

Abstract
Patients with early‐onset Alzheimer's disease (EOAD) are commonly excluded from large‐scale observational and therapeutic studies due to their young age, atypical presentation, or absence of pathogenic mutations. The goals of the Longitudinal EOAD Study (LEADS) are to (1) define the clinical, imaging, and fluid biomarker characteristics of EOAD; (2) develop sensitive cognitive and biomarker measures for future clinical and research use; and (3) establish a trial‐ready network. LEADS will follow 400 amyloid beta (Aβ)‐positive EOAD, 200 Aβ‐negative EOnonAD that meet National Institute on Aging–Alzheimer's Association (NIA‐AA) criteria for mild cognitive impairment (MCI) or AD dementia, and 100 age‐matched controls. Participants will undergo clinical and cognitive assessments, magnetic resonance imaging (MRI), [18F]Florbetaben and [18F]Flortaucipir positron emission tomography (PET), lumbar puncture, and blood draw for DNA, RNA, plasma, serum and peripheral blood mononuclear cells, and post‐mortem assessment. To develop more effective AD treatments, scientists need to understand the genetic, biological, and clinical processes involved in EOAD. LEADS will develop a public resource that will enable future planning and implementation of EOAD clinical trials.
Keywords: Alzheimer's disease, early‐onset, EOAD, LEADS, YOAD, young onset
1. INTRODUCTION
Approximately 5% of the 5.8 million victims of Alzheimer's disease (AD) in the United States (≈200,000 people) develop symptoms at age 64 or younger, and are classified as having early‐onset AD (EOAD). 1 , 2 The onset of dementia at such a young productive age has disproportionately devastating consequences for patients, families, and society. 3 , 4 Individuals with EOAD often face significant delays to diagnosis, or are misdiagnosed with non‐degenerative conditions (eg, other psychiatric or neurologic disorders, hormonal imbalance such as menopause, etc. ). 5 , 6 , 7 , 8 , 9 , 10 Diagnostic delay postpones access to disease education and therapy, as well as social and financial support for people who are in the peak earning years of their life. These delays also result in loss of employment, health insurance, and lasting emotional and financial strain on caregivers.
Although EOAD and late‐onset AD (LOAD; age at onset 65 years or older) share the same pathologic substrate, there are notable differences in their clinical and biological phenotypes. 11 Compared to LOAD, individuals with sporadic EOAD show more rapid clinical decline, 11 , 12 , 13 , 14 , 15 , 16 and lower prevalence of amnestic versus non‐amnestic predominant clinical presentations with greater impairment in non‐memory domains. 11 , 13 , 14 , 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 For the same level of impairment, EOAD is associated with greater baseline cortical atrophy and hypometabolism, less hippocampal atrophy, and more severe tau pathology than LOAD. 21 , 22 , 25 , 28 , 29 , 30 , 31 , 32 , 33 , 34 , 35 , 36 , 37 , 38 , 39 40 , 41 , 42 , 43 , 44 , 45 , 46 , 47 , 48 Pedigree analyses provide evidence for increased heritability in EOAD compared to LOAD. Yet, only a small minority (≈3‐10%) of EOAD carry a known autosomal dominant mutation in the Amyloid Precursor Protein (APP) or Presenilin 1 and 2 (PSEN1/2) genes, 49 , 50 , 51 suggesting that this population may be enriched for novel genetic risk factors. 52 , 53
Despite being highly motivated and having fewer age‐related comorbidities compared to LOAD, patients with EOAD are commonly excluded from clinical research and therapeutic trials—an oversight that has been criticized increasingly as being marginalizing and unethical. 54 Of the two major North American multicenter consortia, the Alzheimer's Disease Neuroimaging Initiative (ADNI) includes only a few EOAD cases all with a “typical” amnestic presentation and the Dominantly Inherited Alzheimer Network (DIAN) is focused solely on autosomal dominant EOAD.
The Longitudinal Early‐onset Alzheimer's disease Study (LEADS, NIA R56057195, NIA U016057195) is a prospective longitudinal multi‐site, observational clinical and biomarker study of EOAD, conducted at key AD research hubs and clinical sites across the United States. The scientific goals of LEADS are to (1) collect clinical, genetic, and biomarker data in this under‐studied AD population; (2) explore the unique features of EOAD yielding novel insights into the mechanisms, heterogeneity, and heritability of AD; (3) develop clinical trial outcome measures sensitive to detect baseline deficits and track longitudinal changes in EOAD; and (4) establish a network of sites that will enable future planning and implementation of clinical trials in EOAD. Herein we describe the study design and methodology.
2. METHODS
2.1. Study design
LEADS (www.leads‐study.org) is a prospective multisite observational clinical and biomarker study registered in clinicaltrails.gov (NCT03507257). LEADS seeks to accomplish the following aims: (1) compare the baseline and longitudinal cognitive and functional characteristics (aim 1) as well as magnetic resonance imaging (MRI), amyloid positron emission tomography (PET), tau PET, and cerebrospinal fluid (CSF) measures (aim 2) of EOAD versus LOAD and identify optimal outcome measures for clinical trials; (2) investigate the influence of apolipoprotein E (APOE) genotype in EOAD (aim 3); and (3) characterize genetic contributions to EOAD (exploratory aim 4).
LEADS leverages existing infrastructure and processes applied in the National Alzheimer's Coordinating Center (NACC) Alzheimer's Disease Center (ADC) Network and the ADNI study. Biomarker collection closely follows the study design employed by ADNI along with the informatics systems provided by the Laboratory of Neuro Imaging (LONI ‐ http://loni.usc.edu/). Similar to ADNI, LEADS links longitudinal clinical and cognitive assessments with multiple imaging and biofluid markers that capture different elements of the AD pathophysiological cascade. The study infrastructure comprises eight cores (Administrative, Clinical, MRI, PET, Genetics and Biorepository, Biostatistics, Informatics, and Neuropathology Cores) and, at present, 19 clinical sites. LEADS sites were carefully selected for their research expertise in EOAD as well as the availability of both 18F‐Florbetaben (FBB) and 18F‐Flortaucipir (FTP) tracer delivery at the site. The study employs an innovative partnership model between federal (NIA), academic (cores and sites), non‐profit (Alzheimer's Association; www.alz.org), and private stakeholders (Life Molecular Imaging and AVID who provide amyloid and tau PET ligands, respectively, at research cost).
RESEARCH IN CONTEXT
Systematic review: We assessed all relevant literature by searching PubMed for papers on early‐onset Alzheimer's disease (EOAD). Despite being highly motivated and having fewer age‐related comorbidities compared to patients with late‐onset AD, patients with EOAD are commonly excluded from clinical research and therapeutic trials.
Interpretation: To fill this gap we launched a multi‐site observational Longitudinal Early‐onset Alzheimer's Disease Study (LEADS) that will collect longitudinal clinical and biomarker data from a large cohort of EOAD participants, as well as develop a clinical trials network.
Future directions: Efforts toward international LEADS expansion and the launch of a LEADS Trials Unit are ongoing. In collaboration with the Advancing Research and Treatment in Frontotemporal Lobar Degeneration (ARTFL)‐Longitudinal Evaluation of Familial Frontotemporal Dementia Subjects (LEFFTDS) Longitudinal Frontotemporal Lobar Degeneration (ALLFTD) study, we are planning a registry for participants interested in enrolling in clinical trials for EOAD or frontotemporal dementia (FTD) with cognitive screening and blood biomarker assessments.
The coordinating center for LEADS is located at the University of Southern California Alzheimer's Therapeutic Research Institute (ATRI). ATRI also serves as the coordinating center for ADNI, the Alzheimer's Clinical Trials Consortium (ACTC), and other observational and therapeutic studies in the field. ATRI provides electronic data capture, regulatory and operational support, data management, central and on‐site clinical monitoring, safety oversight, and reporting to the Data and Safety Monitoring Board.
