

ABSTRACT
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has the largest RNA genome, approximately 30 kb, among RNA viruses. The DDX DEAD box RNA helicase is a multifunctional protein involved in all aspects of RNA metabolism. Therefore, host RNA helicases may regulate and maintain such a large viral RNA genome. In this study, I investigated the potential role of several host cellular RNA helicases in SARS-CoV-2 infection. Notably, DDX21 knockdown markedly accumulated intracellular viral RNA and viral production, as well as viral infectivity of SARS-CoV-2, indicating that DDX21 strongly restricts the SARS-CoV-2 infection. In addition, MOV10 RNA helicase also suppressed the SARS-CoV-2 infection. In contrast, DDX1, DDX5, and DDX6 RNA helicases were required for SARS-CoV-2 replication. Indeed, SARS-CoV-2 infection dispersed the P-body formation of DDX6 and MOV10 RNA helicases as well as XRN1 exonuclease, while the viral infection did not induce stress granule formation. Accordingly, the SARS-CoV-2 nucleocapsid (N) protein interacted with DDX1, DDX3, DDX5, DDX6, DDX21, and MOV10 and disrupted the P-body formation, suggesting that SARS-CoV-2 N hijacks DDX6 to carry out viral replication. Conversely, DDX21 and MOV10 restricted SARS-CoV-2 infection through an interaction of SARS-CoV-2 N with host cellular RNA helicases. Altogether, host cellular RNA helicases seem to regulate the SARS-CoV-2 infection.
IMPORTANCE SARS-CoV-2 has a large RNA genome, of approximately 30 kb. To regulate and maintain such a large viral RNA genome, host RNA helicases may be involved in SARS-CoV-2 replication. In this study, I have demonstrated that DDX21 and MOV10 RNA helicases limit viral infection and replication. In contrast, DDX1, DDX5, and DDX6 are required for SARS-CoV-2 infection. Interestingly, SARS-CoV-2 infection disrupted P-body formation and attenuated or suppressed stress granule formation. Thus, SARS-CoV-2 seems to hijack host cellular RNA helicases to play a proviral role by facilitating viral infection and replication and by suppressing the host innate immune system.
KEYWORDS: SARS-CoV-2, DDX21, RNA helicase, P-body, stress granule, coronavirus, nucleolus, nucleocapsid, DDX1, DDX6
INTRODUCTION
The DDX DEAD box RNA helicase family, which is an ATPase-dependent RNA helicase, is a multifunctional protein involved in all aspects of RNA life cycle, including transcription, mRNA splicing, RNA transport, translation, ribosome biogenesis, RNA decay, and viral infection (1–6). DDX3 RNA helicase has two homologs termed DDX3X and DDX3Y, which were located on the X and Y chromosomes. Actually, DDX3 is involved in translation, transcription, cell cycle, tumorigenesis (oncogenic and tumor suppressor function), viral infection, and innate immunity (7). Indeed, DDX3 is known to be a component of the antiviral innate immune signaling pathway and contributes to induction of antiviral mediators, such as type I interferon and interferon regulatory factor 3 (IRF3). On the other hand, DDX3 is required for both human immunodeficiency virus type 1 (HIV-1) and hepatitis C virus (HCV) infection and replication (7–12). In addition, we identified DDX1, DDX5, DDX17, and DDX21 RNA helicases as HIV-1 Rev-interacting proteins that enhanced the Rev-dependent RNA export function (9). DDX21/RHII/Guα was first isolated as a nucleolar RNA helicase recognized by an autoimmune antibody from a patient with watermelon stomach disease (13). DDX21 interacts with c-Jun and acts as a cofactor for c-Jun-mediated transcription (14). DDX21 regulates transcription and rRNA processing (15). DDX6 (Rck/p54) RNA helicase predominantly localizes in the discrete cytoplasmic foci called the processing body (P-body). DDX6 interacts with initiation factor eIF-4E to repress the translational activity of mRNA (16). DDX6 regulates the activity of the decapping enzymes DCP1 and DCP2 and directly interacts with Argonaute-1 (Ago1) and Ago2 in microRNA (miRNA)-induced silencing complex (miRISC) and is involved in RNA-mediated gene silencing (RNAi). Thus, P-body is an aggregate of translationally repressed mRNA associated with the translation repression and mRNA decay machinery (17, 18). In fact, DDX6 negatively regulates HIV-1 gene expression by preventing HIV-1 mRNA association with polysomes (19). In contrast, DDX6 is required for HCV replication and HIV-1 capsid assembly (12, 20–22). Furthermore, the SF1 RNA helicase Moloney leukemia virus 10 (MOV10) also localizes in P-bodies. MOV10 is incorporated into HIV-1 virion and acts as an antiviral factor (23–25).
In addition, host cells contain another type of RNA granule called the stress granule (SG) (26, 27). SGs are aggregates of untranslating mRNAs in conjunction with translation initiation factors (eIF4E, eIF3, eIF4A, eIFG, and PABP), the 40S ribosomal subunits, and several RNA-binding proteins, including poly(A)-binding protein (PABP), GTPase-activating protein (SH3 domain) binding protein 1 (G3BP1), Ataxin-2 (ATX2), T-cell intracellular antigen-1 (TIA-1), and TIA-R, and SGs regulate mRNA translation and decay, as well as proteins involved in various aspects of mRNA metabolisms. SGs are cytoplasmic phase-dense structures that occur in cells exposed to various environmental stresses, including heat, arsenite, viral infection, oxidative condition, UV irradiation, and hypoxia. SGs and P-bodies physically interact, and mRNPs may shuttle between two compartments (17, 18, 26, 27). On the other hand, several viruses target SGs and P-bodies for the viral replication (12, 17). We previously demonstrated that HCV infection disrupted P-body formation of DDX6, Lsm1, Xrn1, and Ago2 and induced SG formation of G3BP1, PABP, and ATX2 (12). HCV hijacks the P-body and stress granule components around lipid droplets (LDs) to carry out viral replication (7, 12).
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which is the causative agent of coronavirus disease 2019 (COVID-19), has infected more than 100 million people worldwide and remains a global public health problem (28–32). SARS-CoV-2 is an enveloped, positive-stranded RNA virus with the largest RNA genome, ∼30 kb, among RNA viruses and belongs to the family Coronaviridae, including SARS-CoV and Middle East respiratory syndrome coronavirus (MERS-CoV). SARS-CoV-2 encodes 16 nonstructural proteins (Nsp1 to Nsp16), nine putative accessory proteins, and four structural proteins, spike (S), envelope (E), membrane (M), and nucleocapsid (N) proteins (33). The N protein forms a helical ribonucleocapsid complex with viral genomic RNA and interacts with viral membrane protein M during assembly of virion. The S protein allows the virus to attach to and fuse with host cell membrane for viral entry. Angiotensin-converting enzyme 2 (ACE2) acts as the receptor for SARS-CoV-2 (34–36). To regulate and maintain such a large viral RNA genome, host RNA helicases may be involved in SARS-CoV-2 replication. To address this issue, in this study, I investigated the potential effect of host cellular RNA helicases on SARS-CoV-2 infection.
RESULTS
DDX21 RNA helicase restricts SARS-CoV-2 infection.
To study the potential role of host cellular RNA helicases in SARS-CoV-2 infection, I first used lentiviral vector-mediated RNA interference to stably knock down DDX1, DDX3, DDX5, DDX21, or MOV10 RNA helicase in HEK293T ACE2 cells, which constitutively express ACE2, a SARS-CoV-2 receptor (34–36). Then, I used puromycin-resistant pooled cells 10 days after the lentiviral transduction in all experiments. Western blotting showed a very effective knockdown of each RNA helicase in HEK293T ACE2 cells transduced with lentiviral vectors expressing the corresponding short hairpin RNAs (shRNAs) (Fig. 1A). Importantly, shRNAs did not affect cell viabilities (data not shown). I next examined the level of intracellular SARS-CoV-2 RNA in these knockdown cells 72 h after SARS-CoV-2 infection. Real-time LightCycler RT-PCR analysis demonstrated that the accumulation of SARS-CoV-2 RNA was significantly suppressed in DDX1 and DDX5 knockdown cells (Fig. 1B), indicating that both DDX1 and DDX5 are required for SARS-CoV-2 life cycle. In contrast, I noticed that the level of intracellular RNA was strongly enhanced in DDX21 knockdown cells (approximately 4,000-fold) compared with that of the control cells (Fig. 1B), suggesting that DDX21 strongly restricts SARS-CoV-2 infection. In addition, MOV10 RNA helicases also suppressed the SARS-CoV-2 infection. Consistent with these findings, Western blot analysis also showed that intracellular SARS-CoV-2 spike protein expression was markedly enhanced in the DDX21 knockdown cells (Fig. 1C). To further confirm the antiviral effect of DDX21, I examined DDX21 knockdown Caco-2 human colon cancer cells and HepG2 human hepatoma cells (Fig. 1D). Caco-2 cells express endogenous ACE2 and are susceptible to SARS-CoV-2 infection. The DDX21 knockdown demonstrated enhanced intracellular SARS-CoV-2 RNA in both Caco-2 and HepG2 cells (Fig. 1D). Thus, DDX21 seems to restrict SARS-CoV-2 infection.
FIG 1.

DDX21 restricts SARS-CoV-2 infection. (A) Inhibition of host cellular RNA helicase protein expression by the shRNA-producing lentiviral vector. The results of Western blot analysis of cellular lysates with anti-DDX1 (A300-521A), anti-DDX3 (A300-474A), anti-DDX6 (A300-460A), anti-DDX21 (A300-627A), anti-MOV10 (A301-571A), or anti-β-actin antibody are shown. (B) The level of intracellular SARS-CoV-2 RNA in the cells at 72 h postinfection at an MOI of 0.5 was monitored by real-time LightCycler RT-PCR. Results from three independent experiments are shown. The level of SARS-CoV-2 RNA in knockdown cells was calculated relative to the level in HEK293T ACE2 cells transduced with a control lentiviral vector (Con). *, P < 0.05 compared to control cells. Cq, quantitation cycle. (C) SARS-CoV-2 spike protein expression levels in knockdown cells. The results of the Western blot analysis of cellular lysates with anti-SARS-CoV-2 Spike (GTX632604 [1A9]) or anti-β-actin antibody in the SARS-CoV-2-infected HEK293T ACE2 cells at 72 h postinfection at an MOI of 0.5 are shown. (D) DDX21 restricts SARS-CoV-2 infection in Caco-2 and HepG2 cells. Inhibition of endogenous DDX21 protein expression by the shRNA-producing lentiviral vector. The results of Western blot analysis of cellular lysates with anti-DDX21 or anti-β-actin antibody in Caco-2 and HepG2 cells are shown. The level of intracellular SARS-CoV-2 RNA in the cells at 72 h postinfection at an MOI of 0.5 was monitored by real-time LightCycler RT-PCR. Results from three independent experiments are shown. The level of SARS-CoV-2 RNA in DDX21 knockdown cells was calculated relative to the level in HEK293T ACE2 cells transduced with a control lentiviral vector (Con). *, P < 0.05 compared to control cells.
