

Abstract
Hedgehog (Hh) signaling culminates in the conversion of the latent transcription factor Cubitus interruptus (Ci)/Gli from a repressor form (CiR/GliR) into an activator form (CiA/GliA). While sequential phosphorylation of Ci/Gli by protein kinase A(PKA), glycogen synthase kinase 3 (GSK3), and casein kinase 1 (CK1) is essential for its proteolytic processing that generates CiR/GliR, sequential phosphorylation of Ci/Gli by the Fused (Fu)/Unc-51 like kinase (Ulk) family kinases Fu/Ulk3/Stk36 and CK1 contributes to the formation of CiA/GliA. Fu/Ulk3/Stk36-mediated phosphorylation of Ci/Gli is stimulated by Hh, leading to altered interaction between Ci/Gli and the Hh pathway repressor Sufu. Here we describe both in vitro and in vivo assays that determine Ci/Gli phosphorylation by the Fu/Ulk family kinases and its regulation by Hh.
Keywords: Hh, Ci, Gli1, Gli2, Gli3, Fu, CK1, Shh, Ulk3, Stk36, Ulk1, Atg1, Kinase, Phosphorylation, Signaling
1. Introduction
The Hedgehog (Hh) family of secreted glycoproteins governs embryonic development and adult tissue homeostasis in species ranging from insect to mammals and aberrant Hh signaling contributes to a wide range of human diseases [1–4]. The Hh signal is transduced via a largely conserved pathway that culminates in the conversion of the latent transcription factor Ci/Gli from a repressor (CiR/GliR) form into an activator form (CiA/GliA). In the absence of Hh, full-length Ci/Gli (CiF/GliF) is phosphorylated by multiple kinases including protein kinase A (PKA), glycogen synthase kinase 3 (GSK3), and casein kinase 1 (CK1), which targets it for ubiquitination by SCFSlimb/β-TRCP, followed by proteasome-mediated partial degradation to generate CiR/GliR that actively repress a subset of Hh target genes (Fig. 1) [5–13]. In response to Hh, PKA/GSK3/CK1-mediated phosphorylation and subsequently SCFSlimb/β-TRCP-ubiquitination of Ci/Gli is blocked, leading to inhibition of CiR/GliR production [14]. Hh-mediated inhibition of Ci/Gli ubiquitination also depends on the deubiquitinating enzyme USP7 [15]. Meanwhile, Hh converts the accumulated CiF/GliF into a labile CiA/GliA by antagonizing the inhibition imposed by the pathway inhibitor Sufu [16–21]. Furthermore, Hh-induced phosphorylation by CK1 sustains the activity of CiA/GliA by preventing its premature degradation mediated by Cul3-HIB/SPOP [22–24].
Fig. 1.
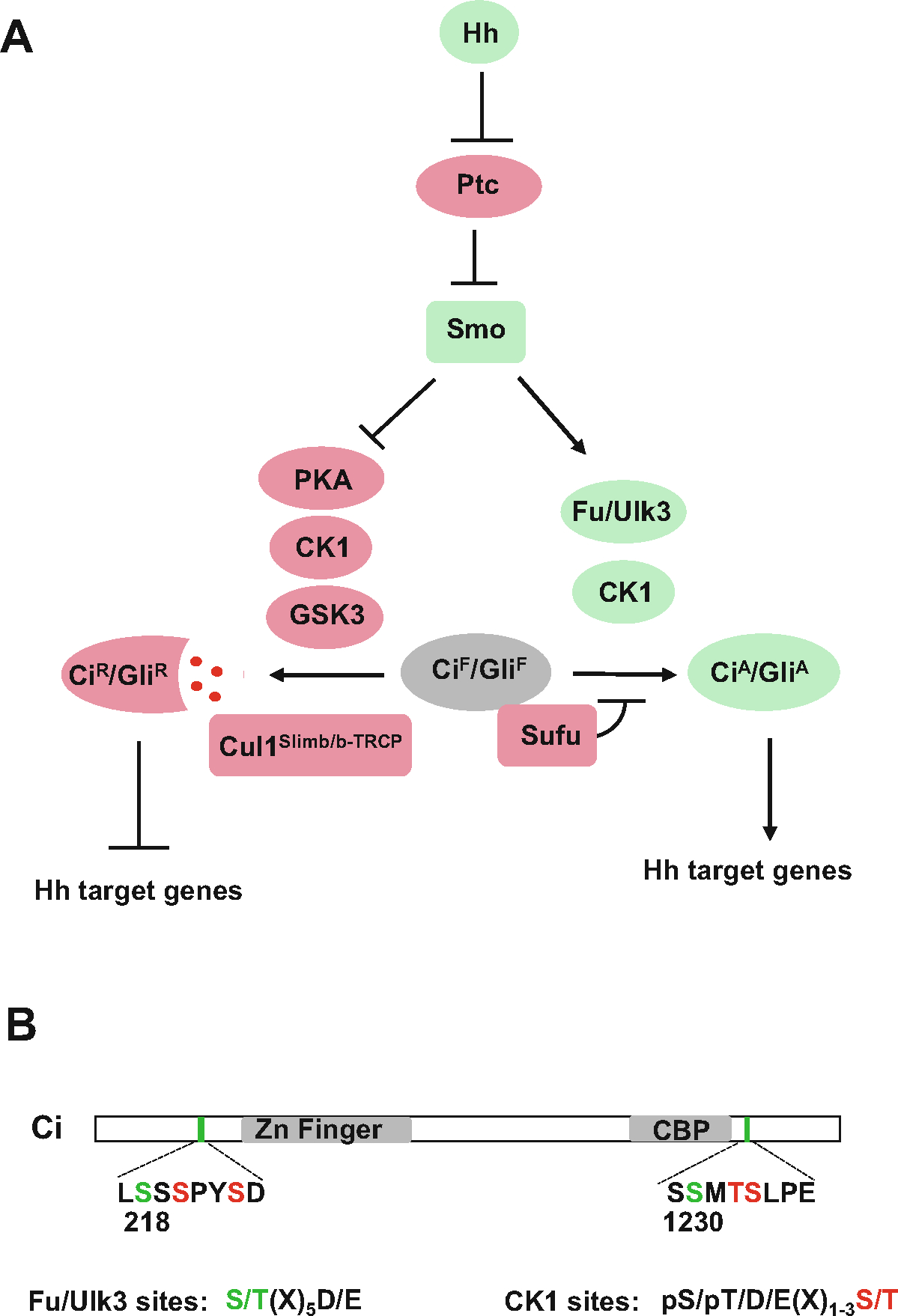
Hh-stimulated phosphorylation of Ci/Gli by the Fu family kinases contributes to its activation. (a) Hh blocks the production of CiR/GliR by inhibiting PKA/GSK3/CK1-mediated phosphorylation of Ci/Gli on Slimb/β-TRCP degrons while converts accumulated CiF/GliF into CiA/GliA through Fu/Ulk3/CK1-mediated phosphorylation of Ci/Gli on conserved sites. (b) Phosphorylation of Ci by Fu on S218 and S1230 primes its further phosphorylation by CK1 on adjacent residues
