

Abstract
Stimulator of interferon genes (STING) plays an important role in infection, autoimmune disease and cancer. STING-mediated type I interferon (IFN) signaling is well recognized and extensively studied. Several IFN-independent activities of STING were also discovered in recent years and their physiological importance has begun to be appreciated. Here, we review recent advance in the evolutionary origin and molecular mechanisms of STING-mediated IFN-independent activities. New insights from these studies suggest that STING functions as a hub that converts multiple environmental cues into diverse cellular responses rather than acts as a simple IFN-producing machine. This expanded view of STING biology should guide future clinical testing of STING agonists and treatment of STING-associated human diseases.
STING-mediated interferon response in infection and autoimmune disease
The Innate immune response to invading pathogens is the first line of defense in mammalian cells. STING plays a vital role in detecting cytosolic pathogens and inducing type I interferon (IFN) response. STING is a transmembrane protein localized on the endoplasmic reticulum (ER). It is classically known as the non-redundant adaptor protein downstream of the cytosolic DNA sensor cyclic GMP-AMP synthase (cGAS). When the mammalian cGAS is exposed to self or microbial DNA, it converts ATP and GTP into 2’3’ cyclic GMP-AMP (cGAMP). Then, cGAMP binds and activates STING and the IFN response. Bacterial cyclic dinucleotides (CDN) can also directly activate STING. Importantly, Sting−/− mice are highly susceptible to viral and bacterial infections, demonstrating the physiological importance of STING in host defense against microbial infection [1–5].
The cGAS-STING pathway has also been implicated in several monogenic autoimmune and autoinflammatory diseases. One classic example is the Aicardi-Goutières syndrome (AGS), which is a congenital neurological disease affecting infant and young children [6]. The best mouse model of AGS is the Trex1−/− mouse (TREX1 is a ER-resident DNase), which exhibits constitutively elevated IFN signaling due to self-DNA sensing by cGAS. Trex1−/− mice develop severe systemic autoinflammatory disease that is dependent on cGAS and STING [7–10]. Targeting the IFN pathway with JAK1/2 inhibitor baricitinib has shown clinical benefit in AGS patients [11]. Therefore, the classical paradigm of the STING-mediated IFN response has clear physiological importance in human diseases.
Evidence for IFN-independent STING function in human disease
The earliest evidence of IFN-independent STING function in human disease is Deoxyribonuclease II (DNase II)-associated polyarthritis. DNase II is a lysosomal endonuclease responsible for digestion of apoptotic cellular DNA taken up by the macrophage. The DNase2−/− mouse is embryonic lethal due to self-DNA mediated activation of the IFN response [12]. The DNase2−/− mouse lacking the type I IFN receptor (DNase2−/−Ifnr1−/−) is viable but suffers from severe early onset polyarthritis. Interestingly, further elimination of STING rescued the polyarthritis phenotype in DNase2−/−Ifnr1−/− mice, suggesting a STING-dependent but IFN-independent function in self-DNA-mediated polyarthritis [13, 14]. Gain-of-function STING mutations cause STING-associated vasculopathy with onset in infancy (SAVI) due to constitutive activation of the STING pathway [15, 16]. In SAVI mouse models, we and others recently showed that SAVI disease develops independently of IFN (also independently of cGAS and IRF3); instead, ER stress and T cell death appear to play a role [17–20]. Clinical treatment of SAVI patients with JAK1/2 inhibitors were partially successful in a subset of patients [21, 22], and the role of IFN-independent activities of STING in humans requires further investigation. The most surprising finding comes from HSV-1 infection. Both STING and IFN are essential for host restriction of HSV-1 infection, but the STING-mediated IFN response is not. Using the StingS365A mouse that carries a point mutation that selectively disrupts the IFN activity of STING, three research groups including ours independently found that STING-mediated IFN-independent activity plays a dominant role in controlling HSV-1 infection [23–25]. The nature of IFN-independent antiviral activity of STING remains unclear.
Other STING-associated human diseases where the role of IFN is unknown
STING has been implicated in several human diseases where an elevated IFN response is observed but whether IFN drives the disease is not known. Stromal Interaction Molecule 1 (STIM1) is an ER retention factor for STING. STIM1 loss-of-function results in constitutive STING trafficking and IFN signaling that are consistent with autoimmune complications observed in STIM1 patients [26, 27]. Whether the STING-mediated IFN response mediates disease pathology in Stim1 deficient mice or STIM1 patients remains to be shown. Loss of COP-I vesicle trafficking also activates STING signaling due to accumulation of STING on the Golgi, which is associated with COPA syndrome [28–30]. Interestingly, clinical presentations of SAVI and COPA disease are highly similar; both are characterized by inflammatory lung disease [31]. We recently showed that STING signaling mediates an early onset neurodegenetive disease Niemann-Pick disease type C (NPC) [32]. Loss of function of the lysosomal protein NPC1 results in activation of STING signaling, increased expression of IFN-stimulated genes (ISGs) in microglia and death of Purkinje neurons [32]. In addition, STING has been implicated in several other neurodegenerative diseases in mice, such as Parkinson’s disease and amyotrophic lateral sclerosis (ALS) [33, 34]. The role of IFN signaling in STING-mediated neurological disease remains unknown.
Evolutionary evidence for IFN-independent activities of STING
STING protein is conserved throughout evolution including many species that do not have the IFN system [35, 36]. STING proteins from most species contain an N-terminal multi-pass transmembrane (TM) domain and a C-terminal CDN-binding domain (CBD) (Figure 1). The C-terminal tail (CTT), which is required for TBK1 binding and IFN signaling, is only present in mammals and certain vertebrate species such as Danio rerio. High conservation of the CBD suggests a common mechanism for STING ligand binding and activation whereas low conservation of the CTT suggests divergent signaling activities amongst various species.
