

Abstract
The prevalence of nonalcoholic steatohepatitis (NASH) and liver cancer is increasing. De novo lipogenesis and fibrosis contribute to disease progression and cancerous transformation. Here, we found that β2-spectrin (SPTBN1) promotes sterol regulatory element (SRE)-binding protein (SREBP)-stimulated lipogenesis and development of liver cancer in mice fed a high-fat diet (HFD) or a western diet (WD). Either hepatocyte-specific knockout of SPTBN1 or siRNA-mediated therapy protected mice from HFD/WD-induced obesity, and fibrosis, lipid accumulation, and tissue damage in the liver. Biochemical analysis suggested that HFD/WD induces SPTBN1 and SREBP1 cleavage by CASPASE-3 and that the cleaved products interact to promote expression of genes with sterol response elements. Analysis of human NASH tissue revealed increased SPTBN1 and CASPASE-3 expression. Thus, our data indicate that SPTBN1 represents a potential target for therapeutic intervention in NASH and liver cancer.
Single sentence summary:
Targeting SBPTN1 with siRNA has the potential to treat NASH and prevent HCC.
Introduction
Nonalcoholic fatty liver diseases (NAFLD) and nonalcoholic steatohepatitis (NASH) arise from obesity and metabolic disorders and affect up to a third of the world’s population (1, 2). These diseases comprise a spectrum including lipid accumulation in the liver (steatosis), injury, inflammation, hepatocyte ballooning (cell death), and progressive fibrosis (cirrhosis), ultimately leading to carcinogenesis (3, 4). Lipid accumulation in the liver promotes chronic oxidative and endoplasmic reticulum (ER) stress, cell death, immune cell infiltration, and fibrogenesis, and these changes ultimately lead to cirrhosis and increased risk of hepatocellular carcinoma (HCC). These effects are exacerbated by factors such as increased intake of free fatty acids (FFAs), sedentary lifestyle, and hyperinsulinemia. Depending on whether NASH occurs with or without cirrhosis, liver cancer incidence can vary from 2.4% (without cirrhosis) to 12.8% (with cirrhosis) (1, 4). Obesity increases the risk of liver cancer mortality twofold, and together with NASH, accounts for the alarming increase in this cancer (5, 6). Despite new therapeutic approaches targeting NASH, few single agents reverse both fibrosis and steatosis, thus NASH presents a major clinical challenge (5, 7). Therefore, understanding the molecular mechanisms that converge on abnormal lipid accumulation, fibrosis, and the fatal switch to hepatocarcinogenesis could lead to new approaches targeting NASH in specific groups of patients who are susceptible to the progression of the disease.
Initiation of NAFLD is considered to involve de novo lipogenesis with abnormal accumulation of free fatty acids, triglycerides (TG), and cholesterol. Activation of hepatocyte death receptor pathways, for example by tumor necrosis factor (TNF), and caspases contribute to tissue injury and steatohepatitis observed in NASH (8, 9). De novo lipogenesis is stimulated by activation of the transcription factors sterol regulatory element (SRE)-binding proteins (SREBPs) and repression of energy-sensing pathways, such as the pathway involving adenosine monophosphate (AMP)-activated protein kinase (AMPK). SREBP1 is a master lipogenic transcription factor that drives fatty acid synthesis and contributes to liver steatosis (10). SREBP proteins are maintained in the ER through interactions with the proteins INSIG (insulin-induced gene) and SCAP (SREBP-cleavage activating protein). SREBP activation in response to sterol depletion or ER stress requires dissociation of INSIG and SCAP-induced cleavage of SREBP by site-1 protease (S1P) followed by a second cleavage by site-2 protease (S2P) to produce the mature form of the SREBP proteins that translocate into the nucleus and regulate target gene transcription (11). In stressed cultured cells with activated CASPASE-3, SREBP1 and SREBP2 are cleaved and activated by CASPASE-3, but the physiological context for this is unknown (12). We refer to the cleaved, nuclear form as n-SREBP and the full-length, ER-localized form as pre-SREBP.
The degree of fibrosis is considered the strongest predictive factor for the progression of NAFLD to NASH and ultimately HCC (1, 13, 14). Critical to hepatic fibrosis is activation of the transforming growth factor β (TGF-β) pathway (15–17). TGF-β1 is the founding member of this family, and this ligand signals through two serine-threonine kinase receptors (TGFBR1 and TGFBR2), which activate the SMAD transcriptional regulators, SMAD2 and SMAD3. SMAD complexes containing SMAD3 are central to the progression of fibrosis by causing excessive extracellular matrix gene expression, such as those encoding collagens COL1A1, COL1A2, COL3A1, COL5A2, COL6A1, and COL6A3, and stimulating genes encoding the protease inhibitors tissue inhibitor of metalloproteinases (TIMP) and plasminogen activator inhibitor-1 (PAI-1) (16). SMAD3 complexes also repress the gene encoding peroxisome proliferator-activated receptor gamma coactivator 1α (PGC-1α) (18); thereby affecting glucose homeostasis and oxidative phosphorylation. Pro-fibrotic effects of TGF-β1 through SMAD3 involve multiple mechanisms and cell types, including enhanced infiltration or proliferation (or both) of tissue-resident fibroblasts, generation of myofibroblasts, induction of epithelial-mesenchymal transition (EMT), and inhibition of collagenolysis (19).
SPTBN1 (also called β2-spectrin, β2SP) is a multidomain adaptor protein with functions in the cytoplasm and nucleus (20–22). In particular, SPTBN1 promotes TGF-β receptor activation of SMAD3 in the cytosol (23) and interacts with SMAD3 in the nucleus to regulate specific target genes (24, 25). SPTBN1 is a dynamic, tetrameric protein consisting of two antiparallel dimers of alpha and beta subunits. SPTBN1 binding partners include ankyrin, which functions to connect proteins at the cell membrane to the spectrin-containing cytoskeleton, and lamins and the chromatin modulator CTCF (CCCTC-binding factor), which function in the nucleus to organize chromatin and regulate gene expression (20, 24, 26, 27). SPTBN1 is a substrate for CASPASE-3 and -7, and cleavage at the SPTBN1 1454DEVD1457 peptide sequence produces two fragments (160 and 80 kDa) with distinct and separate functions in apoptosis and transcription (28). The importance of SPTBN1 in liver disease arises from the finding that mice treated with shRNA targeting SPTBN1 exhibit less acetaminophen-induced hepatotoxicity (28).
Increased amounts of liver SMAD3 and SPTBN1, as well as TGF-β pathway members associated with pro-fibrotic pathways, are observed in ~40% of HCCs, and many HCCs are associated with NASH (25, 29). Here, we investigated the role of SPTBN1 in liver tumorigenesis by generating liver-specific Sptnb1 knockout (LSKO) mice. We showed that LSKO mice or mice treated with Sptbn1-targeted siRNA were protected from detrimental effects of a high-fat diet (HFD), as well as a NASH diet (western diet, WD), through a mechanism involving reduction of the expression of pro-fibrotic genes and genes involved in de novo lipogenesis. The mice did not become obese or develop NASH or HCC. The translational importance of our results was confirmed by analysis of the expression of SPTBN1 in human NASH and HCC and finding that siRNA targeting SPTBN1 reversed transcriptional changes in genes involved in fatty acid metabolism and fibrosis induced in a human 3-dimensional (3D) culture model of NASH. Thus, our results identified a previously unknown role for SPTBN1 in regulating SREBP activity induced by CASPASE-3 in response to stress conditions caused by HFD/WD. These findings suggested potential therapeutic NASH options and HCC prevention strategies in patient populations susceptible to liver disease progression.
Results
Liver-specific Sptbn1 knockout protects mice from HFD-induced NASH
We generated liver-specific Sptbn1 conditional (Sptbn1-flox) knockout mice (LSKO) that are devoid of hepatocyte-specific SPTBN1 (fig. S1A, B) by intercrossing with Albumin-Cre mice. LSKO mice are viable and fertile. We confirmed hepatocyte-specific deletion of SPTBN1 by immunoblot or immunohistochemical (IHC) staining, showing no detectable protein in hepatocytes (fig. S1, B and C), and characterized the liver histology and metabolic status of the LSKO mice fed a normal chow diet (NC) (Fig. 1, A and B, fig. S1D). On NC, the LSKO mice were similar in body weight to the Flox control mice (fig. S1D), but the LSKO displayed significantly decreased concentrations of serum triacylglycerol (TG) (p < 0.05) (Fig. 1A) and blood glucose (p < 0.05) with slightly increased insulin sensitivity in an insulin tolerance test (ITT) (fig. S1D) and improved glucose tolerance test (GTT) (fig.S1D). Histologically, the livers of the LSKO mice appeared to have similar or slightly reduced lipid accumulation (Fig. 1B, Oil Red O staining) compared to the livers of Flox mice. Additionally, the LSKO mice and control mice had similar food and water intake and urine output (fig. S1E, left).
Fig. 1. Liver-specific knockout of SPTBN1 protects mice from HFD-induced obesity, NAFLD, and NASH.
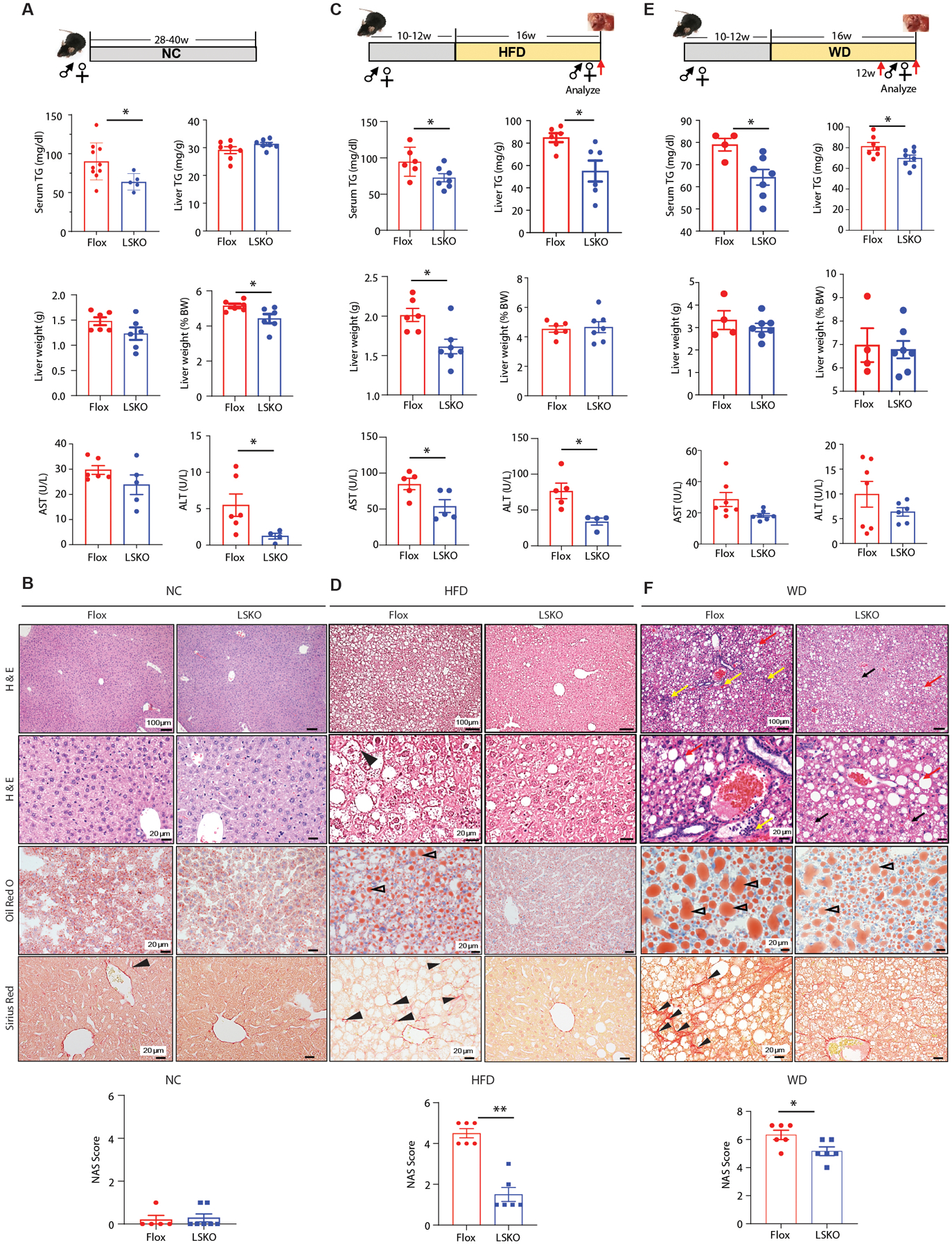
(A, C, E) Serum TG, liver TG, liver weight presented as gram or percentage of body weight, and liver enzyme concentrations in serum in Flox control (n=4 –10) and LSKO mice (n=4 – 8) at 28–40 weeks of normal chow diet (NC) (A), 16 weeks of high-fat-diet (HFD) (C) and 16 weeks of Western diet (WD) (E). AST, aspartate aminotransferase; ALT, alanine aminotransferase.
(B, D, F) Liver histology, lipid content, and NAS score from Flox and LSKO mice after NC (B), HFD (D), WD (F). Top two images are H & E stained images from male age-matched Flox and LSKO mice. An example of a ballooning hepatocyte is indicated by the arrowhead in HFD-fed Flox mice liver (D). Inflammation (yellow arrows) and macro-vesicular lipid (red arrows) are indicated on the WD-fed Flox mouse liver image (F). Micro-vesicular lipid (black arrows) is indicated on the WD fed LSKO mouse liver images. Third row Oil Red O stained images showing fat droplets (red, open arrowheads) are from male mice. Fourth row Sirius Red stained images showing early, fine fibrosis (arrowheads in Flox liver images) are from male mice fed with HFD or WD (D, F). Bottom bar graphs are NAFLD Activity Score (NAS) in Flox and LSKO mice (n = 5 – 7 from each group).
Quantitative data are shown as mean ± SEM. Statistical significance was determined by 2-sided t-test for weekly changes in body weight and for all other quantitative data (*, p < 0.05; **, p < 0.005).
We used the LSKO mice to evaluate the systemic metabolic and liver-specific effects of high-fat diet (HFD) and western diet (WD), another diet-induced NASH model in which NASH was induced by 42% kcal derived from fat together with a high fructose-glucose solution (23.1 g/L d-fructose +18.9 g/L d-glucose) (30) which we refer to as western diet (WD). The HFD derives 60% of calories from fat. We initially selected HFD, following up on similar studies performed with mice deficient in SMAD3, revealing that SMAD3 deficiency protected mice from obesity induced by this diet (HFD) (18, 31, 32), and because LSKO mice revealed reduced serum glucose concentrations compared with control mice fed a NC diet (fig.S1D). In addition, HFD induces a mild form of NASH, enabling the evaluation of not only effects on obesity but also NAFLD and NASH (33–35). We placed 10 to 12-week-old male and female Flox control and LSKO mice on a HFD for 12 to 16 weeks (Fig. 1, C and D). Food and water intake and urine output were similar between the Flox control and LSKO mice after 12 weeks on a HFD (fig. S1E, right). However, after 10 weeks of HFD, LSKO mice gained less weight, had less visceral fat, and were protected from HFD-induced obesity (fig. S2A). Metabolic profiling indicated that the HFD-fed LSKO mice had lower serum TG concentrations (Fig. 1C) but were similar to the HFD-fed Flox control mice for serum glucose and total cholesterol concentrations (fig. S2B). Livers from HFD LSKO mice were on average smaller with significantly reduced amounts of TG (Fig. 1C, p < 0.05) than those of HFD- Flox control mice. The livers of HFD mice of both genotypes had comparable Ki67 labeling, indicating similar numbers of proliferating cells in their livers (fig. S2C). Analysis of liver injury by measuring serum aspartate transaminase (AST) and alanine transaminase (ALT) concentrations showed reduced concentrations in LSKO mice, suggesting lesser liver damage in HFD LSKO mice compared to HFD Flox control mice (Fig. 1C).
Consistent with lesser liver damage in HFD LSKO mice, liver histology of these mice revealed a normal liver architecture, minimal lipid accumulation, and absence of possible ballooning (36) or signs of inflammation in the liver (Fig. 1D, fig. S2D). In the livers of the HFD-fed Flox mice, Sirius red staining (Fig.1D) and electron microscopy (fig. S2E) revealed fibrogenesis, indicated by early, fine collagen deposition at the extracellular matrix, and steatosis, indicated by accumulation of lipid droplets; these changes were not present in livers of LSKO mice fed HFD (Fig. 1D). Compared with livers from the Flox mice fed a HFD for 16 weeks, livers from the LSKO mice had decreased expression of TGF-β/SMAD3-regulated genes associated with liver fibrosis, altered expression of genes associated with inflammation, and reduced expression of genes encoding proteins involved in fatty acid metabolism (fig. S2F), which is consistent with lower NAFLD Activity Score (NAS) score in these mice (Fig. 1D, bottom, fig. S2G, left).
