

ABSTRACT
Next-generation sequencing (NGS) is a powerful tool for detecting and investigating viral pathogens; however, analysis and management of the enormous amounts of data generated from these technologies remains a challenge. Here, we present VPipe (the Viral NGS Analysis Pipeline and Data Management System), an automated bioinformatics pipeline optimized for whole-genome assembly of viral sequences and identification of diverse species. VPipe automates the data quality control, assembly, and contig identification steps typically performed when analyzing NGS data. Users access the pipeline through a secure web-based portal, which provides an easy-to-use interface with advanced search capabilities for reviewing results. In addition, VPipe provides a centralized system for storing and analyzing NGS data, eliminating common bottlenecks in bioinformatics analyses for public health laboratories with limited on-site computational infrastructure. The performance of VPipe was validated through the analysis of publicly available NGS data sets for viral pathogens, generating high-quality assemblies for 12 data sets. VPipe also generated assemblies with greater contiguity than similar pipelines for 41 human respiratory syncytial virus isolates and 23 SARS-CoV-2 specimens.
IMPORTANCE Computational infrastructure and bioinformatics analysis are bottlenecks in the application of NGS to viral pathogens. As of September 2021, VPipe has been used by the U.S. Centers for Disease Control and Prevention (CDC) and 12 state public health laboratories to characterize >17,500 and 1,500 clinical specimens and isolates, respectively. VPipe automates genome assembly for a wide range of viruses, including high-consequence pathogens such as SARS-CoV-2. Such automated functionality expedites public health responses to viral outbreaks and pathogen surveillance.
KEYWORDS: next-generation sequencing (NGS), automated bioinformatics pipeline, viral molecular detection, infectious disease surveillance
INTRODUCTION
Next-generation sequencing (NGS) technologies have become a vital tool for the characterization of microbial pathogens in clinical microbiology and public health laboratories, with numerous applications in infectious disease diagnostics (1), outbreak investigations (2, 3), public health surveillance (4), and discovery of emerging pathogens (5, 6). Compared to standard Sanger sequencing, NGS technologies are cost-effective and enable high-throughput whole-genome sequencing of microbial pathogens from isolates or from clinical specimens containing multiple species (metagenomics) (7–9). However, NGS generates large amounts of sequence data, ranging from 0.03 to 42 gigabases (Gb) per run (10), creating a critical challenge of effectively analyzing, organizing, reporting, and archiving NGS data within a clinically relevant time frame.
The CDC supports public health laboratories (PHLs) in their infectious disease surveillance efforts, which include implementing NGS technologies for molecular detection of viral pathogens (11, 12). Many PHLs have fully adopted NGS technologies (2, 13), yet most PHLs face obstacles in analyzing their sequencing data due to the need for computational bioinformatics infrastructure and/or lack of a dedicated bioinformatics workforce to support analysis pipelines (4, 14). Automated analysis pipelines can help address staffing shortages in bioinformatics analyses, providing rapid data processing and reporting turnaround. Many institutions and research projects have established sophisticated pipelines for the analysis of viral NGS data. Some pipelines focus on assembly of data, often from metagenomes or pathogen host samples, such as InteMAP (15), VirAMP (16), Bio-Docklets (17), MetaVir 2 (18), VirusSeeker (19), drVM (20), VirMAP (21), and LAZYPIPE (22). Other tools, such as DNAscan (23), perform single nucleotide polymorphisms (SNPs) and indel analysis without genome assembly. However, not all of these tools are consistently maintained, or they may require a bioinformatics skillset beyond the capabilities of small public health laboratories. Other viral NGS analysis tools are focused on virus discovery, including SURPI (24), EDGE (25), VIP (26), and Genome Detective (27). However, these packages offer limited capabilities for data storage and species/strain subtyping.
Here, we introduce VPipe, a system that seamlessly combines data management with standardized viral assembly and a graphical user interface (Fig. 1). Following data upload, analysis begins with filtering, trimming, and deduplication of raw fastq reads, followed by de novo assembly and BLAST comparison of contigs against the NCBI GenBank nucleotide database (www.ncbi.nlm.nih.gov/genbank/). In addition, VPipe implements reference recruitment analysis for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), outputting consensus sequences and SNPs for this high-consequence pathogen. VPipe enables free and confidential access to web-based analysis for public health labs. As an additional distinction, VPipe analysis results are permanently available on the VPipe web-based interface, where users can also download filtered reads and assembled contigs.
FIG 1.

VPipe standard analysis pipeline: VPipe takes raw FASTQ data generated by Illumina or Ion Torrent sequencing instruments. Raw reads are processed using the Read Quality and Trimming module prior to de novo assembly using SPAdes and detection of viral contigs via BLASTN. Analysis results are available on the VPipe user interface, accessible through the CDC OAMD portal. For SARS-CoV-2 data sets, reference-based assembly is also run in parallel with the de novo Assembly Module.
RESULTS
VPipe validation for clinical specimens.
