

Abstract
Autophagy mediates cellular quality control mechanisms and energy homeostasis through lysosomal degradation. Autophagy is typically viewed as an adaptive process that allows cells to survive against stress, such as nutrient deprivation and hypoxia. However, autophagy also mediates cell death during development and in response to stress. Cell death accompanied by autophagy activation and accumulation of autophagosomes has been classified as type II programmed cell death. Compared to the wealth of knowledge regarding the adaptive role of autophagy, however, the molecular mechanisms through which autophagy induces cell death and its functional significance are poorly understood. Autophagy is activated excessively under some conditions, causing uncontrolled degradation of cellular materials and cell death. An imbalance between autophagosome formation and lysosomal degradation causes a massive accumulation of autophagosomes, which subsequently causes cellular dysfunction and death. Dysregulation of autophagy induces a unique form of cell death, termed autosis, with defined morphological and biochemical features distinct from other forms of programmed cell death, such as apoptosis and necrosis. In the heart, dysregulated autophagy induces death of cardiomyocytes and actively mediates cardiac injury and dysfunction in some conditions, including reperfusion injury, doxorubicin cardiomyopathy, and lysosomal storage disorders. The goal in this review is to introduce the concept of autophagic cell death and discuss its functional significance in various cardiac conditions.
1. Introduction
Autophagy is a major mechanism of cellular degradation that allows cells to survive against starvation and other stress conditions by facilitating the sequestration and degradation of bulk cytoplasm, damaged organelles, like mitochondria, and protein aggregates [1–3]. Autophagy degrades misfolded or dysfunctional proteins and organelles, thereby serving as a cellular quality control mechanism. In addition, cellular materials degraded by autophagy are recycled to maintain energy homeostasis. Thus, autophagy generally acts as an adaptive mechanism for cellular function and survival. However, dysregulated autophagy can induce cell death under some conditions [4–6]. Cell death accompanied by accumulation of vacuoles induced by autophagy has been classified as type II programmed cell death [7]. Although autophagy affects other forms of cell death, including apoptosis, through competition for Bcl-2 binding between Beclin 1 and Bax, cell death mediated through autophagy utilizes mechanisms that are distinct from those involved in other forms of programmed cell death (Figure 1). Compared to the adaptive role of autophagy, however, the role of autophagy in cell death has been poorly understood. This is in part because proving unequivocally that autophagy plays a direct and causative role in mediating cell death has been challenging. However, autophagy has been shown to actively participate in programmed cell death during Drosophila metamorphosis-mediated destruction of obsolete tissue, including midgut and salivary glands [4–6, 8]. Autophagy-induced cell death also plays an important role in mediating the death of terminally differentiated and apoptosis-resistant skin cells in mice [9]. Furthermore, strong activation or initiation of autophagy induces a defined form of cell death, termed autosis, which is distinct from other forms of cell death, such as apoptosis and necrosis [10, 11]. Cell death accompanied by strong activation of autophagy is also observed in some cardiac conditions [12]. Importantly, inhibition of autophagic initiation or flux prevents cell death and alleviates cardiac injury or dysfunction in some conditions, pointing to the functional significance of cell death induced by autophagy [11, 12]. The goals in this review are to summarize the cardiac conditions in which cell death mediated by autophagy has been observed, to describe the possible underlying mechanisms of cell death induced by autophagy, and to discuss therapeutic options to alleviate cell death mediated by autophagy.
Figure 1. Molecular mechanisms of programmed cell death.
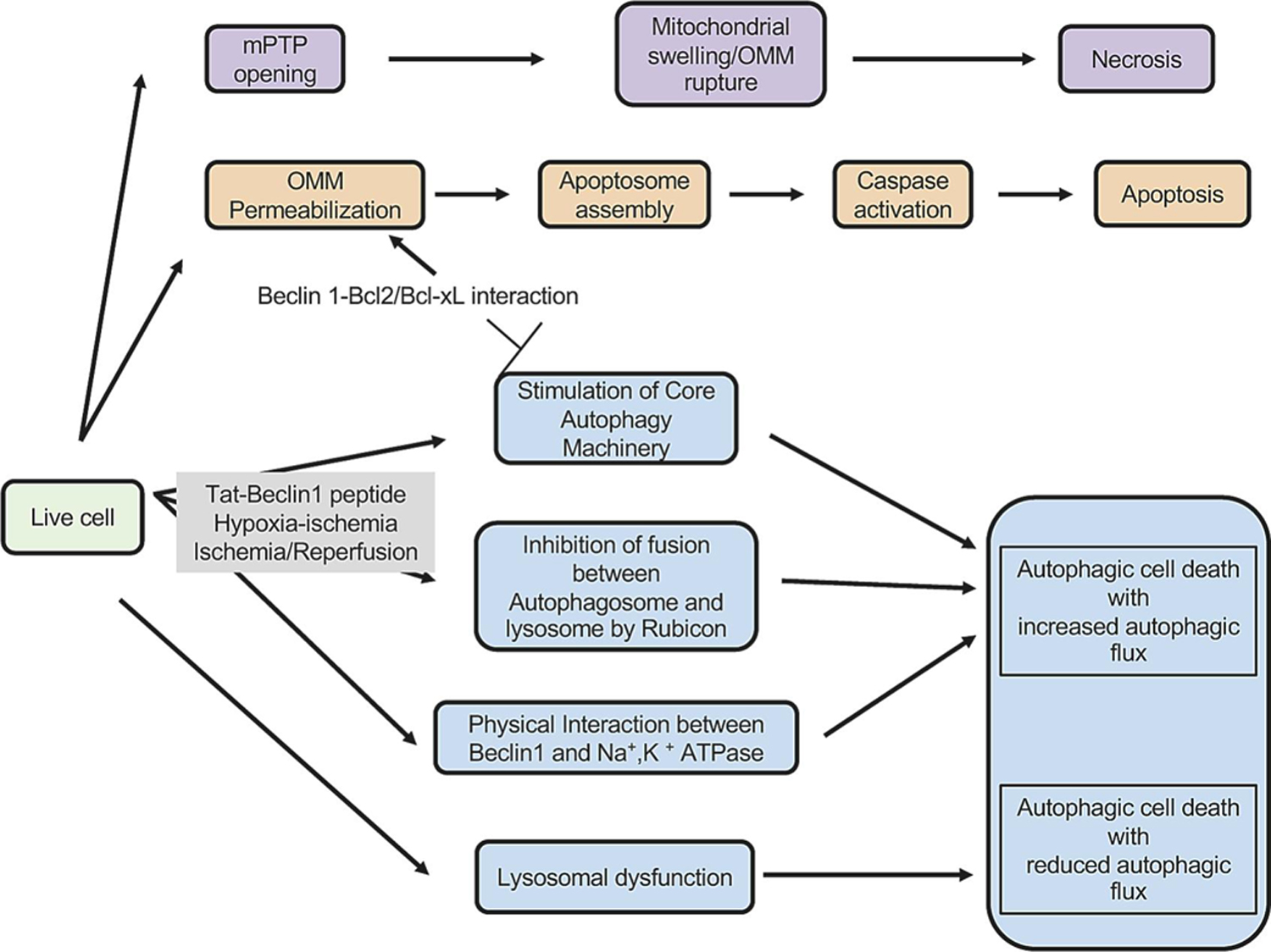
Autophagic cell death (type II programmed cell death, shown in blue) is mediated by mechanisms distinct from those of other forms of cell death (necrosis indicated by purple and apoptosis by orange). Autophagy and apoptosis affect one another through competition for Bcl-2/Bcl-xL binding between Beclin 1 and Bax. The former inhibits autophagy and stimulates apoptosis whereas the latter inhibits apoptosis and stimulates autophagy. Autophagic cell death occurs through multiple mechanisms but it can be broadly classified as either “Autophagic cell death with increased autophagic flux” or “Autophagic cell death with reduced autophagic flux”.
2. Autophagy
Autophagy is an essential catabolic process that delivers cytoplasmic material to lysosomes for degradation [2]. There are three main forms of autophagy, namely macroautophagy (herein referred to as autophagy), microautophagy, and chaperone-mediated autophagy (CMA). In the process of autophagy, cells degrade intracellular organelles and cytosolic materials via double membrane structures called autophagosomes that fuse with lysosomes. In microautophagy, cytosolic cargo components are transferred directly into the lysosome through membrane invaginations. CMA involves the selective translocation of proteins containing the KFERQ-like motif across the lysosomal membrane.
