

Abstract
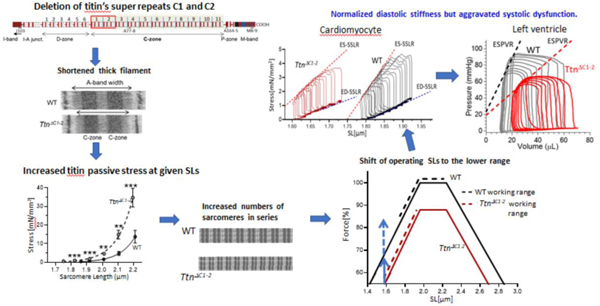
Titin’s C-zone is an inextensible segment in titin, comprised of 11 super-repeats and located in the cMyBP-C-containing region of the thick filament. Previously we showed that deletion of titin’s super-repeats C1 and C2 (TtnΔC1−2 model) results in shorter thick filaments and contractile dysfunction of the left ventricular (LV) chamber but that unexpectedly LV diastolic stiffness is normal. Here we studied the contraction-relaxation kinetics from the time-varying elastance of the LV and intact cardiomyocyte, cellular work loops of intact cardiomyocytes, Ca2+ transients, cross-bridge kinetics, and myofilament Ca2+ sensitivity. Intact cardiomyocytes of TtnΔC1−2 mice exhibit systolic dysfunction and impaired relaxation. The time-varying elastance at both LV and single-cell levels showed that activation kinetics are normal in TtnΔC1−2 mice, but that relaxation is slower. The slowed relaxation is, in part, attributable to an increased myofilament Ca2+ sensitivity and slower early Ca2+ reuptake. Cross-bridge dynamics showed that cross-bridge kinetics are normal but that the number of force-generating cross-bridges is reduced. In vivo sarcomere length (SL) measurements revealed that in TtnΔC1−2 mice the operating SL range of the LV is shifted towards shorter lengths. This normalizes the apparent cell and LV diastolic stiffness but further reduces systolic force as systole occurs further down on the ascending limb of the force-SL relation. We propose that the reduced working SLs reflect titin’s role in regulating diastolic stiffness by altering the number of sarcomeres in series. Overall, our study reveals that thick filament length regulation by titin’s C-zone is critical for normal cardiac function.
Keywords: Diastolic stiffness, contractile function, myofilament function, titin’s C-zone, heart disease
Graphical abstract
Introduction
The contractile unit of muscle, the sarcomere, consists of the actin-based thin filaments anchored in the Z-disk and myosin-based thick filaments that are linked to each other in the middle of the sarcomere, the M-band[1]. These two filament types can interact to cause active force development and shortening[1]. The third myofilament of the sarcomere is titin, with single molecules spanning the half-sarcomere from Z-disk to M-band[2, 3]. The I-band region of titin is extensible and acts as a molecular spring that accounts for the passive stiffness of the sarcomere [4–6]. The A-band region of titin is nearly inextensible[7] and consists mainly of immunoglobulin (Ig)-like and fibronectin (Fn)-like domains. The majority of these domains form 11 super-repeats (each with 11 domains) that comprise the C-zone, so named because it coincides with the region of the thick filament that contains cardiac myosin-binding protein C (cMyBP-C)[8]. We recently generated a TtnΔC1−2 mouse model in which two of titin’s C-zone super-repeats were deleted[9] and found that this results in a reduction in thick filament length by approximately 173 nm, or ~43 nm per deleted super-repeat, revealing that titin regulates the thick filament length (Fig. 1). The shorter thick filaments have ~12% fewer myosin molecules per half thick filament, and thus force (or pressure in the heart) is expected to be reduced by a similar extent. However, systolic function in the TtnΔC1−2 mouse has been found to be depressed by several times more than the expected ~12%[9]. Homozygous TtnΔC1−2 mice were previously shown to manifest reduced contractility and prolonged LV relaxation with increased end-diastolic and end-systolic volumes and thinning of the LV wall, reflecting dilated cardiomyopathy (see [9] and Suppl Fig 1). However, passive stiffness was found to be unaltered [9]. The latter is surprising since shorter thick filaments will, at a given sarcomere length (SL), increase the length of the I-band of the sarcomere and increase the strain on the elastic region of the titin molecule. Diastolic stiffness is therefore expected to be increased in TtnΔC1−2 mice. To better understand the changes that take place in the TtnΔC1−2 mouse, a detailed functional study of the TtnΔC1−2 model was carried out in the present work. Results firmly establish that systolic function is depressed by more than expected based on the shorter thick filaments alone and that diastolic stiffness is unaltered in TtnΔC1−2 mice. We found that in the TtnΔC1−2 mouse, the operating length-range of sarcomeres in the heart is shifted to shorter SLs, and that this contributes to the significant reduction in systolic function and explains why diastolic stiffness is normal in TtnΔC1−2 mice.
Figure 1.

Reduced A-band width in sarcomeres of TtnΔC1−2 mice. Representative electron micrographs showing that compared to WT the A-band is shorter in TtnΔC1−2 mice. By labeling the sarcomere with a cMyBP-C antibody, vertical stripes were obtained that were used to calibrate the image. Using this approach we determined that the A-band width is ~1600 nm in WT mice and ~173 nm shorter in the TtnΔC1−2 mice [9].
Methods
We studied at 2–4 months old wild-type and homozygous TtnΔC1-C2 male mice (for details on this model, see[9]). All procedures were performed according to the NIH Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and University Committee of the University of Arizona.
Time course of LV elastance.
An in-vivo pressure-volume analysis was performed in mice using a SciSense Advantage Admittance Derived Volume Measurement System and 1.2F catheters with 4.5 mm electrode spacing (SciSense). Mice were anesthetized and ventilated with 2% isoflurane, and body temperature was maintained at 37oC. A bilateral subcostal incision was made, and the diaphragm was opened to expose the heart. The catheter was inserted into the LV via the apical approach. Data acquisition and analysis were performed in LabScribe3 (iWorx, Dover NH)[10]. The time-varying elastance of the LV chamber was analyzed from pressure-volume recording as described in [11, 12]. The elastance (E(t)) at a given time point within the cardiac cycle was calculated as E(t) = P(t)/(V(t)-V0), where P(t) and V(t) are the pressure and volume. V0 is the volume intercept of ESPVR [11, 13] and represents the LV volume that does not contribute to pumping function. The E(t) was divided by peak elastance (Ees) to obtain the normalized elastance (En(t)). The elastance during early diastole was fitted with a mono-exponential decay function E(t) = (Ees - plateau)*exp(−K*t) + plateau. The time to 50% relaxation is represented as RT50.The parameters characterizing LV activation and relaxation kinetics were derived.
Intact cardiomyocytes.
Cells were isolated, as described previously[14]. Briefly, mice were heparinized (1,000 U/kg, i.p.) and euthanized using isoflurane. The heart was removed and cannulated via the aorta with a blunted 21-gauge needle for antegrade coronary perfusion. The heart was perfused with perfusion buffer ([in mmol/L] 90 NaCl, 34.7 KCl, 0.6 KH2PO4,0.6 Na2HPO4, 1.2 MgSO4, 12 NaHCO3, 10 KHCO3, 10 HEPES, 10 taurine, 5.5 glucose, 5 BDM,20 Creatine, pH 7.4), followed by 0.05 mg/ml Liberase TM (Roche Applied Science). All intact cell experiments were performed at 37°C in Medium199 (M5017, Sigma-Aldrich) plus 10 μg/mL insulin (I9278, Sigma-Aldrich). An inverted microscope (IX-70; Olympus) was used with a chamber with platinum electrodes to electrically stimulate cells, and a perfusion line with heater control and suction out to maintain a ∼2 ml/min flow rate. Cells were field-stimulated at 2 Hz by MyoPacer stimulator (IonOptix Co, MA). All images were recorded with a 40X objective lens. Data were collected using an IonOptix FSI A/D board and IonWizard 6.3 software (IonOptix Co, MA). Loaded intact cardiomyocytes. A cellular work loop was conducted as described[15, 16]. The glass rods coated with myotak (IonOptix Co) were carefully lowered onto opposite ends of the cell. The myocyte was attached at one end to a glass rod that connected to the force transducer (OFT200, OptiForce transducer, IonOptix LLC). The other end of the cell was attached to a glass rod connected to the piezo translator (Mad City Lab). The cell work loop algorithm was applied through the interface box of the IonOptix system, which contains a field-programmable gate array (FPGA), and the preload and afterload value programmed through the IonWizard software 6.3, as described [15]. The piezo translator adjusted cell length through a feedback control system based on developed force, implementing preload and afterload. The cross-sectional area of the intact cell was obtained from the measured cell width, assuming that the cell’s cross-section was an ellipse [6]. All forces were normalized to stress. Data analysis was performed in LabScribe3 (iWorx, Dover, NH). The ED-SSLR (end-diastolic stress-sarcomere length relation) and ES-SSLR (the end-systolic stress-sarcomere length relation) were fit with linear relation. Time course of cardiomyocyte elastance. The time-varying elastance of the cardiomyocyte was analyzed from cellular work loop recordings. The preload and the afterload values of the work loop clamping algorithm were set at 10% and 50% of the developed force, respectively [15]. The elastance (E(t)) at a given time point within the cardiac cycle was calculated as E(t) = S(t)/(SL(t)-SL0), where S(t) and SL(t) are the stress and sarcomere length, and SL0 is the SL intercept of the ES-SSLR. The beginning of the work loop clamp state 3 was used as Toe, the time of peak elastance was used as Tes. The elastance during work loop clamp state 4 (isometric relaxation) was fit with a mono-exponential decay function to obtain the relaxation constant [15].
Unloaded intact cardiomyocytes.
SL shortening-relengthening in unloaded intact cells were recorded by the IonOptix photometry MultiCell High Throughput system (IonOptix Co, MA). Steady-state twitches (10–20) of isolated cardiomyocytes were averaged, and the transient parameters were obtained from the monotonic transient analysis. Measurement of Ca2+ in unloaded intact cardiomyocytes. Isolated LV cardiac myocytes were incubated with Fura‐2 AM 2 μM (F‐1225, Life Technologies) for 10 min at room temperature and resuspended in Medium199 (M5017, Sigma-Aldrich). Fluorescence was measured ratiometrically. Fura‐2 was excited alternately at 340 and 380 nm, and emission was recorded at 510 nm. Background fluorescence was subtracted for each excitation wavelength. The ratio of fluorescence intensities excited at 340 nm and 380 nm was used as a relative measurement of cytoplasmic Ca2+. Measurement of sarcomeres in series. Cell lengths were recorded by the SoftEdge acquisition module, and the SLs were recorded by SarcLen module based on a fast Fourier transform calculation (IonWizard 6.3 software). The number of sarcomeres in series of the individual cell was calculated from cell length divided by baseline SL.
Echocardiography.
