

Abstract
Introduction.
Tumor- and bone-derived soluble factors have been proposed to participate in the alterations of skeletal muscle size and function in cachexia. We previously showed that mice bearing ovarian cancer (OvCa) exhibit cachexia associated with marked bone loss, whereas bone-targeting agents, such as bisphosphonates, are able to preserve muscle mass in animals exposed to anticancer drugs.
Material and Methods.
De-identified CT images and plasma samples from female patients affected with OvCa were used for body composition assessment and quantification of circulating CTX-I and NFkB ligand RANKL, respectively. Female mice bearing ES-2 tumors were used to characterize cancer- and RANKL-associated effects on muscle and bone. Murine C2C12 and human HSMM myotube cultures were used to determine the OvCa- and RANKL-dependent effects on myofiber size.
Results.
To the extent of isolating new regulators of bone and muscle in cachexia, here we demonstrate that subjects affected with OvCa display evidence of cachexia and increased bone turnover. Similarly, mice carrying OvCa present high RANKL levels. By using in vitro and in vivo experimental models, we found that elevated circulating RANKL is sufficient to cause skeletal muscle atrophy and bone resorption, whereas bone preservation by means of antiresorptive and anti-RANKL treatments concurrently benefits muscle mass and function in cancer cachexia.
Conclusions.
Altogether, our data contribute to identify RANKL as a novel therapeutic target for the treatment of musculoskeletal complications associated with RANKL-expressing non-metastatic cancers.
Keywords: muscle, bone, cachexia, cancer, RANKL, antiresorptive treatments
Graphical Abstract
Introduction
Ovarian cancer (OvCa) represents one of the deadliest gynecologic malignancies (1). Among the most relevant complications of OvCa, accumulation of ascites and development of cachexia contribute to poorer outcomes and increased mortality (2). Advanced OvCa patients often present with loss of body weight, musculoskeletal alteration and reduced response to anticancer treatment, which are all symptoms of cachexia (3,4). It is estimated that musculoskeletal complications affect up to 80% of patients diagnosed with cancer and have been reported to persist for months or years after cancer remission, leading to dramatically decreased quality of life among survivors (5,6). Unfortunately, cachexia cannot be rescued by conventional nutritional support and no cure is available for the cancer-related muscle and bone deficits (7).
Muscle and bone have been shown to cooperate in an endocrine-dependent manner by exchanging myokines and osteokines, which can affect distal organs, including brain, gut, liver and fat (8). Along this line, evidence has emerged that next to their endocrine function, myokines also exert paracrine and autocrine regulatory effects (9). Similarly, Waning et al. have shown that release of transforming growth factor (TGF) β from the bone as a result of bone destruction induced by breast cancer metastases can affect muscle mass by triggering muscle weakness, which is abrogated by treatment with zoledronic acid (ZA), a bisphosphonate and bone-targeting antiresorptive agent (10,11). Our group has previously reported bone loss even in the absence of bone metastases (12,13), as well as following administration of chemotherapy drugs (14,15). Notably, ZA administration in combination with anticancer drugs not only completely preserved bone mass, but also improved skeletal muscle mass and strength (14), thus suggesting that bone soluble factors participate in driving muscle loss in cachexia.
On the other hand, a limited number of studies have examined whether bone resorption factors can be released by the tumor. The receptor activator of NFkB ligand (RANKL or Tnfsf11), a TNF superfamily member and master regulator of bone remodeling, is responsible for osteoclast differentiation and osteoclastic bone resorption (16), and more recently in the control of muscle size and function (17). RANKL expression was shown to be elevated in many solid tumors, including breast, prostate, liver, gastric and ovarian (18–21), and was described to correlate with poor clinical outcomes in patients affected with ovarian tumors (18). RANKL binds to and activates its cognate receptor RANK thereby regulating differentiation, activity and survival of osteoclasts (22). The effects of RANKL are physiologically counterbalanced by the soluble decoy receptor osteoprotegerin (OPG or Tnfrsf11b), which inhibits osteoclastogenesis and bone resorption (23,24).
Dufresne et al. showed RANK is expressed in both C2C12 myotubes and skeletal muscle tissue (17). The same authors also showed that activation of RANK-dependent signaling plays a role in muscle dysfunction (17), while RANKL blockade by full-length OPG-Fc peptide or anti-RANKL antibodies was shown to improve the muscle phenotype in muscular dystrophy (25,26) and in women with osteoporosis (27). Finally, TRAF6, a signal transducer downstream of RANK, was recently shown to mediate TNF-induced muscle atrophy in aging (28), and targeted ablation of TRAF6 in skeletal muscle was reported to improve cachexia in experimental animals (29).
In the present study, we show for the first time that cachectic patients and animals affected with OvCa show elevated circulating RANKL while exhibiting muscle and bone deficits suggesting that tumor-derived RANKL may play a role in the development of the musculoskeletal deficits associated with OvCa. Here, we showed that high circulating RANKL is sufficient to drive bone loss and skeletal muscle atrophy both in vitro and in vivo and validated the use of bisphosphonates or anti-RANKL antibodies, both FDA-approved antiresorptive therapies, to preserve bone and muscle mass in spite of high RANKL expression by non-metastatic tumors.
Methods
Patients
Adult female ovarian cancer patients with active, advanced-stage (FICO Stage III) (30) epithelial ovarian carcinoma were collected at our tertiary referral center from 2010–2019. Patients were included for review if surgical histopathological examination and adequate abdominopelvic computed tomography (CT) imaging at time of diagnosis were available. Patients were excluded if they had any of the following comorbidities which may contribute to cachexia: chronic obstructive pulmonary disease (COPD); congestive heart failure (CHF); chronic renal insufficiency (CKD ≥3); liver failure or cirrhosis; or prior stroke. To serve as a comparative control, healthy adult female patients who underwent abdominal CT imaging in the setting of trauma at our tertiary referral center from 2010–2019 were also included for review. “Healthy” was defined as not having any end-organ failure (as previously denoted), cancer diagnosis, immunosuppression, autoimmune disease (e.g. HIV/AIDS, rheumatoid arthritis, systemic lupus erythematosus, etc.), or physical impairment with activities of daily living. Data were extracted from those patients who subsequently remained for analysis. Information collected included patient demographics, comorbidities, substance use histories, physical performance scores (ECOG) (31), anthropometric measurements, nutrition information (serum albumin, hemoglobin, 6-month weight loss history, 6-month history of anorexia), and cancer information (cancer histology, tumor grade, tumor stage according to FIGO classification, presence of ascites). Cancer cachexia was assigned if patients met criteria as defined by Fearon et al. (32) general cachexia defined by Evans et al. (33) (Table S1). The age, race, and body mass index (BMI, kg/m2) of controls were matched to the ovarian cancer patient group.
Anthropometric Measurements
All subject weights (kg) and heights (cm) were collected at the time of initial CT imaging for cancer diagnosis for ovarian cancer patients and initial trauma for controls. BMIs were calculated. Total skeletal muscle mass at the third lumbar vertebra was measured using SliceOmatic v5.0 software (TomoVision, Magog, Canada). The paraspinal muscles, psoas major, quadratus lumborum, transverse abdominis, internal and external obliques, and recuts abdominis muscle cross-sectional areas (CSA, cm2) were isolated and measured using Hounsfield Units (HU) specific to skeletal muscle (−29 to +150). The CSA was normalized to stature by dividing by patient height in squared meters (m2) to calculate skeletal muscle index (SMI, cm2/m2). A similar strategy was applied to measure adipose tissue using the following HU thresholds: intramuscular adipose (−190 – [−30]); subcutaneous adipose (−190 – [−30]); visceral adipose (−150 – [−50]). Average skeletal muscle densities (SMD, HU) were also recorded. Radiologically-defined sarcopenia was established with the cutoff values used by Paula de Silva et al. (34) with SMI < 38.9 cm2/m2.
Collection of plasma from OvCa patients
De-identified human plasma was obtained from the Tissue Procurement and Distribution Core of the IU Simon Comprehensive Cancer Center. We identified two groups of samples: normal subjects (females, receiving treatment for benign conditions; Control, n=17), and cancer patients affected with OvCa (n=31). To avoid inclusion of potentially osteoporotic subjects only patients younger than 60 years of age were enrolled in the study. A summary of the patients’ demographics is reported in Table S3.
Cell cultures
Human ES-2 cells were purchased from ATCC (Manassas, VA; #CRL-1978) and were cultured in McCoy’s medium supplied with 10% fetal bovine serum, 1% glutamine, 1% sodium pyruvate, 1% penicillin/streptomycin (12). The ES-2 cells have been recently classified as a suitable model for the study of high grade serous ovarian cancer (HGS-OC), which represents the most aggressive subtype of OvCa (35). Upon intraperitoneal inoculation the ES-2 tumors generate ascites and abdominal solid dissemination, both recapitulating the classical clinical presentation of advanced HGS-OC in humans (12,36). Murine C26 cells were originally provided by Donna McCarthy (Ohio State University, Columbus, OH) and cultured in high glucose (4.5 g/L) DMEM supplied with 10% fetal bovine serum, 1% glutamine, 1% sodium pyruvate, and 1% penicillin/streptomycin (37). Murine ID8 cells were a kind gift of Dr. Sumegha Mitra (Indiana University, Indianapolis, IN) and were cultured in RPMI1640 (Corning) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin (38). Human OVCAR3 (ATCC, Manassas, VA) were cultured in high glucose (4.5 g/L) DMEM supplemented with 20% fetal bovine serum and 1% penicillin/streptomycin (39). Murine C2C12 skeletal myoblasts (ATCC, Manassas, VA) were grown in high glucose DMEM supplemented with 10% FBS, 100 U/ml penicillin, 100 mg/ml streptomycin, 100 mg/ml sodium pyruvate, 2 mM L-glutamine, and maintained at 37°C in 5% CO2. Human skeletal muscle myoblast (HSMM; #CC-2580, Lonza, Basel, Switzerland) were propagated in growth medium (CC-3160, Lonza) supplemented with 0.05 ml/ml of fetal calf serum,50μg/ml of fetuin (bovine), 10 ng/ml of epidermal growth factor (recombinant human), 1 ng/ml of basic fibroblast growth factor (recombinant human), and 0.4 μg/ml of dexamethasone (CC-3244, Lonza). Myotubes were generated by exposing the myoblasts to DMEM containing 2% horse serum and replacing the medium every other day for 5 days. All cell lines were maintained in a 5% CO2, 37°C humidified incubator. In order to determine the effects of RANKL on fiber size, murine or human myotubes were exposed to 100 ng/ml recombinant mouse or human RANKL protein for 72 hours (#390-TN and #462-TR murine and human, respectively, Bio-Techne Corporation, Minneapolis, MN). In order to determine the effects of the anti-RANKL on myotubes atrophy induced by ES-2 tumor cells, C2C12 myotubes were co-cultured with ES-2 cells using trans-well permeable inserts (Thermo Fisher Scientific, #12565009) and treated with murine anti-RANKL antibody (CD254, BioxCell, Lebanon, NH) for 48 h. In order to determine the effects of the OvCa cell lines on myotubes, C2C12 myotubes were co-cultured with ID8 and OVCAR3 cells using transwell permeable inserts for 48 h. To overexpress murine RANKL in the C26 adenocarcinoma cell line, pCMV6 plasmid containing the untagged cDNA sequence for the murine Tnfsf11/RANKL (#PS100001, OriGene, Rockville, MD) was transfected in wild type (WT) C26 cell using Lipofectamine 3000 (# L3000–008, Invitrogen Carlsbad, CA) according to the manufacturer instructions. The transfected cells (i.e. C26pR) were selected by adding neomycin (1μg/ml) to the culture medium for up to 2 weeks, tested for overexpression efficiency and used for subsequent in vivo experiments.
