

Abstract
Mitochondrial superoxide (O2.−) production is implicated in aging, neurodegenerative disease, and most recently epilepsy. Yet the specific contribution of neuronal O2.− to these phenomena is unclear. Here, we selectively deleted superoxide dismutase-2 (SOD2) in neuronal basic helix-loop-helix transcription factor (NEX)-expressing cells restricting deletion to a subset of excitatory principle neurons primarily in the forebrain (cortex and hippocampus). This resulted in nSOD2 KO mice that lived into adulthood (2-3 months) with epilepsy, selective loss of neurons, metabolic rewiring and a marked mitohormetic gene response. Surprisingly, expression of an astrocytic gene, glial fibrillary acidic protein (GFAP) was significantly increased relative to WT. Further studies in rat primary neuron-glial cultures showed that increased mitochondrial O2.−, specifically in neurons, was sufficient to upregulate GFAP. These results suggest that neuron-specific mitochondrial O2.− is sufficient to drive a complex and catastrophic epileptic phenotype and highlights the ability of SOD2 to act in a cell-nonautonomous manner to influence an astrocytic response.
INTRODUCTION
Mitochondria are highly dynamic organelles that generate the energy required to drive critical neuronal processes. Due to its high-energy demand, the brain is highly dependent on mitochondria, where they play key roles in redox signaling, neurotransmitter biosynthesis, calcium sequestering and cell death regulation. In addition, mitochondrial energy metabolism is a critical interface between environmental factors (i.e. diet), disease, control of inflammation and optimal neuronal function. A mismatch between the brain’s oxygen consumption (~20%) and per unit body weight (~2%) results in higher oxygen availability for the generation of partially reduced species for redox signaling or oxidative injury. Indeed, brain mitochondria are recognized as major sites of production and detoxification of reactive oxygen species (ROS), particularly the superoxide radical (O2.−). The brain is particularly sensitive to oxidative injury due to a multitude of factors (Cobley, Fiorello, and Bailey 2018). O2.− is only selectively damaging, however as the precursor of more damaging species such as H2O2, hydroxyl radical (OH.) and peroxynitrite (ONOO−), mitochondrial O2.− is a major cause of cellular oxidative injury. In the brain, excessive mitochondrial O2.− and its more toxic metabolites have been consistently demonstrated as a major contributing factor to neurodegenerative diseases, epilepsy and the aging process (Aguiar et al. 2012; Maier and Chan 2002).
The importance of mitochondrial O2.− is highlighted by the observation that homozygous deletion of its major detoxifying enzyme, manganese superoxide dismutase (SOD2) causes embryonic/perinatal lethality, (Huang et al. 2001; Li et al. 1995; Melov et al. 1999), whereas deletion of the cytosolic or extracellular enzymes (SOD1 and SOD3, respectively) results in mild phenotypes (Carlsson et al. 1995; Reaume et al. 1996). Lifespan can be extended in SOD2 deficient mice if bred on different background strains, suggesting the presence of modifier genes and hinting at the complex biology of mitochondrial O2.−. Studies performed on global SOD2 knockout (SOD2KO) or heterozygous mice reveal a host of pathologies across all organs, including cardiomyopathy, (Huang et al. 2001; Lebovitz et al. 1996; Li et al. 1995), metabolic acidosis (Huang et al. 2001; Li et al. 1995), liver steatosis (Li et al. 1995), anemia (Lebovitz et al. 1996) and spongiform encephalopathy (Melov et al. 1998). Neurologically, longer-lived mixed background mice (SOD2/B6D2) exhibit increased brain steady-state O2.− levels, extensive neurodegeneration, and frequent seizures culminating in death at approximately 3 weeks of age (Liang et al. 2012; Lynn et al. 2005). Treatment with brain permeable SOD/catalase mimetics significantly decreased neurodegeneration (Melov et al., 2001) and seizure incidence while prolonging lifespan (Liang et al., 2012). This apparent contribution of increased steady state mitochondrial O2.− to an epilepsy phenotype is supported by data showing increased convulsant-induced seizures and the development of handling-induced and spontaneous seizures with age in mice partially deficient for SOD2 (SOD2+/−) (Liang and Patel 2004).
While global deletion of SOD2 has been useful in understanding the deleterious effects of mitochondrial O2.−, it has several limitations. First, the animals have short lifespans dependent on genetic background and modifier genes, which precludes the study of adult phenotypes. Second, the overall health of the animals is compromised, obscuring some phenotypes and exacerbating others making it difficult to determine the specific role of mitochondrial O2.− in tissues. Recent advances in gene technology have allowed for the cell-type specific deletion of SOD2 which overcomes these limitations and enables the determination of the relative importance of excessive steady state O2.− in specific tissues.
Here, we determined the consequences of increased mitochondrial O2.− in highly metabolically active neurons in adult animals. Using Cre technology we selectively deleted SOD2 in neuronal basic helix-loop-helix transcription factor (NEX) expressing cells which restricts deletion to a subset of principle neurons primarily in the forebrain (cortex and hippocampus). This resulted in mice that lived into adulthood (2-3 months) with epilepsy of either cortical or hippocampal origin, selective loss of neurons in the somatosensory cortex, metabolic rewiring and a marked mitohormetic gene response. Surprisingly, expression of an astrocytic gene and marker of astrogliosis, glial acidic fibrillary protein (GFAP) was significantly increased approximately 4-fold relative to wild-type controls. Further studies showed that GFAP upregulation could be recapitulated in vitro in the absence of seizure activity and neurodegeneration suggesting that mitochondrial O2.− originating in neurons is sufficient to upregulate astrocytic GFAP. These results suggest that neuron-specific mitochondrial O2.− is sufficient to drive a complex, catastrophic epileptic phenotype and highlights the ability of neuronal SOD2 to act in a cell-nonautonomous manner to influence an astrocytic response.
MATERIALS AND METHODS
Mouse breeding and genotyping.
Animal studies were carried out in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 80-23). All procedures were approved by the Institute Animal Care and Use Committee (IACUC) of the University of Colorado Anschutz Medical Campus, which is fully accredited by the American Association for the Accreditation of Laboratory Animal Care. SOD2 P lox C57BL/6CrSlc mice were obtained with permission from the Shirasawa and Shimizu laboratory (Tokyo Metropolitan Institute of Gerontology and National Center for Geriatrics and Gerontology, Japan) (Ikegami et al. 2002). These mice were crossed with NEX-Cre C57BL/6 (Goebbels et al. 2006) to generate the heterozygous F1 generation. The offspring were intercrossed and genotyping was performed on the F2 progeny from ear snips collected at the time of weaning. To identify the p lox copy number, PCR was performed using the following primers: P1= 5’-CGA GGG GCA TCT AGT GGA GAA G-3’, P2=5’-TTA GGG CTC AGG TTT GTC CAG AA- 3’, and P4= 5’-AGC TTG GCT GGA CGT AA-3’ (IDT Technologies). Individuals carrying the NEX-Cre insert were identified as described in (Goebbels et al. 2006). All mice, including breeders, were housed in ventilated caging on a 14/10 light/dark cycle with ad libitum access to standard rodent chow and filtered water. Additionally, cages containing experimental mice were provided with moistened pellets of standard chow daily. All studies utilized two-month-old male and female mice unless otherwise indicated.
Real time PCR.
RNA was isolated from cortical tissue using RNeasy spin columns (Qiagen) and reverse transcribed with the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Custom taqman probes were used in combination with Universal PCR Master Mix to amplify cDNA on a 7500 ABI thermocycler (Life Technologies). Relative gene expression was determined using the ΔΔ Ct comparative method to calculate fold change (RQ). An RT2 PCR array specific for oxidative stress and antioxidant defense was also used in combination with RT2 SYBR Green qPCR mastermix (Qiagen).
SOD2 Activity.
SOD2 enzyme activity was measured by colorimetric detection of formazan dye following production of the superoxide via xanthine oxidase (Sun, Oberley, and Li 1988). Mitochondria were isolated from cortical tissue by homogenizing in cold buffer containing 20 mM Hepes pH 7.2, 1 mM EGTA, 210 mM mannitol, and 70 mM sucrose. Samples were centrifuged first at 1,500 x g followed by 10,000 x g. Residual CuZn-SOD and EC-SOD activity was deactivated via incubation with 3mM potassium cyanide for 30 min. SOD2 enzyme activity was then assessed with a Cayman Chemical superoxide dismutase assay kit and 460 nm absorbance was measured on a BioTek Synergy 4 plate reader. Protein normalization was performed via Coomassie Blue assay (Pierce).
2-hydroxyethidium assay.
2-hydroxyethidium (2-OHDE; GmbH Inc. German, #NOX-14) was measured by a high performance liquid chromatography (HPLC) system with an electrochemical detector following the method previously described with a small modification (Zielonka, Vasquez-Vivar, and Kalyanaraman 2006; Maghzal and Stocker 2007). Mice were injected with 50mg/kg s.c. dihydroethidium (DHE) (Sigma, #37291) and sacrificed at 1 hr. after injection. The brain samples were sonicated in ice cold 0.1N PCA (0.1g/ml, w/v) and centrifuged at 16000g 4°C for 10 min. An aliquot of 20 μl was injected into the HPLC-EC system (ESA Biosciences, Inc.) which consists of two pumps (Model 582), a temperature controlled autosampler (Model 542), and an eight-channel electrochemical CoulArray detector set to the potentials 0, 200, 280, 365, 400, 450, 500, and 600 mV versus the palladium reference electrode. The mobile phase contained 50 mM phosphate buffer (pH 2.6), 2% acetonitrile. HE, 2-OH-E+, and E+ were separated on a Phenomenex Polar-RP 80 A column (4 μm, 250×4.6 mm; Phenomenex). The flow rate was 0.6ml/min and the pressure was approximately 78 bar.
Aconitase and Fumarase Activity.
Aconitase and fumarase activities were measured as described in (Patel et al. 1996). Briefly, mitochondria were isolated from cortical tissue and homogenized in 70 mM sucrose, 210 Mm mannitol, 10 mM Tris HCl, 1 mM EDTA pH=7.4. Mitochondria were isolated from the homogenate following two differential centrifugation steps (3000g followed by 13000g) and snap frozen in liquid nitrogen. 50 uL of the mitochondria were combined with either 100 uL 200 mM isocitrate (aconitase assay) or 50 mM malate (fumarase assay) and placed in a cuvette containing 850 uL reaction buffer (aconitase assay: 50 uL mM Tris HCl and 600 uM MnCl2 and fumarase assay: 30 mM KH2PO4 100 uM EDTA) pH=7.4). The rate of the conversion of cis-aconitate to isocitrate (aconitase assay) or fumarate to L-malate (fumarase assay) was measured spectrophotometrically by detecting the increased rate of absorbance at 240 nm. Protein normalization was performed via Coomassie Blue assay (Pierce).
Western Blotting.