The changes in the Flox control mice indicated that these mice developed steatosis with progression to NASH by 16 weeks of HFD. Furthermore, the LSKO mice were protected from this diet-induced liver condition. Similar results were observed in another diet-induced NASH model in which NASH was induced by western diet (WD). We found that the response of LSKO mice to WD was similar to their response to HFD. LSKO mice gained less weight than control mice fed WD (fig. S2H) and LSKO mice were also protected from WD-induced NASH as indicated by significantly reduced serum TG (p < 0.05) (Fig. 1E) and glucose concentrations (fig. S2I), reduced liver steatosis, inflammation, and fibrosis (Fig. 1F), consistent with the lower NAS score in LSKO mice (Fig.1F, bottom, fig. S2G, right).
SPTBN1 regulates SREBP pathways and fatty acid metabolism
To understand how liver-specific loss of SPTBN1 protected mice from liver disease, we examined key regulators of metabolism and performed global liver transcript analysis of RNA-sequencing (RNA-seq) data to explore differences in gene expression in the livers of Flox and LSKO mice after 16 weeks of HFD (Fig. 2A). We examined transcript and protein abundance of the transcriptional regulator C/EBPα, which regulates adipogenesis (37), and uncoupling protein 2 (UCP2), which reduces mitochondrial ATP production and is associated with NASH (38). Neither of these were different in the livers of the HFD-fed Flox and LSKO mice at the level of transcript or protein abundance (fig. S3A). However, LSKO mice displayed increased body temperatures (fig. S3B). Livers from LSKO mice revealed increased expression of Fgf21, that encodes a liver-produced hormone that protects mice against diet-induced obesity (39), and Ctp1a, encoding an enzyme that is critical for fatty acid oxidation, but no difference from that of livers from control mice for Gdf15, encoding a hormone that limits food intake and is induced by liver injury (40) (fig. S3C).
Fig. 2. SREBP1 signaling is reduced in LSKO mouse livers in both the normal chow and HFD conditions.
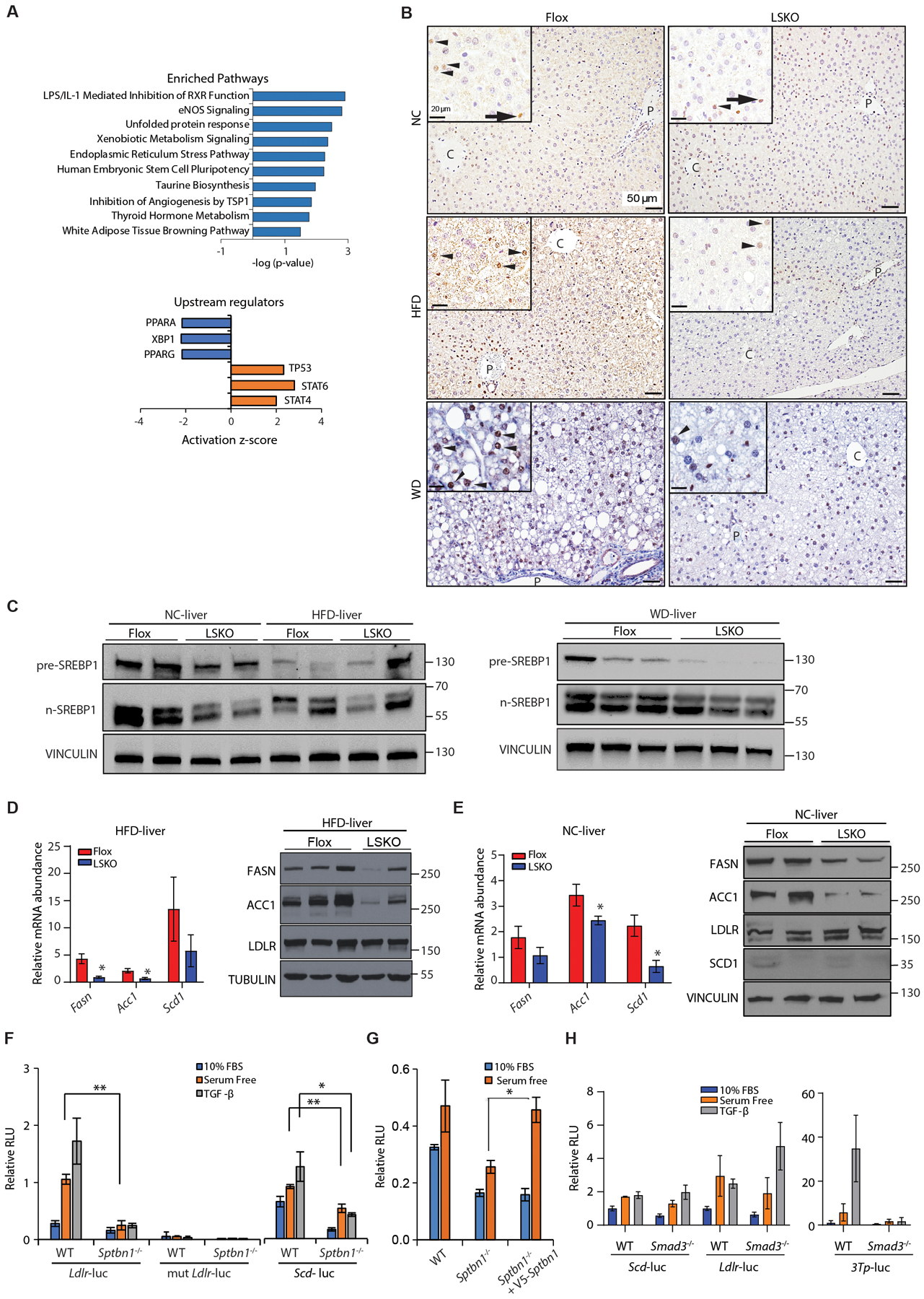
(A) Bioinformatic analysis by Ingenuity Pathway Analysis of differentially regulated genes in livers from LSKO mice compared to Flox mice after 16 weeks of HFD. Differentially regulated genes were determined from RNA-seq data from 5 mice in each group. Upper: Pathway enrichment analysis. Lower: Upstream regulator analysis for regulators with activation (z score ≥2) or inhibition (z score ≤ 2), showing those with p < 0.05.
(B) Immunohistochemistry of SREBP1 in liver sections from Flox and LSKO mice after 16 weeks of feeding with NC, HFD, or WD using anti-SREBP1 from Abcam (ab191857). Black arrowheads indicate hepatocytes nuclear localization, black arrow indicates labeling in non-hepatocytes. C-central vein; P-portal vein.
(C) Analysis of livers from Flox and LSKO mice fed NC, HFD or WD by Western blot for full-length SREBP1 and the active cleaved forms (n-SREBP1) using anti-SREBP1 from Abcam (ab191857). Each lane represents samples from a single mouse (n = 2 or 3).
(D) Relative mRNA and protein abundance for products of SREBP1 target genes from livers of LSKO and Flox mice after 12 to 16 weeks of feeding with HFD. RNA data are normalized to 18S (n = 6 – 7 for each group). Western blot data show results from individual mice.
(E) Relative mRNA and protein abundance for products of SREBP1 target genes (Fasn, Acc1, Scd1) and a SREBP2 target gene (Ldlr) from livers of LSKO and Flox mice fed normal chow (NC). RNA data are normalized to 18S (n = 3 – 5 for each group for). Western blot data show results from individual mice.
(F) SRE-driven luciferase activity from the LDLR-luc, mutant LDLR-luc, and SCD-luc reporters in WT and Sptbn1−/− MEF cells after 24 hours in medium with 10% fetal bovine serum (FBS) or serum-free medium with or without TGF-β1 (200 pM) for 24 hours.
(G) SRE-driven luciferase activity from the LDLR-luc reporter in WT and Sptbn1−/− MEF cells expressing empty vector or SPTBN1 and cultured for 24 hours in 10% FBS or serum-free medium.
(H) Luciferase activity from the SCD-luc and LDLR-luc reporters (SREBP-responsive) and 3TP-luc reporter (SMAD3-responsive) in WT and Smad3−/− MEF cells after 24 hours in medium with 10% fetal bovine serum (FBS) or serum-free medium with or without TGF-β1 (200 pM) for 24 hours.
Quantitative data shown as mean ± SEM in panels D to H are representative performed in triplicate or summarized from 2–3 independent experiments performed in triplicate. Statistical significance was determined by 2-sided t-test or one-way ANOVA (*, p < 0.05; **, p < 0.005).
To gain insight into pathways altered with liver-specific deficiency in SPTBN1, we performed bioinformatic analysis Ingenuity Pathway Analysis (IPA) and Ingenuity “Upstream Regulator” (including only those regulators with Z scores > 2 or < ‐2) comparing livers from LSKO mice and Flox mice. LSKO mice had changes in gene expression that were significantly enriched in pathways or regulators associated with stress or infection—LPS/IL-1 mediated inhibition of retinoid X receptor (RXR) function, unfolded protein response (UPR), and endoplasmic reticulum stress pathway, STAT6, STAT4, and TP53— and with reduced or low activity of transcription factors that control lipid metabolism—lipid sensor protein PPARα and PPARγ (peroxisome proliferator-activated receptor α and γ) (Fig. 2A). We were intrigued by finding a pathway associated with decreased RXR function because RXR is a transcription factor that stimulates SREBP1 expression (41) and we had found SREBP1 coimmunoprecipitated with V5-tagged SPTBN1 in stressed HEK293T cells (table S1) through mass spectrometry analysis. Thus, we hypothesized that SPTBN1 could influence liver physiology by interacting with SREBP1.
Because SPTBN1 has scaffolding functions in both the cytoplasm and the nucleus, we evaluated if SREBP1 distribution or abundance in the liver was altered in LSKO mice either fed NC, HFD or WD. In the livers of Flox mice fed HFD, SREBP1 had an intense punctate distribution with some cells showing nuclear labeling. Zone 1 surrounding the portal tract appeared to demonstrate intense nuclear labeling in non-hepatocytes (Fig. 2B, HFD). In mice fed NC, most hepatocytes in the livers from Flox mice had cytoplasm-localized SREBP1, whereas some hepatocytes and non-hepatocytes showed nuclear SREBP1. Compared to SREBP1 labeling in HFD/WD Flox mice in which most hepatocytes were positive for nuclear SREBP1, SREBP1 staining was less intense in both cytoplasm and nucleus of the liver cells from LSKO mice fed HFD or WD, suggesting reduced SREBP1 activity (Fig. 2B, fig. S3D) in LSKO mice.
There are 2 genes that encode the related proteins SREBP1 and SREBP2. Thus, we evaluated the abundance of pre-SREBP (ER-localized, full-length SREBP) and n-SREBP (cleaved, nuclear SREBP) for both proteins, as well as for their ER-localized regulators SCAP, INSIG1, and INSIG2 in the livers of Flox and LSKO mice fed NC or HFD for 12 to 16 weeks. The abundance of SCAP and both INSIG proteins was similar between Flox and LSKO mice in the NC or HFD groups, although the results were variable among mice within each group (fig.S3, E and F). Consistent with these findings, siRNA-mediated knockdown of SPTBN1 in Huh7 human liver cancer cells did not affect INSIG1 and INSIG2 abundance (fig. S3G). Although variable in the individual mice, when compared with the abundance in the livers of Flox mice, the abundance of n-SREBP1 appeared reduced in the livers of LSKO mice on NC, HFD diet, and to a lesser extent on WD (Fig. 2C). In contrast, the amount of n-SREBP2 appeared similar or slightly less in the LSKO mouse livers (fig.S3E). Thus, these results indicated that the decrease in SREBP1 abundance detected by immunohistochemistry and immunoblot in LSKO mouse livers was not the result of altered abundance of SREBP ER-localized regulators.
To determine whether the reduction in SREBP1 in LSKO mouse livers corresponded to a decrease in SREBP1 target gene expression, we analyzed transcript and protein abundance of products of de novo lipogenesis genes and performed luciferase reporter assays using sterol-responsive element (SRE)-containing promoters. In the livers of mice fed NC or HFD, transcripts of SREBP1 targets— Acc1 (encoding acetyl-CoA carboxylase 1), Scd1 (encoding stearoyl-CoA desaturase-1), and Fasn (encoding fatty acid synthase)— were decreased in LSKO tissues, and these changes were reflected in less protein (Fig. 2, D and E). Thus, we detected reduced expression of SREBP1 target genes in the absence of SPTBN1. The promoter of LDLR contains a sterol-responsive element (SRE) that binds to both SREBP1 and SREBP2 (42–44). However, in liver tissues, LDLR is primarily regulated by SREBP2 (45, 46). We found no difference in the abundance of LDLR in mouse livers for either genotype on either diet (Fig. 2E). The data suggest that SPTBN1 predominantly regulates SREBP1, not SREBP2, in the liver tissues.
Because SREBP1 also binds to the LDLR SRE (42–44), we used SREBP-responsive luciferase reporters containing wild-type SRE from the LDLR promoter region (LDLR-luc), an SREBP-unresponsive mutant SRE (mut-LDLR-luc), and SCD-luc to confirm SREBP-dependent changes in gene expression in mouse embryonic fibroblasts (MEFs) cells isolated from wild-type mice or global null Sptbn1−/− mice. In response to serum starvation, which activates SREBP in cultured cells (44), SRE-dependent LDL-luc activity increased in wild-type MEFs, but little to no SRE-dependent luciferase activity was detected in Sptbn1−/− cells (Fig. 2F). Re-expressing SPTBN1 in Sptbn1−/− MEF cells restored SRE-dependent luciferase activity (Fig. 2G).
SPTBN1 interacts with SMAD3 and influences TGF-β–mediated gene regulation (24). Next, we evaluated TGF-β-stimulated SRE-dependent gene expression. We found that TGF-β stimulated SRE-dependent luciferase activity, but this regulation occurred in Smad3−/− MEFs, indicating that the mechanism was independent of SMAD3 (Fig. 2H). Thus, the data indicated that SPTBN1 regulated de novo lipogenesis independently from its interaction with SMAD3.
SPTBN1 binds to SREBP1
To verify mass spectrometry data showing that SPTBN1 and SREBP1 were part of the same complex (table S1), we immunoprecipitated SPTBN1 from 3 HCC cell lines. Although the signal was weak, we detected pre-SREBP1 and n-SREBP1 in immunoprecipitates from all 3 cell lines (Fig. 3A, fig. S4A).
Fig. 3. SPBTN1 and SREBP1 interact.
