

Abstract
Alcohol use disorder is a chronic debilitated condition adversely affecting the lives of millions of individuals throughout the modern world. Individuals suffering from an alcohol use disorder diagnosis frequently have serious cooccurring conditions, which often further exacerbates problematic drinking behavior. Comprehending the biochemical processes underlying the progression and perpetuation of disease is essential for mitigating maladaptive behavior in order to restore both physiological and psychological health. The range of cellular and biological systems contributing to, and affected by, alcohol use disorder and other comorbid disorders necessitates a fundamental grasp of intricate functional relationships that govern molecular biology. Epigenetic factors are recognized as essential mediators of cellular behavior, orchestrating a symphony of gene expression changes within multicellular environments that are ultimately responsible for directing human behavior. Understanding the epigenetic and transcriptional regulatory mechanisms involved in the pathogenesis of disease is important for improving available pharmacotherapies and reducing the incidence of alcohol abuse and cooccurring conditions.
1. Introduction
Fermentation, the metabolic process of converting basic carbohydrates into alcohol, has been an essential part of civilization throughout different regions of the world spanning much of human history. Since at least the Neolithic period (10,000–4000 BCE), alcohol has been a part of most human cultural and spiritual ceremonies. Capable of killing bacteria and other infectious agents, fermented beverages provided a reliable, portable, and non-contaminated liquid source of energy. The utility of these alcohol-containing beverages helped the domestication of agriculture and due in part to its psychotropic effects fuel social bonds among members of society. Given the rich societal and historical importance afforded to alcohol, it is not surprising that over 2 billion individuals around the world continue to indulge in the consumption of alcohol (GBD 2016 Disease, Injury Incidence, & Prevalence Collaborators, 2017).
Despite the prevalence of alcohol drinking behavior in our modern society, consumption of alcohol may come with certain inherent risks, such as increased risk of developing cancer, cardiovascular disease, liver disease, anxiety, depression, and other psychiatric disorders. Alcohol consumption has remained one of the leading preventable causes of death and disability, as well as being the most preventable of all substance use disorders (GBD 2016 Alcohol & Drug Use Collaborators, 2018). Alcohol, compared to other legal and illegal drugs of abuse, may in fact be the most harmful substance abused by humans (Nutt, King, Phillips, & Independent Scientific Committee on Drugs, 2010). Excessive alcohol abuse has been estimated to contribute a socioeconomic burden of ~$250 billion for the United States of America (Sacks, Gonzales, Bouchery, Tomedi, & Brewer, 2015); however, such costs are likely to be underestimated due to a lack of availability and underreporting of alcohol-related outcomes. In the U.S.A. alone, alcohol abuse has been known to annually account for ~88,000 deaths of citizens (globally accounting for 6% of all deaths); which far surpasses many other substances of abuse such as opioids and psychomotor stimulants. Problematic drinking of alcohol over the past several years has substantially increased, putting many members of society at severe risk of death and disease. Excessive alcohol consumption behavior has particularly increased in older adults, the socioeconomical disadvantaged, women, and racial minorities (Grant et al., 2017). Such widespread, and high-risk, alcohol drinking behavior is also a major factor in other cooccurring human disorders, with evidence for comorbidity of alcohol use disorder with more than 200 other health conditions.
Treatment of alcohol use disorder, and other related disorders, represents a major societal health issue. Evidence-based treatment is available for alcohol use disorder; however, less than 10% of individuals receive any form of therapeutic intervention (Schmidt, 2016). Among individuals fortunate enough to receive treatment, even fewer are prescribed any FDA approved medications. There are currently three medications approved by the FDA for treating alcohol use disorder: disulfiram, acamprosate, and naltrexone (Litten, Allen, & Fertig, 1996). All three of these pharmacotherapies are known to induce widespread changes in gene expression among different cell-types, reversing adaptations that have occurred due to alcohol and other substances of abuse. Although the molecular mechanisms responsible for such system-wide changes in gene expression are not fully known, these changes may suggest a convergence on epigenetic processes and concordant mechanisms of transcriptional regulation.
Addiction to alcohol and other drugs of abuse is the result of harmful uncontrollable substance abuse, pre-existing genetic factors, and cooccurring human disorders. The unchecked and continual misuse of addictive substances is known to cause a number of molecular adaptations throughout different tissues and cellular systems. Persistent alterations of biomolecules controlling cellular activity is dictated by epigenetic and transcriptional regulation of gene expression networks. Substance abuse seizes control of these expression systems, establishing a new biological framework that further perpetuates addictive behavior. Distinguishing all of the disparate but interrelated components from affected biological processes is vital for understanding and helping to reverse the pathophysiology of human disease.
2. Genetics, epigenetics, and epigenomics
Genetic factors account for ~50% of the heritability of the risk for developing alcohol use disorder, with the remaining risk accounted for by the environment and additional non-genetic factors (Ducci & Goldman, 2008). The vast majority of single nucleotide polymorphisms (SNPs) implicated in alcohol use disorder, as well as other human diseases, are located in non-coding regions of the human genome. SNPs located in specific non-coding regions, such as enhancers and promotors, are known to impact transcriptional regulation leading to phenotypic diversity and altered cellular functions (van Arensbergen et al., 2019). Accessibility of genomic mutations associated with human disease may be determined by the epigenome (Polak et al., 2015), the complete set of epigenetic elements present within the cellular environment across the entire genome. This continual crosstalk among genetic and epigenetic cellular adaptations are essential determinants of physiological and behavioral phenotypes.
Epigenetics is broadly defined by the NIH Roadmap Epigenomics Project (http://www.roadmapepigenomics.org/) as “both heritable changes in gene activity and expression (in the progeny of cells or of individuals) and also stable, long-term alterations in the transcriptional potential of a cell that are not necessarily heritable” (Bernstein et al., 2010). The brain is a heterogeneous mixture of cell-types including multiple subtypes of neurons, astrocytes, oligodendrocytes, and microglia. Epigenetic machinery present within each of these diverse cell types function as central intermediaries coordinating cascades of gene expression based on existing genetic variation and in response to external stimuli (Fig. 1). The machinery encompassing the epigenome consists of multiple biochemical constituents, with each requisite part acting in concert with one another to achieve homeostasis.
Fig. 1.
