

Abstract
The human leucocyte antigen (HLA) loci have been widely characterized to be associated with viral infectious diseases using either HLA allele frequency-based association or in silico predicted studies. However, there is less experimental evidence to link the HLA alleles with COVID-19 and other respiratory infectious diseases, particularly in the lung cells. To examine the role of HLA alleles in response to coronavirus and other respiratory viral infections in disease-relevant cells, we designed a two-stage study by integrating publicly accessible RNA-seq data sets, and performed allelic expression (AE) analysis on heterozygous HLA genotypes. We discovered an increased AE pattern accompanied with overexpression of HLA-B gene in SARS-CoV-2-infected human lung epithelial cells. Analysis of independent data sets verified the respiratory virus-induced AE of HLA-B gene in lung cells and tissues. The results were further experimentally validated in cultured lung cells infected with SARS-CoV-2. We further uncovered that the antiviral cytokine IFNβ contribute to AE of the HLA-B gene in lung cells. Our analyses provide a new insight into allelic influence on the HLA expression in association with SARS-CoV-2 and other common viral infectious diseases.
Subject terms: Disease genetics, Viral infection
Introduction
Coronavirus 2019 (COVID-19) is an emerging infectious disease caused by a new coronavirus, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [1]. The wide spectrum of clinical manifestations in patients infected with SARS-CoV-2 has been observed, ranging from a mild syndrome, to severe conditions requiring critical care. In particular, the disease can progress to pneumonia, respiratory failure, and death. This progression is related to hyper-inflammatory response, named as “cytokine storm”. There is growing evidence that the interplay between viral and host factors plays an important role on the varying outcomes of patients with viral infectious diseases [2]. Regarding to COVID-19 risk and severity, genome-wide association studies (GWAS) have uncovered tens of host genetic susceptible loci [3–9]. For example, Ellinghaus et al. reported two risk loci—one is the ABO blood group locus and the other a cluster of genes on chromosome 3, were linked to respiratory failure during SARS-CoV-2 infection [3]. Two studies presented the population-based differences of the allele frequencies of genetic variants in angiotensin converting enzyme 2 (ACE2) [10] and transmembrane protease serine 2 (TMPRSS2) [11], two genes involved in SARS-CoV-2 host cell entry [12]. Using multi-omics approach, Pathak et al. identified 27 genes across 13 genomic regions in association with COVID-19 related hospitalization [5]. However, there is less study about the functional implication of genetic risk factors related to SARS-CoV-2 infection, particularly in the lung respiratory epithelia, a primary entry site for the virus.
The human leukocyte antigen (HLA) class I molecules (HLA-A, -B and -C as representative) play a critical role in the manner that antigen peptides are presented to T cells. Specifically, an effective antiviral immunity requires optimal pathogen-derived peptide presentation by class I on the surface of infected cells for recognition by CD8 + T cells. Genetically, the HLA loci are one of a few hotspots in a strong association with the susceptibility or resistance to infectious diseases, immune disorders and malignancies [13]. Of note, the distinct role of certain HLA-B alleles on viral infections has been shown in previous studies [14, 15]. More specifically, Lin et al. found the association of HLA-B*4601 allele with the severity of SARS [16]. Using in silico analysis of viral peptide-MHC class I binding affinity, Nguyen et al. further proposed the association of this HLA-B allele with the SARS-CoV-2 [17]. However, in current COVID-19 disease GWAS studies [3, 4], this locus does not reach the genome-wide significance. Therefore, it is demanded to understand the genetic impact of SARS-CoV-2 and other respiratory viral infections on expression of HLA class I genes in disease-relevant cells.
Allele-specific expression (AE) is an RNA molecular quantification of a genetic locus by measuring the relative expression on the two alleles at a heterozygous single nucleotide polymorphism. The genes with two copies exhibiting AE can provide a strong foundation for investigating the genetic or epigenetic mechanisms linked to complex traits and diseases [18, 19]. Notably, the AE mapping has become a powerful approach to identify functional cis-regulatory variations [20, 21], genomic imprinting regions [22], X-chromosome inactivation or escaping loci [23, 24]. For this reason, it is possible to utilize AE analysis with proper methods to identify potentially regulatory variants linked with viral respiratory diseases.
Benefitted from data sharing and accessibility in public database, including Gene Expression Omnibus (GEO) database, leveraging functional genomics data-driven strategy to search for functional genetic variants associated with viral diseases becomes practicable. Here, by integrating RNA-sequencing (RNA-seq) data involved in SARS-CoV-2 and other viral respiratory infections in both human lung cell lines and tissue specimens from case-control studies, we performed AE analysis on heterozygous HLA genes to investigate allelic influence on the transcription of HLA genes in association with SARS-CoV-2 and other viral infections. Having results from the independent data sets and experimental validation, our study demonstrated that AE between different HLA-B alleles is associated with SARS-CoV-2 and other viral infections in human lung cells.
Materials and methods
Data collection
By searching the GEO database [25], we collected RNA-seq data sets from three independent studies involved in human lung cell lines infected with SARS-CoV-2 and other respiratory viruses or lung tissue specimens from patients with COVID-19. We designed a two-stage study to analyze these RNA-seq data (see details in Fig. 1).
Fig. 1. Schematic overview of the study design.