A critical mediator of Ci activation in Drosophila is the Fused (Fu) kinase [16, 25]. Hh signaling induced Fu dimerization, auto-phosphorylation, and activation downstream of Smoothened [26–28]. Indeed, forced dimerization using a coiled-coil (CC) dimerization motif or activation loop phospho-mimetic mutations (EE: S151E and T154E) generated constitutively active forms of Fu (CC-Fu, FuEE, and CC-FuEE) [26]. Our recent study has demonstrated that Hh stimulates Ci phosphorylation by Fu on Ser218 and Ser1230, which primes Ci for further phosphorylation by CK1 on adjacent sties, and these phosphorylation events promote Ci activation by altering its binding to Sufu, and consequently increasing its binding to the nuclear transporter such as Transportin (Trn) and the transcriptional co-activator CBP (Fig. 1) [29]. In addition, we have provided evidence that Shh activates Gli family of proteins, including Gli1, Gli2, and Gli3, by stimulating their phosphorylation on conserved Ser residues through the Fu-family kinases Ulk3 and Stk36 in NIH/3T3 cells [29], and that Ulk3/Stk36-mediated Gli2 phosphorylation depends on its ciliary localization, which is mediated by karyopherin β2 [30]. Alignment of Fu/Ulk3 phosphorylation sites in Ci, Gli proteins, and Costal2 identified a consensus sequence: S/T(X)5D/E, in which the acid residue at +6 position is essential for Fu/Ulk3-mediated phosphorylation of S/T at position 0 (Fig. 1) [29]. Uncovering the phosphorylation consensus sites for the Fu/Ulk family kinases will facilitate the identification of Fu/Ulk-regulated events not only in the Hh pathway but also in other cellular processes such as autophagy because the central regulator of autophagy, Ulk1/Atg1, belongs to the same kinase family [29]. Here we describe both the in vitro and in vivo assays that determine Ci/Gli phosphorylation by the Fu family kinases and its regulation by Hh.
2. Materials
2.1. Cell Culture and Transfection
Drosophila S2R+ cells transfected with regular calcium phosphate method in a 25 ° C incubator.
Drosophila S2R+ medium: Schneider’s Drosophila Medium, 10% fetal bovine serum, 100 U/mL penicillin, and 100μg/mL streptomycin.
Hh-conditioned medium: collected from S2 stable cell line expressing HhN with the induction of 0.7 mM copper sulfate for 24 h.
S2 Hh stable cells.
S2 Hh stable cell medium: Schneider’s Drosophila Medium, 10% fetal bovine serum and 500μg/mL hygromycin.
S2 Hh stable cell growth medium: Schneider’s Drosophila Medium, 10% fetal bovine serum.
Sf9 cells.
Cellfectin II reagent.
SF900 III growth medium: 10% fetal bovine serum.
SF900III medium without any serum or antibiotics.
NIH/3T3 cells.
NIH/3T3 cell culture medium: DMEM Medium, 10% bovine calf serum, 100 U/mL penicillin, and 100μg/mL streptomycin.
Cell culture incubator at 37 °C.
Polyjet in vitro DNA transfection reagents.
Transfection-grade plasmids: desired protein coding sequences were subcloned into pcDNA3.1(+) vectors.
Fetal Bovine Serum (FBS).
SDS PAGE gel.
6-Well cell culture plates.
10 cm Cell culture dishes.
2.2. Phosphor Antibodies Production and Purification
Phospho-Ci antibodies using the following phospho-peptides as antigens against: RKRALS(p)SS(p)PYSDS for pS218/220, and QIIDSS(p)MTS(p)LPEL and QIIDSSMT(p)SLPEL for pS1230/1233 and pT1232, respectively. The corresponding non-phosphorylated peptides were synthesized at the same as a negative control for affinity purification.