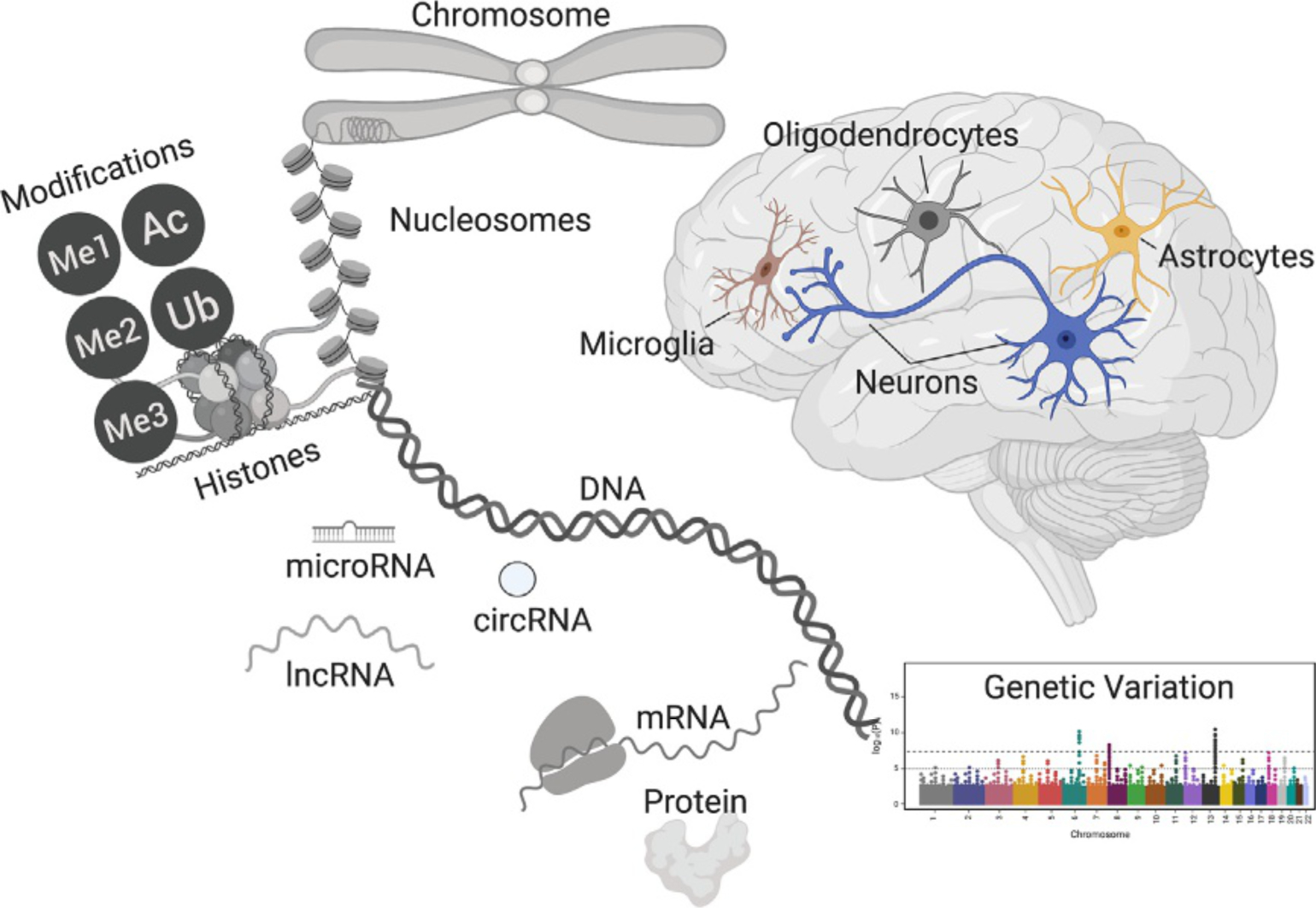
Overview of potential biochemical processes involved in epigenomic regulation of cellular function for the neuropathology of disease. The molecular phenotypes of individual cells in the central nervous system (i.e., neurons, astrocytes, microglia, and oligodendrocytes) are influenced by the continual interaction of single nucleotide variation in DNA and epigenetic regulation. DNA is condensed into chromatin through the association with histone proteins. Post-translational modification of histones may occur through ubiquitination (Ub), acetylation (Ac), mono- (Me1), di- (Me2), and tri-methylation (Me3). The regulation of mRNA and proteins may also be regulated by non-coding RNA transcripts including microRNAs, circular RNAs (circRNAs), and long non-coding RNAs (lncRNAs).
3. Histones and post-translational modifications
Harnessed inside the nucleus of eukaryotic cells nuclear DNA is bundled together by tightly wrapping around histone proteins. Currently five major histone proteins are known to exist, with H2A, H2B, H3 and H4 serving as the core members. Eight histones spooled with DNA form a nucleosome, a highly stable protein-DNA complex. Nucleosomes form compact loops of chromatin, densely wound together to create individual chromosomes. Transcriptional activity within the cell is guided, at least in part, by post-translational modifications creating functional states of condensed heterochromatin and open euchromatin within the nuclear environment. These post-translational modifications occurring at distinct amino acid residues on the terminal tail of histone proteins (mainly H3 and H4) represent a histone code that facilitates chromatin remodeling. Determining the physical abundance and arrangement of these histone modifications is necessary for mapping the dynamic landscape of transcribed features expressed in different cell-types and brain-regions.
Specific enzymes and effector proteins, commonly referred to as readers, writers, and erasers, are ultimately responsible for histone modifications and transcriptional reprogramming. Readers are accountable for recognizing the diverse array of chemical moieties involved in post-translational modifications of histones, determine the sequence context, and assist in the recruitment of additional protein complexes for steering nuclear processes such as DNA repair and transcription. Histone methylation (Me), acetylation (Ac), and ubiquitination (Ub) are examples of epigenetic markers used by molecular readers to convey the general state of transcription at a given genomic locus. The bidirectional transfer of Me, Ac, and Ub to select residues on histone tails by functionally distinct writer and eraser enzymes determines the transitional state of gene activation depending on the type and location of histone modification. For example, histone acetyltransferases (HATs) and histone deacetylases (HDACs) manage the addition and removal of acetyl moieties on lysine charged residues in the N-terminus tail of core histone proteins. This biochemical modification, or epigenetic tag, conferred by addition of an acetyl group helps neutralize the positively charged amino acid, loosening the compact chromatin which is then poised for transcriptional activators of gene expression.
4. Non-coding sequences
Less than 2% of sequences in the human genome are protein-coding (International Human Genome Sequencing Consortium, 2004; Lander et al., 2001; Venter et al., 2001). Remarkably, approximately 90% of genetic variants identified in human genome-wide association studies reside in non-coding regions (Edwards, Beesley, French, & Dunning, 2013; Wojcik et al., 2019). The enrichment of genetic variants in non-coding regions of the genome suggests a potential regulatory role of gene expression and interaction with epigenetic mechanisms involved in cellular function. A comprehensive analysis of 147 human cell types by the Encyclopedia of DNA Elements (ENCODE) Project has demonstrated genetic variants are closely associated with regulatory regions of the genome (The ENCODE Project Consortium, 2012). In these cell-types ENCODE has systematically determined more than 80% of the human genome has some form of biochemical activity, contributing to chromosome architecture, epigenetic inheritance, and long-term regulation of gene expression.
The human genome is pervasively transcribed, with at least 75% of sequence content capable of being actively regulated (Derrien et al., 2012; Djebali et al., 2012). Copied directly from DNA actively regulated loci creates single-stranded ribonucleic acids (RNA) found in all cells. Rendering biological active nucleic acid molecules from the genetic information stored in DNA, RNA is known to be essential to cellular function. In addition to the well-known relationship between DNA and RNA in the formation of proteins, RNA has been shown to participate in biochemical reactions independent of DNA and proteins (Guerrier-Takada, Gardiner, Marsh, Pace, & Altman, 1983; Kruger et al., 1982). These now classical biochemistry studies supported the concept of a RNA as foundational molecules in the origins of life (Gilbert, 1986; Rich, 1962), and sparked a series of investigations demonstrating the potential functional properties of distinct RNA molecules. Since their initial discovery several distinct RNA biotypes have been identified with specialized roles in cellular behavioral (Table 1). Although the precise number of RNAs present in the human transcriptome is still unknown, RNA molecules have been implicated in the regulation of gene expression, alternative splicing, protein synthesis, protein translation, and chromatin structure.
Table 1.
Brief summary of RNA biotypes with known roles in molecular biology and addiction.
| Type of RNA | Description | Biological function |
|---|---|---|
| mRNA | Messenger RNA (e.g., Bdnf) | Translation into proteins |
| tRNA | Transfer RNA | Regulation of protein translation |
| rRNA | Ribosomal RNA | Protein synthesis |
| snoRNA | Small nucleolar RNA | Chemical modification of tRNA and rRNA |
| snRNA | Small nuclear RNA | Processing of pre-mRNA |
| siRNA | Small interfering RNA | Double-stranded RNA regulation of mRNA degradation |
| piRNA | Piwi-interacting RNA | Post-transcriptional regulation of RNA silencing |
| miRNA | MicroRNA (e.g., miR-9) | Anti-sense sequence complementarity regulation of gene expression |
| circRNA | Circular RNA (fused 3′-UTR and 5′-UTR ends, e.g., circ_1639) | Anti-sense sequence regulation of microRNAs |
| lncRNA | Long non-coding RNA (e.g., MALAT1) | Binding and regulation of DNA, RNA, and protein |
Less than 2% of the genome consists of protein-coding (mRNA) genes. A significant fraction of transcribed sequences from the mammalian genome are represented by a diverse set of non-coding genes: tRNA, rRNA, snoRNA, snRNA, siRNA, piRNA, miRNA, circRNA, and lncRNA. Each of class of these non-coding RNAs participates in discrete biochemical functions and may have significant roles in addiction to alcohol and other substances of abuse.
Messenger RNA (mRNA), which is generally the most widely studied type of RNA, are single-stranded RNAs copied directly from the DNA template to create cellular proteins. Alternatively splicing of mRNA is responsible for producing a wide variety of functional proteins within different tissues and cell-types. Both pre- and post-transcriptional mechanisms tightly control the expression of mRNA prior to their translation into cognate proteins. Among the myriad of processes capable of regulating mRNA expression are a tightly orchestrated network of non-coding RNAs (Kleaveland, Shi, Stefano, & Bartel, 2018). Understanding the specialized roles of different types of non-coding RNA is important for basic science research and translating these discoveries into meaningful real-world applications such as improving human health.