I next examined the levels of extracellular SARS-CoV-2 nucleocapsid (N) protein and the level of extracellular SARS-CoV-2 RNA as well as the infectivity of SARS-CoV-2 in the culture supernatants in these knockdown cells 72 h after SARS-CoV-2 infection. The results showed that the release of SARS-CoV-2 N and the accumulation of extracellular SARS-CoV-2 RNA were significantly enhanced in DDX21 knockdown cells (Fig. 2A and B). In this context, the virus titer and the infectivity of SARS-CoV-2 in the culture supernatants were also significantly enhanced in these knockdown cells (Fig. 2C and D). Importantly, I noticed that the viral titer in the culture supernatants of DDX21 knockdown cells was elevated approximately 10,000-fold compared with that of control cells, while the levels of extracellular SARS-CoV-2 N protein and extracellular SARS-CoV-2 RNA in the supernatants of DDX21 knockdown cells were elevated only 5-fold compared with those of control cells (Fig. 2A to D), suggesting that DDX21 suppresses the viral RNA replication, the viral production, and the viral infectivity. Then, I examined the subcellular localization of SARS-CoV-2 N protein in the control and DDX21 knockdown HEK293T ACE2 cells 48 h after inoculation of SARS-CoV-2. The SARS-CoV-2 N protein predominantly localized in cytoplasm; however, there was no clear difference between the control and DDX21 knockdown cells (Fig. 2E). Furthermore, I examined the subcellular localization of endogenous DDX21 and SARS-CoV-2 N in both Vero E6 TMPRSS2 and HEK293T ACE2 cells 24 h after inoculation of SARS-CoV-2. DDX21 predominantly localized in nucleoli and SARS-CoV-2 N localized in cytoplasm in SARS-CoV-2-infected cells (Fig. 2F).
FIG 2.

Characterization of antiviral effect of DDX21. (A) DDX21 suppresses SARS-CoV-2 production. The levels of extracellular SARS-CoV-2 N protein in the culture supernatants from the DDX21 knockdown HEK293T ACE2 cells 72 h after inoculation of SARS-CoV-2 at an MOI of 0.5 were determined by ELISA. Experiments were done in triplicate, and columns show the mean SARS-CoV-2 N protein levels. *, P < 0.05 compared to control cells. (B) DDX21 inhibits the level of extracellular SARS-CoV-2 RNA. The levels of extracellular SARS-CoV-2 RNA in the culture supernatants from the DDX21 knockdown HEK293T ACE2 cells 72 h after inoculation of SARS-CoV-2 at an MOI of 0.5 were monitored by real-time LightCycler RT-PCR. Results from three independent experiments are shown. The level of SARS-CoV-2 RNA in DDX21 knockdown cells was calculated relative to the level in HEK293T ACE2 cells transduced with a control lentiviral vector (Con). *, P < 0.05 compared to control cells. (C) The virus titer of SARS-CoV-2 in the culture supernatants from the DDX21 knockdown HEK293T ACE2 cells 72 h after inoculation of SARS-CoV-2. Naive Vero E6 TMPRSS2 cells were seeded in 24-well plates at 5 × 104 cells per well and then infected the next day with the indicated serial 10-fold dilutions of culture supernatants. The cells were stained with 0.6% Coomassie brilliant blue in 50% methanol and 10% acetate at 72 h postinfection were monitored for cytopathic effect. The virus titer was determined as TCID50/mL. (D) The infectivity of SARS-CoV-2 in the culture supernatants from the control or DDX21 knockdown HEK293T ACE2 cells 72 h after inoculation of SARS-CoV-2 was compared by immunofluorescence. Naive Vero E6 TMPRSS2 cells were plated on Lab-Tek 2-well chamber slides at 2 × 104 cells per well. The next day, 1 μL of culture supernatants of SARS-CoV-2-infected control or DDX21 knockdown HEK293T ACE2 cells was inoculated. The cells were fixed at 24 h postinfection and stained with anti-SARS-CoV-2 nucleocapsid (ab273434 [6H3]). Cells were then stained with donkey anti-mouse IgG (H+L) Alexa Fluor 594-conjugated secondary antibody. Images were visualized using confocal laser scanning microscopy. Nuclei were stained with DAPI (blue). (E) Subcellular localization of SARS-CoV-2 N protein in control or DDX21 knockdown HEK293T ACE2 cells 24 h after inoculation of SARS-CoV-2. The cells were stained with anti-SARS-CoV-2 nucleocapsid. (F) Subcellular localization of endogenous DDX21 and SARS-CoV-2 N protein in Vero E6 TMPRSS2 or HEK293T ACE2 cells 24 h after inoculation of SARS-CoV-2. The cells were stained with anti-SARS-CoV-2 nucleocapsid and anti-DDX21 (A300-627A) antibodies. Cells were then stained with donkey anti-rabbit IgG (H+L) Alexa Fluor 488-conjugated secondary antibody and donkey anti-mouse IgG (H+L) Alexa Fluor 594-conjugated secondary antibody. The two-color overlay images are also exhibited (Merged).
I next attempted to clarify the molecular mechanisms by which DDX21 restricts the viral life cycle of SARS-CoV-2. For this, I compared the time course of the viral growth between the control and DDX21 knockdown cells at 24 h, 48 h, and 72 h postinfection (hpi). Although there remained about a 8-fold enhancement at 24 hpi, the viral growth was markedly enhanced in the DDX21 knockdown cells at 72 hpi (Fig. 3A). In keeping with this, I noticed formation of a few syncytia at 48 hpi and then many evident syncytia in the DDX21 knockdown cells at 72 hpi (Fig. 3B), suggesting that DDX21 inhibits the viral propagation and spread.
FIG 3.
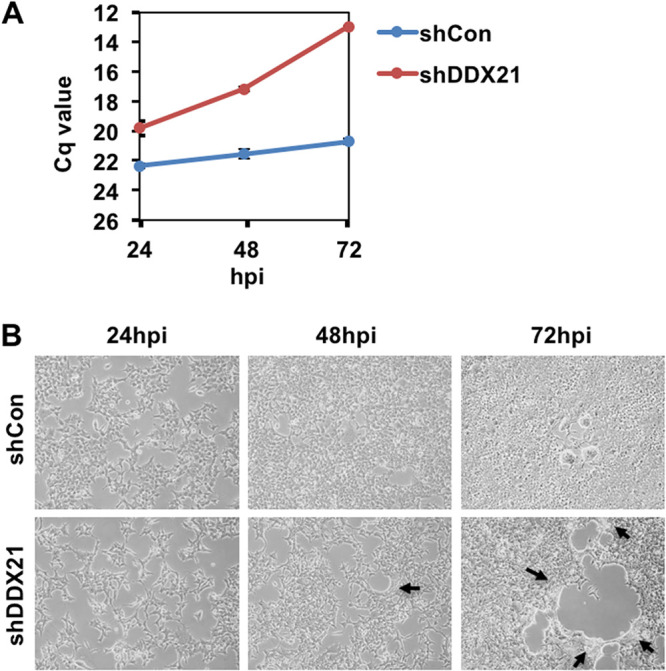
DDX21 knockdown facilitates SARS-CoV-2 propagation and spread. (A) SARS-CoV-2 RNA multiplication in control (shCon) or DDX21 knockdown HEK293T ACE2 cells (shDDX21) (2 × 105 cells/well) at the indicated time postinfection with SARS-CoV-2 (2.7 × 105 TCID50/μL), as determined by real-time RT-PCR. Cq, quantitation cycle. (B) SARS-CoV-2-infected control (shCon) or DDX21 knockdown HEK293T ACE2 cells (shDDX21) at the indicated time postinfection with SARS-CoV-2, as in panel A. Black arrows indicate syncytium formation.
SARS-CoV-2 disrupts P-body formation and hijacks host cellular RNA helicases.
To investigate the potential role of P-body components in SARS-CoV-2 infection, I first examined the alteration of subcellular localization of DDX6 RNA helicase by SARS-CoV-2 infection using confocal laser scanning microscopy. As we previously described (12), DDX6 predominantly localized in the evident cytoplasmic foci termed P-bodies in both uninfected naive Vero E6 TMPRSS2 and HEK293T ACE2 cells (Fig. 4). Notably, SARS-CoV-2 infection disrupted the P-body formation of DDX6 in both cell types at 24 h postinfection (Fig. 4). Indeed, endogenous DDX6 dispersed in the cytoplasm and colocalized with SARS-CoV-2 N in response to the SARS-CoV-2 infection (Fig. 4 and 5A). Therefore, I further examined whether SARS-CoV-2 disrupts the P-body formation of other P-body components, including MOV10 RNA helicase and the 5′-3′ exonuclease Xrn1. As expected, SARS-CoV-2 infection disrupted the P-body formation of MOV10 and XRN1 (Fig. 4). I also observed that most of DDX6 still formed intact P-bodies at an earlier time (6 h postinfection) (Fig. 4A). Then, I noticed that P-body formation of DDX6 began to be disrupted at 24 h postinfection (Fig. 5A).
FIG 4.

SARS-CoV-2 disrupts P-body formation. Uninfected Vero E6 TMPRSS2 or HEK293T ACE2 cells and their SARS-CoV-2-infected cells at 24 h postinfection were stained with anti-SARS-CoV-2 nucleocapsid (ab273434 [6H3]) and anti-DDX6 (A300-460A) antibodies. The cells were also stained with anti-SARS-CoV-2 nucleocapsid and either anti-Xrn1 (A300-443A) or anti-MOV10 (A301-571A) antibodies. Cells were then stained with donkey anti-rabbit IgG (H+L) Alexa Fluor 488-conjugated secondary antibody and donkey anti-mouse IgG (H+L) Alexa Fluor 594-conjugated secondary antibody. Images were visualized using confocal laser scanning microscopy. The two-color overlay images are also exhibited (Merged). Nuclei were stained with DAPI (blue).
FIG 5.