Phospho-Gli2 antibody using the following phospho-peptides as antigens: RKRALS(p)IS(p)PLSDA. The corresponding non-phosphorylated peptides were synthesized at the same as a negative control for affinity purification.
The raw serum from immunized rabbits were sequentially loaded onto Column conjugated with non-phosphorylated and phosphorylated antigen peptides. The final eluted antibodies were collected.
2.3. Detection of Fu-Mediated Ci Phosphorylation with Phospho-Specific Antibodies in S2R + Cells
Lysis Buffer: 20 mM Tris HCl pH 8.0, 150 mM NaCl, 5 mM EDTA, 1% IGPAL CA-630, 10% Glycerol, 1X Complete protease inhibitors cocktail, 1X phosSTOP phosphatase inhibitors cocktail.
SDS loading buffer (4x): 0.25 M Tris–HCl (pH 6.8), 8% SDS, 20% 2-mercaptoethanol, 40% glycerol, and 0.008% bromophenol blue.
Running buffer: 25 mM Tris base, 190 mM glycine, 0.1% SDS.
Transfer buffer: 25 mM Tris base, 190 mM glycine, 20% methanol.
Cell starving medium: DMEM, 0.5% BCS.
Nitrocellulose Membrane.
TBST solution: 20 mM Tris–HCl (pH 7.6), 150 mM NaCl, and 0.1% Tween-20.
Blocking buffer: 5% nonfat dried milk or bovine serum albumin (BSA) in TBST.
IRDye 680LT Donkey anti-Rabbit Secondary Antibody.
Odyssey CLx Imaging System.
Recombinant Protein G Sepharose 4B.
Antibody: Mouse anti-Myc (9E10).
2.4. Detection of Gli2 Phosphorylation with Phospho-Specific Antibody
Recombinant human Sonic Hedgehog N-terminus protein.
Smoothened agonist SAG.
Antibody: mouse anti-Myc (9E10).
Mission shRNA target the 3′UTR of mGli2.
Coding sequence of Myc mGli2 was subcloned into FUXW lentiviral vector; lentiviral packaging vector psPAX2 and envelop vector pMD2.G.
Lysis Buffer: 20 mM Tris HCl pH 8.0, 150 mM NaCl, 5 mM EDTA, 1% IGPAL CA-630, 10% Glycerol, 1X Complete protease inhibitors cocktail, 1X phosSTOP phosphatase inhibitors cocktail.
SDS loading buffer (4x): 0.25 M Tris–HCl (pH 6.8), 8% SDS, 20% 2-mercaptoethanol, 40% glycerol, and 0.008% bromophenol blue.
Running buffer: 25 mM Tris base, 190 mM glycine, 0.1% SDS.
Transfer buffer: 25 mM Tris base, 190 mM glycine, 20% methanol.
Nitrocellulose Membrane.
TBST solution: 20 mM Tris–HCl (pH 7.6), 150 mM NaCl, and 0.1% Tween-20.
Blocking buffer: 5% nonfat dried milk or bovine serum albumin (BSA) in TBST.
IRDye 680LT Donkey anti-Rabbit Secondary Antibody.
Odyssey CLx Imaging System.
Recombinant Protein G Sepharose 4B.
2.5. GST-Ci/Gli2 Fragment Fusion Protein Expression and Purification
All Ci fragments including Fu phosphorylation sites were subcloned into pGEX 4T-1 plasmid.
BL21 Competent E. Coli cells.
Glutathione Sepharose 4B agarose beads.
Amicon Ultra-0.5 centrifugal filter unit with Ultracel-10 membrane.
LB Medium.
IPTG.
Extraction/Binding Buffer: 20 mM Tris HCl pH 8.0, 100 mM NaCl, 5 mM EDTA.
Elution Buffer: 20 mM Tris HCl pH 8.0, 100 mM NaCl, 5 mM EDTA, 10 mM GSH.
Q700 Sonicator.
SimplyBlue SafeStain.
Bovine serum albumin (BSA).
2.6. In Vitro Kinase Assay
CK1δ.
Reaction Buffer (1X): 150 mM Tris–HCl pH 7.5, 0.2 mM Mg2+/ATP.
γ-32P ATP.
pIMAGO-biotin Phosphoprotein Detection Kit (Fluor680 based detection).
Carestream BIOMAX MS Film.
Glutathione Sepharose 4B agarose beads.
HEK293T cells.
Anti-HA agarose.
HA peptide.
2.7. Baculovirus Production and Amplification
Coding sequences of 3XFlag Ci−PKA, 3XHA CC-FuEE, and 3XFlag CC-FuEE were subcloned into pFastBac 1 vector.
MAX Efficiency DH10Bac E. Coli chemically competent cells.
LB Agar plates: LB-Agar medium, 50μg/mL Kanamycin, 7μg/mL Gentamycin, 10μg/mL Tetracycline, 100μg/mL, Bluo-Gal, 40μg/mL IPTG, store in 4 ° C with the protection from light.
LB medium with antibiotics: 50μg/mL Kanamycin, 7μg/mL Gentamycin, 10μg/mL Tetracycline.
SOC medium.
Solution I: 15 mM Tris–HCl (pH 8.0), 10 mM EDTA, 100μg/mL RNase A.
Solution II: 0.2 N NaOH, 1% SDS.
Solution III: 3 M potassium acetate (pH 5.5).
0.7 V Isopropanol.
70% Ethanol.
TE Buffer: 10 mM Tris HCl pH 8.0, 1 mM EDTA.