Although recent scientific research has witnessed an exponential increase in high-throughput biological assays, the exact number of biochemically active non-coding RNAs that exist in nature remains unknown. With a continued emphasis on studying protein-coding genes, the vast majority of experiments that utilize high-throughput genomic and transcriptomic screens are not designed to capture non-coding sequences (Eddy, 2002). Thus, the information gained from such large-scale experiments may be incomplete without the inclusion of non-coding RNA, which is estimated to make up 98–99% of all transcriptional output (Mattick, 2001, 2003). Similar to their protein-coding counterparts non-coding elements of the human genome are often under a similar degree of evolutionary constraint and adaptation (Lindblad-Toh et al., 2011). This may suggest non-coding elements are essential to many aspects of life and possess important functional roles in human health, including regulatory networks that have an effect on the human brain (Lee, Bang, Choi, & Kim, 2020). Continued investigation of such evolutionary conserved non-coding regulatory networks will help paint a more complete picture of fundamental biology and dispel the myth that non-coding DNA/RNA represents junk genetic material.
Crosstalk among protein-coding and non-coding genes within different biological compartments is necessary for cellular homeostasis. Many non-coding RNAs are recognized to have pivotal roles in translation, the process of converting mRNA into specific amino-acids to form macromolecular proteins. Small non-coding RNAs, known as transfer RNAs (tRNAs), work in concert with the ribosome to decode mRNA sequence information and efficiently create functional protein products. Ribosomal and transfer RNAs are actively regulated by small nucleolar RNA (snoRNA), conferring sequence-specific chemical modifications of these non-coding RNAs (Dupuis-Sandoval, Poirier, & Scott, 2015; Kiss, 2002). Selective modification of tRNA sequences are known to affect tRNA biogenesis and the rate of protein translation. Such intricate biochemical relationships among rRNA, tRNA, and snoRNA highlights the remarkable regulatory potential of non-coding RNAs in the production of protein from mRNA during translation. The expression and biochemical activity of these types of non-coding RNAs may also act independently of mRNA expression to meet cellular demands (Torrent, Chalancon, de Groot, Wuster, & Madan Babu, 2018).
Similar to alternative splicing of proteins, intracellular processing of non-coding RNAs contribute to the availability of diverse nucleic acid sequences and their function. Both snoRNAs and tRNAs are capable of being cleaved into smaller fragments of non-coding RNA similar to microRNAs (Maute et al., 2013; Scott & Ono, 2011). MicroRNAs are an abundant class of evolutionary conserved small non-coding RNAs containing approximately 22 nucleotides (Bartel, 2004). Due in part to their small size, microRNAs are generated from larger host genes inside the nucleus forming stable double-stranded RNA. Following export from the nucleus into the cytosol, double-stranded RNA is cleaved by the RNAse III enzyme DICER to produce mature microRNAs (Hutvagner et al., 2001; Ketting et al., 2001). Originally identified in Caenorhabditis elegans (Fire et al., 1998), microRNAs act as potent and selective inhibitors of post-transcriptional gene expression. Through anti-sense sequence complementarity, the canonical function of microRNAs is now recognized as binding to the 3′-untranslated region (3′-UTR) of mRNA to inhibit protein translation and hasten mRNA degradation. Through sequence-specific binding, individual microRNAs can bind and regulate the expression of several hundred mRNAs (Friedman, Farh, Burge, & Bartel, 2009). The large number of mRNAs targeted by microRNAs affords them the opportunity to simultaneously alter the expression of multiple biological pathways and signaling cascades.
Complementary sequence binding of nucleotides by microRNAs is not restricted to protein-coding genes. Additional non-coding RNAs such as transcribed pseudogenes, circular RNA (circRNA) and long non-coding RNA (lncRNA) may also form RNA-RNA binding interactions with microRNAs; creating an elaborate system of competing endogenous RNA networks inside the cell (Cesana & Daley, 2013; Salmena, Poliseno, Tay, Kats, & Pandolfi, 2011). This method of post-transcriptional regulation of coding and non-coding genes facilitates a complex hierarchy of homeostatic interactions, constantly shifting depending on the cellular environment (Bosia et al., 2017; Chiu et al., 2017). Determining the broader equilibrium between these stable and unstable RNA interaction networks may shape the trajectory of developmental and pathological conditions.
Circular RNAs, as the name implies, are single-stranded RNAs with their 3′-UTR and 5′-UTR ends fused together to create a highly stable RNA molecule (Hsu & Coca-Prados, 1979; Sanger, Klotz, Riesner, Gross, & Kleinschmidt, 1976). Processed by the spliceosome machinery inside the nucleus, circRNAs are the byproducts of abnormal alternative splicing from parent genes (Memczak et al., 2013; Nigro et al., 1991). Through abundant localized expression in specific mammalian tissues (Capel et al., 1993; Hansen et al., 2011) circRNAs function as molecular sponges soaking-up sequence matched microRNAs (Hansen et al., 2013). An individual circRNA can harbor multiple conserved binding sites to efficiently target microRNAs and microRNA families, as well as sequester other types of RNA and RNA binding proteins (Wilusz & Sharp, 2013).
Akin to circRNAs, lncRNAs have also been implicated in post-transcriptional regulation of gene expression and miRNA sponges (Militello et al., 2017). Lacking an open reading frame, lncRNAs are biophysically defined as an RNA greater than 200 nucleotides in length that cannot be translated into proteins. Based on their genomic properties lncRNAs may be assigned to particular subcategories (Mattick & Rinn, 2015; Peschansky & Wahlestedt, 2014). The functional annotation of the mammalian genome research consortium has experimentally categorized coding and non-coding transcript models into four separate gene categories: (1) mRNA, (2) divergent promoter lncRNA, (3) intergenic promoter lncRNA, and (4) enhancer lncRNA (Hon et al., 2017). Massively parallel reporter assays have demonstrated each of these gene biotypes have distinctive functional modes of transcriptional regulation and expression patterns (Mattioli et al., 2019). Compared to mRNAs the expression of lncRNAs have greater tissue and cell-type specificity. The total number of lncRNAs found in nature is unknown; however, current evidence suggest at least ~100,000 lncRNAs have important functional roles in human physiology and disease (Fang et al., 2018; Ma et al., 2019). Despite the seemingly ever-growing catalog of lncRNAs, only a small fraction of them have been rigorously studied for cellular function. But due to their structure and chemical composition an individual lncRNA is capable of being tethered to other RNAs, strands of DNA, and proteins. This range of biochemical interactions contributes to the modular ability of lncRNA, serving as molecular scaffolds directing intracellular traffic among vastly different types of biomolecules.
Every eukaryotic cell contains an incredible mixture of coding and non-coding DNA and RNA. It is now increasingly clear that the protein-coding portion of this pool of nucleotides is vastly outnumbered. The non-coding elements of eukaryotic cells are exquisitely interlaced with protein-coding genes, fashioning a highly structured regulatory system equipped with multiple checks and balances. Deciphering all of the biophysical interactions and other functional relationships between coding and non-coding genes is a monumental challenge for scientists. The combination of computational biology and high-throughput biochemical assays has significantly accelerated our ability to systemically examine the interdependence of molecular networks, but with limited resolution. Navigating this burgeoning terrain of RNA biology is bound to discover new principles of organization maintained by living systems. Detailed mechanistic studies of the different non-coding entities traversing the regulome will help ascertain the origins of abnormal behavior and disease.
5. Methylation, acetylation, and ubiquitination
The mammalian genome is a mosaic of highly organized DNA isochores, ranging between 40% and 80% in guanosine cytosine (GC) content (Galtier, Piganeau, Mouchiroud, & Duret, 2001). Variation in GC-content is evolutionary correlated with gene expression and rates of recombination (Jensen-Seaman et al., 2004; Semon, Mouchiroud, & Duret, 2005). Approximately 72% of 5′—C—phosphate—G—3′ (CpG) islands are located in clusters near the known transcription start sites of gene promoter regions (Saxonov, Berg, & Brutlag, 2006), suggesting a correlative role in transcriptional regulation. Methylation patterns of DNA at CpG islands is conserved across species, with recognized roles in development and disease (Greenberg & Bourc’his, 2019; Smith & Meissner, 2013). Methylation of DNA, typically 5-methylation of cytosines (5mC) or 5-hydroxymethylation of cytosine (5-hmC), is associated with gene repression blocking the activity of transcription factors and interacting with other DNA binding bindings.
