

Abstract

Anionic 1,4-dihydro-1,4-diphosphinines were synthesized from tricyclic 1,4-diphosphinines and isolated as blue powdery salts M[2a–2c]. Reaction of solutions of these monoanions with iodomethane led to P-methylated compounds 3a–3c. An oxidation/reduction cycle was examined, starting from solutions of K[2a] via P–P coupled product 4a and back to K[2a], and the recyclability and redox chemistry of this cycle were confirmed by experimental and simulated cyclic voltammetry analysis, which is proposed as a potential 2-electron cathode for rechargeable cells. TD-DFT studies were used to examine species that might be involved in the process.
Short abstract
Synthesis of a set of stable P-anionic 1,4-dihydro-1,4-diphosphinines is described, including an oxidation/reduction cycle of one example, confirmed by experimental and simulated cyclic voltammetry analysis as well as detailed DFT studies. The results indicate that main group phosphorus compounds have unexplored potential for the development of new cathode materials.
Introduction
Since the landmark discovery of 2,4,6-triphenylphosphinine I by Märkl in 1966,1 its reactivity has been studied extensively.2−5 The electrophilic nature of the P center has been exploited to gain access to a variety of compounds, some of which via transformation of anionic derivatives II into III possess a four-coordinate P(V) center (Figure 1).6 A recent report by Müller et al. has also dealt with nucleophilic substitution using Grignard or organolithium reagents, resulting in λ4σ3-phosphinine anions.7 The reason for the selective reaction of (anionic) nucleophiles at the P atom is the large orbital coefficient of the low-lying LUMO.8 So far, the redox chemistry of λ5-phosphinines III has been investigated for optovoltaic applications in the design of organic light-emitting diodes (OLEDs).9 Similarly, PV-phospharhodamines have been extensively investigated for their photovoltaic applications.10 All these six-membered phosphorus heterocycles, mostly dominated by the PV-phospholes, share anodic photovoltaic behavior largely centered in the unsaturated hydrocarbon portion of the molecules with the P(V) centers uninvolved in redox processes.11−15
Figure 1.

First PIII-phosphinine I, anionic derivative II, PV-phosphinines III, first 1,4-diphosphinines IV–VI, and anionic 1,4-diphosphinines VII and VIII.
The chemistry of 1,4-diphosphinines started in 1976 with the first derivative IV, synthesized by Kobayashi et al.,16 but in contrast to I, it could not be isolated.17−19
The advent of stable tricyclic 1,4-diphosphinines fused to heterocyclic-2-thiones, such as V(20) and VI,21 has enabled systematic investigation of their chemistry.22−24 As one example, the reactivity of 1,4-diphosphinines V in [4 + 1] and [4 + 2] cycloaddition reactions has been reported, including unusual reactions with dichalcogenides.23,24 Additionally, the electrophilic P centers of V were explored by the addition of anionic nucleophiles, forming P-anionic species, which were not isolated but could be quenched with iodomethane to yield neutral P-Me substitution products.22 Using a thiazol-2-thione-based 1,4-diphosphinine, first access to an isolable anionic tricyclic 1,4-dihydro-1,4-diphosphinine derivative (VIII) was achieved, which could be oxidized by I2 to give a product with a P–P bond.21 Cyclic voltammetry studies on neutral V and VI disclosed a rich cathodic electrochemistry involving the PIII center, in contrast to λ5-phosphinines (vide supra).20,21
Beyond these rather intensively investigated five- and six-membered P-heterocycles, the redox chemistry of unsaturated three-membered rings, i.e., 4,5,6-triphospha[3]radialene, was studied too, which could be reduced to structurally confirmed dianions. Furthermore, cyclic voltammetry studies showed a reversible initial one-electron reduction and, on further scanning, revealed a second reversible redox couple, but no experiments aimed at demonstrating suitability as electrode materials were performed such as multicycle voltammograms, i.e., the robustness of the cycles was not proven.25−27
Apart from cyclic P-systems, (acyclic) diphosphenes can be easily reduced to form anionic radical species, but again, no further studies were performed.28 Electrochemical investigations of P–P (single) bond formation have been reported from both anodic and cathodic processes involving dimerization of phosphaalkene radical anions, cationic 1-phosphabutadiene radicals, or even R3P•+ centers, going back as far as Märkl et al.29−31 Other main group element redox systems that store energy in element–element bonds such as S–S bonds are the lithium–sulfur and sodium–sulfur storage batteries.32−35 Herein, we report chemical and voltammetric investigations of the anionic 1,4-diphosphinine derivatives VII including their conversion into P–P coupled products and, subsequently, the successful chemical reduction to reform type VII salts, thus closing the redox cycle (also) in solution.
Results and Discussion
When 1,4-diphosphinine 1(20) was treated with KHMDS in diethyl ether, a drastic color change from red to deep blue occurred. Evaporation of the solvent in vacuo yielded K[2a] as a blue-violet powder (Scheme 1). Application of the same protocol, but using LDA and KOtBu, afforded Li[2b] and K[2c] selectively, which, again, were obtained as blue-violet powders. The 31P{1H} NMR data of M[2a–2c] are presented in Table 1.
Scheme 1. Synthesis and Reaction of M[2a–2c] to give P-Methylated Products 3a–3c.