LEADS is one of the first studies of its size and scope in the United States to employ an academic institution (Indiana University [IU]), as the central institutional review board (IRB). The IU central IRB reliance process utilizes the SMART IRB agreement. Determinations to rely on the IU IRB as the single IRB for the study are made by the local Human Research Protection Programs (HRPPs) at all sites and cores locations. The local HRPPs convey all state and local policies that govern the research at their site to the Regulatory Team at ATRI, and the central IRB at IU. Site‐specific documents, such as Informed Consent Forms, HIPAA authorizations, and other regulatory documents for each site, are generated by making all necessary changes to the IU IRB approved study‐wide templates in response to local policies. The IU IRB reviews all site‐specific regulatory documents before approving the site initiation and fully executing the reliance agreements. All LEADS participants provide informed consent according to the Declaration of Helsinki, U.S. federal regulations, local state laws and regulations, and the policies of the IU IRB. Throughout the study, the IU IRB receives reportable event reports from all sites, reviews study‐wide and site‐specific amendments, and facilitates renewals for LEADS. Local site HRPPs maintain responsibility for all other ancillary reviews per the SMART IRB agreement. Storage and distribution of IRB‐approved documents is managed through the online portal IRB Reliance Exchange (IREx).
2.2. Clinical procedures
The original grant application proposed to enroll 400 subjects meeting NIA‐AA criteria for MCI or mild dementia, 55 , 56 ages 40‐64, referred from memory clinics or existing research cohorts across our clinical sites, and 100 cognitively normal age‐matched participants. Additional funding was secured via an administrative supplement to enroll and characterize up to 200 amyloid beta (Aβ)‐negative cognitively impaired participants who were initially presumed to have EOAD (EOnonAD) (U01 AG057195‐02S1), increasing our target enrollment to 700. The Alzheimer's Association facilitates recruitment both through its nationwide chapter network and through the TrialMatch initiative by identifying EOAD participants and connecting them with our enrolling sites. All LEADS participants are offered co‐enrollment in the federally funded Alzheimer's Disease Research Centers (ADRCs), where available.
In 2021, we successfully competed for a Competitive Revision supplement (U01 AG057195‐03S1) and added the following additional research activities:
Addition of mo36 visit with clinical, cognitive, and biomarker assessment for all EOAD and EOnonAD participants
Addition of mo48 visits with clinical, cognitive, and peripheral blood collection for EOAD and EOnonAD
Addition of mo24 visit for cognitively normal (CN) participants
Addition of plasma Aβ40 and 42, P‐tau217, and neurofilament light (NfL) assessments
Addition of 18F‐Fluorodeoxyglucose (FDG) PET for all CN and EOnonAD participants to further characterize neurodegenerative patterns in EOnonAD
Addition of social worker support at our sites
2.3. Inclusion/exclusion criteria
EOAD and EOnonAD participants must meet NIA‐AA criteria for dementia or MCI and have a global Clinical Dementia Rating (CDR) score ≤1. Unlike ADNI and the vast majority of clinical trials, LEADS does not exclude individuals with predominantly non‐amnestic presentations. Individuals meeting criteria for the dysexecutive, logopenic primary progressive aphasia or posterior cortical atrophy variants are eligible to enroll.
Cognitively impaired individuals with two or more first‐degree relatives with EOAD unless mutations in APP, PSEN1, PSEN2, Microtubule Associated Protein Tau gene (MAPT), chromosome 9 open reading frame 72 gene (C9ORF72), and Granulin Precursor aka Progranulin (GRN) have been excluded as well as those with a known mutation in APP, PSEN1, and PSEN2 are ineligible. CN LEADS participants must have a Mini‐Mental State Examination (MMSE) score of ≥24, a global CDR = 0, and score within the cognitively normal range on neuropsychological testing.
In addition, all LEADS participants must be ages 40‐64 at the time of consent, have capacity to consent or, if cognitively impaired, have a legally authorized representative who can provide consent, have a study partner that knows them well, have no contraindications to MRI, be willing and able to complete all study procedures aside from lumbar puncture (optional), not be pregnant or lactating, not have lifetime history of other brain disorder (both neurologic and psychiatric except for seizures thought to be related to EOAD or headaches), have not participated in therapeutic trials targeting Aß and/or tau, moderate or severe substance abuse, or suicidal behaviors or ideations in the past 12 months. Individuals with MRI evidence of infection, focal lesions such as strokes, multiple or strategic lacunes, and/or space‐occupying lesions are also excluded. Our current enrollment statistics as well as the demographic, clinical, and biomarker characteristics for our participants by diagnostic group can be seen in Table 1.
TABLE 1.
Demographic, clinical, and amyloid PET SUVR characteristics of the currently enrolled LEADS participants
| CN (N = 114) | EOAD (N = 170) | EOnonAD (N = 65) | EOAD vs CN, P‐value | EOnonAD vs CN, P‐value | EOAD vs EOnonAD, P‐value | |
|---|---|---|---|---|---|---|
| Age, y, mean (SD) | 55.93 (5.88) | 59.54 (3.77) | 57.91 (6.55) | <.0001 | .08 | .03 |
| Sex, % F | 60.81% | 50.75% | 33.96% | .1630 | .003 | .04 |
| Education, y, mean (SD) | 16.91 (2.12) | 15.34 (2.45) | 15.62 (2.52) | <.0001 | .002 | .47 |
| MMSE, mean (SD) | 29.25 (0.84) | 21.70 (4.68) | 26.10 (3.39) | <.0001 | <.0001 | <.0001 |
| CDR = 0.5, % | N/A | 56 | 81 | N/A | N/A | <.0001 |
| FBB SUVR, mean (SD) | 1.01 (0.07) | 1.54 (0.17) | 0.99 (0.06) | <.0001 | .11 | <0.0001 |
Recruitment for LEADS follows the ADNI model of competitive recruitment. EOAD is a rare AD variant and thus sites enrollment will not be capped.
2.4. Clinical and cognitive assessments
In the originally funded application EOAD and EOnonAD individuals were to be followed for 24 months with a screening/baseline, month 12 and month 24 clinical and cognitive assessments, whereas CN participants were to undergo to screening/baseline and month 12 clinical and cognitive assessments. With the soon to be funded Competitive Revision we were able to add month 36 and 48 visits for all cognitively impaired and month 24 visit for all CN participants. The LEADS schedule of events is shown in Figure 1.
FIGURE 1.
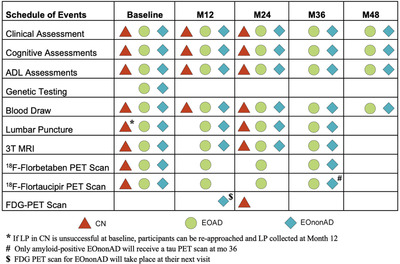
Schedule of events
The LEADS clinical assessments include standardized history of present illness, past medical history, family history, concurrent medication, and detailed general medical and neurological examinations. We also routinely obtain Autoimmune and Early Developmental History questionnaires. LEADS employs the NACC Uniform Data Set cognitive battery, the NACC Frontotemporal Lobar Degeneration module, the Alzheimer's Disease Cooperative Studies ‐ cognitive behavior subscale (ADAS Cog), and several additional cognitive tests tapping into cognitive functions that are commonly impaired in rare AD variants (see Figure 1 and Table 2). Clinical diagnosis is established in a multidisciplinary consensus conference at each clinical site following the NIA‐AA diagnostic criteria for dementia and MCI. Diagnosis of logopenic aphasia and posterior cortical atrophy follow previously published criteria. 57 , 58
TABLE 2.
Clinical and cognitive assessments
| Cognitive Dementia Rating Scale (CDR) | |
|---|---|
| LEADS specific cognitive measures | Mini Mental State Exam (MMSE) |
| Rey Auditory Verbal Learning Test (RAVLT) | |
| Digit Symbol Substitution Test | |
| ADAS Cog | |
| TabCat (Flanker, Line Length, Line Orientation, Match) | |
| NACC UDS MODULE | Montreal Cognitive Assessment (MoCA) |
| Craft Story Immediate and Delayed Recall | |
| Benson Complex Figure Copy and Delayed Recall | |
| Number Span Test: Forward | |
| Number Span Test: Backward | |
| Category Fluency ‐ Animals | |
| Category Fluency ‐ Vegetables | |
| Trails Making Test Part A | |
| Trails Making Test Part B | |
| Multilingual Naming Test (MINT) | |
| Phonemic Fluency, F and L | |
| NACC UDS FTLD MODULE | Regular and Irregular Word Reading |
| Word Picture Matching | |
| Semantic Associates Test | |
| Northwestern Anagram | |
| Sentence Repetition | |
| Noun and Verb Naming | |
| Sentence Reading Test | |
| Social Norms Questionnaire | |
| Social Behavior Observer Checklist | |
| Behavioral Inhibition Scale | |
| Interpersonal Reactivity Index | |
| Revised Self‐monitoring Scale | |
| Behavioral Measures | Geriatric Depression Scale (GDS) |
| Neuropsychiatric Inventory Questionnaire | |
| ADL Measures | Functional Activities Questionnaire (FAQ) |
| Amsterdam IADL measurea | |
Amsterdam IADL was added in February 2020.