Nuclear and cytosplasmic fractions were separated with NE-PER extraction reagents, following the manufacturer’s instructions (Thermo Scientific). Protein yield was quantified via Coomassie Blue assay (Pierce), diluted in PBS, and combined with Laemmli sample buffer/2.5% β mercaptoethanol (1:1) (Bio Rad). Samples were heated to 95°C for 10 minutes and directly placed on ice. 10 ug protein were loaded to Criterion Stain-free gels and electrophoresed for 1 hour at 200v in 1X Tris/Glycine/SDS buffer (Bio Rad). Protein was transferred to PVDF membranes on a Trans-blot turbo transfer system following manufacturer’s instructions (Bio Rad) then blocked for 1 hour in 5% milk and TBST (0.1% tween). Membrane was incubated with primary antibody overnight at 4°C (1:200, Hmox 1: ab13243 (Abcam), actin: a2228 (Sigma) and Nrf2 sc-722 (Santa Cruz Biotechnology)) followed by secondary antibody at 1:10,000 concentration for one hour r.t. (HRP conjugated goat α rabbit, Invitrogen 31460). The membrane was developed with SuperSignal West Pico PLUS Chemiluminescent substrate (Thermo Scientific) and imaged for both total protein and HRP signal on a BioRad ChemiDoc MP imaging system.
Behavioral Assay.
Various parameters of locomotion were assessed 1 h after transport to a dedicated behavioral suite. Separate cohorts of mice were used for each task. A random number generator was used to establish testing order. Behavioral equipment was cleaned between trials using 70% isopropyl alcohol.
Open Field.
Mice explored a novel behavioral area (44 X 44 cm) for 5 minutes. Total distance traveled (cm) was quantified using specialized video tracking software (Noldus Ethovision).
Rotarod Assay.
Mice were placed on 3 cm diameter rotating rods with a height to fall of 16 cm (UGO Basile). Rods were accelerated from 3.0-30 RPM over a total of 300 seconds. The latency to fall was recorded once the mouse fell or failed to remain upright. Each mouse performed a total of three trials and average time to fall was calculated.
Electrode Implantation and EEG analysis.
Subdural and hippocampal surgical implantation and 24/7 video-EEG monitoring was performed in nSOD2 KO mice and WT littermates (6-8 weeks old) as described in (Liang et al. 2012) and (Weitzel et al. 2016). Briefly, mice were anesthetized by continuous inhalation of isoflurane. Depth of anesthesia was ensured by assessing the toe pinch reflex before placement into the stereotaxic apparatus. For cortical analysis, bilateral stainless screws were inserted into craniotomies placed bilaterally at approximately the following coordinates: AP = −0.5 mm from bregma, ML = ±2.0 mm from midline, and DV = below the skull surface and above the cortex. Two stainless steel screws positioned near the lambdoid suture on left and right hemispheres served as a reference and ground. In addition, some mice received bilateral hippocampal electrodes at the following coordinates: anterioposterior (AP) = −2.8 mm from bregma, mediolateral (ML) = ±2.0 mm from midline, and dorsoventral (DV) = 2.0 mm below the skull surface. The electrode assembly was fixed to the head by encasement in dental enamel. Mice were provided with buprenorphine (0.1 mg/kg s.c.) once daily for 2 days, starting immediately after surgery and again 24 h later. 24 h after surgery, synchronized, time-locked video and EEG signals were recorded using the Pinnacle system (Pinnacle Technology, Lawrence, KS). The EEG signals were acquired with acquisition filters set at 0.5 Hz (low) and 500 Hz (high). The signals were digitized at 2000 Hz and stored on a hard disk for offline analysis. EEG files were analyzed with a bandpass filter (1.0 Hz low, 30.0 Hz high) in a semi-automated manner. Areas of the EEG with increased amplitude and frequency (3x baseline and lasting at least 2 seconds) were identified as “putative events” using Sirenia Seizure software. These events were then identified as seizures or noise by an investigator blind to animal genotype. Only convulsive seizures i.e. seizures identified in the EEG with a corresponding behavioral component were considered for quantification. Behavioral seizure severity was assessed and scored using the Racine scale as follows: P3-unilateral forelimb clonus, P4-bilateral forelimb clonus, P5-bilateral forelimb clonus with rearing, falling, or running.
Fluoro-Jade B Staining.
Whole brains were collected and formalin-fixed for 48 h. Following fixation, brains were cut into three coronal blocks, paraffin embedded and sectioned (15 um). Every 6th section was collected and stained with FluoroJade B (FJB) as for degenerating neurons previously described (Schmued and Hopkins 2000; Pearson et al. 2015; Pearson et al. 2017). Sections were deparrafinized by incubating 2X15 min in citrisolv, followed by rehydration in 100% ethanol for 10 min, 70% ethanol for 2 min, and ddH2O for 1 min. Slides were immersed in 1% NaOH 80% ethanol for 5 min, rinsed for 2 min in 70% ethanol, 2 min in ddH2O, followed by 0.06% potassium permanganate solution for 10 min. After 1 min rinse in H2O, slides were incubated in 0.0004% solution of Fluoro-Jade B (Histo-Chem) in 0.1% acetic acid vehicle for 15 min, then rinsed in ddH2O. After drying, slides were cleared in citrisolv and mounted with DPX mounting media (Fluka). Quantification of FJB fluorescence intensity in a given area was measured using Image J software. A preliminary assessment by an investigator blinded to animal genotype revealed that FJB staining, when present, was primarily within the block corresponding to approximately −0.22 to −2.92 mm bregma. This area was then further investigated and imaged using a Nikon Eclipse ti2 Microscope. A total of 7 mice (3 WT, 4 KO) were used in the analysis and results were averaged from 6 sections obtained from each animal. When present, FJB staining was located entirely within the superficial layers of the cortex corresponding to the retrosplenial, motor, and somatosensory areas. Thus, these areas were selected in a viewing window held constant across animal and section. The average fluorescence intensity within this area (10x axis) was quantified using standard ImageJ software (NIH). Thresholds were not applied and all images were obtained from a single plane (i.e. no z-stacking was performed).
Primary Tissue Culture.
Mice.
Mixed cortical cell cultures were established from P1 nSOD2KO and WT mice. Tail snips from tattooed littermates were collected and genotyped within one day of birth. Following genotype identification, nSOD2KO and WT individuals were sacrificed, and cortices were dissected and placed in sterile HBSS+10mM Hepes. Tissue was disassociated into smaller fragments with micro scissors, then trypsinized in a 1% solution of trypsin/HBSS for 25 min at 37°. Cells were then rinsed with HBSS and triturated in a 1% solution of MEM and DNAse (Sigma) while being passed through a flame polished glass pipet several times. Cells were pelleted by centrifugation at 800 rpm for 10 min, followed by re-suspension in NeuroCult plating medium (Stemcell Technologies). 60,000 cells were then plated in PDL treated 96 well XF96 cell culture microplates (Agilent Technologies) and maintained at 37°C, 5% O2, 5% CO2 . After 5 days, one half of the plating medium was replaced with BrainPhys neuronal medium (Stemcell Technologies).
Rats.
E18 rats were harvested via cesarean surgery. Methods were carried out as described above except MEM enriched with 0.3% glucose, 5% FBS and 5% HS was used in lieu of NeuroCult and BrainPhys plating medias. Normoxia cultures were maintained at atmospheric O2. Neuron depleted cultures were achieved through a series of complete media replacements over a course of two weeks (first at 18 hours, then every other day). MitoPQ was administered following two weeks of cell growth.
Immunocytochemistry.
Mice.
Mixed primary cortical cultures obtained from P3-5 nSOD2KO and WT mice were fixed with 4% paraformaldehyde in PBS for 10 min, washed with PBS for 5X3 min, then blocked for 1 hour at r.t. (0.5% BSA, 0.05% Triton x100, 10% normal goat serum). Cells were incubated with primary antibody o/n at 4°C. Primary antibody was removed and washed 3X5 min in PBS. Secondary antibody was applied in blocking buffer for one hour r.t. Cells were covered with 10% glycerol PBS. The following antibodies were used: rabbit α SOD2 (1:80, Enzo ADI-SOD-110), guinea pig α NeuN (1:100 Millipore ABN90) and chicken α GFAP (1:100 Millipore AB5541) followed by goat α rabbit TRITC (1:100 Sigma T6778), goat α guinea pig AlexaFluor 488 (1:100 Invitrogen A16073) and goat α chicken Dylight 405 (1:100 Jackson Immuno Research 103-475-155). These images were acquired on a 3I Marianas spinning disk confocal.
Rats.
Mixed primary rat cortical cultures were stained as described above using the following antibodies: chicken α GFAP (1:500, Millipore AB 5541) and mouse α MAP-2 (1:500, Sigma M1406) followed by goat α chicken Alexa Fluor 594, (1:400, Invitrogen A11001) and goat α mouse Alexa Fluor 488, (1:400 Invitrogen A11042). These images were acquired on a Nikon Eclipse Ti2.
Glycolytic Stress Test.
Extracellular acidification rate was measured in two-week-old primary cortical cells. One day prior to the assay, cells were transferred from 5% O2 to normoxic conditions. Growth media was replaced with glucose and serum free XF base medium (Agilent) enriched with 2mM glutamine. Cells were placed in a CO2 free 37° incubator for one hour. Cells were then placed in an XF24 or XF96 extracellular flux analyzer (Agilent). Three measurements of extracellular acidification rates (ECAR) were performed after each injection of the following: 16 mM final concentration of glucose, 1 uM final concentration of oligomycin, and 50 mM final concentration of 2-DG. Protein normalization was performed via Coomassie Blue assay (Pierce). Additionally, a mitochondrial stress test was performed using 3 ụM FCCP and 0.3 ụM antimycin A.
Glucose Measurements.
Plasma.
Following a 6 hour fast, blood was collected from a facial vein laceration and centrifuged for 15 min at 2500 rpm. Glucose was measured in the separated serum with a DriChem 7000 veterinary chemical analyzer (Heska).
Brain tissue.
Following a 6 hour fast, animals were sacrificed and cortices were removed and flash frozen in PBS. Following thawing, they were then briefly sonicated in ice cold PBS. Samples were centrifuged for 10 minutes at 12,000 rpm and 4° C. Glucose in the supernatant was quantified using a colorimetric assay that utilizes the glucose oxidase-peroxide reaction (Caymen Chemicals). 514 nm absorbance was detected on a BioTek Synergy 4 plate reader. Protein normalization was performed via Coomassie Blue assay (Pierce).
Microarray.
Genome-wide expression profiling was performed on the cortices of 2 month old WT and nSOD2 KO littermates (n=11-12). Single-strand fragmented and biotin-labeled cDNA was hybridized to a Clariom S microarray following the manufacturer’s instructions (Affymetrix). Partek Genomics Suite v6.6 software was used for the microarray data analysis (Partek Inc., St. Louis MO). CEL files were imported including control and interrogating probes, pre-background adjustment was set to adjust for GC content and probe sequence, and Robust Multi-array Average (RMA) background correction was performed. Arrays were normalized using quantile normalization and probe set summarization was done using Median Polish. Probe values were log2 transformed. Differentially expressed genes between groups were analyzed by the unpaired t-test. Contrasts between groups were performed using Fisher’s Least Significant Difference as a contrast method.
H2O2 measurement.
H2O2 production was determined in rat primary cortical cultures that were plated in 96-well plates at a density of 1.5 X 105 cells per well. Growth media was removed after two weeks and replaced with Hank’s buffered saline solution (HBSS) containing a final concentration of 50 uM Amplex Red (Invitrogen), 0.1 U/ml HRP and 1 uM MitoPQ. Resorufin fluorescence (excitation/emission= 530/590 nm) was measured after 24 hours on a Biotek Synergy 4 plate reader. Reaction values were calculated by subtracting the background fluorescence, determined from a non-H2O2 control reaction, from each experimental well.