Table 1. 31P{1H} NMR Data of M[2a–2c] in Et2O-d10 (with and without the Presence of Crown Ethers), CH3CN, and THF (R-P and Anionic P Notations Are in Accordance with Scheme 1).
| δ 31P/ppm | ||||||||
|---|---|---|---|---|---|---|---|---|
|
R-P |
anionic P |
|||||||
| compound | Et2O | CH3CN | THF | Et2O′a | Et2O | CH3CN | THF | Et2O′a |
| K[2a] | –12.1 | –11.3 | –28.1 | –11.8 | –76.1 | –78.2 | –72.1 | –75.7 |
| Li[2b] | –30.8 | –31.8 | –32.2 | –30.6 | –78.0 | –79.4 | –79.5 | –77.6 |
| K[2c] | 18.1 | 11.6 | –28 | 18.3 | –74.1 | –70.1 | –72.1 | –74.3 |
Et2O′ indicates the ethereal solutions in the presence of crown ethers, 18-C-6 ([K(18-C-6)]2a and [K(18-C-6)]2c) or 12-C-4 ([Li(12-C-4)]2b).
The assignment of the two resonances to the P centers was straightforward, but it should be noted that in none of M[2a–2c] do the two inequivalent nuclei show evidence of P–P coupling across the rings. The composition of the product anions was also confirmed via negative ESI-MS experiments (Table S1 in the Supporting Information).
The intense blue colors observed for solids and solutions of M[2a–2c] correspond to single intense absorptions with λmax ≈ 517 nm in Et2O (Figures S5a, S9a, and S13a in the Supporting Information). By contrast, when K[2a] was dissolved in CH3CN for voltammetry (vide infra), the blue color rapidly changed to yellow (λmax ≈ 372 nm, Figure S5b in the Supporting Information).
To gain deeper insight into the electronic structures of 2a–2c–, DFT calculations at the M06-2X/6-311+G** level of theory were performed on models wherein the N-nBu groups are truncated to N-Me (indicated by ′). As expected, the middle rings of 2a′–2c′– exhibit lower aromatic character than in neutral 1a′ (NICS(0) values for 2a′–2c′– varied between −3.4 and −5.2; Tables S4, S8, and S12 in the Supporting Information), while the aromatic character of the outer ring remains high (NICS(0) varied between −8.8 and −9.4, Tables S4, S8, and S12 in the Supporting Information). The shapes and energies of the delocalized π frontier Kohn–Sham molecular orbitals (FMOs) are slightly affected by the variations in substituents (Figure 2). The HOMOs show large coefficients of the unsubstituted phosphorus on the center rings, but the sulfur p lone pairs have non-negligible contributions as well. On the electrostatic potential maps (Figure 2), the negative charges reside mainly on the unsubstituted phosphorus sites and the two sulfur atoms, in accordance with the HOMO coefficients. The TD-DFT calculations on 2a′– (maxima of the lowest excited states at 354 and 326 nm, Table 2) are in reasonably good agreement with the experimentally determined UV/vis absorption value in CH3CN but not with the deep blue colors observed in ether solvents (Et2O or THF). Presuming that the deep colors are from charge transfer (CT) bands associated with the formation of contact ion pairs due to the weaker solvation of K+ cations by ethers compared to CH3CN, additional TD-DFT calculations were performed on the optimized structures of the K+ salts (Table 2), which do indeed indicate transitions deep into the visible region. The charge transfer character of the transitions is clearly seen from the TD-DFT results. The lowest energy excitation of the calculated contact ion pair is the (anion-centered) HOMO-to-(K+ s-type centered) LUMO transition, indicative of CT. The similar 31P NMR shifts measured in Et2O and CH3CN (Table 1) and also the great similarity of the voltammetry results in THF (blue solutions) and CH3CN (yellow, vide infra) point to a small energy difference between the ether-solvated and ion-paired states.
Figure 2.
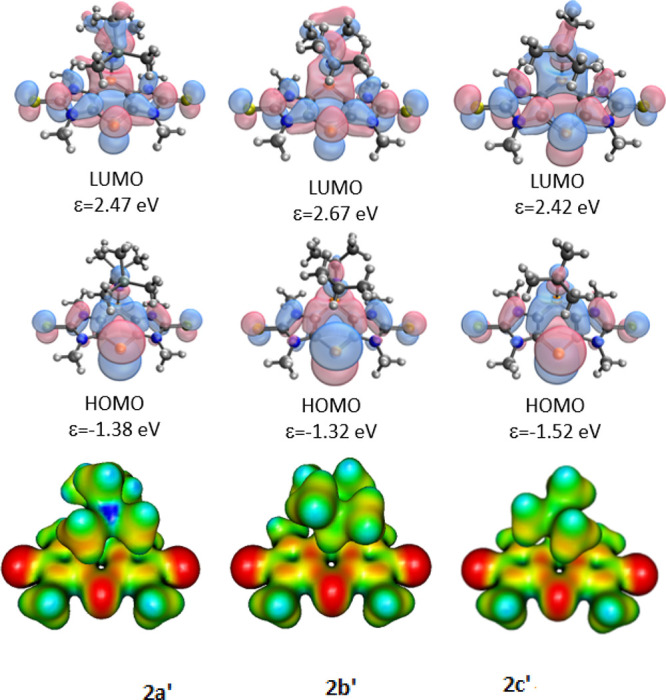
Kohn–Sham frontier orbitals, their energies (top), and electrostatic potential map (bottom; color code of electrostatic potential: red, <−0.1; yellow, −0.1 to −0.05; green, −0.05 to 0.05; light blue, 0.05–0.1; blue, >1.0).