Autophagy is executed through three major steps, namely 1) initiation and nucleation of autophagosome, 2) expansion and completion of autophagosome formation and 3) lysosomal degradation [13]. Autophagy is initiated by activation of the Unc-51-like kinase (ULK) 1/Atg1 complex, whose activity is regulated by mammalian target of rapamycin complex 1 and adenosine monophosphate activated protein kinase, major nutrient and energy sensors in cells. The ULK 1/Atg1 complex in turn phosphorylates Beclin1 and Ambra 1, thereby activating the class III phosphatidylinositol 3 kinase (PI3K) complex, consisting of Vps34, Beclin 1, vacuolar protein sorting 15 (VPS15), and/or additional partners. PI3P produced by Vps34 recruits DFCP1 and WIPI2 to stimulate the formation of isolation membrane, namely the nucleation step. Beclin 1 recruits Beclin 1 binding proteins, including Atg14l, autophagy and Beclin regulator 1 (AMBRA1), ultraviolet resistance associated gene (UVRAG), and Rubicon, to the nucleation site to regulate the lipid kinase activity of the PI3K complex. Beclin 1 contains the BH3 domain and the stimulatory effect of Beclin1 toward PI3K complex is negatively regulated by Bcl-2 family proteins. Beclin 1 is subjected to posttranslational modifications, which in turn affect the activity of Beclin1 toward PI3K. Although endogenous Beclin 1 is anchored at the trans-Golgi membrane, it can be mobilized when autophagy is stimulated [14]. Overall, the activity of Beclin 1 is regulated by multiple mechanisms, and, thus, dysregulation of each mechanism could lead to dysregulation of autophagy [15]. During the process of nucleation, the endoplasmic reticulum (ER) exit sites, mitochondria, ER-mitochondria contact sites, the ER-Golgi intermediate compartment, the Golgi apparatus, and the plasma membrane have been reported as possible sources of isolation membranes [16].
In the second step, expansion of autophagosomes are achieved two conjugated systems, namely Atg12-Atg5-Atg16L and Atg8 (LC3)-Atg3. First, Atg7, an E1-like enzyme, binds to Atg12, an ubiquitin-like protein, and promotes conjugation of Atg12 to Atg5 in the presence of Atg10, an E2-like enzyme, and finally forming the Atg5-Atg12/Atg16 complex. The Atg5-Atg12/Atg16 complex acts as an E3-like ligase to conjugate Atg8 (light chain 3-I), another ubiquitin-like molecule, complexed with Atg3, an E2-like enzyme, to phosphatidylethanolamine (PE) to form LC3-PE (LC-3-II) [13].
At the last step, mature autophagosome fuses with the lysosome to form autolysosome wherein the cargo materials are degraded and recycled by lysosomal enzymes. In particular, the fusion step is achieved by tight coordination of molecular events. During the fusion between autophagosomes and lysosomes, syntaxin 17 (STX17), a SNARE protein localized on the outer autophagosomal membrane and interacting with Atg8, interacts with synaptosomal-associated protein 29 (SNAP29) and lysosomal synaptosomal-associated protein receptor vesicle-associated membrane protein 8 (VAMP8) to enable fusion with the lysosomes [17]. Besides SNARE proteins, many proteins, including the HOPS complex, Atg14, Epg5, Atg8 and Rab7, are involved in the fusion process [17]. Within the lysosomal compartment, membrane components and proteins are degraded by phospholipases and proteolytic enzymes, respectively, and the phospholipid and amino acid degradation products are recycled.
Since the degradation of cargo materials is executed through coordinated activation of multiple steps, dysregulation of even a single step can have profound and diverse effects on both cargo degradation and autophagosome accumulation. For example, upregulation or enhanced mobilization of Beclin 1 can lead to the enhancement of autophagy initiation, namely autophagosome formation [10], which in turn enhances cargo degradation. Rubicon upregulation during stress imposes a block at the step of autophagosome-lysosome fusion [18], thereby leading to the accumulation of autophagosomes. In addition, suppression of autophagy in a mouse model of lysosomal storage disorder (LOS) was observed to induce compensatory upregulation of TFEB, a transcription factor that serves as a master regulator of genes involved in autophagy, which stimulated autophagosome formation despite the presence of a block at the lysosomal level and eventually enhanced the autophagosome buildup [19]. Thus, it is necessary to understand not only the signaling mechanism of each step of autophagy but also the interaction and coordination among multiple steps in order to understand the mechanism and consequences of autophagy dysregulation and the eventual mechanism of cell death.
3. Autophagy in cell death
Autophagic cell death was originally described as massive cytoplasmic vacuolization without nuclear condensation and categorized as type II programmed cell death [7, 20, 21]. A limitation of this purely morphological definition is that the presence of autophagic vacuoles in dying cells, namely cell death with autophagy, does not necessarily signify that autophagy is the cause of cell death, namely cell death by autophagy. Dying cells often activate autophagy in a last bid for survival and, thus, suppression of autophagy promotes cell death in many cases. In order to distinguish cell death by autophagy from that with autophagy, the term “autophagic cell death” should be applied only when the following criteria, showing causative involvement of autophagy in the cell death, are met: (1) cell death occurs without the involvement of other types of cell death, (2) autophagy is activated, and (3) pharmacological or genetic inhibition of autophagy blocks cell death [22]. Currently, causative involvement of autophagy in a given form of cell death is demonstrated using multiple interventions to inhibit autophagy. Importantly, however, none of the chemical inhibitors or genetic interventions currently in use selectively suppress autophagy, so proving the presence of autophagic cell death remains challenging. Given these limitations and the prevailing belief that autophagy is purely adaptive, the concept of autophagic cell death has faced significant challenges from the field, and the underlying mechanism of autophagic cell death remains poorly understood. However, increasing lines of evidence suggest that autophagic cell death plays an important role not only as a physiological mediator of tissue morphogenesis, development, and differentiation but also as a facilitator of tissue damage in postnatal organs, including the heart.
Although autophagy degrades cellular materials in a relatively non-specific fashion, it can also target some molecules through specific interactions between the LC3 receptor and cargos. This is important because most autophagic degradation related to homeostasis is targeted to a specific cargo. Autophagy-dependent cell death may take place when autophagy degrades cell survival molecules, including dBruce, an anti-apoptotic molecule [23], and catalase, an antioxidant [24]. The resultant cell death would be alleviated by suppression of autophagy and, thus, could be broadly categorized as autophagic cell death. However, since this type of cell death may be mediated through activation of apoptosis and/or necrosis, it is not discussed further in this review.
4. Autophagic cell death in the heart
In this section, we review how autophagic cell death occurs in various cardiac conditions. As noted above, we define autophagic cell death as being inhibited by the suppression of autophagy mechanisms. In cardiomyocytes, autophagic cell death takes place through either uncontrolled degradation of cargo materials due to increased autophagic flux or excessive buildup of autophagosomes caused by an imbalance between autophagy initiation and lysosomal degradation. (Figure 2).
Figure 2. Two mechanisms of autophagic cell death.
