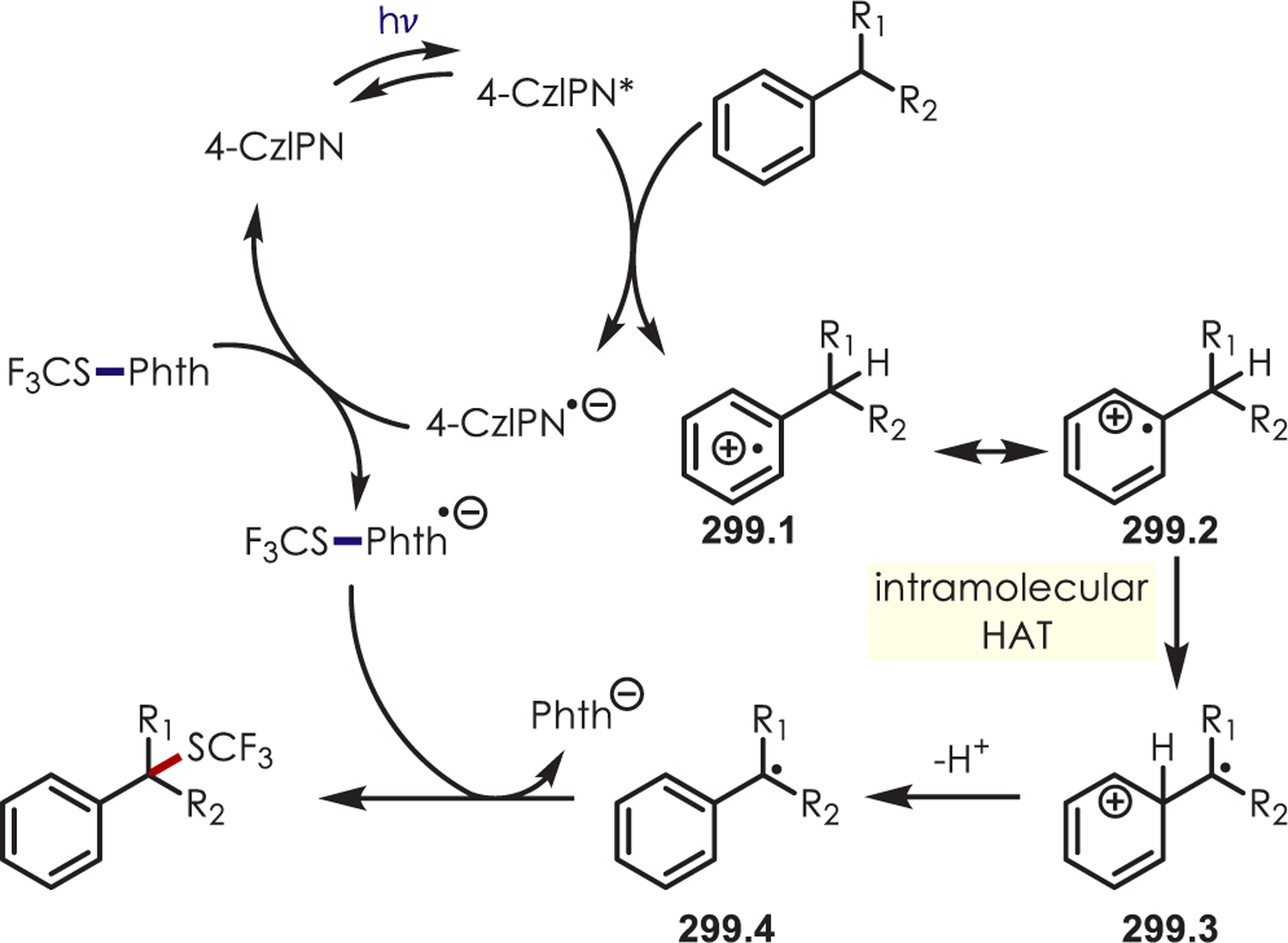
Mechanism for a Benzylic C–H Trifluoromethylation through an Inner-Sphere Hydrogen-Atom-Transfer Mechanism
The addition of photogenerated benzylic radicals to radical acceptors has been reported for benzylic alkylation (Scheme 300).508–510 Initial reports were catalyzed by decatungstate for the alkylation of toluene (employed as solvent) with vinyl ketones, esters, amides, and fumaronitrile as suitable Michael acceptors (300.1).508 Several years later, an acridinium-catalyzed alkylation was reported which required an excess (4–5 equiv) of the arene, rather than solvent levels as the previous example (300.2).509 Later, incorporation of a Lewis acid additive was found to facilitate the acridinium-catalyzed benzylic coupling with enones (300.3 and 300.4).510
Scheme 300.
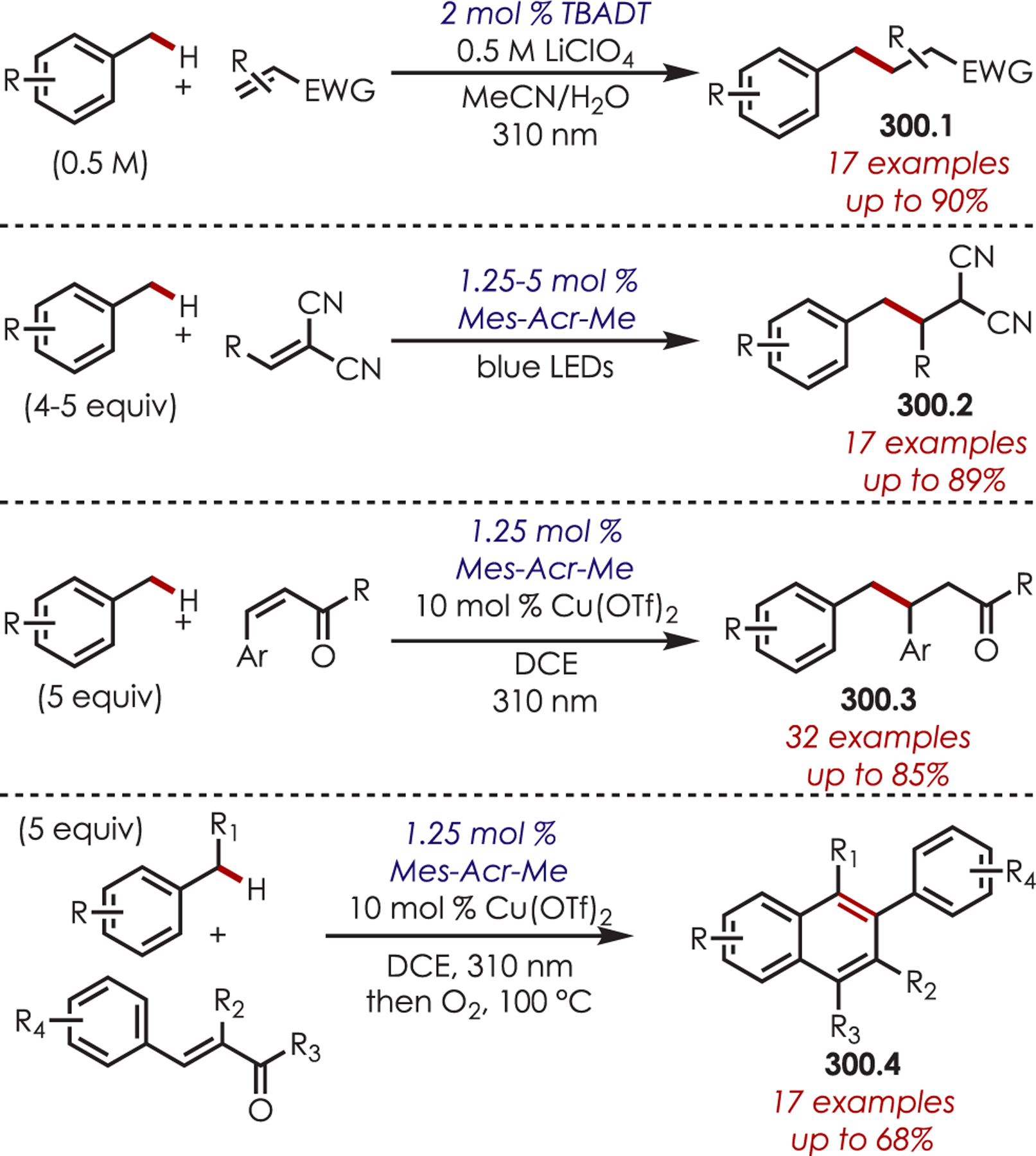
Benzylic C–H Alkylations through Radical Addition to Radical Acceptors
Triplet excited benzophenone has been used in the generation and trapping of benzylic radicals with cyanoarenes (Scheme 301).511 Alkyl (hetero)arenes, including polycyclic derivatives, were successfully coupled with 4-cyanobenzene to afford the desired 4-pyridyl-substituted products.
Scheme 301.
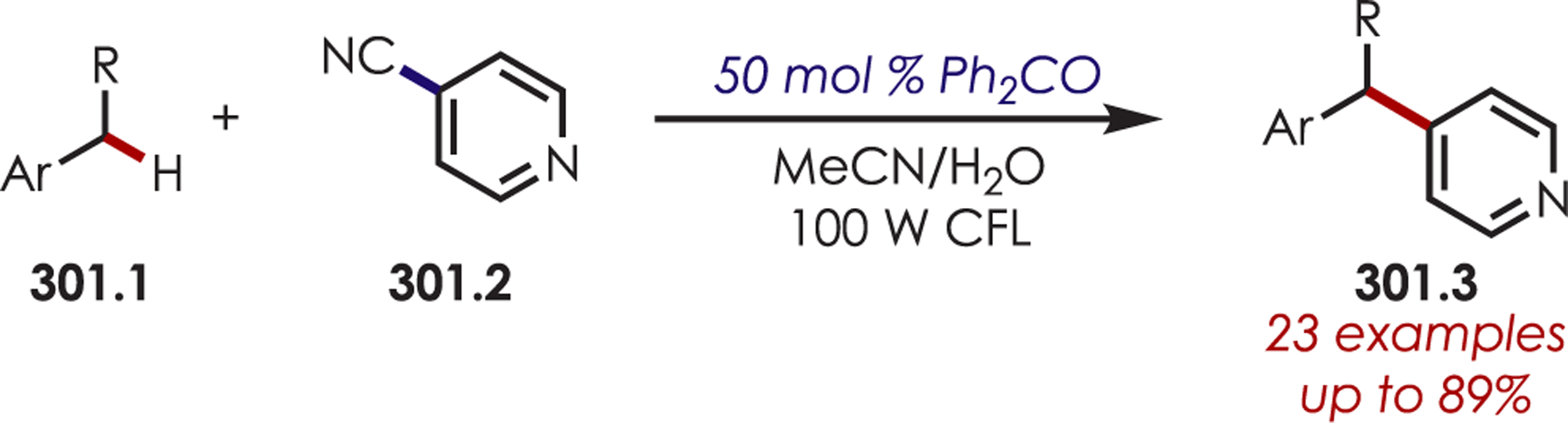
Benzylic C–H Arylation with Cyanoarenes
Benzylic radicals, generated by hydrogen atom abstraction from a triplet excited diaryl ketone, have been successfully intercepted by a nickel catalyst in a benzylic arylation reaction (Scheme 302).512 Electron-rich and electron-poor haloarene coupling partners (302.2) were well-tolerated in this chemistry; however, limited examples of the benzyl arene, which was used as the solvent, were reported.
Scheme 302.
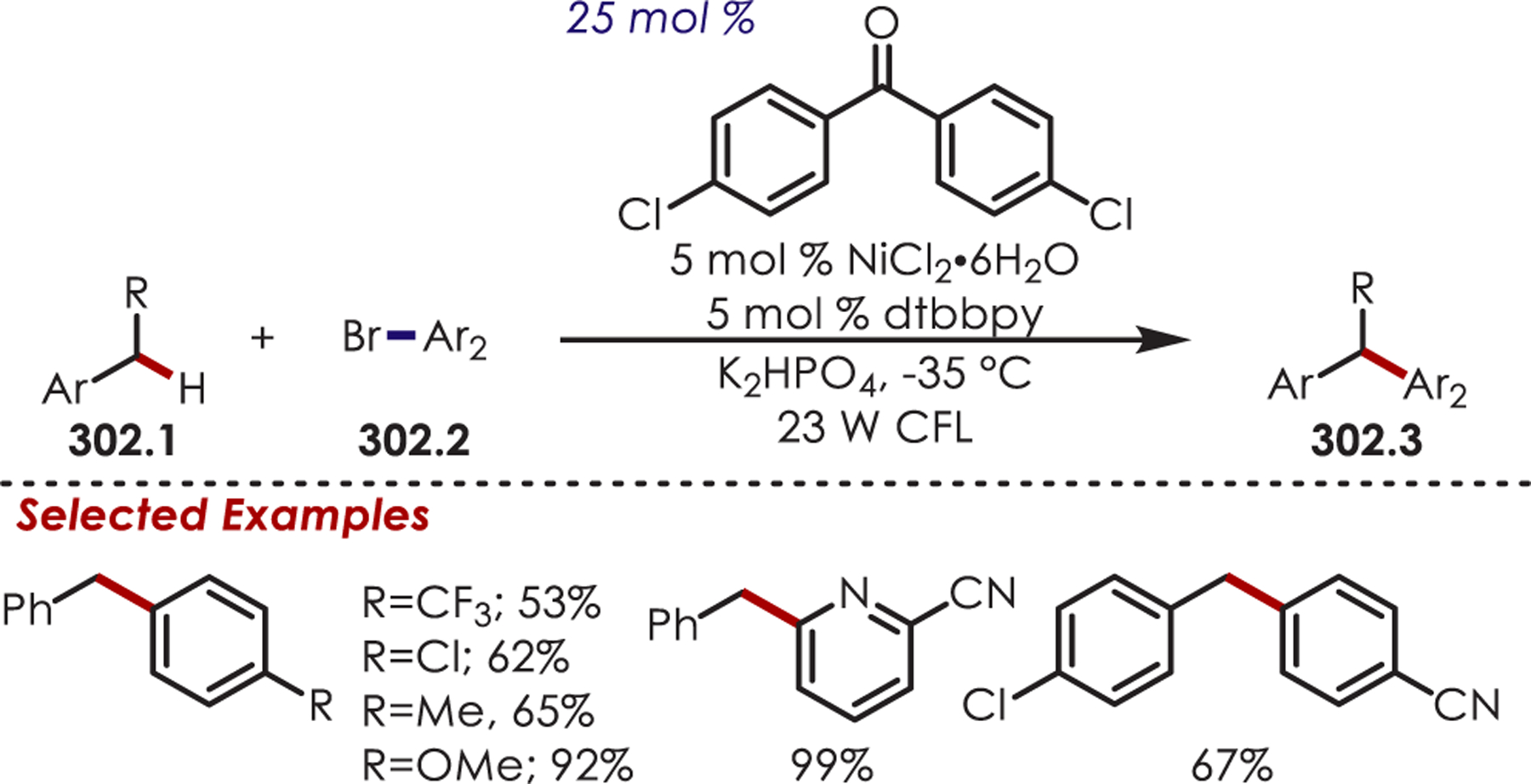
Benzylic C–H Arylation through Dual Triplet Excited Diaryl Ketone and Nickel Catalysis
In addition to benzylic functionalization, Wu and co-workers reported the allylic alkylation of cyclic or acyclic aliphatic and aromatic alkenes (Scheme 303).509 Oxidation by photoexcited Mes-Acr-Me and subsequent deprotonation of the cation radical afforded the key allylic radical intermediate. Di-, tri-, and tetrasubstituted alkenes (303.2), including cyclic derivates, were suitable alkene partners in this alkylation, furnishing the products in moderate to excellent yields.
Scheme 303.
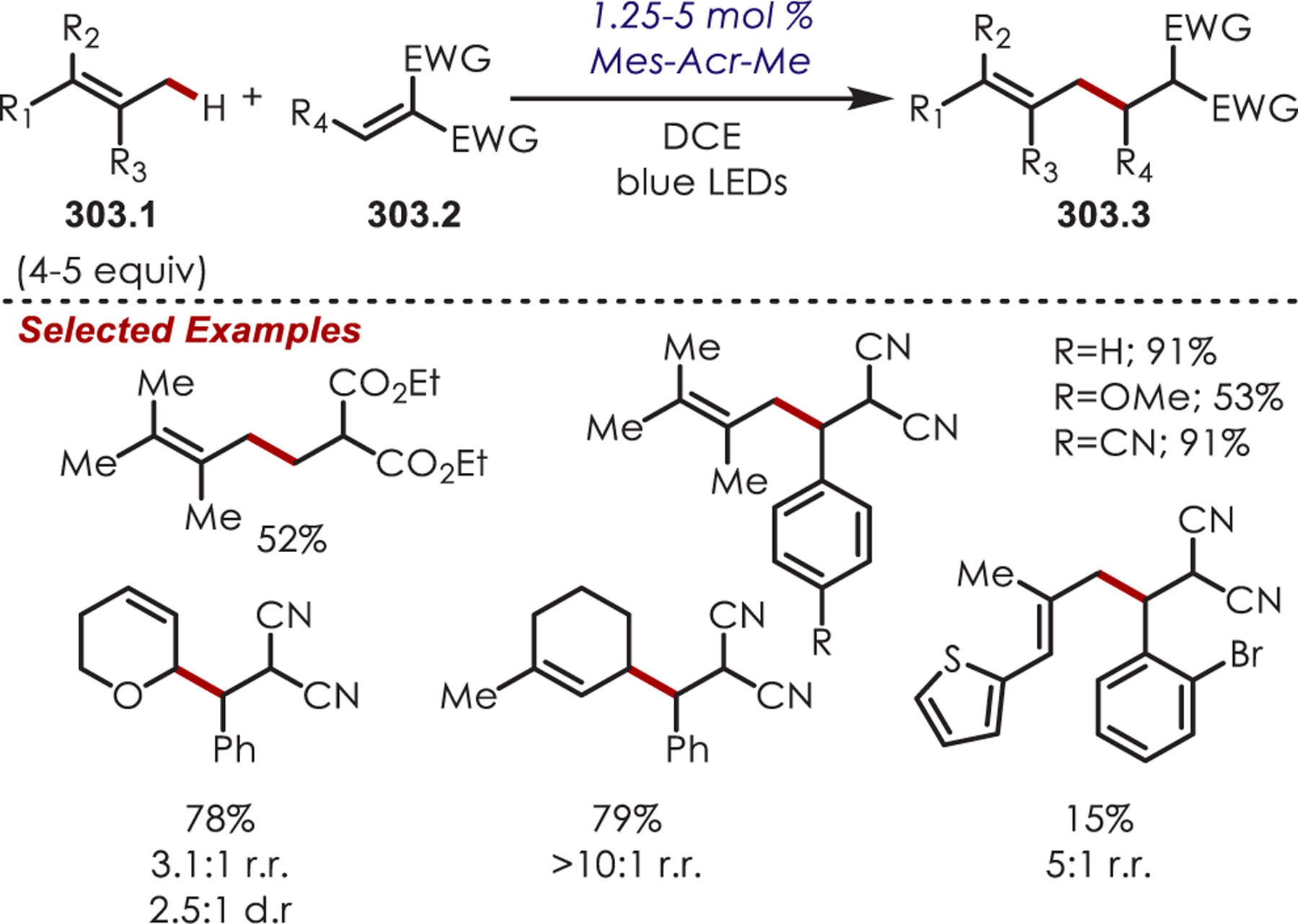
Allylic C–H Alkylation of Alkenes Using an Acridnium Salt as the Photoredox Catalyst
Rueping and co-workers also reported a direct coupling of allylic C–H bonds with aryl and vinyl bromides through a dual nickel and acridinium catalytic system (Scheme 304).513 The protocol was suitable for electron-deficient (hetro)aryl bromides and polycylic arenes (304.2). The olefin coupling covered a range of tri- and tetrasubstituted alkenes (304.1). Notably, this was selective for allylic C–H bonds for substrates that also possessed α-oxy C–H bonds. Additionally, excellent regioselectivity was observed for C–H functionalization of primary C–H bonds over methylene and methane, which is attributed to the HAT event.
Scheme 304.
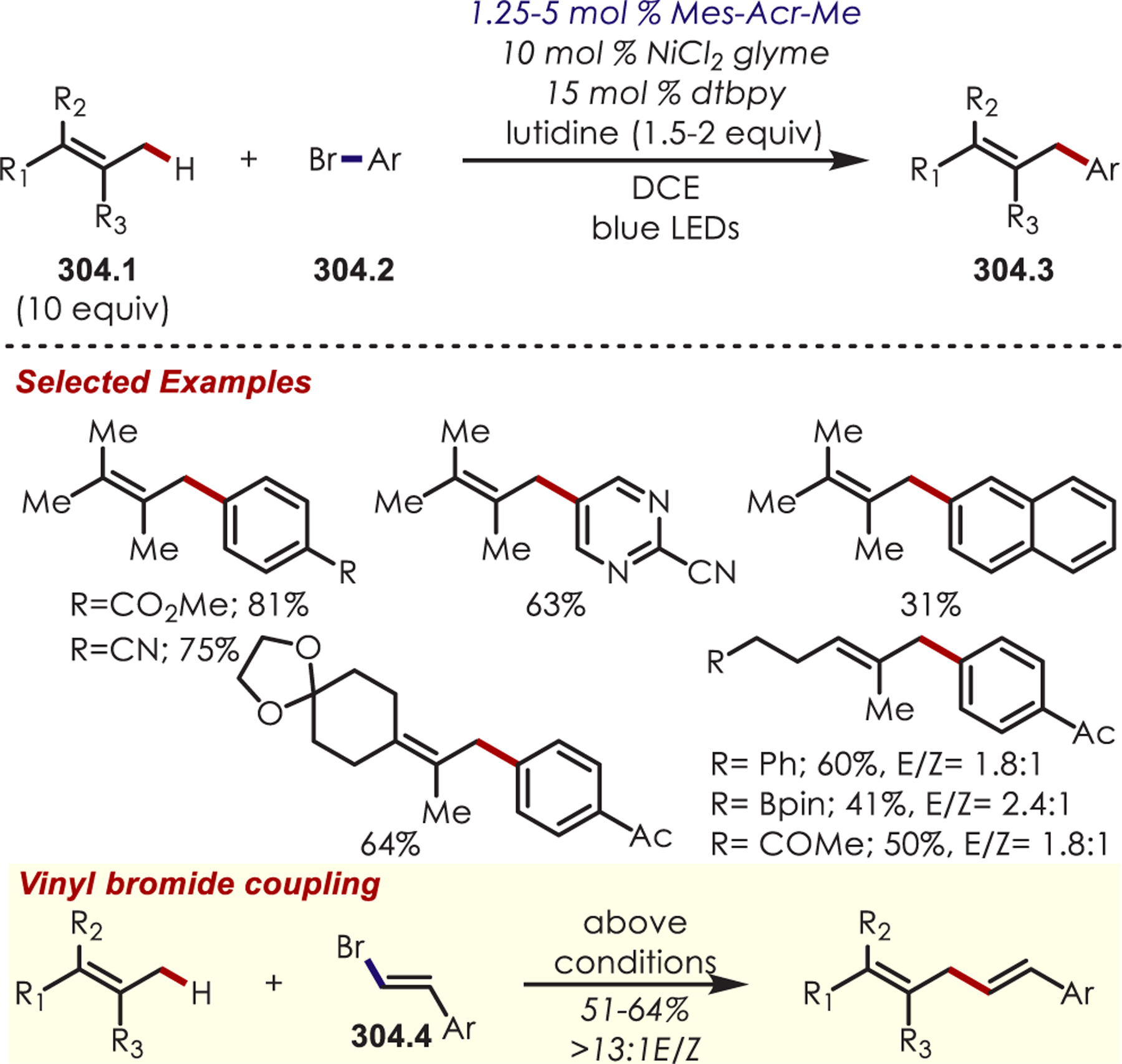
Allylic C–H Arylation and Vinylation through Dual Nickel and Acridinium Photoredox Catalysis
The mechanism was proposed to proceed via a triplet–triplet energy transfer from the excited photoredox catalyst to generate an excited Ni(II) aryl bromide species230 (305.1) which subsequently homolyzes and produces a bromime atom that serves as the hydrogen abstracting agent (Scheme 305). Direct reductive SET is unfavorable (Ered = −0.57 V vs SCE) from the acridine radical and oxidative SET to generate a cation radical is unlikely due the observed regioselectivities and relative oxidation potentials.
Scheme 305.
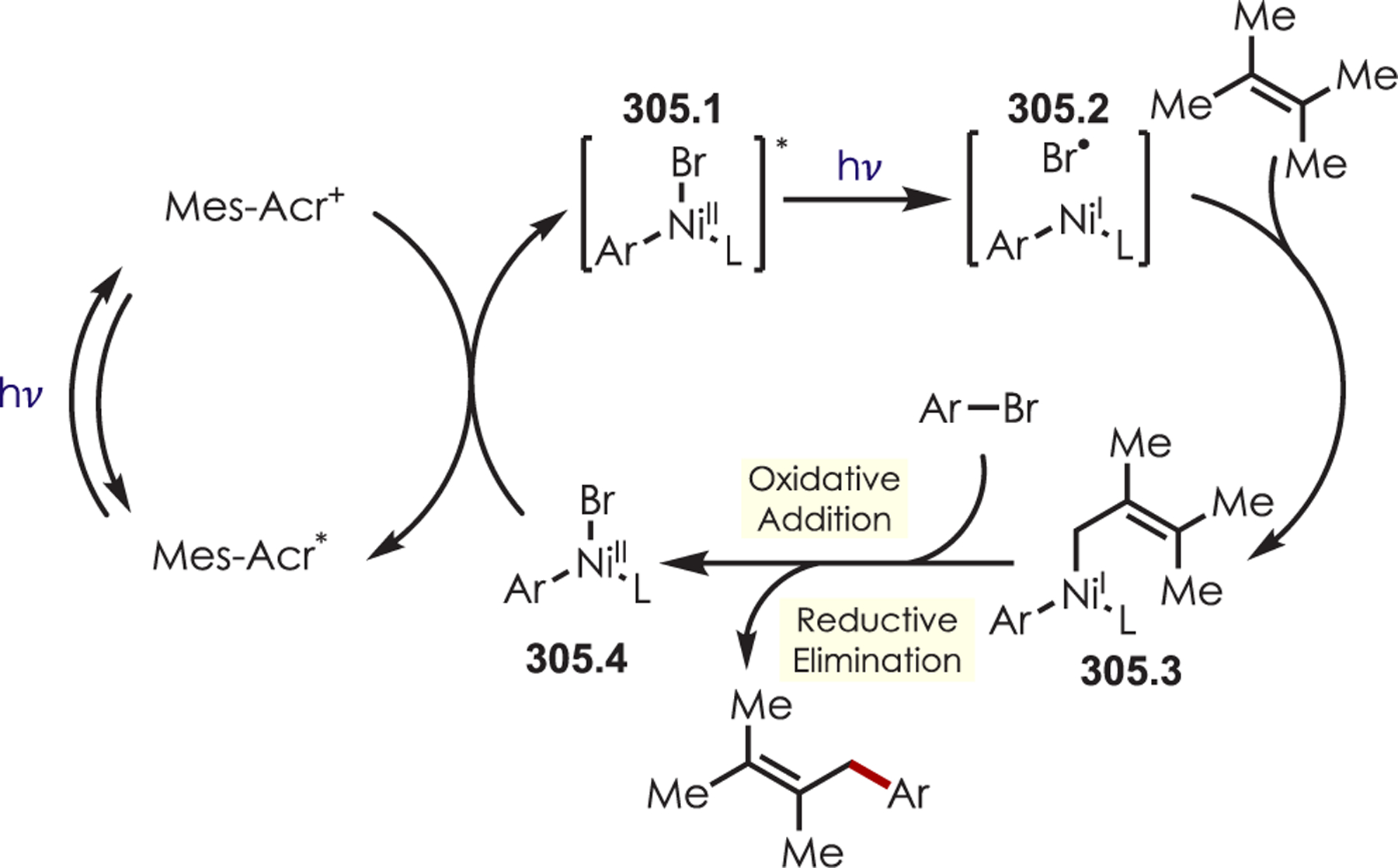
Mechanism of Allylic C–H Arylation and Vinylation through Dual Nickel and Acridinium Photoredox Catalysis
Recently, Scheidt reported a combined photoredox and enzymatic asymmetric benzylic hydroxylation (Scheme 306).514 This system relied on the photogeneration of a benzylic radical (306.3), which reacted with either molecular oxygen or oxygen anion radical to form a hydroperoxidate intermediate (306.4), and subsequent dehydration of this species afforded the corresponding acetophenone derivative (306.5). Reduction from ketoreductase (KRED) with nicotinamide adenine dinucleotide phosphate (NAD(P)H) as the hydride source afforded the hydroxylated products in high enantioselectivities. This method was successful at furnishing electron-rich and poor benzyl alcohols, diarylmethanols, 1,2-amino alcohols, α-hydroxy esters, and γ/δ-lactones with excellent enantioselectivities, and in general, the benzylic hydroxylation occurred at the most electron-rich site on the substrates examined.
Scheme 306.