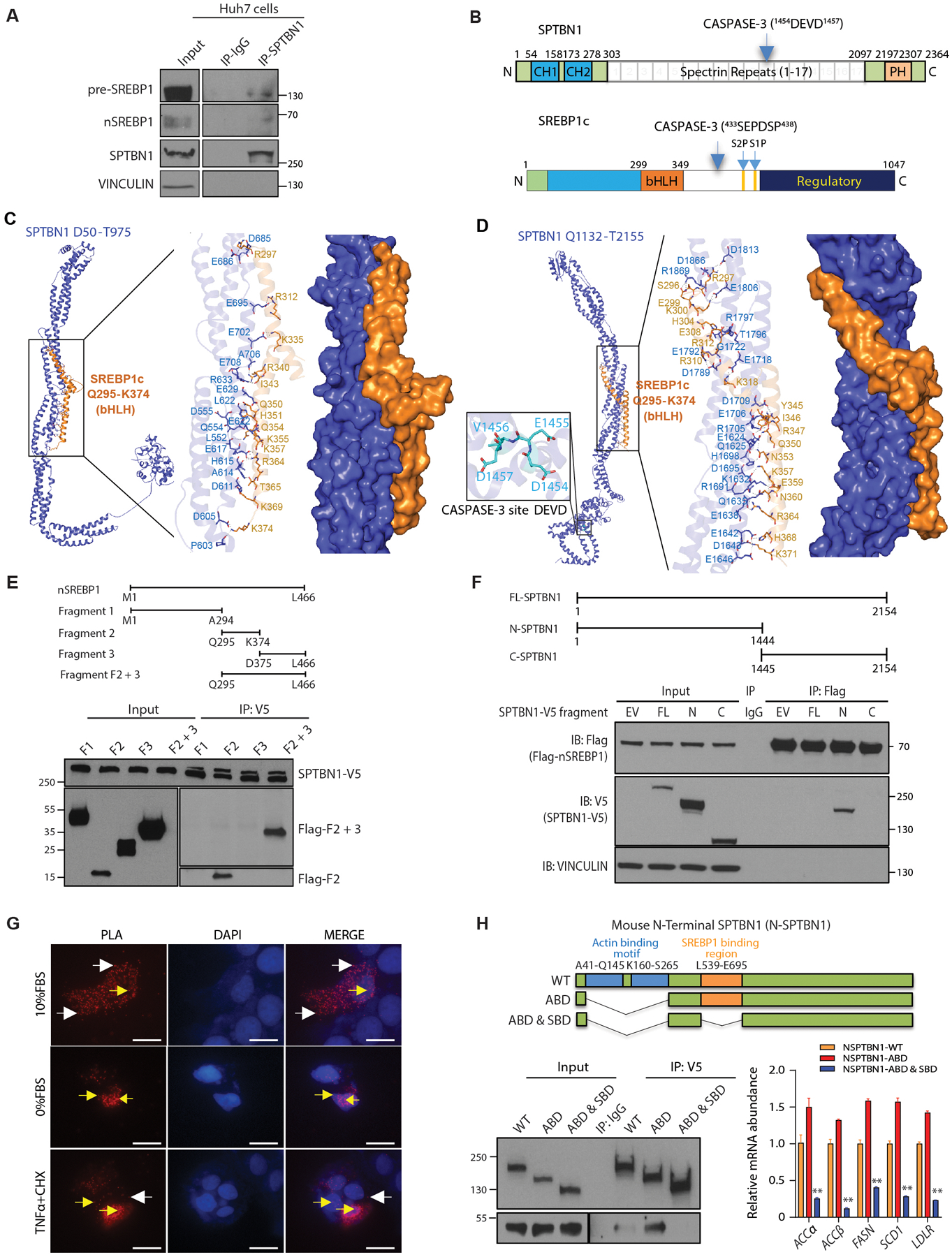
(A) Coimmunoprecipitation of endogenous nSREBP1 and pre-SREBP1 with SPTBN1 from Huh7 cells. IgG served as a negative control for nonspecific binding. Space indicates input samples were analyzed separately because of the need for a shorter exposure. Data are representative of 1 of 3 experiments. IP, immunoprecipitation.
(B) Schematic of the domain organization of SPTBN1 and SREBP1c. CH, calponin homology; PH, pleckstrin homology; CASPASE-3 with cleavage site in parentheses; bHLH, basic helix loop helix; S1P and S2P, intramembrane cleavage sites.
(C) Structural model predicted by molecular docking simulations for the SPTBN1 fragment from amino acid residues D50 – T975 and SREBP1c amino acid residues Q295 – K374, encompassing the bHLH domain. Enlarged region shows predicted interacting residues.
(D) Structural model predicted by molecular docking simulations for the SPTBN1 fragment from amino acid residues Q1132 – T2155 and SREBP1c amino acid residues Q295 – K374, encompassing the bHLH domain. Enlarged region to the right shows predicted interacting residues; enlarged region to the left shows the CASPASE-3 cleavage site in SPTBN1.
(E) Coimmunoprecipitation of the indicated fragments of human nSREBP1 expressed as Flag-tagged peptides in Hep3B cells coexpressing full-length V5-tagged SPTBN1. Upper diagram shows the fragments (F1, fragment 1; F2, fragment 2; F3, fragment 3; F2 +3, fragment containing both parts of fragment 2 and fragment 3). Lower blot shows the input and immunoprecipitated (IP). The immunoprecipitation blot was exposed for longer than the input blot to enable visualization of low abundance fragments. Data are representative of 1 of 3 experiments.
(F) Coimmunoprecipitation of the indicated V5-tagged fragments of mouse SPTBN1 expressed with Flag-tagged peptides in SNU398 cells coexpressing Flag-tagged human nSREBP1. Upper diagram shows the fragments. Lower blot shows the input and immunoprecipitated (IP) proteins by Western blot. EV, empty vector for SPTBN1-V5; FL, full-length SPTBN1; N, N-terminal CASPASE-3 cleavage fragment of SPTBN1; C, C-terminal CASPASE-3-cleavage fragment of SPTBN1. VINCULIN and IgG served as negative controls. Data are representative of 1 of 3 experiments.
(G) Proximity ligation assay (PLA) to show the interaction between NSPTBN1-V5 with Flag-nSREBP1 in Huh7 cells in 10% FBS, 0%FBS, and in 10%FBS but treated with TNFα (20ng/ml) or cycloheximide (10mg/ml) for 3 hrs. Red fluorescent dots indicate NSPTBN1 and nSREBP1 are in close association in both cytoplasm (white arrows) and nucleus (yellow arrows). Scale bar = 20uM.
(H) Coimmunoprecipitation of the indicated V5-tagged fragments of mouse SPTBN1 with a deletion in SREBP1 binding region and/or actin-binding motif in Huh7 cells coexpressing Flag-tagged human nSREBP1 F2+3 fragment. Upper diagram shows the fragments. Lower left blot shows the input and immunoprecipitated (IP) proteins by Western blot, lower right shows the transcripts of SREBP1 target genes (n=3) in cells transfected with indicated N-STPBN1 deletion mutants. WT, wildtype; ABD, Actin binding domain; SDB, SREBP1 binding domain. Data shown as mean ± SEM are representative of 2 independent experiments performed in triplicate. Statistical significance was determined by one-way ANOVA Bonferroni’s multiple comparisons test between WT and mutant (**, p < 0.005).
To explore the structural basis of SPTBN1 and SREBP1 binding and map their potential interaction sites, we modelled three-dimensional structures of SPTBN1 and SREBP1c and then performed structure-based molecular docking simulations. There are 2 splice variants of SREBP1: SREBP1a and SREBP1c (47). We selected SREBP1c for molecular docking simulations because this is the form that specifically stimulates fatty acid synthesis and is associated with liver steatosis (48).
SPTBN1 is comprised of 2,364 amino acid residues and characterized by multiple homologous tandem spectrin repeats, each composed of three antiparallel helices, flanked by a pair of calponin-homology (CH) domains at the N-terminal side and a pleckstrin homology (PH) domain at the C-terminal side (Fig. 3B). SPTBN1 has a CASPASE-3 cleavage site (1454DEVD1457) in the 11th spectrin repeat. SREBP1c is comprised of 1,047 amino acid residues that form an N-terminal DNA binding domain (bHLH), two transmembrane domains each with a cleavage site in the middle, and a regulatory domain at the C-terminus. SREBP1c has a CASPASE-3 cleavage site before the transmembrane domains (433SEPDSP438) (12) (Fig. 3B).
For our modeling, SPTBN1 was examined in three fragments covering D50-T975 (Fig. 3C), Q1132-T2155 (Fig. 3D), and A2198-K2364 (fig. S4B); SREBP1c was modelled in two fragments covering Q295-K374 (Fig. 3, C and D) and P546-S705. We could not dock SREBP1c P546-S705 onto SPTBN1. However, we successfully simulated interactions between the SREBP1c Q295-K374 fragment and SPTBN1 (Fig. 3, C and D, fig. S4B, table S2).
The models predicted that SREBP1c Q295-K374 has a high-affinity conformational fit within a binding cavity on SPTBN1 and that the interactions are stabilized by multiple hydrogen bonds and Van der Waals interactions (table S2). On the basis of the lowest interaction energy values (49, 50), we ranked the predicted models as having the following affinities (table S2): SREBP1c Q295-K374/SPTBN1 Q1132-T2155 (Fig. 3D) > SREBP1c Q295-K374/SPTBN1 D50-T975 (Fig. 3C) >> SREBP1c Q295-K374/SPTBN1 A2198-K2364 (fig. S4B). Overall, the docking analysis predicted SPTBN1 fragments D50-T975 and Q1132-T2155 as preferred SREBP1c Q295-K374 binding sites.
To test predicted models and identify regions required for their interaction biochemically, we generated SREBP1 fragments and SPTBN1 fragments. For n-SREBP1 (amino acids M1 – L466), we generated 4 fragments (Fig. 3E). Fragment 2 of SREBP1 corresponds to the modeled portion (Q295 – K374) and was most effectively coimmunoprecipitated with full-length V5-tagged mouse SPTBN1 (Fig. 3E), which shares 98.3% sequence identity with human SPTBN1. CASPASE-3 cleaves SPTBN1 into two products, N-SPTBN1 (amino acids M1 – D1454) and C-SPTBN1 (E1455 – K2364), which are detected in acetaminophen-damaged livers (28). We found that only N-SPTBN1 coimmunoprecipitated with Flag-tagged n-SREBP1 (Fig. 3F). We found the interaction between N-SPTBN1 and endogenous pre-SREBP1 occurred predominantly in the cytoplasm, whereas the interaction between N-SPTBN1 and endogenous nSREBP1 occurred predominantly in the nucleus (fig. S4C). These data suggest SPTBN1 and SREBP1 can interact directly. We confirmed the interaction between N-SPTBN1 and nSREBP1 through proximity ligation assays (PLA) in Huh7 cells overexpressing tagged forms of N-SPTBN1 and n-SREBP1 (Fig. 3G).
To evaluate functional consequences of interfering with the SPTBN1-SREBP1 interaction, we generated 2 N-SPTBN1 mutants, a mutant with a deletion of the Actin binding motif (NSPTBN1-ABD) and a mutant with deletion of the entire SREBP1 binding interface (SBD; human L552-E708, mouse L539-E695). Because SPTBN1 Actin binding motif seems to negatively regulate SPTBN1 stability (51) and interferes with SPTBN1 binding with other proteins (fig. S4D, Fig.3H), we generated a double mutant with deletions of both SREBP1 binding interface SBD and the ABD (NSPTBN1-ABD & SBD). We compared NSPTBN1-ABD with NSPTBN1-ABD & SBD, a mutant carrying deletions of both the entire SREBP1 binding interface (SBD; human L552-E708, mouse L539-E695) and the ABD (NSPTBN1-ABD & SBD). Compared with either wild-type N-SPTBN1 or NSPTBN1-ABD, the double mutant NSPTBN1-ABD & SBD not only abrogates binding to SREBP1, but also significantly inhibits SREBP1 activity as demonstrated by significantly reduced expression of SREBP1 target genes including SCD1, FASN, and ACC (p < 0.005) in cells with overexpression of different forms of NSPTBN1 (Fig.3H).
From the modeling and fragment interaction analyses, we hypothesized that conditions causing activation of CASPASE-3 in cells result in cleavage of SPTBN1. The resulting N-SPTBN1 interacts with n-SREBP1.
Cell stress induces CASPASE-3-dependent cleavage of both SREBP1 and SPTBN1 in human hepatocytes and HCC cells
Stress-induced caspase activation is linked to the NASH phenotype (8, 9). Our RNA-seq data revealed alteration in ER stress and UPR pathways in the livers of HFD-fed LSKO mice (Fig. 2A). Because both SREBP1 and SPTBN1 are CASPASE-3 substrates (12, 28) and our interaction data suggested that their cleavage products interact (Fig. 3F), we explored the relationship among CASPASE-3, SPTBN1, and SREBP1 in the immortalized human hepatocyte cell line THLE2 and the HCC cell line, Huh7. We used TNFα and cycloheximide or palmitic acid (PA) to induce CASPASE-3 activation. TNFα and PA induce CASPASE-3 activation in liver cells (52, 53) and are associated with NASH development (54, 55). THLE2 cells exposed to TNFα and cycloheximide exhibited CASPASE-3 activation (cleaved CASPASE-3), SREBP1 activation (n-SREBP1), and cleavage of SPTBN1 to produce both N-SPTBN1 and C-SPTBN1 (Fig. 4A). Consistently, we observed increased interaction between NSPTBN1 and nSREBP1 in the nucleus by proximity ligation assays (PLA) in cells treated with TNFα and cycloheximide (Fig. 3G, bottom panel). Analysis of the cytosolic and nuclear distribution of N-SPTBN1 and C-SPTBN1, expressed as V5-tagged proteins in THLE2 cells, showed that N-SPTBN1 was present in both the cytoplasm and nucleus, whereas C-SPTBN1 was only detected in the cytoplasm (Fig. 4B). Knockdown of SPTBN1 in Huh7 cells did not prevent CASPASE-3 activation in cells exposed to TNFα and cycloheximide and appeared to reduce the amount of both preSREBP1 and nSREBP1 under both control and stress conditions (Fig. 4C). However, expression of LDLR and SCD1, SREBP target genes, was reduced (Fig. 4D).
Fig. 4. CASPASE-3-induced SREBP1 activation but not cleavage is reduced in HCC cells with SPTBN1 knockdown.
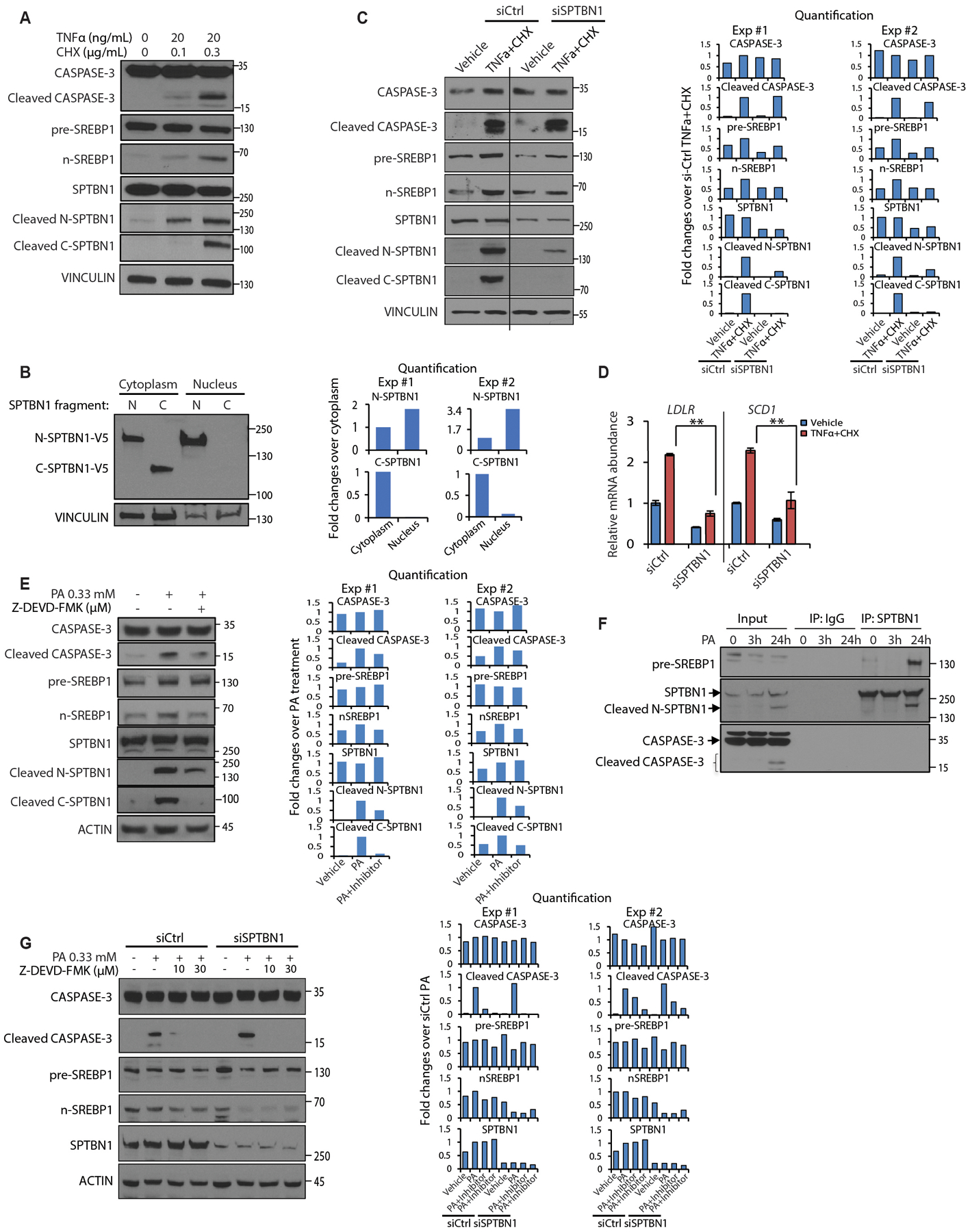
(A) Western blot of the indicated proteins or protein cleavage products in THLE2 cells exposed to the indicated concentrations of TNFα or cycloheximide for 3 hrs.