SARS-CoV-2 hijacks DDX6 for viral replication. (A) Dynamic redistribution of DDX6 in response to SARS-CoV-2 infection. Vero E6 TMPRSS2 cells at the indicated times after inoculation of SARS-CoV-2 were stained with anti-SARS-CoV-2 nucleocapsid (ab273434 [6H3]) and anti-DDX6 (A300-460A) antibodies. (B) SARS-CoV-2 does not induce stress granule formation. Uninfected Vero E6 TMPRSS2 or SARS-CoV-2-infected cells at 24 h postinfection were incubated at 37°C. Uninfected cells were also incubated at 43°C for 45 min. Cells were then stained with anti-SARS-CoV-2 nucleocapsid and anti-G3BP1 (A302-033A) antibodies. (C) Host protein expression levels in response to SARS-CoV-2 infection. The results of the Western blot analysis of cellular lysates with anti-SARS-CoV-2 spike (GTX632604 [1A9]), anti-DDX6, anti-G3BP1, or anti-β-actin antibody in the SARS-CoV-2-infected Vero E6 TMPRSS2 or the HEK293T ACE2 cells at 24 h postinfection at an MOI of 0.5 as well as in the uninfected cells are shown. (D) Requirement of DDX6 for SARS-CoV-2 infection. Inhibition of endogenous DDX6 protein expression by the shRNA-producing lentiviral vector. The results of Western blot analysis of cellular lysates with anti-DDX6 or anti-β-actin antibody are shown. The level of intracellular SARS-CoV-2 RNA in the cells at 72 h postinfection at an MOI of 0.5 was monitored by real-time LightCycler RT-PCR. Results from three independent experiments are shown. The level of SARS-CoV-2 RNA in the DDX6 knockdown cells was calculated relative to the level in HEK293T ACE2 cells transduced with a control lentiviral vector (Con). *, P < 0.05 compared to control cells.
I next examined whether SARS-CoV-2 infection could affect the stress granule formation of G3BP1. The stress granule component G3BP1 dispersed in the cytoplasm at 37°C, while G3BP1 formed stress granules in response to heat shock at 43°C for 45 min in uninfected naive Vero E6 TMPRSS2 cells (Fig. 5B). In contrast, stress granules were not formed in the SARS-CoV-2-infected cells at 24 h postinfection (Fig. 5B), suggesting that SARS-CoV-2 infection does not induce or suppresses stress granule formation. Accordingly, Western blotting clearly demonstrated that the SARS-CoV-2 infection did not affect the level of endogenous DDX6 and G3BP1 protein expression (Fig. 5C), indicating that SARS-CoV-2 does not induce the degradation of DDX6 and G3BP1. To investigate the potential role of DDX6 in SARS-CoV-2 infection, I examined lentiviral-vector-mediated RNA interference to stably knock down DDX6 in HEK293T ACE2 cells. Western blot analysis for DDX6 demonstrated a very effective knockdown of DDX6 in HEK293T ACE2 cells transduced with lentiviral vectors expressing shRNA targeting human DDX6 (Fig. 5D). Importantly, shRNAs did not affect cell viabilities (data not shown). I next examined the level of intracellular SARS-CoV-2 RNA in the DDX6 knockdown cells 72 h after SARS-CoV-2 infection at a multiplicity of infection (MOI) of 0.5. The results showed that the accumulation of SARS-CoV-2 RNA was significantly suppressed in DDX6 knockdown cells (Fig. 5D), indicating that DDX6 is required for SARS-CoV-2 infection. Thus, these results suggested that SARS-CoV-2 disrupts the P-body formation of DDX6 and then hijacks DDX6 for its own replication.
SARS-CoV-2 N disrupts the P-body formation and hijacks host cellular RNA helicases.
Finally, I examined whether SARS-CoV-2 N protein interacts with host cellular RNA helicases and affects their subcellular localization, since SARS-CoV-2 N protein is known to bind to the SARS-CoV-2 genomic RNA (32, 33). The SARS-CoV-2 N protein predominantly localized in the cytoplasm. I also noticed that SARS-CoV-2 N partially localized in the nucleus as well as in nucleoli in 293T cells transfected with pcDNA3.1 SARS-CoV-2 N (37) (Fig. 6A and D). Interestingly, SARS-CoV-2 N disrupted P-body formation of endogenous and ectopically expressed DDX6 (Fig. 6A and B), suggesting that SARS-CoV-2 N interacts with DDX6. Indeed, both endogenous and ectopically expressed DDX6 dispersed in the cytoplasm and colocalized with SARS-CoV-2 N in 293T cells transfected pcDNA3.1 SARS-CoV-2 N (Fig. 6A and B). As well, SARS-CoV-2 N disrupted the P-body formation of hemagglutinin (HA)-tagged MOV10 and colocalized in the cytoplasm in 293T cells cotransfected with pcDNA3.1 SARS-CoV-2 N and pcDNA3-HA-MOV10 (Fig. 6C). Notably, endogenous DDX21 partially colocalized with SARS-CoV-2 N in nucleoli (Fig. 6D), indicating that DDX21 interacts with SARS-CoV-2 N. Furthermore, SARS-CoV-2 N colocalized with HA-tagged DDX1 when both proteins were coexpressed in 293T cells (Fig. 6E). In contrast, SARS-CoV-2 N did not colocalize with HA-tagged DDX3 (Fig. 6F). Therefore, these results suggested that SARS-CoV-2 N interacts with DDX6, DDX21, and MOV10 RNA helicases.
FIG 6.

SARS-CoV-2 nucleocapsid (N) protein disrupts the P-body formation of DDX6 and MOV10. (A) Disruption of P-body formation of endogenous DDX6 by ectopically expressed SARS-CoV-2 N. 293T cells transfected with 200 ng of pcDNA3.1-SARS-CoV-2 N (37) were stained with anti-SARS-CoV-2 nucleocapsid (GTX632269 [6H3]) and anti-DDX6 (A300-460A) antibodies. (B) Disruption of P-body formation of HA-tagged DDX6 by ectopically expressed SARS-CoV-2 N. 293T cells cotransfected with 200 ng of pcDNA3-HA-DDX6 (9) and either 200 ng of pcDNA3.1-SARS-CoV-2 N or pcDNA3 were stained with anti-SARS-CoV-2 nucleocapsid and anti-HA (3F10) antibodies. (C) Disruption of P-body formation of HA-tagged MOV10 by SARS-CoV-2 N and colocalization of SARS-CoV-2 N and HA-MOV10. 293T cells cotransfected with 200 ng of pcDNA3-HA-MOV10 and either 200 ng of pcDNA3.1-SARS-CoV-2 N or pcDNA3 were stained with anti-SARS-CoV-2 nucleocapsid and anti-HA antibodies. (D) Colocalization of endogenous DDX21 and ectopically expressed SARS-CoV-2 N in nucleoli. 293T cells transfected with 200 ng of pcDNA3.1-SARS-CoV-2 N were stained with anti-SARS-CoV-2 nucleocapsid and anti-DDX21 (A300-627A) antibodies. (E) Colocalization of HA-tagged DDX1 and SARS-CoV-2 N. 293T cells cotransfected with 200 ng of pcDNA3-HA-DDX1 (9) and either 200 ng of pcDNA3.1-SARS-CoV-2 N or pcDNA3 were stained with anti-SARS-CoV-2 nucleocapsid and anti-HA antibodies. (F) Subcellular localization of HA-tagged DDX3 and SARS-CoV-2 N. 293T cells cotransfected with 200 ng of pHA-DDX3 (8–12) and either 200 ng of pcDNA3.1-SARS-CoV-2 N or pcDNA3 were stained with anti-SARS-CoV-2 nucleocapsid and anti-HA antibodies.
To examine whether SARS-CoV-2 N binds to DDX21, DDX6, and MOV10, the lysates of either SARS-CoV-2-infected Vero E6 TMPRSS2 cells (Fig. 7A) or 293T cells transfected with pcDNA3.1 SARS-CoV-2 N (Fig. 7B) were immunoprecipitated with anti-SARS-CoV-2 N, anti-DDX21, anti-DDX6, or anti-MOV10 antibody, followed by Western blotting with anti-SARS-CoV-2 N, anti-DDX21, anti-DDX6, or anti-MOV10 antibody. Consequently. SARS-CoV-2 N coimmunoprecipitated with endogenous DDX21, DDX6, and MOV10 in both SARS-CoV-2-infected Vero E6 TMPRSS2 cells (Fig. 7A) and SARS-CoV-2 N-expressing 293T cells (Fig. 7B), indicating that SARS-CoV-2 N binds to DDX21, DDX6, and MOV10. To further confirm this, the lysates of 293T cells cotransfected with pcDNA3.1 SARS-CoV-2 N and/or pcDNA3-HA-DDX1, pHA-DDX3, pcDNA3-HA-DDX5, pcDNA3-HA-DDX6, pcDNA3-HA-DDX21, and pcDNA3-HA-MOV10 were immunoprecipitated with anti-SARS-CoV-2 N antibody, followed by Western blotting with anti-SARS-CoV-2 N and anti-HA antibody. Consequently, SARS-CoV-2 N coimmunoprecipitated with HA-tagged DDX1, DDX3, DDX5, DDX6, DDX21, and MOV10 (Fig. 7C).
FIG 7.

SARS-CoV-2 N binds to host cellular RNA helicases. (A) Vero E6 TMPRSS2 cells (5 × 105 cells/well) were infected with SARS-CoV-2 at an MOI of 0.5. The cell lysates were collected at 24 h postinfection. (B) 293T cells (2 × 105 cells/well) were transfected with 4 μg of pcDNA3.1-SARS-CoV-2 N. The cell lysates were immunoprecipitated with anti-SARS-CoV-2 nucleocapsid (GTX632269 [6H3]), anti-DDX21 (A300-627A), anti-DDX6 (A300-460A), or anti-MOV10 (A301-571A) antibody, followed by immunoblotting analysis using anti-SARS-CoV-2 nucleocapsid, anti-DDX21, anti-DDX6, and anti-MOV10 antibodies, respectively. Both short-exposure and long-exposure images are shown. (C) 293T cells (2 × 106 cells in a 10-cm dish) were cotransfected with 10 μg of pcDNA3-HA-DDX1, pHA-DDX3, pcDNA3-HA-DDX5, pcDNA3-HA-DDX6, pcDNA3-HA-DDX21, or pcDNA3-HA-MOV10 and/or 15 μg of pcDNA3.1-SARS-CoV-2 N. The cell lysates were immunoprecipitated with anti-SARS-CoV-2 nucleocapsid (GTX632269 [6H3]) antibody, followed by immunoblotting analysis using anti-SARS-CoV-2 nucleocapsid and anti-HA (3F10) antibodies, respectively. WCL, whole-cell lysate; IP, immunoprecipitation.