VPipe was validated through analysis of 12 publicly available NGS data sets for clinical specimens, and its performance compared against four similar pipelines. VPipe de novo assembly contig lengths for nonsegmented viruses were similar to or exceeded those of drVM, EDGE, VirMAP, and Genome Detective. VPipe assembly of the Ebola virus data set (SRR1553609) generated a contig 18,756 bp in length (Table 1), equivalent to 99.9% the length of its closest BLASTN match, KM233083 (which is part of BioProject PRJNA257197 along with the SRA data set). The VPipe contig was slightly longer than Ebola contigs from EDGE (18,600 bp) and VirMAP (18,728 bp). For the bovine viral diarrhea virus (BVDV) 2 data set, sequenced using Ion Torrent PGM, VPipe generated a contig of 8,726 bp (Table 1); this aligns to 71.3% the length of the BVDV 2 strain USMARC-60764 (KT832817). By comparison, the metagenomic compositional tools, VirMAP and drVM, constructed longer contigs, aligning with 95.6% and 99.9% the length of the USMARC-60764 genome. For the picornavirus test data sets, VPipe provided nearly complete genomes and robust typing accuracy. The VPipe-generated contigs for enterovirus D94, enterovirus D70, human parechovirus 3, enterovirus A71, and coxsackievirus B5 covered 70.0 to 99.9% of the closest complete genome BLASTN hit, compared to 30.6 to 100% for Genome Detective and 95.6 to 100% for VirMAP (Table S1 in the supplemental material). VPipe successfully typed the six enteroviruses through PiType (Table 1). The other tools compared do not incorporate a specific typing module for this group of viruses. VirMAP identified coxsackievirus B5 as “human echovirus 6” and Genome Detective identified enteroviruses D94 and D70 simply as “enterovirus_D,” and enterovirus A71 as “enterovirus_A.”
TABLE 1.
VPipe compared with previous tools for data sets of predominantly single virus speciesa
| SRA data set, accession no. | Target virus (length, kb)b | Assembly features | Assembly results by pipeline | ||||
|---|---|---|---|---|---|---|---|
| VPipe | drVM (20) | EDGE (25) | VirMAP (21) | Genome Detective (27) | |||
| SRR1553609 | Ebola virus (18.8–19.0) | Contig count | 1 | n/rd | 1 | 1 | 7 |
| Max. contig (bp) | 18,756 | n/r | 18,600 | 18,728 | 7,509 | ||
| SRR1170797 | Bovine viral diarrhea virus 2 (12.3–12.5) | Contig count | 12 | 1 | 4 | 1 | 2 |
| Max. contig (bp) | 8,726 | 12,224 | 1,602 | 11,699 | 8,481 | ||
| SRR13403396 | Enterovirus D94 (7.3–7.4) | Contig count | 1 | n/r | 1 | 1 | 1 |
| Max. contig (bp) | 7,457 | n/r | 670 | 7,573 | 7,320 | ||
| PiType (Vpipe) | EV-D94 | - | Enterovirus_D | ||||
| SRR13402413 | Enterovirus D70 (∼7.4) | Contig count | 1 | n/r | 1 | 1 | 1 |
| Max. contig (bp) | 7,353 | n/r | 7,248 | 7,395e | 7,390 | ||
| PiType (Vpipe) | EV-D70 | - | Enterovirus_D | ||||
| SRR10298816 | Human parechovirus 3 (7.2–7.3) | Contig count | 1 | n/r | 1 | 1 | 1 |
| Max. contig (bp) | 7,153 | n/r | 332 | 7,252 | 7,157 | ||
| PiType (Vpipe) | PEV-A3 | - | - | ||||
| SRR10298815 | Enterovirus A71 (7.4–7.5) | Contig count | 4 | n/r | 123 | 1 | 2 |
| Max. contig (bp) | 6,952 | n/r | 832 | 7,423 | 4,919 | ||
| PiType (Vpipe) | EV-A71 | - | Enterovirus_A | ||||
| SRR10298813 | Human parechovirus 3 (7.2–7.3) | Contig count | 5 | n/r | 6 | 1 | 7 |
| Max. contig (bp) | 6,273 | n/r | 404 | 7,269 | 2,220 | ||
| PiType (Vpipe) | PeV-A3 | - | - | ||||
| SRR10298814 | Coxsackievirus B5 (7.3–7.4) | Contig count | 2 | n/r | 3 | 1 | 1 |
| Max. contig | 5,137 | n/r | 4,294 | 7,361 | 7,165 | ||
| PiType (Vpipe) | CV-B5 | - | Human echovirus 6 | ||||
| DRR049387 | Human rotavirus A (longest segment ∼3.3) | Contig countc | 11 | 13 | 7 | 11 | 11 |
| Max. contig (bp) | 3,462 | 3,375 | 3,338 | 3,349 | 3,299 | ||
| ERR690519 | Influenza A virus (longest segment ∼2.3) | Contig countc | 8 | 8 | 8 | 8 | 9 |
| Max. contig (bp) | 2,340 | 2,340 | 2,370 | 2,297 | 2,285 | ||
| SRR1106548 | HIV-1 (∼9.7) | Contig count | 8 | 12 | 5 | 8 | 4 |
| Max. contig (bp) | 4,956 | 2,819 | 5,195 | 5,163 | 4,319 | ||
| Microviridae (2.7–37.0) | Contig count | 1 | Not detected | Not detected | 1 | Not detected | |
| Max. contig (bp) | 5,211 | 5,129 | |||||
| GB virus C (9.3–9.4) | Contig count | 84 | Not detected | 53 | 8 | 2 | |
| Max. contig (bp) | 1,612 | 1,639 | 2,004 | 9,102 | |||
| SRR1106123 | Hepatitis C virus (9.6–9.8) | Contig count | 2 | 15 | 2 | 1 | 1 |
| Max. contig (bp) | 5,125 | 2,227 | 7,850 | 9,316 | 9,418 | ||
| GB virus C (9.3–9.4) | Contig count | 2 | 159 | 10 | 1 | 2 | |
| Max. contig (bp) | 9,013 | 825 | 5,711 | 9,276 | 9,295 | ||
Strain-typing by PiType shown for VPipe in rows 3 through 8. Comparable presumptive annotation in VirMAP and Genome Detective (rows 3 through 8) shown as ‘-’ unless otherwise indicated.
