

Abstract
TnSeq, or sequencing of transposon-insertion libraries, has proven to be a valuable method for probing the functions of genes in a wide range of bacteria. TnSeq has found many applications for studying genes involved in core functions (such as cell division or metabolism), stress response, virulence, etc., as well as to identify potential drug targets. Two of the most commonly used transposons in practice are Himar1, which inserts randomly at TA dinucleotides, and Tn5, which can insert more broadly throughout the genome. These insertions cause putative gene-function disruption, and clones with insertions in genes that cannot tolerate disruption (in a given condition) are eliminated from the population. Deep-sequencing can be used to efficiently profile the surviving members, with insertions in genes that can be inferred to be non-essential. Data from TnSeq experiments (i.e. transposon insertion counts at specific genomic locations) is inherently noisy, making rigorous statistical analysis (e.g. quantifying significance) challenging. In this paper, we describe Transit, a Python-based software package for analyzing TnSeq data that combines a variety of data processing tools, quality assessment methods, and analytical algorithms for identifying essential (or conditionally essential) genes.
Keywords: essentiality, transposon mutagenesis, TnSeq, statistical analysis, software
1. Introduction
TnSeq, or sequencing of transposon-insertion libraries, has proven to be a valuable method for probing the functions of genes in bacteria [1–3]. Libraries are generated by random insertion of a transposon throughout a genome, creating a pool of mutants. DNA from bacteria grown in a particular condition is extracted, the junctions between transposon and adjacent genomic region are amplified by PCR, the samples are sequenced by high-throughput sequencing, and insertions at individual sites are counted. The pattern of insertion counts can be used to infer the difference between essential (ES) and non-essential (NE) regions; regions that tolerate insertions are generally considered non-essential, while regions lacking insertions are taken as evidence of essentiality. In addition to the binary distinction of essential vs non-essential, some genes can exhibit a partial reduction of insertion counts, which can be interpreted as a growth defect (GD) caused by disruption of the gene, and indeed quantitative changes in insertion counts in has been shown to reflect changes in fitness E. coli [1]. There are several variations of sample preparation protocols currently in use, going by names such as TraDIS [4], HITS [5], and InSeq [6], which differ based on the transposon used, method of DNA fragmentation, and transposon junction amplification. More recently, Barseq [7] has been introduced to exploit barcoding to afford a very high level of multiplexing on next-gen sequencers, allowing the efficient analysis of hundreds of samples in parallel.
TnSeq has found many applications in many different bacteria, including E. coli, Mycobacteria, Pseudomonas, Vibrio, Haemophilus, Salmonella, etc., to study genes invovled in metabolism, stress response, virulence, etc. (see Note 1) TnSeq gives a very different read-out on gene function than RNA-seq (transcriptomics), as essentiality of a gene is orthogonal to (i.e. not necessarily correlated with) the level of expression [8]; one can change without the other. Gene essentiality is especially useful for drug discovery efforts, such as interpreting mechanisms of resistance through mutations found in resistant mutants and identifying vulnerable targets and pathways that are critical for survival in vivo (during infection) [9]. Databases of essential genes, such as DEG [10], have been assembled from curated collections of published results of TnSeq analyses, along with inferences of essential genes extended to other species by homology.