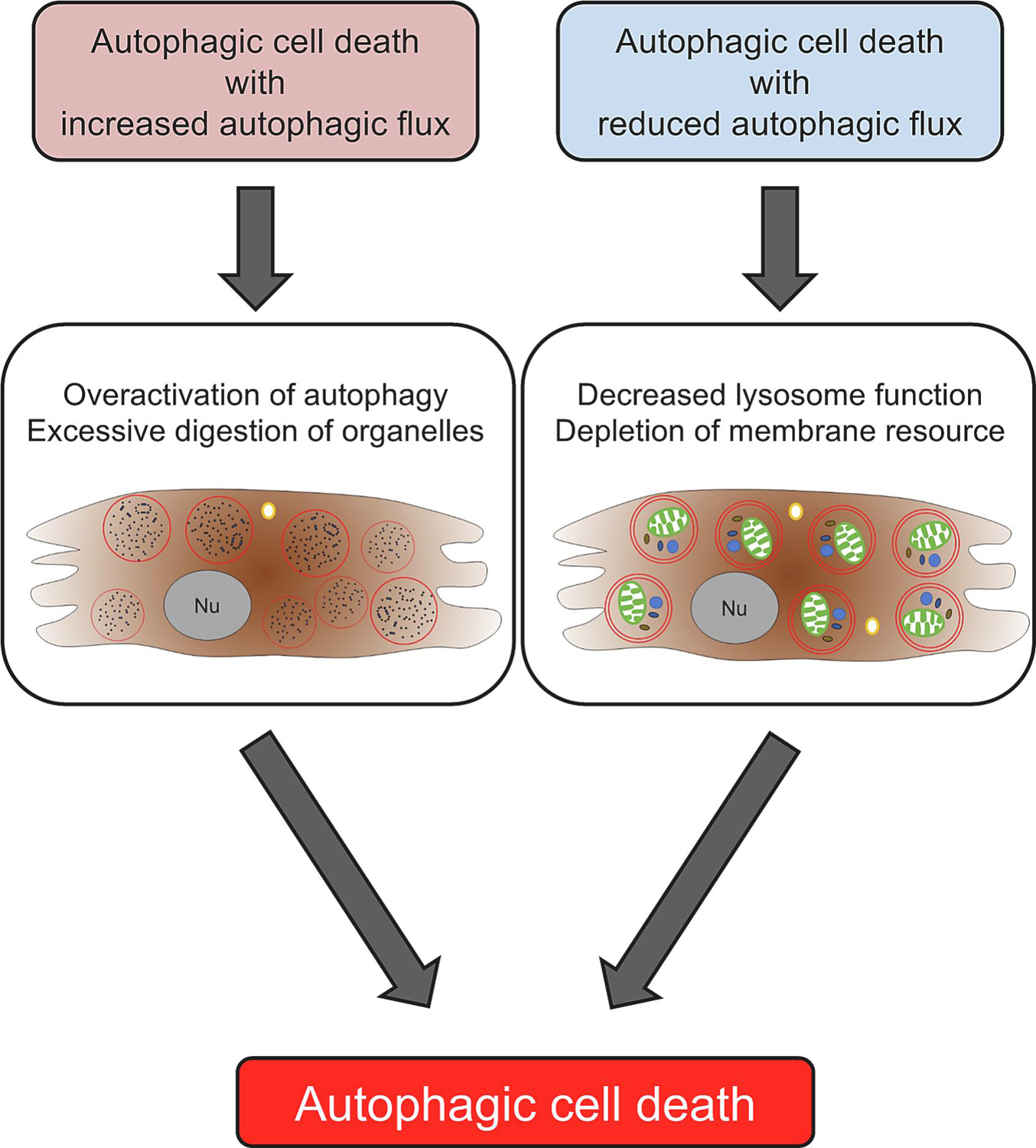
There are two forms of autophagic cell death, one accompanied by increased autophagic flux and the other by reduced autophagic flux. The former is mediated through excessive degradation of cellular materials, the latter through excessive accumulation of autophagosomes and/or consumption/depletion of endomembranes.
4.1. Autophagic cell death with increased autophagic flux
4.1.1. Cell death induced by lysosomal degradation
In adult mammalian skin, keratinocyte lineage cells become highly resistant to apoptosis during differentiation but eventually die by an autophagy-dependent mechanism [9]. When skin cells are differentiating, they exhibit expansion of the Golgi, generation of lysosomes and chromatin condensation. They then show a burst of autophagosome formation, phagocytosis of the ER and the nucleus, and massive degradation of cytoplasmic organelles, and then cell death is induced. This autophagy-mediated cell death effectively removes terminally differentiated cells so that the skin renews constantly and maintains its function. Since the death of differentiated skin cells is accompanied by nearly complete degradation of cytoplasmic organelles, lysosomal degradation appears to play a major role in mediating cell death. Interestingly, the morphology of the dying skin cells presents many similarities to cardiomyocytes undergoing autosis, a mechanism of autophagy-dependent cell death discussed later in this review, during ischemia/reperfusion injury [12]. It should be noted, however, that death by autosis is not alleviated by lysosome inhibitors [10, 12], suggesting that autosis is not caused by massive lysis of cytosolic materials in the lysosomes. Further investigation is required to clarify whether conditions exist in which cardiomyocytes die due to massive autophagy-mediated lysis at lysosomes.
4.1.2. p53-induced cell death
p53 is a tumor suppressor activated by myocardial stresses, including ischemia and angiotensin II stimulation, that promotes cell death. p53 upregulates Bnip3, a BH3 domain-containing protein, in mitochondria, which in turn induces mitochondrial dysfunction, increases in autophagic flux and cell death in cardiomyocytes. Importantly, p53-induced cell death is inhibited by the suppression of autophagy [25]. Furthermore, p53-induced upregulation of autophagy and cell death are Bnip3-dependent. This suggests that p53 and Bnip3 can trigger autophagic cell death. Since both Bnip3 and Beclin 1 are BH3 domain-containing proteins and competition for Bcl-2 binding among BH3 domain-containing proteins affects both apoptosis and autophagy [26], p53-induced upregulation of Bnip3 may be an important mechanism leading to dysregulation of autophagy. Activation of the p53-Bnip3 pathway promotes the complex formation between Bnip3 and cyclophilin D, thereby inducing mPTP opening in a cyclophilin D-dependent manner [27]. Mitochondria-associated Bnip3 is accompanied by Ser616 phosphorylation and excessive mitochondrial fragmentation [28]. Thus, mPTP opening, excessive mitochondrial fragmentation and consequent mitochondrial dysfunction may contribute to mitophagy-dependent cell death induced by the p53-Bnip3 pathway.
4.1.3. Excessive mitophagy
Mitophagy is a mitochondria-specific form of autophagy mediated by mechanisms that mark mitochondria for degradation and adapter/receptor proteins that allow sequestration of mitochondria by autophagosomes. Mitophagy is designed to remove dysfunctional or unnecessary mitochondria. However, dysregulated activation of mitophagy causes mitochondrial depletion, thereby leading to cellular dysfunction and death. Excessive mitophagy has been proposed as a mechanism of neurodegenerative diseases, including Huntington’s disease [29]. Molecular mechanisms through which the elimination of mitochondria is intensified in an uncontrolled manner are not well understood. Although dysregulated activation of mitophagy, mitochondrial dysfunction, and death of cardiomyocytes have been reported in a mouse model of doxorubicin (DOX) cardiomyopathy, its functional significance remains to be determined. Recent evidence suggests that excessive activation of mitophagy is involved in the pathogenesis of DOX-induced cardiac dysfunction [30]. Since the current consensus is that autophagic flux is inhibited during chronic DOX treatment, how mitophagy is activated under such conditions needs to be clarified [31]. Strong activation of Parkin-mediated mitophagy has been reported in cardiac-specific Drp1 knockout mice [32]. Although activation of Parkin appears to be a compensatory mechanism against the loss of key mitochondrial quality control mechanisms, whether this leads to excessive mitophagy has been debated [33, 34]. Since the appropriate balance between mitophagy and mitochondrial biogenesis is critical for the heart to maintain energetics and redox homeostasis, it is important to investigate whether mitophagy can really be dysregulated, and if so, how mitophagy is stimulated in an excessive manner.
4.1.4. Autophagic cell death in the diabetic heart
The number of patients with obesity and metabolic disturbances has risen dramatically over the past few decades and many of them develop type II diabetes. More than half of diabetic patients eventually develop a cardiac abnormality characterized by diastolic dysfunction, cardiac hypertrophy and fibrosis, termed diabetic cardiomyopathy. In severe cases, systolic dysfunction also develops. Many patients suffering from heart failure with preserved ejection fraction (HFpEF) also have obesity and insulin resistance. Autophagic activity in diabetic hearts is regulated by complex mechanisms and differs depending upon the stage and severity of diabetes. Although autophagy is activated transiently in response to high-fat diet consumption in mice, growing lines of evidence suggest that autophagy is inhibited during the chronic phase of type II diabetes [35]. On the other hand, one report showed that autophagy hyperactivation in type II diabetic hearts can cause self-digestion and potentiate reactive oxygen species (ROS) production [36]. Whether hyperactivation of autophagy actually occurs in the heart during the development of type II diabetes and, if so, whether the cell death is directly caused by autophagy remain to be clarified. Interestingly, inhibition of autophagy by downregulating Beclin 1 or Atg 16 improves diabetic cardiomyopathy by activating mitophagy through an unknown mechanism in type I diabetes [37]. Since the unconventional form of mitophagy is activated as a compensatory mechanism when conventional autophagy is suppressed [38], the observation in type I diabetes [37] may be explained by compensatory activation of mitophagy rather than direct suppression of autophagic cell death. Namely, activation of conventional autophagy may promote cell death by negatively affecting mitophagy in type I diabetes hearts. To our knowledge, whether autophagy suppression improves cardiac function in mice with type II diabetes is unknown.