Approximate length of complete viral genomes (kb), or the longest segment for segmented genomes.
Matches and trimming information are shown in Tables S1–, S3.
n/r, no results; only previously published results used for comparison.
Trimmed 137 bp.
For a clinical data set of 41 human respiratory syncytial virus (HRSV) specimens, VPipe generated assemblies of equivalent contig lengths to the manually curated genomes of the original study (28). The distribution of contig lengths generated by VPipe and in the original study was statistically equivalent (pairwise Wilcoxon, P = 0.50), with average maximum contig lengths of 12,575 bp and 13,039 bp, respectively (Fig. 2A). In contrast, the average maximum contig length for EDGE with host read removal was only 2,109 bp (Fig. 2A) (P = 4.55 × 10−13). When EDGE was run on reads preprocessed through VPipe, the average maximum contig length increased to 10,151 bp (Fig. 2A) but was still significantly shorter than VPipe contigs (P = 0.0015). With an average maximum contig length of 10,883 bp, Genome Detective also yielded shorter contigs than VPipe (P = 0.00024).
FIG 2.
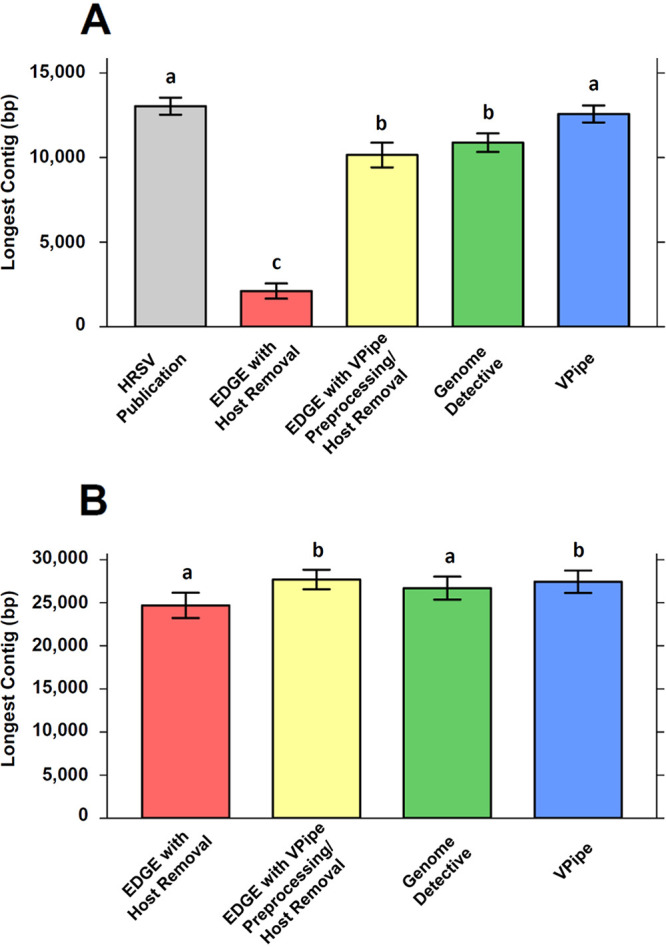
Distribution of longest assembled contigs for HRSV and SARS-CoV-2 clinical data sets. (A) Bar plots indicating the average longest contigs assembled for 41 human respiratory syncytial virus (HRSV) samples. From left to right, bars represent average maximum contig lengths (bp) from Agoti et al. (2015), representing manually curated assemblies (28): EDGE with host sequence removal, EDGE with reads preprocessed through VPipe, Genome Detective, and VPipe. (B) Bar plots indicating the average longest contigs assembled for 23 SARS-CoV-2 specimens using EDGE with host sequence removal, EDGE with reads preprocessed through VPipe, Genome Detective, and VPipe. Whiskers represent standard error of the mean. Bar plots with the same letter are statistically equivalent (pairwise Wilcoxon’s test).
De novo assembly results for segmented viruses were also similar across compared pipelines. For rotavirus A, VPipe, VirMAP, and Genome Detective identified and assembled all 11 genome segments as a single contig (Table 1, Tables S2–S4). Segments assembled through VPipe, VirMAP, and Genome Detective shared an average nucleotide identity of 99.2 to 99.3% with assemblies generated by the researchers who sequenced the corresponding original data set (29, 30) (Tables S2–S4). For influenza virus, each of the compared tools except Genome Detective identified all eight genome segments as a single contig each (Table 1, Tables S5 and S6). Segments assembled by VPipe averaged 1,693 bp in length and shared an average 99.7% identity with their top BLASTN matches, which averaged 1,703 bp in length (Table S5).
VPipe produced assemblies for specimens containing multiple virus species with comparable accuracy to drVM, EDGE, VirMAP, and Genome Detective. For the HIV-1 data set, VPipe produced eight HIV-1 contigs, with a maximum contig length only slightly shorter than the longest contigs assembled using EDGE and VirMAP (by 239 and 207 bp, respectively, Table 1). Also, in the HIV-1 data set, VPipe assembled a set of 84 contigs sharing 86 to 94% BLASTN identity with GB virus C virus isolate R10291 (U45966). In contrast, Genome Detective assembled a 9,102-bp GB virus C contig which aligned to 97.1% of the length of U45966. VPipe and VirMAP assembled and identified a single contig which shared 69% identity (over ∼40% of its length) with Microviridae isolate ctjj553 (MH617085) (Table S1); Genome Detective and EDGE did not report Microviridae-related sequences (Table 1). For the hepatitis C data set, the largest VPipe contig aligned to 54% of the genome length of hepatitis C, isolate HCV-1b/US/BID-V126/1991 (EU234061), whereas for the VirMAP and Genome Detective pipelines, maximum contigs aligned to 99.0% and 100.0% of EU234061, respectively (Table S1). VPipe, VirMAP and Genome Detective also assembled a nearly complete genome of GB virus C, which was also present in the specimen (Table 1, Table S1).