(B) Abundance of V5-tagged N-SPTBN1 or C-SPTBN1 expressed in THLE2 cells in the cytoplasmic or nuclear fractions.
(C) Western blot of the indicated proteins or protein cleavage products in Huh7 cells transfected with the indicated siRNAs and exposed to vehicle or TNFα (20 ng/ml) + cycloheximide (10 μg/ml) for 3 hrs. Black vertical line indicates samples were analyzed on a same blot separated by other samples.
(D) Relative mRNA abundance for the SREBP1 target gene (SCD1) and the SREBP2 target gene (LDLR) in Huh7 cells as described in C. Data are normalized to 18S and shown as mean ± SEM (n = 3). Significance was determined by 2-sided t-test (**, p < 0.005).
(E) Western blot of the indicated proteins or protein cleavage products in Huh7 cells exposed to the indicated concentrations of PA or the caspase inhibitor Z-DEVD-FMK.
(F) Coimmunoprecipitation of SPTBN1 and detection of pre-SREBP1, N-SPTBN1, and SPTBN1 in Huh7 cells exposed to PA (0.33 mM) for the indicated times.
(G) Western blot of the indicated proteins or protein cleavage products in Huh7 cells treated with indicated siRNA and exposed to the indicated concentrations of PA or the caspase inhibitor Z-DEVD-FMK.
Western blot data are representative of 1 of 2–3 experiments. CHX, cycloheximide; IP, immunoprecipitate; siCtrl, control siRNA; siSPTBN1; siRNA against SPTBN1. For B, C, E, G, quantification of 2 independent experiments were shown on the right. Each band was quantified by Image J and normalization to loading control. Treatment group was set as 1 in panel C, E, G; cytoplasm group was set as 1 in panel B.
CASPASE-3 activation in Huh7 cells exposed to PA was reduced by CASPASE-3 inhibitor (Z-DEVD-FMK) (Fig. 4E). PA also triggered cleavage of SPTBN1, which was also reduced by the CASPASE-3 inhibitor, indicating that SPTBN1 cleavage depends on CASPASE-3 activity (Fig. 4E). We found that pre-SREBP1, and SPTBN1 and N-SPTBN1 coimmunoprecipitated from Huh7 cells exposed to PA, consistent with an interaction between SREBP1 and SPTBN1 occurring with the CASPASE-3 cleavage product N-SPTBN1 (Fig. 4F). n-SREBP1 was reduced in PA-treated cells in which SPTBN1 was knocked down even though CASPASE-3 activation was unaffected, suggesting that the interaction with SPTBN1 may stabilize n-SREBP1 (Fig. 4G). Consistent with this, we did not detect any difference in SREBP1 transcript abundance in the livers from Flox control and LSKO mice (fig. S4E), indicating that the reduction in SREBP1 abundance was not mediated at the level of gene expression.
Human NASH is associated with increased SPTBN1 and CASPASE-3 expression and increased TGF-β pathway and SREBP1 activity
To gain insight into the clinical relevance of our findings implicating SPTBN1 and a pathway involving the CASPASE-3-mediated interaction between cleavage products of SPTBN1 and SREBP1 in steatosis, we performed immunohistochemical labeling (IHC) of cleaved CASPASE-3, SREBP1, and SPTBN1 in human liver samples from healthy control and NASH patients. As expected, a subset of NASH liver samples showed increased cleaved CASPASE-3 labeling in hepatocytes where we also observed significantly increased nuclear staining of SPTBN1 (p < 0.05) and SREBP1, that are predominantly cytoplasmic in healthy controls (Fig. 5, A and B).
Fig. 5. SPTBN1 and CASPASE-3 expression and SREBP1 activity are increased in patients with NASH.
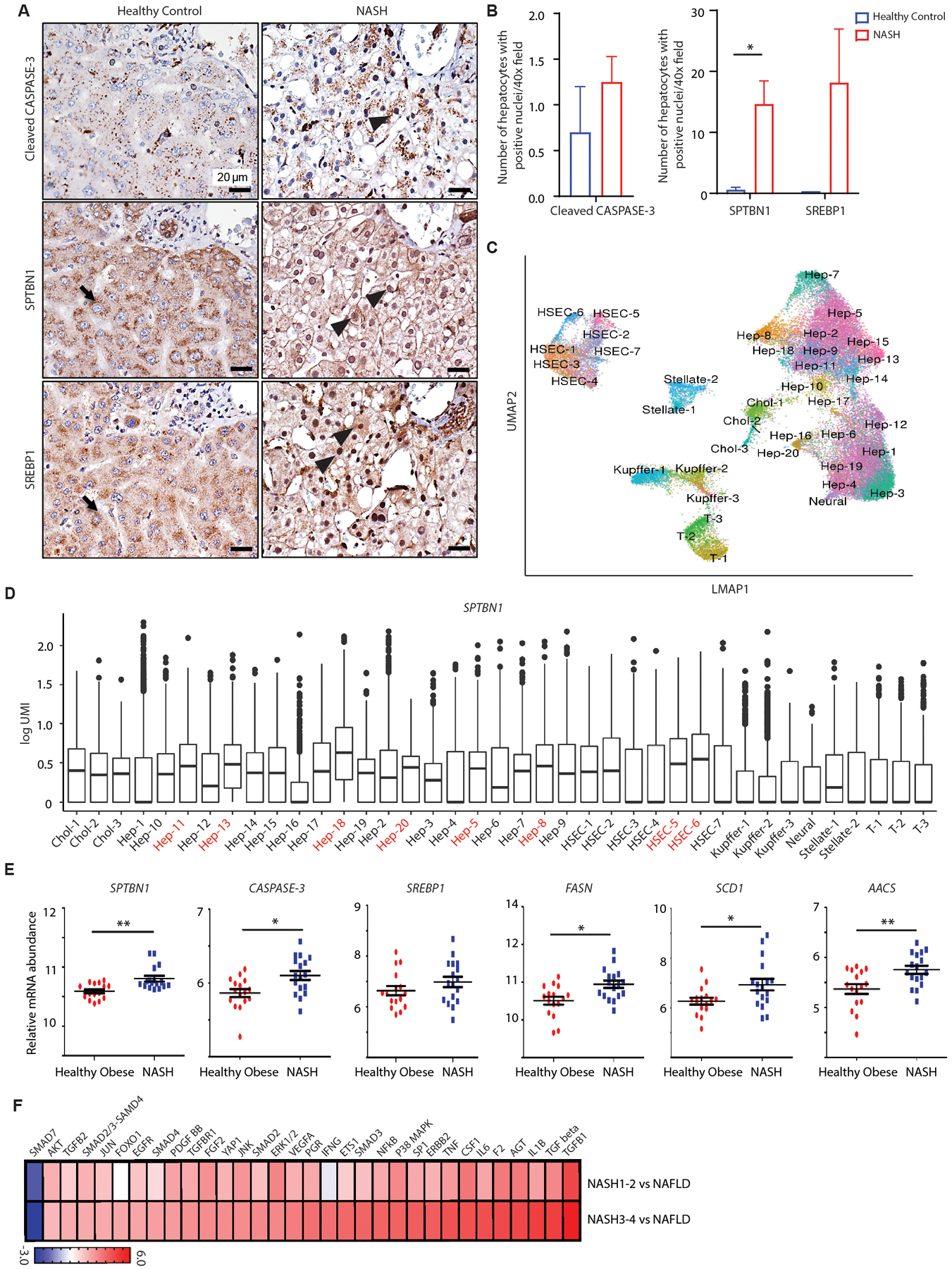
(A) Cleaved CASPASE-3, SPTBN1, and SREBP1 immunohistochemistry in liver tissues from normal healthy subjects and patients with NASH. Black arrow indicates cytoplasmic labeling and black arrowhead indicates nuclear labeling.
(B) Quantification of hepatocytes with positive nuclear labeling of cleaved CASPASE-3, SPTBN1, and SREBP1 in liver tissues from normal healthy individuals (n=2) and NASH patients (n=6 – 12). Average number of hepatocytes with positive nuclear labeling were counted under 40x field from 5 random fields of each sample. Data are shown as mean ± SEM. Significance was determined by 2-sided t-test (*, p < 0.05).
(C) Phenograph (UMAP) representation of the cells identified by single-nucleus sequencing.
(D) Analysis of SPTBN1 expression by single-nucleus sequencing of liver samples (n=6) from 3 patients with NASH mixed with HCC. Hep-1 through 20, hepatocyte subpopulations; HSEC, hepatic sinusoidal endothelial cells; Chol-1 to Chol-3, 3 populations of cholangiocytes; T-1 to T-3, 3 populations of T cells; Kupffer-1 to Kupffer-3, 3 populations of Kupffer cells (a type of macrophage); Stellate-1 to Stellate-2, 2 populations of hepatic stellate cells.
(E) Relative mRNA abundance of SPTBN1, CASPASE-3, SREBP1, and SREBP1 target genes in liver tissue samples from human healthy obese individuals (n=16) and NASH patients (n=17) from a public data set (GSE48452).
(F) Bioinformatic analysis by Ingenuity Pathway Analysis for Upstream Regulators of differentially regulated genes in livers from patients with stage 1 or 2 NASH (NASH1–2) or stage 3 or 4 NASH (NASH3–4) compared to patients with NAFLD. Gene expression was profiled by RNA-seq with differentially regulated genes determined by t-test. Regulators with activation (z score > 0) or inhibition (z score < 0) and p < 0.05 are shown.
Because NASH represents a high risk for progression to HCC (1), we performed single-nucleus RNA-seq (snRNA-seq) for SPTBN1 expression in liver tissue from patients with both NASH and HCC but without cirrhosis. We identified a subset of cells with high SPTBN1 expression (Fig. 5, C and D): 6 subtypes of hepatocytes (Hep-11, Hep-13, Hep-18, Hep-20, Hep-5 and Hep-8), and 2 liver sinusoidal endothelial cells of the portal vein (HSEC-5, and HSEC-6). Hep-18, the cluster with the highest expression of SPTBN1, also showed increased expression of FADS1, an SREBP1 target gene. Analysis of the snRNA-seq data showed that the hepatocytes with high SPTBN1 expression clustered together when plotted on a UMAP (Fig. 5C).
We also analyzed data from publicly available databases by comparing liver tissue data from healthy obese with that from NASH patients for transcripts of SPTBN1, CASPASE-3, SREBP1, as well as for SREBP1 target genes involved in lipogenesis. Both SPTBN1 and CASPASE-3 transcripts were increased in NASH patients, whereas no differences were observed for SREBP1 (Fig. 5E). However, we found significantly increased expression of FASN (p < 0.05), SCD1 (p < 0.05), and AACS (p < 0.005) (Fig. 5E), indicating increased SREBP1 activity.
In addition, we used Ingenuity “Upstream Regulator” analysis to evaluate proteins associated with differences in gene expression between early stages of NASH1–2 (NASH1 and 2) and NAFLD and between late stages of NASH3–4 (NASH3 and 4) and NAFLD. Consistent with the involvement of TGF-β signaling in fibrosis, a key indicator of the progression of NASH from NAFLD (56, 57), the upstream regulators with increased activity included several proteins in the TGF-β pathway, and the only downregulated protein was SMAD7, an inhibitor of TGF-β signaling (Fig. 5F).
Collectively, this evaluation of human NASH liver data is consistent with the findings from our HFD-fed mice that implicate SPTBN1 in progression of this condition. Furthermore, these data confirmed increased SPTBN1 and CASPASE-3, along with increased activity of TGF-β/SMAD signaling and SREBP1-dependent gene expression, in NASH.
Targeting SPTBN1 by siRNA in vivo attenuates HFD-induced NAFLD
Liver diseases are one of the few that have Food and Drug Administration-approved siRNA-based treatments (58). Therefore, we tested if siRNA-mediated knockdown of SPTBN1 in Flox mice protected them from HFD-induced NAFLD. Flox mice (10–12 weeks old) were fed a HFD for 12 weeks. One week after the start of the HFD, mice were injected hydrodynamically with either siRNA targeting Sptbn1 (siSptbn1) or an equivalent volume of siRNA negative control (siCtrl) every two weeks for a total of 3 injections (fig. S5A). We monitored mice weekly for body weight gain and survival. We confirmed the knockdown of SPTBN1 by siSptbn1 in the livers of the Flox mice (fig. S5A, right). For each of the parameters we monitored, the siSptbn1-treated mice responded to HFD similarly to the LSKO mice. Compared to the siCtrl-treated mice, the siSptbn1-treated mice gained less weight (fig. S5B, p < 0.05), accumulated less visceral body fat (fig. S5C, p < 0.05), and had similar blood TG concentrations and blood glucose concentrations (fig. S5D). Expression of pro-fibrotic genes and inflammatory genes in the livers of the siSptbn1-treated mice were significantly less than in the livers from siCtrl-treated mice (fig. S5E) (p < 0.05), and siSptbn1-treated mice had normal liver architecture and low lipid accumulation without any signs of NAFLD or NASH (fig. S5, F and G). We observed the same benefit of siSptbn1 in female Flox mice (fig. S5H).
In addition to disease prevention, we explored the therapeutic effects of siSptbn1 on already established NASH induced by WD containing 42% kcal from fat combined with a high fructose-glucose solution (Fig. 6, A to G). We dissected 2 littermates from each group at 12 weeks of WD feeding and verified the development of NASH. For the remaining animals, we started the siRNA treatment. We found that 4 injections of siSptbn1 ameliorated NASH but did not significantly affect body weight as compared with control siRNA (Fig.6, A and B). siSptbn1 treatment group showed significantly reduced serum TG and glucose concentrations (p < 0.05), but not cholesterol concentrations(Fig.6C). Targeting Sptbn1 with siRNA therapy significantly improved liver histology and inhibited NASH development as demonstrated by significantly reduced liver weight (p < 0.05) (Fig.6D) and NAFLD Activity Score (NAS) (Fig.6E) with reduced hepatic steatosis, lobular inflammation, ballooning, and fibrosis (Fig.6F), blindly assessed by an independent pathologist. Consistently, we observed significantly reduced expression of Mmp2 (p < 0.05), a pro-fibrotic gene, in the livers from mice treated with siSptbn1 (Fig. 6G).
Fig. 6. SPTBN1 siRNA protects mice from WD-induced NAFLD and NASH.
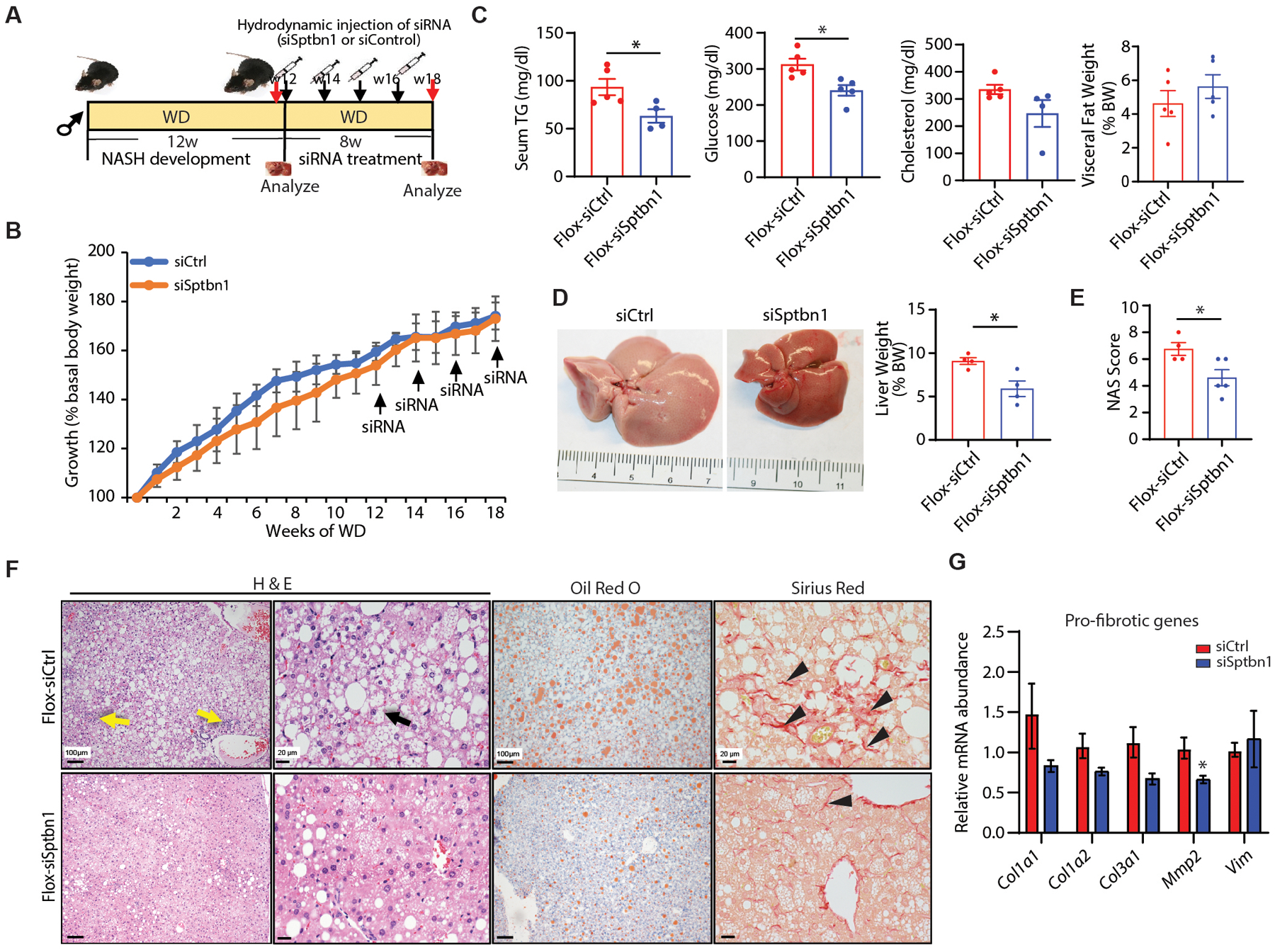
(A) Left: Diagram of the experimental protocol. Flox mice were started on the western diet (WD) and high fructose-glucose water for 12 weeks to induce NASH development, then Flox mice received hydrodynamic injection of either control siRNA (siCtrl) or siRNA targeting Sptbn1 (siSptbn1) at a dosage of 1.25 nmol/mice each time for 4 times every 2 weeks. Body weight was evaluated weekly and mice were euthanized at the end of 20 weeks on the WD.