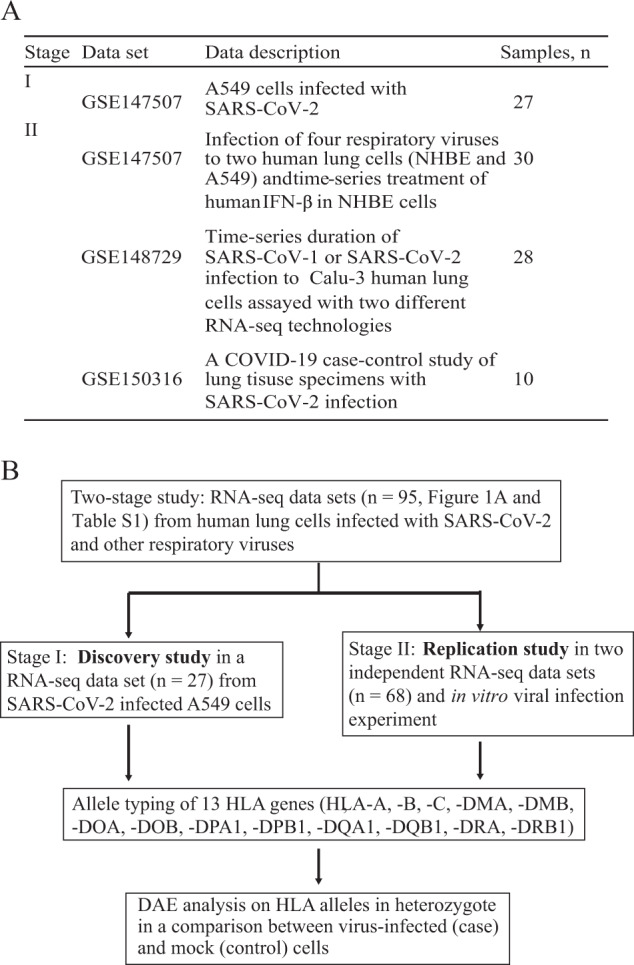
A Summary of RNA-seq data sets used in the study. B On the basis of collected RNA-seq data sets, a two-stage study was designed to identify differential allelic expression of HLA genes in a comparison between cases (virus-infected) and controls.
RNA-seq data processing
We employed similar methods described previously with several modifications [26] to process the RNA-seq data. In brief, all raw sequencing data (FASTQ format) were initially mapped to the human reference genome (hg19) using Hisat2 program [27] with the default setting. Aligned data were processed and converted into BAM files using SAMtools program. To quantify gene expression levels, read count was calculated using the featureCounts program, then implemented in the edgeR package to calculate the count per million (CPM) values.
Allelic expression analysis of HLA genes
We used a similar approach described in previous study to type HLA alleles [28]. With BAM files and the IPD-IMGT/HLA Database [29] as input, thirteen HLA genes (see Fig. 1B) were typed using the SOAP-HLA [30]. Next, we downloaded the corresponding complementary DNA sequence from the IPD-IMGT/HLA database for all typed HLA genes. These sequences were then combined into a single FASTA file as an HLA-personalized genome for each cell type. We then extracted reads mapped on the HLA locus in the human reference genome (chr6:29,651,300–33,160,000, hg19) and unmapped from BAM files. We re-aligned, per sample, those reads to the prepared HLA-personalized genome. We removed PCR duplicates with Picard Tools. For AE analysis of two alleles for a given heterozygous HLA gene, we used counts for the whole cDNA per HLA allele, a method described previously [31]. The allelic fold change (FC) was then compared between two alleles per sample using the following formula:
where allele1 and allele2 denote the two alleles for a given HLA gene.
To determine the association of each heterozygous HLA gene with SARS-CoV-2 infection, we compared the allelic FC of two alleles between two groups (e.g., virus infected and control) in each series using a linear regression model.
The p-values and beta coefficients for the SARS-CoV-2 infection term in our regression models were used to establish the significance of the association at each HLA gene, and to estimate the differences in AE between cases and controls, respectively. Finally, p-values from all series were combined to calculate overall p-value using the Fisher’s Method function in R package MADAM [32].
To quantify HLA gene expression levels per sample, read counts on two alleles per HLA gene were summed and normalized by library size using CPM implemented in the edgeR package. A comparison of HLA expression between two groups with p value was calculated using the exact test of the edgeR package.
Cell cultures infected with SARS-CoV-2, stimulated with IFN and RT-qPCR
The SARS-CoV-2 isolate used in this study, named 8X (our unpublished data), was isolated from a COVID-19 patient by the Zhejiang Provincial Center for Disease Control and Prevention, Zhejiang Province, China. Briefly, cells of human lung epithelial cell line, A549 (Category No. CCL-18, from ATCC) were seeded in 6-well plates with 90% confluence a day before the virus infection. The A549 cells were inoculated with SARS-CoV-2 8X virus at MOI of 0.2 for 1 h. Mock-infected cell cultures were used as negative control. After removing the virus inoculum, the infected cell cultures were washed twice with PBS, fed with DMEM medium (GIBCO) plus 3% FBS and incubated in CO2 5% incubator at 37 °C. The infected cells and mock-infected cells were harvested for RNA extraction at 24, 48 and 72 h.
For IFNβ stimulation, the A549 cells were cultured in 6-well plates and treated with 1000 U/ml human recombinant IFNβ (Category No. 10704-HNAS, from Sino Biological Inc.) and harvested for RNA extraction at 4, 8 and 24 h.
Total RNA was extracted with Qiagen RNeasy extraction kit. 2 μg of total RNA was reverse transcribed (RT) into cDNA using PrimeScript II first-strand cDNA synthesis kit (Takara, 6210 A) and random primers. One-twentieth of the RT reaction was used as a template for real-time PCR using TB Green Premix Ex Taq II kit (Tli RNaseH Plus, Takara, RR820). For each of two HLA-B alleles, the average Ct value from three replicates represented its expression level. Allelic expression level between two alleles was calculated with 2-ΔCt. Then, differential allelic expression was calculated at each time point after SARS-CoV-2 8X infection (or IFNβ treatment) relative to the negative control. Relative expression of HLA-B gene was calculated with 2−ΔΔCt normalized to the average value of housekeeping gene GAPDH. Data are presented as mean ± SD of three replicates. We used the Student’s t-test to test the significance between two conditions. All qPCR primers and pairs are listed in Table S4.