Figure 1. The evolution of STING protein.
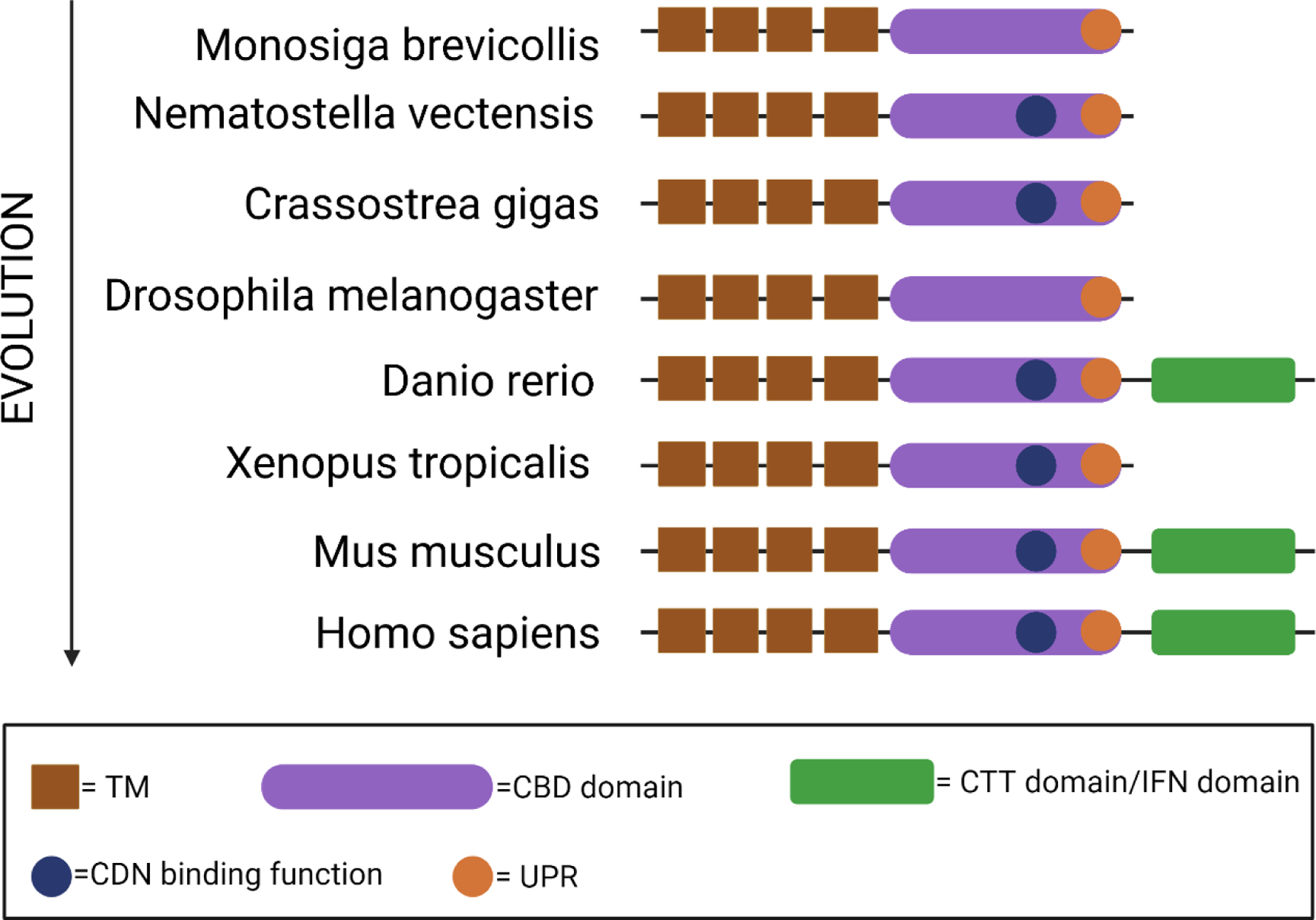
The evolution of STING protein homologs from different species [35, 36] is illustrated by their evolutionary hierarchy. Conserved structure domains and motifs are represented with a diagram on the right. TM, transmembrane; CBD: cyclic dinucleotide binding domain; CDN, cyclic dinucleotide, CTT, C-terminal domain; UPR, unfolded protein response.
The most ancient species expressing STING is starlet sea anemone Nematostella vectensis, living over 600 million years ago [37, 38]. The crystal structure of Nematostella STING (nvSTING) is similar to that of human STING (hSTING), despite only 29% protein sequence homology between the two proteins. Both hSTING and nvSTING bind 2’3’-cGAMP and both induce a similar conformational change. nvSTING does not have the CTT and Nematostella does not have an IFN system. 2’3’-cGAMP induces expression of anti-viral genes that are homologs of vertebrate ISGs, indicating that the IFN response predates the evolution of IFN [39]. 2’3’-cGAMP also induces expression of anti-bacterial genes through NF-κB, which is a main branch of IFN-independent activity of STING.
STING trafficking
A unique feature of STING signaling is the obligatory trafficking of STING protein through the secretory pathway. After ligand binding, STING undergoes a drastic conformational change that is believed to be the trigger for ER-exit. Then, STING translocates to the ER-Golgi intermediate compartment (ERGIC) and the Golgi, where it recruits kinase TBK1 and transcription factor IRF3. TBK1 phosphorylates itself, STING and IRF3. Phosphorylated IRF3 translocates to the nucleus and activates expression of IFN and IFN-stimulated genes (ISG) [40]. Although STING activation occurs on the Golgi, it does not dwell on the Golgi. Instead, STING rapidly moves pass the Golgi to the lysosome where it is degraded. ER-exit is a critical checkpoint for turning on STING signaling, and lysosomal degradation turns off STING signaling [16, 41]. STING trafficking is an incredibly rapid and dynamic process. For example, in fibroblasts stimulated with a STING agonist, nearly all STING protein moves from the ER, across the Golgi, to the lysosome within a few hours, resulting in a spike of IFN signaling and nearly complete degradation of STING protein in the cell.
