

Abstract
Background:
Mutations in GBA1 are a common genetic risk factor for parkinsonism; however, penetrance is incomplete, and biomarkers of future progression to parkinsonism are needed. Both nigral sonography and striatal [18F]-FDOPA PET assay dopamine system health, but their utility and coherence in this context are unclear.
Objectives:
To evaluate the utility and coherence of these modalities in GBA1-associated parkinsonism.
Methods:
Thirty-four patients with GBA1 mutations (seven with parkinsonism), underwent both transcranial studies for substantia nigra echogenicity and [18F]-FDOPA PET to determine striatal tracer specific uptake (Ki).
Results:
Larger nigral echogenic areas and reduced striatal Ki were exclusively seen in parkinsonian subjects. Sonographic and PET measurements showed strong inverse correlations, but solely in individuals with clinical parkinsonism.
Conclusions:
Close correspondence between nigral echogenicity and striatal presynaptic dopamine synthesis capacity seen only in GBA1 carriers with parkinsonism provides validation that these two modalities may conjointly capture aspects of the biology underlying clinical parkinsonism but raises questions about their utility as predictive tools in at-risk subjects.
Keywords: Transcranial sonography, 18F-fluorodopa PET imaging, glucocerebrosidase, parkinsonism, Gaucher disease
Introduction
One anticipated outcome of research identifying genetic variation associated with parkinsonism is an enhanced ability to detect illness at an early stage, when personalized therapeutics or early treatments might be administered.1 However, many of the identified risk alleles for Parkinson disease (PD) have variable penetrance, especially variants in GBA1 and LRRK2, the genes most strongly associated with PD risk.2 Pathologic variants in GBA1, the gene mutated in the lysosomal storage disorder Gaucher disease (GD), have been identified in 2–29% of patients with PD, yet the vast majority of patients with GD and their carrier relatives never develop parkinsonism.3,4 Thus, even in genetically at-risk populations, in order to discover and implement disease-altering therapies, there is an urgency to identify early diagnostic tools that may help identify individuals on a trajectory toward developing parkinsonism. While factors including impaired olfaction, REM sleep behavior disorder, autonomic dysfunction or depression may precede motor involvement in some patients destined to develop parkinsonism, most clinical evaluations are fairly non-specific,5 and hence imaging modalities are being evaluated as potential biomarkers of disease progression.
In PD, loss of midbrain dopaminergic neurons and their nigrostriatal projections leads to deterioration of striatal dopaminergic terminals, which can be reliably measured with [18F]-fluorodopa PET.6 The influx constant, Ki, represents the specific uptake of [18F]-fluorodopa in DOPA decarboxylase-containing cells and reflects presynaptic dopamine synthesis and storage. Marked reduction in striatal Ki has been a well-established, validated indicator of dopaminergic insufficiency in idiopathic PD.6 In prior work, we performed [18F]-fluorodopa PET studies in a cohort of individuals with mutations in GBA1, both with and without clinical evidence of PD, and observed striatal dopamine synthesis deficits in GBA1-associated PD closely resembling that in idiopathic PD.7 A subsequent longitudinal PET study in an overlapping but larger sample again showed significant dopamine loss in those with parkinsonism. However, at baseline and over time, [18F]-fluorodopa specific uptake in at-risk GBA1 carriers without parkinsonism did not differ significantly from controls,8 identifying a notable disconnect between genetic risk and the neurochemical risk phenotype, suggesting that GBA1-mediated genetic risk alone is not sufficient to alter striatal dopaminergic tone. Additionally, these findings raise questions about whether striatal Ki variation within the normal range might reflect nigral neuronal population strength or vulnerability in genetically high-risk groups.