Furthermore, RNase treatment did not affect the interaction of SARS-CoV-2 N with DDX1, DDX6, and DDX21, unlike that with MOV10 (Fig. 8), indicating that SARS-CoV-2 N interacts with DDX1, DDX6, and DDX21 in an RNA-independent manner. In contrast, SARS-CoV-2 N bound to MOV10 in an RNA-dependent manner. Moreover, SARS-CoV-2 N could bind to the DDX21 helicase mutants DEV and SAT, like the wild type (WT) (Fig. 8C). DDX21 contains multiple functional domains, including the N-terminal basic-acidic domain (residues 31 to 153), the central helicase (HEL) domain (residues 186 to 568), dimerization domain (DD) (residues 568 to 620), Gu C-terminal (GUCT) domain (residues 620 to 704), and a C-terminal basic tail (FRGQR/PRGQR repeats) domain (residues 704 to 781). To determine which DDX21 functional domain is required for interaction with SARS-CoV-2 N, I used several DDX21 deletion mutants (proteins containing residues 1 to 704, 1 to 185, 568 to 781, and 621 to 781) (Fig. 9A). Consequently, both 568–781 and 621–781 mutants could bind to SARS-CoV-2 N (Fig. 9B). In contrast, the truncated 1–185 and 1–704 mutants failed to bind it (Fig. 9B), suggesting that the C-terminal FRGQR/PRGQR repeat domain is required for binding with SARS-CoV-2 N. The wild-type DDX21 formed a ring structure in nucleoli (Fig. 9C). Interestingly, the truncated 1–185 mutant did not form the ring structure or localize in nucleoli; however, the other truncated mutants, the 1–704, 568–751, and 621–781 mutants, formed the ring structure in the nucleoli (Fig. 9C). Thus, the C-terminal GUCT domain seems to be required for the nucleolar ring structure formation.
FIG 8.
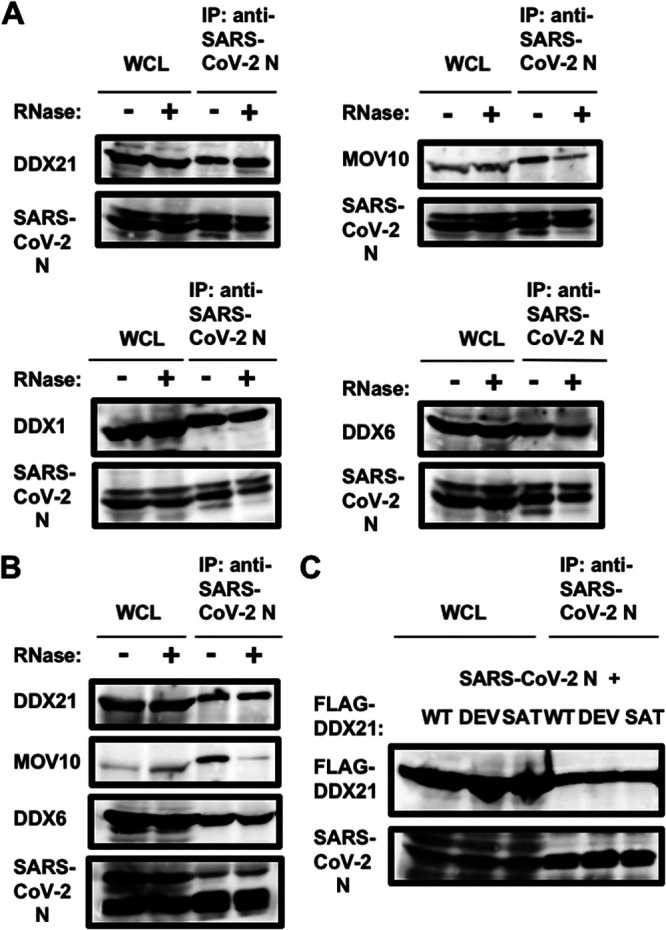
SARS-CoV-2 N binds to DDX1, DDX6, and DDX21 but not MOV10 in an RNA-independent manner. (A) Vero E6 TMPRSS2 cells (5 × 105 cells/well) were infected with SARS-CoV-2 at an MOI of 0.5. The cell lysates were collected at 24 h postinfection. The cell lysates were treated with or without 100 μg of RNase A (Nacalai Tesque) and then immunoprecipitated with anti-SARS-CoV-2 nucleocapsid (GTX632269 [6H3]) antibody, followed by immunoblotting analysis using anti-SARS-CoV-2 nucleocapsid, anti-DDX1 (GTX105205 [N3C2]), anti-DDX21, anti-DDX6, and anti-MOV10 antibodies, respectively. (B) 293T cells (2 × 106 cells in a 10-cm dish) were transfected with 25 μg of pcDNA3.1-SARS-CoV-2 N. The cell lysates were treated with or without 100 μg of RNase A (Nacalai Tesque) and then were immunoprecipitated with anti-SARS-CoV-2 nucleocapsid (GTX632269 [6H3]) antibody, followed by immunoblotting analysis using anti-SARS-CoV-2 nucleocapsid, anti-DDX21, anti-DDX6, and anti-MOV10 antibodies, respectively. (C) 293T cells (2 × 106 cells in a 10-cm dish) were cotransfected with 10 μg of p23-DDX21 WT (FLAG), p23-DDX21 DEV (FLAG), or p23-DDX21 SAT (FLAG) (65) and/or 15 μg of pcDNA3.1-SARS-CoV-2 N. The cell lysates were immunoprecipitated with anti-SARS-CoV-2 nucleocapsid (GTX632269 [6H3]) antibody, followed by immunoblotting analysis using anti-SARS-CoV-2 nucleocapsid and anti-FLAG (M2) antibodies, respectively.
FIG 9.
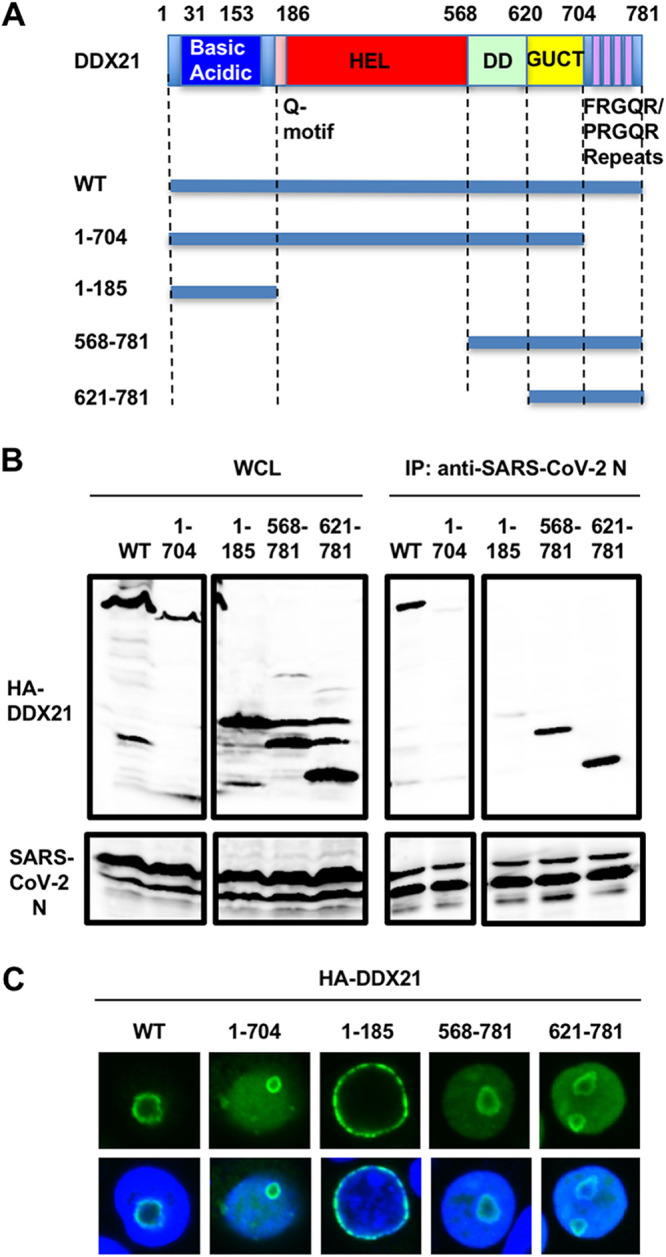
The C-terminal domain on DDX21 is essential for binding with SARS-CoV-2 N. (A) Schematic representation of DDX21 and the deletion mutants used in the present study. The basic acidic domain, Q motif, helicase (HEL), dimerization domain (DD), Gu C terminus (GUCT), and a C-terminal basic tail (FRGQR/PRGQR repeats) are indicated. (B) 293T cells (2 × 106 cells in a 10-cm dish) were cotransfected with 10 μg of pcDNA3-HA-DDX21 WT, 1–704, 1–185, 568–781, and 621–781 and/or 15 μg of pcDNA3.1-SARS-CoV-2 N. The cell lysates were immunoprecipitated with anti-SARS-CoV-2 nucleocapsid (GTX632269 [6H3]) antibody, followed by immunoblotting analysis using anti-SARS-CoV-2 nucleocapsid and anti-HA (3F10) antibodies, respectively. (C) Subcellular localization of DDX21 and the deletion mutants used in this study. 293T cells transfected with 200 ng of pcDNA3-HA-DDX21 (9) or the deletion mutant-expressing plasmid were stained with anti-HA (3F10) antibody. Nuclei were stained with DAPI (blue).
Finally, I compared the amino acid sequences of SARS-CoV-2 N from three different coronaviruses, including SARS-CoV, SARS-CoV-2, and avian infectious bronchitis virus (IBV) (Fig. 10). The SARS-CoV-2 N protein is a homodimer composed of 419 amino acids and contains two functional domains, the N-terminal RNA-binding domain (NTD, also called the RBD; residues 43 to 174) and the C-terminal dimerization domain (CTD, also called the DD; residues 247 to 366) as well as the linker domain (residues 174 to 247) (Fig. 10A). The N proteins of different coronaviruses share a very low degree of sequence homology. Nevertheless, I noticed the conserved sequences among three different coronaviruses in the NTD (Fig. 10B).
FIG 10.

The N-terminal RNA-binding domain of the nucleocapsid protein is conserved among the coronavirus family. (A) Schematic representation of SARS-CoV-2 nucleocapsid. (B) Alignment of the N-terminal RNA-binding and the C-terminal dimerization domains from different coronavirus N proteins: SARS-CoV (P59595, group 2b), SARS-CoV-2 (P0DTC9.1), and avian infectious bronchitis virus (IBV) strain Beaudette (P69596, group 3).
Altogether, SARS-CoV-2 N seems to interact with and hijack several host cellular RNA helicases.
DISCUSSION
Several RNA viruses are known to carry their own RNA helicases to facilitate the replication of their viral genomes, including alphavirus, coronavirus, flavivirus, HCV, and rubella virus; however, HIV-1 does not carry its own RNA helicase (38, 39). Actually, SARS-CoV-2 encodes Nsp13 RNA helicase. Nsp13 RNA helicase is conserved in all coronaviruses, including SARS-CoV, SARS-CoV-2, and MERS-CoV, and is a key player in viral replication. Interestingly, a recent review by Squeglia et al. proposed the structural similarity of SARS-CoV-2 Nsp13 RNA helicase with human DDX helicases (40). SARS-CoV-2 has the largest RNA genome, ∼30 kb, among RNA viruses. To regulate and maintain such a large viral RNA genome, host RNA helicases may be involved in SARS-CoV-2 replication.