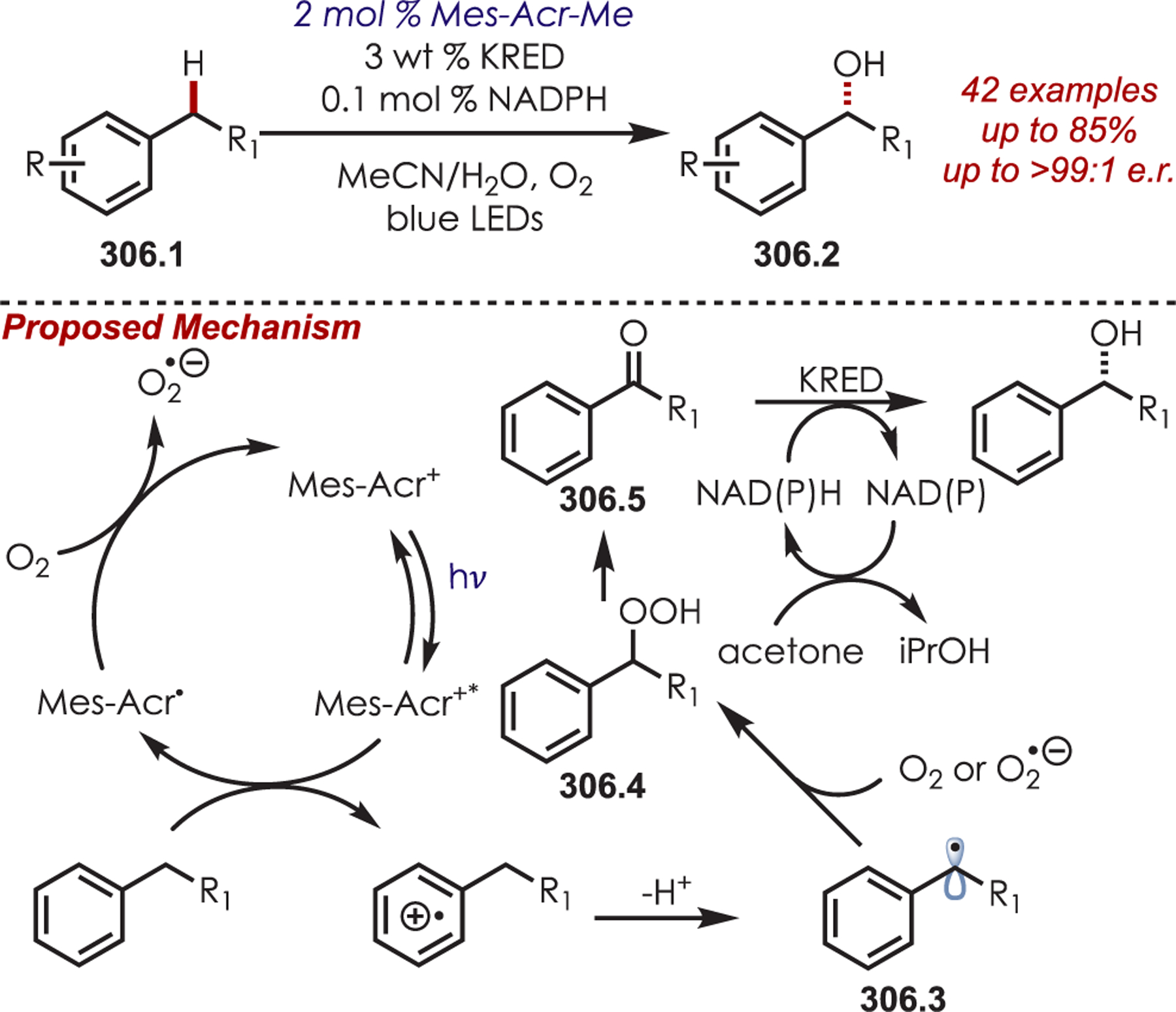
Dual Enzymatic and Photoredox Catalysis for an Asymmetric Benzylic C–H Hydroxylation
3.2.4. Functionalization of Unactivated sp3 C–H Bonds.
Via Direct Hydrogen Atom Abstraction.
The halogenation of unactivated C–H bonds remote from directing functionality has been accomplished through organic photoredox-mediated HAT pathways. Tan first reported a selective bromination515 (307.2) and fluorination516 (307.3) of alkanes using eosin Y and anthraquinone, respectively (Scheme 307). Generation of the alkyl radical, through HAT, and subsequent trapping with CBr4 or Selectfluor furnished the halogenated products. Bromination did not always occur at the weakest C–H bond as might be anticipated but rather was influenced by both electronics and sterics, favoring more accessible and electron-rich sp3 C–H bonds due to the electrophilic cationic N-radical being more electronically paired with these hydridic C–H bonds. Fluorination occurred at the most distal secondary C–H site from an electron-donating group. The selective α-fluorination of ketals has also been accomplished with photoexcited xanthone to generate the amine cation radical of Selectfluor.517
Scheme 307.
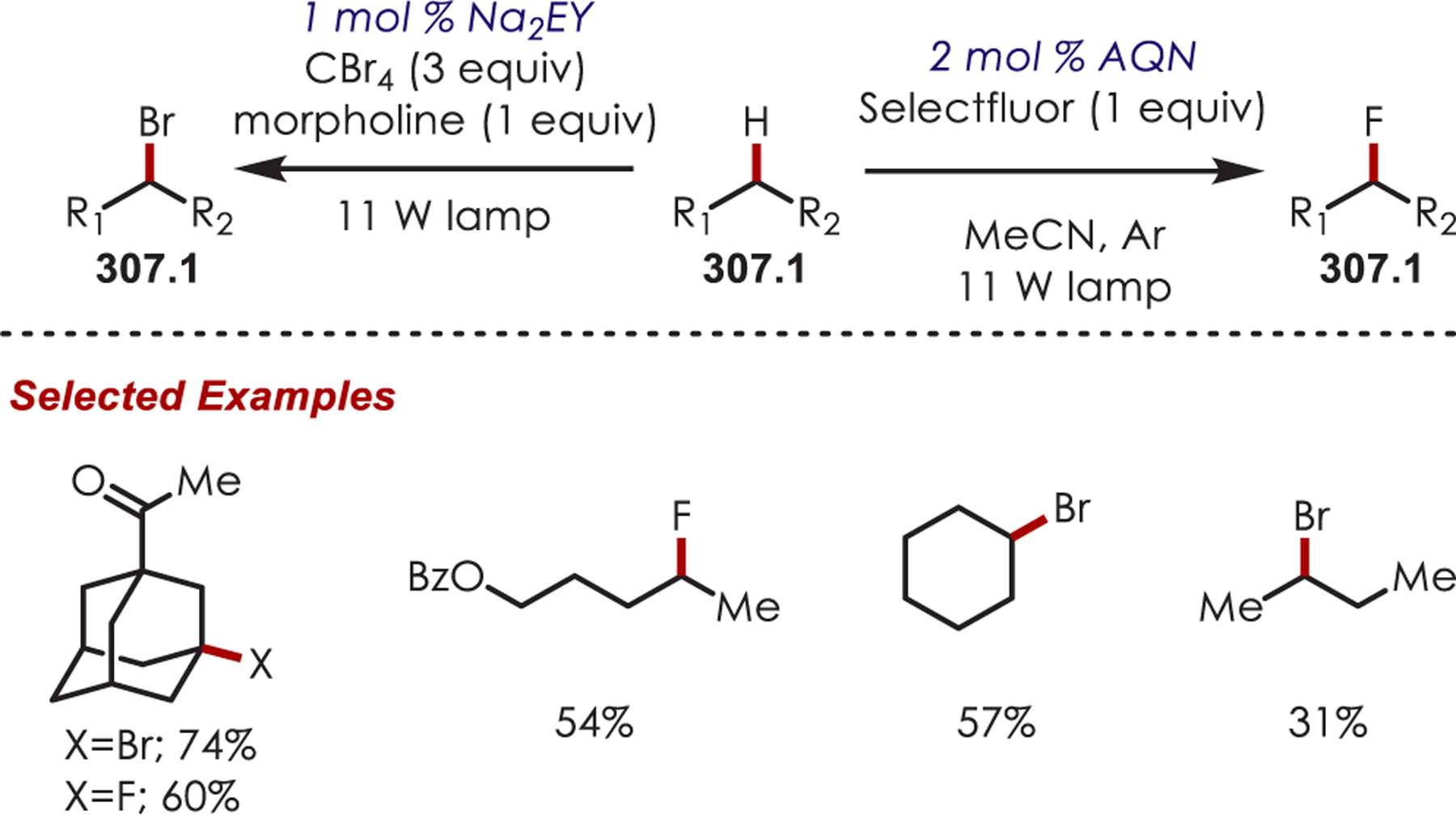
Remote C–H Halogenation with Organic Photoredox Catalysts through HAT Events
Alkyl radicals have been coupled with heteroarenes (308.1) using persulfate radical anions as the hydrogen atom abstractors (Scheme 308).518 Several heteroarenes, including substituted quinolones, quinoline, phthalazine, and pyridine derivatives, were successfully alkylated. However, this transformation required the cycloalkane in excess as the solvent.
Scheme 308.
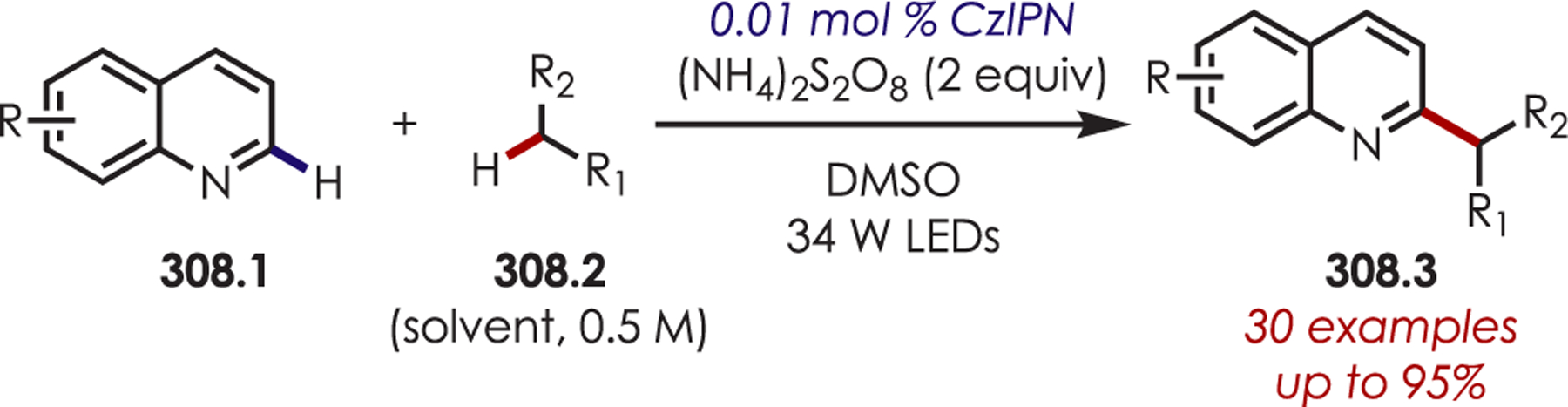
Cycloalkane C–H Functionalization with Persulfate Radical Anions as the Hydrogen Atom Abstracting Agent
Di-tert-butyl peroxide (DTBP) has been utilized as a hydrogen atom abstracting agent in a three-component dialkylation of alkenes with alkanes and 1,3 dicarbonyl compounds (Scheme 309).519 Eosin Y anion radical reduced DTBP to generate the tert-butyl alkoxy radical. Hydrogen atom abstraction of the alkane C–H bonds resulted in an alkyl radical that adds to the electron-deficient alkene. Single electron oxidation of the resultant alkyl radical by Fe(III) generated a carbocation, which underwent an addition of the 1,3-dicarbonyl enolate to furnish the desired products. The scope of the alkane partner was limited to n-hexane, 1,4-dioxane, and cycloalkanes such as cyclopentane, cycloheptane, and adamantane.
Scheme 309.
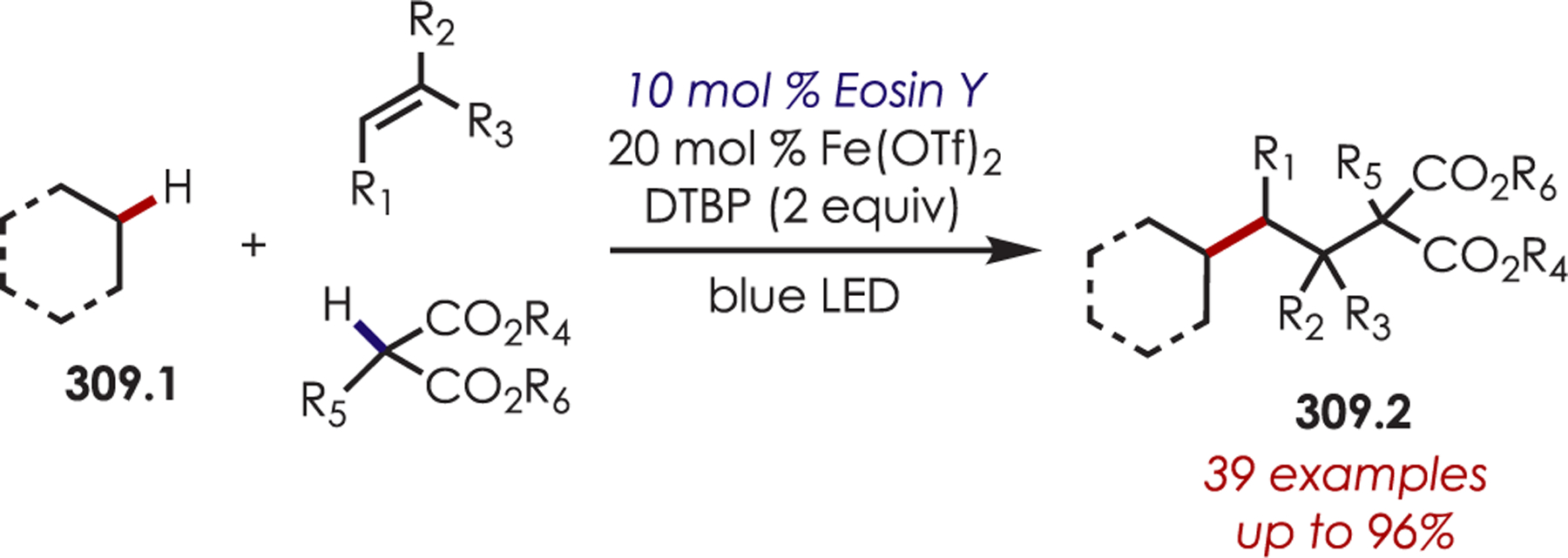
Dialkylation of Alkenes with Two Distinct C–H bonds by Dual Photoredox and Iron Catalysis
In a collaborative effort, Alexanian and Nicewicz have reported an acridinium-catalyzed C–H functionalization employing a phosphate salt as an HAT reagent for the formation of C–N, C–X, C–S, and C–C bonds (Scheme 310).520 PET between the photoexcited acridinium and the phosphate anion generates the oxygen-centered radical, which serves as the hydrogen atom abstracting agent. A variety of radical traps were successful at generating the azidated, halogenated, trifluoromethylthiolated, or alkylated products with moderate to high site selectivity. Additionally, the site-selective functionalization of more complex molecules, including menthol, memantine, differin, ibuprofen, and cholestane-3-one derivatives were reported.
Scheme 310.
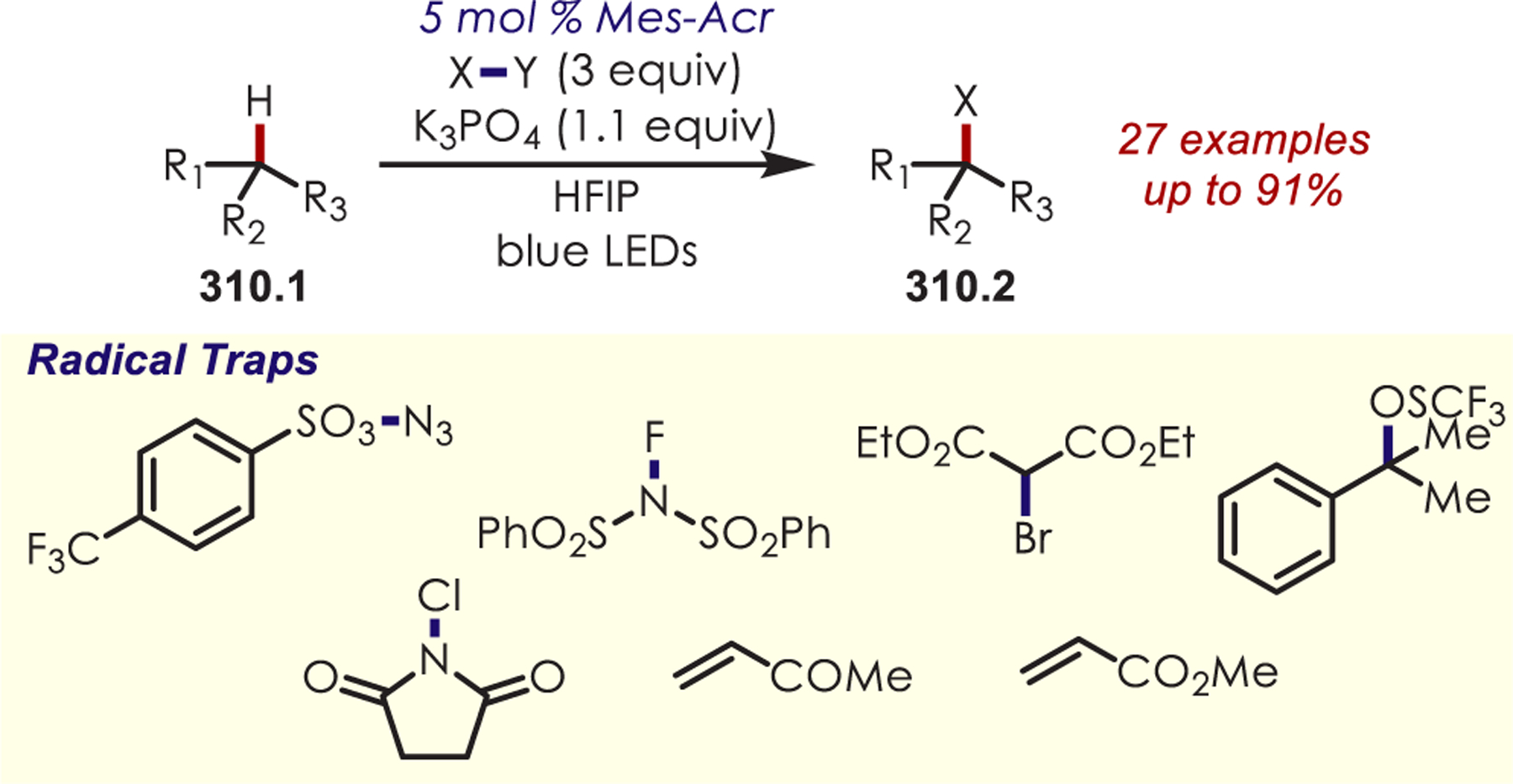
Remote C–H Functionalization with a Phosphate Salt as the Hydrogen Atom Abstracting Agent
The same year, Wu reported an alkylation of C–H bonds remote from functional groups via hydrogen atom abstraction from chlorine atoms generated by an acridinium photooxidant (Scheme 311).521 A microtubing reactor was required to handle the HCl catalyst, thus allowing for the generation of the key abstracting chlorine atom by oxidation from photoexcited acridinium. A range of C–H bonds on cyclic and acylic alkanes, along with aldehyde, α-heteroatom, and benzylic functionalization was demonstrated with methylenemalononitrile radical acceptors (311.3). Other aryl-substituted methylenemalononitrile acceptors including trifluoromethyl, carboxylic acid, halide, and phthalimide-substituted arenes were compatible in the reaction. Moreover, allyl sulfones underwent a subsequent elimination to afford the formal allylated products (311.2).
Scheme 311.
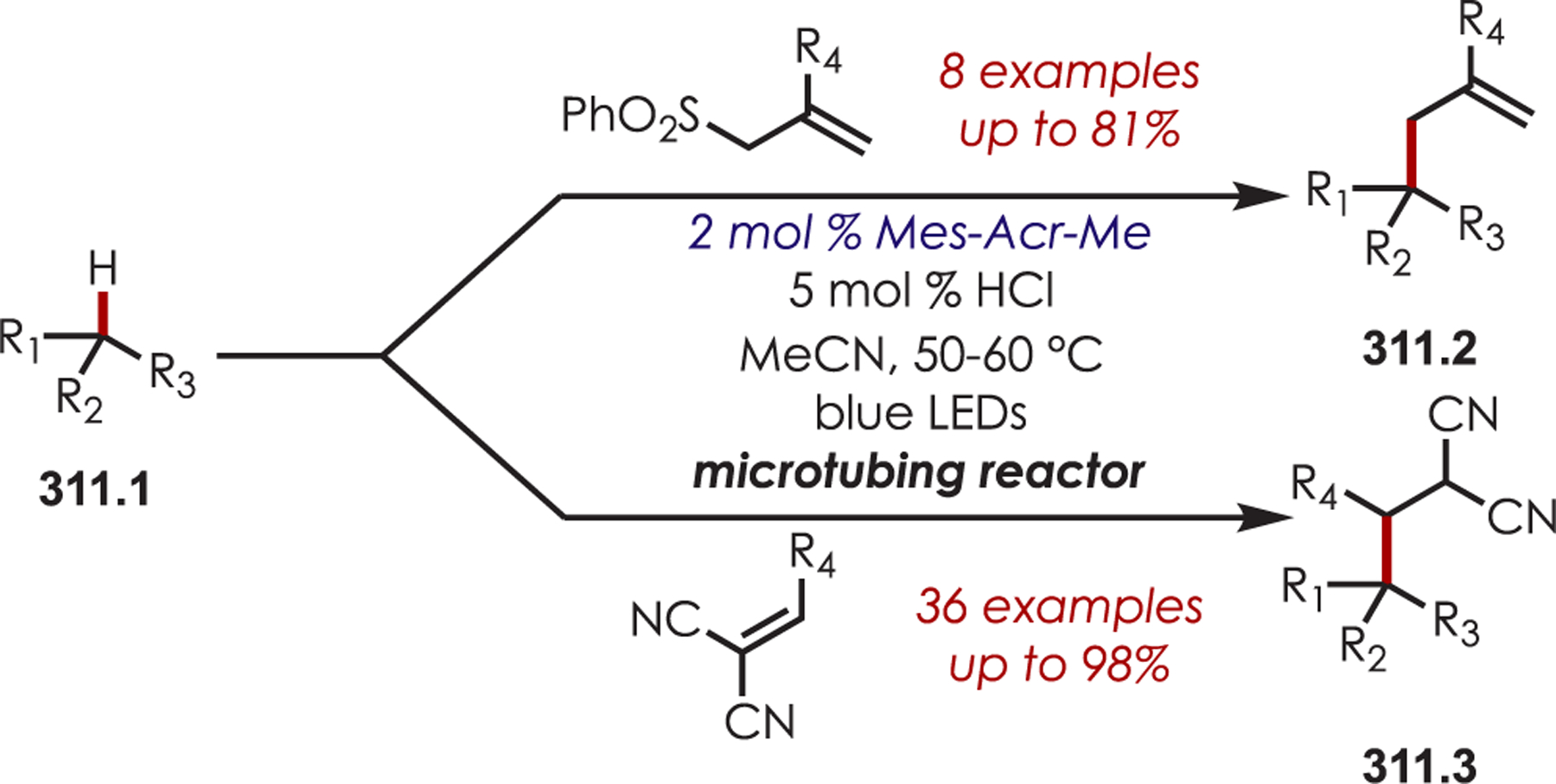
Chlorine Radicals as HAT Catalysts for C–H Alkylations
Via 1,5-Hydrogen Atom Transfers.
An eosin Y-catalyzed γ-vinylation of amides was reported by Yu in 2018 (Scheme 312).522 The scope of this chemistry was applicable to primary, secondary, and tertiary γ-C–H bonds with a range of electron-rich or -poor boronic acid acceptors (312.2). SET by photoexcited eosin Y generated an amidyl radical that underwent 1,5-HAT to form the carbon-centered radical. The newly formed radical was intercepted by the boronic acid, giving rise to an intermediate benzylic radical. A SET to eosin Y cation radical formed a benzylic cation, and subsequent deboronation270,370,523 furnished the desired product.
Scheme 312.
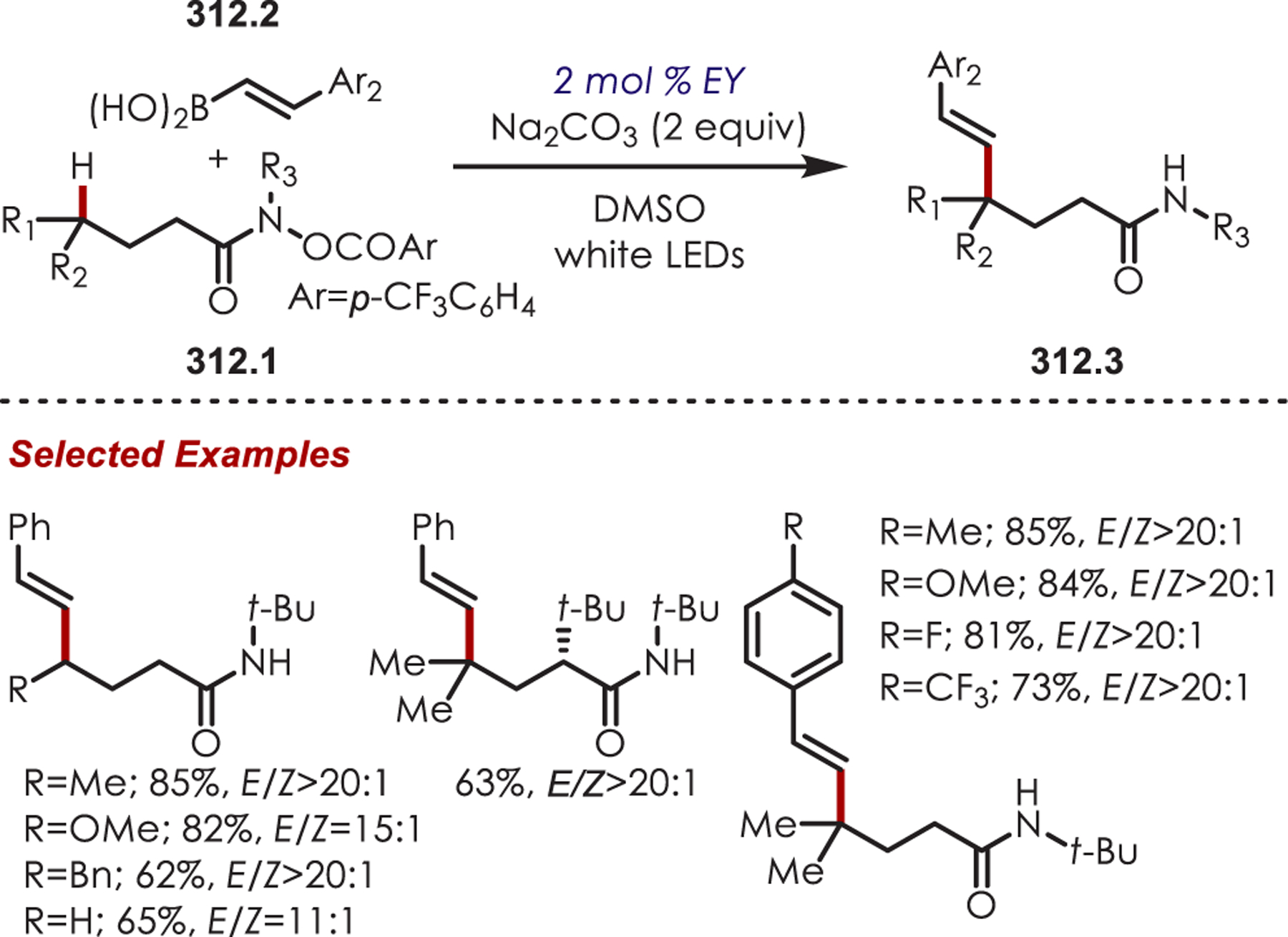
1,5-HAT for the Alkylation of Amide Derivatives with Boronic Acid Acceptors
The following year, Wang and co-workers reported an eosin Y-catalyzed allylation by using allyl sulfones (313.2) as the radical acceptors (Scheme 313).524 Aromatic and aliphatic amides were suitable substrates (313.1) with a range of electron-deficient allyl sulfone acceptors.
Scheme 313.
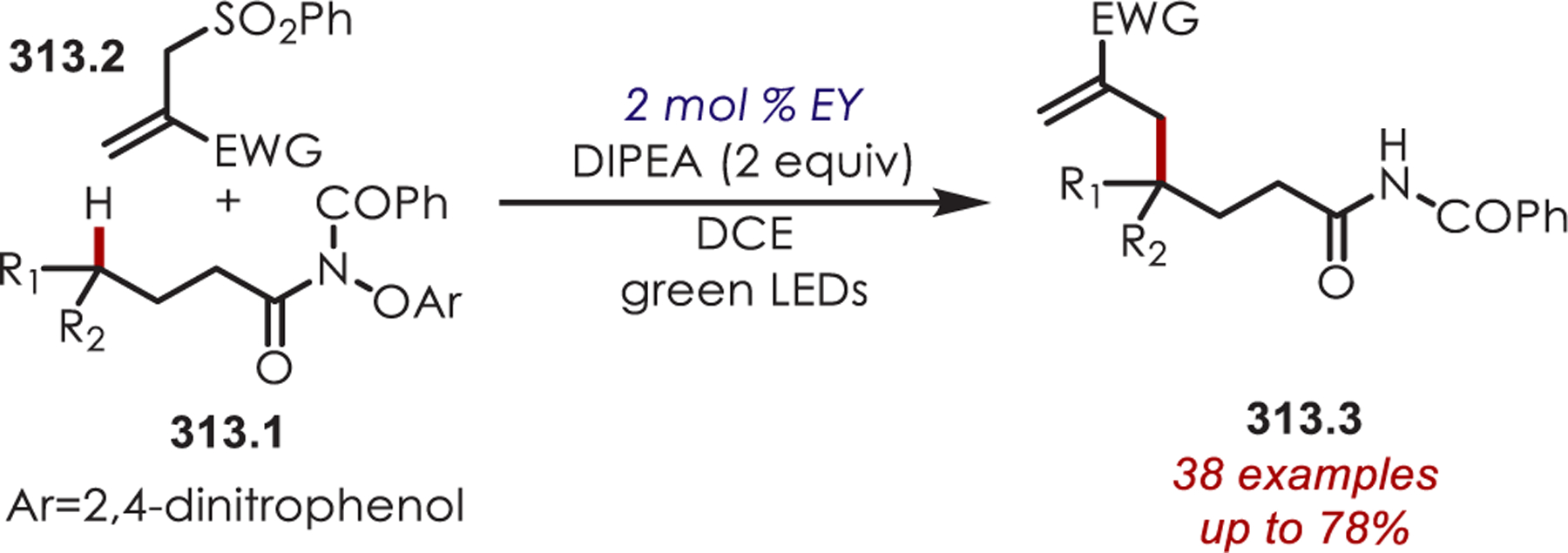
1,5-HAT for the Alkylation of Amide Derivatives with Allyl Sulfone Acceptors
γ-Heteroarylation of amides (314.1) facilitated via a radical translocation process triggered by NCR’s was reported the same year from Yu and co-workers (Scheme 314).525 The carbon-centered radical undergoes a Minisci-type addition with heteroarenes (314.2), including purines, thiazolopyridines, benzoxazole, benzothiazole, benzothiphene, benzofuran, thiazole, and quinoxalines to furnish the alkylated products with high selectivity.
