

Abstract
Platinum-containing drugs such as cisplatin and carboplatin are routinely used for the treatment of many solid tumors including squamous cell carcinoma of the head and neck (SCCHN). However, SCCHN resistance to platinum compounds is well documented. The resistance to platinum has been linked to the activity of divalent transporter ATP7B, which pumps platinum from the cytoplasm into lysosomes, decreasing its concentration in the cytoplasm. Several cancer models show increased expression of ATP7B; however, the reason for such an increase is not known. Here we show a strong positive correlation between mRNA levels of TMEM16A and ATP7B in human SCCHN tumors. TMEM16A overexpression and depletion in SCCHN cell lines caused parallel changes in the ATP7B mRNA levels. The ATP7B increase in TMEM16A-overexpressing cells was reversed by suppression of NADPH oxidase 2 (NOX2), by the antioxidant N-Acetyl-Cysteine (NAC) and by copper chelation using cuprizone and bathocuproine sulphonate (BCS). Pretreatment with either chelator significantly increased cisplatin’s sensitivity, particularly in the context of TMEM16A overexpression. We propose that increased oxidative stress in TMEM16A-overexpressing cells liberates the chelated copper in the cytoplasm, leading to the transcriptional activation of ATP7B expression. This, in turn, decreases the efficacy of platinum compounds by promoting their vesicular sequestration. We think that such a new explanation of the mechanism of SCCHN tumors’ platinum resistance identifies novel approach to treating these tumors.
Introduction
Squamous cell carcinoma of the head and neck (SCCHN) is the sixth leading cause of cancer world-wide. These cancers are strongly associated with a risk to tobacco exposure and alcohol abuse [1]. The mainstay of treatment for SCCHN requires the use of platinum-based drugs (such as cisplatin). However, the survival rate of SCCHN remains at 40–50% largely due to significant resistance to cisplatin. Cisplatin resistance can be acquired through repeated treatment cycles or as inherent characteristic of cancer [2,3]. Molecular mechanisms underlying platinum resistance is attributed to a number of factors including binding to genomic and mitochondrial DNA, cellular proteins and RNA and activation of several signaling pathways that lead to ROS and apoptosis. However, cisplatin fails to exhibit full potential because of its reduced cellular accumulation, cytosolic inactivation by binding to glutathione, and altered DNA repair [4]. Consequently, it is important to find more methodical approaches, like combination therapy, that do not exclusively rely on cisplatin.
Copper is an indispensable trace element that serves a variety of physiological functions that also regulates cellular process as proliferation, angiogenesis, and motility. Studies have demonstrated the role of copper in tumor growth, epithelial-mesenchymal transition, in the formation of the tumor microenvironment and pre-metastatic niche. Hence, the copper reduction has emerged as a novel therapeutic strategy in the treatment of metastatic cancer [5]. Copper transporters, specifically ATP7B, a P-type ATPase that uses ATP to export, have been implicated in cellular import and export of platinating agents.
Increased ATP7B expression in poorly differentiated tumors of ovary [6], gastric [7,8], colorectum [9], bladder [10], breast [11], lung [12] and oral cavity [13,14] has been reported. At basal copper levels, ATP7B is located in the trans-Golgi network (TGN). As the intracellular copper increases, ATP7B translocates to the cytoplasmic vesicles, fusing with the plasma membrane and releasing copper ions out of the cells. ATP7B can mediate resistance to platinum drugs in various tumor types including ovarian cancer, non-small cell lung carcinoma (NSCLC), colorectal carcinoma, endometrial cancer, oral squamous cell carcinoma, and esophageal cancer [9,12–16].
In the present work, we show that ATP7B expression is modulated by ROS in TMEM16A overexpressing cells. Second, copper chelators like cuprizone and bathocuproine sulphonate ostensibly ‘mop up’ the copper to sensitize the cells to cisplatin. We propose that this evidence is first in kind to model the use of copper chelation as a therapy in SCCHN.
Methods
Reagents
Cuprizone (Bis(cyclohexanone)oxaldihydrazone; Cuz), bathocuproinedisulfonic acid (BCS) and N-acetyl-L-cysteine (NAC) were purchased from Sigma–Aldrich (St. Louis, MO). Apocynin was purchased from Santa Cruz Biotechnology (Dallas, TX). Cisplatin was from EMD Millipore (now part of Millipore Sigma, Burlington, MA). Cuprizone was solubilized in a minimum volume of 50% ethanol and the final volume was made up in distilled water. Stock cuprizone was made fresh for each experiment and a final concentration of 2.5 or 5 μM was used in experiments. Stock solution of BCS was made in distilled water, stored at −20°C and diluted in culture media before use. ATP7B siRNA was purchased from Sigma–Aldrich (St. Louis, MO). Primers were bought from IDT Technologies (Coralville, IA). RNeasy kit for RNA isolation and QIAshredder were purchased from Qiagen (Valencia, CA). iScript RT supermix for RT-qPCR and iQ™ SYBR® Green Supermix for qPCR was from Bio-Rad Laboratories (Hercules, CA).
Cell lines
OSC19, Cal33, HN30 and FaDu cells were maintained in DMEM; Te1 and Te9 were maintained in RPMI media supplemented with 10%FBS and 1% penicillin/streptomycin mixture. All cells were used for 10 passages and then discarded. Cell lines were authenticated using human cancer cell line STR profiles. Vector Control or TMEM16A overexpressing cells were engineered by transducing viral pBABE-puromycin control or TMEM16A plasmid as previously described by Duvvuri et al. [17]. FaDu cells were engineered to express non-targeted shRNA or 2 independent TMEM16A-targeting shRNA (#12 and #1018) in doxycycline-inducible manner as previously described [23]. These cells were cultured in DMEM containing 10% tetracycline-free serum. For experimental use, cells were cultured in 10 ng/ml doxycycline containing media for 72 h, to achieve induction of shRNA. Each cell line was maintained at 37°C in a humidified atmosphere of 95% air and 5% CO2.
ATP7B siRNA transfection
OSC19 and FaDu cells were plated in six-well plates and grown to 70% confluency. A total of 10 μM siRNA was transfected with Lipofectamine 3000™ reagent (Invitrogen) for 48 h before re-plating and analyzing for qPCR or cell proliferation.
Quantitative real-time PCR (qRT-PCR)
Human tumor samples were obtained from the University of Pittsburgh Medical Center in accordance with established University of Pittsburgh IRB guidelines. Tumors were disrupted and homogenized in appropriate volume of lysis buffer provided in the RNeasy kit. The tissue lysate was loaded onto the QIAshredder homogenizer after which RNA was isolated using the manufacturer’s protocol. Total RNA from OSC19-VC/TMEM16A or FaDu-NT/shTMEM16A cells were isolated using RNeasy kit. First-strand cDNA was synthesized using iScript RT and qPCR was done using a suitable dilution of cDNA. Primer sequences for target genes are as follows:
-
ATP7B Forward 5′-GTGGGCAATGACACCACTTT-3′
Reverse 5′-TGGGTGCCTTTGACATCTGA-3′
-
HMOX` Forward 5′-GAGACGGCTTCAAGCTGGTGAT-3′
Reverse 5′-CCGTACCAGAAGGCCAGGTC-3′
-
NQO1 Forward 5′-CGC AGA CCT TGT GAT ATT CCA G-3′
Reverse 5′-CGT TTC TTC CAT CCT TCC AGG-3′
RT conditions: 15 s denaturation/95°C, 30 s annealing/60°C, 30 s extension/72°C for 40 cycles. Relative quantification was performed using the 2−ΔΔCq method [18].
Cell proliferation assay
2.5–5 × 104 OSC19 and FaDu were plated in 96-well plates and allowed to attach overnight. After indicated treatments, 10 μl premix WST-1 cell proliferation reagent (Takara Bio Inc, Clontech Laboratories, Inc.) was added to each well and the plate was returned to the incubator for 2 h after which absorbance was read at 450 nm in microplate reader from BioTek Instruments Inc.
Amplex Red assay
Extracellular H2O2 was determined from intact cells using 10-acetyl-3,7-dihydroxyphenoxazine (Amplex Red reagent) using the reagents and protocols provided in the Amplex Red Hydrogen Peroxide Assay Kit (Molecular Probes Inc., Eugene, OR). 25 000cells/well were allowed to attach in 96-well culture plates for overnight. Next day, after cuprizone (6 h, 2.5 μM)/NAC (20 mM)/apocynin (100 μM) treatment for 3 h, cells were washed once in PBS, then incubated for 30 min at 37°C in reaction buffer containing 0.2 U/ml horseradish per-oxidase and 100 μM amplex Red; background fluorescence was measured in parallel in wells containing all reactants except samples. Fluorescence was expressed as H2O2 levels using a standard curve generated with known concentrations of H2O2 stabilized solution using Synergy H1 Hybrid microplate reader from BioTek at excitation of 530 nm and emission of 590 nm. Cells were then counterstained with crystal violet to correct the mM H2O2 values for variations in cell culture densities.
