

Improving statistical power for genome-wide association studies (GWAS) in admixed populations is imperative since more and larger genomic studies in admixed populations are desperately needed to accelerate genomic medicine and reduce health inequities1. Recently, Atkinson et al.2 introduced a statistical framework (Tractor) for GWAS in admixed populations (for example, African Americans) that corrects for population structure through the use of local ancestry and concluded that GWAS in admixed populations increases discovery power over traditional GWAS only in the presence of allelic effect size heterogeneity by ancestry; a decrease in power is expected when allelic effects at tested variants are similar across ancestries. However, the conclusion reached by Atkinson et al.2 is specific to their particular choice of statistical association test, which prioritizes allelic effect size heterogeneity by ancestry and does not hold for other existing tests for GWAS in admixed populations. Existing association tests attain increased power over traditional GWAS in admixed populations, even when the causal variant has similar allelic effects across ancestries3–5. Therefore GWAS in admixed populations increase the power for discovery over homogeneous populations in either scenario—similar or different ancestry-specific allelic effects.
Powerful GWAS in admixed populations when the causal variant has similar allelic effects across ancestries are performed either through explicit modeling of the relationships between allelic and local ancestry effects5–8 or implicit inclusion of the admixture signal in tests that do not correct for local ancestry3,9. In all approaches, population structure is appropriately controlled by correcting for global ancestry3. The gain in power stems from the differentiation of causal allele frequencies by ancestry that induces heterogeneity in the standardized ancestry-specific effects, which in turn induces a local ancestry effect on the trait. Therefore, larger power gains over traditional GWAS are expected for causal variants with higher degrees of frequency differentiation between ancestral populations3,5. Most importantly, by using such tests, GWAS in individuals with African American ancestry attain superior power relative to GWAS in ancestrally homogeneous populations, such as Europeans or Africans3–5. Therefore, when allelic effects are similar across ancestries, correcting for local ancestry is expected to impair statistical power for GWAS discovery compared to global ancestry adjustment9 and is more useful as a localization tool in post-GWAS fine-mapping3.
We used simulations to compare the test proposed by Atkinson et al.2 (Tractor) to existing methods for GWAS in admixed populations when the causal allelic effects are similar across ancestries4. Starting from 1000 Genomes Project genotypes10, we simulated 40,000 admixed individuals assuming admixture fractions of 80% African and 20% European followed by 7 generations of random mating (Fig. 1a). We simulated a phenotype with 10% prevalence under the Tractor logistic model with a single causal variant with the same allelic effect across ancestries2; variability in causal variant frequencies across ancestries induces heterogeneity by ancestry in the marginal standardized effects. We compared the following tests for disease mapping in admixed populations: Cochran–Armitage trend test with correction for global ancestry (ATT); logistic regression with genotypic effects only (ATT-logit)–this test is similar to that used by the Population Architecture using Genomics and Epidemiology (PAGE) study9; case-only admixture mapping (ADM); case–control admixture mapping (ADM-logit), similar to the M1 model of Atkinson et al.2; SNP1 (association conditioned on local ancestry; similar to the M2 model referred to as ‘traditional GWAS’ in Atkinson et al.2); combined case-only admixture and SNP case–control association (MIX)5; sum of case–control SNP association and case-only admixture association (SUM); and Tractor (logistic regression assuming independent effects across ancestries with correction for local ancestry)2. All tests correct for global ancestry; SUM and Tractor are two d.f. tests while all others are one d.f. tests.
Fig. 1 |.