In this study, I have demonstrated that several host cellular RNA helicases, including DDX1, DDX5, DDX6, DDX21, and MOV10, facilitate or restrict SARS-CoV-2 infection. Indeed, the DDX21 knockdown markedly accumulated intracellular viral RNA in the SARS-CoV-2-infected HEK293T ACE2 cells (approximately 4,000-fold) compared with that of control cells (Fig. 1B), suggesting that DDX21 strongly restricts SARS-CoV-2 infection. The SARS-CoV-2 N protein binds to the viral RNA genome and plays a multifunctional role in the SARS-CoV-2 viral life cycle, from regulation of viral replication and transcription and viral genome packaging to modulation of host cell processes. Recent interactome analysis demonstrated that infectious bronchitis virus (IBV) N binds to DDX1, DDX3, DDX5, DDX6, DDX21, and MOV10 (41). The avian IBV also belongs to the family Coronaviridae and causes avian infectious bronchitis, a highly contagious disease. IBV N protein localizes in the cytoplasm and nucleoli (41). Consistent with this, SARS-CoV-2 N localized in cytoplasm and nucleoli and colocalized with DDX21 in nucleoli (Fig. 6D). SARS-CoV-2 N also coimmunoprecipitated with DDX1, DDX3, DDX5, DDX6, DDX21, and MOV10 (Fig. 7). Indeed, SARS-CoV-2 N bound to DDX1, DDX6, and DDX21 in an RNA-independent manner (Fig. 8). In contrast, SARS-CoV-2 N bound to MOV10 in an RNA-dependent manner (Fig. 8). Accordingly, the C-terminal FRGQR/PRGQR repeat domain on DDX21 is required for the binding with SARS-CoV-2 N (Fig. 9B). Furthermore, recent interactome analysis also supports my finding that SARS-CoV-2 N binds to DDX21 (Fig. 7) (42). However, the effect of this interaction on SARS-CoV-2 replication was unable to be determined by interactome analysis alone.
In this study, I clarified the multiple roles of DDX21 in SARS-CoV-2 infection. DDX21 is involved in multiple functions, including transcription, processing, and modification of pre-rRNA as well as innate immunity. Goodier et al. reported that MOV10 associates with the LINE-1 ribonucleoprotein (RNP), along with other RNA helicases, including DDX5, DHX9, DDX17, DDX21, and DDX39 (43). Moreover, Zhang et al. reported that DDX1, DDX21, and DHX36 helicases form a complex with the adaptor molecule TRIF to sense double-strand RNA (dsRNA) in dendritic cells (44). Thus, the DDX1-DDX21-DHX36 complex participates in innate immunity. Therefore, DDX21 may act as an antiviral protein. In fact, it has been reported that DDX21 restricts infection and replication of several viruses, including influenza virus, dengue virus, Borna disease virus (BDV), and human cytomegalovirus (HCMV) (45–48). DDX21 regulates viral replication through various mechanisms, such as suppressing viral genome replication, inhibiting virion assembly and release, and modulating antiviral innate immune responses (45–48). In this regard, I noticed that the DDX21 knockdown accumulated intracellular viral RNA (Fig. 1B), extracellular SARS-CoV-2 N protein, and extracellular SARS-CoV-2 RNA and that the viral titer and viral infectivity were elevated in the supernatants of DDX21 knockdown cells (Fig. 2A to D), with induction of many evident syncytia at 72 hpi (Fig. 3B). Therefore, DDX21 may be involved in multiple steps of the SARS-CoV-2 life cycle, including intracellular viral RNA replication, viral production, viral infectivity, viral propagation, and viral spread.
Importantly, the SARS-CoV-2 N interactors DDX1, DDX3, DDX5, DDX6, DDX21, and MOV10 overlapped with IBV N interactors, suggesting that these cellular RNA helicases regulate both SARS-CoV-2 and IBV replication. In this regard, I found conserved sequences among three coronaviruses, SARS-CoV, SARS-CoV-2, and IBV, in the NTD of the N protein (Fig. 10B), suggesting that SARS-CoV-2 N binds to the host cellular RNA helicase through the conserved sequences in the NTD. However, it has not been shown that SARS-CoV-2 N protein interacts with these helicases in the other two reported interactome analyses of SARS-CoV-2 (49, 50). The discrepancy may be explained by differences in experimental materials and methods, such as the difference in the cells used. I used HEK293T ACE2 cells for the knockdown studies; however, these two groups used Vero E6 monkey kidney cells and Huh7.5 hepatoma cells for CRISPR screening (49, 50). Consistent with my methods, Gordon et al., who reported that SARS-CoV-2 N binds to DDX21 as well as MOV10 by a proteomic analysis, used HEK-293T/17 cells (42). Furthermore, the interactome analysis by Emmott et al. showing that IBV N bound to DDX1, DDX3, DDX5, DDX6, DDX21, and MOV10 (41) used 293T cells. There are SARS-CoV-2 protein interactors that are essential for one or more of the other coronaviruses but not essential for SARS-CoV-2 (51). For example, DDX10 interacted with SARS-CoV-2 NSP8, while DDX10 was apparently not essential for SARS-CoV-2 replication (42, 51). In this study, I found that SARS-CoV-2 N bound to DDX3 by coimmunoprecipitation analysis (Fig. 7C); however, DDX3 was not essential for the SARS-CoV-2 replication in HEK293T ACE2 cells (Fig. 1B). In contrast, a recent proteomic study reported that DDX3X is required for the SARS-CoV-2 replication in Vero E6 cells (52).
So far, P-bodies and stress granules (SGs) have been implicated in all aspects of RNA life cycle, including RNA translation, RNA silencing, and RNA degradation as well as viral infection (16–18, 26, 27). P body and SG components can facilitate or limit viral infection, and some viral RNAs and viral proteins accumulate in P-bodies and/or SGs. Indeed, P-body components, including DDX6, GW182, Lsm1, and Xrn1, negatively regulate HIV-1 gene expression by preventing viral mRNA association with polysomes (19). In contrast, miRNA effectors, such as DDX6, Lsm1, PatL1, and Ago2, positively regulate HCV replication (12, 20–22), since the liver-specific and abundant miR-122 interacts with the 5′ untranslated region (UTR) of the HCV RNA genome and facilitates HCV replication (53). In this study, I have found, for the first time, that SARS-CoV-2 infection disrupted the P-body formation of DDX6 and MOV10 RNA helicases (Fig. 4). Then, SARS-CoV-2 N colocalized with DDX6 and MOV10 in the cytoplasm (Fig. 6). Importantly, DDX6 was required for SARS-CoV-2 infection (Fig. 5D). In contrast, MOV10 restricted SARS-CoV-2 infection (Fig. 1B). On the other hand, SARS-CoV-2 infection did not induce SGs of G3BP1 (Fig. 5B). Consistent with these findings, recent interactome analyses indicated that SARS-CoV-2 and IBV N bind to G3BP1 (41, 42, 52). Thus, SARS-CoV-2 infection may attenuate or disturb the SGs formation through the interaction of SARS-CoV-2 with G3BP1. Similarly, we previously demonstrated that HCV hijacks the P-body and stress granule components around lipid droplets (LDs) to carry out viral replication (7, 12). LDs have been involved in an important cytoplasmic organelle for HCV production (54). In this regard, Dias et al. recently reported that SARS-CoV-2 infection upregulates lipid metabolism and induces LDs, and LDs are required for the production of infectious SARS-CoV-2 viral particles, indicating that LDs are sites for SARS-CoV-2 replication, as for HCV (55). Therefore, SARS-CoV-2 infection may sequester host cellular RNA helicases around LDs and carry out viral replication.
Altogether, SARS-CoV-2 seems to hijack host cellular RNA helicases to play a proviral role by facilitating viral infection and replication and by suppressing the host innate immune system. Viral infection triggers host innate immune responses through activation of the transcription factors NF-κB and interferon (IFN) regulatory factor 3 (IRF-3), leading to type I IFN production and an antiviral state (56). Viruses must overcome the host antiviral innate immunity. HCVNS3-4A protease cleaves IPS-1/Cardif to block IFN-β induction (57) In addition, HCV core protein can disrupt the DDX3-IPS-1/MAVS/Cardif/VISA interaction and act as a viral immune evasion protein preventing IFN-β induction (58). Actually, SARS-CoV-2 suppresses the type I IFN signaling pathway (59, 60). The N, NSP1, NSP3, NSP12, NSP13, NSP14, ORF3, ORF6, ORF8, and M proteins inhibit type I IFN responses, whereas NSP2 and S proteins exert opposite effects (59, 60). On the other hand, some host cellular RNA helicases, including DDX21 and MOV10, limit viral infection and replication as the guardians of host cells. Viruses evolve a counteracting system against such host restriction factors. For example, APOBEC3G cytidine deaminase strongly restricts HIV-1 infection during reverse transcription. HIV-1 Vif protein overcomes the antiviral effect of APOBEC3G by degradation through the ubiquitin-proteasome pathway (61–63). In this study, I could not demonstrate that SARS-CoV-2 protein counteracts the antiviral effects of DDX21 and MOV10. Further studies are needed to identify the SARS-CoV-2 viral proteins counteracting host restriction factors, if they exist.
MATERIALS AND METHODS
Cell culture.
293T, HepG2, HEK293T ACE2 (SL221; GeneCopoeia Inc., Rockville, MD, USA), and Vero E6 TMPRSS2 cells (a gift from Shutoku Matsuyama and Makoto Takeda, National Institute of Infectious Diseases, Tokyo, Japan) (64) were cultured in Dulbecco's modified Eagle's medium (DMEM; Life Technology, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS). CACO-2 human colon carcinoma cells (RCB0988; RIKEN BioResource Research Center, Tsukuba, Ibaraki, Japan) were cultured in DMEM supplemented with 20% FBS.
Plasmid construction.