Gene-based association analysis
For gene-based analysis, we first downloaded three GWAS meta-analysis datasets (round 6, https://www.covid19hg.org/results/r6/) from the COVID-19 Host Genetics Initiative (COVID-19 HGI) [4]. The three datasets are implicated the genetic susceptibility with SARS-CoV-2 infection and severity of COVID-19 disease [6]. Then we extracted p values for variants across the HLA-B locus (chr6:31353875-31357179, GRCh38.p13) and calculated the overall p value using Fisher’s Method function implemented in R package MADAM [32].
Prediction of peptide binding difference in SARS-CoV-2 proteins presented by HLA-B alleles
Sixteen SARS-CoV-2 protein sequences were downloaded from the UniProt database (https://www.uniprot.org/). By running the MHCflurry 2.0 with the default setting [33], we scanned the binding affinity of HLA-B alleles on peptides in 16 SARS-CoV-2 proteins to predict peptide presentation difference for different HLA-B alleles.
Statistical analysis
Statistical analyses were presented above for single or multiple comparisons, except for several analyses that were noted in the figure legend. P values < 0.05 were considered statistically significant. All values were expressed as means ± SD (standard deviation).
Results
Data summary
RNA-seq data sets of 95 samples from three independent studies (Accession ID: GSE147507, GSE148729 and GSE150316) were collected in the current study (Fig. 1A, Table S1 and S2). All these data were from experiments involved in either human lung cells infected with SARS-CoV-2 or other respiratory viruses, or lung tissues from patients with COVID-19 and healthy controls.
Allelic expression of HLA-B in association with SARS-CoV-2 infection
We next designed a two-stage study (Fig. 1B) to identify the HLA genes showing differences in allelic expression (DAE) in association with SARS-CoV-2 infection (see Methods in detail). In stage I, also termed as the discovery stage, an RNA-seq dataset (Accession ID: GSE147507, Table S1) comprised 27 samples from experiments with four series (biologically independent experiments) in A549 lung cells infected with SARS-CoV-2 was analyzed. Based on the genotyping result, seven of 13 HLA loci are consistently typed in RNA-seq data across the four series (Table S3). Of seven HLA loci, three HLA class I genes (HLA-A, -B and -C) show both alleles expressed in heterozygous in A549 cells. Then, by using the linear regression model, we analyzed the DAE on these three HLA loci in a comparison between infected and uninfected samples for each of four series, and subsequently combined results from the four series. We found that two alleles (B*18:01:01:01 and B*44:03:01:01) in HLA-B gene showed a significant increase of allelic fold change (FC) with beta coefficients ranging from 0.14 to 1.17 (combined P < 10−4, Fig. 2A) in A549 cells infected with SARS-CoV-2, relative to control cells. We also observed a positive correlation between allelic FCs between two HLA-B alleles and overall expression levels of HLA-B gene (Fig. 2B). In addition, we did not detect a significant change between two alleles in another two HLA class I genes (HLA-A and -C, combined P > 0.05). Collectively, these results indicate that two alleles in HLA-B locus show distinct responses to the SARS-CoV-2 infection in human lung cells.
Fig. 2. Elevated AE signals on two HLA-B alleles in SARS-CoV-2 infected A549 lung cells.

(A) Forest plot presenting the increased allelic fold change (FC) of two HLA-B alleles in A549 cells infected with SARS-CoV-2 in four biologically independent experiments (series), compared to mock as control. P value for each series and combined P value are calculated using linear regression model and the Fisher’s Method, respectively. (B) Correlation of HLA-B expression levels (y-axis) with allelic FC between HLA-B two alleles (x-axis) for the SARS-CoV-2 infected and uninfected conditions. P value is based on the Pearson’s correlation test.
Validation in independent datasets
We next analyzed an independent mRNA-seq data set from experiments with duplicate in Calu-3 lung cells harvested at time 0, 4 and 24 h after infection with SARS-CoV-2 or SARS-CoV-1 (Accession ID: GSE148729, Table S1). We identified a heterozygous genotype (HLA-B*51:01:01:01 and HLA-B*07:02:01:01) at the HLA-B gene in Calu-3 cells, enabling us to examine the temporal AE changes on this gene. The result showed that allelic FCs between two alleles (HLA-B*51:01:01:01 versus HLA-B*07:02:01:01) were gradually increased with the time course of SARS-CoV-2 infection in Calu-3 cells, albeit the unavailability of statistical tests due to only two biological replicates for each time point (Fig. 3A). For example, at 24 h after the viral infection, there was approximately 1.2-fold increase of allelic FC between two HLA-B alleles, compared with the baseline at 4 h. Similar trend towards the increase of allelic expression of HLA-B gene was observed for the SARS-CoV-1 infection (Fig. 3A). In contrast, there was no such obvious change in control cells under mock treatment at 4 and 24 h, relative to the starting point. In addition, we also analyzed another RNA-seq data set using total RNA-seq technology (Accession ID: GSE148729, Table S1), from the biologically independent experiments in the same cell line. Identical result was observed in SARS-CoV-2 infected Calu-3 cells, relative to the control with mock treatment (Figure S1). Similarly, we observed a positive correlation between allelic FCs of two HLA-B alleles and overall expression levels of HLA-B gene (Fig. 3B). The results were reproducibly observed in another total RNA-seq data set (Figure S2). Together, these findings suggest the AE difference at HLA-B locus in response to the coronavirus infection in human lung cells.