MitoSOX
Detection of mitochondrial superoxide in live cells was done using MitoSOX™ Red (Molecular Probes Inc., Eugene, OR). 25 000cells cultured in 96-well plate were treated with cuprizone/NAC/apocynin as indicated in amplex red assay. Cells were washed once in PBS, then incubated with 5 μM MitoSOX in PBS for 30 min at 37°C. Following incubation in dark, MitoSOX was removed, cells were washed again with PBS and then plates read in Synergy H1 Hybrid microplate reader from BioTek at excitation of 510 nm and emission of 595 nm.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 8. A paired t-test or ANOVA with Dunnet or Tukey’s adjustment was used to test significance as appropriate. P < 0.05 was considered statistically significant. Each experiment was repeated 3–4 times and the combined graph of mean ± SEM is shown unless stated otherwise.
Results
We and others have previously shown an up-regulation of TMEM16A mRNA in a significant fraction of human SCCHN tumors [17–19]. Also, many SCCHN tumors show resistance to platinum [20–22]. ATP7B up-regulation has been shown in many tumors and was proposed to drive the resistance to platinum compounds [9,12–16]. To answer whether TMEM16A overexpression in SCCHN entails ATP7B up-regulation, we performed qPCR analysis of human SCCHN tumors isolated from 13 patients. Interestingly, these samples segregate into two subsets, one which plays a stronger correlation coefficient. To illuminate the mechanistic connection between TMEM16A mRNA and ATP7B mRNA correlation, we used the SCCHN cell lines OSC19 and FaDu. ATP7B mRNA levels increased more than threefold following TMEM16A overexpression in the natively low-expressing tongue squamous cell carcinoma cell line OSC19 (Figure 1B). TMEM16A overexpression was accomplished by stable transfection as before [17]. The TMEM16A-rich cell line FaDu dropped ATP7B mRNA levels after TMEM16A levels knockdown using the inducible shRNA system described before [23] (Figure 1B). The control remained untransfected. siRNA assays in Figure 1C show the specificity of the observed effects on ATP7B mRNA. These data show that TMEM16A expression impacts ATP7B mRNA levels.
Figure 1. TMEM16A up-regulates ATP7B.
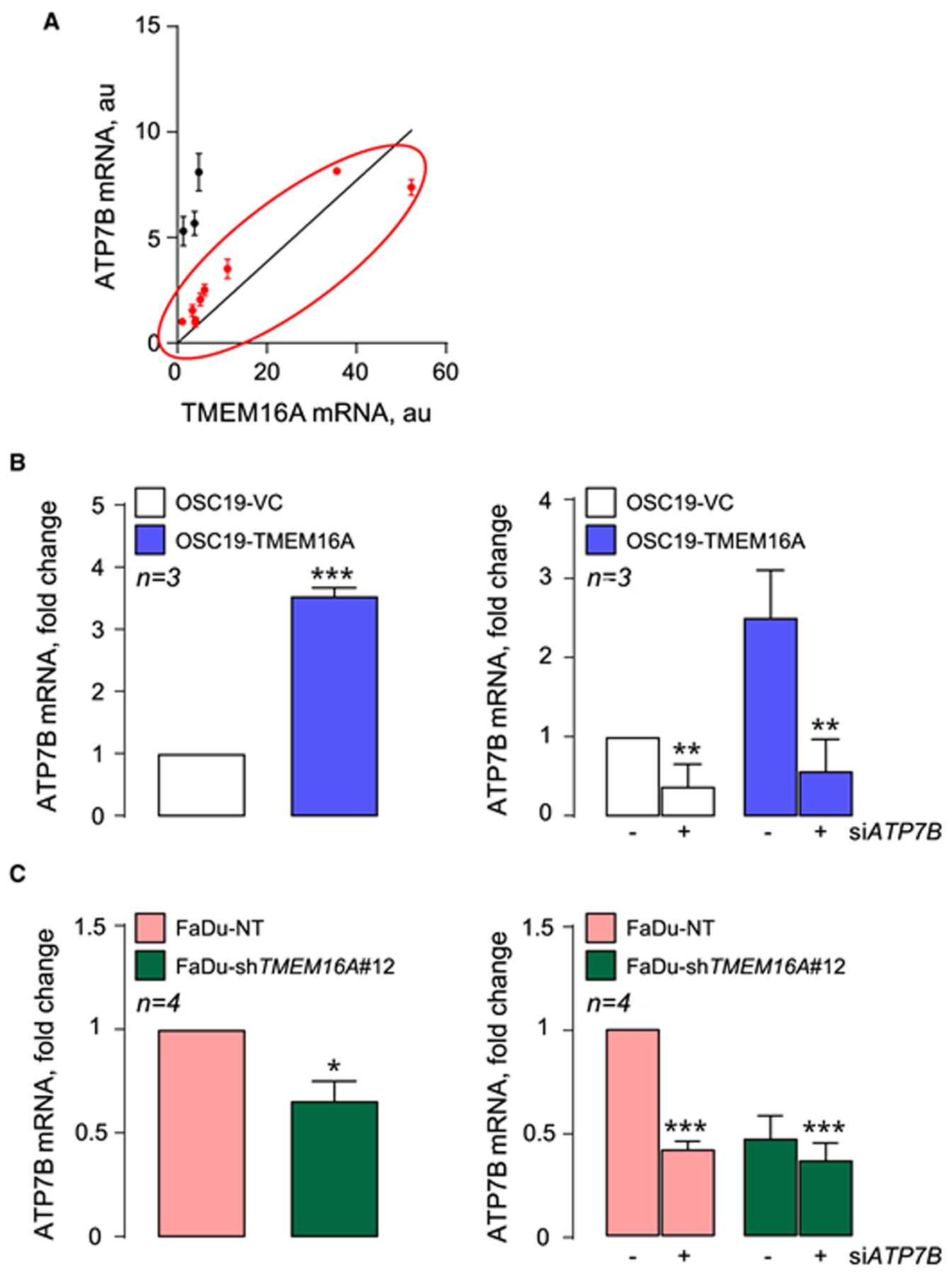
ATP7B mRNA qRT-PCR in: (A) Human tumors shows linear correlation of ATP7B with TMEM16A mRNA. The red data points represent subset showing correlation significant at P < 0.05 with Spearman’s and P < 0.0001 with Pearson’s adjustment. (B) OSC19 cells (left panel) and after siATP7B treatment (right panel). (C) FaDu cells (left panel) and after siATP7B treatment (right panel). *** P < 0.0001, ** P < 0.001 and * P < 0.05 with t-test.
ATP7B expression has been linked to increased resistance to platinum compounds, ostensibly by facilitating platinum sequestration in the lysosomes or related vesicles and its expulsion by exocytosis [24–26]. TMEM16A-overexpressing OSC19 cells have higher proliferation and resistance to cisplatin than wild-type, low-TMEM16A cells, and native high-TMEM16A expressing FaDu cells have higher proliferation and cisplatin resistance than the same cells in which TMEM16A was deleted (Figure 2A). This trend was reversed by ATP7B depletion. TMEM16A-overexpressing ATP7B-deficient cells lost proliferative advantage over low-TMEM16A expressing cells and become significantly more sensitive to platinum. Specifically, proliferation rate of TMEM16A-overexpressing OSC19 cells was ~150% of vector control cells (145.3 ± 14.72%, n = 3). ATP7B knockdown dropped the OSC19-TMEM16A cells to the levels undistinguishable from control cells. ATP7B knockdown induced nearly 30% drop in proliferation rate of TMEM-overexpressing cells (proliferation rate decreased to 72.85 ± 7.03% (n = 3) of control cells. Treatment with 5 μM cisplatin (CDDP) induced 20% drop (78.88 ± 10.52%, n = 3). Overall, a combination of ATP7B knockdown and CDDP caused 50% drop in the proliferation rate of TMEM16A-overexpressing OSC19 (52.38 ± 0.96%, n = 3), and only 25% drop in the proliferation rate of control, natively TMEM16A-low OSC19. Cell proliferation in OSC19-VC subjected to the combination of ATP7B knockdown and CDDP was 75.77 ± 6.57% (n = 3) of its value in control cells.
Figure 2. ATP7B knockdown increases the sensitivity of cancer cell models to cisplatin.