Scheme 314.
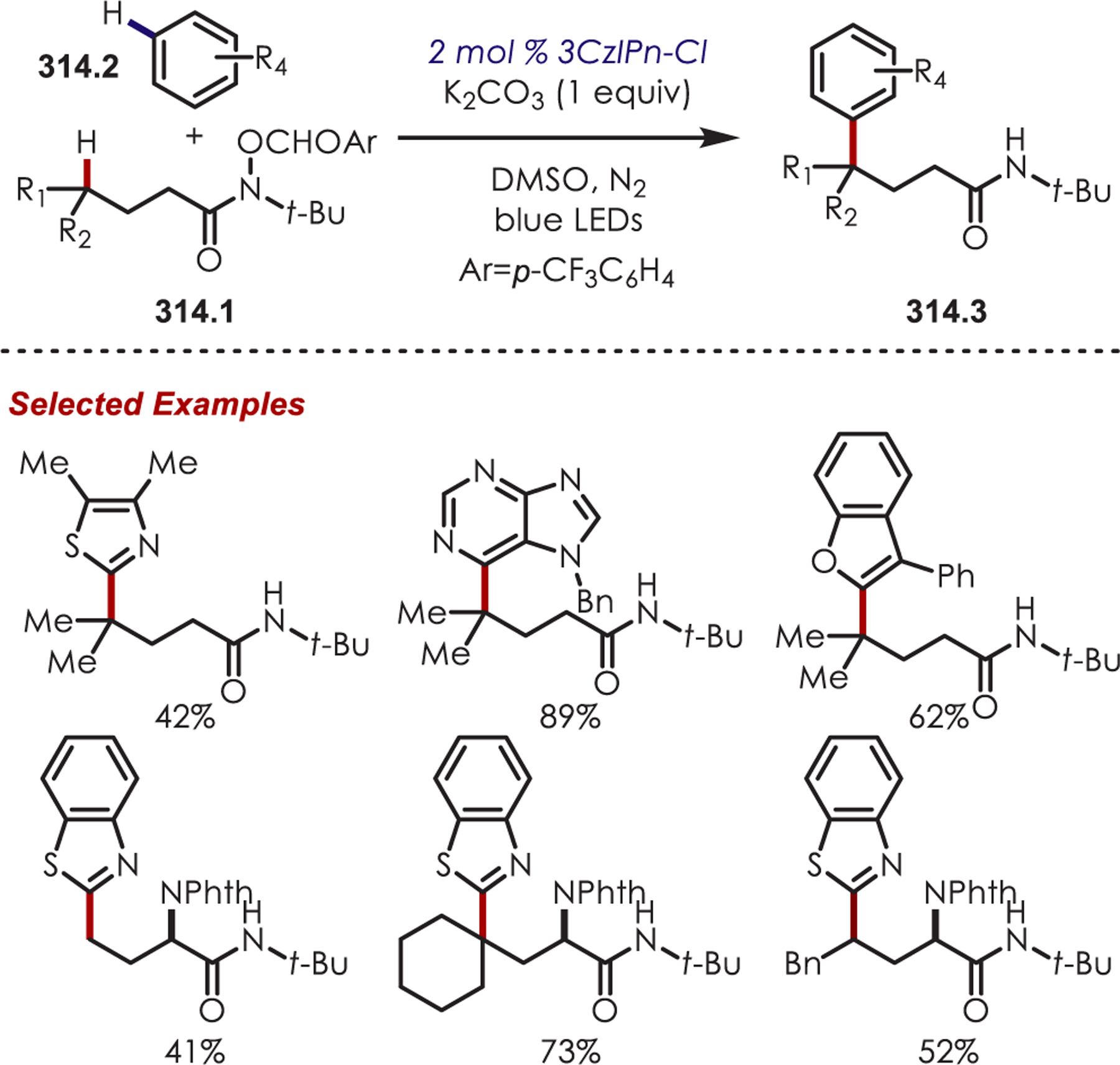
Aryl Radical Translocation by a 1,5-HAT Event for the Arylation of Amide Derivatives
Leonori has recently disclosed a C–H functionalization protocol remote from functionality for fluorination, chlorination, thioetherification, cyanation, and alkynlation of amides and amines (315.1) was accomplished using an acridinium salt to generate a nitrogen-centered radical intermediate (Scheme 315).526 Following a 1,5-HAT, the resulting carbon-centered radical undergoes an SH2 functionalization to install the γ-functional groups.
Scheme 315.
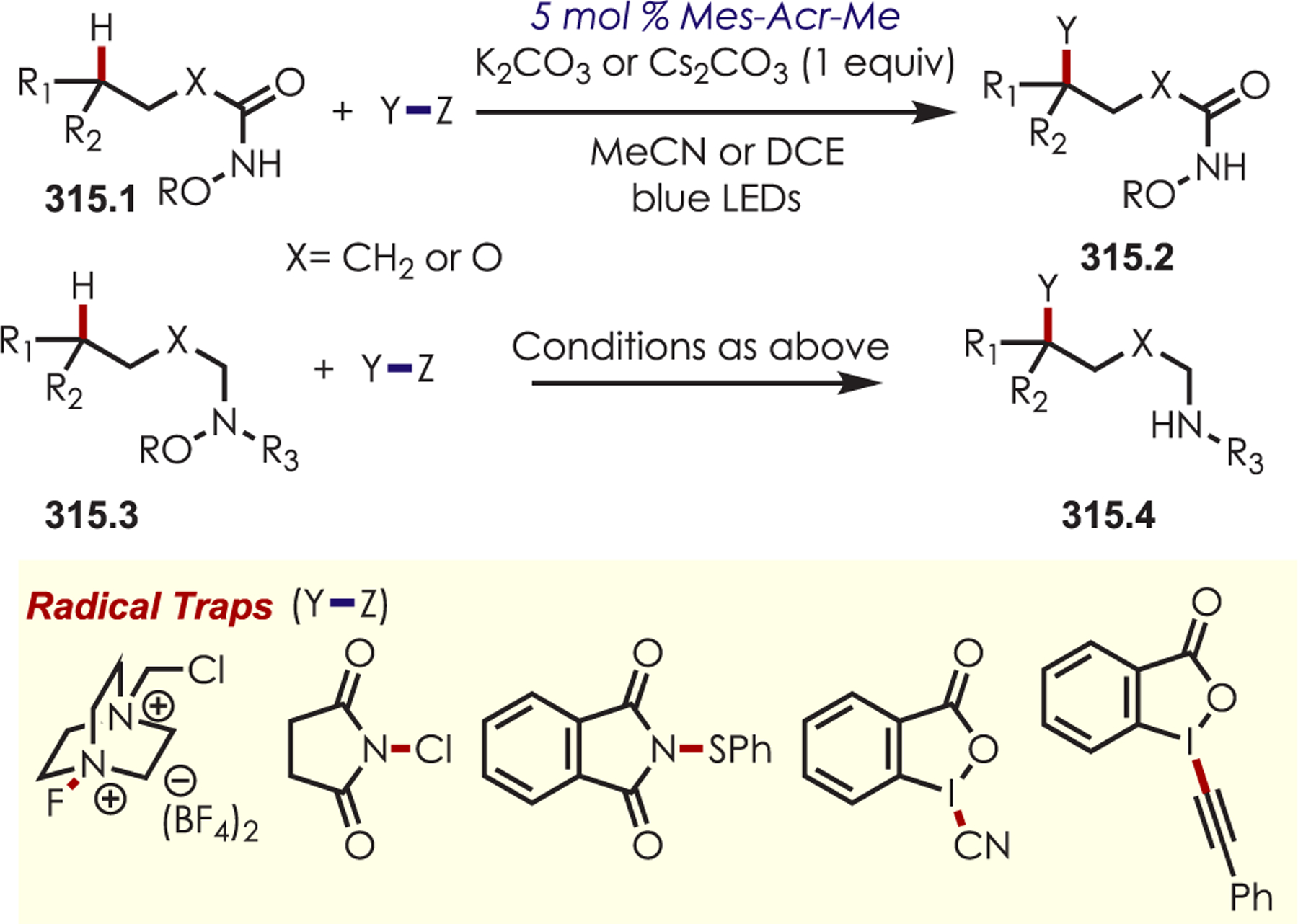
1,5-HAT for the Functionalization of Amides and Amines with Various Radical Traps
Oximes, which are known NCR precursors, (316.1 and 316.3), have been utilized under oxidative conditions in a remote γ-chlorination and fluorination to afford the functionalized N-chloro imine and ketone products (Scheme 316).527 This reaction was tolerant of electron-rich and -poor heteroaromatics, nitrile, azide, ester, hydroxy, thioether, and N-Boc functional groups.
Scheme 316.
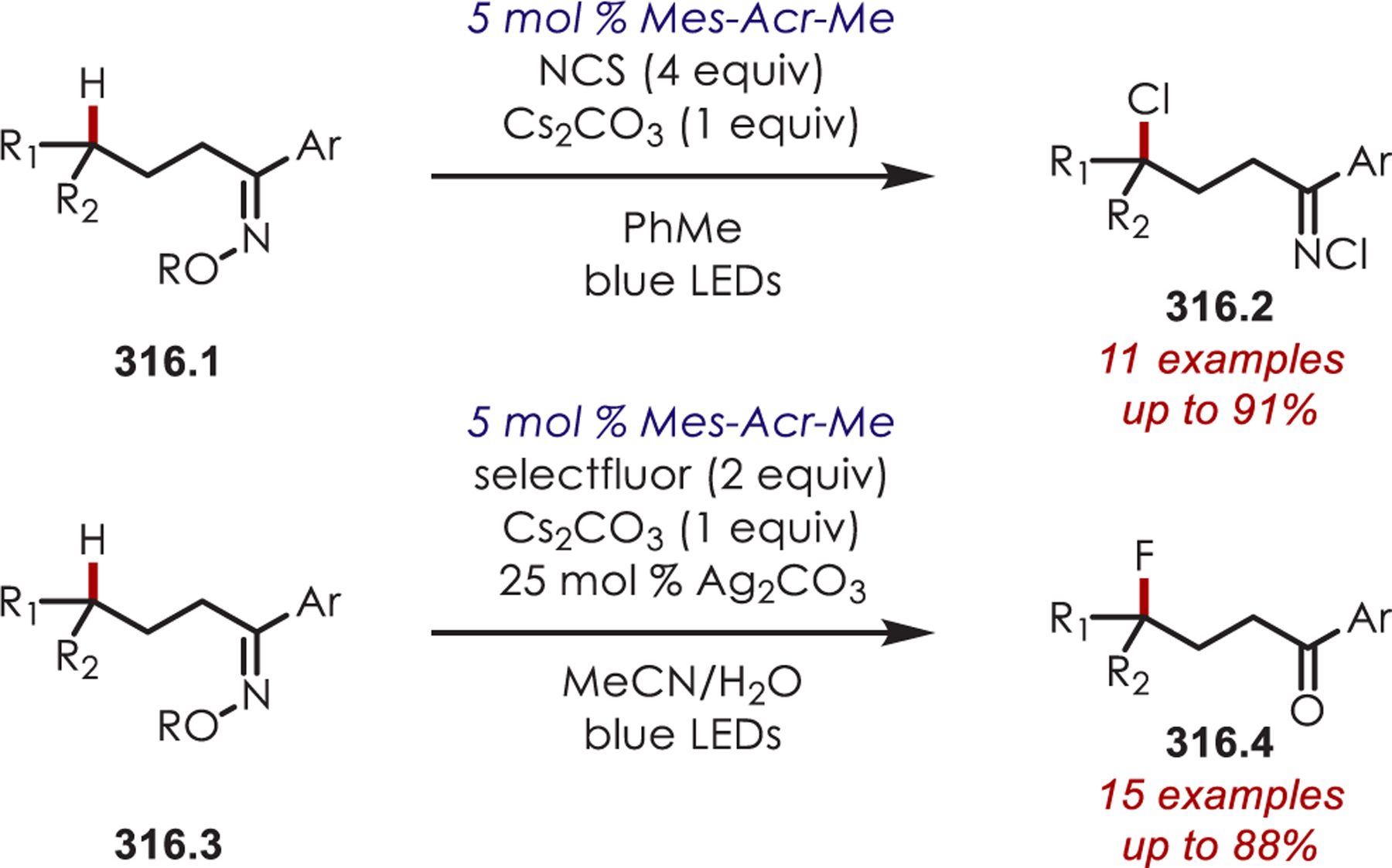
1,5-HAT for the Remote Halogenation of Imine and Ketone Derivatives
Reductive SET to N-alkoxypyridinium salts (317.1) by photoexcited quinolinones have been reported to initiate 1,5-HAT for the pyridinylation of remote sp3 C–H bonds (Scheme 317).528 Following reduction from the photoexcited catalyst, the resulting alkoxy radical undergoes a 1,5-HAT to generate a carbon-centered radical. This radical is intercepted by another equivalent of substrate in a pyridylation reaction to afford the desired product. Importantly, this reaction proceeds via a radical chain pathway, evidenced by a quantum yield >1 (Φ = 4.4), where the product was formed after a N–O bond cleavage event that generated a new alkoxy radical. Substitution on the heteroarene was well tolerated, including the addition of electron-donating or -withdrawing substituents. Both 2- and 4-pyridyl heteroarenes were also suitable. The scope of the aliphatic portion of the substrate was limited to aliphatic hydroxy derivatives for the formation of tertiary or quaternary carbons. Zhu later reported a similar heteroarylation of amides initiated by photolysis of an Ni(III) complex.529
Scheme 317.
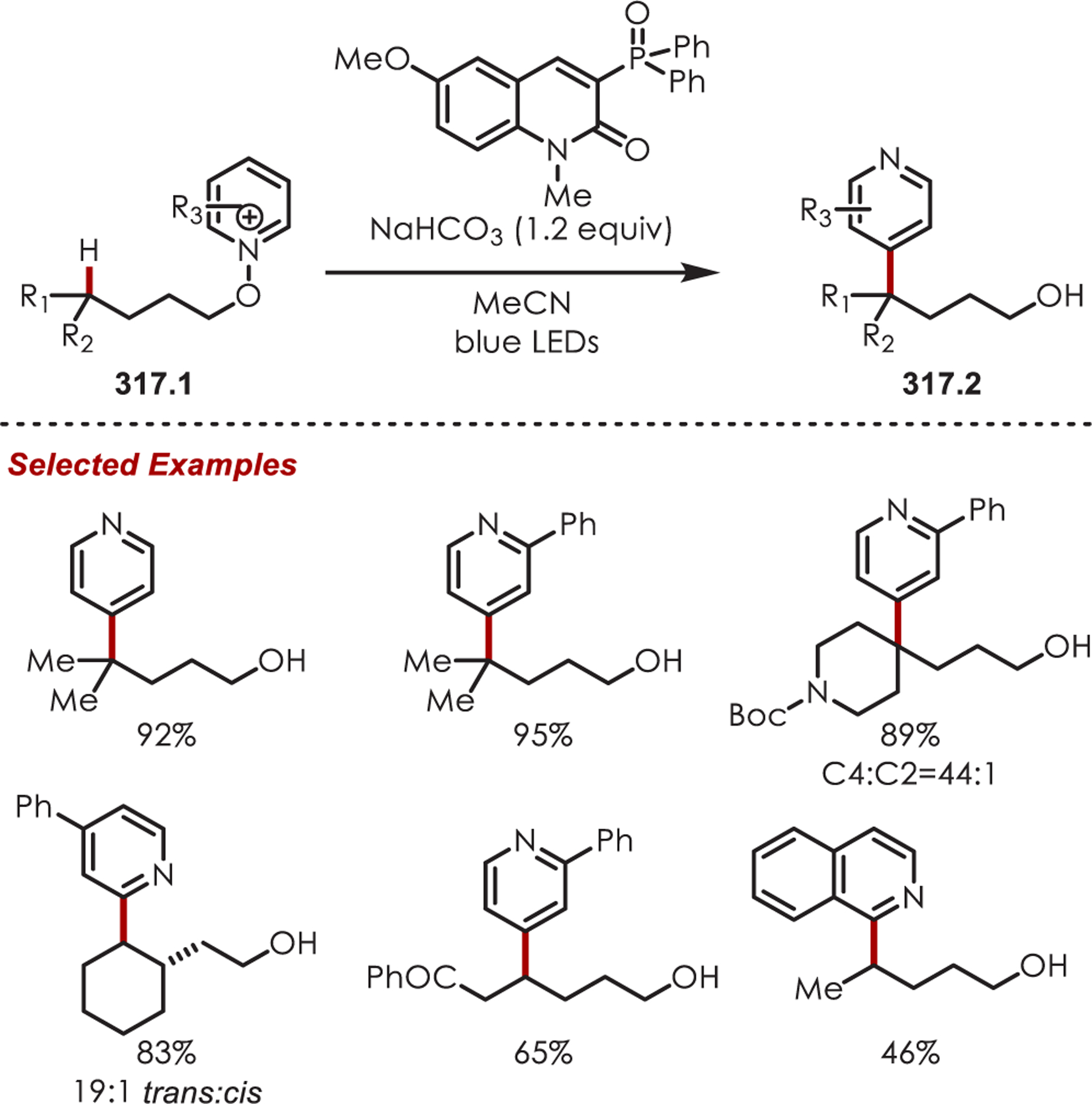
Remote Pyridiylation via a 1,5-HAT Radical Translocation of N-Alkoxypyridinium Salts
An intramolecular amination of sulfonamides via 1,5-HAT was reported with triphenylpyrylium (TPT) as the oxidant (Scheme 318).530 First, N-iodination of the starting material afforded an intermediate that homolyzed under irradiation to generate an amidyl radical. This amidyl radical facilitated a 1,5-HAT and intramolecular cyclization to afford the cyclized products. The organic photoredox catalyst is proposed to be involved in the regeneration of molecular I2 by oxidation of two HI species.
Scheme 318.
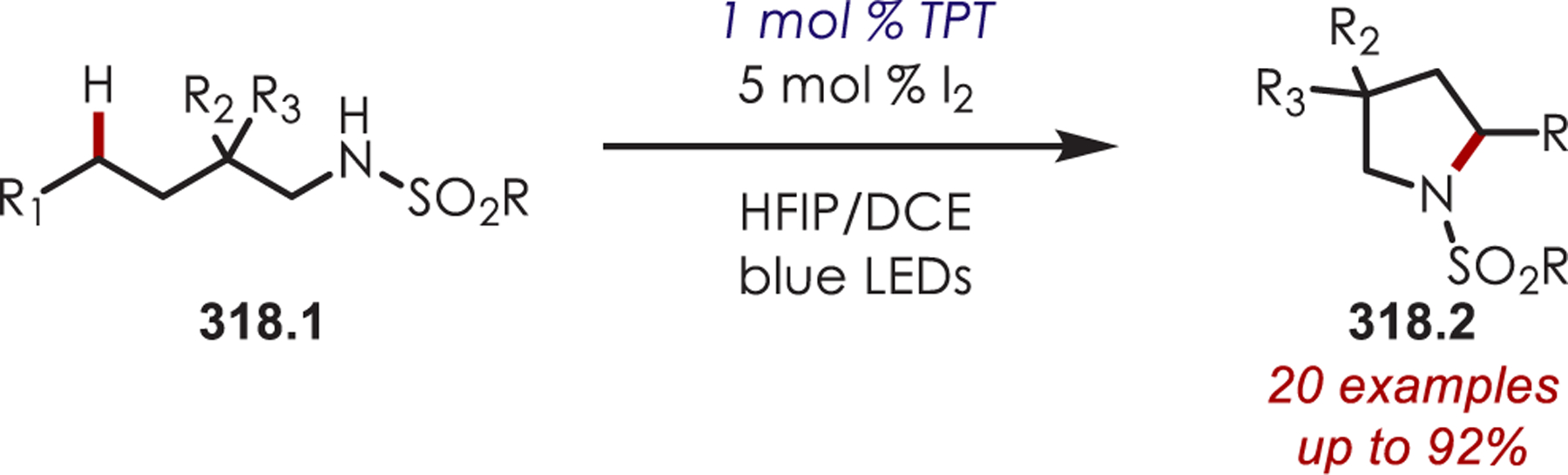
Intramolecular Amination through a 1,5-HAT event for the Synthesis of Pyrrolidines
4. CONCLUSIONS
The myriad of C–H functionalization reactions now possible using photoredox catalysis is truly impressive and will undoubtedly continue to grow. Collectively, these transformations represent new tools for synthetic practitioners in many different fields including medicinal chemistry, agrochemicals, natural product synthesis and materials. The strategies employed in photoredox manifolds for C–H functionalization such as hydrogen atom abstraction, single-electron oxidation/deprotonation, and proton-coupled electron transfer are truly well-suited for the class of catalysis. These reactive intermediates have been intercepted in a range of C–C and C–X bond-forming reactions either directly or via the intervention of cross-coupling co-catalysts with inorganic and organic photoredox catalysts (Scheme 319). In summary, arene C–H bond functionalization is more commonly accomplished through sp2 and sp3 carbon-centered radical intermediates with inorganic catalysts and through aryl cation radicals with organic catalysts. The generation of iminiums and α-hetero radicals for α-heteroatom C–H bond functionalization is established with both classes of catalysts. Carbonyl C–H bond functionalization is explored to a greater extent with inorganic photoredox catalysts, including β-carbonyl C–H bond functionalization, which has yet to be accomplished with organic catalysts. Benzylic, allylic and unactivated C–H bond functionalization is established with both classes of catalysts. Applications to natural product synthesis, radioisotope labeling, and late-stage functionalization reactions have only just begun.
Scheme 319.
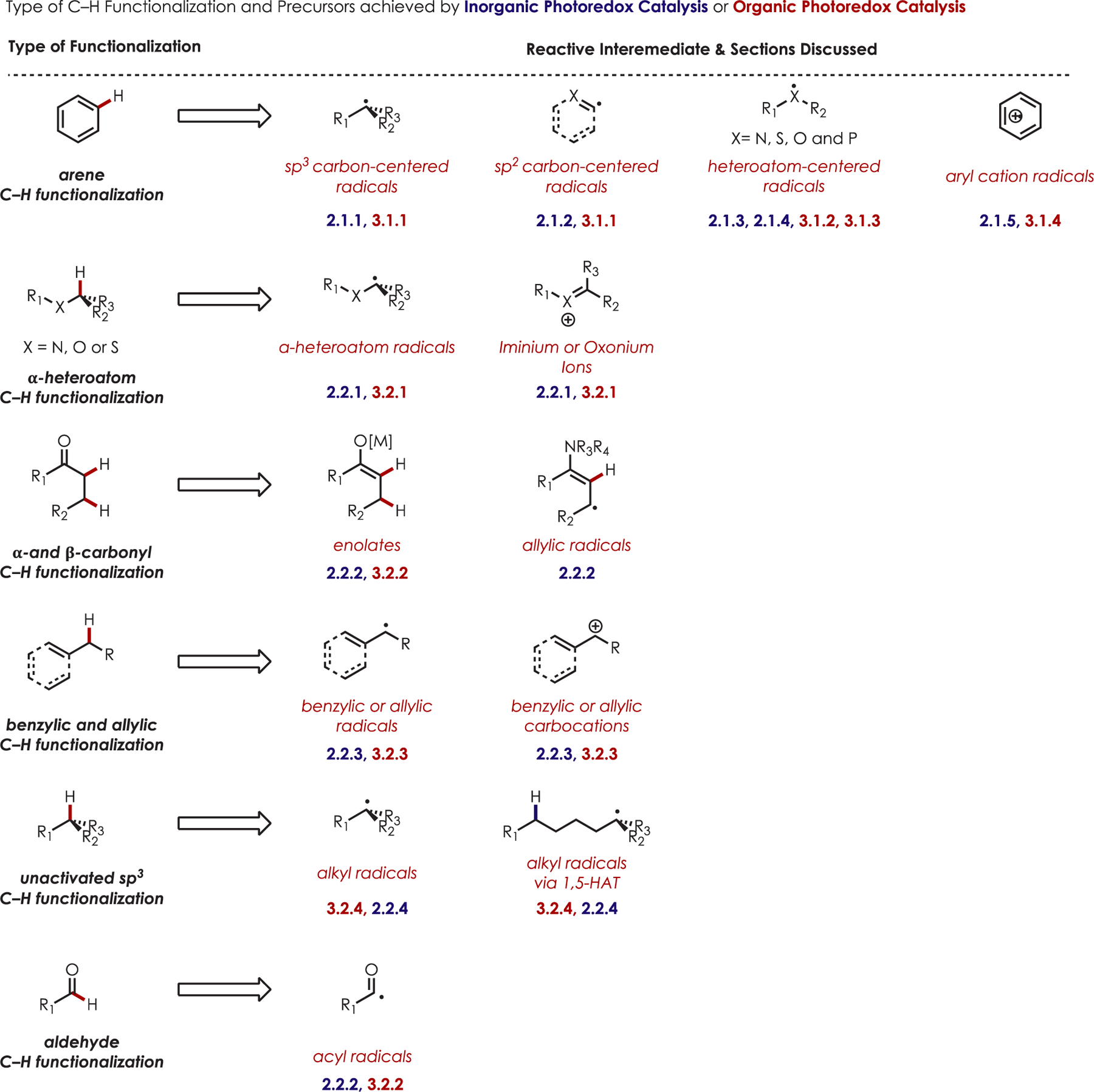
Summary of the Reactive Intermediates Accessed via Inorganic and Organic Photoredox Catalysis for C–H Bond Functionalizations
Going forward, we anticipate that new photoredox catalytic systems will be designed that are able to target specific stronger C–H bonds, such as 1° or 2°, in substrates containing far weaker C–H bonds. New catalyst design will continue to improve efficiency and could continue to push the absorption profile of the catalysts to the near-IR region.50,531 As ever more selective enzymatic systems develop, many opportunities to employ photoredox catalysis abound and offer fertile ground for ever more selective C–H functionalization reactions, particularly enantioselective in nature. Lastly, will we begin to see the first photoredox-catalyzed C–H functionalization on an industrial-scale process? Only time will tell, but the future does appear bright.
ACKNOWLEDGMENTS
We would like to thank several members of the Nicewicz lab for their help and support with editing this paper including Susanna Liang, Nicholas Onuska, Vincent Pistritto, Marcel Schlegel, and Nicholas Venditto. We also thank Alex Boley, Roukaya El Mokadem, Tanya Lazarus, and Katharine Toll for their help in literature searches.
Funding
NIGMS (1R35GM136330-01)
ABBREVIATIONS
- Acac
acetylacetone
- Acr
acridinium
- Ad
adamantyl
- Alk
alkyl
- Ar
aryl
- AQ
anthraquinone
- BArF
3,5-bis(trifluoromethyl)-phenyl)borate
- BDE
bond dissociation enthalpy
- Bn
benzyl
- Boc
tert-butyloxycarbonyl
- Box
bisoxazoline
- BPO
benzoyl peroxide
- BPY
2,2′-bipyridine
- BPZ
2,2′-bipyrazyl
- Bz
benzoyl
- Cbz
carboxybenzyl
- CFL
compact fluorescenct lamp
- Cod
cyclooctadiene
- CT
charge transfer
- Cy
cyclohexyl
- DABCO
1,4-diazabicyclo[2.2.2]octane
- DBA
dibenzylideneacetone
- DBU
1,8-diazabicyclo[5.4.0]undec-7-ene
- DCA
dichloroacetic acid
- DCE
1,2-dichloroethane
- DCM
dichloromethane
- DCN
dicyanobenzene
- DDQ
2,3-dichloro-5,6-dicyano-1,4-benzoquinone
- DFT
density functional theory
- DIPEA
N,N-diisopropylethylamine
- DMA
N,N-dimethylacetamide
- DME
dimethoxyethane
- DMF
dimethylformamide
- DMG
dimethylglyoxime
- DMPU
N,N′-dimethylpropyleneurea
- DMPPY
5-methyl-2- (4-methyl-2-pyridinyl)
- DMSO
dimethyl sulfoxide
- DPPM
bis(diphenylphosphino)methane
- DPPP
1,3-bis(diphenylphosphino)propane
- DPZ
5,6-bis(5-methoxythiophen-2-yl)pyrazine-2,3-dicarbonitrile
- DTBBPY
4,4′-di-tert-butyl-2,2′-dipyridyl
- EY
eosin Y
- Fc
ferrocene
- FMOC
fluorenylmethyloxycarbonyl
- GO
graphene oxide
- HAT
hydrogen atom transfer
- HFIP
hexafluoroisopropanol
- ICD
isocupreidine
- KRED
ketoreductase
- LED
light-emitting diode
- MeCN
acetonitrile
- Mes
mesityl
- Ms
methanesulfonyl
- MS
molecular sieve
- NaDPH
nicotinamide adenine dinucleotide phosphate
- NCS
N-chlorosuccinimide
- NBS
N-bromosuccinimide
- NHE
normal hydrogen electrode
- NMA
N-methylaniline
- NMP
N-methyl-2-pyrrolidone
- PCET
proton-coupled electron transfer
- Phen
phenanthroline
- Phth
phthalimide
- PMP
p-methoxyphenyl
- PPY
2-phenylpyridine
- Py
pyridine
- QuCN
3-cyano-1-methylquinolinium
- RB
rose bengal
- SCE
saturated calomel electrode
- SET
single-electron transfer
- SHE
standard hydrogen electrode
- SOMO
singly occupied molecular orbital
- TBADT
tetra-n-butylammonium decatungstate
- TBHP
tert-butyl hydroperoxide
- TBS
tert-butyldimethylsilyl
- TBN
tert-butyl nitrite
- TEA
triethylamine
- TEMPO
(2,2,6,6-tetramethyl-piperidin-1-yl)oxyl
- Tf
trifluoromethylsulfonyl
- TFA
trifluoroacetic acid
- TFE
trifluoroethanol
- TMEDA
tetramethyl ethylenediamine
- TMS
trimethylsilyl
- TPT
triphenyl pyrylium
- Ts
p-toluenesulfonyl
Biographies
Natalie Holmberg-Douglas received her B.S. in biochemistry and animal science from California State University of Chico. There she carried out undergraduate research under the supervision of Prof. Carolynn Arpin. In 2016, Natalie began graduate studies at the University of North Carolina at Chapel Hill where she joined the Nicewicz group. Her interests were on method development for arene and aza-heterocycle functionalization enabled by organic photoredox catalysis, and she obtained her Ph.D. in Chemistry in 2021.