To construct pcDNA3-HA-MOV10, a DNA fragment encoding MOV10 was amplified from HuH-7 cDNA by PCR using KOD-Plus DNA polymerase (Toyobo, Osaka, Japan) and the primer pair 5′-CGGGATCCAAGATGCCCAGTAAGTTCAGCTG-3′ (forward) and 5′-CCGCTCGAGTCAGAGCTCATTCCTCCACTCTG-3′ (reverse). The obtained DNA fragments were subcloned into the BamHI-NotI sites of the pcDNA3-HA vector, and the nucleotide sequences were determined by Sanger sequencing. To construct the DDX21 deletion mutants (1–704, 1–185, 568–781, and 621–781 mutants), a DNA fragment encoding DDX21 was amplified from pcDNA3-HA-DDX21 (9) by PCR using KOD-Plus DNA polymerase (Toyobo, Osaka, Japan) and the following pairs of primers: 1–704, 5′-CGGGATCCAAGATGCCGGGAAAACTCCGTA-3′ (forward) and 5′-CGCTCGAGTTACTGCCAGCGTCGTGAATCA-3′ (reverse); 1–185, 5′-CGGGATCCAAGATGCCGGGAAAACTCCGTA-3′ (forward) and 5′-CGCTCGAGTTATTCTTTTTGTTCCACAGGT-3′ (reverse); 568–781, 5′-CGGGATCCGCAACAGAAATAATAAAAGCTT-3′ (forward) and 5′-CGCTCGAGTTATTGACCAAATGCTTTACTG-3′ (reverse); 621–781, 5′-CGGGATCCGCCACGTCCGTAGACCAGCGCT-3′ (forward) and 5′-CGCTCGAGTTATTGACCAAATGCTTTACTG-3′ (reverse). The obtained DNA fragments were subcloned into either the BamHI-XhoI sites of the pcDNA3-HA vector. We previously described pHA-DDX3 (8–12), pcDNA3-HA-DDX1 (9), and pcDNA3-HA-DDX6 (9). pcDNA3.1 SARS-CoV-2 N was a gift from Jeremy Luban (Addgene plasmid 158079; http://n2t.net/addgene:158079; RRID: Addgene_158079) (37). p23-DDX21 WT, p23-DDX21 DEV, and p23-DDX21 SAT were gifts from Ling-Ling Chen (WT, Addgene plasmid 128803 [http://n2t.net/addgene:128803], RRID Addgene_128803; DEV, Addgene plasmid 128805 [http://n2t.net/addgene:128805], RRID Addgene_128805; SAT, Addgene plasmid 128804 [http://n2t.net/addgene:128804], RRID Addgene_128804) (65).
Western blot analysis.
Cells were lysed in buffer containing 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 4 mM EDTA, 1% Nonidet P-40 (NP-40), 0.1% sodium dodecyl sulfate (SDS), 1 mM dithiothreitol (DTT), and 1 mM phenylmethylsulfonyl fluoride (PMSF). Supernatants from these lysates were subjected to SDS-polyacrylamide gel electrophoresis, followed by immunoblot analysis using anti-SARS-CoV-2 spike (GTX632604 [1A9]; GeneTex, Irvine, CA, USA), anti-DDX3 (A300-474A; Bethyl Lab, Inc., Montgomery, TX, USA), anti-DDX1 (A300-521A; Bethyl Lab, GTX105205 [N3C2]; GeneTex), anti-DDX5 (A300-523A; Bethyl Lab), anti-DDX6 (A300-460A; Bethyl Lab), anti-DDX21 (A300-627A; Bethyl Lab), anti-MOV10 (A301-571A; Bethyl Lab), anti-G3BP1 (A302-033A; Bethyl Lab), or anti-β-actin (A5441; Sigma, St. Louis, MO, USA). I used peroxidase-conjugated donkey anti-rabbit IgG (heavy plus light chain [H+L]) (Jackson ImmunoResearch, West Grove, PA, USA) and Amersham enhanced chemiluminescence (ECL) peroxidase-linked sheep anti-mouse IgG (GE Healthcare Bio-Sciences, Uppsala, Sweden) as secondary antibodies. The proteins were detected by using Western Lightning Plus ECL substrate (PerkinElmer, Waltman, MA) and an ImageQuant LAS4000 system (GE Healthcare Bio-Sciences).
RNA interference.
Oligonucleotides with the following sequences were used for the cloning of short hairpin RNA (shRNA)-encoding sequences targeted to DDX1, DDX21, or MOV10 in a lentiviral vector: DDX1, 5′-GATCCCCGGAGATGTAAGATTCTTGATTCAAGAGATCAAGAATCTTACATCTCCTTTTTGGAAA-3′ (sense) and 5′-AGCTTTTCCAAAAAGGAGATGTAAGATTCTTGATCTCTTGAATCAAGAATCTTACATCTCCGGG-3′ (antisense); DDX21, 5′-GATCCCCGGAGCATCTGGCTATTAAGTTCAAGAGACTTAATAGCCAGATGCTCCTTTTTGGAAA-3′ (sense) and 5′-AGCTTTTCCAAAAAGGAGCATCTGGCTATTAAGTCTCTTGAACTTAATAGCCAGATGCTCCGGG-3′ (antisense); MOV10, 5′-GATCCCCGCTGACCTTCAAGGTGAACTTCAAGAGAGTTCACCTTGAAGGTCAGCTTTTTGGAAA-3′ (sense) and 5′-AGCTTTTCCAAAAAGCTGACCTTCAAGGTGAACTCTCTTGAAGTTCACCTTGAAGGTCAGCGGG-3′ (antisense). The oligonucleotides above were annealed and subcloned into the BglII-HindIII site, downstream from an RNA polymerase III promoter of pSUPER (66), to generate pSUPER-DDX1, pSUPER-DDX21, and pSUPER-MOV10, respectively. To construct pLV-shDDX1, pLV-shDDX21, or pLV-shMOV10, the BamHI-SalI fragments of the corresponding pSUPER plasmids were subcloned into the BamHI-SalI site of pRDI292, an HIV-1-derived self-inactivating lentiviral vector containing a puromycin resistance marker allowing the selection of transduced cells (67). We previously described pLV-shDDX3, pLV-shDDX5, and pLV-shDDX6 (11, 12, 68).
Lentiviral vector production.
The vesicular stomatitis virus G (VSV-G)-pseudotyped HIV-1-based vector system has been described previously (69, 70). The lentiviral vector particles were produced by transient transfection of the second-generation packaging construct pCMVΔR8.74 (69, 70) and the VSV-G-envelope-expressing plasmid pMD2G as well as pRDI292 into 293T cells with TransIT-LT1 transfection reagent (Mirus Bio LLC, Madison, WI, USA).
Immunoprecipitation.
Cells were lysed in buffer containing 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 4 mM EDTA, 0.1% NP-40, 10 mM NaF, protease inhibitor cocktail (Nacalai Tesque, Kyoto, Japan). Lysates were precleared with 30 μL of protein-G-Sepharose (GE Healthcare Bio-Sciences). Precleared supernatants were incubated with 5 μg of anti-SARS-CoV-2 nucleocapsid antibody mixture (GTX135357 and GTX632269 [6H3]; GeneTex) or anti-DDX21 (A300-627A; Bethyl Lab), anti-DDX6 (A300-460A; Bethyl Lab), or anti-MOV10 (A301-571A; Bethyl Lab) antibody at 4°C for 1 h. Following absorption of the precipitates on 30 μL of protein G-Sepharose resin for 1 h, the resin was washed four times with 700 μL of lysis buffer. Proteins were eluted by boiling the resin for 5 min in 2× Laemmli sample buffer. The proteins were then subjected to SDS-PAGE, followed by immunoblot analysis using anti-SARS-CoV-2 nucleocapsid (GTX632269 [6H3]), anti-DDX21, anti-DDX6, or anti-MOV10 antibody.
Immunofluorescence and confocal microscopic analysis.
Cells were grown on Lab-Tek 2-well chamber slides (Nunc, Thermo) at 2 × 104 cells per well. The cells were fixed in 3.6% formaldehyde in phosphate-buffered saline (PBS), permeabilized in 0.1% NP-40 in PBS at room temperature, and incubated with anti-HA antibody (3F10; F. Hoffmann-La Roche AG, Basel, Switzerland) at a 1:300 dilution in PBS containing 3% bovine serum albumin (BSA) at 37°C for 30 min. I also used anti-DDX6 (A300-460A; Bethyl Lab), anti-XRN1 (A300-443A; Bethyl Lab), anti-G3BP1 (A302-033A; Bethyl), anti-DDX21 (A300-627A; Bethyl Lab), anti-MOV10 (A301-571A; Bethyl Lab), anti-DDX5 (A300-523A; Bethyl Lab), and anti-SARS-CoV-2 nucleocapsid (ab273434 [6H3]; Abcam or GTX632269 [6H3]; GeneTex) antibodies as primary antibodies. Cells were then stained with donkey anti-mouse IgG (H+L) Alexa Fluor 594-conjugated secondary antibody and/or donkey anti-rabbit or anti-rat IgG (H+L) Alexa Fluor 594-conjugated secondary antibody (Thermo Fisher Scientific Inc., Waltham, MA, USA), at a 1:300 dilution in PBS containing 3% BSA at 37°C for 30 min. Nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole). Following washing 3 times in PBS, the cover slides were mounted on slides using SlowFade Gold antifade reagent (Life Technology). Samples were analyzed under a confocal laser-scanning microscope (FV1200; Olympus, Tokyo, Japan).
Virus infection.
In this study, I used SARS-CoV-2 hCoV-19/Japan/TY-WK-521/2020 (GISAID accession ID: EPI_ISL_408667) isolated from a patient in Japan. The virus was amplified in Vero E6 TMPRSS2 cells in high-glucose DMEM supplemented with 10% FBS and incubated at 37°C in 5% CO2 for 2 days of infection. Virus titers were determined as 50% tissue culture infectious doses (TCID50) per milliliter, and the virus stocks (2.7 × 108 TCID50/mL) were kept in a −80°C freezer until use. All virus infection studies were performed in a biosafety level 3 (BSL3) facility. For infection experiments with SARS-CoV-2, HEK293T ACE2 cells (2 × 105 cells/well) were plated onto 6-well plates and cultured for 24 h. Then, cells were infected at a multiplicity of infection (MOI) of 0.5. One microliter of culture supernatants of SARS-CoV-2-infected Vero E6 TMPRSS2 cells (2.7 × 108 TCID50/mL) was inoculated. The culture supernatants were collected at 72 h postinfection, and the levels of the nucleocapsid (N) protein were determined by enzyme-linked immunosorbent assay (ELISA) using a RayBio COVID-19/SARS-CoV-2 nucleocapsid protein ELISA kit (RayBiotech, Norcross, GA, USA) and xMark microplate spectrometer (Bio-Rad). Total RNA was also isolated from the infected cellular lysates using an RNeasy minikit (Qiagen, Hilden, Germany) for analysis of intracellular SARS-CoV-2 RNA. The extracellular SARS-CoV-2 RNA in the culture supernatant was analyzed using a SARS-CoV-2 direct detection reverse transcription quantitative PCR (RT-qPCR) kit (RC300A; TaKaRa-Bio, Shiga, Japan). The infectivity of SARS-CoV-2 in the culture supernatants was determined at 48 h postinfection. The SARS-CoV-2-infected cells were detected using anti-SARS-CoV-2 spike (GTX632604 [1A9]; GeneTex, Irvine, CA, USA) or anti-SARS-CoV-2 nucleocapsid (ab273434 [6H3]; Abcam or GTX632269 [6H3]; GeneTex) antibodies.
Real-time RT-PCR.
