

SUMMARY
Colorectal cancers (CRCs) arise from precursor polyps whose cellular origins, molecular heterogeneity, and immunogenic potential may reveal diagnostic and therapeutic insights when analyzed at high resolution. We present a single-cell transcriptomic and imaging atlas of the two most common human colorectal polyps, conventional adenomas and serrated polyps, and their resulting CRC counterparts. Integrative analysis of 128 datasets from 62 participants reveals adenomas arise from WNT-driven expansion of stem cells, while serrated polyps derive from differentiated cells through gastric metaplasia. Metaplasia-associated damage is coupled to a cytotoxic immune microenvironment preceding hypermutation, driven partly by antigen-presentation differences associated with tumor cell-differentiation status. Microsatellite unstable CRCs contain distinct non-metaplastic regions where tumor cells acquire stem cell properties and cytotoxic immune cells are depleted. Our multi-omic atlas provides insights into malignant progression of colorectal polyps and their microenvironment, serving as a framework for precision surveillance and prevention of CRC.
In brief
A single-cell resolution atlas of human colorectal polyps maps out distinct paths for pre-cancer to cancer transformation, accompanied by differential immune microenvironment features.
Graphical Abstract
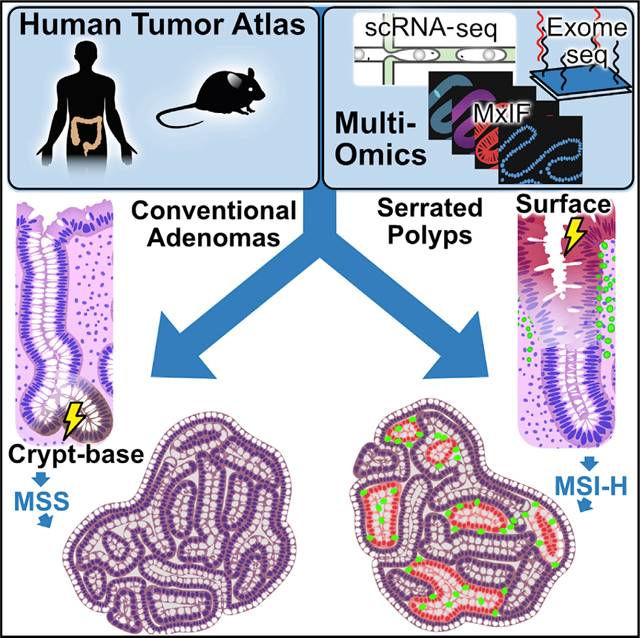
INTRODUCTION
Classification schemes for human colorectal cancer (CRC) focus largely on intrinsic features of tumor cells, including histopathology, bulk gene expression (Consensus Molecular Subtypes or CMS), chromosomal instability (CIN), hypermethylation (CpG Island Methylator Phenotype or CIMP), and microsatellite-instability (MSI) (Guinney et al., 2015; Ogino and Goel, 2008). The tumor immune microenvironment is also critical to CRC pathogenesis (Pelka et al., 2021). Hypermutated MSI-high (MSI-H) tumors exhibit a neoantigen-triggered cytotoxic immune infiltration that contributes to their responsiveness to immunotherapy (Le et al., 2015; Llosa et al., 2015). However, a significant subset of low mutation burden CRCs appears to exhibit an activated immune microenvironment via ill-defined mechanisms (Mlecnik et al., 2016). We hypothesize that mapping the routes toward tumorigenesis in precursors of MSI-H and MSS CRCs will uncover mechanisms that define the CRC cellular landscape and identify targets with diagnostic or therapeutic utility.
Most MSS and MSI-H CRCs develop from pre-cancerous conventional adenomas (ADs) and sessile serrated lesions (SSLs; formerly sessile serrated adenomas/polyps), respectively. As proposed by Vogelstein and co-workers, ADs arise from truncating mutations in APC, which result in activation of the WNT pathway and CIN (Fearon and Vogelstein, 1990). ADs subsequently accumulate gain-of-function mutations in oncogenes (chiefly KRAS) and loss-of-function mutations in tumor suppressor genes such as TP53, ultimately forming MSS CRCs. Conversely, SSLs resemble MSI-H CRCs molecularly and are distinct from ADs in that tumorigenesis is not initiated by genetic disruptions of APC (Crockett and Nagtegaal, 2019; Thorstensen et al., 2005). Instead, they have epigenetic disruptions, including MLH1 hypermethylation and a 40%–75% prevalence of CIMP (Leggett and Whitehall, 2010; Yang et al., 2004). These tumors harbor BRAF mutations in contrast to KRAS mutations commonly present in ADs. Mirroring the relatively lower incidence of MSI-H CRCs and their prevalence in the proximal colon, SSLs represent only 10%–20% of polyps and are also found in the proximal colon more often, unlike the more frequently distal ADs (Crockett and Nagtegaal, 2019; Markowitz and Bertagnolli, 2009).
We present a multi-omic human pre-cancer atlas integrating single-cell transcriptomics, genomics, and immunohistopathology describing the two most common pathways toward CRC. We identify and functionally validate distinct origins and molecular processes that establish divergent tumor landscapes. Notably, this clearer understanding of advanced and highly heterogeneous cancers was enabled only by looking at CRCs through the lens of their originating lesions, paving a path to new strategies for precision prevention, surveillance, and therapeutics.
RESULTS
Distinct histopathologic and molecular features define colonic pre-cancer subtypes
Polyps, as well as matching normal biopsies, were collected from COLON MAP study participants. Most polyps were small (median diameter ≤5 mm) and were bisected for multi-assay analysis. Single-cell RNA sequencing (scRNA-seq), multiplex immunofluorescence (MxIF), and multiplex immunohistochemistry (MxIHC) were performed on two independent sets of specimens collected approximately 1 year apart. The discovery (DIS) set consisted of 65 specimens analyzed including 30 tumors. The validation (VAL) set consisted of 63 specimens analyzed including 32 tumors (Figure 1A). Overall, 128 independent scRNA-seq datasets on 62 tumors were generated (Table S1). Specimens were collected from diverse sex, racial, and age groups (Table S2). In addition, we performed bulk RNA-seq and targeted gene sequencing on an orthogonal set of 66 and 281 polyps, respectively (Figure S1A; Table S2). Polyps were histologically categorized by two pathologists into two subtypes: ADs consisting of tubular ADs (TAs) and tubulovillous ADs (TVAs), or serrated polyps (SERs) consisting of hyperplastic polyps (HPs) and SSLs (Figure 1B). While standard histological features were observed for polyps, HPs were further subdivided into goblet cell-rich HPs (GCHPs) and microvesicular HPs (MVHPs), with MVHPs appearing more advanced and may progress to SSLs (Crockett and Nagtegaal, 2019). Epithelial serrations of GCHPs, if present, were subtle and confined to the mucosal surface, while, for MVHPs, serrations extended from the surface to two-thirds down the crypt, sparing the morphologically normal crypt base. In contrast, SSLs showed epithelial serrations that extended to the base of crypts, which were dilated and spread laterally above the muscularis mucosae.
Figure 1. Features of human colonic pre-cancers.

(A) Experimental design for profiling tumor subtypes across multiple datasets.
(B) Haematoxylin and eosin (H&E) images of normal colonic tissue and polyp subtypes. Green brackets, crypt portions occupied by neoplastic cells.
(C) Oncoplot of somatic mutations by WES for polyps. (Top) Mutation burden represented by bar plot. (Dark-gray boxes) CRC driver genes are grouped into pathways. (Right) Percentage of mutations within subtypes summarized as a table.
We characterized the mutational profiles of ADs and SERs by conducting whole-exome sequencing (WES) and somatic mutation calling (Figure 1C). Due to small polyp sizes and the prioritization of fresh tissue for single-cell assays, we used the clinical formalin-fixed paraffin-embedded (FFPE) material for WES. About half generated sufficient sequence quality for analysis, and the predominant mutational patterns were consistent with published literature. APC mutations were detected in 85% of the TAs and in both TVAs. Only one (8%) TA had a KRAS mutation, while both TVAs did, consistent with TVAs being more histologically advanced. All but one SSL (89%) had the oncogenic BRAFV600E mutation; none of the three GCHPs harbored BRAF mutations, but two (67%) MVHPs did, consistent with MVHPs being SSLs in evolution. Neither APC nor KRAS mutations were detected in SSLs, and none of the ADs had BRAF mutations. Somewhat surprisingly, none of the SSLs exhibited a hypermutation phenotype, while a portion of TA/TVAs did. Whereas MLH1 expression is usually lost in MSI-H CRCs due to promoter methylation, MLH1 protein and gene expression in SSLs were comparable to ADs, both higher than the mean MSI-H CRC level (Figures S1B and S1C). Biallelic loss in mismatch repair genes was not detected in any polyp, further supporting that these SSLs had not yet acquired a hypermutation phenotype.
We validated this mutational analysis using targeted gene sequencing of a separate set of 281 premalignant tumors (Figure S1A). General trends were consistent, with mutations in APC increasing from 67% to 91%, and KRAS from 5% to 42% between TAs and TVAs. BRAF mutations were enriched in SSLs (67%) compared to TAs (1%) and TVAs (4%). Again, none of the SSLs exhibited a high mutation load, where several TA/TVAs did, confirming WES results. Non-APC mutations in WNT pathway genes, such as RNF43 or ZNRF43, were uncommon in SSLs from either dataset. Signaling pathway analysis from combined mutational analysis paints a picture of WNT-driven tumorigenesis in TA and TVAs but not in SSLs.
Single-cell analysis identifies neoplastic cells that arose from subtype-specific tumorigenic processes
We generated scRNA-seq data on 70,691 (DIS dataset) and 71,374 cells (VAL dataset) (total: 142,065), after filtering for high-quality barcodes using dropkick (Heiser et al., 2021), and cells from specimens with unconfirmed histology (labeled UNC) were transcriptomically classified (Table S3). We conducted UMAP dimension reduction on raw scRNA-seq data and observed intermixing of epithelial cells from normal colonic biopsies and immune cells from different participants, indicating the absence of batch effects (Figure S1D). However, neoplastic cells clustered by sample, demonstrating intertumoral variability consistent with unique tumorigenic processes.
Since transcription factor (TF)-defined regulon activities are considered to be a determinant of cell identity, we used SCENIC (single-cell regulatory network inference and clustering), which is a regulon-based, batch-robust feature extraction tool, to adjust for polyp-specific effects (Aibar et al., 2017; Van de Sande et al., 2020). Clustering and co-embedding epithelial cells from the DIS dataset in regulon space revealed seven normal, canonical epithelial cell populations using normal biopsy datasets as reference landmarks (Figures 2A and 2B; Figure S2A). Polyp specimens also contained substantial numbers of normal cells consistent with their histopathology (Figures 1B, 2B; Figure S2B; Table S3). However, two cell populations were overwhelmingly represented in polyp samples, as determined by sample-by-sample breakdown of proportional cluster representation (Figures 2B and 2C; Figure S2C). One population was enriched in TA and TVA, hereafter referred to as ASCs (AD-specific cells, p < 1E-4 Mann-Whitney U [MWU] test) (Tables S3 and S4). The second neoplastic population was enriched in SSLs and HPs, hereafter referred to as SSCs (serrated-specific cells, p < 1E-4 MWU test). Importantly, these results, as well as others below, were consistent across DIS and VAL datasets (Figures 2A–2C; Figures S2A–S2D), demonstrating rigor and reproducibility.
Figure 2. Single-cell gene expression and regulatory network landscape of pre-cancers.

(A) Heatmap of top biologically relevant and differentially expressed genes for (left) DIS (n = 62) and (right) VAL (n = 59) epithelial datasets. The inset circle indicates prevalence and intensity represents scaled expression.
(B) Regulon-based UMAPs of (top) DIS and (bottom) VAL epithelial datasets color overlaid with (left) tissue or (right) cell type.
(C) Scatterplots of normalized (left) ASC or (right) SSC representation per tissue subtype. Points represent individual specimens. Error bars represent SEM of n = 29 for AD, n = 19 for SER, and n = 66 for NL.
(D) Stem, metaplasia, and fetal signature scores overlaid onto UMAPs of (C).
(E) Ridge plots of CytoTRACE score distributions for ASC, SSC, and NL cell populations across (top) DIS and (bottom) VAL datasets.
(F and G) TF target network created from normal and pre-cancer cells, organized into super-regulons for (F) ASCs and (G) SSCs. Color overlays are regulon enrichment scores, while edge opacities are the inferred TF-target weightings. ***p < 0.001.
We identified gene programs and pathways differentially activated in ASCs and SSCs compared to normal epithelial cells. ASCs resembled colonic stem and progenitor cells, expressed genes indicative of WNT pathway activation (LGR5, OLFM4, ASCL2, AXIN2, RNF43, and EPHB2) (Figure 2A), and possessed a stem cell signature greater than normal stem cells from the same individuals (Figure 2D; Figure S2D; Tables S4, S5, and S6). Because ASCs resembled normal stem cells, we used CytoTRACE to infer their stem potential (Gulati et al., 2020). Normal stem cells had high CytoTRACE scores and transitioned into differentiated cells with lower scores (Figure 2E; Figure S2E), forming a score distribution that was relatively uniform between stem, transitioning, and differentiated cells. In contrast, CytoTRACE analysis of ASCs yielded a distribution skewed toward cells with high predicted stem potential (Figure 2E; Table S4). This variation in stemness suggests the presence of tumor stem cells, supported by the enrichment of WNT-driven stemness GO terms in specific ASC subclusters (Figure S2F; Table S6). These analyses describe a model wherein WNT-dependent stem cell expansion initiates tumorigenesis in ADs most often driven by loss-of-function mutations in APC.
In marked contrast to ASCs, SSCs did not exhibit WNT pathway activation nor a stem cell signature (Figures 2A and 2D; Figures S2A and S2D). CytoTRACE scores of SSCs skewed toward a lower predicted stem potential, opposite to ASCs (Figure 2E; Figure S2E), although heterogeneity in stemness was still observed (Figure S2F; Table S6). The transcriptomic profiles of SSCs resembled absorptive-lineage cells, but SSCs also expressed functional goblet cell genes, including TFF3 and MUC2, but surprisingly without the master secretory cell TF ATOH1 and the ATOH1 regulon, suggesting SSCs harbor a mixed cellular identity (Figure 2A; Figure S2G). To this point, SSCs highly expressed genes not normally observed in the colon (MUC5AC, AQP5, TACSTD2 [TROP2], TFF2, MUC17, and MSLN) but rather found in other endodermal organs, most notably, the gastric epithelium (Figures 2A and 2D; Figure S2D; Tables S4, S5, and S6). This surprising finding, along with the expression of differentiated cell gene signatures in SSC, led us to hypothesize metaplasia may underlie the pathogenesis of SSLs.
Metaplasia is a process by which differentiated cells transdifferentiate to non-native cell types, often occurring as a regenerative mechanism after damage. Loss of CDX2, a hindgut homeobox TF, in the colon is associated with an imperfect pyloric-type gastric metaplasia and a shift toward expression of genes more rostral in the rostral-caudal gradient (Balbinot et al., 2018; Tong et al., 2017). CDX2 was expressed in most colonic cell types, including ASCs; however, it was downregulated in SSCs, supporting a loss of regional identity in these cells (Figure 2A). This loss of caudal identity in SSCs was accompanied by a reversion to an embryonic stage, supported by a fetal gene-expression signature, including the MDK gene, which encodes a heparin-binding growth factor only transiently expressed in early colonic development (Figures 2A and 2D; Figures S2A and S2D; Tables S4 and S5) (Park et al., 2005). Luminal retinoic acid-induced absorptive cell-differentiation genes (RXRA/RARA/ALDOB) were increased in SSCs (Lukonin et al., 2020), while rostral identity genes suppressed in absorptive cell differentiation (ANAX10/ANXA1) were also paradoxically increased (Figure 2A). These gene signatures depict a loss of colonic identity and provide further evidence that SSCs arise from a metaplastic process.
We used TF target similarity to create a common TF regulatory network depicting the coordinated regulation of genes as programs and pathways. Some coordinated clusters of regulons, which we referred to as super-regulons, were overrepresented in ASCs versus SSCs, including WNT- and Hippo-driven super-regulons marked by MYC, ASCL2, TCF7, and TEAD1 activities (Figure 2F; Figure S2G), consistent with the role of these programs in the regeneration and renewal of intestinal stem cells (Ayyaz et al., 2019; Murata et al., 2020). For SSCs, supporting the role of a damage-induced metaplastic process, a super-regulon indicating interleukin signaling and microbiota interaction was observed (Figure 2G; Figure S2G). Specifically, upregulated transcription factor activities for SSCs included RELB (nuclear factor κB [NF-κB] signaling), IRF1, IRF6, and IRF7, reflecting an immunogenic state (Figures 2A and 2G), which was corroborated by gene set enrichment for microbial infection response, innate immune activation, and epithelial wound-healing pathways (Table S6) (Raudvere et al., 2019). Supporting the activation of interferon response elements, coordinated upregulation of inflammasome-related genes such as IL18 and gasdermins further implicated responses to external pathogens as triggers of metaplasia (Figure 2A) (Man, 2018). Similarly, regulons related to FOSL2, KLF4, and ATF3 were enriched (Figure 2G; Figure S2G), drawing parallels to recent work documenting increased chromatin accessibility of these TF targets in a mouse model of microbiota-driven colitis (Ansari et al., 2020). Gene signatures classifying polyp subtypes were validated with bulk RNA-seq on an additional 58 ADs (36 TAs, 22 TVAs) and eight SSLs (Figure S2H). These results confirmed our findings of a WNT-activated program of stem cell expansion in ADs, and a program of gastric metaplasia, likely arising from a committed cell lineage, in SSLs.
Serrated polyps arise from a cellular origin distinct from adenomas
Because SSCs may arise from metaplasia of differentiated cells, we hypothesized that SERs originate from differentiated cells in a “top-down” model of tumorigenesis, compared to ADs arising from proliferative stem cells in a “bottom-up” fashion. To provide histological evidence of tumor origins, we mapped the location of neoplastic cells by multiplex imaging. Stem cell markers, OLFM4 and SOX9, were abundant in ADs but were significantly reduced in HPs and SSLs (Figures 3A and 3B; Figures S3A and S3B). Nuclear CDX2 was detected in the normal colon and in ADs but was decreased in HPs and absent in SSLs (Figure 3C; Figure S3C). MUC5AC, a marker of SSCs, was highly expressed in HPs and SSLs but not in normal biopsies and ADs (Figure 3D; Figure S3D). Interestingly, MUC5AC-positive, neoplastic cells were often observed at the top of the crypt with normal-appearing MUC5AC-negative cells at the crypt bottom, implying a non-crypt origin of SERs. MUC5AC-positive cells first appeared at the luminal surface in GCHPs and then extended further to the crypt base in MVHPs and SSLs (Figures S3D and S3E), consistent with the histopathological progression of these SERs (Figure 1B) and supporting the luminal surface origin of SSCs. MUC5AC-positive cells were detected in the majority of abnormal crypts from SERs (Figure S3F) but were largely absent in the normal colon. However, occasional MUC5AC staining was detected, again, at the luminal surface in a few specimens, and was further increased in ulcerative colitis patients (Figure S3G). Luminal surface colonic cells appear susceptible to damage-induced metaplasia that may elicit serrated polyp formation if the damage is not resolved.
Figure 3. Inferred origins of pre-cancers.

(A–D) Multiplex images of colonic polyps and normal tissues for (A) SOX9, (B) OLFM4, (C) CDX2, and (D) MUC5AC. (Right) Image quantification (n = 20 polyps per subtype).
(E) p-Creode analysis on epithelial regulon landscapes, for (top) DIS and (bottom) VAL datasets. For gene overlays, node size represents cell proportion and intensity represents scaled expression.
(F) RNA velocity for representative NL, TA, and SSL overlaid on combined UMAP embedding for DIS. Vectors inferring average transitions shown as black arrows. Colored points are cells derived from the representative specimen, and gray points are all other cells in the dataset.
*p < 0.05, **p < 0.01, ****p < 0.0001. See also Figure S3.
We inferred transition trajectories from epithelial cells using p-Creode on batch-robust SCENIC regulons, which produced a stereotypical colonic differentiation hierarchy (Herring et al., 2018). CytoTRACE score and WNT target gene overlays identified the stem cell branch, which was shared with ASCs, suggesting aberrantly expanded stem cells as the origin of ADs (Figure 3E; Figure S3H). In marked contrast, SSCs were inferred to develop from absorptive progenitors and colonocytes. RNA velocity analysis on individual tumors largely confirmed these findings (Figure 3F; Figure S3I) (Bergen et al., 2020; La Manno et al., 2018). In normal specimens, velocity vectors originated from stem cells and flowed into differentiated cell types. ASCs were implicated to develop from stem cells, but the velocity vectors were reversed for SSCs, suggesting the origin of these cells to be non-stem cells.
To further infer shared origins, we determined phylogenetic distances from genetic variants between normal and neoplastic cells. We used DENDRO (DNA-based evolutionary tree prediction by scRNA-seq technology), a phylogenetic reconstruction algorithm on scRNA-seq data that adjusts for inherent data sparsity (Zhou et al., 2020). We improved on DENDRO’s robustness, and exonic variants detected were further validated through WES of paired FFPE tissues. DENDRO reconstruction of 34 polyps showed that ASCs were more genetically related to crypt base stem cells than SSCs (p < 5E-02 MWU test) (Figures S3J and S3K; Table S1). In fact, SSCs often clustered genetically with differentiated colonocytes and absorptive progenitors (Figure S3J). Orthogonal methodologies produced histological, transcriptomic, and genetic evidence to support the hypothesis that ADs arise from dysregulation of the stem cell compartment, but SSLs appear to arise from a developmentally committed cell.
Subtype-specific features are altered during malignant progression from pre-cancer to cancer
We performed scRNA-seq on seven (two MSI-H, five MSS) fresh CRC specimens and procured a CRC scRNA-seq dataset (n = 60; 32 MSI-H, 28 MSS) from the Broad Institute for validation. Furthermore, we analyzed whole tumor blocks from 26 additional CRC patients (14 MSI-H, 12 MSS) (Table S7). WES of CRC specimens revealed expected mutational features in MSS CRCs following the conventional tumorigenesis pathway with APC (100%), KRAS (35%), and TP53 (71%) mutations (Figure S4A). MSI-H CRCs had fewer of these conventional mutations (33%, 0%, 7%, respectively) but more BRAF mutations (53% in MSI-H versus 0% in MSS). All MSI-H CRCs were hyper-mutated compared to MSS CRCs. Histologically, all CRCs showed invasive adenocarcinoma with cribriform architecture (Figure S4B), with MSI-H CRCs exhibiting mucinous features.
scRNA-seq data of malignant CRC cells revealed substantial tumor-to-tumor variability, as seen by others (Lee et al., 2020; Pelka et al., 2021) even after regulon-based embedding (Figure 4A). Combined with our pre-cancer data, an increase in intertumoral heterogeneity was observed as epithelial cells transition from normal to pre-cancer to malignant cells. We considered that the intrinsic complexity and heterogeneity of CRC transcriptomics might be reduced by looking at CRC cells through the lens of pre-cancerous polyps. By using pre-identified gene sets from ADs and SERs, we observed that both MSS and MSI-H CRC cells retained aspects of their respective precursors. Comparing the two subtypes, MSS CRC cells overexpressed a signature of regenerative crypt base stem cells, and MSI-H CRC cells retained a metaplastic signature (Figures 4B and 4C; Figure S4C; Tables S4 and S5). These patterns were observed using another scRNA-seq dataset (Lee et al., 2020) (Figure S4D). To further support commonalities between pre-cancer and cancer, we classified ASCs, SSCs, and CRC cells by consensus molecular subtype (CMS) (Eide et al., 2017; Guinney et al., 2015) (Figure 4D; Table S4). ASCs and MSS CRC cells scored highly for CMS2, the subtype most often associated with WNT pathway dysregulation. In contrast, both SSCs and MSI-H CRC cells scored low for CMS2, but high for CMS1 and CMS3, which feature immunogenic and RAS pathway activation, respectively (Chi et al., 2009; Feng et al., 2011; Liao et al., 2018). None of the examined cells enriched strongly for CMS4, consistent with previous reports (Chang et al., 2018; Komor et al., 2018). Shared features between malignant cells and pre-cancerous cells provide additional evidence of precursor-cancer relationships.
Figure 4. Analysis of CRCs through the lens of pre-cancers.

(A) Regulon-based UMAPs for tumor-specific cells overlaid with (top) subtypes and (bottom) specimen for the (left) VUMC and (right) Broad datasets.
(B) Stem, metaplasia, and fetal signature scores overlaid onto UMAPs in (A).
(C) Heatmap representation of pre-cancer-derived gene sets for VUMC (n = 55 specimens) and Broad (n = 60 specimens) tumor-specific cells.
(D) Single-cell CMS scoring based on single sample predictor for tumor-specific cells.
(E) Ridge plots of CytoTRACE score distributions for tumor-specific cells.
(F–I) TF target network created from tumor-specific cells, organized into super-regulons for (F) ASC, (G) MSS, (H) SSC, and (I) MSI-H.
We also examined the characteristics acquired or lost during the transition from pre-cancer to malignancy. MSI-H CRC cells showed relatively decreased metaplastic and fetal features compared to SSCs. However, key genes within the WNT-activated stem cell program were increased relative to SSCs (Figure 4C; Figure S4C; Tables S4 and S5). Supporting reactivation of stemness, CytoTRACE analysis demonstrated MSI-H CRC cells had higher inferred stem potential than SSCs, while scores of MSS CRC cells also were higher than ASCs (Figure 4E; Table S4). Gene regulatory network analysis more clearly demonstrated how molecular pathways were either maintained or altered during malignant transition, supported through GSEA (Figures 4F–4I; Figure S4E; Table S7). Both CRC subtypes activated their proliferative super-regulon compared to polyps, with enrichment of DNA synthesis and repair programs (Figures 4F–4I). The WNT signaling super-regulon was consistently upregulated in ASCs and MSS CRC cells (Figures 4F and 4G). For MSI-H CRC cells, the super-regulon describing pathogen damage response in SSCs was suppressed, but the WNT signaling super-regulon, previously suppressed in SERs, was activated (Figures 4H and 4I). The differences in super-regulon enrichment were maintained in the Broad dataset (Figure S4F). Activation of the WNT pathway was supported by acquisition of activating mutations in non-APC WNT pathway components in MSI-H CRCs, including RNF43 (60%), TCF7L2 (53%), ZNRF3 (33%), APC2 (27%), AXIN2 (20%), FAT1 (33%), FAT2 (47%), and FAT4 (40%) (Figure S4A). TCGA WES data also showed enrichment of non-APC WNT pathway gene mutations in MSI-H CRC (Figure S5A) (Cancer Genome Atlas Network, 2012). These results suggest MSI-H CRC acquired metaplasia-independent events by transitioning into more aggressive stem-like cells.