VPipe validation for SARS-CoV-2.
VPipe produced both de novo and reference-based assemblies of SARS-CoV-2 with quality comparable to that of the leading pipelines. For de novo assembly of 23 SARS-CoV-2 specimens, VPipe produced significantly longer contigs than EDGE with host removal and Genome Detective (pairwise Wilcoxon’s, P = 0.00603 and P = 0.00282, respectively; Fig. 2B), whereas there was no significant difference in the contig length distribution between VPipe and EDGE with VPipe preprocessing (P = 0.862). For the reference-based assembly of SARS-CoV-2, VPipe generated 22 consensus genomes that were 29,903 bp in length (matching the length of the reference, MN908947, Table S7) plus one consensus which was 29,904 bp in length due to an extra masked base (which was manually removed). The majority of SNPs were found to be concordant between the original consensus available in GenBank and the consensus generated with VPipe and Genome Detective. For 22 of 23 sequences, the number of SNPs detected in VPipe consensus (with 25× minimum read cutoff filter) and Genome Detective consensus sequences differed from the original consensus by no more than 3 SNPs (Table S8). Discordant SNPs typically occurred within 100 bp of the 5′ or 3′ end of the SARS-CoV-2 genomes (not likely changing protein function) and reflected SNPs with low read coverage. In VPipe consensus with the 25× filter, 98.5% of SNPs were concordant with Genome Detective.
DISCUSSION
The VPipe de novo assembly analysis pipeline comprises three stages: Read Quality and Trimming, de novo Assembly, and Virus Detection (Fig. 1). EDGE and Genome Detective similarly implement three stages and were employed in comparative analyses with VPipe across all data sets in the current study. Whereas VPipe uses Cutadapt (31) and a custom Python script (dedup.py [32]) to perform trimming and de-duplication/masking, EDGE and Genome Detective use FaQCs (33) and Trimmomatic (34), respectively. As an optional host-read removal step, EDGE implements BWA (25) while host removal (non-optional in VPipe) is implemented by Bowtie2 (35). VPipe, Genome Detective, and EDGE (depending on user settings) all employ SPAdes, which previous studies have shown to produce the most consistent assemblies for viral data (36, 37). Use of SPAdes likely led to similar performance for 9 of the 12 specimens shown in Table 1. In some instances, VPipe produced less fragmented assemblies. This may be due to the Read Quality and Trimming module employed by VPipe, since using Vpipe-preprocessed reads as the input for EDGE led to longer assembled contigs for HRSV genomes (Fig. 2A). Only VirMAP consistently outperformed VPipe, likely due to the implementation of reference-based assembly in the default VirMAP pipeline.
The VPipe reference recruitment module compared favorably to current reference-based pipelines for SARS-CoV-2 and identified comparable SNPs to Genome Detective. SNPs which were not concordant corresponded to regions with lower read coverage, where differences in quality, read trimming, and coverage cutoffs between pipelines likely led to slight differences between consensus genome results. The 25× coverage cutoff for VPipe reference recruitment is intermediate between less stringent 10× (38) and more stringent 75× (39) cutoffs employed in other SARS-CoV-2 analysis pipelines. Since accurate identification of SNPs is important for classifying SARS-CoV-2 variants and accurate phylogenies (38–40), the >98% SNP concordance between VPipe (25× filter consensus) and Genome Detective supports the results generated by the VPipe reference recruitment module.
VPipe enables users to perform quality control, de novo assembly, virus discovery, strain typing for picornaviruses, and reference-recruitment analysis (for SARS-CoV-2 data). VPipe utilizes open-source software which performs consistently well for viral data as shown through internal testing and previous studies (25, 37, 41, 42). The system allows standardized, whole-genome assemblies, making it easier to reproduce and publish NGS results (Fig. 1). VPipe simplifies bioinformatics analyses by providing a user-friendly, web-based system that can be operated by users ranging from experienced bioinformaticians to laboratorians with little or no scientific computing experience. To date, VPipe has been utilized for NGS analysis in 20 studies published from 2016 to 2020 to identify 16 different species of viral pathogens in multiple specimen types (Table S9). VPipe can also reconstruct viral genomes from specimens containing multiple viruses, as demonstrated by the detection of additional viruses in the HIV-1 and hepatitis C data sets (Table 1). To facilitate NGS data analysis and archiving, VPipe possesses a set of data management and query functions not commonly found in other freely available NGS pipelines. For example, the advanced sorting and filtering options in VPipe make it easy to identify contigs of interest from multiple samples within an analysis run (Fig. S1). Finally, VPipe provides a permanent display of analysis results, including visualizations. Future development of VPipe will involve the expansion and addition of modules in VPipe to address ever-changing public health needs. This includes implementation of the reference recruitment module for other high-consequence viral pathogens, integrating additional typing tools, and developing a module to analyze Oxford Nanopore sequence data.
MATERIALS AND METHODS
Pipeline structure and organization.