2.5. Genetic screening and counseling
The LEADS Genetics and Biorepository Core oversees genetic screening and genetic counseling for the study. During the consenting process, cognitively impaired participants are given the option to learn the results of their genetic screening performed as part of the LEADS. The participant watches a 7‐minute video, which discusses the potential benefits, risks, and limitations of genetic testing for mutations in genes known to be important in the risk of EOAD (PSEN1, PSEN2, and APP) and EOnonAD (GRN, MAPT, and C9ORF72) and is given the option of speaking to a genetic counselor to address any questions or concerns prior to consenting to receive the genetic screening results. The participants are given a link to the video so that they may review it at a later time or discuss it with other family members if they wish to do so. Participants can opt in or out of genetic disclosure and can withdraw consent at any point prior to disclosure.
DNA samples are collected from all LEADS participants for research purposes. The DNA samples from all cognitively impaired LEADS participants are screened for genetic mutations in early‐onset dementia‐related genes APP, PSEN1, PSEN2, GRN, and MAPT. Data are processed and quality controlled following the GATK best practices workflow. 59 Data are annotated using Annovar, and then filtered using a list of known pathogenic mutations for EOAD and frontotemporal dementia. These mutations were collated and curated from the Human Gene Mutation Database, 60 ClinVar, 61 the Leiden Open Variation Database, 62 and the pathogenic mutations list curated by the DIAN study, 63 which is used to determine if DIAN participants are eligible for clinical trials. Participants with known pathogenic mutations in APP, PSEN1, PSEN2, GRN, and MAPT are identified. Screening is also conducted to identify repeat expansions of the C9ORF72 gene. DNA is screened with repeat primed PCR to identify samples showing evidence of pathogenic repeat expansion. A separate 6 mL vial of blood is also collected and stored for cognitively impaired participants for the purpose of DNA extraction and genetic testing by a Clinical Laboratory Improvement Amendments (CLIA)–certified genetic laboratory.
Participants who do not show evidence of pathogenic mutations in the six screened genes are considered mutation negative. Negative mutation status is not verified by CLIA testing. Those who elect to receive their genetic results and are found to not carry a pathogenic variant in the research screen are informed of this by the site PI, study physician, or genetic counselor in person or by phone. They also receive a letter explaining the results and the limitations of that testing, and disclosing that this testing was conducted in a research laboratory settings.
For participants in whom a known pathogenic mutation is identified and who indicated that they wish to learn the results of genetic testing, the separate blood sample collected specifically for verification of research screening is sent by the National Centralized Repository for Alzheimer's Disease and Related Dementias (NCRAD) to a CLIA‐certified laboratory for confirmatory testing. Once CLIA results are returned to the LEADS Genetics and Biorepository Core, the consent status for each participant is verified that the consent to receive genetic results is current and participants wish to receive genetic results. The participants who are positive for pathogenic mutations meet with the site's genetic counselor for disclosure. They are given a copy of the test report describing CLIA confirmation of the variant identified in research testing. They are no longer eligible to continue in LEADS but they are referred to other studies (ie, Dominantly Inherited Alzheimer Network [DIAN], ARTFL‐LEFFTDS Longitudinal Frontotemporal Lobar Degeneration [ALLFTD]). To avoid inadvertent disclosure, mutation carriers who decline result disclosure or withdraw consent prior to disclosure continue through the study per protocol but are excluded from analyses.
3. BIOMARKER DATA COLLECTION
3.1. MRI methods
The administrative organization of the LEADS MRI Core includes two teams, one based at the Mayo Clinic and one based at Massachusetts General Hospital. The LEADS MRI Core is charged with supporting each of the sites in acquiring high‐quality MRI scans with standardized procedures at the timepoints described above. We have employed the 3 Tesla ADNI‐3 acquisition protocol, using the advanced protocol at as many sites as possible (http://adni.loni.usc.edu/methods/documents/mri‐protocols/). The typical scan parameters are provided below; these vary slightly by vendor and system type. The sequences acquired using 3 Tesla MRI scanners at all LEADS sites are (1) three plane/tri‐planar auto‐alignment scout scan, yielding orthogonal orientation and AC‐PC alignment; (2) sagittal 3D accelerated MPRAGE/IRSPGR T1‐weighted sequence: TR/TE/TI = 2300/3/900 ms, flip angle 9°, sagittal orientation, FOV = 256 × 240 mm with 208 slices, 1 × 1 × 1 mm resolution, and 2x acceleration; (3) sagittal 3D fluid‐attenuated inversion recovery (FLAIR) TR/TE/TI = 4800/119/1650 ms, FOV = 256 × 256 mm with 160 slices, and 1.2 × 1 × 1 mm resolution; (4) axial T2*/gradient echo (GRE) for cerebral microbleed assessment with TR/TE = 650/20 ms, FOV = 220 × 220 mm, and 176 slides; (5) diffusion tensor imaging (DTI) TR/TE = 3300/71 ms, FOV = 232 × 232 × 160 mm, three shells b = 500, 1000, 2000 s/mm2, 112 directions, and 2 × 2 × 2 mm resolution; (6) 3D Pseudo Continuous Arterial Spin Labeled (pCASL) perfusion imaging TR/TE 4885/10.5, FOV = 240 × 240 × 160 mm, and 1.9 × 1.9 × 4 mm resolution; (7) task‐free functional MRI, T2*‐weighted gradient echo–echo planar sequence with TR/TE = 600/30 ms, flip angle 53°, FOV = 220 × 220 × 160 mm, 2.5 × 2.5 × 2.5 mm resolution, 64 slices, SMS = 8, CAIPI shift = 4. Subjects are instructed to remain awake with their eyes closed, (8) High‐resolution hippocampal sequence acquired obliquely to the long axis of the hippocampi, TR/TE 8020/50, FOV 175 × 60 × 175 mm, and 0.39 × 3 × 0.39 mm resolution. Total exam duration is under 1 hour.
Each site receives electronic files from the LEADS MRI Core, with the protocol to be loaded on their system. For site qualification, each site scans the ADNI phantom or other similar local phantom using the electronically loaded LEADS Phantom QC protocols and LEADS Human Scan protocols. Achieving a reproducible phantom placement position is a key element to the system performance analysis that is done at initial site certification, when there are software/hardware upgrades, or when substantial maintenance is performed. The MRI core has developed a scanning procedures manual that supports sites in performing the procedures as similarly as possible.
Each site captures information about the scan session for the electronic data capture record and uploads MRI DICOM images to LONI. De‐identified data are downloaded from LONI by the LEADS MRI Core Mayo site, which employs the ADNI QC pipeline, including (1) visual inspection for artifacts (eg, subject motion) and evaluation of image quality; (2) verification of adherence to all sequence protocols (protocol consistency check) based on DICOM header fields; (3) verification of hardware and software versions; (4) immediate site contact in the case of a deviation; (5) logging of MR QC with the Informatics Core and the coordinating center at ATRI. The LEADS MRI Core MGH site performs FreeSurfer reconstruction and processing for regional estimates of subcortical gray matter (GM) tissue volume and cortical GM morphometrics including volume and thickness measures. The reconstructed MRI images are also used by the PET core for co‐registration. The LEADS MRI Core Mayo site performs microhemorrhage assessments, infarct grading, white matter hyperintensity volumes, and also computes longitudinal MRI change assessment using Symmetric Diffeomorphic Image Normalization tensor‐based morphometry method (TBM‐SyN). 64 The MRI Core labs have monthly phone conferences and interact closely to ensure efficiency of data flow and to resolve issues that may arise with MRI scanning or processing.