Table 2. Important TD-DFT Results at the B3LYP/6-311G**//M06-2X/6-311+G** Level of Theory Calculated for 2a′–2c′– and the CAM-B3LYP/6-31G*//M06-2X/6-311+G** Level of Theory Calculated for the Contact Ion Pair M[2a′–2c′].
| model | excited state | wavelength | oscillator strength | transition | contribution |
|---|---|---|---|---|---|
| 2a′– | 1 | 354 nm | 0.1137 | HOMO-LUMO | 0.69524 |
| 2 | 326 nm | 0.1735 | HOMO-1-LUMO | 0.10945 | |
| HOMO-LUMO+1 | 0.22399 | ||||
| HOMO-LUMO+2 | 0.65285 | ||||
| K[2a′] | 1 | 523 nm | 0.0171 | HOMO-LUMO | 0.68460 |
| HOMO-LUMO+3 | 0.13519 | ||||
| 2b′– | 1 | 366 nm | 0.1501 | HOMO-LUMO | 0.69593 |
| 4 | 319 nm | 0.0774 | HOMO-LUMO+3 | 0.69343 | |
| Li[2b′] | 1 | 393 nm | 0.0231 | HOMO-4-LUMO | 0.15155 |
| HOMO-LUMO | 0.68258 | ||||
| 2c′– | 1 | 354 nm | 0.1235 | HOMO-LUMO | 0.69214 |
| 4 | 294 nm | 0.4978 | HOMO-1-LUMO | 0.65771 | |
| HOMO-LUMO+1 | 0.17021 | ||||
| HOMO-LUMO+2 | 0.12292 | ||||
| K[2c′] | 1 | 419 nm | 0.0178 | HOMO-LUMO | 0.67546 |
| HOMO-LUMO+4 | –0.14105 |
To better support the notion that the color changes can be attributed to the presence/absence of CT bands, the encapsulation of K+ or Li+ by crown ether (18-crown-6 and 12-crown-4) was attempted. Indeed, in the presence of 18-crown-6 and 12-crown-4, compounds [K(18-C-6)]2a and [K(18-C-6)]2c could be isolated from Et2O as orange solids, but [Li(12-C-4)]2b remained purple and apparently showcased insignificant 31P NMR chemical shift changes in the cases of CH3CN and Et2O (Table 1).
Reactions of salts M[2a–2c] with iodomethane at low temperatures yielded products 3a–3c as clearly revealed by their 31P{1H} NMR spectra. All products were isolated as white powders (see the Supporting Information), and their constitutions were confirmed by NMR experiments (selected data are given in Table 3). While, for 3b and 3c, mixtures of cis/trans isomers (Scheme 1) were obtained, in the case of 3a (see Table 3; for the structure of trans-3a, see Figure 3), only the trans product was detected. While it was surmised that the steric demand of the bis(trimethylsilyl)amino group leads to the selective formation of trans-3a, DFT calculations reveal the energy difference between the cis and trans isomers, ΔE (cis/trans values are 0.2, 1.1, and 0.9 kcal/mol for 3a′, 3b′, and 3c′, respectively). In addition, very high and similar inversion barriers were determined (Figure S38 and Table S20 in the Supporting Information).
Table 3. 31P{1H} NMR Data (C6D6) of 3a–3c.
| δ 31P/ppm | |||||
|---|---|---|---|---|---|
| R-P | CH3-P | 3JP,P/Hz | cis and trans ratio | ΔE(cis-trans)/kcal·mol–1 | |
| 3a | –4.7 | –72.3 | 16.6 | only trans isomer | 0.2 |
| 3b | –17.2 and −19.7 | –75.2 and −69.3 | 9.1, 11.1 | 1:3.1 | 1.1 |
| 3c | 25.6 and 26.5 | –79.6 and −69.4 | 7.2, 13.4 | 1:4.1 | 0.9 |
Figure 3.

Molecular structure of trans-3a; hydrogen atoms are omitted for clarity (50% probability level). Selected bond lengths [Å] and angles [°]: P1-N5, 1.7161(16); P2-C29, 1.846(3); P1-C1, 1.8240(19); P1-C12, 1.816(2); P2-C3, 1.803(2); P2-C14, 1.8033(19); Σ < °P1 307.86 and Σ < °P2 297.08.
Since all of these energy differences are small, it is possible that the selective formation of the trans isomer in the case of 3a is due to kinetic control. This supposition is supported also by the calculated structures as salts K[2a′], K[2b′], and K[2c′], which are presented in the Supporting Information. Clearly, the bulky N(SiMe3)2 group occupies much more space below the central ring than the other two substituents, raising a barrier for the formation of the cis isomer. Single crystals of compound 3a, suitable for X-ray diffraction analysis, were grown from a saturated diethyl ether solution.The structure confirmed the trans position of the amino and methyl groups at the central ring (Figure 3) having sums of angles at P1 and P2 of 307.9° and 297.1°, respectively. The endocyclic angle at C1-P2-C12 of 95.19(9)° and that at C3-P1-C14 of 96.11(9)° are rather acute.
Compound K[2a] was then selected for further chemical redox reactions because it has shown promising robustness under various reaction conditions. When an Et2O solution of I2 was added dropwise at −80 °C to a freshly prepared solution of K[2a] in Et2O (Scheme 2), a dark green color appeared and rapidly disappeared (within a few seconds) to finally give an orange solution containing product 4a (42% yield).
Scheme 2. Oxidation of K[2a] to 4a and Subsequent Reduction.

The attribution of the transient green color to an intermediate free radical is supported by TD-DFT calculations on 2a′• (Table S14 in the Supporting Information; lowest transition calculated at 1005 nm), while the orange color of 4 fits with calculations on 4a′ (computed as 509 nm).