Two of the most commonly used transposons in practice are Himar1 [11, 12] and Tn5 [4, 13]. While Himar1, a member of the mariner family, is restricted to inserting at TA dinucleotides [14], Tn5 can insert effectively at any site in the genome, which has consequences for statistical analysis. The magnitude of counts at an individual site is highly dependent on the library, as insertion into the chromosome is a stochastic process, which is a major factor contributing to variability. Although the magnitude of insertion counts can vary significantly between adjacent TA sites in non-essential regions, no strong sequence-dependent bias has yet been found for insertion of various transposons. (see Note 2) Thus insertion counts are usually treated as a random variable, and analysis methods rely on averaging over multiple TA sites in a gene to make a statistical assessment of essentiality. Even a GC-rich organism like M. tuberculosis has 3 or more TA sites for most genes (median is 13 TA sites; only 2.7% of genes have less than 3 TA sites); analysis of genes with only 1-2 TA sites is typically not reliable (analysis of short genes becomes highly dependent on the degree of saturation), and of course genes with 0 TA sites are unanalyzable by TnSeq. However, intergenic regions and ncRNAs are often short enough to have only 0-2 TA sites, and are difficult to anlayze using TnSeq. Many of the analytical methods for Himar1 can be extended to Tn5 data, provided that the saturation is high enough and the assuming the locations and magnitudes of insertions can effectively be treated as random [2]. It is important to note that essential genes, which typically lack insertions throughout the body of the ORF, have often been observed to tolerate insertions at the N- and C-termini, as well as in linkers between domains, and can even contain non-essential domains, which also pose challenges for rigorous identification of essentials. (see Note 3)
Data from TnSeq experiments (i.e. insertion counts) is inherently noisy, making rigorous statistical analysis challenging. Variability can come from a variety of sources, including representation (abundance) in the library, stochastic differences between identically-treated samples (plates, cultures, amimals), and the sequencing process. Reducing raw counts to template counts using barcodes can help ameliorate PCR jackpotting [15]. Loss of diversity (especially in animal infections) and amplification of fitness differences due to differences in growth time (generations) can also lead to significant artifacts such as skewing of read-count distributions [16]. Larger genes with more TA sites ease this because the impact of isolated deviations (e.g. missing or outlier counts) can be mitgated by averaging over multiple observations. Conversely, higher noise and lower saturation can make statistical calls for smaller genes uncertain. Hence, normalization and rigorous statistical analysis are critical to identifying significant genes (e.g. essential or conditionally essential) Also, collection of multiple replicates (2-3 replicates per sample is recommended) is important for increasing statistical certainty through increasing the number of observations per gene. (Biological replicates, i.e. samples from different plates or animals, are more important than technical replicates, i.e. resequencing, for giving a fair picture of stochastic variability of insertion counts, which is critical assessing statistical confidence in observed differences.) Several software packages are available for analysis of TnSeq data, each based on different theoretical framework: ESSENTIALS [17], TnSeq-Explorer [18], TraDIS-Toolkit [19], TnSeqDiff [20] fitness ratios [21], ARTIST [22], and others.
In this paper, we describe Transit [23], a Python-based software package for analyzing TnSeq data. Transit combines implementations of a range of previously published statistical analysis methods. Transit was originally designed for statistical analysis of Himar1 TnSeq datasets (in which insertions are assumed to be restricted to TA sites), though some of the methods have been adapted for Tn5 data (for which the typical lower saturation causes challenges). Transit has a pre-processor (TPP) for extracting insertion counts from raw sequence (.fastq) files that encodes best practices accumulated from experience in multiple labs over the years. Transit also incorporates tools for quality control (QC analysis) for assessing the quality of datasets. Transit has a graphical user interface (GUI) to make it easy for users to to perform most tasks, though this review will focus on running tasks from the command line. The methods are also accessible as a Python library for programmers to call in their own scripts (the source code can be downloaded from Github, https://github.com/mad-lab/transit). Further details can be found in the online documentation, https://transit.readthedocs.io/en/latest/.
The analytical tools in Transit are oriented around addressing three types of questions:
identifying essential genes in a single (e.g. reference) condition
comparative evaluation of conditional essentiality between two conditions
analysis of genes showing variability across multiple conditions
Each of these has different use cases. (see Note 4) This paper describes how to do these analyses using Transit, and includes comments about the impact of (and guidelines on) data quality, file formats, and other practical information.