LDH Assay.
The rate of conversion of NADH and pyruvate to lactate and NAD+ through LDH was measured by loss of absorbance at 340 nm. 20uL of cell media was combined with an equal volume of sodium pyruvate (8.7 mM). 110 uL of 256.2 uM NADH was added. Both sodium pyruvate and NADH were prepared in Tris NaCl (81 mM Tris, 203.6 mM NaCl, pH 7.2). Rate of absorbance was determined over 4 min on a Molecular Devises Versa Max microplate reader.
Statistical Analysis.
Investigators blinded to experimental condition carried out all experiments and analyses. A two-tailed unpaired Student’s T-test was used to evaluate differences between WT and nSOD2 KO mice unless otherwise indicated. Number of samples (n) represents the number of individual animals. In the case of cell culture studies, n represents the number of times an experiment was repeated with technical replicates nested within each biological replicate. Results were considered significant when p <0.05. Data are presented as mean ± SEM unless otherwise noted. Analyses were performed using Prism 8 software (GraphPad Software, San Diego, CA).
RESULTS
NEX driven Cre recombinase selectively and efficiently targets forebrain neurons for SOD2 gene deletion and accumulation of intracellular O2−..
Global SOD2 deletion in various mouse strains is generally incompatible with life and results in perinatal or early life lethality (Huang et al. 2001; Li et al. 1995; Lebovitz et al. 1996). Compared to global SOD2KO mice in the C57B16 and DBA/2J (Huang et al. 2001), CD1 (Li et al. 1995) or mixed backgrounds (Lebovitz et al. 1996) the NEX-Cre forebrain neuron-specific nSOD2 KO mice showed increased longevity with a typical lifespan between 2 and 3 months, and litters maintained normal Mendelian birth ratios. NEX-Cre mice drive expression of the Cre recombinase protein primarily in pyramidal, hilar mossy cells, and dentate gyrus granule cells of the neocortex and hippocampus (Goebbels et al. 2006). In situ hybridization reveals an expression pattern of NEX that demonstrates an association with later stages of neurogenesis, i.e., after neuronal determination (Schwab et al. 2000). In mice, this occurs at E11.5 (Goebbels et al. 2006). Therefore its expression is absent from proliferating precursor cells. Within this NEX-cre line, upon cross-breeding to either floxLacZ or R26R mice, β-galactosidase activity overlapped with neuronal markers (NeuN and IL-beta tubulin) but was not found to overlap with APC or CNP (oligodendrocytes) or GFAP (astrocytes) (Goebbels et al. 2006).
The efficiency and specificity of the brain specific SOD2 gene deletion was confirmed using multiple methods. We first verified the brain specificity of the SOD2 gene deletion by RTPCR in cortex and liver among WT and nSOD2 KO littermates. As expected, SOD2 expression was significantly decreased in the cortex of nSOD2 KO mice (0.4227 fold change, p<0.001: Figure 1A) but remained unchanged in the liver (Figure 1B), consistent with brain-specific deletion of SOD2. We performed immunocytochemistry on cultured cells derived from nSOD2 KO pups to verify the cell-type specificity of SOD2 deletion. Consistent with neuronal-specific deletion of SOD2, only astrocytes (i.e. GFAP-labeled cells) were found to overlap with SOD2 (Supplemental Figure 1B–D). This is in contrast to WT cell preparations, in which SOD2 was found to co-stain with both NeuN and GFAP (Supplemental Figure 1F–H) (60X magnification). Imaging at 20X reveals a similar pattern of overlap with GFAP and SOD2 but not NeuN and SOD2 within KO cultures (Supplemental Figure 1L). However, imaging the WT cultures reveals a more robust staining of SOD2 in general and co-staining within both neuronal and astrocytic cell populations (Supplemental Figure 1O,P).
Figure 1.
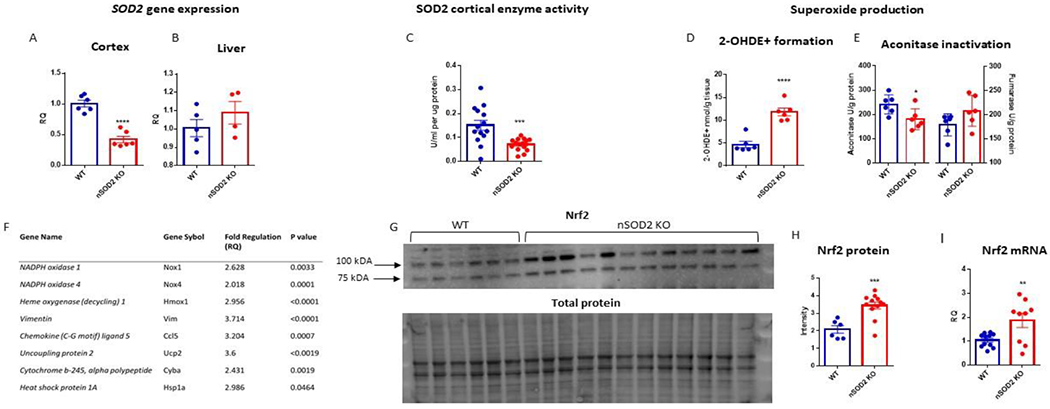
Nex cre P lox deletion of SOD2 effectively targets neurons, results in measurable superoxide production and induces Nrf2 signaling in the cortex at 2 months of age. (A) RTPCR analysis of cortex (unpaired t test, n=6, p<0.0001) and (B) liver (unpaired t test, n=4-5, p= 0.3064). (C) SOD2 enzyme activity is reduced in nSOD2 KO mice (unpaired t test, n=5-6, p=0.0007). (D) Formation of 2-hydroxyethidium was measured via HPLC separation (unpaired t test, n=6, p<0.0001). (E) Aconitase and fumarase activity was measured in isolated cortical mitochondria (unpaired t test, n=6, p=0.0290 and 0.1017 respectively). (F) RTPCR of cDNA isolated from the forebrain of 2 month old nSOD2 KO and WT littermates identified the upregulation of 8 genes previously implicated in oxidative stress and inflammation. (G) Band densitometry analysis in the nuclear fraction revealed a significant increase in the 100 kDA band of Nrf2 (unpaired t test, n=6-12, p=0.006). Total protein was quantified via stain-free technology (BioRad) for loading normalization. (I) RTPCR reveals an increase in gene expression of Nrf2 in cortical tissue (unpaired t test, n=9-10, p=0.0092).
To confirm that this targeted deletion was sufficient to produce increased steady state mitochondrial O2.−, we first assessed SOD2 enzymatic activity in forebrain mitochondria of 2 month old mice via the xanthine/xanthine oxidase assay (Sun, Oberley, and Li 1988). SOD2 activity was significantly decreased in nSOD2 KO mice (mean ± SEM 0.07 U/ml/ug protein ± 0.006567) compared to WT mice (mean ± SEM= 0.15 U/ml/ug protein ± 0.02070) (Figure 1C). To ensure that decreased SOD2 activity corresponded with increased O2.− we measured intracellular O2.− using two distinct assays. Given the ephemeral and reactive nature of O2.−, we chose two well established assays that reliably and specifically detect O2.− in biological tissue i.e. formation of 2-OH-E and the mitochondrial aconitase enzymatic assay.
The formation of 2-OHDE upon reaction between O2.− and hydroethidium (HE) with subsequent HPLC detection is the gold standard method to quantify the production of O2.−. Because other reactive species such as H2O2, hydroxyl radical, ONOO-, and hypochlorous acid do not oxidize HE, it is specific to the measurement of O2.−. Additionally, this method allows for the distinction between the formation of 2-OH-E+ and the ethidium (E+)/DNA complex (Zhao et al. 2005; Maghzal and Stocker 2007).
Fluorescent detection revealed a >2.5 fold increase in the formation of 2-OH-E+ in nSOD2 KO mice (mean ± SEM 11.81 nmol 2-OH-E+/g tissue ± 0.8199) as compared to WT littermates (mean ± SEM 4.63 ± 0.6906 nmol 2-OH-E+/g tissue) indicative of increased O2.− production in cortical tissue 1h following peripheral injection of HE (50 mg/kg s.c.; Figure 1D).
To confirm that increased O2.− production was mitochondrial in origin, we assessed the activity of the iron-sulphur (Fe-S)-containing TCA cycle enzyme aconitase in mitochondrial lysates. Only ~15% of total aconitase activity in brain is due to its cytosolic form (Liang, Ho, and Patel 2000). The active [4Fe-4S]2+ form of aconitase is converted to the inactive [3Fe-4S]+ form in the presence of O2.− making it a specific indicator of mitochondrial-derived oxidative stress (Patel et al. 1996; Gardner et al. 1995). Aconitase activity was significantly decreased in nSOD2 KO mice relative to WT mice by ~25% (Figure 1E). Fumarase is another TCA enzyme, however, it lacks an iron-sulfur center making it insensitive to oxidative inactivation (Patel et al. 1996). Fumarase activity was not altered between groups (Figure 1E) suggesting that decreased aconitase activity was not the result of overall decreased activity of TCA enzymes. Additionally, immunoblot analysis of mitochondrial aconitase apoprotein levels revealed no alteration in its levels (data not shown) suggesting that inactivation of mitochondrial aconitase occurred by an oxidative post-translational mechanism.
These findings demonstrate that conditional deletion of SOD2 in forebrain neurons is sufficient to inhibit SOD2 expression and enzymatic activity by approximately 50% resulting in significantly increased steady-state mitochondrial O2.− levels in whole cortex. Despite a reduced lifespan relative to WT mice, SOD2 deficiency restricted to forebrain neurons produces mice that live up to 3 months. This provides a unique model in which to investigate the role of mitochondrial O2.− in neuronal processes.
Increased mitochondrial O2.− in a subset of neurons alters cortical expression of oxidative stress response genes.
To determine if increased steady-state O2.− levels arising from a subset of neurons in nSOD2 KO mice was sufficient to elicit an oxidative stress gene response, 84 genes previously reported to be involved in mouse oxidative stress and antioxidant defense pathways were examined by PCR array (Qiagen, Supplemental Table 1). Eight genes were found to be upregulated (>two-fold change with p< 0.05) and independently confirmed with specific Taqman probes using GAPDH as a normalizer control (Figure 1F). Of this panel, vimentin (Vim) was found to have the greatest increase in expression (3.714 fold change, p<0.0001) followed by uncoupling protein 2 (3.6 fold change, p<0.0019). Pro-inflammatory genes were upregulated including the chemotactic gene chemokine ligand 5 (3.204 fold change, p=0.0007), and heat shock protein 1a (2.986 fold change, p=0.0464). Both nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 1 and 4 (Nox 1 and 4) showed an increase (2.628 fold change, p=0.0033 and 2.018 fold change, p=0.0001 respectively), as did the NADPH oxidase 2 subunit cytochrome b-245 alpha polypeptide (2.431 fold change, p=0.0019). This provides an interesting example of increased O2−. resulting in an increase in a second source of O2−., perhaps pathologically contributing to the overall load of oxidative stress in these mice. Heme oxygenase 1 (Hmox1) is involved in both the inflammatory and antioxidant response, and its expression was increased by 2.956 fold (p<0.0001). Together, these data suggest that neuronal SOD2 deficiency is sufficient to induce a pro-oxidative and pro-inflammatory gene response.