4.2. Autophagic cell death with reduced autophagic flux
4.2.1. Autophagic cell death in DOX-induced cardiomyopathy
DOX, a non-selective class I anthracycline antibiotic, is a potent chemotherapeutic agent that is used for the treatment of numerous cancers. However, the use of DOX is limited due to serious side effects. DOX induces the formation of DNA double-strand breaks in a topoisomerase IIβ-dependent manner, which can lead to DNA damage responses and ROS formation [39] and induce apoptotic and necrotic cell death [31]. DOX treatment activates autophagy in cardiomyocytes very rapidly. However, this activation of autophagy is transient, as DOX impairs the final step of autophagy through inhibition of lysosome acidification [31]. Although DOX suppresses autophagic flux chronically, DOX-induced cardiotoxicity is alleviated by inhibition of autophagy initiation in Beclin 1+/− mice [40]. On the other hand, downregulation of Rubicon, a negative regulator of autophagosome-lysosome fusion and endosomal trafficking, also ameliorates DOX-induced cardiotoxicity [41]. These results are consistent with the notion that chronic DOX treatment exacerbates the accumulation of autophagosomes by inhibiting autophagic flux, which in turn induces autophagic cell death in cardiomyocytes and cardiac dysfunction. Importantly, however, these reports did not examine the status of the cardiomyocytes, including the degree of autophagosome accumulation. Thus, further investigation is required to elucidate the mechanism through which loss of Beclin 1 or Rubicon function improves cardiac function in DOX-treated mice. In particular, whether DOX promotes autophagic cell death of cardiomyocytes in the chronic phase remains to be clarified. It should be noted that DOX-induced suppression of autophagic flux occurs in a time-dependent manner [42]. DOX has been shown to promote cardiomyocyte death by exacerbating autophagic flux under some conditions [43]. suggesting that there may be conditions under which DOX induces autophagic cell death and promotes cardiomyopathy by stimulating autophagic flux. In contrast, increased initiation of autophagy by Atg7 rescues DOX cardiomyopathy in some conditions [42]. We speculate that DOX-induced autophagic cell death may occur in a time-dependent manner and may also depend upon the presence of an autophagic block at the lysosomal level.
4.2.2. Autosis during ischemia/reperfusion
Death of cardiomyocytes during reperfusion after a period of myocardial ischemia is a major health issue since myocardial infarction is a major risk factor for the development of heart failure. Although percutaneous coronary interventions successfully reduce the size of myocardial infarcts caused by ischemia, interventions effectively preventing reperfusion injury remain poorly developed. Autophagy is activated in response to myocardial ischemia but activation of autophagy during reperfusion is complex and appears to be time-dependent. Although an initial study focusing on the early phase of reperfusion reported that autophagy is strongly activated, other studies have shown that autophagic flux is inhibited during the late phase of reperfusion [44]. Whether autophagy prevents or promotes the death of cardiomyocytes during reperfusion has been debated [45, 46]. However, recent evidence suggests that the role of autophagy is time-dependent and that the effect of intervention differs depending upon how autophagy is modulated [18]. Since we have already discussed the detailed role of autophagy during myocardial reperfusion in other review articles [2], we here focus on how myocardial reperfusion induces autophagic cell death.
In a mouse model of ischemia/reperfusion (I/R), autophagy is activated during reperfusion, accompanied by prominent upregulation of Beclin 1 and time-dependent accumulation of autophagosomes and autolysosomes [47]. Accumulation of autophagosomes, but not autolysosomes, becomes more prominent 6 hours after reperfusion. Assessment by multiple methods indicated that autophagic flux fell below baseline during the late stage of reperfusion [12]. Thus, I/R induces a condition in which autophagosomes accumulate due to the blockade of autophagic flux. Electron microscopic analyses of cardiomyocytes showed typical morphological features of autosis, a novel form of autophagic cell death, including marked accumulation of autophagosomes, autolysosomes and vacuoles, ballooning of the perinuclear space (PNS), condensed mitochondria and disappearance of intracellular organelles. Interestingly, both the morphological features of autosis and the myocardial infarct size were decreased when mice were treated with ouabain, a cardiac glycoside and known inhibitor of autosis, suggesting that autosis is induced by I/R and contributes to I/R injury [12]. These results are consistent with the fact that I/R injury is attenuated by Beclin 1 haploinsufficiency [47], which also prevents the marked accumulation of autophagosomes during reperfusion.
Autosis was originally described by Beth Levine’s group in fibroblast cell lines treated with high doses of TAT-Beclin 1, an autophagy-inducing peptide, and in mice after starvation and permanent brain ischemia, conditions in which autophagy is strongly activated [10, 11, 48]. Since then, autosis has also been observed in other cell types and organs under stress [10, 12, 48–50] (Table 2). Although autosis may not be the sole form of autophagy-dependent cell death, it is certainly the most well-defined form of cell death in this category. Autosis is rescued by inhibitors of autophagy, but not by inhibitors of apoptosis or necrosis. Thus, autosis clearly fulfills the criteria of autophagy-dependent cell death. Furthermore, the fact that inhibition of autosis by cardiac glycoside attenuates organ damage/dysfunction suggests that it has functional significance.
Table 2.
Autosis models confirmed to date
| Experiment type | Animals/Cells | Model | References |
|---|---|---|---|
| Vivo | Rat | Perinatal cerebral hypoxia-ischemia | Liu Y, et al. [10] Fernández AF, et al. [48] |
| Vivo | Mice | Kidneys of mice subjected to renal ischemia/reperfusion injury (IRI) | Fernández AF, et al. [48] |
| Vivo | Human | Liver of patients with severe anorexia nervosa | Kheloufi M, et al. [49] |
| Vivo | Mice | Hearts and Livers of starved mice | Fernández AF, et al. [48] |
| Vivo | Mice | Hearts in late phase ischemia-reperfusion(I/R) | Nah J, et al. [12] |
| Vitro | Cells | Hela cells treated with Tat-Beclin1 Hela cells after starvation Neonatal cardiomyocytes treated with Tat-Beclin1 |
Liu Y, et al. [10] Nah J, et al. [12] |
Autosis is characterized by the presence of unique morphological and biochemical features, and the morphological features are induced in a time-dependent manner (Table 1 and Figure 3). In phase 1a, dilated and fragmented ER and an increased number of autophagosomes, autolysosomes, and empty vacuoles are observed. In phase 1b, a swollen PNS containing cytoplasmic materials and electron-dense mitochondria can be observed by transmission electron microscopy. In phase 2, the last step of autosis, cytoplasmic organelles are drastically decreased and focal nuclear concavity and focal ballooning of the PNS are observed. Cells that have died by autosis are generally more firmly attached to the culture dishes than those that do not undergo autosis, and the increased adherence appears to be another morphological feature of autosis in vitro. To date, the best method for defining autosis is electron microscopy analysis. Alternatively, immunofluorescence assays can also help distinguish autotic cells by detecting fragmented ER or mitochondria with nuclear concavity. Although autosis can be inhibited by chemical inhibitors of autophagy, including 3-methyladenine, and down-regulation of the autophagic machinery, such as Beclin 1 and Atg7, it cannot be inhibited by inhibitors of apoptosis, necrosis, or any other form of programmed cell death. Autosis can be inhibited interventions that block phagophore assembly (e.g., 3-methyladenine) but not lysosome fusion (e.g., Bafilomycin A1). In a high throughput screen, it was found that inhibitors of Na+, K+ ATPase, like the cardiac glycosides ouabain and digoxin, can also inhibit autosis, which is diagnostic for autosis [10, 12].
Table 1.