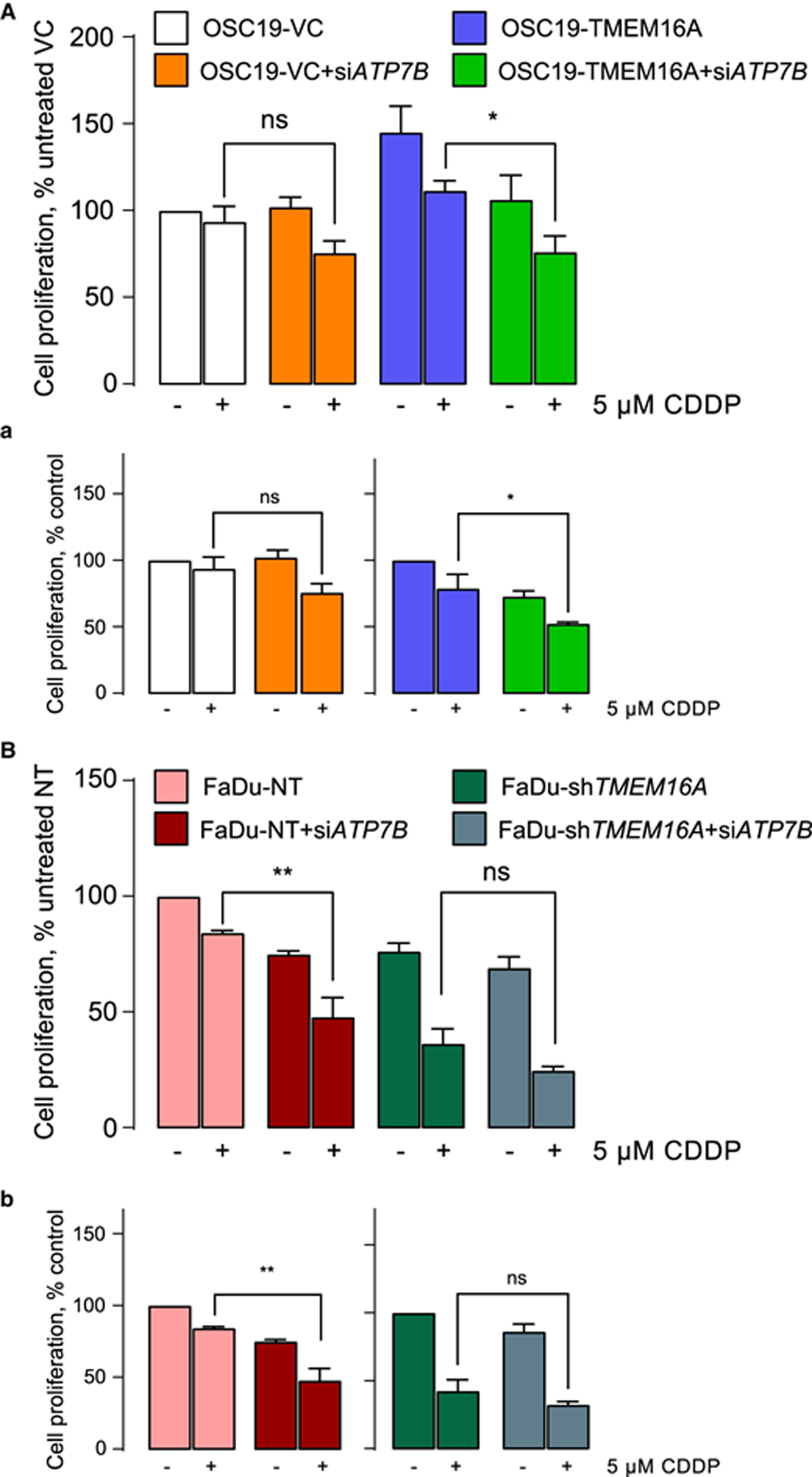
Cell proliferation measured in OSC19 (A) and FaDu (B) after treatment with ATP7B siRNA. These panels represent change in proliferation relative to untreated transfected controls. Panels a and b show the change in proliferation recalculated by taking OSC19-VC (a, left panel), OSC19-TMEM16A (a, right panel), FaDu-NT (b, left panel) and FaDu-shTMEM16A (b, right panel) as 100%. * P < 0.05 and ** P < 0.001 with one-way ANOVA after Tukey’s multiple comparison test.
ATP7B transcription was shown to respond to oxidative stress [27,28], in a mechanism that was shown, by different groups, to involve the metal-sensitive transcription factor MTF-1 [29]. Oxidative stress is known to liberate copper from cytoplasmic chelating proteins such as metallothioneins, which is likely to accelerate oxidative damage due to the oxidizing nature of free copper ions [30]. The runaway damage is countered by binding of the liberated copper MTF-1, leading to the transcriptional activation of genes coding for metallothioneins and divalent transporters including ATP7B. Such a feedback loop appears to limit the cellular damage induced by oxidative stress. To answer whether this mechanism is involved in ATP7B up-regulation in the TMEM16A-overexpressing cancer models, we measured oxidative stress in these models using mRNA levels of heme oxygenase 1 (HMOX1) and NAD(P)H Quinone Dehydrogenase 1 (NQO1) as in our recent studies [31,32]. The expression of these genes is regulated by the transcription factor NRF-2, which is activated by oxidative stress [33,34]. This is a robust and highly quantitative readout of oxidative stress. Figure 3 shows increased oxidative stress associated with high levels of TMEM16A expression: intrinsic (FaDu) or induced by recombinant expression (OSC19-TMEM16A). In OSC19 cells, TMEM16A expression increased HMOX1 and NQO1 mRNA levels 4- and 3-fold, respectively (mRNA fold change relative to VC cells averaged 4.09 ± 0.96 and 3.13 ± 0.58, n = 3). At the same time, TMEM16A knockdown in TMEM16A-rich FaDu cells, decreased HMOX1 and NQO1 mRNA levels decreased by 30%, respectively (mRNA fold change relative to NT cells: 0.77 ± 0.03 and 0.66 ± 0.03; n = 3).
Figure 3. Cuprizone (Cuz) and BCS inhibit ROS production.
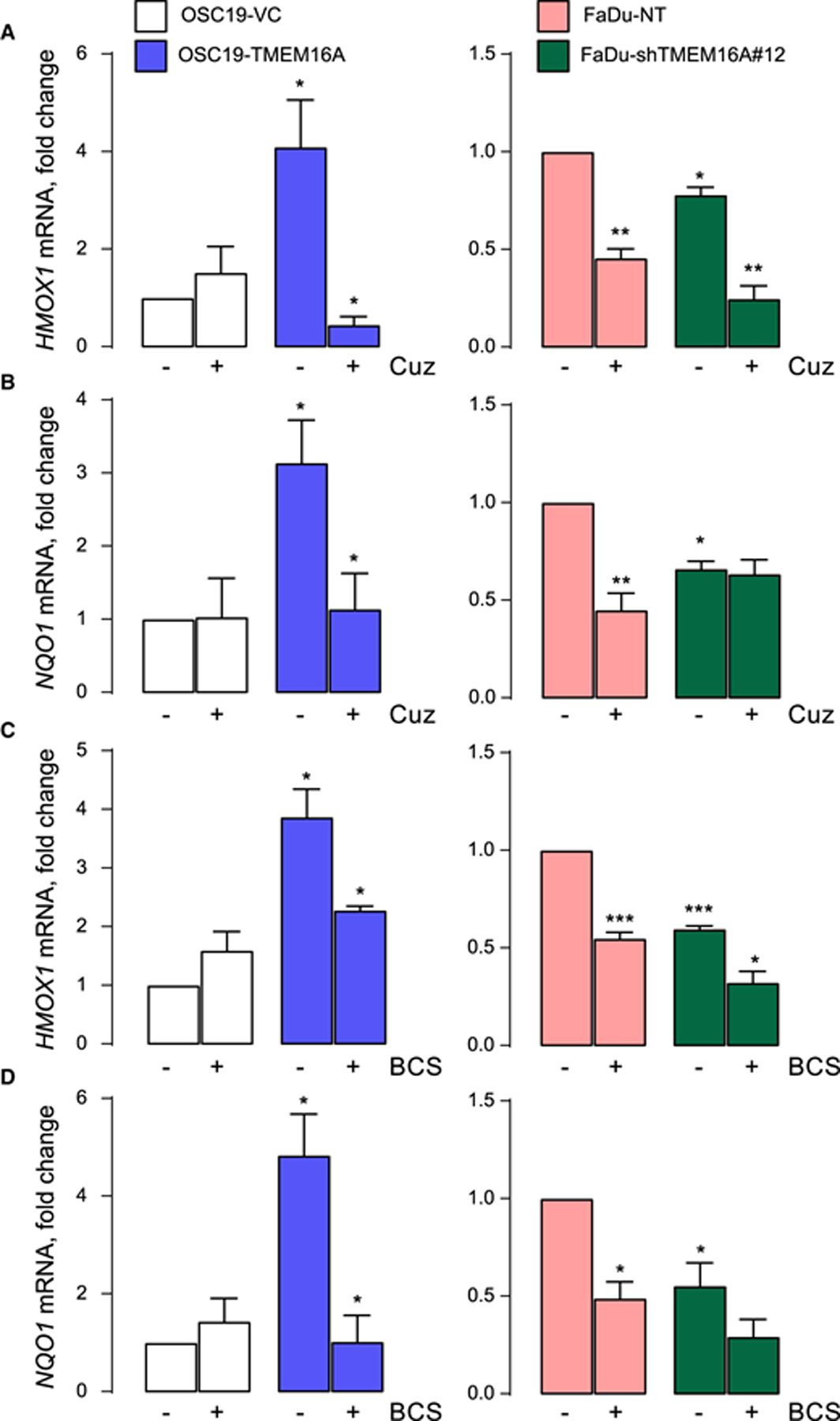
(A and B) HMOX1 and NQO1 mRNA levels analyzed using qPCR after 6 h-long treatment with 2.5 μM Cuz in OSC19 (left panel) and FaDu (right panel). (C and D) HMOX1 and NQO1 mRNA levels analyzed using qPCR after 6 h-long treatment with 100 μM BCS in OSC19 (left panel) and FaDu (right panel). Data is normalized to OSC19-VC or FaDu-NT. Significance is shown as one-way ANOVA after Tukey’s multiple comparison test.