(B) Body weight measurements are presented as percent increase from basal weight before start of the WD (n = 4 – 5 mice per group).
(C) Serum TG, glucose, cholesterol concentrations, and visceral fat of mice as % of body weight (BW) in mice receiving the indicated treatments (n = 4 – 5 per group).
(D) Gross picture of liver tissue and liver weight as % of BW from mice treated with siCtrl or siSptbn1(n=4). Quantification on right.
(E) NAFLD Activity Score (NAS) in Flox mice treated with siCtrl or siSptbn1 (n=4 – 5 from each group).
(F) Liver histology (H &E), lipid accumulation (Oil Red O), and fibrosis (Sirius Red) in Flox mice receiving the indicated treatment. Yellow arrow indicates inflammation; black arrow indicates ballooning cell; black arrowhead indicates fine fibrosis.
(G) Relative mRNA abundance of pro-fibrotic genes in mice treated with siCtrl or siSptbn1 (n=3 – 4 from each group).
SPTBN1 knockdown in a human 3D-culture NASH model reduces transcriptional changes associated with NASH
Our mouse experiments showed that SPTBN1 knockdown prevented HFD-induced NAFLD and progression to NASH. We also tested this potential therapy in a newly developed 3D perfused microphysical system that enables the co-culture of primary human hepatocytes, hepatic Kupffer cells, and stellate cells in medium enriched in fatty acids, sugars, and insulin for 2 weeks as a culture model of human NASH (59, 60). The 3D cultures were exposed to different concentrations of siSPTBN1 and equivalent concentrations of siCtrl, applying the siRNA every 2 days concurrent with medium changes from day 4 (Fig. 7A). We observed > 90% decrease in SPTBN1 transcripts after 96 h (day 8) with the 25 nM siRNA treatment without significant changes in cell viability (LDH activity) or hepatic function (albumin production) (Fig. 7B). Oil-Red O staining showed a slight but not significant decrease in lipid accumulation at day 8 (4 days after the addition of the siRNA (Fig. 7B).
Figure 7. SPBTN1 siRNA reverses transcriptional changes associated with NASH in a human 3D culture model.
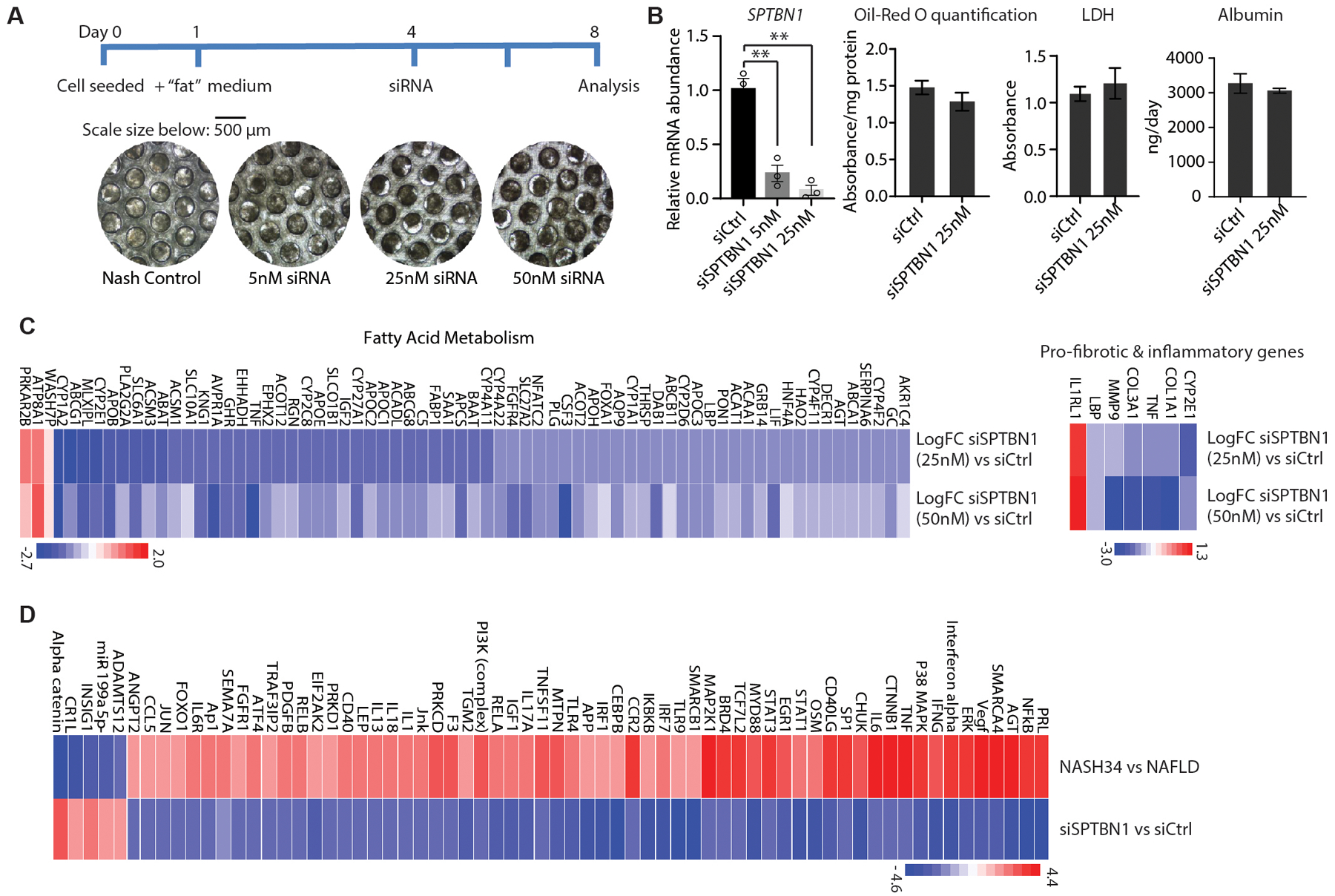
(A) Diagram and representative brightfield microscopy images of the cells scaffolds from 3D human NASH co-culture model system used to evaluate the effect of siSPTBN1 treatment.
(B) Left: Relative mRNA abundance of SPTBN1 in 3D human NASH co-culture model 96 h after the second addition of the indicated siRNAs. Middle: Quantification of Oil-Red O staining on day 8 in cultures treated with the indicated siRNAs. Right: LDH (lactate dehydrogenase) activity and albumin production in the medium of cultures treated with indicated siRNA (n=3).
(C) Heat map of changes in the expression of genes in fatty acid metabolism and genes associated with fibrosis and inflammation in the 3D culture treated with 25 nM siSPTBN1 or 50 nM siSPTBN1 compared to siCtrl. Cultures were collected 96 h after siRNA treatment and transcripts were evaluated by RNA-seq. Blue, downregulated; red, upregulated.
(D) Heat map of changes in upstream regulators significantly associated with differentially expressed genes in human NASH stages 3 and 4 compared with human NAFLD and the changes in these regulators in the 3D culture for siSPTBN1 compared to siCtrl. Cultures were collected 96 h after siRNA treatment and transcripts were evaluated by RNA-seq. Upstream regulator activity was determined with Ingenuity Pathway Analysis of “Upstream Regulators”.
Quantitative data are shown as mean ± SEM. Statistical significance was determined by 2-sided t-test for weekly changes in body weight and for all other quantitative data (*, p < 0.05; **, p < 0.005).
RNA-seq analysis of cultures exposed to siSPTBN1 (25 nM or 50 nM) or siCtrl was performed on samples collected 96 h after the siRNA treatment. Pathway analysis revealed significant decreases in the siSPTBN1-treated cultures in transcripts encoding proteins involved in fatty acid metabolism, including those involved in lipid transport, triglyceride and glycogen metabolism, and lipoprotein catabolism as well as SREBP1 target genes (fig. S6A). We also found a reduction in the transcripts encoding proteins involved in fibrosis and altered inflammatory gene expression in the siSptbn1-treated cultures (Fig. 7C). These changes indicated that siSPTBN1 treatment reduced de novo lipogenesis, inflammation, and fibrosis. As expected, we noted a marked decrease in a gene expression signature associated with TGF-β signaling in siSPTBN1-treated cultures (fig. S6B), consistent with the mouse models, showing that this profile was reduced in LSKO and was activated in mouse (HFD-induced NASH) and human NASH (fig. S6B).
“Upstream Regulators” analysis with IPA indicated that regulators with higher activity in human NASH compared to NAFLD were uniformly associated with lower activity in the siSPTBN1-treated cultures compared to the siCtrl-treated cultures (Fig. 7D). Furthermore, upstream regulators with lower activity in NASH versus NAFLD had higher activity in the siSPTBN1-treated cultures versus the siCtrl-treated cultures (Fig. 7D). Thus, siSPTBN1 appeared to reverse or counter NASH-associated regulatory changes, further supporting the concept of siSPTBN1 as a potential therapy.
LSKO mice are protected from HCC
Our data indicated that the complete liver-specific loss of SPTBN1 protects mice from HFD-induced NASH, a well-established risk factor for HCC (1). Here, we evaluated the impact of the homozygous loss of hepatic SPTBN1 on the occurrence of HCC. First, we explored the HCC incidence in LSKO mice and control mice including both Flox and liver Sptbn1 heterozygous mice fed a HFD for 28 weeks. Whereas we found two tumor nodules in one of four HFD-fed control mice, we did not find any liver tumors in any of the age-matched, HFD-fed LSKO mice (Fig. 8A).
Fig. 8. LSKO mice develop fewer liver tumors.
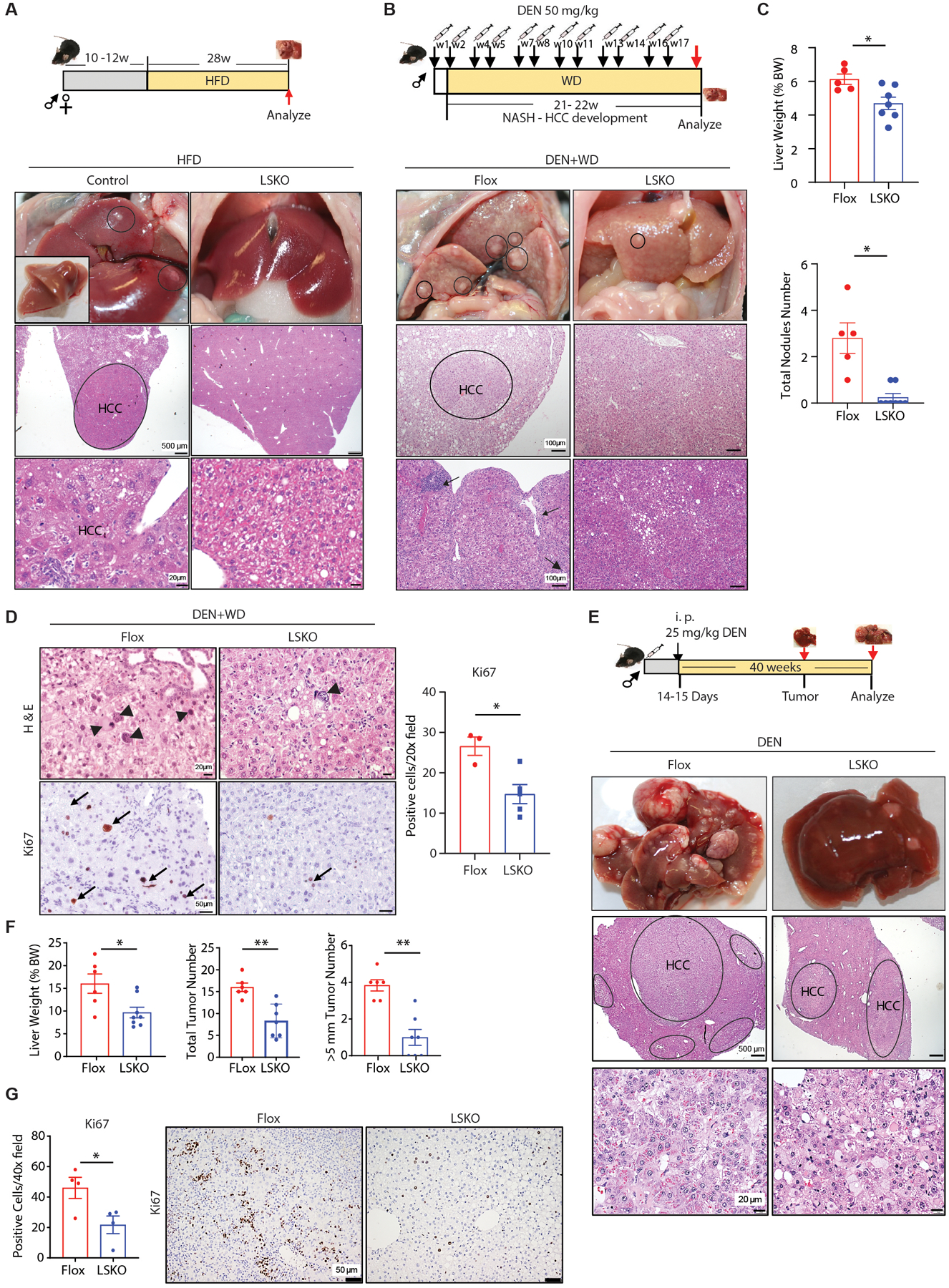
(A) Diagram of the experimental procedure of HFD-induced HCC (top). One of 4 control female control mice developed liver tumors after 28 weeks of HFD. Top: Gross morphology of the liver of that animal and morphology of the liver from an LSKO female littermate fed HFD for 28 weeks are shown. Inset shows excised liver. Circles show tumors. Middle and bottom: Liver histology by H & E staining. Tumor (HCC) is circled and bottom shows enlargement of tumor tissue.
(B) Diagram of the experimental procedure of chemical (DEN) plus WD-induced HCC (top). Gross morphology and H & E stained sections of livers in Flox or LSKO mice 22 weeks after DEN and WD. Inflammation (black arrows) sites are indicated.
(C) Top: Liver weight as percentage of body weight (BW) from Flox or LSKO mice 22 of weeks after DEN and WD (top). Bottom: Number of visible liver nodules observed in the liver tissues from Flox and LSKO mice. Flox, n=5; LSKO, n=7–8.
(D) Representative H & E stained sections and Ki67 immunohistochemistry and quantification of positive cells in liver sections from Flox and LSKO mice under 20X magnification. Arrowheads indicate abnormal hepatocytes with evidence of dysplasia Arrows indicate cells with positive staining of Ki67 staining. Flox, n=3; LSKO, n=5.
(E) Diagram of the experimental procedure of DEN-induced HCC (top). Gross pictures and H & E of liver tumors from Flox and LSKO mice 40 weeks after DEN injection. Upper: gross liver morphology. Middle: H & E stained sections with HCC tissue circled. Lower: H & E stained sections through tumor tissue circled.