The formation of 4a′ from two radicals was calculated to be exergonic (298 K, 1 bar), and the rather high reaction Gibbs free energy (28.7 kcal/mol) is in good agreement with the rapid changes in color. The reaction mixture was filtered via cannula to remove the KI salt and product 4a isolated as an orange powder. The 31P{1H} NMR spectrum of 4a (CDCl3) displays a pseudo-triplet signal at −0.4 (3/4JP,P = 25.6 Hz, P-N(SiMe3)2) and −50.9 (3/4JP,P = 25.6 Hz, P–P) ppm.
Clear orange crystals of compound 4a, suitable for X-ray diffraction analysis, were grown from a saturated diethyl ether solution (Figure 4). The analysis revealed a monoclinic crystal system with the space group C2/c. The structure shows a twisted arrangement along the P–P single bond. The C2-P2-P2′-C3′ torsion angle of 96.3° between the two tricyclic units is greater than the torsion angle observed previously for the sterically less demanding thiazol-2-thione-based tricycle.21 Most probably, also in this case, dispersion force-induced orientation of the two tricyclic units is present, which may also help in the aggregation and preorganization during the formation of the P–P bond.
Figure 4.
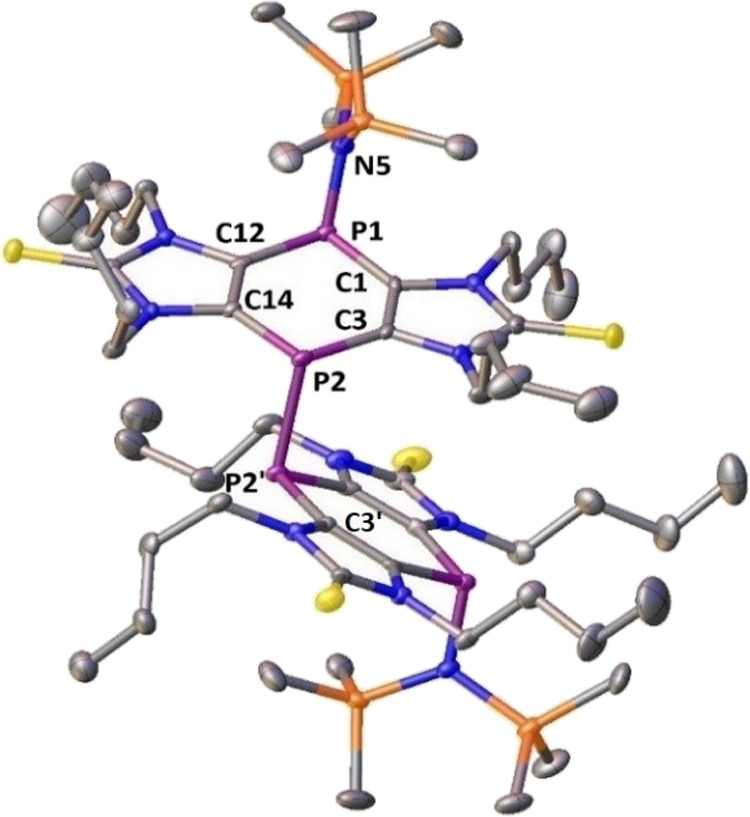
Molecular structure of compound 4a; hydrogen atoms are omitted for clarity (50% probability level). Selected bond lengths [Å] and angles [°]: P1-C1, 1.807(4); P1-C12, 1.817(4); P2-C3, 1.791(4); P2-C14, 1.806(4); C12-C14, 1.360(5); C1-C3, 1.376(5); P1-N5, 1.718(3); P2-P2′, 2.303(2); C1-P1-C12, 94.43(18); C3-P2-C14, 96.40(18); Σ < °P1 304.48 and Σ < °P2 300.79.
To check if compound 4a can be used to reform 2 equiv of K[2a] in a clean fashion, i.e., to formally reverse the oxidation with I2, compound 4a was treated with an excess of potassium in Et2O at room temperature to avoid desulfurization of the thione functionality, which usually takes place at higher temperatures (Scheme 2). After 10 min of stirring, the color of the solution turned dark blue, indicating that the anionic species K[2a] was formed, which was additionally confirmed by the 31P{1H} NMR spectrum of the reaction mixture showing two singlets at −13 and −78 ppm.
The redox chemistry of K[2a] and 4a was subsequently investigated electrochemically via interfacial voltammetry at ceramic screen-printed Pt composite electrodes (also incorporating counter and Ag/AgCl solid-dot reference electrodes; details in the Supporting Information). Cyclic voltammetry (CV) on K[2a] in CH3CN/[nBu4N][PF6] identifies a chemically irreversible (IRR) oxidation process EpIa = −0.90 V and a similarly chemically irreversible reduction process Ep = −1.63 V (Figure 5, blue trace) vs the ferrocene/ferrocenium redox couple (Fc+/0). But, when the initial scan direction was cathodic, no reduction signal occurs on the first cycle (Figure 5, red trace). Thus, the species responsible for EpIIIc appears to be an electrolysis product of the process Ep.
Figure 5.
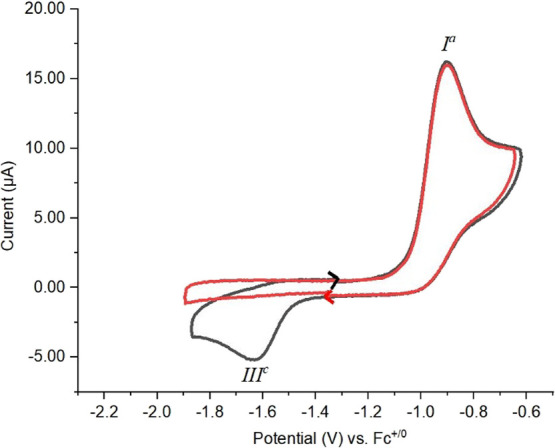
Cyclic voltammograms of K[2a] (2.59 mM) at a Pt electrode in a 0.1 M nBu4NPF6/CH3CN solution; red solid line, cathodic initial scan direction; black solid line, anodic initial scan direction; scan rates = 200 mV/s.