Neuronal SOD2 deficiency activates the Nrf2 antioxidant response pathway.
The increase in Hmox1 expression in nSOD2 KO mice suggested the possible induction of the Nrf2 antioxidant response pathway. To investigate this further, we examined if increased Hmox1 gene expression corresponded to increased protein expression via western blot. Hmox1 protein levels were increased at one month of age (WT=5.735 units of intensity ± 0.3811 SEM, and KO=8.125 units of intensity ± 0.4074 SEM, (n=4, p=0.0052) and trending but not significant at 2 months of age (Supplemental Figure 2A,B). Next, western blot was performed on nuclear fractions from whole cortex in 2 month old mice and probed for Nrf2 (Figure 1G). The ~100 kDa band was selected for densitometry because it has been demonstrated to be the most biologically relevant (Lau et al. 2013), and was found to be significantly increased in nSOD2 KO nuclear fractions (Figure 1H; WT mean ± SEM =2.075 units of intensity ± 0.2132, and KO = 3.438 units of intensity ± 0.1998). Nrf2 activation is not limited to changes in post-transcriptional regulation pathways (Li, Jia, and Zhu 2019). Therefore we examined RNA transcript levels via RTPCR. nSOD2 KO mice demonstrated a 1.87 fold change increase in cortical gene expression (p=0.0092) at 2 months of age (Figure 1I). Several other well-established targets of Nrf2 were not found to be induced when examined by RTPCR, including NAD(P)H dehydrogenase, quinone 1 (Nquo1), glutamate-cysteine ligase, catalytic subunit (Gclc), glutamate-cysteine ligase, modifier subunit (Gclm), and sulfiredoxin 1 (Srxn1). However, given that Nrf2 regulation is intricately associated with Nox enzymes, particularly Nox 4 our observation of Nox4 upregulation further supports Nrf2 activation (Brewer et al. 2011; Papaiahgari et al. 2004; Kovac et al. 2015; Pendyala and Natarajan 2010). Taken together, these data suggest that although neuronal SOD2 deficiency and resultant mitochondrial O2.− activates the Nrf2 pathway, this response is incomplete, transient, and unable to effectively counter the progressive accumulation of oxidative damage.
Forebrain neuron-specific SOD2 deficiency results in motor impairment and epilepsy.
By approximately 1 m old, nSOD2 KO mice were visually distinguishable from their WT littermates based on body weight (mean ± SEM 11.07g ± 0.5226 and 14.27g ± 0.7946 respectively) despite the availability of moistened chow in all housing cages starting at weaning. Additionally, nSOD2 KO mice were observed to possess a kyphotic body posture and progressive motor dysfunction including forelimb tremors and gait abnormalities predominated by hind limb ataxia. This distinctive phenotype was present in both male and female nSOD2 KO mice.
To characterize motor deficits, we performed open field and rotarod behavioral testing in 6 week old mice. nSOD2 KO mice were able to ambulate in the open field and readily explored the environment. However, spontaneous locomotion measured as the total distance traveled was significantly decreased in nSOD2 KO mice (Figure 2A; nSOD2 KO: mean ± SEM=1193 cm ± 187.1 compared to WT: mean ± SEM=2135 cm ± 394.7). Similarly, motor coordination and balance measured by latency to fall from a rotating rod was found to be highly impaired in nSOD2 KO mice (Figure 2B; mean ± SEM=31.73 sec ± 5.414) relative to their WT littermates (mean ± SEM=248.3 sec ± 37.59).
Figure 2.
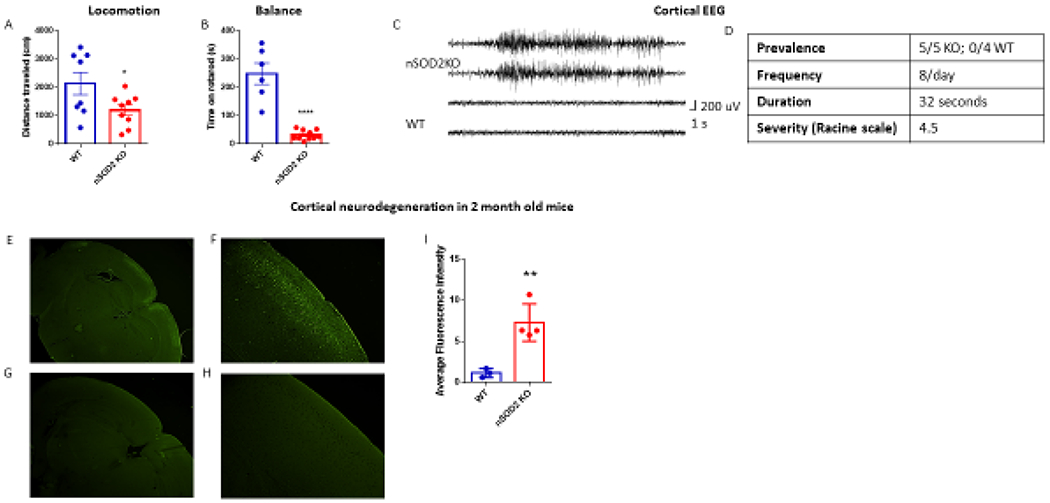
Behavioral characterization of nSOD2 KO mice and region specific neuronal degeneration. (A) Locomotive behavior was assessed with the open field test (unpaired t test, n=8-9, p=0.0408). (B) Motor performance was assessed with the rotarod test (unpaired t test, n=6-10, p<0.0001). (C) Representative electrographic seizure in 2 month old nSOD2 KO mouse corresponding to a class 5 behavioral seizure. (D) Average frequency, duration, and severity of seizures. No seizures were observed in WT littermates. (E-I) Fluorojade B staining in 2 month old nSOD2 KO mice reveals degenerating neurons in the somatosensory and motor cortical regions (E 2x objective, F 10x objective) but not in WT littermates (G 2x objective, H 10x objective). (I) Fluorescence intensity was quantified and analyzed via unpaired t test, n=3-4, p=0.0067.
Starting at approximately 1 month of age, nSOD2 KO mice were observed to display seizure-like behaviors including Straub tail reaction, wet dog shaking, forelimb clonus, and hind limb clonus with rearing, falling and running. To confirm that these behaviors were seizures, bilateral subdural electroencephalogram (EEG) electrodes were surgically implanted over the motor cortex of 6-8 week old mice. A representative electrographic seizure of a nSOD2 KO mouse is shown in figure 2C, which corresponded to a class 5 convulsive behavior including fore- and hind-limb tonic-clonic activity. All nSOD2 KO mice (5/5, 100%) exhibited similar seizure activity during the observation period whereas no electrographic or behavioral abnormalities were noted in any WT mice (0/4, 0%, Figure 2D). nSOD2 KO mice exhibited approximately 8 seizure events per day, with a duration of approximately 32 seconds and an average severity score of 4.5 (based on a modified Racine scale where 1=non-convulsive and 5=most severe seizure) (Figure 2D).
Additional bilateral depth electrodes were implanted directly into the hippocampus in a subset of mice to further ascertain seizure origin. In 2 out of 3 mice implanted with depth electrodes, seizure activity was first detected in the cortical electrodes and hippocampal seizure activity lagged cortical activity for several seconds. However, in the third mouse, seizure activity was first apparent on hippocampal electrodes, which would then generalize to the contralateral hippocampus, and often then become cortical. Thus, seizure activity is apparent in both hippocampus and motor cortex. Together, these data indicate that increased steady state mitochondrial O2.− in a subset of neurons is sufficient to drive significant motor impairment and a robust epileptic phenotype.
Neuronal SOD2 deficiency induces region-specific neuronal degeneration.
Both oxidative stress and seizures can cause neuronal degeneration. Indeed, global SOD2 deficient mice on a mixed genetic background display severe neurodegeneration known as spongiform encephalopathy (Melov et al. 1998). To determine if loss of SOD2 in forebrain neurons resulted in neuronal death, we quantified neurodegeneration using Fluorojade B staining (FJB) (Figure 2E–I). FJB fluorescence was significantly increased in nSOD2 KO mice (Figure 2E, 2F; mean ± SEM=7.287 a.u. ± 1.143) relative to WT mice (Figure 2G,H) (mean ± SEM= 1.148 a.u. ± 0.3128), indicative of increased neuronal degeneration (Figure 2I). Remarkably, staining was limited to the retrosplenial, motor, and somatosensory cortex in nSOD2 KO mice (Figure 2E, 2F), whereas WT littermates were devoid of fluorojade B staining in all regions examined (Figure 2G, 2H). Moreover, staining in nSOD2 KO mice was comprised of neurons located entirely within layer II/III of the cortex. This distinct pattern of neuronal degeneration cannot be explained by NEX expression alone, as NEX expression and therefore SOD2 deletion is robust in hilar mossy cells, dentate gyrus granule cells and pyramidal neurons throughout the dorsal telencephalon (Goebbels et al. 2006). The fact that staining was entirely absent in the hippocampus of nSOD2 KO mice was surprising, given the observation of seizure activity within the hippocampus and the relative sensitivity of hippocampal neurons to seizure-induced degeneration. This could suggest that the effects of increased steady-state mitochondrial O2.− are highly dependent on cell-type. Alternatively, it could suggest that these degenerating cortical neurons were under some additional metabolic stress.
Neuronal SOD2 deficiency induces a metabolic switch toward aerobic glycolysis in neuron-glial cultures and alters plasma and cortical glucose levels in mice.
Seizures represent a state of heightened metabolic demand as evidenced by enhanced cerebral blood flow and increases in cerebral glucose uptake and oxygen consumption (Blennow et al. 1979). If seizures are persistent enough however, energy demand exceeds what aerobic metabolism can provide thus shunting energy production towards glycolysis (Federico et al. 2005; Sacktor, Wilson, and Tiekert 1966). Given the pervasiveness of seizures in nSOD2 KO mice, we examined how this may result in altered bioenergetics within primary neuronal-glial cultures derived from nSOD2 KO and WT littermates. Glycolytic function in these cells was assessed by measuring extracellular acidification rates (ECAR; XF96 extracellular flux analyzer, Seahorse Bioscience). One hour prior to the analysis, the cells were deprived of glucose. A representative glycolytic stress test profile is shown in Figure 3A, which enables the examination of four glycolytic parameters: glycolysis (the level of ECAR reached following the addition of a saturation amount of glucose), glycolytic capacity (the maximum ECAR reached following disablement of oxidative phosphorylation), glycolytic reserve (the capability to respond to an energetic demand, and how close the glycolytic function is to the cell’s theoretical maximum) and non-glycolytic acidification (other sources of extracellular acidification not attributed to glycolysis) (Wu et al. 2007), (Seahorse Bioscience). Of those parameters, only non-glycolytic acidification was unaltered in nSOD2 KO cell preparations. Glycolysis was found to be increased 133.6% (p=0.0088), glycolytic capacity was increased 150.1% (P=0.0047) and glycolytic reserve was increased 138.0% (P=0.0265) (Figure 3B–D). It remains unclear whether this increase in aerobic glycolysis is due to mitochondrial dysfunction or overall increased metabolic demand due to underlying hyperexcitability in these cells. However, mitochondrial function was unaltered in nSOD2 KO cell preparations as measured by XF Cell Mito Stress Test (Seahorse Bioscience) (Supplemental Figure 3). Additionally, no observable change in basal oxygen consumption was observed among any of the ECAR studies performed (data not shown).