(F) Liver weight as % of total body weight (BW), total number of tumors number, and number of tumors with a size > 5 mm in livers from Flox and LSKO mice at 40 weeks after DEN injection (n = 6 – 8).
(G) Representative Ki67 immunohistochemistry and quantification of positive cells in liver sections from Flox and LSKO mice under 20X magnification (n = 4 mice per group).
Quantitative data are shown as mean ± SEM. Statistical significance was determined by 2-sided t-test (*, p < 0.05; **, p < 0.005).
Because HCC development did not occur with sufficient frequency in this experimental condition, we switched to a chemically induced HCC model known to result in a greater frequency of HCC using diethylnitrosamine (DEN) to induce HCC with WD feeding (WD+DEN) or without WD feeding (DEN alone) (Fig.8, B to G). We evaluated liver tumor development within 22 weeks and found that DEN injections with WD feeding induced liver tumors in Flox mice but not in LSKO mice (Fig. 8C). Flox mice demonstrated an increase in liver weight, visible tumor nodules, and features of severe liver injury that included steatosis, inflammation, and regenerative nodules with bridges of fibrosis. These features were much less evident in the LSKO mice with far fewer visible liver nodules (Fig. 8, B and C, fig.S7A). Liver histology in the Flox mice compared to LSKO mice revealed evidence of increased cellular proliferation in the regenerative nodules, as demonstrated by immunolabeling with Ki67 (Fig. 8D). There was also evidence of preneoplastic changes in the form of hepatocellular dysplasia in all Flox mice, including enlarged cells and nuclei with high nuclear: cytoplasmic ratio, multinucleated cells, and nuclear pleomorphism with focal nuclear inclusions (Fig.8D, fig.S7A). Thus, LSKO mice are protected against DEN and WD-induced liver steatosis, fibrosis, and tumor development.
Analyzing 24 weeks or 40 weeks after a single injection of DEN without WD, we found that all mice developed tumors in this model. However, LSKO mice had less severe disease (Fig. 8, E to G). We found small nodules in the livers 24 weeks after DEN injection (fig. S7B), and tumors developed by 40 weeks (Fig. 8E). Most of the LSKO mice had a lower tumor burden indicated by lower tumor numbers, smaller tumor sizes, and improved liver pathology when compared with the Flox control mice (Fig. 8, E and F). Furthermore, we observed fewer proliferating cells, indicated by Ki67 labeling, in LSKO mouse livers compared with livers of Flox control mice (Fig. 8G). However, no significant differences were observed in cleaved CASPASE-3 labeling, a marker for apoptosis, suggesting that the reduced tumor burden in LSKO mice is not due to increased apoptosis (fig. S7C). In agreement to the LSKO HCC protection phenotype, STAT4, an inhibitor of HCC (61, 62), was identified as an activated upstream regulator of the differentially regulated genes in livers of LSKO mice fed a HFD (Fig. 2A).
To validate whether complete loss of SPTBN1 has a protective effect in human HCC, we first analyzed snRNA-seq data to find differences in cell type-specific mRNA abundance of SPTBN1 between matched tumor and adjacent non-tumor tissue in the three individuals. We found SPTBN1 present in all cell types and showed a downward trend in the NASH-HCC tumors in a hepatocyte-specific manner, but this was not significant (p = 0.14; fig. S7D). We also analyzed the frequency of SPTBN1 homozygous loss (deep deletion) in human HCC. In data from The Cancer Genome Atlas (TCGA), only one HCC patient (1/440, 0.2%) had a homozygous loss of SPTBN1; whereas the heterozygous deletion occurred in 7.8% (30/440) of HCC patients. The human data of the heterozygous deletion of SPTBN1 is not surprising to us because mice with whole body Sptbn1 heterozygous deletion (Sptbn1+/−) are more susceptible to tumor development (24). To better understand the homozygous deletion of SPTBN1 in human cancer, we evaluated copy number alterations in SPTBN1 in 30 different cancer types within TCGA data and found that homozygous loss of SPTBN1 is rare with an overall frequency of 0.1% across 30 cancer types, representing 33,039 cancer patients (fig. S7E). Because of the regulation between SREBP1 and SPTBN1, we also stratified HCC patients in TCGA by SREBP1 activity and found that increased SREBP1 target gene expression in HCC patients correlated with significantly reduced overall survival as well as disease-free survival (p < 0.05, fig. S7F). Thus, our results showing that LSKO mice (homozygous deletion of Sptnb1 in the liver) were protected against HCC are consistent with the lack of homozygous deletion of SPTBN1 in the TCGA human cancer database.
Discussion
Obesity and HFD are associated with fatty liver, hepatocyte death, and liver fibrosis (63–65). With the increase in obesity throughout the world, liver disease and HCC are also increasing (63). Treatments to prevent the progression from NAFLD to NASH and the development of HCC (66) are urgently needed. Collectively, our results indicate that SPTBN1 is a molecular target for therapeutic intervention both in NASH development and treatment. First, we found hepatocyte-specific loss of SPTBN1 prevents NASH development in response to 2 different diet-induced models in mice. Targeting SPTBN1 by siRNA in mice prevented HFD–induced obesity and the development of NASH. Finally, we showed the therapeutic effects of siSptbn1 on established NASH in mice. Mechanistically, our data indicated that these effects related to reducing SPTBN1-mediated promotion of SREBP activity induced by CASPASE-3 in response to stress conditions caused by HFD (fig. S8). These findings can lead to potential therapeutic options for NASH and HCC prevention strategies in patient populations susceptible to progression. Indeed, through single-nucleus analysis, we identified subsets of hepatocytes with increased SPTBN1 expression, indicating that patients with NASH and HCC and this biomarker could benefit from targeting SPTBN1. Our results with a human 3D NASH model strongly supported the beneficial effects of siRNA targeting of SPTBN1, which reversed the NASH transcriptomic signature. Additionally, our results provide a mechanism for the predictive association between increased plasma concentrations of cytokeratin-18, which is cleaved by activated hepatocyte CASPASE-3, and NASH in human subjects (67, 68).
In particular, we found that the LSKO mice fed a HFD were protected from development of obesity and NAFLD and progression to NASH, as well as the development of HCC. Furthermore, treatment of mice fed a HFD with siRNA targeting Sptbn1 also protected the mice from obesity and NAFLD/NASH. Not only was the expression of pro-fibrotic TGF-β-SMAD3-regulated genes reduced by loss of SPTBN1 function, but also SRE-regulated genes involved in de novo lipogenesis and responsive to SREBP1 were decreased in expression. We determined that, in stressed hepatocytes or hepatocytes cultured with high amounts of PA, CASPASE-3 cleaved both SREBP1 and SPTBN1 and that the cleaved products interacted. We proposed that the interaction between the cleaved products stabilized the nuclear form of SREBP1, thereby promoting de novo lipogenesis. We cannot rule out an effect of cleaved SPTBN1 on the recruitment of nSREBP1 to target gene promoters or the interaction with other transcriptional regulators or with ubiquitin ligases. Those potential effects remain for future study.
Our results placed SPTBN1 as a central player in both steatosis and fibrosis in response to HFD: As a participant in TGF-β/SMAD3 signaling, SPTBN1 promotes fibrosis and, as a participant in stress-activated SREBP1 signaling, SPTBN1 promotes de novo lipogenesis and steatosis. With regard to HCC, the effect of SPTBN1 on the liver may be dose dependent or may involve cells other than hepatocytes. Studies of mice with global heterozygosity of Sptbn1 exhibit increased inflammatory signaling (69) and increased susceptibility to HCC through effects on cyclin-dependent kinase 4 (70), genome stability (71), and β-catenin activity (72). Encouragingly, hepatocyte-specific absence of SPTBN1 had no obvious detrimental effects on liver morphology or function, and siRNA targeting Sptbn1 was an effective therapeutic in a preclinical mouse NASH model and reduced HCC development in a chemically induced mouse model.
Additionally, acetaminophen is a liver-toxic, commonly used over-the-counter medication (73). Low doses of acetaminophen induce phosphorylation of TGFBR2 and SMAD signaling with higher toxic doses promoting caspase activation and leading to production of caspase-cleaved SPTBN1 and severe hepatotoxicity (28). Our data suggest that targeting SPTBN1 could be a useful therapy to prevent acetaminophen liver toxicity, although we did not investigate this in this study.
Our biochemical data provide a mechanistic link between nonapoptotic CASPASE-3 activity (74, 75) and de novo lipogenesis through formation of nSREBPs and stabilization of nSREBP1 by interaction with caspase-cleaved N-SPTBN1. This provides a second pathway, in addition to ER stress-induced activation of SREBPs, for aberrant activation of de novo lipogenesis that results in steatosis and development of NAFLD (fig. S8). Our data suggest that caspase-mediated activation of SREBP1 is independent of changes in SCAP and INSIG, the ER-localized regulators of SREBP activation, further supporting previous findings by others (64, 76); thereby bypassing the normal controls that limit de novo lipogenesis under conditions of sufficient or excess lipids.
Our result that siSptbn1 ameliorated NASH without affecting body weight suggests that loss of SPTBN1 protects mice against HFD-induced NASH mainly through body weight-independent effects, largely through SREBP1-dependent effects. A limitation of our study is that the mechanism by which LSKO mice are protected from weight gain induced by HFD remains a puzzle. Our data suggested involvement of an increased metabolism that is linked with increased body temperature, and increased expression of genes involved in fatty acid oxidation and energy expenditure (Ctp1a and Fgf21, respectively). Another limitation is that the therapeutic analysis was performed with mice that had liver-specific knockout of SPTBN1 in hepatocytes from birth, which could have produced adaptive changes that differ from the effects of an inducible system. Finally, our study also does not include analysis of potential therapeutic benefit in human subjects with NAFLD or NASH.
Future studies will explore the link between SPTBN1 and other aspects of metabolism, especially that outside of liver tissues. Other areas of ongoing investigation include evaluation of SPTBN1 depletion in reversing steatosis and continued exploration into the crosstalk between TGF-β signaling and de novo lipogenesis. Because some siRNA therapies are approved for diseases of the liver (77), we anticipate that our preclinical findings in mice and human 3D culture NASH model system can be translated for clinical application to help prevent the progression of NAFLD to NASH in patients.
Materials and Methods
Study design
The goal of this study was to evaluate the role of SPTBN1 specifically in the liver and determine its roles in diet-induced liver disease and HCC. We used hepatocyte-specific knockout of SPTBN1 in mice exposed to a normal diet, a HFD, or a Western diet and evaluated the effect on the development of NAFLD, NASH, and HCC. We also evaluated the effects of siRNA-targeted therapy (siSPTBN1) on diet-induced NAFLD/NASH mouse models. The relevance to human disease was determined with a human 3D-NASH culture model, analysis of HCC data in TCGA, and evaluation of NASH, NAFLD, and HCC patient liver tissues or gene expression data. Molecular mechanisms of the functional effects of SPTBN1 on de novo lipogenesis were examined by RNA-seq analysis and bioinformatic-based molecular pathway analysis in liver tissues as well as in the 3D culture model. Interactions and binding sites between SPTBN1 and SREBP1 were examined by biochemical studies in cultured cells. All experimental data were evaluated in two or more individual biological replicates. Sample sizes, biological replicates, and statistical methods are provided in the corresponding figure legends.
Study population (patients with NASH)
The Cambridge cohort consisted of 58 NAFLD patients (NAFL: 19; NASH F0–2: 24; NASH F3–4: 15) recruited at NASH Service at the Cambridge University Hospital. All the patients had a clinical and biopsy-proven diagnosis of NAFLD (patients with alternate diagnoses and fatty liver from different etiologies were excluded), histology scored by a trained human pathologist according to the NASH CRN Scoring System (NAS1), and snap-frozen tissue for research purposes (Gene Expression by Next Generations Sequencing, see below). All the comparisons have been carried out against the NAFLD group. This study was approved by the local Ethics Committee; the principles of the Declaration of Helsinki were followed. All patients gave their informed consent for the use of clinical/omics data and samples for research purposes.
Single-nucleus RNA-seq of human liver
The study was approved by the Institutional Review Board (#20–001319) of the University of California, Los Angeles. All participants provided informed consent and the study was approved by the local ethics committee. Archived liver biopsies were obtained from tumor and adjacent non-tumor tissue at the time of surgical resection in three patients with hepatocellular carcinoma (HCC). The samples were snap-frozen, placed in an Optimal Cutting Temperature compound, and kept at −80°C until nuclei extraction. Clinical characteristics of these patients are presented in table S3. Frozen biopsies were then processed for nuclei isolation and single-nucleus RNA-seq (snRNA-seq). Briefly, frozen biopsies were homogenized in ice-cold lysis buffer consisting of 0.1% IGEPAL, 10 mM Tris-HCl, 10 mM NaCl, and 3 mM MgCl2. After a 10 minute incubation period, the lysate was gently homogenized using a Dounce homogenizer and filtered through a 70 μm MACS smart strainer (Miltenyi Biotec #130-098-462) to remove debris. Nuclei were centrifuged at 500 × g for 5 minutes at 4°C and resuspended in a wash buffer consisting of 1X PBS, 1.0% BSA, and 0.2 U/μl RNase inhibitor. We further filtered nuclei using a 30 μm MACS smart strainer (Miltenyi Biotec #130-098-458) and centrifuged at 500 × g for 5 minutes at 4°C. Pelleted nuclei were resuspended in wash buffer and immediately processed with the 10X Chromium platform (78) following the Single Cell 3' v3 protocol. Libraries were then sequenced on the Illumina platform targeting 300–400 million reads per sample.
Before alignment, we trimmed template switch oligos, primers, and polyA sequences greater than 20 base pairs from the fastq reads using cutadapt: Reads were aligned to the GRCh38 human genome reference and Gencode (79) v26 gene annotations using STARSolo in STAR v2.7.3a (80). Gene counts were taken from the full pre-mRNA transcript using the “—soloFeatures GeneFull” option. Empty and contaminated droplets were filtered out using DIEM (81) where we further adapted estimation of the multinomial mixture model parameters by adding a prior count of 1 to the gene mean estimates and the cluster membership estimates to avoid overfitting. To further remove doublets and contaminated clusters, we separately clustered parenchymal hepatocytes and non-parenchymal nuclei. Clusters showing evidence of doublet markers were removed. The parenchymal and non-parenchymal nuclei were then re-clustered separately to identify cell subtypes with higher resolution. After cell type assignment, the parenchymal and non-parenchymal nuclei and their assignments were merged. All clustering was performed with Seurat v3 (82) using SCT normalization (83) and CCA integration using the top 3,000 variable genes and 30 principal components. Clustering was performed with the Seurat FindClusters function with the resolution parameter set to 1. For cell type marker and tumor/non-tumor differential expression, we normalized the counts by scaling the total droplet counts to 1,000, as this was the approximate median total across the six samples, and log-transformed. Differential expression was run with the Seurat FindMarkers function using a logistic regression test. Cell types were assigned based on known marker genes.
Mouse models and histology
All animal experiments were performed according to the guidelines for the care and use of laboratory animals and were approved by the Institutional Biomedical Research Ethics Committee of The George Washington University for Biomedical Research. C57BL/6 mice were purchased from The Jackson Laboratories and were engineered. Both male and female mice were used in our study. To generate liver-specific deletion of Sptbn1 mice, Flox sites were inserted into the flanks of exon 24 to 26 of Sptbn1 gene locus, with Neo cassette. Neo cassettes were then removed by intercrossing with Flp mice. Sptbn1-Flox mice were then intercrossed with Albumin-Cre to generate liver-specific deletion of Sptbn1 mice. Blood glucose, TG, and cholesterol concentrations were measured by a glucometer, CardioChek PA analyzer, and PTS Panels Lipid Panel test strips (pts Diagnostics, #1710). For glucose tolerance testing, mice were fasted overnight and measure the concentration of blood glucose before glucose injection as time 0. After intraperitoneal injection of 2 g/kg of glucose, blood glucose concentrations were measure at 15, 30, 60, 90, 120 min after injection. An infrared Thermometer (Animal Care Equipment & Services, #TEMP-1) was used to measure the resting body temperature of mice and at least 3 times measurements were performed in each mouse. We measured food intake by weight difference, water intake, and urine amount by volume and weight for 24h by housing a single mouse in a metabolic cage. Liver tissues were excised and weighed. Routine tissue processing and histologic techniques were used for tissue sections stained with H&E and Sirius red and for frozen sections stained with Oil-Red-O. Liver triglycerides were quantified by colorimetric measurements using a Triglyceride Quantification kit (BioVision #K622) according to the manufacturer's instructions. Colorimetric measurements were performed at 570 nm using a Biotek Synergy 2 plate reader and liver TG concentrations were normalized against protein concentration.