In comparison to glial cells, DNA methylation patterns within the brain are markedly more robust in neuronal cells (Lister et al., 2013). The overall configuration of DNA methylation, and the activity of DNA metabolizing enzymes, is associated with size of the mammalian brain and cognitive ability (Sousa, Meyer, Santpere, Gulden, & Sestan, 2017). Thus, altering patterns of DNA methylation within different brain-regions and cell-types at distinct genomic loci could have an impact on the susceptibility for neurological and neuropsychiatric disorders. Chronic stress, during different stages of development and throughout adulthood, is recognized as a prominent feature of deteriorating mental health and well-being. Epigenome-wide association studies and candidate gene studies of DNA methylation have revealed several potential gene × environment interactions underlying chronic stress (Gottschalk, Domschke, & Schiele, 2020). Using bisulfite sequencing an early life stress model has shown DNA hypermethylation of tyrosine hydroxylase in dopaminergic neurons, which is functionally dependent on glucocorticoid receptor signaling (Niwa et al., 2013). Genome-wide methylation studies of postmortem human brain tissue for alcohol use disorder have demonstrated differential DNA methylation and gene expression of multiple stress-related genes, including the glucocorticoid receptor (Gatta et al., 2019). In addition to the stress responsive and dopaminergic system, multiple genes and biological pathways have been reported to exhibit alterations in DNA methylation for addiction and comorbid disorders (Bredy, 2017; Brown & Feng, 2017). These DNA modifications consequently play a part in coordinately regulating gene expression networks, leading to maladaptive CNS function and behavior.
In addition to methylation of DNA molecules, biochemical modifications of DNA binding proteins can finely tune the flow of biological information within the nucleus. Responding to synaptic signaling, the neurotransmitters serotonin and dopamine have recently been shown to cause monoaminyl modifications of histones that account for changes in transcriptional plasticity and behavior (Farrelly et al., 2019; Lepack et al., 2020). Such post-translational modifications of histones are important mediators of chromatin structure and successive regulation of gene expression. Each post-translational modification of histones may collectively integrate information from the environment to carefully finely-tune cellular and behavioral plasticity (Campbell & Wood, 2019). Genome-wide changes in H3K4me3 are not significantly correlated with overall changes gene expression due cocaine addiction or alcohol abuse (Zhou, Yuan, Mash, & Goldman, 2011); however, layering these H3K4me3 and gene expression measurements identifies coherent biological networks relevant to each condition (Farris, Harris, & Ponomarev, 2015). Unlike the canonical role of H3K4me3 in the activation of transcription, tri-methylation of the 27th lysine residue of histone H3 (H3K27me3) is associated with transcriptional repression. H3K27me3 methylation is down-regulated in the brain of alcohol-dependent rats, leading to increased gene expression of H3K27me3 associated binding regions (Johnstone et al., 2019). Acetylation of H3K27 (H3K27ac) prevents H3K27 tri-methylation (Tie et al., 2009), typically coinciding with sites of H3K4me3 to promote activation of gene expression. The bidirectional control of transcription by specific post-translational modification of histone residues proposes a set of molecular mechanisms capable of redirecting transcription, and reshaping control of behavior.
Specific enzymes are responsible for the biochemical modification of histone tails. Histone acetyltransferases (HATs) and histone deacetylases (HDACs) respectively add and remove acetyl groups to the N-terminus of histone tails. Systemic administration of HDAC inhibitors trichostatin A (TSA), suberanilohydroxamic acid (SAHA), and entinostat (MSG-275) decreases binge-like alcohol consumption in mice (Warnault, Darcq, Levine, Barak, & Ron, 2013). Treatment of alcohol-dependent rats with the HDAC inhibitor SAHA alleviates withdrawal, improving behavioral signs of depression (Chen et al., 2019). Acute ethanol exposure can inhibit endogenous HDAC activity, altering histone acetylation, gene expression, and the anxiolytic properties of ethanol (Pandey, Ugale, Zhang, Tang, & Prakash, 2008; Sakharkar, Zhang, Tang, Shi, & Pandey, 2012). Patterns of histone acetylation induced by ethanol regulate critical genes involved in alcohol tolerance (Ghezzi et al., 2013; Wang, Krishnan, Ghezzi, Yin, & Atkinson, 2007). The ethanol-induced changes in HDAC activity may be particularly sensitive during periods of development, affecting long-lasting behaviors into adulthood (Sakharkar et al., 2016). Examining the acute and chronic effects of ethanol on the CNS strongly supports a functional role for molecular enzymes that can remodel chromatin structure and alter the transcriptional landscape of cellular states linked to behavioral adaptations involved in addiction.
Determining the epigenetic state of CNS activity is challenging due to considerable heterogeneity among cell-types and the sheer number of potential post-translational histone modifications. In order to effectively coordinate transcriptional regulation throughout the genome, many of these epigenetic marks are capable of functionally coalescing based on cellular and environmental demands. The cooperative actions of multiple biological catalysts and scaffolds redefines the accessibility of specific gene response elements necessary for the succeeding waves of intracellular biogenesis. Our ability to functionally map out the epigenomic blueprint of individual cells and cellular networks is essential for explaining the molecular basis of behavior.
6. The role of non-coding RNAs in addiction
Non-coding RNAs offer an additional layer of security for balancing the inner workings of cells. Every bit as diverse and complex as proteins, non-coding RNAs participate in a broad spectrum of biochemical processes. Acting at both the transcriptional and post-transcriptional level of regulation non-coding RNAs are a rich source of sequences to manipulate the overall population of stably expressed genes inside the nuclear and cytoplasmic environments. It is still a matter of considerable debate as to how much of non-coding RNA is indeed functional; however, the confluence of multiple types of non-coding RNAs participating in a wide variety of molecular functions underscores their utility within diverse cell-types (Gomes, Nolasco, & Soares, 2013). The versatility of non-coding RNAs are well suited to negotiate the complex interplay of genetic and environmental factors in the emergence of substance abuse and related human disorders.
Since they were initially discovered in the 1950s and 1960s due to their high expression and valuable contribution to protein synthesis, a growing number of non-coding RNAs have been formally classified (Brosius & Raabe, 2016). Small non-coding RNAs are processed transcripts measuring less than 200 bases in length, which includes small nucleolar RNA (snoRNA), small nuclear RNA (snRNA), small interfering RNA (siRNA), microRNA (miRNA), piwi-interacting RNA (piRNA), and transfer RNA (tRNA) (Table 1). Each of these small non-coding RNAs have recognized intrinsic biological functions necessary for cellular viability and the pathogenesis of disease. Sussing out their overlapping and non-overlapping roles in communicating cellular activity is pertinent for establishing connections with the neurobiology of behavior.
MicroRNAs are presently the most well studied non-coding RNA in addiction to alcohol and other drugs of abuse. Through sequence-specific binding, particularly of the 3′-UTR of mRNA, miRNAs post-transcriptionally regulate nearly all of expressed protein-coding genes (Friedman et al., 2009). All drugs of abuse can induce changes in the expression of miRNAs and their downstream targets (Smith & Kenny, 2018). Microarray studies of the superior prefrontal gyrus from human postmortem brain tissue has shown an up-regulation of ~48 miRNAs due to chronic alcohol abuse (Lewohl et al., 2011). A comparable list of alcohol-induced changes in miRNA expression has also been reported for human neuroblastoma cells (Yadav et al., 2011). Many of these changes in miRNA expression are evolutionary conserved in an animal models of alcohol dependence, showing expected changes in mRNA and protein expression networks (Gorini, Nunez, & Mayfield, 2013; Nunez et al., 2013; Tapocik et al., 2013). Experimental perturbation of specific microRNAs have shown a causal correlation in their ability to alter alcohol consumption (Bahi & Dreyer, 2013; Darcq et al., 2015; Tapocik et al., 2014), anxiety-like behavior (Bahi, 2017; Teppen, Krishnan, Zhang, Sakharkar, & Pandey, 2016), tolerance (Pietrzykowski et al., 2008), and neurotoxicity (Coleman Jr., Zou, & Crews, 2017; Yadav et al., 2011). The phenotypic effect determined for each microRNA has generally been attributed to their regulatory capacity of individual protein-coding genes with the identical corresponding phenotypes. Expression profiling studies have consistently demonstrated changes in multiple microRNAs with a number of predicted molecular targets, including shared sets of redundant microRNA response elements. The pleotropic action of miRNAs and additional transcripts contends that the expression for an entire legion of coding and non-coding genes are innately aligned in response to internal and external stimuli.