Animals
All animal studies were approved by the Institutional Animal Care and Use Committee at Indiana University School of Medicine and were in compliance with the National Institutes of Health Guidelines for Use and care of Laboratory Animals. All animals were maintained on a regular dark-light cycle (light from 8 a.m. to 8 p.m.), with free access to food and water during the whole experimental period. For the ES-2 experiments 8-week old female NOD-scid/IL2Rgnull (NSG) immunodeficient mice (In Vivo Therapeutics Core Facility, IU Simon Cancer Center, Indianapolis, IN) were used and housed in a pathogen-free facility at IU LARC (up to 5 per cage). 1×107 ES-2 cells were inoculated intraperitoneally (i.p.) in sterile saline (12). To investigate whether bisphosphonates could preserve both bone and muscle, similar to our previously published observations (14), in an initial experiment mice were stratified according to the body weight into four groups: control mice receiving vehicle (sterile saline; C), control mice treated with Zoledronic acid (ZA, 5μg/Kg, s.c., as previously described (11)), ES-2 tumor-bearing mice receiving vehicle (ES-2) and ES-2 tumor-bearing mice treated with ZA (ES-2+ZA). The treatment was administered three times per week for the entire duration of the experiment, starting the day after tumor injection. Zoledronic acid was provided by Mylan Institutional LLC (Rockford, IL). To determine whether anti-RANKL strategies could preserve muscle and bone in cachexia, in a second experiment, mice were divided into four groups: control mice receiving anti-IgG2a (IgG2a; 250μg/mouse, i.p.), control mice receiving anti-RANKL antibodies (R Ab; 250μg/mouse, i.p.), ES-2 tumor-bearing mice receiving anti-IgG2a (ES-2 + IgG2a) and ES-2 hosts treated with anti-RANKL antibodies (ES-2 + R Ab). The treatment was administered every three days for the entire duration of the experiment, starting three days after tumor implantation. IgG2a (clone 2A3; #BE0089) and anti-RANKL antibodies (clone IK22/5; #BE0191) were provided by Bio-X-Cell (Lebanon, NH). To study the effects resulting from in vivo overexpression of RANKL, 8-week old CD2F1 male mice (Envigo, Indianapolis, IN) were infected with serotype 6 recombinant replication-deficient adeno-associated virus (AAV6) vectors, characterized by tropism for skeletal muscle, and injected within the right gastrocnemius muscle to augment RANKL with AAV6-CMV-RANKL (AAV-R; #AAV-274722; n=5) at both local level and systemically, as shown in (40). Control animals (n=3) received the same amount of AAV6-CMV-Null (AAV-N; #AAV-7029; n=3). To perform the infection, the animals were anesthetized with isoflurane. The right hind limb was shaved and 100 μl of viral suspension containing 1×1012 vg was injected into the tibialis anterior and gastrocnemius muscle belly, 4 mm beneath the aponeurosis using 31-gauge syringes (Becton–Dickinson, Franklin Lakes, NJ). The AAV6 constructs were purchased from Vector Biolabs (Malvern, PA). To study the effects resulting from tumor-derived RANKL in the C26 tumor-bearing mice, 8-week old CD2F1 male mice (Envigo, Indianapolis, IN) were divided into two groups, namely C26 or C26pR, and inoculated subcutaneously (s.c.) with 1×106 C26 or C26pR cells in sterile saline. For all the in vivo experiments, the mice were identified with a code, whereas the investigators were blinded during allocation, animal handling, and endpoint measurements. The animals were weighed daily and at time of sacrifice several tissues, including skeletal muscles, were collected, weighed, frozen in liquid nitrogen and stored at −80°C for further analyses. The tibialis anterior muscles were mounted in OCT and frozen in liquid nitrogen-cooled isopentane for histology, as previously described (41). The femurs and tibiae were accurately cleaned, scraped and flushed to eliminate the bone marrow, then frozen in liquid nitrogen and stored at −80°C for further analyses. The mouse carcasses were fixed for 2 days in 10% neutral buffered formalin, and then transferred into 70% ethanol for storage of bone tissues and subsequent histological analyses.
Grip strength measurement
The evaluation of the whole body strength in mice was assessed as described in (42). The absolute grip strength (peak force, expressed in grams) was recorded by means of a grip strength meter (Columbus Instruments, Columbus, OH). Five measurements were completed and the top three measurements were included in the analysis.
In Vivo Muscle Contractility
Animals (n = 3–5) underwent in vivo plantarflexion torque assessment (Aurora Scientific Inc, Canada), as previously described. Briefly, the left hind foot was taped to the force transducer and positioned to where the foot and tibia were aligned at 90°. The knee was then clamped at the femoral condyles, avoiding compression of the fibular nerve. Two disposable monopolar electrodes (Natus Neurology, Middleton, WI) were placed subcutaneously posterior/medial to the knee in order to stimulate the tibial nerve. Maximum twitch torque was first determined using supramaximal stimulations (0.2 ms square wave pulse). Peak plantarflexion torque was then assessed following a supramaximal square wave stimulation (0.2 ms) delivered at 100Hz stimulation frequency(42).
Assessment of muscle cross sectional area (CSA)
To assess skeletal muscle atrophy, 10-μm-thick cryosections of tibialis anterior muscles taken at the mid-belly were processed for immunostaining as described previously (42). Briefly, sections were blocked for 1 h at room temperature and incubated overnight at 4°C with a dystrophin primary antibody [1:50, #MANDRA1(7A10), Developmental Studies Hybridoma Bank, Iowa City, Iowa, USA;], followed by a 1 h secondary antibody (AlexaFluor 555, 1:1000, A21127, Thermo Fisher Scientific) incubation at room temperature. Entire dystrophin-stained sections were analyzed for CSA using a Lionheart LX automated microscope (BioTek Instruments).
Assessment of myotube size
Cell layers were fixed in ice-cold acetone-methanol (50/50) and incubated with an anti-Myosin Heavy Chain antibody (MF-20, 1:100; Developmental Studies Hybridoma Bank, Iowa City, IA) and AlexaFluor 594 or 488-labeled secondary antibodies (Invitrogen, Grand Island, NY). Analysis of myotube size was performed by measuring the average diameter of long, multi-nucleated fibers (n=50–100 per condition) avoiding regions of clustered nuclei on a calibrated image using the Image J 1.43 software.
Body composition assessment by dual-energy X-ray absorptiometry
Body bone mineral density (BMD) and bone mineral content (BMC) were assessed by means of dual-energy X-ray absorptiometry (DXA) scanning of formalin-fixed carcasses. According to the manufacturer’s guidelines, in order to calibrate and validate the apparatus for its performance, a spine phantom was scanned using the Lunar PIXImus densitometer (PIXImus, Fitchburg, WI) before scanning the first carcass. Animal carcasses were placed in a prone position with the limbs outstretched. From the whole-body scans, areal BMD and BMC were calculated for the entire body minus head region of interest (13).
Micro computed tomography (μCT) analysis of femurs and vertebrae bone morphometry
MicroCT (μCT) scanning was performed to measure morphological indices of metaphyseal regions of femurs, as described in Huot et al.(42). After euthanasia, the mouse carcasses were fixed for 2 days in 10% neutral buffered formalin, transferred into 70% ethanol, the right femurs dissected, and prepared for μCT scanning on a high-throughput μCT specimen scanner Bruker Skyscan 1176 (Bruker, Kontich, Belgium). Bone samples were rotated around their long axes and images were acquired with the following parameters: pixel size = 9 μm3; peak tube potential = 50 kV; X-ray intensity = 500 μA; 0.3° rotation step. Raw images were reconstructed using SkyScan reconstruction software (NRecon; Bruker, Kontich, Belgium) to 3-dimensional cross-sectional image data sets using a 3-dimensional cone beam algorithm. Structural indices were calculated on reconstructed images using the Skyscan CT Analyzer software (CTAn; Bruker, Kontich, Belgium). Trabecular bone was separated using a custom processing algorithm in CTAn, based on the different thicknesses of the structures. Trabecular bone was analyzed between 1.5 mm to 2.5 mm under the femoral distal growth plate using a threshold of 80 – 255. Trabecular parameters included bone volume fraction (Tb.BV/TV), number (Tb.N), thickness (Tb.Th), separation (Tb.Sp), and connectivity (Conn.Dn).
Western Blotting
Total protein extracts were obtained by lysing cell layers in RIPA buffer (150 mM NaCl, 1.0% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, and 50 mM Tris, pH 8.0) completed with protease (Roche, Indianapolis, IN) and phosphatase (Thermo Scientific, Rockford, IL) inhibitor cocktails. After 10 minutes incubation on ice, cell debris were removed by centrifugation (15 min, 14000 g) and the supernatant collected and stored at −80°C. Protein concentration was determined using the BCA protein assay method (Thermo Scientific, Rockford, IL). Protein extracts (30 μg) were then electrophoresed in 4–15% gradient SDS Criterion TGX precast gels (Bio-Rad, Hercules, CA). Gels were transferred to nitrocellulose membranes (Bio-Rad, Hercules, CA). Membranes were blocked with SEA BLOCK blocking reagent (Thermo Scientific, Rockford, IL) at room temperature for 1 h, followed by an overnight incubation with diluted antibody in SEA BLOCK buffer containing 0.2% Tween-20 at 4°C with gentle shaking. After washing with PBS containing 0.2% Tween-20 (PBST), the membrane was incubated at room temperature for 1 h with either Anti-rabbit IgG (H+L) DyLight 800 or Anti-mouse IgG (H+L) DyLight 600 (Cell Signaling Technologies, Danvers, MA). Blots were then visualized with Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE). Antibodies used were anti-RANKL (#ab9957) from Abcam and anti-GAPDH (#5174S) from Cell Signaling technology.
Real-time quantitative polymerase chain reaction (qRT-PCR)
Long bones were dissected, the epiphyses removed, and flushed to remove bone marrow. Bones or quadriceps muscle were homogenized in TRIzol Reagent. RNA from bones was isolated using the miRNeasy Mini kit (Qiagen, Valencia, CA, USA) and following the protocol provided by the manufacturer. RNA was quantified by using a Synergy H1 spectrophotometer (Biotek, Winooski, VT, USA). Total RNA was reverse transcribed to cDNA by using the Verso cDNA kit (Thermo Fisher Scientific, Waltham, MA, USA). Transcript levels were measured by Real-Time PCR (Light Cycler 96, Roche, Indianapolis, IN, USA) taking advantage of the TaqMan gene expression assay system (Life Technologies, Carlsbad, CA). Expression levels for Tnfsf11 (Mm00441906m1) and Tnfrsf11b (Mm00435454_m1) were quantitated in the flushed bone. Expression levels for Fbxo32 (Mm00499523_m1) and Trim63 (Mm01185221_m1), were quantitated in the gastrocnemius muscle. Gene expression was normalized to Gapdh (Mm01277042_m1) or TATA-binding protein (Tbpl1) (Mm01277042_m1) levels for bone and muscle respectively, using the standard 2−ΔΔCT methods.
RNA Sequencing
Total RNA was first evaluated for quantity and quality by using the Agilent Bioanalyzer 2100. For RNA quality, a RIN number of 7 or higher was considered acceptable. One hundred nanograms of total RNA were used. cDNA library preparation included mRNA purification/enrichment, RNA fragmentation, cDNA synthesis, ligation of index adaptors, and amplification following the KAPA mRNA Hyper Prep Kit Technical Data Sheet, KR1352 – v4.17 (Roche Corporate). Each resulting indexed library was quantified and its quality accessed by Qubit and Agilent Bioanalyzer, and multiple libraries were pooled in equal molarity. Five microliters of 2 nM pooled libraries per lane were then denatured, neutralized and applied to the cBot for flow cell deposition and cluster amplification, before loading to HiSeq 4000 for 75b paired-end sequencing (Illumina, Inc.). Approximately 30M reads per library were generated. A Phred quality score (Q score) was used to measure the quality of sequencing. More than 90% of the sequencing reads reached Q30 (99.9% base call accuracy). The sequencing data were first assessed using FastQC (Babraham Bioinformatics, Cambridge, UK) for quality control. Then, all sequenced libraries were mapped to the mouse genome (mm10) using STAR RNA-seq aligner (43) with the following parameter: “--outSAMmapqUnique 60”. The reads distribution across the genome was assessed using bamutils (from ngsutils) (44). Uniquely mapped sequencing reads were assigned to mm10 refGene genes using featureCounts (from subread) (45) with the following parameters: “-s 2 –p –Q 10”. Quality control of sequencing and mapping results was summarized using MultiQC (46). Genes with read count per million (CPM) < 0.5 in more than 4 of the samples were removed. The data was normalized using the TMM (trimmed mean of M values) method. Differential expression analysis was performed using edgeR (47). False discovery rate (FDR) was computed from p-values using the Benjamini-Hochberg procedure. Differentially expressed genes were identified at 1.4 fold change and FDR of 0.05. The heatmap was generated using Partek Flow genomic analysis software (St. Louis, Missouri, USA). Ingenuity pathway analysis (IPA) software was used to identify the canonical pathways and upstream regulators. Pathways with p<0.05 were considered significant. Only the upstream molecules that were differentially expressed in our dataset were considered, along with the pathways which had any z-score. The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number (GSE149606) (48).
ELISA for RANKL, OPG and CTX-I
The levels of RANKL were measured in mouse centrifuged platelet-poor plasma and ascites from control and ES-2 bearers, in mice infected intramuscularly with AAV-RANKL or AAV-NULL, and in the conditioned media (CM) from ES-2, C26 and C2C12 cell lines by using a specific ELISA kit (#D6050; Bio-Techne Corporation, Minneapolis, MN), according to the manufacturer’s protocol. The circulating levels of human RANKL were measured in human serum from ovarian cancer patients and in control by using a specific ELISA kit (#BI-20462, Eagle biosciences Inc, Columbia, NH). The circulating levels of human CTX-I were measured in human serum from ovarian cancer patients and in control by using a specific Serum CrossLaps ELISA kit (#AC02F1, Thermo Fisher Scientific, Suwanee, GA, USA). The levels of OPG were measured in plasma from control and ES-2 bearers and in mice infected intramuscularly with AAV-RANKL or AAV-NULL by using a specific ELISA kit (#MOP00; Bio-Techne Corporation, Minneapolis, MN).
Immunohistochemistry
To perform the immunohistochemical analysis of human OvCa tissue, a commercially available paraffin-embedded tissue microarray of ovarian carcinomas and normal tissue (#OV8069, Biomax Inc, US) was used. Deparaffinization of the slides was performed through a series of incubations in xylene, 100% ethanol, 95% ethanol, 70% ethanol, and deionized water. Antigen retrieval was performed by treatment in 10 mM citrate buffer solution for 30 min at sub-boiling temperature. Endogenous peroxidase activity was inhibited by a 10 min incubation in 3% H2O2. Sections were subsequently blocked in Tris-buffered saline (TBS)-Tween buffer containing 5% goat serum for 1 h at room temperature. A primary rabbit anti-RANKL antibody (#ab9957, Abcam, Cambridge, MA, USA) was added (1:50) and incubated overnight in a humidified box at 4°C. Normal rabbit IgG were used as negative controls. Sections were then incubated with the ‘EnVision+System-HRP-labelled polymer secondary antibody’ (DAKO, Carpinteria, CA, USA) for 1 h at room temperature. The reaction was then visualized with the DAKO ‘Liquid DAB+substrate chromogen system’ according to the manufacturer’s instructions. Slides were counterstained with Methyl green for up to 5 min, dehydrated with incubations in 70% ethanol, 95% ethanol, 100% ethanol, and xylene, and mounted with xylene-based Cytoseal XYL mounting medium (Thermo Fisher Scientific, Suwanee, GA, USA).