(i) Bioinformatics pipeline. The VPipe standard analysis pipeline (Fig. 1) processes raw FASTQ data generated on Illumina (Illumina, San Diego, CA), and Ion Torrent platforms by user request (Thermo Fisher Scientific, Waltham, MA). The raw FASTQ sequencing reads (paired-end reads) are first filtered to remove human sequences using the Bowtie2 aligner (version 2.3.5.1) (35), based on recruitment to the hg19 reference genome (43). Sequence reads are then trimmed to remove primers and adapters using Cutadapt (version 2.3) (31), which also filters out any reads shorter than 50 bp or with a Phred quality score of less than 20. To prevent biased coverage of genomic regions, duplicate reads are removed with the Python program dedup.py (32). The resultant FASTQ reads are assembled de novo using SPAdes (version 3.15.0) (44) with the kmer parameter “-k 21,33,55,77,99,121” for data sets with read lengths of >130 bp; otherwise, “-k 21,33,55,77,99.” Assembled contigs are compared against the NCBI nonredundant database by BLASTN (version 2.9.0) (45). Nucleotide alignment scores are used to categorize contigs according to their similarity to viral pathogens.
For selected viral groups, filtered FASTQ reads are processed through a reference recruitment assembly module in addition to de novo assembly. At present, only SARS-CoV-2 data are processed through the reference recruitment module, where a guided assembly is conducted using the SARS-CoV-2 reference strain Wuhan-Hu-1 (MN908947). A hard trim of 30 nucleotides is applied on each end of sequencing reads using Cutadapt (version 2.3) to remove sequence over potential primer binding regions (since many labs are employing amplicon-based strategies for targeted SARS-CoV-2 sequencing). Trimmed reads are then aligned using Bowtie2 aligner (version 2.1.0). Samtools (version 1.10) is then used to generate sorted .bam files, which are then used to generate bedGraph files. Freebayes (version 1.0.2) is then used to generate .vcf files, and BEDTools (version 2.27.1) is used to assemble a consensus fasta sequence file for the SARS-CoV-2 specimens. Reference and new consensus sequences are aligned using kalign (version 1.04).
(ii) Pipeline architecture. Data flow in VPipe is coordinated by four major components which control access, data analysis, storage, and visualization, respectively (Fig. S2). The Data Transfer tool, a supporting application from CDC OAMD, enables batch uploads from multiple directories corresponding to different NGS runs to be processed through VPipe. The Data Processing Layer monitors sequencing data upload to the Data Transfer tool (accessible to external users with a VPipe account) and initializes automation of the “Bioinformatics Analysis Pipeline.” The VPipe Software Modules (Fig. 1) which encompass the Data Processing and Database layers (Fig. S2), are hidden from the user and run automatically without user interaction. The VPipe Database layer utilizes MongoDB, which provides a centralized database for NGS analysis results. The Data Visualization component utilizes the MERN architecture (MongoDB, ExpressJS, ReactJS, NodeJS) to modulate the flow of information from the database to a webpage to display the final results.
(iii) VPipe interface. Analysis results are viewed on the VPipe web interface, accessible through the CDC Secure Access Management System (SAMS) (https://sams.cdc.gov) and OAMD portal. The 20 most recently analyzed runs are listed on the Run page (i.e., landing page); older runs can be accessed using the “Search by Run ID” feature at the top of the page (Fig. S3A). Selecting a run and clicking the “View Samples” button (Fig. S3B) opens the “Samples” page (Fig. S3C), providing basic information on the specimens analyzed and the top virus detected (i.e., BLASTN hit for the longest contig with a viral match). FastQC results for each specimen are also accessible from this page by clicking “Expand” at the bottom of each sample tile (Fig. S3C). Users select the tiles for specimens to view, and then select “View Contigs.” This opens the “Contigs” results page, displaying the BLASTN results for assembled contigs and allowing selection of contigs for download (Fig. S1). Contig results can be sorted based on contig length, sample ID (when reviewing results for multiple specimens), minimum percent identity, species, and genus, and can be filtered based on the taxonomy and percent identity of the BLASTN results. By default, a classification filter for “viruses” is applied, but this filter can be removed to view contigs with non-viral BLASTN matches. A blue “Type” button is activated for contigs with a BLASTN hit to picornaviruses. This pushes the contig to PiType, a web-based typing tool for picornaviruses (https://pitype.cdc.gov/).
For SARS-CoV-2 data, on the Samples page for a given run, users can choose to view either de novo assembly results or reference-based recruitment results. The reference-based recruitment output displays an alignment of the consensus sequences and reference genome, and a table summarizing SNPs and their quality, computed internally by FreeBayes. The output also includes a section for downloading the filtered reads, consensus sequences, alignments, and variant/SNP calls.
VPipe benchmarking.