The repeatability of the cyclic voltammograms was examined by carrying out multicycle experiments. Even after 50 cycles, the oxidation peak and reduction peak positions remain invariant and hardly any attenuation in peak intensities is observed (Figure S31a in the Supporting Information). The scan rate dependence was examined from 0.05 to 2.5 V/s with the expected current increase with scan rate and incremental increases in the potential peak positions as expected for IRR processes (Figure S31b in the Supporting Information and Table 4). The current ratios, moreover, remain quite similar over all the scan rates.
Table 4. Peak Potentials and Currents for Cyclic Voltammograms of K[2a] at Different Scan Ratesa.
| scan rate (mV/s) | EpIIIc (V) | EpIa (V) | IpIIIc (μA) | IpIa (μA) | |IpIIIc/Ip| |
|---|---|---|---|---|---|
| 2500 | –1.81 | –0.92 | –19.52 | 57.28 | 0.34 |
| 1000 | –1.79 | –0.94 | –10.37 | 30.17 | 0.34 |
| 500 | –1.77 | –0.95 | –6.65 | 20.46 | 0.32 |
| 200 | –1.63 | –0.90 | –5.20 | 16.20 | 0.32 |
| 50 | –1.62 | –0.91 | –1.70 | 6.65 | 0.25 |
Potentials are in V vs the Fc+/0 redox couple.
CV experiments (Figure 6) were also conducted on solutions of 4a under similar experimental conditions to K[2a]. The results appear as almost an inverse of the latter trace: a large, IRR reduction peak labeled EpIIIc is found at −1.80 V and an equally IRR oxidation process labeled Ep occurs at −1.08 V (Figure 6, blue trace) when the initial scan direction is cathodic. Scans starting in the anodic direction do not display EpIa in the first cycle (Figure 6, red trace). Notably, however, the current ratios of the two processes are distinctly different, with the relative size of Ip compared to the anodic peak appearing much larger in Figure 6 compared to Figure 5. Here too, the multicycle experiments corroborated the robust repeatability of the CV processes (Figure S32a in the Supporting Information), while variable scan rate experiments from 0.05 to 2.5 V/s also fit expectations for increased currents with scan rates and incrementing of the potentials with faster scans (Figure S32b in the Supporting Information and Table 5).
Figure 6.

Cyclic voltammograms of 4a (2.59 mM) at a Pt electrode in a 0.1 M nBu4NPF6/CH3CN solution; red solid line, anodic initial scan direction; black solid line, cathodic initial scan direction; scan rates = 200 mV/s.
Table 5. Peak Potentials and Currents for Cyclic Voltammograms of 4a at Different Scan Ratesa.
| scan rate (mV/s) | EpIIIc (V) | EpIa (V) | IpIIIc (μA) | IpIa (μA) | |IpIa/Ip| |
|---|---|---|---|---|---|
| 2500 | –1.82 | –0.85 | –65.42 | 42.22 | 0.64 |
| 1000 | –1.88 | –0.90 | –39.71 | 20.50 | 0.51 |
| 500 | –1.85 | –0.91 | –27.60 | 13.99 | 0.50 |
| 200 | –1.80 | –1.08 | –19.55 | 10.96 | 0.56 |
| 50 | –1.73 | –0.99 | –8.45 | 3.69 | 0.43 |
Potentials are in V vs the Fc+/0 redox couple.
These CV experiments, as voltammetric monitors, are fully consistent with the redox interconversion of K[2a] and 4a already demonstrated in the chemical oxidation with I2 and reduction by elemental potassium. A plausible mechanism for the electrochemical processes based on the CV results is:
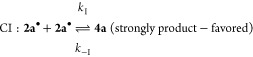 |
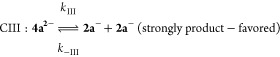 |
Here (according to the standard notation), E are electrochemical steps and C are (rapid, following) chemical steps. Process I represents the oxidation of K[2a] to form 4a via dimerization of a short-lived P-centered radical species 2a•. The reduction of 4a involves cleavage of a P–P bond and is almost certainly a two-step process, denoted II and III. There is some evidence for process II in very fast scan rate cyclic voltammograms at ∼−1.6 V (see the Supporting Information), but under most CV conditions, II and III appear as a merged peak of double intensity, which accounts for the larger relative size of IpIIIc when 4a is the bulk analyte at the electrode interface.
Digital simulations of the cyclic voltammograms were undertaken using this common mechanism applied to K[2a] and 4a, modifying only the analyte concentrations and the starting points and initial scan directions of the cyclic voltammograms (Figures S34 and S35 in the Supporting Information). Satisfactory agreement is obtained from such simulations when the forward rate constants for product formation, both kIII for the cleavage of 4a2– and kI for fusing two 2a• to form the P–P bond, are much (106 times) larger than the respective reverse reactions, at the relative applied potentials. Simulation, along with chemical redox cycling, provides strong support for robust redox shuttling between K[2a] and 4a.
There is a strong need for new battery technologies to enable modern culture to address the impacts of energy and co-related climate challenges.36 The explosive growth of lithium-ion battery production and the ongoing strong demand for such devices have placed the sustainability of this technology under scrutiny.37,38 Lithium itself is a relatively scarce element—relative to the other alkali metals—but of major concern are the metals (Mn, Fe, Co, and Ni), especially Co, required for battery cathode construction.39−41 To the best of our knowledge, organophosphorus materials have not been considered for battery design. The low theoretical charge density of the 4a/K[2a] redox shuttle and the rather negative cathode potential are obvious disadvantages, but further research in this direction may open the door to other phosphorus compounds with better properties.