Alongside the [18F]-FDOPA PET PD literature, accumulated evidence has supported the diagnostic and potentially predictive value of transcranial sonography (TCS) of the substantia nigra in PD.9 Postmortem evaluations have shown nigral echogenicity is associated with co-localized iron stores,10 yet the how this measurement relates to hallmark dopaminergic pathology in PD is incompletely understood. Studies employing both ultrasonography and molecular neuroimaging to examine relationships between nigral echogenicity and dopamine-related parameters have varied in methodology and results. Some studies of PD have identified inverse relationships between nigral echogenicity and monoaminergic transporter SPECT assays,11, 12 whereas others have not.13–16 [18F]-FDOPA PET investigations in individuals with incidentally discovered nigral hyperechogenicity,17 and in Parkin mutation carriers,18 suggest an inverse correlation between presynaptic dopamine synthesis capacity and nigral echogenicity although these studies have limitation in size and design. No prior reports have compared TCS and molecular neuroimaging results in cohorts with GBA1 mutations. Anticipating that nigral echogenicity would be expanded in individuals with clinical parkinsonism and would be inversely related to striatal presynaptic dopamine synthesis capacity in groups with parkinsonism and at-risk for parkinsonism, we studied a cohort with homozygous and heterozygous GBA1 mutations with and without clinical parkinsonism using both TCS and [18F]-FDOPA PET.
Methods
Thirty-four individuals of European descent with pathological GBA1 variants (20 with GD and 14 heterozygous carriers) underwent both [18F]-FDOPA PET neuroimaging and TCS nigral measurements. Participants provided informed consent under an NHGRI Institutional Review Board-approved clinical protocol (NCT00302146). Sanger sequencing of GBA1, performed on blood samples,19 confirmed the presence of pathologic variants associated with GD.20, 21,22 The UK Brain Bank Criteria was used to establish the diagnosis of PD23 identifying seven individuals, five with GD and two GBA1 heterozygotes, with parkinsonism. The two imaging assessments were conducted independently by two separate teams during the same patient visit. [18F]-FDOPA PET imaging was performed in a fasting state with a GE Advance tomograph operating in 3D mode as reported.8 In brief, after tapering any confounding medications, suspending antiparkinsonian medications for at least 12 hours, abstaining from caffeine and nicotine for at least four hours, and receiving a single oral dose of carbidopa 200 mg one hour prior to [18F]-FDOPA injection, participants completed scanning, which included a transmission scan and, immediately after injection, a 90-minute emission scan series. Filtered-back-projection-based reconstruction and corrections, including attenuation correction and realignment for interframe motion, were applied, and data (voxel size 1.5×1.5×1.5mm3) were spatially warped to an [18F]-FDOPA-specific template, smoothed (10mm Gaussian kernel), and modeled voxelwise in PMOD using a graphical linearization model with cerebellar activity as the input function.24 TCS was performed using a phased-array ultrasound system with a 2.5 MHz transducer (Acuson Antares; Siemens).25 Planimetric measurements of the maximal area of echogenicity at the anatomical location of the SN were obtained bilaterally and averaged. Statistical comparisons of TCS group differences (using R software https://www.r-project.org/) and voxelwise associations between neuroimaging measurements (using SPM software https://www.fil.ion.ucl.ac.uk/spm/) were performed using standard general linear models (Student’s t-tests/ANOVA for group mean comparisons and linear regression analyses for Ki-echogenicity and Ki-echogenicity-by-group interaction association tests), with p<0.05 (FDR-corrected for voxelwise analyses conducted exclusively within the striatum) considered significant. The striatal search region included the caudate, putamen and subcomissural ventral striatum (including nucleus accumbens) and was delineated with the help of Freesurfer software, which was applied to an MNI space template derived from T1 weighted MRI data from 240 healthy adults.
Results
Individuals in this study were clinically monitored for up to twelve years after neuroimaging (see Table 1 for demographics, genetic, and clinical information). Only one participant, an N370S carrier without parkinsonism at the time of neuroimaging, developed parkinsonism during the follow-up interval. Relative to a TCS cutoff of 20 mm2 from prior literature26 and a putamen Ki reference value of 0.0065 min−1 representing two standard deviations below the mean in a previously studied healthy cohort using the same PET methods,8 this individual’s nigral echogenicity (16.7 mm2) and striatal Ki (mean putamen [18F]-FDOPA Ki 0.00877 min−1) were both unremarkable one year prior to symptom onset. In the entire cohort, most individuals with parkinsonism were outside these thresholds for TCS (5/7) and PET (6/7), whereas most individuals without parkinsonism (4/27 and 0/27, respectively) were not.