Expressed alongside mRNA and small non-coding RNAs are large non-coding RNAs typically above 200 nucleotides in length, such as transcribed pseudogenes, circular non-coding RNA (circRNA) and long non-coding RNA (lncRNA). Unlike small non-coding RNA, all three of these non-coding RNAs have been suggested to contain regions capable of synthesizing small peptides (Ji, Song, Regev, & Struhl, 2015; Kim et al., 2014; Pamudurti et al., 2017). It currently remains unknown how stable these peptides are, and if they have any functional significance. In spite of this controversy, many of these well-established non-coding RNAs have genuine biological roles in regulation of gene expression and remodeling of the eukaryotic genome. Yet the overwhelming majority of large non-coding RNAs remain to be studied, with likely many new and unforeseen functions yet to be discovered.
CircRNAs are reported to be significantly enriched in the mammalian brain with the potential to regulate synaptic plasticity (Rybak-Wolf et al., 2015; You et al., 2015). Exposure to the psychomotor stimulants cocaine and methamphetamine alters the expression of several circRNAs in specific brain-regions and cortical neurons, respectively (Bu et al., 2019; Li et al., 2019). Bioinformatics analysis of differentially expressed circRNAs further implicated an RNA interaction network with several miRNAs and the regulation of candidate mRNAs. Selective knockdown of the top circRNA for cocaine treatment (mmu_circRNA_002381) using siRNA in Neuro2a cells confirmed down-regulation of brain-related transcripts Limk1 and Bdnf (Bu et al., 2019). To the best of our knowledge there are no other published reports on circRNA expression profiling in the CNS following exposure to substances of abuse. Chronic alcohol exposure in mice has revealed changes in distinct profiles of circRNA expression for heart and liver tissue (Dou et al., 2020; Meng, Wang, You, Huang, & Li, 2019; Yang et al., 2018). Mice chronically exposed to alcohol using the Lieber-DeCarli liquid diet to model alcoholic-liver disease demonstrate increased expression of circ_1639 in Kupffer cells (Lu et al., 2019), resident liver macrophages known to contribute to alcohol-induced inflammation. Transfection of circ_1639 inhibited the expression of miR-122, and led to an increase in several pro-inflammatory molecules (Lu et al., 2019). Taken together these studies and others may indicate that circRNAs may be novel biomarkers for alcohol-related tissue damage (Chien et al., 2020). Additional studies are certainly needed to determine the mechanism of action for individual circRNAs; however, their endogenous structure and stable exosome expression within human serum may further advocate for their feasibility as biomarkers of disease (Li et al., 2015).
The largest known class of linear RNA molecules with biochemical function in a sundry of tissues and cell-types are long non-coding RNAs (Quinn & Chang, 2016; St Laurent, Wahlestedt, & Kapranov, 2015). Strongly expressed during embryogenesis the lncRNA H19, named after a library screen of cDNA clones from 19-day old fetal liver samples, was the first lncRNA to be discovered (Pachnis, Belayew, & Tilghman, 1984). Since their initial discovery, H19 and other lncRNAs have been linked to a number of chronic diseases (Bao et al., 2019; Delas & Hannon, 2017; Shi, Sun, Liu, Yao, & Song, 2013). The exponential rise in RNA-sequencing has spurred the analysis and investigation of lncRNAs, with at least 58,648 lncRNAs experimentally verified in the human transcriptome (Iyer et al., 2015). Predisposition for the risk developing an alcohol use disorder may be related to genetic variation of specific lncRNAs (Adkins et al., 2017; Gelernter et al., 2014; Meyers et al., 2020; Peng, Bizon, Gizer, Wilhelmsen, & Ehlers, 2019; Polimanti et al., 2017; Procopio et al., 2013). A network of coordinately expressed lncRNAs in the human brain, inversely associated with protein-coding genes, are correlated with lifetime consumption of alcohol abuse (Farris, Arasappan, Hunicke-Smith, Harris, & Mayfield, 2015; Farris, Harris, et al., 2015). Expression of lncRNA MALAT1 is significantly increased in multiple brain-regions of human alcoholics and rats following alcohol withdrawal (Kryger, Fan, Wilce, & Jaquet, 2012). Another well annotated lncRNA that is evolutionary conserved known as BDNF anti-sense lncRNA (BDNF-AS) (Modarresi et al., 2012), is significantly upregulated in human amygdala during early onset of alcohol abuse (Bohnsack, Teppen, Kyzar, Dzitoyeva, & Pandey, 2019). The grand total of lncRNAs and their biological involvement in the alcohol use disorder and other human conditions is still unknown; however, it is unquestionably an emerging area of academic research. Uncovering the molecular essence for all of the lncRNAs in the genome is a daunting task. Tackling this enormous problem will require the use of sophisticated computational models and detailed methodical studies of affected molecular circuitry.
7. Conclusion and future directions
An inordinate amount of DNA and RNA sequence data currently being produced is pushing the boundaries of genomics and epigenomics. Expanded access to these libraries of information is beginning to meticulously catalog a reservoir of unchartered territories in biology. Intensive investigation of these mounting resources is helping to outline the molecular basis of human health and disease. For several decades the protein-coding genome has been the main fulcrum of cellular biology; however, an appreciation for the functional roles of non-coding RNA is gaining ground within the scientific community. The compulsory interdependence of protein-coding and non-coding RNA is a fundamental property of most living systems. Exploring the latent biological processes that occur at this interface of genetics and epigenetics will continue to be a bedrock of innovation. Technological advancements, such as those under way in the fields of single-cell sequencing, spatial transcriptomics, and bioinformatics, are forcing researchers to reexamine existing hypotheses and forge new frontiers of scientific knowledge. This newfound wealth of information will be transformative not only to basic science, but also translational research. A rapidly growing division of clinical trials and medication development is aimed at therapeutics that are capable of uniquely targeting RNA and epigenetic networks (Dowdy, 2017; Wang, Zuroske, & Watts, 2020). Future treatments for human diseases such as alcohol use disorder may thus depend upon the ability to functionally characterize non-coding genes once ascribed to be junk.
Acknowledgment
This work was supported by NIH/NIAAA U01 AA020926 (rdm), R01 AA012404 (rdm), and R00 AA024836 (spf).
Footnotes
Disclosure
The authors declare no conflict of interest.