3.2. PET methods
The LEADS PET Core teams are based at the University of California San Francisco (UCSF) and University of Michigan. The UCSF team is responsible for overall PET Core activities, amyloid, tau, and FDG PET central reads for cohort assignment (impaired participants only) and PET quantification and analyses. The University of Michigan team is responsible for scanner qualification, image quality control, and standardization. PET scans are stored at the Laboratory of Neuroimaging (LONI) at the University of Southern California. PET procedures in LEADS, including choice of radiotracers, image standardization, and quantification are by design aligned with ADNI‐3 to enhance comparability (http://adni.loni.usc.edu/wp‐content/uploads/2012/10/ADNI3_PET‐Tech‐Manual_V2.0_20161206.pdf).
All LEADS participants undergo amyloid PET with FBB PET and tau PET with FTP PET(formerly known as 18F‐AV1451 and 18F‐T807). These radiotracers have been validated as sensitive and specific for detecting moderate‐frequent neuritic plaque and Braak V‐VI neurofibrillary tangle pathology, respectively, 65 and are approved for clinical use by the U.S. Food and Drug Administration.
In anticipation that the EOnonAD group includes a variety of nonAD etiologies, with the soon to be funded Competitive Revision we are adding FDG PET assessments to further characterize the EOnonAD participants. Some EOnonAD participants will likely harbor other neurodegenerative pathologies, whereas others may have psychiatric or medical etiologies for cognitive impairment. Although MRI may offer insights into the specific nonAD etiology through regional atrophy patterns, FDG PET provides complimentary data to MRI regarding both the extent and pattern of neurodegeneration, as demonstrated in FTLD 66 and in amyloid‐negative clinical AD. 67 LEADS CN will also receive one FDGPET assessments and will serve as age‐matched comparison group.
All PET scanners are standardized with a 18F‐filled Hoffman brain phantom. 68 FBB PET is acquired via intravenous injection of ≈8 mCi of FBB, followed by acquisition of 4 × 5 minute PET frames at t = 90‐110 minute post‐injection. FTP PET is acquired via intravenous injection of ≈10 mCi of FTP followed by acquisition of 6 × 5 minute PET frames at 75‐105 minutes post‐injection. FDG PET will be acquired via intravenous injection of ≈5 mCi, followed by acquisition of 6 × 5 minute PET frames at 30‐60 minutes post‐injection. 68 Attenuation correction for each scan is performed using either CT or PET transmission data, and reconstruction uses site‐specific iterative algorithms developed for ADNI. Raw PET images undergo quality control procedures, which include statistical noise check, motion assessment across temporal frames, checking for full coverage of the brain, visual check to look for common PET artifacts as well as visual and image header check to assure that the study protocol has been followed.
PET data flow in LEADS is shown in Figure 2. Raw images are set to a standard orientation, intensity‐normalized and smoothed to standard resolution using procedures developed for ADNI. 68 Fully pre‐processed FBB PET, FTP PET, and FDG PET images are co‐registered to the participants’ respective T1 structural MRI studies. Reference regions for FBB PET (whole cerebellum) and FTPPET (inferior cerebellar gray matter) are obtained via Freesurfer (FBB PET) and Freesurfer plus the SUIT template (FTP PET). 69 , 70 FDG PET standardized uptake value ratio (SUVR) images are created using mean activity in FreeSurfer parcellated pons as reference region. Images are scaled to the average binding in the reference regions to obtain SUVR images utilizing the full acquisition time: FBB PET SUVR90‐110 and FTP PET SUVR75‐105. Regional average PET SUVR values are extracted in native MRI space from regions defined in the Desikan‐Killiani Atlas labeled by Freesurfer 71 For FBB, a composite neocortical SUVR is computed and converted to Centiloid units 72 using the ADNI formula. For FTP PET, average SUVR values from an AD meta‐region of interest 73 and composite Braak stage‐like regions 69 are computed. Summary and regional PET measures are shared via LONI.
FIGURE 2.
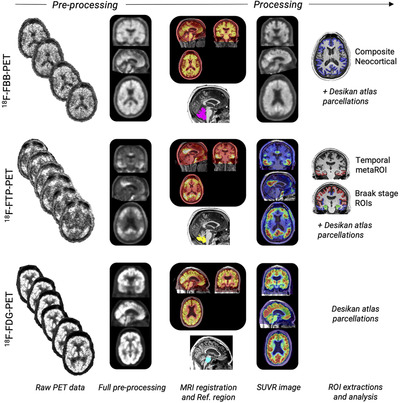
PET data processing
Central reads of baseline FBB PET scans are performed in cognitively impaired participants using a hybrid approach that includes visual reads and global SUVR quantification. First, scans are visually read as “amyloid‐positive" or "amyloid‐negative” using validated criteria 65 by a PET Core physician who has completed the manufacturer online training program and is certified to read FBB PET for clinical purposes. Visual reads are performed blinded to a participant's clinical information and scan quantification. SUVR quantification is performed using a PET‐only processing pipeline that closely mirrors the MRI‐based pipeline described above. A PET‐only pipeline is used for this step because study MRI scans are not always available at the time of central read. A global SUVR≥1.18 (corresponding to 39.2 Centiloids) is used as a quantitative threshold for amyloid PET positivity. If the visual read and quantitative assessment agree that a scan is Aß‐positive, the participant is assigned to the EOAD cohort. If visual read and quantification agree that a scan is Aß‐negative, the participant is assigned to the EOnonAD cohort. If there is discordance between the visual read and quantification, a “tie breaker” visual read is provided by an additional reader (also blinded to quantification results), and this is considered the consensus read for cohort assignment. Central amyloid PET reads are returned to the site principal investigators and disclosed to the cognitively impaired participants by the study physicians following best practices. 74 The central read report includes a binary read outcome, a narrative description of tracer uptake intensity and distribution, the global neocortical SUVR, and the results of consensus review (if applicable).
An analogous approach will be adapted to provide central clinical reads of FTP PET scans. Scans will be visually interpreted as negative or positive for an AD pattern using the autopsy‐validated method introduced by Fleisher and colleagues. 75 Scans will be quantitatively classified as tau‐positive or tau‐negative based on an SUVR threshold of 1.27 76 using a temporal “meta region‐of‐interest.” 73 In instances when the visual read and quantification are discordant, a “tie breaker” visual read will be performed. The central read report will include the binary read outcome, a narrative description of tracer uptake intensity and distribution, the temporal meta region‐of‐interest SUVR, and the results of consensus review (if applicable).
Central reads of FDG PET will also be provided to the sites and the LEADS participants. Using the CN group and a voxelwise W‐score approach 77 we will generate single‐subject statistical FDG PET hypometabolism maps (W‐maps), corrected for age. Individual W‐maps and FDG PET SUVR images will be visually rated by the PET core blind to FTP PET and clinical/neuropsychological data.
3.3. Genetics methods
The LEADS Genetics and Biorepository Core includes a genetics and biorepository team at IU and the biofluids team at Washington University. The Genetics and Biorepository Core oversees the genetic and counseling for the study, as already discussed in the Genetic Screening and Counseling section.
As part of LEADS biospecimen collection protocol, blood collected at baseline for all participants is shipped to NCRAD, which extracts and stores DNA for genetic screening and genomic analyses. Following DNA extraction at NCRAD, in‐house genotyping is done with a custom 96‐SNP fingerprint panel, to check DNA quality and to verify that reported and genetic sex match. This panel is also used to generate genotypes for APOE, as it includes assays for rs429358 and rs7412. APOE genotypes for all participants are collected, and de‐identified APOE genotypes are uploaded to LONI. APOE genotype results are not returned to participants.