Table 1:
Demographic, Genetic, and Clinical Data
| No Parkinsonism, N = 27 | Parkinsonism, N = 7 | |
|---|---|---|
| GBA1 Status | ||
| Carrier | 12 (44%) | 2 (29%) |
| Gaucher disease | 15 (56%) | 5 (71%) |
| Age | ||
| Sex | 57 (11, 34–78) | 57 (11, 40–72) |
| Female | 13 (48%) | 2 (29%) |
| Male | 14 (52%) | 5 (71%) |
| Genotype | ||
| c.84insG/wt | 1 (3.7%) | 0 (0%) |
| C342Y/R496H | 1 (3.7%) | 0 (0%) |
| L444P/wt | 4 (15%) | 0 (0%) |
| N370S/55bpdel | 0 (0%) | 1 (14%) |
| N370S/c.84insG | 2 (7.4%) | 0 (0%) |
| N370S/L444P | 2 (7.4%) | 1 (14%) |
| N370S/N370S | 9 (33%) | 2 (29%) |
| N370S/R257Q | 0 (0%) | 1 (14%) |
| N370S/V394L | 1 (3.7%) | 0 (0%) |
| N370S/wt | 6 (22%) | 1 (14%) |
| R120W/wt | 0 (0%) | 1 (14%) |
| RecTL/wt | 1 (3.7%) | 0 (0%) |
| UPDRS-III | 1 (2, 0–8) | 26 (13, 7–39) |
| Duration of Follow-up | 6.68 (1.37, 5.26–10.13) | 7.43 (1.89, 5.36–11.40) |
n (%); Mean (SD, Minimum-Maximum)
The participants with parkinsonism had larger areas of maximal nigral echogenicity than those without parkinsonism (t(30)=3.92;p<0.0005). However, neither the effect of GBA1 status (carrier v. GD; t(30)=0.66, p=0.51) nor the clinical parkinsonism-GBA1 status interaction (t(30)=1.66,p=0.11) reached significance (Figure 1A). Voxelwise interrogation of the striatum revealed a robust inverse relationship between [18F]-FDOPA specific uptake (Ki) and nigral echogenic areas across individuals with clinical parkinsonism (p<0.05, FDR corrected, Figure 1B). However, there was no relationship between Ki and echogenicity in the at-risk GBA1 mutation carriers without parkinsonism. Direct comparison of Ki-echogenicity relationships in the two groups (with and without parkinsonism) showed these differences to constitute a significant interaction (p<0.05, FDR corrected; see Figure 1C, D).
Figure 1.
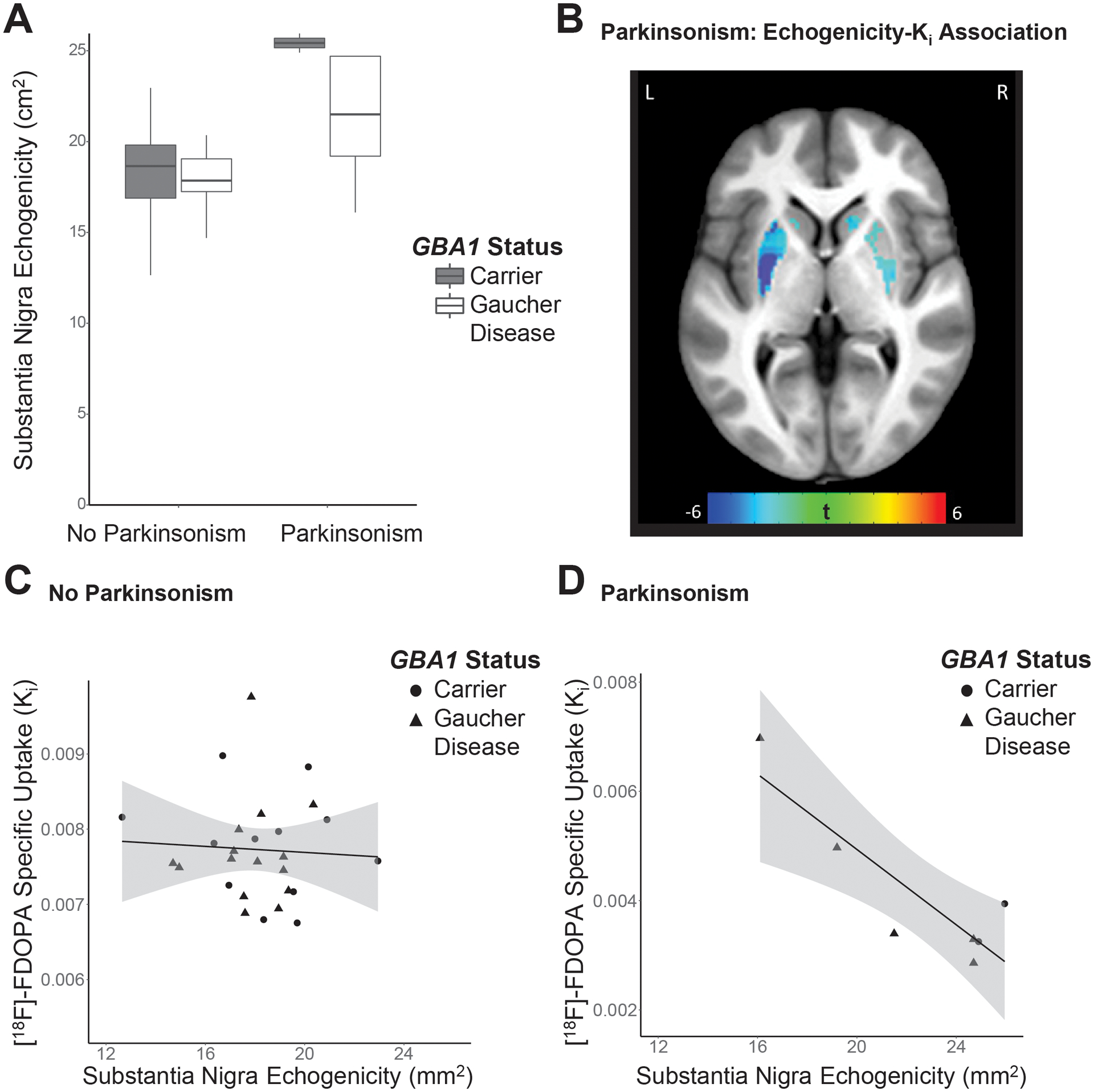
Substantia nigra echogenicity and striatal presynaptic dopamine synthesis in individuals with GBA1 mutations. A. Boxplots show size of substantia nigra echogenic area by clinical parkinsonism and GBA1 mutation group. Centrality lines, hinges and whiskers represent the median, interquartile range, and the farthest datapoint no greater than 1.5 times the interquartile range, respectively. B. Overlying a grayscale anatomical T1-weighted MRI axial section, a colorized t-value statistical