Interestingly, these oxidative stress markers were suppressed by incubation of the cells with copper chelators cuprizone and BCS. Cuprizone caused 9- and 2.8-fold drop in HMOX1 and NQO1 mRNA levels in TMEM16A-overexpressing OSC19 cells. The drops for both genes in FaDu cells averaged 2.2-fold. The drops were TMEM16A-specific as they were absent or not significant in OSC19 cells and in FaDu cells expressing TMEM16A shRNA. Therefore, free copper is likely increased in the cytoplasm, contributing to oxidative stress, of TMEM16A-overexpressing cells.
Additional tests of oxidative stress included measuring hydrogen peroxide (H2O2) release and MitoSOX fluorescence as a function of TMEM16A status and the presence of antioxidants. Hydrogen peroxide release was measured using Amplex Red (10-acetyl-3,7-dihydroxyphenoxazine). Figure 4A shows ≈30% increased hydrogen peroxide release in cells with higher TMEM16A levels: intrinsic (FaDu) or induced by recombinant expression (OSC19-TMEM16A). In TMEM16A-overexpressing cells hydrogen peroxide release averaged 1.3-fold (128.2 ± 7.2%, n = 3) that of natively TMEM16A-poor control OSC19 cells. This was reversed by apocynin, the inhibitor of NOX2, which is one of the main producers of reactive oxygen species [34,35], and by the antioxidant NAC [36]. In TMEM16A-overexpressing OSC19 cells apocynin induced a 2.3-fold drop in hydrogen peroxide release (to 44.83 ± 11.2% of control values, n = 3); NAC caused a 1.6-fold drop (to 61.15 ± 2.40% of control values, n = 3). The drop was not significant in control OSC19 cells. In FaDu cells, apocynin induced a 1.3-fold in hydrogen peroxide release (to 78.79 ± 9.44% of control values, n = 3) and NAC caused a 1.2-fold drop (to 83.37 ± 7.05% of control values, n = 3, Figure 5A). The drops were absent or not significant in FaDu cells expressing TMEM16A shRNA (Figure 5A).
Figure 4. Cuprizone selectively inhibits H2O2 release and mitochondrial oxidative stress in OSC19-TMEM16A cells.
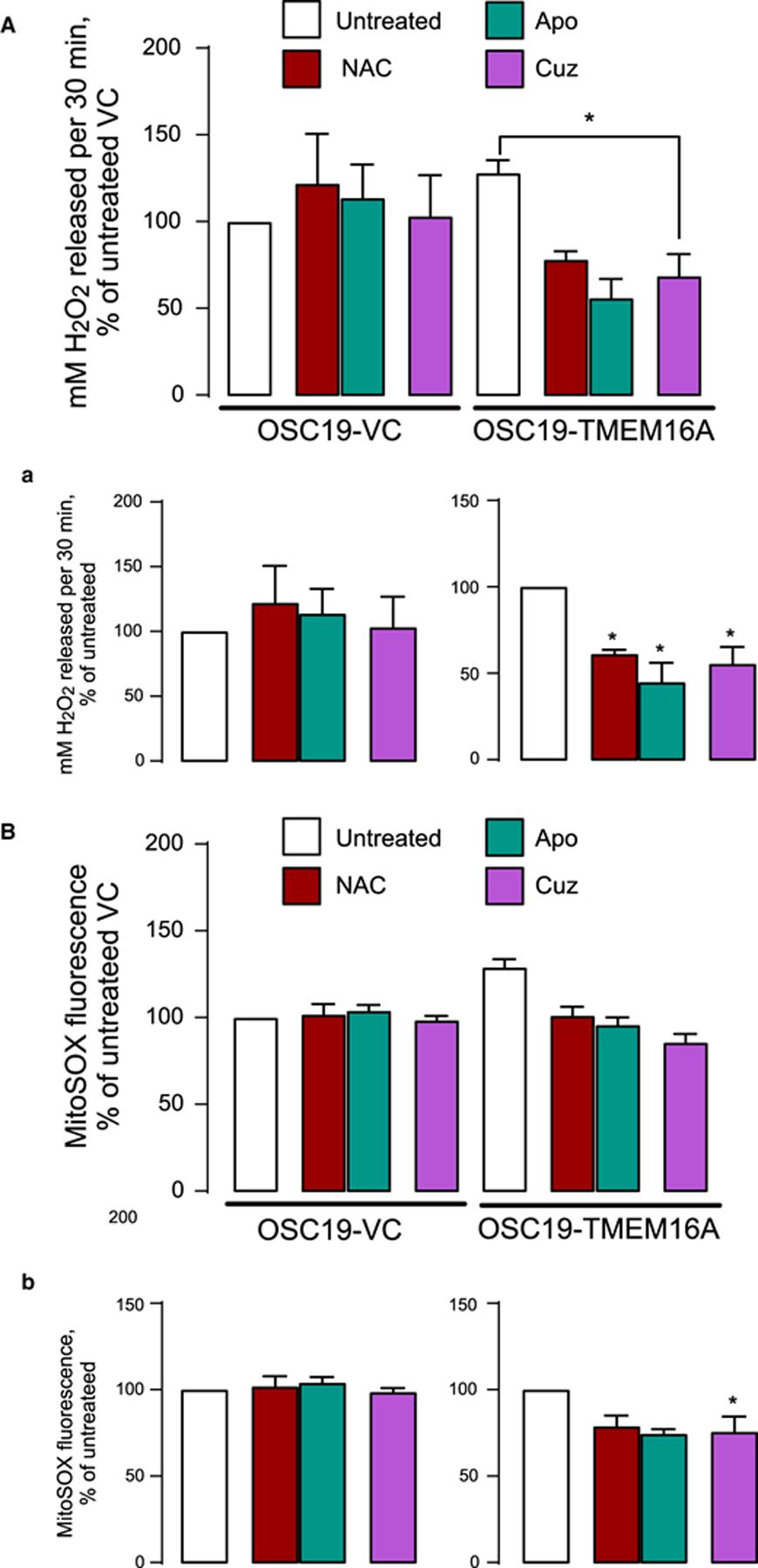
(A) Amplex Red is measured in OSC19-VC and TMEM16A cells treated with 5 μM Cuz for 6 h. H2O2 released is represented as % change of untreated VC (a, left panel) and as % change of untreated TMEM16A (a, right panel). (B) MitoSOX is measured in OSC19-VC and TMEM16A cells after treatment with 5 μM Cuz for 6 h. MitoSOX fluorescence is shown as % change of untreated VC (b, left panel) and as % change of untreated TMEM16A (b, right panel). Significance is shown at * P < 0.05 using unpaired t-test.
Figure 5. Cuprizone selectively inhibits H2O2 release and mitochondrial oxidative stress in FaDu cells.
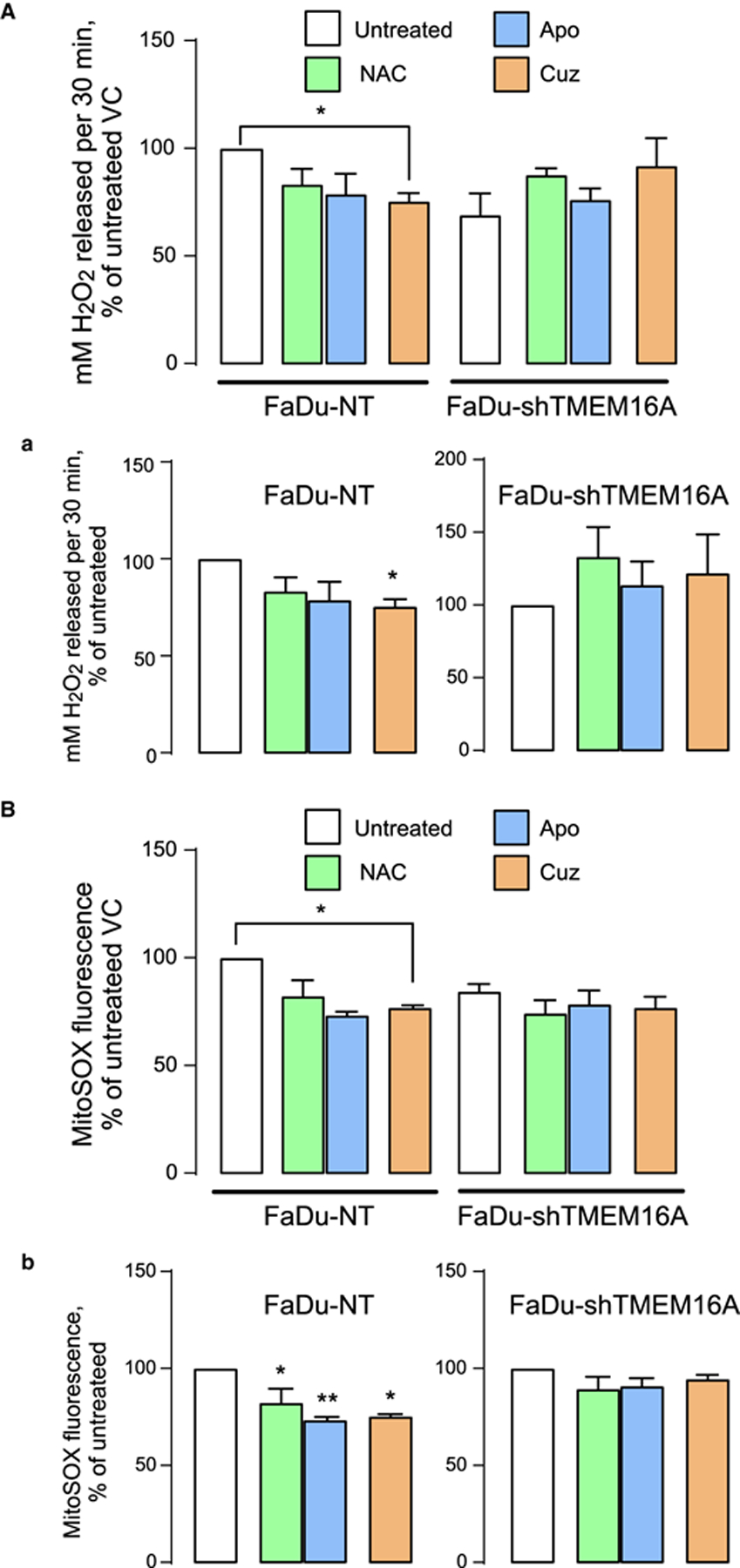
(A) Amplex Red is measured in FaDu-NT and FaDu-shTMEM16A cells. H2O2 released is represented as % change of untreated NT (a, left panel) or % change of untreated shTMEM16A (a, right panel). (B) MitoSOX is measured in FaDu-NT and FaDu-shTMEM16A cells. MitoSOX fluorescence is shown as % change of untreated NT (b, left panel) and as % change of untreated shTMEM16A (b, right panel). Significance is shown at * P < 0.05 and ** P < 0.001 when compared with untreated control by one-way ANOVA with Dunnet’s adjustment.