References
- Adkins AE, Hack LM, Bigdeli TB, Williamson VS, McMichael GO, Mamdani M, et al. (2017). Genomewide association study of alcohol dependence identifies risk loci altering ethanol-response behaviors in model organisms. Alcoholism, Clinical and Experimental Research, 41, 911–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahi A (2017). Hippocampal BDNF overexpression or microR124a silencing reduces anxiety- and autism-like behaviors in rats. Behavioural Brain Research, 326, 281–290. [DOI] [PubMed] [Google Scholar]
- Bahi A, & Dreyer JL (2013). Striatal modulation of BDNF expression using microRNA124a-expressing lentiviral vectors impairs ethanol-induced conditioned-place preference and voluntary alcohol consumption. The European Journal of Neuroscience, 38, 2328–2337. [DOI] [PubMed] [Google Scholar]
- Bao Z, Yang Z, Huang Z, Zhou Y, Cui Q, & Dong D (2019). LncRNADisease 2.0: An updated database of long non-coding RNA-associated diseases. Nucleic Acids Research, 47, D1034–D1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP (2004). MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell, 116, 281–297. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, et al. (2010). The NIH roadmap epigenomics mapping consortium. Nature Biotechnology, 28, 1045–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnsack JP, Teppen T, Kyzar EJ, Dzitoyeva S, & Pandey SC (2019). The lncRNA BDNF-AS is an epigenetic regulator in the human amygdala in early onset alcohol use disorders. Translational Psychiatry, 9, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosia C, Sgro F, Conti L, Baldassi C, Brusa D, Cavallo F, et al. (2017). RNAs competing for microRNAs mutually influence their fluctuations in a highly non-linear microRNA-dependent manner in single cells. Genome Biology, 18, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredy TW (2017). DNA modifications in the brain: Neuroepigenetic regulation of gene expression. London, United Kingdom: Academic Press, is an imprint of Elsevier. [Google Scholar]
- Brosius J, & Raabe CA (2016). What is an RNA? A top layer for RNA classification. RNA Biology, 13, 140–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AN, & Feng J (2017). Drug addiction and DNA modifications. Advances in Experimental Medicine and Biology, 978, 105–125. [DOI] [PubMed] [Google Scholar]
- Bu Q, Long H, Shao X, Gu H, Kong J, Luo L, et al. (2019). Cocaine induces differential circular RNA expression in striatum. Translational Psychiatry, 9, 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell RR, & Wood MA (2019). How the epigenome integrates information and reshapes the synapse. Nature Reviews. Neuroscience, 20, 133–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capel B, Swain A, Nicolis S, Hacker A, Walter M, Koopman P, et al. (1993). Circular transcripts of the testis-determining gene Sry in adult mouse testis. Cell, 73, 1019–1030. [DOI] [PubMed] [Google Scholar]
- Cesana M, & Daley GQ (2013). Deciphering the rules of ceRNA networks. Proceedings of the National Academy of Sciences of the United States of America, 110, 7112–7113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WY, Zhang H, Gatta E, Glover EJ, Pandey SC, & Lasek AW (2019). The histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) alleviates depression-like behavior and normalizes epigenetic changes in the hippocampus during ethanol withdrawal. Alcohol, 78, 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien Y, Tsai PH, Lai YH, Lu KH, Liu CY, Lin HF, et al. (2020). CircularRNA as novel biomarkers in liver diseases. Journal of the Chinese Medical Association, 83, 15–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu HS, Martinez MR, Bansal M, Subramanian A, Golub TR, Yang X, et al. (2017). High-throughput validation of ceRNA regulatory networks. BMC Genomics, 18, 418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman LG Jr., Zou J, & Crews FT (2017). Microglial-derived miRNA let-7 and HMGB1 contribute to ethanol-induced neurotoxicity via TLR7. Journal of Neuroinflammation, 14, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darcq E, Warnault V, Phamluong K, Besserer GM, Liu F, & Ron D (2015). MicroRNA-30a-5p in the prefrontal cortex controls the transition from moderate to excessive alcohol consumption. Molecular Psychiatry, 20, 1219–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delas MJ, & Hannon GJ (2017). lncRNAs in development and disease: From functions to mechanisms. Open Biology, 7, 170121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, et al. (2012). The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Research, 22, 1775–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, et al. (2012). Landscape of transcription in human cells. Nature, 489, 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou X, Feng L, Ying N, Ding Q, Song Q, Jiang F, et al. (2020). RNA sequencing reveals a comprehensive circular RNA expression profile in a mouse model of alcoholic liver disease. Alcoholism, Clinical and Experimental Research, 44, 415–422. [DOI] [PubMed] [Google Scholar]
- Dowdy SF (2017). Overcoming cellular barriers for RNA therapeutics. Nature Biotechnology, 35, 222–229. [DOI] [PubMed] [Google Scholar]
- Ducci F, & Goldman D (2008). Genetic approaches to addiction: Genes and alcohol. Addiction, 103, 1414–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuis-Sandoval F, Poirier M, & Scott MS (2015). The emerging landscape of small nucleolar RNAs in cell biology. Wiley Interdisciplinary Reviews. RNA, 6, 381–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy SR (2002). Computational genomics of noncoding RNA genes. Cell, 109, 137–140. [DOI] [PubMed] [Google Scholar]
- Edwards SL, Beesley J, French JD, & Dunning AM (2013). Beyond GWASs: Illuminating the dark road from association to function. American Journal of Human Genetics, 93, 779–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang S, Zhang L, Guo J, Niu Y, Wu Y, Li H, et al. (2018). NONCODEV5: A comprehensive annotation database for long non-coding RNAs. Nucleic Acids Research, 46, D308–D314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrelly LA, Thompson RE, Zhao S, Lepack AE, Lyu Y, Bhanu NV, et al. (2019). Histone serotonylation is a permissive modification that enhances TFIID binding to H3K4me3. Nature, 567, 535–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farris SP, Arasappan D, Hunicke-Smith S, Harris RA, & Mayfield RD (2015). Transcriptome organization for chronic alcohol abuse in human brain. Molecular Psychiatry, 20, 1438–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farris SP, Harris RA, & Ponomarev I (2015). Epigenetic modulation of brain gene networks for cocaine and alcohol abuse. Frontiers in Neuroscience, 9, 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, & Mello CC (1998). Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature, 391, 806–811. [DOI] [PubMed] [Google Scholar]
- Friedman RC, Farh KK, Burge CB, & Bartel DP (2009). Most mammalian mRNAs are conserved targets of microRNAs. Genome Research, 19, 92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galtier N, Piganeau G, Mouchiroud D, & Duret L (2001). GC-content evolution in mammalian genomes: The biased gene conversion hypothesis. Genetics, 159, 907–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatta E, Grayson DR, Auta J, Saudagar V, Dong E, Chen Y, et al. (2019). Genome-wide methylation in alcohol use disorder subjects: Implications for an epigenetic regulation of the cortico-limbic glucocorticoid receptors (NR3C1). Molecular Psychiatry, 10.1038/s41380-019-0449-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GBD 2016 Alcohol, & Drug Use Collaborators. (2018). The global burden of disease attributable to alcohol and drug use in 195 countries and territories, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Psychiatry, 5, 987–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GBD 2016 Disease, Injury Incidence, & Prevalence Collaborators. (2017). Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet, 390, 1211–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelernter J, Kranzler HR, Sherva R, Almasy L, Koesterer R, Smith AH, et al. (2014). Genome-wide association study of alcohol dependence: Significant findings in African- and European-Americans including novel risk loci. Molecular Psychiatry, 19, 41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghezzi A, Krishnan HR, Lew L, Prado FJ 3rd, Ong DS, & Atkinson NS (2013). Alcohol-induced histone acetylation reveals a gene network involved in alcohol tolerance. PLoS Genetics, 9, e1003986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert W (1986). Origin of life: The RNA world. Nature, 319, 618. [Google Scholar]
- Gomes AQ, Nolasco S, & Soares H (2013). Non-coding RNAs: Multi-tasking molecules in the cell. International Journal of Molecular Sciences, 14, 16010–16039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorini G, Nunez YO, & Mayfield RD (2013). Integration of miRNA and protein profiling reveals coordinated neuroadaptations in the alcohol-dependent mouse brain. PLoS One, 8, e82565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottschalk MG, Domschke K, & Schiele MA (2020). Epigenetics underlying susceptibility and resilience relating to daily life stress, work stress, and socioeconomic status. Frontiers in Psychiatry, 11, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant BF, Chou SP, Saha TD, Pickering RP, Kerridge BT, Ruan WJ, et al. (2017). Prevalence of 12-month alcohol use, high-risk drinking, and DSM-IV alcohol use disorder in the United States, 2001–2002 to 2012–2013: Results from the National Epidemiologic Survey on Alcohol and Related Conditions. JAMA Psychiatry, 74, 911–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg MVC, & Bourc’his D (2019). The diverse roles of DNA methylation in mammalian development and disease. Nature Reviews. Molecular Cell Biology, 20, 590–607. [DOI] [PubMed] [Google Scholar]