2.8. Sf9 Cell Infection, Protein Expression, Purification
Lysis Buffer: 20 mM Tris HCl pH 8.0, 150 mM NaCl, 5 mM EDTA, 1% IGPAL CA-630, 10% Glycerol, 1X Complete protease inhibitors cocktail, 1X phosSTOP phosphatase inhibitors cocktail.
Flag M2 affinity agarose.
3X Flag peptide.
Sephadex 200 10/300 GL column.
AKTA system.
Gel filtration running buffer: 50 mM NH4HCO3.
Fused Kinase storage buffer: 20 mM Tris HCl pH 7.0, 100 mM NaCl, 1 mM DTT, 1 mM EDTA, 1 mM EGTA, 0.1% Triton X-100.
PBS.
8% SDS-PAGE gel.
3. Methods
3.1. Hh-Conditioned Medium Preparation
Seed S2 Hh stable cell line at one million/mL.
72 h later, wash cells with S2 Hh stable cell growth medium to remove residual hygromycin and resuspend in S2 Hh stable cell growth medium.
Induce expression of Hh by adding copper sulfate with the final concentration at 0.7μM.
After 24 h, remove the cells by centrifugation and collect the conditioned medium, store at 4 °C with protection from light.
3.2. Detection of Fu-Mediated Ci Phosphorylation with Phosphor-Specific Antibodies in S2R+ Cells
Seed S2R+ cells at one million/mL.
After 24 h, transfect 5μg of both Ub-Gal4 and pUAST Myc Ci−PKA plasmids by using regular calcium phosphate method.
24 h after the transfection, replace S2 Hh stable cell growth medium with 10 mL of new medium composed of 60% Hh-conditioned medium and 40% Drosophila S2R+ medium (see Note 1).
24 h after the Hh treatment, collect and lyse cells. Add 2–3μg of Myc 9E10 antibody to resultant cell lysate. Incubate with primary antibody at 4 °C with agitation for 1–2 h.
Add 15–20μL (bed volume) protein G beads, incubate cell lysate at 4 °C for 1 h.
After centrifugation, collect protein G beads and wash 2–3 times with cold lysis buffer. Elute the bound protein with 2X SDS loading buffer at 95 °C for 5 min.
Load the eluted Ci proteins onto an 8% SDS-PAGE and analyze by standard western blot procedure. In cases when accurate quantification is needed, employment of a fluorogenic secondary antibody combined with membrane scanning with Odyssey Imaging system is carried out. Basically, after incubation with primary Ci phosphor-specific antibody, the membrane is washed and incubated with IRDye LT680 donkey at rabbit secondary antibody at 1:10,000 for 1 h at room temperature as a replacement of regular HRP-conjugated secondary antibody incubation. The membrane is then scanned, and immune-reactive signal is quantified by Odyssey Imaging system (see Note 2).
3.3. Detection of Gli2 Phosphorylation with Phosphor-Specific Antibodies in NIH/3T3 Cells
3.3.1. Knock Down and Reintroduce mGli2 in NIH/3T3 with Lentivirus
Seed one million NIH/3T3 cells into one well of a 6-well plate.
The next day, transfect the cells with 0.5μg of pLKO.1 mGli2 shRNA, 0.375μg psPAX2, and 0.125μg pMD2.G with polyJet in vitro DNA transfection reagent.
Seed appropriate number of NIH/3T3 cells into one well of a 6-well plate on the third day.
48 h post transfection, collect the medium from transfected cells by passing through a 0.45μM membrane filter.
Replace the culture medium of the cells seeded on the third day with 1 mL lentivirus containing medium plus 1 mL fresh NIH/3T3 culture medium.
Incubate the cells for 48 h and verify the knockdown by western blot or RT-PCR.
The reintroduction of exogenous Myc mGli2 use similar protocol only except for replacing pLKO.1 mGli2 shRNA vector with a lentiviral mGli2 expressing vector FUXW Myc mGli2.
3.3.2. Detection of Gli2 Phosphorylation with Phosphor-Specific Antibodies
Seed the above lentivirus transduced NIH/3T3 cells (with endogenous mGli2 replaced with Myc-tagged exogenous mGli2) (see Note 3).
Until the seeded cells almost reach confluency, serum starve them using cell starving medium for 16–24 h.
Add 0.1μg/mL Shh-N or 100 nM SAG for 12–16 h.
Wash the cells with cold PBS once and collect the cells with centrifugation.
Incubate the cells with cold lysis buffer before centrifugation.
Remove cell debris and incubate resultant lysate with Myc antibody at 4 °C for 1–2 h.
Add pre-washed Protein G beads and rock for another hour at 4 °C.
Wash the beads three times with lysis buffer for 5 min each.
Elute the absorbed protein with 2X SDS loading buffer for 5 min at 95 °C.
Load the elute onto an SDS PAGE gel and transfer the protein to a Nitrocellulose membrane.
After blocking, incubate the membrane with mGli2 phosphor-specific antibody for 2 h at room temperature.
After wash, incubate the membrane using a fluorogenic secondary antibody at room temperature for 1 h.
Scan the membrane with an Odyssey CLx imaging system.
3.4. GST-Ci/Gli2 Fragment Fusion Protein Expression and Purification
Transform BL21 chemically competent cells with pGEX 4T-1 and pGEX 4T-1 Ci/Gli2 fragment plasmids.
Inoculate single colonies and incubate overnight in a 37 °C shaker at 250 rpm.