Fig. 3. Increased AE pattern of two HLA-B alleles in coronavirus infected Calu-3 lung cells in an independent dataset.

(A) Fold change between two HLA-B alleles (B*51:01:01:01 versus B*07:02:01:01) in Calu-3 cells infected with either SARS-CoV-1 or SARS-CoV-2 at the indicated time point after virus inoculation, relative to starting time point. (B) Correlation of HLA-B expression levels (y-axis) with allelic FC between HLA-B two alleles (x-axis) for the coronavirus infected and uninfected conditions. P value is based on the Pearson’s correlation test.
Validation in a case-control study
The data thus far have demonstrated the functional importance of increased AE profile of HLA-B locus in vitro cell experiments after challenge with SARS-CoV-2 virus. To test the effects of this locus on the primary lung tissues associated with COVID-19 disease, we analyzed another independent RNA-seq dataset in lung tissues from five patients infected with COVID-19 and five healthy individuals from a case-control study (Accession ID: GSE150316). Based on HLA-B alleles called across ten individuals, six samples (four cases and two controls) showed heterozygous in HLA-B gene. In a comparison of allelic FCs between cases and controls for HLA-B alleles, we found that HLA-B gene exhibited evidence of significant DAE between two groups with P < 0.05 (Figure S3).
Validation in cultured cells infected with SARS-CoV-2
To further provide evidence of DAE on the HLA-B gene in response to SARS-CoV-2 infection, we performed experiments in cultured A549 epithelial cells infected with SARS-CoV-2 assayed with allelic RT-qPCR. Our results confirmed that allelic expression pattern of the two HLA-B alleles was gradually increased when the A549 cells were infected with SARS-CoV-2, showing the most significant allelic fold change at the 72 h after viral infection (Fig. 4). Collectively, these data demonstrate that the increased HLA-B allelic expression associates with the SARS-CoV-2 infection in lung cells.
Fig. 4. Validation of the association of HLA-B allelic expression with SARS-CoV-2 infection in A549 cells assayed by allelic quantitative RT-PCR.
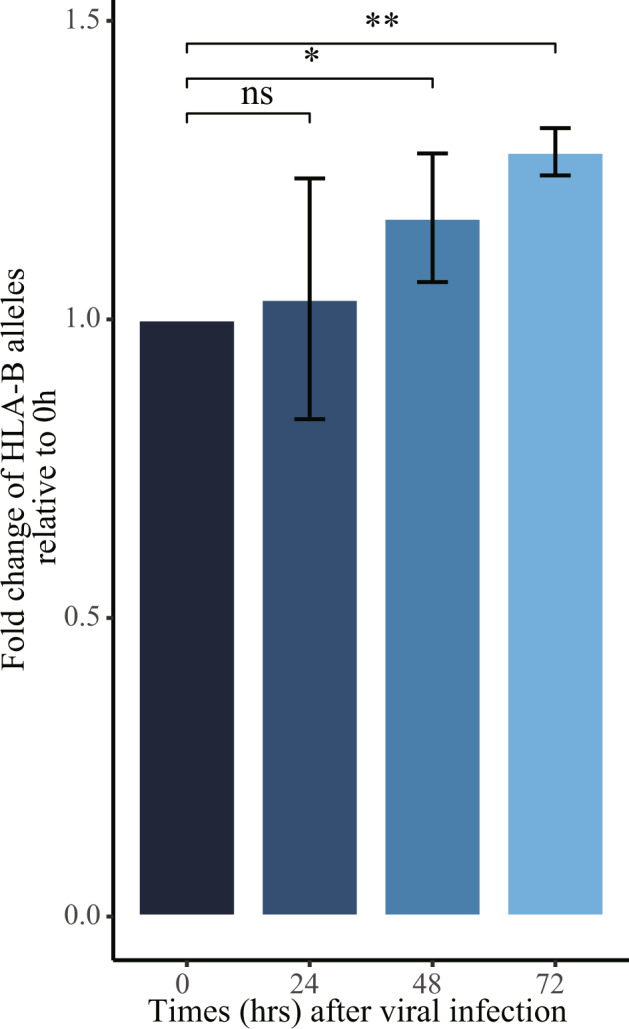
Barplot showing the time course of allelic fold changes of HLA-B two alleles relative to the starting time point. For the allelic qPCR assay, we conducted at least two independent viral infection experiments, where one representative experiment with triplicate is shown. *P < 0.05, **P < 0.01, compared with control cells.
HLA-B allelic expression in association with other viral respiratory infections
To determine the generality and robustness of the HLA-B locus in association with viral infection in lung cells, we analyzed another RNA-seq dataset (Accession ID: GSE147507, Table S1 and S2) comprised 24 samples from Blanco-Melo et al. study [34]. Concordant with the results for SARS-CoV-2 infection, we found a similar increase of allelic FC in the HLA-B gene in two human lung cells (A549 and NHBE cells) 24 h post infection with four respiratory viruses, in comparison with the mock treatment controls (Fig. 5A). A significant positive correlation between the expression levels of HLA-B gene and the allelic FCs of two HLA-B alleles was also observed (Fig. 5B).
Fig. 5. Increased allelic expression profiles of two HLA-B alleles in respiratory virus infected two lung cell lines, A549 and NHBE cells (Accession ID: GSE147507).

(A) Forest plot presenting the increased allelic FC between two HLA-B alleles in lung epithelial cells infected with four types of respiratory viruses, compared to mock as control. (B) Correlation of HLA-B expression levels (y-axis) with allelic FC between HLA-B two alleles (x-axis) for the respiratory viruses infected and uninfected conditions.