Transition from metaplasia to stemness contributes to tumor heterogeneity in MSI-H CRCs
We further queried 63 bulk RNA-seq datasets from the TCGA and validated the association between CMS subtypes and stem/metaplastic signatures (Figure S5B). However, the data were noisier than scRNA-seq data on an individual tumor basis, likely due to poor data quality and/or additional intratumoral heterogeneity. This led us to perform spatial profiling using whole slide scanning of entire CRC specimens. Strikingly, none of the MSS CRCs (0/17) stained positive for MUC5AC, but most MSI-H CRCs (13/14) did (Figures 5A and 5B). However, the amount of tumor area stained by MUC5AC was variable within the positive MSI-H CRCs. CDX2 staining followed the inverse trend; virtually all tumor cells in MSS CRCs were CDX2 positive, and MSI-H CRCs had variably decreased CDX2 staining. Stem cell markers (OLFM4, SOX9) were expressed throughout MSS CRCs, and they uniformly lacked MUC5AC expression (Figures 5C and 5D; Figure S5C). In contrast, MSI-H CRCs displayed considerable intratumoral MUC5AC heterogeneity, with low staining in certain regions of MSI-H CRCs; these regions were positive for OLFM4 and to some degree CDX2 (Figures 5E–5H). SOX9 was generally overexpressed in MSI-H CRCs, suggesting all malignant cells gained some level of stemness (Figure 5H). Focused analysis of a single scRNA-seq dataset validated these results. Positive MUC5AC and MSLN expression, coupled to loss of CDX2 expression, distinguished metaplastic cells from LGR5/β-catenin-expressing proliferative stem cells within the same tumor (Figures 5I, 4C, and 4E). We further confirmed heterogeneity of CDX2 and MUC5AC expression in a CRC tissue microarray using MLH1 staining to infer the microsatellite status of cells (Figure S5D). In multiple instances of MSI-H CRCs, we observed intratumoral heterogeneity characterized by mutual exclusivity of stem-like cells and metaplastic cells.
Figure 5. Heterogeneity of CRCs with metaplastic and stem-like features.

(A) IHC scans for MUC5AC and CDX2 of CRCs.
(B) Image quantification of n = 17 MSS and n = 14 MSI-H CRCs.
(C and D) (C) Low-mag. view and (D) high-mag. view of a MSS CRC with protein markers.
(E) Low-mag. view of a MSI-H CRC.
(F) High-mag. view of MUC5AC high and low areas for metaplasia markers of the CRC in (E).
(G and H) Same as in (E) and (F) but for stem cell markers. Black rectangles in the restitched image represent fields of views that were not scanned.
(I) UMAP of scRNA-seq data of the MSI-H CRC in (E) overlaid with markers and cell cycle signatures.
*p < 0.05, **p < 0.01.
Serrated polyps associate with a cytotoxic microenvironment prior to hypermutation
Because SERs did not demonstrate hypermutation and MSI-H CRCs did, we sought to determine whether SERs possess a distinct tumor microenvironment at this early stage. We combined analyses of the non-epithelial scRNA-seq data from pre-cancers and CRCs and identified different cell types based on marker gene expression and their compositional changes between tumor subtypes (Figures 6A and 6B; Figures S6A–S6C). Most immune cell types were increased in polyps compared to normal tissues, including CD4+ T cells, although many were not different between polyp subtypes (Figure 6C; Figure S6D; Tables S3 and S4). Strikingly, CD8+ T cells, natural killer (NK) cells, and γδT cells (labeled cytotoxic cells) were significantly increased in SERs compared to ADs (Figure 6C). The overrepresentation of cytotoxic, but not CD4+ T cells, was also observed in MSI-H CRCs compared to MSS CRCs, suggesting a consistent dichotomy in the adaptive microenvironment between subtypes regardless of hypermutation.
Figure 6. The immune landscape of colonic tumor subtypes.

(A) Regulon-based UMAP representation of non-epithelial cells.
(B) Heatmap of marker genes defining each cell type in (A). T - T cell, PLA - Plasma B cell, MYE - Myeloid, MAS - Mast, FIB - Fibroblast cell, END - Endothelial cell, B - B cell.
(C) Scatterplots of cell type representation of (top) polyp and (bottom) CRC subtypes. Points represent individual specimens. Error bars represent SEM of n = 28 for AD, n = 17 for SER, n = 66 for NL, n = 33 for MSS, and n = 34 for MSI-H.
(D and E) Scatterplots of (D) CD4+ T cell and (E) tumor cell-specific signature scores, with each point representing a single cell. Error bars depict SEM of single cells.
(F) MxIF images of CD8+ cells in polyps.
(G) Image quantification of intraepithelial CD8+ cells for n = 20 polyps per type.
(H) MxIF images of CD68+ and MUC5AC+ in cells in polyps.
(I) MxIF scans of intratumoral heterogeneous regions within CRCs (OLFM4+ stem regions versus MUC5AC+ metaplastic regions). MSS CRC only has stem regions. MxIF images of CD8 and CD3 within stem and metaplastic regions. The inset is the quantification of CD8-positive pixels in these regions from MxIF scans of n = 15 MSS and n = 10 MSI-H CRCs.
*p < 0.05, **p < 0.01, ***p < 0.001. See also Figure S6 and Tables S3, S4, and S5.
Gene signatures related to cytotoxicity and exhaustion within CD8+ T cells did not differ between ADs and SERs, but they were intensified in CD8+ T cells of MSI-H compared to MSS CRC (Figure S6E; Tables S4 and S5), signifying neoantigen hyper-interaction in the malignant, but not pre-malignant, microenvironment. FOXP3 regulon activity was higher in AD-derived versus normal colonic CD4+ T cells, consistent with a degree of Treg-dependent immunosuppression (Figure 6D). ASCs expressed a monocyte-attracting chemokine signature, while SSCs expressed a lymphocyte-attracting cytokine signature important for establishing an adaptive immune environment (Figure 6E; Tables S4 and S5) (Hieshima et al., 1997; Nelson et al., 2001). An antigen-processing and presentation gene signature (Lee et al., 2020; Pelka et al., 2021) was significantly higher in SSCs relative to ASCs, which was also increased in MSI-H CRC cells relative to MSS CRC cells (Figure 6E; Tables S4 and S5). These data illustrate the persistence of some adaptive immunity regulation mechanisms from pre-cancer to cancer that appear independent of hypermutation.
Multiplex imaging showed that SERs had a higher number of T cells, CD8+ T cells, and a higher ratio of CD8+ to CD4+ T cells compared to ADs (Figures 6F and 6G; Figures S6F and S6G), while other immune cell populations were not significantly different. CD8+ T cells infiltrated into the epithelial compartments of SERs. More CD8+ T cells were observed in ADs with higher mutational loads, although our analysis was underpowered statistically (Figure S6H). Myeloid cell abundance was not different by both scRNA-seq and imaging, but CD68+ macrophages were distributed throughout the AD stroma, while they were concentrated at the luminal surfaces of SERs, coinciding with the surface location of MUC5AC+ metaplastic cells (Figure 6H; Figure S6I). A similar striking distribution of CD68+ macrophages was reported after fecal transplant and successful immunotherapy response (Baruch et al., 2021), supporting the influence of epithelial-microbial interactions on cytotoxic immune responses. MSI-H CRCs had a heterogeneous distribution of CD8+ T cells mirroring the observed tumor cell heterogeneity. There was a significant enrichment of CD8+ T cells in MUC5AC+ metaplastic regions and reduced numbers in OLFM4+ stem-like regions (Figure 6I; Figures S6J–S6L). In contrast, MSS CRCs had fewer T cells throughout the tumors, which were homogeneously composed of OLFM4+ stem-like cells (Figure 6I; Figures S6J–S6L). These results strengthen the association between the metaplastic origin of SERs and the cytotoxic immune microenvironment and implicate immune suppression as tumor cells gain stemness.
Tumor cell-differentiation status shapes the adaptive immune microenvironment
To determine whether the cytotoxic response in serrated tumorigenesis is intrinsic to tumor cell state prior to hypermutation, we used genetically engineered mice that model the earliest tumorigenic events. The Lrig1CreERT2/+;Apc2lox14/+ is a model of the AD pathway, resulting in adenomatous tumors in the distal colon (Powell et al., 2012). Driving a Braf-activating mutation (Lrig1CreERT2/+;BrafLSL-V600E/+) did not result in macroscopic tumors but induced villiform metaplasias in the proximal colon (Figure 7A). Apc mutant tumors had elevated β-catenin staining and a reduced number of CD8+ T cells compared to control normal colon, consistent with human ADs and MSS CRCs. In contrast, Braf mutant lesions were associated with increased CD8+ T cell infiltration, strikingly, only in the differentiated cell compartment and not in mutant crypts (Figures 7B and 7C). Similar results were observed in a parallel Kras-activating mouse model (Lrig1CreERT2/+;KrasLSL-G12D/+) (Figures S7A–S7C). Thus, mutant differentiated cells in lesions, but not stem cells, drive the cytotoxic immune microenvironment.
Figure 7. Functional validation of the tumor cell-differentiation status and the effects on cytotoxic immunity.

(A) IF images of Apc-driven colonic tumor and Braf-driven proximal colon villiform metaplasia (white arrows).
(B) Quantification of CD8-positive pixels from IF. Red line denotes the mean level detected in adjacent normal colon in Braf mice. Error bars represent SEM from n = 3 animals per group.
(C) IF images of CD8+ T cells in tumor, villiform metaplasia (white arrows), and control colon. Dotted line demarcates border between villus and crypt compartments.
(D and E) H&E (D) and β-catenin IHC (E) of colonic tissues and tumors of tamoxifen-induced Lrig1 or Mist1 tumor mice 28 days after DSS.
(F–H) UMAP of epithelial scRNA-seq data generated from mouse colonic tissues and tumors, with overlays indicating (F) cell type, (G) gene overlays, and (H) biological replicates.
(I) Heatmap of genes defining human metaplastic and cell signatures in specified epithelial populations from mouse scRNA-seq.
(J and K) Combined UMAP of immune cell scRNA-seq data from mouse colonic tissues and tumors, with overlays indicating (J) conditions and (K) cell type.
(L and M) Quantification of (L) general immune cell types and (M) specific lymphocyte populations from Lrig1 (left) and Mist1 (right) scRNA-seq data.
(N) UMAP overlays of genes related to immunosuppression or cytotoxicity in myeloid and lymphoid cells.
(O) MxIF images of T cells in tumors.
(P) Image quantification of T cells. Each dot represents a field of view. Error bars represent SEM from n = 3 animals per group.
(Q) CytoTRACE score for TSCs from scRNA-seq.
(R) Organoid formation efficiency of single cells isolated from tumors and control colons. Each dot represents data from a well with representative images shown in insets. Error bars represent SEM from n = 4 animals per tumor, 2 for control.
(S) SCI, II, and III metagene signatures for TSCs from scRNA-seq.
(T) Heatmap of individual antigen-presentation genes at single-cell level.
(U) MHCII metagene signature expression for TSCs.
(V) Quantification of DQ-OVA+/I-AI-E+ epithelial tumoroid cells from flow plots. Error bars represent SEM from n = 6 animals per condition.
(W) Percentage of proliferating T cells determined by CellTrace Violet assay when co-cultured with organoids derived from colonic tumors or normal tissues (+DSS) +/— 50 mg/mL OVA peptide. Error bars represent SEM of organoids from n = 5 mice for tumors and two for normal.
*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. See also Figure S7 and Table S5.
To determine how a differentiated cell versus stem cell state influences the immune microenvironment, we normalized the genetic event by driving the same Apc mutation from stem (Lrig1CreERT2) versus non-stem (Mist1CreERT2) cells. While Lrig1+ cells are bona fide stem cells (Powell et al., 2012), lineage-tracing studies showed Mist1+ cells are non-stem cells in the proximal colon under both homeostasis and DSS-induced damage (Figures S7D–S7F). Using immunostaining and transcriptomics, we determined Mist1+ cells represent a subset of committed (goblet/enteroendocrine) cells outside the colonic crypt base (Figures S7G–S7K).
Importantly, Mist1+ cells initiated colonic tumors (abbreviated as Mist1 tumors) with biallelic recombination of Apc (Mist1CreERT2/+; Apc2lox14/2lox14) followed by 2.5% DSS damage, representing a non-stem-driven tumor model. At most, one or two Mist1 tumors developed per mouse in the proximal versus distal colon by a 7:1 ratio (Figures 7D and 7E; Figures S7L and S7M), which differs from the distal colon predominance of tumors in the Lrig1CreERT2/+; Apc2lox14/+ model (Powell et al., 2012). We developed a stem cell-driven tumor model (abbreviated as Lrig1 tumors) for comparison, using Lrig1CreERT2/+; Apc2lox14/2lox14 mice and focal Cre activation, followed by DSS. Blinded histological assessment revealed that Lrig1 tumors were high-grade dysplastic tumors, but Mist1 tumors were low grade (Figure S7N). To decipher the molecular landscape of the two tumor types, we performed scRNA-seq on tumor tissues along with control colons and identified cells specific to tumors, including abnormal Paneth cells (Figures 7F–7H; Figure S7O). Due to a common WNT-driven mutational process, tumor-specific cells (TSCs) from both tumor types formed an Lgr5-overexpressing cell population without a metaplastic gene signature (Figures 7G–7I). Moreover, both tumor types exhibited elevated β-catenin staining reflecting WNT activation (Figure 7E; Figure S7M).
While the mutational processes between the tumor types were identical, we revealed marked differences in the immune microenvironments. Mist1 tumors, similar to SERs, harbored higher proportions of CD8+ T cells (Figures 7J–7M; Figures S7P and S7Q). These cells expressed markers of active cytotoxicity and killing effectors (Figure 7N; Figure S7Q; Table S5). Lrig1 tumors possessed a distinct population of dysfunctional CD4+ T cells that may have transitioned into anergy or exhaustion (Figures 7J–7M; Figure S7P). These cells expressed immunosuppressive markers, such as Pdcd1 (PD1), Ctla4, Prdm1, and Havcr2 (TIM3), as well as genes of the Foxp3 regulon, implicating dysfunctional T cells exhibiting regulatory characteristics (Figure 7N; Figure S7Q; Table S5). Strikingly, Lrig1 tumors, but not Mist1 tumors, had a large infiltration of myeloid cells that include tumor-associated macrophages and myeloid derived suppressive-like cells, and distinct neutrophils expressing Cd274 (PDL1) (Figures 7J–7N; Figures S7P and S7Q; Table S5). Multiplex imaging showed a significantly higher number of tumor-infiltrating CD8+ T cells but not CD4+ T cells in Mist1 tumors compared to Lrig1 tumors (Figures 7O and 7P; Figures S7R and S7S). In separate mouse models with identical Apc mutations, tumors originating from differentiated cells promote a cytotoxic microenvironment, while tumors driven by stem cells associate with a suppressive immune microenvironment.
To relate epithelial stemness to microenvironmental differences, we applied CytoTRACE to show that Lrig1 TSCs had significantly higher inferred stem potential, and expressed more stem and less differentiated cell genes than Mist1 TSCs (Figure 7Q; Figure S7T). In turn, Lrig1 tumor cells were significantly more successful in forming organoids than Mist1 tumor cells (Figure 7R). Gleaning from previous work defining a gradient of stemness (ISCI > ISCII > ISCIII) in normal intestinal stem cells associated with immune cell interactions (Biton et al., 2018), we found Lrig1 TSCs exhibited a higher ISCI score while Mist1 TSCs exhibited higher ISCII and ISCIII scores (Figure 7S). Consistent with ISCII’s and ISCIII’s increased antigen-presentation capacities, Mist1 TSCs also had increased expression of antigen-presentation machinery (Figures 7T and 7U). Lrig1CreERT2/+;BrafLSL-V600E/+ villiform metaplasias also exhibited increased epithelial expression of antigen-presentation machinery compared to Lrig1CreERT2/+;Apc2lox14/+ tumors, but only in the differentiated and not in the stem compartment (Figure S7U). GSEA demonstrated Mist1 TSCs were significantly enriched for genes associated with immune-mediated processes, with antigen presentation being the most significant (Figure S7V). These results demonstrate how the degree of stemness within neoplastic compartments, as dictated by cellular origins, is linked to the tumor immune microenvironment.
To validate expression of antigen-presentation machinery actually reflects function, we assayed for antigen processing and presentation in Lrig1 and Mist1 tumor-derived tumoroids using the class 2 antigen ovalbumin (OVA). Mist1 tumoroids processed and presented more antigen than Lrig1 tumoroids, reflected by endocytosis and proteolysis of DQ-OVA coupled to I-A/I-E staining indicating surface antigen presentation (Figure 7V; Figure S7W). In support of this observation, Mist1 tumoroids had an increased ability to stimulate T cell proliferation upon presentation of OVA peptide compared to Lrig1 tumoroids (Figure 7W; Figure S7X); suppression of this effect was observed in Lrig1 tumoroids compared to normal distal colonoids. Human tumoroid assays revealed a decrease in stem capacity alongside an increased antigen-presentation gene signature in human SERs compared to ADs (Figure S7Y; Figure 6E). Between tumors, cytotoxic cell infiltration positively correlated with metaplastic signatures in SERs (Figure S7Z). Differentiation media, interferon-γ (IFN-γ) (representative of type 1 immune environment found in SERs), or the two combined were used to induce human AD tumoroids. All three conditions increased expression of antigen-presentation machinery, although the effect of IFN-γ was greater (Figure S7A′). In the human colon epithelium, expression of the antigen-presentation machinery was inversely proportional to stemness (Figure S7B′; Tables S4 and S5). Our data implicate how stemness influence antigen-presentation ability, which may partly underlie the differential stimulation of a cytotoxic immune response.
DISCUSSION
By definition, metaplasia is a process by which differentiated cells transition into cell types non-native to the tissue. Metaplasia often arises in response to damage of the epithelium, which activates a regenerative program to direct the conversion to reparative mucous-secreting lineages resembling those of pyloric glands (Goldenring, 2018). Metaplastic programs have been observed in other organs of the GI tract (Goldenring and Mills, 2021). In SERs, we observed misexpression of genes found in the gastric pylorus, reversion to a fetal gene program, and loss of regional identity with reduced CDX2 expression. It is important to distinguish metaplastic transitions from dedifferentiation of committed cells into stem cells (Buczacki et al., 2013; van Es et al., 2012; Schonhoff et al., 2004; Tetteh et al., 2016), because the latter still retains the identity of the original organ. We propose a new paradigm in which damage to the proximal colon, possibly from microbiota, initiates a metaplastic cascade that may eventually select for survival/proliferative pathways, such as activating BRAF mutations. Reversion to a fetal developmental identity is a feature of WNT-independent tumorigenesis found in recent mouse models (Han et al., 2020), which can be triggered by MAPK activation either via Braf-activating mutations, epithelial damage response, or stress triggered by mismatch repair deficiency (Bommi et al., 2021; Leach et al., 2021). Critically, Braf mutations in mouse models must be accompanied by a “second hit,” such as perturbation of transforming growth factor-β (TGF-β) signaling, for tumor induction (Han et al., 2020; Leach et al., 2021; Tong et al., 2017). This “second hit” may be provided by microenvironmental signals.
Methylation of the CDX2 locus has been frequently observed in serrated tumors, potentially leading to its downregulation, and loss of Cdx2 can provide the “second hit” in a serrated tumorigenesis model (Tong et al., 2017). Increased methylation has been found to be dependent on extrinsic factors such as aging (Tao et al., 2019), consistent with the preponderance of BRAFV600E mutations in MSI-H CRCs in older individuals (Lieu et al., 2019). Shown more recently, microbial dysbiosis can also be an environmental trigger for hypermethylation (DeStefano Shields et al., 2021). Antibiotic suppression of the microbiota reduces colonic tumorigenesis in a Braf mutant model (Leach et al., 2021), whereas in another study, enterotoxigenic Bacteroides fragilis (ETBF) infection is a required trigger for tumorigenesis in the proximal mid-colon in a Braf mutant mouse model (DeStefano Shields et al., 2021). In the latter report, the earliest events of the ETBF response in epithelial cells prior to tumor formation occur at the colonic mucosal surface, where colonic epithelial cells and luminal contents interact. The importance of the microbiota to this type of tumorigenesis is underscored by the co-occurrence of polymicrobial biofilms in ~90% of right-sided CRCs, which are enriched for serrated tumors, versus ~12% biofilm-positive left-sided CRCs (Dejea et al., 2014). Considering the crypt-to-lumen vertical axis of the colonic mucosa, differentiated cells at the luminal surface are exposed to the microbiota, are more susceptible to damage, and utilize repair mechanisms reliant on cellular plasticity. Conversely, stem cells residing in the crypt base are more protected from luminal stressors (Kaiko et al., 2016). We speculate that adenomatous and serrated tumorigenesis originate from fundamentally different mechanisms: the former from DNA replication-induced mutations in continually renewing stem cells and the latter from damage and repair at the colonic surface triggered and maintained by foreign stressors in the luminal environment. Distinct origins of neoplastic cells then select for different mutational pathways required for tumorigenesis.
Several of our findings have clinical value. SSLs can be challenging to identify as the diagnosis is based on the presence of a single “architecturally distorted serrated crypt” as defined by the recently revised WHO classification (Kim and Kang, 2020). Our results suggest biomarkers, such as MUC5AC staining coupled to the absence of CDX2, may confirm the diagnosis of lesions suspicious for SSLs. In addition, the cytotoxic immune response in SSLs precedes hypermutation in human tumors, which is consistent with recent mouse modeling showing the same order of events (DeStefano Shields et al., 2021). Hypermutation is a characteristic of MSI-H CRCs, and the resulting high neoantigen load is thought to be the critical driver of the cytotoxic microenvironment. What then drives the cytotoxic immune response without hypermutation? Our data implicate that tumor cells with a differentiated state, by virtue of their previous exposure to the luminal microenvironment, are more adept at antigen presentation and setting up an active immune environment. Differentiated goblet cells that potentially develop from Mist1+ precursors have shown capacity for luminal antigen passage (Knoop et al., 2015). How tumor cells with a differentiated phenotype acquire and maintain immuno-stimulating properties remains to be determined. In contrast, acquisition of stem cell characteristics by MSI-H CRCs contributes to spatial intratumoral heterogeneity: metaplastic compartments retain their association with cytotoxic immune cells, and stem cell compartments become associated with immunosuppressive cells and signals. In addition to mutations, transition to stemness can also be modulated by recruitment of fibroblasts that express stem cell niche factors (Pelka et al., 2021). Colon cancer stem-like cells have been shown to downregulate their antigen-presentation machinery (Tallerico et al., 2013; Volonté et al., 2014). The degree to which MSI-H CRCs acquire stem-like properties is variable; future studies will be needed to determine whether acquisition of stemness in these cancers impacts the likelihood of an immunotherapeutic response. The top-down spatial organization, differentiated and metaplastic transcriptional program, and cytotoxic immune environment associated with SSLs may open novel strategies for interception of cancer progression, including better informed interval guidelines for surveillance, chemoprevention, or pre- and pro-biotic therapies.
LIMITATIONS OF THE STUDY
Since our study profiled largely small polyps, the material for multi-omic analyses was limiting, as seen from our inability to obtain high-quality DNA from a number of samples. Enriching for specific cell populations was not performed due to potential material loss, which contributed to the heavy epithelial representation in our scRNA-seq data, and non-comprehensive characterization of some non-epithelial cell populations. Longitudinal analysis of polyps was not possible due to complete colonoscopic removal of polyps identified. Finally, while we performed functional validation experiments in vitro and in vivo, the exact molecular pathway(s) by which tumor cells maintain the characteristics of their origins and when the immune system engages tumor neoantigens remain undefined.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by Lead Contact: Ken S. Lau, PhD at ken.s.lau@vanderbilt.edu.
Materials availability
This study did not generate any unique reagents.