Statistical analyses
For the human studies, including the in vitro and in vivo experiments, statistical analyses were performed using SPSS v25.0 (IBM Inc., Armonk, NY). Nominal data were displayed as proportions of whole (percentage) and analyzed with Pearson’s χ2 test (two-way test). The human data were analyzed by applying a non-parametric test, whereas the murine data were considered with a normal distribution and analyzed with parametric test, as specified below. Ordinal data were written as mean ± SD and analyzed using a Mann-Whitney non-parametric test followed by post hoc test as appropriate for the human data, a two-way Student’s t-test for the mouse data comparing no more than two groups, a one-way analysis of variance (ANOVA) test followed by Tukey’s post-test for the murine data including more than two groups. Differences were considered significant when p<0.05.
Results
Human OvCa associates with cachexia, bone loss and elevated RANKL.
We showed that NOD-scid/IL2Rgnull (NSG) female mice bearing ES-2 human OvCa cells present loss of body weight and skeletal muscle, as well as bone (12). To validate these findings in humans, similar to previous studies (34) we performed a retrospective analysis to compare body composition between healthy women (n=16) and patients with epithelial OvCa (n=27). By stratifying cancer patients based on evidence of cachexia according to the definitions by Fearon et al. and Evans et al. (32,33) (Table S1), we found that 74.1% and 63.0% of subjects with OvCa presented with cachexia, respectively. As shown by the quantification of CT scan images taken at L3 vertebra (Figure 1A), similar to previous work (49), the skeletal muscle index (SMI) was reduced in the OvCa cachectic patients compared to both control and OvCa without cachexia groups (Table S2). Additionally, the visceral adipose cross-sectional areas and the visceral adipose to subcutaneous adipose ratios were reduced in the cachectic patients (Table S2). To validate elevated bone turnover in OvCa patients similar to rodents, C-telopeptide of type I collagen (CTX-I) was measured in serum from 17 control patients and 31 women diagnosed with OvCa (Table S3) and found to be markedly increased (Figure 1B), suggesting evidence of bone loss in this population. Since RANKL is the most important regulator of bone resorption, we measured the levels of soluble RANKL in the same set of samples and observed increased RANKL in the OvCa patients (Figure 1C). Notably, CTX-I and RANKL were positively correlated (r=0.40, p<0.01) (Figure 1D), thus directly supporting the evidence of bone loss in the OvCa patients. Since several types of tumors can secrete RANKL, we investigated the expression of RANKL in different OvCa histological types by performing an immunohistochemical analysis in a tissue microarray including 80 OvCas and normal ovarian tissue, and discovered marked positivity for RANKL expression among clear cell carcinomas, transitional cell carcinomas and serous adenocarcinomas (Figure 1F). In a TCGA-based analysis, OvCa patients with low-Tnfsf11 tumor expression also presented improved overall survival (Figure 1E).
Figure 1. Human OvCa associates with cachexia, bone loss and elevated RANKL.
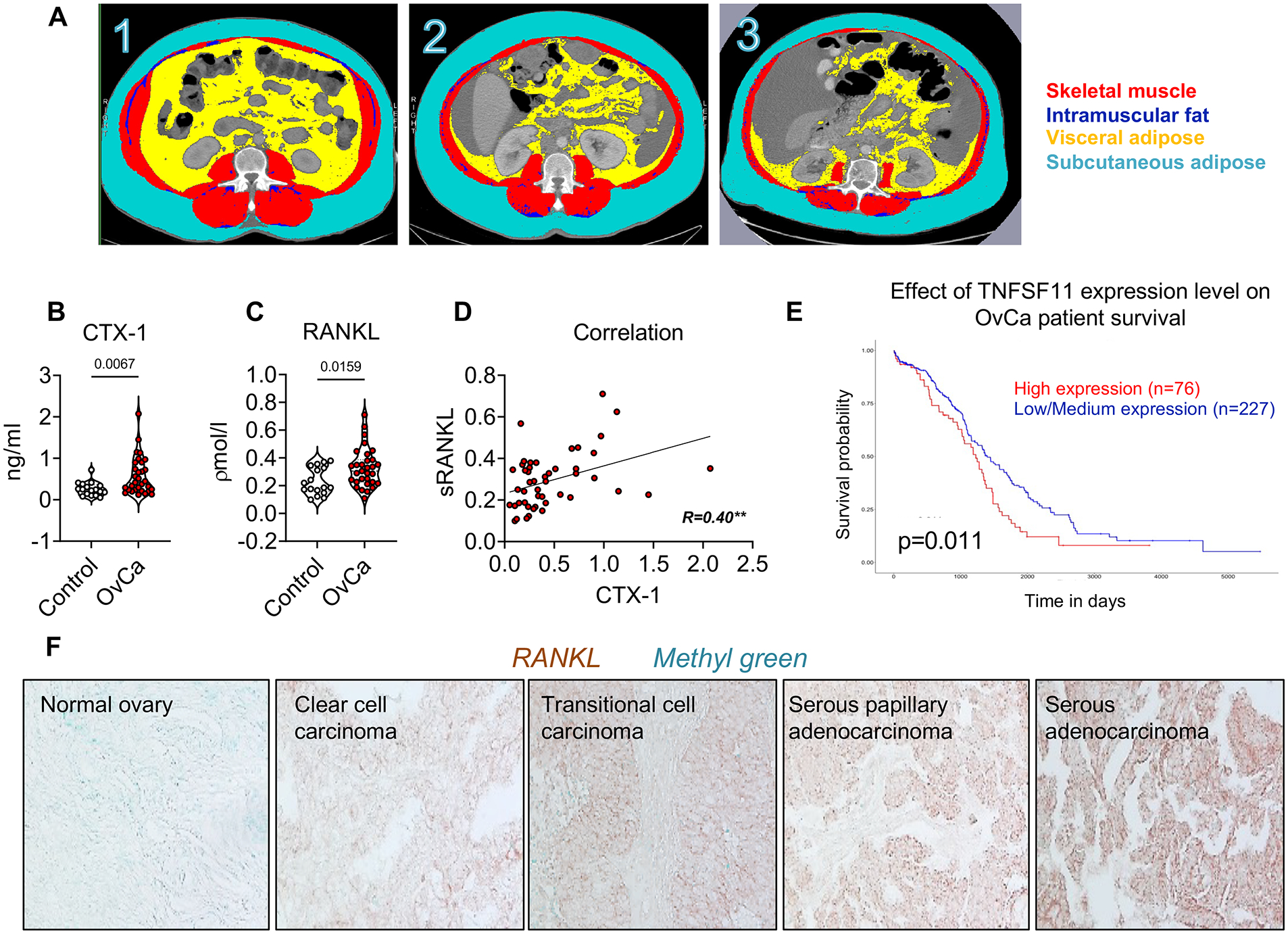
A: 1- 54-year-old control patient, BMI = 33.6 kg/m2, SMI = 63.1 cm2/m2, visceral adipose = 266.6 cm2. 2- 55-year-old patient with ovarian cancer (OvCa), without cachexia, BMI = 37.2 kg/m2, SMI = 55.5 cm2/m2, visceral adipose = 139.2 cm2. 3- 77-year-old patient with ovarian cancer and cachexia, BMI = 31.6 kg/m2, SMI = 33.7 cm2/m2, visceral adipose = 86.6 cm2. Red: skeletal muscle; dark blue: intramuscular adipose tissue; indigo: subcutaneous adipose tissue; yellow: visceral adipose tissue. B: C-telopeptide of type I collagen (CTX-I) levels in plasma from human OvCa patients. C: sRANKL levels in plasma from human OvCa patients. D: Correlation between RANKL levels and CTX-I levels in plasma from human OvCa patients. E: TCGA-Kaplan-Meier analysis estimating survival in patients with low vs. high-RANKL OvCa. Significance of the differences *p<0.05 (long-rank test) (82). F: IHC for RANKL in a human OvCa tissue microarray. Data were expressed as means ± SD. Significance of the differences *p<0.05, **p<0.01, ***p<0.001 (unpaired t-test).
Pharmacologic bone preservation improves muscle mass and strength in murine OvCa hosts.
In order to understand if bisphosphonate treatment was sufficient to prevent bone loss associated with OvCa-induced cachexia, ES-2 tumor-bearing mice were treated with ZA. Similar to our previous observations (12), the ES-2 hosts displayed dramatic loss of trabecular bone (Figure 2A–B), whereas ZA significantly preserved the trabecular bone in the tumor-bearing mice, as suggested by ameliorated BV/TV, Tb.N and Conn.Dn, and by reduced Tb.Sp and Tb.Pf. The altered bone microarchitecture in the ES-2 hosts was also associated with changes in the mRNA expression of osteokines previously involved in the regulation of bone mass (22,50), including the Tnfsf11/Tnfrsf11b (RANKL/OPG) ratio (Figure 2C). Notably, ZA was able to normalize the expression levels of Tnfsf11 (Figure S1A), thus also correcting the Tnfsf11/Tnfrsf11b ratio (Figure 2C). Furthermore, the circulating levels of RANKL were also reduced upon administration of ZA (Figure 2D). To determine if OvCa directly participates in promoting RANKL elevations, we assessed RANKL levels in the conditioned medium (CM) of ES-2 cultures, compared to C2C12 myotubes and C26 colorectal adenocarcinoma cells. In the ES-2 medium RANKL was markedly higher vs. C2C12 (Figure 2E). This observation was further corroborated by the detection of high RANKL (Figure 2E) in the ascites collected from ES-2 bearers. In order to verify whether ZA was also able to preserve muscle mass in a model of cancer cachexia, similar to our previous observations in a model of cisplatin-derived muscle and bone loss (14) we investigated the ZA-dependent effects in the muscle of ES-2 bearers. The ES-2 growth led to ascites accumulation (Figure 2F), reduced tumor-free body weight (Figure S1C) and marked loss of quadriceps (Quad), gastrocnemius (GSN) and tibialis (TA) muscles mass compared to the vehicle-treated animals (Figure 2G). Moreover, the white adipose tissue (WAT) was severely depleted in the ES-2 hosts (Figure S1C), whereas liver and spleen where unchanged (Figure S1C). Conversely, despite no effects on tumor size (Figure 2F) and organ size (Figure S1C), ZA administration was able to partially protect muscle mass in the ES-2 tumor-bearing mice, as suggested by larger Quad, GSN and TA (Figure 2G), whereas no changes were reported in the control group treated with ZA (Figure 2G). Consistently, growth of the ES-2 tumor was accompanied by reduced whole body muscle strength, which was improved by ZA administration (Figure 2H).
Figure 2. Pharmacologic bone preservation improves muscle mass and strength in murine OvCa hosts.