- Guerrier-Takada C, Gardiner K, Marsh T, Pace N, & Altman S (1983). The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell, 35, 849–857. [DOI] [PubMed] [Google Scholar]
- Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, et al. (2013). Natural RNA circles function as efficient microRNA sponges. Nature, 495, 384–388. [DOI] [PubMed] [Google Scholar]
- Hansen TB, Wiklund ED, Bramsen JB, Villadsen SB, Statham AL, Clark SJ, et al. (2011). miRNA-dependent gene silencing involving Ago2-mediated cleavage of a circular antisense RNA. The EMBO Journal, 30, 4414–4422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon CC, Ramilowski JA, Harshbarger J, Bertin N, Rackham OJ, Gough J, et al. (2017). An atlas of human long non-coding RNAs with accurate 5′ ends. Nature, 543, 199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu MT, & Coca-Prados M (1979). Electron microscopic evidence for the circular form of RNA in the cytoplasm of eukaryotic cells. Nature, 280, 339–340. [DOI] [PubMed] [Google Scholar]
- Hutvagner G, McLachlan J, Pasquinelli AE, Balint E, Tuschl T, & Zamore PD (2001). A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science, 293, 834–838. [DOI] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium. (2004). Finishing the euchromatic sequence of the human genome. Nature, 431, 931–945. [DOI] [PubMed] [Google Scholar]
- Iyer MK, Niknafs YS, Malik R, Singhal U, Sahu A, Hosono Y, et al. (2015). The landscape of long noncoding RNAs in the human transcriptome. Nature Genetics, 47, 199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen-Seaman MI, Furey TS, Payseur BA, Lu Y, Roskin KM, Chen CF, et al. (2004). Comparative recombination rates in the rat, mouse, and human genomes. Genome Research, 14, 528–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Z, Song R, Regev A, & Struhl K (2015). Many lncRNAs, 5’UTRs, and pseudogenes are translated and some are likely to express functional proteins. eLife, 4, e08890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone AL, Andrade NS, Barbier E, Khomtchouk BB, Rienas CA, Lowe K, et al. (2019). Dysregulation of the histone demethylase KDM6B in alcohol dependence is associated with epigenetic regulation of inflammatory signaling pathways. Addiction Biology, e12816. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketting RF, Fischer SE, Bernstein E, Sijen T, Hannon GJ, & Plasterk RH (2001). Dicer functions in RNA interference and in synthesis of small RNA involved in developmental timing in C. elegans. Genes & Development, 15, 2654–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Pinto SM, Getnet D, Nirujogi RS, Manda SS, Chaerkady R, et al. (2014). A draft map of the human proteome. Nature, 509, 575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss T (2002). Small nucleolar RNAs: An abundant group of noncoding RNAs with diverse cellular functions. Cell, 109, 145–148. [DOI] [PubMed] [Google Scholar]
- Kleaveland B, Shi CY, Stefano J, & Bartel DP (2018). A network of noncoding regulatory RNAs acts in the mammalian brain. Cell, 174, 350–62 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger K, Grabowski PJ, Zaug AJ, Sands J, Gottschling DE, & Cech TR (1982). Self-splicing RNA: Autoexcision and autocyclization of the ribosomal RNA intervening sequence of tetrahymena. Cell, 31, 147–157. [DOI] [PubMed] [Google Scholar]
- Kryger R, Fan L, Wilce PA, & Jaquet V (2012). MALAT-1, a non protein-coding RNA is upregulated in the cerebellum, hippocampus and brain stem of human alcoholics. Alcohol, 46, 629–634. [DOI] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. (2001). Initial sequencing and analysis of the human genome. Nature, 409, 860–921. [DOI] [PubMed] [Google Scholar]
- Lee KS, Bang H, Choi JK, & Kim K (2020). Accelerated evolution of the regulatory sequences of brain development in the human genome. Molecules and Cells, 43, 331–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepack AE, Werner CT, Stewart AF, Fulton SL, Zhong P, Farrelly LA, et al. (2020). Dopaminylation of histone H3 in ventral tegmental area regulates cocaine seeking. Science, 368, 197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewohl JM, Nunez YO, Dodd PR, Tiwari GR, Harris RA, & Mayfield RD (2011). Up-regulation of microRNAs in brain of human alcoholics. Alcoholism, Clinical and Experimental Research, 35, 1928–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Shi Q, Wang Q, Tan X, Pang K, Liu X, et al. (2019). Profiling circular RNA in methamphetamine-treated primary cortical neurons identified novel circRNAs related to methamphetamine addiction. Neuroscience Letters, 701, 146–153. [DOI] [PubMed] [Google Scholar]
- Li Y, Zheng Q, Bao C, Li S, Guo W, Zhao J, et al. (2015). Circular RNA is enriched and stable in exosomes: A promising biomarker for cancer diagnosis. Cell Research, 25, 981–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindblad-Toh K, Garber M, Zuk O, Lin MF, Parker BJ, Washietl S, et al. (2011). A high-resolution map of human evolutionary constraint using 29 mammals. Nature, 478, 476–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND, et al. (2013). Global epigenomic reconfiguration during mammalian brain development. Science, 341, 1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litten RZ, Allen J, & Fertig J (1996). Pharmacotherapies for alcohol problems: A review of research with focus on developments since 1991. Alcoholism, Clinical and Experimental Research, 20, 859–876. [DOI] [PubMed] [Google Scholar]
- Lu X, Liu Y, Xuan W, Ye J, Yao H, Huang C, et al. (2019). Circ_1639 induces cells inflammation responses by sponging miR-122 and regulating TNFRSF13C expression in alcoholic liver disease. Toxicology Letters, 314, 89–97. [DOI] [PubMed] [Google Scholar]
- Ma L, Cao J, Liu L, Du Q, Li Z, Zou D, et al. (2019). LncBook: A curated knowledgebase of human long non-coding RNAs. Nucleic Acids Research, 47, D128–D134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattick JS (2001). Non-coding RNAs: The architects of eukaryotic complexity. EMBO Reports, 2, 986–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattick JS (2003). Challenging the dogma: The hidden layer of non-protein-coding RNAs in complex organisms. BioEssays, 25, 930–939. [DOI] [PubMed] [Google Scholar]
- Mattick JS, & Rinn JL (2015). Discovery and annotation of long noncoding RNAs. Nature Structural & Molecular Biology, 22, 5–7. [DOI] [PubMed] [Google Scholar]
- Mattioli K, Volders PJ, Gerhardinger C, Lee JC, Maass PG, Mele M, et al. (2019). High-throughput functional analysis of lncRNA core promoters elucidates rules governing tissue specificity. Genome Research, 29, 344–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maute RL, Schneider C, Sumazin P, Holmes A, Califano A, Basso K, et al. (2013). tRNA-derived microRNA modulates proliferation and the DNA damage response and is down-regulated in B cell lymphoma. Proceedings of the National Academy of Sciences of the United States of America, 110, 1404–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, et al. (2013). Circular RNAs are a large class of animal RNAs with regulatory potency. Nature, 495, 333–338. [DOI] [PubMed] [Google Scholar]
- Meng H, Wang L, You H, Huang C, & Li J (2019). Circular RNA expression profile of liver tissues in an EtOH-induced mouse model of alcoholic hepatitis. European Journal of Pharmacology, 862, 172642. [DOI] [PubMed] [Google Scholar]
- Meyers JL, Zhang J, Chorlian DB, Pandey AK, Kamarajan C, Wang JC, et al. (2020). A genome-wide association study of interhemispheric theta EEG coherence: Implications for neural connectivity and alcohol use behavior. Molecular Psychiatry. 10.1038/s41380-020-0777-6. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Militello G, Weirick T, John D, Doring C, Dimmeler S, & Uchida S (2017). Screening and validation of lncRNAs and circRNAs as miRNA sponges. Briefings in Bioinformatics, 18, 780–788. [DOI] [PubMed] [Google Scholar]
- Modarresi F, Faghihi MA, Lopez-Toledano MA, Fatemi RP, Magistri M, Brothers SP, et al. (2012). Inhibition of natural antisense transcripts in vivo results in gene-specific transcriptional upregulation. Nature Biotechnology, 30, 453–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigro JM, Cho KR, Fearon ER, Kern SE, Ruppert JM, Oliner JD, et al. (1991). Scrambled exons. Cell, 64, 607–613. [DOI] [PubMed] [Google Scholar]
- Niwa M, Jaaro-Peled H, Tankou S, Seshadri S, Hikida T, Matsumoto Y, et al. (2013). Adolescent stress-induced epigenetic control of dopaminergic neurons via glucocorticoids. Science, 339, 335–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunez YO, Truitt JM, Gorini G, Ponomareva ON, Blednov YA, Harris RA, et al. (2013). Positively correlated miRNA-mRNA regulatory networks in mouse frontal cortex during early stages of alcohol dependence. BMC Genomics, 14, 725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nutt DJ, King LA, Phillips LD, & Independent Scientific Committee on Drugs. (2010). Drug harms in the UK: A multicriteria decision analysis. Lancet, 376, 1558–1565. [DOI] [PubMed] [Google Scholar]