David Nicewicz completed his Bachelor’s (2000) and Master’s (2001) degrees in Chemistry at the University of North Carolina at Charlotte with Professor Craig A. Ogle. He then moved to the University of North Carolina at Chapel Hill where he completed his Ph.D. in 2006 with Professor Jeffrey S. Johnson. Following his graduate education, Nicewicz was a Ruth L. Kirschstein Postdoctoral Fellow in the laboratories of Professor David W. C. MacMillan. It was during this time that Nicewicz pioneered the use of ruthenium photoredox catalysis in combination with chiral amine organocatalysis to develop a general method for enantioselective aldehyde alkylation. In July 2009, Dave began his independent career at the UNC Chapel Hill, where his laboratory has focused on organic photoredox catalysis for the development of novel chemical reactivity. He is currently the Royce Murray Term Professor of Chemistry at UNC Chapel Hill.
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.chemrev.1c00311
The authors declare no competing financial interest.
Contributor Information
Natalie Holmberg-Douglas, Department of Chemistry, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599-3290, United States.
David A. Nicewicz, Department of Chemistry, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599-3290, United States;.
REFERENCES
- (1).Arndtsen BA; Bergman RG; Mobley TA; Peterson TH Selective Intermolecular Carbon-Hydrogen Bond Activation by Synthetic Metal Complexes in Homogeneous Solution. Acc. Chem. Res 1995, 28, 154–162. [Google Scholar]
- (2).Alberico D; Scott ME; Lautens M Aryl-Aryl Bond Formation by Transition-Metal-Catalyzed Direct Arylation. Chem. Rev 2007, 107, 174–238. [DOI] [PubMed] [Google Scholar]
- (3).Davies HML; Manning JR Catalytic C-H Functionalization by Metal Carbenoid and Nitrenoid Insertion. Nature 2008, 451, 417–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Colby DA; Bergman RG; Ellman JA Rhodium-Catalyzed C-C Bond Formation via Heteroatom-Directed C-H Bond Activation. Chem. Rev 2010, 110, 624–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Mkhalid IAI; Barnard JH; Marder TB; Murphy JM; Hartwig JF C-H Activation for the Construction of C-B Bonds. Chem. Rev 2010, 110, 890–931. [DOI] [PubMed] [Google Scholar]
- (6).Doyle MP; Duffy R; Ratnikov M; Zhou L Catalytic Carbene Insertion into C-H Bonds. Chem. Rev 2010, 110, 704–724. [DOI] [PubMed] [Google Scholar]
- (7).Gutekunst WR; Baran PS C-H Functionalization Logic in Total Synthesis. Chem. Soc. Rev 2011, 40, 1976–1991. [DOI] [PubMed] [Google Scholar]
- (8).Engle KM; Mei T-S; Wasa M; Yu J-Q Weak Coordination as a Powerful Means for Developing Broadly Useful C-H Functionalization Reactions. Acc. Chem. Res 2012, 45, 788–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Yamaguchi J; Yamaguchi AD; Itami K C-H Bond Functionalization: Emerging Synthetic Tools for Natural Products and Pharmaceuticals. Angew. Chem., Int. Ed 2012, 51, 8960–9009. [DOI] [PubMed] [Google Scholar]
- (10).Zhang F; Spring DR Arene C-H Functionalisation Using a Removable/Modifiable or a Traceless Directing Group Strategy. Chem. Soc. Rev 2014, 43, 6906–6919. [DOI] [PubMed] [Google Scholar]
- (11).Hartwig JF; Larsen MA Undirected, Homogeneous C-H Bond Functionalization: Challenges and Opportunities. ACS Cent. Sci 2016, 2, 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Davies HML; Morton D Recent Advances in C-H Functionalization. J. Org. Chem 2016, 81, 343–350. [DOI] [PubMed] [Google Scholar]
- (13).Murakami K; Yamada S; Kaneda T; Itami K C-H Functionalization of Azines. Chem. Rev 2017, 117, 9302–9332. [DOI] [PubMed] [Google Scholar]
- (14).Davies HML; Morton D Collective Approach to Advancing C-H Functionalization. ACS Cent. Sci 2017, 3, 936–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Abrams DJ; Provencher PA; Sorensen EJ Recent Applications of C-H Functionalization in Complex Natural Product Synthesis. Chem. Soc. Rev 2018, 47, 8925–8967. [DOI] [PubMed] [Google Scholar]
- (16).Cernak T; Dykstra KD; Tyagarajan S; Vachal P; Krska SW The Medicinal Chemist’s Toolbox for Late Stage Functionalization of Drug-like Molecules. Chem. Soc. Rev 2016, 45, 546–576. [DOI] [PubMed] [Google Scholar]
- (17).Yang Y; Nishiura M; Wang H; Hou Z Metal-Catalyzed C H Activation for Polymer Synthesis and Functionalization. Coord. Chem. Rev 2018, 376, 506–532. [Google Scholar]
- (18).Williamson JB; Lewis SE; Johnson RR; Manning IM; Leibfarth FA C-H Functionalization of Commodity Polymers. Angew. Chem., Int. Ed 2019, 58, 8654–8668. [DOI] [PubMed] [Google Scholar]
- (19).Cannalire R; Pelliccia S; Sancineto L; Novellino E; Tron GC; Giustiniano M Visible Light Photocatalysis in the Late-Stage Functionalization of Pharmaceutically Relevant Compounds. Chem. Soc. Rev 2021, 50, 766–897. [DOI] [PubMed] [Google Scholar]
- (20).Zeitler K Photoredox Catalysis with Visible Light. Angew. Chem., Int. Ed 2009, 48, 9785–9789. [DOI] [PubMed] [Google Scholar]
- (21).Yoon TP; Ischay MA; Du J Visible Light Photocatalysis as a Greener Approach to Photochemical Synthesis. Nat. Chem 2010, 2, 527–532. [DOI] [PubMed] [Google Scholar]
- (22).Narayanam JMR; Stephenson CRJ Visible Light Photoredox Catalysis: Applications in Organic Synthesis. Chem. Soc. Rev 2011, 40, 102–113. [DOI] [PubMed] [Google Scholar]
- (23).Teplý F Photoredox Catalysis by [Ru(bpy)3]2+ to Trigger Transformations of Organic Molecules. Organic Synthesis Using Visible-Light Photocatalysis and Its 20th Century Roots. Collect. Czech. Chem. Commun 2011, 76, 859–917. [Google Scholar]
- (24).Ravelli D; Fagnoni M; Albini A Photoorganocatalysis. What For? Chem. Soc. Rev 2013, 42, 97–113. [DOI] [PubMed] [Google Scholar]
- (25).Xuan J; Xiao W-J Visible-Light Photoredox Catalysis. Angew. Chem., Int. Ed 2012, 51, 6828–6838. [DOI] [PubMed] [Google Scholar]
- (26).Tucker JW; Stephenson CRJ Shining Light on Photoredox Catalysis: Theory and Synthetic Applications. J. Org. Chem 2012, 77, 1617–1622. [DOI] [PubMed] [Google Scholar]
- (27).Shi L; Xia W Photoredox Functionalization of C-H Bonds Adjacent to a Nitrogen Atom. Chem. Soc. Rev 2012, 41, 7687–7697. [DOI] [PubMed] [Google Scholar]
- (28).Prier CK; Rankic DA; MacMillan DWC Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev 2013, 113, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Hopkinson MN; Sahoo B; Li J-L; Glorius F Dual Catalysis Sees the Light: Combining Photoredox with Organo-, Acid, and Transition-Metal Catalysis. Chem. - Eur. J 2014, 20, 3874–3886. [DOI] [PubMed] [Google Scholar]
- (30).Xie J; Jin H; Xu P; Zhu C When C-H Bond Functionalization Meets Visible-Light Photoredox Catalysis. Tetrahedron Lett. 2014, 55, 36–48. [Google Scholar]
- (31).Nicewicz DA; Nguyen TM Recent Applications of Organic Dyes as Photoredox Catalysts in Organic Synthesis. ACS Catal. 2014, 4, 355–360. [Google Scholar]
- (32).Koike T; Akita M Visible-Light Radical Reaction Designed by Ru- and Ir-Based Photoredox Catalysis. Inorg. Chem. Front 2014, 1, 562–576. [Google Scholar]
- (33).Zhang G; Bian C; Lei A Advances in Visible Light-Mediated Oxidative Coupling Reactions. Chin. J. Catal 2015, 36, 1428–1439. [Google Scholar]
- (34).Angnes RA; Li Z; Correia CRD; Hammond GB Recent Synthetic Additions to the Visible Light Photoredox Catalysis Toolbox. Org. Biomol. Chem 2015, 13, 9152–9167. [DOI] [PubMed] [Google Scholar]
- (35).Romero NA; Nicewicz DA Organic Photoredox Catalysis. Chem. Rev 2016, 116, 10075–10166. [DOI] [PubMed] [Google Scholar]
- (36).Arias-Rotondo DM; McCusker JK The Photophysics of Photoredox Catalysis: A Roadmap for Catalyst Design. Chem. Soc. Rev 2016, 45, 5803–5820. [DOI] [PubMed] [Google Scholar]
- (37).Tóth BL; Tischler O; Novák Z Recent Advances in Dual Transition Metal-Visible Light Photoredox Catalysis. Tetrahedron Lett. 2016, 57, 4505–4513. [Google Scholar]
- (38).Skubi KL; Blum TR; Yoon TP Dual Catalysis Strategies in Photochemical Synthesis. Chem. Rev 2016, 116, 10035–10074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Shaw MH; Twilton J; MacMillan DWC Photoredox Catalysis in Organic Chemistry. J. Org. Chem 2016, 81, 6898–6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Levin MD; Kim S; Toste FD Photoredox Catalysis Unlocks Single-Electron Elementary Steps in Transition Metal Catalyzed Cross-Coupling. ACS Cent. Sci 2016, 2, 293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Srivastava V; Singh PP Eosin Y Catalysed Photoredox Synthesis: A Review. RSC Adv. 2017, 7, 31377–31392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Qin Q; Jiang H; Hu Z; Ren D; Yu S Functionalization of C-H Bonds by Photoredox Catalysis. Chem. Rec 2017, 17, 754–774. [DOI] [PubMed] [Google Scholar]
- (43).Kärkäs MD Photochemical Generation of Nitrogen-Centered Amidyl, Hydrazonyl, and Imidyl Radicals: Methodology Developments and Catalytic Applications. ACS Catal. 2017, 7, 4999–5022. [Google Scholar]
- (44).Wang C-S; Dixneuf PH; Soulé J-F Photoredox Catalysis for Building C-C Bonds from C(Sp2)-H Bonds. Chem. Rev 2018, 118, 7532–7585. [DOI] [PubMed] [Google Scholar]
- (45).Sideri IK; Voutyritsa E; Kokotos CG Photoorganocatalysis, Small Organic Molecules and Light in the Service of Organic Synthesis: The Awakening of a Sleeping Giant. Org. Biomol. Chem 2018, 16, 4596–4614. [DOI] [PubMed] [Google Scholar]
- (46).Banerjee A; Lei Z; Ngai M-Y Acyl Radical Chemistry via Visible-Light Photoredox Catalysis. Synthesis 2019, 51, 303–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Uygur M; Garcia Mancheño O Visible Light-Mediated Organophotocatalyzed C-H Bond Functionalization Reactions. Org. Biomol. Chem 2019, 17, 5475–5489. [DOI] [PubMed] [Google Scholar]
- (48).Siddiqui R; Ali R Recent Developments in Photoredox-Catalyzed Remote Ortho and Para C-H Bond Functionalizations. Beilstein J. Org. Chem 2020, 16, 248–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Amos SGE; Garreau M; Buzzetti L; Waser J Photocatalysis with Organic Dyes: Facile Access to Reactive Intermediates for Synthesis. Beilstein J. Org. Chem 2020, 16, 1163–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Ravetz BD; Tay NES; Joe CL; Sezen-Edmonds M; Schmidt MA; Tan Y; Janey JM; Eastgate MD; Rovis T Development of a Platform for Near-Infrared Photoredox Catalysis. ACS Cent. Sci 2020, 6, 2053–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).MacKenzie IA; Wang L; Onuska NPR; Williams OF; Begam K; Moran AM; Dunietz BD; Nicewicz DA Discovery and Characterization of an Acridine Radical Photoreductant. Nature 2020, 580, 76–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Yu Y; Guo P; Zhong J-S; Yuan Y; Ye K-Y Merging Photochemistry with Electrochemistry in Organic Synthesis. Org. Chem. Front 2020, 7, 131–135. [Google Scholar]
- (53).Weinberg DR; Gagliardi CJ; Hull JF; Murphy CF; Kent CA; Westlake BC; Paul A; Ess DH; McCafferty DG; Meyer TJ Proton-Coupled Electron Transfer. Chem. Rev 2012, 112, 4016–4093. [DOI] [PubMed] [Google Scholar]
- (54).Knowles R Light-Driven PCET in Organic Synthesis. Chem. Rev 2021. [Google Scholar]
- (55).Roth H; Romero N; Nicewicz D Experimental and Calculated Electrochemical Potentials of Common Organic Molecules for Applications to Single-Electron Redox Chemistry. Synlett 2016, 27, 714–723. [Google Scholar]
- (56).McTiernan CD; Morin M; McCallum T; Scaiano JC; Barriault L Polynuclear Gold(I) Complexes in Photoredox Catalysis: Understanding Their Reactivity through Characterization and Kinetic Analysis. Catal. Sci. Technol 2016, 6, 201–207. [Google Scholar]
- (57).Seiple IB; Su S; Rodriguez RA; Gianatassio R; Fujiwara Y; Sobel AL; Baran PS Direct C-H Arylation of Electron-Deficient Heterocycles with Arylboronic Acids. J. Am. Chem. Soc 2010, 132, 13194–13196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Fujiwara Y; Domingo V; Seiple IB; Gianatassio R; Del Bel M; Baran PS Practical C-H Functionalization of Quinones with Boronic Acids. J. Am. Chem. Soc 2011, 133, 3292–3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Vallée F; Mousseau JJ; Charette AB Iron-Catalyzed Direct Arylation through an Aryl Radical Transfer Pathway. J. Am. Chem. Soc 2010, 132, 1514–1516. [DOI] [PubMed] [Google Scholar]
- (60).Tucker JW; Narayanam JMR; Krabbe SW; Stephenson CRJ Electron Transfer Photoredox Catalysis: Intramolecular Radical Addition to Indoles and Pyrroles. Org. Lett 2010, 12, 368–371. [DOI] [PubMed] [Google Scholar]
- (61).Furst L; Matsuura BS; Narayanam JMR; Tucker JW; Stephenson CRJ Visible Light-Mediated Intermolecular C-H Functionalization of Electron-Rich Heterocycles with Malonates. Org. Lett 2010, 12, 3104–3107. [DOI] [PubMed] [Google Scholar]
- (62).Swift EC; Williams TM; Stephenson CRJ Intermolecular Photocatalytic C-H Functionalization of Electron-Rich Heterocycles with Tertiary Alkyl Halides. Synlett 2016, 27, 754–758. [Google Scholar]
- (63).Cheng J; Deng X; Wang G; Li Y; Cheng X; Li G Intermolecular C-H Quaternary Alkylation of Aniline Derivatives Induced by Visible-Light Photoredox Catalysis. Org. Lett 2016, 18, 4538–4541. [DOI] [PubMed] [Google Scholar]
- (64).Fry AJ; Krieger RL Electrolyte Effects upon the Polarographic Reduction of Alkyl Halides in Dimethyl Sulfoxide. J. Org. Chem 1976, 41, 54–57. [Google Scholar]
- (65).McCallum T; Barriault L Direct Alkylation of Heteroarenes with Unactivated Bromoalkanes Using Photoredox Gold Catalysis. Chem. Sci 2016, 7, 4754–4758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Iqbal N; Choi S; Ko E; Cho EJ Trifluoromethylation of Heterocycles via Visible Light Photoredox Catalysis. Tetrahedron Lett. 2012, 53, 2005–2008. [Google Scholar]
- (67).Wang L; Wei X-J; Jia W-L; Zhong J-J; Wu L-Z; Liu Q Visible-Light-Driven Difluoroacetamidation of Unactive Arenes and Heteroarenes by Direct C-H Functionalization at Room Temperature. Org. Lett 2014, 16, 5842–5845. [DOI] [PubMed] [Google Scholar]
- (68).Zhu M; Han X; Fu W; Wang Z; Ji B; Hao X-Q; Song M-P; Xu C Regioselective 2,2,2-Trifluoroethylation of Imidazopyridines by Visible Light Photoredox Catalysis. J. Org. Chem 2016, 81, 7282–7287. [DOI] [PubMed] [Google Scholar]
- (69).Hagmann WK The Many Roles for Fluorine in Medicinal Chemistry. J. Med. Chem 2008, 51, 4359–4369. [DOI] [PubMed] [Google Scholar]
- (70).Purser S; Moore PR; Swallow S; Gouverneur V Fluorine in Medicinal Chemistry. Chem. Soc. Rev 2008, 37, 320–330. [DOI] [PubMed] [Google Scholar]
- (71).Nagib DA; MacMillan DWC Trifluoromethylation of Arenes and Heteroarenes by Means of Photoredox Catalysis. Nature 2011, 480, 224–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Chen L; Wu L; Duan W; Wang T; Li L; Zhang K; Zhu J; Peng Z; Xiong F Photoredox-Catalyzed Cascade Radical Cyclization of Ester Arylpropiolates with CF3SO2Cl To Construct 3-Trifluoromethyl Coumarin Derivatives. J. Org. Chem 2018, 83, 8607–8614. [DOI] [PubMed] [Google Scholar]
- (73).Jin J; MacMillan DWC Direct α-Arylation of Ethers through the Combination of Photoredox-Mediated C-H Functionalization and the Minisci Reaction. Angew. Chem., Int. Ed 2015, 54, 1565–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Zhang S-Y; Zhang F-M; Tu Y-Q Direct Sp3 α-C-H Activation and Functionalization of Alcohol and Ether. Chem. Soc. Rev 2011, 40, 1937–1949. [DOI] [PubMed] [Google Scholar]
- (75).McCallum T; Jouanno L-A; Cannillo A; Barriault L Persulfate-Enabled Direct C-H Alkylation of Heteroarenes with Unactivated Ethers. Synlett 2016, 27, 1282–1286. [Google Scholar]
- (76).Jin J; MacMillan DWC Alcohols as Alkylating Agents in Heteroarene C-H Functionalization. Nature 2015, 525, 87–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Huff CA; Cohen RD; Dykstra KD; Streckfuss E; DiRocco DA; Krska SW Photoredox-Catalyzed Hydroxymethylation of Heteroaromatic Bases. J. Org. Chem 2016, 81, 6980–6987. [DOI] [PubMed] [Google Scholar]
- (78).Garza-Sanchez RA; Tlahuext-Aca A; Tavakoli G; Glorius F Visible Light-Mediated Direct Decarboxylative C-H Functionalization of Heteroarenes. ACS Catal. 2017, 7, 4057–4061. [Google Scholar]
- (79).Chawla OP; Fessenden RW Electron Spin Resonance and Pulse Radiolysis Studies of Some Reactions of Peroxysulfate (SO4.1,2). J. Phys. Chem 1975, 79, 2693–2700. [Google Scholar]
- (80).Huie RE; Clifton CL Temperature Dependence of the Rate Constants for Reactions of the Sulfate Radical, SO4−, with Anions. J. Phys. Chem 1990, 94, 8561–8567. [Google Scholar]
- (81).Rustgi SM; Riesz P An E.S.R. and Spin-Trapping Study of the Reactions of the •SO4− Radical with Protein and Nucleic Acid Constituents. Int. J. Radiat. Biol. Relat. Stud. Phys., Chem. Med 1978, 34, 301–316. [DOI] [PubMed] [Google Scholar]
- (82).DiRocco DA; Dykstra K; Krska S; Vachal P; Conway DV; Tudge M Late-Stage Functionalization of Biologically Active Heterocycles Through Photoredox Catalysis. Angew. Chem., Int. Ed 2014, 53, 4802–4806. [DOI] [PubMed] [Google Scholar]
- (83).Xie J; Xu P; Li H; Xue Q; Jin H; Cheng Y; Zhu C A Room Temperature Decarboxylation/C-H Functionalization Cascade by Visible-Light Photoredox Catalysis. Chem. Commun 2013, 49, 5672–5674. [DOI] [PubMed] [Google Scholar]
- (84).Yang B; Yu D; Xu X-H; Qing F-L Visible-Light Photoredox Decarboxylation of Perfluoroarene Iodine(III) Trifluoroacetates for C-H Trifluoromethylation of (Hetero)Arenes. ACS Catal. 2018, 8, 2839–2843. [Google Scholar]
- (85).Cheng W-M; Shang R; Fu Y Photoredox/Brønsted Acid Co-Catalysis Enabling Decarboxylative Coupling of Amino Acid and Peptide Redox-Active Esters with N-Heteroarenes. ACS Catal. 2017, 7, 907–911. [Google Scholar]
- (86).Wayner DDM; Dannenberg JJ; Griller D Oxidation Potentials of α-Aminoalkyl Radicals: Bond Dissociation Energies for Related Radical Cations. Chem. Phys. Lett 1986, 131, 189–191. [Google Scholar]
- (87).Nakajima K; Miyake Y; Nishibayashi Y Synthetic Utilization of α-Aminoalkyl Radicals and Related Species in Visible Light Photoredox Catalysis. Acc. Chem. Res 2016, 49, 1946–1956. [DOI] [PubMed] [Google Scholar]
- (88).Proctor RSJ; Davis HJ; Phipps RJ Catalytic Enantioselective Minisci-Type Addition to Heteroarenes. Science 2018, 360, 419–422. [DOI] [PubMed] [Google Scholar]
- (89).Hari DP; König B The Photocatalyzed Meerwein Arylation: Classic Reaction of Aryl Diazonium Salts in a New Light. Angew. Chem., Int. Ed 2013, 52, 4734–4743. [DOI] [PubMed] [Google Scholar]
- (90).Andrieux CP; Pinson J The Standard Redox Potential of the Phenyl Radical/Anion Couple. J. Am. Chem. Soc 2003, 125, 14801–14806. [DOI] [PubMed] [Google Scholar]
- (91).Cano-Yelo H; Deronzier A Photocatalysis of the Pschorr Reaction by Tris-(2,2’-Bipyridyl)Ruthenium(II) in the Phenanthrene Series. J. Chem. Soc., Perkin Trans 2 1984, No. 6, 1093–1098. [Google Scholar]
- (92).Cano-Yelo H; Deronzier A Photocatalysis of the Pschorr Reaction by Ru(Bpy)32+. J. Photochem 1987, 37, 315–321. [Google Scholar]
- (93).Kalyani D; McMurtrey KB; Neufeldt SR; Sanford MS Room-Temperature C-H Arylation: Merger of Pd-Catalyzed C-H Functionalization and Visible-Light Photocatalysis. J. Am. Chem. Soc 2011, 133, 18566–18569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Neufeldt SR; Sanford MS Combining Transition Metal Catalysis with Radical Chemistry: Dramatic Acceleration of Palladium-Catalyzed C-H Arylation with Diaryliodonium Salts. Adv. Synth. Catal 2012, 354, 3517–3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Xue D; Jia Z-H; Zhao C-J; Zhang Y-Y; Wang C; Xiao J Direct Arylation of N-Heteroarenes with Aryldiazonium Salts by Photoredox Catalysis in Water. Chem. - Eur. J 2014, 20, 2960–2965. [DOI] [PubMed] [Google Scholar]
- (96).Zhang Y-P; Feng X-L; Yang Y-S; Cao B-X Metal-Free, C-H Arylation of Indole and Its Derivatives with Aryl Diazonium Salts by Visible-Light Photoredox Catalysis. Tetrahedron Lett. 2016, 57, 2298–2302. [Google Scholar]
- (97).Maity P; Kundu D; Ranu BC Visible-Light-Photocatalyzed Metal-Free C-H Heteroarylation of Heteroarenes at Room Temperature: A Sustainable Synthesis of Biheteroaryls: A Sustainable Synthesis of Biheteroaryls. Eur. J. Org. Chem 2015, 2015, 1727–1734. [Google Scholar]
- (98).Rybicka-Jasińska K; König B; Gryko D Porphyrin-Catalyzed Photochemical C-H Arylation of Heteroarenes. Eur. J. Org. Chem 2017, 2017, 2104–2107. [Google Scholar]
- (99).Gomes F; Narbonne V; Blanchard F; Maestri G; Malacria M Formal Base-Free Homolytic Aromatic Substitutions via Photoredox Catalysis. Org. Chem. Front 2015, 2, 464–469. [Google Scholar]
- (100).Zhang J; Chen J; Zhang X; Lei X Total Syntheses of Menisporphine and Daurioxoisoporphine C Enabled by Photoredox-Catalyzed Direct C-H Arylation of Isoquinoline with Aryldiazonium Salt. J. Org. Chem 2014, 79, 10682–10688. [DOI] [PubMed] [Google Scholar]
- (101).Liang L; Xie M-S; Wang H-X; Niu H-Y; Qu G-R; Guo H-M Visible-Light-Mediated Monoselective Ortho C-H Arylation of 6-Arylpurine Nucleosides with Diazonium Salts. J. Org. Chem 2017, 82, 5966–5973. [DOI] [PubMed] [Google Scholar]
- (102).Gauchot V; Sutherland DR; Lee A-L Dual Gold and Photoredox Catalysed C-H Activation of Arenes for Aryl-Aryl Cross Couplings. Chem. Sci 2017, 8, 2885–2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Liang Y-F; Steinbock R; Yang L; Ackermann L Continuous Visible-Light Photoflow Approach for a Manganese-Catalyzed (Het)-Arene C-H Arylation. Angew. Chem., Int. Ed 2018, 57, 10625–10629. [DOI] [PubMed] [Google Scholar]
- (104).Jiang J; Zhang W-M; Dai J-J; Xu J; Xu H-J Visible-Light-Promoted C-H Arylation by Merging Palladium Catalysis with Organic Photoredox Catalysis. J. Org. Chem 2017, 82, 3622–3630. [DOI] [PubMed] [Google Scholar]
- (105).Zhang S; Tang Z; Bao W; Li J; Guo B; Huang S; Zhang Y; Rao Y Perylenequinonoid-Catalyzed Photoredox Activation for the Direct Arylation of (Het)Arenes with Sunlight. Org. Biomol. Chem 2019, 17, 4364–4369. [DOI] [PubMed] [Google Scholar]
- (106).Huang L; Zhao J Iodo-Bodipys as Visible-Light-Absorbing Dual-Functional Photoredox Catalysts for Preparation of Highly Functionalized Organic Compounds by Formation of C-C Bonds via Reductive and Oxidative Quenching Catalytic Mechanisms. RSC Adv. 2013, 3, 23377–23388. [Google Scholar]
- (107).Ouyang X-H; Cheng J; Li J-H 1,2-Diarylation of Alkenes with Aryldiazonium Salts and Arenes Enabled by Visible Light Photoredox Catalysis. Chem. Commun 2018, 54, 8745–8748. [DOI] [PubMed] [Google Scholar]
- (108).Wang X; Han Y-F; Ouyang X-H; Song R-J; Li J-H The Photoredox Alkylarylation of Styrenes with Alkyl N-Hydroxyphthalimide Esters and Arenes Involving C-H Functionalization. Chem. Commun 2019, 55, 14637–14640. [DOI] [PubMed] [Google Scholar]
- (109).Klauck FJR; Yoon H; James MJ; Lautens M; Glorius F Visible-Light-Mediated Deaminative Three-Component Dicarbofunctionalization of Styrenes with Benzylic Radicals. ACS Catal. 2019, 9, 236–241. [Google Scholar]
- (110).Deng G-B; Wang Z-Q; Xia J-D; Qian P-C; Song R-J; Hu M; Gong L-B; Li J-H Tandem Cyclizations of 1,6-Enynes with Arylsulfonyl Chlorides by Using Visible-Light Photoredox Catalysis. Angew. Chem., Int. Ed 2013, 52, 1535–1538. [DOI] [PubMed] [Google Scholar]
- (111).Natarajan P; Bala A; Mehta SK; Bhasin KK Visible-Light Photocatalyzed Synthesis of 2-Aryl N-Methylpyrroles, Furans and Thiophenes Utilizing Arylsulfonyl Chlorides as a Coupling Partner. Tetrahedron 2016, 72, 2521–2526. [Google Scholar]
- (112).Nguyen JD; D’Amato EM; Narayanam JMR; Stephenson CRJ Engaging Unactivated Alkyl, Alkenyl and Aryl Iodides in Visible-Light-Mediated Free Radical Reactions. Nat. Chem 2012, 4, 854–859. [DOI] [PubMed] [Google Scholar]