map identifies locales of relationships between average nigral maximal echogenicity area and striatal dopamine synthesis capacity measured with [18F]-FDOPA PET in those with both GBA1 mutations and parkinsonism. All colored regions meet a voxelwise statistical threshold of p<0.05, FDR corrected, and represent two large clusters (peak1 [left posterior putamen]=(−27,−13.5, 6), k1=1809, t1=9.59, pFDR=0.015; peak2 [right caudate head]=(15,16.5,−7.5), k2=1204, t2=5.14, pFDR=0.02) C,D. Relationships between substantia nigra echogenicity and bilateral putamen mean Ki are plotted for subjects without (C) and with clinical parkinsonism (D).
Discussion
The larger echogenic areas observed in the patients with parkinsonism is consistent with the broader literature describing ultrasonography in PD.27 Based on a reference cutoff of 20 mm2 from prior literature,26 TCS measurements from participants without parkinsonism – including one individual who subsequently developed parkinsonism – were largely in the normal range, while most of those with parkinsonism showed abnormally elevated values. The current data do not rule out a subtle statistical increase in overall mean nigral echogenicity in individuals with GBA1 mutation carriers without parkinsonism relative to healthy genetic controls, as seen in one recent report that did not include post-sonography clinical follow-up;28 however, they do suggest a paucity of clinically significant TCS-measured pathology in this group. This finding parallels the group differences observed in our prior study with [18F]-FDOPA PET, where significantly reduced striatal dopamine synthesis capacity in the GBA1 mutation carriers relative to controls was seen exclusively in those with clinical parkinsonism.8 Despite the hypothesis that TCS and PET assessments might conjointly identify those motorically-intact GBA1 mutation carriers at greatest risk of developing parkinsonism, no genetically at-risk individuals without parkinsonism in the current study had both abnormally larger nigral echogenic areas and relatively lowered striatal Ki. Thus, excepting the possibility no non-parkinsonism participants in the sample had a heightened risk (at odds with the one converter identified), parkinsonism risk in this group may not necessarily be captured by the conjunction of these two imaging metrics.