Type I Interferon induced allelic expression of HLA-B
In the same RNA-seq data set, we also analyzed the DAE in six RNA-seq samples from Blanco-Melo et al. experiments in NHBE cells stimulated with IFNβ in time course [34]. In this analysis, we found an increase of AE at the HLA-B locus (Fig. 6A), and a positive correlation between them in response to treatment of the IFNβ in NHBE cells (Fig. 6B). To further validate the findings from bioinformatics analysis, we conducted an experiment in cultured A549 lung cells stimulated with IFNβ as reported in Blanco-Melo et al. study [34]. The result showed that the type I Interferon induced DAE in the HLA-B locus in a time-course manner assayed by RT-qPCR (Fig. 6C).
Fig. 6. Increased AE patterns between HLA-B alleles under IFNβ treatment condition in human lung cells.

(A) Barplot showing the time course of allelic FCs of HLA-B two alleles in a time-course way. Dotted line plots shown in the upper parts correspond to fold changes (y-axis on the right) that are normalized to the time point on treatment initiation. The p value is unavailable due to only two biological replicates for each time point. (B) Correlation of HLA-B expression levels (y-axis) with allelic FC between HLA-B two alleles (x-axis) for the IFNβ treatment and untreated conditions in NHBE cells. P value is based on the Pearson’s correlation test. (C) Barplot showing the time course of allelic fold changes of HLA-B two alleles relative to the time point on treatment initiation. For the allelic qPCR assay, we conducted at least two independent viral infection experiments, where one representative experiment with triplicate is shown. *P < 0.05, compared with controls.
In silico prediction of SARS-CoV-2 proteins presented by HLA-B alleles
We performed in silico analysis of peptide binding affinity across sixteen SARS-CoV-2 proteins potentially presented by two HLA-B alleles (B*18:01 and B*44:03). The result indicates that three out of sixteen SARS-CoV-2 proteins, namely spike protein and two replicase polyproteins that are crucial for viral entry and replication in host cells [35], show a significant peptide binding difference between the two HLA-B alleles (binomial test P < 0.05, Table S5).
HLA-B gene-based analysis of COVID-19
Finally, we performed an HLA-B gene-based association analysis with the susceptibility to SARS-CoV-2 and severity of COVID-19 (see Methods). First, collapsing 94 genetic variants within HLA-B gene suggested HLA-B locus associated with the SARS-CoV-2 infection at P < 0.01 (the C2 study in Table S6), the result that is comparable to that of Ellinghaus et al. report [3]. Meanwhile, HLA-B locus also shows an association with the severity of COVID-19 in a comparison of hospitalized (moderate or severe COVID-19) cases with non-hospitalized cases (P = 1.42 × 10−5), and with controls (P = 1.22 × 10−10, Table S6).
Discussion
Taking advantage of numerous RNA-seq data from virus-infected experiments in both cultured lung cells and tissue specimens, our AE analysis reveals that the HLA-B locus is transcribed in an allelic manner that is reproducibly associated with the COVID-19. Consequently, up-regulation of the HLA-B gene in SARS-CoV-2 infected-cultured lung cells and lung tissues from patients with COVID-19 is attributed to the preferred expression of one haplotype. We further validated the DAE in HLA-B gene in vitro cell models. Meanwhile, the same direction of AE bias is uncovered in RNA-seq data from experiments in cultured epithelial cells infected with other respiratory viruses. Collectively, these findings indicate that the allelic expression performance of HLA-B gene may be a common feature for the COVID-19, as well as for other respiratory infectious diseases.
We note that gene-based association analysis identified a strong association of HLA-B locus with COVID-19, particularly the severity of COVID-19, expanding the knowledge on the host genetic factor conferring the susceptibility to SARS-CoV-2 and severity of COVID-19. Meanwhile, using RNA-seq data from functional experiments in human lung cells and tissue specimens, our AE study shows the potential functional importance of the HLA locus in response to the infection of SARS-CoV-2 and other respiratory viruses. It implies that, for this specific locus, the control of HLA gene haplotype expression, rather than allele or haplotype frequencies, may have more directly functional implication to the pathogenesis of COVID-19 and other infectious disease, particularly in disease relevant tissues. As a result, the current study further provides another proof-of-principle example presenting the allelic imbalance analysis as one of the powerful methods in the post-GWAS era [21, 36].
One of the most intriguing findings of our study is the over-expression of one of the HLA-B alleles occurs preferentially in non-immune cells and tissues infected with SARS-CoV-2 and other viruses. Previous reports point toward an important role of HLA-B alleles in mediating the most effective antiviral immunity [14, 37]. Lenna et al. also reported that HLA-B alleles have distinct capability in inducing the expression of some immune-related genes in lymphocytes [38]. Meanwhile, our and other in silico studies [17] have shown that HLA-B proteins carrying different alleles may have different antigen binding affinity. Collectively, we speculate that the HLA-B may act a defense role in human epithelial cells during acute SARS-CoV-2 or other viral infection. According to this logic, the differences in the expression of HLA-B alleles indicate the differences in the response of different individuals to the COVID-19 infection, a mechanism that may have a distinct ability to tag infected cells for destruction by the innate or adaptive immune system. Accordingly, allelic expression of HLA-B gene product represents a distinct response to present viral epitopes on the surface of infected epithelia, which results in the variation and genetic control of host cells against the respiratory viruses.
Mechanically, although the precise causes of the DAE in HLA-B locus remain undefined, there are, at least, three biologically plausible explanations during the virus infection. One possibility is that the expression of one allele is altered (activation or silence) due to a pathological defect. The second possibility is that the transcriptional activity of each haplotype is regulated independently. The third possibility is that the neighboring linked regions may bear regulatory element(s) performing cis-acting function in an allelic manner [39]. Therefore, a better understanding of the mechanism of allelic regulation exerting on the alleles of HLA-B locus awaits further investigation.