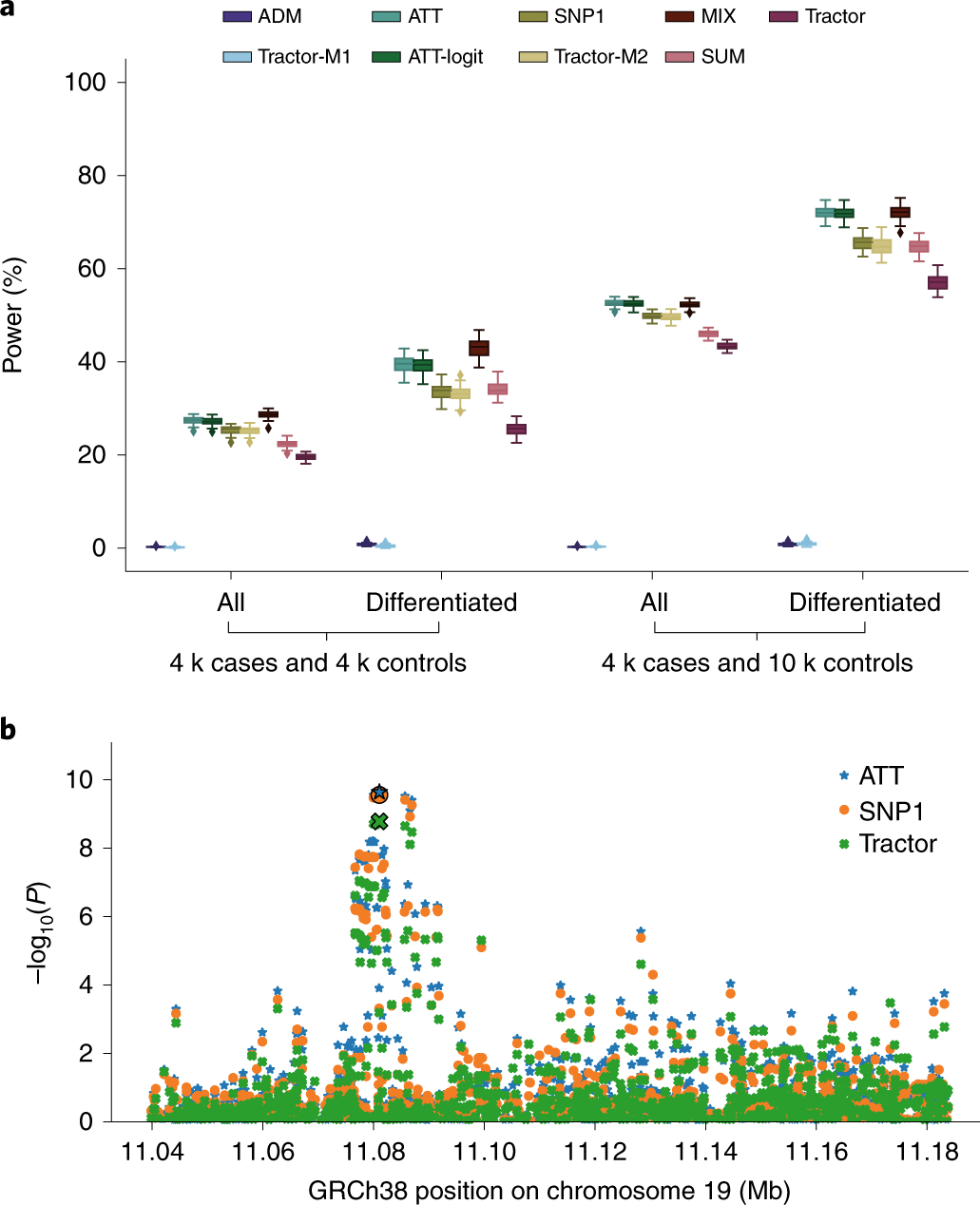
a, Comparison of the power of GWAS tests in admixed populations in simulations. ‘All’ represents distributions of power estimates from 50 simulation replicates and 3,000 causal SNPs uniformly drawn from the set of all SNPs, while ‘Differentiated’ distributions are restricted to the subset of SNPs (904 out of 3,000) with an absolute allele frequency difference >0.2 between Europeans and Africans (50 points per box plot). For box plots, the central lines correspond to the medians. The boxes represent the first and third quartiles of the points. The whiskers represent the minimum and maximum points located within 1.5× interquartile range from the first and third quartiles, respectively. In this study, we present results for an OR of 1.2; additional results, including null simulations, can be found in Supplementary Fig. 2. b, −log10(P) of SNP associations with LDL in the LDLR locus. The SNP with the strongest Tractor association P value has been framed and enlarged. Results at other considered GWAS regions for lipids (APOE, PCSK9, SORT1) showed similar patterns (Table 1).