3.4. Fluid biomarkers
All LEADS participants are asked to consent for lumbar puncture (LP) for the evaluation of CSF biomarkers, although refusal is not exclusionary for the study. CSF acquisition, processing and analysis conform to standardized protocols applied in ADNI. 78 , 79 CSF samples (15‐20 mL) are collected at baseline for all participants as well as longitudinally (at 12 and 24 months) in EOAD and EOnonAD participants. All CSF samples are sent directly to NCRAD at IU for processing and storage according to standard operating procedures (SOPs). At defined study time points, NCRAD will provide aliquots of baseline and follow‐up CSF samples to the Fagan Fluid Biomarker Laboratory at Washington University in St. Louis for the evaluation of established and emerging biomarkers of several AD pathologies, including (1) Aß40 and Aß42 (ß‐amyloid plaques), total tau (neuronal injury), and ptau181 (neurofibrillary tangles) using the high‐performance, automated LUMIPULSE assay platform (Fujirebio, Malverne, PA); (2) neurofilament light chain (NfL) (axonal damage) via commercial enzyme‐linked immunosorbent assay (ELISA) (Uman Diagnostics Umeå, Sweden); (3) visinin‐like protein 1 (VILIP‐1) (tau‐independent neuronal injury/death) and neurogranin (Ng) (post‐synaptic dysfunction/injury) by microparticle‐based immunoassays using Single Molecule Counting (SMC) technology employing antibodies developed in the laboratory of Dr. Jack Ladenson at Washington University in St. Louis; and (4) YKL‐40 (astrogliosis/neuroinflammation) via commercial ELISA (Quidel, San Diego, CA). In a separately funded study, CSF samples will also be assayed for Aß40, Aß42, total tau, and ptau181 using the high‐performance, automated Elecsys assay platform (Roche Diagnostics, Basel, Switzerland) for method‐evaluation purposes. Baseline CSF samples are expected to be analyzed in year 4 and again with longitudinal follow‐up samples in year 5 of the grant or order to minimize potential assay lot‐to‐lot variability. All assays are performed with strict adherence to SOPs so to maximize rigor and reproducibility. Remaining sample aliquots are banked at NCRAD for future analyses of novel biomarkers.
In addition to CSF, blood is obtained and processed for analysis of a variety of blood products: (1) red top for serum biomarkers; (2) EDTA for plasma biomarkers and buffy coat for DNA; (3) PAXgene for RNA; and (4) sodium heparin for peripheral blood mononuclear cells (PBMCs). Samples are shipped to NCRAD for banking and eventual distribution to qualified investigators for future analyses.
Plasma biomarkers have in recent years shown tremendous promise as diagnostic biomarkers for AD. 80 , 81 , 82 , 83 , 84 Serial measures from blood are easier to obtain, safer, and more affordable than either amyloid PET or lumbar punctures. With the soon to be funded Competitive Revision in collaboration with Dr. Randall Bateman's group from Washington University and Eli Lilly we will be able to add longitudinal plasma Aβ40 and Aβ42 (liquid chromatography–mass spectrometry approach 84 , 85 ), as well as P‐tau217 (P‐tau217 Meso Scale Discovery optimized Lilly protocol) and NfL (Quanterix single‐molecule array assay ‐ SiMOA) to LEADS.
3.5. Neuropathology core
The LEADS Neuropathology core (NPC) coordinates autopsies at participating sites and performs clinicopathologic analyses on EOAD and EOnonAD brain tissue. The NPC will fulfill the following goals: (1) facilitate brain procurement at the time of death from all LEADS participants; (2) conduct thorough, uniform post‐mortem neuropathologic examinations and assign all appropriate diagnoses for each case and contribute neuropathologic data to the LEADS database (including diagnoses, accurate staging, and semiquantitative assessments of distribution and density of neuropathologic lesions); (3) maintain a resource of formalin‐fixed brain tissue blocks for LEADS investigators and outside investigators with an updated database of frozen tissue location and availability; and (4) review tissue requests by committee and provide samples and data to qualified investigators.
LEADS NPC comprises three hub sites that will oversee participating clinical sites. The three hub sites are located at IU, the UCSF, and the Mayo Clinic Jacksonville (MCJ) and are led by Dr. Bernardino Ghetti, Dr. Lea T. Grinberg, and Dr. Melissa E. Murray, respectively. The NPC leaders at IU, UCSF, and MCJ hubs will support the clinical sites, collaborate with LEADS investigators, organize and participate in consensus‐building activities, and lead tissue‐sharing efforts.
Brain procurement and processing will follow site‐specific protocols to allow each site to maintain methodological continuity within cohorts. Although these protocols differ slightly between sites, all sites will follow NIA‐AA guidelines for regional sampling and staining. 86 These guidelines focus on regions relevant to AD, Lewy body disease, TDP‐43 proteinopathies, and vascular brain injury. To harmonize methods and data elements across centers, each site will complete the NACC Neuropathology Form, following guidelines provided in the NACC Neuropathology Guidebook.
Sites will be supported and encouraged to donate intact fixed hemibrains from each case. If a site elects to retain brain tissue locally, a standard set of formalin‐fixed tissue blocks will be shipped to the appropriate NPC hub. Although we recognize that high quality frozen tissue is essential for research purposes, after careful consideration and thoughtful discussions with non‐hub clinical sites, the NPC has devised an approach in which the sites will keep frozen tissue and the NPC will maintain a centralized inventory list to facilitate tissue location and distribution to investigators. This approach will avoid possible compromise of tissue integrity while in transfer from the site to the hub. Requests for LEADS samples can be made via an online request portal (see section Data Sharing Procedures). Recipients of LEADS samples will be encouraged to make data from their tissue studies widely available to the research community to support studies utilizing LEADS tissue samples.
4. STATISTICAL METHODOLOGY
The Biostatistics Core for LEADS is located at the Center for Statistical Sciences, Brown University School of Public Health, which also serves a similar role in the IDEAS studies. The data flow in LEADS is depicted in Figure 3.
FIGURE 3.
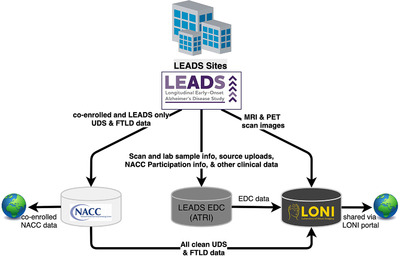
LEADS data flow
The main goals of our study are to compare the baseline and longitudinal cognitive and functional characteristics as well as MRI, amyloid PET, tau PET, and CSF biomarkers of EOAD versus LOAD, and to identify optimal outcome measures for EOAD clinical trials. We plan to use ADNI LOAD subjects in our comparisons. To allow for this comparison, our clinical protocol, scanner qualification, imaging protocols, fluid biomarker acquisition, and processing have been aligned to those in ADNI. Our Biostatistics Core will work with ADNI's Biostatistics Core to select a comparison group with LOAD that will allow us to achieve our scientific goals. We will select a matching cohort from ADNI LOAD of the same size as the LEADS EOAD participant group, matching for global CDR, MMSE ± 2 points, education ± 2 years, sex, and APOE genotype. For our main comparison analyses we will use linear mixed models including subject‐specific random intercepts and slopes controlling for covariates. We will also implement clustering analyses and machine learning techniques 87 to develop composite measures that are sensitive to cognitive change over time. In addition, we will implement unsupervised latent variable analysis techniques to identify latent features in the psychometric data predictive of cognitive and functional decline in EOAD. K‐fold cross‐validation will be used for tuning the parameters in the machine‐learning models. Additional analyses in the imaging space will be conducted by the LEADS PET and MRI cores.
5. DATA SHARING AND PUBLICATIONS
LEADS Data Sharing Policy follows the principles of Productivity, Transparency, Fairness, and Inclusiveness. The full policy can be reviewed at https://leads‐study.medicine.iu.edu/researchers/. Analyses that are specified in the specific aims of the project from the original grant application and any subsequent revisions and renewals will be led by the LEADS Principal Investigators and Core Leaders. Analyses that are not proposed in the specific aims of the main grant and its subsequent revisions and renewals can be conducted by investigators within and outside of LEADS after their proposals are reviewed and approved by the Data Sharing Committee. Requestors will be asked to specify the principal hypotheses, the materials needed (variables, imaging data, biospecimens, and so on), the analytic plan, and assurance of non‐overlap with planned LEADS analyses. Data requests will be reviewed based on scientific merit, feasibility, and appropriateness of the investigator's qualifications and resources to protect the data. An IRB approval will be required prior to releasing LEADS data. After a request is approved and IRB approval of the proposed analyses is verified, de‐identified data will be made available through the LONI interactive data portal to investigators to conduct analyses. Analyses will be based on frozen data sets that have been quality controlled and cleaned. Investigators will be asked to return any leftover DNA or fluid biomarker samples. New data generated through analyses of LEADS data sets will need to be provided to the Publications Committee prior to submission of the manuscript for publication for review for possible inclusion in the project database or into another NIH‐approved government database such as dbGap or NIAGADS. A 6‐month embargo will be placed on returned data to allow publication of results. The Publications Committee will review all manuscripts ensuring proper description of informed consent, approach to confidentiality, acknowledgement of LEADS investigators and funding sources, and disclosure of potential and actual conflicts of interest. Acceptance of LEADS data obligates the recipient to cite/reference all LEADS funding sources in presentations or publications that may result from this research. No sharing of data with a third party will be allowed without the permission of the LEADS Executive Committee.