2. Materials
Transit is designed to run on Linux, Macs, and Windows machines. Transit is written in python3, and requires wxPython4, R, and many other packages as dependencies. Transit can be installed in two ways: 1) via ‘pip3 install’, which will downloaded and install the compiled code from the repository on PyPi along with all dependencies (so that it can be run directly from the command line as ‘transit’), or 2) by cloning the source code from GitHub, and then manually installing the required packages (see Installation Instructions in the online documentation, https://transit.readthedocs.io/en/latest/). The current version is 3.1.0 as of this writing; for future changes, see the online documentation. Note that BWA must be installed for TPP, and some of the more advanced functions (like ZINB) require R to be installed, along with some specific packages.
In the sections below, we will give examples based on a study of gene requirements for growth of M. tuberculosis H37Rv on cholesterol compared to glycerol [24]. In this study, there are 5 TnSeq datasets collection, 2 for glycerol and 3 for cholestrol. For brevity, we will refer to these wig files as G1.wig, G2.wig, C1.wig, C2.wig, and C3.wig.
If installed from GitHub, running Transit would require command sequences like ‘python3 $TRANSITDIR/src/transit.py …’, where $TRANSITDIR is path where Transit is installed. However, for simplicity, we will simply use the command ‘transit’ in the examples below.
Users can get help on most commands (e.g. reminders of arguments and flags) by running Transit without any arguments, or with ‘--help’.
Most of the analyses in Transit generate tab-separated output files as a convention, which can be opened as spreadsheets in Excel. Lines prefixed with ‘#’ in the output files are comments.
All the input and output files for the examples in this chapter can be accessed online at: http://orca1.tamu.edu/essentiality/transit/examples/index.html
3. Methods
3.1. Pre-Processing (TPP)
Transit has a pre-processing step called TPP (Transit Pre-Processor) that maps reads from sequencing a Tn library (files in .fastq or .fasta format) into a genome (.fasta format) and tabulates insertion counts at TA sites. The counts are output in a file format called ‘.wig’ files, which simply have two values on each line - coordinate and insertion counts for each TA site. Wig files have two header lines, the second of which indicates the name of the genome sequence that was used as a reference (‘variableStep chrom=H37Rv’ indicating H37Rv.fna as the reference sequence, for example). In subsequent analyses, it is critical to use the annotation file corresponding to the genomes sequence used in TPP, to ensure consistency of the coordinate system.
TPP uses BWA [25] (which must be installed separately) to map reads into the genome. (see Note 5)
An example of running TPP is as follow:
> tpp -bwa /home/bwa/bwa-0.7.12/bwa -ref H37Rv.fna -reads1 G1_R1.fastq
-reads2 G1_R2.fastq -output G1
The inputs are the sequencing data (fastq files for read 1 and 2) and the reference genome sequence (H37Rv.fna, nucleotide fasta). Multiple intermediate files will be generated for the base filename given by -output. The primary output file would be the wig file, G1.wig, which contains the insertion counts at TA sites.
TPP can also map reads to genome sequences containing multiple contigs (or replicons), such as when an organism contains multiple chromosomes and/or plasmids. Separate reference sequences can be provides to TPP as a comma-separated list.
Another important output file from TPP is the .tn_stats file that gets generated. It contains important parameters and diagnostics from the run. As a quick check, it is useful to examine the saturation (fraction of TA sites with insertions, ideally > 30%) and NZmean (mean over non-zero sites, ideally > 10). The .tn_stats files also reports the total reads, number of reads mapped, and related statistics which can give insight into the degree of attrition and possible reasons for it (for example, whether reads lacked the expected prefix, mapped predominantly to a single site, or matched known sequences related to the transposon vector or primers).
3.1.1. Making Combined Wig Files
When working with a large collection of datasets, it can be cumbersome to have to type multiple wig filenames as inputs to subsequent commands. In such cases, it is convenient to make a ‘combined_wig’ file, which combines the counts at the same TA sites from multiple wig files on the same line (and appends on the ORF id and gene name from the annotation). To aid in comparing insertion counts between datasets, the counts are normalized using TTR normalization (by default, see below), though other normalization methods (including ‘nonorm’ which preserves raw counts) can be applied using the -n flag. The “#File:” header lines cocument the datasets that were combined.