Data and code availability
The raw single-cell RNA sequencing (scRNA-seq), final QC-filtered data for analysis, as well as all raw imaging data generated from this study are available on the HTAN data portal: https://data.humantumoratlas.org/. This paper also analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
All original code used to process scRNA-seq data has been deposited at https://github.com/Ken-Lau-Lab/STAR_Protocol and is described in detail in (Chen et al., 2021). A code repository containing the analysis of post-processed sequencing data, as performed in this study, can be found at https://github.com/Ken-Lau-Lab/STAR_Methods. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| Reagent or resource | Source | Identifier |
|---|---|---|
| Biological samples | ||
|
| ||
| COLON MAP (Polyp) | See Experimental Model and Subject Details; Data and Code Availability | Synapse: syn23564801 Synapse: syn23630431 Synapse: syn23520239 HTAN Data Portal: HTA11; https://humantumoratlas.org/HTA11 |
| CHTN TMA (CRC) | See Experimental Model and Subject Details; Data and Code Availability | syn21050481: https://doi.org/10.7303/syn21050481 |
| TCPS (Polyp) | See Experimental Model and Subject Details; Data and Code Availability | syn21050481: https://doi.org/10.7303/syn21050481 |
|
| ||
| Critical commercial reagents | ||
|
| ||
| Muc2, F-2 clone, A488 dye, antibody | Santa Cruz | Catalog: sc-515032 AF488; RRID: AB_2815005 |
| Collagen Peptide, R-CHP clone, Cy3 dye, antibody | 3Helix | Catalog: RED300 |
| SNA, Lectin clone, Cy5 dye, antibody | Vector | Catalog: CL-1305-1 |
| CD11B, C67F154 clone, A488 dye, antibody |
Thermo Fisher | Catalog: 53-0196-82; RRID: AB_2637196 |
| CD45, 2D-1 clone, A546 dye, antibody | Santa Cruz | Catalog: sc-1187 AF546; RRID: AB_627073 |
| CD20, D-10 clone, A647 dye, antibody | Santa Cruz | Catalog: sc-393894 AF647 |
| PCNA, PC-10 clone, A488 dye, antibody | Cell Signaling | Catalog: 8580S; RRID: AB_11178664 |
| B-catenin, 12F751 clone, 550 dye, antibody | Vanderbilt Antibody and Protein Resource | Catalog: In-House |
| p-STAT3, D3A7 clone, A647 dye, antibody | Cell Signaling | Catalog: 4324S; RRID: AB_10694637 |
| pEGFR, EP774Y clone, A488 dye, antibody | Abcam | Catalog: ab205827 |
| CgA, C-12 clone, A546 dye, antibody | Santa Cruz | Catalog: sc-393941; RRID: AB_2801371 |
| CD4, EPR6855 clone, A647 dye, antibody | Abcam | Catalog: ab196147 |
| Cox2, D5H5 clone, A488 dye, antibody | Cell Signaling | Catalog: 13596S; RRID: AB_2798270 |
| CD3d, EP4426 clone, A555 dye, antibody | Abcam | Catalog: ab208514; RRID: AB_2728789 |
| HLA-A, EP1395Y clone, A647 dye, antibody | Abcam | Catalog: ab199837; RRID: AB_2728798 |
| PanCK, AE1/AE3 clone, A488 dye, antibody | Thermo Fisher | Catalog: 53-9003-82; RRID: AB_1834350 |
| OLFM4, D1E4M clone, A555 dye, antibody | Cell Signaling | Catalog: 14369S; RRID: AB_2798465 |
| CD8, C8/114B clone, A647 dye, antibody | Biolegend | Catalog: 372906; RRID: AB_2650712 |
| Alpha-actinin, EPR2533(2) clone, A488 dye, antibody | Abcam | Catalog: ab198608 |
| CD68, KP1 clone, A546 dye, antibody | Santa Cruz | Catalog: sc-20060 AF546; RRID: AB_2891106 |
| NaKATPase, EP1845Y clone, A647 dye, antibody | Abcam | Catalog: ab198367 |
| Vimentin, E-5 clone, A488 dye, antibody | Santa Cruz | Catalog: sc-373717 AF488; RRID: AB_10917747 |
| Sox9, EPR14335 clone, A555 dye, antibody | Abcam | Catalog: ab202516 |
| FOXP3, 206D clone, A647 dye, antibody | Biolegend | Catalog: 320114; RRID: AB_439754 |
| Lysozyme, E-5 clone, A488 dye, antibody | Santa Cruz | Catalog: sc-518012 AF488; RRID: AB_2889359 |
| SMA, 1A4 clone, Cy3 dye, antibody | Millipore Sigma | Catalog: C6198-100UL; RRID: AB_476856 |
| ERBB2, EPR19547 clone, A647 dye, antibody | Abcam | Catalog: ab225510; RRID: AB_2889201 |
| P-p44/42 MAPK, Rabbit Monoclonal antibody | Cell Signaling | Catalog: 4370; RRID: AB_2315112 |
| MUC5AC, Rabbit Monoclonal antibody | Cell Signaling | Catalog: 61193; RRID: AB_2799603 |
| CDX2, Rabbit Monoclonal antibody | Cell Signaling | Catalog: 12306; RRID: AB_2797879 |
| Midkine, Rabbit Monoclonal antibody | Abcam | Catalog: ab52637; RRID: AB_880698 |
| YAP, Rabbit Monoclonal antibody | Cell Signaling | Catalog: 14074; RRID: AB_2650491 |
| MLH1, Rabbit Monoclonal antibody | Abcam | Catalog: Ab92312; RRID: AB_2049968 |
| EPCAM antibody | Santa Cruz | Catalog: Sc-53532; RRID: AB_2277892 |
| GFP antibody | Novus | Catalog: NB600-308SS; RRID: AB_10005904 |
| DCAMKL1 antibody | Abcam | Catalog: ab109029; RRID: AB_10864128 |
| CHGA (C20) antibody | Santa Cruz | Catalog: sc1488; RRID: AB_2276319 |
| MUCIN2 (H-330) antibody | Santa Cruz | Catalog: sc15334; RRID: AB_2146667 |
| CD3 (Sp7) antibody | Thermo Fisher | Catalog: RM-9107-50 |
| CD8 (4SM15) antibody | Invitrogen | Catalog: 14-0808-80; RRID: AB_2572860 |
| CD4 (4SM95) antibody | Invitrogen | Catalog: 14-9766-80; RRID: AB_2573007 |
| CD11b-AF647 antibody | Abcam | Catalog: ab204471; RRID: AB_204471 |
| CD11c antibody | Biolegend | Catalog: 117301; RRID: AB_313770 |
| CD45/B220-AF647 antibody | Biolegend | Catalog: 103228 |
| Hoechst 33342 | Invitrogen | Catalog: H3570 |
| Ia/le (M5/114.15.2) | Biolegend | Catalog: 107601 |
| DQ-Ovalbumin | Thermo Fisher | Catalog: D12053 |
| Ovalbumin peptide | Anaspec | Catalog: OVA323-339 |
| CD3-PerCP/Cy5.5, clone 145-2C11 | Biolegend | Catalog: 100328; RRID: AB_893318 |
| CD4-Apc-Cy7, clone GK1.5 | Biolegend | Catalog: 100414; RRID: AB_312699 |
| MHCII-PE-Cy7, clone M5/144.15.2 | Biolegend | Catalog: 107629; RRID: AB_2290801 |
| CD69-FITC, clone H1.2F3 | eBioscience | Catalog: 11-0691-82; RRID: AB_465119 |
| CD45-BV785, clone 30-F11 | Biolegend | Catalog: 103149; RRID: AB_2564590 |
| Ia/Ie-AF647 | Biolegend | Catalog: 10760 |
| ROCK inhibitor | STEMCELL Technologies | Catalog: Y-27632 |
| Matrigel | Corning | Catalog: 356231 |
| RNase A | Thermo Fisher | Catalog: EN0531 |
| Lysing Matrix E | MP Bio | Catalog: 116914100 |
| Mouse Intesticult | STEMCELL Technologies | Catalog: 06005 |
| Gastrin I | Sigma-Aldrich | Catalog: 39024-57-2 |
| TrypLE Express | Thermo Fisher | Catalog: 12604013 |
| N-acetyl-L-cysteine | Sigma-Aldrich | Catalog: A9165 |
| H2O2 | Sigma-Aldrich | Catalog: 216763 |
| A83-01 | Tocris | Catalog: 2939 |
| IGF-1 | Biolegend | Catalog: 590904 |
| FGF-2 | Thermo Fisher | Catalog: PHG0024 |
| Primocin | InvivoGen | Catalog: am-pm-05 |
| Human IntestiCult (OGM) | STEMCELL Technologies | Catalog: 06010 |
| Human IntestiCult (ODM) | STEMCELL Technologies | Catalog: 100-0214 |
| Human IFN gamma | Biolegend | Catalog: 570206 |
| CD8-alpha, 53-6.7 clone, antibody | Biolegend | Catalog: 100711; RRID: AB_312750 |
| MHC class I antibody, clone ER-HR 52 | Abcam | Catalog: ab15681; RRID: AB_302030 |
| MHC class II (I-A/I-E) antibody, clone (M5/114.15.2) | Thermofisher | Catalog: 14-5321-82; RRID: AB_467561 |
| RT Probe: HLA_A_F | Sigma | AGATACACCTGCCATGTGCAGC |
| RT Probe: HLA_A_R | Sigma | GATCACAGCTCCAAGGAGAACC |
| RT Probe: HLA_B_F | Sigma | CTGCTGTGATGTGTAGGAGGAAG |
| RT Probe: HLA_B_R | Sigma | GCTGTGAGAGACACATCAGAGC |
| RT Probe: HLA_C_F | Sigma | GGAGACACAGAAGTACAAGCGC |
| RT Probe: HLA_C_R | Sigma | ACATCCTCTGGAGGGTGTGAGA |
| RT Probe: CD74_F | Sigma | AAGCCTGTGAGCAAGATGCGCA |
| RT Probe: CD74_R | Sigma | AGCAGGTGCATCACATGGTCCT |
| RT Probe: C2TA_F | Sigma | CTACTTCAGGCAGCAGAGGAGA |
| RT Probe: C2TA_R | Sigma | GCTGTGTCTTCCGAGGAACTTC |
| RT Probe: HLA-DRB1_F | Sigma | GAGCAAGATGCTGAGTGGAGTC |
| RT Probe: HLA-DRB1_R | Sigma | CTGTTGGCTGAAGTCCAGAGTG |
| RT Probe: GAPDH_F | Sigma | GTCTCCTCTGACTTCAACAGCG |
| RT Probe: GAPDH_R | Sigma | ACCACCCTGTTGCTGTAGCCAA |
|
| ||
| Deposited data | ||
|
| ||
| TCGA (CRC) | (Cancer Genome Atlas Network, 2012) | TCGA GDAC Firehose: COADREAD |
| SMC (CRC) | (Lee et al., 2020) |
GEO: GSE132465 |
| Broad (CRC) | (Pelka et al., 2021) | HTAN Data Portal: HTA1 |
|
| ||
| Software and algorithms | ||
|
| ||
| pCreode | (Herring et al., 2018) | https://github.com/Ken-Lau-Lab/pCreode |
| Scanpy | (Wolf et al., 2018) | https://github.com/theislab/scanpy |
| Pegasus | Klarman Cell Observatory | https://github.com/klarman-cell-observatory/pegasus |
| pySCENIC | (Aibar et al., 2017) | https://github.com/aertslab/pySCENIC |
| CMScaller | (Eide et al., 2017) | https://github.com/peterawe/CMScaller |
| CytoTRACE | (Gulati et al., 2020) | https://cytotrace.stanford.edu |
| CMSclassifier | (Guinney et al., 2015) | https://github.com/Sage-Bionetworks/CMSclassifier |
| Seaborn | Seaborn | https://github.com/mwaskom/seaborn |
| cBioPortal | (Cerami et al., 2012; Gao et al., 2013) | https://www.cbioportal.org/ |
| Matplotlib | Matplotlib | https://github.com/matplotlib/matplotlib |
| GATK4 | (Poplin et al., 2017) | https://gatk.broadinstitute.org/hc/en-us |
| DENDRO | (Zhou et al., 2020) | https://github.com/zhouzilu/DENDRO |
| dropkick | (Heiser et al., 2021) | https://github.com/Ken-Lau-Lab/dropkick |
| DropEst | (Petukhov et al., 2018) | https://github.com/kharchenkolab/dropEst |
| STAR | (Dobin et al., 2013) | https://github.com/alexdobin/STAR |
| Cytoscape | (Shannon et al., 2003) | https://cytoscape.org/ |
| g:Profiler | (Raudvere et al., 2019) | https://biit.cs.ut.ee/gprofiler/ |
| Scipy | (Virtanen et al., 2020) | https://scipy.org/ |
| Sinto | Sinto | https://github.com/timoast/sinto |
| Dendextend | (Galili, 2015) | https://github.com/talgalili/dendextend |
| Numpy | (Harris et al., 2020) | https://numpy.org/ |
| Pandas | Pandas | https://pandas.pydata.org/ |
| BWA | (Li and Durbin, 2009) | https://sourceforge.net/projects/maq/ |
| ANNOVAR | (Wang et al., 2010; Yang and Wang, 2015) | https://github.com/WGLab/doc-ANNOVAR |
| Picard | Broad Institute | https://broadinstitute.github.io/picard/ |
| Sambamba | (Tarasov et al., 2015) | https://github.com/biod/sambamba |
| lme4 | (Bates et al., 2015) | https://github.com/lme4/lme4 |
| lmerTest | (Kuznetsova et al., 2017) | https://github.com/runehaubo/lmerTestR |
| emmeans | emmeans | https://github.com/rvlenth/emmeans |
| Cytobank | (Kotecha et al., 2010) | https://www.cytobank.org/ |
| MANDO | (McKinley et al., 2019) | https://github.com/Coffey-Lab/CellSegmentation |
| UMAP | (McInnes et al., 2018) | https://github.com/lmcinnes/umap |
| Dask | Dask | https://dask.org/ |
| Harmony | (Korsunsky et al., 2019) | https://github.com/immunogenomics/harmony |
| Scikit-posthoc | (Terpilowski, 2019) | https://scikit-posthocs.readthedocs.io/en/latest/ |
| GSEA Webapp | (Mootha et al., 2003; Subramanian et al., 2005) | https://www.gsea-msigdb.org/gsea/index.jsp |
EXPERIMENTAL MODEL AND SUBJECTS DETAILS
Colorectal Molecular Atlas Project (COLON MAP)
COLON MAP participants were recruited from adults undergoing routine screening or surveillance colonoscopy or surgery for resection of a polyp at Vanderbilt University Medical Center in Nashville, TN, USA that began in March 2019 and is still on-going, participant characteristics are shown in Table S2. The participants included in this study are the first 56 participants from COLON MAP with polyps collected for analysis by scRNA-seq. All participants provided written informed consent approved by the Vanderbilt University Medical Center Institutional Review Board.
Eligibility criteria for COLON MAP include ability to provide informed consent, free-living (not a resident of an institution), ability to speak and understand English, aged 40 to 75 years, permanent residence or telephone, and no personal confirmed or suspected histories of hereditary polyposis syndromes, familial or genetic colorectal cancer syndromes, inflammatory bowel disease, primary sclerosing cholangitis, colon resection or colectomy, cancer, neoadjuvant therapy, or cystic fibrosis. Eligible individuals were first identified from the schedule within the electronic health record (EHR) and assigned a random number. Potential participants undergoing colonoscopy were further selected using a stratified weighted random sampling design to increase the inclusion of non-White or Latinx participants in the study. Within strata of colonoscopy appointment day and time, random sampling was weighted by EHR-derived racial/ethnic category (White non-Latinx versus all other races and ethnicities) such that non-White or Latinx patients were first selected at random within colonoscopy day and time. White, non-Latinx patients were then selected at random within remaining time slots.
Following selection, study staff conducted a manual review of the EHR to confirm study eligibility. The majority of eligible individuals were mailed a letter to introduce the study and a few days later were attempted to be reached by telephone to discuss their willingness to participate in the study. Individuals who were willing to participate completed an additional screening form to confirm eligibility, and eligible and willing individuals completed an interviewer-administered, computer-assisted telephone interview to solicit information on personal health history, family history of cancer and polyps, lifestyle factors, and other risk factors for colorectal polyps and cancer. When the schedule of the study staff would allow, individuals who were not reached by telephone were approached in the colonoscopy waiting room or at the surgical appointment to determine eligibility and willingness as well as some individuals who did not receive a mailing.
For histopathological diagnosis, standard clinical histology was performed. Information on the colonoscopy or surgery and diagnosis was initially abstracted from the EHR colonoscopy, surgery, and pathology reports by study staff including in vivo size and polyp location. Two study pathologists additionally reviewed each case to standardize diagnoses and identify HP subtypes which are not part of routine clinical practice. For polyps which were partial due to the sampling for this study, the portion which had been reserved for clinical diagnosis was reviewed. SSLs were defined using the World Health Organization criteria of at least one distorted, dilated, or horizontally branched crypt within the polyp (Rex et al., 2012). Subtypes of ADs were identified using standard diagnostic criteria based on the villous component (tubular (< 25% villous component), tubulovillous (25%–74% villous component), and villous (≥75%)). HPs were classified as microvesicular HP or goblet cell HP (Leggett and Whitehall, 2010). In this analysis, participants were classified based upon the diagnosis of their index polyps but may have had synchronous polyps with the same or different histopathologies as shown in Table S1.
Cooperative Human Tumor Network
Tissue was collected for COLON MAP from 33 colorectal cancer (CRC) patients via the CHTN Western Division. These participants were aged between 21 and 82 years of age from both sexes (51.5% male, 48.5% female) and were white (75.8%), Black (21.2%), or Asian (3.0%). De-identified clinical metadata from each patient was extracted from clinical pathology reports in accordance with policies from CHTN. Tumors were classified by grade and staging, ranging from G1 to G3 and I to IV, respectively. The majority (75.6%) of the tumors were classified as G2, or moderately differentiated, and staged primarily as IIA (30.3%) and IIIB (33.3%). Additionally, 51.5% were microsatellite stable (MSS) and 49.5% were microsatellite-high (MSI-H). Participant characteristics of the 33 CRC patients obtained from the CHTN Western Division are shown in Table S7.
A colorectal carcinoma progression tissue microarray (TMA) was also provided by the CHTN Mid-Atlantic Division which included cores from 54 individuals. The mean (standard deviation) age of the individuals included on the TMA was 56.9 (14.7), 56.9% were men, and 43.1% were women. Race and ethnicity were not provided. Information on the TMA is available at https://chtn.sites.virginia.edu/chtn-crc2
Tennessee Colorectal Polyp Study
The TCPS was a large colonoscopy-based case-control study among individuals undergoing colonoscopy in Nashville, Tennessee, USA between February 2003 and October 2010. Institutional approval for human subjects research was provided by the VUMC and VA Institutional Review Boards and the VA Research and Development Committee. TCPS participants were aged between 40 to 75 years of age and had no personal history of colon resection, cancer, polyposis syndrome, inflammatory bowel disease, hereditary colorectal cancer syndromes, or previous adenoma. In TCPS, the diagnostic criteria for polyps were identical to the criteria used for COLON MAP. Additionally, all polyps were reviewed by one of the COLON MAP pathologists. Features of these archived participants and polyps included are shown in Table S2.
Detailed methods have been previously published (Davenport et al., 2018). In this analysis, a subset of TCPS formalin-fixed paraffin-embedded polyps which were previously analyzed by bulk RNA-seq were included to validate findings from the COLON MAP scRNA-seq analysis. In addition, a subset of fresh frozen polyps which were selected for targeted gene sequencing were also included.
Mouse models
All animal experiments were performed under protocols approved by the Vanderbilt University Animal Care and Use Committee and in accordance with NIH guidelines. Mice were 8 weeks old at the start of experiments and were humanely euthanized at the end of experiments according to approved guidelines. Animal weights were recorded at initiation of experiment and at the time of euthanasia. All animals used in this study were predominantly of the C57BL/6J background and both sexes were used. Littermate controls were used for experiments when possible. All animals were housed 2 to 5 per cage in a controlled environment in standard bedding with a standard 12-hour daylight cycle, cessation of light at 6 PM, and free access to standard chow diet and water. Experiments were conducted during the light cycle, excluding continuous dietary interventions.
Human organoids
Polyps were dissociated and washed as described in the COLON MAP scRNA-seq, Encapsulation and Library Generation section. After dissociation, cells were washed 3 times with PBS containing 10 μM ROCK inhibitor (STEMCELL Technologies) and pelleted by quick-pulse centrifugation for 7 s. Human organoid models were generated from COLON MAP individuals of both sexes (70% female, 30% male). Polyp-derived cells were grown with Human IntestiCult organoid growth media (STEMCELL Technologies) supplemented with 10 μM Y-27632, 10 nM Gastrin I (Sigma-Aldrich), 1 mM N-acetyl-L-cysteine (Sigma-Aldrich), 500 nM A83–01 (Tocris), 50 ng/mL FGF-2 (Thermo Fisher), 100 ng/mL IGF-1 (BioLegend), 100 μg/mL Primocin (InvivoGen), and Matrigel (Corning) in a 3:1 ratio of Matrigel to media. Media was replaced every 2–3 days, and passaging was performed by dissociating the organoids in TrypLE Express (Thermo Fisher) with 10 μM Y-27632 for 15 minutes at 37°C while shaking and triturating.
Mouse organoids
Mouse organoids were generated from the same pool of mice used in mouse model experiments, with both sexes being used. Mouse tumors were dissociated using TrypLE Express, and cell pellets were resuspended in Matrigel and seeded in 25 μL droplets in a 24-well or 12-well plate. Once solidified, samples were incubated in 1 mL Mouse IntestiCult culture medium (STEMCELL Technologies) with 100 μg/mL Primocin for 5 days. Fresh media was replaced on day 3. Passaging was performed similarly to human organoids.
METHOD DETAILS
COLON MAP biological specimen collection and processing, blood and oral rinse
Prior to the procedure, an oral mouthwash rinse sample was collected from participants. Blood was also collected through the IV line, prior to colonoscopy, in EDTA and serum tubes. The EDTA and serum samples were spun at 1,500 g for 10 minutes, using a refrigerated centrifuge (at 4°C). The plasma was pipetted into four sterile 2ml cryovials, white blood cells were aliquoted into two 2ml vials, and red blood cells were stored in two 2ml vials after being washed two times with cold saline solution. Serum was pipetted into four 2ml vials and the blood clot into two 2ml vials. The mouth rinse samples were centrifuged, and the pellets were suspended using TE buffer, then aliquoted into a 2ml vial. All samples were placed into —80°C freezers for storage until use.
COLON MAP biological specimen collection and processing, colorectal tissue
During the colonoscopy, the gastroenterologist used biopsy forceps to collect normal appearing mucosa samples from the ascending and descending colon for all participants. One of the biopsies from each colon segment was placed into RPMI. Any polyps were removed during the colonoscopy per standard clinical practice. In this analysis, the first polyp which was removed from a participant that was larger than 0.5 cm was selected for scRNA-seq analysis (index polyp). Polyps which were removed intact were bisected along the vertical axis using a sterile razor blade and half was placed in RPMI. For polyps which were removed piecemeal, the second largest piece was placed in RPMI. The other portions of the polyps were placed into formalin for diagnosis and fixed and processed using standard clinical practice in the Vanderbilt Pathology Laboratory. All polyps which were placed in RPMI were immediately transported to the research lab for use in scRNA-seq analysis.
COLON MAP bulk DNA extraction
For germline, DNA was isolated from thawed buffy coat or mouth rinse samples using a QIAmp DNA kit (QIAGEN). For tumors, DNA for whole exome sequencing (WES) was purified with the truXTRAC FFPE microTUBE DNA Kit-Column Purification kit (Covaris). In brief, tumor tissues were scraped from 1–5 of 10 mm FFPE sections, deparaffinized using xylene, and lysed in an optimized lysis buffer that contains proteinase K. Following the proteinase K digestion to release DNA from the tissue, a higher temperature was used incubation to reverse formalin crosslinking alongside RNase treatment using RNase A (Thermo Fisher). The DNA and RNA samples were stored at −80°C before being used for assays.
COLON MAP whole exome sequencing and alignment
Standard WES was performed on S4 flow cells on NovaSeq6000 (PE150) to the targeted coverage. WES reads were aligned to the human reference genome hg19 using BWA (Li and Durbin, 2009), sorted and indexed by Sambamba (Tarasov et al., 2015). Duplicated reads were removed by the mark duplicates function with Picard. Somatic mutations were called using sequenced DNA extracted from specimens detailed in the COLON MAP Biological Specimen Collection and Processing, Blood and Oral Rinse section. These somatic mutations were then called using GATK4 Mutect2 in “normal-tumor” paired mode (Van der Auwera et al., 2013).
COLON MAP scRNA-seq, single-cell encapsulation and library generation
Colonic biopsy samples were first placed into RPMI solution, minced to approximately 4mm2, and washed with 1x DPBS. These samples were then incubated in chelation buffer (4mM EDTA, 0.5 mM DTT) at 4 °C for 1 h 15 min. Then, the resulting tissue suspension was dissociated with cold protease and DNase I for 25 minutes (Banerjee et al., 2020; Liu et al., 2018). This suspension was titurated throughout the process, every 10 minutes, then washed three times with 1x DPBS before encapsulation. Cells were encapsulated using a modified inDrop platform (Klein et al., 2015), and sequencing libraries were prepared using the TruDrop protocol (Southard-Smith et al., 2020). Libraries were sequenced in a S4 flow cell using a PE150 kit on an Illumina NovaSeq 6000 to a target of 150 million reads.
COLON MAP scRNA-seq, alignment and droplet matrix generation
We demultiplexed, aligned, and corrected the detected read counts of these libraries with the DropEst pipeline (Petukhov et al., 2018), using the STAR aligner with the Ensembl reference genome (Dobin et al., 2013), GRCh38 release 25. This was paired with the corresponding GTF annotations. The protocol for running this pipeline is described by (Chen et al., 2021).
COLON MAP scRNA-seq, droplet matrix quality control
We identified high-quality, cell-containing droplets and their respective barcodes through the joint application of cumulative sum inflection point thresholding, our dropkick QC algorithm (Heiser et al., 2021), and prior-knowledge gene expression profiling. This droplet matrix was processed as an AnnData object using our preprocessing pipeline which utilizes the Scanpy toolkit (Wolf et al., 2018). First, we ran dropkick with 5-fold cross validation on the unprocessed droplet matrix, which assigned each barcode a probability of being a high-quality cell. Second, the droplet matrix was preprocessed for low dimensional analysis through finding the inflection point of the cumulative sum curve, and droplets with low information content were removed. Third, the remaining cells were normalized to the median number of counts per single-cell library per dataset, inverse hyperbolic sine transformed, and then scaled as a Z-score. Fourth, normalized matrices were projected into 2 dimensions by using its 50 principal component decomposition to initialize a UMAP (McInnes et al., 2018). Fifth, gene expression and dropkick probability scores were overlaid and checked for consistency. The genes overlaid were based on prior knowledge of the colonic epithelial markers, deferring to dropkick scores when no markers were found. Sixth, the selection of the final set of high-quality cell-containing droplets were determined by setting a binarization threshold on the dropkick probability scores, given concordance to marker gene expression and other general quality metrics such as total counts, mitochondrial count percentage, and transcriptional diversity. The full protocol for running this QC pipeline is described by (Chen et al., 2021).
CHTN bulk DNA extraction of fresh frozen samples
Fresh frozen samples were stored in Tissue-Tek O.C.T. (Fisher Scientific) compound until ready for processing. These samples were washed in cold 1x PBS followed by centrifugation before using the QIAGEN DNeasy Blood and Tissue kits (QIAGEN) for DNA extraction. All following processing was performed according to the manufacturer’s guidelines. The DNA extract collected from these samples were sequenced and aligned as detailed in the COLON MAP Whole Exome Sequencing (WES) and Alignment section.