- Pachnis V, Belayew A, & Tilghman SM (1984). Locus unlinked to alpha-fetoprotein under the control of the murine raf and Rif genes. Proceedings of the National Academy of Sciences of the United States of America, 81, 5523–5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pamudurti NR, Bartok O, Jens M, Ashwal-Fluss R, Stottmeister C, Ruhe L, et al. (2017). Translation of circRNAs. Molecular Cell, 66, 9–21 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey SC, Ugale R, Zhang H, Tang L, & Prakash A (2008). Brain chromatin remodeling: A novel mechanism of alcoholism. The Journal of Neuroscience, 28, 3729–3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Q, Bizon C, Gizer IR, Wilhelmsen KC, & Ehlers CL (2019). Genetic loci for alcohol-related life events and substance-induced affective symptoms: Indexing the “dark side” of addiction. Translational Psychiatry, 9, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peschansky VJ, & Wahlestedt C (2014). Non-coding RNAs as direct and indirect modulators of epigenetic regulation. Epigenetics, 9, 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrzykowski AZ, Friesen RM, Martin GE, Puig SI, Nowak CL, Wynne PM, et al. (2008). Posttranscriptional regulation of BK channel splice variant stability by miR-9 underlies neuroadaptation to alcohol. Neuron, 59, 274–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polak P, Karlic R, Koren A, Thurman R, Sandstrom R, Lawrence M, et al. (2015). Cell-of-origin chromatin organization shapes the mutational landscape of cancer. Nature, 518, 360–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polimanti R, Zhang H, Smith AH, Zhao H, Farrer LA, Kranzler HR, et al. (2017). Genome-wide association study of body mass index in subjects with alcohol dependence. Addiction Biology, 22, 535–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Procopio DO, Saba LM, Walter H, Lesch O, Skala K, Schlaff G, et al. (2013). Genetic markers of comorbid depression and alcoholism in women. Alcoholism, Clinical and Experimental Research, 37, 896–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn JJ, & Chang HY (2016). Unique features of long non-coding RNA biogenesis and function. Nature Reviews. Genetics, 17, 47–62. [DOI] [PubMed] [Google Scholar]
- Rich A (1962). On the problems of evolution and biochemical information transfer. In Kasha M, & B Pullman (Eds.), Horizons in Biochemistry (pp. 103–126). New York: Academic Press. [Google Scholar]
- Rybak-Wolf A, Stottmeister C, Glazar P, Jens M, Pino N, Giusti S, et al. (2015). Circular RNAs in the mammalian brain are highly abundant, conserved, and dynamically expressed. Molecular Cell, 58, 870–885. [DOI] [PubMed] [Google Scholar]
- Sacks JJ, Gonzales KR, Bouchery EE, Tomedi LE, & Brewer RD (2015). 2010 National and state costs of excessive alcohol consumption. American Journal of Preventive Medicine, 49, e73–e79. [DOI] [PubMed] [Google Scholar]
- Sakharkar AJ, Vetreno RP, Zhang H, Kokare DM, Crews FT, & Pandey SC (2016). A role for histone acetylation mechanisms in adolescent alcohol exposure-induced deficits in hippocampal brain-derived neurotrophic factor expression and neurogenesis markers in adulthood. Brain Structure & Function, 221, 4691–4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakharkar AJ, Zhang H, Tang L, Shi G, & Pandey SC (2012). Histone deacetylases (HDAC)-induced histone modifications in the amygdala: A role in rapid tolerance to the anxiolytic effects of ethanol. Alcoholism, Clinical and Experimental Research, 36, 61–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmena L, Poliseno L, Tay Y, Kats L, & Pandolfi PP (2011). A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell, 146, 353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanger HL, Klotz G, Riesner D, Gross HJ, & Kleinschmidt AK (1976). Viroids are single-stranded covalently closed circular RNA molecules existing as highly base-paired rod-like structures. Proceedings of the National Academy of Sciences of the United States of America, 73, 3852–3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxonov S, Berg P, & Brutlag DL (2006). A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proceedings of the National Academy of Sciences of the United States of America, 103, 1412–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt LA (2016). Recent developments in alcohol services research on access to care. Alcohol Research: Current Reviews, 38, 27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott MS, & Ono M (2011). From snoRNA to miRNA: Dual function regulatory non-coding RNAs. Biochimie, 93, 1987–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semon M, Mouchiroud D, & Duret L (2005). Relationship between gene expression and GC-content in mammals: Statistical significance and biological relevance. Human Molecular Genetics, 14, 421–427. [DOI] [PubMed] [Google Scholar]
- Shi X, Sun M, Liu H, Yao Y, & Song Y (2013). Long non-coding RNAs: A new frontier in the study of human diseases. Cancer Letters, 339, 159–166. [DOI] [PubMed] [Google Scholar]
- Smith ACW, & Kenny PJ (2018). MicroRNAs regulate synaptic plasticity underlying drug addiction. Genes, Brain, and Behavior, 17, e12424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ZD, & Meissner A (2013). DNA methylation: Roles in mammalian development. Nature Reviews. Genetics, 14, 204–220. [DOI] [PubMed] [Google Scholar]
- Sousa AMM, Meyer KA, Santpere G, Gulden FO, & Sestan N (2017). Evolution of the human nervous system function, structure, and development. Cell, 170, 226–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Laurent G, Wahlestedt C, & Kapranov P (2015). The landscape of long noncoding RNA classification. Trends in Genetics, 31, 239–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapocik JD, Barbier E, Flanigan M, Solomon M, Pincus A, Pilling A, et al. (2014). microRNA-206 in rat medial prefrontal cortex regulates BDNF expression and alcohol drinking. The Journal of Neuroscience, 34, 4581–4588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapocik JD, Solomon M, Flanigan M, Meinhardt M, Barbier E, Schank JR, et al. (2013). Coordinated dysregulation of mRNAs and microRNAs in the rat medial prefrontal cortex following a history of alcohol dependence. The Pharmacogenomics Journal, 13, 286–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teppen TL, Krishnan HR, Zhang H, Sakharkar AJ, & Pandey SC (2016). The potential role of amygdaloid microRNA-494 in alcohol-induced anxiolysis. Biological Psychiatry, 80, 711–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium. (2012). An integrated encyclopedia of DNA elements in the human genome. Nature, 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tie F, Banerjee R, Stratton CA, Prasad-Sinha J, Stepanik V, Zlobin A, et al. (2009). CBP-mediated acetylation of histone H3 lysine 27 antagonizes Drosophila Polycomb silencing. Development, 136, 3131–3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrent M, Chalancon G, de Groot NS, Wuster A, & Madan Babu M (2018). Cells alter their tRNA abundance to selectively regulate protein synthesis during stress conditions. Science Signaling, 11(546), eaat6409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Arensbergen J, Pagie L, FitzPatrick VD, de Haas M, Baltissen MP, Comoglio F, et al. (2019). High-throughput identification of human SNPs affecting regulatory element activity. Nature Genetics, 51, 1160–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, et al. (2001). The sequence of the human genome. Science, 291, 1304–1351. [DOI] [PubMed] [Google Scholar]
- Wang Y, Krishnan HR, Ghezzi A, Yin JC, & Atkinson NS (2007). Drug-induced epigenetic changes produce drug tolerance. PLoS Biology, 5, e265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Zuroske T, & Watts JK (2020). RNA therapeutics on the rise. Nature Reviews. Drug Discovery, 19, 441–442. [DOI] [PubMed] [Google Scholar]
- Warnault V, Darcq E, Levine A, Barak S, & Ron D (2013). Chromatin remodeling—A novel strategy to control excessive alcohol drinking. Translational Psychiatry, 3, e231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilusz JE, & Sharp PA (2013). Molecular biology. A circuitous route to noncoding RNA. Science, 340, 440–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik GL, Graff M, Nishimura KK, Tao R, Haessler J, Gignoux CR, et al. (2019). Genetic analyses of diverse populations improves discovery for complex traits. Nature, 570, 514–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav S, Pandey A, Shukla A, Talwelkar SS, Kumar A, Pant AB, et al. (2011). miR-497 and miR-302b regulate ethanol-induced neuronal cell death through BCL2 protein and cyclin D2. The Journal of Biological Chemistry, 286, 37347–37357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Chen H, Ding N, Wang S, Duan Z, Birnbaum Y, et al. (2018). Expression profiling of circular RNAs and micrornas in heart tissue of mice with alcoholic cardiomyopathy. Cellular Physiology and Biochemistry, 46, 2284–2296. [DOI] [PubMed] [Google Scholar]
- You X, Vlatkovic I, Babic A, Will T, Epstein I, Tushev G, et al. (2015). Neural circular RNAs are derived from synaptic genes and regulated by development and plasticity. Nature Neuroscience, 18, 603–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Yuan Q, Mash DC, & Goldman D (2011). Substance-specific and shared transcription and epigenetic changes in the human hippocampus chronically exposed to cocaine and alcohol. Proceedings of the National Academy of Sciences of the United States of America, 108, 6626–6631. [DOI] [PMC free article] [PubMed] [Google Scholar]