For HFD-induced liver steatosis, 10- to 12-week-old male and female mice were fed the control diet or HFD (ENVIGO, Cat. TD.06414) for 12 to 28 weeks. For the Western- diet (WD)–induced NASH model, 10- to 12-week-old male and female Flox mice or LSKO mice were fed a diet containing 42% kcal from fat (Harlan TD.88137) together with a high fructose-glucose solution (SW, 23.1 g/L d-fructose +18.9 g/L d-glucose) for 12 to 20 weeks (30). For DEN-induced liver cancer models, 25 mg/kg DEN was injected in 14 – 15 day old male and female mice. Tumor development was analyzed 6 – 10 months later. For DEN plus WD diet-induced NASH associated HCC, 10 – 12 week old male mice were injected weekly with 50 mg/kg DEN for two consecutive weeks followed by a one week break for a total of 6 cycles over a total of 18 weeks. WD was started 1 week after the first DEN injection and animals received WD for a total of 21 – 22 weeks.
Cell lines and cell culture
Sptbn1−/−, Sptbn1+/−, and WT MEFs were generated from Sptbn1 intercrossed mice. Human liver cancer cell lines, HepG2, Huh7, and Hep3B, as well as the mouse immortalized liver cell line, AML12, were purchased from ATCC and cultured in a complete culture medium, DMEM/F12 medium (Corning, Cat. 10–090-CV) supplemented with 1% Streptomycin-Penicillin and 10% Fetal Bovine Serum (FBS) (Hyclone, Cat. SH30396.03). The human immortalized liver cell line THLE-2, purchased from ATCC, was cultured in BEGM medium (Lonza/Clonetics Corporation, Cat. CC3170) supplemented with 10% FBS (Sigma-Aldrich, Cat. F2442), 40 ug phosphor-ethanolamine (Sigma-Aldrich, Cat. P0503), and 3 ug human recombinant EGF (Corning, Cat. 354052) in addition to BPE (Bovine Pituitary Extract), hydrocortisone, human recombinant EGF (Corning, Cat. 354052), insulin, triiodothyronine, transferrin, and retinoic acid from the BEGM culture medium kit (Lonza/Clonetics Corporation, Cat. CC3170). THLE-2 cells were cultured in collagen-coated flasks (Cat. 132707) or plates pre-coated with a mixture of 0.01 mg/ml fibronectin and 0.01 mg/ml bovine serum albumin.
Targeting SPTBN1 by siRNA in mice and human 3D NASH model
For hydrodynamic injection of siRNA to mouse tails, siSptbn1 and siCtrl were resuspended in Nuclease-free Water (Life Technologies) to 0.25 mM, and incubated with RNAiMAX transfection reagent (Invitrogen) at a 1:1 ratio at room temperature for 20 minutes. The complexes were diluted in TransIT-QR hydrodynamic delivery solution (Mirus to 0.625 μM. Mice received 2.0 mL of diluted siSptbn1 (1.25 nmol) or siCtrl through hydrodynamic tail vein injection as previously described (84, 85). For the prevention experiment, 1 week after HFD, mice received the tail vein injection of siRNA 3 times with two weeks intervals. The experiment proceeded for 12 weeks total under HFD. For the therapeutic experiment, mice were on 12 weeks of WD containing 42% kcal from fat (Harlan TD.88137) together with a high fructose-glucose solution [23.1 g/L d-fructose (Sigma) +18.9 g/L d-glucose (Sigma)] to induce NASH (30). Then these mice received the tail vein injection of siRNA 4 times with two-week intervals; the experiment proceeded for 22 weeks total under the WD diet and fructose-glucose drinking.
For the human 3D NASH model, a perfused microphysical system composed of primary human hepatocytes, hepatic Kupffer cells, and stellate cells was used. The cells were co-cultured in the PhysioMimix OOC platform (CN-Bio Innovations) in a medium enriched in fatty acids, sugars, and insulin to mimic NASH conditions. After 4 days of culture in NASH conditions, the cells were transfected with siSPTBN1 and siCtrl in a 1:3 ratio with RNAiMAX transfection reagent (Invitrogen). RT-PCR and RNA sequencing were performed on samples taken after 8 days in NASH conditions, and lipid accumulation was measured using Oil Red O and normalized to a total protein on samples taken at day 8 and day 13.
Statistics
Differences between 2 groups were evaluated using 2-tailed Student’s t-tests using GraphPad Prism. We performed a pairwise comparison for body weight gain measured weekly. For multiple comparisons, one-way ANOVA with posthoc Bonferroni’s test was used. In vitro experiments were performed 2 – 4 times. All luciferase experiments were performed in triplicate. Results are expressed as mean ± SEM unless otherwise indicated. For all statistical analyses, p < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments:
We thank Rong Li, Robert Miller, Nancy R. Gough, Anton Sidawy, Tomasz Kostrzewski (CN-Bio Innovations) for critical review of the manuscript; Nancy R. Gough for editing the manuscript; Ka Bian for technical support with animal studies and Jameson McShea for the technical support for the ki67 labeling.
Funding:
This research was supported by the National Institute of Health grants R01AA023146 (L. Mishra), R01CA236591-01 (L. Mishra), U01 CA230690-01 (L. Mishra), I01BX003732 (L. Mishra), Elaine H. Snyder Cancer Research Award (L. Mishra). Sn-RNA seq was supported by American Association of the Study of Liver Diseases (AASLD) Advanced Transplant Hepatology award; NIH/NIDDK (P30DK41301) Pilot and Feasibility award (JN Benhammou).
Footnotes
Competing interests: S.R., K.O., S.Z., B-N.N., B.M. and L.M. have a patent pending related to this work (application #: 63/147,141; Title: β-SPECTRIN (SPTBN1) DEFICIENCY PROTECTS MICE FROM HIGH-FAT DIET-INDUCED LIVER DISEASE AND CANCER DEVELOPMENT). All other authors declare no competing interests.
Data and materials availability:
All data associated with this study are present in the paper or the Supplementary Materials. snRNA seq data are available upon request.
References:
- 1.Anstee QM, Reeves HL, Kotsiliti E, Govaere O, Heikenwalder M, From NASH to HCC: current concepts and future challenges. Nat Rev Gastroenterol Hepatol 16, 411–428 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Liu Z, Suo C, Mao X, Jiang Y, Jin L, Zhang T, Chen X, Global incidence trends in primary liver cancer by age at diagnosis, sex, region, and etiology, 1990–2017. Cancer 126, 2267–2278 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Yarchoan M, Agarwal P, Villanueva A, Rao S, Dawson LA, Llovet JM, Finn RS, Groopman JD, El-Serag HB, Monga SP, Wang XW, Karin M, Schwartz RE, Tanabe KK, Roberts LR, Gunaratne PH, Tsung A, Brown KA, Lawrence TS, Salem R, Singal AG, Kim AK, Rabiee A, Resar L, Hoshida Y, He AR, Ghoshal K, Ryan PB, Jaffee EM, Guha C, Mishra L, Coleman CN, Ahmed MM, Recent Developments and Therapeutic Strategies against Hepatocellular Carcinoma. Cancer Res 79, 4326–4330 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, Harrison SA, Brunt EM, Sanyal AJ, The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 67, 328–357 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ, Mechanisms of NAFLD development and therapeutic strategies. Nat Med 24, 908–922 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cotter TG, Rinella M, Nonalcoholic Fatty Liver Disease 2020: The State of the Disease. Gastroenterology 158, 1851–1864 (2020). [DOI] [PubMed] [Google Scholar]
- 7.Pedrosa M, Seyedkazemi S, Francque S, Sanyal A, Rinella M, Charlton M, Loomba R, Ratziu V, Kochuparampil J, Fischer L, Vaidyanathan S, Anstee QM, A randomized, double-blind, multicenter, phase 2b study to evaluate the safety and efficacy of a combination of tropifexor and cenicriviroc in patients with nonalcoholic steatohepatitis and liver fibrosis: Study design of the TANDEM trial. Contemp Clin Trials 88, 105889 (2020). [DOI] [PubMed] [Google Scholar]
- 8.Kim JY, Garcia-Carbonell R, Yamachika S, Zhao P, Dhar D, Loomba R, Kaufman RJ, Saltiel AR, Karin M, ER Stress Drives Lipogenesis and Steatohepatitis via Caspase-2 Activation of S1P. Cell 175, 133–145 e115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao P, Sun X, Chaggan C, Liao Z, In Wong K, He F, Singh S, Loomba R, Karin M, Witztum JL, Saltiel AR, An AMPK-caspase-6 axis controls liver damage in nonalcoholic steatohepatitis. Science 367, 652–660 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shimano H, Sato R, SREBP-regulated lipid metabolism: convergent physiology - divergent pathophysiology. Nat Rev Endocrinol 13, 710–730 (2017). [DOI] [PubMed] [Google Scholar]
- 11.DeBose-Boyd RA, Brown MS, Li WP, Nohturfft A, Goldstein JL, Espenshade PJ, Transport-dependent proteolysis of SREBP: relocation of site-1 protease from Golgi to ER obviates the need for SREBP transport to Golgi. Cell 99, 703–712 (1999). [DOI] [PubMed] [Google Scholar]
- 12.Wang X, Zelenski NG, Yang J, Sakai J, Brown MS, Goldstein JL, Cleavage of sterol regulatory element binding proteins (SREBPs) by CPP32 during apoptosis. EMBO J 15, 1012–1020 (1996). [PMC free article] [PubMed] [Google Scholar]
- 13.Ekstedt M, Hagstrom H, Nasr P, Fredrikson M, Stal P, Kechagias S, Hultcrantz R, Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology 61, 1547–1554 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Taylor RS, Taylor RJ, Bayliss S, Hagstrom H, Nasr P, Schattenberg JM, Ishigami M, Toyoda H, Wai-Sun Wong V, Peleg N, Shlomai A, Sebastiani G, Seko Y, Bhala N, Younossi ZM, Anstee QM, McPherson S, Newsome PN, Association Between Fibrosis Stage and Outcomes of Patients With Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Gastroenterology 158, 1611–1625 e1612 (2020). [DOI] [PubMed] [Google Scholar]
- 15.Sanderson N, Factor V, Nagy P, Kopp J, Kondaiah P, Wakefield L, Roberts AB, Sporn MB, Thorgeirsson SS, Hepatic expression of mature transforming growth factor beta 1 in transgenic mice results in multiple tissue lesions. Proc Natl Acad Sci U S A 92, 2572–2576 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Flanders KC, Smad3 as a mediator of the fibrotic response. Int J Exp Pathol 85, 47–64 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bissell DM, Roulot D, George J, Transforming growth factor beta and the liver. Hepatology 34, 859–867 (2001). [DOI] [PubMed] [Google Scholar]
- 18.Yadav H, Quijano C, Kamaraju AK, Gavrilova O, Malek R, Chen W, Zerfas P, Zhigang D, Wright EC, Stuelten C, Sun P, Lonning S, Skarulis M, Sumner AE, Finkel T, Rane SG, Protection from obesity and diabetes by blockade of TGF-beta/Smad3 signaling. Cell Metab 14, 67–79 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Majumdar A, Curley SA, Wu X, Brown P, Hwang JP, Shetty K, Yao ZX, He AR, Li S, Katz L, Farci P, Mishra L, Hepatic stem cells and transforming growth factor beta in hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol 9, 530–538 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bennett V, Spectrin-based membrane skeleton: a multipotential adaptor between plasma membrane and cytoplasm. Physiol Rev 70, 1029–1065 (1990). [DOI] [PubMed] [Google Scholar]
- 21.Bennett V, Lorenzo DN, An Adaptable Spectrin/Ankyrin-Based Mechanism for Long-Range Organization of Plasma Membranes in Vertebrate Tissues. Curr Top Membr 77, 143–184 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Viel A, Branton D, Spectrin: on the path from structure to function. Curr Opin Cell Biol 8, 49–55 (1996). [DOI] [PubMed] [Google Scholar]
- 23.Tang Y, Katuri V, Dillner A, Mishra B, Deng CX, Mishra L, Disruption of transforming growth factor-beta signaling in ELF beta-spectrin-deficient mice. Science 299, 574–577 (2003). [DOI] [PubMed] [Google Scholar]
- 24.Chen J, Yao ZX, Chen JS, Gi YJ, Munoz NM, Kundra S, Herlong HF, Jeong YS, Goltsov A, Ohshiro K, Mistry NA, Zhang J, Su X, Choufani S, Mitra A, Li S, Mishra B, White J, Rashid A, Wang AY, Javle M, Davila M, Michaely P, Weksberg R, Hofstetter WL, Finegold MJ, Shay JW, Machida K, Tsukamoto H, Mishra L, TGF-beta/beta2-spectrin/CTCF-regulated tumor suppression in human stem cell disorder Beckwith-Wiedemann syndrome. J Clin Invest 126, 527–542 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen J, Zaidi S, Rao S, Chen JS, Phan L, Farci P, Su X, Shetty K, White J, Zamboni F, Wu X, Rashid A, Pattabiraman N, Mazumder R, Horvath A, Wu RC, Li S, Xiao C, Deng CX, Wheeler DA, Mishra B, Akbani R, Mishra L, Analysis of Genomes and Transcriptomes of Hepatocellular Carcinomas Identifies Mutations and Gene Expression Changes in the Transforming Growth Factor-beta Pathway. Gastroenterology 154, 195–210 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Matteis MA, Morrow JS, Spectrin tethers and mesh in the biosynthetic pathway. J Cell Sci 113 ( Pt 13), 2331–2343 (2000). [DOI] [PubMed] [Google Scholar]
- 27.Bennett V, Lorenzo DN, Spectrin- and ankyrin-based membrane domains and the evolution of vertebrates. Curr Top Membr 72, 1–37 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Baek HJ, Lee YM, Kim TH, Kim JY, Park EJ, Iwabuchi K, Mishra L, Kim SS, Caspase-3/7-mediated Cleavage of beta2-spectrin is Required for Acetaminophen-induced Liver Damage. Int J Biol Sci 12, 172–183 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Korkut A, Zaidi S, Kanchi RS, Rao S, Gough NR, Schultz A, Li X, Lorenzi PL, Berger AC, Robertson G, Kwong LN, Datto M, Roszik J, Ling S, Ravikumar V, Manyam G, Rao A, Shelley S, Liu Y, Ju Z, Hansel D, de Velasco G, Pennathur A, Andersen JB, O'Rourke CJ, Ohshiro K, Jogunoori W, Nguyen BN, Li S, Osmanbeyoglu HU, Ajani JA, Mani SA, Houseman A, Wiznerowicz M, Chen J, Gu S, Ma W, Zhang J, Tong P, Cherniack AD, Deng C, Resar L, Weinstein JN, Mishra L, Akbani R, A Pan-Cancer Analysis Reveals High-Frequency Genetic Alterations in Mediators of Signaling by the TGF-beta Superfamily. Cell Syst 7, 422–437 e427 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Asgharpour A, Cazanave SC, Pacana T, Seneshaw M, Vincent R, Banini BA, Kumar DP, Daita K, Min HK, Mirshahi F, Bedossa P, Sun X, Hoshida Y, Koduru SV, Contaifer D Jr., Warncke UO, Wijesinghe DS, Sanyal AJ, A diet-induced animal model of non-alcoholic fatty liver disease and hepatocellular cancer. J Hepatol 65, 579–588 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee JH, Mellado-Gil JM, Bahn YJ, Pathy SM, Zhang YE, Rane SG, Protection from beta-cell apoptosis by inhibition of TGF-beta/Smad3 signaling. Cell Death Dis 11, 184 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tan CK, Leuenberger N, Tan MJ, Yan YW, Chen Y, Kambadur R, Wahli W, Tan NS, Smad3 deficiency in mice protects against insulin resistance and obesity induced by a high-fat diet. Diabetes 60, 464–476 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gutierrez-Cuevas J, Sandoval-Rodriguez A, Monroy-Ramirez HC, Vazquez-Del Mercado M, Santos-Garcia A, Armendariz-Borunda J, Prolonged-release pirfenidone prevents obesity-induced cardiac steatosis and fibrosis in a mouse NASH model. Cardiovasc Drugs Ther, (2020). [DOI] [PubMed] [Google Scholar]