STING trafficking allows more dynamic control of an otherwise linear signaling cascade (Figure 2). This feature is likely a double-edged sword, which allows more precise control of the cGAS-STING pathway but also is vulnerable for microbial antagonism. Many regulators of STING signaling are in fact regulators of STING trafficking. As mentioned previously, the ER calcium sensor STIM1 moonlights as an ER retention factor that keeps STING on the ER [27, 42]. Inactive rhomboid protein 2 (iRhom2) recruits the translocon-associated protein (TRAPβ) to STING to facilitate ER-to-Golgi trafficking [43]. YIPF5 binds to STING and facilitate STING sorting into the COP-II vesicle for translocation [44]. Cytoplasmic coat protein complex II (COP-II) and ADP-ribosylation factor (ARF) GTPases mediate STING ER-to-Golgi trafficking [45]. After arriving on the Golgi, STING is palmitoylated which is required for oligomerization and IFN activation [46]. Defective retrograde COP-I vesicle trafficking in COPA syndrome causes STING to accumulate on the Golgi leading to overactivation of STING signaling [28–30].
Figure 2. STING-mediated IFN-independent activities.
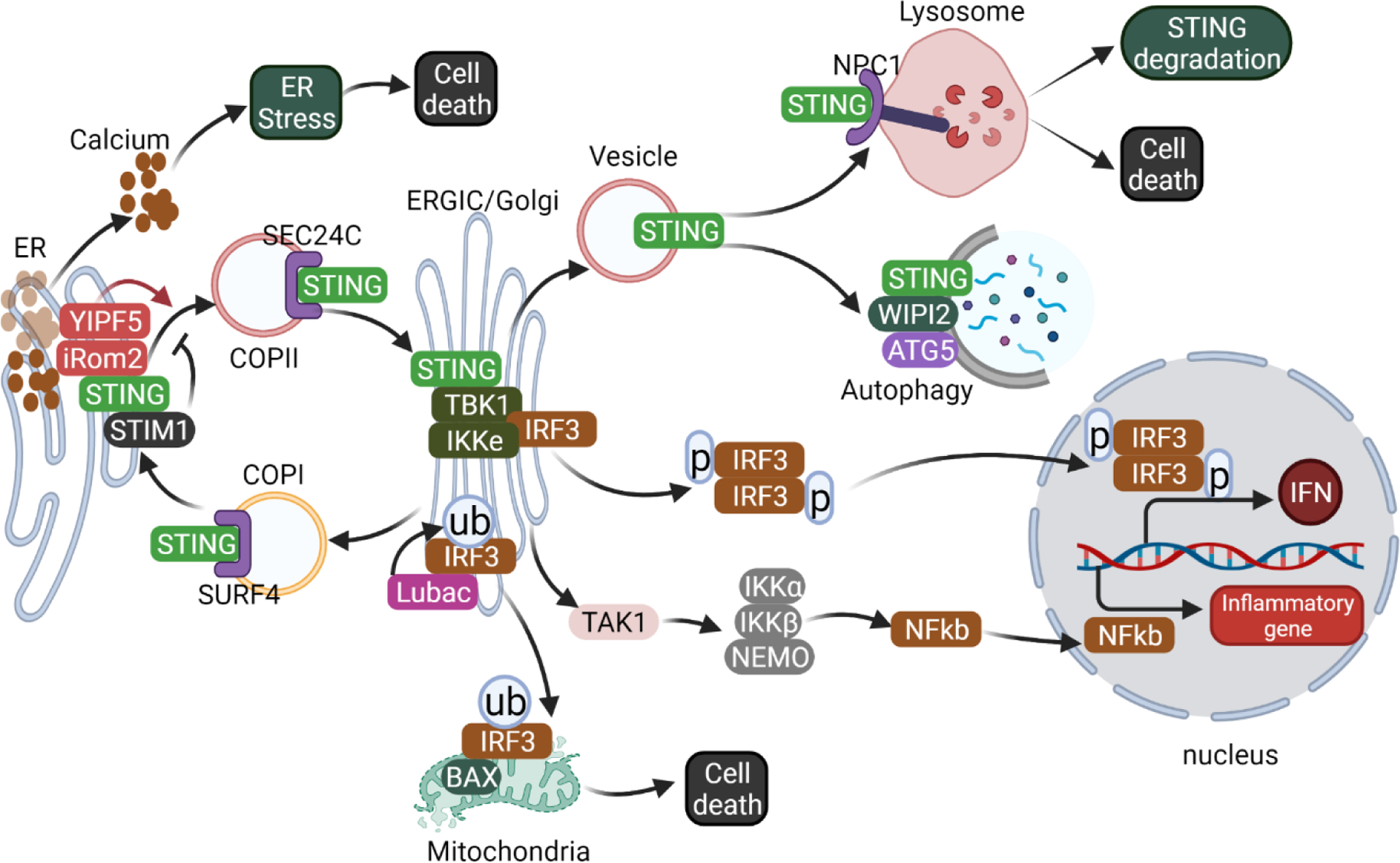
STING localized to the ER through retention by the ER calcium sensor STIM1 [27]. Upon activation, STING is recruited to COP-II vesicles to exit the ER in a process that is regulated by many cofactors such as iROM2, YIPF5, SEC24C [43, 44]. STING ER-exit triggers ER calcium efflux, inducing ER stress and the UPR, which can further lead to cell death in T cells [18]. Upon arrival on the Golgi, STING forms a complex with TBK1, IKKε and IRF3, leading to activation of downstream IFN and NF-κb signaling pathways [67]. STING is also transported from the Golgi back to the ER through COP-I vesicles regulated by COPA and SURF4 [28, 73]. IRF3 can also be ubiquitinated by LUBAC, then translocates to mitochondria and induces Bax-mediated cell death [58]. STING can initiated autophagy from the ERGIC membrane in an non-classical process that is dependent on WIPI2 and ATG5 but not ULK1 or Beclin1 [45]. Post-Golgi STING vesicles are recruited to the lysosome by NPC1, promoting STING degradation [32]. STING trafficking to the lysosome can also induce lysosome rupture-mediated cell death in human myeloid cells [74].