Figure 3.
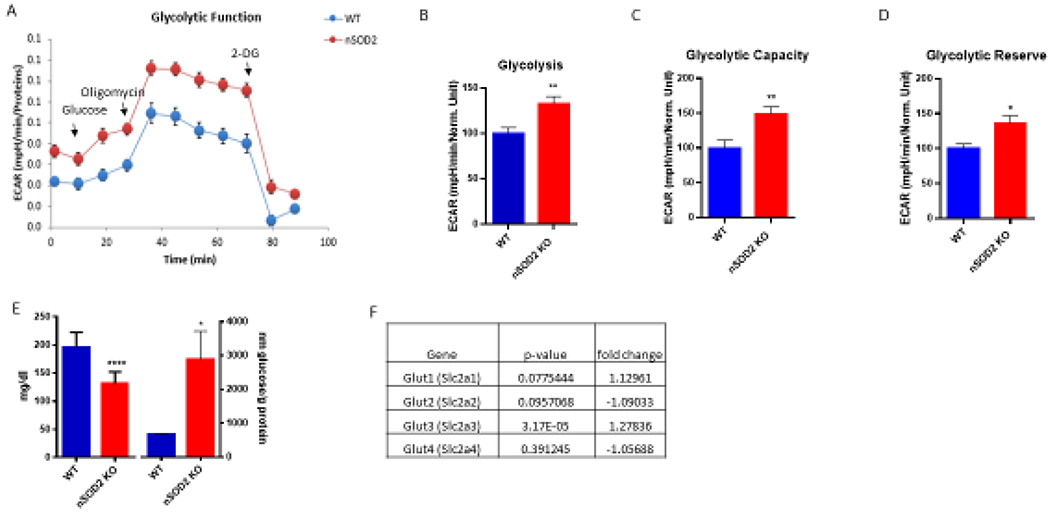
Neuronal SOD2 deficiency induces a metabolic switch toward aerobic glycolysis. (A) Representative trace of a glycolytic stress test performed on primary cortical cells derived from pnd 2-3 nSOD2 KO and WT littermates (n=10 wells/animal, representing 6 WT, 10 KO, across 3 experiments). (B-D) Extracellular acidification rates measured key parameters of glycolytic flux, including (B) glycolysis (p=0.0088), (C) glycolytic capacity (p=0.0047), and (D) glycolytic reserve (p=0.0265). (E) Glucose levels were measured in 2 month old mice and found to be significantly decreased in plasma (unpaired t test, n=9-12, p<0.0001) but increased in cortical tissue (unpaired t test, n=11-12, p=0.0104). (F) Gene expression analysis of glucose transporters in cortical tissue of 2 m old nSOD2 mice (unpaired t test, n=12/group).
This observed increase in glycolysis in the presence of oxygen prompted the examination of glucose levels in both plasma and cortical tissue of WT and nSOD2 KO mice. Following a 6 h fast, nSOD2 KO mice were found to have a significant decrease in plasma glucose (mean ± SEM 131.2 mg/dl ± 6.158 as compared to 195.4 mg/dl ± 9.025). This is in contrast with what is observed in cortical tissue, in which glucose levels were found to be significantly increased (WT= mean ± SEM 665.7 nm/g protein ± 33.05, KO= 2889.0 ± 826.7) (Figure 3E).
Transcriptome wide gene expression profiling uncovered a modest but highly significant increase in the primary neuronal glucose transporter Glut3 (Figure 3F, fold change=1.27835, p=3.17X10−5). No other glucose transporter was found to have significantly altered gene expression in these mice.
Increased neuronal mitochondrial O2.− elevates expression of intermediate filaments and promotes astrogliosis.
In order to gain a more comprehensive view of alterations to cortical gene expression in these mice we employed a mouse Clariom S genome microarray (ThermoFisher). Of the ~22,000 genes examined, 3,807 were found to be differentially expressed in nSOD2 KO mice. The 20 most significantly altered genes are listed in Supplemental Table 2 (fold change > 2, p<0.05). A closer examination of specific genes upregulated revealed robust expression of GFAP and was, in accordance with our earlier finding, accompanied by an almost equally robust upregulation of vimentin (Vim) (4.4327 fold increase, p=5.3X10−17 and 3.32499 fold increase, p=6.36X10−14 respectively; Supplemental Table 2). In fact, GFAP was found to have the highest level of increased gene expression in the entire transcriptome. Both GFAP and Vim results were confirmed by RTPCR (Figure 4A, 1F). The induction of astrogliosis was developmentally dependent, and was not observed in mice at one and three weeks of age (data not shown). However, by 2 months of age, astroglial activation sharply increased in nSOD2 KO mice which could be observed by RTPCR methods (4.936 fold increase, p<0.0001; Figure 4A).
Figure 4.
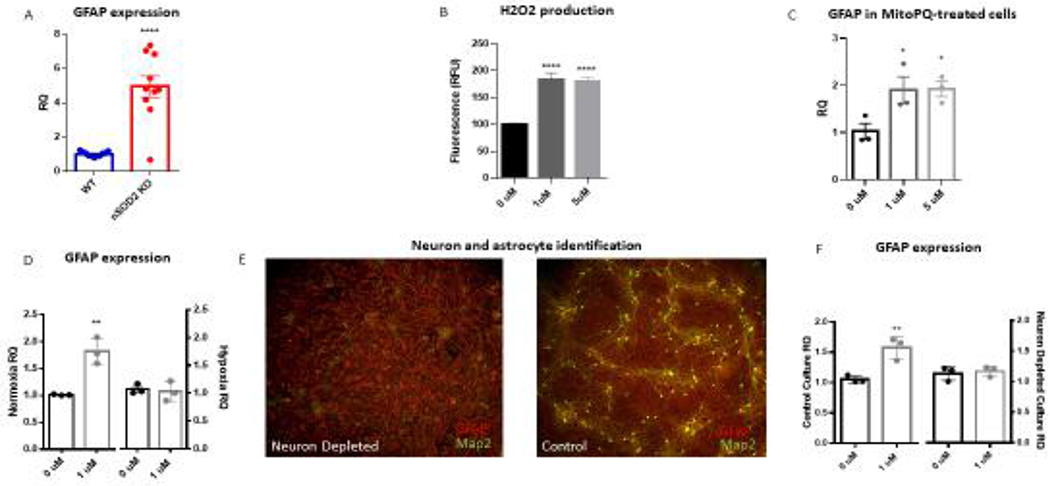
Neuronal SOD2 deficiency in vivo and neuronal mitochondrial oxidative stress in vitro induces astrogliosis. (A) GFAP transcription was found to be increased in the cortex of nSOD2 KO mice at 2 months of age as compared to WT littermates in a whole genome array and this finding was verified via RT-PCR (unpaired t test, n=4-5, p<0.0001). (B) The selective mitochondria-target redox cycler MitoPQ increases ROS production in rat primary cortical cells at 1 and 5 μM within 24 hours as measured by amplex red (one-way ANOVA with Dunnett’s multiple comparison, p<0.0001). (C) Following a one week incubation, 1 and 5 uM MitoPQ induces astroglyosis as measured by GFAP gene expression (three independent trials, one-way ANOVA with Dunnett’s multiple comparison, p=0.0463 (1uM) and p=0.0413 (5uM)). (D) This induction of GFAP is abated when cells are grown under hypoxic conditions (unpaired t test, n=3 independent trials, p=0.0008 (normoxia) p=0.5213 (hypoxia). (E) ICC of GFAP (red) and Map2 (green) confirms the establishment of neuronally-depleted cortical cultures as compared to side-by-side controls. (F) Neuronal depletion prevents MitoPQ-induced astrogliosis as evidenced by GFAP gene expression (unpaired t test, n=3 independent trials, p=0.028 (control), p=0.9186 (neuron depleted)
In order to explore the mechanistic underpinnings of astroglial activation in these mice, we examined GFAP expression in rat primary cortical cultures following treatment with the mitochondria targeted redox-cycler MitoPQ. MitoPQ is a compound well suited to mimic the redox status in nSOD2 KO mice because it selectively induces production of O2.− within the mitochondrial matrix (Robb et al. 2015). We first wanted to discover a concentration of drug that would effectively increase O2.− production as indicated by H2O2 production without causing cytotoxicity. Both 1 and 5 uM were shown to increase H2O2 production within 24 hours (83.4% and 80.22% respective increases) (Figure 4B) and the 5 μM, but not the 1μM were shown to cause cytotoxicity after one week via LDH assay (data not shown). Next, mixed cortical cells were grown for two weeks then treated with MitoPQ for one week under normoxic conditions. Both 1 and 5 μM significantly induced astrogliosis as measured by GFAP RTPCR analysis. The 1 μM samples increased GFAP expression by 1.862 fold change (p=0.0306) and the 5 μM samples were increased by 1.913 fold change (p=0.0249; Figure 4C).
MitoPQ reacts with atmospheric O2 to produce O2.−. Therefore we surmised that decreasing available O2 would reduce GFAP expression, further strengthening O2.− production and astrogliosis causation. To that end, cells were grown in hypoxic conditions (5% O2) during a one week treatment with MitoPQ. MitoPQ-induced astrogliosis was abated when cells were grown under hypoxic conditions, as demonstrated by GFAP RTPCR analysis (Figure 4D).
The discovery that a neuronal deletion of SOD2 and subsequent neuron-derived oxidative stress was sufficient to induce astrogliosis in our nSOD2 KO mice was both surprising and intriguing. In order to corroborate this finding in vitro, neuron-depleted primary cell preparations were established through multiple complete media replacements over a two week course (Souza et al. 2013). Neuron depletion was confirmed via ICC (Figure 4E) and astrogliosis was measured via GFAP RTPCR following MitoPQ treatment (Figure 4F). Neuronal depletion abated MitoPQ-induced GFAP upregulation (RQ=1.145, control and RQ=1.169 1 μM MitoPQ, p=0.9186) as compared to cells derived from the same preparations and grown concurrently but lacking the media changes (RQ=1.055, control and RQ=1.561, 1uM MitoPQ, p=0.0273). The experiment was repeated 3 times and combined results are reported.
DISCUSSION
Here we describe conditional inactivation of SOD2 in forebrain neurons which improved longevity relative to constitutive SOD2 deletion and allowed for a more focused examination of neuron-specific pathological outcomes. Selectively depleting SOD2 in NEX- expressing excitatory principal neurons decreased total cortical SOD2 expression and activity to approximately 50% that of WT littermates. This was associated with significantly elevated cortical O2.− and oxidative modification of mitochondrial proteins. nSOD2 KO mice exhibited a mitohormetic gene response including the induction of the Nrf2 pathway and the upregulation of several antioxidant response genes. Despite attempts to mitigate increased steady state O2.− through mitohormetic adaptation, nSOD2 KO mice exhibited progressive motor impairment and spontaneous seizures. Cortical cultures derived from these mice demonstrated a metabolic switch to aerobic glycolysis to meet increased energetic demand. Neuronal degeneration was evident in somatosensory and motor regions of the cortex. Additionally, GFAP, a marker of astrocytes was strikingly upregulated in nSOD2 KO. Further in vitro studies suggested this induction was primarily driven by increased neuronal O2.− indicating that SOD2 deletion and neuronal O2.− can function in a cell nonautonomous manner to influence astrocytes. Together, these studies elucidate novel cell-type specific effects of SOD2 and provide a mouse model of epilepsy originating from a perpetual state of mitochondrial oxidative stress.