- 34.Sandoval-Rodriguez A, Monroy-Ramirez HC, Meza-Rios A, Garcia-Banuelos J, Vera-Cruz J, Gutierrez-Cuevas J, Silva-Gomez J, Staels B, Dominguez-Rosales J, Galicia-Moreno M, Vazquez-Del Mercado M, Navarro-Partida J, Santos-Garcia A, Armendariz-Borunda J, Pirfenidone Is an Agonistic Ligand for PPARalpha and Improves NASH by Activation of SIRT1/LKB1/pAMPK. Hepatol Commun 4, 434–449 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Valdecantos MP, Pardo V, Ruiz L, Castro-Sanchez L, Lanzon B, Fernandez-Millan E, Garcia-Monzon C, Arroba AI, Gonzalez-Rodriguez A, Escriva F, Alvarez C, Ruperez FJ, Barbas C, Konkar A, Naylor J, Hornigold D, Santos AD, Bednarek M, Grimsby J, Rondinone CM, Valverde AM, A novel glucagon-like peptide 1/glucagon receptor dual agonist improves steatohepatitis and liver regeneration in mice. Hepatology 65, 950–968 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Liang W, Menke AL, Driessen A, Koek GH, Lindeman JH, Stoop R, Havekes LM, Kleemann R, van den Hoek AM, Establishment of a general NAFLD scoring system for rodent models and comparison to human liver pathology. PLoS One 9, e115922 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lefterova MI, Zhang Y, Steger DJ, Schupp M, Schug J, Cristancho A, Feng D, Zhuo D, Stoeckert CJ Jr., Liu XS, Lazar MA, PPARgamma and C/EBP factors orchestrate adipocyte biology via adjacent binding on a genome-wide scale. Genes Dev 22, 2941–2952 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Serviddio G, Bellanti F, Tamborra R, Rollo T, Capitanio N, Romano AD, Sastre J, Vendemiale G, Altomare E, Uncoupling protein-2 (UCP2) induces mitochondrial proton leak and increases susceptibility of non-alcoholic steatohepatitis (NASH) liver to ischaemia-reperfusion injury. Gut 57, 957–965 (2008). [DOI] [PubMed] [Google Scholar]
- 39.Kharitonenkov A, Shiyanova TL, Koester A, Ford AM, Micanovic R, Galbreath EJ, Sandusky GE, Hammond LJ, Moyers JS, Owens RA, Gromada J, Brozinick JT, Hawkins ED, Wroblewski VJ, Li DS, Mehrbod F, Jaskunas SR, Shanafelt AB, FGF-21 as a novel metabolic regulator. J Clin Invest 115, 1627–1635 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Assadi A, Zahabi A, Hart RA, GDF15, an update of the physiological and pathological roles it plays: a review. Pflugers Arch 472, 1535–1546 (2020). [DOI] [PubMed] [Google Scholar]
- 41.Yoshikawa T, Shimano H, Amemiya-Kudo M, Yahagi N, Hasty AH, Matsuzaka T, Okazaki H, Tamura Y, Iizuka Y, Ohashi K, Osuga J, Harada K, Gotoda T, Kimura S, Ishibashi S, Yamada N, Identification of liver X receptor-retinoid X receptor as an activator of the sterol regulatory element-binding protein 1c gene promoter. Mol Cell Biol 21, 2991–3000 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yokoyama C, Wang X, Briggs MR, Admon A, Wu J, Hua X, Goldstein JL, Brown MS, SREBP-1, a basic-helix-loop-helix-leucine zipper protein that controls transcription of the low density lipoprotein receptor gene. Cell 75, 187–197 (1993). [PubMed] [Google Scholar]
- 43.Seo YK, Chong HK, Infante AM, Im SS, Xie X, Osborne TF, Genome-wide analysis of SREBP-1 binding in mouse liver chromatin reveals a preference for promoter proximal binding to a new motif. Proc Natl Acad Sci U S A 106, 13765–13769 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bertolio R, Napoletano F, Mano M, Maurer-Stroh S, Fantuz M, Zannini A, Bicciato S, Sorrentino G, Del Sal G, Sterol regulatory element binding protein 1 couples mechanical cues and lipid metabolism. Nat Commun 10, 1326 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brown MS, Goldstein JL, The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 89, 331–340 (1997). [DOI] [PubMed] [Google Scholar]
- 46.Horton JD, Goldstein JL, Brown MS, SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 109, 1125–1131 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brown MS, Goldstein JL, A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood. Proc Natl Acad Sci U S A 96, 11041–11048 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Knebel B, Haas J, Hartwig S, Jacob S, Kollmer C, Nitzgen U, Muller-Wieland D, Kotzka J, Liver-specific expression of transcriptionally active SREBP1c is associated with fatty liver and increased visceral fat mass. PLoS One 7, e31812 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kozakov D, Hall DR, Xia B, Porter KA, Padhorny D, Yueh C, Beglov D, Vajda S, The ClusPro web server for protein–protein docking. Nature protocols 12, 255 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kozakov D, Brenke R, Comeau SR, Vajda S, PIPER: an FFT‐based protein docking program with pairwise potentials. Proteins: Structure, Function, and Bioinformatics 65, 392–406 (2006). [DOI] [PubMed] [Google Scholar]
- 51.Ghisleni A, Galli C, Monzo P, Ascione F, Fardin MA, Scita G, Li Q, Maiuri P, Gauthier NC, Complementary mesoscale dynamics of spectrin and acto-myosin shape membrane territories during mechanoresponse. Nat Commun 11, 5108 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ricchi M, Odoardi MR, Carulli L, Anzivino C, Ballestri S, Pinetti A, Fantoni LI, Marra F, Bertolotti M, Banni S, Lonardo A, Carulli N, Loria P, Differential effect of oleic and palmitic acid on lipid accumulation and apoptosis in cultured hepatocytes. J Gastroenterol Hepatol 24, 830–840 (2009). [DOI] [PubMed] [Google Scholar]
- 53.Urbanik T, Kohler BC, Boger RJ, Worns MA, Heeger S, Otto G, Hovelmeyer N, Galle PR, Schuchmann M, Waisman A, Schulze-Bergkamen H, Down-regulation of CYLD as a trigger for NF-kappaB activation and a mechanism of apoptotic resistance in hepatocellular carcinoma cells. Int J Oncol 38, 121–131 (2011). [PubMed] [Google Scholar]
- 54.Tomita K, Tamiya G, Ando S, Ohsumi K, Chiyo T, Mizutani A, Kitamura N, Toda K, Kaneko T, Horie Y, Han JY, Kato S, Shimoda M, Oike Y, Tomizawa M, Makino S, Ohkura T, Saito H, Kumagai N, Nagata H, Ishii H, Hibi T, Tumour necrosis factor alpha signalling through activation of Kupffer cells plays an essential role in liver fibrosis of non-alcoholic steatohepatitis in mice. Gut 55, 415–424 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wandrer F, Liebig S, Marhenke S, Vogel A, John K, Manns MP, Teufel A, Itzel T, Longerich T, Maier O, Fischer R, Kontermann RE, Pfizenmaier K, Schulze-Osthoff K, Bantel H, TNF-Receptor-1 inhibition reduces liver steatosis, hepatocellular injury and fibrosis in NAFLD mice. Cell Death Dis 11, 212 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Calzadilla Bertot L, Adams LA, The Natural Course of Non-Alcoholic Fatty Liver Disease. Int J Mol Sci 17, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Singh S, Allen AM, Wang Z, Prokop LJ, Murad MH, Loomba R, Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: a systematic review and meta-analysis of paired-biopsy studies. Clin Gastroenterol Hepatol 13, 643–654 e641–649; quiz e639–640 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Balwani M, Sardh E, Ventura P, Peiro PA, Rees DC, Stolzel U, Bissell DM, Bonkovsky HL, Windyga J, Anderson KE, Parker C, Silver SM, Keel SB, Wang JD, Stein PE, Harper P, Vassiliou D, Wang B, Phillips J, Ivanova A, Langendonk JG, Kauppinen R, Minder E, Horie Y, Penz C, Chen J, Liu S, Ko JJ, Sweetser MT, Garg P, Vaishnaw A, Kim JB, Simon AR, Gouya L, Investigators E, Phase 3 Trial of RNAi Therapeutic Givosiran for Acute Intermittent Porphyria. N Engl J Med 382, 2289–2301 (2020). [DOI] [PubMed] [Google Scholar]
- 59.Kostrzewski T, Cornforth T, Snow SA, Ouro-Gnao L, Rowe C, Large EM, Hughes DJ, Three-dimensional perfused human in vitro model of non-alcoholic fatty liver disease. World J Gastroenterol 23, 204–215 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kostrzewski T, Maraver P, Ouro-Gnao L, Levi A, Snow S, Miedzik A, Rombouts K, Hughes D, A Microphysiological System for Studying Nonalcoholic Steatohepatitis. Hepatol Commun 4, 77–91 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jiang DK, Sun J, Cao G, Liu Y, Lin D, Gao YZ, Ren WH, Long XD, Zhang H, Ma XP, Wang Z, Jiang W, Chen TY, Gao Y, Sun LD, Long JR, Huang HX, Wang D, Yu H, Zhang P, Tang LS, Peng B, Cai H, Liu TT, Zhou P, Liu F, Lin X, Tao S, Wan B, Sai-Yin HX, Qin LX, Yin J, Liu L, Wu C, Pei Y, Zhou YF, Zhai Y, Lu PX, Tan A, Zuo XB, Fan J, Chang J, Gu X, Wang NJ, Li Y, Liu YK, Zhai K, Hu Z, Liu J, Yi Q, Xiang Y, Shi R, Ding Q, Zheng W, Shu XO, Mo Z, Shugart YY, Zhang XJ, Zhou G, Shen H, Zheng SL, Xu J, Yu L, Genetic variants in STAT4 and HLA-DQ genes confer risk of hepatitis B virus-related hepatocellular carcinoma. Nat Genet 45, 72–75 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li J, Liang L, Liu Y, Luo Y, Liang X, Luo D, Feng Z, Dang Y, Yang L, Chen G, Clinicopathological significance of STAT4 in hepatocellular carcinoma and its effect on cell growth and apoptosis. Onco Targets Ther 9, 1721–1734 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hassan MM, Abdel-Wahab R, Kaseb A, Shalaby A, Phan AT, El-Serag HB, Hawk E, Morris J, Singh Raghav KP, Lee JS, Vauthey JN, Bortus G, Torres HA, Amos CI, Wolff RA, Li D, Obesity Early in Adulthood Increases Risk but Does Not Affect Outcomes of Hepatocellular Carcinoma. Gastroenterology 149, 119–129 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Neuschwander-Tetri BA, Non-alcoholic fatty liver disease. BMC Med 15, 45 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sun B, Karin M, Obesity, inflammation, and liver cancer. J Hepatol 56, 704–713 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gough NR, Xiang X, Mishra L, TGF-β Signaling in Liver, Pancreas, and Gastrointestinal Diseases and Cancer. Gastroenterology 161, 434–452.e415 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cao W, Zhao C, Shen C, Wang Y, Cytokeratin 18, alanine aminotransferase, platelets and triglycerides predict the presence of nonalcoholic steatohepatitis. PLoS One 8, e82092 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Feldstein AE, Wieckowska A, Lopez AR, Liu YC, Zein NN, McCullough AJ, Cytokeratin-18 fragment levels as noninvasive biomarkers for nonalcoholic steatohepatitis: a multicenter validation study. Hepatology 50, 1072–1078 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lin L, Chen S, Wang H, Gao B, Kallakury B, Bhuvaneshwar K, Cahn K, Gusev Y, Wang X, Wu Y, Marshall JL, Zhi X, He AR, SPTBN1 inhibits inflammatory responses and hepatocarcinogenesis via the stabilization of SOCS1 and downregulation of p65 in hepatocellular carcinoma. Theranostics 11, 4232–4250 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Baek HJ, Pishvaian MJ, Tang Y, Kim TH, Yang S, Zouhairi ME, Mendelson J, Shetty K, Kallakury B, Berry DL, Shin KH, Mishra B, Reddy EP, Kim SS, Mishra L, Transforming growth factor-β adaptor, β2-spectrin, modulates cyclin dependent kinase 4 to reduce development of hepatocellular cancer. Hepatology 53, 1676–1684 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen J, Shukla V, Farci P, Andricovich J, Jogunoori W, Kwong LN, Katz LH, Shetty K, Rashid A, Su X, White J, Li L, Wang AY, Blechacz B, Raju GS, Davila M, Nguyen BN, Stroehlein JR, Chen J, Kim SS, Levin H, Machida K, Tsukamoto H, Michaely P, Tzatsos A, Mishra B, Amdur R, Mishra L, Loss of the transforming growth factor-β effector β2-Spectrin promotes genomic instability. Hepatology 65, 678–693 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen Y, Meng L, Shang H, Dou Q, Lu Z, Liu L, Wang Z, He X, Song Y, β2 spectrin-mediated differentiation repressed the properties of liver cancer stem cells through β-catenin. Cell Death Dis 9, 424 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Larson AM, Polson J, Fontana RJ, Davern TJ, Lalani E, Hynan LS, Reisch JS, Schiodt FV, Ostapowicz G, Shakil AO, Lee WM, Acute G Liver Failure Study, Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 42, 1364–1372 (2005). [DOI] [PubMed] [Google Scholar]
- 74.Aram L, Yacobi-Sharon K, Arama E, CDPs: caspase-dependent non-lethal cellular processes. Cell Death Differ 24, 1307–1310 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Akbari-Birgani S, Hosseinkhani S, Mollamohamadi S, Baharvand H, Delay in apoptosome formation attenuates apoptosis in mouse embryonic stem cell differentiation. J Biol Chem 289, 16905–16913 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yang T, Espenshade PJ, Wright ME, Yabe D, Gong Y, Aebersold R, Goldstein JL, Brown MS, Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell 110, 489–500 (2002). [DOI] [PubMed] [Google Scholar]
- 77.Hu B, Zhong L, Weng Y, Peng L, Huang Y, Zhao Y, Liang XJ, Therapeutic siRNA: state of the art. Signal Transduct Target Ther 5, 101 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zheng GX, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, Gregory MT, Shuga J, Montesclaros L, Underwood JG, Masquelier DA, Nishimura SY, Schnall-Levin M, Wyatt PW, Hindson CM, Bharadwaj R, Wong A, Ness KD, Beppu LW, Deeg HJ, McFarland C, Loeb KR, Valente WJ, Ericson NG, Stevens EA, Radich JP, Mikkelsen TS, Hindson BJ, Bielas JH, Massively parallel digital transcriptional profiling of single cells. Nat Commun 8, 14049 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, Barnes I, Bignell A, Boychenko V, Hunt T, Kay M, Mukherjee G, Rajan J, Despacio-Reyes G, Saunders G, Steward C, Harte R, Lin M, Howald C, Tanzer A, Derrien T, Chrast J, Walters N, Balasubramanian S, Pei B, Tress M, Rodriguez JM, Ezkurdia I, van Baren J, Brent M, Haussler D, Kellis M, Valencia A, Reymond A, Gerstein M, Guigo R, Hubbard TJ, GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res 22, 1760–1774 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR, STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Alvarez M, Rahmani E, Jew B, Garske KM, Miao Z, Benhammou JN, Ye CJ, Pisegna JR, Pietilainen KH, Halperin E, Pajukanta P, Enhancing droplet-based single-nucleus RNA-seq resolution using the semi-supervised machine learning classifier DIEM. Sci Rep 10, 11019 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, Satija R, Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 e1821 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hafemeister C, Satija R, Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol 20, 296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Song E, Lee SK, Wang J, Ince N, Ouyang N, Min J, Chen J, Shankar P, Lieberman J, RNA interference targeting Fas protects mice from fulminant hepatitis. Nat Med 9, 347–351 (2003). [DOI] [PubMed] [Google Scholar]
- 85.Tompkins SM, Lo CY, Tumpey TM, Epstein SL, Protection against lethal influenza virus challenge by RNA interference in vivo. Proc Natl Acad Sci U S A 101, 8682–8686 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ishibashi H, Suzuki T, Suzuki S, Moriya T, Kaneko C, Takizawa T, Sunamori M, Handa M, Kondo T, Sasano H, Sex steroid hormone receptors in human thymoma. J Clin Endocrinol Metab 88, 2309–2317 (2003). [DOI] [PubMed] [Google Scholar]
- 87.Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp-Arida A, Yeh M, McCullough AJ, Sanyal AJ, Nonalcoholic N Steatohepatitis Clinical Research, Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 41, 1313–1321 (2005). [DOI] [PubMed] [Google Scholar]
- 88.Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ, The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 10, 845–858 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fiser A, Sali A, ModLoop: automated modeling of loops in protein structures. Bioinformatics 19, 2500–2501 (2003). [DOI] [PubMed] [Google Scholar]
- 90.Guex N, Peitsch MC, SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18, 2714–2723 (1997). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper or the Supplementary Materials. snRNA seq data are available upon request.