Mice were anesthetized under 2% isoflurane (USP, Phoenix) in an oxygen mixture. Transthoracic echo images were obtained with a Vevo 2100 High-Resolution Imaging System (Visual-Sonics, Toronto, Canada). The images of the LV in parasternal short-axis view in B-mode were used for LV strain analysis.
Length dependence of activation (LDA) of force development.
LDA studies were conducted as described in[17, 18]. Briefly, mouse LV papillary muscles were dissected and permeabilized (skinned) in a relaxing solution with 1% Triton X-100. Skinned papillary muscles were clipped with aluminum t-clips and attached at one end to a force transducer (model 406, Aurora Scientific) and the other end to a length controller (model 322C, Aurora Scientific). The muscles were imaged with a CCD camera, and SL was measured using the first-order diffraction band from a He-Ne laser[19]. The experimental temperature was 15°C. The thickness and width of the preparation were measured, and CSA was calculated assuming an elliptical cross-section. Prior to determining the stress-pCa relationship, the passive stress and slack lengths for each fiber tested were determined. Fibers were stretched to SL 1.95 um then held for 5 min to allow for stress relaxation to occur. Then fibers were activated in a sequence of progressively increasing pCa activating solutions and then relaxed and a released back to the slack length. Note that SL was not controlled during activation. The measurements were then repeated at SL 2.1 um. Measured stresses at each submaximal activation were normalized by the maximal active stress, and the normalized stresses were plotted against the pCa to determine the stress–pCa curve. The stress–pCa curves were fit to the Hill equation: normalized stress = [Ca2+]nH /(K+[Ca2+]nH), where nH is the Hill coefficient, and pCa50=(−logK)/nH. The pCa50 was used as an indicator of Ca2+ sensitivity. The differences between pCa50 measured at SL 1.95 and 2.1 μm were used to index the length-dependent activation (i.e., Δ pCa50). Two preparations were studied per heart, and the average was obtained for each mouse.
Cross-bridge dynamics.
A step-response protocol was applied to maximally activated skinned LV papillary muscle at SL 1.95 µm, using the approach of Campbell and Chandra [20] [21]. Briefly, thin permeabilized fibers were clipped with aluminum t-clips and attached to a force transducer and motor arm, and stretched to an SL of 1.95 ± 0.02 µm, using the video SL system (802D from Aurora Scientific, ASI 25 Industry St., Aurora, ON, Canada, L4G 1X6). After equilibrating the fibers in a low EGTA solution, the fibers were rapidly moved to a bath containing pCa 4.0 solution, and force was allowed to develop. Once force reached a steady-state (FSS), a step length change was imposed, followed by a 5-second hold and then a return to the base length. A series of steps were applied during a single activation (each with a 5-second hold in-between) with amplitude +0.5%, +1.5%, −0.5%, and −1.5% baseline muscle length. The developed force responses were corrected for passive stress measured in pCa 9.0 using the same series of steps. The obtained force response was fit to nonlinear distortion recruitment (NLDR) model[21] to provide the rate of cross-bridge detachment (c) and the rate of strong cross-bridge recruitment (b), as well as the instantaneous fiber force (F1) and new steady-state force (FNSS). Two preparations were studied per heart, and the average was obtained for each mouse.
In situ diastolic SL measurement of LV free wall’s mid-layer.
The hearts were fixed and dissected as described in [22, 23]. In brief, the hearts were removed and cannulated via the aorta with a blunted 21-gauge needle for anterograde coronary perfusion, then were arrested with a buffer containing KCL and 2,3-butanedione monoxide (BDM) ([in mmol/L] 90 NaCl, 34.7 KCl, 0.6 KH2PO4,0.6 Na2HPO4, 1.2 MgSO4, 12 NaHCO3, 10 KHCO3, 10 HEPES, 10 taurine, 30 BDM, pH 7.4), and the LV apex was vented to ensure zero cavity pressure. Then the hearts were perfusion-fixed with 3.5 % glutaraldehyde and stored in PBS solution. The hearts were cut into 2 mm thick equatorial rings. Lateral wall sections of the full wall thickness (approximately 2 mm wide along the circumference) were dissected; thin muscle strips were carefully dissected from the LV free wall’s mid-layer. The SL was measured via laser diffraction. To understand how the shift in the operating SLs is predicted to impact the active force level, the in situ diastolic SLs were plotted relative to the predicted force –SL relations, which was based on the thick filament lengths of 1.6 µm in WT and 1.43 µm in TtnΔC1−2 mice[9], the 0.14 µm width of the thick filament bare zone, the Z-disc width of 0.1 µm, and the thin filament length of 1.0 µm[24–26]. The plateau of the force –SL relation in both WT and TtnΔC1−2 sarcomeres is predicted to be at SL 1.96 to 2.24 µm (the SL at the beginning of the plateau is equal to twice the thin filament length + Z disc width minus the bare zone, and the width of the plateau is similar to the twice the bare zone width (=2 × 0.14 µm)). The SL on the descending limb with zero force is predicted to be 3.7 and 3.53 µm.
Measurement of skinned cardiomyocyte dimension.
Mouse cells, isolated as explained above, were skinned for 7 mins in relaxing solution ([in mmol/L] 40 BES, 10 EGTA, 6.56 MgCl2, 5.88 Na-ATP, 1.0 DTT, 46.35 K-propionate, 15 creatine phosphate, pH 7.0) with protease inhibitors ([in mmol/L] 0.4 leupeptin, 0.1 E64, and 0.5 PMSF) and 0.3% Triton X-100 (Ultrapure; Thermo Fisher Scientific). Cells were washed with relaxing solution (pCa ~9) and stored on ice. Myocytes were added to a room temperature chamber mounted on the stage of an inverted microscope (Diaphot 200; Nikon). The images of skinned cells were digitized and cellular dimensions were determined by ImageJ 1.41 software (National Institutes of Health).
Statistics.
Statistical analysis was performed in Graphpad Prism 8 (GraphPad Software, Inc). Data are shown as mean ± SEM. Statistical significance was set at p<0.05. * p≤0.05 ** p≤0.01 ***p≤0.001 ****p≤0.0001. Differences between groups were assessed by the unpaired t-test or Mann-Whitney U test (for 2 groups); and the two-way ANOVA with a Tukey test for multiple comparisons (for data with 2 controlled variables). A nested t-test was used for intact cardiomyocyte experiments[27].
Results
Contraction-relaxation characteristics of the LV chamber
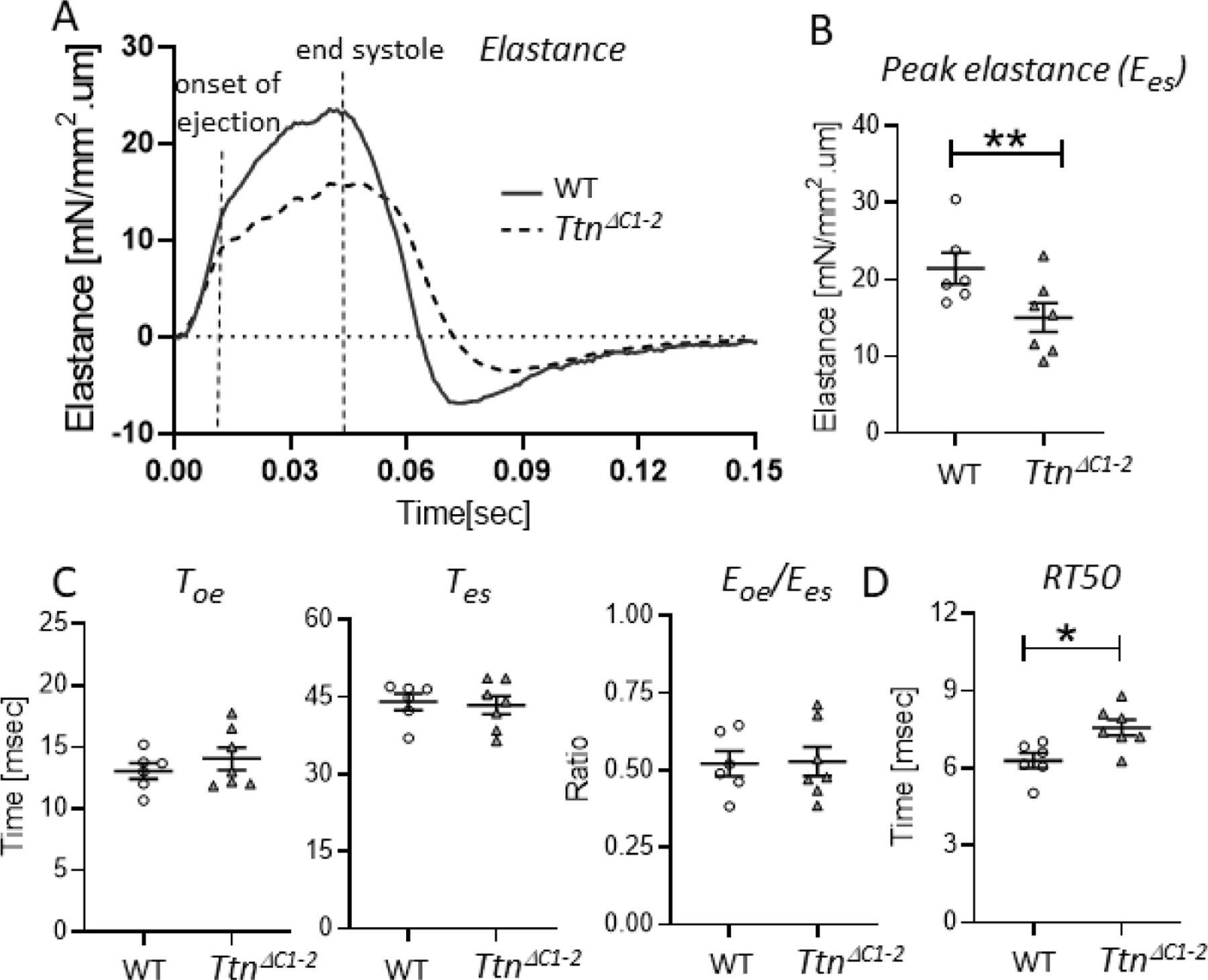
In the earlier characterization of the TtnΔC1−2 model[9], the end systolic pressure-volume relation (ESPVR) was found to be reduced, indicating contractile dysfunction. However, systolic function at the LV chamber level relies not only on the peak pressure but also on the kinetics of ventricular contraction[11, 28]. For instance, the duration of ejection will also determine the stroke volume. Hence, to gain insights into LV contraction-relaxation kinetics, the time-varying elastance (E(t);the LV pressure change relative to the volume change determined throughout the cardiac cycle) was obtained from the PV recording. The timevarying elastance reveals the precise timing of the various phases in the cardiac cycle and the kinetics of LV activation and relaxation[28], Fig. 2A. The peak elastance at end-systole (Ees) was reduced in the TtnΔC1−2 mice by ~56% (Fig. 2B). We determined the time to the onset of ejection (Toe, time to aortic valve opening), the time to end-systole (Tes, time to aortic valve closure), and the ratio of elastance at the onset of ejection relative to at the end-systole (Eoe/Ees). These parameters were unaltered in the TtnΔC1−2 mice (Fig. 2C). However, the elastance analysis revealed that relaxation was slowed in TtnΔC1−2 mice, as shown by the increased time required for elastance to decay by 50% (Fig. 2D). Consistent with these findings, when the elastance curves were normalized to their maximum, the contraction phase was largely the same in both genotypes, while the slowed relaxation in TtnΔC1−2 mice was apparent (Fig. 2A, right curve). Thus, in TtnΔC1−2 mice, the contraction kinetics are unaltered, but relaxation is slow.