Mitochondrial O2.− is produced as a natural consequence of aerobic metabolism. Neurons which comprise approximately 50% of the cell types in brain account for 80-90% of ATP consumption making them prime generators of O2.− (Herculano-Houzel 2014). Moreover, O2.− production varies across neuronal sub-type suggesting that perturbations to antioxidant defenses may render some neurons more vulnerable to oxidative insults than others (Hoegger, Lieven, and Levin 2008). SOD2 is ubiquitously expressed in mitochondria where it converts O2.− to less reactive molecules. In contrast to other isoforms of SOD, constitutive genetic deletion of SOD2 is neonatally lethal highlighting the devastating effects of mitochondrial O2.− production (Li et al. 1995; Lebovitz et al. 1996; Huang et al. 2001; Melov et al. 1999). Longer lived, mixed-background strain SOD2 deficient mice present with significant cardiac and neurological impairments supporting the hypothesis that mitochondrial O2.− is uniquely damaging to highly metabolically active tissues (Huang et al. 1999; Liang et al. 2012). Organ and cell-type specific deletion of SOD2 has allowed for an even more detailed examination of this hypothesis. Indeed SOD2 deletion in liver, muscle or kidney results in mild phenotypes with normal lifespan (Ikegami et al. 2002; Kuwahara et al. 2010; Lustgarten et al. 2011; Parajuli et al. 2011), whereas ubiquitous deletion of SOD2 in brain recapitulates many aspects of the constitutive knockout including reduced lifespan and severe neurodegeneration (Izuo et al. 2015). Even within the brain, mitochondrial O2.− appears to have differential effects based on cell type. SOD2 deletion in motor neurons has no observable effect on health or lifespan (Misawa et al. 2006). Yet, as we report here, its deletion in principal forebrain neurons is sufficient to drive selective neurodegeneration, seizures, and death by 3 months of age. This demonstrates that deletion of SOD2 from a specific subset of neurons can recapitulate the deleterious effects of constitutive SOD2 removal. Moreover, it suggests that effects of persistently increased steady state mitochondrial O2.− varies as a function of neuronal type.
Measurement of O2.− in biological systems is a complex task owing to its high reactivity and rapid dismutation to other reactive molecules. As such, common measurement methodologies such as fluorescent probes are often plagued with issues of sensitivity or non-specificity (Wardman 2007). In our model, detection of O2.− and resulting oxidative damage is further complicated by the presence of functional SOD2 in astrocytes, oligodendrocytes, microglia, and other neuronal subtypes. We therefore opted to measure cortical O2.− using two independent methods. First, we assessed O2.− production in cortical tissue using HPLC based detection of O2.− sensitive 2-OHDE following peripheral injection of DHE. Because other reactive species such as H2O2 hydroxyl radical, ONOO-, and hypochlorous acid do not oxidize DHE in water, 2-OHDE is formed from the specific oxidation of DHE by O2.− (Fink et al. 2004; Zhao et al. 2005; Zhao et al. 2003). Additionally, HPLC based detection allows for the distinction between the formation of 2-OHDE and the ethidium (E+)/DNA complex (Zhao et al. 2005; Maghzal and Stocker 2007). Using this method we found significantly increased levels of O2.− in cortical tissue of nSOD2 KO mice. Importantly, since SOD2 deletion is cell-type specific in this model, regional O2.− levels, particularly in forebrain, are likely greater than those detected. All biochemical measurements were performed in bulk cortical tissues and not cells sorted according to SOD2 deletion, so it is unknown if SOD2 depletion, which is present in a subset of neurons, affected only those neurons or also resulted in changes to other neurons by indirect mechanisms.
HPLC detection of 2-OHDE is specific to O2.− formation, however it does not implicate a mitochondrial source. Therefore we employed the well-validated aconitase activity assay to confirm mitochondrial O2.− origin (Gardner et al. 1995; Patel et al. 1996). Aconitase is unique among TCA enzymes due to the presence of a cubane [4Fe-4S]2+ cluster containing a labile iron vulnerable to oxidant-mediated loss, thus rendering the enzyme inactive (Verniquet et al. 1991). O2.− is especially, but not exclusively reactive to [4Fe-4S]2+; other oxidants such as H2O2. and ONOO- are capable of aconitase inactivation (Castro, Rodriguez, and Radi 1994). Thus combining both methods of O2.− detection elucidates both the species and source of oxidants occurring in this mouse model.
An antioxidant response gene array revealed a significant increase in both Nox1 and Nox4, along with the Nox2 complex subunit Cyba. This finding is curious, since Nox enzymes are major cellular sources of O2.− and increased expression is associated with elevated oxidative stress (Tarafdar and Pula 2018). Indeed, Nox enzymes have been implicated in neurodegenerative diseases including Parkinson’s and Alzheimer’s (Hernandes and Britto 2012) and have been proposed as potential therapeutic targets to reduce seizure susceptibility (Huang et al. 2018; Malkov et al. 2019). Nox 1 and 4 upregulation contributes not only to the overall load of oxidative stress but also seizure pathology through glutamate induced and NMDA-mediated activation of Nox (Malkov et al. 2019). Indeed we are not the first to report this seemingly paradoxical induction of Nox4 following oxidative stress. Vallet et al., (Vallet et al. 2005) reported an increase in Nox 4 following ischemia-induced neuronal damage, which is known to be associated with a chronic induction of ROS, and (Kleinschnitz et al. 2010) was able to demonstrate neuroprotective outcomes by using a Nox inhibitor. Interestingly, recent studies suggest that Nox4 is localized to neuronal mitochondria (Case et al. 2013) suggesting its regulation by mitochondrially-generated O2−..
Despite its contribution to pathology when produced in excess, O2.− at physiological levels serves as an important signaling molecule involved in homeostatic and adaptive responses (Shadel and Horvath 2015). The accumulation of H2O2 following spontaneous dismutation due to mitochondrial aconitase inactivation (Cantu et al. 2011; Liochev and Fridovich 1994) may also drive the Nrf2 response. Consistent with this, transient SOD2 depletion during embryonic development produces a mitohormetic gene response in liver including induction of the Nrf2 pathway, increased antioxidant gene expression and reduced production of reactive species (Cox et al. 2018). This response is sustained suggesting that transient mitochondrial O2.− can be beneficial. In our study, cortical SOD2 expression and activity was decreased to approximately half that of WT control, in a specific brain cell population i.e. cortical neurons. This persistent, yet spatially restricted O2.− production yielded a similar attempt at mitohormetic adaptation through gene expression in the brain which ultimately failed to prevent the observed pathological phenotypes including a catastrophic epilepsy syndrome. Also, it has been shown that Nox4 is a lesser-known target gene of Nrf2 and that when Nrf2 is constitutively activated, Nox4 is upregulated (Pendyala et al. 2010; Kovac et al. 2015). Thus there is a complex and reciprocal interaction between Nrf2 and Nox enzymes, and may explain our observed activation of the Nrf2 pathway.
The role of mitochondrial dysfunction in the etiology of epilepsies has lagged despite knowledge that metabolic functions can impact neuronal excitability and a subset of inherited epilepsies such as myoclonic epilepsy with ragged red fibers (MERRF) and mitochondrial encephalopathy (MELAS) directly arise due to mitochondrial DNA mutations and consequent dysfunction. Here we describe a further example of combined genetic and mitochondrial causation in epilepsy. While we report a mixed (cortical/hippocampal) origin of seizures, we did not find a complete overlap between seizure activity and neuronal death, which was limited to cortical layers II/III in the retrosplenial, motor, and somatosensory regions. Certainly, seizures were found to be long in duration and repetitive; characteristics found to predispose the brain toward neuronal death (Fujikawa 1996). Furthermore, NEX-Cre mediated knock down is robust in hippocampus, so it is intriguing that this prominently sensitive brain region would be immune to necrotic cell death in this model.
Human studies clearly demonstrate hypo- and hyper-metabolic alterations associated with interictal and ictal phases in epilepsy patients. Several key features of epilepsies suggest glycolytic exhaustion and mitochondrial decline in their genesis (Pitkanen and Sutula 2002; Hauser and Annegers 1991; Leiderman et al. 1992). The observation that cortical cultures derived from nSOD2 KO mice display a preferential upregulation of glycolytic function, suggest an adaptive response secondary to chronic mitochondrial oxidative stress reminiscent of a classical Warburg effect. Increase in network activity in the brain relies on aerobic glycolysis as a rapid, albeit comparatively low yield source of ATP (Dienel and Cruz 2016). Although we did not observe a change in basal oxidative phosphorylation in cortical cultures, mitochondrial dysfunction as a cause of increased glycolysis cannot be ruled out. SOD2 deletion in mouse embryonic fibroblasts displayed a reduction in mitochondrial spare respiratory capacity (Cox et al. 2018). When SOD2 was knocked down in either neurons or cells of neurogenic lineage, there was a selective loss of enzymatic activity of ETC complex II (Izuo et al. 2015; Maity-Kumar et al. 2015). Previously, we reported that synaptosomal isolates from these mice exhibited severe mitochondrial deficits in reserve and maximal respiratory capacity (Rowley et al. 2015). Whether we are observing a Warburg-type effect to meet increased energetic demand or a compensatory adaptation due to ROS-driven mitochondrial dysfunction is an area for future discovery. In addition, decreased levels of plasma glucose and increased brain glucose, which was accompanied by an increase in neuronal glucose transporter Glut3 expression in nSOD2 KO mice indicates that neurons are directly increasing their uptake of glucose and glycolysis as opposed to relying on astrocytic lactate as a source of fuel. The observed plasma hypoglycemia could be brought on by increased glucose delivery to the brain as a fuel for ongoing seizure activity. However, no other glucose transporters were upregulated, including Glut1 the major glucose transporter across the mammalian blood-brain barrier. Rather, it is likely that neuronal deletion of SOD2 results in plasma hypoglycemia due to impaired central regulation of glucose homeostasis, as evidenced by (Maity-Kumar et al. 2015).
In addition to the well-known roles of mitochondria in the production of ATP, metabolites, ionic regulation, cell death and redox homeostasis, they are now recognized to play an important role in establishing and maintaining various immune responses (Angajala et al. 2018). In many instances, mitochondrial-generated ROS provide the necessary signaling component to launch these responses. In our nSOD2 KO mice, we found a striking age-dependent upregulation of GFAP and vim, and through a series of in vitro experiments identified neuronal mitochondrial O2.− as a sufficient signal for astrogliosis since GFAP failed to upregulate following depletion of neurons or under low oxygen conditions. The observation that neuronal-specific mitochondrial O2.− initiates astrocyte-specific GFAP mRNA and protein expression suggests a redox dependent pathway. Several mediators including nitric oxide have been shown to increase GFAP expression in the brain, however to date, the role of mitochondria and/or mitochondrial O2.− has not been associated with the upregulation of GFAP (McKeon and Benarroch 2018). Redox modification of GFAP and Vim, which are both type III intermediate filament structural proteins and highly upregulated in the nSOD2 KO mice has been recently observed. GFAP and Vim share a conserved cysteine (Cys294 in GFAP and Cys328 in Vim) which can be modified by electrophiles (Cy-prostaglandins) or oxidants (Duarte et al. 2018; Viedma-Poyatos et al. 2018). The resulting self-polymerization of GFAP or cross-polymerization with vimentin could contribute to the upregulation in gene expression resulting in an astrogliotic response. The precise mechanism underlying this intriguing effect remains to be explored.