Total RNA was isolated using an RNeasy minikit (Qiagen), and cDNAs were synthesized using SARS-CoV-2 nucleocapsid (N) reverse primer N660R (5′-AGCAAGAGCAGCATCACCGCCATTGCCAGC-3′) and Moloney murine leukemia virus (M-MLV) reverse transcriptase (Invitrogen). Then, real-time RT-PCR was performed with SARS-CoV-2 N primer sets and SYBR Premix Ex Taq II (TaKaRa-Bio) using a LightCycler Nano (Roche) with 40 cycles. Primer sequences are 5′-ATGCTGCAATCGTGCTACAA-3′ (forward) and 5′-GACTGCCGCCTCTGCTC-3′ (reverse) for SARS-CoV-2 N.
Statistical analysis.
A statistical comparison of the RNA levels of SARS-CoV-2 between the knockdown cells and the control cells was performed by using Student's t test. Two-sided P values of less than 0.05 were considered statistically significant. Data are shown as means and standard errors of the means (SEM) from three independent experiments.
ACKNOWLEDGMENTS
I thank Didier Trono, Priscilla Turelli, Hiroyuki Oshiumi, Jeremy Luban, Ling-Ling Chen, Shutoku Matsuyama, Makoto Takeda, National Institute of Infectious Diseases, and RIKEN BioResource Research Center for reagents. I also thank Kunitada Shimotohno and Masafumi Takiguchi for valuable discussion and encouragement.
This work was supported by the Research Program on Hepatitis B grant JP17929672 from Japan Agency for Medical Research and Development, AMED, and by an Amabie Research grant from Kumamoto University.
Y.A. contributed to the design of experiments, the conduction of experiments, manuscript writing, and editing.
We have no conflicts of interest to disclose.
Contributor Information
Yasuo Ariumi, Email: ariumi@kumamoto-u.ac.jp.
Tom Gallagher, Loyola University Chicago.
REFERENCES
- 1.Linder P. 2008. mRNA export: RNP remodeling by DEAD-box proteins. Curr Biol 18:R297–R299. 10.1016/j.cub.2008.02.027. [DOI] [PubMed] [Google Scholar]
- 2.Linder P, Lasko P. 2006. Bent out of shape: RNA unwinding by the DEAD-box helicase Vasa. Cell 125:219–221. 10.1016/j.cell.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 3.Rocak S, Linder P. 2004. DEAD-box proteins: the driving forces behind RNA metabolism. Nat Rev Mol Cell Biol 5:232–241. 10.1038/nrm1335. [DOI] [PubMed] [Google Scholar]
- 4.Cordin O, Banroques J, Tanner NK, Linder P. 2006. The DEAD-box protein family of RNA helicases. Gene 367:17–37. 10.1016/j.gene.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 5.Jankowsky E. 2011. RNA helicases at work: binding and rearranging. Trends Biochem Sci 36:19–29. 10.1016/j.tibs.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fuller-Pace FV. 2006. DExD/H box RNA helicases: multifunctional proteins with important roles in transcriptional regulation. Nucleic Acids Res 34:4206–4215. 10.1093/nar/gkl460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ariumi Y. 2014. Multiple functions of DDX3 RNA helicase in gene regulation, tumorigenesis, and viral infection. Front Genet 5:423. 10.3389/fgene.2014.00423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yedavalli VSRK, Neuveut C, Chi Y-h, Kleiman L, Jeang K-T. 2004. Requirement of DDX3 DEAD box RNA helicase for HIV-1 Rev-RRE export function. Cell 119:381–392. 10.1016/j.cell.2004.09.029. [DOI] [PubMed] [Google Scholar]
- 9.Yasuda-Inoue M, Kuroki M, Ariumi Y. 2013. Distinct DDX DEAD-box RNA helicases cooperate to modulate the HIV-1 Rev function. Biochem Biophys Res Commun 434:803–808. 10.1016/j.bbrc.2013.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yasuda-Inoue M, Kuroki M, Ariumi Y. 2013. DDX3 RNA helicase is required for HIV-1 Tat function. Biochem Biophys Res Commun 441:607–611. 10.1016/j.bbrc.2013.10.107. [DOI] [PubMed] [Google Scholar]
- 11.Ariumi Y, Kuroki M, Abe K, Dansako H, Ikeda M, Wakita T, Kato N. 2007. DDX3 DEAD-box RNA helicase is required for hepatitis C virus RNA replication. J Virol 81:13922–13926. 10.1128/JVI.01517-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ariumi Y, Kuroki M, Kushima Y, Osugi K, Hijikata M, Maki M, Ikeda M, Kato N. 2011. Hepatitis C virus hijacks P-body and stress granule components around lipid droplets. J Virol 85:6882–6892. 10.1128/JVI.02418-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Valdez BC, Henning D, Busch RK, Woods K, Flores-Rozas H, Hurwitz J, Perlaky L, Busch H. 1996. A nucleolar RNA helicase recognized by autoommune antibodies from a patient with watermelon stomach disease. Nucleic Acids Res 24:1220–1224. 10.1093/nar/24.7.1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Westermarck J, Weiss C, Saffrich R, Kast J, Musti AM, Wessely M, Ansorge W, Séraphin B, Wilm M, Valdez BC, Bohmann D. 2002. The DEXD/H-box RNA helicase RHII/Gu is a co-factor for c-Jun-activated transcription. EMBO J 21:451–460. 10.1093/emboj/21.3.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Calo E, Flynn RA, Martin L, Spitale RC, Chang HY, Wysocka J. 2015. RNA helicase DDX21 coordinates transcription and ribosomal RNA processing. Nature 518:249–253. 10.1038/nature13923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weston A, Sommerville J. 2006. Xp54 and related (DDX6-like) RNA helicase: roles in messenger RNP assembly, translation regulation and RNA degradation. Nucleic Acids Res 34:3082–3094. 10.1093/nar/gkl409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beckham CJ, Parker R. 2008. P bodies, stress granules, and viral life cycles. Cell Host Microbe 3:206–212. 10.1016/j.chom.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parker R, Sheth U. 2007. P bodies and the control of mRNA translation and degradation. Mol Cell 25:635–646. 10.1016/j.molcel.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 19.Chable-Bessia C, Meziane O, Latreille D, Triboulet R, Zamborlini A, Wagschal A, Jacquet JM, Reynes J, Levy Y, Saib A, Bennasser Y, Benkirane M. 2009. Suppression of HIV-1 replication by microRNA effector. Retrovirology 6:26. 10.1186/1742-4690-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jangra RK, Yi M, Lemon SM. 2010. DDX6 (Rck/p54) is required for efficient hepatitis C virus replication but not for internal ribosome entry site-directed translation. J Virol 84:6810–6824. 10.1128/JVI.00397-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scheller N, Mina LB, Galão RP, Chari A, Giménez-Barcons M, Noueiry A, Fischer U, Meyerhans A, Díez J. 2009. Translation and replication of hepatitis C virus genomic RNA depends on ancient cellular proteins that control mRNA fates. Proc Natl Acad Sci USA 106:13517–13522. 10.1073/pnas.0906413106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reed JC, Molter B, Geary CD, McNevin J, McElrath J, Giri S, Klein KC, Lingappa JR. 2012. HIV-1 Gag co-opts a cellular complez containing DDX6, a helicase that facilitates capsid assembly. J Cell Biol 198:439–456. 10.1083/jcb.201111012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burdick R, Smith JL, Chaipan C, Friew Y, Chen J, Venkatachari NJ, Delviks-Frankenberry KA, Hu WS, Pathak VK. 2010. P-body-associated protein Mov10 inhibits HIV-1 replication at multiple stages. J Virol 84:10241–10253. 10.1128/JVI.00585-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Furtak V, Mulky A, Rawlings SA, Kozhaya L, Lee K, Kewalramani VN, Unutmaz D. 2010. Perturbation of the P-body component Mov10 inhibits HIV-1 infectivity. PLoS One 5:e9081. 10.1371/journal.pone.0009081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang X, Han Y, Dang Y, Fu W, Zhou T, Ptak RG, Zheng YH. 2010. Moloney leukemia virus 10 (MOV10) protein inhibits retrovirus replication. J Biol Chem 285:14346–14355. 10.1074/jbc.M110.109314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anderson P, Kedersha N. 2008. Stress granules: the Tao of RNA triage. Trends Biochem Sci 33:141–150. 10.1016/j.tibs.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 27.Kedersha N, Anderson P. 2007. Mammalian stress granules and processing bodies. Methods Enzymol 431:61–81. 10.1016/S0076-6879(07)31005-7. [DOI] [PubMed] [Google Scholar]
- 28.Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, Wang W, Song H, Huang B, Zhu N, Bi Y, Ma X, Zhan F, Wang L, Hu T, Zhou H, Hu Z, Zhou W, Zhao L, Chen J, Meng Y, Wang J, Lin Y, Yuan J, Xie Z, Ma J, Liu WJ, Wang D, Xu W, Holmes EC, Gao GF, Wu G, Chen W, Shi W, Tan W. 2020. Genomic characterization and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 395:565–574. 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu F, Zhao S, Yu B, Chen YM, Wang W, Song ZG, Hu Y, Tao ZW, Tian JH, Pei YY, Yuan ML, Zhang YL, Dai FH, Liu Y, Wang QM, Zheng JJ, Xu L, Holmes EC, Zhang YZ. 2020. A new coronavirus associated with human respiratory disease in China. Nature 579:265–269. 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, Si HR, Zhu Y, Li B, Huang C, Chen HD, Chen J, Luo Y, Guo H, Jiang RD, Liu MQ, Chen Y, Shen XR, Wang X, Zheng XS, Zhao K, Chen QJ, Deng F, Liu LL, Yan B, Zhan FX, Wang YY, Xiao GF, Shi ZL. 2020. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579:270–273. 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, Zhao X, Huang B, Shi W, Lu R, Niu P, Zhan F, Ma X, Wang D, Xu W, Wu G, Gao GF, Tan W, China Novel Coronavirus Investigating and Research Team. 2020. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med 382:727–733. 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tang D, Comish P, Kang R. 2020. The hallmarkers of COVID-19 disease. PLoS Pathog 16:e1008536. 10.1371/journal.ppat.1008536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.V'kovski P, Kratzel A, Steiner S, Stalder H, Thiel V. 2021. Coronavirus biology and replication: implications for SARS-CoV-2. Nat Rev Microbiol 19:155–170. 10.1038/s41579-020-00468-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Letko M, Marzi A, Munster V. 2020. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat Microbiol 5:562–569. 10.1038/s41564-020-0688-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu N-H, Nitsche A, Müller MA, Drosten C, Pöhlmann S. 2020. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181:271–280. 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. 2020. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181:281–292. 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yurkovetskiy L, Wang X, Pascal KE, Tomkins-Tinch C, Nyalile TP, Wang Y, Baum A, Diehl WE, Dauphin A, Carbone C, Veinotte K, Egri SB, Schaffner SF, Lemieux JE, Munro JB, Rafique A, Barve A, Sabeti PC, Kyratsous CA, Dudkina NV, Shen K, Luban J. 2020. Structural and functional analysis of the D614G SARS-CoV-2 spike protein variants. Cell 183:739–751. 10.1016/j.cell.2020.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kwong AD, Rao BG, Jeang KT. 2005. Viral and cellular RNA helicases as antiviral targets. Nat Rev Drug Discov 4:845–853. 10.1038/nrd1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Utama A, Shimizu H, Hasebe F, Morita K, Igarashi A, Shoji I, Matsuura Y, Hatsu M, Takamizawa K, Hagiwara A, Miyamura T. 2000. Role of the DExH motif of the Japanese encephalitis virus and hepatitis C virus NS3 proteins in the ATPase and RNA helicase activities. Virology 273:316–324. 10.1006/viro.2000.0417. [DOI] [PubMed] [Google Scholar]