Figure 2.
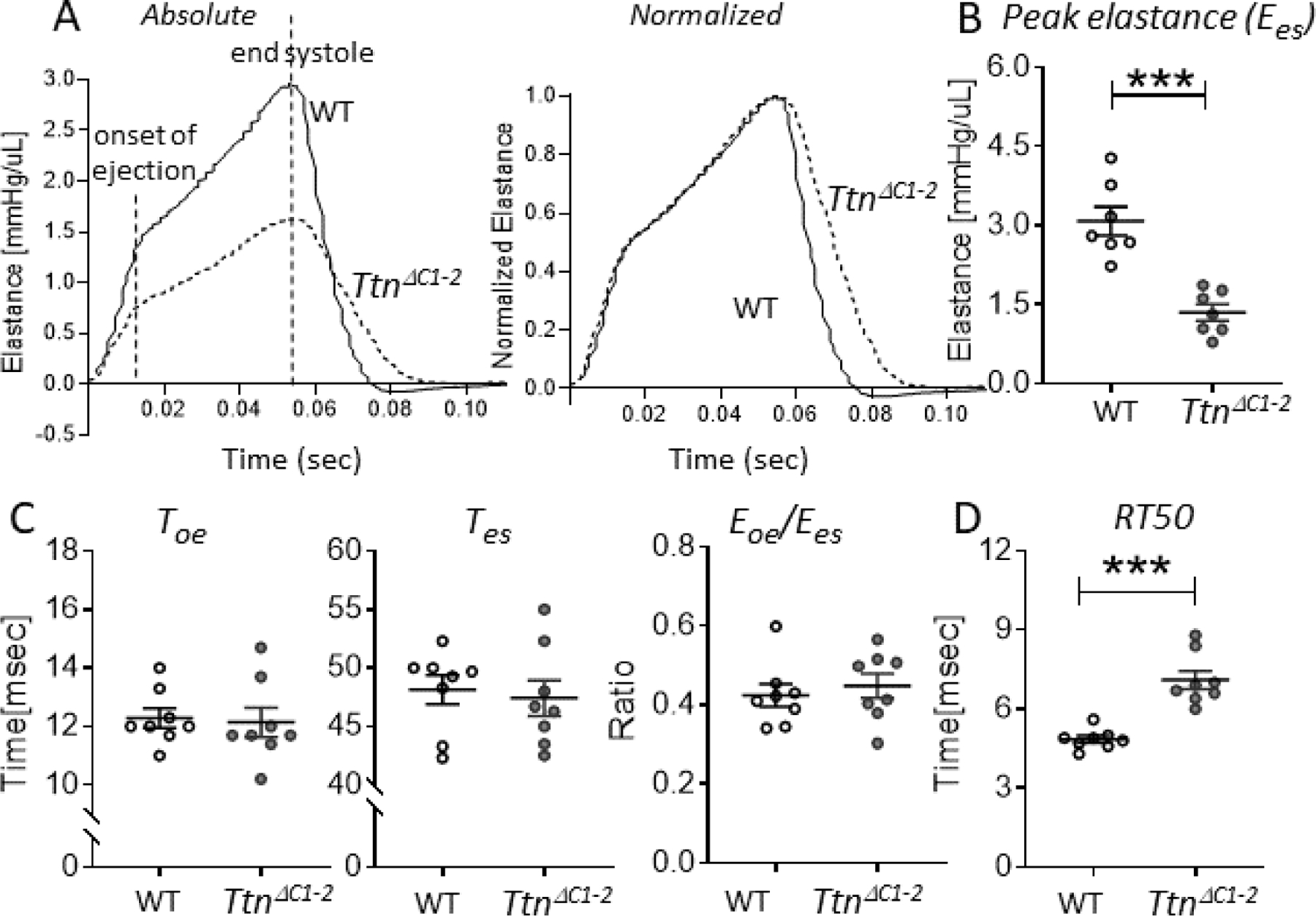
Contraction-relaxation kinetics of the LV chamber. A) Representative examples of an absolute time-varying elastance (left) and an amplitude normalized time-varying elastance (right) of WT (solid lines) and TtnΔC1−2 (broken lines) mice. The onset of ejection and the end-systole are indicated. B) The peak elastance at end systole (Ees) is reduced in the TtnΔC1−2 mice. C) The time to onset of ejection (Toe:left),the time to end systole (Tes:middle), and the ratio of elastance at the onset of ejection relative to at the end systole (Eoe/Ees:right) are not different between genotypes. D) The time to 50% elastance decay is prolonged in TtnΔC1–2. n= 7–8 mice per group. Each data point represents a single mouse. Mean ± S.E.M are shown. Mann Whitney test: ***P < 0.001.
Characterization of TtnΔC1−2 cardiac function at the cellular level.
To study the cellular basis of the chamber level findings, intact cardiomyocytes were isolated from the LV. Both loaded and unloaded cells were studied at 37°C and using 2 Hz field stimulation.
Loaded intact cardiomyocytes.
Single-cell force measurements were performed using a work loop protocol, equivalent to PV analysis at the level of the LV chamber [15]. Cardiomyocytes were glued at their base length to a force transducer at one end and a piezo length controller at the other end. Measured forces were divided by the cross-sectional area of the cell to determine stress. An example of a baseline cellular work loop is shown in Fig. 3A. The cell work loop algorithm, driven by a feedback control system, was applied through the piezo controller to adjust cell length and generate 4 phases that mimic the cardiac cycle, phase I: isovolumic contraction, phase II: systolic ejection, phase III: isovolumic relaxation, and phase IV: diastolic filling [15]. It was found that at baseline, the maximal rate of stress rise (dS/dtmax) during isometric contraction was not different (Fig. 3B, left). In contrast, the maximal rate of stress decline (dS/dtmin) during isometric relaxation was reduced (Fig. 3B, right).
Figure 3.
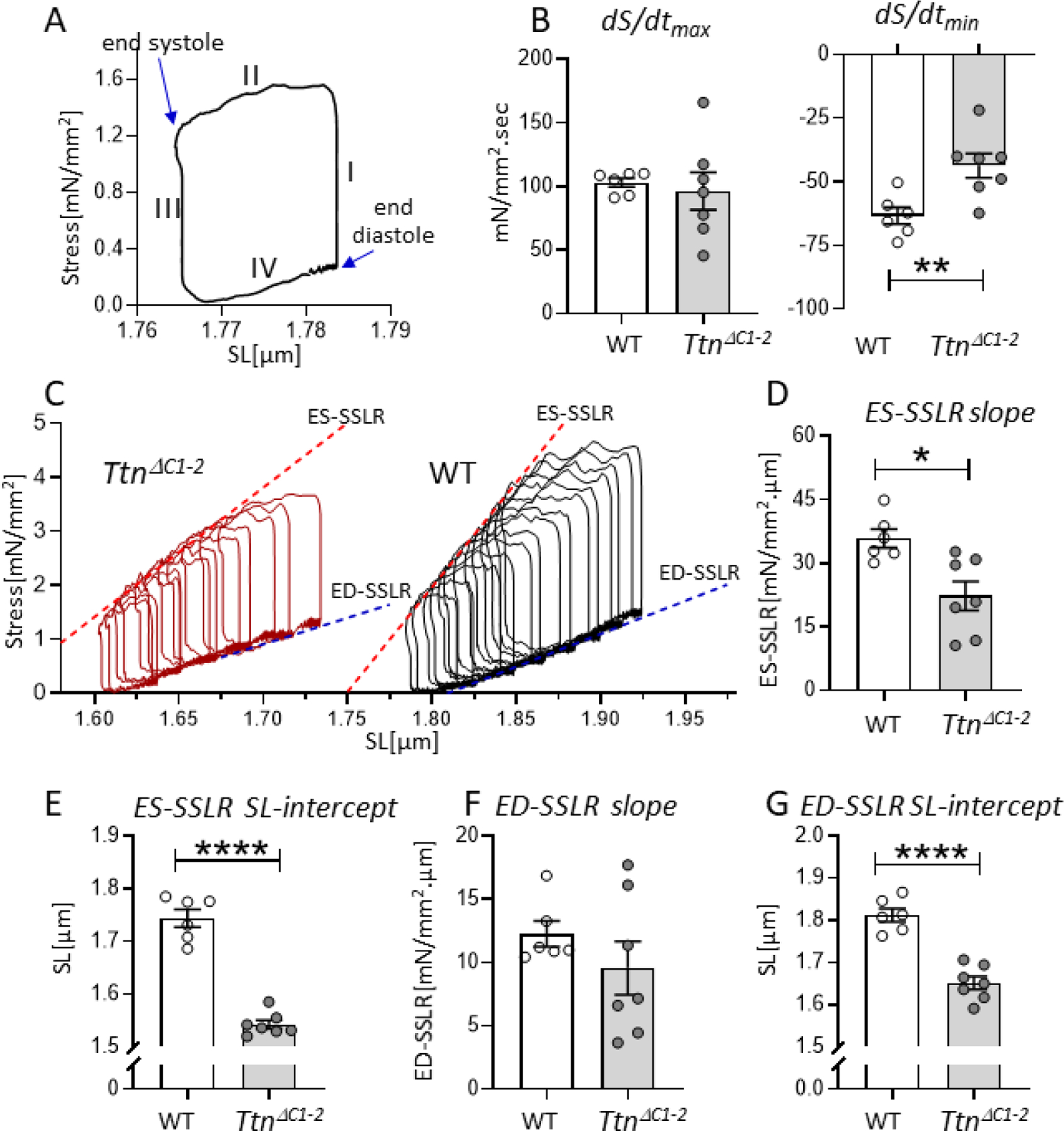
Loaded intact cardiomyocyte mechanics. A) The cellular work loop example shows 4 phases that mimic isovolumic contraction (phase I), systolic ejection (II), isovolumic relaxation (III), and diastolic filling (IV) of the cardiac cycle. B) At baseline, the maximal rate of stress rise (dS/dtmax: left) during isometric contraction is not different in TtnΔC1−2 cells compared to WT. However, the maximal rate of stress decline (dS/dtmin: right) is reduced. C) Representative examples of work loop series of WT and TtnΔC1−2 cells. D) The slope of ES-SSLR, which reflects cellular contractility, is reduced in TtnΔC1−2 cells. E) The SL-intercept of the ES-SSLR is reduced. F) The slope of ED-SSLR, which reflects cellular diastolic stiffness, is not different. G) The SL-intercept of the ED-SSLR of TtnΔC1−2 is reduced. n= 6–7 mice per group, and 3–12 cells per mouse. Each data point represents the mean value of all cells of a single mouse. Mean ± S.E.M are shown in the bar graphs. Nested t-test: *P < 0.05 **P < 0.01****P < 0.0001.