In summary, these studies illuminate novel consequences of neuron-specific elevation of mitochondrial O2.− which include a unique pattern of cortical neurodegeneration, epileptic phenotype, metabolic reprogramming and a cell-nonautonomous astrocytic response. Together they highlight the complex and important role of mitochondrial O2.− in neurons.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by grants R01NS039587, RO1NS039587S1 and R01 NS086423 from NINDS and the ALSAM Therapeutic Innovation Award (M.P.) and T32 AG000279 (J.P.S.). The authors appreciate the contribution to this research made by E. Erin Smith, HTL (ASCP)CMQIHC, Allison Quador, HTL(ASCP)CM and Jessica Arnold, HTL(ASCP)CM of the University of Colorado Biorepository Core Facility, Research Histology Shared Resource, and Dr. Linda Johnson, DVM, MPH, DACVP and Susan Tousey, CVT (NAVTA) MLT (ASCP) of the Comparative Pathology Shared Resources Clinical Laboratory, CU Denver Anschutz for clinical pathology support. Additionally, we would like to thank Katrina Diener, Etienne Danis, and Ted Shade of the Microarray and Genomics Core within the Cancer Center at CU Denver Anschutz, and Marianne Alexis. The authors are grateful to Dr. Amy Brooks-Kayal and Dr. Yogendra Raol for assistance in verifying origin of seizure onset. Research support was provided by the Behavior and In Vivo Neurophysiology core, a member of the NeuroTechnology Center at the University of Colorado-School of Medicine, funded in part by the National Institute of Neurological Disorders and Stroke of the National Institutes of Health under award number P30NS048154. This resource is supported in part by the Cancer Center Support Grant.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Aguiar CCT, Almeida AB, Araujo PVP, de Abreu RNDC, Chaves EMC, do Vale OC, Macedo DS, Woods DJ, Fonteles MMD, and Vasconcelos SMM. 2012. ‘Oxidative Stress and Epilepsy: Literature Review’, Oxidative medicine and cellular longevity, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angajala A, Lim SB, Phillips JB, Kim JH, Yates C, You ZB, and Tan M. 2018. ‘Diverse Roles of Mitochondria in Immune Responses: Novel Insights Into Immuno-Metabolism’, Frontiers in Immunology, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blennow G, Folbergrova J, Nilsson B, and Siesjo BK. 1979. ‘Cerebral metabolic and circulatory changes in the rat during sustained seizures induced by DL-homocysteine’, Brain Res, 179: 129–46. [DOI] [PubMed] [Google Scholar]
- Brewer AC, Murray TV, Arno M, Zhang M, Anilkumar NP, Mann GE, and Shah AM. 2011. ‘Nox4 regulates Nrf2 and glutathione redox in cardiomyocytes in vivo’, Free Radic Biol Med, 51: 205–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantu D, Fulton RE, Drechsel DA, and Patel M. 2011. ‘Mitochondrial aconitase knockdown attenuates paraquat-induced dopaminergic cell death via decreased cellular metabolism and release of iron and H2O2’, Journal of Neurochemistry, 118: 79–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson LM, Jonsson J, Edlund T, and Marklund SL. 1995. ‘Mice lacking extracellular superoxide dismutase are more sensitive to hyperoxia’, Proc Natl Acad Sci U S A, 92: 6264–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case AJ, Li SM, Basu U, Tian J, and Zimmerman MC. 2013. ‘Mitochondrial-localized NADPH oxidase 4 is a source of superoxide in angiotensin II-stimulated neurons’, American Journal of Physiology-Heart and Circulatory Physiology, 305: H19–H28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro L, Rodriguez M, and Radi R. 1994. ‘Aconitase Is Readily Inactivated by Peroxynitrite, but Not by Its Precursor, Nitric-Oxide’, Journal of Biological Chemistry, 269: 29409–15. [PubMed] [Google Scholar]
- Cobley James Nathan, Fiorello Maria Luisa, and Bailey Damian Miles. 2018. ‘13 reasons why the brain is susceptible to oxidative stress’, Redox Biology, 15: 490–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox CS, McKay SE, Holmbeck MA, Christian BE, Scortea AC, Tsay AJ, Newman LE, and Shadel GS. 2018. ‘Mitohormesis in Mice via Sustained Basal Activation of Mitochondrial and Antioxidant Signaling’, Cell Metab, 28: 776–86 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dienel GA, and Cruz NF. 2016. ‘Aerobic glycolysis during brain activation: adrenergic regulation and influence of norepinephrine on astrocytic metabolism’, Journal of Neurochemistry, 138: 14–52. [DOI] [PubMed] [Google Scholar]
- Duarte S, Viedma-Poyatos A, Monico A, and Perez-Sala D. 2018. ‘The conserved cysteine residue of type III intermediate filaments serves as a structural element and redox sensor’, Free Radical Biology and Medicine, 120: S84–S84. [DOI] [PubMed] [Google Scholar]
- Federico P, Abbott DF, Briellmann RS, Harvey AS, and Jackson GD. 2005. ‘Functional MRI of the pre-ictal state’, Brain, 128: 1811–7. [DOI] [PubMed] [Google Scholar]
- Fink B, Laude K, McCann L, Doughan A, Harrison DG, and Dikalov S. 2004. ‘Detection of intracellular superoxide formation in endothelial cells and intact tissues using dihydroethidium and an HPLC-based assay’, American Journal of Physiology-Cell Physiology, 287: C895–C902. [DOI] [PubMed] [Google Scholar]
- Fujikawa DG 1996. ‘The temporal evolution of neuronal damage from pilocarpine-induced status epilepticus’, Brain Res, 725: 11–22. [DOI] [PubMed] [Google Scholar]
- Gardner PR, Raineri I, Epstein LB, and White CW. 1995. ‘Superoxide radical and iron modulate aconitase activity in mammalian cells’, J Biol Chem, 270: 13399–405. [DOI] [PubMed] [Google Scholar]
- Goebbels S, Bormuth I, Bode U, Hermanson O, Schwab MH, and Nave KA. 2006. ‘Genetic targeting of principal neurons in neocortex and hippocampus of NEX-Cre mice’, Genesis, 44: 611–21. [DOI] [PubMed] [Google Scholar]
- Hauser WA, and Annegers JF. 1991. ‘Risk factors for epilepsy’, Epilepsy Research - Supplement, 4: 45–52. [PubMed] [Google Scholar]
- Herculano-Houzel S 2014. ‘The Glia/Neuron Ratio: How it Varies Uniformly Across Brain Structures and Species and What that Means for Brain Physiology and Evolution’, Glia, 62: 1377–91. [DOI] [PubMed] [Google Scholar]
- Hernandes MS, and Britto LRG. 2012. ‘NADPH Oxidase and Neurodegeneration’, Current Neuropharmacology, 10: 321–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoegger MJ, Lieven CJ, and Levin LA. 2008. ‘Differential production of superoxide by neuronal mitochondria’, BMC Neurosci, 9: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang TT, Carlson EJ, Kozy HM, Mantha S, Goodman SI, Ursell PC, and Epstein CJ. 2001. ‘Genetic modification of prenatal lethality and dilated cardiomyopathy in Mn superoxide dismutase mutant mice’, Free Radic Biol Med, 31: 1101–10. [DOI] [PubMed] [Google Scholar]
- Huang TT, Carlson EJ, Raineri M, Gillespie AM, Kozy H, and Epstein CJ. 1999. ‘The use of transgenic and mutant mice to study oxygen free radical metabolism’, Oxidative/Energy Metabolism in Neurodegenerative Disorders, 893: 95–112. [DOI] [PubMed] [Google Scholar]
- Huang WY, Lin SK, Chen HY, Chen YP, Chen TY, Hsu KS, and Wu HM. 2018. ‘NADPH oxidases as potential pharmacological targets against increased seizure susceptibility after systemic inflammation’, Journal of Neuroinflammation, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikegami T, Suzuki Y, Shimizu T, Isono K, Koseki H, and Shirasawa T. 2002. ‘Model mice for tissue-specific deletion of the manganese superoxide dismutase (MnSOD) gene’, Biochem Biophys Res Commun, 296: 729–36. [DOI] [PubMed] [Google Scholar]
- Izuo N, Nojiri H, Uchiyama S, Noda Y, Kawakami S, Kojima S, Sasaki T, Shirasawa T, and Shimizu T. 2015. ‘Brain-Specific Superoxide Dismutase 2 Deficiency Causes Perinatal Death with Spongiform Encephalopathy in Mice’, Oxidative medicine and cellular longevity, 2015: 238914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinschnitz C, Grund H, Wingler K, Armitage ME, Jones E, Mittal M, Barit D, Schwarz T, Geis C, Kraft P, Barthel K, Schuhmann MK, Herrmann AM, Meuth SG, Stoll G, Meurer S, Schrewe A, Becker L, Gailus-Durner V, Fuchs H, Klopstock T, de Angelis MH, Jandeleit-Dahm K, Shah AM, Weissmann N, and Schmidt HH. 2010. ‘Post-stroke inhibition of induced NADPH oxidase type 4 prevents oxidative stress and neurodegeneration’, PLoS Biol, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovac S, Angelova PR, Holmstrom KM, Zhang Y, Dinkova-Kostova AT, and Abramov AY. 2015. ‘Nrf2 regulates ROS production by mitochondria and NADPH oxidase’, Biochim Biophys Acta, 1850: 794–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwahara H, Horie T, Ishikawa S, Tsuda C, Kawakami S, Noda Y, Kaneko T, Tahara S, Tachibana T, Okabe M, Melki J, Takano R, Toda T, Morikawa D, Nojiri H, Kurosawa H, Shirasawa T, and Shimizu T. 2010. ‘Oxidative stress in skeletal Muscle causes severe disturbance of exercise activity without muscle atrophy’, Free Radical Biology and Medicine, 48: 1252–62. [DOI] [PubMed] [Google Scholar]
- Lau A, Tian W, Whitman SA, and Zhang DD. 2013. ‘The predicted molecular weight of Nrf2: it is what it is not’, Antioxid Redox Signal, 18: 91–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebovitz RM, Zhang H, Vogel H, Cartwright J Jr., Dionne L, Lu N, Huang S, and Matzuk MM. 1996. ‘Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice’, Proc Natl Acad Sci U S A, 93: 9782–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiderman DB, Balish M, Sato S, Kufta C, Reeves P, Gaillard WD, and Theodore WH. 1992. ‘Comparison of PET measurements of cerebral blood flow and glucose metabolism for the localization of human epileptic foci’, Epilepsy Res, 13: 153–7. [DOI] [PubMed] [Google Scholar]
- Li R, Jia Z, and Zhu H. 2019. ‘Regulation of Nrf2 Signaling’, React Oxyg Species (Apex), 8: 312–22. [PMC free article] [PubMed] [Google Scholar]
- Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ, Yoshimura MP, Berger C, Chan PH, Wallace DC, and Epstein CJ. 1995. ‘Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase’, Nat Genet, 11: 376–81. [DOI] [PubMed] [Google Scholar]