> transit export combined_wig G1.wig,G2.wig,C1.wig,C2.wig,C3.wig H37Rv.prot_table
glyc_chol_combined_wig.txt # TTR norm is implicit
> transit export combined_wig G1.wig,G2.wig,C1.wig,C2.wig,C3.wig H37Rv.prot_table
glyc_chol_combined_wig_raw.txt -n nonorm
A recommended practice is to use the combined_wig file for examining and comparing normalized insertion counts in a gene or locus that is indicated to be conditionally essential, to confirm whether the effect is genuine or perhaps influenced by a few outlier insertion counts.
The ‘.prot_table’ file contains the coordinates and annotations of genes in the genome (see Section 3.2.2 below).
It is often useful to reduce the data in each sample to the mean insertion counts for each gene, which can be done with the ‘export mean_counts’ command:
> transit export mean_counts G1.wig,G2.wig,C1.wig,C2.wig,C3.wig H37Rv.prot_table
glyc_chol_gene_means.txt
The output file, glyc_chol_gene_means.txt can be opened as a spreadsheet.
An individual wig file may be normalized itself using the following command,
> transit normalize <input.wig> <output.wig> -n [method]
where ‘method’ is TTR, betageom, or several others (see documentation). Normalization can also be applied simultaneous to multiple datasets in a combined_wig file by adding the ‘-c’ flag before the filename.
3.1.2. Evaluting Quality of TnSeq Data
To summarize the statistics of multiple samples in a combined_wig file, one may use the ‘tnseq_stats’ command (given either a list of individual wig files, or a combined_wig file):
> transit tnseq_stats G1.wig G2.wig C1.wig C2.wig C3.wig -o glyc_chol_tnseq_stats.txt
or
> transit tnseq_stats -c glyc_chol_combined_wig_raw.txt -o glyc_chol_tnseq_stats.txt
The output file is a tab-separated file that can be opened as a spreadsheet. It contains important information, including the saturation, total counts, mean, NZmean, and max count, for each dataset, along with several statistics on the read-count distribution (skewness, etc.). While the saturation will not change, it is best to run tnseq_stats on a combined_wig file to which no normalization has been applied (‘-n nonorm’), since otherwise, the mean counts will be reflect the results of TTR normalization. If datasets with especially low saturation (e.g. < 15%) or low NZmean (e.g. < 1) are observed, the researcher might consider excluding them from further analyses.
Users can assess the quality of the TnSeq datasets by generating and examining the tnseq_stats table mentioned above. The primary metrics are saturation and mean insertion count (specifically, NZmean). While there are not rigorous criteria for defining “bad” datasets, rules of thumb I use for “good” datasets are: density> 30% (ideally > 50%) and NZmean>10 (ideally >50). In addition, I look at MaxReadCount and Skewness as indicators. Typically, MaxReadCount will be in the range of a few thousand to tens-of-thousands. If you see individual sites with counts in the range of 105 – 106, it might mean you have some positive selection at a site (e.g. biological (fitness advantage), or an artifiact due to things like PCR jackpotting), and this can have the effect of reducing counts and influencing the distribution at all the other sites. If MaxReadCount<100, that is also probably problematic (either not enough reads, or possibly skewing). Also, skewness>30 often (but not always) signals a problem. The reason it is not easy to boil all these down to a simple set of criteria is that some some of the metrics interact with each other.
A useful tool when evaluating the quality of a collection of TnSeq datasets is to make a correlation plot of the mean insertion counts (averaged at the gene-level). While, it is difficult to state how much correlation there should be between conditions (or even between replicates of the same condition), the corrplot can often reveal individual samples which stand out as being far less correlated with all the others (which subsequently might be excluded from analyses).