MitoSOX Red is a fluorescent mitochondrial superoxide indicator that reports mitochondrial superoxide in live cells. MitoSOX Red studies showed increased oxidative stress in TMEM16A-overexpressing cells, which was reversed by the antioxidants. Oxidative stress in TMEM16A-overexpressing cells reported by hydrogen peroxide release and by MitoSOX signal appears to be at least partially driven by copper because cell incubation with low micromolar concentrations of cuprizone decreased it to the levels observed in cells expressing low TMEM16A levels (Figures 4B and 5B). We conclude that increased free copper in the cytoplasm of TMEM16A-overexpressing cells contributes to oxidative stress. We do acknowledge the increased hydrogen peroxide release in NAC-treated group of low-TMEM16A expressing OSC19 and FaDu cell-lines (Figures 4A and 5A). While more in-depth experiments are needed to elucidate the exact cause of this phenomena, we propose that low TMEM16A cells have minimal levels of ROS. The addition of NAC might in fact be increasing ROS production by inducing reductive stress and inhibiting mitochondrial function [35], partly because there is no role of NAC as antioxidant.
The increase in ATP7B mRNA in TMEM16A-overexpressing cells shown in Figures 1 and 2 is at least partially driven by oxidative stress because it was suppressed by apocynin (NOX2 inhibitor) and by NAC (ROS inhibitor) (Figure. 6A). Figures 6B,C shows that ATP7B mRNA increase in TMEM16A-overexpressing cells was reversed by cuprizone and BCS as well. ATP7B mRNA levels decreased from 4.01 ± 0.66-fold change over control in OSC19-TMEM16A overexpressing cells to 1.22 ± 0.43 after cuprizone treatment. In BCS treatment the decrease was from 6.70 ± 1.86 to 1.37 ± 0.45 (n = 3). TMEM16A-rich FaDu cells showed levels similar to those in FaDu cells expressing TMEM16A shRNA (Figures 6B,C). Nether control OSC19 nor TMEM16A-deficient FaDu cells responded to copper chelators by decreasing ATP7B mRNA to a significant extent.
Figure 6. Cu-chelators Cuprizone and BCS down-regulate ATP7B mRNA expression.
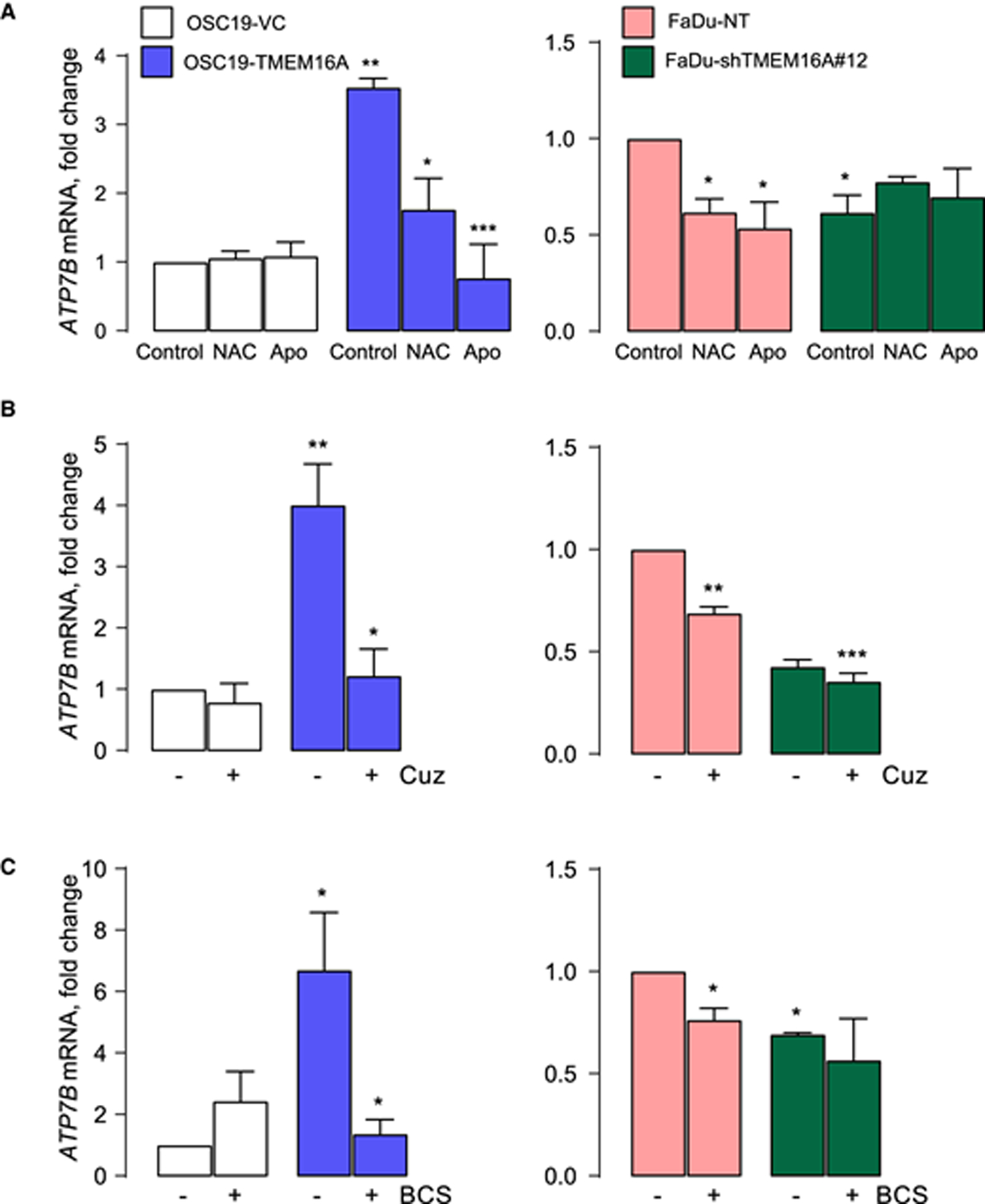
The results of qPCR for ATP7B mRNA in OSC19 (left panel) and FaDu (right panel) after: (A) Three-hours long treatment with 20 mM NAC or 100 μM Apo treatment. Significance is calculated using unpaired t-test. (B) Six-hours long treatment with 2.5 μM Cuz. (C) Six-hours long treatment with 100 μM BCS. * P < 0.05, ** P < 0.001 and *** P < 0.0001 when compared with untreated VC or NT by one-way ANOVA with Tukey’s adjustment.
This model was directly tested using cell proliferation assays. Figure 7A shows that TMEM16A-overexpressing OSC19 cells are significantly more resistant to 5 μM CDDP than control OSC19. However, in contrast with control OSC19 cells, the proliferation of TMEM16A-overexpressing cells was suppressed by low-micromolar cuprizone. Furthermore, a when the cells were pretreated with cuprizone, CDDP treatment was significantly more effective, decreasing proliferation rate to ~50% of that in control or CDDP-treated cells. The same trend persisted in FaDu cells: control FaDu cells were significantly more sensitive to cuprizone, and the latter has additive effects with platinum in these cells rather than in FaDu cells transfected with TMEM16A shRNA (Figure 7B).