- 40.Squeglia F, Romano M, Ruggiero A, Maga G, Berisio R. 2020. Host DDX helicases as possible SARS-CoV-2 proviral factors: a structural overview of their hijacking through multiple viral proteins. Front Chem 8:602162. 10.3389/fchem.2020.602162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Emmott E, Munday D, Bickerton E, Britton P, Rodgers MA, Whitehouse A, Zhou EM, Hiscox JA. 2013. The cellular interactome of coronavirus infectious bronchitis virus nucleocapsid protein and functional implications for virus biology. J Virol 87:9486–9500. 10.1128/JVI.00321-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM, O'Meara MJ, Rezelj VV, Guo JZ, Swaney DL, Tummino TA, Hüttenhain R, Kaake RM, Richards AL, Tutuncuoglu B, Foussard H, Batra J, Haas K, Modak M, Kim M, Haas P, Polacco BJ, Braberg H, Fabius JM, Eckhardt M, Soucheray M, Bennett MJ, Cakir M, McGregor MJ, Li Q, Meyer B, Roesch F, Vallet T, Mac Kain A, Miorin L, Moreno E, Naing ZZC, Zhou Y, Peng S, Shi Y, Zhang Z, Shen W, Kirby IT, Melnyk JE, Chorba JS, Lou K, Dai SA, Barrio-Hernandez I, Memon D, Hernandez-Armenta C, et al. 2020. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 583:459–468. 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goodier JL, Cheung LE, Kazazian JHH. 2012. MOV10 RNA helicase is a potent inhibitor of retrotransposition in cells. PLoS Genet 8:e1002941. 10.1371/journal.pgen.1002941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Z, Kim T, Bao M, Facchinetti V, Jung SY, Ghaffari AA, Qin J, Cheng G, Liu YJ. 2011. DDX1, DDX21, and DHX36 helicases form a complex with the adaptor molecule TRIF to sense dsRNA in dendritic cells. Immunity 34:866–878. 10.1016/j.immuni.2011.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen G, Liu CH, Zhou L, Krug RM. 2014. Cellular DDX21 RNA helicase inhibits influenza A virus replication but is counteracted by the viral NS1 protein. Cell Host Microbe 15:484–493. 10.1016/j.chom.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dong Y, Ye W, Yang J, Han P, Wang Y, Ye C, Weng D, Zhang F, Xu Z, Lei Y. 2016. DDX21 translocates from nucleus to cytoplasm and stimulates the innate immune response due to dengue virus infection. Biochem Biophys Res Commun 473:648–653. 10.1016/j.bbrc.2016.03.120. [DOI] [PubMed] [Google Scholar]
- 47.Hao H, Han T, Xuan B, Sun Y, Tang S, Yue N, Qian Z. 2019. Dissecting the role of DDX21 in regulating human cytomegalovirus replication. J Virol 93:e01222-19. 10.1128/JVI.01222-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Watanabe Y, Ohtaki N, Hayashi Y, Ikuta K, Tomonaga K. 2009. Autogenous translational regulation of the borna disease virus negative control factor X from polycistronic mRNA using host RNA helicases. PLoS Pathog 5:e1000654. 10.1371/journal.ppat.1000654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wei J, Alfajaro MM, DeWeirdt PC, Hanna RE, Lu-Culligan WJ, Cai WL, Strine MS, Zhang S-M, Graziano VR, Schmitz CO, Chen JS, Mankowski MC, Filler RB, Ravindra NG, Gasque V, de Miguel FJ, Patil A, Chen H, Oguntuyo KY, Abriola L, Surovtseva YV, Orchard RC, Lee B, Lindenbach BD, Politi K, van Dijk D, Kadoch C, Simon MD, Yan Q, Doench JG, Wilen CB. 2021. Genome-wide CRISPR screens reveal host factors critical for SARS-CoV-2 infection. Cell 184:76–91. 10.1016/j.cell.2020.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Flynn RA, Belk JA, Qi Y, Yasumoto Y, Wei J, Alfajaro MM, Shi Q, Mumbach MR, Limaye A, DeWeirdt PC, Schmitz CO, Parker KR, Woo E, Chang HY, Horvath TL, Carette JE, Bertozzi CR, Wilen CB, Satpathy AT. 2021. Discovery and functional interrogation of SARS-CoV-2 RNA-host protein interactions. Cell 184:2394–2411. 10.1016/j.cell.2021.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hoffmann HH, Sánchez-Rivera FJ, Schneider WM, Luna JM, Soto-Feliciano YM, Ashbrook AW, Le Pen J, Leal AA, Ricardo-Lax I, Michailidis E, Hao Y, Stenzel AF, Peace A, Zuber J, Allis CD, Lowe SW, MacDonald MR, Poirier JT, Rice CM. 2021. Functional interrogation of a SARS-CoV-2 host protein interactome identifies unique and shared coronavirus host factors. Cell Host Microbe 29:267–280.E5. 10.1016/j.chom.2020.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ciccosanti F, Di Rienzo M, Romagnoli A, Colavita F, Refolo G, Castilletti C, Agrati C, Brai A, Manetti F, Botta L, Capobianchi MR, Ippolito G, Piacentini M, Fimia GM. 2021. Proteomic analysis identifies the RNA helicase DDX3X as a host target against SARS-CoV-2 infection. Antiviral Res 190:105064. 10.1016/j.antiviral.2021.105064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. 2005. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 309:1577–1581. 10.1126/science.1113329. [DOI] [PubMed] [Google Scholar]
- 54.Miyanari Y, Atsuzawa K, Usuda N, Watashi K, Hishiki T, Zayas M, Bartenschlager R, Wakita T, Hijikata M, Shimotohno K. 2007. The lipid droplet is an important organelle for hepatitis C virus production. Nat Cell Biol 9:1089–1097. 10.1038/ncb1631. [DOI] [PubMed] [Google Scholar]
- 55.Dias SSG, Soares VC, Ferreira AC, Sacramento CQ, Fintelman-Rodrigues N, Temerozo JR, Teixeira L, da Silva MAN, Barreto E, Mattos M, de Freitas CS, Azevedo-Quintanilha IG, Manso PPA, Miranda MD, Siqueira MM, Hottz ED, Päo CRR, Bou-Habib DC, Barreto-Vieira DF, Bozza FA, Souza TML, Bozza PT. 2020. Lipid droplets fuel SARS-CoV-2 replication and production of inflammatory mediators. PLoS Pathog 16:e1009127. 10.1371/journal.ppat.1009127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gale MJr, Foy EM. 2005. Evasion of intracellular host defence by hepatitis C virus. Nature 436:939–945. 10.1038/nature04078. [DOI] [PubMed] [Google Scholar]
- 57.Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J. 2005. Cardif is an adaptor protein in the RIG-1 antiviral pathway and is targeted by hepatitis C virus. Nature 437:1167–1172. 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 58.Oshiumi H, Ikeda M, Matsumoto M, Watanabe A, Takeuchi O, Akira S, Kato N, Shimotohno K, Seya T. 2010. Hepatitis C virus core protein abrogates the DDX3 function that enhances IPS-1-mediated IFN-beta induction. PLoS One 5:e14258. 10.1371/journal.pone.0014258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li JY, Liao CH, Wang Q, Tan YJ, Luo R, Qiu Y, Ge XY. 2020. The ORF6, ORF8 and nucleocapsid proteins of SARS-CoV-2 inhibit type I interferon signaling pathway. Virus Res 286:198074. 10.1016/j.virusres.2020.198074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lei X, Dong X, Ma R, Wang W, Xiao X, Tian Z, Wang C, Wang Y, Li L, Ren L, Guo F, Zhao Z, Zhou Z, Xiang Z, Wang J. 2020. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat Commun 11:3810. 10.1038/s41467-020-17665-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sheehy AM, Gaddis NC, Choi JD, Malim MH. 2002. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral vif protein. Nature 418:646–650. 10.1038/nature00939. [DOI] [PubMed] [Google Scholar]
- 62.Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, Trono D. 2003. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 424:99–103. 10.1038/nature01709. [DOI] [PubMed] [Google Scholar]
- 63.Zhang H, Yang B, Pomerantz RJ, Zhang C, Arunachalam SC, Gao L. 2003. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature 424:94–98. 10.1038/nature01707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Matsuyama S, Nao N, Shirato K, Kawase M, Saito S, Takayama I, Nagata N, Sekizuka T, Katoh H, Kato F, Sakata M, Tahara M, Kutsuna S, Ohmagari N, Kuroda M, Suzuki T, Kageyama T, Takeda M. 2020. Enhanced isolation of SARS-CoV-2 by TMPRSS2-expressing cells. Proc Natl Acad Sci USA 117:7001–7003. 10.1073/pnas.2002589117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xing YH, Yao RW, Zhang Y, Guo CJ, Jiang S, Xu G, Dong R, Yang L, Chen LL. 2017. SLERT regulates DDX21 rings associated with Pol I transcription. Cell 169:664–678. 10.1016/j.cell.2017.04.011. [DOI] [PubMed] [Google Scholar]
- 66.Brummelkamp TR, Bernard R, Agami R. 2002. A system for stable expression of short interfering RNAs in mammalian cells. Science 296:550–553. 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- 67.Bridge AJ, Pebernard S, Ducraux A, Nicoulaz AL, Iggo R. 2003. Induction of an interferon response by RNAi vectors in mammalian cells. Nat Genet 34:263–264. 10.1038/ng1173. [DOI] [PubMed] [Google Scholar]
- 68.Kuroki M, Ariumi Y, Hijikata M, Ikeda M, Dansako H, Wakita T, Shimotohno K, Kato N. 2013. PML tumor suppressor protein is required for HCV production. Biochem Biophys Res Commun 430:592–597. 10.1016/j.bbrc.2012.11.108. [DOI] [PubMed] [Google Scholar]
- 69.Naldini L, Blömer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D. 1996. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272:263–267. 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- 70.Zufferey R, Nagy D, Mandel RJ, Naldini L, Trono D. 1997. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat Biotechnol 15:871–875. 10.1038/nbt0997-871. [DOI] [PubMed] [Google Scholar]