(i) Validation of the de novo analysis pipeline. VPipe was validated through analysis of publicly available NGS data sets for clinical specimens. The performance of the pipeline was benchmarked against similar tools, including drVM (20), EDGE (25), VirMAP (21), and Genome Detective (27). For comparative analysis using EDGE, both raw reads and VPipe-preprocessed reads were analyzed with human/host read removal and SPAdes assembly. Raw reads were run through VirMAP and Genome Detective using the default settings. Assembly results for drVM were taken from published comparisons (20). Contigs constructed in VPipe, VirMAP, and EDGE which exceeded the target virus genome or segment size were trimmed in Geneious 11.1.5 (https://www.geneious.com/). For rotavirus (DRR049387), up to 167 bp, 667 bp, and 17 bp was trimmed from ends of contigs constructed using VPipe, VirMAP, and Genome Detective, respectively (Tables S2–S4). For influenza virus (ERR690519), VPipe contigs and VirMAP contigs both required up to 20-bp trimming from ends (Tables S5 and S6); Genome Detective contigs required no trimming. For enterovirus D70, the VirMAP contig required 137 bp to be trimmed from the 5′ end (Table 1). For nonsegmented viruses, genome coverage of contigs was estimated by comparing them against selected complete GenBank genomes through NCBI BLASTN (46) (Table S1). For SARS-CoV-2 analysis in VPipe, 23 de novo assemblies were manually corrected in Geneious 11.1.5, due to either misassembled regions or mismatching regions relative to the Wuhan reference (MN908947.3), and had an average of 281 bp trimmed (Table S7). These specimens are the same data set used for validation of the reference recruitment module (see Methods). For the human respiratory syncytial virus (HRSV) and SARS-CoV-2 data sets, maximum contig lengths were compared across VPipe, EDGE, and Genome Detective using R (version 3.6.1). Run times in VPipe for the SRA samples analyzed ranged from 23.2 min to 41.6 h (median = 59.5 min), depending upon the size and complexity of the data set.
(ii) Validation of the reference recruitment module. Twenty SARS-CoV-2 specimens and three control isolates were previously sequenced at the CDC in January 2021 and the consensus genomes submitted to NCBI (MZ348310, MZ348328-MZ348329, MZ391030-MZ391046). Briefly, SARS-CoV-2 amplicons were generated using a multiplex PCR (4-pool procedure was derived from the 6-pool procedure as previously described [47]), followed by Illumina DNA Prep and sequencing on a MiSeq 300 cycle v2 run (2 × 150 bp). The original consensus genomes uploaded to NCBI were generated using a pipeline at the CDC, employing IRMA with settings customized for SARS-CoV-2 (48). Consensus sequences from NCBI GenBank were aligned with VPipe (unmasked consensus and consensus masked at positions with coverage of <25×) and Genome Detective consensus using MAFFT in Geneious 11.1.5 to detect and compare SNPs.
(iii) VPipe availability. VPipe is hosted on the CDC's Office of Advanced Molecular Detection (OAMD) Scientific Computing and Bioinformatics Support and high-performance cluster. The application is available to CDC partner public health laboratories, and access can be requested by emailing vpipe@cdc.gov.
Supplementary Material
ACKNOWLEDGMENTS
We thank Yan Li, Anna Montmayeur, Ying Tao, and Anna Uehara for their work preparing/analyzing the SARS-CoV-2 specimens utilized in this study.
This work was made possible through support from the CDC NCIRD Office of Informatics and Office of Advanced Molecular Detection.
The findings and conclusions are those of the authors and do not represent the official position of the CDC.
Footnotes
Supplemental material is available online only.
Contributor Information
Margaret Okomo-Adhiambo, Email: gfv3@cdc.gov.
Wei-Hua Chen, Huazhong University of Science and Technology.
REFERENCES
- 1.Dunne WM, Jr., Westblade LF, Ford B. 2012. Next-generation and whole-genome sequencing in the diagnostic clinical microbiology laboratory. Eur J Clin Microbiol Infect Dis 31:1719–1726. doi: 10.1007/s10096-012-1641-7. [DOI] [PubMed] [Google Scholar]
- 2.Maljkovic Berry I, Melendrez MC, Bishop-Lilly KA, Rutvisuttinunt W, Pollett S, Talundzic E, Morton L, Jarman RG. 2020. Next generation sequencing and bioinformatics methodologies for infectious disease research and public health: approaches, applications, and considerations for development of laboratory capacity. J Infect Dis 221:S292–S307. doi: 10.1093/infdis/jiz286. [DOI] [PubMed] [Google Scholar]
- 3.Charre C, Ginevra C, Sabatier M, Regue H, Destras G, Brun S, Burfin G, Scholtes C, Morfin F, Valette M, Lina B, Bal A, Josset L. 2020. Evaluation of NGS-based approaches for SARS-CoV-2 whole genome characterisation. Virus Evol 6:veaa075. doi: 10.1093/ve/veaa075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oakeson KF, Wagner JM, Mendenhall M, Rohrwasser A, Atkinson-Dunn R. 2017. Bioinformatic analyses of whole-genome sequence data in a public health laboratory. Emerg Infect Dis 23:1441–1445. doi: 10.3201/eid2309.170416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Firth C, Lipkin WI. 2013. The genomics of emerging pathogens. Annu Rev Genomics Hum Genet 14:281–300. doi: 10.1146/annurev-genom-091212-153446. [DOI] [PubMed] [Google Scholar]