First, we found that all tests appropriately controlled false positive rates under the null hypothesis (Supplementary Fig. 1). Second, as reported previously, we found that one d.f. methods that only correct for global ancestry (ATT, ATT-logit, MIX) attained superior power over methods that correct for both global and local ancestry (SNP1/Tractor-M2). As expected, a larger gain in power was observed at SNPs with higher frequency differentiation by ancestry. Since SNP1 and Tractor-M2 are analogous to disease mapping in ancestrally homogeneous populations3,5, it follows that admixed populations can offer increased power for disease mapping compared to ancestrally homogeneous populations. For example, when the odds ratio (OR) = 1.2 for a causal variant uniformly drawn from the genome, in a GWAS of 4,000 cases and 4,000 controls, ATT and MIX yielded approximately 27% power compared to 25% for SNP1/ Tractor-M2 and 20% for Tractor (Fig.1a). A larger gain in power was observed at causal variants with frequency differentiation >0.2 between ancestries (28% of all variants), where we observed a power of 43% for MIX, 33% for SNP1/Tractor-M2 and 26% for Tractor (Fig. 1a). Tractor had reduced power in these simulations because it requires some degree of heterogeneity in allelic effects to improve power (for example, >60% difference in allelic effects when frequency is fixed across ancestries2). Similar results were observed at other effect sizes or when the causal variant was untyped and missing from the data, thus confirming that GWAS in admixed populations outperform traditional GWAS when the causal variant has similar allelic effects across ancestries (Fig. 1a and Supplementary Figs. 2 and 3).
Next we analyzed the GWAS data of real lipid phenotypes–total cholesterol and low-density lipoprotein (LDL)–in individuals of African–European ancestries within the UK Biobank (n = 4,327). We focused on four well-known regions containing GWAS signals for lipid traits (APOE, LDLR, PCSK9, SORT1). Like the simulations, we observed that the association with correction for genome-wide ancestry-only (ATT) yielded the strongest signal, followed by tests that correct for both local and global ancestry (SNP1). Tractor, which also models heterogeneous effects, yielded the weakest association signal (Table 1). For example, at the LDLR region ATT attained P = 2.3 × 10-10 followed by 2.76 × 10-10 for SNP1 and 1.64 × 10-9 for Tractor (Fig. 1b). Notably, averaging across the four regions, Tractor yielded approximately 11% decreased effective sample size compared to ATT. For an extensive evaluation of admixture-aware tests at risk regions under strong admixture peaks, we refer the reader to Pasaniuc et al.5.
Table 1 |.
−log10(P) association statistics for the top Tractor SNP at known risk loci
| trait | Locus | ATT | SNP1 | Tractor |
|---|---|---|---|---|
|
| ||||
| Total cholesterol | APOE | 30.6 | 30.3 (−1.0%) | 28.9 (−5.6%) |
| LDL | APOE | 50 | 49.8 (−0.5%) | 47.5 (−5.1%) |
| Total cholesterol | LDLR | 8.3 | 8.2 (−0.9%) | 7.6 (−8.4%) |
| LDL | LDLR | 9.6 | 9.6 (−0.8%) | 8.8 (−8.9%) |
| Total cholesterol | PCSK9 | 9.4 | 8.5 (−9.9%) | 7.7 (−18.3%) |
| LDL | PCSK9 | 9.6 | 9.4 (−1.3%) | 8.5 (−10.9%) |
| Total cholesterol | SORT1 | 5.1 | 5.0 (−0.9%) | 4.3 (−15.7%) |
| LDL | SORT1 | 7.1 | 7.1 (−0.5%) | 6.3 (−11.7%) |
| Average relative difference | −2.0% | −10.6% | ||
We considered three GWAS tests with correction for global ancestry (ATT), global and local ancestry (SNP1) and global and local ancestry while allowing for heterogeneous effects (Tractor). The index SNP was selected based on the strongest Tractor association P value. Relative differences to the ATT score are shown in parentheses and in the last row.
In conclusion, GWAS in admixed populations attain improved power for discovery over homogeneous populations in either scenario—similar or different ancestry-specific allelic effects—thus further supporting the need for larger genomic studies in such populations. In this study, we showed that disease mapping in admixed populations is well powered when allelic effects are similar across ancestries, whereas Atkinson et al.2 showcased the power gains from two d.f. tests in the presence of effect size heterogeneity by ancestry2,3,5. Since the true extent of heterogeneity in causal allelic effects across ancestries is currently unknown11–15, we recommend careful consideration of the balance between expected allelic effect size heterogeneity across ancestries and association power when selecting a statistical test for GWAS in admixed populations. A further consideration should be given to linkage disequilibrium-induced heterogeneity at tagging variants, which can occur even when causal allelic effects are similar across ancestries2,3,5; in this scenario, there is an expected loss of power due to imperfect tagging, although preliminary results suggest that the loss in power is small, particularly when genotype imputation is employed (Supplementary Fig. 3 and refs. 3,5). Properly aligned statistical tests will enable new discoveries in admixed populations that have long been understudied and underserved.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41588-021-00953-5.