6. PUBLIC PRIVATE PARTNERSHIP
LEADS represents an innovative partnership between federal, academic, and private stakeholders. LEADS investigators are partnering with Life Molecular Imaging and Eli Lily/AVID Radiopharmaceuticals, who provide the amyloid and tau PET tracers at reduced cost. LEADS investigators are working with industry partners such as Eli Lilly, Roche, and Araclon Biotech, as well as investigators from Washington University to expand the fluid biomarker analyses to include CSF and plasma Aβ and tau biomarkers in addition to other promising disease‐associated markers such as neurofilament light (NfL). Further collaborations with the private sector will be leveraged to expand LEADS research.
LEADS received a $1 million grant from the Alzheimer's Association to expand the genetic analyses of the study to include whole genome sequencing, DNA methylation, and copy number variant studies. LEADS also received a $235,000 Diversity Recruitment Grant by the Alzheimer's Association to support minority recruitment.
In an ongoing collaboration with researchers from the School of Health Sciences and the Purdue Institute for Integrative Neuroscience at Purdue University we will produce the first induced pluripotent stem cell lines (iPSCs) from the PBMCs collected from LEADS participants.
7. FUTURE PLANS
The over‐arching goals of LEADS are (1) to advance our knowledge about disease mechanisms and heterogeneity, (2) to develop sensitive composite clinical and biomarker tools that capture disease progression in this unique cohort, (3) to establish a network of sites that will enable future planning and implementation of clinical trials in EOAD. Additional efforts for international expansion and the development of a LEADS Trials Unit are ongoing. In collaboration with the Alzheimer's Association we are also planning to develop educational webinars and support groups for EOAD participants and their families at the local site level, as well as a Site Ambassador Program that will liaise between study participants and LEADS researchers.
CONFLICT OF INTEREST
Dr. Apostolova served as a paid consultant for Biogen and Two Labs, serves on a DSMB for IQVIA, and receives research support from the NIH, the Alzheimer's Association, Roche, Life Molecular Imaging, and Eli Lilly. Dr. Aisen reports grants from NIA, FNIH, the Alzheimer's Association, Janssen, Lilly, and Eisai, and personal fees from Merck, Roche, Biogen, Lundbeck, ImmunoBrain Checkpoint, and Samus. Dr. Eloyan has nothing to disclose. Dr. Fagan has received research support from Biogen, Fujirebio, and Roche Diagnostics. She is a member of the scientific advisory boards for Roche Diagnostics, Genentech, and AbbVie and also consults for Araclon/Griffols, DiademRes, and Otsuka Pharmaceuticals. Dr. Fargo has nothing to disclose. Dr. Foroud has nothing to disclose. Dr. Gatsonis has nothing to disclose. Dr. Grinberg has nothing to disclose. Dr. Jack serves on an independent data monitoring board for Roche and has consulted for and served as a speaker for Eisai, but he receives no personal compensation from any commercial entity. He receives research support from the NIH and the Alexander Family Alzheimer's Disease Research Professorship of the Mayo Clinic. Dr. Kramer has nothing to disclose. Dr. Koeppe has nothing to disclose. Dr. Kukull is supported by the NIA. Dr. Murray served as a paid consultant for AVID Radiopharmaceuticals and receives research support from the NIH, the Alzheimer's Association, and the state of Florida. Dr. Nudelman has nothing to disclose. Mrs. Rumbaugh has nothing to disclose. Dr. Toga has nothing to disclose. Dr. Vemuri has nothing to disclose. Mrs. Trullinger has nothing to disclose. Dr. Iaccarino has nothing to disclose. Dr. Day is supported by the NIA; he serves as a topic editor on dementia for DynaMed Plus (EBSCO Industries, Inc), is the clinical director for the Anti‐NMDA Receptor Encephalitis Foundation and holds stocks in ANI Pharmaceuticals. Dr. Graff‐Radford receives research support from Lilly, AbbVie, and Biogen. Dr. Honig has nothing to disclose. Dr. Jones has nothing to disclose. Dr. Masdeu served on the speaker's bureau and consulted for Biogen. He received research support from the NIH, Avanir, Abbvie, Biogen, Eli Lilly, Esai, and Novartis. Dr. Mendez receives grant support from NIA and has received support from Biogen. Dr. Musiek received research funding from Eisai Pharmaceuticals Inc. Dr. Onyike has nothing to disclose. Dr. Rogalski has nothing to disclose. Dr. Salloway received consultation fees and research support from Biogen, Eisai, Lilly, Genentech, and Roche. Dr. Wolk received grants from Eli Lilly/Avid Radiopharmaceuticals, Merck, and Biogen, and consulted for Merck, Janssen, and GE Healthcare. Dr. Wingo has nothing to disclose. Dr. Carrillo has nothing to disclose. Dr. Dickerson receives research support from the NIH and the Alzheimer's Drug Discovery Foundation. He consulted for Arkuda, Axovant, Lilly, Biogen, Merck, Novartis, and Wave LifeSciences. He is editor for Neuroimage: Clinical and Cortex. He receives royalties from Oxford University Press and Cambridge University Press. Dr. Rabinovici receives research support from the NIH, Alzheimer's Association, American College of Radiology, Rainwater Charitable Foundation, Avid Radiopharmaceuticals, Eli Lilly, GE Healthcare, Life Molecular Imaging, Genentech, and Roche. He has served as a paid consultant for Axon Neurosciences, Eisai, GE Healthcare, Genentech, Roche, and Johnson & Johnson, and is an Associate Editor for JAMA Neurology.
ACKNOWLEDGEMENTS
This study is generously supported by R56 AG057195, U01AG6057195, U24AG021886, Alzheimer's Association LEADS GENETICS‐19‐639372, U01 AG016976, P30 AG010133, P50 AG008702, P50 AG025688, P50 AG005146, P30 AG062421, P30 AG062422, P50 AG023501, P30 AG010124, P30 AG013854, and P50 AG005681.