Comparison of biochemical and morphological characteristics of the types of cell death
| Autophagy dependent cell death(autosis) | Apoptosis | Necrosis | |
|---|---|---|---|
| Morphological features | Focal ballooning of PNS, focal concavity of the nuclear surface, mild chromatin condensation Extensive cytoplasmic vacuolization Enhanced cell substrate adhesion Focal plasma membrane rupture |
Nuclear compaction and fragmentation Marked chromatin condensation Cell shrinkage, membrane blebbing Cell rounding up, detachment from substrate |
Swelling of organelles Breakdown of plasma membrane and nucleus Karyolysis and caspase-independent DNA fragmentation, lysis of nucleolus, dilation of nuclear membrane |
| Mechanism of execution | Occurs independently of apoptotic pathways Requires core autophagy machinery Occurs independently of apoptotic pathways Depends on Na+, K+ ATPase |
Requires apoptotic pathways Occurs independently of core autophagy machinery |
ATP depletion mPTP opening |
| Blocked by | Pharmacological inhibition or genetic ablation of key components of core autophagy machinery(not inhibitors of apoptosis and necrosis) Pharmacological inhibition of genetic ablation of Na+, K+ ATPase and Rubicon |
Caspase inhibitors Genetic ablation of proapoptotic factors |
Necrosis Inhibitors |
| Disposal of cell corpses | Phagocytic uptake and lysosomal degradation | Phagocytic uptake and lysosomal degradation | Leakage of organelle contents |
Figure 3. Morphological features of autosis.
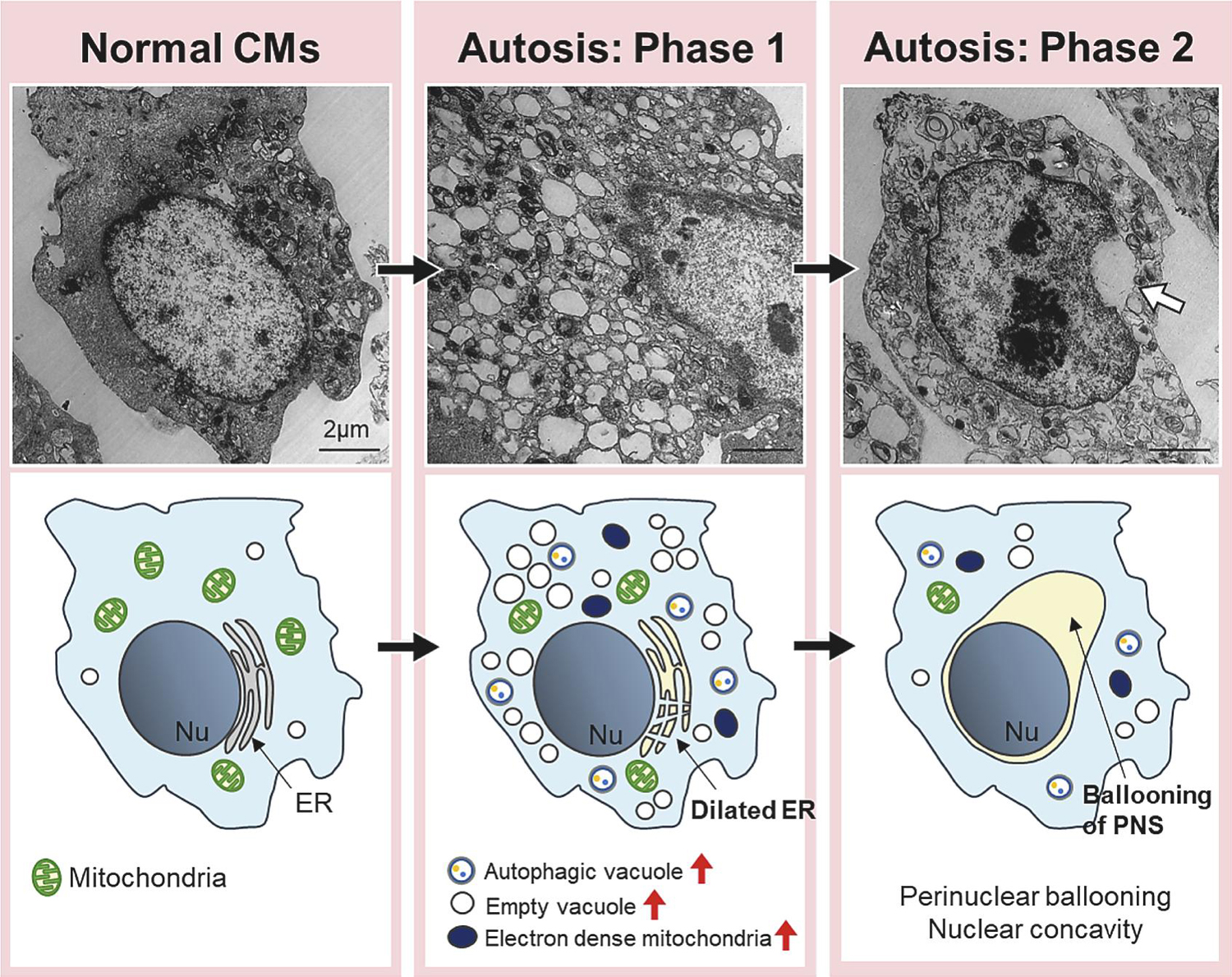
During the early phase (Phase 1) of autosis, the number of vacuoles, including empty vacuoles and autophagic vacuoles, is increased. Dilated and fragmented ER is observed. Electron dense mitochondria are observed under electron microscopy. In the later phase (Phase 2) of autosis, CMs show ballooning of perinuclear space (PNS), one of the most representative features of autosis. Nu= nucleus, ER= endoplasmic reticulum. White arrow indicates ballooning of PNS. Scale bars = 2 µm.
Since autosis is not inhibited by preventing lysosomal degradation, it is unlikely that death is mediated by excessive degradation of cargos. This property distinguishes autosis from other forms of autophagic cell death, including those induced by excessive activation of mitophagy, which is accompanied by selective depletion of mitochondria. Autosis may be initiated by excessive accumulation of autophagosomes rather than lysosomal degradation. In fact, suppression of autophagosome maturation due to Rubicon upregulation induces marked accumulation of autophagic vacuoles and facilitates autosis. In addition, it has been suggested that autosis is accompanied by a shortage of essential cytosolic membranes [12]. It has been speculated that excessive production of autophagosomes in the presence of Rubicon prevents the degradation and recycling of autophagosomes by lysosomes. Importantly, cardiomyocyte-specific conditional knockout of Rubicon restores autophagic flux and reduces the autotic cell death rate during the late phase (over 6 hours) of reperfusion in the heart, suggesting that inhibiting autosis through inhibition of Rubicon may be a unique and promising approach to reduce the extent of myocardial injury in patients with I/R [12, 18].
Na+, K+ ATPase physically interacts with Beclin 1 when autosis occurs. One could speculate that this interaction dysregulates Beclin 1 function to induce autophagy. However, this hypothesis remains to be tested and the role of Na+, K+ ATPase in autosis may not necessarily be limited to the process of autophagosome formation. Interestingly, blockade of endogenous ligands of Na+, K+ ATPase, called digitalis-like substances, with Digibind was shown to increase cardiomyocyte autosis in the mouse heart during exercise [48]. Currently, how digitalis-like substances affect Na+, K+ ATPase remains unclear. However, these results suggest that the heart is protected against autosis by endogenous mechanisms, without which autosis in the heart might be more common than previously thought.
Although more lines of evidence are required, endogenous digitalis-like substrates have been implicated in some forms of hypertension [51]. It is fascinating to speculate that it may be possible to control the level of autosis by stimulating the production of endogenous digitalis-like substrates. More investigation is needed to determine the effectiveness of this approach.
In the heart, strong activation of autophagy is also observed during the acute phase of myocardial infarction caused by permanent coronary artery ligation [26] and in response to severe pressure overload [52]. Since suppression of autophagy initiation due to Beclin 1 haploinsufficiency attenuates cardiac injury and cardiac dysfunction [52], autophagic cell death of cardiomyocytes may occur in these conditions. However, whether autosis is involved in myocardial injury in these conditions is currently unknown.
By definition, autosis should not be suppressed by inhibition of apoptosis or necrosis. Although autosis may share upstream mechanisms with apoptosis and necrosis, unique mechanisms not shared by apoptosis or necrosis appear essential for the induction of autosis. Whether cell death initiated by autosis partially utilizes downstream mechanisms common to other forms of cell death remains to be clarified.