A series of work loops were generated next by gradually increasing the preload and afterload thresholds to mimic an increased venous return at the LV chamber level (Fig. 3C). The ES-SSLR (end-systolic stress-SL relation) and the ED-SSLR (end-diastolic stress-SL relation) were determined. The ES-SSLR, which reflects contractility, had a reduced slope (Fig. 3D) and a much shorter SL intercept in TtnΔC1−2 cells (Fig. 3E). The ED-SSLR, which reflects the cellular diastolic stiffness, had a slope that was not different (Fig. 3F) with an SL intercept that was much reduced in TtnΔC1−2 cells (Fig. 3G). Thus similar to what was found at the level of the LV chamber, cardiac myocytes of TtnΔC1−2 mice have reduced contractility, slowed relaxation, but no change in contraction velocity and diastolic stiffness. Interestingly, while the PV relation of the TtnΔC1−2 revealed a slight right-shift along the volume axis[9], the cellular work loops of the TtnΔC1−2 mice showed a large left-shift along the SL axis.
To determine the intact cardiomyocyte’s contraction-relaxation kinetics, the time course of elastance was analyzed at a diastolic SL of 1.98 um in WT and 1.75 um in TtnΔC1−2, which are in situ diastolic SLs of each genotype (described in Fig 8A). The Toe was determined from the beginning of isotonic contraction or the beginning of phase 2 of the cell work loop algorithm (Figs 3A and 4A)). The Tes was defined as the time of peak elastance (Fig 4A). The peak elastance (Ees) was found to be reduced in TtnΔC1−2 myocyte by 30% (Fig 4B). The Toe,Tes, and the ratio of Eoe/Ees were unaltered (Fig 4C). The time required for elastance to decay by 50% was prolonged (Fig 4D). Thus, consistent with the analysis at the LV level, in TtnΔC1−2 cardiomyocytes the contraction kinetics are unaltered, but relaxation has slowed.
Figure 8.
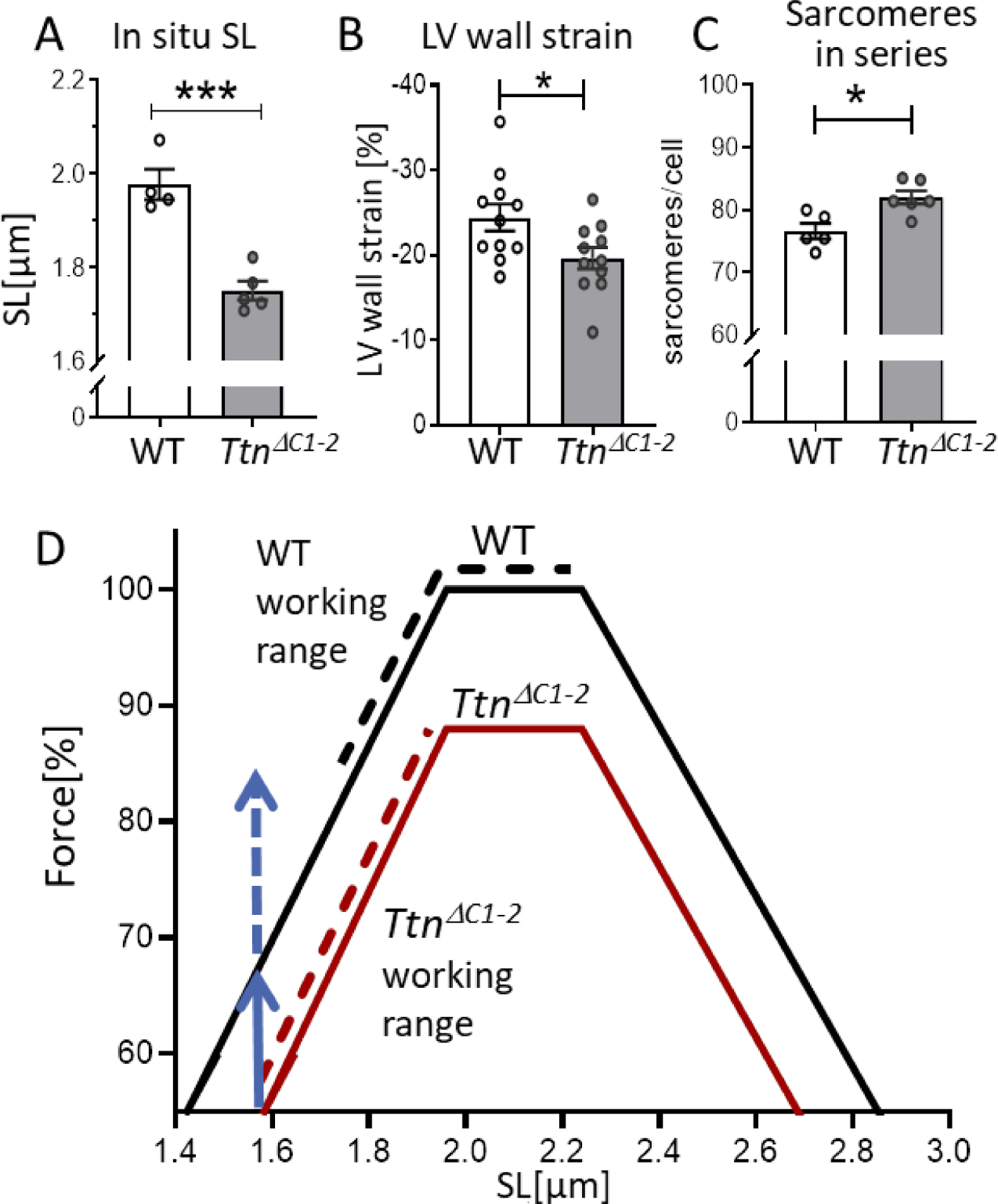
In situ diastolic SL and predicted operating SL ranges (A) The in situ diastolic SLs of the LV mid-wall at Veq is shorter in TtnΔC1−2 hearts. B) The LV circumferential wall strain measured by echocardiography. C) The number of sarcomeres in series per cell is increased in TtnΔC1–2. D) The predicted force–SL relations (solid lines), and the predicted operating SL ranges (broken lines). The vertical blue arrows indicate the predicted force reduction due to the shorter thick filaments (solid blue) and due to the shorter end-systolic SL (broken blue line). See text for details. n= 4–5 mice per group, 80–100 measurements per mouse for A. n=11 mice per group for B. n= 5–6 mice per group, and 50–75 cells per mouse for C. Each data point in A and C represents the mean values of all measurements from a single mouse. Mean ± S.E.M are shown in the bar graphs. Mann-Whitney test, *P < 0.05 ***P < 0.001.
Figure 4.
Intact cardiomyocyte time course of elastance. (A) Representative examples of an absolute time-varying elastance of WT (solid lines) and TtnΔC1−2 (broken lines) cardiomyocytes. The onset of ejection and the end-systole are indicated. B) The peak elastance at end systole (Ees) is reduced in the TtnΔC1−2 myocytes. C) The time to onset of ejection (Toe:left),the time to end systole (Tes:middle), and the ratio of elastance at the onset of ejection relative to at the end systole (Eoe/Ees:right) are not different between genotypes. D) The time to 50% elastance decay is prolonged in TtnΔC1–2. n= 6,7 mice, 3–12 cells per mouse. Each data point represents the mean value of all cells from a single mouse. Mean ± S.E.M are shown. Nested t-test: *P < 0.05 **P < 0.01.
Unloaded intact cardiomyocytes.
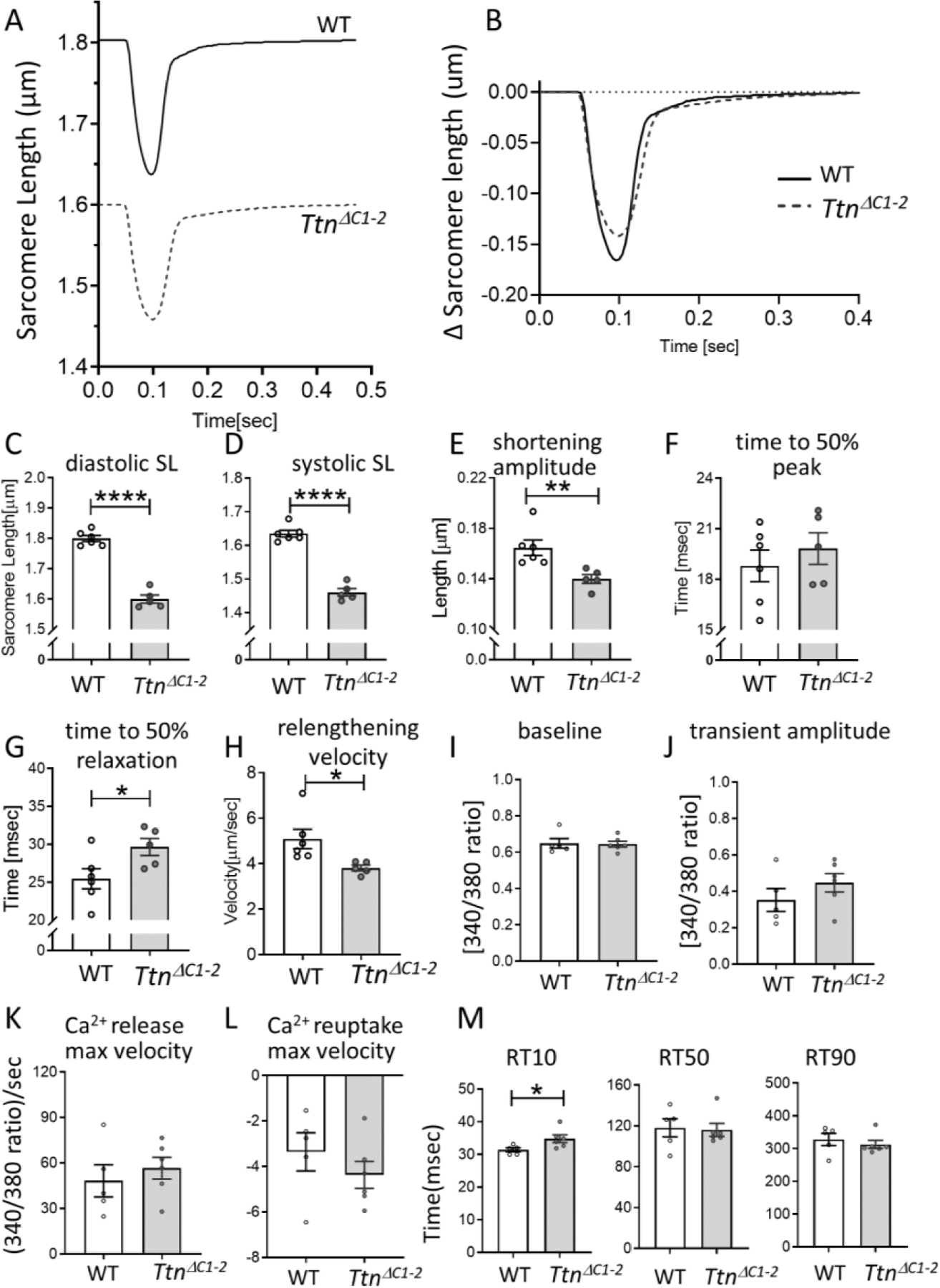
To gain additional insights into intact cellular behavior under baseline conditions, we studied unloaded myocytes (i.e., not attached to force and length probes) that were twitch-activated while SL was measured. Representative SL traces are shown in Fig. 5A and changes in SL amplitude during twitch activation in Fig. 5B. Diastolic and systolic SLs were shorter in TtnΔC1−2 cells than WT (Figs. 5C and D), and the shortening amplitude was reduced (Fig. 5E). The time to 50% of peak shortening was unaltered (Fig. 5F). However, the time from peak shortening to 50% relaxation (Fig. 5G) was prolonged, and the maximal re-lengtening velocity was reduced (Fig. 5H).