COLON MAP and CHTN TMA MxIHC
MxIHC was performed by iterative antibody staining and chromogen removal based on the protocol in (Tsujikawa et al., 2017). Chromogen was removed between sequential rounds through sequential alcohol baths, and antibody was stripped by high temperature (95°C for 15 minutes). Single antibody stains using 3,3′-Diaminobenzidine were performed using standard protocols. Incubation and detection conditions are listed in the methods github repository: https://github.com/Ken-Lau-Lab/STAR_Methods/blob/main/Methods_Tables.xlsx (Methods Table 1)
COLON MAP and CHTN TMA MxIF
Cyclical antibody staining, detection, and dye inactivation was performed as described previously by (Gerdes et al., 2013). Briefly, fluorescence imaging was performed on a GE IN Cell Analyzer 2500 using the Cell DIVE platform. Images were acquired at x200 magnification with exposure times determined for each antibody. Antibody reagents are listed in the Key Resources Table. Staining sequence, conditions, and exposure times are listed in tables found in the methods github repository: https://github.com/Ken-Lau-Lab/STAR_Methods/blob/main/Methods_Tables.xlsx (Methods Table 2). For each round of staining, DAPI images were aligned using rigid transformations to the first imaging round. The registered images were corrected for uneven illumination and autofluorescence was removed for each channel.
TCPS bulk DNA and RNA extraction
DNA was extracted from FFPE tissue sections using QIAamp DNA FFPE Tissue Kit (QIAGEN), following the manufacturer’s instructions. Briefly, tumor tissues were scraped from 1–5 of 10 μm FFPE sections, deparaffinized using xylene, and lysed under denaturing conditions with proteinase K. The sample lysate was incubated at 90°C to reverses formalin crosslinking and then applied to a QIAamp MinElute spin column, where DNA was captured on a silica membrane. The genomic DNA was then washed and eluted from the membrane.
DNA and total RNA were extracted from fresh frozen polyps and purified using QIAGEN’s AllPrep DNA/RNA/miRNA Universal Kit (QIAGEN), following the manufacturer’s instructions. Briefly, the frozen tissue samples were first disrupted and homogenized using Lysing Matrix E (MP Bio) by shaking the tubes on a bead-beater at 5.5 m/sec for 30 s. The lysate was then passed through an AllPrep DNA Mini spin column. This column allows selective and efficient binding of genomic DNA. Following on-column Proteinase K digestion, the column was then washed and pure, ready-to-use DNA was eluted. Flow-through from the DNA Mini spin column was then digested by Proteinase K in the presence of ethanol and applied to the RNeasy Mini spin column, where the total RNA binds to the membrane. Following DNase I digestion, contaminants were efficiently washed away and high-quality RNA was eluted in RNase-free water. The quantity and quality of the DNA/RNA samples were checked by Nanodrop (E260/E280 and E260/E230 ratio) and by separation on an Agilent BioAnalyzer.
TCPS targeted DNA sequencing and alignment
The list of candidate genes included in the targeted sequencing was developed from a literature review of candidate mutations which showed 1) evidence that mutation is common in adenoma (> 5% of adenomas), 2) evidence that the mutation is associated with or predictive of adenoma recurrence in previous studies, 3) evidence that mutation is associated with clinically more significant adenoma (i.e., advanced adenoma or multiplicity), 4) evidence that mutation is associated with colorectal field carcinogenesis, and 5) evidence that mutation is associated with colorectal cancer aggressiveness and survival. In addition, additional candidate mutations were identified from potential mutations observed in Lrig1-Cre:Apc adenomas. All primer development and next-generation sequencing were conducted by Covance. Sequencing depth was 500X. Targeted sequencing reads were aligned to the human reference genome hg19 using BWA (Li and Durbin, 2009),and then were sorted and indexed by Sambamba (Tarasov et al., 2015). Alignments were further refined, and variants were called using GATK Best Practices tools (Van der Auwera et al., 2013), including mark duplicates with Picard, base quality-score recalibration, and variant calling with HaplotypeCaller and GenotypeGVCFs (Poplin et al., 2017). SNPs were filtered using GATK VariantFiltration function with the parameters “QD < 2.0 ‖ Qual < 30.0 ‖ FS > 60.0 ‖ SOR>3.0 ‖ MQ < 40.0 ‖ MQRankSum < —12.5 ‖ ReadPosRankSum < —8.0,” while indels were filtered with the parameters “QD < 2.0 ‖ Qual < 30.0 ‖ FS > 200.0 ‖ ReadPosRankSum < —20.0.” The variants with a minor allele frequency > 0.1% in ExAC, gnomAD, TOPMed or 1000 Genomes were also removed. The functional effects of variants were annotated by ANNOVAR (Wang et al., 2010; Yang and Wang, 2015).
TCPS bulk RNA sequencing and alignment
Bulk RNA-sequencing was performed by Aros Applied Biotechnology A/S. This process involves the initial QC on an Agilent Bioanalyzer, with a minimum quality threshold of the DV200 at 30%. Total RNA-seq libraries which pass this QC threshold are prepared alongside a high-quality human reference RNA control. 100ng of RNA per sample is input to an Illumina TruSeq RNA Access Library Prep Kit, with protocol version 0.2. The yielded libraries undergo another round of QC through qPCR and quantified with a Qubit 2.0 Fluorometer, using its corresponding DNA BR Assay kit (Qubit), and size profiled on an Agilent Bioanalyzer. Pools of 4 libraries in equimolar amounts are created and undergo a final round of QC. These pools are loaded onto paired-end flow cells of a HiSeq2500 equipped with a cBot for sequencing at: 101 read cycles, 7 index cycles, and 101. The samples will be sequenced on a HiSeq2500 using 101 cycles for read 1, 7 index reads, and another 101 cycles for read 2. Following sequencing data generation, the reads are demultiplexed through Illumina’s Genome Studio CASAVA software, which detected an average of 120 million reads per 4 sample pool.
COLON MAP pre-cancer organoid replating efficiency assay
COLON MAP samples that successfully formed organoids were dissociated and counted using a Bio-RAD TC20 automated cell counter and plated at 1,000 cells/well in 5 μL Matrigel domes in a 96-well plate. Organoids were imaged and counted using an inverted microscope (Fisherbrand) after 8 days in culture. Patient IDs were matched to histopathology results after compilation and tabulation of results. GraphPad Prism 9 was used for plotting and statistical analysis using unpaired t-tests.
COLON MAP pre-cancer organoid differentiation assay
COLON MAP organoids were cultured in appropriately supplemented Human IntestiCult organoid growth media (OGM) for 3 days. They either remained in OGM for control or switched to supplemented Human IntestiCult organoid differentiation media (ODM) (STEMCELL Technologies) for 3 more days. For IFN gamma treatment, human recombinant IFN-gamma (Biolegend) was added to each media condition at 100 ng/mL for 24 hours prior to harvesting.
Murine lineage tracing
For homeostatic lineage tracing studies, Lrig1CreERT2/+;Rosa26LSL-EYFP/+ mice were injected intraperitoneally (i.p.) for 3 consecutive days with 2.5 mg tamoxifen (Sigma-Aldrich; T5648) in corn oil, while Mist1CreERT2/+;Rosa26LSL-EYF/+P were injected i.p. for 3 consecutive days with 5 mg tamoxifen. Mice were euthanized 24 h, 10 days, and 28 days later. For damage-induced lineage tracing, Mist1CreERT2/+;Rosa26LSL-EYFP/+ and Mist1CreERT2/+;Rosa26mT/mG/+ mice were injected i.p. for 3 consecutive days with 5 mg tamoxifen, and were then administered 2.5% DSS (TdB Consultancy; Batches DB001–37, DB001–42) in drinking water for the following 6 days. After cessation of DSS, mice were euthanized 24 h and 28 days later.
Murine induction of recombination using different promoters
To recombine genes, Lrig1CreERT2/+;BrafLSL-V600E/+ and Lrig1CreERT2/+;Apc2lox14/+ mice were induced and had their tissues harvested using established protocols (Kondo et al., 2020; Powell et al., 2012). Tissues were harvested from these mice approximately 12 weeks after induction of recombination. Lrig1CreERT2/+;KrasLSL-G12D/+ mice were anesthetized and induced with 100 μL of 10 mg/mL 4-hydroxytamoxifen (Sigma-Aldrich) in ethanol delivered with an enema using a gavage feeding needle, and tissues were harvested around 8 weeks later.
For generating tumors, Mist1CreERT2/+;Apc2lox14/2lox14 were injected intraperitoneally for 3 consecutive days with 5 mg tamoxifen in corn oil. They were administered 2.5% DSS in drinking water for the following 6 days, followed by a 9-day rest period, and a second round of DSS. Lrig1CreERT2/+;Apc2lox14/2lox14 were injected with 0.01mM 4-hydroxytamoxifen through colonoscopy-guided orthotopic injections into the mucosal lining of the distal colon (Roper et al., 2017), and were administered 2.5% DSS in drinking water for the following 6 days. Control mice received PBS injections followed by DSS. Mice were euthanized approximately 28 days following Cre induction.
Murine immunofluorescence and histological imaging
Upon euthanasia of an animal, colonic tissue was removed, washed with 1X DPBS, spread longitudinally onto Whatman filter paper and fixed in 4% PFA (Thermo Scientific) overnight. Fixed tissues were washed with 1X DPBS, swiss-rolled, and stored in 70% EtOH until processing and paraffin embedding. Tissues were sectioned at 5 mm thick onto glass slides. Slides were processed for deparaffinization, rehydration, and antigen retrieval using citrate buffer (pH 6.0; Dako) for 20 minutes in a pressure cooker at 105°C followed by a 20-minute bench cool down. Endogenous background signal was reduced by incubating slides in 1% H2O2 (Sigma-Aldrich) for 10 minutes, before blocking for 30 minutes in 2.5% Normal Donkey Serum in 1X DPBS prior to antibody staining. Primary antibodies against selected markers were incubated on the slides in a humidity chamber overnight, followed by three washes in PBS, and 1 hour incubation in Hoechst 33342 (Invitrogen), and compatible secondaries (1:500) conjugated to Invitrogen AlexaFluor-488 (AF-488) or Invitrogen AF-647. Slides were washed in 1X DPBS, mounted in Prolong Gold (Invitrogen) and imaged using a Zeiss Axio Imager M2 microscope with Axiovision digital imaging system (Zeiss; Jena GmBH). Multiplexed imaging using an immune cell-based antibody panel was performed by using a multiplex iterative staining and fluorescence-inactivation protocol, as previously described (McKinley et al., 2017, 2019), and imaged on an Olympus X81 inverted microscope (20X magnification) with a motorized stage. For histological analysis, slides were processed and stained for hematoxylin and eosin and beta-catenin using standard approaches. Blind scoring was conducted by a pathologist (Dr. Kay Washington) using brightfield microscopy and a standard grading scale for dysplasia. Antibodies, working concentrations, and incubations can be found in the methods github repository: https://github.com/Ken-Lau-Lab/STAR_Methods/blob/main/Methods_Tables.xlsx (Methods Table 3)
Murine organoid formation assay
Organoids derived from Lrig1 and Mist1 tumors were dissociated using TrypLE Express. Cell pellets were resuspended in matrigel and seeded in 25 μL/well in a 24-well plate with 500 mL of Mouse Intesticult (STEMCELL Technologies) media. After one week, the number of organoids was counted using the GelCount system (Oxford Optronix). The number of organoids formed in each well was normalized to the number of single cells plated to determine organoid formation rate. Results were tabulated and plotted using Prism 9 (GraphPad) with unpaired t test.
Murine organoid antigen processing and presentation assay
Organoids were formed and cultured for one week in Matrigel and Mouse Intesticult media. They were collected and reseeded without Matrigel in media with 100 μg/mL DQ-Ovalbumin (Thermo Fisher Scientific) for approximately 24 hours. After 24 hours, organoids were fixed, stained overnight with antibodies against GFP and Ia/Ie-AF647 (1:100; Biolegend), and analyzed using a BD LSRII 5-laser flow cytometer. Flow data were analyzed using Cytobank (Kotecha et al., 2010).
Murine T cell activation assay
Naive OTII cells were isolated from the spleen of 8–10-week-old OT-II mice. Cells were purified using the naive CD4+ T Cell Isolation Kit (STEMCELL Technologies) following manufacturer’s protocol. CD11c+ DCs were isolated using MagniSort Mouse CD11c Positive Selection Kit (Thermo Fisher) per manufacturer’s recommendations. Murine Organoids were dissociated with TrypLE containing 10 μM Y-27632 for 15 minutes at 37°C while shaking. Cells were counted using Bio-RAD TC20 automated cell counter for use in the antigen presentation assay.
To track T cell proliferation, naive CD4+ OTII T cells were labeled using 5 mM CellTrace Violet (Thermo Fisher) by incubating for 20 minutes at 37°C, 5% CO2 in PBS and then an equal volume of T cell media containing serum was added and incubated an additional 5 minutes at 37°C, 5% CO2 to quench free dye. 5×104 labeled OTII CD4+ T cells were plated in a 96-well round bottom plate with 2.5×105 organoid-dissociated single cells (without Matrigel) or 2.5×105 CD11c+ DCs and in the presence or absence of 50 μg/mL ovalbumin peptide (Anaspec), spun at 350 x g for 5 minutes and then incubated at 37°C, 5% CO2 for 72 hours. Following co-culture, cells were analyzed. Wells containing cells were pipetted up and down to resuspend all cells and placed in 5mL Falcon Round-Bottom Polystyrene Tubes. These were centrifuged briefly at 350 x g for 3 minutes at 4°C, washed in FACS buffer (PBS w/o Ca2+Mg2+, 2% FBS, 2 mM EDTA), and resuspended in 100 μL FACS buffer containing the antibody cocktail: https://github.com/Ken-Lau-Lab/STAR_Methods/blob/main/Methods_Tables.xlsx (Methods Table 4), and stained for 15 min at 4°C. Cells were spun down as before, washed in FACS buffer, and were resuspended in 250 μL of FACS buffer and kept on ice until acquired on a 4-laser Fortessa. Cytometry data analysis was done using FlowJo v10 software and T cell proliferation results were tabulated and plotted in GraphPad Prism 9 using ANOVA with post hoc Tukey tests. This protocol was adapted from (Biton et al., 2018).
MARIS bulk sequencing of reporter expressing murine cells
After chelation but prior to single-cell dissociation, tissue was processed with a modified fixation/dissociation protocol (Scurrah et al., 2019). Briefly, tissue was fixed for 15 min (0.1% Saponin in 4% PFA, RNase-inhibitor), washed (0.1% Saponin in 1X DPBS RNase-inhibitor), and stained overnight with primary antibodies against GFP and EPCAM (1:100; Santa Cruz Biotech) in wash buffer. The following day, samples were washed with 1X DPBS, followed by a 1-hour incubation with compatible secondary antibodies in wash buffer. Samples were subsequently fixed followed by mechanical disassociation into single cells before flow sorting using BD FACSAria III.
After sorting, total RNA was isolated from the flow sorted cells using the RecoverAll Total Nucleic Acid Isolation kit (Ambion), starting at the protease digestion stage of the manufacturer-recommended protocol similarly to Hrvatin et al. (Hrvatin et al., 2014). The initial protease digest was scaled to the number of the cells post-sorting. Complementary DNA (cDNA) was generated from 160 ng of total RNA with Poly A priming using Maxima H minus reverse transcriptase (Thermo Fisher). The poly A capture primers used were the identical to unconjugated primers used for inDrop scRNA-seq (Klein et al., 2015) in order to generate cDNA libraries comparable to the reference scRNA-seq datasets for downstream integrative analysis. RNA-seq libraries were prepared as in (Southard-Smith et al., 2020) and sequenced on an Illumina NextSeq 500 as described below. To integrate with scRNA-seq datasets, the resulting bulk RNA-seq dataset was treated as a single cell datapoint, and normalized and processed accordingly (Heiser et al., 2021).
RNA isolation and qPCR
Total RNA was isolated using the RNeasy Plus Mini Kit (QIAGEN) and concentration was quantified using nanodrop (Thermo Fisher Scientific). cDNA was synthesized using the QuantiTect Reverse Transcription Kit (QIAGEN) according to the manufacturer’s instructions. Gene-specific primers for SYBR Green real-time PCR were designed by PrimerBLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast/) and published sequences, and synthesized by Sigma Genosis. Real-time PCR was performed and analyzed using CFX96 Real-Time PCR Detection System (Bio-Rad) and using SsoAdvanced Universal SYBR Green Supermix (Bio-Rad) according to the manufacturer’s instructions. PCR conditions are 95°C for 3 min and followed by 40 cycle amplification (95°C for 15 s, 60°C 15 s, 72°C for 30 s). Relative mRNA expression was determined by normalizing to GAPDH expression, which served as an internal control. See Key Resources Table for primers used for qPCR.
QUANTIFICATION AND STATISTICAL ANALYSIS
scRNA-seq, regulon network prediction, activity inference, and visualization
The Single-Cell rEgulatory Network Inference and Clustering or SCENIC pipeline was used to integrate cancer, pre-cancer, and their corresponding normal tissue datasets (Aibar et al., 2017; Van de Sande et al., 2020). For each group of integrated datasets, we concatenated the individual target datasets with an outer join and generated a combined AnnData object (Wolf et al., 2018). This AnnData object underwent further gene filtering, selecting only those that were expressed in at least 1% of all cells, primarily for the sake of speedup in running the module inference step of SCENIC. The resulting cumulative count matrix was input, without normalization, into the first step of SCENIC with default parameters, as suggested by the published protocol. We used a Dask client to parallelize the grnboost2 version of this step on an AMD Threadripper 2990WX CPU. Subsequently, cisTarget was performed using default parameters and three hg19 .feather ranking databases, comparing 10 species: tss-centered-5kb, tss-centered-10kb, and 500bp-upstream.
Further, this cisTarget step produced a list of detected regulons, their driving TFs, and their corresponding weights for the prediction of individual gene expression. These weights were used to build a feature matrix defining each regulon by its predicted targets. This feature matrix was then used to generate an adjacency matrix per SCENIC integration run, which was the basis of the regulon-regulon target network. This target network was based on a k-nearest neighbors graph (with k equal to the square root of the number of total regulons) of the adjacency matrix. For each of these target networks, the Louvain community detection algorithm was run at a resolution of 2, defining super-regulons (Traag et al., 2019). This regulon-regulon target network (along with its cluster labels and average enrichment per regulon) was exported as a weighted adjacency matrix for visualization in Cytoscape (Shannon, 2003).
Finally, we performed AUCell with default recommended parameters across 64 threads to generate a regulon activity enrichment matrix, which was jointly analyzed with the count-based matrix. Additional regulon activity enrichment scores were calculated for the Broad cohort by performing AUCell with regulon definitions learned from VUMC pre-cancer and CRC datasets. For visualization, target-network heatmaps featuring these regulon enrichment values were Z-score transformed, color scaled in a regulon-wise manner, and standardized to jointly integrated normal biopsies or polyp-derived normal cells when possible.
scRNA-seq, count matrix normalization and heatmap generation
Using scanpy and numpy functions, raw count data were normalized by median library size, log-like transformed with Arcsinh, and Z-score standardized per gene (Harris et al., 2020; Wolf et al., 2018). This yielded interpretable unit variance scaled and centered values. Heatmaps featuring individual gene expression depict this normalized, transformed, and standardized data with color scaling in a gene-wise manner.
scRNA-seq, UMAP and t-SNE visualization
Three modes of UMAP visualization were used in this study based on regulons, feature-selected counts, or Harmony-corrected components. All human epithelial UMAP visualizations were generated using the “scanpy.tl.umap” function with a min_dist parameter of 0.15. The input to this function was Z-score standardized AUCell values, their 50-principal component decompositions with no feature selection, and a subsequent KNN graph with k equal to the square root of the number cells projected. Human nonepithelial UMAP visualizations that included all nonepithelial subtypes were performed the same way. To finely resolve T cell subtypes with UMAP, we generated a KNN based on the PCA of a feature-selected set of genes after normalizing, log-like transforming with Arcsinh, and Z-score standardizing raw counts. Finally, murine validation experiments were integrated with the Harmony algorithm, generating adjusted principal components with default parameters (Korsunsky et al., 2019). These components were used as the basis for KNN and UMAP generation with the same parameters as used in the human data. For t-SNE visualizations, the perplexity was set to the same as the k used in the UMAP KNN graph. The bootstrapped variant of t-SNE visualization was performed by running t-SNE with the same parameters 100 times to ensure qualitatively robust embeddings, given the algorithms inherent stochasticity.
scRNA-seq, gene signature scoring
We used a gene signature scoring method implemented in scanpy and first detailed by (Satija et al., 2015). This method scores a defined gene set by finding the difference between its average expression against the average expression of randomly sampled sets of reference genes, corresponding to matched and binned expression levels. Each signature in this study was calculated on normalized, transformed, and standardized data (as described in the scRNA-seq, Count Matrix Normalization and Heatmap Generation section) using a reference sample size of 2000 genes across 25 bins. The x axis range of scatterplots featuring these signature data was set by excluding single-cell outliers beyond the 1.5x interquartile range. Statistical tests of these score distributions encompass an initial Kruskal-Wallis test. If the null hypothesis was rejected, these tests were followed by post hoc Mann-Whitney U tests and appropriate p value adjustments (Terpilowski, 2019; Varoquaux et al., 2015). The resulting statistics are found in Table S4. Genes comprising each human gene signature calculated are listed in Table S5 for: exhaustion, cytokines, chemokines, MHC I&II processing and presentation, fetal, WNT and stem cell, and metaplasia and damage response (Barker et al., 2007; Cadigan and Waterman, 2012; Clevers and Batlle, 2006; Du et al., 2008; Fife et al., 2009; van der Flier et al., 2009; Hieshima et al., 1997; Imajo et al., 2015; Lee et al., 2020, 2021; Lili et al., 2016; Mustata et al., 2013; Nelson et al., 2001; Park et al., 2005; Pelka et al., 2021; Zhang et al., 2015). Gene signatures for murine TSC scRNA-seq were calculated for ISCI, ISCII, and ISCIII as described by Biton et al., with the same method applied to calculating the murine MHCII signature (Biton et al., 2018). Genes used for the identification of cell populations or expression programs are detailed in Table S5 for: CD8 T cell cytotoxicity, CD8 T cell activation, CD8 T cell effectors, CD8 T cell homing and memory, suppressed CD4 T cells, CD4 T cell RORa-dependent/tumor-promoting inflammation, CD4 T cell dysfunction, T cell immunosuppressive markers, CD4 T regulatory cells, myeloid TAMs, myeloid suppressive TAMs, myeloid MDSC-like, MDSC-like IL6 signaling, MDSC-like cytokine suppression, MDSC-like inflammation, MDSC-like secretion, myeloid immunosuppressive, M1 cells, M2 cells, ISCI cells, ISCII cells, and ISCIII cells (de Almeida Nagata et al., 2019; Alshetaiwi et al., 2020; Anderson et al., 2016; Baitsch et al., 2011; Biton et al., 2018; Blackburn et al., 2009; Bronte et al., 2016; Carlin et al., 2005; Castello et al., 2017; Duckworth et al., 2014; Fife et al., 2009; Greenwald et al., 2001; Hwang et al., 2018; Jiang et al., 2017; Jin et al., 2017; Joshi et al., 2007; Lee et al., 2020; Lesokhin et al., 2012; Marigo et al., 2010; McDonald et al., 2018; Moon et al., 2015; Movahedi et al., 2008; Schwartz, 2003; Trikha and Carson, 2014; Utting et al., 2000; Wang and Denhardt, 2008; Wells et al., 2000; Yamada et al., 2009; Youn et al., 2008; Zhang et al., 2020; Zhu et al., 2015).
scRNA-seq, unsupervised clustering and cell type labeling
The labeling of single-cell subpopulations was done through the Leiden algorithm, as part of the Scanpy toolkit. We performed Leiden clustering based on the KNN derived from the distances calculated in the principal component space of Z-score transformed regulon enrichment scores, as these represented cell-cell transcriptional states in a more batch-robust manner. The resolution of this clustering was based on the detection of rarer populations such as enteroendocrine cells, at 2. Since this algorithm detected discrete clusters in a continuum of cell states, we aggregated multiple discrete clusters by the observation of marker gene expression. Similarly, these methods were applied to nonepithelial datasets given their regulon or feature-selected matrices, depending on the subtypes of interest. This Leiden algorithm was also used to determine clusters for murine scRNA-seq validation experiments. Higher resolution subclustering was also done by performing k-means clustering after the initial Leiden clustering. Importantly, some subclusters were identified as a result of patient-to-patient variation originating from mitochondrial read enrichment, as evidenced by mitochondrial read percentage distributions and GO terms. These subclusters were identified and statements regarding their relative, subpopulational variation were excluded. These patient-to-patient variations did not affect overall comparisons between tumor-specific and normal cell types. For example, after excluding these mitochondrially-enriched subclusters, the SSC subpopulational analysis focused on GO terms related to intercellular communication and stromal interactions.
scRNA-seq, differential gene-expression testing and gene set enrichment analysis
The differential testing of gene expression was performed based on cluster labels (as defined by the scRNA-seq, Unsupervised Clustering and Cell Type Labeling section), both in the context of raw gene counts and regulon enrichment values. For both cases, we used Mann-Whitney U tests with Benjamini-Hochberg corrections, on the raw values, implemented through the “scanpy.tl.rank_ genes_groups” function, identifying the top 200 genes and top 50–100 regulons (Wolf et al., 2018). Further, biological insight was gathered through scanpy’s integration of g:profiler gene set enrichment framework (Raudvere et al., 2019). The full differential expression, GSEA tables, and their respective statistics generated through g:Profiler are available in Tables S6 and S7. This process was also performed on the stem and TSC components of the murine scRNA-seq datasets using the GSEA webapp (Mootha et al., 2003; Subramanian et al., 2005).
scRNA-seq, proportional cell type representation and identifying polyp-specific populations
Given the detected clusters (as described in the scRNA-seq, Unsupervised Clustering and Cell Type Labeling section), we calculated the proportional cell type representations of each individual sample. We counted the raw number of epithelial and nonepithelial cells as well as the raw number of cells falling into any given cell cluster. These results were cross-tabulated as contingency tables, summarizing how many cells were observed in each category and for which samples using pandas. Proportional values were then calculated by normalizing cluster counts to the number of epithelial cells per sample (Figures 2C and S2C) or to the cumulative number of cells per sample (Figures 6C and S6D). Clusters were designated as polyp-specific populations if, proportionally, they were significantly overrepresented in polyp samples and not normal samples, which was indicated by post hoc statistical tests following Kruskal-Wallis null hypothesis rejection. The resulting statistics are found in Table S4. The x axis range of scatterplots featuring these proportional representation data was set by excluding samples with values beyond the 1.5x interquartile range. In the context of the murine scRNA-seq datasets, the proportional representation of cell types was calculated by normalizing to the total number of epithelial or immune cell subtypes for each Mist1 and Lrig1 tumor sample.