Dilute with the fresh LB medium at the ratio 1:200 and continue the incubation until OD600 reaches around 0.6.
Induce the GST/GST-fusion protein expression with 0.1 M IPTG and grow the cells at 30 °C for 3–4 h.
Collect the cells by centrifugation at 5000 ×g for 15 min.
Resuspend the bacteria in Lysis or Binding buffer and break the cells with a Sonicator on ice in short burst (15 s on, 15 s off and 15–20 min in total, 30–40% output) (see Note 4).
Centrifuge at 12000 ×g for 15 min at 4 ° C and collect the supernatant.
Incubate the supernatant with Glutathione Sepharose at 4 ° C overnight with shaking.
Wash the beads three times with fresh Lysis/Binding buffer.
Elute GST/GST-fusion proteins with addition of elute buffer and further incubate for 1–2 h at 4 °C.
Load a small aliquot of eluted protein along with BSA standard (for quantification purpose) on an SDS-PAGE gel and determine the protein yield and purity by a Coomassie Blue staining.
Concentrate the protein by ultracentrifugation to final concentration to 0.5–1 mg/mL.
3.5. Bacmid Preparation
Thaw DH10Bac chemical competent cells on ice.
Add 1–10 ng of pFastBac1 vector containing the coding sequence of Ci−PKA and CC FuEE into DH10Bac chemical competent cells and incubate on ice for 30 min.
Heat shock the cells at 42 °C for 45 s.
Cool down the cells on ice for 2 min.
Add 900μL SOC medium and incubate the cells at 37 °C for 4 h with shaking.
Dilute the mixture with SOC medium at 1:10 and 1:100 (namely, 1μL mixture with 99μL SOC medium and 10μL mixture with 90μL SOC medium), for each plasmid, spread 100μL of undiluted mixture along with the above two dilutions (100μL each) on one LB Agar plate.
Grow the cells at 37 °C protected from light for at least 48 h (see Note 5).
Pick up two white colonies for each plasmid and grow them in 1.5 mL LB medium with antibiotics at 37 °C overnight (see Note 6).
Collect cells by centrifugation and resuspend in 300μL Solution I.
Mix with 300μL Solution II gently and incubate at room temperature for 2–3 min.
Neutralize pH by adding 300μL of Solution III and gently invert the tubes 4–6 times.
Sediment the precipitation by centrifugation at highest speed for 10 min.
Carefully withdraw around 800μL supernatant (avoid disturbing the white pellet) and mix gently with 560μL (0.7 V) of isopropanol. Let the mixture sit on ice for 10 min.
Collect the Bacmid DNA by centrifugation at highest speed for 10–15 min and wash with 500μL of 70% ethanol.
Desiccate the Bacmid DNA at room temperature in open air for 10 min (can be done alternatively at a 37 °C incubator).
Dissolve the Bacmid DNA in 40μL TE buffer and store at −20 °C (see Note 7).
3.6. Bacmid DNA Transfection and Baculovirus Amplification
Determine the DNA concentration of recombinant Bacmid.
Seed one million sf9 cells into one well of a 6-well plate (see Note 8).
Allow cells grow in a 27 °C incubator for at least 1–2 h (see Note 8).
Replace the growth medium with 2 mL SF900III medium with 1.5% FBS.
For each transfection, dilute 8μL Cellfectin II reagent (pre-warm to room temperature and gently vortex 1–2 s) in 100μL SF900III medium without any serum or antibiotics.
For each transfection, diluted 1–3μg Bacmid DNA (usually 2–3μL/40μL Bacmid DNA solution) in 100μL SF900III medium without any serum or antibiotics (see Note 9).
Combine diluted DNA and Cellfectin II and incubate at room temperature for 15 min.
Add the above mixture to sf9 cells dropwise.
Let the cells sit at 27 °C for 3 h.
Change medium to regular sf9 growth medium (see Note 10).
Incubate cells at 27 °C for 72 h before collecting medium as P1 virus. Store the P1 virus at 4 °C with protection from light (see Note 11).
Seed one to two million sf9 cells into one well of 6-well plate and let the cells grow 1–2 h at 27 °C.
Add 1–2μL P1 virus into each well and let the cell grow 2–3 days. Collect the medium as P2 virus and verify the expression of intended protein in cells with standard western blot procedure (see Note 12).
For large-scale virus amplification (P3 virus), seed 50–100 mL of sf9 cells at the density of 1 × 106/mL. The next day, add 1/2000 volume of P2 virus and incubate for 3 days at 27 °C.
Collect P3 virus and store at 4 °C for future infection (see Note 13).
3.7. Phosphorylation Mapping of Ci Protein Co-expressed with Active Form of Fu Using Baculovirus System
Seed 2 L of sf9 cells at 1 × 106/mL and infect the cells with 3XFlag-Ci−PKA (20 mL) and HA-CC-FuEE or HA-CC-FuGV (10 mL) P3 virus 24 h later.
48 h after infection, collect cells by centrifugation at 5000 ×g for 20 min.
Resuspend the cell pellet with 100 mL lysis buffer and incubate at 4 °C for 10 min.
Remove the cell debris by centrifugation at 12000 ×g for 20 min.
Incubate the resultant cell lysate with 200μL (bed volume) Flag M2 affinity agarose at 4 °C overnight with shaking.
Wash the beads with fresh cold lysis buffer three times at 4 °C for 5 min each.
Elute the bound protein by boiling the beads for 5 min using 400μL 2X SDS loading buffer.