- (113).Kim H; Lee C Visible-Light-Induced Photocatalytic Reductive Transformations of Organohalides. Angew. Chem., Int. Ed 2012, 51, 12303–12306. [DOI] [PubMed] [Google Scholar]
- (114).Cheng Y; Gu X; Li P Visible-Light Photoredox in Homolytic Aromatic Substitution: Direct Arylation of Arenes with Aryl Halides. Org. Lett 2013, 15, 2664–2667. [DOI] [PubMed] [Google Scholar]
- (115).Paria S; Reiser O Visible Light Photoredox Catalyzed Cascade Cyclizations of α-Bromochalcones or α-Bromocinnamates with Heteroarenes. Adv. Synth. Catal 2014, 356, 557–562. [Google Scholar]
- (116).Karki BS; Pramanik MMD; Kant R; Rastogi N Visible Light Catalyzed Reaction of α-Bromochalcones with Chalcones: Direct Access to the Urundeuvine Scaffold. Org. Biomol. Chem 2018, 16, 7152–7156. [DOI] [PubMed] [Google Scholar]
- (117).Nagode SB; Kant R; Rastogi N Synthesis of Phenanthrenes through Visible-Light Photoredox Catalyzed Intramolecular Cyclization of α-Bromochalcones. Eur. J. Org. Chem 2018, 2018 (13), 1533–1537. [Google Scholar]
- (118).Paria S; Kais V; Reiser O Visible Light-Mediated Coupling of α-Bromochalcones with Alkenes. Adv. Synth. Catal 2014, 356, 2853–2858. [Google Scholar]
- (119).Banerjee A; Lei Z; Ngai M-Y Acyl Radical Chemistry via Visible-Light Photoredox Catalysis. Synthesis 2019, 51, 303–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).Cheng P; Qing Z; Liu S; Liu W; Xie H; Zeng J Regiospecific Minisci Acylation of Phenanthridine via Thermolysis or Photolysis. Tetrahedron Lett. 2014, 55, 6647–6651. [Google Scholar]
- (121).White HS; Bard AJ Electrogenerated Chemiluminescence and Chemiluminescence of the Ru(2,2′-bpy)32+-S2O82− System in Acetonitrile-Water Solutions. J. Am. Chem. Soc 1982, 104, 6891–6895. [Google Scholar]
- (122).Sharma UK; Gemoets HPL; Schröder F; Noël T; Van der Eycken EV Merger of Visible-Light Photoredox Catalysis and C-H Activation for the Room-Temperature C-2 Acylation of Indoles in Batch and Flow. ACS Catal. 2017, 7, 3818–3823. [Google Scholar]
- (123).Jung S; Lee H; Moon Y; Jung H-Y; Hong S Site-Selective C-H Acylation of Pyridinium Derivatives by Photoredox Catalysis. ACS Catal. 2019, 9, 9891–9896. [Google Scholar]
- (124).Chu L; Lipshultz JM; MacMillan DWC Merging Photoredox and Nickel Catalysis: The Direct Synthesis of Ketones by the Decarboxylative Arylation of α-Oxo Acids. Angew. Chem., Int. Ed 2015, 54, 7929–7933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Gu L; Jin C; Liu J; Zhang H; Yuan M; Li G Acylation of Indoles via Photoredox Catalysis: A Route to 3- Acylindoles. Green Chem 2016, 18, 1201–1205. [Google Scholar]
- (126).Ruzi R; Zhang M; Ablajan K; Zhu C Photoredox-Catalyzed Deoxygenative Intramolecular Acylation of Biarylcarboxylic Acids: Access to Fluorenones. J. Org. Chem 2017, 82, 12834–12839. [DOI] [PubMed] [Google Scholar]
- (127).Bergonzini G; Cassani C; Wallentin C-J Acyl Radicals from Aromatic Carboxylic Acids by Means of Visible-Light Photoredox Catalysis. Angew. Chem., Int. Ed 2015, 54, 14066–14069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Sultan S; Rizvi MA; Kumar J; Shah BA Acyl Radicals from Terminal Alkynes: Photoredox-Catalyzed Acylation of Heteroarenes. Chem. - Eur. J 2018, 24, 10617–10620. [DOI] [PubMed] [Google Scholar]
- (129).Zard SZ Recent Progress in the Generation and Use of Nitrogen-Centred Radicals. Chem. Soc. Rev 2008, 37, 1603–1618. [DOI] [PubMed] [Google Scholar]
- (130).Sutcliffe R; Griller D; Lessard J; Ingold KU The Structure of Amidyl Radicals. Evidence for the. Pi.-Electronic Ground State and for Twist about the Acyl-Nitrogen Bond by Electron Paramagnetic Resonance Spectroscopy. J. Am. Chem. Soc 1981, 103, 624–628. [Google Scholar]
- (131).Allen LJ; Cabrera PJ; Lee M; Sanford MS N-Acyloxyphthalimides as Nitrogen Radical Precursors in the Visible Light Photocatalyzed Room Temperature C-H Amination of Arenes and Heteroarenes. J. Am. Chem. Soc 2014, 136, 5607–5610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Kim H; Kim T; Lee DG; Roh SW; Lee C Nitrogen-Centered Radical-Mediated C-H Imidation of Arenes and Heteroarenes via Visible Light Induced Photocatalysis. Chem. Commun 2014, 50, 9273–9276. [DOI] [PubMed] [Google Scholar]
- (133).Tong K; Liu X; Zhang Y; Yu S Visible-Light-Induced Direct Oxidative C-H Amidation of Heteroarenes with Sulfonamides. Chem. - Eur. J 2016, 22, 15669–15673. [DOI] [PubMed] [Google Scholar]
- (134).Ito E; Fukushima T; Kawakami T; Murakami K; Itami K Catalytic Dehydrogenative C-H Imidation of Arenes Enabled by PhotoGenerated Hole Donation to Sulfonimide. Chem. 2017, 2, 383–392. [Google Scholar]
- (135).Qin Q; Yu S Visible-Light-Promoted Redox Neutral C-H Amidation of Heteroarenes with Hydroxylamine Derivatives. Org. Lett 2014, 16, 3504–3507. [DOI] [PubMed] [Google Scholar]
- (136).Brachet E; Ghosh T; Ghosh I; König B Visible Light C-H Amidation of Heteroarenes with Benzoyl Azides. Chem. Sci 2015, 6, 987–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (137).Davies J; Svejstrup TD; Fernandez Reina D; Sheikh NS; Leonori D Visible-Light-Mediated Synthesis of Amidyl Radicals: Transition-Metal-Free Hydroamination and N-Arylation Reactions. J. Am. Chem. Soc 2016, 138, 8092–8095. [DOI] [PubMed] [Google Scholar]
- (138).Svejstrup TD; Ruffoni A; Juliá F; Aubert VM; Leonori D Synthesis of Arylamines via Aminium Radicals. Angew. Chem., Int. Ed 2017, 56, 14948–14952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (139).Greulich TW; Daniliuc CG; Studer A N-Aminopyridinium Salts as Precursors for N-Centered Radicals - Direct Amidation of Arenes and Heteroarenes. Org. Lett 2015, 17, 254–257. [DOI] [PubMed] [Google Scholar]
- (140).Ruffoni A; Juliá F; Svejstrup TD; McMillan AJ; Douglas JJ; Leonori D Practical and Regioselective Amination of Arenes Using Alkyl Amines. Nat. Chem 2019, 11, 426–433. [DOI] [PubMed] [Google Scholar]
- (141).Wang J-D; Liu Y-X; Xue D; Wang C; Xiao J Amination of Benzoxazoles by Visible-Light Photoredox Catalysis. Synlett 2014, 25, 2013–2018. [Google Scholar]
- (142).Moon Y; Jang E; Choi S; Hong S Visible-Light-Photocatalyzed Synthesis of Phenanthridinones and Quinolinones via Direct Oxidative C-H Amidation. Org. Lett 2018, 20, 240–243. [DOI] [PubMed] [Google Scholar]
- (143).Jiang H; An X; Tong K; Zheng T; Zhang Y; Yu S Visible-Light-Promoted Iminyl-Radical Formation from Acyl Oximes: A Unified Approach to Pyridines, Quinolines, and Phenanthridines. Angew. Chem., Int. Ed 2015, 54, 4055–4059. [DOI] [PubMed] [Google Scholar]
- (144).An X-D; Yu S Visible-Light-Promoted and One-Pot Synthesis of Phenanthridines and Quinolines from Aldehydes and O-Acyl Hydroxylamine. Org. Lett 2015, 17, 2692–2695. [DOI] [PubMed] [Google Scholar]
- (145).Rao H; Wang P; Li C-J Visible-Light-Triggered Direct Benzoyloxylation of Electron-Rich Arenes at Room Temperature without Chelation Assistance. Eur. J. Org. Chem 2012, 2012, 6503–6507. [Google Scholar]
- (146).Wimmer A; König B Photocatalytic Formation of Carbon-Sulfur Bonds. Beilstein J. Org. Chem 2018, 14, 54–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (147).Zhang G; Liu C; Yi H; Meng Q; Bian C; Chen H; Jian J-X; Wu L-Z; Lei A External Oxidant-Free Oxidative Cross-Coupling: A Photoredox Cobalt-Catalyzed Aromatic C-H Thiolation for Constructing C-S Bonds. J. Am. Chem. Soc 2015, 137, 9273–9280. [DOI] [PubMed] [Google Scholar]
- (148).Cheng Y; Yang J; Qu Y; Li P Aerobic Visible-Light Photoredox Radical C-H Functionalization: Catalytic Synthesis of 2-Substituted Benzothiazoles. Org. Lett 2012, 14, 98–101. [DOI] [PubMed] [Google Scholar]
- (149).Chen M; Huang Z-T; Zheng Q-Y Visible Light-Induced 3-Sulfenylation of N-Methylindoles with Arylsulfonyl Chlorides. Chem. Commun 2012, 48, 11686–11688. [DOI] [PubMed] [Google Scholar]
- (150).Bai P; Sun S; Li Z; Qiao H; Su X; Yang F; Wu Y; Wu Y Ru/Cu Photoredox or Cu/Ag Catalyzed C4-H Sulfonylation of 1-Naphthylamides at Room Temperature. J. Org. Chem 2017, 82, 12119–12127. [DOI] [PubMed] [Google Scholar]
- (151).Pagire SK; Hossain A; Reiser O Temperature Controlled Selective C-S or C-C Bond Formation: Photocatalytic Sulfonylation versus Arylation of Unactivated Heterocycles Utilizing Aryl Sulfonyl Chlorides. Org. Lett 2018, 20, 648–651. [DOI] [PubMed] [Google Scholar]
- (152).Pandey G; Singh D; Laha R Selective C(sp2)-H Functionalization of Arenes for Amination Reactions by Using Photoredox Catalysis. Asian J. Org. Chem 2017, 6, 469–474. [Google Scholar]
- (153).Shaikh RS; Ghosh I; König B Direct C-H Phosphonylation of Electron-Rich Arenes and Heteroarenes by Visible-Light Photoredox Catalysis. Chem. - Eur. J 2017, 23, 12120–12124. [DOI] [PubMed] [Google Scholar]
- (154).Rössler SL; Jelier BJ; Tripet PF; Shemet A; Jeschke G; Togni A; Carreira EM Pyridyl Radical Cation for C-H Amination of Arenes. Angew. Chem., Int. Ed 2019, 58, 526–531. [DOI] [PubMed] [Google Scholar]
- (155).Ham WS; Hillenbrand J; Jacq J; Genicot C; Ritter T Divergent Late-Stage (Hetero)Aryl C-H Amination by the Pyridinium Radical Cation. Angew. Chem., Int. Ed 2019, 58, 532–536. [DOI] [PubMed] [Google Scholar]
- (156).Morofuji T; Ikarashi G; Kano N Photocatalytic C-H Amination of Aromatics Overcoming Redox Potential Limitations. Org. Lett 2020, 22, 2822–2827. [DOI] [PubMed] [Google Scholar]
- (157).Schlesener CJ; Kochi JK Stoichiometry and Kinetics of P-Methoxytoluene Oxidation by Electron Transfer. Mechanistic Dichotomy between Side Chain and Nuclear Substitution. J. Org. Chem 1984, 49, 3142–3150. [Google Scholar]
- (158).Jarrige L; Levitre G; Masson G Visible-Light Photoredox-Catalyzed Coupling Reaction of Azoles with α-Carbamoyl Sulfides. J. Org. Chem 2016, 81, 7230–7236. [DOI] [PubMed] [Google Scholar]
- (159).Lebée C; Languet M; Allain C; Masson G α-Carbamoylsulfides as N-Carbamoylimine Precursors in the Visible Light Photoredox-Catalyzed Synthesis of α,α-Disubstituted Amines. Org. Lett 2016, 18, 1478–1481. [DOI] [PubMed] [Google Scholar]
- (160).Wang Z-Q; Hu M; Huang X-C; Gong L-B; Xie Y-X; Li J-H Direct α-Arylation of α-Amino Carbonyl Compounds with Indoles Using Visible Light Photoredox Catalysis. J. Org. Chem 2012, 77, 8705–8711. [DOI] [PubMed] [Google Scholar]
- (161).Fabry DC; Zoller J; Raja S; Rueping M Combining Rhodium and Photoredox Catalysis for C-H Functionalizations of Arenes: Oxidative Heck Reactions with Visible Light. Angew. Chem., Int. Ed 2014, 53, 10228–10231. [DOI] [PubMed] [Google Scholar]
- (162).Fabry DC; Ronge MA; Zoller J; Rueping M C-H Functionalization of Phenols Using Combined Ruthenium and Photoredox Catalysis: In Situ Generation of the Oxidant. Angew. Chem., Int. Ed 2015, 54, 2801–2805. [DOI] [PubMed] [Google Scholar]
- (163).Zoller J; Fabry DC; Ronge MA; Rueping M Synthesis of Indoles Using Visible Light: Photoredox Catalysis for Palladium-Catalyzed C-H Activation. Angew. Chem., Int. Ed 2014, 53, 13264–13268. [DOI] [PubMed] [Google Scholar]
- (164).Choi S; Chatterjee T; Choi WJ; You Y; Cho EJ Synthesis of Carbazoles by a Merged Visible Light Photoredox and Palladium-Catalyzed Process. ACS Catal. 2015, 5, 4796–4802. [Google Scholar]
- (165).Sagadevan A; Greaney MF Meta-Selective C-H Activation of Arenes at Room Temperature Using Visible Light: Dual-Function Ruthenium Catalysis. Angew. Chem 2019, 131, 9931–9935. [DOI] [PubMed] [Google Scholar]
- (166).Fumagalli F; Warratz S; Zhang S-K; Rogge T; Zhu C; Stückl AC; Ackermann L Arene-Ligand-Free Ruthenium(II/III) Manifold for Meta-C-H Alkylation: Remote Purine Diversification. Chem. - Eur. J 2018, 24, 3984–3988. [DOI] [PubMed] [Google Scholar]
- (167).Wayner DDM; Clark KB; Rauk A; Yu D; Armstrong DA C-H Bond Dissociation Energies of Alkyl Amines: Radical Structures and Stabilization Energies. J. Am. Chem. Soc 1997, 119, 8925–8932. [Google Scholar]
- (168).Dhar D; Tolman WB Hydrogen Atom Abstraction from Hydrocarbons by a Copper(III)-Hydroxide Complex. J. Am. Chem. Soc 2015, 137, 1322–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (169).Bordwell FG; Harrelson JA Jr Acidities and Homolytic Bond Dissociation Energies of the AC—H Bonds in Ketones in DMSO. Can. J. Chem 1990, 68, 1714–1718. [Google Scholar]
- (170).Ellison GB; Davico GE; Bierbaum VM; DePuy CH Thermochemistry of the Benzyl and Allyl Radicals and Ions. Int. J. Mass Spectrom. Ion Processes 1996, 156, 109–131. [Google Scholar]
- (171).Tsang W Heats of Formation of Organic Free Radicals by Kinetic Methods. In Energetics of Organic Free Radicals; Martinho Simões JA, Greenberg A, Liebman JF, Eds.; Structure Energetics and Reactivity in Chemistry Series (SEARCH series); Springer Netherlands: Dordrecht, 1996; pp 22–58. [Google Scholar]
- (172).da Silva G; Bozzelli JW Enthalpies of Formation, Bond Dissociation Energies, and Molecular Structures of the n-Aldehydes (Acetaldehyde, Propanal, Butanal, Pentanal, Hexanal, and Heptanal) and Their Radicals. J. Phys. Chem. A 2006, 110, 13058–13067. [DOI] [PubMed] [Google Scholar]
- (173).Hu J; Wang J; Nguyen TH; Zheng N The Chemistry of Amine Radical Cations Produced by Visible Light Photoredox Catalysis. Beilstein J. Org. Chem 2013, 9, 1977–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (174).Morris SA; Wang J; Zheng N The Prowess of Photogenerated Amine Radical Cations in Cascade Reactions: From Carbocycles to Heterocycles. Acc. Chem. Res 2016, 49, 1957–1968. [DOI] [PubMed] [Google Scholar]
- (175).Beatty JW; Stephenson CRJ Amine Functionalization via Oxidative Photoredox Catalysis: Methodology Development and Complex Molecule Synthesis. Acc. Chem. Res 2015, 48, 1474–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (176).Yoshida J; Suga S; Suzuki S; Kinomura N; Yamamoto A; Fujiwara K Direct Oxidative Carbon-Carbon Bond Formation Using the “Cation Pool” Method. 1. Generation of Iminium Cation Pools and Their Reaction with Carbon Nucleophiles. J. Am. Chem. Soc 1999, 121, 9546–9549. [Google Scholar]
- (177).Shono T; Matsumura Y; Tsubata K Electroorganic Chemistry. 46. A New Carbon-Carbon Bond Forming Reaction at the. Alpha.-Position of Amines Utilizing Anodic Oxidation as a Key Step. J. Am. Chem. Soc 1981, 103, 1172–1176. [Google Scholar]
- (178).Murahashi S Synthetic Aspects of Metal-Catalyzed Oxidations of Amines and Related Reactions. Angew. Chem., Int. Ed. Engl 1995, 34, 2443–2465. [Google Scholar]
- (179).DeLaive PJ; Sullivan BP; Meyer TJ; Whitten DG Applications of Light-Induced Electron-Transfer Reactions. Coupling of Hydrogen Generation with Photoreduction of Ruthenium(II) Complexes by Triethylamine. J. Am. Chem. Soc 1979, 101, 4007–4008. [Google Scholar]
- (180).Condie AG; González-Gómez JC; Stephenson CRJ Visible-Light Photoredox Catalysis: Aza-Henry Reactions via C-H Functionalization. J. Am. Chem. Soc 2010, 132, 1464–1465. [DOI] [PubMed] [Google Scholar]
- (181).Li Z; Li C-J Highly Efficient Copper-Catalyzed Nitro-Mannich Type Reaction: Cross-Dehydrogenative-Coupling between Sp3 C-H Bond and Sp3 C-H Bond. J. Am. Chem. Soc 2005, 127, 3672–3673. [DOI] [PubMed] [Google Scholar]
- (182).Freeman DB; Furst L; Condie AG; Stephenson CRJ Functionally Diverse Nucleophilic Trapping of Iminium Intermediates Generated Utilizing Visible Light. Org. Lett 2012, 14, 94–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (183).Narayanam JMR; Tucker JW; Stephenson CRJ Electron-Transfer Photoredox Catalysis: Development of a Tin-Free Reductive Dehalogenation Reaction. J. Am. Chem. Soc 2009, 131, 8756–8757. [DOI] [PubMed] [Google Scholar]
- (184).Tucker JW; Zhang Y; Jamison TF; Stephenson CRJ Visible-Light Photoredox Catalysis in Flow. Angew. Chem., Int. Ed 2012, 51, 4144–4147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (185).Rueping M; Vila C; Koenigs RM; Poscharny K; Fabry DC Dual Catalysis: Combining Photoredox and Lewis Base Catalysis for Direct Mannich Reactions. Chem. Commun 2011, 47, 2360–2362. [DOI] [PubMed] [Google Scholar]
- (186).Zhao G; Yang C; Guo L; Sun H; Chen C; Xia W Visible Light-Induced Oxidative Coupling Reaction: Easy Access to Mannich-Type Products. Chem. Commun 2012, 48, 2337. [DOI] [PubMed] [Google Scholar]
- (187).Rueping M; Zhu S; Koenigs RM Visible-Light Photoredox Catalyzed Oxidative Strecker Reaction. Chem. Commun 2011, 47, 12709–12711. [DOI] [PubMed] [Google Scholar]
- (188).Rueping M; Zhu S; Koenigs RM Photoredox Catalyzed C-P Bond Forming Reactions—Visible Light Mediated Oxidative Phosphonylations of Amines. Chem. Commun 2011, 47, 8679–8681. [DOI] [PubMed] [Google Scholar]
- (189).Rueping M; Koenigs RM; Poscharny K; Fabry DC; Leonori D; Vila C Dual Catalysis: Combination of Photocatalytic Aerobic Oxidation and Metal Catalyzed Alkynylation Reactions—C-C Bond Formation Using Visible Light. Chem. - Eur. J 2012, 18, 5170–5174. [DOI] [PubMed] [Google Scholar]
- (190).Barham JP; John MP; Murphy JA One-Pot Functionalisation of N-Substituted Tetrahydroisoquinolines by Photo-oxidation and Tunable Organometallic Trapping of Iminium Intermediates. Beilstein J. Org. Chem 2014, 10, 2981–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (191).Feng Z-J; Xuan J; Xia X-D; Ding W; Guo W; Chen J-R; Zou Y-Q; Lu L-Q; Xiao W-J Direct Sp3 C-H Acroleination of N-Aryl-Tetrahydroisoquinolines by Merging Photoredox Catalysis with Nucleophilic Catalysis. Org. Biomol. Chem 2014, 12, 2037–2040. [DOI] [PubMed] [Google Scholar]
- (192).Kwon SJ; Gil MG; Kim DY Visible Light Mediated Photocatalytic Oxidative Coupling Reaction of N-Phenyl Tetrahydroisoquinoline with β-Keto Acids. Tetrahedron Lett. 2017, 58, 1592–1594. [Google Scholar]
- (193).Ji J; Chen L-Y; Qiu Z-B; Ren X; Li Y Visible-Light Photoredox-Catalyzed Cross-Dehydrogenative Coupling of Tetrahydroisoquinolines with 3-Fluorooxindoles. Asian J. Org. Chem 2019, 8, 1436–1440. [Google Scholar]
- (194).To W-P; Tong GS-M; Lu W; Ma C; Liu J; Chow AL-F; Che C-M Luminescent Organogold(III) Complexes with Long-Lived Triplet Excited States for Light-Induced Oxidative C-H Bond Functionalization and Hydrogen Production. Angew. Chem., Int. Ed 2012, 51, 2654–2657. [DOI] [PubMed] [Google Scholar]
- (195).Xue Q; Xie J; Jin H; Cheng Y; Zhu C Highly Efficient Visible-Light-Induced Aerobic Oxidative C-C, C-P Coupling from C-H Bonds Catalyzed by a Gold(III)-Complex. Org. Biomol. Chem 2013, 11, 1606–1609. [DOI] [PubMed] [Google Scholar]
- (196).Yu J; Cai C Photocatalytic Oxidative Cyclization of α-Halo Hydrazones with Tetrahydroisoquinoline for Construction of Isoquino[3,4-a][1,2,4]-Triazines. Catal. Commun 2018, 109, 60–64. [Google Scholar]
- (197).Tiwari DK; Maurya RA; Nanubolu JB Visible-Light/Photoredox-Mediated Sp3 C-H Functionalization and Coupling of Secondary Amines with Vinyl Azides in Flow Microreactors. Chem. - Eur. J 2016, 22, 526–530. [DOI] [PubMed] [Google Scholar]
- (198).Zou Y-Q; Lu L-Q; Fu L; Chang N-J; Rong J; Chen J-R; Xiao W-J Visible-Light-Induced Oxidation/[3 + 2] Cycloaddition/Oxidative Aromatization Sequence: A Photocatalytic Strategy To Construct Pyrrolo[2,1-a]Isoquinolines. Angew. Chem., Int. Ed 2011, 50, 7171–7175. [DOI] [PubMed] [Google Scholar]
- (199).Rueping M; Leonori D; Poisson T Visible Light Mediated Azomethine Ylide Formation—Photoredox Catalyzed [3 + 2] Cycloadditions. Chem. Commun 2011, 47, 9615–9617. [DOI] [PubMed] [Google Scholar]
- (200).Xuan J; Feng Z-J; Duan S-W; Xiao W-J Room Temperature Synthesis of Isoquino[2,1-a][3,1]Oxazine and Isoquino-[2,1-a]Pyrimidine Derivatives via Visible Light Photoredox Catalysis. RSC Adv. 2012, 2, 4065–4068. [Google Scholar]
- (201).Mathis CL; Gist BM; Frederickson CK; Midkiff KM; Marvin CC Visible Light Photooxidative Cyclization of Amino Alcohols to 1,3-Oxazines. Tetrahedron Lett. 2013, 54, 2101–2104. [Google Scholar]
- (202).DiRocco DA; Rovis T Catalytic Asymmetric α-Acylation of Tertiary Amines Mediated by a Dual Catalysis Mode: N-Heterocyclic Carbene and Photoredox Catalysis. J. Am. Chem. Soc 2012, 134, 8094–8097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (203).Moore JL; Rovis T Carbene Catalysts. Top. Curr. Chem 2009, 291, 77–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (204).Vora HU; Rovis T Asymmetric N-Heterocyclic Carbene (NHC) Catalyzed Acyl Anion Reactivity. Aldrichimica Acta 2011, 44, 3–11. [PMC free article] [PubMed] [Google Scholar]
- (205).Bergonzini G; Schindler CS; Wallentin C-J; Jacobsen EN; Stephenson CRJ Photoredox Activation and Anion Binding Catalysis in the Dual Catalytic Enantioselective Synthesis of β-Amino Esters. Chem. Sci 2014, 5, 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (206).Querard P; Perepichka I; Zysman-Colman E; Li C-J Copper-Catalyzed Asymmetric Sp3 C-H Arylation of Tetrahydroisoquinoline Mediated by a Visible Light Photoredox Catalyst. Beilstein J. Org. Chem 2016, 12, 2636–2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (207).Yang Q; Zhang L; Ye C; Luo S; Wu L-Z; Tung C-H Visible-Light-Promoted Asymmetric Cross-Dehydrogenative Coupling of Tertiary Amines to Ketones by Synergistic Multiple Catalysis. Angew. Chem., Int. Ed 2017, 56, 3694–3698. [DOI] [PubMed] [Google Scholar]
- (208).Dai C; Meschini F; Narayanam JMR; Stephenson CRJ Friedel-Crafts Amidoalkylation via Thermolysis and Oxidative Photocatalysis. J. Org. Chem 2012, 77, 4425–4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (209).Zhu S; Rueping M Merging Visible-Light Photoredox and Lewis Acid Catalysis for the Functionalization and Arylation of Glycine Derivatives and Peptides. Chem. Commun 2012, 48, 11960–11962. [DOI] [PubMed] [Google Scholar]
- (210).Xuan J; Cheng Y; An J; Lu L-Q; Zhang X-X; Xiao W-J Visible Light-Induced Intramolecular Cyclization Reactions of Diamines: A New Strategy to Construct Tetrahydroimidazoles. Chem. Commun 2011, 47, 8337–8339. [DOI] [PubMed] [Google Scholar]
- (211).Yilmaz O; Oderinde MS; Emmert MH Photoredox-Catalyzed Cα-H Cyanation of Unactivated Secondary and Tertiary Aliphatic Amines: Late-Stage Functionalization and Mechanistic Studies. J. Org. Chem 2018, 83, 11089–11100. [DOI] [PubMed] [Google Scholar]
- (212).Hu B; Dong W; Feng Z; Gao X; Gao H; Xie X; Zhang Z Tandem Photocatalysis: An Efficient Synthesis of Multisubstituted Benzimidazoles by Visible-Light-Induced Intramolecular Cyclization and Deprotection. Asian J. Org. Chem 2016, 5, 1467–1470. [Google Scholar]
- (213).Hou H; Zhu S; Atodiresei I; Rueping M Asymmetric Organocatalysis and Photoredox Catalysis for the α-Functionalization of Tetrahydroisoquinolines. Eur. J. Org. Chem 2018, 2018, 1277–1280. [Google Scholar]
- (214).Yang X; Xie Z; Li Y; Zhang Y Enantioselective Aerobic Oxidative Cross-Dehydrogenative Coupling of Glycine Derivatives with Ketones and Aldehydes via Cooperative Photoredox Catalysis and Organocatalysis. Chem. Sci 2020, 11, 4741–4746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (215).Orgren LR; Maverick EE; Marvin CC Synthesis of (±)-Tetrabenazine by Visible Light Photoredox Catalysis. J. Org. Chem 2015, 80, 12635–12640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (216).Benimana SE; Cromwell N; Meer H; Marvin C Visible Light Photoredox and Polonovski-Potier Cyclizations for the Synthesis of (±)-5-Epi-Cermizine C and (±)-Epimyrtine. Tetrahedron Lett. 2016, 57, 5062–5064. [Google Scholar]