scRNA-seq, predicting differentiation potential with CytoTRACE
CytoTRACE is a relative scoring method dependent on included datasets for inferring developmental potential. CytoTRACE was performed based on the default recommended settings after concatenating the batches of interest using an outer join (Gulati et al., 2020). We performed CytoTRACE with five separate groupings of single-cell libraries. First, the discovery cohort (Figures 2E and S2E), including all epithelial cells from both its normal biopsies and polyps. Second, the validation cohort (Figures 2E and S2E), including all epithelial cells from both its normal biopsies and polyps. Third, the epithelial VUMC polyp-specific cells (Figure 4E), including only tumor-specific cells from VUMC AD, MSS, SER, and MSI-H samples. Fourth, the epithelial Broad cohort (Figures 4E), including MSS, MSI-H, and Normal samples. The Broad cohort (including n = 32 normal samples) distribution was only calculated from 50% random sample of the total cells detected due tomemory constraints. Fifth, CytoTRACE was performed on the stemand TSC component ofthe murine scRNA-seq datasets. Statistical tests of these score distributions encompass an initial Kruskal-Wallis test. If the null hypothesis was rejected, these tests were followed by post hoc Mann-Whitney U tests and appropriate p value adjustments. The resulting statistics are found in Table S4.
scRNA-seq, CMS scoring at single-cell resolution
The single-cell distributions of CMS scores were calculated on the VUMC ASC, MSS, SSC, and MSI-H and the Broad MSI and MSI-H libraries using the CMSclassifier R package as described by (Eide et al., 2017; Guinney et al., 2015). To accommodate the heterogeneity of the single-cell landscape, the single sample predictor or SSP mode of the software was used after converting gene symbols to Entrez IDs. This SSP mode calculated the median correlation distance between each single cell to established, standard centroids derived from CMS1, CMS2, CMS3, and CMS4 CRC subtypes. Further, these score distributions were visualized through a normalized kernel density estimation implemented in the Seaborn python package. Statistical tests of these score distributions encompass an initial Kruskal-Wallis test. If the null hypothesis was rejected, these tests were followed by post hoc Mann-Whitney U tests and appropriate p value adjustments. The resulting statistics are found in Table S4.
scRNA-seq, trajectory inference
pCreode was used to map the developmental state transitions of the single-cell transcriptional landscape of our Discovery cohort pre-cancer and normal COLON MAP samples (Herring et al., 2018). This algorithm was generalized to process regulon-based principal components, inheriting its batch-robust properties. By examining the variation captured by the principal components, we selected the first 4 components based on their capture of rare cell populations, such as Tuft and enteroendocrine cells. We developed this algorithm to traverse a density weighted KNN generated from the pairwise distances between each single cell; subsequently, we used a histogram thresholding method to estimate the neighborhood distance cutoff for calculating local densities. These densities were used as input to a supervised variant of pCreode, which established developmental endstates through K-means clustering and marker-defined labels. The downsampling and noise parameters were both set to 4, resulting in samples of around 6,000 cells per run, and repeated 50 times. Each of these runs was scored by the minimization of the Gromov–Hausdorff distance, resulting in a single, most representative graph layout. Overlays were generated based on pre-computed single-cell observation vectors, such as a CytoTRACE score, or the normalized, transformed, and z-scored gene expression values.
scRNA-seq, RNA velocity
RNA velocity analysis was performed using velocyto CLI version 0.17 (La Manno et al., 2018). Individual sample BAM files were used as input to the “run-dropest” command along with a human gene annotation file (GTF) for GRCh38.85, and a tab-delimited text file containing dropkick-filtered cell barcodes from the corresponding sample as the “—bcfile” flag. Then, scVelo version 0.2.3 was used to build models of splicing kinetics to estimate and visualize RNA velocity vector fields in SCENIC integrated UMAP space (Bergen et al., 2020). Each sample was individually filtered to the top 2,000 genes expressed in a minimum of 20 cells using the function “scvelo.pp.filter_and_normalize.” The moments of all RNA velocity vectors were calculated with 30 principal components and 30 nearest neighbors using the function “scvelo.pp.moments” prior to estimating velocities using “scvelo.tl.velocity” with default parameters. Finally, velocity UMAP embeddings were plotted using the function “scvelo.pl.velocity_embedding_stream” and the subset of SCENIC master UMAP coordinates for each sample.
sc-RNA-seq, subclone phylogeny estimation
We used DENDRO, an algorithm designed to reconstruct subclonal phylogenies within scRNA-seq datasets (Zhou et al., 2020). Specifically, information of both somatic and germline single nucleotide variations (SNVs) are used in this reconstruction, differing from purely somatic variant-based methods. Since our sequencing libraries are generated through the tag-based in-Drop method, the short, 3′-biased reads necessitated the aggregation of single-cell transcriptomes. For each of the 34 sequencing libraries we performed this analysis on (24 ADs, 11 SERs), we defined 20 aggregate populations through regulon-based K-means clustering (Hartigan and Wong, 1979). Thus, we predicted the average genotypic representation of multiple single-cells, or pseudo-bulk RNA-seq libraries, and created a phylogenetic tree between the defined clusters. DropEst produced a filtered and sorted .bam file, which we derived read information from, and split into 20 distinct .bam files using the sinto python package. These bam files were then processed with GATK4 (Van der Auwera et al., 2013), according to guidelines detailed by Zhou et al. The GATK4 steps of this pipeline involved the following: adding read groups, marking duplicates, splitting N-cigar reads, applying base quality recalibration with known single nucleotdie polymorphisms (SNPs), haplotype calling (with the GATK4 HaplotypeCaller and an hg38 reference) to generate VCF files, consolidating these VCFs into genomicsdb databases, and then genotyping these data.
Because the measurement of SNVs within transcriptomes is dependent on dynamic expression patterns, we used a beta-binomial framework, as described by Zhou et al., to model genetic divergence between each pseudo-bulk, cell aggregate. Standard genetic divergence frameworks, such as those comparing DNA-derived genomic variants, do not consider the varying levels of low nor high gene expression between pseudo-bulk populations. Examples of this transcription-specific variation would be stochastic bursts of gene expression captured in a minority of populations, yielding low average expression across all populations, and constitutively expressed genes, yielding high average expression across all populations. These bursty loci will more likely represent genes dropped out from the majority of pseudo-bulk populations, so including its respective variants would yield an inflated genetic divergence value. Conversely, variants in loci that are expressed and observed in the vast majority of pseudo-bulk populations would be uninformative in terms of phylogenetic discrimination. The genetic divergence d between each possible cell aggregate pair c and c’ at loci g is represented formally as:
Where c and c’ represent two different cell aggregates, while I and I’ represent their originating clonal groups. Correspondingly, Xcg is the alternative allele read count for cell aggregate c at loci g, while Ncg is the respective total read count. Thus, d is a function of five derived probabilities:
Which, first, represents the alternative allele frequency across cell aggregates estimated by the above GATK4 pipeline.
Which, second and third, represent detected variants due to rare editing and technical sequencing error events in c and c’ at g. Here, Zcg is set as 0, modeling scenarios lacking SNVs, which can be approximated as the following binomial distribution with ε set to 0.001 or 0.1%.
ε, representing the combined error rate, was used according to our sequencing platform, a NovaSeq 6000 System. This is in line with empirical studies of Illumina sequencing instruments as detailed by Stoler et al., observing a median error rate of 0.109% across 239 samples on a NovaSeq 6000 device (Stoler and Nekrutenko, 2021). Another previous study by Fox et al. had similar estimates for sequencing-by-synthesis platforms, including the Illumina MiSeq and HiSeq2000, with an error frequency of 10–3 (or 0.1%) attributed to single nucleotide substitutions (Fox et al., 2014).
Which, fourth and fifth, represent detected variants due to the presence of SNVs in c and c’ at g. In this case, Zcg is set as 1, modeling scenarios with SNVs present.
This can be approximated as a beta-binomial distribution, as previously described by Jiang et al. and Skelly et al. in the context of single-cell and bulk RNA sequencing (Skelly et al., 2011; Xiong et al., 2019). Qcg is the proportion of alternative alleles in cell aggregate c at g, using a beta distribution prior, approximated as q. q is parameterized by αg and βg, as estimated gene activation and deactivation rates respectively.
Before performing genetic divergence calculations based on these probabilistic models, two filters were applied to minimize the inclusion of stochastically or constitutively expressed variants:
The first filter is dependent on the observed variant allele frequencies (VAFs) across each set of cell aggregates. VAFs were visualized as histograms representing the number of times each unique variant was observed across each set of cell aggregates. We observed that these VAF distributions were unimodal and positively skewed, with the vast majority of variants being detected in very few cell aggregates, which was in line with stochastic gene expression. To remove these stochastically expressed variants, we heuristically determined a cutoff at observed convex elbow/knee points of the curve, at 10%. This cutoff was symmetrically applied to the top 10% of the most pervasive variants as well, as these represented constitutively expressed variants.
The second filter is dependent on α and β parameter estimations. If either the α or β parameters of the beta prior were estimated to be 0 or 1, it meant that the activation and deactivation rates were completely on or off. Akin to the rationale for our first filter, these variants would not be informative in the genetic divergence calculation since they likely represent genes with a tendency to dropout/be stochastically expressed or be constitutively active. These cases would inflate or deflate genetic divergence metrics, respectively. The quality of the filtered variants, consisting of about 5.07% (std. 1.46%) of the initially detected variants, met appropriate QD and DP levels suggested by GATK4 guidelines and were also located within genomic regions characteristic of the inDrop barcoding chemistry (https://github.com/Ken-Lau-Lab/STAR_Methods/blob/main/Supplemental_Table_Variant_Type_Func.refGene_Distribution.xlsx). Exonic variants detected through this method were validated through the exome sequencing of paired FFPE tissue and respective GATK HaplotypeCaller pipeline. If the exact exonic genomic loci and genotypes were detected in both the exome and scRNA-seq pseudo-bulk aggregates, the variants were flagged as validated. An average of 53.9% (std. 12.9%, max > 75%) of the exonic variants detected through scRNA-seq were validated with this orthogonal exome sequencing. Tables of these detected variants per cell population and their exome-seq statuses are shown at (https://github.com/Ken-Lau-Lab/STAR_Methods/tree/main/Tables).
After filtering, the genetic divergence is calculated for all possible pairs of cell aggregates, and a phylogenetic tree is constructed. The leaves of these trees represent the previously defined cell aggregates, which were assigned cell type labels accordingly. For each set of pseudo-bulk cell aggregates, we also calculated the minimum genetic divergence between tumor-specific cell aggregates (ASCs and SSCs) and canonical stem cell aggregates (STM). These values were normalized to the maximum distances observed per tumor sample, yielding a value between 0 and 1. This metric was interpolated with a value of 1 in samples which lacked measurable canonical stem cell aggregates.
MxIF, single-cell segmentation and image analysis
Cell segmentation was accomplished using the MANDO pipeline (McKinley et al., 2019). Briefly, random forest pixel classification on manually annotated images was used to define tissue and subcellular regions in each image. An initial watershed segmentation using cell nuclei as seed points and the learned cell membranes as boundaries was followed by re-segmentation of objects containing internal cell membranes. For every identified cell, image intensities for each marker were then calculated as well as morphological features such as cell size and location. For quantifying marker positive cells in MxIF, we fitted linear mixed effects models on the logit transformed cell proportions within epithelial or stromal tissue compartments. We estimated the proportion of marker positive cells within each compartment, by dividing the number of marker positive cells by the total number of cells within the tissue compartment. We added 1/2 to the numerator and denominator of the proportion to accommodate zero proportions; this is equivalent to a Bayesian estimator for the proportions using a noninformative beta prior. We fit the logit transformed proportions using a linear mixed effects model with an interaction between tissue compartment (epithelium/stromal), tissue type (AD/SSL), and a random effect for slide to account for the correlation between regions on a slide (Bates et al., 2015). We estimated differences between tumor types within each tissue compartment using emmeans. We computed false discovery rate (FDR) adjusted p values using Benjamini-Hochberg. For murine tissue, tumor areas were established by a beta-Catenin mask and cell counts for image quantification were determined the same way as human tissues.
MxIF and MxIHC, pixel-based image quantification
MATLAB was initially used to create masks to mark positive pixels of each cell type marker from MxIF images. The tumor region was divided into an epithelial region (masked by beta-Catenin, pan-Cytokeratin, and NaKATPase expression) and a stromal region (tumor mask minus the epithelial mask). An overlay of OLFM4, MUC5AC, and PANCK was used as a guide for identifying stem (OLFM4+) and metaplastic (MUC5AC+) epithelial (PANCK+) regions, which were then manually demarcated. Each region was validated by quantifying MUC5AC and OLFM4 positive pixels within the regions. Cell types were defined by combinations of marker masks; for example, CD4+ T cells were defined by intersecting CD4 and CD3 pixel masks. On the other hand, CD8+ T cells were defined using the CD8 marker. We then calculated the fraction of pixels occupied by each cell type, normalized to the number of pixels of each tumor region. For example, a ratio of intraepithelial CD8+ cells to stromal CD4+ T cells was calculated from two sets of values calculated in this way. The measurements from all regions of the same type within each tumor was used to calculate a mean value; thus, each patient is a biological replicate. One-way ANOVAs with Dunnett post-tests were used for statistical testing. For IHC images, a similar process was used, with whole tumor regions demarcated by tissue morphology using hematoxylin nuclear counterstain. Antibody stains (3,3′-diaminobenzidine - DAB or 3-amino-9-ethylcarbazole - AEC) were spectrally unmixed such that individual marker masks can be generated and quantified as above.
Supplementary Material
(A) Oncoplot representation of the mutational landscape of 281 polyp specimens detected through bulk targeted sequencing and mutational variant calling with layout similar to Figure 1C. (B) Representative IHC for MLH1 in AD and SER. (C) Quantification of MLH1 gene expression in AD and SER from scRNA-seq of polyp-specific cells compared to the same analysis of MSI-H CRC cells (red line - mean). n = 29 for AD and 19 for SER. Each point represents a polyp, averaged over normalized and scaled Arcsinh-transformed single-cell counts. (D) UMAP representation of epithelial scRNA-seq data, color coded by specimen to depict interspecimen heterogeneity, examining (top) DIS and (bottom) VAL sets, and (left) raw count-based and (right) SCENIC regulon-based.
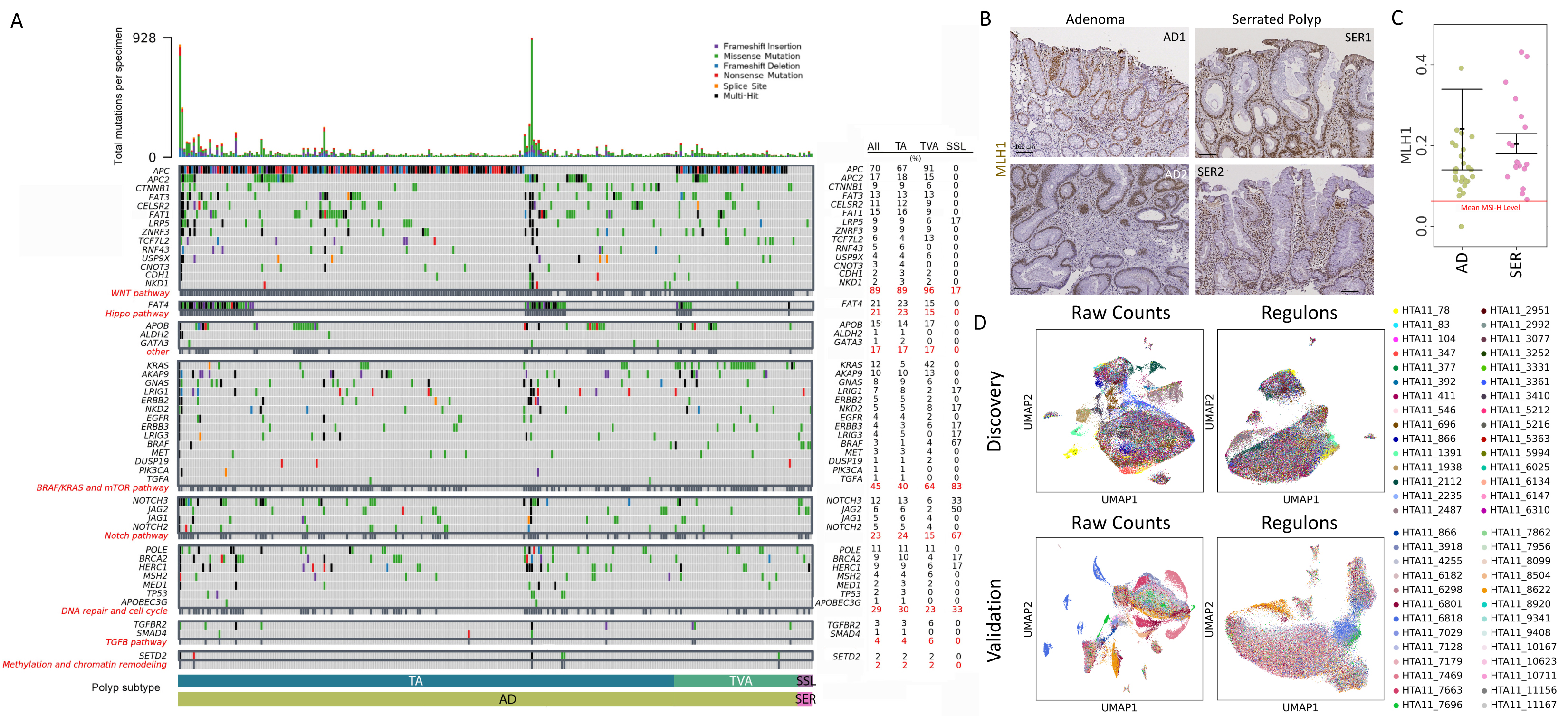{kind=link}
(A) UMAP of regulon-based epithelial cell scRNA-seq data overlaid with cell type-specific genes. Color intensity represents scaled and standardized Arcsinh gene expression. (B) UMAP of (left) DIS and (right) VAL datasets color overlaid with HP subtypes. (C) Scatterplots of normalized (left) ASC or (right) SSC representation per tissue and polyp subtype. Points represent individual specimens. Error bars represent SEM of n = 29 for AD, n = 19 for SER, and n = 66 for NL. Coloring of points indicates (top) polyp subtype or (bottom) DIS or VAL dataset. (D) Scatterplots of signature scores by cell type with each point representing a single cell for (top) DIS and (bottom) VAL datasets. Error bars depict SEM of single cells. Not all post hoc tests shown. (E) Regulon-based UMAP of epithelial cell scRNA-seq data overlaid with CytoTRACE scores for (top) DIS and (bottom) VAL datasets. (F) Regulon-based UMAPs of (top) ASC and (bottom) SSC-gated scRNA-seq data with colored label overlays indicating Leiden subcluster, cohort, polyp subtype, and specimen ID. ASC Leiden subcluster 0 is relatively enriched for WNT stemness terms. SSC Leiden subcluster 2 exhibits variation attributed to mitochondrial gene expression and lower quality. (G) Expanded TF target network created from normal and pre-cancer cells organized into super-regulons derived from shared targets for AD-derived normals, ASCs, SER-derived normals, and SSCs. Color overlays for each TF node are averaged and normalized regulon enrichment scores, while edge opacities are the inferred TF-target weightings. (H) Heatmap representation of gene sets derived from the scRNA-seq applied to bulk RNA-seq data of colonic polyps (n = 36 tubular, 22 tubulovillous, 8 SSLs). Inset circle indicates the fraction of specimens presented with gene expression and color intensity represents mean scaled and standardized Arcsinh gene expression. *p < 0.05, **p < 0.01, ***p < 0.001.
{kind=link}
(A-D) Additional representative multiplex images of colonic polyp subtypes for (A) SOX9, (B) OLFM4, (C) CDX2, and (D) MUC5AC. White outline demarcates HP location. (E) MUC5AC staining for different GCHPs. (F) Low-magnification view of MVHPs and SSLs for MUC5AC staining. (G) MUC5AC staining of non-neoplastic colonic epithelium: (row 1) normal, (row 2) tumor adjacent, and (row 3) ulcerative colitis. (H) p-Creode trajectories inferred from epithelial scRNA-seq data, for both (top) DIS and (bottom) VAL cohorts. Overlay of genes relevant to developmental lineages. Node size represents the proportion of cells and overlay color intensity represents mean scaled and standardized Arcsinh gene expression. (I) Additional RNA velocity maps for normal colonic tissues and polyps on combined UMAP embedding for (top) DIS and (bottom) VAL datasets. Vectors inferring average cell state transitions within each specimen are shown as black arrows. Colored points represent cells derived from the individual specimen, with gray points representing all other cells. (J) Representative genetic phylogenies inferred through DENDRO with each tree representing a specimen for colonic polyp subtypes. Hierarchical clusters demarcated by dotted boxes and cell clusters denoted by leaf node color. Tree height is derived from a beta-binomial adjusted genetic divergence between single cells and shows the evolutionary time before splits in phylogeny. ABS – absorptive, ASC – adenoma specific cells, CT – crypt top, EE – enteroendocrine, GOB – goblet, STM – stem, SSC – serrated specific cells, TAC – transit amplifying cells, TUF – tuft. (K) Summary metric for phylogenetic analysis depicting the minimum genetic divergence distance between polyp-specific cells (ASC or SSC) and stem cells. n = 23 AD, and 11 SER specimens. Error bars depict SEM of individual libraries. *p < 0.05.
{kind=link}
(A) Oncoplot representation of the mutational landscape of CRCs detected through exome sequencing, and (left) mutational variant calling or (right) somatic mutation calling. Total number of mutations detected per specimen represented as bar plot (top) and different type of mutations color-coded. Important genes to CRC are presented and grouped into pathways (dark gray boxes). Proportion of gene and pathway mutation within CRC subtypes (combined) summarized as table. (B) Representative H&E of each CRC subtype at low and high magnification views of the corresponding colored insets. (C) Scatterplots of signature scores by tumor-specific cells per subtype, with each point representing a single cell, for (top) VUMC and (bottom) Broad datasets. Error bars depict SEM of single cells. (D) Heatmap representation of top biologically relevant and differentially expressed genes, for Broad (MSS, n = 28; MSI-H, n = 32) and Samsung Medical Center (SMC) (CMS1, n = 5; CMS2, n = 8; CMS3, n = 4; CMS4, n = 6) single-cell datasets. Inset circle indicates prevalence within defined single-cell populations and color intensity represents scaled and standardized Arcsinh gene expression. (E,F) Expanded TF target network created from (E) VUMC or (F) Broad tumor-specific cells, organized into super-regulons derived from clustering of shared targets. Color overlays for each TF node are averaged and normalized regulon enrichment scores, while edge opacities are the inferred TF-target weightings. VUMC regulon values are normalized to tumor-derived normal cells while Broad values are unnormalized due to lack of detected tumor-resident normal cells. *p < 0.05, **p < 0.01, ***p < 0.001.
{kind=link}
(A) Oncoplot visualizing genomic tracks for 276 assayed TCGA CRC specimens. Each row represents unsorted mutational status of key pathway genes. Each column represents an individual specimen with hypermutation status noted. (B) Heatmap representation of gene signatures identified by scRNA-seq of pre-cancers applied to bulk RNA-seq of TCGA CRC specimens (CMS1, n=8; CMS2, n=26; CMS3, n=10; CMS4, n=19). Inset circle on summary heatmap (left) indicates the fraction of specimens presented with gene expression and color intensity represents mean (left) or individual (right) scaled and standardized Arcsinh gene expression. (C) Representative stitched MxIF scans of OLFM4 in 3 MSS and 3 MSI-H CRCs. Black rectangles in the restitched image represent fields of views that were not scanned, due to these areas being empty (glass) or of low tissue quality. (D) Staining for various metaplastic markers of 4 CRCs from a tissue microarray. Red outline represents the MLH1 low area in CRC 4.