Furthermore, the close correspondence of nigral echogenicity and striatal presynaptic dopamine synthesis capacity in subjects with parkinsonism, but not in those without, suggests that pathologic GBA1 variants alone do not necessarily confer strongly coherent neuroimaging indicators of dopaminergic system aberrancy. Rather, interindividual variability of presynaptic dopamine synthesis capacity in the at-risk group is likely driven by factors unrelated to mesencephalic iron-bound stores associated with PD. Identifying these additional factors may help refine and amplify the diagnostic and prognostic utility of molecular imaging in high-risk groups and poses an important challenge ahead.
Prior longitudinal studies of TCS in PD have reported relative stability of nigral echogenic areas in affected adults,29 though significantpu-related effects have been noted.30 If largely stable, hyperechogenicity associated with parkinsonism may be more valuable as a trait marker of disease, rather than for tracking nigral health at any given timepoint. Some reports suggest a correlation between echogenicity and clinical symptoms burden,29, 30 hypothesizing that TCS measurements may have prognostic value with respect to severity,29 although this is controversial.31 In contrast, striatal Ki is directly dependent on the cumulative dopamine synthetic capacity of nigral cell populations, and shows clear decline with disease progression in both sporadic PD32 and GBA1-associated parkinsonism.8 If nigral hyperechogenicity is exclusively a trait measure of illness, the demonstrated association with striatal Ki deficit in those with clinical parkinsonism may reflect interindividual differences in disease aggressiveness or other characteristics, as opposed to both neuroimaging measures equivalently indexing illness course per se.
As discussed in the longitudinal [18F]-FDOPA PET study,8 one limitation in our cohort is the dearth of individuals who developed parkinsonism during our period of follow-up. On one hand, this ensures that findings in the at-risk group were not substantially confounded by an imminent premorbid parkinsonian state. On the other hand, this makes it challenging to determine whether the observation that our at-risk group did not show unequivocally convergent imaging indications of parkinsonism across modalities reflects an inherent limitation of these complementary tools as long-term predictive measures or is due to the very low penetrance of parkinsonism in this population. Longer follow-up and larger dual-imaging longitudinal studies in GBA1 mutation carriers are needed to disambiguate these possibilities and further elucidate disease pathology.
Acknowledgements
This work was supported by the National Institutes of Health Intramural Research Programs of the National Human Genome Research Institute and the National Institute of Mental Health (ZIAMH002652). Some of this work utilized the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov).
Financial Disclosures
The Sidransky lab has received funding from the Michael J Fox Foundation, the Aligning Science Across Parkinson’s Initiative and from F.Hoffmann-La Roche Ltd. under a Cooperative Research and Development Agreement with the NHGRI and NCATS. The other authors have nothing to disclose.
Funding sources:
This work was supported by the intramural research programs of the National Human Genome Research Institute, the National Institute of Mental Health and the National Institutes of Health.
Footnotes
Disclosures: The authors have no conflicts of interest or financial disclosures to report.