- 6.Chiu CY. 2013. Viral pathogen discovery. Curr Opin Microbiol 16:468–478. doi: 10.1016/j.mib.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dark MJ. 2013. Whole-genome sequencing in bacteriology: state of the art. Infect Drug Resist 6:115–123. doi: 10.2147/IDR.S35710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koboldt DC, Steinberg KM, Larson DE, Wilson RK, Mardis ER. 2013. The next-generation sequencing revolution and its impact on genomics. Cell 155:27–38. doi: 10.1016/j.cell.2013.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sboner A, Mu XJ, Greenbaum D, Auerbach RK, Gerstein MB. 2011. The real cost of sequencing: higher than you think! Genome Biol 12:125. doi: 10.1186/gb-2011-12-8-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deurenberg RH, Bathoorn E, Chlebowicz MA, Couto N, Ferdous M, García-Cobos S, Kooistra-Smid AM, Raangs EC, Rosema S, Veloo AC, Zhou K, Friedrich AW, Rossen JW. 2017. Application of next generation sequencing in clinical microbiology and infection prevention. J Biotechnol 243:16–24. doi: 10.1016/j.jbiotec.2016.12.022. [DOI] [PubMed] [Google Scholar]
- 11.Armstrong GL, MacCannell DR, Carleton HA, Neuhaus EB, Bradbury RS, Posey JE, Taylor J, Gwinn M. 2019. Pathogen genomics in public health. N Engl J Med 381:2569–2580. doi: 10.1056/NEJMsr1813907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gwinn M, MacCannell DR, Khabbaz RF. 2017. Integrating advanced molecular technologies into public health. J Clin Microbiol 55:703–714. doi: 10.1128/JCM.01967-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gargis AS, Kalman L, Lubin IM. 2016. Assuring the quality of next-generation sequencing in clinical microbiology and public health laboratories. J Clin Microbiol 54:2857–2865. doi: 10.1128/JCM.00949-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Association of Public Health Laboratories (APHL). 2015. Next generation sequencing in public health laboratories. AHPL. [Google Scholar]
- 15.Lai B, Wang F, Wang X, Duan L, Zhu H. 2015. InteMAP: integrated metagenomic assembly pipeline for NGS short reads. BMC Bioinformatics 16:244. doi: 10.1186/s12859-015-0686-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wan Y, Renner DW, Albert I, Szpara ML. 2015. VirAmp: a galaxy-based viral genome assembly pipeline. GigaScience 4:19. doi: 10.1186/s13742-015-0060-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim B, Ali T, Lijeron C, Afgan E, Krampis K. 2017. Bio-Docklets: virtualization containers for single-step execution of NGS pipelines. GigaScience 6:1–7. doi: 10.1093/gigascience/gix048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roux S, Tournayre J, Mahul A, Debroas D, Enault F. 2014. Metavir 2: new tools for viral metagenome comparison and assembled virome analysis. BMC Bioinformatics 15:76. doi: 10.1186/1471-2105-15-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao G, Wu G, Lim ES, Droit L, Krishnamurthy S, Barouch DH, Virgin HW, Wang D. 2017. VirusSeeker, a computational pipeline for virus discovery and virome composition analysis. Virology 503:21–30. doi: 10.1016/j.virol.2017.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin H-H, Liao Y-C. 2017. drVM: a new tool for efficient genome assembly of known eukaryotic viruses from metagenomes. GigaScience 6:1–10. doi: 10.1093/gigascience/gix003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ajami NJ, Wong MC, Ross MC, Lloyd RE, Petrosino JF. 2018. Maximal viral information recovery from sequence data using VirMAP. Nat Commun 9:3205. doi: 10.1038/s41467-018-05658-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Plyusnin I, Kant R, Jääskeläinen AJ, Sironen T, Holm L, Vapalahti O, Smura T. 2020. Novel NGS pipeline for virus discovery from a wide spectrum of hosts and sample types. Virus Evol 6:veaa091. doi: 10.1093/ve/veaa091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iacoangeli A, Al Khleifat A, Sproviero W, Shatunov A, Jones AR, Morgan SL, Pittman A, Dobson RJ, Newhouse SJ, Al-Chalabi A. 2019. DNAscan: personal computer compatible NGS analysis, annotation and visualisation. BMC Bioinformatics 20:213. doi: 10.1186/s12859-019-2791-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Naccache SN, Federman S, Veeraraghavan N, Zaharia M, Lee D, Samayoa E, Bouquet J, Greninger AL, Luk KC, Enge B, Wadford DA, Messenger SL, Genrich GL, Pellegrino K, Grard G, Leroy E, Schneider BS, Fair JN, Martinez MA, Isa P, Crump JA, DeRisi JL, Sittler T, Hackett J, Jr., Miller S, Chiu CY. 2014. A cloud-compatible bioinformatics pipeline for ultrarapid pathogen identification from next-generation sequencing of clinical samples. Genome Res 24:1180–1192. doi: 10.1101/gr.171934.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li PE, Lo CC, Anderson JJ, Davenport KW, Bishop-Lilly KA, Xu Y, Ahmed S, Feng S, Mokashi VP, Chain PS. 2017. Enabling the democratization of the genomics revolution with a fully integrated web-based bioinformatics platform. Nucleic Acids Res 45:67–80. doi: 10.1093/nar/gkw1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Y, Wang H, Nie K, Zhang C, Zhang Y, Wang J, Niu P, Ma X. 2016. VIP: an integrated pipeline for metagenomics of virus identification and discovery. Sci Rep 6:23774. doi: 10.1038/srep23774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vilsker M, Moosa Y, Nooij S, Fonseca V, Ghysens Y, Dumon K, Pauwels R, Alcantara LC, Vanden Eynden E, Vandamme A-M, Deforche K, de Oliveira T. 2019. Genome Detective: an automated system for virus identification from high-throughput sequencing data. Bioinformatics 35:871–873. doi: 10.1093/bioinformatics/bty695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Agoti CN, Otieno JR, Munywoki PK, Mwihuri