An interesting feature of our system is its compatability with potassium, an attractive alternative to the current overdemand on Li.42 Considerable progress has already been reported for K/C8 (i.e., graphite intercalated) anodes.43−45 A consideration of cell potentials indicates that such anodes are at approximately −3.4 V vs Fc+/0, which, coupled with the average voltage of the 4a/K[2a] redox cycle of −1.3 V, indicates an attractive nominal cell voltage approaching 2 V for the proposed cells, albeit that the whole redox cycle would operate at strongly negative potentials.46 Moreover, secondary cells based on this technology should be able to operate efficiently at ambient temperatures, unlike the elevated temperatures contemplated for sodium/sulfur battery technologies.35
Conclusions
In this study, we have demonstrated the chemical two-electron switching between the anionic imidazole-2-thione-fused 1,4-dihydro-1,4-diphosphinine K[2a] and the P–P bonded oxidized dimeric form 4a. Both K[2a] and 4a show robust responses in multicycle cyclic voltammetry, which agree well with digital simulations. Based on the facile synthesis of the starting material (also in larger batches including tunability of N- and P-substituents) and the quantitative formation of their respective anions, these findings prompt us to propose this organophosphorus system as a potential cathode material for further research on the suitability of phosphorus compounds for secondary battery engineering.
Acknowledgments
R.K.S. and M.M. are grateful to the Deutsche Forschungsgemeinschaft (STR 441/52-1) for financial support of this work. The re-invitation grants for R.T.B. and L. N. from the Alexander von Humboldt Foundation are acknowledged. Z.K. is grateful for the general support of Hungarian Academy of Science under the Premium Postdoctoral Research Program 2019. This research was supported for L.N. and Z.K. from the Hungarian National Research and Development and Innovation Fund under the TKP2021/BME-NVA-02 program. The authors would also like to acknowledge Prof. Dr. A. C. Filippou and Prof. D. Menche for the use of X-ray facilities and C. Rödde for the X-ray diffraction studies.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.1c03620.
General synthetic methods, NMR spectra, UV–Vis spectra, mass spectra, X-ray diffraction studies, electrochemistry experiments, digital CV simulations, and results of the TD-DFT calculations of compounds M[2a′–2c′] , anions 2a′–2c′– , compounds 4a′ and 2b′, and free radical intermediates 2a′•–2c′• (PDF)
Accession Codes
CCDC 2001346–2001347 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Märkl G. 2,4,6-Triphenylphosphabenzene. Angew. Chem., Int. Ed. 1966, 5, 846–847. 10.1002/anie.196608463. [DOI] [Google Scholar]
- Le Floch P. in Phosphorus-Carbon Heterocyclic Chemistry, ed. F., Mathey. Elsevier Science Ltd, Oxford: 2001, 485–533. [Google Scholar]
- Dillon K. B.; Mathey F.; Nixon J. F.. Phosphorus: the carbon copy: from organophosphorus to phospha-organic chemistry. John Wiley and Sons, Chichester: 2001. [Google Scholar]
- Streubel R. 1λ5-Phosphinines. Sci. Synth. 2005, 1157–1179. 10.1055/sos-SD-015-01896. [DOI] [Google Scholar]
- Black D. S. C.; Ihmels H.; Alvarez M.; Bergsträßer U.; Joule J. A.. Science of Synthesis: Houben-Weyl Methods of Molecular Transformations Vol. 15: Six-Membered Hetarenes with One Nitrogen or Phosphorus Atom, Thieme. Thieme 2014. [Google Scholar]
- Märkl G.; Merz A. Ein Beitrag zum Problem der “nichtklassischen” Phosphabenzole. Tetrahedron Lett. 1968, 9, 3611–3614. 10.1016/S0040-4039(00)75513-4. [DOI] [Google Scholar]
- Bruce M.; Meissner G.; Weber M.; Wiecko J.; Müller C. Lithium Salts of 2,4,6-Triaryl-λ4 -phosphinine Anions - A Comparison Study. Eur. J. Inorg. Chem. 2014, 2014, 1719–1726. 10.1002/ejic.201301259. [DOI] [Google Scholar]
- Müller C.; Broeckx L. E. E.; de Krom I.; Weemers J. J. M. Developments in the Coordination Chemistry of Phosphinines. Eur. J. Inorg. Chem. 2013, 2013, 187–202. 10.1002/ejic.201200912. [DOI] [Google Scholar]
- Pfeifer G.; Chahdoura F.; Papke M.; Weber M.; Szűcs R.; Geffroy B.; Tondelier D.; Nyulászi L.; Hissler M.; Müller C. Synthesis, Electronic Properties and OLED Devices of Chromophores Based on λ5-Phosphinines. Chem. Eur. J. 2020, 26, 10534–10543. 10.1002/chem.202000932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regulska E.; Hindenberg P.; Romero-Nieto C. From Phosphaphenalenes to Diphosphahexaarenes: An Overview of Linearly Fused Six-Membered Phosphorus Heterocycles. Eur. J. Inorg. Chem. 2019, 2019, 1519–1528. 10.1002/ejic.201801340. [DOI] [Google Scholar]