References
- 1.Schneider SA, Alcalay RN. Precision medicine in Parkinson’s disease: emerging treatments for genetic Parkinson’s disease. J Neurol. Mar 2020;267(3):860–869. doi: 10.1007/s00415-020-09705-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sidransky E, Nalls MA, Aasly JO, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med. Oct 22 2009;361(17):1651–61. doi: 10.1056/NEJMoa0901281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Do J, McKinney C, Sharma P, Sidransky E. Glucocerebrosidase and its relevance to Parkinson disease. Mol Neurodegener. Aug 29 2019;14(1):36. doi: 10.1186/s13024-019-0336-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lopez G, Steward A, Ryan E, et al. Clinical evaluation of sibling pairs with gaucher disease discordant for parkinsonism. Mov Disord. Feb 2020;35(2):359–365. doi: 10.1002/mds.27916 [DOI] [PubMed] [Google Scholar]
- 5.Heinzel S, Roeben B, Ben-Shlomo Y, et al. Prodromal Markers in Parkinson’s Disease: Limitations in Longitudinal Studies and Lessons Learned. Front Aging Neurosci. 2016;8:147. doi: 10.3389/fnagi.2016.00147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu ZY, Liu FT, Zuo CT, Koprich JB, Wang J. Update on Molecular Imaging in Parkinson’s Disease. Neurosci Bull. Apr 2018;34(2):330–340. doi: 10.1007/s12264-017-0202-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goker-Alpan O, Masdeu JC, Kohn PD, et al. The neurobiology of glucocerebrosidase-associated parkinsonism: a positron emission tomography study of dopamine synthesis and regional cerebral blood flow. Brain. Aug 2012;135(Pt 8):2440–8. doi: 10.1093/brain/aws174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lopez G, Eisenberg DP, Gregory MD, et al. Longitudinal Positron Emission Tomography of Dopamine Synthesis in Subjects with GBA1 Mutations. Ann Neurol. Apr 2020;87(4):652–657. doi: 10.1002/ana.25692 [DOI] [PubMed] [Google Scholar]
- 9.Li DH, He YC, Liu J, Chen SD. Diagnostic Accuracy of Transcranial Sonography of the Substantia Nigra in Parkinson’s disease: A Systematic Review and Meta-analysis. Sci Rep. Feb 16 2016;6:20863. doi: 10.1038/srep20863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zecca L, Berg D, Arzberger T, et al. In vivo detection of iron and neuromelanin by transcranial sonography: A new approach for early detection of substantia nigra damage. Movement Disorders. 2005/October/01 2005;20(10):1278–1285. doi: 10.1002/mds.20550 [DOI] [PubMed] [Google Scholar]
- 11.Bártová P, Kraft O, Bernátek J, et al. Transcranial Sonography and 123I-FP-CIT Single Photon Emission Computed Tomography in Movement Disorders. Ultrasound in Medicine & Biology. 2014/October/01/ 2014;40(10):2365–2371. doi: 10.1016/j.ultrasmedbio.2014.05.014 [DOI] [PubMed] [Google Scholar]
- 12.Weise D, Lorenz R, Schliesser M, Schirbel A, Reiners K, Classen J. Substantia nigra echogenicity: A structural correlate of functional impairment of the dopaminergic striatal projection in Parkinson’s disease. Movement Disorders. 2009/August/15 2009;24(11):1669–1675. doi: 10.1002/mds.22665 [DOI] [PubMed] [Google Scholar]
- 13.Li D-h, Zhang L-y, Hu Y-y, et al. Transcranial sonography of the substantia nigra and its correlation with DAT-SPECT in the diagnosis of Parkinson’s disease. Parkinsonism & Related Disorders. 2015/August/01/ 2015;21(8):923–928. doi: 10.1016/j.parkreldis.2015.05.024 [DOI] [PubMed] [Google Scholar]
- 14.Bor-Seng-Shu E, Pedroso JL, Felicio AC, et al. Substantia nigra echogenicity and imaging of striatal dopamine transporters in Parkinson’s disease: A cross-sectional study. Parkinsonism & Related Disorders. 2014/May/01/ 2014;20(5):477–481. doi: 10.1016/j.parkreldis.2014.01.015 [DOI] [PubMed] [Google Scholar]
- 15.Spiegel J, Hellwig D, Möllers M-O, et al. Transcranial sonography and [123I]FP-CIT SPECT disclose complementary aspects of Parkinson’s disease. Brain. 2006;129(5):1188–1193. doi: 10.1093/brain/awl042 [DOI] [PubMed] [Google Scholar]