Figure 7. Combination of Cuprizone and CDDP sensitizes cell death in TMEM16A-overexpressing cells.
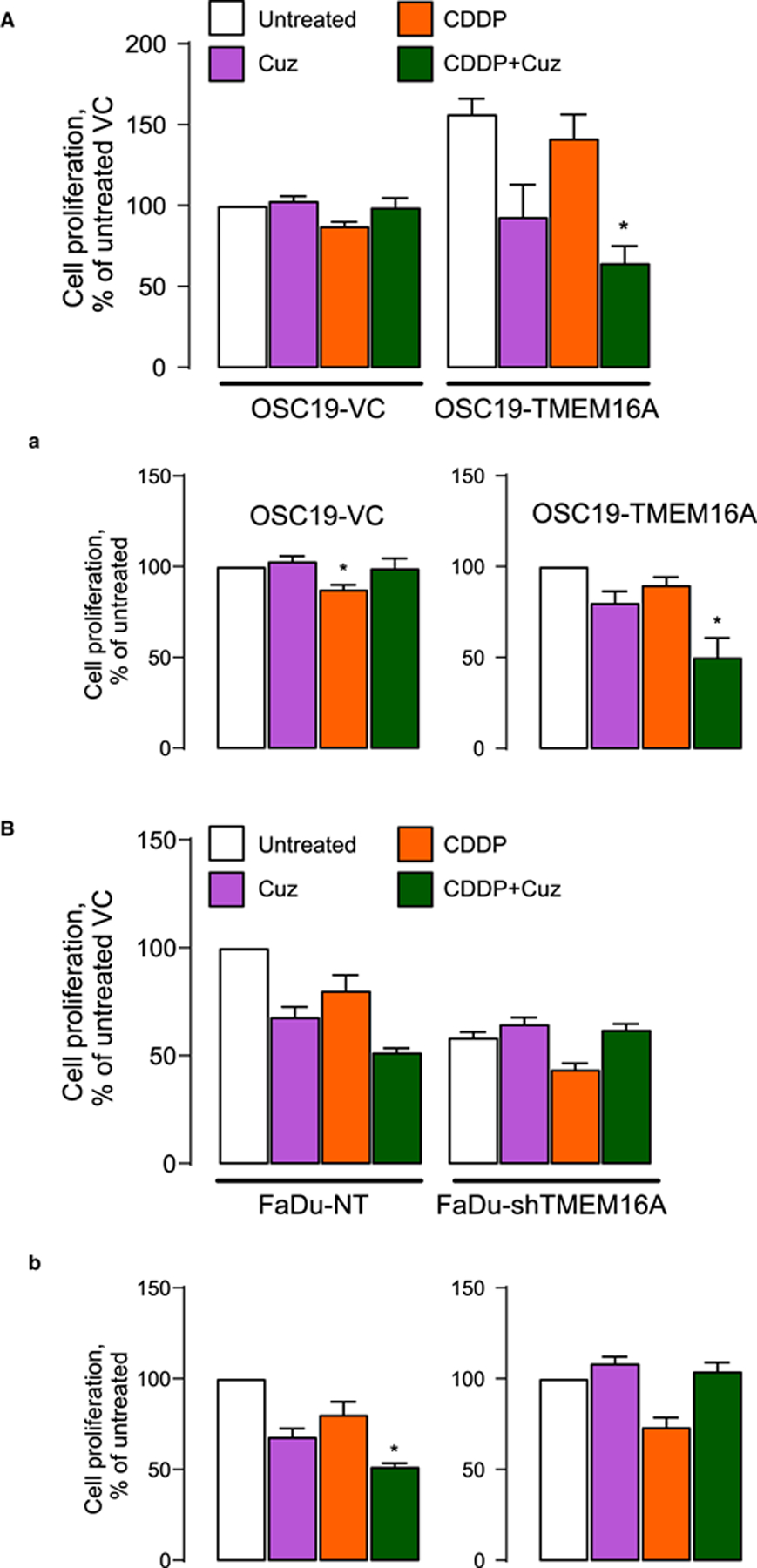
Cell proliferation was measured by WST-1 after 24-h long exposure to 10 μM CDDP with and without 6-h long pretreatment with 1 μM Cuz. (A) OSC19-VC and TMEM16A cells. Drug effects are shown as % change of proliferation relative to VC (A and a, left panel) and TMEM16A (a, right panel) cells. (B) FaDu-NT and FaDu-shTMEM16A cells. Drug effects are shown as % change of proliferation relative to NT (B and b, left panel) and shTMEM16A (b, right panel) cells.
It is important to note that the observed effects of cuprizone and BCS on both oxidative stress and ATP7B expression are TMEM16A-specific, as they only manifested in cells with high TMEM16A expression. These data suggest that oxidative stress and the subsequent liberation of copper drive ATP7B overexpression in TMEM16A-overexpressing cancer models. ATP7B was previously shown to facilitate platinum sequestration in the lysosomes, decreasing its cytoplasmic concentration and, likely, access to genomic DNA [23–25]. We propose that ATP7B up-regulation caused by ROS and copper liberation in TMEM16A-overexpressing cancer models increases the resistance of these cells to platinum (model in Figure 8). Therefore, ATP7B down-regulation by cuprizone exposes cells to pro-apoptotic effects of CDDP. This provides a mechanistic explanation for the observed effects of cuprizone and for the acquisition of platinum resistance by the TMEM16A-overexpressing cells before their exposure to platinum.
Figure 8. A model of increased CDDP resistance of TMEM16A-overexpressing cells.
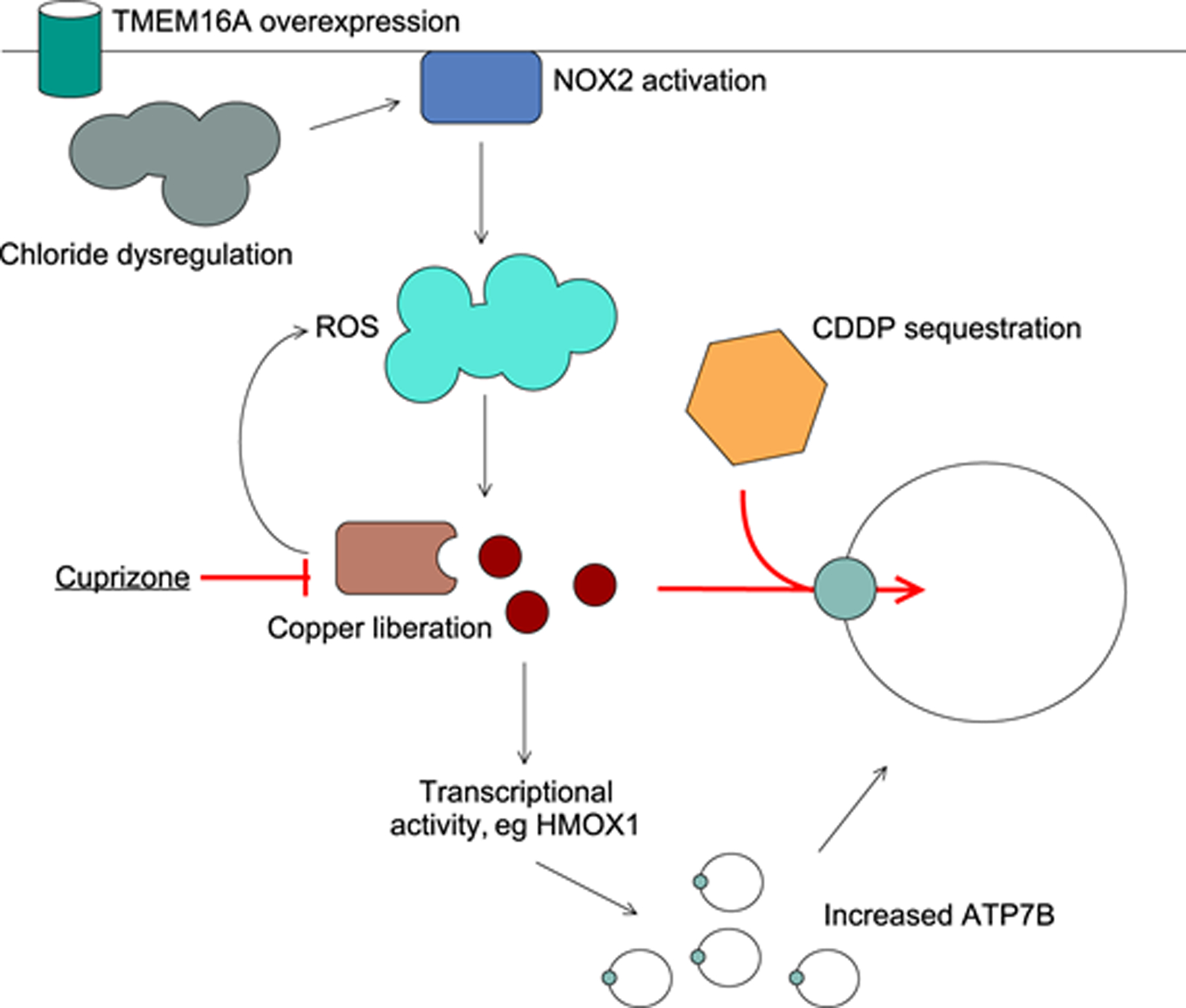
TMEM16A up-regulation affects intracellular chloride and/or activates NOX2 as shown before. NOX2 activation increases ROS leading to copper release from the cytoplasmic copper-chelating proteins. Copper stimulates ATP7B expression, increasing platinum intake into the lysosomes.