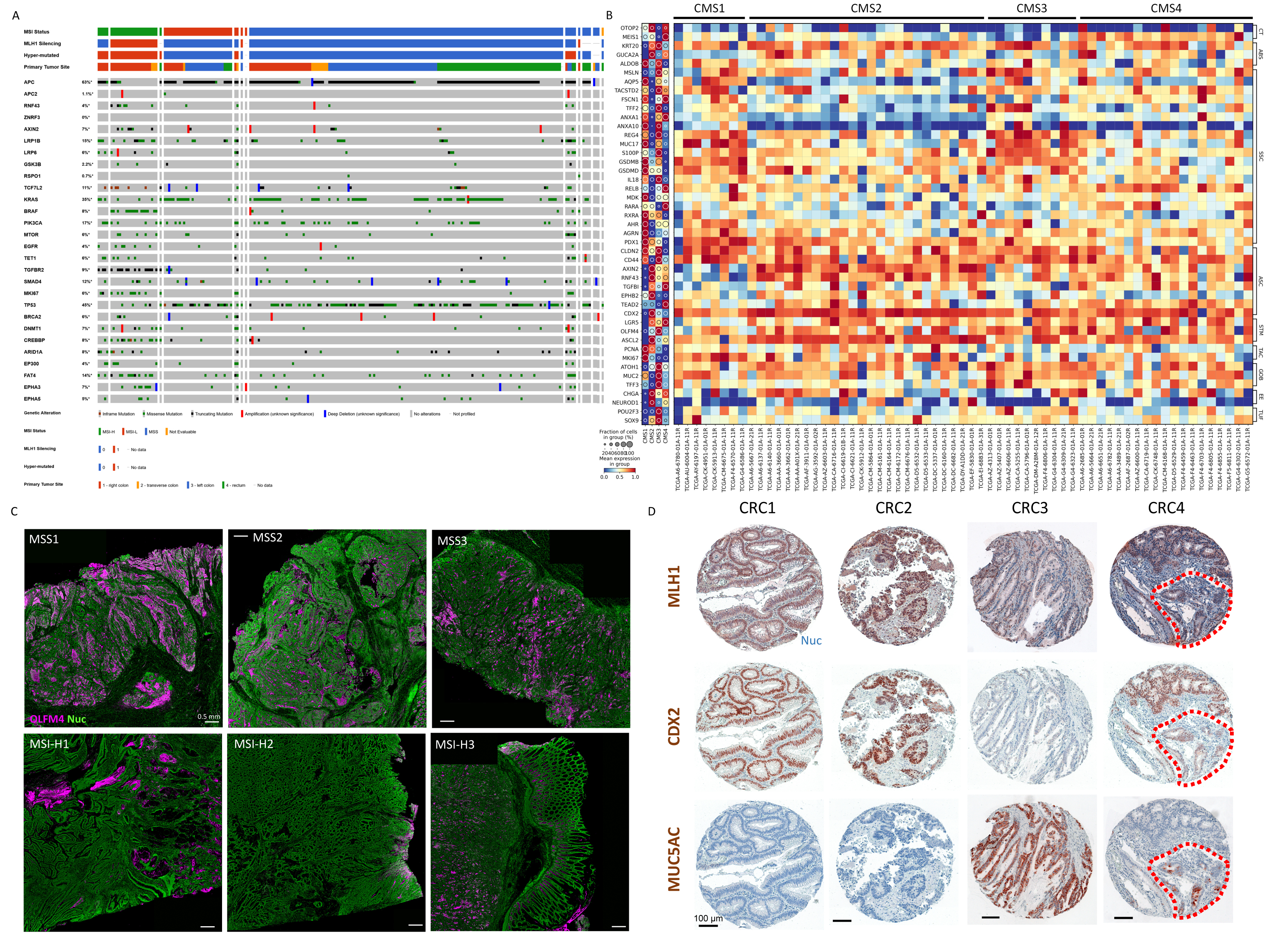{kind=link}
(A) Regulon-based UMAP representation of non-epithelial cell populations color overlaid with tissue and tumor subtype. (B) Feature-selected, count-based UMAP representation of lymphocyte subtypes, color overlaid with (left) tissue and tumor subtype and (right) cell type defined by clustering and marker gene expression. CD4 – CD4+ T cells, CD8 – CD8+ T cells, CYC – cycling T cells, GD – Gamma delta (γδ) T cells, NK – natural killer cells, T REG – regulatory T cells. (C) Heatmap representation of feature-selected genes used to generate UMAP in B. Inset circle indicates prevalence within defined single-cell populations and color intensity represents scaled and standardized Arcsinh gene expression. (D) Comprehensive scatterplots of normalized cell type representation per tissue, (top) polyp and (middle) cancer subtypes. Points represent individual specimens. Error bars represent SEM of n = 28 for AD, n = 17 for SER, n = 66 for NL, n = 33 for MSS, and n = 34 for MSI-H. Plots in the third row colored by VUMC or Broad datasets. (E) Scatterplots of CD8+ T cell cytotoxicity and exhaustion signatures, with each point representing a single cell for (top) polyp and (bottom) CRC. Error bars depict SEM of single cells. (F) Additional representative MxIF images of CD8+ cells in multiple ADs and SERs. (G) Quantification of MxIF images of marker positive cells in epithelial or stromal compartments normalized to total number of cells in each compartment. Violin plots with median as white circles and quartiles as candles for n = 20 polyps per type. (H) Quantification as in G for CD8+ T cells in the epithelial compartment of TAs separated by mutational burden for n = 6 high (> 1500 mutations) versus n = 7 low (< 1500 mutations). (I) Additional representative MxIF images of CD68+ cells and MUC5AC+ in multiple ADs and SERs. (J,K) Additional representative MxIF scans of intratumoral heterogeneous regions within CRCs (OLFM4+ stem regions versus MUC5AC+ metaplastic regions), and additional representative MxIF images of CD8+ and CD3+ cells within each of the stem and metaplastic regions of (J) MSI-H CRCs and (K) MSS CRCs with only stem regions. (L) Quantification of marker-positive pixels per tumor area in stem versus metaplastic regions in MxIF scans of n = 15 MSS and n = 10 MSI-H CRCs, presented as violin plots with median as solid lines and quartiles as dotted lines. *p < 0.05, **p < 0.01, ***p < 0.001.
{kind=link}
(A) Representative macroscopic IF images depicting a Lrig1CreERT2/+; Apc2lox14/+ tumor and Lrig1CreERT2/+; KrasLSL-G12D/+ proximal villiform metaplasia (white arrows). (B) Quantification of CD8-positive pixels per nuclear area in Apc-driven tumor and the villus and crypt compartments of Kras-driven villiform metaplasias. Red line denotes the mean level detected in adjacent normal colon in Kras mice. Error bars represent SEM from n = 3 animals per group. (C) IF images of CD8+ T cells in an Apc-driven tumor overexpressing β-catenin, Kras-driven villiform metaplasias (white arrows), and control colon. Dotted line demarcates border between villus and crypt compartments. (D) Representative IF images of short-term and long-term (rows: 24 hours and 28 days, respectively) lineage tracing in the colon using Lrig1CreERT2/+;R26RLSL-EYFP/+, Mist1CreERT2/+;R26RLSL-EYFP/+, and Mist1CreERT2/+;R26RLSL-EYFP/+ with DSS mouse models (columns). (E) Quantification of lineage-traced glands at 28 days at homeostasis or with DSS-damage from stem (Lrig1) and non-stem (Mist1) cells using different reporters. Error bars represent SEM from n = 3 animals for each condition. (F) Representative IF images of long-term (28 days) lineage tracing in the proximal colon of Mist1CreERT2/+; R26RLSL-mTmG/+ with or without DSS. (G) Representative IF images of reporter co-expression with cell-type-specific markers, DCLK1 for tuft cells, MUC2 for goblet cells, and CHGA for enteroendocrine cells, after 24 hours of lineage tracing in Mist1CreERT2/+;R26RLSL-EYFP/+ mouse colons. Arrows point to single positive (green/red) or double positive (yellow) cells. (H) Quantification of double-positive cells compared to single positive cells for each marker. Each dot represents a field of view. Error bars represent SEM from n = 3 animals. (I) Representative IF image of reporter co-expression with MUC2 in Mist1CreERT2/+; R26RmTmG/+ 10 days after initiation of lineage tracing at homeostasis. (J) Combined t-SNE embedding of bulk RNA sequenced Mist1 reporter (mTmG – left or EYFP – right)-expressing cells (solid circle in red) with n = 3 reference murine colonic scRNA-seq dataset (colored dots from different mice). Goblet (green) and enteroendocrine (magenta) cell populations are delineated by dotted lines. (K) Quantification of Mist1 reporter (EYFP or mTmG)-expressing cells co-embedding with different cell types using boot-strapped t-SNE runs in J. n = 3 independent experiments. (L,M) Representative H&E (L) and b-catenin IHC (M) of control (uninduced) normal colonic tissues as in Figures 7D and 7E. (N) Histological scoring of tissues of multiple biological replicates of Mist1 and Lrig1 tumors along with uninduced controls. 0: none; 1: unicrypt; 2: low; 3: high, 4: advanced. (O) Combined UMAP embedding as in Figure 7F with overlays of key marker gene expression delineating epithelial cell types and states. Color intensity represents scaled and standardized Arcsinh gene expression. (P,Q) Combined UMAP embedding as in Figure 7J with overlay of (P) marker gene expression delineating immune cell types and (Q) expression of genes involved in immunosuppression, cytotoxicity, and effector function. Color intensity represents scaled and standardized Arcsinh gene expression. (R) Representative MxIF images for tumor-infiltrating immune cells in Lrig1 versus Mist1 tumors. (S) Quantification of immune cells from MxIF. Error bars represent SEM from n = 3 animals. Dots represent fields of view. (T) Gene expression of Myc, Ctnnb1, Krt18, and Krt8 for Lrig1 and Mist1 TSCs from scRNA-seq. Library size-normalized and Arcsinh-scaled. (U) Representative IF images of MHC class I and II staining of colonic tissues from mouse lesions in the conditions of Figure 7A, compared with wild-type proximal and distal colon. n = 3 mice analyzed but n = 2 shown. Villiform metaplasias are denoted by white arrows, and dotted line demarcates border between villus and crypt compartments. (V) Selected GSEA enrichment plots generated using KEGG and Gene Ontology of Mist1 TSCs against Lrig1 TSCs. (W) Representative flow cytometry plots of DQ-OVA+/I-AI-E+ epithelial cells comparing antigen processing and presentation abilities between Lrig1 and Mist1 tumoroids. (X) Representative flow cytometry plots of CellTrace Violet peaks that depicts proliferating T cells when co-cultured with organoids derived from colonic tumors or normal tissues (+DSS) treated with or without 50 ug/ml OVA peptide. Positive control is dendritic cells used for antigen presentation. Gates denote proliferating cells plotted in Figure 7W. (Y) Organoid replating efficiency presented as number of organoids formed from 1000 single cells isolated from human ADs and SERs. Each dot represents data from a well. Error bars represent SEM from n = 7 ADs and 6 SERs. (Z) Linear regression analysis of individual SERs regressing cytotoxic cell proportions to metaplastic signature derived from scRNA-seq. (A’) qPCR analysis of antigen presentation machinery genes in 2 human AD organoid lines cultured in organoid growth media (OGM), organoid differentiating media (ODM), with or without IFN gamma. Heatmap represents log fold change in expression compared to OGM controls. n = 3 replicates performed and p values are listed in Table S4. (B’) Scatterplots of antigen presentation signature scores for normal colonic epithelial cell types, with each point representing a single cell, for (left) DIS and (right) VAL datasets. Error bars depict SEM of single cells. * < p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
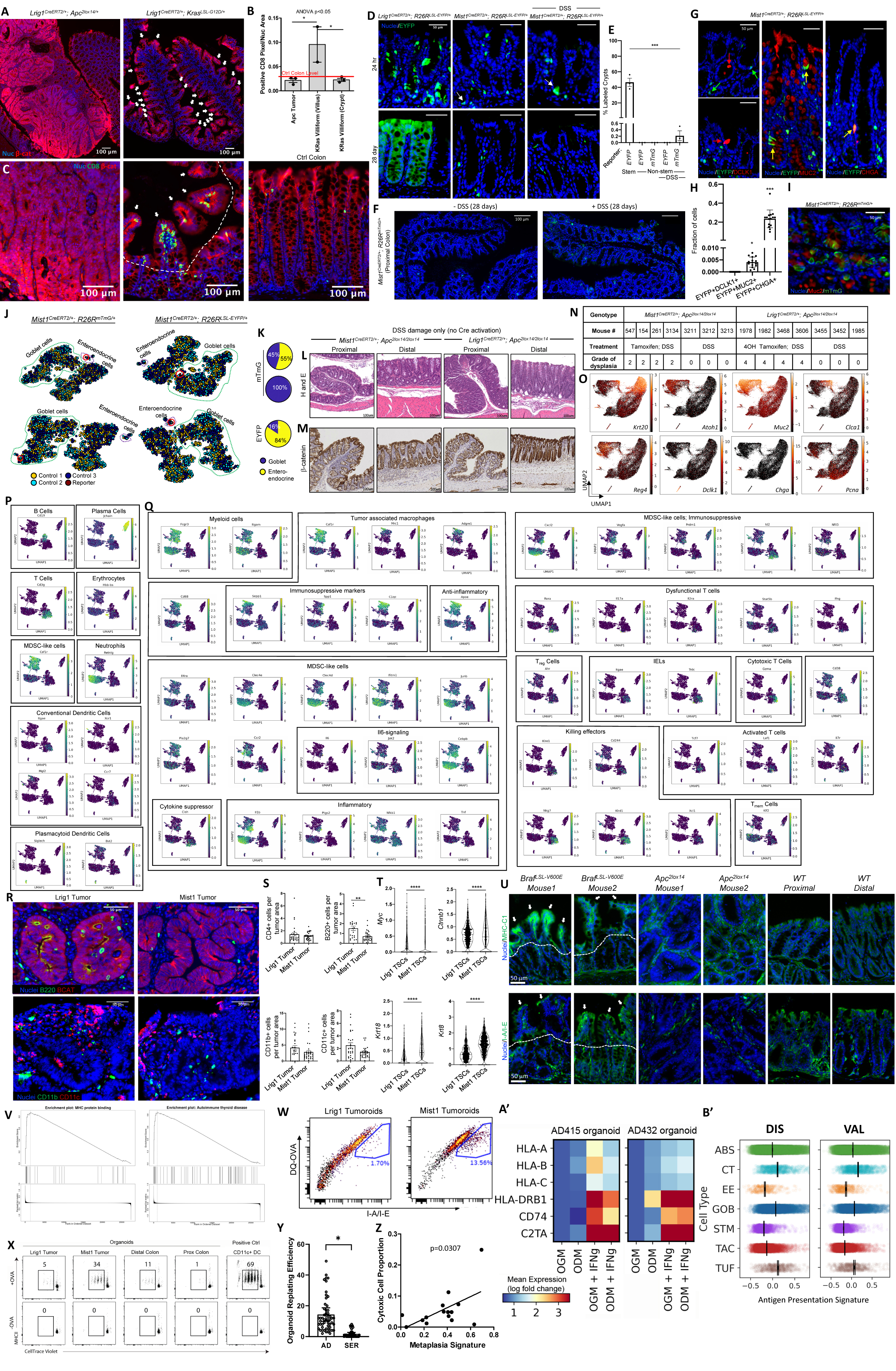{kind=link}
Highlights.
A single-cell resolution atlas of human adenomas and serrated polyps
Serrated polyps arise from metaplasia as opposed to stem cell expansion
Cytotoxic immunity in serrated polyps occurs independently of hypermutation
Distinct immune microenvironments track tumor cell-differentiation states
ACKNOWLEDGMENTS
This publication is part of the HTAN (Human Tumor Atlas Network) Consortium paper package. A list of HTAN members is available at https://humantumoratlas.org/htan-authors/. The authors wish to thank the study participants and other contributing investigators, including Jumpei Kondo, Emily Poulin, Eunyoung Choi, Reid Ness, Yu Shyr, Harvey Murff, David Pocalyko, Jeffrey Rathmell, Mary Philip, Nancy Zhang, Joke Reumers, Harrison Kiang, and Eric Eisenberg. We apologize in advance to those we have failed to acknowledge due to space constraints. This study was supported by the Human Tumor Atlas Network grant U2CCA233291 (to R.J.C., K.S.L., and M.J.S.), R01CA97386 (to W.Z.), R35CA197570 and P50CA236733 (to R.J.C.), R01DK103831 (to K.S.L.), K07CA122451 (to M.J.S.), T32LM012412 (in support of B.C.), DK123489 (to J.A.G.), U01CA215798 (in support of C.R.S.), F31DK127687 (to P.N.V.), NCI task order HHSN261100039 under contract HHSN261201500003I (to A. Regev), VA IBX000930, DOD CA160479 and DK101332, and Cancer UK 29075 (to J.R.G.) and P30CA068485 (to Vanderbilt-Ingram Cancer Center). Polyp RNA-seq funding was provided by Janssen (to M.J.S.). Cores used by this study included Survey and Biospecimen Shared Resource, TPSR (U24DK059637), DHSR, the CHTN (UM1CA183727), VANTAGE, and REDCap (UL1TR000445). R.J.C. acknowledges the generous support of the Nicholas Tierney GI Cancer Memorial Fund. A portion of the participants were studied as the result of resources and the use of facilities at the Veterans Affairs Tennessee Valley Healthcare System.
M.J.S., C.L.S., W.M.G., and K.N. receive funding from Janssen. C.L.S., M.G., and K.N. receive funding from Bristol Myers Squibb. W.M.G. receives funding from Tempus and Pavmed technologies; is a board member for Freenome, Guardant Health, and SEngine; and consults for DiaCarta. A.J.A. receives funding from Mirati Therapeutics, Deerfield, and Novo Ventures and consults for Oncorus, Arrakis Therapeutics, and Merck. G.M.B. receives funding from Palleon Pharmaceuticals, Olink Proteomics, and Takeda Oncology and is a board member for Novartis and Nektar Therapeutics. A.C.A. is a board member for Tizona Therapeutics, Compass Therapeutics, Zumutor Biologics, and ImmuneOncia and consults for iTeos Therapeutics. M.G. and K.N. receive funding from Merck and Servier. K.N. receives funding from Revolution Medicines, Evergrande Group, Pharmavite, and Merck; is a board member for Seattle Genetics and BiomX; and consults for X-Biotix Therapeutics. A. Regev is a founder of and equity holder in Celsius Therapeutics and holds equity in Immunitas Therapeutics. O.R.-R. and A. Regev are employees of Genentech. N.H. holds equity in BioNTech and consults for Related Sciences/Danger Bio.
Footnotes
DECLARATION OF INTERESTS
All other authors declare no competing interests.
INCLUSION AND DIVERSITY
We worked to ensure gender balance in the recruitment of human subjects. We worked to ensure ethnic or other types of diversity in the recruitment of human subjects. We worked to ensure that the study questionnaires were prepared in an inclusive way. We worked to ensure sex balance in the selection of non-human subjects. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper received support from a program designed to increase minority representation in science.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.cell.2021.11.031.
REFERENCES
- Aibar S, González-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, Rambow F, Marine JC, Geurts P, Aerts J, et al. (2017). SCENIC: single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alshetaiwi H, Pervolarakis N, McIntyre LL, Ma D, Nguyen Q, Rath JA, Nee K, Hernandez G, Evans K, Torosian L, et al. (2020). Defining the emergence of myeloid-derived suppressor cells in breast cancer using single-cell transcriptomics. Sci. Immunol 5, eaay6017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson AC, Joller N, and Kuchroo VK (2016). Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 44, 989–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari I, Raddatz G, Gutekunst J, Ridnik M, Cohen D, Abu-Remaileh M, Tuganbaev T, Shapiro H, Pikarsky E, Elinav E, et al. (2020). The microbiota programs DNA methylation to control intestinal homeostasis and inflammation. Nat. Microbiol 5, 610–619. [DOI] [PubMed] [Google Scholar]
- Ayyaz A, Kumar S, Sangiorgi B, Ghoshal B, Gosio J, Ouladan S, Fink M, Barutcu S, Trcka D, Shen J, et al. (2019). Single-cell transcriptomes of the regenerating intestine reveal a revival stem cell. Nature 569, 121–125. [DOI] [PubMed] [Google Scholar]
- Baitsch D, Bock HH, Engel T, Telgmann R, Müller-Tidow C, Varga G, Bot M, Herz J, Robenek H, von Eckardstein A, and Nofer JR (2011). Apolipoprotein E induces antiinflammatory phenotype in macrophages. Arterioscler. Thromb. Vasc. Biol 31, 1160–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balbinot C, Armant O, Elarouci N, Marisa L, Martin E, De Clara E, Onea A, Deschamps J, Beck F, Freund J-N, and Duluc I. (2018). The Cdx2 homeobox gene suppresses intestinal tumorigenesis through non-cell-autonomous mechanisms. J. Exp. Med 215, 911–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee A, Herring CA, Chen B, Kim H, Simmons AJ, Southard-Smith AN, Allaman MM, White JR, Macedonia MC, Mckinley ET, et al. (2020). Succinate Produced by Intestinal Microbes Promotes Specification of Tuft Cells to Suppress Ileal Inflammation. Gastroenterology 159, 2101–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, and Clevers H. (2007). Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449, 1003–1007. [DOI] [PubMed] [Google Scholar]
- Baruch EN, Youngster I, Ben-Betzalel G, Ortenberg R, Lahat A, Katz L, Adler K, Dick-Necula D, Raskin S, Bloch N, et al. (2021). Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science 371, 602–609. [DOI] [PubMed] [Google Scholar]
- Bates D, Mächler M, Bolker B, and Walker S. (2015). Fitting Linear Mixed-Effects Models Using lme4. J. Stat. Softw 67, 1–48. [Google Scholar]
- Bergen V, Lange M, Peidli S, Wolf FA, and Theis FJ (2020). Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol 38, 1408–1414. [DOI] [PubMed] [Google Scholar]
- Biton M, Haber AL, Rogel N, Burgin G, Beyaz S, Schnell A, Ashenberg O, Su C-W, Smillie C, Shekhar K, et al. (2018). T Helper Cell Cytokines Modulate Intestinal Stem Cell Renewal and Differentiation. Cell 175, 1307–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, Betts MR, Freeman GJ, Vignali DAA, and Wherry EJ (2009). Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol 10, 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bommi PV, Bowen CM, Reyes-Uribe L, Wu W, Katayama H, Rocha P, Parra ER, Francisco-Cruz A, Ozcan Z, Tosti E, et al. (2021). The transcriptomic landscape of mismatch repair-deficient intestinal stem cells. Cancer Res. 81, 2760–2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronte V, Brandau S, Chen S-H, Colombo MP, Frey AB, Greten TF, Mandruzzato S, Murray PJ, Ochoa A, Ostrand-Rosenberg S, et al. (2016). Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun 7, 12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buczacki SJA, Zecchini HI, Nicholson AM, Russell R, Vermeulen L, Kemp R, and Winton DJ (2013). Intestinal label-retaining cells are secretory precursors expressing Lgr5. Nature 495, 65–69. [DOI] [PubMed] [Google Scholar]
- Cadigan KM, and Waterman ML (2012). TCF/LEFs and Wnt signaling in the nucleus. Cold Spring Harb. Perspect. Biol 4, a007906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network (2012). Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlin LM, Yanagi K, Verhoef A, Nolte-’t Hoen EN, Yates J, Gardner L, Lamb J, Lombardi G, Dallman MJ, and Davis DM (2005). Secretion of IFN-γ and not IL-2 by anergic human T cells correlates with assembly of an immature immune synapse. Blood 106, 3874–3879. [DOI] [PubMed] [Google Scholar]
- Castello LM, Raineri D, Salmi L, Clemente N, Vaschetto R, Quaglia M, Garzaro M, Gentilli S, Navalesi P, Cantaluppi V, et al. (2017). Osteopontin at the Crossroads of Inflammation and Tumor Progression. Mediators Inflamm. 2017, 4049098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang K, Willis JA, Reumers J, Taggart MW, San Lucas FA, Thirumurthi S, Kanth P, Delker DA, Hagedorn CH, Lynch PM, et al. (2018). Colorectal premalignancy is associated with consensus molecular subtypes 1 and 2. Ann. Oncol 29, 2061–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Ramirez-Solano MA, Heiser CN, Liu Q, and Lau KS (2021). Processing single-cell RNA-seq data for dimension reduction-based analyses using open-source tools. STAR Protoc 2, 100450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi X-Z, Kim J, Lee Y-H, Lee J-W, Lee K-S, Wee H, Kim W-J, Park W-Y, Oh B-C, Stein GS, et al. (2009). Runt-related transcription factor RUNX3 is a target of MDM2-mediated ubiquitination. Cancer Res. 69, 8111–8119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H, and Batlle E. (2006). EphB/EphrinB receptors and Wnt signaling in colorectal cancer. Cancer Res. 66, 2–5. [DOI] [PubMed] [Google Scholar]
- Crockett SD, and Nagtegaal ID (2019). Terminology, Molecular Features, Epidemiology, and Management of Serrated Colorectal Neoplasia. Gastroenterology 157, 949–966. [DOI] [PubMed] [Google Scholar]
- Davenport JR, Su T, Zhao Z, Coleman HG, Smalley WE, Ness RM, Zheng W, and Shrubsole MJ (2018). Modifiable lifestyle factors associated with risk of sessile serrated polyps, conventional adenomas and hyperplastic polyps. Gut 67, 456–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Almeida Nagata DE, Chiang EY, Jhunjhunwala S, Caplazi P, Arumugam V, Modrusan Z, Chan E, Merchant M, Jin L, Arnott D, et al. (2019). Regulation of Tumor-Associated Myeloid Cell Activity by CBP/EP300 Bromodomain Modulation of H3K27 Acetylation. Cell Rep. 27, 269–281. [DOI] [PubMed] [Google Scholar]
- Dejea CM, Wick EC, Hechenbleikner EM, White JR, Mark Welch JL, Rossetti BJ, Peterson SN, Snesrud EC, Borisy GG, Lazarev M, et al. (2014). Microbiota organization is a distinct feature of proximal colorectal cancers. Proc. Natl. Acad. Sci. USA 111, 18321–18326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeStefano Shields CE, White JR, Chung L, Wenzel A, Hicks JL, Tam AJ, Chan JL, Dejea CM, Fan H, Michel J, et al. (2021). Bacterial-driven inflammation and mutant braf expression combine to promote murine colon tumorigenesis that is sensitive to immune checkpoint therapy. Cancer Discov. 11, 1792–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du L, Wang H, He L, Zhang J, Ni B, Wang X, Jin H, Cahuzac N, Mehrpour M, Lu Y, and Chen Q. (2008). CD44 is of functional importance for colorectal cancer stem cells. Clin. Cancer Res 14, 6751–6760. [DOI] [PubMed] [Google Scholar]
- Duckworth A, Glenn M, Slupsky JR, Packham G, and Kalakonda N. (2014). Variable induction of PRDM1 and differentiation in chronic lymphocytic leukemia is associated with anergy. Blood 123, 3277–3285. [DOI] [PubMed] [Google Scholar]
- Eide PW, Bruun J, Lothe RA, and Sveen A. (2017). CMScaller: an R package for consensus molecular subtyping of colorectal cancer pre-clinical models. Sci. Rep 7, 16618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearon ER, and Vogelstein B. (1990). A genetic model for colorectal tumorigenesis. Cell 61, 759–767. [DOI] [PubMed] [Google Scholar]
- Feng X, Wang H, Takata H, Day TJ, Willen J, and Hu H. (2011). Transcription factor Foxp1 exerts essential cell-intrinsic regulation of the quiescence of naive T cells. Nat. Immunol 12, 544–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fife BT, Pauken KE, Eagar TN, Obu T, Wu J, Tang Q, Azuma M, Krummel MF, and Bluestone JA (2009). Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat. Immunol 10, 1185–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox EJ, Reid-Bayliss KS, Emond MJ, and Loeb LA (2014). Accuracy of Next Generation Sequencing Platforms. Next Gener. Seq. Appl 1, 1000106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galili T. (2015). dendextend: an R package for visualizing, adjusting and comparing trees of hierarchical clustering. Bioinformatics 31, 3718–3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdes MJ, Sevinsky CJ, Sood A, Adak S, Bello MO, Bordwell A, Can A, Corwin A, Dinn S, Filkins RJ, et al. (2013). Highly multiplexed single-cell analysis of formalin-fixed, paraffin-embedded cancer tissue. Proc. Natl. Acad. Sci. USA 110, 11982–11987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldenring JR (2018). Pyloric metaplasia, pseudopyloric metaplasia, ulcer-associated cell lineage and spasmolytic polypeptide-expressing metaplasia: reparative lineages in the gastrointestinal mucosa. J. Pathol 245, 132–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldenring JR, and Mills JC (2021). Cellular Plasticity, Reprogramming, and Regeneration: Metaplasia in the Stomach and Beyond. Gastroenterology, S0016-5085(21)03708-2. [DOI] [PMC free article] [PubMed]
- Greenwald RJ, Boussiotis VA, Lorsbach RB, Abbas AK, and Sharpe AH (2001). CTLA-4 regulates induction of anergy in vivo. Immunity 14, 145–155. [DOI] [PubMed] [Google Scholar]
- Guinney J, Dienstmann R, Wang X, de Reyniès A, Schlicker A, Soneson C, Marisa L, Roepman P, Nyamundanda G, Angelino P, et al. (2015). The consensus molecular subtypes of colorectal cancer. Nat. Med 21, 1350–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulati GS, Sikandar SS, Wesche DJ, Manjunath A, Bharadwaj A, Berger MJ, Ilagan F, Kuo AH, Hsieh RW, Cai S, et al. (2020). Single-cell transcriptional diversity is a hallmark of developmental potential. Science 367, 405–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han T, Goswami S, Hu Y, Tang F, Zafra MP, Murphy C, Cao Z, Poirier JT, Khurana E, Elemento O, et al. (2020). Lineage reversion drives wnt independence in intestinal cancer. Cancer Discov. 10, 1590–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris CR, Millman KJ, van der Walt SJ, Gommers R, Virtanen P, Cournapeau D, Wieser E, Taylor J, Berg S, Smith NJ, et al. (2020). Array programming with NumPy. Nature 585, 357–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiser CN, Wang VM, Chen B, Hughey JJ, and Lau KS (2021). Automated quality control and cell identification of droplet-based single-cell data using dropkick. Genome Res. 31, 1742–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herring CA, Banerjee A, McKinley ET, Simmons AJ, Ping J, Roland JT, Franklin JL, Liu Q, Gerdes MJ, Coffey RJ, and Lau KS (2018). Unsupervised Trajectory Analysis of Single-Cell RNA-Seq and Imaging Data Reveals Alternative Tuft Cell Origins in the Gut. Cell Syst. 6, 37–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hieshima K, Imai T, Opdenakker G, Van Damme J, Kusuda J, Tei H, Sakaki Y, Takatsuki K, Miura R, Yoshie O, and Nomiyama H. (1997). Molecular cloning of a novel human CC chemokine liver and activation-regulated chemokine (LARC) expressed in liver. Chemotactic activity for lymphocytes and gene localization on chromosome 2. J. Biol. Chem 272, 5846–5853. [DOI] [PubMed] [Google Scholar]