> transit corrplot glyc_chol_combined.wig.txt glyc_chol_corrplot.png
Although saturation and NZmean are the easiest to assess, a more complicated aspect of data quailty is the insertion-count distribution. (see Note 6)
3.2. Analyses for Single Conditions
3.2.1. Gumbel Analysis
Analyses of individual conditions might occur when a new strain is being evaluated in a reference condition (rich growth medium, for example), and the goal is to make a preliminary catalog of essential genes. There are two principle methods in Transit: Gumbal analysis, and a Hidden Markov Model (HMM). Gumbel analysis focuses on genes with statistically significant gaps, or consecutive sequences of TA sites lacking insertions (empty sites, with counts of 0) [26] Since most libraries are sub-saturated, empty sites will occur in non-essential regions at random, but long sequences of empty sites are statistically unlikely, and genes containing such gaps are taken as essential. The Gumbel method uses the Extreme Value Distribution to quantify the certainty, and a posterior probability is reported in the output file (with separate thresholds for Essential and Non-essential genes, and Uncertain genes in between). An important advantage of this gap-based approach is that it is tolerant of insertions which sometimes occur at the N- and C-termini of ORFs. A disadvantage is that it works less well with less-saturated libraries (< 40%), and can yield many more Uncertain calls, especially for shorter genes (with < 10 TA sites). Note that the magnitude of insertion counts (and hence normalization) do not matter for Gumbel analysis.
> transit gumbel G1.wig,G2.wig ref.prot_table gumbel_H37Rv_glycerol.txt
Gumbel can be run on multiple replicates. (see Note 7)
A typical expectation is that around 10-15% of genes will be essential for growth in vitro in most bacterial genomes, as has been observed across many organisms [10], though of course it depends on size of genome and growth condition.
3.2.2. Prot Tables
An important input file to Gumbel, like many other methods in Transit, is a “prot_table,” which contains the annotation of the genome (including start and stop coordinates of genes). The prot_table format is derived from an old format that could be downloaded from GenBank years ago. It is a tab-separated file with the following fields for each gene: function/description, start coord, end coord, strain (‘+’ or ‘−’), gene length (in amino acids; unused), 3 unused fields, gene name, and ORF id. More recently, the gff (or gff3) format is being used for genome annotations. Transit has a method to convert a gff file into a prot_table. However, there is greater flexibility in the gff format (especially, varying use of keywords), which is difficult to anticipate, so if the Transit command does not work, users might have to write their own script to convert their gff file to a prot_table.
> transit convert gff_to_prot_table <ref.gff> <ref.prot_table>
See Notes on including other types of genes in prot_tables. (see Note 8)
3.2.3. Hidden Markov Model
The HMM can also be used to assess TnSeq data in a single condition. It works a different principle - using a probabilistic model to estimate the state at each TA based on the counts and consistency with adjacent sites [27]. This allows the HMM to smooth over individual outlier values (such as an isolated insertion in any otherwise empty region, or empty sites scattered among insertion in a non-essential region) and make a call for a region/gene that integrates information over multiple sites. The important difference from Gumbel analysis is that the HMM takes into account the magnitudes of insertion counts, which can also carry information about the growth requirement (or fitness effect) of a gene. This allows the HMM to make finer distinctions, utilizing 4 states for individual sites: ES (essental), GD (growth-defect), NE (non-essential), and GA (growth-advantaged). One might see a GD call for a gene whose disruption (by the transposon) impairs growth, so counts are suppressed (compared to the global average, but not all the way to 0), while a gene might be called GA if transposon insertions actually confer a growth advantage, resulting in inflated counts [21]. The gene-level calls are made based on the majority call among the TA sites within each gene. The HMM automatically tunes its internal parameters (e.g. transition probabilities) to the characterisitics of the input datasets (saturation and mean insertion counts), and can work over a broad range of saturation levels (as low as 20%, [27]).