Another interesting finding is the IFNβ-inducible DAE in the HLA-B locus. Extensive studies have revealed that type I IFN (IFN-I) plays a master to elicit effective antiviral responses in both infected cells and immune cells [40, 41]. Meanwhile, studies also reported that HLA-B gene can be induced by cytokines including IFNβ in both immune and non-immune cells [42–44]. Together, these studies emphasized a key connection between HLA-B and IFN-I in response to rival infection in host cells, thereby providing evidence that DAE in HLA-B alleles may be directly (or indirectly) mediated by putative factor(s) in the IFN-I signaling cascades.
Another interesting, but beyond the current topic, is the immune response of lung epithelial cells to viral infection. Numerous studies have shown that two types of lymphocytes, cytotoxic CD8+ T lymphocytes (CTLs) and natural killer (NK) cells, are able to recognize viral antigens on the cell surface presented by HLA class-I molecules. The functional exhaustion of CTLs and NK cells is also reported in the peripheral blood of patients infected with SARS-CoV-2 [45]. During acute SARS-CoV-2 infection, two possible disconcordant results may be present–one is the enhanced expression of HLA-B that will present more viral antigens at the cell surface, the other is, due to the functional exhaustion, CTLs and NK cells are likely to be less capable to recognize those infected cells. Therefore, future investigation is warranted to decompose the complexity of the interaction between lung epithelial cells and cytotoxic lymphocytes in the lung epithelial microenvironment of COVID-19 patients.
Our quantitative analysis of genetic variants at the transcription level indicates that allelic biased expression could be induced by environmental exposures, including the virus infection in the current study. On the one hand, as shown in this study, the effect of polymorphisms affecting the cis-regulatory expression of the HLA-B gene was reproducibly detected in RNA-seq datasets from different batches and independent labs, suggesting the differential AE analysis is a robust method to unravel the inter-relationship between the environmental and genetic factors. On the other hand, some reports have shown that AE at a specific locus may exhibit tissue-specific [46, 47]. We observed the DAE pattern in lung tissues or cells infected with SARS-CoV-2 and other respiratory viruses, highlighting the virus-host interaction in infectious disease relevant tissues. Because multiple tissues may be also affected in the COVID-19 disease, as observed from clinical investigation [48]. Given the constitutive expression pattern of HLA-B gene in almost all human cells, it will be attractive to determine whether the DAE could be present in other virus-infected tissues.
Several limitations preclude us draw further conclusions. One limitation is the small sample size of case-control group, including the association of the severity of COVID-19 with the expression levels and allelic performance of HLA-B haplotypes in infectious lung tissues. Testing the correlation of HLA-B expression levels and its allelic expression pattern with the proposed cytokine storm is also warranted to further understand the pathogenesis of COVID-19 disease in patients with severe syndrome. Because of the tremendous polymorphism of HLA loci, haplotype-based AE analysis on HLA genes on far larger sample size may comprehensively understand the truly molecular association and immune response to human infectious diseases. Secondly, due to the limited number of HLA loci bearing expressed heterozygous alleles and relatively small sample size in this study, it is likely that more HLA alleles associated with COVID-19 remain to be discovered, although it is unclear how many. Another limitation is no functional investigation at the HLA-B protein levels. In particular, it is unknown whether there is a robust link between HLA-B genetics and its protein abundance in epithelial cells infected by respiratory viruses, a topic, namely protein quantitative trait locus (pQTL) [49] that remain to be explored.
In summary, integrated functional genomics data using reliable statistical methods is a robust approach to elucidate functional genetic variants or genes associated with human infectious diseases.
Supplementary information
Figure legends for supplementary material
Acknowledgements
We thank Dr. Le Su (HudsonAlpha Institute for biotech.) for helpful discussions and critical reading of the manuscript. This study was supported by the Natural Science Foundation of Guangdong Province (No. 2019A1515010190) to HH, and supported by the Guangdong Innovative and Entrepreneurial Research Team Program (No. 2013Y113) to HXL. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author contributions
YFZ conceived and designed the study with intellectual contribution from YXZ and HXL. YFZ collected the data, performed the bioinformatics analyses and designed the experiments. HXL, YXZ, JMJ and PPY coordinated the study. YXZ, HPZ and YSS designed and performed the experiments. HXL, JMJ, HPZ and HH provided essential resources. YFZ wrote and revised the manuscript with significant contributions from YXZ, YSS, HXL and HH. All authors read, reviewed and approved the final content of the paper.
Data availability
The datasets presented in this study are included in the article/Supplementary Material.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Yuanxu Zhang, Yisheng Sun.
Contributor Information
Pingping Yao, Email: ppyao@cdc.zj.cn.
Huaxin Liao, Email: larryhliao@163.com.
Yanfeng Zhang, Email: yanfengzhang1984@outlook.com.
Supplementary information
The online version contains supplementary material available at 10.1038/s41431-022-01070-5.