Methods
For Fig. 1a, 3,000 causal SNPs with a frequency >1% in individuals with both European and African ancestry in the 1000 Genomes Project were randomly sampled from chromosome 2. Each causal SNP had the same allelic effect size in both ancestral populations. Phenotypes were simulated under the logistic model of Atkinson et al.2 assuming 10% prevalence and no global ancestry effect. We defined power as the proportion of causal SNPs with an association P < 5 × 10-8 (for all tests except ADM and Tractor-M1) or P < 1 × 10-5 (for ADM and Tractor-M1) (ref. 4). We compared results for the GWAS of 4,000 cases/4,000 controls and 4,000 cases/10,000 controls.
Data availability
This research was conducted using the UK Biobank Resource under application 33297. We thank the participants of UK Biobank for making this work possible. The UK Biobank genotype and phenotype data are available by application from https://www.ukbiobank.ac.uk/. Extended results can be accessed at our Zenodo repository https://doi.org/10.5281/zenodo.5308562.
Code availability
Software and extended results, including an implementation of the Tractor association models, can be found at our Zenodo repository. (The Tractor software currently does not include logistic models for association; https://github.com/eatkinson/Tractor accessed 22 February 2021.)
Supplementary Material
Footnotes
Competing interests
The authors declare no competing interests.
Additional information
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41588-021-00953-5.
Peer review information Nature Genetics thanks Loïc Yengo and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Reprints and permissions information is available at www.nature.com/reprints.
References
- 1.Martin AR et al. Human demographic history impacts genetic risk prediction across diverse populations. Am. J. Hum. Genet. 100, 635–649 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Atkinson EG et al. Tractor uses local ancestry to enable the inclusion of admixed individuals in GWAS and to boost power. Nat. Genet. 53, 195–204 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang J & Stram DO The role of local ancestry adjustment in association studies using admixed populations. Genet. Epidemiol. 38, 502–515 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seldin MF, Pasaniuc B & Price AL New approaches to disease mapping in admixed populations. Nat. Rev. Genet. 12, 523–528 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pasaniuc B et al. Enhanced statistical tests for GWAS in admixed populations: assessment using African Americans from CARe and a Breast Cancer Consortium. PLoS Genet. 7, e1001371 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tang H, Siegmund DO, Johnson NA, Romieu I & London SJ Joint testing of genotype and ancestry association in admixed families. Genet. Epidemiol. 34, 783–791 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shriner D, Adeyemo A & Rotimi CN Joint ancestry and association testing in admixed individuals. PLoS Comput. Biol. 7, e1002325 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yorgov D, Edwards KL & Santorico SA Use of admixture and association for detection of quantitative trait loci in the Type 2 Diabetes Genetic Exploration by Next-Generation Sequencing in Ethnic Samples (T2D-GENES) study. BMC Proc. 8, S6 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wojcik GL et al. Genetic analyses of diverse populations improves discovery for complex traits. Nature 570, 514–518 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Auton A et al. A global reference for human genetic variation. Nature 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Candia TR et al. Additive genetic variation in schizophrenia risk is shared by populations of African and European descent. Am. J. Hum. Genet. 93, 463–470 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lam M et al. Comparative genetic architectures of schizophrenia in East Asian and European populations. Nat. Genet. 51, 1670–1678 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu JZ et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 47, 979–986 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shi H et al. Population-specific causal disease effect sizes in functionally important regions impacted by selection. Nat. Commun. 12, 1098 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van Rheenen W, Peyrot WJ, Schork AJ, Lee SH & Wray NR Genetic correlations of polygenic disease traits: from theory to practice. Nat. Rev. Genet. 20, 567–581 (2019). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This research was conducted using the UK Biobank Resource under application 33297. We thank the participants of UK Biobank for making this work possible. The UK Biobank genotype and phenotype data are available by application from https://www.ukbiobank.ac.uk/. Extended results can be accessed at our Zenodo repository https://doi.org/10.5281/zenodo.5308562.