- (217).McNally A; Prier CK; MacMillan DWC Discovery of an α-Amino C-H Arylation Reaction Using the Strategy of Accelerated Serendipity. Science 2011, 334, 1114–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (218).Walker MM; Koronkiewicz B; Chen S; Houk KN; Mayer JM; Ellman JA Highly Diastereoselective Functionalization of Piperidines by Photoredox-Catalyzed α-Amino C-H Arylation and Epimerization. J. Am. Chem. Soc 2020, 142, 8194–8202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (219).Kohls P; Jadhav D; Pandey G; Reiser O Visible Light Photoredox Catalysis: Generation and Addition of N-Aryltetrahydroisoquinoline-Derived α-Amino Radicals to Michael Acceptors. Org. Lett 2012, 14, 672–675. [DOI] [PubMed] [Google Scholar]
- (220).Miyake Y; Nakajima K; Nishibayashi Y Visible-Light-Mediated Utilization of α-Aminoalkyl Radicals: Addition to Electron-Deficient Alkenes Using Photoredox Catalysts. J. Am. Chem. Soc 2012, 134, 3338–3341. [DOI] [PubMed] [Google Scholar]
- (221).Miyake Y; Nakajima K; Nishibayashi Y Direct Sp3 C-H Amination of Nitrogen-Containing Benzoheterocycles Mediated by Visible-Light-Photoredox Catalysts. Chem. - Eur. J 2012, 18, 16473–16477. [DOI] [PubMed] [Google Scholar]
- (222).Ruiz Espelt L; Wiensch EM; Yoon TP Brønsted Acid Cocatalysts in Photocatalytic Radical Addition of α-Amino C-H Bonds across Michael Acceptors. J. Org. Chem 2013, 78, 4107–4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (223).Dai X; Cheng D; Guan B; Mao W; Xu X; Li X The Coupling of Tertiary Amines with Acrylate Derivatives via Visible-Light Photoredox Catalysis. J. Org. Chem 2014, 79, 7212–7219. [DOI] [PubMed] [Google Scholar]
- (224).Dai X; Mao R; Guan B; Xu X; Li X Visible Light Photoredox Catalysis: Regioselective Radical Addition of Aminoalkyl Radicals to 2,3-Allenoates. RSC Adv. 2015, 5, 55290–55294. [Google Scholar]
- (225).Xie J; Shi S; Zhang T; Mehrkens N; Rudolph M; Hashmi ASK A Highly Efficient Gold-Catalyzed Photoredox α-C(sp3)-H Alkynylation of Tertiary Aliphatic Amines with Sunlight. Angew. Chem., Int. Ed 2015, 54, 6046–6050. [DOI] [PubMed] [Google Scholar]
- (226).Xu J-T; Xu G-Q; Wang Z-Y; Xu P-F Visible Light Photoredox-Catalyzed α-Alkylation of Cyclic Tertiary Arylamines. J. Org. Chem 2019, 84, 14760–14769. [DOI] [PubMed] [Google Scholar]
- (227).Zuo Z; Ahneman DT; Chu L; Terrett JA; Doyle AG; MacMillan DWC Merging Photoredox with Nickel Catalysis: Coupling of α-Carboxyl Sp3-Carbons with Aryl Halides. Science 2014, 345, 437–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (228).Shaw MH; Shurtleff VW; Terrett JA; Cuthbertson JD; MacMillan DWC Native Functionality in Triple Catalytic Cross-Coupling: sp3 C-H Bonds as Latent Nucleophiles. Science 2016, 352, 1304–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (229).Twilton J; Christensen M; DiRocco DA; Ruck RT; Davies IW; MacMillan DWC Selective Hydrogen Atom Abstraction through Induced Bond Polarization: Direct α-Arylation of Alcohols through Photoredox, HAT, and Nickel Catalysis. Angew. Chem., Int. Ed 2018, 57, 5369–5373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (230).Heitz DR; Tellis JC; Molander GA Photochemical Nickel-Catalyzed C-H Arylation: Synthetic Scope and Mechanistic Investigations. J. Am. Chem. Soc 2016, 138, 12715–12718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (231).Yu J; Zhao C; Zhou R; Gao W; Wang S; Liu K; Chen S; Hu K; Mei L; Yuan L; et al. Visible-Light-Enabled C-H Functionalization by a Direct Hydrogen Atom Transfer Uranyl Photocatalyst. Chem. - Eur. J 2020, 26, 16521–16529. [DOI] [PubMed] [Google Scholar]
- (232).Le C; Liang Y; Evans RW; Li X; MacMillan DWC Selective sp3 C-H Alkylation via Polarity-Match-Based Cross-Coupling. Nature 2017, 547, 79–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (233).Ahneman DT; Doyle AG C-H Functionalization of Amines with Aryl Halides by Nickel-Photoredox Catalysis. Chem. Sci 2016, 7, 7002–7006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (234).Shields BJ; Doyle AG Direct C(sp3)-H Cross Coupling Enabled by Catalytic Generation of Chlorine Radicals. J. Am. Chem. Soc 2016, 138, 12719–12722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (235).Nielsen MK; Shields BJ; Liu J; Williams MJ; Zacuto MJ; Doyle AG Mild, Redox-Neutral Formylation of Aryl Chlorides through the Photocatalytic Generation of Chlorine Radicals. Angew. Chem., Int. Ed 2017, 56, 7191–7194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (236).Rohe S; Morris AO; McCallum T; Barriault L Hydrogen Atom Transfer Reactions via Photoredox Catalyzed Chlorine Atom Generation. Angew. Chem., Int. Ed 2018, 57, 15664–15669. [DOI] [PubMed] [Google Scholar]
- (237).Jiang J; Ramozzi R; Moteki S; Usui A; Maruoka K; Morokuma K Mechanism of Metal-Free C-H Activation of Branched Aldehydes and Acylation of Alkenes Using Hypervalent Iodine Compound: A Theoretical Study. J. Org. Chem 2015, 80, 9264–9271. [DOI] [PubMed] [Google Scholar]
- (238).Loh YY; Nagao K; Hoover AJ; Hesk D; Rivera NR; Colletti SL; Davies IW; MacMillan DWC Photoredox-Catalyzed Deuteration and Tritiation of Pharmaceutical Compounds. Science 2017, 358, 1182–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (239).Vasilopoulos A; Krska SW; Stahl SS C(sp3)-H Methylation Enabled by Peroxide Photosensitization and Ni-Mediated Radical Coupling. Science 2021, 372, 398–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (240).Ohmatsu K; Suzuki R; Furukawa Y; Sato M; Ooi T Zwitterionic 1,2,3-Triazolium Amidate as a Catalyst for Photoinduced Hydrogen-Atom Transfer Radical Alkylation. ACS Catal. 2020, 10, 2627–2632. [Google Scholar]
- (241).Joe CL; Doyle AG Direct Acylation of C(Sp3)-H Bonds Enabled by Nickel and Photoredox Catalysis. Angew. Chem., Int. Ed 2016, 55, 4040–4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (242).Sun Z; Kumagai N; Shibasaki M Photocatalytic α-Acylation of Ethers. Org. Lett 2017, 19, 3727–3730. [DOI] [PubMed] [Google Scholar]
- (243).Shu X; Huan L; Huang Q; Huo H Direct Enantioselective C(sp3)-H Acylation for the Synthesis of α-Amino Ketones. J. Am. Chem. Soc 2020, 142, 19058–19064. [DOI] [PubMed] [Google Scholar]
- (244).Šakić D; Zipse H Radical Stability as a Guideline in C-H Amination Reactions. Adv. Synth. Catal 2016, 358, 3983–3991. [Google Scholar]
- (245).Jeffrey JL; Terrett JA; MacMillan DWC O-H Hydrogen Bonding Promotes H-Atom Transfer from α C-H Bonds for C-Alkylation of Alcohols. Science 2015, 349, 1532–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (246).Sakai K; Oisaki K; Kanai M Identification of Bond-Weakening Spirosilane Catalyst for Photoredox α-C-H Alkylation of Alcohols. Adv. Synth. Catal 2020, 362, 337–343. [Google Scholar]
- (247).Dimakos V; Su HY; Garrett GE; Taylor MS Site-Selective and Stereoselective C-H Alkylations of Carbohydrates via Combined Diarylborinic Acid and Photoredox Catalysis. J. Am. Chem. Soc 2019, 141, 5149–5153. [DOI] [PubMed] [Google Scholar]
- (248).Wan ICS; Witte MD; Minnaard AJ Site-Selective Carbon-Carbon Bond Formation in Unprotected Monosaccharides Using Photoredox Catalysis. Chem. Commun 2017, 53, 4926–4929. [DOI] [PubMed] [Google Scholar]
- (249).Deng H-P; Fan X-Z; Chen Z-H; Xu Q-H; Wu J Photoinduced Nickel-Catalyzed Chemo- and Regioselective Hydroalkylation of Internal Alkynes with Ether and Amide α-Hetero C(Sp3)-H Bonds. J. Am. Chem. Soc 2017, 139, 13579–13584. [DOI] [PubMed] [Google Scholar]
- (250).Maity B; Zhu C; Yue H; Huang L; Harb M; Minenkov Y; Rueping M; Cavallo L Mechanistic Insight into the Photoredox-Nickel-HAT Triple Catalyzed Arylation and Alkylation of α-Amino Csp3-H Bonds. J. Am. Chem. Soc 2020, 142, 16942–16952. [DOI] [PubMed] [Google Scholar]
- (251).Xie J; Yu J; Rudolph M; Rominger F; Hashmi ASK Monofluoroalkenylation of Dimethylamino Compounds through Radical-Radical Cross-Coupling. Angew. Chem., Int. Ed 2016, 55, 9416–9421. [DOI] [PubMed] [Google Scholar]
- (252).Wang J; Huang B; Gao Y; Yang C; Xia W Direct C-H Multifluoroarylation of Ethers through Hydrogen Atom Transfer Using Photoredox Catalysis. J. Org. Chem 2019, 84, 6895–6903. [DOI] [PubMed] [Google Scholar]
- (253).Ye J; Kalvet I; Schoenebeck F; Rovis T Direct α-Alkylation of Primary Aliphatic Amines Enabled by CO2 and Electrostatics. Nat. Chem 2018, 10, 1037–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (254).Pagire SK; Reiser O Tandem Cyclisation of Vinyl Radicals: A Sustainable Approach to Indolines Utilizing Visible-Light Photoredox Catalysis. Green Chem. 2017, 19, 1721–1725. [Google Scholar]
- (255).Ashley MA; Yamauchi C; Chu JCK; Otsuka S; Yorimitsu H; Rovis T Photoredox-Catalyzed Site-Selective α-C(sp3)-H Alkylation of Primary Amine Derivatives. Angew. Chem., Int. Ed 2019, 58, 4002–4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (256).Wakaki T; Sakai K; Enomoto T; Kondo M; Masaoka S; Oisaki K; Kanai M C(sp3)-H Cyanation Promoted by Visible-Light Photoredox/Phosphate Hybrid Catalysis. Chem. - Eur. J 2018, 24, 8051–8055. [DOI] [PubMed] [Google Scholar]
- (257).Meanwell M; Lehmann J; Eichenberger M; Martin RE; Britton R Synthesis of Acyl Fluorides via Photocatalytic Fluorination of Aldehydic C-H Bonds. Chem. Commun 2018, 54, 9985–9988. [DOI] [PubMed] [Google Scholar]
- (258).Ide T; Barham JP; Fujita M; Kawato Y; Egami H; Hamashima Y Regio- and Chemoselective Csp3-H Arylation of Benzylamines by Single Electron Transfer/Hydrogen Atom Transfer Synergistic Catalysis. Chem. Sci 2018, 9, 8453–8460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (259).Rand AW; Yin H; Xu L; Giacoboni J; Martin-Montero R; Romano C; Montgomery J; Martin R Dual Catalytic Platform for Enabling Sp3 α C-H Arylation and Alkylation of Benzamides. ACS Catal. 2020, 10, 4671–4676. [Google Scholar]
- (260).Wang C; Qin J; Shen X; Riedel R; Harms K; Meggers E Asymmetric Radical-Radical Cross-Coupling through Visible-Light-Activated Iridium Catalysis. Angew. Chem., Int. Ed 2016, 55, 685–688. [DOI] [PubMed] [Google Scholar]
- (261).Uraguchi D; Kinoshita N; Kizu T; Ooi T Synergistic Catalysis of Ionic Brønsted Acid and Photosensitizer for a Redox Neutral Asymmetric α-Coupling of N-Arylaminomethanes with Aldimines. J. Am. Chem. Soc 2015, 137, 13768–13771. [DOI] [PubMed] [Google Scholar]
- (262).Thullen SM; Rovis T A Mild Hydroaminoalkylation of Conjugated Dienes Using a Unified Cobalt and Photoredox Catalytic System. J. Am. Chem. Soc 2017, 139, 15504–15508. [DOI] [PubMed] [Google Scholar]
- (263).Zheng J; Breit B Regiodivergent Hydroaminoalkylation of Alkynes and Allenes by a Combined Rhodium and Photoredox Catalytic System. Angew. Chem., Int. Ed 2019, 58, 3392–3397. [DOI] [PubMed] [Google Scholar]
- (264).Xuan J; Zeng T-T; Feng Z-J; Deng Q-H; Chen J-R; Lu L-Q; Xiao W-J; Alper H Redox-Neutral α-Allylation of Amines by Combining Palladium Catalysis and Visible-Light Photoredox Catalysis. Angew. Chem., Int. Ed 2015, 54, 1625–1628. [DOI] [PubMed] [Google Scholar]
- (265).Feng Z; Zeng T; Xuan J; Liu Y; Lu L; Xiao W-J C-H Allylation of N-Aryl-Tetrahydroisoquinolines by Merging Photoredox Catalysis with Iodide Catalysis. Sci. China: Chem 2016, 59, 171–174. [Google Scholar]
- (266).Zhang H-H; Zhao J-J; Yu S Enantioselective α-Allylation of Anilines Enabled by a Combined Palladium and Photoredox Catalytic System. ACS Catal. 2020, 10, 4710–4716. [Google Scholar]
- (267).Leitch JA; Rossolini T; Rogova T; Maitland JAP; Dixon DJ α-Amino Radicals via Photocatalytic Single-Electron Reduction of Imine Derivatives. ACS Catal. 2020, 10, 2009–2025. [Google Scholar]
- (268).Beeson TD; Mastracchio A; Hong J-B; Ashton K; MacMillan DWC Enantioselective Organocatalysis Using SOMO Activation. Science 2007, 316, 582–585. [PubMed] [Google Scholar]
- (269).Jang H-Y; Hong J-B; MacMillan DWC Enantioselective Organocatalytic Singly Occupied Molecular Orbital Activation: The Enantioselective α-Enolation of Aldehydes. J. Am. Chem. Soc 2007, 129, 7004–7005. [DOI] [PubMed] [Google Scholar]
- (270).Kim H; MacMillan DWC Enantioselective Organo-SOMO Catalysis: The α-Vinylation of Aldehydes. J. Am. Chem. Soc 2008, 130, 398–399. [DOI] [PubMed] [Google Scholar]
- (271).Nicewicz DA; MacMillan DWC Merging Photoredox Catalysis with Organocatalysis: The Direct Asymmetric Alkylation of Aldehydes. Science 2008, 322, 77–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (272).Zhu Y; Zhang L; Luo S Asymmetric α-Photoalkylation of β-Ketocarbonyls by Primary Amine Catalysis: Facile Access to Acyclic All-Carbon Quaternary Stereocenters. J. Am. Chem. Soc 2014, 136, 14642–14645. [DOI] [PubMed] [Google Scholar]
- (273).Nagib DA; Scott ME; MacMillan DWC Enantioselective α-Trifluoromethylation of Aldehydes via Photoredox Organocatalysis. J. Am. Chem. Soc 2009, 131, 10875–10877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (274).Shih H-W; Vander Wal MN; Grange RL; MacMillan DWC Enantioselective α-Benzylation of Aldehydes via Photoredox Organocatalysis. J. Am. Chem. Soc 2010, 132, 13600–13603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (275).Welin ER; Warkentin AA; Conrad JC; MacMillan DWC Enantioselective α-Alkylation of Aldehydes by Photoredox Organocatalysis: Rapid Access to Pharmacophore Fragments from β-Cyanoaldehydes. Angew. Chem., Int. Ed 2015, 54, 9668–9672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (276).Li M; Sang Y; Xue X-S; Cheng J-P Origin of Stereocontrol in Photoredox Organocatalysis of Asymmetric α-Functionalizations of Aldehydes. J. Org. Chem 2018, 83, 3333–3338. [DOI] [PubMed] [Google Scholar]
- (277).Nacsa ED; MacMillan DWC Spin-Center Shift-Enabled Direct Enantioselective α-Benzylation of Aldehydes with Alcohols. J. Am. Chem. Soc 2018, 140, 3322–3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (278).Arceo E; Jurberg ID;Álvarez-Fernández A; Melchiorre P Photochemical Activity of a Key Donor-Acceptor Complex Can Drive Stereoselective Catalytic α-Alkylation of Aldehydes. Nat. Chem 2013, 5, 750–756. [DOI] [PubMed] [Google Scholar]
- (279).Arceo E; Bahamonde A; Bergonzini G; Melchiorre P Enantioselective Direct α-Alkylation of Cyclic Ketones by Means of Photo-Organocatalysis. Chem. Sci 2014, 5, 2438–2442. [Google Scholar]
- (280).Silvi M; Arceo E; Jurberg ID; Cassani C; Melchiorre P Enantioselective Organocatalytic Alkylation of Aldehydes and Enals Driven by the Direct Photoexcitation of Enamines. J. Am. Chem. Soc 2015, 137, 6120–6123. [DOI] [PubMed] [Google Scholar]
- (281).Bahamonde A; Melchiorre P Mechanism of the Stereoselective α-Alkylation of Aldehydes Driven by the Photochemical Activity of Enamines. J. Am. Chem. Soc 2016, 138, 8019–8030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (282).Huo H; Shen X; Wang C; Zhang L; Röse P; Chen L-A; Harms K; Marsch M; Hilt G; Meggers E Asymmetric Photoredox Transition-Metal Catalysis Activated by Visible Light. Nature 2014, 515, 100–103. [DOI] [PubMed] [Google Scholar]
- (283).Huo H; Wang C; Harms K; Meggers E Enantioselective, Catalytic Trichloromethylation through Visible-Light-Activated Photoredox Catalysis with a Chiral Iridium Complex. J. Am. Chem. Soc 2015, 137, 9551–9554. [DOI] [PubMed] [Google Scholar]
- (284).Huang X; Webster RD; Harms K; Meggers E Asymmetric Catalysis with Organic Azides and Diazo Compounds Initiated by Photoinduced Electron Transfer. J. Am. Chem. Soc 2016, 138, 12636–12642. [DOI] [PubMed] [Google Scholar]
- (285).Liang H; Xu G-Q; Feng Z-T; Wang Z-Y; Xu P-F Dual Catalytic Switchable Divergent Synthesis: An Asymmetric Visible-Light Photocatalytic Approach to Fluorine-Containing γ-Keto Acid Frameworks. J. Org. Chem 2019, 84, 60–72. [DOI] [PubMed] [Google Scholar]
- (286).Tan Y; Yuan W; Gong L; Meggers E Aerobic Asymmetric Dehydrogenative Cross-Coupling between Two C-H Groups Catalyzed by a Chiral-at-Metal Rhodium Complex. Angew. Chem., Int. Ed 2015, 54, 13045–13048. [DOI] [PubMed] [Google Scholar]
- (287).Capacci AG; Malinowski JT; McAlpine NJ; Kuhne J; MacMillan DWC Direct, Enantioselective α-Alkylation of Aldehydes Using Simple Olefins. Nat. Chem 2017, 9, 1073–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (288).Larionov E; Mastandrea MM; Pericàs MA Asymmetric Visible-Light Photoredox Cross-Dehydrogenative Coupling of Aldehydes with Xanthenes. ACS Catal. 2017, 7, 7008–7013. [Google Scholar]
- (289).Pham PV; Nagib DA; MacMillan DWC Photoredox Catalysis: A Mild, Operationally Simple Approach to the Synthesis of α-Trifluoromethyl Carbonyl Compounds. Angew. Chem 2011, 123, 6243–6246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (290).Koike T; Akita M Photoinduced Oxyamination of Enamines and Aldehydes with TEMPO Catalyzed by [Ru(bpy)3]2+. Chem. Lett 2009, 38, 166–167. [Google Scholar]
- (291).Sibi MP; Hasegawa M Organocatalysis in Radical Chemistry. Enantioselective α-Oxyamination of Aldehydes. J. Am. Chem. Soc 2007, 129, 4124–4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (292).Rybicka-Jasińska K; Ciszewski ŁW; Gryko D Photocatalytic Reaction of Diazo Compounds with Aldehydes. Adv. Synth. Catal 2016, 358, 1671–1678. [Google Scholar]
- (293).Franchino A; Rinaldi A; Dixon DJ α-Alkylation of Ketimines Using Visible Light Photoredox Catalysis. RSC Adv. 2017, 7, 43655–43659. [Google Scholar]
- (294).Daniel M; Fensterbank L; Goddard J-P; Ollivier C Visible-Light Photocatalytic Oxidation of 1,3-Dicarbonyl Compounds and Carbon-Carbon Bond Formation. Org. Chem. Front 2014, 1, 551–555. [Google Scholar]
- (295).Pirnot MT; Rankic DA; Martin DBC; MacMillan DWC Photoredox Activation for the Direct β-Arylation of Ketones and Aldehydes. Science 2013, 339, 1593–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (296).Terrett JA; Clift MD; MacMillan DWC Direct β-Alkylation of Aldehydes via Photoredox Organocatalysis. J. Am. Chem. Soc 2014, 136, 6858–6861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (297).Petronijević FR; Nappi M; MacMillan DWC Direct β-Functionalization of Cyclic Ketones with Aryl Ketones via the Merger of Photoredox and Organocatalysis. J. Am. Chem. Soc 2013, 135, 18323–18326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (298).Ma J; Rosales AR; Huang X; Harms K; Riedel R; Wiest O; Meggers E Visible-Light-Activated Asymmetric β-C-H Functionalization of Acceptor-Substituted Ketones with 1,2-Dicarbonyl Compounds. J. Am. Chem. Soc 2017, 139, 17245–17248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (299).Liu J; Ding W; Zhou Q-Q; Liu D; Lu L-Q; Xiao W-J Enantioselective Di-/Perfluoroalkylation of β-Ketoesters Enabled by Cooperative Photoredox/Nickel Catalysis. Org. Lett 2018, 20, 461–464. [DOI] [PubMed] [Google Scholar]
- (300).Czyz ML; Lupton DW; Polyzos A Auxiliary-Directed C(Sp3)-H Arylation by Synergistic Photoredox and Palladium Catalysis. Chem. - Eur. J 2017, 23, 14450–14453. [DOI] [PubMed] [Google Scholar]
- (301).Iqbal N; Choi S; You Y; Cho EJ Aerobic Oxidation of Aldehydes by Visible Light Photocatalysis. Tetrahedron Lett. 2013, 54, 6222–6225. [Google Scholar]
- (302).Dinda M; Bose C; Ghosh T; Maity S Cross Dehydrogenative Coupling (CDC) of Aldehydes with N-Hydroxyimides by Visible Light Photoredox Catalysis. RSC Adv. 2015, 5, 44928–44932. [Google Scholar]
- (303).Iqbal N; Cho EJ Visible-Light-Mediated Synthesis of Amides from Aldehydes and Amines via in Situ Acid Chloride Formation. J. Org. Chem 2016, 81, 1905–1911. [DOI] [PubMed] [Google Scholar]
- (304).Mukherjee S; Patra T; Glorius F Cooperative Catalysis: A Strategy To Synthesize Trifluoromethyl-Thioesters from Aldehydes. ACS Catal. 2018, 8, 5842–5846. [Google Scholar]
- (305).Zhang X; MacMillan DWC Direct Aldehyde C-H Arylation and Alkylation via the Combination of Nickel, Hydrogen Atom Transfer, and Photoredox Catalysis. J. Am. Chem. Soc 2017, 139, 11353–11356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (306).Wang L; Wang T; Cheng G-J; Li X; Wei J-J; Guo B; Zheng C; Chen G; Ran C; Zheng C Direct C-H Arylation of Aldehydes by Merging Photocatalyzed Hydrogen Atom Transfer with Palladium Catalysis. ACS Catal. 2020, 10, 7543–7551. [Google Scholar]
- (307).Vu MD; Das M; Liu X-W Direct Aldehyde Csp2-H Functionalization through Visible-Light-Mediated Photoredox Catalysis. Chem. - Eur. J 2017, 23, 15899–15902. [DOI] [PubMed] [Google Scholar]
- (308).Mukherjee S; Garza-Sanchez RA; Tlahuext-Aca A; Glorius F Alkynylation of C (O)-H Bonds Enabled by Photoredox-Mediated Hydrogen-Atom Transfer. Angew. Chem., Int. Ed 2017, 56, 14723–14726. [DOI] [PubMed] [Google Scholar]
- (309).Xu P; Wang G; Zhu Y; Li W; Cheng Y; Li S; Zhu C Visible-Light Photoredox-Catalyzed C-H Difluoroalkylation of Hydrazones through an Aminyl Radical/Polar Mechanism. Angew. Chem., Int. Ed 2016, 55, 2939–2943. [DOI] [PubMed] [Google Scholar]
- (310).Cheng J; Xu P; Li W; Cheng Y; Zhu C The Functionalization of a Cascade of C(Sp2)-H/C(Sp3)-H Bonds: Synthesis of Fused Dihydropyrazoles via Visible-Light Photoredox Catalysis. Chem. Commun 2016, 52, 11901–11904. [DOI] [PubMed] [Google Scholar]
- (311).Xie J; Zhang T; Chen F; Mehrkens N; Rominger F; Rudolph M; Hashmi ASK Gold-Catalyzed Highly Selective Photoredox C(Sp2)-H Difluoroalkylation and Perfluoroalkylation of Hydrazones. Angew. Chem., Int. Ed 2016, 55, 2934–2938. [DOI] [PubMed] [Google Scholar]
- (312).Khursan SL; Mikhailov DA; Yanborisov VM; Borisov DI AM1 Calculations of Bond Dissociation Energies. Allylic and Benzylic C-H Bonds. React. Kinet. Catal. Lett 1997, 61, 91–95. [Google Scholar]
- (313).Qvortrup K; Rankic DA; MacMillan DWC A General Strategy for Organocatalytic Activation of C-H Bonds via Photoredox Catalysis: Direct Arylation of Benzylic Ethers. J. Am. Chem. Soc 2014, 136, 626–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (314).Hager D; MacMillan DWC Activation of C-H Bonds via the Merger of Photoredox and Organocatalysis: A Coupling of Benzylic Ethers with Schiff Bases. J. Am. Chem. Soc 2014, 136, 16986–16989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (315).Cuthbertson JD; MacMillan DWC The Direct Arylation of Allylic Sp 3 C-H Bonds via Organic and Photoredox Catalysis. Nature 2015, 519, 74–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (316).Vu MD; Das M; Guo A; Ang Z-E; Dōkić M; Soo HS; Liu X-W Visible-Light Photoredox Enables Ketone Carbonyl Alkylation for Easy Access to Tertiary Alcohols. ACS Catal. 2019, 9, 9009–9014. [Google Scholar]
- (317).Nakashima T; Ohmatsu K; Ooi T Mannich-Type Allylic C-H Functionalization of Enol Silyl Ethers under Photoredox-Thiol Hybrid Catalysis. Org. Biomol. Chem 2021, 19, 141. [DOI] [PubMed] [Google Scholar]
- (318).Jia J; Kancherla R; Rueping M; Huang L Allylic C(Sp3)-H Alkylation via Synergistic Organo- and Photoredox Catalyzed Radical Addition to Imines. Chem. Sci 2020, 11, 4954–4959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (319).Tanaka H; Sakai K; Kawamura A; Oisaki K; Kanai M Sulfonamides as New Hydrogen Atom Transfer (HAT) Catalysts for Photoredox Allylic and Benzylic C-H Arylations. Chem. Commun 2018, 54, 3215–3218. [DOI] [PubMed] [Google Scholar]
- (320).Zhong L-J; Wang H-Y; Ouyang X-H; Li J-H; An D-L Benzylic C-H Heteroarylation of N -(Benzyloxy)Phthalimides with Cyanopyridines Enabled by Photoredox 1,2-Hydrogen Atom Transfer. Chem. Commun 2020, 56, 8671–8674. [DOI] [PubMed] [Google Scholar]
- (321).Wang H; Zhang D; Bolm C Sulfoximidations of Benzylic C-H Bonds by Photocatalysis. Angew. Chem., Int. Ed 2018, 57, 5863–5866. [DOI] [PubMed] [Google Scholar]
- (322).Gong X; Chen J; Lai L; Cheng J; Sun J; Wu J Benzylic C(Sp3)-H Bond Sulfonylation of 4-Methylphenols with the Insertion of Sulfur Dioxide under Photocatalysis. Chem. Commun 2018, 54, 11172–11175. [DOI] [PubMed] [Google Scholar]