Figure 5.
Shortening-relengthening analysis and Ca2+ transients of unloaded intact cardiomyocytes. (A) Representative examples of sarcomere length (SL) shortening-relengthening traces and B) change in SL during twitch activation of WT and TtnΔC1−2 cells, stimulated at 2 Hz, C) Diastolic SL, D) systolic SL, and E) shortening amplitude are all reduced in TtnΔC1−2 cells. F) The time to 50% of peak shortening is not different. G) The time from peak to 50% relaxation is prolonged, and H) the relenghtening velocity is reduced in TtnΔC1−2 myocytes. n= 5–6 mice per group, and 10–20 cells per mouse. I-M, Ca2+ transient of isolated intact cardiomyocytes, measured with Fura-2 340/380 signal. I) Diastolic baseline signal, J) transient amplitude, K) maximal velocity of rising phase of the transient, and L) maximal velocity of Ca2+ reuptake are not different between WT and TtnΔC1–2. M) The time from peak transient to 10%, 50% and 90% signal decay shows that RT10 is prolonged in TtnΔC1−2 cells, however, RT50 and RT90 are not different from WT. n= 5–6 mice per group, and 50–75 cells per mouse. Each data point represents the mean value of all cells from a single mouse. Mean ± S.E.M are shown in the bar graphs. Nested t-test: *P < 0.05 **P < 0.01 ****P < 0.0001.
Ca2+ release-reuptake kinetics.
To determine whether the contractile dysfunction and delayed relaxation were (in part) due to defective Ca2+ handling in the TtnΔC1−2 mice, Ca2+ transients were measured in intact cardiomyocytes, using the ratiometric fluorescent dye Fura-2. The result showed that the diastolic baseline signal (Fig. 5I), the transient amplitude (Fig. 5J), the maximal Ca2+ release velocity (Fig. 5K), and the maximal velocity of Ca2+ reuptake (Fig. 5L) were not different between WT and TtnΔC1−2 cells. These results suggest that altered Ca2+ handling is unlikely to contribute to systolic dysfunction in TtnΔC1−2 mice. The time from peak to 10%, 50%, and 90% signal decay (RT10, RT50, and RT90) showed that RT10 was prolonged in TtnΔC1−2, while RT50 and RT90 were not different from WT (Fig. 5M).
Myofilament function
We focused on the myofilaments’ intrinsic properties and studied whether they are altered in the TtnΔC1−2 model. We examined thin filament-based regulation of contraction and additionally focused on cross-bridge cycling kinetics.
Ca2+ sensitivity of force production in permeabilized papillary muscle.
For each muscle strip, we measured first their passive stress-SL relations, which revealed that passive stress was higher in TtnΔC1−2 mice and that the slack SL was shorter (Fig. 6A), consistent with the single-cell studies. Stress-pCa relations measured at an SL of either 1.95 µm or 2.1 µm are shown in Fig. 6B, and the maximal active stress (pCa 4.0) in Fig. 6C. The maximal active stress was lower in TtnΔC1−2 at both SLs (31% and 23% lower than WT at SLs 1.95 and 2.1 µm, respectively). The stress-pCa relations were fit to the Hill equation to provide the Hill coefficient (nH) and the pCa50. The Hill coefficient was not different (Fig. 6D), suggesting that the cooperative activation of TtnΔC1−2 was normal. The pCa50 revealed that the Ca2+ sensitivity of TtnΔC1−2 muscle was increased at SL 1.95 µm but was similar to WT at SL 2.15 µm (Fig. 6E). Note that when Ca2+ sensitivity is compared not at the same SL but at a more or less comparable level of passive stress (SL 2.1 um in WT vs. SL 1.95 um in TtnΔC1−2 ), the Ca2+ sensitivity of TtnΔC1−2 muscle is lower than in WT muscle (Fig. 6E). The ΔpCa50 (pCa50 at SL 2.1µm - pCa50 at SL 1.95 µm) was reduced in the TtnΔC1−2 muscle (Fig. 6F).
Figure 6.
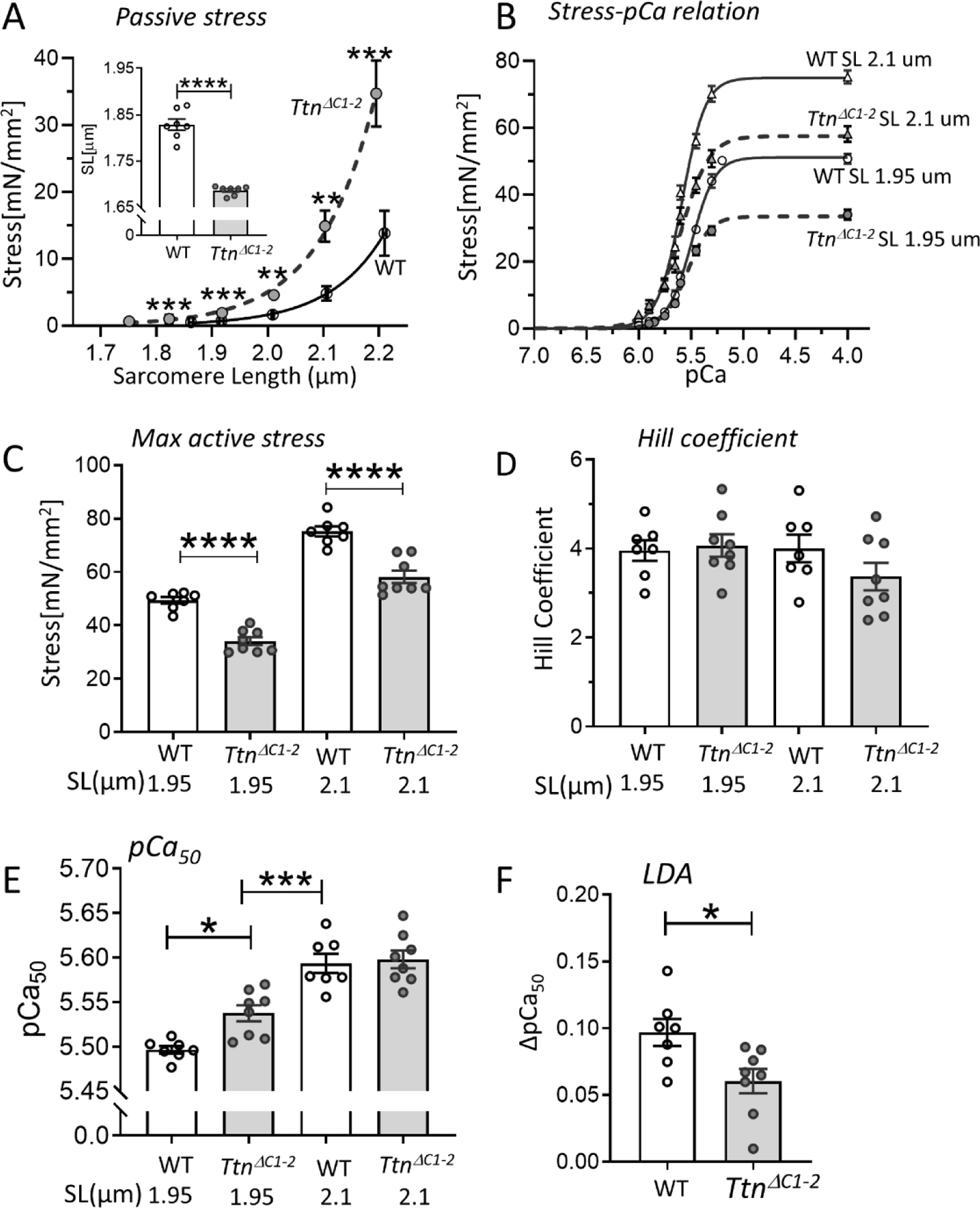
Myofilament Ca2+ sensitivity and length dependence of activation (LDA). (A) Passive stress-SL relation of skinned LV myocardium of WT and TtnΔC1−2 shows a higher passive stress and a shorter slack SL (inset) in TtnΔC1−2 mice. B) Average stress-pCa curves at SL 1.95 µm and 2.1 µm for both genotypes. C) The maximal active stress is lower in TtnΔC1−2 at both SLs. D) The Hill coefficient (nH), which reflects cooperative activation is not different. E) The pCa50, which reflects the myofilament Ca2+ sensitivity, is increased in TtnΔC1−2 at SL 1.95 µm but is not different at 2.1 µm. The pCa50 of TtnΔC1−2 at SL 1.95 um is lower than WT at SL 2.1 um. F) The LDA as reflected by the pCa50 2.1 µm – pCa50 1.95 µm is reduced in TtnΔC1–2. n=7–8 mice per group, 2 preparations per mouse. Each data point represents the mean value of all muscle preparations from a single mouse. Mean ± S.E.M are shown in the bar graphs. Mann-Whitney test for A and F, Two-way ANOVA with Tukey’s multiple comparison test for C-E: *P < 0.05 ***P < 0.001 ****P < 0.0001.
Cross-bridge dynamics.