Discussion
The mechanism by which cells accumulate cisplatin is poorly understood in the current scenario and only a few kinds of transporters are known to influence the uptake and efflux of cisplatin [15]. ATP7B has been proposed to be a significant biomarker for predicting platinum-resistance in different types of cancer [7,36]. Our data demonstrates that ATP7B expression correlates with TMEM16A expression in SCCHN cell lines as well as in patient-derived xenografts.
A link between ATP7B and CDDP resistance has been known since Komatsu et al. [37] showed that prostate cells resistant to CDDP expressed higher levels of ATP7B, which affects the efflux of CDDP; and that cells transfected with ATP7B are resistant to copper and CDDP. In the current set of experiments, we show that SCCHN cells are sensitized to as low as 5 μM CDDP after ATP7B knockdown. The dynamics of cisplatin-induced reduced proliferation may be attributed to a number of factors like transport of cisplatin from extracellular space to trans-Golgi network through cytosol, lysosomal sequestration and exocytosis from the cell. However, cisplatin and its derivatives are known to be shuttled in and out of the cells through proteins involved in the metabolism of copper, most notably ATP7A and ATP7B. ATP7B has the ability to transport not only copper, but other metals. Binding of multiple metal ions like Zn2+, Hg2+, Au3+ and Cd2+ at the copper-binding domain of ATP7B has been confirmed [38]; however, the exact binding site for cisplatin on ATP7B is so far unknown.
Evidence has shown that high copper levels are correlated with rapid cell proliferation, inflammation, oxidative stress and cancer growth and metastasis [39–41] and several copper chelators, formerly intended for other purposes are being routed towards cancer therapy. Intracellular elevation of free copper is toxic because of their redox reactivity and participation in ROS metabolism. Here, we show that this detrimental effect of copper can be used to selectively eliminate high TMEM16A cells. First, cuprizone and BCS, both target the depletion of HMOX1 and NQO1, both of which are models for redox-regulated gene expression. The mechanism of action of copper agents cuprizone and BCS may stem from their high affinity for metals and eventually stripping off the Cu from cellular proteins. Cuprizone is a copper chelating agent, known for producing demyelination within the central nervous system and also affecting iron metabolism [42]. It has not yet been used to intervene or target cancer therapy. Second, cuprizone and BCS participate in redox-recycling and in elimination of hydrogen peroxide or superoxide species. Finally, combination of cisplatin and cuprizone is selectively synergistic in cells that have endogenous high TMEM16A levels. However, the anti-cancer activity of cuprizone seems to be beyond than just redox-recycling and copper chelation, since cuprizone alone is not toxic in low-TMEM16A cells.
The other metal chelator bathocuproinedisulfonic acid (BCS) seems to show a different mechanism of action compared with cuprizone, in the sense that while it might participate in redox recycling, it does not necessarily show synergistic or additive effect in cell proliferation assay along with cisplatin. Hence it is safe to hypothesize that while cuprizone has a role in regulation of Cu-ATPase ATP7B towards resistance of human cells to cisplatin, the same cannot be concluded for BCS. Our results demonstrate a more complex relationship between the resistance of cells to cisplatin and ATP7B than is currently thought.
Cancer cells exhibit increased levels of ROS and altered redox status, allowing to preferentially eliminate these cells by traditional antioxidants. However, the up-regulation of intrinsic antioxidant capacity in response to the increased ROS levels might confer resistance to pharmacological inhibitors of ROS. Hence, modulating the redox mechanism in cancer cells is a suggested target to eliminate these cells [43].
In summary, the selective killing of cisplatin-resistant high-TMEM16A SCCHN cells can be achieved using cuprizone, with the mechanism of action directed to redox cycling and elimination of ROS.
Acknowledgements
We thank Dr. L. Alex Gaither for the kind gift of the FaDU shTMEM16A cells. Human subjects: Human tumor samples were obtained from the University of Pittsburgh Medical Center in accordance with established University of Pittsburgh IRB guidelines and with the World Medical Association Declaration of Helsinki. All subjects provided written informed consent.
Funding
This work was supported in part by grants from the Department of Veterans Affairs (IO1-002345) and the National Institutes of Health (RO1-DE028343), the Myers Family foundation, PNC Foundation and the Eye & Ear Foundation (to U.D.).
Abbreviations
- BCS
bathocuproine sulphonate
- CDDP
cisplatin
- HMOX1
heme oxygenase 1
- MTF
metal-sensitive transcription factor
- NAC
N-Acetyl-Cysteine
- NOX2
NADPH oxidase 2
- NQO1
NAD(P)H Quinone Dehydrogenase 1
- NSCLC
non-small cell lung carcinoma
- SCCHN
squamous cell carcinoma of the head and neck
- TGN
trans-Golgi network
Footnotes
Competing Interests
The authors declare that there are no competing interests associated with the manuscript.
References
- 1.Marur S and Forastiere AA (2016) Head and neck squamous cell carcinoma: update on epidemiology, diagnosis, and treatment. Mayo Clin. Proc 91, 386–396 10.1016/j.mayocp.2015.12.017 [DOI] [PubMed] [Google Scholar]
- 2.Canning M, Guo G, Yu M, Myint C, Groves MW, Byrd JK et al. (2019) Heterogeneity of the head and neck squamous cell carcinoma immune landscape and its impact on immunotherapy. Front. Cell Dev. Biol 7, 52 10.3389/fcell.2019.00052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abuzeid WM, Davis S, Tang AL, Saunders L, Brenner JC, Lin J et al. (2011) Sensitization of head and neck cancer to cisplatin through the use of a novel curcumin analog. Arch. Otolaryngol. Head Neck Surg 137, 499–507 10.1001/archoto.2011.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amable L (2016) Cisplatin resistance and opportunities for precision medicine. Pharmacol. Res 106, 27–36 10.1016/j.phrs.2016.01.001 [DOI] [PubMed] [Google Scholar]
- 5.Lopez J, Ramchandani D and Vahdat L (2019) Copper depletion as a therapeutic strategy in cancer. Met. Ions Life Sci 19, 303–330 10.1515/9783110527872-018 [DOI] [PubMed] [Google Scholar]
- 6.Nakayama K, Kanzaki A, Terada K, Mutoh M, Ogawa K, Sugiyama T et al. (2004) Prognostic value of the Cu-transporting ATPase in ovarian carcinoma patients receiving cisplatin-based chemotherapy. Clin. Cancer Res 10, 2804–2811 10.1158/1078-0432.CCR-03-0454 [DOI] [PubMed] [Google Scholar]
- 7.Li YQ, Zhang XY, Chen J, Yin JY and Li XP (2018) ATP7B rs9535826 is associated with gastrointestinal toxicity of platinum-based chemotherapy in nonsmall cell lung cancer patients. J. Cancer Res. Ther 14, 881–886 10.4103/jcrt.JCRT_890_17 [DOI] [PubMed] [Google Scholar]
- 8.Wu DL, Yi HX, Sui FY, Jiang XH, Jiang XM and Zhao YY (2006) Expression of ATP7B in human gastric cardiac carcinomas in comparison with distal gastric carcinomas. World J. Gastroenterol 12, 7695–7698 10.3748/wjg.v12.i47.7695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martinez-Balibrea E, Martinez-Cardus A, Musulen E, Gines A, Manzano JL, Aranda E et al. (2009) Increased levels of copper efflux transporter ATP7B are associated with poor outcome in colorectal cancer patients receiving oxaliplatin-based chemotherapy. Int. J. Cancer 124, 2905–2910 10.1002/ijc.24273 [DOI] [PubMed] [Google Scholar]
- 10.Schmid SC, Schuster T, Horn T, Gschwend J, Treiber U and Weirich G (2013) Utility of ATP7B in prediction of response to platinum-based chemotherapy in urothelial bladder cancer. Anticancer Res. 33, 3731–3737 [PubMed] [Google Scholar]
- 11.Karginova O, Weekley CM, Raoul A, Alsayed A, Wu T, Lee SS et al. (2019) Inhibition of copper transport induces apoptosis in triple-negative breast cancer cells and suppresses tumor angiogenesis. Mol. Cancer Ther 18, 873–885 10.1158/1535-7163.MCT-18-0667 [DOI] [PubMed] [Google Scholar]