- 16.Doepp F, Plotkin M, Siegel L, et al. Brain parenchyma sonography and 123I-FP-CIT SPECT in Parkinson’s disease and essential tremor. Movement Disorders. 2008/February/15 2008;23(3):405–410. doi: 10.1002/mds.21861 [DOI] [PubMed] [Google Scholar]
- 17.Berg D, Becker G, Zeiler B, et al. Vulnerability of the nigrostriatal system as detected by transcranial ultrasound. Neurology. Sep 22 1999;53(5):1026–31. doi: 10.1212/wnl.53.5.1026 [DOI] [PubMed] [Google Scholar]
- 18.Hagenah JM, König IR, Becker B, et al. Substantia nigra hyperechogenicity correlates with clinical status and number of Parkin mutated alleles. Journal of Neurology. 2007/October/15 2007;254(10):1407. doi: 10.1007/s00415-007-0567-y [DOI] [PubMed] [Google Scholar]
- 19.Tayebi N, Reissner KJ, Lau EK, et al. Genotypic heterogeneity and phenotypic variation among patients with type 2 Gaucher’s disease. Pediatr Res. May 1998;43(5):571–8. doi: 10.1203/00006450-199805000-00003 [DOI] [PubMed] [Google Scholar]
- 20.Sidransky E, Lopez G. The link between the GBA gene and parkinsonism. Lancet Neurol. Nov 2012;11(11):986–98. doi: 10.1016/S1474-4422(12)70190-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beutler E, Gelbart T, Scott CR. Hematologically important mutations: Gaucher disease. Blood Cells Mol Dis. Nov-Dec 2005;35(3):355–64. doi: 10.1016/j.bcmd.2005.07.005 [DOI] [PubMed] [Google Scholar]
- 22.Stone DL, Tayebi N, Orvisky E, Stubblefield B, Madike V, Sidransky E. Glucocerebrosidase gene mutations in patients with type 2 Gaucher disease. Hum Mutat. 2000;15(2):181–8. doi: 10.1002/(SICI)1098-1004(200002)15:2<181::AID-HUMU7>3.0.CO;2-S [DOI] [PubMed] [Google Scholar]
- 23.Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry. Mar 1992;55(3):181–4. doi: 10.1136/jnnp.55.3.181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Patlak CS, Blasberg RG. Graphical evaluation of blood-to-brain transfer constants from multiple-time uptake data. Generalizations. J Cereb Blood Flow Metab. Dec 1985;5(4):584–90. doi: 10.1038/jcbfm.1985.87 [DOI] [PubMed] [Google Scholar]
- 25.Walter U How to measure substantia nigra hyperechogenicity in Parkinson disease: detailed guide with video. J Ultrasound Med. Oct 2013;32(10):1837–43. doi: 10.7863/ultra.32.10.1837 [DOI] [PubMed] [Google Scholar]
- 26.Gaenslen A, Unmuth B, Godau J, et al. The specificity and sensitivity of transcranial ultrasound in the differential diagnosis of Parkinson’s disease: a prospective blinded study. Lancet Neurol. May 2008;7(5):417–24. doi: 10.1016/S1474-4422(08)70067-X [DOI] [PubMed] [Google Scholar]
- 27.Xu R, Chen G, Mao Z, Gao H, Deng Y, Tao A. Diagnostic Performance of Transcranial Sonography for Evaluating Substantia Nigra Hyper-echogenicity in Patients with Parkinson’s Disease. Ultrasound Med Biol. May 2020;46(5):1208–1215. doi: 10.1016/j.ultrasmedbio.2020.01.019 [DOI] [PubMed] [Google Scholar]
- 28.Arkadir D, Dinur T, Becker Cohen M, et al. Prodromal substantia nigra sonography undermines suggested association between substrate accumulation and the risk for GBA-related Parkinson’s disease. Eur J Neurol. Jul 2019;26(7):1013–1018. doi: 10.1111/ene.13927 [DOI] [PubMed] [Google Scholar]
- 29.Berg D, Merz B, Reiners K, Naumann M, Becker G. Five-year follow-up study of hyperechogenicity of the substantia nigra in Parkinson’s disease. Mov Disord. Mar 2005;20(3):383–5. doi: 10.1002/mds.20311 [DOI] [PubMed] [Google Scholar]
- 30.Yu SY, Cao CJ, Zuo LJ, et al. Clinical features and dysfunctions of iron metabolism in Parkinson disease patients with hyper echogenicity in substantia nigra: a cross-sectional study. BMC Neurol. Jan 17 2018;18(1):9. doi: 10.1186/s12883-018-1016-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walter U Substantia nigra hyperechogenicity is a risk marker of Parkinson’s disease: no. J Neural Transm (Vienna). Apr 2011;118(4):607–12. doi: 10.1007/s00702-010-0564-7 [DOI] [PubMed] [Google Scholar]
- 32.Gallagher CL, Oakes TR, Johnson SC, et al. Rate of 6-[18F]fluorodopa uptake decline in striatal subregions in Parkinson’s disease. Mov Disord. Mar 2011;26(4):614–20. doi: 10.1002/mds.23503 [DOI] [PMC free article] [PubMed] [Google Scholar]