To gain insights into cross-bridge kinetics in TtnΔC1−2 and WT mice, a step-response protocol was applied to a maximally activated skinned LV papillary muscle at SL 1.95 µm, using the approach of Campbell and Chandra [20] [21]. Muscles were bathed in pCa 4.0 activating solution, and when force reached a steady-state (FSS), a step length change was imposed, followed by a 5-second hold and then a return to the base length. A series of steps were applied during a single activation (each with a 5-second hold in-between) with amplitude +0.5%, +1.5%,−0.5%, and-1.5% baseline muscle length. A schematic diagram of a step increase and its ensuing force response is shown in Fig. 7A. The force shows a characteristic increase that coincides with the step itself and reaches a maximum at the end of the step (F1 in 7A). This is immediately followed by a rapid partial recovery towards the base level of force, attaining a minimum force (Fmin), followed by a final force increase to a new steady-state (FNSS) that differs from the old steady-state (FSS), reflecting cross-bridge recruitment. The slope (ED) of the linear fit to the magnitude of change in force during the step (F1- FSS) and the imposed length change (fraction of muscle length change) reflects the number of the strongly bound cross-bridges[20, 21]. Fig. 7B shows that ED is significantly reduced in the TtnΔC1−2 mice, as expected from the reduced number of cross-bridges per thick filament. The step-induced change in steady-state force (FNSS - FSS) varies linearly with the step size, and the slope of the linear fit (ER), which reflects the number of recruited strong cross-bridges, is reduced in the TtnΔC1−2 mice (Fig. 7C). This finding is also consistent with the reduced number of cross-bridges per thick filament.
Figure 7.
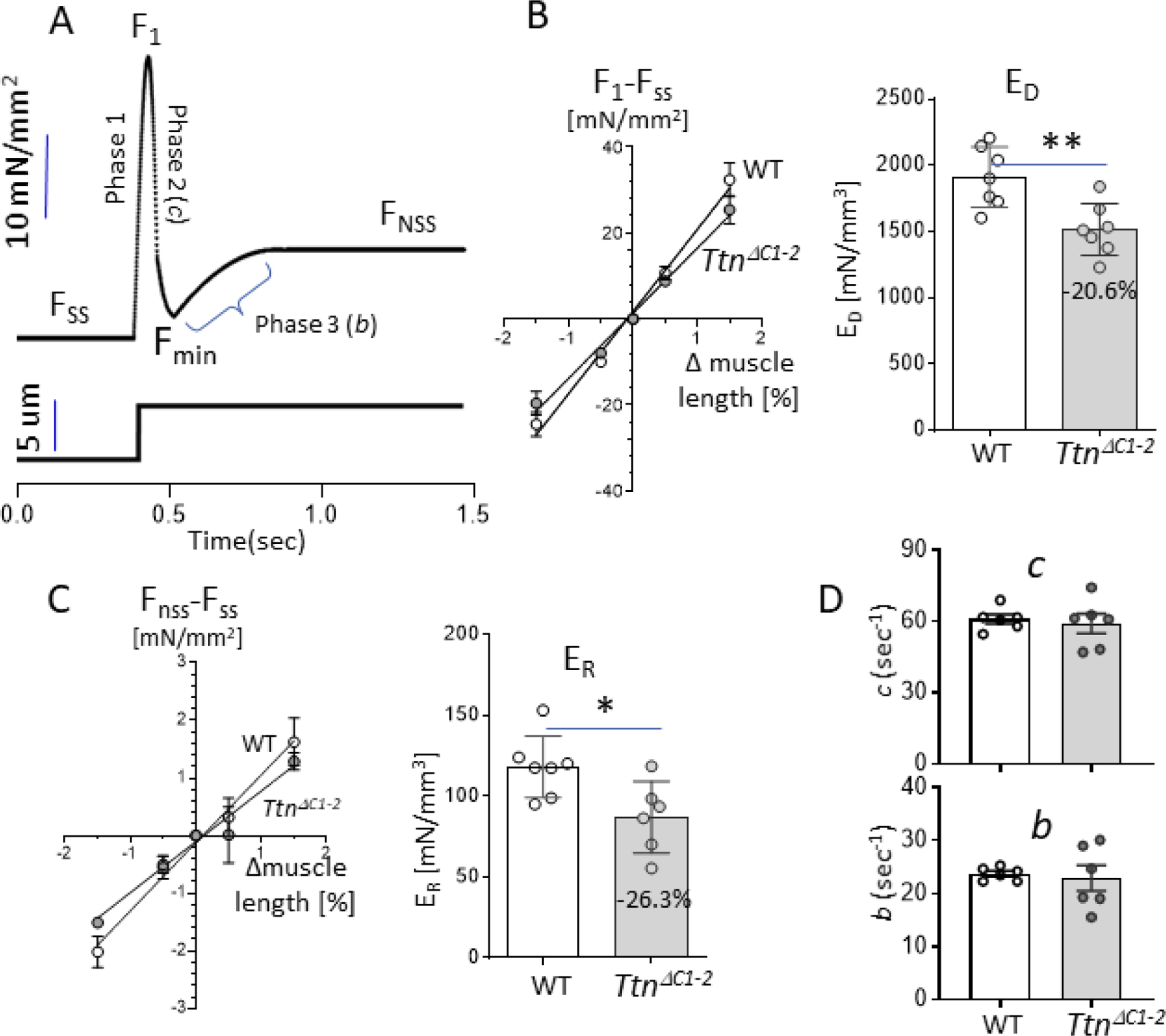
Step response analysis. (A) A schematic diagram describing the features of the force response to a small amplitude step length change imposed on a maximally activated papillary muscle. See text for technical details. B) Left: magnitude of the change in force during the step (F1-Fss) vs. the amplitude of the step (% muscle length change) can be fit with a linear regression line (R2 0.99). Right: the slope of the linear regression fit is reduced in TtnΔC1−2 mice. C) Left: magnitude of the change in steady-state force (FNSS-Fss) vs. the amplitude of the step (% muscle length change) can be fit with a linear regression line (R2 0.99). Right: the slope of the linear fit is reduced in the TtnΔC1−2 mice. D) The rate of cross-bridge detachment (c; top) and the rate of cross-bridge recruitment (b; bottom) are unchanged in the TtnΔC1−2 mice. n=7 mice per group, 2 preparations per mouse. Each data point represents the mean value of all muscle strips from a single mouse. Bar graphs show the mean ± S.E.M. Mann-Whitney test, *P < 0.05 ***P < 0.001 ****P < 0.0001.
The obtained force response to all steps was fit to nonlinear distortion recruitment (NLDR) model[21] to provide the rate of cross-bridge detachment (c) and the rate of strong cross-bridge recruitment (b), see schematic in Fig. 7A. The rate constant, c, was found unaltered in the TtnΔC1−2 mice, and unchanged was also the rate constant b (Fig. 7D, top, and bottom). Thus the step-response analysis shows that the number of cross-bridges is reduced in the TtnΔC1−2 mice and that the cross-bridge dynamics are unchanged.
Operating sarcomere-length range.
Since isolated intact cardiomyocytes had a much shorter SL at baseline, we measured the diastolic SLs of cells in intact hearts that were chemically fixed in the passive state and at their equilibrium volume (Veq, volume at which transmural pressure is zero[29]). (See Methods for details.) Muscle strips were dissected from the LV free wall’s mid-layer, and SL was determined using laser diffraction. The obtained diastolic SLs were 1.98± 0.06 µm in WT and 1.75 ± 0.05 µm in TtnΔC1−2 mice (Fig. 8A), suggesting that the sarcomeres of TtnΔC1−2 operate at lower SLs. The operating SL ranges were estimated based on the echocardiography-based change in the LV circumferential wall strain during systole (Fig. 8B) and assuming that the diastolic SL at Veq reflects the SL in the middle of the operating range. The obtained operating SL ranges were 1.74–2.22 µm in WT and 1.58–1.93 µm in TtnΔC1−2 mice. To gain insights into the mechanistic basis for the reduced operating SL range we measured cell dimensions and the number of sarcomeres in series in isolated cardiomyocytes (see Methods for details). There was no difference in cell dimension between the two genotypes (Supplemental Fig 3). However, the number of sarcomeres in series was increased in TtnΔC1−2(Fig. 8C).
Fig. 8D shows the predicted maximal force-SL relations for WT and TtnΔC1−2 myocytes based on their known thin and thick filaments lengths (for details, see Methods). The predicted force-SL relation of the TtnΔC1−2 mice has a lower plateau due to the ~12% reduction in the length of the cross-bridge bearing part of the thick filament[9]. The broken lines indicated the estimated operating ranges of the two genotypes, revealing that the TtnΔC1−2 operates further down on the ascending limb.
Discussion
The present study on the TtnΔC1−2 model establishes that reducing the thick filament length greatly lowers systolic function, without impacting contraction kinetics, that it slows relaxation kinetics, and does not affect diastolic stiffness. The mechanistic basis of these findings likely includes a shift in the working sarcomere length range to shorter lengths. Below we discuss these findings and focus on 1) systolic dysfunction, 2) slowed relaxation, 3) unaltered diastolic stiffness, and 4) the increased number of sarcomeres in series.
1.1). Reduced number of crossbridges per thick filament.
The TtnΔC1−2 model expresses titin with only 9 titin C-zone repeats (11 in WT titin) and contains thick filaments that are well defined in length but that are shorter than in WT mice [9]. The reduction in thick filament length is ~86 nm per half thick filament, which corresponds to a ~12% reduction in the number of myosin molecules (WT: 49 layers of myosin heads; TtnΔC1−2 : 43 layers of myosin heads, see also [9]) and a similarly predicted reduction in maximal active force [9]. However, the negative impact on systolic performance in TtnΔC1−2 mice is much larger than predicted, e.g., the LV peak systolic elastance is reduced by ~56% (Fig. 2B). A possible explanation for the larger than expected effect is that the missing myosin molecules in the TtnΔC1−2 model contribute more than average to active force production. Indeed it has been proposed[30] that myosin motors in the C-zone of the thick filament are more likely to be activated than those in the D-zone (distal ~160 nm segment of the thick filament). However, making the extreme assumption that only myosin molecules in the C-zone contribute to force development and none in the D-zone, results in a predicted force reduction that is still only ~18% (loss of 2 of the 11 C-zone repeats per half thick filament). Thus although a reduction in myosin reduces force, additional factors must exist to explain the measured large reduction in systolic function.
1.2). Systolic Ca2+.
A reduction in systolic sarcoplasmic Ca2+ would also lower peak elastance of the TtnΔC1−2 LV chamber. However, measured Ca2+ transients of unloaded cardiac myocytes had an unaltered baseline level, peak amplitude, and Ca2+ release velocity (Figs. 5I–K). Although these findings do not definitely exclude changes in the intact ventricle, these available data do not support that a reduced Ca2+ level contributes to the observed large systolic dysfunction. Consistent with this are the unaltered contraction kinetics at the LV chamber and cardiac myocyte levels (Figs. 2 – 5). Hence it appears unlikely that reduced systolic Ca2+ levels are responsible for the much larger than expected reduction in systolic function.
1.3). Sarcomere length working range.