- (323).Nodwell MB; Bagai A; Halperin SD; Martin RE; Knust H; Britton R Direct Photocatalytic Fluorination of Benzylic C-H Bonds with N-Fluorobenzenesulfonimide. Chem. Commun 2015, 51, 11783–11786. [DOI] [PubMed] [Google Scholar]
- (324).Pandey G; Laha R; Singh D Benzylic C(Sp3)-H Functionalization for C-N and C-O Bond Formation via Visible Light Photoredox Catalysis. J. Org. Chem 2016, 81, 7161–7171. [DOI] [PubMed] [Google Scholar]
- (325).Rabet PTG; Fumagalli G; Boyd S; Greaney MF Benzylic C-H Azidation Using the Zhdankin Reagent and a Copper Photoredox Catalyst. Org. Lett 2016, 18, 1646–1649. [DOI] [PubMed] [Google Scholar]
- (326).Pandey G; Laha R; Mondal PK Heterocyclization Involving Benzylic C(Sp3)-H Functionalization Enabled by Visible Light Photoredox Catalysis. Chem. Commun 2019, 55, 9689–9692. [DOI] [PubMed] [Google Scholar]
- (327).Lee BJ; DeGlopper KS; Yoon TP Site-Selective Alkoxylation of Benzylic C-H Bonds by Photoredox Catalysis. Angew. Chem., Int. Ed 2020, 59, 197–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (328).Kawasaki T; Ishida N; Murakami M Dehydrogenative Coupling of Benzylic and Aldehydic C-H Bonds. J. Am. Chem. Soc 2020, 142, 3366–3370. [DOI] [PubMed] [Google Scholar]
- (329).Lei Z; Banerjee A; Kusevska E; Rizzo E; Liu P; Ngai M-Y β-Selective Aroylation of Activated Alkenes by Photoredox Catalysis. Angew. Chem., Int. Ed 2019, 58, 7318–7323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (330).Kim J; Kang B; Hong SH Direct Allylic C(Sp3)-H Thiolation with Disulfides via Visible Light Photoredox Catalysis. ACS Catal. 2020, 10, 6013–6022. [Google Scholar]
- (331).Xia J-D; Deng G-B; Zhou M-B; Liu W; Xie P; Li J-H Reusable Visible Light Photoredox Catalysts; Catalyzed Benzylic C(Sp3)-H Functionalization/Carbocyclization Reactions. Synlett 2012, 23, 2707–2713. [Google Scholar]
- (332).Schwarz JL; Schäfers F; Tlahuext-Aca A; Lückemeier L; Glorius F Diastereoselective Allylation of Aldehydes by Dual Photoredox and Chromium Catalysis. J. Am. Chem. Soc 2018, 140, 12705–12709. [DOI] [PubMed] [Google Scholar]
- (333).Schäfers F; Quach L; Schwarz JL; Saladrigas M; Daniliuc CG; Glorius F Direct Access to Monoprotected Homoallylic 1,2-Diols via Dual Chromium/Photoredox Catalysis. ACS Catal. 2020, 10, 11841–11847. [Google Scholar]
- (334).Ohmatsu K; Nakashima T; Sato M; Ooi T Direct Allylic C-H Alkylation of Enol Silyl Ethers Enabled by Photoredox-Brønsted Base Hybrid Catalysis. Nat. Commun 2019, 10, 2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (335).Blanksby SJ; Ellison GB Bond Dissociation Energies of Organic Molecules. Acc. Chem. Res 2003, 36, 255–263. [DOI] [PubMed] [Google Scholar]
- (336).Liu W; Huang X; Cheng M-J; Nielsen RJ; Goddard WA; Groves JT Oxidative Aliphatic C-H Fluorination with Fluoride Ion Catalyzed by a Manganese Porphyrin. Science 2012, 337, 1322–1325. [DOI] [PubMed] [Google Scholar]
- (337).Bloom S; Pitts CR; Miller DC; Haselton N; Holl MG; Urheim E; Lectka T A Polycomponent Metal-Catalyzed Aliphatic, Allylic, and Benzylic Fluorination. Angew. Chem., Int. Ed 2012, 51, 10580–10583. [DOI] [PubMed] [Google Scholar]
- (338).Amaoka Y; Nagatomo M; Inoue M Metal-Free Fluorination of C(sp3)-H Bonds Using a Catalytic N-Oxyl Radical. Org. Lett 2013, 15, 2160–2163. [DOI] [PubMed] [Google Scholar]
- (339).Halperin SD; Fan H; Chang S; Martin RE; Britton R A Convenient Photocatalytic Fluorination of Unactivated C-H Bonds. Angew. Chem., Int. Ed 2014, 53, 4690–4693. [DOI] [PubMed] [Google Scholar]
- (340).Halperin SD; Kwon D; Holmes M; Regalado EL; Campeau L-C; DiRocco DA; Britton R Development of a Direct Photocatalytic C-H Fluorination for the Preparative Synthesis of Odanacatib. Org. Lett 2015, 17, 5200–5203. [DOI] [PubMed] [Google Scholar]
- (341).Nodwell MB; Yang H;Čolović M; Yuan Z; Merkens H; Martin RE; Bénard F; Schaffer P; Britton R 18F-Fluorination of Unactivated C-H Bonds in Branched Aliphatic Amino Acids: Direct Synthesis of Oncological Positron Emission Tomography Imaging Agents. J. Am. Chem. Soc 2017, 139, 3595–3598. [DOI] [PubMed] [Google Scholar]
- (342).Yuan Z; Yang H; Malik N;Čolović M; Weber DS; Wilson D; Bénard F; Martin RE; Warren JJ; Schaffer P; et al. Electrostatic Effects Accelerate Decatungstate-Catalyzed C-H Fluorination Using [18F]- and [19F]NFSI in Small Molecules and Peptide Mimics. ACS Catal. 2019, 9, 8276–8284. [Google Scholar]
- (343).West JG; Bedell TA; Sorensen EJ The Uranyl Cation as a Visible-Light Photocatalyst for C(sp3)-H Fluorination. Angew. Chem., Int. Ed 2016, 55, 8923–8927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (344).Yamada K; Fukuyama T; Fujii S; Ravelli D; Fagnoni M; Ryu I Cooperative Polar/Steric Strategy in Achieving Site-Selective Photocatalyzed C(sp3)-H Functionalization. Chem. - Eur. J 2017, 23, 8615–8618. [DOI] [PubMed] [Google Scholar]
- (345).Fukuyama T; Yamada K; Nishikawa T; Ravelli D; Fagnoni M; Ryu I Site-Selectivity in TBADT-Photocatalyzed C(Sp3)-H Functionalization of Saturated Alcohols and Alkanes. Chem. Lett 2018, 47, 207–209. [Google Scholar]
- (346).Fukuyama T; Nishikawa T; Ryu I Site-Selective C(Sp3)-H Functionalization of Fluorinated Alkanes Driven by Polar Effects Using a Tungstate Photocatalyst. Eur. J. Org. Chem 2020, 2020, 1424–1428. [Google Scholar]
- (347).Capaldo L; Merli D; Fagnoni M; Ravelli D Visible Light Uranyl Photocatalysis: Direct C-H to C-C Bond Conversion. ACS Catal. 2019, 9, 3054–3058. [Google Scholar]
- (348).Hu A; Guo J-J; Pan H; Zuo Z Selective Functionalization of Methane, Ethane, and Higher Alkanes by Cerium Photocatalysis. Science 2018, 361, 668–672. [DOI] [PubMed] [Google Scholar]
- (349).An Q; Wang Z; Chen Y; Wang X; Zhang K; Pan H; Liu W; Zuo Z Cerium-Catalyzed C-H Functionalizations of Alkanes Utilizing Alcohols as Hydrogen Atom Transfer Agents. J. Am. Chem. Soc 2020, 142, 6216–6226. [DOI] [PubMed] [Google Scholar]
- (350).Laudadio G; Deng Y; Wal K; van der Ravelli D; Nuño M; Fagnoni M; Guthrie D; Sun Y; Noël T C(Sp3)-H Functionalizations of Light Hydrocarbons Using Decatungstate Photocatalysis in Flow. Science 2020, 369, 92–96. [DOI] [PubMed] [Google Scholar]
- (351).Perry IB; Brewer TF; Sarver PJ; Schultz DM; DiRocco DA; MacMillan DWC Direct Arylation of Strong Aliphatic C-H Bonds. Nature 2018, 560, 70–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (352).Ackerman LKG; Martinez Alvarado JI; Doyle AG Direct C-C Bond Formation from Alkanes Using Ni-Photoredox Catalysis. J. Am. Chem. Soc 2018, 140, 14059–14063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (353).Sarver PJ; Bacauanu V; Schultz DM; DiRocco DA; Lam Y; Sherer EC; MacMillan DWC The Merger of Decatungstate and Copper Catalysis to Enable Aliphatic C(Sp3)-H Trifluoromethylation. Nat. Chem 2020, 12, 459–467. [DOI] [PubMed] [Google Scholar]
- (354).Ravelli D; Fagnoni M; Fukuyama T; Nishikawa T; Ryu I Site-Selective C-H Functionalization by Decatungstate Anion Photocatalysis: Synergistic Control by Polar and Steric Effects Expands the Reaction Scope. ACS Catal. 2018, 8, 701–713. [Google Scholar]
- (355).Yahata K; Sakurai S; Hori S; Yoshioka S; Kaneko Y; Hasegawa K; Akai S Coupling Reaction between Aldehydes and Non-Activated Hydrocarbons via the Reductive Radical-Polar Crossover Pathway. Org. Lett 2020, 22, 1199–1203. [DOI] [PubMed] [Google Scholar]
- (356).Dai Z-Y; Nong Z-S; Wang P-S Light-Mediated Asymmetric Aliphatic C-H Alkylation with Hydrogen Atom Transfer Catalyst and Chiral Phosphoric Acid. ACS Catal. 2020, 10, 4786–4790. [Google Scholar]
- (357).Huang C; Wang J-H; Qiao J; Fan X-W; Chen B; Tung C-H; Wu L-Z Direct Arylation of Unactivated Alkanes with Heteroarenes by Visible-Light Catalysis. J. Org. Chem 2019, 84, 12904–12912. [DOI] [PubMed] [Google Scholar]
- (358).Yang H-B; Feceu A; Martin DBC Catalyst-Controlled C-H Functionalization of Adamantanes Using Selective H-Atom Transfer. ACS Catal. 2019, 9, 5708–5715. [Google Scholar]
- (359).Kruppa GH; Beauchamp JL Energetics and Structure of the 1- and 2-Adamantyl Radicals and Their Corresponding Carbonium Ions by Photoelectron Spectroscopy. J. Am. Chem. Soc 1986, 108, 2162–2169. [DOI] [PubMed] [Google Scholar]
- (360).Weigel WK; Dang HT; Yang H-B; Martin DBC Synthesis of Amino-Diamondoid Pharmacophores via Photocatalytic C-H Aminoalkylation. Chem. Commun 2020, 56, 9699–9702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (361).Mukherjee S; Maji B; Tlahuext-Aca A; Glorius F Visible-Light-Promoted Activation of Unactivated C(sp3)-H Bonds and Their Selective Trifluoromethylthiolation. J. Am. Chem. Soc 2016, 138, 16200–16203. [DOI] [PubMed] [Google Scholar]
- (362).Chu JCK; Rovis T Complementary Strategies for Directed C(Sp3)-H Functionalization: A Comparison of Transition-Metal-Catalyzed Activation, Hydrogen Atom Transfer, and Carbene/Nitrene Transfer. Angew. Chem., Int. Ed 2018, 57, 62–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (363).Qin Q; Yu S Visible-Light-Promoted Remote C(Sp3)-H Amidation and Chlorination. Org. Lett 2015, 17, 1894–1897. [DOI] [PubMed] [Google Scholar]
- (364).Zhang J; Li Y; Zhang F; Hu C; Chen Y Generation of Alkoxyl Radicals by Photoredox Catalysis Enables Selective C(Sp3)-H Functionalization under Mild Reaction Conditions. Angew. Chem., Int. Ed 2016, 55, 1872–1875. [DOI] [PubMed] [Google Scholar]
- (365).Bordwell FG; Zhang S; Zhang X-M; Liu W-Z Homolytic Bond Dissociation Enthalpies of the Acidic H-A Bonds Caused by Proximate Substituents in Sets of Methyl Ketones, Carboxylic Esters, and Carboxamides Related to Changes in Ground State Energies. J. Am. Chem. Soc 1995, 117, 7092–7096. [Google Scholar]
- (366).Bordwell FG; Ji GZ Effects of Structural Changes on Acidities and Homolytic Bond Dissociation Energies of the Hydrogen-Nitrogen Bonds in Amidines, Carboxamides, and Thiocarboxamides. J. Am. Chem. Soc 1991, 113, 8398–8401. [Google Scholar]
- (367).Chen D-F; Chu JCK; Rovis T Directed γ-C(sp3)-H Alkylation of Carboxylic Acid Derivatives through Visible Light Photoredox Catalysis. J. Am. Chem. Soc 2017, 139, 14897–14900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (368).Jiang H; Studer A α-Aminoxy-Acid-Auxiliary-Enabled Intermolecular Radical γ-C(sp3)-H Functionalization of Ketones. Angew. Chem., Int. Ed 2018, 57, 1692–1696. [DOI] [PubMed] [Google Scholar]
- (369).Xu B; Tambar UK Remote Allylation of Unactivated C(Sp3)-H Bonds Triggered by Photogenerated Amidyl Radicals. ACS Catal. 2019, 9, 4627–4631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (370).Shen X; Zhao J-J; Yu S Photoredox-Catalyzed Intermolecular Remote C-H and C-C Vinylation via Iminyl Radicals. Org. Lett 2018, 20, 5523–5527. [DOI] [PubMed] [Google Scholar]
- (371).Wang C; Harms K; Meggers E Catalytic Asymmetric C -H Functionalization under Photoredox Conditions by Radical Translocation and Stereocontrolled Alkene Addition. Angew. Chem., Int. Ed 2016, 55, 13495–13498. [DOI] [PubMed] [Google Scholar]
- (372).Yuan W; Zhou Z; Gong L; Meggers E Asymmetric Alkylation of Remote C(Sp3)-H Bonds by Combining Proton-Coupled Electron Transfer with Chiral Lewis Acid Catalysis. Chem. Commun 2017, 53, 8964–8967. [DOI] [PubMed] [Google Scholar]
- (373).Huo H; Harms K; Meggers E Catalytic, Enantioselective Addition of Alkyl Radicals to Alkenes via Visible-Light-Activated Photoredox Catalysis with a Chiral Rhodium Complex. J. Am. Chem. Soc 2016, 138, 6936–6939. [DOI] [PubMed] [Google Scholar]
- (374).Nakafuku KM; Zhang Z; Wappes EA; Stateman LM; Chen AD; Nagib DA Enantioselective Radical C-H Amination for the Synthesis of β-Amino Alcohols. Nat. Chem 2020, 12, 697–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (375).Chen H; Jin W; Yu S Enantioselective Remote C(Sp3)-H Cyanation via Dual Photoredox and Copper Catalysis. Org. Lett 2020, 22, 5910–5914. [DOI] [PubMed] [Google Scholar]
- (376).Cheng Z; Chen P; Liu G Enantioselective Cyanation of Remote C—H Bonds via Cooperative Photoredox and Copper Catalysis. Huaxue Xuebao 2019, 77, 856–860. [Google Scholar]
- (377).Bao X; Wang Q; Zhu J Dual Photoredox/Copper Catalysis for the Remote C(Sp3)-H Functionalization of Alcohols and Alkyl Halides by N-Alkoxypyridinium Salts. Angew. Chem., Int. Ed 2019, 58, 2139–2143. [DOI] [PubMed] [Google Scholar]
- (378).Hari DP; Schroll P; König B Metal-Free, Visible-Light-Mediated Direct C-H Arylation of Heteroarenes with Aryl Diazonium Salts. J. Am. Chem. Soc 2012, 134, 2958–2961. [DOI] [PubMed] [Google Scholar]
- (379).Xiao T; Dong X; Tang Y; Zhou L Phenanthrene Synthesis by Eosin Y-Catalyzed, Visible Light-Induced [4 + 2] Benzannulation of Biaryldiazonium Salts with Alkynes. Adv. Synth. Catal 2012, 354, 3195–3199. [Google Scholar]
- (380).Martinez-Haya R; Miranda MA; Marin ML Metal-Free Photocatalytic Reductive Dehalogenation Using Visible-Light: A Time-Resolved Mechanistic Study. Eur. J. Org. Chem 2017, 2017, 2164–2169. [Google Scholar]
- (381).Matsubara R; Yabuta T; Md Idros U; Hayashi M; Ema F; Kobori Y; Sakata K UVA- and Visible-Light-Mediated Generation of Carbon Radicals from Organochlorides Using Nonmetal Photocatalyst. J. Org. Chem 2018, 83, 9381–9390. [DOI] [PubMed] [Google Scholar]
- (382).Gu L; Jin C; Liu J; Ding H; Fan B Transition-Metal-Free, Visible-Light Induced Cyclization of Arylsulfonyl Chlorides with 2-Isocyanobiphenyls to Produce Phenanthridines. Chem. Commun 2014, 50, 4643–4645. [DOI] [PubMed] [Google Scholar]
- (383).Cui L; Matusaki Y; Tada N; Miura T; Uno B; Itoh A Metal-Free Direct C-H Perfluoroalkylation of Arenes and Heteroarenes Using a Photoredox Organocatalyst. Adv. Synth. Catal 2013, 355, 2203–2207. [Google Scholar]
- (384).Zhou C; Li P; Zhu X; Wang L Merging Photoredox with Palladium Catalysis: Decarboxylative Ortho-Acylation of Acetanilides with α-Oxocarboxylic Acids under Mild Reaction Conditions. Org. Lett 2015, 17, 6198–6201. [DOI] [PubMed] [Google Scholar]
- (385).Bao P; Liu F; Lv Y; Yue H; Li J-S; Wei W Visible-Light-Promoted Acridine Red Catalyzed Aerobic Oxidative Decarboxylative Acylation of α-Oxo-Carboxylic Acids with Quinoxalin-2(1H)-Ones. Org. Chem. Front 2020, 7, 492–498. [Google Scholar]
- (386).Cao H; Jiang H; Feng H; Kwan JMC; Liu X; Wu J Photo-Induced Decarboxylative Heck-Type Coupling of Unactivated Aliphatic Acids and Terminal Alkenes in the Absence of Sacrificial Hydrogen Acceptors. J. Am. Chem. Soc 2018, 140, 16360–16367. [DOI] [PubMed] [Google Scholar]
- (387).Aukland MH;Šiaučiulis M; West A; Perry GJP; Procter DJ Metal-Free Photoredox-Catalysed Formal C-H/C-H Coupling of Arenes Enabled by Interrupted Pummerer Activation. Nat. Chem 2020, 3, 163–169. [Google Scholar]
- (388).Xiong T; Zhang Q New Amination Strategies Based on Nitrogen-Centered Radical Chemistry. Chem. Soc. Rev 2016, 45, 3069–3087. [DOI] [PubMed] [Google Scholar]
- (389).Tripathi CB; Ohtani T; Corbett MT; Ooi T Photoredox Ketone Catalysis for the Direct C-H Imidation and Acyloxylation of Arenes. Chem. Sci 2017, 8, 5622–5627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (390).Yamaguchi T; Yamaguchi E; Itoh A Cross-Dehydrogenative C-H Amination of Indoles under Aerobic Photo-Oxidative Conditions. Org. Lett 2017, 19, 1282–1285. [DOI] [PubMed] [Google Scholar]
- (391).Liu X; Qing Z; Cheng P; Zheng X; Zeng J; Xie H Metal-Free Photoredox Catalyzed Cyclization of O-(2,4-Dinitrophenyl)-Oximes to Phenanthridines. Molecules 2016, 21, 1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (392).Zhao Y; Huang B; Yang C; Li B; Gou B; Xia W Photocatalytic Cross-Dehydrogenative Amination Reactions between Phenols and Diarylamines. ACS Catal. 2017, 7, 2446–2451. [Google Scholar]
- (393).Wei W; Wang L; Bao P; Shao Y; Yue H; Yang D; Yang X; Zhao X; Wang H Metal-Free C(sp2)-H/N-H Cross-Dehydrogenative Coupling of Quinoxalinones with Aliphatic Amines under Visible-Light Photoredox Catalysis. Org. Lett 2018, 20, 7125–7130. [DOI] [PubMed] [Google Scholar]
- (394).He Y; Wu H; Toste FD A Dual Catalytic Strategy for Carbon-Phosphorus Cross-Coupling via Gold and Photoredox Catalysis. Chem. Sci 2015, 6, 1194–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (395).Xuan J; Zeng T-T; Chen J-R; Lu L-Q; Xiao W-J Room Temperature C-P Bond Formation Enabled by Merging Nickel Catalysis and Visible-Light-Induced Photoredox Catalysis. Chem. - Eur. J 2015, 21, 4962–4965. [DOI] [PubMed] [Google Scholar]
- (396).Luo K; Chen Y-Z; Yang W-C; Zhu J; Wu L Cross-Coupling Hydrogen Evolution by Visible Light Photocatalysis Toward C(Sp2)-P Formation: Metal-Free C-H Functionalization of Thiazole Derivatives with Diarylphosphine Oxides. Org. Lett 2016, 18, 452–455. [DOI] [PubMed] [Google Scholar]
- (397).Peng P; Peng L; Wang G; Wang F; Luo Y; Lei A Visible Light Mediated Aerobic Radical C-H Phosphorization toward Arylphosphonates. Org. Chem. Front 2016, 3, 749–752. [Google Scholar]
- (398).Qiao H; Sun S; Zhang Y; Zhu H; Yu X; Yang F; Wu Y; Li Z; Wu Y Merging Photoredox Catalysis with Transition Metal Catalysis: Site-Selective C4 or C5-H Phosphonation of 8-Aminoquinoline Amides. Org. Chem. Front 2017, 4, 1981–1986. [Google Scholar]
- (399).Su X; Yang F; Wu Y; Wu Y Direct C4-H Phosphonation of 8-Hydroxyquinoline Derivatives Employing Photoredox Catalysis and Silver Catalysis. Org. Biomol. Chem 2018, 16, 2753–2756. [DOI] [PubMed] [Google Scholar]
- (400).Ramirez NP; Bosque I; Gonzalez-Gomez JC Photocatalytic Dehydrogenative Lactonization of 2-Arylbenzoic Acids. Org. Lett 2015, 17, 4550–4553. [DOI] [PubMed] [Google Scholar]
- (401).Zhang M; Ruzi R; Li N; Xie J; Zhu C Photoredox and Cobalt Co-Catalyzed C(Sp 2)-H Functionalization/C-O Bond Formation for Synthesis of Lactones under Oxidant- and Acceptor-Free Conditions. Org. Chem. Front 2018, 5, 749–752. [Google Scholar]
- (402).Srivastava V; Singh PK; Singh PP Eosin Y Catalyzed Visible-Light-Promoted One-Pot Facile Synthesis of 1,3,4- Thiadiazole. Croat. Chem. Acta 2015, 88, 59–65. [Google Scholar]
- (403).Srivastava V; Singh PK; Singh PP Eosin Y Catalyzed Visible-Light-Promoted Aerobic Oxidative Cyclization of 2-Aminobenzothiazole. Croat. Chem. Acta 2015, 88, 227–233. [Google Scholar]
- (404).Guo W; Tan W; Zhao M; Tao K; Zheng L-Y; Wu Y; Chen D; Fan X-L Photocatalytic Direct C-S Bond Formation: Facile Access to 3-Sulfenylindoles via Metal-Free C-3 Sulfenylation of Indoles with Thiophenols. RSC Adv. 2017, 7, 37739–37742. [Google Scholar]
- (405).Rahaman R; Das S; Barman P Visible-Light-Induced Regioselective Sulfenylation of Imidazopyridines with Thiols under Transition Metal-Free Conditions. Green Chem. 2018, 20, 141–147. [Google Scholar]
- (406).Fan W; Yang Q; Xu F; Li P A Visible-Light-Promoted Aerobic Metal-Free C-3 Thiocyanation of Indoles. J. Org. Chem 2014, 79, 10588–10592. [DOI] [PubMed] [Google Scholar]
- (407).Mitra S; Ghosh M; Mishra S; Hajra A Metal-Free Thiocyanation of Imidazoheterocycles through Visible Light Photoredox Catalysis. J. Org. Chem 2015, 80, 8275–8281. [DOI] [PubMed] [Google Scholar]
- (408).Sun P; Yang D; Wei W; Jiang M; Wang Z; Zhang L; Zhang H; Zhang Z; Wang Y; Wang H Visible Light-Induced C-H Sulfenylation Using Sulfinic Acids. Green Chem. 2017, 19, 4785–4791. [Google Scholar]
- (409).Kitaguchi H; Ohkubo K; Ogo S; Fukuzumi S Electron-Transfer Oxidation Properties of Unsaturated Fatty Acids and Mechanistic Insight into Lipoxygenases. J. Phys. Chem. A 2006, 110, 1718–1725. [DOI] [PubMed] [Google Scholar]
- (410).Arnold DR; Maroulis AJ Radical Ions in Photochemistry. 3. Photosensitized (Electron Transfer) Cleavage of. Beta.-Phenethyl Ethers. J. Am. Chem. Soc 1976, 98, 5931–5937. [Google Scholar]
- (411).Davis HF; Das PK; Griffin GW Electron-Transfer Sensitized Carbon-Carbon Bond Cleavage. Facile Homolytic Fission via Geminate Back Electron Transfer in Photogenerated Ion Pairs. J. Am. Chem. Soc 1984, 106, 6968–6973. [Google Scholar]
- (412).Reichel LW; Griffin GW; Muller AJ; Das PK; Ege SN Photoinduced Electron Transfer C—C Bond Cleavage Reactions; Oxidations and Isomerizations. Can. J. Chem 1984, 62, 424. [Google Scholar]
- (413).Albini A; Fasani E; Mella M Photochemical Reaction of 1,4-Naphthalenedicarbonitrile with Alkylbenzenes and Bibenzyls. J. Am. Chem. Soc 1986, 108, 4119–4125. [Google Scholar]
- (414).Albini A; Fasani E; Oberti R The Photochemical Reaction between 1,4-Dicyanonaphtalene and Methylbenzenes: Electron Transfer and Formation of Benzylic Radicals. Tetrahedron 1982, 38, 1027–1034. [Google Scholar]
- (415).Pandey G; Krishna A; Rao JM Single Electron Transfer Initiated Photocyclization of Substituted Cinnamic Acids to Corresponding Coumarins. Tetrahedron Lett. 1986, 27, 4075–4076. [Google Scholar]
- (416).Pandey G; Krishna A Synthetic Application of Photoinduced Single Electron Transfer Reactions: A Convenient Synthetic Approach for the 2,2-Dimethyl-2H-Chromene System. J. Org. Chem 1988, 53, 2364–2365. [Google Scholar]
- (417).Pandey G; Krishna A; Bhalerao UT A One Step Synthesis of 2-Substituted Benzofurans from 2-Aryl-1-Substituted Ethane-1-Ones by Photoinduced Set Reactions. Tetrahedron Lett. 1989, 30, 1867–1870. [Google Scholar]
- (418).Pandey G; Karthikeyan M; Murugan A New Intramolecular α-Arylation Strategy of Ketones by the Reaction of Silyl Enol Ethers to Photosensitized Electron Transfer Generated Arene Radical Cations: Construction of Benzannulated and Benzospiroannulated Compounds. J. Org. Chem 1998, 63, 2867–2872. [Google Scholar]
- (419).Pandey G; Krishna A; Girija K; Karthikeyan M Intramolecular Nucleophilic Addition of Silylenol Ether to Photosensitized Electron Transfer (PET) Generated Arene Radical Cations: A Novel Non-Reagent Based Carboannulation Reaction: Tetrahedron Lett. 1993, 34, 6631–6634. [Google Scholar]
- (420).Pandey G; Murugan A; Balakrishnan M A New Strategy towards the Total Synthesis of Phenanthridone Alkaloids: Synthesis of (+)-2,7-Dideoxypancratistatin as a Model Study. Chem. Commun 2002, 624–625. [DOI] [PubMed] [Google Scholar]
- (421).Pandey G; Sridhar M; Bhalerao UT Regiospecfic Dihydroindoles Directly from β-Arylethylamines by Photoinduced Set Reaction: One Pot “Wavelength Switch” Approach to Benzopyrrolizidines Related to Mitomycin. Tetrahedron Lett. 1990, 31, 5373–5376. [Google Scholar]
- (422).Xu J; He J; Qian Y Photoinduced Coupling Reaction of Benzotriazole with Aromatic Hydrocarbons Sensitized by 9,10-Dicyanoanthracene (DCA)via an Electron-Transfer Mechanism. A Simple Synthesis of 1-Arylbenzotriazole. J. Chem. Soc., Chem. Commun 1991, 714–715. [Google Scholar]
- (423).Ohkubo K; Kobayashi T; Fukuzumi S Direct Oxygenation of Benzene to Phenol Using Quinolinium Ions as Homogeneous Photocatalysts. Angew. Chem., Int. Ed 2011, 50, 8652–8655. [DOI] [PubMed] [Google Scholar]
- (424).Ohkubo K; Fujimoto A; Fukuzumi S Visible-Light-Induced Oxygenation of Benzene by the Triplet Excited State of 2,3-Dichloro-5,6-Dicyano-p-Benzoquinone. J. Am. Chem. Soc 2013, 135, 5368–5371. [DOI] [PubMed] [Google Scholar]
- (425).Ohkubo K; Kobayashi T; Fukuzumi S Photocatalytic Alkoxylation of Benzene with 3-Cyano-1-Methylquinolinium Ion. Opt. Express 2012, 20, A360. [DOI] [PubMed] [Google Scholar]
- (426).Ohkubo K; Fujimoto A; Fukuzumi S Photocatalytic Monofluorination of Benzene by Fluoride via Photoinduced Electron Transfer with 3-Cyano-1-Methylquinolinium. J. Phys. Chem. A 2013, 117, 10719–10725. [DOI] [PubMed] [Google Scholar]
- (427).Ohkubo K; Mizushima K; Iwata R; Fukuzumi S Selective Photocatalytic Aerobic Bromination with Hydrogen Bromidevia an Electron-Transfer State of 9-Mesityl-10-Methylacridinium Ion. Chem. Sci 2011, 2, 715–722. [Google Scholar]