- Hrvatin S, Deng F, O’Donnell CW, Gifford DK, and Melton DA (2014). MARIS: method for analyzing RNA following intracellular sorting. PLoS ONE 9, e89459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang S-M, Sharma G, Verma R, Byun S, Rudra D, and Im S-H (2018). Inflammation-induced Id2 promotes plasticity in regulatory T cells. Nat. Commun 9, 4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imajo M, Ebisuya M, and Nishida E. (2015). Dual role of YAP and TAZ in renewal of the intestinal epithelium. Nat. Cell Biol 17, 7–19. [DOI] [PubMed] [Google Scholar]
- Jiang M, Chen J, Zhang W, Zhang R, Ye Y, Liu P, Yu W, Wei F, Ren X, and Yu J. (2017). Interleukin-6 Trans-Signaling Pathway Promotes Immunosuppressive Myeloid-Derived Suppressor Cells via Suppression of Suppressor of Cytokine Signaling 3 in Breast Cancer. Front. Immunol 8, 1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin G, Sakitani K, Wang H, Jin Y, Dubeykovskiy A, Worthley DL, Tailor Y, and Wang TC (2017). The G-protein coupled receptor 56, expressed in colonic stem and cancer cells, binds progastrin to promote proliferation and carcinogenesis. Oncotarget 8, 40606–40619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, and Kaech SM (2007). Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity 27, 281–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiko GE, Ryu SH, Koues OI, Collins PL, Solnica-Krezel L, Pearce EJ, Pearce EL, Oltz EM, and Stappenbeck TS (2016). The Colonic Crypt Protects Stem Cells from Microbiota-Derived Metabolites. Cell 165, 1708–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, and Kang GH (2020). Evolving pathologic concepts of serrated lesions of the colorectum. J. Pathol. Transl. Med 54, 276–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein AM, Mazutis L, Akartuna I, Tallapragada N, Veres A, Li V, Peshkin L, Weitz DA, and Kirschner MW (2015). Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 161, 1187–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoop KA, McDonald KG, McCrate S, McDole JR, and Newberry RD (2015). Microbial sensing by goblet cells controls immune surveillance of luminal antigens in the colon. Mucosal Immunol. 8, 198–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komor MA, Bosch LJ, Bounova G, Bolijn AS, Delis-van Diemen PM, Rausch C, Hoogstrate Y, Stubbs AP, de Jong M, Jenster G, et al. ; NGS-ProToCol Consortium (2018). Consensus molecular subtype classification of colorectal adenomas. J. Pathol 246, 266–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo J, Huh WJ, Franklin JL, Heinrich MC, Rubin BP, and Coffey RJ (2020). A smooth muscle-derived, Braf-driven mouse model of gastrointestinal stromal tumor (GIST): evidence for an alternative GIST cell-of-origin. J. Pathol 252, 441–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh PR, and Raychaudhuri S. (2019). Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotecha N, Krutzik PO, and Irish JM (2010). Web-Based Analysis and Publication of Flow Cytometry Experiments. Curr. Protoc. Cytom 53, Unit10.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuznetsova A, Brockhoff PB, and Christensen RHB (2017). lmerTest Package: Tests in Linear Mixed Effects Models. J. Stat. Softw 82, 1–26. [Google Scholar]
- La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, Lidschreiber K, Kastriti ME, Lönnerberg P, Furlan A, et al. (2018). RNA velocity of single cells. Nature 560, 494–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, et al. (2015). PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med 372, 2509–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach JDG, Vlahov N, Tsantoulis P, Ridgway RA, Flanagan DJ, Gilroy K, Sphyris N, Vázquez EG, Vincent DF, Faller WJ, et al. ; S:CORT consortium (2021). Oncogenic BRAF, unrestrained by TGFβ-receptor signalling, drives right-sided colonic tumorigenesis. Nat. Commun 12, 3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H-O, Hong Y, Etlioglu HE, Cho YB, Pomella V, Van den Bosch B, Vanhecke J, Verbandt S, Hong H, Min J-W, et al. (2020). Lineage-dependent gene expression programs influence the immune landscape of colorectal cancer. Nat. Genet 52, 594–603. [DOI] [PubMed] [Google Scholar]
- Lee S-H, Jang B, Min J, Contreras-Panta EW, Presentation KS, Delgado AG, Piazuelo MB, Choi E, and Goldenring JR (2021). Up-regulation of Aquaporin 5 Defines Spasmolytic Polypeptide-Expressing Metaplasia and Progression to Incomplete Intestinal Metaplasia. Cell. Mol. Gastroenterol. Hepatol Published online August 25, 2021. 10.1016/J.JCMGH.2021.08.017. [DOI] [PMC free article] [PubMed]
- Leggett B, and Whitehall V. (2010). Role of the serrated pathway in colorectal cancer pathogenesis. Gastroenterology 138, 2088–2100. [DOI] [PubMed] [Google Scholar]
- Lesokhin AM, Hohl TM, Kitano S, Cortez C, Hirschhorn-Cymerman D, Avogadri F, Rizzuto GA, Lazarus JJ, Pamer EG, Houghton AN, et al. (2012). Monocytic CCR2(+) myeloid-derived suppressor cells promote immune escape by limiting activated CD8 T-cell infiltration into the tumor microenvironment. Cancer Res. 72, 876–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao G-B, Li X-Z, Zeng S, Liu C, Yang S-M, Yang L, Hu C-J, and Bai J-Y (2018). Regulation of the master regulator FOXM1 in cancer. Cell Commun. Signal 16, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieu CH, Golemis EA, Serebriiskii IG, Newberg J, Hemmerich A, Connelly C, Messersmith WA, Eng C, Eckhardt SG, Frampton G, et al. (2019). Comprehensive genomic landscapes in early and later onset colorectal cancer. Clin. Cancer Res 25, 5852–5858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lili LN, Farkas AE, Gerner-Smidt C, Overgaard CE, Moreno CS, Parkos CA, Capaldo CT, and Nusrat A. (2016). Claudin-based barrier differentiation in the colonic epithelial crypt niche involves Hopx/Klf4 and Tcf7l2/Hnf4α cascades. Tissue Barriers 4, e1214038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Herring CA, Sheng Q, Ping J, Simmons AJ, Chen B, Banerjee A, Li W, Gu G, Coffey RJ, et al. (2018). Quantitative assessment of cell population diversity in single-cell landscapes. PLoS Biol. 16, e2006687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llosa NJ, Cruise M, Tam A, Wicks EC, Hechenbleikner EM, Taube JM, Blosser RL, Fan H, Wang H, Luber BS, et al. (2015). The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 5, 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukonin I, Serra D, Challet Meylan L, Volkmann K, Baaten J, Zhao R, Meeusen S, Colman K, Maurer F, Stadler MB, et al. (2020). Phenotypic landscape of intestinal organoid regeneration. Nature 586, 275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man SM (2018). Inflammasomes in the gastrointestinal tract: infection, cancer and gut microbiota homeostasis. Nat. Rev. Gastroenterol. Hepatol 15, 721–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, Ugel S, Sonda N, Bicciato S, Falisi E, et al. (2010). Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity 32, 790–802. [DOI] [PubMed] [Google Scholar]
- Markowitz SD, and Bertagnolli MM (2009). Molecular origins of cancer: Molecular basis of colorectal cancer. N. Engl. J. Med 361, 2449–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald BD, Jabri B, and Bendelac A. (2018). Diverse developmental pathways of intestinal intraepithelial lymphocytes. Nat. Rev. Immunol 18, 514–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInnes L, Healy J, Saul N, and Großberger L. (2018). UMAP: Uniform Manifold Approximation and Projection. J. Open Source Softw 3, 861. [Google Scholar]
- McKinley ET, Sui Y, Al-Kofahi Y, Millis BA, Tyska MJ, Roland JT, Santamaria-Pang A, Ohland CL, Jobin C, Franklin JL, et al. (2017). Optimized multiplex immunofluorescence single-cell analysis reveals tuft cell heterogeneity. JCI Insight 2, e93487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinley ET, Roland JT, Franklin JL, Macedonia MC, Vega PN, Shin S, Coffey RJ, and Lau KS (2019). Machine and deep learning single-cell segmentation and quantification of multi-dimensional tissue images. BioRxiv. [DOI] [PMC free article] [PubMed]
- Mlecnik B, Bindea G, Angell HK, Maby P, Angelova M, Tougeron D, Church SE, Lafontaine L, Fischer M, Fredriksen T, et al. (2016). Integrative Analyses of Colorectal Cancer Show Immunoscore Is a Stronger Predictor of Patient Survival Than Microsatellite Instability. Immunity 44, 698–711. [DOI] [PubMed] [Google Scholar]
- Moon YW, Hajjar J, Hwu P, and Naing A. (2015). Targeting the indoleamine 2,3-dioxygenase pathway in cancer. J. Immunother. Cancer 3, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson K-F, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, et al. (2003). PGC-1a-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet 34, 267–273. [DOI] [PubMed] [Google Scholar]
- Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, De Baetselier P, and Van Ginderachter JA (2008). Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood 111, 4233–4244. [DOI] [PubMed] [Google Scholar]
- Murata K, Jadhav U, Madha S, van Es J, Dean J, Cavazza A, Wucherpfennig K, Michor F, Clevers H, and Shivdasani RA (2020). Ascl2-Dependent Cell Dedifferentiation Drives Regeneration of Ablated Intestinal Stem Cells. Cell Stem Cell 26, 377–390.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustata RC, Vasile G, Fernandez-Vallone V, Strollo S, Lefort A, Libert F, Monteyne D, Pérez-Morga D, Vassart G, and Garcia M-I (2013). Identification of Lgr5-independent spheroid-generating progenitors of the mouse fetal intestinal epithelium. Cell Rep. 5, 421–432. [DOI] [PubMed] [Google Scholar]
- Nelson RT, Boyd J, Gladue RP, Paradis T, Thomas R, Cunningham AC, Lira P, Brissette WH, Hayes L, Hames LM, et al. (2001). Genomic organization of the CC chemokine mip-3a/CCL20/larc/exodus/SCYA20, showing gene structure, splice variants, and chromosome localization. Genomics 73, 28–37. [DOI] [PubMed] [Google Scholar]
- Ogino S, and Goel A. (2008). Molecular classification and correlates in colorectal cancer. J. Mol. Diagn 10, 13–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Y-K, Franklin JL, Settle SH, Levy SE, Chung E, Jeyakumar LH, Shyr Y, Washington MK, Whitehead RH, Aronow BJ, and Coffey RJ (2005). Gene expression profile analysis of mouse colon embryonic development. Genesis 41, 1–12. [DOI] [PubMed] [Google Scholar]
- Pelka K, Hofree M, Chen JH, Sarkizova S, Pirl JD, Jorgji V, Bejnood A, Dionne D, Ge WH, Xu KH, et al. (2021). Spatially organized multicellular immune hubs in human colorectal cancer. Cell 184, 4734–4752.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petukhov V, Guo J, Baryawno N, Severe N, Scadden DT, Samsonova MG, and Kharchenko PV (2018). dropEst: pipeline for accurate estimation of molecular counts in droplet-based single-cell RNA-seq experiments. Genome Biol. 19, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poplin R, Ruano-Rubio V, DePristo MA, Fennell TJ, Carneiro MO, Van der Auwera GA, Kling DE, Gauthier LD, Levy-Moonshine A, Roazen D, et al. (2017). Scaling accurate genetic variant discovery to tens of thousands of samples. BioRxiv.
- Powell AE, Wang Y, Li Y, Poulin EJ, Means AL, Washington MK, Higginbotham JN, Juchheim A, Prasad N, Levy SE, et al. (2012). The pan-ErbB negative regulator Lrig1 is an intestinal stem cell marker that functions as a tumor suppressor. Cell 149, 146–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H, and Vilo J. (2019). g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 47 (W1), W191–W198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rex DK, Ahnen DJ, Baron JA, Batts KP, Burke CA, Burt RW, Goldblum JR, Guillem JG, Kahi CJ, Kalady MF, et al. (2012). Serrated lesions of the colorectum: review and recommendations from an expert panel. Am. J. Gastroenterol 107, 1315–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roper J, Tammela T, Cetinbas NM, Akkad A, Roghanian A, Rickelt S, Almeqdadi M, Wu K, Oberli MA, Sánchez-Rivera FJ, et al. (2017). In vivo genome editing and organoid transplantation models of colorectal cancer and metastasis. Nat. Biotechnol 35, 569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satija R, Farrell JA, Gennert D, Schier AF, and Regev A. (2015). Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol 33, 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonhoff SE, Giel-Moloney M, and Leiter AB (2004). Neurogenin 3-expressing progenitor cells in the gastrointestinal tract differentiate into both endocrine and non-endocrine cell types. Dev. Biol 270, 443–454. [DOI] [PubMed] [Google Scholar]
- Schwartz RH (2003). T cell anergy. Annu. Rev. Immunol 21, 305–334. [DOI] [PubMed] [Google Scholar]
- Scurrah CR, Simmons AJ, and Lau KS (2019). Single-Cell Mass Cytometry of Archived Human Epithelial Tissue for Decoding Cancer Signaling Pathways. Methods Mol. Biol 1884, 215–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skelly DA, Johansson M, Madeoy J, Wakefield J, and Akey JM (2011). A powerful and flexible statistical framework for testing hypotheses of allele-specific gene expression from RNA-seq data. Genome Res. 21, 1728–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southard-Smith AN, Simmons AJ, Chen B, Jones AL, Ramirez Solano MA, Vega PN, Scurrah CR, Zhao Y, Brenan MJH, Xuan J, et al. (2020). Dual indexed library design enables compatibility of in-Drop single-cell RNA-sequencing with exAMP chemistry sequencing platforms. BMC Genomics 21, 456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoler N, and Nekrutenko A. (2021). Sequencing error profiles of Illumina sequencing instruments. NAR Genomics Bioinforma 3, lqab019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallerico R, Todaro M, Di Franco S, Maccalli C, Garofalo C, Sottile R, Palmieri C, Tirinato L, Pangigadde PN, La Rocca R, et al. (2013). Human NK cells selective targeting of colon cancer-initiating cells: a role for natural cytotoxicity receptors and MHC class I molecules. J. Immunol 190, 2381–2390. [DOI] [PubMed] [Google Scholar]
- Tao Y, Kang B, Petkovich DA, Bhandari YR, In J, Stein-O’Brien G, Kong X, Xie W, Zachos N, Maegawa S, et al. (2019). Aging-like Spontaneous Epigenetic Silencing Facilitates Wnt Activation, Stemness, and BrafV600E-Induced Tumorigenesis. Cancer Cell 35, 315–328.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, and Prins P. (2015). Sambamba: fast processing of NGS alignment formats. Bioinformatics 31, 2032–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terpilowski M. (2019). scikit-posthocs: Pairwise multiple comparison tests in Python. J. Open Source Softw 4, 1169. [Google Scholar]
- Tetteh PWW, Basak O, Farin HFF, Wiebrands K, Kretzschmar K, Begthel H, van den Born M, Korving J, de Sauvage F, van Es JH, et al. (2016). Replacement of Lost Lgr5-Positive Stem Cells through Plasticity of Their Enterocyte-Lineage Daughters. Cell Stem Cell 18, 203–213. [DOI] [PubMed] [Google Scholar]
- Thorstensen L, Lind GE, Løvig T, Diep CB, Meling GI, Rognum TO, and Lothe RA (2005). Genetic and epigenetic changes of components affecting the WNT pathway in colorectal carcinomas stratified by microsatellite instability. Neoplasia 7, 99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong K, Pellón-Cárdenas O, Sirihorachai VR, Warder BN, Kothari OA, Perekatt AO, Fokas EE, Fullem RL, Zhou A, Thackray JK, et al. (2017). Degree of Tissue Differentiation Dictates Susceptibility to BRAF-Driven Colorectal Cancer. Cell Rep. 21, 3833–3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traag VA, Waltman L, and van Eck NJ (2019). From Louvain to Leiden: guaranteeing well-connected communities. Sci. Rep 9, 5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trikha P, and Carson WE 3rd. (2014). Signaling pathways involved in MDSC regulation. Biochim. Biophys. Acta 1846, 55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujikawa T, Kumar S, Borkar RN, Azimi V, Thibault G, Chang YH, Balter A, Kawashima R, Choe G, Sauer D, et al. (2017). Quantitative Multiplex Immunohistochemistry Reveals Myeloid-Inflamed Tumor-Immune Complexity Associated with Poor Prognosis. Cell Rep. 19, 203–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utting O, Teh S-J, and Teh H-S (2000). A population of in vivo anergized T cells with a lower activation threshold for the induction of CD25 exhibit differential requirements in mobilization of intracellular calcium and mitogen-activated protein kinase activation. J. Immunol 164, 2881–2889. [DOI] [PubMed] [Google Scholar]
- Van de Sande B, Flerin C, Davie K, De Waegeneer M, Hulselmans G, Aibar S, Seurinck R, Saelens W, Cannoodt R, Rouchon Q, et al. (2020). A scalable SCENIC workflow for single-cell gene regulatory network analysis. Nat. Protoc 15, 2247–2276. [DOI] [PubMed] [Google Scholar]
- Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, et al. (2013). From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinforma 43, 11.10.1–11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Flier LG, Haegebarth A, Stange DE, van de Wetering M, and Clevers H. (2009). OLFM4 is a robust marker for stem cells in human intestine and marks a subset of colorectal cancer cells. Gastroenterology 137, 15–17. [DOI] [PubMed] [Google Scholar]
- van Es JH, Sato T, van de Wetering M, Lyubimova A, Yee Nee AN, Gregorieff A, Sasaki N, Zeinstra L, van den Born M, Korving J, et al. (2012). Dll1+ secretory progenitor cells revert to stem cells upon crypt damage. Nat. Cell Biol 14, 1099–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varoquaux G, Buitinck L, Louppe G, Grisel O, Pedregosa F, and Mueller A. (2015). Scikit-learn. GetMobile Mob. Comput. Commun 19, 29–33. [Google Scholar]
- Virtanen P, Gommers R, Oliphant TE, Haberland M, Reddy T, Cournapeau D, Burovski E, Peterson P, Weckesser W, Bright J, et al. ; SciPy 1.0 Contributors (2020). SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods 17, 261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volonté A, Di Tomaso T, Spinelli M, Todaro M, Sanvito F, Albarello L, Bissolati M, Ghirardelli L, Orsenigo E, Ferrone S, et al. (2014). Cancer-initiating cells from colorectal cancer patients escape from T cell-mediated immunosurveillance in vitro through membrane-bound IL-4. J. Immunol 192, 523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang KX, and Denhardt DT (2008). Osteopontin: role in immune regulation and stress responses. Cytokine Growth Factor Rev. 19, 333–345. [DOI] [PubMed] [Google Scholar]
- Wang K, Li M, and Hakonarson H. (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells AD, Walsh MC, Sankaran D, and Turka LA (2000). T cell effector function and anergy avoidance are quantitatively linked to cell division. J. Immunol 165, 2432–2443. [DOI] [PubMed] [Google Scholar]
- Wolf FA, Angerer P, and Theis FJ (2018). SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong L, Xu K, Tian K, Shao Y, Tang L, Gao G, Zhang M, Jiang T, and Zhang QC (2019). SCALE method for single-cell ATAC-seq analysis via latent feature extraction. Nat. Commun 10, 4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Park CS, Mamonkin M, and Lacorazza HD (2009). Transcription factor ELF4 controls the proliferation and homing of CD8+ T cells via the Krüppel-like factors KLF4 and KLF2. Nat. Immunol 10, 618–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, and Wang K. (2015). Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat. Protoc 10, 1556–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Farraye FA, Mack C, Posnik O, and O’Brien MJ (2004). BRAF and KRAS Mutations in hyperplastic polyps and serrated adenomas of the colorectum: relationship to histology and CpG island methylation status. Am. J. Surg. Pathol 28, 1452–1459. [DOI] [PubMed] [Google Scholar]
- Youn J-I, Nagaraj S, Collazo M, and Gabrilovich DI (2008). Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J. Immunol 181, 5791–5802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YG, Wu S, Lu R, Zhou D, Zhou J, Carmeliet G, Petrof E, Claud EC, and Sun J. (2015). Tight junction CLDN2 gene is a direct target of the vitamin D receptor. Sci. Rep 5, 10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Li Z, Skrzypczynska KM, Fang Q, Zhang W, O’Brien SA, He Y, Wang L, Zhang Q, Kim A, et al. (2020). Single-Cell Analyses Inform Mechanisms of Myeloid-Targeted Therapies in Colon Cancer. Cell 181, 442–459.e29. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Xu B, Minn A, and Zhang NR (2020). DENDRO: genetic heterogeneity profiling and subclone detection by single-cell RNA sequencing. Genome Biol. 21, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C, Sakuishi K, Xiao S, Sun Z, Zaghouani S, Gu G, Wang C, Tan DJ, Wu C, Rangachari M, et al. (2015). An IL-27/NFIL3 signalling axis drives Tim-3 and IL-10 expression and T-cell dysfunction. Nat. Commun 6, 6072. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Oncoplot representation of the mutational landscape of 281 polyp specimens detected through bulk targeted sequencing and mutational variant calling with layout similar to Figure 1C. (B) Representative IHC for MLH1 in AD and SER. (C) Quantification of MLH1 gene expression in AD and SER from scRNA-seq of polyp-specific cells compared to the same analysis of MSI-H CRC cells (red line - mean). n = 29 for AD and 19 for SER. Each point represents a polyp, averaged over normalized and scaled Arcsinh-transformed single-cell counts. (D) UMAP representation of epithelial scRNA-seq data, color coded by specimen to depict interspecimen heterogeneity, examining (top) DIS and (bottom) VAL sets, and (left) raw count-based and (right) SCENIC regulon-based.
(A) UMAP of regulon-based epithelial cell scRNA-seq data overlaid with cell type-specific genes. Color intensity represents scaled and standardized Arcsinh gene expression. (B) UMAP of (left) DIS and (right) VAL datasets color overlaid with HP subtypes. (C) Scatterplots of normalized (left) ASC or (right) SSC representation per tissue and polyp subtype. Points represent individual specimens. Error bars represent SEM of n = 29 for AD, n = 19 for SER, and n = 66 for NL. Coloring of points indicates (top) polyp subtype or (bottom) DIS or VAL dataset. (D) Scatterplots of signature scores by cell type with each point representing a single cell for (top) DIS and (bottom) VAL datasets. Error bars depict SEM of single cells. Not all post hoc tests shown. (E) Regulon-based UMAP of epithelial cell scRNA-seq data overlaid with CytoTRACE scores for (top) DIS and (bottom) VAL datasets. (F) Regulon-based UMAPs of (top) ASC and (bottom) SSC-gated scRNA-seq data with colored label overlays indicating Leiden subcluster, cohort, polyp subtype, and specimen ID. ASC Leiden subcluster 0 is relatively enriched for WNT stemness terms. SSC Leiden subcluster 2 exhibits variation attributed to mitochondrial gene expression and lower quality. (G) Expanded TF target network created from normal and pre-cancer cells organized into super-regulons derived from shared targets for AD-derived normals, ASCs, SER-derived normals, and SSCs. Color overlays for each TF node are averaged and normalized regulon enrichment scores, while edge opacities are the inferred TF-target weightings. (H) Heatmap representation of gene sets derived from the scRNA-seq applied to bulk RNA-seq data of colonic polyps (n = 36 tubular, 22 tubulovillous, 8 SSLs). Inset circle indicates the fraction of specimens presented with gene expression and color intensity represents mean scaled and standardized Arcsinh gene expression. *p < 0.05, **p < 0.01, ***p < 0.001.