STING trafficking from the Golgi to lysosomes is much less understood. Since this segment of trafficking down-regulates STING signaling, loss of function of a cofactor would increase STING signaling, which is usually more difficult to capture than decreased IFN response in in vitro assays. We showed that inhibiting lysosome with Bafilomycin A1 increases STING-mediated IFN response and anti-tumor immunity [41]. We further identified lysosomal protein NPC1 as a key cofactor that recruits STING to the lysosome for degradation [32, 47]. Whether STING vesicles fuse with or are engulfed by lysosomes remains unclear. Additional mediators on both sides of the membrane interface likely also exist and remain to be determined.
STING-mediated autophagy
HSV-1 infection or exogenous DNA stimulation directly activates cGAS- and STING-dependent autophagy in mammals. This promotes the degradation of viral DNA as an alternative antiviral response in additional to the IFN response [48]. STING-mediated autophagy is mechanistically distinct from conventional autophagy, in that it does not require ULK1 and Beclin1. Instead, STING directly recruits LC3 to the ERGIC membrane and promotes LC3 lipidation as well as autophagosome formation through WIPI2 and ATG5 [45]. STING contains a LC3 interacting region (LIR) and directly binds to LC3 to drive autophagy [49]. In addition to viral infection, STING:LC3 interaction and autophagy are also important for host defense against bacterial infection such as Mycobacterium tuberculosis (Mtb)[3–5].
Autophagy is likely one of the most primordial activities of STING. Both Nematostella vectensis STING and choanoflagellate Monosiga brevicollis STING induce autophagy in response to stimulation by cGAMP [45, 50]. The physiological function of STING-mediated autophagy in these organisms has not been fully demonstrated yet, largely due to lack of molecular tools for these species. Drosophila STING also lacks the CTT required for IFN signaling, and STING-mediated autophagy protects Drosophila against Zika virus infection in the brain [51].
STING-mediated autophagy in mammals does not require recruitment of TBK1, IRF3 or IFN signaling. This is recently demonstrated in vivo using genetically engineered STING mutant mice. StingS365A blocks IRF3 binding and StingL373A blocks TBK1 binding; both mutants are inactive for IFN signaling but active for autophagy [23, 24, 52]. StingΔCTT lacks the entire C-terminal tail and can still induce autophagy, albeit at a lower level compared to wild-type STING [23, 24].
STING-mediated T cell death and UPR
STING-mediated cell death was first reported in malignant B cells, where STING agonist treatment induces mitochondria-mediated B cell apoptosis [53]. STING agonist treatment also induces T cell death [54]. The underlying mechanism of STING-mediated T cell death is not completely understood. IRF3 and p53 each plays a partial role [55]. Interestingly, the level of STING protein expression in T cells is much higher than that in macrophages, even though macrophages have been the benchmark for the majority of STING signaling studies. STING agonist stimulation of myeloid cells does not induce cell death [52, 55]. Although, viral infection could invoke STING-dependent cell death in monocytes. For example, human T cell leukemia virus type 1 (HTLV-1) infection of monocytes activates STING-TBK1-IRF3 signaling; IRF3 then translocates to mitochondria and induces Bax-mediated apoptosis [56]. STING can also induce lysosomal cell death. In human myeloid cells, STING trafficking to the lysosome causes membrane permeabilization and LCD; this can further lead to potassium efflux and activation of NLRP3-mediated pyroptosis [57].
The STING-mediated cell death mechanism is likely cell type- and context-dependent. It remains an open question how STING balances inflammation and cell death when the cell is facing different external threats. One possibility is through distinct post-translational modifications (PTM) of IRF3. IRF3 phosphorylation by TBK1 promotes nuclear translocation and activation of IFN signaling. IRF3 ubiquitination by linear ubiquitin chain assembly complex (LUBAC) promotes interaction with BAX, localization to mitochondria and induction of apoptosis [58]. Other mechanisms and contextual ques are also likely and remain to be uncovered.
The physiological importance of STING-mediated cell death, at least in T cells, was shown in the gain-of-function StingN153S mutant mouse. StingN153S mice suffer from T cell cytopenia, a condition that is genetically independent of cGAS, IRF3 or IFNAR [17, 19, 59]. The N153S mutation (N154S in human STING) causes STING to constitutively translocate from the ER to Golgi, resulting in altered ER calcium homeostasis as well as elevated ER stress and unfolded protein response (UPR). This new activity of STING was mapped to the α-helix between amino acid 322 and 343, which is a highly evolutionarily conserved motif located at the C-terminal end of the ligand binding domain CBD (we named it the UPR motif) [18]. Key residues in this motif are also required for STING-mediated autophagy [45]. Therefore, this motif could mediate multiple IFN-independent activities of STING.