- (428).Fukuzumi S; Ohkubo K; Suenobu T; Kato K; Fujitsuka M; Ito O Photoalkylation of 10-Alkylacridinium Ion via a Charge-Shift Type of Photoinduced Electron Transfer Controlled by Solvent Polarity. J. Am. Chem. Soc 2001, 123, 8459–8467. [DOI] [PubMed] [Google Scholar]
- (429).Fukuzumi S; Ohkubo K Organic Synthetic Transformations Using Organic Dyes as Photoredox Catalysts. Org. Biomol. Chem 2014, 12, 6059–6071. [DOI] [PubMed] [Google Scholar]
- (430).Ohkubo K; Mizushima K; Fukuzumi S Oxygenation and Chlorination of Aromatic Hydrocarbons with Hydrochloric Acid Photosensitized by 9-Mesityl-10-Methylacridinium under Visible Light Irradiation. Res. Chem. Intermed 2013, 39, 205–220. [Google Scholar]
- (431).Romero NA; Margrey KA; Tay NE; Nicewicz DA Site-Selective Arene C-H Amination via Photoredox Catalysis. Science 2015, 349, 1326–1330. [DOI] [PubMed] [Google Scholar]
- (432).Pan X-M; Schuchmann MN; von Sonntag C Oxidation of Benzene by the OH Radical. A Product and Pulse Radiolysis Study in Oxygenated Aqueous Solution. J. Chem. Soc., Perkin Trans 2 1993, 289–297. [Google Scholar]
- (433).Pan X-M; Nien Schuchmann M; von Sonntag C Hydroxyl-Radical-Induced Oxidation of Cyclohexa-1,4-Diene by O2 in Aqueous Solution. A Pulse Radiolysis and Product Study. J. Chem. Soc., Perkin Trans 2 1993, 1021–1028. [Google Scholar]
- (434).Fang X; Pan X; Rahmann A; Schuchmann H-P; von Sonntag C Reversibility in the Reaction of Cyclohexadienyl Radicals with Oxygen in Aqueous Solution. Chem. - Eur. J 1995, 1, 423–429. [Google Scholar]
- (435).Margrey KA; McManus JB; Bonazzi S; Zecri F; Nicewicz DA Predictive Model for Site-Selective Aryl and Heteroaryl C-H Functionalization via Organic Photoredox Catalysis. J. Am. Chem. Soc 2017, 139, 11288–11299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (436).Zheng Y-W; Chen B; Ye P; Feng K; Wang W; Meng Q-Y; Wu L-Z; Tung C-H Photocatalytic Hydrogen-Evolution Cross-Couplings: Benzene C-H Amination and Hydroxylation. J. Am. Chem. Soc 2016, 138, 10080–10083. [DOI] [PubMed] [Google Scholar]
- (437).McManus JB; Nicewicz DA Direct C-H Cyanation of Arenes via Organic Photoredox Catalysis. J. Am. Chem. Soc 2017, 139, 2880–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (438).Margrey KA; Levens A; Nicewicz DA Direct Aryl C-H Amination with Primary Amines Using Organic Photoredox Catalysis. Angew. Chem., Int. Ed 2017, 56, 15644–15648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (439).Chen W; Huang Z; Tay NES; Giglio B; Wang M; Wang H; Wu Z; Nicewicz D; Li Z Direct Arene C-H Fluorination with 18F-via Organic Photoredox Catalysis. Science 2019, 364, 1170–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (440).Wang L; White AR; Chen W; Wu Z; Nicewicz DA; Li Z Direct Radiofluorination of Arene C-H Bonds via Photoredox Catalysis Using a Peroxide as the Terminal Oxidant. Org. Lett 2020, 22, 7971–7975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (441).Samanta S; Ravi C; Rao SN; Joshi A; Adimurthy S Visible-Light-Promoted Selective C-H Amination of Heteroarenes with Heteroaromatic Amines under Metal-Free Conditions. Org. Biomol. Chem 2017, 15, 9590–9594. [DOI] [PubMed] [Google Scholar]
- (442).You G; Wang K; Wang X; Wang G; Sun J; Duan G; Xia C Visible-Light-Mediated Nickel(II)-Catalyzed C-N Cross-Coupling in Water: Green and Regioselective Access for the Synthesis of Pyrazole-Containing Compounds. Org. Lett 2018, 20, 4005–4009. [DOI] [PubMed] [Google Scholar]
- (443).Sun M; Wang L; Zhao L; Wang Z; Li P Visible-Light Photoredox Catalyzed C-N Coupling of Quinoxaline-2(1H)-Ones with Azoles without External Photosensitizer. ChemCatChem 2020, 12, 5261–5268. [Google Scholar]
- (444).Chen H; Yi H; Tang Z; Bian C; Zhang H; Lei A External Oxidant-Free Regioselective Cross Dehydrogenative Coupling of 2-Arylimidazoheterocycles and Azoles with H2 Evolution via Photoredox Catalysis. Adv. Synth. Catal 2018, 360, 3220–3227. [Google Scholar]
- (445).Niu L; Yi H; Wang S; Liu T; Liu J; Lei A Photo-Induced Oxidant-Free Oxidative C-H/N-H Cross-Coupling between Arenes and Azoles. Nat. Commun 2017, 8, 14226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (446).Neogi S; Ghosh AK; Majhi K; Samanta S; Kibriya G; Hajra A Organophotoredox-Catalyzed Direct C-H Amination of 2H-Indazoles with Amines. Org. Lett 2020, 22, 5605–5609. [DOI] [PubMed] [Google Scholar]
- (447).Hu X; Zhang G; Bu F; Luo X; Yi K; Zhang H; Lei A Photoinduced Oxidative Activation of Electron-Rich Arenes: Alkenylation with H2 Evolution under External Oxidant-Free Conditions. Chem. Sci 2018, 9, 1521–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (448).Holmberg-Douglas N; Onuska NPR; Nicewicz DA Regioselective Arene C-H Alkylation Enabled by Organic Photoredox Catalysis. Angew. Chem., Int. Ed 2020, 59, 7425–7429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (449).Griffin JD; Zeller MA; Nicewicz DA Hydrodecarboxylation of Carboxylic and Malonic Acid Derivatives via Organic Photoredox Catalysis: Substrate Scope and Mechanistic Insight. J. Am. Chem. Soc 2015, 137, 11340–11348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (450).Song C; Yi H; Dou B; Li Y; Singh AK; Lei A Visible-Light-Mediated C2-Amination of Thiophenes by Using DDQ as an Organophotocatalyst. Chem. Commun 2017, 53, 3689–3692. [DOI] [PubMed] [Google Scholar]
- (451).Lämmermann H; Sudau A; Rackl D; Weinmann H; Collins K; Wortmann L; Candish L; Hog DT; Meier R Late-Stage Sulfoximidation of Electron-Rich Arenes by Photoredox Catalysis. Synlett 2018, 29, 2679–2684. [Google Scholar]
- (452).Meyer AU; Berger AL; Konig B Metal-Free C-H Sulfonamidation of Pyrroles by Visible Light Photoredox Catalysis. Chem. Commun 2016, 52, 10918–10921. [DOI] [PubMed] [Google Scholar]
- (453).Wimmer A; König B Visible-Light-Mediated Photoredox-Catalyzed N-Arylation of NH-Sulfoximines with Electron-Rich Arenes. Adv. Synth. Catal 2018, 360, 3277–3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (454).Petzold D; König B Photocatalytic Oxidative Bromination of Electron-Rich Arenes and Heteroarenes by Anthraquinone. Adv. Synth. Catal 2018, 360, 626–630. [Google Scholar]
- (455).Das S; Natarajan P; König B Teaching Old Compounds New Tricks: DDQ-Photocatalyzed C-H Amination of Arenes with Carbamates, Urea, and N-Heterocycles. Chem. - Eur. J 2017, 23, 18161–18165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (456).Düsel SJS; König B Visible-Light-Mediated Nitration of Protected Anilines. J. Org. Chem 2018, 83, 2802–2807. [DOI] [PubMed] [Google Scholar]
- (457).Niu L; Liu J; Yi H; Wang S; Liang X-A; Singh AK; Chiang C-W; Lei A Visible-Light-Induced External Oxidant-Free Oxidative Phosphonylation of C(Sp2)-H Bonds. ACS Catal. 2017, 7, 7412–7416. [Google Scholar]
- (458).Li X; Gu X; Li Y; Li P Aerobic Transition-Metal-Free Visible-Light Photoredox Indole C-3 Formylation Reaction. ACS Catal. 2014, 4, 1897–1900. [Google Scholar]
- (459).Rogers DA; Brown RG; Brandeburg ZC; Ko EY; Hopkins MD; LeBlanc G; Lamar AA Organic Dye-Catalyzed, Visible-Light Photoredox Bromination of Arenes and Heteroarenes Using N-Bromosuccinimide. ACS Omega 2018, 3, 12868–12877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (460).Kalsi D; Dutta S; Barsu N; Rueping M; Sundararaju B Room-Temperature C-H Bond Functionalization by Merging Cobalt and Photoredox Catalysis. ACS Catal. 2018, 8, 8115–8120. [Google Scholar]
- (461).Guo J-D; Yang X-L; Chen B; Tung C-H; Wu L-Z Photoredox/Cobalt-Catalyzed C(sp3)-H Bond Functionalization toward Phenanthrene Skeletons with Hydrogen Evolution. Org. Lett 2020, 22, 9627–9632. [DOI] [PubMed] [Google Scholar]
- (462).Düsel SJS; König B Oxidative Photochlorination of Electron-Rich Arenes via in Situ Bromination. Eur. J. Org. Chem 2020, 2020, 1491–1495. [Google Scholar]
- (463).Pan Y; Kee CW; Chen L; Tan C-H Dehydrogenative Coupling Reactions Catalysed by Rose Bengal Using Visible Light Irradiation. Green Chem. 2011, 13, 2682–2685. [Google Scholar]
- (464).Liu Q; Li Y-N; Zhang H-H; Chen B; Tung C-H; Wu L-Z Reactivity and Mechanistic Insight into Visible-Light-Induced Aerobic Cross-Dehydrogenative Coupling Reaction by Organophotocatalysts. Chem. - Eur. J 2012, 18, 620–627. [DOI] [PubMed] [Google Scholar]
- (465).Yadav AK; Yadav LDS Visible-Light-Induced Direct α-C(sp3)-H Thiocyanation of Tertiary Amines. Tetrahedron Lett. 2015, 56, 6696–6699. [Google Scholar]
- (466).Xiao T; Li L; Lin G; Mao Z; Zhou L Metal-Free Visible-Light Induced Cross-Dehydrogenative Coupling of Tertiary Amines with Diazo Compounds. Org. Lett 2014, 16, 4232–4235. [DOI] [PubMed] [Google Scholar]
- (467).Pan Y; Wang S; Kee CW; Dubuisson E; Yang Y; Loh KP; Tan C-H Graphene Oxide and Rose Bengal: Oxidative C-H Functionalisation of Tertiary Amines Using Visible Light. Green Chem. 2011, 13, 3341–3344. [Google Scholar]
- (468).Fu W; Guo W; Zou G; Xu C Selective Trifluoromethylation and Alkynylation of Tetrahydroisoquinolines Using Visible Light Irradiation by Rose Bengal. J. Fluorine Chem 2012, 140, 88–94. [Google Scholar]
- (469).Okamura I; Park S; Han JH; Notsu S; Sugiyama H A Combination of Visible-Light Photoredox and Metal Catalysis for the Mannich-Type Reaction of N-Aryl Glycine Esters. Chem. Lett 2017, 46, 1597–1600. [Google Scholar]
- (470).Wang J; Li L; Guo Y; Li S; Wang S; Li Y; Zhang Y Visible-Light-Enabled Aerobic Oxidative Csp3-H Functionalization of Glycine Derivatives Using an Organic Photocatalyst: Access to Substituted Quinoline-2-Carboxylates. Org. Biomol. Chem 2020, 18, 8179–8185. [DOI] [PubMed] [Google Scholar]
- (471).Yang X; Li L; Li Y; Zhang Y Visible-Light-Induced Photocatalytic Aerobic Oxidative Csp3-H Functionalization of Glycine Derivatives: Synthesis of Substituted Quinolines. J. Org. Chem 2016, 81, 12433–12442. [DOI] [PubMed] [Google Scholar]
- (472).Rusch F; Unkel L-N; Alpers D; Hoffmann F; Brasholz M A Visible Light Photocatalytic Cross-Dehydrogenative Coupling/Dehydrogenation/6π-Cyclization/Oxidation Cascade: Synthesis of 12-Nitroindoloisoquinolines from 2-Aryltetrahydroisoquinolines. Chem. - Eur. J 2015, 21, 8336–8340. [DOI] [PubMed] [Google Scholar]
- (473).Wei G; Zhang C; Bureš F; Ye X; Tan C-H; Jiang Z Enantioselective Aerobic Oxidative C(Sp3)-H Olefination of Amines via Cooperative Photoredox and Asymmetric Catalysis. ACS Catal. 2016, 6, 3708–3712. [Google Scholar]
- (474).Xiang M; Meng Q-Y; Li J-X; Zheng Y-W; Ye C; Li Z-J; Chen B; Tung C-H; Wu L-Z Activation of C-H Bonds through Oxidant-Free Photoredox Catalysis: Cross-Coupling Hydrogen-Evolution Transformation of Isochromans and β-Keto Esters. Chem. - Eur. J 2015, 21, 18080–18084. [DOI] [PubMed] [Google Scholar]
- (475).Pandey G; Tiwari SK; Singh B α-Alkylation of Tertiary Amines by C(sp3)-C(sp3) Cross-Coupling under Redox Neutral Photocatalysis. Tetrahedron Lett. 2016, 57, 4480–4483. [Google Scholar]
- (476).Liu X; Ye X; Bureš F; Liu H; Jiang Z Controllable Chemoselectivity in Visible-Light Photoredox Catalysis: Four Diverse Aerobic Radical Cascade Reactions. Angew. Chem., Int. Ed 2015, 54, 11443–11447. [DOI] [PubMed] [Google Scholar]
- (477).McManus JB; Onuska NPR; Nicewicz DA Generation and Alkylation of α-Carbamyl Radicals via Organic Photoredox Catalysis. J. Am. Chem. Soc 2018, 140, 9056–9060. [DOI] [PubMed] [Google Scholar]
- (478).McManus JB; Onuska NPR; Jeffreys MS; Goodwin NC; Nicewicz DA Site-Selective C-H Alkylation of Piperazine Substrates via Organic Photoredox Catalysis. Org. Lett 2020, 22, 679–683. [DOI] [PubMed] [Google Scholar]
- (479).Fan X-Z; Rong J-W; Wu H-L; Zhou Q; Deng H-P; Tan JD; Xue C-W; Wu L-Z; Tao H-R; Wu J Eosin Y as a Direct Hydrogen-Atom Transfer Photocatalyst for the Functionalization of C-H Bonds. Angew. Chem., Int. Ed 2018, 57, 8514–8518. [DOI] [PubMed] [Google Scholar]
- (480).Hou J; Ee A; Cao H; Ong H-W; Xu J-H; Wu J Visible-Light-Mediated Metal-Free Difunctionalization of Alkenes with CO2 and Silanes or C(sp3)-H Alkanes. Angew. Chem., Int. Ed 2018, 57, 17220–17224. [DOI] [PubMed] [Google Scholar]
- (481).Si X; Zhang L; Hashmi ASK Benzaldehyde- and Nickel-Catalyzed Photoredox C(sp3)-H Alkylation/Arylation with Amides and Thioethers. Org. Lett 2019, 21, 6329–6332. [DOI] [PubMed] [Google Scholar]
- (482).Shen Y; Gu Y; Martin R Sp3 C-H Arylation and Alkylation Enabled by the Synergy of Triplet Excited Ketones and Nickel Catalysts. J. Am. Chem. Soc 2018, 140, 12200–12209. [DOI] [PubMed] [Google Scholar]
- (483).Twilton J; Le C; Zhang P; Shaw MH; Evans RW; MacMillan DW C. The Merger of Transition Metal and Photocatalysis. Nat. Rev. Chem 2017, 1, 1–19. [Google Scholar]
- (484).Zhang L; Si X; Yang Y; Zimmer M; Witzel S; Sekine K; Rudolph M; Hashmi ASK The Combination of Benzaldehyde and Nickel-Catalyzed Photoredox C(Sp3)-H Alkylation/Arylation. Angew. Chem., Int. Ed 2019, 58, 1823–1827. [DOI] [PubMed] [Google Scholar]
- (485).Paul S; Guin J Radical C(sp3)-H Alkenylation, Alkynylation and Allylation of Ethers and Amides Enabled by Photocatalysis. Green Chem. 2017, 19, 2530–2534. [Google Scholar]
- (486).Zhang Q; Huang Y; Zhan L-W; Tang W-Y; Hou J; Li B-D Photoredox-Catalyzed α-C(sp3)-H Activation of Unprotected Secondary Amines: Facile Access to 1,4-Dicarbonyl Compounds. Org. Lett 2020, 22, 7460–7464. [DOI] [PubMed] [Google Scholar]
- (487).Ryder ASH; Cunningham WB; Ballantyne G; Mules T; Kinsella AG; Turner-Dore J; Alder CM; Edwards LJ; McKay BSJ; Grayson MN; et al. Photocatalytic α-Tertiary Amine Synthesis via C-H Alkylation of Unmasked Primary Amines. Angew. Chem., Int. Ed 2020, 59, 14986–14991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (488).Ma Z-Y; Li M; Guo L-N; Liu L; Wang D; Duan X-H Sulfonamide as Photoinduced Hydrogen-Atom Transfer Catalyst for Regioselective Alkylation of C(sp3)-H Bonds Adjacent to Heteroatoms. Org. Lett 2021, 23, 474–479. [DOI] [PubMed] [Google Scholar]
- (489).Neumann M; Füldner S; König B; Zeitler K Metal-Free, Cooperative Asymmetric Organophotoredox Catalysis with Visible Light. Angew. Chem., Int. Ed 2011, 50, 951–954. [DOI] [PubMed] [Google Scholar]
- (490).Fidaly K; Ceballos C; Falguières A; Veitia MS-I; Guy A; Ferroud C Visible Light Photoredox Organocatalysis: A Fully Transition Metal-Free Direct Asymmetric α-Alkylation of Aldehydes. Green Chem. 2012, 14, 1293–1297. [Google Scholar]
- (491).Kawaai K; Yamaguchi T; Yamaguchi E; Endo S; Tada N; Ikari A; Itoh A Photoinduced Generation of Acyl Radicals from Simple Aldehydes, Access to 3-Acyl-4-Arylcoumarin Derivatives, and Evaluation of Their Antiandrogenic Activities. J. Org. Chem 2018, 83, 1988–1996. [DOI] [PubMed] [Google Scholar]
- (492).de Souza GFP; Bonacin JA; Salles AG Visible-Light-Driven Epoxyacylation and Hydroacylation of Olefins Using Methylene Blue/Persulfate System in Water. J. Org. Chem 2018, 83, 8331–8340. [DOI] [PubMed] [Google Scholar]
- (493).Leow D Phenazinium Salt-Catalyzed Aerobic Oxidative Amidation of Aromatic Aldehydes. Org. Lett 2014, 16, 5812–5815. [DOI] [PubMed] [Google Scholar]
- (494).Leung FK-C; Cui J-F; Hui T-W; Kung KK-Y; Wong M-K Photooxidative Amidation of Aldehydes with Amines Catalyzed by Rose Bengal. Asian J. Org. Chem 2015, 4, 533–536. [Google Scholar]
- (495).Mitsunuma H; Tanabe S; Fuse H; Ohkubo K; Kanai M Catalytic Asymmetric Allylation of Aldehydes with Alkenes through Allylic C(Sp3)-H Functionalization Mediated by Organophotoredox and Chiral Chromium Hybrid Catalysis. Chem. Sci 2019, 10, 3459–3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (496).Tanabe S; Mitsunuma H; Kanai M Catalytic Allylation of Aldehydes Using Unactivated Alkenes. J. Am. Chem. Soc 2020, 142, 12374–12381. [DOI] [PubMed] [Google Scholar]
- (497).Monga A; Pandey AP; Sharma A Visible-Light Mediated Photooxidative Synthesis of α-Keto Amides. Adv. Synth. Catal 2019, 361, 3554–3559. [Google Scholar]
- (498).Kuang Y; Wang K; Shi X; Huang X; Meggers E; Wu J Asymmetric Synthesis of 1,4-Dicarbonyl Compounds from Aldehydes by Hydrogen Atom Transfer Photocatalysis and Chiral Lewis Acid Catalysis. Angew. Chem., Int. Ed 2019, 58, 16859–16863. [DOI] [PubMed] [Google Scholar]
- (499).Pandey G; Laha R Visible-Light-Catalyzed Direct Benzylic C(Sp3)-H Amination Reaction by Cross-Dehydrogenative Coupling. Angew. Chem., Int. Ed 2015, 54, 14875–14879. [DOI] [PubMed] [Google Scholar]
- (500).Xia J-B; Zhu C; Chen C Visible Light-Promoted Metal-Free C-H Activation: Diarylketone-Catalyzed Selective Benzylic Mono- and Difluorination. J. Am. Chem. Soc 2013, 135, 17494–17500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (501).Xiang M; Xin Z-K; Chen B; Tung C-H; Wu L-Z Exploring the Reducing Ability of Organic Dye (Acr+−Mes) for Fluorination and Oxidation of Benzylic C(sp3)-H Bonds under Visible Light Irradiation. Org. Lett 2017, 19, 3009–3012. [DOI] [PubMed] [Google Scholar]
- (502).Xiang M; Zhou C; Yang X-L; Chen B; Tung C-H; Wu L-Z Visible Light-Catalyzed Benzylic C-H Bond Chlorination by a Combination of Organic Dye (Acr+−Mes) and N-Chlorosuccinimide. J. Org. Chem 2020, 85, 9080–9087. [DOI] [PubMed] [Google Scholar]
- (503).Yan D-M; Zhao Q-Q; Rao L; Chen J-R; Xiao W-J Eosin Y as a Redox Catalyst and Photosensitizer for Sequential Benzylic C-H Amination and Oxidation. Chem. - Eur. J 2018, 24, 16895–16901. [DOI] [PubMed] [Google Scholar]
- (504).Meng Q-Y; Schirmer TE; Berger AL; Donabauer K; König B Photocarboxylation of Benzylic C-H Bonds. J. Am. Chem. Soc 2019, 141, 11393–11397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (505).Pandey G; Pal S; Laha R Direct Benzylic C-H Activation for C-O Bond Formation by Photoredox Catalysis. Angew. Chem., Int. Ed 2013, 52, 5146–5149. [DOI] [PubMed] [Google Scholar]
- (506).Im H; Kang D; Choi S; Shin S; Hong S Visible-Light-Induced C-O Bond Formation for the Construction of Five- and Six-Membered Cyclic Ethers and Lactones. Org. Lett 2018, 20, 7437–7441. [DOI] [PubMed] [Google Scholar]
- (507).Xu W; Wang W; Liu T; Xie J; Zhu C Late-Stage Trifluoromethylthiolation of Benzylic C-H Bonds. Nat. Commun 2019, 10, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (508).Qrareya H; Ravelli D; Fagnoni M; Albini A Decatungstate Photocatalyzed Benzylation of Alkenes with Alkylaromatics. Adv. Synth. Catal 2013, 355, 2891–2899. [Google Scholar]
- (509).Zhou R; Liu H; Tao H; Yu X; Wu J Metal-Free Direct Alkylation of Unfunctionalized Allylic/Benzylic Sp 3 C-H Bonds via Photoredox Induced Radical Cation Deprotonation. Chem. Sci 2017, 8, 4654–4659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (510).Liu H; Ma L; Zhou R; Chen X; Fang W; Wu J One-Pot Photomediated Giese Reaction/Friedel-Crafts Hydroxyalkylation/Oxidative Aromatization To Access Naphthalene Derivatives from Toluenes and Enones. ACS Catal. 2018, 8, 6224–6229. [Google Scholar]
- (511).Hoshikawa T; Inoue M Photoinduced Direct 4-Pyridination of C(Sp3)-H Bonds. Chem. Sci 2013, 4, 3118–3123. [Google Scholar]
- (512).Dewanji A; Krach PE; Rueping M The Dual Role of Benzophenone in Visible-Light/Nickel Photoredox-Catalyzed C-H Arylations: Hydrogen-Atom Transfer and Energy Transfer. Angew. Chem., Int. Ed 2019, 58, 3566–3570. [DOI] [PubMed] [Google Scholar]
- (513).Huang L; Rueping M Direct Cross-Coupling of Allylic C(sp3)-H Bonds with Aryl- and Vinylbromides by Combined Nickel and Visible-Light Catalysis. Angew. Chem., Int. Ed 2018, 57, 10333–10337. [DOI] [PubMed] [Google Scholar]
- (514).Betori RC; May CM; Scheidt KA Combined Photoredox/Enzymatic C-H Benzylic Hydroxylations. Angew. Chem 2019, 131, 16642–16646. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- (515).Kee CW; Chan KM; Wong MW; Tan C-H Selective Bromination of Sp3 C-H Bonds by Organophotoredox Catalysis. Asian J. Org. Chem 2014, 3, 536–544. [Google Scholar]
- (516).Kee CW; Chin KF; Wong MW; Tan C-H Selective Fluorination of Alkyl C-H Bonds via Photocatalysis. Chem. Commun 2014, 50, 8211–8214. [DOI] [PubMed] [Google Scholar]
- (517).Capilato JN; Pitts CR; Rowshanpour R; Dudding T; Lectka T Site-Selective Photochemical Fluorination of Ketals: Unanticipated Outcomes in Selectivity and Stability. J. Org. Chem 2020, 85, 2855–2864. [DOI] [PubMed] [Google Scholar]
- (518).Tian H; Yang H; Tian C; An G; Li G Cross-Dehydrogenative Coupling of Strong C(sp3)-H with N-Heteroarenes through Visible-Light-Induced Energy Transfer. Org. Lett 2020, 22, 7709–7715. [DOI] [PubMed] [Google Scholar]
- (519).Ouyang X-H; Li Y; Song R-J; Hu M; Luo S; Li J-H Intermolecular Dialkylation of Alkenes with Two Distinct C(sp3)-H Bonds Enabled by Synergistic Photoredox Catalysis and Iron Catalysis. Sci. Adv 2019, 5, No. eaav9839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (520).Margrey KA; Czaplyski WL; Nicewicz DA; Alexanian EJ A General Strategy for Aliphatic C-H Functionalization Enabled by Organic Photoredox Catalysis. J. Am. Chem. Soc 2018, 140, 4213–4217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (521).Deng H-P; Zhou Q; Wu J Microtubing-Reactor-Assisted Aliphatic C-H Functionalization with HCl as a Hydrogen-Atom-Transfer Catalyst Precursor in Conjunction with an Organic Photoredox Catalyst. Angew. Chem., Int. Ed 2018, 57, 12661–12665. [DOI] [PubMed] [Google Scholar]
- (522).Chen H; Guo L; Yu S Primary, Secondary, and Tertiary γ-C(sp3)-H Vinylation of Amides via Organic Photoredox-Catalyzed Hydrogen Atom Transfer. Org. Lett 2018, 20, 6255–6259. [DOI] [PubMed] [Google Scholar]
- (523).Fernandez Reina D; Ruffoni A; Al-Faiyz YSS; Douglas JJ; Sheikh NS; Leonori D Visible-Light-Mediated Reactions of Electrophilic Radicals with Vinyl and Allyl Trifluoroborates. ACS Catal. 2017, 7, 4126–4130. [Google Scholar]
- (524).Wu K; Wang L; Colón-Rodríguez S; Flechsig G-U; Wang T Amidyl Radical Directed Remote Allylation of Unactivated sp3 C-H Bonds by Organic Photoredox Catalysis. Angew. Chem., Int. Ed 2019, 58, 1774–1778. [DOI] [PubMed] [Google Scholar]
- (525).Chen H; Fan W; Yuan X-A; Yu S Site-Selective Remote C(sp3)-H Heteroarylation of Amides via Organic Photoredox Catalysis. Nat. Commun 2019, 10, 4743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (526).Morcillo SP; Dauncey EM; Kim JH; Douglas JJ; Sheikh NS; Leonori D Photoinduced Remote Functionalization of Amides and Amines Using Electrophilic Nitrogen Radicals. Angew. Chem., Int. Ed 2018, 57, 12945–12949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (527).Dauncey EM; Morcillo SP; Douglas JJ; Sheikh NS; Leonori D Photoinduced Remote Functionalisations by Iminyl Radical Promoted C-C and C-H Bond Cleavage Cascades. Angew. Chem., Int. Ed 2018, 57, 744–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (528).Kim I; Park B; Kang G; Kim J; Jung H; Lee H; Baik M-H; Hong S Visible-Light-Induced Pyridylation of Remote C(sp3)-H Bonds by Radical Translocation of N-Alkoxypyridinium Salts. Angew. Chem., Int. Ed 2018, 57, 15517–15522. [DOI] [PubMed] [Google Scholar]
- (529).Tang N; Wu X; Zhu C Practical, Metal-Free Remote Heteroarylation of Amides via Unactivated C(sp3)-H Bond Functionalization. Chem. Sci 2019, 10, 6915–6919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (530).Becker P; Duhamel T; Stein CJ; Reiher M; Muñiz K Cooperative Light-Activated Iodine and Photoredox Catalysis for the Amination of C -H Bonds. Angew. Chem., Int. Ed 2017, 56, 8004–8008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (531).Mei L; Veleta JM; Gianetti TL Helical Carbenium Ion: A Versatile Organic Photoredox Catalyst for Red-Light-Mediated Reactions. J. Am. Chem. Soc 2020, 142, 12056–12061. [DOI] [PubMed] [Google Scholar]