Another possible mechanism for depressing systolic function is the shift in the working sarcomere length (SL) range that was found, with an estimated end-systolic SL of 1.74 µm in WT and 1.58 µm in the TtnΔC1−2 model. Fig. 8D shows the predicted maximal force – SL curves based on filament overlap in WT sarcomeres and assuming a 12% reduction in maximal force of sarcomeres in the TtnΔC1–2. The curves show that at end systole, WT sarcomeres produce ~85% of maximal WT force (SL 1.74 µm) and TtnΔC1−2 sarcomeres ~55% of maximal WT force (SL 1.58 µm). Of this ~35% lower force, ~12–18% can be explained by the shorter thick filaments (solid vertical arrow in Fig. 8D) and the remaining level by the shorter end-systolic SL (broken arrow in 8D). Thus, the altered sarcomere length range in TtnΔC1−2 mice is a likely major contributor to systolic dysfunction. This conclusion is supported by the muscle stiffness (ED) measurements on skinned papillary. Since these measurements were at maximal activation and at the same SL, the obtained 20% lower ED (Fig 7B) in TtnΔC1−2 muscle reflects the true myofilament based deficit due to shorter thick filaments. The 56% reduction in the LV peak elastance (Fig. 2B) reflects not only the deficit due to shorter thick filaments but also the multiple effects due to the shorter lengths at which sarcomeres operate. Thus, the altered SL working range in TtnΔC1−2 mice is a major source of systolic dysfunction.
1.4). Calcium sensitivity of force generation.
Another factor to consider is the SL dependence of Ca2+ sensitivity. The predicted force-SL relations based on filament overlap (Fig 8D) assume that force production is maximal at all SLs. However, it is well established that in reality the Ca2+ level in twitch-activated cardiac muscle is submaximal and that its effectiveness to generate force is progressively less as sarcomeres shorten farther on the ascending limb, due to the phenomenon of length-dependence of activation (LDA) [19, 31]. Earlier mechanical studies on skinned cardiac muscle have shown that the higher titin-based passive forces at long SLs enhances Ca2+ sensitivity of force generation [17, 32] and structural analyses that passive stress mediates rearrangement of the myosin motors, although it is unclear whether this only happens in the presence of Ca2+ [33] or also in the absence of Ca2+ [34, 35]. Based on these previous studies we expected that at a given SL, TtnΔC1−2 sarcomeres have a higher Ca2+ sensitivity than WT sarcomeres. This expected increase was indeed present at the short SL (1.95 µm) that was studied but not at SL 2.1 µm (Fig. 6E). As a result, the LDA (pCa50 at SL 2.1µm - pCa50 at SL 1.95 µm) was reduced in the TtnΔC1−2 muscle (Fig. 6F). We speculate that due to the DCM pathology of the TtnΔC1−2 mouse, changes in post-transcriptional and posttranslational modification of myofilament proteins, which are common in DCM [36, 37], might negate at long SL the expected increase in Ca2+ sensitivity in the TtnΔC1−2 mouse. In addition, deletion of titin’s C1 and C2 super repeats not only causes a reduction in thick filament length but also eliminates one axial stripe of cMyBP-C[9], the sarcomeric protein that regulates contraction-relaxation kinetics and activates thin filament in a strain-dependent manner [28, 38]. Thus, a partial loss of cMyBP-C may contribute to attenuated LDA in TtnΔC1−2 [33, 39–42]. It is important to note that the increased Ca2+ sensitivity that was found at the short SL in TtnΔC1−2 mice is functionally most relevant as in TtnΔC1−2 mice the heart operates at short SL (~1.6–1.9 µm). Assuming that the increased Ca2+ sensitivity at 1.95 µm that was found extrapolates to shorter SL, then this phenomenon is expected to offset at least partially the expected reduction in Ca2+ sensitivity due to the much shorter end-systolic SL in TtnΔC1−2 mice compared to WT mice (Fig 8D).
In summary, the compromised systolic function in TtnΔC1−2 mice is likely to have several sources that include the lower number of myosin molecules per thick filament, and the shorter systolic SL range of TtnΔC1−2 mice. Our work highlights the importance of knowing the operating SL range in animal disease models and in heart disease patients.
2). Mechanistic basis of slowed relaxation.
Pressure-volume studies on the LV revealed that relaxation is slowed (Fig 2D) and the intact twitching myocytes showed that re-lengthening of the myocytes is significantly slower than in WT mice (Figs 3B,4D,5G). The Ca2+ release-reuptake studied in unloaded intact myocytes showed that only the early reuptake (R10) was altered (Fig 5M). Although this might contribute to the slower relaxation it seems unlikely that slowed Ca2+ uptake is the sole explanation. A possible additional explanation is the increased Ca2+ sensitivity of force development that was found at short SL (see above). In addition, since cMyBP-C null mice have slowed relaxation kinetics [43], the reduction in cMyBP-C in TtnΔC1−2 might also contribute to the slow relaxation found at the LV and intact myocyte levels.
3). Why is diastolic stiffness unaffected in TtnΔC1−2 mice?
Diastolic LV chamber stiffness was found to be unaltered in TtnΔC1−2 mice, despite the increase in titin-based passive force at a given SL that is predicted to increased diastolic stiffness (the shorter thick filaments result in a longer I-band and thus a higher strain of titin’s spring region for a given degree of sarcomere stretch). The shorter lengths at which sarcomeres function in the TtnΔC1−2 mice provides a natural explanation. This point can be illustrated by plotting the passive tension of skinned trabeculae against SL using either the same SL scale for both genotypes (as we did in Fig. 6A and reproduced in supplemental Fig 4A) or a different scale that for each genotype reflects its working sarcomere-length range. The results shown in Supplemental Fig 4B reveal that within the physiological SL range of each genotype, passive tension differences are small with a slightly lower passive tension in the TtnΔC1−2 muscle. Thus operating at shorter SLs negatively impacts systolic function but preserves in vivo diastolic function. A similar conclusion was drawn in a recent study on skeletal muscle using the TtnΔ112−158 mouse [44] a model in which titin’s spring is shortened by deleting PEVK exons. Even though this increases passive force at a given sarcomere stretch, also in this model an increased number of sarcomeres in series shifts the sarcomere working range to shorter lengths, which normalizes passive stiffness [44]. Thus, findings in both cardiac and skeletal muscles both show that when titin’s stiffness is experimentally increased, a shift in the physiological SL range ensues that prevents the in vivo passive stiffness from increasing beyond that of WT mice.
4). Sarcomeres in series.
The shift in the SL working range in TtnΔC1−2 mice towards shorter lengths can be explained by an increase in the number of sarcomeres in series (Fig. 8C). As already mentioned above, this feature is also evident in skeletal muscle where increasing the stiffness of titin by genetically shortening the spring region of titin results in an increase in the number of sarcomeres in series [44, 45], while reducing the stiffness of titin has the opposite effect [46]. These responses are not only seen in genetic models but can also be experimentally invoked. For example, using a novel surgery model of unilateral diaphragm denervation, Van der Pijl [46] has recently shown that cyclic stretch of passive muscle is a strong stimulus for increasing the number of sarcomeres in series. In WT mice, a ~25% increase in sarcomeres in series was detected within days of initiating passive stretch and, furthermore, the effect was shown to scale with the level of titin-based passive stiffness. Although the mechanism by which titin-based passive force alters the number of sarcomeres in series is unknown, protein expression studies revealed that during passive stretch, titin-binding proteins such as muscle ankyrin repeat proteins (MARPs) are 10–100 fold upregulated and they might play a role [46]. Clearly, the mechanistic basis of altering the number of sarcomeres in series is an important area of future study as it strongly affects the SL working range and thereby muscle function.
In summary, our results reveal that maintaining thick filament length is critical for normal cardiac function. The reduction in thick filament length in TtnΔC1−2 mice causes a deficit in peak systolic elastance that far exceeds the expected level due to the reduced number of cross-bridges contained within the shortened thick filaments. This larger than expected deficit is likely due to the shift in the operating SL range that was found in the present study, a shift that in TtnΔC1−2 mice moves the sarcomeres further down the ascending limb of the force-SL relation. A delayed relaxation was also found in TtnΔC1−2 mice, possibly due to increased myofilament Ca2+ sensitivity at short SL. Although detrimental for systolic function, the shift in SL working range benefits diastole as it lowers the diastolic stress of TtnΔC1−2 sarcomeres and this offsets the increase in titin-based passive force that would have occurred otherwise. We propose that the shift in the SL working range reflects titin’s role as a mechanosensor that senses the elevated titin-based force and triggers signaling pathways that increase the number of serially-linked sarcomeres. This shortens the length of the sarcomeres and normalizes titin-based force. Whether this mechanism is relevant for disease states where the titin-based passive force is altered [47–49]remains to be established and is an important area for future research.
Supplementary Material
Highlights.
Deletion of titin’s super-repeats C1 and C2 (TtnΔC1−2 mice) results in shorter thick filaments and contractile dysfunction that is more pronounced than expected based on reduced thick filament lengths per se.
The diastolic stress is expected to be increased in TtnΔC1−2 mice, but instead was found to be unaltered.
These findings can be explained by the discovered shift in the operating sarcomere-length range of TtnΔC1−2 mice, which further depresses contractility but normalizes titin-based stress. We propose that titin regulates passive stiffness at the detriment of contractile function.
Acknowledgments.
This research was supported by National Institutes of Health R35HL144998 (H.G.), American Heart Association 19CDA34660099 (M.M.), and a Novel Research Project Awards in the area of Cardiovascular Disease and Medicine, Sarver Heart Center, the University of Arizona (M.M.). We kindly acknowledge our lab members who assisted in this work and the animals that made this research possible.