STING-mediated T cell death also has important implications in anti-tumor immunity. Tumor infiltrating T cells (TILs) undergo cell death through multiple pathways, including the STING pathway [52]. Many tumors secrete cGAMP to the extracellular space [60]. Thus, it is possible that tumor-secreted cGAMP can be taken up by surrounding T cells and induce T cell death. This could be another way for tumors to antagonize the T cell response that would be analogous to the PD-1/PD-L1 checkpoint mechanism.
An interesting conundrum arises with using STING agonist to treat tumors. STING agonist treatment induces favorable anti-tumor response through activation of dendritic cells (DCs) and cross-priming of T cells in multiple pre-clinical tumor models [61, 62]. However, when administered at a higher dose, STING agonist causes substantial unintended T cell death that compromises anti-tumor immunity [63]. As more STING agonists go into human clinical trials, dosing optimization to balance the pros (e.g. IFN response, DC:T cell priming) and the cons (e.g. T cell death, tissue toxicity) of STING activation should be considered.
STING-mediated NF-κB signaling
STING-mediated NF-κB signaling was recognized as soon as mammalian STING was discovered [64, 65]. This signaling activity is evolutionarily conserved, and even nvSTING induces anti-bacterial gene expression via NF-κB [39]. TBK1 appears to be required for STING-mediated NF-κB signaling in mouse embryonic fibroblasts (MEFs) [66] but is functionally redundant with IKKε in primary macrophages [67]. The StingL374A mouse carrying a point mutation that disrupts TBK1 binding fails to activate NF-κB signaling [68], supporting a role for TBK1. StingS365A disrupts STING-mediated IFN but not NF-κB signaling in mice, suggesting that NF-κB signaling occurs independently of IFN.
NF-κB signaling promotes inflammation, which is commonly observed in STING-associated diseases. STING-mediated NF-κB signaling also has anti-viral and anti-tumor activities that are traditionally attributed to IFN. For example, both wild-type and StingS365A mice can successfully control HSV-1 infection while both Sting−/− and StingL373A mice fail to do so. The only known difference between StingS365A and StingL373A is that S365A induces NF-κB and L373A does not [23, 24, 52]. Moreover, StingS365A, but not StingL373A, can promote a cGAMP-mediated anti-tumor response against Lewis lung carcinoma (LL2) in mice, demonstrating that STING-mediated NF-κB signaling also has anti-tumor activity [24]. Taken together, although IFN and NF-κB are two independent branches of STING signaling, they are also intertwined and contribute to similar physiological responses in disease. Therefore, an important consideration in treating STING-mediated inflammatory disease is to distinguish contributions of IFN and NF-κB activities, then target each pathway or both accordingly.
Other IFN-independent activities of STING
Anti-proliferation.
STING-mediated anti-tumor immunity through activating DC:T cell cross priming is well recognized. STING also has an anti-proliferation activity intrinsic to cancer cells. For example, STING gene deletion in several human and mouse cancer cell lines results in increased proliferation in vitro and increased tumor growth in nude mice [69]. The underlying mechanism involves NF-κB- and p53-driven activation of p21. Cancer cells lacking STING prematurely enter the S-phase of the cell cycle, induce activation of CDK1 and increase chromosome instability. This tumor intrinsic suppressive activity of STING is consistent with widespread downregulation of STING expression in many type of cancers.
Translational repression.
Although RNA does not directly activate mammalian cGAS or STING, several RNA viruses activate STING signaling during infection through indirect mechanisms such as membrane fusion and mitochondrial damage [70, 71]. Sting-deficient cells and mice are also more susceptible to RNA virus infection [65, 72]. One mechanism for STING-mediated restriction of RNA virus infection is through translational repression [72]. Viral RNA is sensed by cytosolic RNA sensor RIG-I, which then signals to STING independently of MAVS. This RIG-I-STING axis inhibits translational initiation of viral and host proteins, representing a unique crosstalk between cytosolic DNA and RNA sensing pathways during host defense.
Other uncharacterized pathways.
Whole transcriptome analysis comparing STING signaling activities in wild-type, Sting−/− and StingS365A mice uncovered many IFN-independent activities of STING in different cell types [52]. For example, in macrophages, STING activates antigen presentation, MAPK signaling and HMGB1 signaling, all independently of IFN signaling. Most STING activities in T cells are IFN-independent, including TH17 signaling, NFAT signaling, TCR pathway and others. How STING mediates these activities remain uncharacterized and their physiological relevance requires further investigation.
Future Perspectives
The classical paradigm of STING activation by cGAMP followed by induction of type I IFN response is clearly important for infection, autoimmune disease and anti-tumor immunity. However, STING function extends beyond IFN, and several IFN-independent activities are clearly physiologically important. Many of these new STING functions came from studies of non-mammalian species or non-myeloid cell types in mammals. As the exploration of STING biology widens in the field, a new paradigm emerges where STING can be activated by multiple environmental cues and that STING mediates many cellular responses. An increasingly diverse array of human diseases is being associated with STING, ranging from autoimmune and autoinflammatory diseases to neurodegenerative diseases and cancer. Depending on which STING activity is engaged in a particular disease, solely targeting the IFN pathway may not be sufficient (or effective at all) for treating that disease. As we begin to appreciate the evolutionary origin of STING and its many functions, the perception of STING signaling is shifting from ‘a simple IFN machine’ to ‘a multi-functional hub’ that converts different environmental cues into diverse cellular responses.
Research highlights:
STING is an evolutionarily conserved protein that activates signaling beyond IFN.
STING trafficking is a unique feature that is dynamically regulated and is associated with STING-mediated IFN-independent activities.
STING-mediated IFN-independent activities, such autophagy, UPR and cell death, are physiologically important in various diseases.