References
- 1.Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565–74. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Casanova JL, Abel L. The human genetic determinism of life-threatening infectious diseases: genetic heterogeneity and physiological homogeneity? Hum Genet. 2020;139:681–94. doi: 10.1007/s00439-020-02184-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Severe COVID-19 GWAS Group. Genomewide Association study of severe Covid-19 with respiratory failure. New Engl J Med. 2020;383:1522–34. [DOI] [PMC free article] [PubMed]
- 4.COVID-19 Host Genetics Initiative. Mapping the human genetic architecture of COVID-19. Nature. 2021;600:472–7. [DOI] [PMC free article] [PubMed]
- 5.Pathak GA, Singh K, Miller-Fleming TW, Wendt FR, Ehsan N, Hou K, et al. Integrative genomic analyses identify susceptibility genes underlying COVID-19 hospitalization. Nat Commun. 2021;12:4569. doi: 10.1038/s41467-021-24824-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Initiative C-HG. The COVID-19 Host Genetics Initiative, a global initiative to elucidate the role of host genetic factors in susceptibility and severity of the SARS-CoV-2 virus pandemic. Eur J Hum Genet. 2020;28:715–8. doi: 10.1038/s41431-020-0636-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shelton JF, Shastri AJ, Ye C, Weldon CH, Filshtein-Sonmez T, Coker D, et al. Trans-ancestry analysis reveals genetic and nongenetic associations with COVID-19 susceptibility and severity. Nat Genet. 2021;53:801–8. doi: 10.1038/s41588-021-00854-7. [DOI] [PubMed] [Google Scholar]
- 8.Downes DJ, Cross AR, Hua P, Roberts N, Schwessinger R, Cutler AJ, et al. Identification of LZTFL1 as a candidate effector gene at a COVID-19 risk locus. Nat Genet. 2021;53:1606–15. doi: 10.1038/s41588-021-00955-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pairo-Castineira E, Clohisey S, Klaric L, Bretherick AD, Rawlik K, Pasko D, et al. Genetic mechanisms of critical illness in COVID-19. Nature. 2021;591:92–8. doi: 10.1038/s41586-020-03065-y. [DOI] [PubMed] [Google Scholar]
- 10.Cao Y, Li L, Feng Z, Wan S, Huang P, Sun X, et al. Comparative genetic analysis of the novel coronavirus (2019-nCoV/SARS-CoV-2) receptor ACE2 in different populations. Cell Discov. 2020;6:11. doi: 10.1038/s41421-020-0147-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Irham LM, Chou W-H, Calkins MJ, Adikusuma W, Hsieh S-L, Chang W-C. Genetic variants that influence SARS-CoV-2 receptor TMPRSS2 expression among population cohorts from multiple continents. Biochem Biophys Res Commun. 2020;529:263–9. doi: 10.1016/j.bbrc.2020.05.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–80.e278. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blackwell JM, Jamieson SE, Burgner D. HLA and infectious diseases. Clin Microbiol Rev. 2009;22:370–85. doi: 10.1128/CMR.00048-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bihl F, Frahm N, Di Giammarino L, Sidney J, John M, Yusim K, et al. Impact of HLA-B alleles, epitope binding affinity, functional avidity, and viral coinfection on the immunodominance of virus-specific CTL responses. J Immunol. 2006;176:4094–101. doi: 10.4049/jimmunol.176.7.4094. [DOI] [PubMed] [Google Scholar]
- 15.Pretti MAM, Galvani RG, Vieira GF, Bonomo A, Bonamino MH, Boroni M. Class I HLA Allele predicted restricted antigenic coverages for spike and nucleocapsid proteins are associated with deaths related to COVID-19. Front Immunol. 2020;11:565730. [DOI] [PMC free article] [PubMed]
- 16.Lin M, Tseng HK, Trejaut JA, Lee HL, Loo JH, Chu CC, et al. Association of HLA class I with severe acute respiratory syndrome coronavirus infection. BMC Med Genet. 2003;4:9. doi: 10.1186/1471-2350-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nguyen A, David JK, Maden SK, Wood MA, Weeder BR, Nellore A, et al. Human leukocyte antigen susceptibility map for severe acute respiratory syndrome Coronavirus 2. J Virol. 2020;94:e00510–20. [DOI] [PMC free article] [PubMed]
- 18.Pastinen T, Hudson TJ. Cis-acting regulatory variation in the human genome. Science. 2004;306:647–50. doi: 10.1126/science.1101659. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Y, Li X, Gibson A, Edberg J, Kimberly RP, Absher DM. Skewed allelic expression on X chromosome associated with aberrant expression of XIST on systemic lupus erythematosus lymphocytes. Hum Mol Genet. 2020;29:2523–34. doi: 10.1093/hmg/ddaa131. [DOI] [PubMed] [Google Scholar]
- 20.Cowper-Sal lari R, Zhang X, Wright JB, Bailey SD, Cole MD, Eeckhoute J, et al. Breast cancer risk-associated SNPs modulate the affinity of chromatin for FOXA1 and alter gene expression. Nat Genet. 2012;44:1191–8. doi: 10.1038/ng.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Q, Seo JH, Stranger B, McKenna A, Pe’er I, Laframboise T, et al. Integrative eQTL-based analyses reveal the biology of breast cancer risk loci. Cell. 2013;152:633–41. doi: 10.1016/j.cell.2012.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pollard KS, Serre D, Wang X, Tao H, Grundberg E, Hudson TJ, et al. A genome-wide approach to identifying novel-imprinted genes. Hum Genet. 2008;122:625–34. doi: 10.1007/s00439-007-0440-1. [DOI] [PubMed] [Google Scholar]