4.2.3. Autophagic cell death in lysosomal storage disorder
Lysosomal storage disorder (LSD) is caused by genetic mutations in genes involved in lysosomal functions and is characterized by increased storage of materials as a result of inhibition of lysosomal degradation. Importantly, lysosomal dysfunction attenuates autophagy, which in turn induces secondary accumulation of damaged proteins and organelles in cells and functional abnormalities in major organs, including the heart, skeletal muscle and liver [53]. In LSDs, autophagy is inhibited at the last step of the degradation process, namely lysosomal degradation, whereas autophagosome formation is preserved or even stimulated. Uncoupling between autophagosome formation and lysosomal degradation leads to a prominent accumulation of autophagosomes. We have shown recently that suppressing autophagosome formation by downregulating Atg7 attenuates the excessive accumulation of autophagosomes and cardiac dysfunction in cardiac-specific RagA/B knock-out mice, a mouse model of LSD caused by the failure of lysosomal acidification [19]. Since stimulation of autophagy by TAT-Beclin 1 exacerbates autophagosome accumulation and cardiac dysfunction, it is likely that accumulation of autophagosomes promotes organ malfunction in this model. In the presence of severe or permanent lysosomal dysfunction, a feedback mechanism may be activated. In fact, YAP, a transcription factor co-factor that is degraded by autophagy, is activated in RagA/B cKO mice, and stimulates the activity of TFEB, a master transcription factor that promotes autophagy and lysosomal biogenesis, thereby exacerbating the accumulation of autophagosomes [19]. Interestingly, loss of YAP function alleviated autophagosome accumulation and cardiac dysfunction in RagA/B cKO mice. These results suggest that cardiomyopathy in LSD may be due to excessive accumulation of autophagosomes and consequent cellular dysfunction rather than accumulation of unmetabolized substrates of lysosomal enzymes. Although autophagosome accumulation is caused by simultaneous inhibition of autophagic flux and activation of autophagosome formation, as during the late phase of ischemia/reperfusion when autosis is induced, typical morphological features of autosis, including a swollen perinuclear space, were not observed in RagA/B cKO mice [19]. Thus, although the triggering mechanism of cardiomyocyte death in LSD is similar to that in autosis, the form of death in these conditions does not appear to be identical. Further investigation is needed to elucidate the causes of cardiomyocyte death in LSDs. It should be noted that interventions to restore autophagy have been shown to alleviate cardiomyopathy in some LSDs [54, 55]. Where lysosomal dysfunction is reversible, restoring autophagic flux may reduce the accumulation of autophagosomes and lysosome enzyme substrates. Thus, although the goal for treatment of LSD in general would be to alleviate the massive buildup of autophagosomes, strategies to reduce autophagosome accumulation could differ depending upon the reversibility of lysosomal function. A cautionary note is that normalization of autophagosome buildup by targeting autophagy may not be sufficient to normalize cardiac function in LSDs, and additional problems, including accumulation of ubiquitinated proteins, oxidative stress, and muscle atrophy, have been noted. Thus, a therapy minimizing side effects needs to be explored [56].
5. Therapeutic interventions
As discussed above, autophagic cell death can be separated into two groups, namely that induced by increased cargo degradation at lysosomes and that mediated by excessive accumulation of autophagosomes (Figure 2). Whereas the former is caused by increased autophagic flux, the latter results from decreased autophagic flux due to a blockage in the late stages of autophagy. When considering the prevention of autophagic cell death of cardiomyocytes as a therapeutic intervention, it is essential to clarify which category the death falls into. Various interventions to inhibit autophagy at the level of autophagosome formation, lysosomal degradation, or cargo-autophagosome interaction may be considered when the problem is lysosomal degradation. On the other hand, therapeutic options for cell death induced by excessive autophagosome accumulation appear to be more complex. For example, interventions to alleviate blocked autophagosome-lysosome fusion or rescue lysosomal dysfunction, including trehalose [57] and 3,4-dimethoxychalcone [58], may improve autophagic flux and alleviate excessive accumulation of autophagosomes. When lysosomal dysfunction is irreversible, however, interventions to prevent autophagosome formation should be considered. Although inhibition of autophagosome formation in the presence of severe or permanent blockage of lysosomal degradation may alleviate a pathological buildup of autophagosomes, additional interventions to improve cellular quality control mechanisms independent of autophagy may be necessary, particularly in the presence of stress.
Autosis is blocked by cardiac glycosides [10]. At present, the molecular mechanism through which cardiac glycosides inhibit autosis remains unclear. Whether cardiac glycosides prevent excessive accumulation of autophagosomes by regulating Beclin 1 remains to be tested. Interestingly, Empagliflozin, an SGLT2 inhibitor, inhibits autosis in response to glucose deprivation or myocardial ischemia through inhibition of Na+H+ exchanger 1 in cardiomyocytes [50]. It is tempting to speculate that changes in the ionic environment near the membrane may affect autophagosome formation. Alternatively, empagliflozin may directly affect the mechanism of cell death. Aside from the characteristic morphological features, including the large perinuclear space and the disappearance of intracellular organelles [11], little is known about how excessive autophagy leads to cardiomyocyte death by autosis. A better understanding of the underlying mechanism of autoptic cell death should allow for the development of a unique intervention to prevent autosis.
6. Unsolved questions
Despite the increasing number of publications supporting the existence of autophagic cell death, unresolved issues remain. Autophagic cell death is considered adaptive if it is activated during organ development or when apoptosis, another mechanism of programmed cell death, is inhibited [8, 9]. On the other hand, it is maladaptive and leads to organ malfunction when autophagy mechanisms are dysregulated [12]. Current knowledge regarding the physiological function of autophagic cell death, particularly in the adult heart, is insufficient. For example, it is of great interest to determine whether excessive mitophagy selectively kills dysfunctional cardiomyocytes in order to prevent increases in oxidative stress. Investigating the molecular mechanism involved in physiological autophagic cell death during development may provide us with hints to better understand how autophagic cell death controls cellular function. How autophagy is dysregulated and becomes pathological should also be investigated. For example, blockade at the level of autophagosome-lysosome fusion plays an important role in mediating autosis in the heart during the late phase of reperfusion [12]. However, blockade of autophagosome-lysosome fusion alone is usually insufficient to induce autosis. We speculate that mechanisms to stimulate autophagosome formation are activated in the presence of the blockade of autophagic flux in order to respond to the cellular demand to remove managed proteins and organelles during reperfusion. Thus, a two-hit mechanism seems to be required for autophagy dysregulation and stimulation of autophagic cell death. The superimposing signaling mechanisms facilitating dysregulated autophagy need to be clarified. Although existing evidence suggests that excessive accumulation of autophagosomes leads to autophagic cell death, the detailed mechanism through which cell death occurs is unknown. Densely packed autophagosomes could interfere with cellular functions by mechanically damaging organelles and disrupting cellular transport. We observed that excessive accumulation of autophagosomes is accompanied by decreases in the endomembrane systems, including endoplasmic reticulum, and organelle dysfunction [12]. However, these observations provide only correlative evidence and, thus, more mechanistic proof directly connecting excessive autophagosomes and cellular dysfunction is needed. An important feature of autosis is that autotic cell death cannot be inhibited by suppression of other forms of cell death, including apoptosis and necrosis [11]. Thus, in theory, one could expect that blockade of autosis should have an additive effect on top of the current regimens of treatment for reperfusion injury targeting other forms of cell death. If so, it would be important to explore effective combinations and timing of interventions to reduce multiple forms of cell death during cardiovascular disease, including ischemia/reperfusion injury. Cardiac glycosides were identified through screenings of natural compounds inhibiting autosis [10]. A more extensive search for small molecules affecting autophagic cell death may allow the discovery of novel interventions to reduce death of cardiomyocytes.
Highlights.
Dysregulated autophagy can induce death in cardiomyocytes.
Autophagic cell death is characterized by strong activation of autophagy and attenuation of cell death by suppression of autophagy but not other forms of cell death, such as apoptosis and necrosis.
Autophagic cell death can occur through either excessive autophagic flux or a block of autophagic flux at the level of lysosomes.