- 12.Miyashita H, Nitta Y, Mori S, Kanzaki A, Nakayama K, Terada K et al. (2003) Expression of copper-transporting P-type adenosine triphosphatase (ATP7B) as a chemoresistance marker in human oral squamous cell carcinoma treated with cisplatin. Oral Oncol. 39, 157–162 10.1016/S1368-8375(02)00038-6 [DOI] [PubMed] [Google Scholar]
- 13.Yoshizawa K, Nozaki S, Kitahara H, Ohara T, Kato K, Kawashiri S et al. (2007) Copper efflux transporter (ATP7B) contributes to the acquisition of cisplatin-resistance in human oral squamous cell lines. Oncol. Rep 18, 987–991 [PubMed] [Google Scholar]
- 14.Dmitriev OY (2011) Mechanism of tumor resistance to cisplatin mediated by the copper transporter ATP7B. Biochem. Cell Biol 89, 138–147 10.1139/O10-150 [DOI] [PubMed] [Google Scholar]
- 15.Leonhardt K, Gebhardt R, Mossner J, Lutsenko S and Huster D (2009) Functional interactions of Cu-ATPase ATP7B with cisplatin and the role of ATP7B in the resistance of cells to the drug. J. Biol. Chem 284, 7793–7802 10.1074/jbc.M805145200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Safaei R, Otani S, Larson BJ, Rasmussen ML and Howell SB (2008) Transport of cisplatin by the copper efflux transporter ATP7B. Mol. Pharmacol 73, 461–468 10.1124/mol.107.040980 [DOI] [PubMed] [Google Scholar]
- 17.Duvvuri U, Shiwarski DJ, Xiao D, Bertrand C, Huang X, Edinger RS et al. (2012) TMEM16A induces MAPK and contributes directly to tumorigenesis and cancer progression. Cancer Res. 72, 3270–3281 10.1158/0008-5472.CAN-12-0475-T [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shiwarski DJ, Shao C, Bill A, Kim J, Xiao D, Bertrand CA et al. (2014) To “grow” or “go”: TMEM16A expression as a switch between tumor growth and metastasis in SCCHN. Clin. Cancer Res 20, 4673–4688 10.1158/1078-0432.CCR-14-0363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Godse NR, Khan NI, Yochum ZA, Gomez-Casal R, Kemp C, Shiwarski DJ et al. (2017) TMEM16A/ANO1 inhibits apoptosis via down-regulation of Bim expression. Clin. Cancer Res 23, 7324–7332 10.1158/1078-0432.CCR-17-1561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamano Y, Uzawa K, Saito K, Nakashima D, Kasamatsu A, Koike H et al. (2010) Identification of cisplatin-resistance related genes in head and neck squamous cell carcinoma. Int. J. Cancer 126, 437–449 10.1002/ijc.24704 [DOI] [PubMed] [Google Scholar]
- 21.Bauer JA, Kumar B, Cordell KG, Prince ME, Tran HH, Wolf GT et al. (2007) Targeting apoptosis to overcome cisplatin resistance: a translational study in head and neck cancer. Int. J. Radiat. Oncol. Biol. Phys 69, S106–S108 10.1016/j.ijrobp.2007.05.080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li H, Wheeler S, Park Y, Ju Z, Thomas SM, Fichera M et al. (2016) Proteomic characterization of head and neck cancer patient-derived xenografts. Mol. Cancer Res 14, 278–286 10.1158/1541-7786.MCR-15-0354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Britschgi A, Bill A, Brinkhaus H, Rothwell C, Clay I, Duss S et al. (2013) Calcium-activated chloride channel ANO1 promotes breast cancer progression by activating EGFR and CAMK signaling. Proc. Natl Acad. Sci. U.S.A 110, E1026–E1034 10.1073/pnas.1217072110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Polishchuk EV and Polishchuk RS (2016) The emerging role of lysosomes in copper homeostasis. Metallomics 8, 853–862 10.1039/C6MT00058D [DOI] [PubMed] [Google Scholar]
- 25.Moinuddin FM, Shinsato Y, Komatsu M, Mitsuo R, Minami K, Yamamoto M et al. (2016) ATP7B expression confers multidrug resistance through drug sequestration. Oncotarget 7, 22779–22790 10.18632/oncotarget.8059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kalayda GV, Wagner CH, Buss I, Reedijk J and Jaehde U (2008) Altered localisation of the copper efflux transporters ATP7A and ATP7B associated with cisplatin resistance in human ovarian carcinoma cells. BMC Cancer 8, 175 10.1186/1471-2407-8-175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Agnihotry S, Dhusia K, Srivastav AK, Upadhyay J, Verma V, Shukla PK et al. (2019) Biochemical regulation and structural analysis of copper-transporting ATPase in a human hepatoma cell line for Wilson disease. J. Cell Biochem 120, 18826–18844 10.1002/jcb.29199 [DOI] [PubMed] [Google Scholar]
- 28.Nunes EA, Manieri TM, Matias AC, Bertuchi FR, da Silva DA, Lago L et al. (2018) Protective effects of neocuproine copper chelator against oxidative damage in NSC34 cells. Mutat. Res. Genet. Toxicol. Environ. Mutagen 836, 62–71 10.1016/j.mrgentox.2018.06.019 [DOI] [PubMed] [Google Scholar]
- 29.Zhang B, Georgiev O, Hagmann M, Gunes C, Cramer M, Faller P et al. (2003) Activity of metal-responsive transcription factor 1 by toxic heavy metals and H2O2 in vitro is modulated by metallothionein. Mol. Cell Biol 23, 8471–8485 10.1128/MCB.23.23.8471-8485.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peroza EA, dos Santos Cabral A, Wan X and Freisinger E (2013) Metal ion release from metallothioneins: proteolysis as an alternative to oxidation. Metallomics 5, 1204–1214 10.1039/c3mt00079f [DOI] [PubMed] [Google Scholar]
- 31.Peña KA and Kiselyov K (2015) Transition metals activate TFEB in overpexpressing cells. Biochem. J 470, 65–76 10.1042/BJ20140645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Coblentz J, St Croix C and Kiselyov K (2014) Loss of TRPML1 promotes production of reactive oxygen species: is oxidative damage a factor in mucolipidosis type IV? Biochem. J 457, 361–368 10.1042/BJ20130647 [DOI] [PubMed] [Google Scholar]
- 33.Loboda A, Damulewicz M, Pyza E, Jozkowicz A and Dulak J (2016) Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: an evolutionarily conserved mechanism. Cell. Mol. Life Sci 73, 3221–3247 10.1007/s00018-016-2223-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li L, Dong H, Song E, Xu X, Liu L and Song Y (2014) Nrf2/ARE pathway activation, HO-1 and NQO1 induction by polychlorinated biphenyl quinone is associated with reactive oxygen species and PI3K/AKT signaling. Chem. Biol. Interact 209, 56–67 10.1016/j.cbi.2013.12.005 [DOI] [PubMed] [Google Scholar]
- 35.Singh F, Charles AL, Schlagowski AI, Bouitbir J, Bonifacio A, Piquard F et al. (2015) Reductive stress impairs myoblasts mitochondrial function and triggers mitochondrial hormesis. Biochim. Biophys. Acta 1853, 1574–1585 10.1016/j.bbamcr.2015.03.006 [DOI] [PubMed] [Google Scholar]
- 36.Li YQ, Chen J, Yin JY, Liu ZQ and Li XP (2018) Gene expression and single nucleotide polymorphism of ATP7B are associated with platinum-based chemotherapy response in non-small cell lung cancer patients. J. Cancer 9, 3532–3539 10.7150/jca.26286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Komatsu M, Sumizawa T, Mutoh M, Chen ZS, Terada K, Furukawa T et al. (2000) Copper-transporting P-type adenosine triphosphatase (ATP7B) is associated with cisplatin resistance. Cancer Res. 60, 1312–1316 [PubMed] [Google Scholar]
- 38.DiDonato M, Narindrasorasak S, Forbes JR, Cox DW and Sarkar B (1997) Expression, purification, and metal binding properties of the N-terminal domain from the wilson disease putative copper-transporting ATPase (ATP7B). J. Biol. Chem 272, 33279–33282 10.1074/jbc.272.52.33279 [DOI] [PubMed] [Google Scholar]
- 39.Zhang M, Shi M and Zhao Y (2018) Association between serum copper levels and cervical cancer risk: a meta-analysis. Biosci. Rep 38,BSR20180161 10.1042/BSR20180161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stepien M, Jenab M, Freisling H, Becker NP, Czuban M, Tjonneland A et al. (2017) Pre-diagnostic copper and zinc biomarkers and colorectal cancer risk in the European Prospective Investigation into Cancer and Nutrition cohort. Carcinogenesis 38, 699–707 10.1093/carcin/bgx051 [DOI] [PubMed] [Google Scholar]
- 41.Wang F, Jiao P, Qi M, Frezza M, Dou QP and Yan B (2010) Turning tumor-promoting copper into an anti-cancer weapon via high-throughput chemistry. Curr. Med. Chem 17, 2685–2698 10.2174/092986710791859315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pandur E, Pap R, Varga E, Janosa G, Komoly S, Forizs J et al. (2019) Relationship of iron metabolism and short-term cuprizone treatment of C57BL/6 mice. Int. J. Mol. Sci 20, E2257 10.3390/ijms20092257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Trachootham D, Alexandre J and Huang P (2009) Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat. Rev. Drug Discov 8, 579–591 10.1038/nrd2803 [DOI] [PubMed] [Google Scholar]