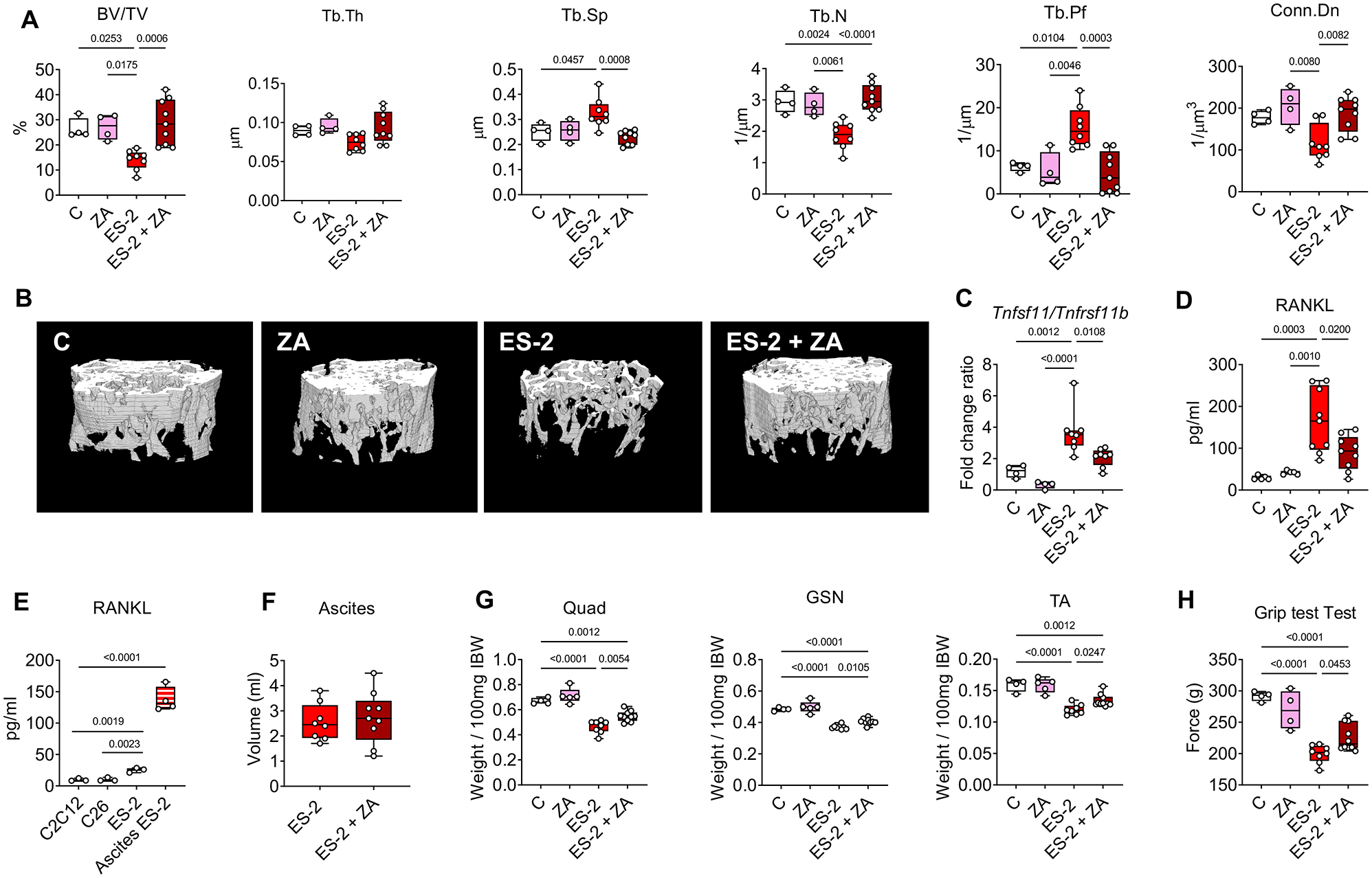
A: Assessment of trabecular bone volume fraction (BV/TV; expressed as %), trabecular thickness (Tb.Th; expressed as μm), trabecular separation (Tb.Sp; expressed as μm), trabecular number (Tb.N; expressed as 1/μm), trabecular pattern factor (Tb.Pf; expressed as 1/μm) and trabecular connectivity density (Conn.Dn; expressed as 1/μm 3) in the femurs from mice receiving vehicle (C, n=4) or zoledronic acid (ZA, n=4) and from ES-2 tumor-bearing mice (ES-2, n=8) and ES-2 hosts receiving ZA (ES-2+ZA, n=10). B: Representative microCT-based three-dimensional rendering of trabecular bone in mouse femurs. C: mRNA expression for Tnfsf11/Tnfrsf11b ratio in the marrow flushed-bones of C (n=4), ZA (n=4), ES-2 (n=8) and ES-2+ZA (n=8). Gene expression was normalized to Gapdh levels and expressed as fold change vs. vehicle (C). D: Plasma RANKL levels in C (n=5), ZA (n=5), ES-2 (n=9) and ES-2+ZA (n=9). E: RANKL levels in the conditioned medium (CM) of ES-C2C12, C26 and ES-2 cultures and in the ascites collected from ES-2 tumor-bearing mice. F: Tumor volume in ES-2 (n=8) and ES-2+ZA (n=9). G: Skeletal muscle weights in C (n=4), ZA (n=5), ES-2 (n=8) and ES-2+ZA (n=10). Muscle weight were normalized to the initial body weight (IBW) and expressed as weight/100mg IBW. H: Whole body grip strength (expressed in grams of force) in C (n=4), ZA (n=5), ES-2 (n=8) and ES-2+ZA (n=10). Data were expressed as means ± SD. Significance of the differences: *p<0.05, **p<0.01, ***p<0.001 vs. C, #p<0.05, ##p<0.01, ###p<0.001 vs. ZA, $p<0.05, $$p<0.01, $$$p<0.001 vs. ES-2 (one-way ANOVA with Tukey’s test).
RANKL causes myotube atrophy and modulation of inflammatory signaling pathways.
Previous reports indicate that RANKL-RANK signaling participates in regulating muscle mass and strength in denervated animals (17), while RANKL inhibition by OPG-Fc peptide improves the muscle phenotype in dystrophic mdx mice (25). To determine the role of RANKL treatment in the regulation of fiber size, fully differentiated murine C2C12 and human HSMM myotubes were exposed to recombinant mouse or human RANKL, respectively. Interestingly, both treatment with recombinant RANKL were able to induce myotube atrophy in both models (Figure 3A–B). To understand whether RANKL was driving changes in gene expression that may be account for the atrophic phenotype, we performed Next-generation RNA Sequencing (RNA-Seq) in C2C12 myotubes exposed to recombinant mouse RANKL and observed changes in 283 genes (Figure 3C). The pathway analysis revealed modulation of muscle-regulatory signaling pathways, including acute phase response, Tnfrsf1a/Tnfrsf1b, Hif1α, Toll-like receptor, Tnfsf12, Nfkb, and Prkag2 (Figure 3D). Moreover, the network analysis conducted on our RNA-Seq identified some of the well-known upstream regulatory molecules involved in muscle wasting such as Fgf21, Nfkbia, Cxcl2 and Relb, further supporting a role of RANKL in muscle wasting (Figure S2). Consistently, we also found increased levels of Traf6, previously involved in cachexia (29). On the other hand, the roles of other upstream regulators, such as Zc3h12a, Edn1, have not been studied in the context of muscle wasting, thus requiring further investigations.
Figure 3. RANKL causes myotube atrophy, along with modulation of inflammatory signaling pathways.
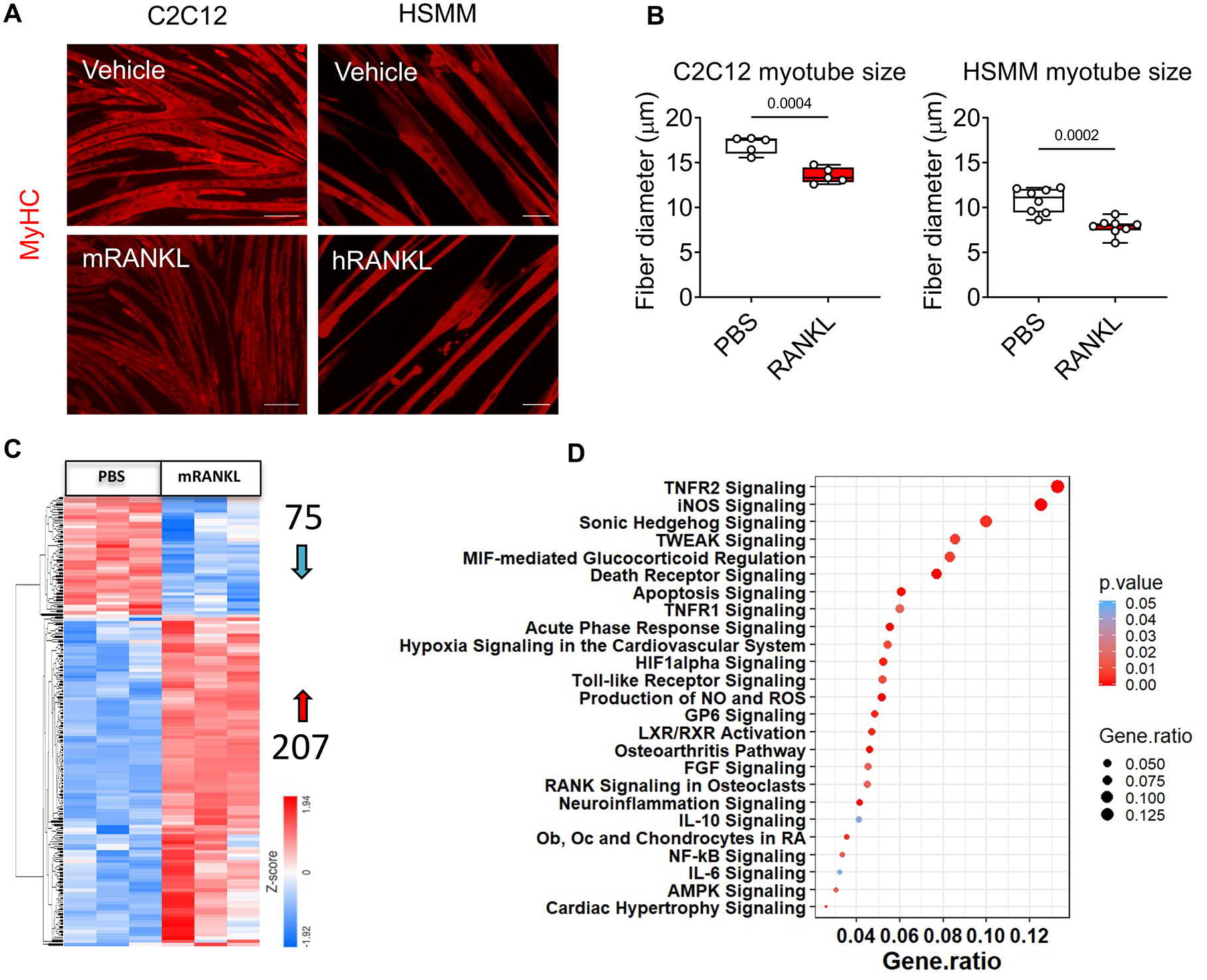
A-B: Representative images and quantification of size in murine C2C12 and human HSMM myotubes exposed to PBS or recombinant RANKL (mouse or human, respectively; 200 pg/ml) for 48h. Red staining: myosin heavy chain. Up to 400 myotubes per experimental condition were measured, in triplicate. Next-Generation RNA-Sequencing in C2C12 myotubes treated with recombinant RANKL (200 pg/ml) for 48h (n=3). C: Heatmap represents the differentially expressed genes (red= 207 genes upregulated and blue= 75 genes downregulated). D: The dot plot graph represents canonical pathways from IPA. The size of the dot represents the gene ratio, while the color indicates p-value. IPA pathway analysis using a p<0.05 discovery cutoff. Data were expressed as means ± SD. Significance of the differences *p<0.05, **p<0.01, ***p<0.001 vs. PBS (unpaired t-test).
In vivo elevated RANKL induces murine muscle atrophy, muscle weakness and bone loss.
To clarify if elevated RANKL was sufficient to induce muscle atrophy and weakness also in in vivo conditions, heathy mice were infected intramuscularly with either AAV-NULL (AAV-N) or AAV-RANKL (AAV-R). After 4 weeks from infection, the levels of circulating RANKL in the mice infected with AAV-R were markedly elevated in comparison to the animals receiving AAV-N (Figure 4A). Conversely, no changes were observed in the levels of OPG, altogether contributing to marked elevations in the RANKL/OPG ratio (Figure 4A). Similar to the alterations observed in vitro, the mice infected with AAV-R displayed generalized and significant skeletal muscle atrophy (Figure 4B), which was more prominent in the right vs. left hindlimb (data not shown), thus suggesting a systemic effect of RANKL overexpression, in line with the elevation in circulating RANKL. The same animals also exhibited reduced muscle fiber size (Figure 4C–D), consistent with decreased muscle strength (Figure 4E). The RANKL-associated effects on bone mass were monitored by means of DXA and revealed progressively decreased total body BMD and BMC (Figure 4F). The bone microarchitecture analysis in the femur of AAV-R mice exhibited severe loss of trabecular bone (Figure 4G), as suggested by reduced BV/TV and Tb.N, as well as by increased Tb.Sp (Figure 4H).
Figure 4. In vivo elevated RANKL induces murine muscle atrophy, muscle weakness and bone loss.
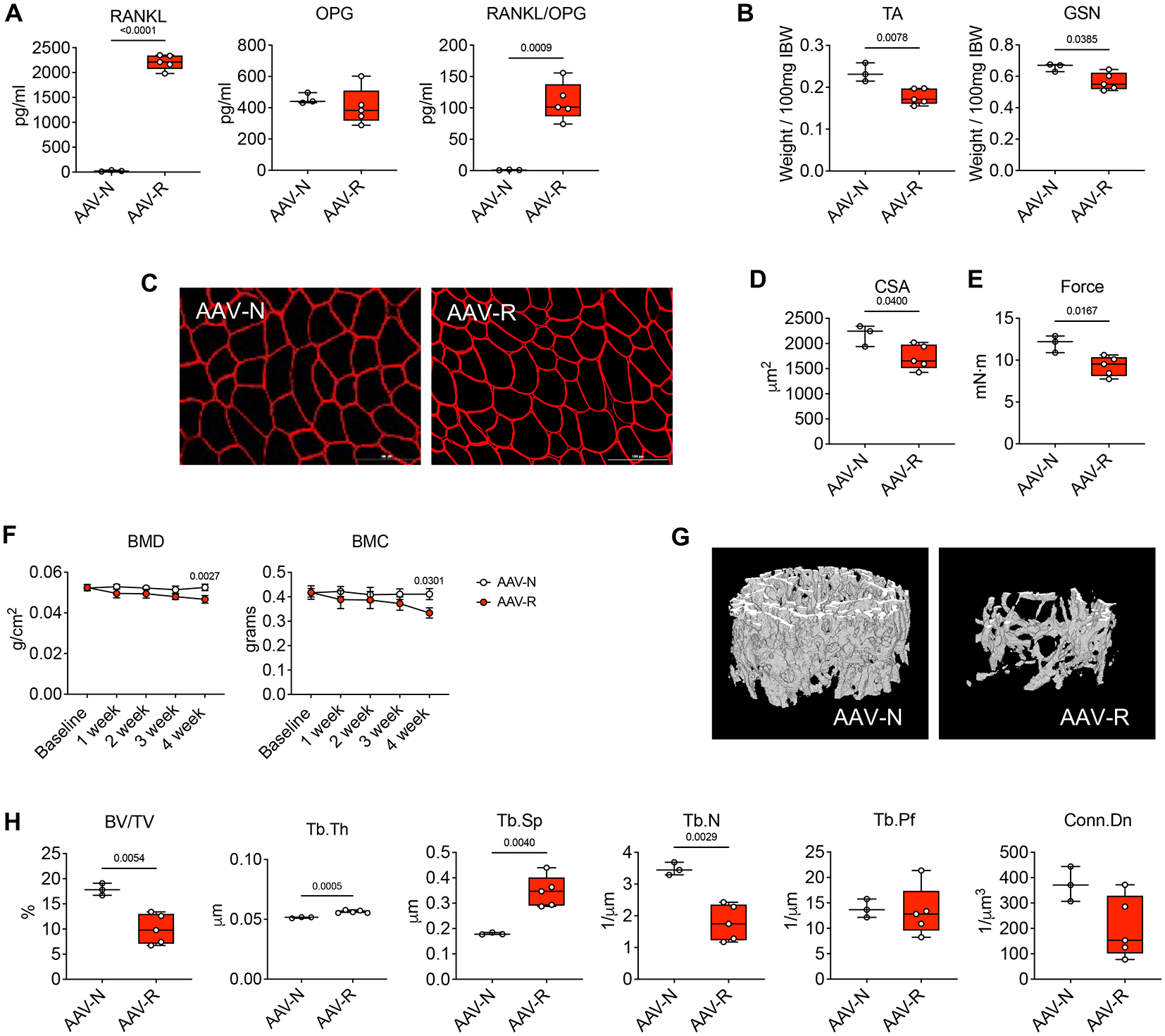
A: Plasma levels of RANKL and OPG, as well as RANKL/OPG ratio in healthy mice infected with AAV-NULL (AAV-N; n=3) or with AAV-RANKL (AAV-R; n=5). B: Skeletal muscle weights in tibialis anterior (TA) and gastrocnemius (GSN) muscles. Muscle weights were expressed as weight/100mg IBW. C-D: Representative images of TA muscle cross sections stained with anti-dystrophin antibody and cross-sectional area (CSA) of entire TA muscles from AAV-N (n=3) or AAV-R (n=5) mice. Scale bar: 100 μm. E: Total force recorded at 100 Hertz measured in the limbs of AAV-N (n=3) or AAV-R (n=5) mice. Data (means ± SD) are expressed as mN-m. F: Changes in total bone mineral density (BMD) and bone mineral content (BMC) in AAV-N (n=3) or AAV-R (n=5) mice. Data (means ± SD) are expressed as g (BMC) or g/cm2 (for BMD). (two-way repeated measure ANOVA with Tukey’s test). Interaction p values: 0.0363 and 0.2911 for BMD and BMC, respectively. G: Representative microCT-based three-dimensional rendering of trabecular bone in femurs from AAV-N (n=3) or AAV-R (n=5) mice. H: Assessment of trabecular bone volume fraction (BV/TV; expressed as %), trabecular thickness (Tb.Th; expressed as μm), trabecular separation (Tb.Sp; expressed as μm), trabecular number (Tb.N; expressed as 1/μm), trabecular pattern factor (Tb.Pf; expressed as 1/μm) and trabecular connectivity density (Conn.Dn; expressed as 1/μm 3) in the femurs from AAV-N (n=3) or AAV-R (n=5) mice. Data (means ± SD). Significance of the differences *p<0.05, **p<0.01, ***p<0.001 vs. PBS (unpaired t-test).