Abbreviation
- LV
left ventricle
- SL
sarcomere length
- cMyBP-C
cardiac myosin-binding protein C
- ESPVR
end-systolic pressure-volume relation
- EDPVR
end-diastolic pressure-volume relation
- E(t)
elastance
- En(t)
normalized elastance
- Toe
time to the onset of ejection
- Tes
time to the end-systole
- Eoe
elastance at the onset of ejection
- Ees
elastance at end-systole
- dS/dtmax
maximal rate of stress rise
- dS/dtmin
maximal rate of stress decline
- ES-SSLR
end-systolic stress-sarcomere length relation
- ED-SSLR
end-diastolic stress-sarcomere length relation
- LDA
length dependence of activation
- IVRT
Isovolumic relaxation time
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Disclosures
None
References
- 1.Huxley HE. Fifty years of muscle and the sliding filament hypothesis. Eur J Biochem 2004;271(8):1403–15. [DOI] [PubMed] [Google Scholar]
- 2.Granzier HL, Labeit S. The giant protein titin: a major player in myocardial mechanics, signaling, and disease. Circ Res 2004;94(3):284–95. [DOI] [PubMed] [Google Scholar]
- 3.Furst DO, Osborn M, Nave R, Weber K. The organization of titin filaments in the half-sarcomere revealed by monoclonal antibodies in immunoelectron microscopy: a map of ten nonrepetitive epitopes starting at the Z line extends close to the M line. The Journal of cell biology 1988;106(5):1563–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Watanabe K, Muhle-Goll C, Kellermayer MS, Labeit S, Granzier H. Different molecular mechanics displayed by titin’s constitutively and differentially expressed tandem Ig segments. Journal of structural biology 2002;137(1–2):248–58. [DOI] [PubMed] [Google Scholar]
- 5.Li H, Linke WA, Oberhauser AF, Carrion-Vazquez M, Kerkvliet JG, Lu H, et al. Reverse engineering of the giant muscle protein titin. Nature 2002;418(6901):998–1002. [DOI] [PubMed] [Google Scholar]
- 6.Granzier HL, Irving TC. Passive tension in cardiac muscle: contribution of collagen, titin, microtubules, and intermediate filaments. Biophys J 1995;68(3):1027–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Granzier HL, Hutchinson KR, Tonino P, Methawasin M, Li FW, Slater RE, et al. Deleting titin’s I-band/A-band junction reveals critical roles for titin in biomechanical sensing and cardiac function. Proc Natl Acad Sci U S A 2014;111(40):14589–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bang ML, Centner T, Fornoff F, Geach AJ, Gotthardt M, McNabb M, et al. The complete gene sequence of titin, expression of an unusual approximately 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ Res 2001;89(11):1065–72. [DOI] [PubMed] [Google Scholar]
- 9.Tonino P, Kiss B, Strom J, Methawasin M, Smith JE 3rd, Kolb J, et al. The giant protein titin regulates the length of the striated muscle thick filament. Nat Commun 2017;8(1):1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burkhoff D, Mirsky I, Suga H. Assessment of systolic and diastolic ventricular properties via pressure-volume analysis: a guide for clinical, translational, and basic researchers. American journal of physiology. Heart and circulatory physiology 2005;289(2):H501–12. [DOI] [PubMed] [Google Scholar]
- 11.Nagayama T, Takimoto E, Sadayappan S, Mudd JO, Seidman JG, Robbins J, et al. Control of in vivo left ventricular [correction] contraction/relaxation kinetics by myosin binding protein C: protein kinase A phosphorylation dependent and independent regulation. Circulation 2007;116(21):2399–408. [DOI] [PubMed] [Google Scholar]
- 12.Davidson S, Pretty C, Pironet A, Kamoi S, Balmer J, Desaive T, et al. Minimally invasive, patient specific, beat-by-beat estimation of left ventricular time varying elastance. Biomed Eng Online 2017;16(1):42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Georgakopoulos D, Mitzner WA, Chen CH, Byrne BJ, Millar HD, Hare JM, et al. In vivo murine left ventricular pressure-volume relations by miniaturized conductance micromanometry. Am J Physiol 1998;274(4):H1416–22. [DOI] [PubMed] [Google Scholar]
- 14.O’Connell TD, Rodrigo MC, Simpson PC. Isolation and culture of adult mouse cardiac myocytes. Methods Mol Biol 2007;357:271–96. [DOI] [PubMed] [Google Scholar]
- 15.Helmes M, Najafi A, Palmer BM, Breel E, Rijnveld N, Iannuzzi D, et al. Mimicking the cardiac cycle in intact cardiomyocytes using diastolic and systolic force clamps; measuring power output. Cardiovasc Res 2016;111(1):66–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Methawasin M, Strom J, Borkowski T, Hourani Z, Runyan R, Smith JE 3rd, et al. Phosphodiesterase 9a Inhibition in Mouse Models of Diastolic Dysfunction. Circ Heart Fail 2020;13(5):e006609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee EJ, Peng J, Radke M, Gotthardt M, Granzier HL. Calcium sensitivity and the Frank-Starling mechanism of the heart are increased in titin N2B region-deficient mice. J Mol Cell Cardiol 2010;49(3):449–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Farman GP, Walker JS, de Tombe PP, Irving TC. Impact of osmotic compression on sarcomere structure and myofilament calcium sensitivity of isolated rat myocardium. Am J Physiol Heart Circ Physiol 2006;291(4):H1847–55. [DOI] [PubMed] [Google Scholar]
- 19.Kentish JC, ter Keurs HE, Ricciardi L, Bucx JJ, Noble MI. Comparison between the sarcomere length-force relations of intact and skinned trabeculae from rat right ventricle. Influence of calcium concentrations on these relations. Circ Res 1986;58(6):755–68. [DOI] [PubMed] [Google Scholar]
- 20.Ford SJ, Chandra M, Mamidi R, Dong W, Campbell KB. Model representation of the nonlinear step response in cardiac muscle. J Gen Physiol 2010;136(2):159–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gollapudi SK, Reda SM, Chandra M. Omecamtiv Mecarbil Abolishes Length-Mediated Increase in Guinea Pig Cardiac Myofiber Ca(2+) Sensitivity. Biophys J 2017;113(4):880–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Methawasin M, Hutchinson KR, Lee EJ, Smith JE, 3rd, Saripalli C, Hidalgo CG, et al. Experimentally increasing titin compliance in a novel mouse model attenuates the Frank-Starling mechanism but has a beneficial effect on diastole. Circulation 2014;129(19):1924–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chung CS, Granzier HL. Contribution of titin and extracellular matrix to passive pressure and measurement of sarcomere length in the mouse left ventricle. J Mol Cell Cardiol 2011;50(4):731–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sosa H, Popp D, Ouyang G, Huxley HE. Ultrastructure of skeletal muscle fibers studied by a plunge quick freezing method: myofilament lengths. Biophys J 1994;67(1):283–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rassier DE, MacIntosh BR, Herzog W. Length dependence of active force production in skeletal muscle. J Appl Physiol (1985) 1999;86(5):1445–57. [DOI] [PubMed] [Google Scholar]
- 26.Gordon AM, Huxley AF, Julian FJ. The variation in isometric tension with sarcomere length in vertebrate muscle fibres. J Physiol 1966;184(1):170–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eisner DA. Pseudoreplication in physiology: More means less. J Gen Physiol 2021;153(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Palmer BM, Georgakopoulos D, Janssen PM, Wang Y, Alpert NR, Belardi DF, et al. Role of cardiac myosin binding protein C in sustaining left ventricular systolic stiffening. Circ Res 2004;94(9):1249–55. [DOI] [PubMed] [Google Scholar]
- 29.Bell SP, Nyland L, Tischler MD, McNabb M, Granzier H, LeWinter MM. Alterations in the determinants of diastolic suction during pacing tachycardia. Circ Res 2000;87(3):235–40. [DOI] [PubMed] [Google Scholar]
- 30.Brunello E, Fusi L, Ghisleni A, Park-Holohan SJ, Ovejero JG, Narayanan T, et al. Myosin filament-based regulation of the dynamics of contraction in heart muscle. Proc Natl Acad Sci U S A 2020;117(14):8177–8186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Campbell KS. Impact of myocyte strain on cardiac myofilament activation. Pflugers Archiv : European journal of physiology 2011;462(1):3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee EJ, Nedrud J, Schemmel P, Gotthardt M, Irving TC, Granzier HL. Calcium sensitivity and myofilament lattice structure in titin N2B KO mice. Arch Biochem Biophys 2013;535(1):76–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Park-Holohan SJ, Brunello E, Kampourakis T, Rees M, Irving M, Fusi L. Stress-dependent activation of myosin in the heart requires thin filament activation and thick filament mechanosensing. Proc Natl Acad Sci U S A 2021;118(16). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ait-Mou Y, Hsu K, Farman GP, Kumar M, Greaser ML, Irving TC, et al. Titin strain contributes to the Frank-Starling law of the heart by structural rearrangements of both thin- and thick-filament proteins. Proc Natl Acad Sci U S A 2016;113(8):2306–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Farman GP, Gore D, Allen E, Schoenfelt K, Irving TC, de Tombe PP. Myosin head orientation: a structural determinant for the Frank-Starling relationship. Am J Physiol Heart Circ Physiol 2011;300(6):H2155–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schwinger RH, Bohm M, Koch A, Schmidt U, Morano I, Eissner HJ, et al. The failing human heart is unable to use the Frank-Starling mechanism. Circ Res 1994;74(5):959–69. [DOI] [PubMed] [Google Scholar]
- 37.Bollen IAE, Schuldt M, Harakalova M, Vink A, Asselbergs FW, Pinto JR, et al. Genotype-specific pathogenic effects in human dilated cardiomyopathy. J Physiol 2017;595(14):4677–4693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moss RL, Fitzsimons DP, Ralphe JC. Cardiac MyBP-C regulates the rate and force of contraction in mammalian myocardium. Circ Res 2015;116(1):183–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kampourakis T, Yan Z, Gautel M, Sun YB, Irving M. Myosin binding protein-C activates thin filaments and inhibits thick filaments in heart muscle cells. Proc Natl Acad Sci U S A 2014;111(52):18763–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mun JY, Previs MJ, Yu HY, Gulick J, Tobacman LS, Beck Previs S, et al. Myosin-binding protein C displaces tropomyosin to activate cardiac thin filaments and governs their speed by an independent mechanism. Proc Natl Acad Sci U S A 2014;111(6):2170–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mamidi R, Gresham KS, Stelzer JE. Length-dependent changes in contractile dynamics are blunted due to cardiac myosin binding protein-C ablation. Front Physiol 2014;5:461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kumar M, Govindan S, Zhang M, Khairallah RJ, Martin JL, Sadayappan S, et al. Cardiac Myosin-binding Protein C and Troponin-I Phosphorylation Independently Modulate Myofilament Length-dependent Activation. J Biol Chem 2015;290(49):29241–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pohlmann L, Kroger I, Vignier N, Schlossarek S, Kramer E, Coirault C, et al. Cardiac myosin-binding protein C is required for complete relaxation in intact myocytes. Circ Res 2007;101(9):928–38. [DOI] [PubMed] [Google Scholar]
- 44.Brynnel A, Hernandez Y, Kiss B, Lindqvist J, Adler M, Kolb J, et al. Downsizing the molecular spring of the giant protein titin reveals that skeletal muscle titin determines passive stiffness and drives longitudinal hypertrophy. Elife 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.de Winter JM, Molenaar JP, Yuen M, van der Pijl R, Shen S, Conijn S, et al. KBTBD13 is an actin-binding protein that modulates muscle kinetics. J Clin Invest 2020;130(2):754–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van der Pijl R, Strom J, Conijn S, Lindqvist J, Labeit S, Granzier H, et al. Titin-based mechanosensing modulates muscle hypertrophy. J Cachexia Sarcopenia Muscle 2018;9(5):947–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zile MR, Baicu CF, Ikonomidis JS, Stroud RE, Nietert PJ, Bradshaw AD, et al. Myocardial stiffness in patients with heart failure and a preserved ejection fraction: contributions of collagen and titin. Circulation 2015;131(14):1247–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hopf AE, Andresen C, Kotter S, Isic M, Ulrich K, Sahin S, et al. Diabetes-Induced Cardiomyocyte Passive Stiffening Is Caused by Impaired Insulin-Dependent Titin Modification and Can Be Modulated by Neuregulin-1. Circ Res 2018;123(3):342–355. [DOI] [PubMed] [Google Scholar]
- 49.Guo W, Schafer S, Greaser ML, Radke MH, Liss M, Govindarajan T, et al. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat Med 2012;18(5):766–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.