- Hay C.; Hissler M.; Fischmeister C.; Rault-Berthelot J.; Toupet L.; Nyulászi L.; Réau R. Phosphole-Containing π-Conjugated Systems: From Model Molecules to Polymer Films on Electrodes. Chem. Eur. J. 2001, 7, 4222–4236. . [DOI] [PubMed] [Google Scholar]
- Baumgartner T.; Réau R. Organophosphorus π-conjugated materials. Chem. Rev. 2006, 106, 4681–4727. 10.1021/cr040179m. [DOI] [PubMed] [Google Scholar]
- Joly D.; Tondelier D.; Deborde V.; Delaunay W.; Thomas A.; Bhanuprakash K.; Geffroy B.; Hissler M.; Réau R. White Organic Light-Emitting Diodes Based on Quench-Resistant Fluorescent Organophosphorus Dopants. Adv. Funct. Mater. 2012, 22, 567–576. 10.1002/adfm.201102005. [DOI] [Google Scholar]
- Shameem M. A.; Orthaber A. Organophosphorus Compounds in Organic Electronics. Chem. Eur. J. 2016, 22, 10718–10735. 10.1002/chem.201600005. [DOI] [PubMed] [Google Scholar]
- Larrañaga O.; Romero-Nieto C.; de Cózar A. Dismantling the Hyperconjugation of π-Conjugated Phosphorus Heterocycles. Chem. – Eur. J. 2019, 25, 9035–9044. 10.1002/chem.201900225. [DOI] [PubMed] [Google Scholar]
- Kobayashi Y.; Kumadaki I.; Ohsawa A.; Hamana H. 2,3,5,6-Tetrakis (trifluoromethyl)-1,4-diphosphabenzene. Tetrahedron Lett. 1976, 17, 3715–3716. 10.1016/S0040-4039(00)93089-2. [DOI] [Google Scholar]
- Downie I. M.; Lee J. B.; Matough M. F. S. The reaction of alcohols with carbon tetrachloride and phosphorus trisdimethylamide. Chem. Commun. 1968, 1350b. 10.1039/C1968001350B. [DOI] [Google Scholar]
- Kobayashi Y.; Hamana H.; Fujino S.; Ohsawa A.; Kumadaki I. Studies on organic fluorine compounds. 28. Synthesis and some reactions of tetrakis(trifluoromethyl)-1,4-diphosphabenzene. J. Am. Chem. Soc. 1980, 102, 252–255. 10.1021/ja00521a039. [DOI] [Google Scholar]
- Kobayashi Y.; Fujino S.; Kumadaki I. Syntheses of trifluoromethylated thiadiphosphanorbornadiene and thiadiphosphole. J. Am. Chem. Soc. 1981, 103, 2465–2466. 10.1021/ja00399a079. [DOI] [Google Scholar]
- Koner A.; Pfeifer G.; Kelemen Z.; Schnakenburg G.; Nyulászi L.; Sasamori T.; Streubel R. 1,4-Diphosphinines from Imidazole-2-thiones. Angew. Chem., Int. Ed. 2017, 56, 9231–9235. 10.1002/anie.201704070. [DOI] [PubMed] [Google Scholar]
- Begum I.; Schnakenburg G.; Kelemen Z.; Nyulászi L.; Boeré R. T.; Streubel R. Expanding the chemistry of ring-fused 1,4-diphosphinines by stable mono anion formation. Chem. Commun. 2018, 54, 13555–13558. 10.1039/C8CC08158A. [DOI] [PubMed] [Google Scholar]
- Koner A.; Kunz M.; Schnakenburg G.; Streubel R. The Quest for Twofold Reductive P-C Bond Cleavage of P-Ph Substituted 1,4-Dihydro-1,4-diphosphinine Derivatives. Eur. J. Inorg. Chem. 2018, 2018, 3778–3784. 10.1002/ejic.201800753. [DOI] [Google Scholar]
- Koner A.; Kelemen Z.; Schnakenburg G.; Nyulászi L.; Streubel R. 1,4-Additions of tricyclic 1,4-diphosphinines - a novel system to study σ-bond activation and π-π dispersion interactions. Chem. Commun. 2018, 54, 1182–1184. 10.1039/C7CC09349G. [DOI] [PubMed] [Google Scholar]
- Koner A.; Gabidullin B. M.; Kelemen Z.; Nyulászi L.; Nikonov G. I.; Streubel R. 7-Metalla-1,4-diphosphanorbornadienes: cycloaddition of monovalent group 13 NacNac complexes to a stable 1,4-diphosphinine. Dalton Trans. 2019, 48, 8248–8253. 10.1039/C9DT01425J. [DOI] [PubMed] [Google Scholar]
- Miyake H.; Sasamori T.; Wu J. I.-C.; Schleyer P. v. R.; Tokitoh N. The 4,5,6-triphospha[3]radialene dianion: a phosphorus analogue of the deltate dianion. A NICS(0)πzz examination of their aromaticity. Chem. Commun. 2012, 48, 11440–11442. 10.1039/c2cc35978b. [DOI] [PubMed] [Google Scholar]
- Miyake H.; Sasamori T.; Tokitoh N. Synthesis and properties of 4,5,6-triphospha[3]radialene. Angew. Chem., Int. Ed. 2012, 51, 3458–3461. 10.1002/anie.201200374. [DOI] [PubMed] [Google Scholar]
- Sasamori T.; Tokitoh N.; Streubel R.. π-Electron Redox Systems of Heavier Group 15 Elements, in Organic Redox Systems, Synthesis, Properties and Applications (ed. T., Nishinaga). Wiley, Hoboken, New Jersey: 2016, 563–578. [Google Scholar]