Load the elute onto a Sephadex 200 10/300 column and collect all Ci-containing fractions, snap freeze with liquid nitrogen, and lyophilize.
Redissolve desalted protein in 40μL of PBS and load (20μL) on an 8% SDS-PAGE gel.
Stain the gel with Coomassie Blue and Ci band (around lug) and cut for further PTM analysis with Mass Spec (see Note 14).
3.8. Expression and Purification of Active Form of Fu from sf9 Cells Using Baculovirus System
Seed 600 mL sf9 cells at the density of 1 × 106/mL.
24 h later, infect the cells with 6 mL Flag-CC-FuEE P3 virus.
48 h after infection, collect cells by centrifugation at 5000 ×g for 20 min.
Resuspend the cell pellet with 40 mL lysis buffer and incubate at 4 °C for 10 min.
Remove the cell debris by centrifugation at 12000 ×g for 20 min.
Incubate the resultant cell lysate with 100μL (bed volume) Flag M2 affinity agarose at 4 °C overnight with shaking.
Wash the beads with fresh cold lysis buffer three times at 4 °C for 5 min each.
Elute the bound protein by incubation with 2X Kinase storage buffer containing 200μg/mL 3X Flag peptide at 4 °C for 2 h with shaking.
Aliquot a small amount of eluted protein for SDS-PAGE/Coomassie Blue Staining to determine the purity and quantity of Fused kinase.
Concentrate kinase to around 0.2μg/μL in 2X Kinase storage buffer and reduce 3X Flag peptide to minimal level at the same time.
Dilute Fused kinase with equal volume of glycerol, aliquot and freeze, store at −80 °C.
3.9. Expression and Purification of Ulk3 Kinase from HEK293T Cells
Seed 2–3 × 106 HEK293T cells into a 10 cm culture dish.
Transfect the cells with 5μg of HA-tagged Ulk3 plasmid using Polyjet in vitro DNA transfection reagent.
48 h after transfection, collect the cells with scrape and centrifugation.
Incubate cells with cold lysis buffer (the same as S2R+ cells) for 10 min at 4 °C with shaking.
After centrifugation, incubate the cell lysate with anti-HA affinity agarose for 2–3 h at 4 °C with agitation.
Wash beads with cold lysis buffer three times at 4 °C 5 min each time.
Elute Ulk3 protein with 2X kinase storage buffer with 500μg/mL HA peptide.
Concentrate eluted protein with Ultracentrifugation (HA peptide is removed at the same time).
Mix the final solution with equal volume of Glycerol and store at −80 °C.
3.10. In Vitro Kinase Assay
We employed three different protein phosphorylation detection approaches after the initial in vitro kinase assay either with Fused or Ulk3 alone or sequential Fused/Ck1 or Ulk3/CK1, namely, radioactive 32P autoradiograph, standard immunoblot procedure with phosphor-specific antibodies, and pIMAGO phosphoprotein detection kit.
3.10.1. In Vitro Kinase Assay with Fu/Ulk3 Alone
For each reaction, set up a 25μL mixture 1X in vitro kinase buffer, 1–2μL sf9 cell derived purified active form of fused kinase or HEK293T cell derived Ulk3 kinase and around 1ug of GST Ci/Gli2 fragment fusion protein as substrate. In case of 32P based assay, add 1ul of γ-32P ATP.
Incubate the reaction mixture at 30 °C for 30 min.
Stop the reaction by adding equal volume of 2X SDS loading buffer and boil for 5 min.
3.10.2. In Vitro Kinase Assay with Sequential Phosphorylation by Fu and CK1
After the first step of in vitro kinase assay with fused, instead of terminating the reaction by 2X SDS loading buffer, add 20μL Glutathione beads (pre-absorbed) to pull down GST fusion protein substrates.
Wash the beads with 1X kinase buffer three times.
The washed beads are used as substrate in final kinase assay with CK1. Everything is similar to the first step except for using CK1 as kinase.
Stop the reaction by adding equal volume of 2X SDS loading buffer and boil for 5 min.
3.10.3. In Vitro Kinase Assay with Sequential Phosphorylation by Ulk3 and CK1
After the first step of in vitro kinase assay with Ulk3, instead of terminating the reaction by 2X SDS loading buffer, add 20μL Glutathione beads (pre-absorbed) to pull down GST fusion protein substrates.
Wash the beads with 1X kinase buffer three times.
The washed beads are used as substrate in final kinase assay with CK1. Everything is similar to the first step except for using CK1 as kinase.
Stop the reaction by adding equal volume of 2X SDS loading buffer and boil for 5 min.
3.10.4. 32P Based Autoradiograph
Load a small aliquot of reaction mixture on an SDS-PAGE and transfer proteins to a PVDF membrane using standard western blot procedure.
Carefully wrap up the membrane in plastic wrap and expose to a film in a film cassette for appropriate time.
Reveal the radioactive signal by a film developer.
3.10.5. Immunoblot with Ci Phosphor-Specific Antibodies
Load a small aliquot of reaction mixture on an SDS-PAGE and transfer proteins to a nitrocellulose membrane using standard western blot procedure.
Block the membrane in blocking buffer for 1 h at room temperature.
Incubate the membrane sequentially with Ci phosphor-specific antibodies (primary) and fluorogenic donkey anti-rabbit antibody (secondary).
Finally, scan the membrane by an Odyssey CLx imaging system.
3.10.6. Immunoblot with pIMAGO Biotin Phosphoprotein Detection Kit
Add 5X IAA solution (10μL) to quenched reaction mixture (40μL) and incubate in the dark for 15 min.