Tumor-derived RANKL exacerbates muscle and bone loss in murine cancer cachexia.
To further investigate the role of tumor-derived RANKL in driving muscle and bone wasting we used mice bearing C26 colorectal adenocarcinomas, a well-established model of cancer cachexia characterized by marked muscle wasting, absence of bone loss (13,37) and low RANKL (Figure 2E), and generated C26 cells stably overexpressing RANKL (C26pR). As shown in Figure S3A, RANKL, normally undetectable in the wild-type C26 cells (C26), was expressed in the C26pR clones, consistent with high RANKL levels in the CM (Figure S3B). To determine whether RANKL was sufficient to worsen cachexia and drive bone loss in the presence of non-metastatic tumors, male mice were injected with C26 or C26pR cells. After 11 days from tumor injection, levels of circulating RANKL in mice bearing the C26pR were markedly elevated in comparison to mice bearing C26 tumors (Figure 5A). Conversely, no changes were observed in the levels of OPG, altogether contributing to marked elevations in the RANKL/OPG ratio (Figure 5A). The loss of body weight, as well as final body weight, tumor-free body weight and carcass weight following growth of C26pR tumors were worsened compared to C26 (Figures 5B, S3C). Consistently, high circulating RANKL was able to exacerbate skeletal muscle atrophy induced by the C26 tumors (Figures 5C–D, S3D). Of note, the size of heart, liver, spleen and white adipose tissue (WAT) was unchanged in the C26pR hosts (Figure S3E). Similarly, no effects on tumor growth in the presence of high RANKL expression were observed (Figure 5E). Notably, the C26pR tumor growth also resulted in decreased whole body BMD and BMC (Figure 5F), in line with the loss of trabecular bone in the femur (Figure 5G), as suggested by reduced BV/TV and Tb.N, and by increased Tb.Sp (Figure 5H).
Figure 5. High RANKL exacerbates muscle and bone loss in murine cancer cachexia.
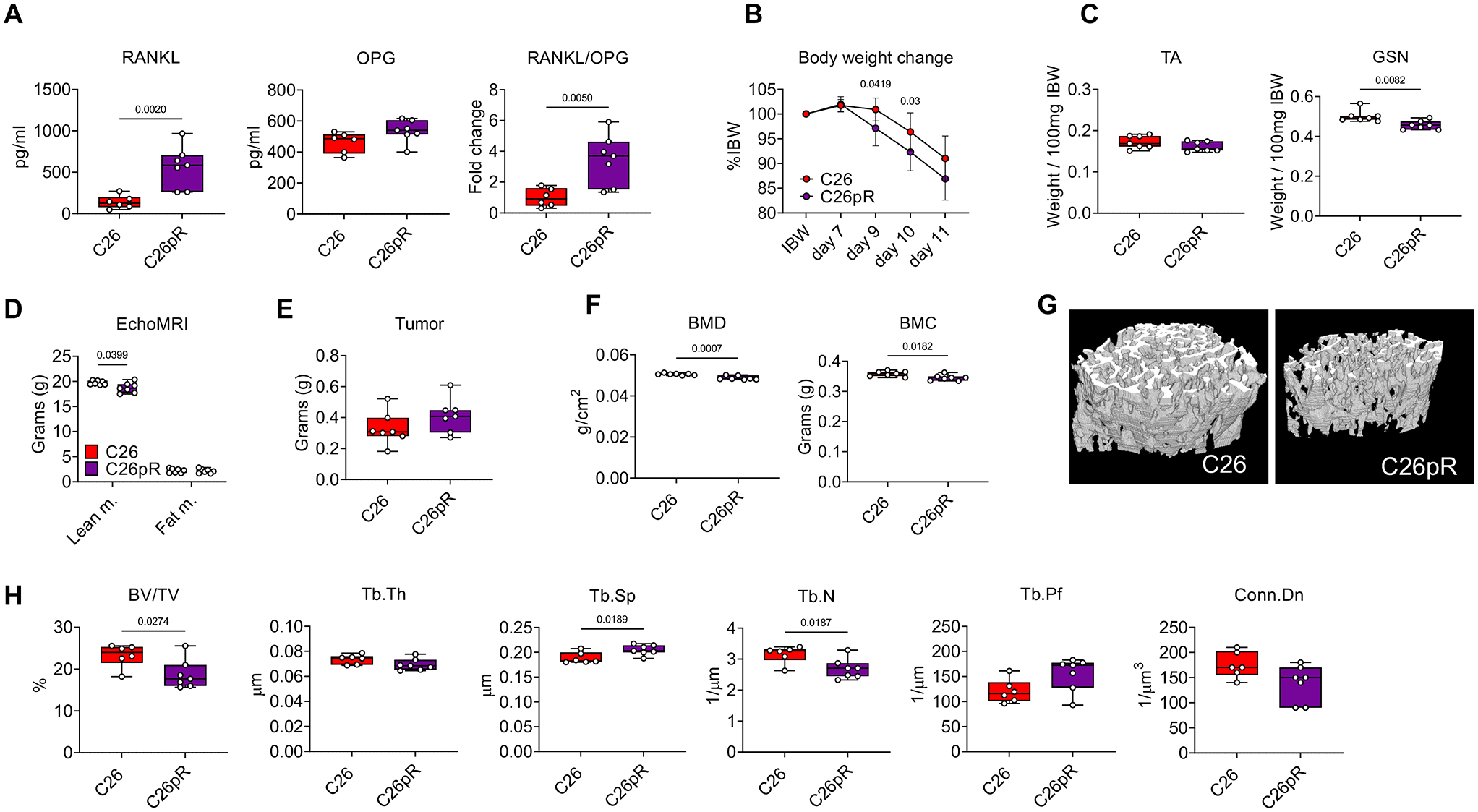
A: Plasma levels of RANKL and OPG, as well as RANKL/OPG ratio in C26 (n=6) or C26pR (n=7) barers. B: Body weight change curves during 11 days of tumor development in C26 (n=7) or C26pR (n=7) tumor-bearing mice. (two-way repeated measure ANOVA with Tukey’s test). Interaction p value: 0.1098. C: Skeletal muscle weights in tibialis anterior (TA) and gastrocnemius (GSN) muscles in C26 (n=7) or C26pR (n=7) tumor-bearing mice. Muscle weights were expressed as weight/100mg IBW. D: Lean and fat mass on the day of the sacrifice were assessed in C26 (n=7) or C26pR (n=7) tumor-bearing mice by using EchoMRI. Data (means ± SEM) are expressed in grams. E: Tumor mass in C26 (n=6) or C26pR (n=7) tumor-bearing mice. F: Changes in total bone mineral density (BMD) and bone mineral content (BMC) in C26 (n=6) or C26pR (n=7) tumor-bearing mice. Data (means ± SD) are expressed as g/cm2 (for BMD) or g (BMC). G: Representative microCT-based three-dimensional rendering of trabecular bone in femurs from C26 (n=6) or C26pR (n=6) tumor-bearing mice. H: Assessment of trabecular bone volume fraction (BV/TV; expressed as %), trabecular thickness (Tb.Th; expressed as μm), trabecular separation (Tb.Sp; expressed as μm), trabecular number (Tb.N; expressed as 1/μm), trabecular pattern factor (Tb.Pf; expressed as 1/μm) and trabecular connectivity density (Conn.Dn; expressed as 1/μm 3) in the femurs from C26 (n=6) or C26pR (n=6) tumor-bearing mice. Data (means ± SD). Significance of the differences *p<0.05, **p<0.01, ***p<0.001 vs. C26 (unpaired t-test).
Anti-RANKL treatments reduce myotube atrophy induced by ES-2 cells, improve muscle mass and strength, preserve bone mass and normalize RANKL/OPG expression in the OvCa hosts.
RANKL blockade was shown to improve the dystrophic muscle phenotype in mdx mice (25). We previously showed that C2C12 myotubes exposed to ES-2 CM present significantly reduced diameter. Hence, to determine whether tumor-derived RANKL was directly involved in causing myofiber atrophy, we co-cultured C2C12 myotubes with ES-2 cells and co-treated with anti-RANKL neutralizing antibodies and observed preservation of myotube size (Figure 6A), thereby suggesting that blockade of tumor-derived RANKL ameliorates myofiber size. To further validate our findings with different OvCa types, C2C12 myotubes were co-cultured with murine ID8 and human OVCAR3 cancer cells. Both cells lines were able to induce myotube atrophy (Figure S4A–B), in line with high Tnfsf11 mRNA expression (Figure S4C) and with previous reports (18). In order to verify whether the neutralization of RANKL was also able to improve the negative effect of OvCa growth on the musculoskeletal system, mice bearing ES-2 tumors were treated with neutralizing anti-RANKL antibodies (R Ab). In line with our initial results, the ES-2 progression caused significant loss of skeletal muscle mass, which was partially counteracted by the neutralization of RANKL, as suggested by improved Quad, GSN and TA size (Figure 6B). The administration of R Ab also partially preserved muscle strength in the ES-2 hosts (Figure 6C), whereas no effects of R Ab on muscle mass and strength were reported in the control group (Figure 6C–D). In line with improved skeletal muscle mass, treatment with R Ab was able to partially correct the expression of E3 ubiquitin ligases Fbxo32 and Trim63 (Figure 6D), which we previously showed increased in the skeletal muscle of ES-2 hosts (12), despite no changes in tumor growth (Figure 6H), as well as body weight, WAT, liver and spleen (Figure S5B). Similar to our previous data, the ES-2 bearers showed marked bone loss (Figure 6E–F), which was completely prevented by the administration of R Ab, as shown by ameliorated BV/TV, Tb.Th, Tb.N and Conn.Dn, as well as by reduced Tb.Sp, Tb.Pf (Figure 6E–F). In line with the data shown in Figure 2, also in this case the bones from the tumor-bearing mice exhibited altered Tnfsf11/Tnfrsf11b ratio (Figure 6G), whereas the treatment with R Ab was able to re-equilibrate the Tnfsf11/Tnfrsf11b ratio (Figure 6G), in line with increased Tnfrsf11 expression (Figure S5A).
Figure 6. Anti-RANKL treatments reduce myotube atrophy induced by ES-2 cells, improve muscle mass and strength, preserve bone mass and normalize RANKL/OPG expression in the OvCa hosts.