> transit hmm G1.wig,G2.wig H37Rv.prot_table hmm_H37Rv_glycerol.txt
The HMM command has flags for normalization (-n) and also how to handle merging of replicates (-r), though defaults are usually fine. There are actually two output files generated by the HMM, one named on the command line, which will contain the analysis (e.g. state probabilities and call) at each individual TA site, and another, hmm_H37Rv_glycerol_genes.txt (‘genes’ added as suffix to filename), which contains the call for each gene. There is no “probability” associated with the call for each gene. It is important to keep in mind that the call for a given gene can be influenced by insertions in the adjacent region, which is part of the design of HMMs.
The ‘-l’ flag may be used to apply a LOESS correction to adjust for large-scale variations in insertion counts across the genome. (see Note 9)
Because essential and growth-defect regions are close (both have suppressed counts), the difference in call by the HMM (ES vs GD) can be affected by noise, so we often combine the two categories. Typically, 15-20% of genes in a bacterial genome are called by the HMM as ES or GD, suggesting they are either absolutely required (ES) or contribute to fitness (GD) in the growth condition evaluated.
3.3. Pairwise Comparisons: Resampling
Comparisons between two conditions can be used to identify conditionally-essential genes. In addition to binary cases where an essential becomes non-essential or vice versa, we also include genes with quantitative changes in insertion counts, reflecting apparent fitness changes for mutants (for example, genes in which the mean insertion count decreases by 2-fold, though not going all the way to 0). The primary tool in Transit used for pairwise comparisons is “resampling.” It is equivalent to a permutation test on the difference of the mean counts between the two conditions for each gene. Of course, because of stochasticity, almost every gene will exhibit some difference in mean insertion counts between any pair of conditions. What matters is whether the difference is statistically significant.
Resampling is a frequentist approach that compares the observed differences of mean insertion counts to a null distribution to determine whether the difference is larger than would be expected by chance (given the same counts but without knowledge of the condition). (see Note 10)
Normalization is critically important for resampling. If the individual datasets are not properly normalized, it can lead to the appearance of an artifically inflated number of conditional essentials (i.e. false positives, artifacts). The default normalization used in Transit is called TTR (Trimmed Total Read-count). (see Note 11)
Resampling can be run on 2 sets of wig files (comma-separated) as follows:
> transit resampling G1.wig,G2.wig C1.wig,C2.wig,C3.wig H37Rv.prot_table
resampling_glyc_chol.txt -a
The full list of options for the resampling command is:
> transit resampling
usage:
transit resampling <comma-separated .wig control files> <comma-separated .wig experimental files>
<annotation .prot_table or GFF3> <output file> [Optional Arguments]
or
transit resampling -c <combined_wig> <samples_metadata> <ctrl condition name> <exp condition name>
<annotation .prot_table> <output file> [Optional Arguments]
NB: The ctrl and exp condition names should match Condition names in samples_metadata file.
Optional Arguments:
-s <int> := Number of samples. Default: -s 10000
-n <string> := Normalization method. (Default: -n TTR)
-h := Output histogram of the permutations for each gene. (Default: Turned Off)
-a := Perform adaptive resampling. (Default: Turned Off)
-ez := Exclude rows with zero across conditions. (Default: Turned Off)
-PC <float> := Pseudocounts used in calculating LFC. (default: 1)
-l := Perform LOESS Correction (Default: Turned Off)
-iN <int> := Ignore TAs occuring within given percent (as int) of the N terminus. (Def: 0)
-iC <int> := Ignore TAs occuring within given percent (as int) of the C terminus. (Def: 0)
--ctrl_lib := String of letters representing library of control files in order
e.g. ’AABB’. Default empty. Letters used must also be used in --exp_lib
If non-empty, resampling will limit permutations to within-libraries.
--exp_lib := String of letters representing library of experimental files in order
where typically the experimental condition represents a treatment and the control condition representation the untreated or reference condition. The samples_metadata used with combined_wig inputs will be explained in the next section.
The input files are implicitly normalized by TTR (though this can be changed using ‘-n’).
TA sites near the termini of ORFs can be trimmed using -iC and -iN. For example, to ignore insertion in the first or last 5% of an ORF, use ‘-iC 5 -iN 5.’