(A-D) Additional representative multiplex images of colonic polyp subtypes for (A) SOX9, (B) OLFM4, (C) CDX2, and (D) MUC5AC. White outline demarcates HP location. (E) MUC5AC staining for different GCHPs. (F) Low-magnification view of MVHPs and SSLs for MUC5AC staining. (G) MUC5AC staining of non-neoplastic colonic epithelium: (row 1) normal, (row 2) tumor adjacent, and (row 3) ulcerative colitis. (H) p-Creode trajectories inferred from epithelial scRNA-seq data, for both (top) DIS and (bottom) VAL cohorts. Overlay of genes relevant to developmental lineages. Node size represents the proportion of cells and overlay color intensity represents mean scaled and standardized Arcsinh gene expression. (I) Additional RNA velocity maps for normal colonic tissues and polyps on combined UMAP embedding for (top) DIS and (bottom) VAL datasets. Vectors inferring average cell state transitions within each specimen are shown as black arrows. Colored points represent cells derived from the individual specimen, with gray points representing all other cells. (J) Representative genetic phylogenies inferred through DENDRO with each tree representing a specimen for colonic polyp subtypes. Hierarchical clusters demarcated by dotted boxes and cell clusters denoted by leaf node color. Tree height is derived from a beta-binomial adjusted genetic divergence between single cells and shows the evolutionary time before splits in phylogeny. ABS – absorptive, ASC – adenoma specific cells, CT – crypt top, EE – enteroendocrine, GOB – goblet, STM – stem, SSC – serrated specific cells, TAC – transit amplifying cells, TUF – tuft. (K) Summary metric for phylogenetic analysis depicting the minimum genetic divergence distance between polyp-specific cells (ASC or SSC) and stem cells. n = 23 AD, and 11 SER specimens. Error bars depict SEM of individual libraries. *p < 0.05.
(A) Oncoplot representation of the mutational landscape of CRCs detected through exome sequencing, and (left) mutational variant calling or (right) somatic mutation calling. Total number of mutations detected per specimen represented as bar plot (top) and different type of mutations color-coded. Important genes to CRC are presented and grouped into pathways (dark gray boxes). Proportion of gene and pathway mutation within CRC subtypes (combined) summarized as table. (B) Representative H&E of each CRC subtype at low and high magnification views of the corresponding colored insets. (C) Scatterplots of signature scores by tumor-specific cells per subtype, with each point representing a single cell, for (top) VUMC and (bottom) Broad datasets. Error bars depict SEM of single cells. (D) Heatmap representation of top biologically relevant and differentially expressed genes, for Broad (MSS, n = 28; MSI-H, n = 32) and Samsung Medical Center (SMC) (CMS1, n = 5; CMS2, n = 8; CMS3, n = 4; CMS4, n = 6) single-cell datasets. Inset circle indicates prevalence within defined single-cell populations and color intensity represents scaled and standardized Arcsinh gene expression. (E,F) Expanded TF target network created from (E) VUMC or (F) Broad tumor-specific cells, organized into super-regulons derived from clustering of shared targets. Color overlays for each TF node are averaged and normalized regulon enrichment scores, while edge opacities are the inferred TF-target weightings. VUMC regulon values are normalized to tumor-derived normal cells while Broad values are unnormalized due to lack of detected tumor-resident normal cells. *p < 0.05, **p < 0.01, ***p < 0.001.
(A) Oncoplot visualizing genomic tracks for 276 assayed TCGA CRC specimens. Each row represents unsorted mutational status of key pathway genes. Each column represents an individual specimen with hypermutation status noted. (B) Heatmap representation of gene signatures identified by scRNA-seq of pre-cancers applied to bulk RNA-seq of TCGA CRC specimens (CMS1, n=8; CMS2, n=26; CMS3, n=10; CMS4, n=19). Inset circle on summary heatmap (left) indicates the fraction of specimens presented with gene expression and color intensity represents mean (left) or individual (right) scaled and standardized Arcsinh gene expression. (C) Representative stitched MxIF scans of OLFM4 in 3 MSS and 3 MSI-H CRCs. Black rectangles in the restitched image represent fields of views that were not scanned, due to these areas being empty (glass) or of low tissue quality. (D) Staining for various metaplastic markers of 4 CRCs from a tissue microarray. Red outline represents the MLH1 low area in CRC 4.
(A) Regulon-based UMAP representation of non-epithelial cell populations color overlaid with tissue and tumor subtype. (B) Feature-selected, count-based UMAP representation of lymphocyte subtypes, color overlaid with (left) tissue and tumor subtype and (right) cell type defined by clustering and marker gene expression. CD4 – CD4+ T cells, CD8 – CD8+ T cells, CYC – cycling T cells, GD – Gamma delta (γδ) T cells, NK – natural killer cells, T REG – regulatory T cells. (C) Heatmap representation of feature-selected genes used to generate UMAP in B. Inset circle indicates prevalence within defined single-cell populations and color intensity represents scaled and standardized Arcsinh gene expression. (D) Comprehensive scatterplots of normalized cell type representation per tissue, (top) polyp and (middle) cancer subtypes. Points represent individual specimens. Error bars represent SEM of n = 28 for AD, n = 17 for SER, n = 66 for NL, n = 33 for MSS, and n = 34 for MSI-H. Plots in the third row colored by VUMC or Broad datasets. (E) Scatterplots of CD8+ T cell cytotoxicity and exhaustion signatures, with each point representing a single cell for (top) polyp and (bottom) CRC. Error bars depict SEM of single cells. (F) Additional representative MxIF images of CD8+ cells in multiple ADs and SERs. (G) Quantification of MxIF images of marker positive cells in epithelial or stromal compartments normalized to total number of cells in each compartment. Violin plots with median as white circles and quartiles as candles for n = 20 polyps per type. (H) Quantification as in G for CD8+ T cells in the epithelial compartment of TAs separated by mutational burden for n = 6 high (> 1500 mutations) versus n = 7 low (< 1500 mutations). (I) Additional representative MxIF images of CD68+ cells and MUC5AC+ in multiple ADs and SERs. (J,K) Additional representative MxIF scans of intratumoral heterogeneous regions within CRCs (OLFM4+ stem regions versus MUC5AC+ metaplastic regions), and additional representative MxIF images of CD8+ and CD3+ cells within each of the stem and metaplastic regions of (J) MSI-H CRCs and (K) MSS CRCs with only stem regions. (L) Quantification of marker-positive pixels per tumor area in stem versus metaplastic regions in MxIF scans of n = 15 MSS and n = 10 MSI-H CRCs, presented as violin plots with median as solid lines and quartiles as dotted lines. *p < 0.05, **p < 0.01, ***p < 0.001.
(A) Representative macroscopic IF images depicting a Lrig1CreERT2/+; Apc2lox14/+ tumor and Lrig1CreERT2/+; KrasLSL-G12D/+ proximal villiform metaplasia (white arrows). (B) Quantification of CD8-positive pixels per nuclear area in Apc-driven tumor and the villus and crypt compartments of Kras-driven villiform metaplasias. Red line denotes the mean level detected in adjacent normal colon in Kras mice. Error bars represent SEM from n = 3 animals per group. (C) IF images of CD8+ T cells in an Apc-driven tumor overexpressing β-catenin, Kras-driven villiform metaplasias (white arrows), and control colon. Dotted line demarcates border between villus and crypt compartments. (D) Representative IF images of short-term and long-term (rows: 24 hours and 28 days, respectively) lineage tracing in the colon using Lrig1CreERT2/+;R26RLSL-EYFP/+, Mist1CreERT2/+;R26RLSL-EYFP/+, and Mist1CreERT2/+;R26RLSL-EYFP/+ with DSS mouse models (columns). (E) Quantification of lineage-traced glands at 28 days at homeostasis or with DSS-damage from stem (Lrig1) and non-stem (Mist1) cells using different reporters. Error bars represent SEM from n = 3 animals for each condition. (F) Representative IF images of long-term (28 days) lineage tracing in the proximal colon of Mist1CreERT2/+; R26RLSL-mTmG/+ with or without DSS. (G) Representative IF images of reporter co-expression with cell-type-specific markers, DCLK1 for tuft cells, MUC2 for goblet cells, and CHGA for enteroendocrine cells, after 24 hours of lineage tracing in Mist1CreERT2/+;R26RLSL-EYFP/+ mouse colons. Arrows point to single positive (green/red) or double positive (yellow) cells. (H) Quantification of double-positive cells compared to single positive cells for each marker. Each dot represents a field of view. Error bars represent SEM from n = 3 animals. (I) Representative IF image of reporter co-expression with MUC2 in Mist1CreERT2/+; R26RmTmG/+ 10 days after initiation of lineage tracing at homeostasis. (J) Combined t-SNE embedding of bulk RNA sequenced Mist1 reporter (mTmG – left or EYFP – right)-expressing cells (solid circle in red) with n = 3 reference murine colonic scRNA-seq dataset (colored dots from different mice). Goblet (green) and enteroendocrine (magenta) cell populations are delineated by dotted lines. (K) Quantification of Mist1 reporter (EYFP or mTmG)-expressing cells co-embedding with different cell types using boot-strapped t-SNE runs in J. n = 3 independent experiments. (L,M) Representative H&E (L) and b-catenin IHC (M) of control (uninduced) normal colonic tissues as in Figures 7D and 7E. (N) Histological scoring of tissues of multiple biological replicates of Mist1 and Lrig1 tumors along with uninduced controls. 0: none; 1: unicrypt; 2: low; 3: high, 4: advanced. (O) Combined UMAP embedding as in Figure 7F with overlays of key marker gene expression delineating epithelial cell types and states. Color intensity represents scaled and standardized Arcsinh gene expression. (P,Q) Combined UMAP embedding as in Figure 7J with overlay of (P) marker gene expression delineating immune cell types and (Q) expression of genes involved in immunosuppression, cytotoxicity, and effector function. Color intensity represents scaled and standardized Arcsinh gene expression. (R) Representative MxIF images for tumor-infiltrating immune cells in Lrig1 versus Mist1 tumors. (S) Quantification of immune cells from MxIF. Error bars represent SEM from n = 3 animals. Dots represent fields of view. (T) Gene expression of Myc, Ctnnb1, Krt18, and Krt8 for Lrig1 and Mist1 TSCs from scRNA-seq. Library size-normalized and Arcsinh-scaled. (U) Representative IF images of MHC class I and II staining of colonic tissues from mouse lesions in the conditions of Figure 7A, compared with wild-type proximal and distal colon. n = 3 mice analyzed but n = 2 shown. Villiform metaplasias are denoted by white arrows, and dotted line demarcates border between villus and crypt compartments. (V) Selected GSEA enrichment plots generated using KEGG and Gene Ontology of Mist1 TSCs against Lrig1 TSCs. (W) Representative flow cytometry plots of DQ-OVA+/I-AI-E+ epithelial cells comparing antigen processing and presentation abilities between Lrig1 and Mist1 tumoroids. (X) Representative flow cytometry plots of CellTrace Violet peaks that depicts proliferating T cells when co-cultured with organoids derived from colonic tumors or normal tissues (+DSS) treated with or without 50 ug/ml OVA peptide. Positive control is dendritic cells used for antigen presentation. Gates denote proliferating cells plotted in Figure 7W. (Y) Organoid replating efficiency presented as number of organoids formed from 1000 single cells isolated from human ADs and SERs. Each dot represents data from a well. Error bars represent SEM from n = 7 ADs and 6 SERs. (Z) Linear regression analysis of individual SERs regressing cytotoxic cell proportions to metaplastic signature derived from scRNA-seq. (A’) qPCR analysis of antigen presentation machinery genes in 2 human AD organoid lines cultured in organoid growth media (OGM), organoid differentiating media (ODM), with or without IFN gamma. Heatmap represents log fold change in expression compared to OGM controls. n = 3 replicates performed and p values are listed in Table S4. (B’) Scatterplots of antigen presentation signature scores for normal colonic epithelial cell types, with each point representing a single cell, for (left) DIS and (right) VAL datasets. Error bars depict SEM of single cells. * < p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Data Availability Statement
The raw single-cell RNA sequencing (scRNA-seq), final QC-filtered data for analysis, as well as all raw imaging data generated from this study are available on the HTAN data portal: https://data.humantumoratlas.org/. This paper also analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
All original code used to process scRNA-seq data has been deposited at https://github.com/Ken-Lau-Lab/STAR_Protocol and is described in detail in (Chen et al., 2021). A code repository containing the analysis of post-processed sequencing data, as performed in this study, can be found at https://github.com/Ken-Lau-Lab/STAR_Methods. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| Reagent or resource | Source | Identifier |
|---|---|---|
| Biological samples | ||
|
| ||
| COLON MAP (Polyp) | See Experimental Model and Subject Details; Data and Code Availability | Synapse: syn23564801 Synapse: syn23630431 Synapse: syn23520239 HTAN Data Portal: HTA11; https://humantumoratlas.org/HTA11 |
| CHTN TMA (CRC) | See Experimental Model and Subject Details; Data and Code Availability | syn21050481: https://doi.org/10.7303/syn21050481 |
| TCPS (Polyp) | See Experimental Model and Subject Details; Data and Code Availability | syn21050481: https://doi.org/10.7303/syn21050481 |
|
| ||
| Critical commercial reagents | ||
|
| ||
| Muc2, F-2 clone, A488 dye, antibody | Santa Cruz | Catalog: sc-515032 AF488; RRID: AB_2815005 |
| Collagen Peptide, R-CHP clone, Cy3 dye, antibody | 3Helix | Catalog: RED300 |
| SNA, Lectin clone, Cy5 dye, antibody | Vector | Catalog: CL-1305-1 |
| CD11B, C67F154 clone, A488 dye, antibody |
Thermo Fisher | Catalog: 53-0196-82; RRID: AB_2637196 |
| CD45, 2D-1 clone, A546 dye, antibody | Santa Cruz | Catalog: sc-1187 AF546; RRID: AB_627073 |
| CD20, D-10 clone, A647 dye, antibody | Santa Cruz | Catalog: sc-393894 AF647 |
| PCNA, PC-10 clone, A488 dye, antibody | Cell Signaling | Catalog: 8580S; RRID: AB_11178664 |
| B-catenin, 12F751 clone, 550 dye, antibody | Vanderbilt Antibody and Protein Resource | Catalog: In-House |
| p-STAT3, D3A7 clone, A647 dye, antibody | Cell Signaling | Catalog: 4324S; RRID: AB_10694637 |
| pEGFR, EP774Y clone, A488 dye, antibody | Abcam | Catalog: ab205827 |
| CgA, C-12 clone, A546 dye, antibody | Santa Cruz | Catalog: sc-393941; RRID: AB_2801371 |
| CD4, EPR6855 clone, A647 dye, antibody | Abcam | Catalog: ab196147 |
| Cox2, D5H5 clone, A488 dye, antibody | Cell Signaling | Catalog: 13596S; RRID: AB_2798270 |
| CD3d, EP4426 clone, A555 dye, antibody | Abcam | Catalog: ab208514; RRID: AB_2728789 |
| HLA-A, EP1395Y clone, A647 dye, antibody | Abcam | Catalog: ab199837; RRID: AB_2728798 |
| PanCK, AE1/AE3 clone, A488 dye, antibody | Thermo Fisher | Catalog: 53-9003-82; RRID: AB_1834350 |
| OLFM4, D1E4M clone, A555 dye, antibody | Cell Signaling | Catalog: 14369S; RRID: AB_2798465 |
| CD8, C8/114B clone, A647 dye, antibody | Biolegend | Catalog: 372906; RRID: AB_2650712 |
| Alpha-actinin, EPR2533(2) clone, A488 dye, antibody | Abcam | Catalog: ab198608 |
| CD68, KP1 clone, A546 dye, antibody | Santa Cruz | Catalog: sc-20060 AF546; RRID: AB_2891106 |
| NaKATPase, EP1845Y clone, A647 dye, antibody | Abcam | Catalog: ab198367 |
| Vimentin, E-5 clone, A488 dye, antibody | Santa Cruz | Catalog: sc-373717 AF488; RRID: AB_10917747 |
| Sox9, EPR14335 clone, A555 dye, antibody | Abcam | Catalog: ab202516 |
| FOXP3, 206D clone, A647 dye, antibody | Biolegend | Catalog: 320114; RRID: AB_439754 |
| Lysozyme, E-5 clone, A488 dye, antibody | Santa Cruz | Catalog: sc-518012 AF488; RRID: AB_2889359 |
| SMA, 1A4 clone, Cy3 dye, antibody | Millipore Sigma | Catalog: C6198-100UL; RRID: AB_476856 |
| ERBB2, EPR19547 clone, A647 dye, antibody | Abcam | Catalog: ab225510; RRID: AB_2889201 |
| P-p44/42 MAPK, Rabbit Monoclonal antibody | Cell Signaling | Catalog: 4370; RRID: AB_2315112 |
| MUC5AC, Rabbit Monoclonal antibody | Cell Signaling | Catalog: 61193; RRID: AB_2799603 |
| CDX2, Rabbit Monoclonal antibody | Cell Signaling | Catalog: 12306; RRID: AB_2797879 |
| Midkine, Rabbit Monoclonal antibody | Abcam | Catalog: ab52637; RRID: AB_880698 |
| YAP, Rabbit Monoclonal antibody | Cell Signaling | Catalog: 14074; RRID: AB_2650491 |
| MLH1, Rabbit Monoclonal antibody | Abcam | Catalog: Ab92312; RRID: AB_2049968 |
| EPCAM antibody | Santa Cruz | Catalog: Sc-53532; RRID: AB_2277892 |
| GFP antibody | Novus | Catalog: NB600-308SS; RRID: AB_10005904 |
| DCAMKL1 antibody | Abcam | Catalog: ab109029; RRID: AB_10864128 |
| CHGA (C20) antibody | Santa Cruz | Catalog: sc1488; RRID: AB_2276319 |
| MUCIN2 (H-330) antibody | Santa Cruz | Catalog: sc15334; RRID: AB_2146667 |
| CD3 (Sp7) antibody | Thermo Fisher | Catalog: RM-9107-50 |
| CD8 (4SM15) antibody | Invitrogen | Catalog: 14-0808-80; RRID: AB_2572860 |
| CD4 (4SM95) antibody | Invitrogen | Catalog: 14-9766-80; RRID: AB_2573007 |
| CD11b-AF647 antibody | Abcam | Catalog: ab204471; RRID: AB_204471 |
| CD11c antibody | Biolegend | Catalog: 117301; RRID: AB_313770 |
| CD45/B220-AF647 antibody | Biolegend | Catalog: 103228 |
| Hoechst 33342 | Invitrogen | Catalog: H3570 |
| Ia/le (M5/114.15.2) | Biolegend | Catalog: 107601 |
| DQ-Ovalbumin | Thermo Fisher | Catalog: D12053 |
| Ovalbumin peptide | Anaspec | Catalog: OVA323-339 |
| CD3-PerCP/Cy5.5, clone 145-2C11 | Biolegend | Catalog: 100328; RRID: AB_893318 |
| CD4-Apc-Cy7, clone GK1.5 | Biolegend | Catalog: 100414; RRID: AB_312699 |
| MHCII-PE-Cy7, clone M5/144.15.2 | Biolegend | Catalog: 107629; RRID: AB_2290801 |
| CD69-FITC, clone H1.2F3 | eBioscience | Catalog: 11-0691-82; RRID: AB_465119 |
| CD45-BV785, clone 30-F11 | Biolegend | Catalog: 103149; RRID: AB_2564590 |
| Ia/Ie-AF647 | Biolegend | Catalog: 10760 |
| ROCK inhibitor | STEMCELL Technologies | Catalog: Y-27632 |
| Matrigel | Corning | Catalog: 356231 |
| RNase A | Thermo Fisher | Catalog: EN0531 |
| Lysing Matrix E | MP Bio | Catalog: 116914100 |
| Mouse Intesticult | STEMCELL Technologies | Catalog: 06005 |
| Gastrin I | Sigma-Aldrich | Catalog: 39024-57-2 |
| TrypLE Express | Thermo Fisher | Catalog: 12604013 |
| N-acetyl-L-cysteine | Sigma-Aldrich | Catalog: A9165 |
| H2O2 | Sigma-Aldrich | Catalog: 216763 |
| A83-01 | Tocris | Catalog: 2939 |
| IGF-1 | Biolegend | Catalog: 590904 |
| FGF-2 | Thermo Fisher | Catalog: PHG0024 |
| Primocin | InvivoGen | Catalog: am-pm-05 |
| Human IntestiCult (OGM) | STEMCELL Technologies | Catalog: 06010 |
| Human IntestiCult (ODM) | STEMCELL Technologies | Catalog: 100-0214 |
| Human IFN gamma | Biolegend | Catalog: 570206 |
| CD8-alpha, 53-6.7 clone, antibody | Biolegend | Catalog: 100711; RRID: AB_312750 |
| MHC class I antibody, clone ER-HR 52 | Abcam | Catalog: ab15681; RRID: AB_302030 |
| MHC class II (I-A/I-E) antibody, clone (M5/114.15.2) | Thermofisher | Catalog: 14-5321-82; RRID: AB_467561 |
| RT Probe: HLA_A_F | Sigma | AGATACACCTGCCATGTGCAGC |
| RT Probe: HLA_A_R | Sigma | GATCACAGCTCCAAGGAGAACC |
| RT Probe: HLA_B_F | Sigma | CTGCTGTGATGTGTAGGAGGAAG |
| RT Probe: HLA_B_R | Sigma | GCTGTGAGAGACACATCAGAGC |
| RT Probe: HLA_C_F | Sigma | GGAGACACAGAAGTACAAGCGC |
| RT Probe: HLA_C_R | Sigma | ACATCCTCTGGAGGGTGTGAGA |
| RT Probe: CD74_F | Sigma | AAGCCTGTGAGCAAGATGCGCA |
| RT Probe: CD74_R | Sigma | AGCAGGTGCATCACATGGTCCT |
| RT Probe: C2TA_F | Sigma | CTACTTCAGGCAGCAGAGGAGA |
| RT Probe: C2TA_R | Sigma | GCTGTGTCTTCCGAGGAACTTC |
| RT Probe: HLA-DRB1_F | Sigma | GAGCAAGATGCTGAGTGGAGTC |
| RT Probe: HLA-DRB1_R | Sigma | CTGTTGGCTGAAGTCCAGAGTG |
| RT Probe: GAPDH_F | Sigma | GTCTCCTCTGACTTCAACAGCG |
| RT Probe: GAPDH_R | Sigma | ACCACCCTGTTGCTGTAGCCAA |
|
| ||
| Deposited data | ||
|
| ||
| TCGA (CRC) | (Cancer Genome Atlas Network, 2012) | TCGA GDAC Firehose: COADREAD |
| SMC (CRC) | (Lee et al., 2020) |
GEO: GSE132465 |
| Broad (CRC) | (Pelka et al., 2021) | HTAN Data Portal: HTA1 |
|
| ||
| Software and algorithms | ||
|
| ||
| pCreode | (Herring et al., 2018) | https://github.com/Ken-Lau-Lab/pCreode |
| Scanpy | (Wolf et al., 2018) | https://github.com/theislab/scanpy |
| Pegasus | Klarman Cell Observatory | https://github.com/klarman-cell-observatory/pegasus |
| pySCENIC | (Aibar et al., 2017) | https://github.com/aertslab/pySCENIC |
| CMScaller | (Eide et al., 2017) | https://github.com/peterawe/CMScaller |
| CytoTRACE | (Gulati et al., 2020) | https://cytotrace.stanford.edu |
| CMSclassifier | (Guinney et al., 2015) | https://github.com/Sage-Bionetworks/CMSclassifier |
| Seaborn | Seaborn | https://github.com/mwaskom/seaborn |
| cBioPortal | (Cerami et al., 2012; Gao et al., 2013) | https://www.cbioportal.org/ |
| Matplotlib | Matplotlib | https://github.com/matplotlib/matplotlib |
| GATK4 | (Poplin et al., 2017) | https://gatk.broadinstitute.org/hc/en-us |
| DENDRO | (Zhou et al., 2020) | https://github.com/zhouzilu/DENDRO |
| dropkick | (Heiser et al., 2021) | https://github.com/Ken-Lau-Lab/dropkick |
| DropEst | (Petukhov et al., 2018) | https://github.com/kharchenkolab/dropEst |
| STAR | (Dobin et al., 2013) | https://github.com/alexdobin/STAR |
| Cytoscape | (Shannon et al., 2003) | https://cytoscape.org/ |
| g:Profiler | (Raudvere et al., 2019) | https://biit.cs.ut.ee/gprofiler/ |
| Scipy | (Virtanen et al., 2020) | https://scipy.org/ |
| Sinto | Sinto | https://github.com/timoast/sinto |
| Dendextend | (Galili, 2015) | https://github.com/talgalili/dendextend |
| Numpy | (Harris et al., 2020) | https://numpy.org/ |
| Pandas | Pandas | https://pandas.pydata.org/ |
| BWA | (Li and Durbin, 2009) | https://sourceforge.net/projects/maq/ |
| ANNOVAR | (Wang et al., 2010; Yang and Wang, 2015) | https://github.com/WGLab/doc-ANNOVAR |
| Picard | Broad Institute | https://broadinstitute.github.io/picard/ |
| Sambamba | (Tarasov et al., 2015) | https://github.com/biod/sambamba |
| lme4 | (Bates et al., 2015) | https://github.com/lme4/lme4 |
| lmerTest | (Kuznetsova et al., 2017) | https://github.com/runehaubo/lmerTestR |
| emmeans | emmeans | https://github.com/rvlenth/emmeans |
| Cytobank | (Kotecha et al., 2010) | https://www.cytobank.org/ |
| MANDO | (McKinley et al., 2019) | https://github.com/Coffey-Lab/CellSegmentation |
| UMAP | (McInnes et al., 2018) | https://github.com/lmcinnes/umap |
| Dask | Dask | https://dask.org/ |
| Harmony | (Korsunsky et al., 2019) | https://github.com/immunogenomics/harmony |
| Scikit-posthoc | (Terpilowski, 2019) | https://scikit-posthocs.readthedocs.io/en/latest/ |
| GSEA Webapp | (Mootha et al., 2003; Subramanian et al., 2005) | https://www.gsea-msigdb.org/gsea/index.jsp |