Load a small aliquot of reaction mixture onto an SDS-PAGE gel and transfer proteins to a nitrocellulose membrane (see Note 15).
Block the membrane with 1X blocking buffer at room temperature for 1 h.
Incubate the membrane with 1X pIMAGO buffer supplemented with 1:1000 pIMAGO reagent at room temperature for 1 h.
Wash the membrane with 1X wash buffer three times for 5 min each.
Wash the membrane with TBST for 5 min.
Incubate the membrane with 1X blocking buffer supplemented with 1:1000 Avidin-Fluor at room temperature for 1 h.
Wash the membrane with TBST three times for 5 min each.
Scan the membrane with an Odyssey CLx imaging system.
Acknowledgments
This work was supported by grants from the National Institutes of Health (R35GM118063) and Welch Foundation (I-1603) to J.J.
Footnotes
Ci phosphorylation can be detected as low as 1 mL added Hh-conditioned medium, in certain cases Hh-conditioned medium could be increased to 80%.
We usually added 1μL 10% SDS to reduce the background in fluorogenic secondary antibody incubation.
Endogenous Gli2 was diminished by an shRNA targeting its 3′ UTR and an N-terminal 6XMyc-tagged exogenous Gli2 was introduced in the same NIH/3T3 cell line. The expression level of exogenous Myc-tagged Gli2 was comparable to endogenous protein based on an immunoblot with Gli2 antibody.
The evidence of cell disruption is partial clearing of the cell suspension and avoid protein denaturation caused by overheating.
DH10Bac cells grows slow on these plates with multiple antibiotics, only small colonies can be seen around 24 h.
Try to pick up those big and white colonies surrounded by blue colonies, for optimal contrast between blue and white colonies, plates can be kept at 4 °C protected from light for 1–2 days before final colonies inoculation.
Bacmid DNA store at −20 °C should be good for sf9 cell transfection at least 6–12 months.
Best transfection and baculovirus production can only be achieved with sf9 at the mid-log phase which can be monitored if 95% sf9 cells attach to the bottom of well within 5–10 min.
Avoid transfecting too much Bacmid DNA into sf9 cells which may cause significant cell death.
Sf9 is extremely sensitive to desiccation.
At this point, it is extremely hard to judge if virus is successfully generated and usually the titer of P1 virus is around 106 pfu/mL.
The titer of P2 virus is usually 108 pfu/mL which is no different to that of P3 virus.
For baculovirus amplification, the most critical thing is to keep the MOI as low as 0.01–0.1 and do not over amplify the virus; high MOI and extended incubation will cause accumulated mutations in the genome of virus or appearance of wild-type baculovirus and never amplify virus from P3 virus but always from P2 virus; the most decisive factor of virus shelf life is light protection.
The expression level of Ci protein derived from sf9 cells was estimated to be 2–4μg/L.
To reduce the contamination from the transfer system which might cause high background, a secondary nitrocellulose membrane was added before the gel during the transfer procedure.
Contributor Information
Yuhong Han, Department of Molecular Biology, UT Southwestern Medical Center at Dallas, Dallas, TX, USA.
Jin Jiang, Department of Molecular Biology, UT Southwestern Medical Center at Dallas, Dallas, TX, USA.
References
- 1.Jiang J, Hui CC (2008) Hedgehog signaling in development and cancer. Dev Cell 15 (6):801–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Briscoe J, Therond PP (2013) The mechanisms of Hedgehog signalling and its roles in development and disease. Nat Rev Mol Cell Biol 14 (7):418–431. 10.1038/nrm3598 [DOI] [PubMed] [Google Scholar]
- 3.Nieuwenhuis E, Hui CC (2005) Hedgehog signaling and congenital malformations. Clin Genet 67(3):193–208 [DOI] [PubMed] [Google Scholar]
- 4.Jia J, Jiang J (2006) Decoding the Hedgehog signal in animal development. Cell Mol Life Sci 63(11):1249–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jiang J, Struhl G (1995) Protein kinase A and hedgehog signaling in Drosophila limb development. Cell 80(4):563–572. 10.1016/0092-8674(95)90510-3 [DOI] [PubMed] [Google Scholar]
- 6.Li W, Ohlmeyer JT, Lane ME, Kalderon D (1995) Function of protein kinase A in hedghehog signal transduction and Drosophila imaginal disc development. Cell 80:553–562 [DOI] [PubMed] [Google Scholar]
- 7.Jiang J, Struhl G (1998) Regulation of the Hedgehog and wingless signalling pathways by the F- box/WD40-repeat protein Slimb. Nature 391(6666):493–496 [DOI] [PubMed] [Google Scholar]
- 8.Wang B, Fallon JF, Beachy PA (2000) Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell 100 (4):423–434 [DOI] [PubMed] [Google Scholar]
- 9.Jia J, Amanai K, Wang G, Tang J, Wang B, Jiang J (2002) Shaggy/GSK3 antagonizes Hedgehog signalling by regulating cubitus interruptus. Nature 416(6880):548–552 [DOI] [PubMed] [Google Scholar]
- 10.Price MA, Kalderon D (2002) Proteolysis of the Hedgehog signaling effector cubitus interruptus requires phosphorylation by glycogen synthase kinase 3 and casein kinase 1. Cell 108(6):823–835 [DOI] [PubMed] [Google Scholar]