AG, Cane PA, Nokes DJ, Kellam P, Cotten M. 2015. Local evolutionary patterns of human respiratory syncytial virus derived from whole-genome sequencing. J Virol 89:3444–3454. doi: 10.1128/JVI.03391-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shintani T, Ghosh S, Wang Y-H, Zhou X, Zhou D-J, Kobayashi N. 2012. Whole genomic analysis of human G1P[8] rotavirus strains from different age groups in China. Viruses 4:1289–1304. doi: 10.3390/v4081289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Y-H, Pang B-B, Ghosh S, Zhou X, Shintani T, Urushibara N, Song Y-W, He M-Y, Liu M-Q, Tang W-F, Peng J-S, Hu Q, Zhou D-J, Kobayashi N. 2014. Molecular epidemiology and genetic evolution of the whole genome of G3P[8] human rotavirus in Wuhan, China, from 2000 through 2013. PLoS One 9:e88850. doi: 10.1371/journal.pone.0088850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17:10–12. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 32.Deng X, Naccache SN, Ng T, Federman S, Li L, Chiu CY, Delwart EL. 2015. An ensemble strategy that significantly improves de novo assembly of microbial genomes from metagenomic next-generation sequencing data. Nucleic Acids Res 43:e46. doi: 10.1093/nar/gkv002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lo C-C, Chain PS. 2014. Rapid evaluation and quality control of next generation sequencing data with FaQCs. BMC Bioinformatics 15:366. doi: 10.1186/s12859-014-0366-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Castro CJ, Marine RL, Ramos E, Ng TFF. 2020. The effect of variant interference on de novo assembly for viral deep sequencing. BMC Genomics 21:421. doi: 10.1186/s12864-020-06801-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sutton TDS, Clooney AG, Ryan FJ, Ross RP, Hill C. 2019. Choice of assembly software has a critical impact on virome characterisation. Microbiome 7:12. doi: 10.1186/s40168-019-0626-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sapoval N, Mahmoud M, Jochum MD, Liu Y, Leo Elworth RA, Wang Q, Albin D, Ogilvie HA, Lee MD, Villapol S, Hernandez KM, Berry IM, Foox J, Beheshti A, Ternus K, Aagaard KM, Posada D, Mason CE, Sedlazeck FJ, Treangen TJ. 2021. SARS-CoV-2 genomic diversity and the implications for qRT-PCR diagnostics and transmission. Genome Res 31:635–644. doi: 10.1101/gr.268961.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Popa A, Genger J-W, Nicholson MD, Penz T, Schmid D, Aberle SW, Agerer B, Lercher A, Endler L, Colaço H, Smyth M, Schuster M, Grau ML, Martínez-Jiménez F, Pich O, Borena W, Pawelka E, Keszei Z, Senekowitsch M, Laine J, Aberle JH, Redlberger-Fritz M, Karolyi M, Zoufaly A, Maritschnik S, Borkovec M, Hufnagl P, Nairz M, Weiss G, Wolfinger MT, von Laer D, Superti-Furga G, Lopez-Bigas N, Puchhammer-Stöckl E, Allerberger F, Michor F, Bock C, Bergthaler A. 2020. Genomic epidemiology of superspreading events in Austria reveals mutational dynamics and transmission properties of SARS-CoV-2. Sci Transl Med 12:eabe2555. doi: 10.1126/scitranslmed.abe2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Simonetti M, Zhang N, Harbers L, Milia MG, Brossa S, Nguyen TTH, Cerutti F, Berrino E, Sapino A, Bienko M, Sottile A, Ghisetti V, Crosetto N. 2021. COVseq is a cost-effective workflow for mass-scale SARS-CoV-2 genomic surveillance. Nat Commun 12:3903. doi: 10.1038/s41467-021-24078-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.García-López R, Vázquez-Castellanos JF, Moya A. 2015. Fragmentation and coverage variation in viral metagenome assemblies, and their effect in diversity calculations. Front Bioeng Biotechnol 3:141–115. doi: 10.3389/fbioe.2015.00141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ji H, Enns E, Brumme CJ, Parkin N, Howison M, Lee ER, Capina R, Marinier E, Avila-Rios S, Sandstrom P, Van Domselaar G, Harrigan R, Paredes R, Kantor R, Noguera-Julian M. 2018. Bioinformatic data processing pipelines in support of next-generation sequencing-based HIV drug resistance testing: the Winnipeg Consensus. J Int Aids Soc 21:e25193. doi: 10.1002/jia2.25193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, Stange-Thomann Y, Stojanovic N, Subramanian A, Wyman D, Rogers J, Sulston J, Ainscough R, Beck S, Bentley D, Burton J, Clee C, Carter N, Coulson A, Deadman R, Deloukas P, Dunham A, Dunham I, Durbin R, French L, Grafham D, International Human Genome Sequencing Consortium, et al. 2001. Initial sequencing and analysis of the human genome. Nature 409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 44.Nurk S, Bankevich A, Antipov D, Gurevich AA, Korobeynikov A, Lapidus A, Prjibelski AD, Pyshkin A, Sirotkin A, Sirotkin Y, Stepanauskas R, Clingenpeel SR, Woyke T, McLean JS, Lasken R, Tesler G, Alekseyev MA, Pevzner PA. 2013. Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J Comput Biol 20:714–737. doi: 10.1089/cmb.2013.0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 47.Paden CR, Tao Y, Queen K, Zhang J, Li Y, Uehara A, Tong S. 2020. Rapid, sensitive, full-genome sequencing of severe acute respiratory syndrome coronavirus 2. Emerg Infect Dis 26:2401–2405. doi: 10.3201/eid2610.201800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shepard SS, Meno S, Bahl J, Wilson MM, Barnes J, Neuhaus E. 2016. Viral deep sequencing needs an adaptive approach: IRMA, the iterative refinement meta-assembler. BMC Genomics 17:708. doi: 10.1186/s12864-016-3030-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material. Download SPECTRUM02564-21_Supp_1_seq1.pdf, PDF file, 0.5 MB (1.1MB, pdf)