Acknowledgments
Research in the Wu and Yan labs are supported by grants from NIH (AI161708 to N.Y.) and start-up funds from Cleveland Clinic (J.W.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Reference:
- [1].Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Zhang Y, Yeruva L, Marinov A, Prantner D, Wyrick PB, Lupashin V, et al. The DNA sensor, cyclic GMP-AMP synthase, is essential for induction of IFN-beta during Chlamydia trachomatis infection. J Immunol. 2014;193:2394–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Collins AC, Cai H, Li T, Franco LH, Li XD, Nair VR, et al. Cyclic GMP-AMP Synthase Is an Innate Immune DNA Sensor for Mycobacterium tuberculosis. Cell Host Microbe. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wassermann R, Gulen MF, Sala C, Perin SG, Lou Y, Rybniker J, et al. Mycobacterium tuberculosis Differentially Activates cGAS- and Inflammasome-Dependent Intracellular Immune Responses through ESX-1. Cell Host Microbe. 2015. [DOI] [PubMed] [Google Scholar]
- [5].Watson RO, Bell SL, MacDuff DA, Kimmey JM, Diner EJ, Olivas J, et al. The Cytosolic Sensor cGAS Detects Mycobacterium tuberculosis DNA to Induce Type I Interferons and Activate Autophagy. Cell Host Microbe. 2015;17:811–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Motwani M, Pesiridis S, Fitzgerald KA. DNA sensing by the cGAS–STING pathway in health and disease. Nature Reviews Genetics. 2019:1–18. [DOI] [PubMed] [Google Scholar]
- [7].Mackenzie KJ, Carroll P, Lettice L, Tarnauskaite Z, Reddy K, Dix F, et al. Ribonuclease H2 mutations induce a cGAS/STING-dependent innate immune response. EMBO J. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Pokatayev V, Hasin N, Chon H, Cerritelli SM, Sakhuja K, Ward JM, et al. RNase H2 catalytic core Aicardi-Goutieres syndrome-related mutant invokes cGAS-STING innate immune-sensing pathway in mice. J Exp Med. 2016;213:329–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gao D, Li T, Li XD, Chen X, Li QZ, Wight-Carter M, et al. Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. Proc Natl Acad Sci U S A. 2015;112:E5699–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gray EE, Treuting PM, Woodward JJ, Stetson DB. Cutting Edge: cGAS Is Required for Lethal Autoimmune Disease in the Trex1-Deficient Mouse Model of Aicardi-Goutieres Syndrome. J Immunol. 2015;195:1939–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Vanderver A, Adang L, Gavazzi F, McDonald K, Helman G, Frank DB, et al. Janus Kinase Inhibition in the Aicardi–Goutières Syndrome. New England Journal of Medicine. 2020;383:986–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Yoshida H, Okabe Y, Kawane K, Fukuyama H, Nagata S. Lethal anemia caused by interferon-beta produced in mouse embryos carrying undigested DNA. Nat Immunol. 2005;6:49–56. [DOI] [PubMed] [Google Scholar]
- [13].Baum R, Sharma S, Carpenter S, Li QZ, Busto P, Fitzgerald KA, et al. Cutting edge: AIM2 and endosomal TLRs differentially regulate arthritis and autoantibody production in DNase II-deficient mice. J Immunol. 2015;194:873–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ahn J, Gutman D, Saijo S, Barber GN. STING manifests self DNA-dependent inflammatory disease. Proc Natl Acad Sci U S A. 2012;109:19386–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Sanchez GAM, et al. Activated STING in a vascular and pulmonary syndrome. The New England journal of medicine. 2014;371:507 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Dobbs N, Burnaevskiy N, Chen D, Gonugunta VK, Alto NM, Yan N. STING Activation by Translocation from the ER Is Associated with Infection and Autoinflammatory Disease. Cell Host Microbe. 2015;18:157–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Warner JD, Irizarry-Caro RA, Bennion BG, Ai TL, Smith AM, Miner CA, et al. STING-associated vasculopathy develops independently of IRF3 in mice. J Exp Med. 2017;214:3279–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wu J, Chen YJ, Dobbs N, Sakai T, Liou J, Miner JJ, et al. STING-mediated disruption of calcium homeostasis chronically activates ER stress and primes T cell death. J Exp Med. 2019;216:867–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bouis D, Kirstetter P, Arbogast F, Lamon D, Delgado V, Jung S, et al. Severe combined immunodeficiency in stimulator of interferon genes (STING) V154M/wild-type mice. J Allergy Clin Immunol. 2019;143:712–25 e5. [DOI] [PubMed] [Google Scholar]
- [20].Motwani M, Pawaria S, Bernier J, Moses S, Henry K, Fang T, et al. Hierarchy of clinical manifestations in SAVI N153S and V154M mouse models. Proc Natl Acad Sci U S A. 2019;116:7941–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Volpi S, Insalaco A, Caorsi R, Santori E, Messia V, Sacco O, et al. Efficacy and Adverse Events During Janus Kinase Inhibitor Treatment of SAVI Syndrome. J Clin Immunol. 2019;39:476–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Banday AZ, Jindal AK, Singh S. Correspondence on ‘Points to consider for the treatment of immune-mediated inflammatory diseases with Janus kinase inhibitors: a consensus statement’. Ann Rheum Dis. 2021. [DOI] [PubMed] [Google Scholar]