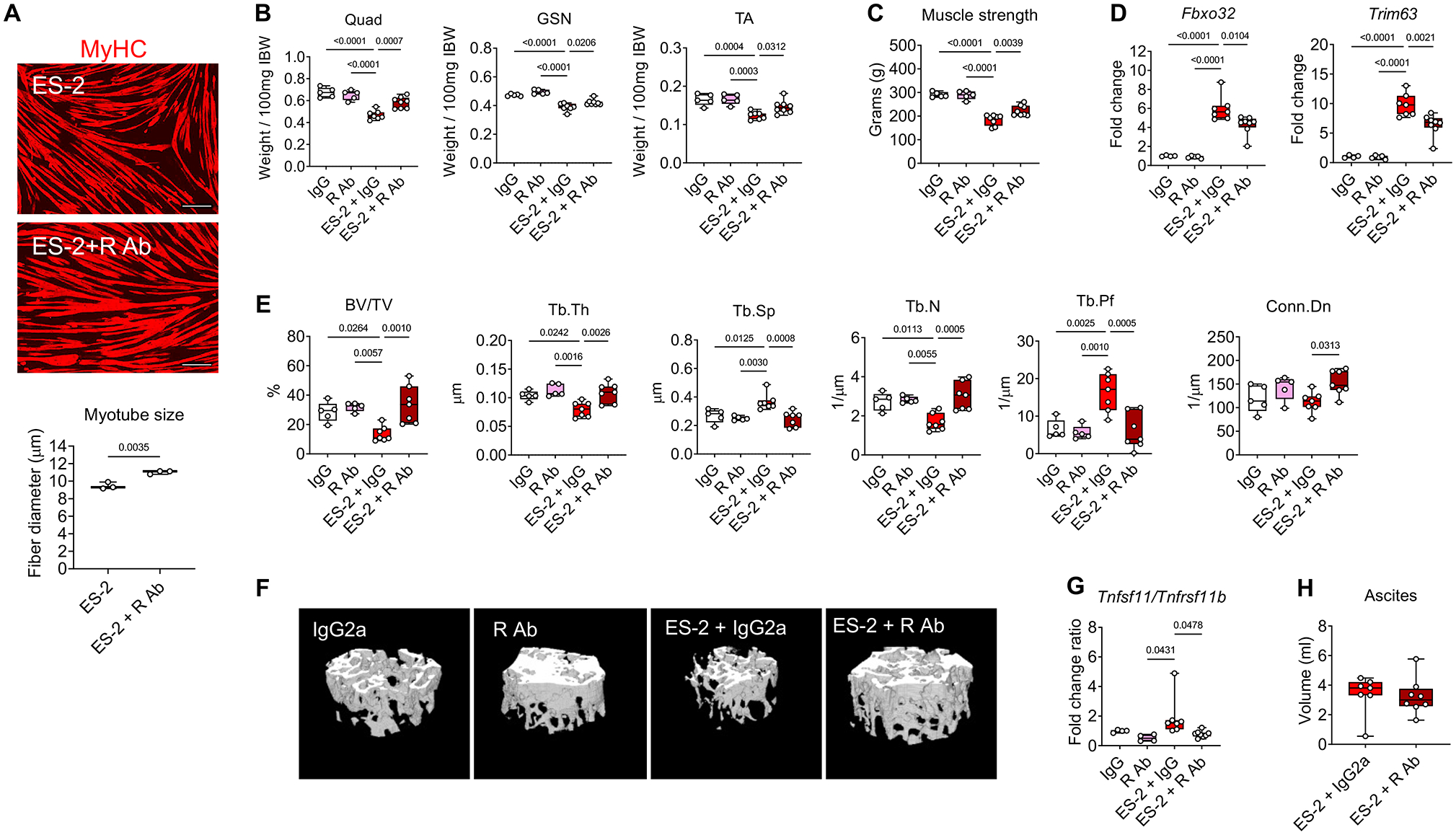
A: Representative images and quantification of C2C12 myotubes size co-cultured with ES-2 cells and exposed to anti-RANKL antibodies. Red staining: myosin heavy chain. Scale bar: 100 μm. B-C: Skeletal muscle weights and whole-body grip strength (expressed in grams) in IgG2 (n=5), R Ab (n=5), ES-2+IgG2 (n=7) or R Ab+IgG2 (n=8). Muscle weights were expressed as weight/100mg IBW. D: Gene expression levels for Fbxo32 and Trim63 (normalized to Tbpl1) in the quadriceps muscle of IgG2a (n=4), R Ab (n=5), ES-2+IgG2 (n=7) or ES-2+R Ab (n=8). Gene expression is expressed as fold change vs. IgG2a. E: Assessment of trabecular bone volume fraction (BV/TV; expressed as %), trabecular thickness (Tb.Th; expressed as μm), trabecular separation (Tb.Sp; expressed as μm), trabecular number (Tb.N; expressed as 1/μm), trabecular pattern factor (Tb.Pf; expressed as 1/μm) and trabecular connectivity density (Conn.Dn; expressed as 1/μm3) in the femurs from IgG (n=5), R Ab (n=5), ES-2+IgG2 (n=7) or ES-2+R Ab (n=7). F: Representative microCT-based three-dimensional rendering of trabecular bone in mouse femurs. G: mRNA expression levels for the Tnfsf11/Tnfrsf11b ratio in the marrow flushed-bone of IgG2a (n=4), R Ab (n=4), ES-2+IgG2 (n=7) or R Ab+IgG2 (n=8). Gene expression was normalized to Gapdh levels and expressed as fold change vs. IgG2. H: Tumor (ascites) volume in ES-2+IgG2 (n=7) or R Ab+IgG2 (n=8). Data (means ± SD). Significance of the differences: *p<0.05, **p<0.01, ***p<0.001 vs. IgG2a, ##p<0.01, ###p<0.001 vs. R Ab, $p<0.05, $$p<0.01, $$$p<0.001 vs. ES-2 (one-way ANOVA with Tukey’s test).
Discussion
Cachexia has been defined as a multi-organ comorbidity of cancer, though dysregulations of muscle-bone crosstalk, especially in the absence of tumor metastases to bone or as a result of abnormal release of tumor- or host-derived soluble factors, have been reported only recently (10,12,14,51,52). Despite difficulties in diagnosing changes in muscle mass due to ascites formation and bloating, sarcopenia is now known to play a critical role as a prognostic factor for survival in OvCa patients (2,53,54). Here we showed that individuals affected with OvCa present skeletal muscle atrophy and white adipose tissue wasting, both co-morbidities of cachexia which negatively impact quality of life and reduce efficacy of and tolerance to anticancer treatments (7). Only a small number of studies conducted in chemotherapy-naïve malignancies, including prostate and breast cancers (55,56), as well as cervical, ovarian and endometrial tumors (57,58), have shown evidence of bone derangements, e.g. reduced BMD, in the absence of bone metastases potentially as a consequence of tumor-derived factors, including prostaglandin, parathyroid hormone analogue and high-molecular-weight transforming growth factor activity (59–61). Keeping in mind that reduced BMD is frequently associated with increased risk of bone fractures and higher mortality rates, being able to diagnose enhanced bone resorption and osteoporosis in the absence of bone metastases or before bone lesions occur has become a relevant clinical problem. Here we showed that RANKL was elevated in multiple OvCa types and experimental cell lines. Along with previous evidence supporting a role for RANKL-RANK signaling in OvCa growth (18), this suggests that tumor-derived RANKL may play a role in driving the systemic effects of OvCa on muscle and bone. This idea is also supported by our data showing high circulating RANKL in the plasma of OvCa patients, along with increased levels of CTX-I, emphasizes negative effects on bone homeostasis from non-metastatic OvCa. Altogether, our observations showing reduced skeletal muscle mass, as well as a positive correlation between RANKL and CTX-I levels in subjects affected with OvCa strongly suggest the occurrence of musculoskeletal derangements from OvCa, which may ultimately worsen patient survival.
It has been shown that bisphosphonates represent effective treatment strategies to counteract bone loss in various disease states (62–64). Bisphosphonates attenuate muscle mass loss in burn patients, decrease muscle weakness in mice with breast cancer-induced bone metastases and counteract chemotherapy-induced musculoskeletal alterations (10,14,65). We showed that ZA not only blocked bone loss caused by the ES-2 tumors, but also partially prevented skeletal muscle atrophy and weakness. Our results suggest that abnormal muscle-bone crosstalk plays a role in cancer cachexia, and that bone-derived factors may drive the skeletal muscle derangements in OvCa hosts. In this regard, the correction of bone RANKL/OPG ratio by ZA treatment suggests that bone preservation may positively impact muscle in cancer also by preventing the release of RANKL from bone. We recently showed that ES-2 tumor-derived factor(s) directly induce osteocyte death and increase the RANKL/OPG ratio in osteocytes, both in vivo and in vitro (66). Similarly, immobilization-induced osteocyte apoptosis was reported in association with increased osteocytic RANKL(67). Hence, in our animal model the ZA treatment could reduce the release of osteocyte-derived RANKL mainly by preventing osteocyte death (68), thereby also normalizing the RANKL/OPG ratio systemically, as shown by the reduction of the circulation RANKL. Notably, body weight loss in cancer cachexia is also due to WAT wasting. As previously shown (12), cachexia induced by ES-2 tumors or by chemotherapeutics is also associated with severe WAT wasting. Along the same line, we previously reported that circulating IL-6 is elevated in ES-2 hosts, consistent with depletion of skeletal muscle and adipose tissue mass (12). Interestingly, Rupert et al. recently showed that tumor-derived IL-6 and trans-signaling among tumor, fat and muscle mediate cachexia in pancreatic cancer, therefore highlighting an IL-6 direct impact on adipose tissue wasting (69). However, contrarily to the positive effects exerted by antiresorptive treatments in skeletal muscle, in this study we did not detect preservation of WAT following administration of ZA or anti-RANKL antibodies, thus further suggesting that, at least in this animal model, the ZA-dependent reduction of RANKL directly impacts a bone-muscle crosstalk. This observation is also in line with our previous study showing that cisplatin-induced loss of WAT was not prevented in animals co-treated with bisphosphonates (14).
Even though osteocytes are the major source of RANKL (70), several different cell types, including lymphocytes and endothelial cell (71,72), as well as several cancer types (73) can contribute to RANKL levels. We showed that different OvCa cell lines are able to produce RANKL and concomitantly induce muscle atrophy, strengthening the concept that RANKL may act in a paracrine manner on distal organs and contribute to the onset of cachexia in OvCa. In our in vitro models, recombinant RANKL drove myofiber atrophy in both murine and human cultured myotubes, supporting RANKL’s ability to directly induce pro-cachectic effects on skeletal muscle, in line with recent data generated in C2C12 cultures (74). To further support the ability of RANKL to directly induce pro-atrophic effects on skeletal muscle, we showed that AAV-mediated RANKL overexpression was sufficient to drive local and systemic muscle fiber atrophy and weakness, along with increases in circulating RANKL. Consistent with previous data obtained in mice hyperexpressing human RANKL (75), the animals infected with AAV-R in this study exhibited dramatic bone turnover. Next, we investigated whether overexpression of RANKL per se was sufficient to exacerbate cachexia in conditions normally not associated with bone loss and high RANKL levels, such as mice bearing the C26 colorectal cancer (13). Interestingly, mice bearing RANKL-overexpressing C26 tumors showed exacerbated muscle atrophy and bone loss, therefore supporting the idea that tumor-derived RANKL actively participates in the pathogenesis of muscle and bone derangements in cachexia.
The only FDA-approved IgG2 monoclonal antibody targeting human RANKL with high selectivity and affinity is Denosumab (76). This treatment is currently used to correct the imbalance between bone formation and resorption in osteoporosis (77). Anti-RANKL therapies are also prescribed to handle radio/chemotherapy-induced bone loss and cancer-derived metastases to bone (78), as well as to maintain bone mass in patients undergoing hormone ablation therapy for the treatment of breast and prostate cancers (79). Due to the high levels of circulating RANKL in our OvCa patients, as well as in the ES-2 hosts, we sought to test the effects of antibody-mediated RANKL neutralization on bone and muscle in a mouse model of cancer cachexia. Treatment with anti-RANKL monoclonal antibodies completely preserved the trabecular bone and was also able to partially protect skeletal muscle mass and strength in OvCa hosts, consistent with in vitro observations that RANKL blockade prevented a reduction in myotube size of C2C12 myotubes co-cultured with ES-2 cells.
Our findings show for the first time the efficacy of anti-RANKL neutralizing strategies to ameliorate some of the most important derangements of cancer cachexia, i.e. muscle atrophy and weakness. Our results are also reinforced by recent results conducted in a cohort of osteoporotic women revealing increased appendicular lean mass and handgrip strength upon treatment with Denosumab (27). The anti-RANKL treatment was also shown to improve not only muscle integrity and function in a Duchenne muscular dystrophy preclinical model, but also to increase bone stiffness (26).
In light of this, the positive effects obtained by inhibiting RANKL signaling supports our hypothesis that tumor-derived RANKL plays a detrimental role in the regulation of muscle mass and function. The most relevant signaling cascade activated by RANKL-RANK interaction involves the recruitment of TRAF6, followed by activation of NF-kB (80). TRAF6 has been previously implicated in triggering muscle atrophy during cancer cachexia, primarily by regulating the proteolytic machinery (29). In our in vitro system, myotube atrophy induced by RANKL was associated with several changes in the expression of genes related to inflammatory and pro-atrophic pathways, such as NFkB, Tnfrsf12a, Tnfrsf1a/Tnfrsf1b and Toll-like Receptor. This is also in line with previous data showing increased NF-kB phosphorylation in Ser563 upon RANKL stimulation in C2C12 myotubes, thus strengthening the idea that the RANKL-RANK- NF-kB pathway may be involved in the regulation of myofiber size also in our in vitro models (74). Moreover, we showed partially corrected Fbxo32 and Trim63 expression in the skeletal muscle of ES-2 hosts treated with R Ab, suggesting that blockade of the RANKL-RANK interaction may inhibit the activation of NF-kB-dependent proteolysis.