- 23.Tukiainen T, Villani AC, Yen A, Rivas MA, Marshall JL, Satija R, et al. Landscape of X chromosome inactivation across human tissues. Nature. 2017;550:244–8. doi: 10.1038/nature24265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y, Castillo-Morales A, Jiang M, Zhu Y, Hu L, Urrutia AO, et al. Genes that escape X-Inactivation in Humans Have High Intraspecific Variability in Expression, Are Associated with Mental Impairment but Are Not Slow Evolving. Mol Biol Evol. 2013;30:2588–601. doi: 10.1093/molbev/mst148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, et al. NCBI GEO: archive for functional genomics data sets-update. Nucleic Acids Res. 2013;41:D991–995. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Y, Wagner EK, Guo X, May I, Cai Q, Zheng W, et al. Long intergenic non-coding RNA expression signature in human breast cancer. Sci Rep. 2016;6:37821. doi: 10.1038/srep37821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 2019;37:907–15. doi: 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Y, Song J, Day K, Absher D. dCATCH-Seq: improved sequencing of large continuous genomic targets with double-hybridization. BMC Genom. 2017;18:811. doi: 10.1186/s12864-017-4159-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robinson J, Barker DJ, Georgiou X, Cooper MA, Flicek P, Marsh SGE. IPD-IMGT/HLA Database. Nucleic Acids Res. 2019;48:D948–D955. doi: 10.1093/nar/gkz950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cao H, Wu J, Wang Y, Jiang H, Zhang T, Liu X, et al. An integrated tool to study MHC region: accurate SNV detection and HLA genes typing in human MHC region using targeted high-throughput sequencing. PLOS ONE. 2013;8:e69388. doi: 10.1371/journal.pone.0069388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gutierrez-Arcelus M, Baglaenko Y, Arora J, Hannes S, Luo Y, Amariuta T, et al. Allele-specific expression changes dynamically during T cell activation in HLA and other autoimmune loci. Nat Genet. 2020;52:247–53. doi: 10.1038/s41588-020-0579-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kugler KG, Mueller LAJ, Graber A. MADAM - An open source meta-analysis toolbox for R and Bioconductor. Source Code Biol Med. 2010;5:3. doi: 10.1186/1751-0473-5-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O’Donnell TJ, Rubinsteyn A, Laserson U. MHCflurry 2.0: improved Pan-Allele prediction of MHC Class I-presented peptides by incorporating antigen processing. Cell Syst. 2020;11:418–9. doi: 10.1016/j.cels.2020.09.001. [DOI] [PubMed] [Google Scholar]
- 34.Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Moller R, et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. 2020;181:1036–45 e1039. doi: 10.1016/j.cell.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Malone B, Urakova N, Snijder EJ, Campbell EA. Structures and functions of coronavirus replication-transcription complexes and their relevance for SARS-CoV-2 drug design. Nat Rev Mol Cell Biol. 2022;23:21–39. [DOI] [PMC free article] [PubMed]
- 36.Gallagher MD, Chen-Plotkin AS. The Post-GWAS era: from association to function. Am J Hum Genet. 2018;102:717–30. doi: 10.1016/j.ajhg.2018.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kiepiela P, Leslie AJ, Honeyborne I, Ramduth D, Thobakgale C, Chetty S, et al. Dominant influence of HLA-B in mediating the potential co-evolution of HIV and HLA. Nature. 2004;432:769–75. doi: 10.1038/nature03113. [DOI] [PubMed] [Google Scholar]
- 38.Lenna S, Assassi S, Farina GA, Mantero JC, Scorza R, Lafyatis R, et al. The HLA-B*35 allele modulates ER stress, inflammation and proliferation in PBMCs from Limited Cutaneous Systemic Sclerosis patients. Arthritis Res Ther. 2015;17:363. doi: 10.1186/s13075-015-0881-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gobin SJ, van Zutphen M, Woltman AM, van den Elsen PJ. Transactivation of classical and nonclassical HLA class I genes through the IFN-stimulated response element. J Immunol. 1999;163:1428–34. [PubMed] [Google Scholar]
- 40.McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. Type I interferons in infectious disease. Nat Rev Immunol. 2015;15:87–103. doi: 10.1038/nri3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arunachalam PS, Wimmers F, Mok CKP, Perera RAPM, Scott M, Hagan T, et al. Systems biological assessment of immunity to mild versus severe COVID-19 infection in humans. Science. 2020;369:1210–20. doi: 10.1126/science.abc6261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Girdlestone J. Synergistic induction of HLA class I expression by RelA and CIITA. Blood. 2000;95:3804–8. doi: 10.1182/blood.V95.12.3804. [DOI] [PubMed] [Google Scholar]
- 43.Johnson DR. Locus-specific constitutive and cytokine-induced HLA class I gene expression. J Immunol. 2003;170:1894–902. doi: 10.4049/jimmunol.170.4.1894. [DOI] [PubMed] [Google Scholar]
- 44.Schmidt H, Bühring H-J, Blum GE, Reichmann U, Müller C, Weiss E, et al. Differential regulation of HLA B antigen expression by interferon; in: Dupont B (ed): Immunobiology of HLA: Volume II: immunogenetics and histocompatibility. Berlin, Heidelberg: Springer Berlin Heidelberg, 1989, pp 155–6.
- 45.Zheng M, Gao Y, Wang G, Song G, Liu S, Sun D, et al. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell Mol Immunol. 2020;17:533–5. doi: 10.1038/s41423-020-0402-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wilkins JM, Southam L, Price AJ, Mustafa Z, Carr A, Loughlin J. Extreme context specificity in differential allelic expression. Hum Mol Genet. 2007;16:537–46. doi: 10.1093/hmg/ddl488. [DOI] [PubMed] [Google Scholar]
- 47.Zhang K, Li JB, Gao Y, Egli D, Xie B, Deng J, et al. Digital RNA allelotyping reveals tissue-specific and allele-specific gene expression in human. Nat Methods. 2009;6:613–8. doi: 10.1038/nmeth.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li X, Xu S, Yu M, Wang K, Tao Y, Zhou Y, et al. Risk factors for severity and mortality in adult COVID-19 inpatients in Wuhan. J Allergy Clin Immunol. 2020;146:110–8. [DOI] [PMC free article] [PubMed]
- 49.Wu L, Candille SI, Choi Y, Xie D, Jiang L, Li-Pook-Than J, et al. Variation and genetic control of protein abundance in humans. Nature. 2013;499:79–82. doi: 10.1038/nature12223. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure legends for supplementary material
Data Availability Statement
The datasets presented in this study are included in the article/Supplementary Material.