One form of autophagic cell death includes autosis characterized by unique morphological and biochemical features, including the sensitivity to cardiac glycosides.
Cardiomyocyte autosis occurs at the late phase of myocardial ischemia/reperfusion and contributes to myocardial injury.
Acknowledgments:
This work was supported in part by U.S. Public Health Service Grants HL67724, HL91469, HL102738, HL112330, HL138720, HL144626, HL150881, and AG23039 (J.S.), the American Heart Association Merit Award 20 Merit 35120374 (J.S.), and by the Fondation Leducq Transatlantic Network of Excellence 15CVD04 (J.S.).
Abbreviations:
- AMBRA1
autophagy and Beclin regulator 1
- ATG
autophagy-related
- CMA
chaperone-mediated autophagy
- DOX
Doxorubicin
- ER
endoplasmic reticulum
- HFpEF
heart failure with preserved ejection fraction
- I/R
ischemia/reperfusion
- LSD
lysosomal storage disorder
- ULK
Unc-51-like kinase
- PI3K
phosphatidylinositol 3 kinase
- PNS
perinuclear space
- ROS
reactive oxygen species
- STX17
syntaxin 17
- SNAP29
synaptosomal-associated protein 29
- TFEB
transcription factor EB
- UVRAG
ultraviolet resistance associated gene
- VAMP8
vesicle-associated membrane protein 8
- VPS15
vacuolar protein sorting 15
- YAP
Yes-associated protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures:
None.
Reference
- [1].Ohsumi Y, Historical landmarks of autophagy research, Cell research 24(1) (2014) 9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Sciarretta S, Maejima Y, Zablocki D, Sadoshima J, The Role of Autophagy in the Heart, Annu Rev Physiol 80 (2018) 1–26. [DOI] [PubMed] [Google Scholar]
- [3].Mizushima N, Levine B, Autophagy in Human Diseases, N Engl J Med 383(16) (2020) 1564–1576. [DOI] [PubMed] [Google Scholar]
- [4].Denton D, Kumar S, Autophagy-dependent cell death, Cell Death Differ 26(4) (2019) 605–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Jung S, Jeong H, Yu SW, Autophagy as a decisive process for cell death, Exp Mol Med 52(6) (2020) 921–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Schwartz LM, Autophagic Cell Death During Development - Ancient and Mysterious, Front Cell Dev Biol 9 (2021) 656370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Clarke PG, Developmental cell death: morphological diversity and multiple mechanisms, Anatomy and embryology 181(3) (1990) 195–213. [DOI] [PubMed] [Google Scholar]
- [8].Denton D, Shravage B, Simin R, Mills K, Berry DL, Baehrecke EH, Kumar S, Autophagy, not apoptosis, is essential for midgut cell death in Drosophila, Curr Biol 19(20) (2009) 1741–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Koenig U, Robenek H, Barresi C, Brandstetter M, Resch GP, Groger M, Pap T, Hartmann C, Cell death induced autophagy contributes to terminal differentiation of skin and skin appendages, Autophagy (2019) 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Liu Y, Shoji-Kawata S, Sumpter RM Jr., Wei Y, Ginet V, Zhang L, Posner B, Tran KA, Green DR, Xavier RJ, Shaw SY, Clarke PG, Puyal J, Levine B, Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia, Proc Natl Acad Sci U S A 110(51) (2013) 20364–20371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Nah J, Zablocki D, Sadoshima J, Autosis: A New Target to Prevent Cell Death, JACC Basic Transl Sci 5(8) (2020) 857–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Nah J, Zhai P, Huang CY, Fernandez AF, Mareedu S, Levine B, Sadoshima J, Upregulation of Rubicon promotes autosis during myocardial ischemia/reperfusion injury, J Clin Invest 130(6) (2020) 2978–2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Yang Z, Klionsky DJ, An overview of the molecular mechanism of autophagy, Curr Top Microbiol Immunol 335 (2009) 1–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Shoji-Kawata S, Sumpter R, Leveno M, Campbell GR, Zou Z, Kinch L, Wilkins AD, Sun Q, Pallauf K, MacDuff D, Huerta C, Virgin HW, Helms JB, Eerland R, Tooze SA, Xavier R, Lenschow DJ, Yamamoto A, King D, Lichtarge O, Grishin NV, Spector SA, Kaloyanova DV, Levine B, Identification of a candidate therapeutic autophagy-inducing peptide, Nature 494(7436) (2013) 201–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Maejima Y, Isobe M, Sadoshima J, Regulation of autophagy by Beclin 1 in the heart, J Mol Cell Cardiol 95 (2016) 19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Melia TJ, Lystad AH, Simonsen A, Autophagosome biogenesis: From membrane growth to closure, J Cell Biol 219(6) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Li Y, Cheng X, Li M, Wang Y, Fu T, Zhou Z, Wang Y, Gong X, Xu X, Liu J, Pan L, Decoding three distinct states of the Syntaxin17 SNARE motif in mediating autophagosome-lysosome fusion, Proc Natl Acad Sci U S A 117(35) (2020) 21391–21402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Nah J, Zablocki D, Sadoshima J, The roles of the inhibitory autophagy regulator Rubicon in the heart: A new therapeutic target to prevent cardiac cell death, Exp Mol Med 53(4) (2021) 528–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ikeda S, Nah J, Shirakabe A, Zhai P, Oka SI, Sciarretta S, Guan KL, Shimokawa H, Sadoshima J, YAP plays a crucial role in the development of cardiomyopathy in lysosomal storage diseases, J Clin Invest 131(5) (2021) e143173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Klionsky DJ, Autophagy: from phenomenology to molecular understanding in less than a decade, Nat Rev Mol Cell Biol 8(11) (2007) 931–937. [DOI] [PubMed] [Google Scholar]
- [21].Levine B, Yuan J, Autophagy in cell death: an innocent convict?, J Clin Invest 115(10) (2005) 2679–2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Shen HM, Codogno P, Autophagic cell death: Loch Ness monster or endangered species?, Autophagy 7(5) (2011) 457–465. [DOI] [PubMed] [Google Scholar]
- [23].Nezis IP, Shravage BV, Sagona AP, Lamark T, Bjorkoy G, Johansen T, Rusten TE, Brech A, Baehrecke EH, Stenmark H, Autophagic degradation of dBruce controls DNA fragmentation in nurse cells during late Drosophila melanogaster oogenesis, J Cell Biol 190(4) (2010) 523–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Yu L, Wan F, Dutta S, Welsh S, Liu Z, Freundt E, Baehrecke EH, Lenardo M, Autophagic programmed cell death by selective catalase degradation, Proc Natl Acad Sci U S A 103(13) (2006) 4952–4957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wang EY, Gang H, Aviv Y, Dhingra R, Margulets V, Kirshenbaum LA, p53 mediates autophagy and cell death by a mechanism contingent on Bnip3, Hypertension 62(1) (2013) 70–77. [DOI] [PubMed] [Google Scholar]
- [26].Maejima Y, Kyoi S, Zhai P, Liu T, Li H, Ivessa A, Sciarretta S, Del Re DP, Zablocki DK, Hsu CP, Lim DS, Isobe M, Sadoshima J, Mst1 inhibits autophagy by promoting Beclin1-Bcl-2 interaction Nature Med 19(11) (2013) 1478–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Dhingra R, Guberman M, Rabinovich-Nikitin I, Gerstein J, Margulets V, Gang H, Madden N, Thliveris J, Kirshenbaum LA, Impaired NF-kappaB signalling underlies cyclophilin D-mediated mitochondrial permeability transition pore opening in doxorubicin cardiomyopathy, Cardiovasc Res 116(6) (2020) 1161–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Dhingra A, Jayas R, Afshar P, Guberman M, Maddaford G, Gerstein J, Lieberman B, Nepon H, Margulets V, Dhingra R, Kirshenbaum LA, Ellagic acid antagonizes Bnip3-mediated mitochondrial injury and necrotic cell death of cardiac myocytes, Free Radic Biol Med 112 (2017) 411–422. [DOI] [PubMed] [Google Scholar]