Overall, we provided novel insights on the role of RANKL in muscle atrophy and in particular on the notion that cancer cachexia in OvCa is associated with increased bone turnover due to elevated tumor- and bone-derived RANKL. Despite the importance and the novelty of such results, we are aware of some limitations affecting our study. First, here we characterized cachexia in a model of OvCa induced by human ES-2 tumors. Despite the fact that the ES-2 cells were originally derived from clear cell ovarian carcinomas, this cell line was later classified as a suitable model for the study of HGS-OC based on copy number alterations, TP53 mutations and low frequency of somatic mutations in protein-coding regions (35) and on the in vivo phenotype resembling the clinical presentation of HGS-OC (12). Quite surprisingly, recent studies described the ES-2 cells similar to endometrial carcinoma (81) or even suspected to be skin cutaneous melanoma (SKCM). Nevertheless, independent the origin of the ES-2 cells, they remain an excellent model for the study of musculoskeletal effects associated with RANKL-expressing tumors. Second, the cohort of patients used for the body composition assessment based on CT imaging was different from the one used for the measurement of the CTX-I and RANKL levels, thus only allowing to speculate that skeletal muscle atrophy occurs in concomitance with increased bone turnover. We also do not have information on whether these OvCa patients presented with evidence of osteoporosis or were receiving concurrent treatments with antiresorptive agents. However, in order to overcome such limitation, we carefully chose a cutoff of 60 years of age to avoid the inclusion of potentially osteoporotic subjects in our study. Lastly, due to the retrospective nature of our human studies, we were not able to provide information on functional status in the patients affected with OvCa. Further analyses are warranted to determine whether bone mass is directly affected in patients with non-bone metastatic OvCa, along with randomized clinical trials aimed to validate the effect of RANKL on cachexia induced by different types of cancer to the extent of elucidating whether alterations of muscle function ultimately correlate with loss of muscle mass and with enhanced bone turnover.
In summary, here we report evidence that OvCa patients present underlying cachexia. The detection of high plasma CTX-I and RANKL suggested for the first time that bone turnover can be accelerated in subjects affected with OvCa, even in the absence of bone metastases. Notably, RANKL can be expressed by different OvCa types, and high circulating RANKL frequently associates with dismal prognosis. By taking advantage of multiple in vitro and in vivo approaches, we showed that the RANKL-RANK axis plays a role in driving muscle atrophy and weakness during cancer cachexia, consistent with evidence that RANKL originating from non-bone metastatic tumors is sufficient to induce bone loss and muscle wasting, even without direct tumor dissemination to bone. Our observations showing beneficial effects resulting from the treatment of ES-2 hosts with ZA or anti-RANKL antibodies opens up new opportunities for the testing of safe, well-characterized and FDA-approved antiresorptive drugs as standard of care for the management of muscle and bone defects in OvCa.
Supplementary Material
Acknowledgments
This study was supported by the Department of Surgery and the Department of Otolaryngology – Head & Neck Surgery at Indiana University, by grants from the National Cancer Institute (R01CA122596, R01CA194593), the Veterans Administration (1I01-BX004177-01), the Lustgarten Foundation, the IU Simon Cancer Center (NIH P30CA082709) to TAZ, by grants from the National Institute of Aging PO1AG039355 to LFB, and by grants from the Showalter Research Trust, the V Foundation for Cancer Research (V2017-021), the American Cancer Society (Research Scholar Grant 132013-RSG-18-010-01-CCG) and the National Institute of Arthritis and Musculoskeletal and Skin Diseases (R01AR079379) to AB. The #MF-20 anti-Myosin Heavy Chain monoclonal antibody (developed by Fischman DA at Cornell University) and the #MANDRA1(7A10) anti-Dystrophin monoclonal antibody (developed by Morris GE at NE Wales Institute) were obtained from the Developmental Studies Hybridoma Bank, created by the NICHD of the NIH and maintained at The University of Iowa, Department of Biology, Iowa City, IA. We thank the Simon Comprehensive Cancer Center at Indiana University School of Medicine funded by the IU Simon Cancer Center Support Grant P30 CA082709 for the use of the In Vivo Therapeutic Core, which provided the NSG mice, and the Tissue Procurement and Distribution Core, which provided the plasma samples from OvCa patients. The RNA Sequencing was carried out in the Center for Medical Genomics at Indiana University School of Medicine, which is partially supported by the Indiana Genomic Initiative at Indiana University (INGEN); INGEN is supported in part by the Lilly Endowment, Inc. The survival data in the OvCa patients were generated thanks to the Human Protein Atlas database, available from http://www.proteinatlas.org.
Footnotes
Conflicts of interest
The authors have declared that no conflict of interest exists.
Data Availability
The datasets produced in this study are available in the following databases:
RNA-Seq data: Gene Expression Omnibus GSE149606 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE149606)
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. Jan 2020;70(1):7–30. Epub 2020/01/09. [DOI] [PubMed] [Google Scholar]
- 2.Bronger H, Hederich P, Hapfelmeier A, Metz S, Noel PB, Kiechle M, et al. Sarcopenia in Advanced Serous Ovarian Cancer. Int J Gynecol Cancer. Feb 2017;27(2):223–32. Epub 2016/11/22. [DOI] [PubMed] [Google Scholar]
- 3.Gadducci A, Cosio S, Fanucchi A, Genazzani AR. Malnutrition and cachexia in ovarian cancer patients: pathophysiology and management. Anticancer Res. Jul–Aug 2001;21(4B):2941–7. Epub 2001/11/20. [PubMed] [Google Scholar]
- 4.Huang CY, Yang YC, Chen TC, Chen JR, Chen YJ, Wu MH, et al. Muscle loss during primary debulking surgery and chemotherapy predicts poor survival in advanced-stage ovarian cancer. J Cachexia Sarcopenia Muscle. Apr 2020;11(2):534–46. Epub 2020/01/31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hayes S, Battistutta D, Newman B. Objective and subjective upper body function six months following diagnosis of breast cancer. Breast Cancer Res Treat. Nov 2005;94(1):1–10. [DOI] [PubMed] [Google Scholar]
- 6.Meeske K, Smith AW, Alfano CM, McGregor BA, McTiernan A, Baumgartner KB, et al. Fatigue in breast cancer survivors two to five years post diagnosis: a HEAL Study report. Qual Life Res. Aug 2007;16(6):947–60. Epub 2007/04/26. [DOI] [PubMed] [Google Scholar]
- 7.Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, et al. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol. May 2011;12(5):489–95. Epub 2011/02/08. [DOI] [PubMed] [Google Scholar]
- 8.Guntur AR, Rosen CJ. Bone as an endocrine organ. Endocr Pract. Sep–Oct 2012;18(5):758–62. Epub 2012/07/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Severinsen MCK, Pedersen BK. Muscle-Organ Crosstalk: The Emerging Roles of Myokines. Endocr Rev. Aug 1 2020;41(4). Epub 2020/05/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Waning DL, Mohammad KS, Reiken S, Xie W, Andersson DC, John S, et al. Excess TGF-beta mediates muscle weakness associated with bone metastases in mice. Nature medicine. Nov 2015;21(11):1262–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wright LE, Harhash AA, Kozlow WM, Waning DL, Regan JN, She Y, et al. Aromatase inhibitor-induced bone loss increases the progression of estrogen receptor-negative breast cancer in bone and exacerbates muscle weakness in vivo. Oncotarget. Jan 31 2017;8(5):8406–19. Epub 2017/01/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pin F, Barreto R, Kitase Y, Mitra S, Erne CE, Novinger LJ, et al. Growth of ovarian cancer xenografts causes loss of muscle and bone mass: a new model for the study of cancer cachexia. Journal of cachexia, sarcopenia and muscle. Aug 2018;9(4):685–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bonetto A, Kays JK, Parker VA, Matthews RR, Barreto R, Puppa MJ, et al. Differential Bone Loss in Mouse Models of Colon Cancer Cachexia. Front Physiol. 2016;7:679. Epub 2017/01/27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Essex AL, Pin F, Huot JR, Bonewald LF, Plotkin LI, Bonetto A. Bisphosphonate Treatment Ameliorates Chemotherapy-Induced Bone and Muscle Abnormalities in Young Mice. Front Endocrinol (Lausanne). 2019;10:809. Epub 2019/12/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barreto R, Kitase Y, Matsumoto T, Pin F, Colston KC, Couch KE, et al. ACVR2B/Fc counteracts chemotherapy-induced loss of muscle and bone mass. Sci Rep. Oct 31 2017;7(1):14470. Epub 2017/11/02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fuller K, Wong B, Fox S, Choi Y, Chambers TJ. TRANCE is necessary and sufficient for osteoblast-mediated activation of bone resorption in osteoclasts. J Exp Med. Sep 7 1998;188(5):997–1001. Epub 1998/09/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dufresne SS, Dumont NA, Boulanger-Piette A, Fajardo VA, Gamu D, Kake-Guena SA, et al. Muscle RANK is a key regulator of Ca2+ storage, SERCA activity, and function of fast-twitch skeletal muscles. Am J Physiol Cell Physiol. Apr 15 2016;310(8):C663–72. Epub 2016/01/31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wieser V, Sprung S, Tsibulak I, Haybaeck J, Hackl H, Fiegl H, et al. Clinical Impact of RANK Signalling in Ovarian Cancer. Cancers. Jun 8 2019;11(6). Epub 2019/06/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones DH, Nakashima T, Sanchez OH, Kozieradzki I, Komarova SV, Sarosi I, et al. Regulation of cancer cell migration and bone metastasis by RANKL. Nature. Mar 30 2006;440(7084):692–6. Epub 2006/03/31. [DOI] [PubMed] [Google Scholar]
- 20.Santini D, Perrone G, Roato I, Godio L, Pantano F, Grasso D, et al. Expression pattern of receptor activator of NFkappaB (RANK) in a series of primary solid tumors and related bone metastases. J Cell Physiol. Mar 2011;226(3):780–4. Epub 2010/09/22. [DOI] [PubMed] [Google Scholar]
- 21.Zhang X, Song Y, Song N, Zhang L, Wang Y, Li D, et al. Rankl expression predicts poor prognosis in gastric cancer patients: results from a retrospective and single-center analysis. Braz J Med Biol Res. Jan 11 2018;51(3):e6265. Epub 2018/01/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Theoleyre S, Wittrant Y, Tat SK, Fortun Y, Redini F, Heymann D. The molecular triad OPG/RANK/RANKL: involvement in the orchestration of pathophysiological bone remodeling. Cytokine Growth Factor Rev. Dec 2004;15(6):457–75. Epub 2004/11/25. [DOI] [PubMed] [Google Scholar]
- 23.Leibbrandt A, Penninger JM. RANK/RANKL: regulators of immune responses and bone physiology. Ann N Y Acad Sci. Nov 2008;1143:123–50. Epub 2008/12/17. [DOI] [PubMed] [Google Scholar]
- 24.Luan X, Lu Q, Jiang Y, Zhang S, Wang Q, Yuan H, et al. Crystal structure of human RANKL complexed with its decoy receptor osteoprotegerin. J Immunol. Jul 1 2012;189(1):245–52. Epub 2012/06/06. [DOI] [PubMed] [Google Scholar]
- 25.Dufresne SS, Boulanger-Piette A, Bosse S, Argaw A, Hamoudi D, Marcadet L, et al. Genetic deletion of muscle RANK or selective inhibition of RANKL is not as effective as full-length OPG-fc in mitigating muscular dystrophy. Acta Neuropathol Commun. Apr 24 2018;6(1):31. Epub 2018/04/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hamoudi D, Marcadet L, Piette Boulanger A, Yagita H, Bouredji Z, Argaw A, et al. An anti-RANKL treatment reduces muscle inflammation and dysfunction and strengthens bone in dystrophic mice. Hum Mol Genet. Sep 15 2019;28(18):3101–12. Epub 2019/06/11. [DOI] [PubMed] [Google Scholar]
- 27.Bonnet N, Bourgoin L, Biver E, Douni E, Ferrari S. RANKL inhibition improves muscle strength and insulin sensitivity and restores bone mass. J Clin Invest. May 23 2019;129(8):3214–23. Epub 2019/05/24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li J, Yi X, Yao Z, Chakkalakal JV, Xing L, Boyce BF. TNF Receptor-Associated Factor 6 Mediates TNFalpha-Induced Skeletal Muscle Atrophy in Mice During Aging. J Bone Miner Res. Apr 8 2020. Epub 2020/04/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paul PK, Gupta SK, Bhatnagar S, Panguluri SK, Darnay BG, Choi Y, et al. Targeted ablation of TRAF6 inhibits skeletal muscle wasting in mice. J Cell Biol. Dec 27 2010;191(7):1395–411. Epub 2010/12/29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Amin MB, Greene FL, Edge SB, Compton CC, Gershenwald JE, Brookland RK, et al. The Eighth Edition AJCC Cancer Staging Manual: Continuing to build a bridge from a population-based to a more “personalized” approach to cancer staging. CA Cancer J Clin. Mar 2017;67(2):93–9. Epub 2017/01/18. [DOI] [PubMed] [Google Scholar]