Apostolova LG, Aisen P, Eloyan A, et al. the LEADS Consortium . The Longitudinal Early‐onset Alzheimer's Disease Study (LEADS): Framework and methodology. Alzheimer's Dement. 2021;17:2043–2055. 10.1002/alz.12350
REFERENCES
- 1. Alzheimer Association . 2019 Alzheimer's disease facts and figures. Alzheimers Dement 2019;15(3):321‐387. [Google Scholar]
- 2. Alzheimer Association . 2020 Alzheimer's disease facts and figures. Alzheimers Dement 2020;16(3):391‐460. [Google Scholar]
- 3. van Vliet D, de Vugt ME, Bakker C, et al. Impact of early onset dementia on caregivers: a review. Int J Geriatr Psychiatry. 2010;25(11):1091‐1100. [DOI] [PubMed] [Google Scholar]
- 4. Werner P, Stein‐Shvachman I, Korczyn AD. Early onset dementia: clinical and social aspects. Int Psychogeriatr. 2009;21(4):631‐636. [DOI] [PubMed] [Google Scholar]
- 5. Beach TG, Monsell SE, Phillips LE, et al. Accuracy of the clinical diagnosis of Alzheimer disease at National Institute on Aging Alzheimer Disease Centers, 2005‐2010. J Neuropathol Exp Neurol. 2012;71(4):266‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Crutch SJ, Lehmann M, Schott JM, et al. Posterior cortical atrophy. Lancet Neurol. 2012;11(2):170‐178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Alladi S, Xuereb J, Bak T, et al. Focal cortical presentations of Alzheimer's disease. Brain. 2007;130(Pt 10):2636‐2645. [DOI] [PubMed] [Google Scholar]
- 8. Forman MS, Farmer J, Johnson JK, et al. Frontotemporal dementia: clinicopathological correlations. Ann Neurol. 2006;59(6):952‐962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kertesz A, McMonagle P, Blair M, et al. The evolution and pathology of frontotemporal dementia. Brain. 2005;128(Pt 9):1996‐2005. [DOI] [PubMed] [Google Scholar]
- 10. Varma AR, Snowden JS, Lloyd JJ, et al. Evaluation of the NINCDS‐ADRDA criteria in the differentiation of Alzheimer's disease and frontotemporal dementia. J Neurol Neurosurg Psychiatry. 1999;66(2):184‐188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Seltzer B, Sherwin I. A comparison of clinical features in early‐ and late‐onset primary degenerative dementia. One entity or two?. Arch Neurol. 1983;40(3):143‐146. [DOI] [PubMed] [Google Scholar]
- 12. Barclay LL, Zemcov A, Blass JP, et al. Factors associated with duration of survival in Alzheimer's disease. Biol Psychiatry. 1985;20(1):86‐93. [DOI] [PubMed] [Google Scholar]
- 13. Jacobs D, Sano M, Marder K, et al. Age at onset of Alzheimer's disease: relation to pattern of cognitive dysfunction and rate of decline. Neurology. 1994;44:1215‐1220. [DOI] [PubMed] [Google Scholar]
- 14. Koss E, Edland S, Fillenbaum G, et al. Clinical and neuropsychological differences between patients with earlier and later onset of Alzheimer's disease: a CERAD analysis, Part XII. Neurology. 1996;46(1):136‐141. [DOI] [PubMed] [Google Scholar]
- 15. Mortimer JA, Ebbitt B, Jun SP, et al. Predictors of cognitive and functional progression in patients with probable Alzheimer's disease. Neurology. 1992;42(9):1689‐1696. [DOI] [PubMed] [Google Scholar]
- 16. Heyman A, Wilkinson WE, Hurwitz BJ, et al. Early‐onset Alzheimer's disease: clinical predictors of institutionalisation and death. Neurology. 1987;27:980‐984. [DOI] [PubMed] [Google Scholar]
- 17. Loring DW, Largen JW. Neuropsychological patterns of presenile and senile dementia of the Alzheimer type. Neuropsychologia. 1985;23(3):351‐357. [DOI] [PubMed] [Google Scholar]
- 18. Filley CM, Kelly J, Heaton RK. Neuropsychologic features of early‐ and late‐onset Alzheimer's disease. Arch Neurol. 1986;43(6):574‐576. [DOI] [PubMed] [Google Scholar]
- 19. Imamura T, Takatsuki Y, Fujimori M, et al. Age at onset and language disturbances in Alzheimer's disease. Neuropsychologia. 1998;36(9):945‐949. [DOI] [PubMed] [Google Scholar]
- 20. Fujimori M, Imamura T, Yamashita H, et al. Age at onset and visuocognitive disturbances in Alzheimer disease. Alzheimer Dis Assoc Disord. 1998;12(3):163‐166. [DOI] [PubMed] [Google Scholar]
- 21. Frisoni GB, Pievani M, Testa C, et al. The topography of grey matter involvement in early and late onset Alzheimer's disease. Brain. 2007;130(Pt 3):720‐730. [DOI] [PubMed] [Google Scholar]
- 22. Kim EJ, Cho SS, Jeong Y, et al. Glucose metabolism in early onset versus late onset Alzheimer's disease: an SPM analysis of 120 patients. Brain. 2005;128(Pt 8):1790‐1801. [DOI] [PubMed] [Google Scholar]
- 23. Snowden JS, Stopford CL, Julien CL, et al. Cognitive phenotypes in Alzheimer's disease and genetic risk. Cortex. 2007;43(7):835‐845. [DOI] [PubMed] [Google Scholar]
- 24. Binetti G, Magni E, Padovani A, et al. Neuropsychological heterogeneity in mild Alzheimer's disease. Dementia. 1993;4(6):321‐326. [DOI] [PubMed] [Google Scholar]
- 25. Frisoni GB, Testa C, Sabattoli F, et al. Structural correlates of early and late onset Alzheimer's disease: voxel based morphometric study. J Neurol Neurosurg Psychiatry. 2005;76(1):112‐114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Koedam EL, Lauffer V, van der Vlies AE, et al. Early‐versus late‐onset Alzheimer's disease: more than age alone. J Alzheimers Dis. 2010;19(4):1401‐1408. [DOI] [PubMed] [Google Scholar]
- 27. Mendez MF, Lee AS, Joshi A, et al. Nonamnestic presentations of early‐onset Alzheimer's disease. Am J Alzheimers Dis Other Demen. 2012;27(6):413‐420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sakamoto S, Ishii K, Sasaki M, et al. Differences in cerebral metabolic impairment between early and late onset types of Alzheimer's disease. J Neurol Sci. 2002;200(1‐2):27‐32. [DOI] [PubMed] [Google Scholar]
- 29. Shiino A, Watanabe T, Kitagawa T, et al. Different atrophic patterns in early‐ and late‐onset Alzheimer's disease and evaluation of clinical utility of a method of regional z‐score analysis using voxel‐based morphometry. Dement Geriatr Cogn Disord. 2008;26(2):175‐186. [DOI] [PubMed] [Google Scholar]
- 30. Shiino A, Watanabe T, Maeda K, et al. Four subgroups of Alzheimer's disease based on patterns of atrophy using VBM and a unique pattern for early onset disease. Neuroimage. 2006;33(1):17‐26. [DOI] [PubMed] [Google Scholar]
- 31. Karas G, Scheltens P, Rombouts S, et al. Precuneus atrophy in early‐onset Alzheimer's disease: a morphometric structural MRI study. Neuroradiology. 2007;49(12):967‐976. [DOI] [PubMed] [Google Scholar]
- 32. Ishii K, Kawachi T, Sasaki H, et al. Voxel‐based morphometric comparison between early‐ and late‐onset mild Alzheimer's disease and assessment of diagnostic performance of z score images. AJNR Am J Neuroradiol. 2005;26(2):333‐340. [PMC free article] [PubMed] [Google Scholar]
- 33. Grady CL, Hazby JV, Horwitz B, et al. Neuropsychological and cerebral metabolic function in early vs late onest dementia of the Alzheimer type. Neuropsychologia. 1987;25(5):807‐816. [DOI] [PubMed] [Google Scholar]
- 34. Yasuno F, Imamura T, Hirono N, et al. Age at onset and regional cerebral glucose metabolism in Alzheimer's disease. Dement Geriatr Cogn Disord. 1998;9(2):63‐67. [DOI] [PubMed] [Google Scholar]
- 35. Salmon E, Collette F, Degueldre C, et al. Voxel‐based analysis of confounding effects of age and dementia severity on cerebral metabolism in Alzheimer's disease. Hum Brain Mapp. 2000;10(1):39‐48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jagust WJ, Reed BR, Seab JP, et al. Alzheimer's disease: age at onset and single‐photon emission computed tomographic patterns of regional cerebral blood flow. Arch Neurol. 1990;47:628‐633. [DOI] [PubMed] [Google Scholar]
- 37. Kemp PM, Holmes C, Hoffmann SM, et al. Alzheimer's disease: differences in technetium‐99m HMPAO SPECT scan findings between early onset and late onset dementia. J Neurol Neurosurg Psychiatry. 2003;74(6):715‐719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rabinovici GD, Furst AJ, Alkalay A, et al. Increased metabolic vulnerability in early‐onset Alzheimer's disease is not related to amyloid burden. Brain. 2010;133(Pt 2):512‐528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Moller C, Vrenken H, Jiskoot L, et al. Different patterns of gray matter atrophy in early‐ and late‐onset Alzheimer's disease. Neurobiol Aging. 2013;34(8):2014‐2022. [DOI] [PubMed] [Google Scholar]
- 40. Mann DM, Yates PO, Marcyniuk B. Alzheimer's presenile dementia, senile dementia of Alzheimer type and Down's syndrome in middle age form an age related continuum of pathological changes. Neuropathol Appl Neurobiol. 1984;10(3):185‐207. [DOI] [PubMed] [Google Scholar]
- 41. Hansen LA, DeTeresa R, Davies P, et al. Neocortical morphometry, lesion counts, and choline acetyltransferase levels in the age spectrum of Alzheimer's disease. Neurology. 1988;38(1):48‐54. [DOI] [PubMed] [Google Scholar]