- [29].Subramaniam S, Exaggerated mitophagy: a weapon of striatal destruction in the brain?, Biochem Soc Trans 48(2) (2020) 709–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Catanzaro MP, Weiner A, Kaminaris A, Li C, Cai F, Zhao F, Kobayashi S, Kobayashi T, Huang Y, Sesaki H, Liang Q, Doxorubicin-induced cardiomyocyte death is mediated by unchecked mitochondrial fission and mitophagy, FASEB J 33(10) (2019) 11096–11108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Koleini N, Kardami E, Autophagy and mitophagy in the context of doxorubicin-induced cardiotoxicity, Oncotarget 8(28) (2017) 46663–46680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Song M, Mihara K, Chen Y, Scorrano L, Dorn GW 2nd, Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts, Cell Metab 21(2) (2015) 273–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J, Nomura M, Mihara K, Egashira K, Ohishi M, Abdellatif M, Sadoshima J, Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress, Circ Res 116(2) (2015) 264–278. [DOI] [PubMed] [Google Scholar]
- [34].Kageyama Y, Hoshijima M, Seo K, Bedja D, Sysa-Shah P, Andrabi SA, Chen W, Hoke A, Dawson VL, Dawson TM, Gabrielson K, Kass DA, Iijima M, Sesaki H, Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain, EMBO J 33(23) (2014) 2798–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Jia G, Hill MA, Sowers JR, Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity, Circ Res 122(4) (2018) 624–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zhang D, He Y, Ye X, Cai Y, Xu J, Zhang L, Li M, Liu H, Wang S, Xia Z, Activation of autophagy inhibits nucleotide-binding oligomerization domain-like receptor protein 3 inflammasome activation and attenuates myocardial ischemia-reperfusion injury in diabetic rats, J Diabetes Investig 11(5) (2020) 1126–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Xu X, Kobayashi S, Chen K, Timm D, Volden P, Huang Y, Gulick J, Yue Z, Robbins J, Epstein PN, Liang Q, Diminished autophagy limits cardiac injury in mouse models of type 1 diabetes, J Biol Chem 288(25) (2013) 18077–18092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Tong M, Saito T, Zhai P, Oka SI, Mizushima W, Nakamura M, Ikeda S, Shirakabe A, Sadoshima J, Alternative Mitophagy Protects the Heart Against Obesity-Associated Cardiomyopathy, Circ Res (2021). [DOI] [PubMed] [Google Scholar]
- [39].Zhang S, Liu X, Bawa-Khalfe T, Lu LS, Lyu YL, Liu LF, Yeh ET, Identification of the molecular basis of doxorubicin-induced cardiotoxicity, Nat Med 18(11) (2012) 1639–1642. [DOI] [PubMed] [Google Scholar]
- [40].Li DL, Wang ZV, Ding G, Tan W, Luo X, Criollo A, Xie M, Jiang N, May H, Kyrychenko V, Schneider JW, Gillette TG, Hill JA, Doxorubicin Blocks Cardiomyocyte Autophagic Flux by Inhibiting Lysosome Acidification, Circulation 133(17) (2016) 1668–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Liu X, Zhang S, An L, Wu J, Hu X, Lai S, Mazhar H, Zou Y, He L, Zhu H, Loss of Rubicon ameliorates doxorubicin-induced cardiotoxicity through enhancement of mitochondrial quality, Int J Cardiol 296 (2019) 129–135. [DOI] [PubMed] [Google Scholar]
- [42].Wang Y, Lu X, Wang X, Qiu Q, Zhu P, Ma L, Ma X, Herrmann J, Lin X, Wang W, Xu X, atg7-Based Autophagy Activation Reverses Doxorubicin-Induced Cardiotoxicity, Circ Res 129(8) (2021) e166–e182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kobayashi S, Volden P, Timm D, Mao K, Xu X, Liang Q, Transcription factor GATA4 inhibits doxorubicin-induced autophagy and cardiomyocyte death, J Biol Chem 285(1) (2010) 793–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Ma X, Godar RJ, Liu H, Diwan A, Enhancing lysosome biogenesis attenuates BNIP3-induced cardiomyocyte death, Autophagy 8(3) (2012) 297–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Sciarretta S, Hariharan N, Monden Y, Zablocki D, Sadoshima J, Is autophagy in response to ischemia and reperfusion protective or detrimental for the heart?, Pediatr Cardiol 32(3) (2011) 275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Gustafsson AB, Gottlieb RA, Recycle or die: the role of autophagy in cardioprotection, J Mol Cell Cardiol 44(4) (2008) 654–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J, Distinct Roles of Autophagy in the Heart During Ischemia and Reperfusion. Roles of AMP-Activated Protein Kinase and Beclin 1 in Mediating Autophagy, Circ Res 100 (2007) 914–922. [DOI] [PubMed] [Google Scholar]
- [48].Fernandez AF, Liu Y, Ginet V, Shi M, Nah J, Zou Z, Zhou A, Posner BA, Xiao G, Tanguy M, Paradis V, Sadoshima J, Rautou PE, Puyal J, Hu MC, Levine B, Interaction between the autophagy protein Beclin 1 and Na+,K+-ATPase during starvation, exercise, and ischemia, JCI Insight 5(1) (2020) e133282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kheloufi M, Boulanger CM, Codogno P, Rautou PE, Autosis occurs in the liver of patients with severe anorexia nervosa, Hepatology 62(2) (2015) 657–658. [DOI] [PubMed] [Google Scholar]
- [50].Jiang K, Xu Y, Wang D, Chen F, Tu Z, Qian J, Xu S, Xu Y, Hwa J, Li J, Shang H, Xiang Y, Cardioprotective mechanism of SGLT2 inhibitor against myocardial infarction is through reduction of autosis, Protein Cell (2021) (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Kelly RA, O’Hara DS, Mitch WE, Steinman TI, Goldszer RC, Solomon HS, Smith TW, Endogenous digitalis-like factors in hypertension and chronic renal insufficiency, Kidney Int 30(5) (1986) 723–729. [DOI] [PubMed] [Google Scholar]
- [52].Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, Richardson JA, Le V, Levine B, Rothermel BA, Hill JA, Cardiac autophagy is a maladaptive response to hemodynamic stress, J Clin Invest 117(7) (2007) 1782–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Myerowitz R, Puertollano R, Raben N, Impaired autophagy: The collateral damage of lysosomal storage disorders, EBioMedicine 63 (2021) 103166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Chi C, Leonard A, Knight WE, Beussman KM, Zhao Y, Cao Y, Londono P, Aune E, Trembley MA, Small EM, Jeong MY, Walker LA, Xu H, Sniadecki NJ, Taylor MR, Buttrick PM, Song K, LAMP-2B regulates human cardiomyocyte function by mediating autophagosome-lysosome fusion, Proc Natl Acad Sci U S A 116(2) (2019) 556–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Kakhlon O, Vaknin H, Mishra K, D’Souza J, Marisat M, Sprecher U, Wald-Altman S, Dukhovny A, Raviv Y, Da’adoosh B, Engel H, Benhamron S, Nitzan K, Sweetat S, Permyakova A, Mordechai A, Akman HO, Rosenmann H, Lossos A, Tam J, Minassian BA, Weil M, Alleviation of a polyglucosan storage disorder by enhancement of autophagic glycogen catabolism, EMBO molecular medicine (2021) e14554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Lim JA, Sun B, Puertollano R, Raben N, Therapeutic Benefit of Autophagy Modulation in Pompe Disease, Mol Ther 26(7) (2018) 1783–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Sciarretta S, Yee D, Nagarajan N, Bianchi F, Saito T, Valenti V, Tong M, Del Re DP, Vecchione C, Schirone L, Forte M, Rubattu S, Shirakabe A, Boppana VS, Volpe M, Frati G, Zhai P, Sadoshima J, Trehalose-Induced Activation of Autophagy Improves Cardiac Remodeling After Myocardial Infarction, J Am Coll Cardiol 71(18) (2018) 1999–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Chen G, Xie W, Nah J, Sauvat A, Liu P, Pietrocola F, Sica V, Carmona-Gutierrez D, Zimmermann A, Pendl T, Tadic J, Bergmann M, Hofer SJ, Domuz L, Lachkar S, Markaki M, Tavernarakis N, Sadoshima J, Madeo F, Kepp O, Kroemer G, 3,4-Dimethoxychalcone induces autophagy through activation of the transcription factors TFE3 and TFEB, EMBO molecular medicine 11(11) (2019) e10469. [DOI] [PMC free article] [PubMed] [Google Scholar]