- 31.Oken MM, Creech RH, Tormey DC, Horton J, Davis TE, McFadden ET, et al. Toxicity and response criteria of the Eastern Cooperative Oncology Group. Am J Clin Oncol. Dec 1982;5(6):649–55. Epub 1982/12/01. [PubMed] [Google Scholar]
- 32.Fearon KC, Voss AC, Hustead DS, Cancer Cachexia Study G. Definition of cancer cachexia: effect of weight loss, reduced food intake, and systemic inflammation on functional status and prognosis. Am J Clin Nutr. Jun 2006;83(6):1345–50. Epub 2006/06/10. [DOI] [PubMed] [Google Scholar]
- 33.Evans WJ, Morley JE, Argiles J, Bales C, Baracos V, Guttridge D, et al. Cachexia: a new definition. Clinical nutrition. Dec 2008;27(6):793–9. [DOI] [PubMed] [Google Scholar]
- 34.Silva de Paula N, de Aguiar Bruno K, Azevedo Aredes M, Villaca Chaves G. Sarcopenia and Skeletal Muscle Quality as Predictors of Postoperative Complication and Early Mortality in Gynecologic Cancer. Int J Gynecol Cancer. Feb 2018;28(2):412–20. Epub 2017/12/22. [DOI] [PubMed] [Google Scholar]
- 35.Domcke S, Sinha R, Levine DA, Sander C, Schultz N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat Commun. 2013;4:2126. Epub 2013/07/11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mitra AK, Davis DA, Tomar S, Roy L, Gurler H, Xie J, et al. In vivo tumor growth of high-grade serous ovarian cancer cell lines. Gynecol Oncol. Aug 2015;138(2):372–7. Epub 2015/06/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bonetto A, Rupert JE, Barreto R, Zimmers TA. The Colon-26 Carcinoma Tumor-bearing Mouse as a Model for the Study of Cancer Cachexia. J Vis Exp. Nov 30 2016(117). Epub 2016/12/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roby KF, Taylor CC, Sweetwood JP, Cheng Y, Pace JL, Tawfik O, et al. Development of a syngeneic mouse model for events related to ovarian cancer. Carcinogenesis. Apr 2000;21(4):585–91. Epub 2000/04/07. [DOI] [PubMed] [Google Scholar]
- 39.Hamilton TC, Young RC, McKoy WM, Grotzinger KR, Green JA, Chu EW, et al. Characterization of a human ovarian carcinoma cell line (NIH:OVCAR-3) with androgen and estrogen receptors. Cancer Res. Nov 1983;43(11):5379–89. Epub 1983/11/01. [PubMed] [Google Scholar]
- 40.Chen JL, Walton KL, Winbanks CE, Murphy KT, Thomson RE, Makanji Y, et al. Elevated expression of activins promotes muscle wasting and cachexia. FASEB J. Apr 2014;28(4):1711–23. [DOI] [PubMed] [Google Scholar]
- 41.Fontes-Oliveira CC, Busquets S, Toledo M, Penna F, Paz Aylwin M, Sirisi S, et al. Mitochondrial and sarcoplasmic reticulum abnormalities in cancer cachexia: altered energetic efficiency? Biochim Biophys Acta. Mar 2013;1830(3):2770–8. Epub 2012/12/04. [DOI] [PubMed] [Google Scholar]
- 42.Huot JR, Pin F, Narasimhan A, Novinger LJ, Keith AS, Zimmers TA, et al. ACVR2B antagonism as a countermeasure to multi-organ perturbations in metastatic colorectal cancer cachexia. J Cachexia Sarcopenia Muscle. Dec 2020;11(6):1779–98. Epub 2020/11/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. Jan 1 2013;29(1):15–21. Epub 2012/10/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Breese MR, Liu Y. NGSUtils: a software suite for analyzing and manipulating next-generation sequencing datasets. Bioinformatics. Feb 15 2013;29(4):494–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. Apr 01 2014;30(7):923–30. [DOI] [PubMed] [Google Scholar]
- 46.Ewels P, Magnusson M, Lundin S, Kaller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. Oct 1 2016;32(19):3047–8. Epub 2016/06/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. Jan 1 2010;26(1):139–40. Epub 2009/11/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. Jan 1 2002;30(1):207–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mourtzakis M, Prado CM, Lieffers JR, Reiman T, McCargar LJ, Baracos VE. A practical and precise approach to quantification of body composition in cancer patients using computed tomography images acquired during routine care. Appl Physiol Nutr Metab. Oct 2008;33(5):997–1006. Epub 2008/10/17. [DOI] [PubMed] [Google Scholar]
- 50.Cheung WY, Simmons CA, You L. Osteocyte apoptosis regulates osteoclast precursor adhesion via osteocytic IL-6 secretion and endothelial ICAM-1 expression. Bone. Jan 2012;50(1):104–10. Epub 2011/10/12. [DOI] [PubMed] [Google Scholar]
- 51.Pin F, Bonetto A, Bonewald LF, Klein GL. Molecular Mechanisms Responsible for the Rescue Effects of Pamidronate on Muscle Atrophy in Pediatric Burn Patients. Front Endocrinol (Lausanne). 2019;10:543. Epub 2019/08/27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huot JR, Novinger LJ, Pin F, Narasimhan A, Zimmers TA, O’Connell TM, et al. Formation of colorectal liver metastases induces musculoskeletal and metabolic abnormalities consistent with exacerbated cachexia. JCI Insight. May 7 2020;5(9). Epub 2020/04/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Torres ML, Hartmann LC, Cliby WA, Kalli KR, Young PM, Weaver AL, et al. Nutritional status, CT body composition measures and survival in ovarian cancer. Gynecologic oncology. Jun 2013;129(3):548–53. [DOI] [PubMed] [Google Scholar]
- 54.Rutten IJ, Ubachs J, Kruitwagen RF, van Dijk DP, Beets-Tan RG, Massuger LF, et al. The influence of sarcopenia on survival and surgical complications in ovarian cancer patients undergoing primary debulking surgery. Eur J Surg Oncol. Apr 2017;43(4):717–24. [DOI] [PubMed] [Google Scholar]
- 55.Berruti A, Dogliotti L, Terrone C, Cerutti S, Isaia G, Tarabuzzi R, et al. Changes in bone mineral density, lean body mass and fat content as measured by dual energy x-ray absorptiometry in patients with prostate cancer without apparent bone metastases given androgen deprivation therapy. J Urol. Jun 2002;167(6):2361–7; discussion 7. Epub 2002/05/07. [PubMed] [Google Scholar]
- 56.Wei JT, Gross M, Jaffe CA, Gravlin K, Lahaie M, Faerber GJ, et al. Androgen deprivation therapy for prostate cancer results in significant loss of bone density. Urology. Oct 1999;54(4):607–11. Epub 1999/10/08. [DOI] [PubMed] [Google Scholar]
- 57.Lee JE, Park CY, Lee E, Ji YI. Effect of gynecological cancer and its treatment on bone mineral density and the risk of osteoporosis and osteoporotic fracture. Obstet Gynecol Sci. Jul 2020;63(4):470–9. Epub 2020/07/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hung YC, Yeh LS, Chang WC, Lin CC, Kao CH. Prospective study of decreased bone mineral density in patients with cervical cancer without bone metastases: a preliminary report. Jpn J Clin Oncol. Oct 2002;32(10):422–4. Epub 2002/11/27. [DOI] [PubMed] [Google Scholar]
- 59.Seyberth HW, Segre GV, Morgan JL, Sweetman BJ, Potts JT Jr., Oates JA. Prostaglandins as mediators of hypercalcemia associated with certain types of cancer. N Engl J Med. Dec 18 1975;293(25):1278–83. Epub 1975/12/28. [DOI] [PubMed] [Google Scholar]
- 60.Sherwin SA, Twardzik DR, Bohn WH, Cockley KD, Todaro GJ. High-molecular-weight transforming growth factor activity in the urine of patients with disseminated cancer. Cancer Res. Jan 1983;43(1):403–7. Epub 1983/01/01. [PubMed] [Google Scholar]
- 61.Suva LJ, Winslow GA, Wettenhall RE, Hammonds RG, Moseley JM, Diefenbach-Jagger H, et al. A parathyroid hormone-related protein implicated in malignant hypercalcemia: cloning and expression. Science. Aug 21 1987;237(4817):893–6. Epub 1987/08/21. [DOI] [PubMed] [Google Scholar]
- 62.Agarwala S, Vijayvargiya M. Single Dose Therapy of Zoledronic Acid for the Treatment of Transient Osteoporosis of Hip. Ann Rehabil Med. Jun 2019;43(3):314–20. Epub 2019/07/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.O’Carrigan B, Wong MH, Willson ML, Stockler MR, Pavlakis N, Goodwin A. Bisphosphonates and other bone agents for breast cancer. Cochrane Database Syst Rev. Oct 30 2017;10:CD003474. Epub 2017/10/31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.von Moos R, Costa L, Gonzalez-Suarez E, Terpos E, Niepel D, Body JJ. Management of bone health in solid tumours: From bisphosphonates to a monoclonal antibody. Cancer Treat Rev. Jun 2019;76:57–67. Epub 2019/05/29. [DOI] [PubMed] [Google Scholar]
- 65.Borsheim E, Herndon DN, Hawkins HK, Suman OE, Cotter M, Klein GL. Pamidronate attenuates muscle loss after pediatric burn injury. J Bone Miner Res. Jun 2014;29(6):1369–72. Epub 2013/12/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pin F, Prideaux M, Huot JR, Essex AL, Plotkin LI, Bonetto A, et al. Non-bone metastatic cancers promote osteocyte-induced bone destruction. Cancer Lett. Nov 1 2021;520:80–90. Epub 2021/07/08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Plotkin LI, Gortazar AR, Davis HM, Condon KW, Gabilondo H, Maycas M, et al. Inhibition of osteocyte apoptosis prevents the increase in osteocytic receptor activator of nuclear factor kappaB ligand (RANKL) but does not stop bone resorption or the loss of bone induced by unloading. J Biol Chem. Jul 31 2015;290(31):18934–42. Epub 2015/06/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Plotkin LI, Weinstein RS, Parfitt AM, Roberson PK, Manolagas SC, Bellido T. Prevention of osteocyte and osteoblast apoptosis by bisphosphonates and calcitonin. J Clin Invest. Nov 1999;104(10):1363–74. Epub 1999/11/24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rupert JE, Narasimhan A, Jengelley DHA, Jiang Y, Liu J, Au E, et al. Tumor-derived IL-6 and trans-signaling among tumor, fat, and muscle mediate pancreatic cancer cachexia. J Exp Med. Jun 7 2021;218(6). Epub 2021/04/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xiong J, Piemontese M, Onal M, Campbell J, Goellner JJ, Dusevich V, et al. Osteocytes, not Osteoblasts or Lining Cells, are the Main Source of the RANKL Required for Osteoclast Formation in Remodeling Bone. PLoS One. 2015;10(9):e0138189. Epub 2015/09/24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kanematsu M, Sato T, Takai H, Watanabe K, Ikeda K, Yamada Y. Prostaglandin E2 induces expression of receptor activator of nuclear factor-kappa B ligand/osteoprotegrin ligand on pre-B cells: implications for accelerated osteoclastogenesis in estrogen deficiency. J Bone Miner Res. Jul 2000;15(7):1321–9. Epub 2000/07/14. [DOI] [PubMed] [Google Scholar]
- 72.Collin-Osdoby P, Rothe L, Anderson F, Nelson M, Maloney W, Osdoby P. Receptor activator of NF-kappa B and osteoprotegerin expression by human microvascular endothelial cells, regulation by inflammatory cytokines, and role in human osteoclastogenesis. J Biol Chem. Jun 8 2001;276(23):20659–72. Epub 2001/03/29. [DOI] [PubMed] [Google Scholar]
- 73.Renema N, Navet B, Heymann MF, Lezot F, Heymann D. RANK-RANKL signalling in cancer. Biosci Rep. Aug 2016;36(4). Epub 2016/06/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hamoudi D, Bouredji Z, Marcadet L, Yagita H, Landry LB, Argaw A, et al. Muscle weakness and selective muscle atrophy in osteoprotegerin-deficient mice. Hum Mol Genet. Feb 1 2020;29(3):483–94. Epub 2020/01/17. [DOI] [PubMed] [Google Scholar]
- 75.Rinotas V, Niti A, Dacquin R, Bonnet N, Stolina M, Han CY, et al. Novel genetic models of osteoporosis by overexpression of human RANKL in transgenic mice. J Bone Miner Res. 2014;29(5):1158–69. Epub 2013/10/16. [DOI] [PubMed] [Google Scholar]
- 76.Kostenuik PJ, Nguyen HQ, McCabe J, Warmington KS, Kurahara C, Sun N, et al. Denosumab, a fully human monoclonal antibody to RANKL, inhibits bone resorption and increases BMD in knock-in mice that express chimeric (murine/human) RANKL. J Bone Miner Res. Feb 2009;24(2):182–95. [DOI] [PubMed] [Google Scholar]
- 77.Zhang N, Zhang ZK, Yu Y, Zhuo Z, Zhang G, Zhang BT. Pros and Cons of Denosumab Treatment for Osteoporosis and Implication for RANKL Aptamer Therapy. Front Cell Dev Biol. 2020;8:325. Epub 2020/06/02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McClung MR. Inhibition of RANKL as a treatment for osteoporosis: preclinical and early clinical studies. Curr Osteoporos Rep. Mar 2006;4(1):28–33. [DOI] [PubMed] [Google Scholar]
- 79.Lipton A, Smith MR, Ellis GK, Goessl C. Treatment-induced bone loss and fractures in cancer patients undergoing hormone ablation therapy: efficacy and safety of denosumab. Clin Med Insights Oncol. 2012;6:287–303. Epub 2012/08/31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Franzoso G, Carlson L, Xing L, Poljak L, Shores EW, Brown KD, et al. Requirement for NF-kappaB in osteoclast and B-cell development. Genes Dev. Dec 15 1997;11(24):3482–96. Epub 1998/02/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Anglesio MS, Wiegand KC, Melnyk N, Chow C, Salamanca C, Prentice LM, et al. Type-specific cell line models for type-specific ovarian cancer research. PloS one. 2013;8(9):e72162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chandrashekar DS, Bashel B, Balasubramanya SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi B, et al. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia. Aug 2017;19(8):649–58. Epub 2017/07/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets produced in this study are available in the following databases:
RNA-Seq data: Gene Expression Omnibus GSE149606 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE149606)