- 11.Jia J, Zhang L, Zhang Q, Tong C, Wang B, Hou F, Amanai K, Jiang J (2005) Phosphorylation by double-time/CKIepsilon and CKIalpha targets cubitus interruptus for Slimb/beta-TRCP-mediated proteolytic processing. Dev Cell 9(6):819–830 [DOI] [PubMed] [Google Scholar]
- 12.Chen Y, Jiang J (2013) Decoding the phosphorylation code in Hedgehog signal transduction. Cell Res 23(2):186–200. 10.1038/cr.2013.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiang J (2002) Degrading Ci: who is Cul-pable? Genes Dev 16(18):2315–2321 [DOI] [PubMed] [Google Scholar]
- 14.Zhang W, Zhao Y, Tong C, Wang G, Wang B, Jia J, Jiang J (2005) Hedgehog-regulated costal2-kinase complexes control phosphorylation and proteolytic processing of cubitus interruptus. Dev Cell 8(2):267–278 [DOI] [PubMed] [Google Scholar]
- 15.Zhou Z, Yao X, Li S, Xiong Y, Dong X, Zhao Y, Jiang J, Zhang Q (2015) Deubiquitination of Ci/Gli by Usp7/HAUSP regulates Hedgehog signaling. Dev Cell 34(1):58–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ohlmeyer JT, Kalderon D (1998) Hedgehog stimulates maturation of cubitus interruptus into a labile transcriptional activator. Nature 396(6713):749–753. 10.1038/25533 [DOI] [PubMed] [Google Scholar]
- 17.Wang G, Amanai K, Wang B, Jiang J (2000) Interactions with Costal2 and suppressor of fused regulate nuclear translocation and activity of cubitus interruptus. Genes Dev 14 (22):2893–2905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Y, Fu L, Qi X, Zhang Z, Xia Y, Jia J, Jiang J, Zhao Y, Wu G (2013) Structural insight into the mutual recognition and regulation between suppressor of fused and Gli/Ci. Nat Commun 4:2608. 10.1038/ncomms3608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shi Q, Han Y, Jiang J (2014) Suppressor of fused impedes Ci/Gli nuclear import by opposing Trn/Kapbeta2 in Hedgehog signaling. J Cell Sci 127(Pt 5):1092–1103. 10.1242/jcs.142828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han Y, Shi Q, Jiang J (2015) Multisite interaction with Sufu regulates Ci/Gli activity through distinct mechanisms in Hh signal transduction. Proc Natl Acad Sci U S A 112 (20):6383–6388. 10.1073/pnas.1421628112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu C, Zhou Z, Yao X, Chen P, Sun M, Su M, Chang C, Yan J, Jiang J, Zhang Q (2014) Hedgehog signaling downregulates suppressor of fused through the HIB/SPOP-Crn axis in Drosophila. Cell Res 24(5):595–609. 10.1038/cr.2014.29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shi Q, Li S, Li S, Jiang A, Chen Y, Jiang J (2014) Hedgehog-induced phosphorylation by CK1 sustains the activity of Ci/Gli activator. Proc Natl Acad Sci U S A 111(52): E5651–E5660. 10.1073/pnas.1416652111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Q, Zhang L, Wang B, Ou CY, Chien CT, Jiang J (2006) A hedgehog-induced BTB protein modulates hedgehog signaling by degrading Ci/Gli transcription factor. Dev Cell 10(6):719–729 [DOI] [PubMed] [Google Scholar]
- 24.Zhang Q, Shi Q, Chen Y, Yue T, Li S, Wang B, Jiang J (2009) Multiple Ser/Thr-rich degrons mediate the degradation of Ci/Gli by the Cul3-HIB/SPOP E3 ubiquitin ligase. Proc Natl Acad Sci U S A 106(50):21191–21196. 10.1073/pnas.0912008106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Preat T, Therond P, Limbourg-Bouchon B, Pham A, Tricoire H, Busson D, Lamour-Isnard C (1993) Segmental polarity in Drosophila melanogaster: genetic dissection of fused in a suppressor of fused background reveals interaction with costal-2. Genetics 135(4):1047–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shi Q, Li S, Jia J, Jiang J (2011) The Hedgehog-induced smoothened conformational switch assembles a signaling complex that activates fused by promoting its dimerization and phosphorylation. Development 138 (19):4219–4231. . dev.067959 [pii]. 10.1242/dev.067959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou Q, Kalderon D (2011) Hedgehog activates fused through phosphorylation to elicit a full spectrum of pathway responses. Dev Cell 20(6):802–814. S1534–5807(11)00174–2 [pii]. 10.1016/j.devcel.2011.04.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Y, Mao F, Lu Y, Wu W, Zhang L, Zhao Y (2011) Transduction of the Hedgehog signal through the dimerization of fused and the nuclear translocation of cubitus interruptus. Cell Res 21(10):1436–1451. 10.1038/cr.2011.136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Han Y, Wang B, Cho YS, Zhu J, Wu J, Chen Y, Jiang J (2019) Phosphorylation of Ci/Gli by fused family kinases promotes Hedgehog signaling. Dev Cell 50(5):610–626.e614. 10.1016/j.devcel.2019.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Han Y, Xiong Y, Shi X, Wu J, Zhao Y, Jiang J (2017) Regulation of Gli ciliary localization and Hedgehog signaling by the PY-NLS/karyopherin-beta2 nuclear import system. PLoS Biol 15(8):e2002063. 10.1371/journal.pbio.2002063 [DOI] [PMC free article] [PubMed] [Google Scholar]