- 42. Nochlin D, van Belle G, Bird TD, et al. Comparison of the severity of neuropathologic changes in familial and sporadic Alzheimer's disease. Alzheimer Dis Assoc Disord. 1993;7(4):212‐222. [PubMed] [Google Scholar]
- 43. Bigio EH, Hynan LS, Sontag E, et al. Synapse loss is greater in presenile than senile onset Alzheimer disease: implications for the cognitive reserve hypothesis. Neuropathol Appl Neurobiol. 2002;28(3):218‐227. [DOI] [PubMed] [Google Scholar]
- 44. Ho GJ, Hansen LA, Alford MF, et al. Age at onset is associated with disease severity in Lewy body variant and Alzheimer's disease. Neuroreport. 2002;13(14):1825‐1828. [DOI] [PubMed] [Google Scholar]
- 45. Berg L, McKeel DW Jr, Miller JP, et al. Clinicopathologic studies in cognitively healthy aging and Alzheimer's disease: relation of histologic markers to dementia severity, age, sex, and apolipoprotein E genotype. Arch Neurol. 1998;55(3):326‐335. [DOI] [PubMed] [Google Scholar]
- 46. Marshall GA, Fairbanks LA, Tekin S, et al. Early‐onset Alzheimer's disease is associated with greater pathologic burden. J Geriatr Psychiatry Neurol. 2007;20(1):29‐33. [DOI] [PubMed] [Google Scholar]
- 47. Bird TD, Stranahan S, Sumi SM, et al. Alzheimer's disease: choline acetyltransferase activity in brain tissue from clinical and pathological subgroups. Ann Neurol. 1983;14(3):284‐293. [DOI] [PubMed] [Google Scholar]
- 48. Rossor MN, Iversen LL, Reynolds GP, et al. Neurochemical characteristics of early and late onset types of Alzheimer's disease. Br Med J (Clin Res Ed). 1984;288(6422):961‐964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Campion D, Dumanchin C, Hannequin D, et al. Early‐onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. Am J Hum Genet. 1999;65(3):664‐670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Janssen JC, Beck JA, Campbell TA, et al. Early onset familial Alzheimer's disease ‐ Mutation frequency in 31 families. Neurology. 2003;60(2):235‐239. [DOI] [PubMed] [Google Scholar]
- 51. Signorini S, Ghidoni R, Barbiero L, et al. Prevalence of pathogenic mutations in an italian clinical series of patients with familial dementia. Curr Alzheimer Res. 2004;1(3):215‐218. [DOI] [PubMed] [Google Scholar]
- 52. Cacace R, Sleegers K, Van Broeckhoven C. Molecular genetics of early‐onset Alzheimer's disease revisited. Alzheimers Dement. 2016;12(6):733‐748. [DOI] [PubMed] [Google Scholar]
- 53. Wingo TS, Lah JJ, Levey AI, et al. Autosomal recessive causes likely in early‐onset Alzheimer disease. Arch Neurol. 2012;69(1):59‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Szigeti K, Doody RS. Should EOAD patients be included in clinical trials?. Alzheimers Res Ther. 2011;8(3):1‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7(3):270‐279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7(3):263‐269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Crutch SJ, Schott JM, Rabinovici GD, et al. Consensus classification of posterior cortical atrophy. Alzheimers Dement. 2017;13(8):870‐884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gorno‐Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology. 2011;76(11):1006‐1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Van der Auwera GA, Carneiro MO, Hartl C, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Stenson PD, Mort M, Ball EV, et al. The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next‐generation sequencing studies. Hum Genet. 2017;136(6):665‐677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Landrum MJ, Lee JM, Benson M, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46(D1):D1062‐D1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Fokkema IF, Taschner PE, Schaafsma GC, et al. LOVD v.2.0: the next generation in gene variant databases. Hum Mutat. 2011;32(5):557‐563. [DOI] [PubMed] [Google Scholar]
- 63. Mills SM, Mallmann J, Santacruz AM, et al. Preclinical trials in autosomal dominant AD: implementation of the DIAN‐TU trial. Rev Neurol (Paris). 2013;169(10):737‐743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Vemuri P, Senjem ML, Gunter JL, et al. Accelerated vs. unaccelerated serial MRI based TBM‐SyN measurements for clinical trials in Alzheimer's disease. Neuroimage. 2015;113:61‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sabri O, Sabbagh MN, Seibyl J, et al. Florbetaben PET imaging to detect amyloid beta plaques in Alzheimer's disease: phase 3 study. Alzheimers Dement. 2015;11(8):964‐974. [DOI] [PubMed] [Google Scholar]
- 66. Bejanin A, Tammewar G, Marx G, et al. Longitudinal structural and metabolic changes in frontotemporal dementia. Neurology. 2020;95(2):e140‐e154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Chetelat G, Ossenkoppele R, Villemagne VL, et al. Atrophy, hypometabolism and clinical trajectories in patients with amyloid‐negative Alzheimer's disease. Brain. 2016;139(Pt 9):2528‐2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Jagust WJ, Landau SM, Koeppe RA, et al. The Alzheimer's disease neuroimaging initiative 2 PET core: 2015. Alzheimers Dement. 2015;11(7):757‐771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Baker SL, Maass A, Jagust WJ. Considerations and code for partial volume correcting [(18)F]‐AV‐1451 tau PET data. Data Brief. 2017;15:648‐657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Diedrichsen J. A spatially unbiased atlas template of the human cerebellum. Neuroimage. 2006;33(1):127‐138. [DOI] [PubMed] [Google Scholar]
- 71. Desikan RS, Ségonne F, Fischl B, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage. 2006;31(3):968‐980. [DOI] [PubMed] [Google Scholar]
- 72. Klunk WE, Koeppe RA, Price JC, et al. The Centiloid Project: standardizing quantitative amyloid plaque estimation by PET. Alzheimers Dement. 2015;11(1):1‐15.e1‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Jack CR Jr, Wiste HJ, Weigand SD, et al. Defining imaging biomarker cut points for brain aging and Alzheimer's disease. Alzheimers Dement. 2017;13(3):205‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Rabinovici GD, Karlawish J, Knopman D, et al. Testing and disclosures related to amyloid imaging and Alzheimer's disease: common questions and fact sheet summary. Alzheimers Dement. 2016;12(4):510‐515. [DOI] [PubMed] [Google Scholar]
- 75. Fleisher AS, Pontecorvo MJ. Positron emission tomography imaging with [18F]flortaucipir and postmortem assessment of Alzheimer disease neuropathologic changes. JAMA Neurol. 2020;77(7):829‐839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ossenkoppele R, Rabinovici GD, Smith R, et al. Discriminative accuracy of [18F]flortaucipir positron emission tomography for Alzheimer disease vs other neurodegenerative disorders. JAMA. 2018;320(11):1151‐1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. La Joie R, Perrotin A, Barre L, et al. Region‐specific hierarchy between atrophy, hypometabolism, and beta‐amyloid (Abeta) load in Alzheimer's disease dementia. J Neurosci. 2012;32(46):16265‐16273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Bateman RJ, Xiong C, Benzinger TL, et al. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med. 2012;367(9):795‐804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Shaw LM, Vanderstichele H, Knapik‐Czajka M, et al. Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann Neurol. 2009;65(4):403‐413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Nakamura A, Kaneko N, Villemagne VL, et al. High performance plasma amyloid‐beta biomarkers for Alzheimer's disease. Nature. 2018;554(7691):249‐254. [DOI] [PubMed] [Google Scholar]
- 81. Janelidze S, Stomrud E, Palmqvist S, et al. Plasma beta‐amyloid in Alzheimer's disease and vascular disease. Sci Rep. 2016;6:26801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Li D, Mielke MM. An update on blood‐based markers of Alzheimer's disease using the SiMoA platform. Neurol Ther. 2019;8(Suppl 2):73‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Palmqvist S, Janelidze S, Stomrud E, et al. Performance of fully automated plasma assays as screening tests for Alzheimer disease‐related beta‐amyloid status. JAMA Neurol. 2019;76(9):1060‐1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Schindler SE, Bollinger JG, Ovod V, et al. High‐precision plasma beta‐amyloid 42/40 predicts current and future brain amyloidosis. Neurology. 2019;93(17):e1647‐e1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Chatterjee P, Elmi M, Goozee K, et al. Ultrasensitive detection of plasma Amyloid‐beta as a biomarker for cognitively normal elderly individuals at risk of Alzheimer's disease. J Alzheimers Dis 2019;71(3):775‐83 [DOI] [PubMed] [Google Scholar]
- 86. Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement. 2012;8(1):1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Hastie T, Tibshirani R, Friedman J. The Elements of Statistical Learning: Data Mining, Inference, and Prediction. NY: Springer; 2009. [Google Scholar]