- Liang LP, Ho YS, and Patel M. 2000. ‘Mitochondrial superoxide production in kainate-induced hippocampal damage’, Neuroscience, 101: 563–70. [DOI] [PubMed] [Google Scholar]
- Liang LP, and Patel M. 2004. ‘Mitochondrial oxidative stress and increased seizure susceptibility in Sod2(−/+) mice’, Free Radic Biol Med, 36: 542–54. [DOI] [PubMed] [Google Scholar]
- Liang LP, Waldbaum S, Rowley S, Huang TT, Day BJ, and Patel M. 2012. ‘Mitochondrial oxidative stress and epilepsy in SOD2 deficient mice: attenuation by a lipophilic metalloporphyrin’, Neurobiol Dis, 45: 1068–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liochev SI, and Fridovich I. 1994. ‘The role of O2.- in the production of HO.: in vitro and in vivo’, Free Radic Biol Med, 16: 29–33. [DOI] [PubMed] [Google Scholar]
- Lustgarten MS, Jang YC, Liu YH, Qi WB, Qin YJ, Dahia PL, Shi Y, Bhattacharya A, Muller FL, Shimizu T, Shirasawa T, Richardson A, and Van Remmen H. 2011. ‘MnSOD deficiency results in elevated oxidative stress and decreased mitochondrial function but does not lead to muscle atrophy during aging’, Aging Cell, 10: 493–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynn S, Huang EJ, Elchuri S, Naeemuddin M, Nishinaka Y, Yodoi J, Ferriero DM, Epstein CJ, and Huang TT. 2005. ‘Selective neuronal vulnerability and inadequate stress response in superoxide dismutase mutant mice’, Free Radic Biol Med, 38: 817–28. [DOI] [PubMed] [Google Scholar]
- Maghzal GJ, and Stocker R. 2007. ‘Improved analysis of hydroethidine and 2-hydroxyethidium by HPLC and electrochemical detection’, Free Radic Biol Med, 43: 1095–6. [DOI] [PubMed] [Google Scholar]
- Maier CM, and Chan PH. 2002. ‘Role of superoxide dismutases in oxidative damage and neurodegenerative disorders’, Neuroscientist, 8: 323–34. [DOI] [PubMed] [Google Scholar]
- Maity-Kumar G, Thal DR, Baumann B, Scharffetter-Kochanek K, and Wirth T. 2015. ‘Neuronal redox imbalance results in altered energy homeostasis and early postnatal lethality’, FASEB J, 29: 2843–58. [DOI] [PubMed] [Google Scholar]
- Malkov A, Ivanov AI, Latyshkova A, Bregestovski P, Zilberter M, and Zilberter Y. 2019. ‘Activation of nicotinamide adenine dinucleotide phosphate oxidase is the primary trigger of epileptic seizures in rodent models’, Annals of Neurology, 85: 907–20. [DOI] [PubMed] [Google Scholar]
- McKeon A, and Benarroch EE. 2018. ‘Glial fibrillary acid protein Functions and involvement in disease’, Neurology, 90: 925–30. [DOI] [PubMed] [Google Scholar]
- Melov S, Coskun P, Patel M, Tuinstra R, Cottrell B, Jun AS, Zastawny TH, Dizdaroglu M, Goodman SI, Huang TT, Miziorko H, Epstein CJ, and Wallace DC. 1999. ‘Mitochondrial disease in superoxide dismutase 2 mutant mice’, Proc Natl Acad Sci U S A, 96: 846–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melov S, Schneider JA, Day BJ, Hinerfeld D, Coskun P, Mirra SS, Crapo JD, and Wallace DC. 1998. ‘A novel neurological phenotype in mice lacking mitochondrial manganese superoxide dismutase’, Nat Genet, 18: 159–63. [DOI] [PubMed] [Google Scholar]
- Misawa H, Nakata K, Matsuura J, Monwaki Y, Kawashima K, Shimizu T, Shirasawa T, and Takahashi R. 2006. ‘Conditional knockout of Mn superoxide dismutase in postnatal motor neurons reveals resistance to mitochondrial generated superoxide radicals’, Neurobiology of Disease, 23: 169–77. [DOI] [PubMed] [Google Scholar]
- Papaiahgari S, Kleeberger SR, Cho HY, Kalvakolanu DV, and Reddy SP. 2004. ‘NADPH oxidase and ERK signaling regulates hyperoxia-induced Nrf2-ARE transcriptional response in pulmonary epithelial cells’, J Biol Chem, 279: 42302–12. [DOI] [PubMed] [Google Scholar]
- Parajuli N, Marine A, Simmons S, Saba H, Mitchell T, Shimizu T, Shirasawa T, and MacMillan-Crow LA. 2011. ‘Generation and characterization of a novel kidney-specific manganese superoxide dismutase knockout mouse’, Free Radical Biology and Medicine, 51: 406–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel M, Day BJ, Crapo JD, Fridovich I, and McNamara JO. 1996. ‘Requirement for superoxide in excitotoxic cell death’, Neuron, 16: 345–55. [DOI] [PubMed] [Google Scholar]
- Pearson Jennifer N., Rowley Shane, Liang Li-Ping, White Andrew M., Day Brian J., and Patel Manisha. 2015. ‘Reactive oxygen species mediate cognitive deficits in experimental temporal lobe epilepsy’, Neurobiology of Disease, 82: 289–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson Jennifer N., Warren Eric, Liang Li-Ping, Roberts L. Jackson, and Patel Manisha. 2017. ‘Scavenging of highly reactive gamma-ketoaldehydes attenuates cognitive dysfunction associated with epileptogenesis’, Neurobiology of Disease, 98: 88–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendyala S, Moitra J, Kalari S, Gorshkova IA, Zhao Y, He D, Garcia JG, and Natarajan V. 2010. ‘Transcriptional Regulation of Hyperoxia- Induced Nox4 Expression in Lung Endothelium by Nrf2-Antioxidant Response Element’, Journal of Investigative Medicine, 58: 657–58. [Google Scholar]
- Pendyala S, and Natarajan V. 2010. ‘Redox regulation of Nox proteins’, Respir Physiol Neurobiol, 174: 265–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitkanen A, and Sutula TP. 2002. ‘Is epilepsy a progressive disorder? Prospects for new therapeutic approaches in temporal-lobe epilepsy’, Lancet Neurol, 1: 173–81. [DOI] [PubMed] [Google Scholar]
- Reaume AG, Elliott JL, Hoffman EK, Kowall NW, Ferrante RJ, Siwek DF, Wilcox HM, Flood DG, Beal MF, Brown RH, Scott RW, and Snider WD. 1996. ‘Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury’, Nature Genetics, 13: 43–47. [DOI] [PubMed] [Google Scholar]
- Robb EL, Gawel JM, Aksentijevic D, Cocheme HM, Stewart TS, Shchepinova MM, Qiang H, Prime TA, Bright TP, James AM, Shattock MJ, Senn HM, Hartley RC, and Murphy MP. 2015. ‘Selective superoxide generation within mitochondria by the targeted redox cycler MitoParaquat’, Free Radical Biology and Medicine, 89: 883–94. [DOI] [PubMed] [Google Scholar]
- Rowley S, Liang LP, Fulton R, Shimizu T, Day B, and Patel M. 2015. ‘Mitochondrial respiration deficits driven by reactive oxygen species in experimental temporal lobe epilepsy’, Neurobiol Dis, 75: 151–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacktor B, Wilson JE, and Tiekert CG. 1966. ‘Regulation of glycolysis in brain, in situ, during convulsions’, J Biol Chem, 241: 5071–5. [PubMed] [Google Scholar]
- Schmued Larry C., and Hopkins Keri J.. 2000. ‘Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration’, Brain Research, 874: 123–30. [DOI] [PubMed] [Google Scholar]
- Schwab MH, Bartholomae A, Heimrich B, Feldmeyer D, Druffel-Augustin S, Goebbels S, Naya FJ, Zhao S, Frotscher M, Tsai MJ, and Nave KA. 2000. ‘Neuronal basic helix-loop-helix proteins (NEX and BETA2/Neuro D) regulate terminal granule cell differentiation in the hippocampus’, J Neurosci, 20: 3714–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shadel GS, and Horvath TL. 2015. ‘Mitochondrial ROS signaling in organismal homeostasis’, Cell, 163: 560–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza Débora Guerini, Bellaver Bruna, Souza Diogo Onofre, and Quincozes-Santos André. 2013. ‘Characterization of adult rat astrocyte cultures’, PLOS ONE, 8: e60282–e82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Oberley LW, and Li Y. 1988. ‘A simple method for clinical assay of superoxide dismutase’, Clin Chem, 34: 497–500. [PubMed] [Google Scholar]
- Tarafdar A, and Pula G. 2018. ‘The Role of NADPH Oxidases and Oxidative Stress in Neurodegenerative Disorders’, Int J Mol Sci, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallet P, Charnay Y, Steger K, Ogier-Denis E, Kovari E, Herrmann F, Michel JP, and Szanto I. 2005. ‘Neuronal expression of the NADPH oxidase NOX4, and its regulation in mouse experimental brain ischemia’, Neuroscience, 132: 233–8. [DOI] [PubMed] [Google Scholar]
- Verniquet F, Gaillard J, Neuburger M, and Douce R. 1991. ‘Rapid Inactivation of Plant Aconitase by Hydrogen-Peroxide’, Biochemical Journal, 276: 643–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viedma-Poyatos A, de Pablo Y, Pekny M, and Perez-Sala D. 2018. ‘The cysteine residue of glial fibrillary acidic protein is a critical target for lipoxidation and required for efficient network organization’, Free Radical Biology and Medicine, 120: 380–94. [DOI] [PubMed] [Google Scholar]
- Wardman P 2007. ‘Fluorescent and luminescent probes for measurement of oxidative and nitrosative species in cells and tissues: Progress, pitfalls, and prospects’, Free Radical Biology and Medicine, 43: 995–1022. [DOI] [PubMed] [Google Scholar]
- Weitzel LR, Sampath D, Shimizu K, White AM, Herson PS, and Raol YH. 2016. ‘EEG power as a biomarker to predict the outcome after cardiac arrest and cardiopulmonary resuscitation induced global ischemia’, Life Sciences, 165: 21–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Neilson A, Swift AL, Moran R, Tamagnine J, Parslow D, Armistead S, Lemire K, Orrell J, Teich J, Chomicz S, and Ferrick DA. 2007. ‘Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells’, American Journal of Physiology-Cell Physiology, 292: C125–C36. [DOI] [PubMed] [Google Scholar]
- Zhao H, Joseph J, Fales HM, Sokoloski EA, Levine RL, Vasquez-Vivar J, and Kalyanaraman B. 2005. ‘Detection and characterization of the product of hydroethidine and intracellular superoxide by HPLC and limitations of fluorescence’, Proc Natl Acad Sci U S A, 102: 5727–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao HT, Kalivendi S, Zhang H, Joseph J, Nithipatikom K, Vasquez-Vivar J, and Kalyanaraman B. 2003. ‘Superoxide reacts with hydroethidine but forms a fluorescent product that is distinctly different from ethidium: Potential implications in intracellular fluorescence detection of superoxide’, Free Radical Biology and Medicine, 34: 1359–68. [DOI] [PubMed] [Google Scholar]
- Zielonka J, Vasquez-Vivar J, and Kalyanaraman B. 2006. ‘The confounding effects of light, sonication, and Mn(III)TBAP on quantitation of superoxide using hydroethidine’, Free Radic Biol Med, 41: 1050–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.