- [23].Yamashiro LH, Wilson SC, Morrison HM, Karalis V, Chung JJ, Chen KJ, et al. Interferon-independent STING signaling promotes resistance to HSV-1 in vivo. Nat Commun. 2020;11:3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Yum S, Li M, Fang Y, Chen ZJ. TBK1 recruitment to STING activates both IRF3 and NF-kappaB that mediate immune defense against tumors and viral infections. Proc Natl Acad Sci U S A. 2021;118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wu JJ, Li W, Shao Y, Avey D, Fu B, Gillen J, et al. Inhibition of cGAS DNA Sensing by a Herpesvirus Virion Protein. Cell Host Microbe. 2015;18:333–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lacruz RS, Feske S. Diseases caused by mutations in ORAI1 and STIM1. Ann N Y Acad Sci. 2015;1356:45–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Srikanth S, Woo JS, Wu B, El-Sherbiny YM, Leung J, Chupradit K, et al. The Ca(2+) sensor STIM1 regulates the type I interferon response by retaining the signaling adaptor STING at the endoplasmic reticulum. Nat Immunol. 2019;20:152–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Deng Z, Chong Z, Law CS, Mukai K, Ho FO, Martinu T, et al. A defect in COPI-mediated transport of STING causes immune dysregulation in COPA syndrome. J Exp Med. 2020;217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Mukai K, Ogawa E, Uematsu R, Kuchitsu Y, Uemura T, Waguri S, et al. Homeostatic regulation of STING by Golgi-to-ER membrane traffic. bioRxiv. 2020:2020.05.20.107664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lepelley A, Martin-Niclós MJ, Bihan ML, Marsh JA, Uggenti C, Rice GI, et al. Mutations in COPA lead to abnormal trafficking of STING to the Golgi and interferon signaling. The Journal of experimental medicine. 2020;217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Frémond M-L, Crow YJ. STING-Mediated Lung Inflammation and Beyond. Journal of Clinical Immunology. 2021:1–14. [DOI] [PubMed] [Google Scholar]
- [32].Chu T-T, Tu X, Yang K, Wu J, Repa JJ, Yan N. Tonic prime-boost of STING signalling mediates Niemann–Pick disease type C. Nature. 2021:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Sliter DA, Martinez J, Hao L, Chen X, Sun N, Fischer TD, et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature. 2018;302:89 262. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [34].Yu C-H, Davidson S, Harapas CR, Hilton JB, Mlodzianoski MJ, Laohamonthonkul P, et al. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wu X, Wu FH, Wang X, Wang L, Siedow JN, Zhang W, et al. Molecular evolutionary and structural analysis of the cytosolic DNA sensor cGAS and STING. Nucleic Acids Res. 2014;42:8243–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Margolis SR, Wilson SC, Vance RE. Evolutionary Origins of cGAS-STING Signaling. Trends Immunol. 2017;38:733–43. [DOI] [PubMed] [Google Scholar]
- [37].Kranzusch PJ, Wilson SC, Lee AS, Berger JM, Doudna JA, Vance RE. Ancient Origin of cGAS-STING Reveals Mechanism of Universal 2’,3’ cGAMP Signaling. Mol Cell. 2015;59:891–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Putnam NH, Srivastava M, Hellsten U, Dirks B, Chapman J, Salamov A, et al. Sea anemone genome reveals ancestral eumetazoan gene repertoire and genomic organization. Science. 2007;317:86–94. [DOI] [PubMed] [Google Scholar]
- [39].Margolis SR, Dietzen PA, Hayes BM, Wilson SC, Remick BC, Chou S, et al. The STING ligand 2′3′-cGAMP induces an NF-κB-dependent anti-bacterial innate immune response in the starlet sea anemone Nematostella vectensis. BioRxiv. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science. 2015;347:aaa2630. [DOI] [PubMed] [Google Scholar]
- [41].Gonugunta VK, Sakai T, Pokatayev V, Yang K, Wu J, Dobbs N, et al. Trafficking-Mediated STING Degradation Requires Sorting to Acidified Endolysosomes and Can Be Targeted to Enhance Anti-tumor Response. Cell Rep. 2017;21:3234–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wu J, Yan N. STIM1 moonlights as an anchor for STING. Nat Immunol. 2019;20:112–4. [DOI] [PubMed] [Google Scholar]
- [43].Luo WW, Li S, Li C, Lian H, Yang Q, Zhong B, et al. iRhom2 is essential for innate immunity to DNA viruses by mediating trafficking and stability of the adaptor STING. Nat Immunol. 2016. [DOI] [PubMed] [Google Scholar]
- [44].Ran Y, Xiong MG, Xu ZS, Luo WW, Wang SY, Wang YY. YIPF5 Is Essential for Innate Immunity to DNA Virus and Facilitates COPII-Dependent STING Trafficking. J Immunol. 2019;203:1560–70. [DOI] [PubMed] [Google Scholar]
- [45].Gui X, Yang H, Li T, Tan X, Shi P, Li M, et al. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature. 2019;567:262–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Mukai K, Konno H, Akiba T, Uemura T, Waguri S, Kobayashi T, et al. Activation of STING requires palmitoylation at the Golgi. Nat Commun. 2016;7:11932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Chu T-TT Xintao; Yang Kun; Wu Jianjun; Repa Joyce J; Yan Nan. Tonic Prime-boost of STING signalling mediates Niemann-Pick disease type C. Nature. 2021;in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Liang Q, Seo GJ, Choi YJ, Kwak MJ, Ge J, Rodgers MA, et al. Crosstalk between the cGAS DNA sensor and Beclin-1 autophagy protein shapes innate antimicrobial immune responses. Cell Host Microbe. 2014;15:228–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Liu D, Wu H, Wang C, Li Y, Tian H, Siraj S, et al. STING directly activates autophagy to tune the innate immune response. Cell Death Differ. 2019;26:1735–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Woznica A, Kumar A, Sturge CR, Xing C, King N, Pfeiffer JK. STING mediates immune responses in a unicellular choanoflagellate. [DOI] [PMC free article] [PubMed]