I recommend using the ‘-a’ flag to apply the adaptive version of resampling, which is much faster and generally outputs p-values close to those obtained by performing all 10,000 samples (within a factor of 2-3). It truncates the resampling early for genes when it is clear that they are not going to be significant. Almost all significant genes will be still be significant, except for possibly a few marginal differences very close to the 0.05 threshold on adjusted p-value.
If the -h flag is given, histograms of the null distribution from resampling for each gene, along with a demarcation for the observed difference in mean insertion count between the conditions, will be generated in a sub-directory. These images can be examined to determine whether the null distribution for a gene of interest looks appropriately bell-shaped (versus bimodal, for example).
The output file contains the various statistics on the resampling for each gene and a Padj column at the end. Users can open the tab-separated file as a spreadsheet and sort by Padj. The conditionally-essential genes are those with Padj < 0.05.
It is difficult to say what the expectations would be for number of conditional-essentials, since it depends on the impact of the biological effects of the selection conditions. (see Note 12)
The output also reports the log-fold-change (LFC, base 2) of the mean counts. It can be used to sort the genes to identify those showing the greatest increase or decrease in counts (relatively less or more essential). However, only genes with Padj < 0.05 should be considered significant. But these could be uncertain, and should be ignored if they have a Padj above 0.05. Frequently, genes with the largest-magnitude LFC are often smaller genes (with fewer TA sites) or genes with very low insertion counts, which are often not statistically significant. Of course, as with other frequentist statistical tests, genes with Padj above 0.05 should not be over-interpreted as evidence that they are unaffected by the condition.
Pseudo-counts are used in the calculation of LFCs to help reduce noise. (see Note 13)
See Notes for analyzing datasets from different libraries using resampling. (see Note 14)
3.3.1. Pathway Analysis
Typically, the significant genes do not all come from the same pathway, but often represent a variety of pathways in which genes experience apparent increases or decreases in fitness. In order to get a sense of which pathways might be enriched among the conditionally essential genes, the ‘pathway_enrichment’ command may be used:
> transit pathway_enrichment <resampling_file> <associations> <pathways>
<output_file> [-M <FET|GSEA|ONT>] [-PC <int>]
The 〈associations〉 file lists mappings of ORF ids to pathway ids in a two-column format. Three example systems of funtional categories are: Sanger roles [28] (123 roles), COG categories (Clusters of Orthologous Genes, [29], 20 categories), and GO terms (Gene Ontology, http://www.geneontology.org/; there are ~4000 GO terms with at least 1 gene in H37Rv). The corresponding pathway associations files for genes in Mtb H37Rv are provided in the Transit data directory ($TRANSIT/src/pytransit/data/) are: H37Rv_sanger_roles.dat, H37Rv_COG_roles.dat, and H37Rv_GO_terms.txt.
There may be multiple roles/pathway associations for each gene (listed on separate lines), or none (e.g. for hypothetical genes not yet functionally annotated). Note that these files have been expanded to include associations of each gene with all parents of each GO term or role (since these are hierarchical systems). The 〈pathways〉 file gives the functional descriptions for each pathway_id/role/GO term (e.g. sanger_roles.dat, COG_roles.dat, and GO_term_names.dat in the data directory). These will be the same for all organisms.
There are three alternative methods used to evaluate significance of pathway enrichment:
The first is to use Fisher’s exact test, where the p-value based on hypergeometric distribution of observed counts of category members among the hits compared the expected number based on the whole genome [30]. This is the default method (‘-M FET’). The output file contains a list of significant pathways, sorted by adjusted p-value. In addition, the enrichment for each gene is reported. Enrichment is defined as the ratio of observed pathway members among the significant genes to the expected number based on the background proportion of category members in the overall genome. To mitigate the impact small pathways with only a few genes (which might spuriously appear as enriched), pseudocounts of 2 are incorporated in the numerator and denominator. This can be modified with the ‘-PC’ flag. However, this analysis can be of limited use if the total number of significant (conditionally essential) genes is small (< 10).