- Shah S.; Burdette S. C.; Swavey S.; Urbach F. L.; Protasiewicz J. D. Alkali Metal Induced Rupture of a Phosphorus–Phosphorus Double Bond. Electrochemical and EPR Investigations of New Sterically Protected Diphosphenes and Radical Anions [ArPPAr]•-. Organometallics 1997, 16, 3395–3400. 10.1021/om970025f. [DOI] [Google Scholar]
- Tohmé A.; Grelaud G.; Argouarch G.; Roisnel T.; Labouille S.; Carmichael D.; Paul F. Redox-induced reversible P-P bond formation to generate an organometallic σ4λ4-1,2-biphosphane dication. Angew. Chem., Int. Ed. 2013, 52, 4445–4448. 10.1002/anie.201208682. [DOI] [PubMed] [Google Scholar]
- Lejeune M.; Grosshans P.; Berclaz T.; Sidorenkova H.; Besnard C.; Pattison P.; Geoffroy M. Role of the aromatic bridge on radical ions formation during reduction of diphosphaalkenes. New J. Chem. 2011, 35, 2510. 10.1039/c1nj20314b. [DOI] [Google Scholar]
- Märkl G.; Kreitmeier P.; Daffner R. Oxidation von 4-N,N-dimethylaminophenyl-1-phosphabutatrienen zu diphosphanen mit “Malachitgrün”-Chromophoren. Tetrahedron Lett. 1993, 34, 7045–7048. 10.1016/S0040-4039(00)61593-9. [DOI] [Google Scholar]
- Manan N. S. A.; Aldous L.; Alias Y.; Murray P.; Yellowlees L. J.; Lagunas M. C.; Hardacre C. Electrochemistry of sulfur and polysulfides in ionic liquids. J. Phys. Chem. B 2011, 115, 13873–13879. 10.1021/jp208159v. [DOI] [PubMed] [Google Scholar]
- Evans A.; Montenegro M. I.; Pletcher D. The mechanism for the cathodic reduction of sulphur in dimethylformamide: low temperature voltammetry. Electrochem. Commun. 2001, 3, 514–518. 10.1016/S1388-2481(01)00203-X. [DOI] [Google Scholar]
- Yamin H.; Gorenshtein A.; Penciner J.; Sternberg Y.; Peled E. Lithium Sulfur Battery: Oxidation/Reduction Mechanisms of Polysulfides in THF Solutions. J. Electrochem. Soc. 1988, 135, 1045–1048. 10.1149/1.2095868. [DOI] [Google Scholar]
- Steudel R.; Chivers T. The role of polysulfide dianions and radical anions in the chemical, physical and biological sciences, including sulfur-based batteries. Chem. Soc. Rev. 2019, 48, 3279–3319. 10.1039/C8CS00826D. [DOI] [PubMed] [Google Scholar]
- Armand M.; Tarascon J.-M. Building better batteries. Nature 2008, 451, 652–657. 10.1038/451652a. [DOI] [PubMed] [Google Scholar]
- Dai Q.; Kelly J. C.; Gaines L.; Wang M. Life Cycle Analysis of Lithium-Ion Batteries for Automotive Applications. Batteries 2019, 5, 48. 10.3390/batteries5020048. [DOI] [Google Scholar]
- Aichberger C.; Jungmeier G. Environmental Life Cycle Impacts of Automotive Batteries Based on a Literature Review. Energies 2020, 13, 6345. 10.3390/en13236345. [DOI] [Google Scholar]
- Massé R. C.; Liu C.; Li Y.; Mai L.; Cao G. Energy storage through intercalation reactions: electrodes for rechargeable batteries. Natl. Sci. Rev. 2017, 4, 26–53. 10.1093/nsr/nww093. [DOI] [Google Scholar]
- Liu C.; Neale Z. G.; Cao G. Understanding electrochemical potentials of cathode materials in rechargeable batteries. Mater. Today 2016, 19, 109–123. 10.1016/j.mattod.2015.10.009. [DOI] [Google Scholar]
- Tarascon J. M.; Armand M. Issues and challenges facing rechargeable lithium batteries. Nature 2001, 414, 359–367. 10.1038/35104644. [DOI] [PubMed] [Google Scholar]
- Bhide A.; Hofmann J.; Dürr A. K.; Janek J.; Adelhelm P. Electrochemical stability of non-aqueous electrolytes for sodium-ion batteries and their compatibility with Na(0.7)CoO2. Phys. Chem. Chem. Phys. 2014, 16, 1987–1998. 10.1039/C3CP53077A. [DOI] [PubMed] [Google Scholar]
- Kapaev R. R.; Troshin P. A. Organic-based active electrode materials for potassium batteries: status and perspectives. J. Mater. Chem. A 2020, 8, 17296–17325. 10.1039/D0TA04741D. [DOI] [Google Scholar]
- Carboni M.; Naylor A. J.; Valvo M.; Younesi R. Unlocking high capacities of graphite anodes for potassium-ion batteries. RSC Adv. 2019, 9, 21070–21074. 10.1039/C9RA01931F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komaba S.; Hasegawa T.; Dahbi M.; Kubota K. Potassium intercalation into graphite to realize high-voltage/high-power potassium-ion batteries and potassium-ion capacitors. Electrochem. Commun. 2015, 60, 172–175. 10.1016/j.elecom.2015.09.002. [DOI] [Google Scholar]
- Hodge S. A.; Tay H. H.; Anthony D. B.; Menzel R.; Buckley D. J.; Cullen P. L.; Skipper N. T.; Howard C. A.; Shaffer M. S. P. Probing the charging mechanisms of carbon nanomaterial polyelectrolytes. Faraday Discuss. 2014, 172, 311–325. 10.1039/C4FD00043A. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.