- [51].Liu Y, Gordesky-Gold B, Leney-Greene M, Weinbren NL, Tudor M, Cherry S. Inflammation-Induced, STING-Dependent Autophagy Restricts Zika Virus Infection in the Drosophila Brain. Cell Host Microbe. 2018;24:57–68 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Wu J, Dobbs N, Yang K, Yan N. Interferon-Independent Activities of Mammalian STING Mediate Antiviral Response and Tumor Immune Evasion. Immunity. 2020;53:115–26 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Tang CH, Zundell JA, Ranatunga S, Lin C, Nefedova Y, Del Valle JR, et al. Agonist-Mediated Activation of STING Induces Apoptosis in Malignant B Cells. Cancer Res. 2016;76:2137–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Larkin B, Ilyukha V, Sorokin M, Buzdin A, Vannier E, Poltorak A. Cutting Edge: Activation of STING in T Cells Induces Type I IFN Responses and Cell Death. J Immunol. 2017;199:397–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Gulen MF, Koch U, Haag SM, Schuler F, Apetoh L, Villunger A, et al. Signalling strength determines proapoptotic functions of STING. Nat Commun. 2017;8:427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Sze A, Belgnaoui SM, Olagnier D, Lin R, Hiscott J, van Grevenynghe J. Host restriction factor SAMHD1 limits human T cell leukemia virus type 1 infection of monocytes via STING-mediated apoptosis. Cell Host Microbe. 2013;14:422–34. [DOI] [PubMed] [Google Scholar]
- [57].Gaidt MM, Ebert TS, Chauhan D, Ramshorn K, Pinci F, Zuber S, et al. The DNA Inflammasome in Human Myeloid Cells Is Initiated by a STING-Cell Death Program Upstream of NLRP3. Cell. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Chattopadhyay S, Kuzmanovic T, Zhang Y, Wetzel JL, Sen GC. Ubiquitination of the Transcription Factor IRF-3 Activates RIPA, the Apoptotic Pathway that Protects Mice from Viral Pathogenesis. Immunity. 2016;44:1151–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Motwani M, Pawaria S, Bernier J, Moses S, Henry K, Fang T, et al. Hierarchy of clinical manifestations in SAVI N153S and V154M mouse models. Proceedings of the National Academy of Sciences of the United States of America. 2019;74:201818281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Carozza JA, Böhnert V, Nguyen KC, Skariah G, Shaw KE, Brown JA, et al. Extracellular cGAMP is a cancer-cell-produced immunotransmitter involved in radiation-induced anticancer immunity. Nature Cancer. 2020;1:184–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Wang H, Hu S, Chen X, Shi H, Chen C, Sun L, et al. cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc Natl Acad Sci U S A. 2017;114:1637–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Sivick KE, Desbien AL, Glickman LH, Reiner GL, Corrales L, Surh NH, et al. Magnitude of Therapeutic STING Activation Determines CD8(+) T Cell-Mediated Anti-tumor Immunity. Cell Rep. 2019;29:785–9. [DOI] [PubMed] [Google Scholar]
- [63].Sivick KE, Desbien AL, Glickman LH, Reiner GL, Corrales L, Surh NH, et al. Magnitude of Therapeutic STING Activation Determines CD8(+) T Cell-Mediated Anti-tumor Immunity. Cell Rep. 2018;25:3074–85 e5. [DOI] [PubMed] [Google Scholar]
- [64].Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Abe T, Barber GN. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-kappaB activation through TBK1. J Virol. 2014;88:5328–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Balka KR, Louis C, Saunders TL, Smith AM, Calleja DJ, D’Silva DB, et al. TBK1 and IKKepsilon Act Redundantly to Mediate STING-Induced NF-kappaB Responses in Myeloid Cells. Cell Rep. 2020;31:107492. [DOI] [PubMed] [Google Scholar]
- [68].Zhao B, Du F, Xu P, Shu C, Sankaran B, Bell SL, et al. A conserved PLPLRT/SD motif of STING mediates the recruitment and activation of TBK1. Nature. 2019;569:718–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Ranoa DRE, Widau RC, Mallon S, Parekh AD, Nicolae CM, Huang X, et al. STING Promotes Homeostasis via Regulation of Cell Proliferation and Chromosomal Stability. Cancer Res. 2019;79:1465–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Aguirre S, Luthra P, Sanchez-Aparicio MT, Maestre AM, Patel J, Lamothe F, et al. Dengue virus NS2B protein targets cGAS for degradation and prevents mitochondrial DNA sensing during infection. Nat Microbiol. 2017;2:17037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Holm CK, Jensen SB, Jakobsen MR, Cheshenko N, Horan KA, Moeller HB, et al. Virus-cell fusion as a trigger of innate immunity dependent on the adaptor STING. Nat Immunol. 2012;13:737–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Franz KM, Neidermyer WJ, Tan YJ, Whelan SPJ, Kagan JC. STING-dependent translation inhibition restricts RNA virus replication. Proc Natl Acad Sci U S A. 2018;115:E2058–E67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Mukai K, Ogawa E, Uematsu R, Kuchitsu Y, Kiku F, Uemura T, et al. Homeostatic regulation of STING by retrograde membrane traffic to the ER. Nat Commun. 2021;12:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Gaidt MM, Ebert TS, Chauhan D, Ramshorn K, Pinci F, Zuber S, et al. The DNA Inflammasome in Human Myeloid Cells Is Initiated by a STING-Cell Death Program Upstream of NLRP3. Cell. 2017;171:1110–24 e18. [DOI] [PMC free article] [PubMed] [Google Scholar]