An alternative approach is Gene Set Enrichment Analysis (GSEA, [31]) (using the ‘-M GSEA’ flag). GSEA takes into account the ranking of all the genes, without regard to a significance cutoff. First, the genes are sorted by LFC. Alternatively, they can be ranked by the signed log-P-value (SLPV = sign(LFC) * log10(pval)), which effectively ranks the genes by significance from largest increase in insertion counts to largest decrease (with insignificant genes falling in the middle of the ranking). Then the GSEA algorithm calculates a score reflecting the mean rank of a given set of genes and performs a simulation to determine the signifance (p-value) by comparing to a null distribution of scores derived from random shuffling of the order. The closer the mean rank of a group of genes is to the top of the entire ranked list, or the closer to the bottom, the more significant. The potential advantage of GSEA over Fisher’s exact test is that all the genes in a pathway can contribute to its enrichment, even if only a few (or none) are above the significance cutoff, as long as there is a systematic trend of increased or decreased counts shared by many of them.
Finally, the Ontologizer method [32] has been implemented and is available via the ‘-M ONT’ flag. The Ontologizer method acknowledges the hierarchical nature of the Gene Ontology and is designed to take advantage of parent-child relationships among GO terms by computing a conditional version of Fisher’s exact test for a node conditioned on the genes in its parents. This can help focus the analysis on nodes in the GO hierarchy showing the most specific enrichment.
Here are some examples of using these various options.
# uses Fisher’s exact test by default (with PC=2 as pseudocounts)
> transit pathway_enrichment resampling_glyc_chol.txt $DATA/H37Rv_sanger_roles.dat
$DATA/sanger_roles.dat pathways_glyc_chol_Sanger.txt
# can do this with GO terms too
> transit pathway_enrichment resampling_glyc_chol.txt $DATA/H37Rv_GO_terms.txt
$DATA/GO_term_names.dat pathways_glyc_chol_GO.txt
# with COG categories
> transit pathway_enrichment resampling_glyc_chol.txt $DATA/H37Rv_COG_roles.dat
$DATA/COG_roles.dat pathways_glyc_chol_COG.txt
# can also do GSEA method (on any system of functional categories)
> transit pathway_enrichment resampling_glyc_chol.txt $DATA/H37Rv_sanger_roles.dat
$DATA/sanger_roles.dat pathways_Sanger_GSEA.txt -M GSEA
# Ontologizer is a specialized method for GO terms
> transit pathway_enrichment resampling_glyc_chol.txt $DATA/H37Rv_GO_terms.txt
$DATA/GO_term_names.dat pathways_Ontologizer.txt -M ONT
where $DATA refers to the path to the Transit data directory noted above. In this dataset, 2 COG and 6 Sanger categories related to secondary-metabolite/small-molecule metabolism and lipid metabolism are identified as significant. The most significant GO term identified (among 26 significant terms, though many overlap) is GO:0008202, ‘steroid metabolic process’ (Padj = 0.0000089; 15 out of 75 conditionally essential genes from resampling), which is consistent with the experiment (growth on cholesterol versus glycerol).
3.4. Analyses of Multiple Conditions
Recently, researchers have begun conducting more complex experiments using Tn libraries where a library is assessed across a large number of treatment conditions. For example, one might evaluate responses (conditional essentiality) to treatment with a panel of different antibiotics, possibly at different concentrations. One might assess the library in different stress conditions (heat, cold, hypoxia, SDS, nutrient starvation, iron limitation, etc.), or with media supplemented in various ways (e.g. multiple sources of carbon, nitrogen, sulfur, or phosphorous). Or one might compare survival of a bacterial transposon mutants in animal models with different genetic backgrounds, or for different durations of infection, etc. Analysis of such TnSeq datasets goes well beyond simple pairwise comparisons between conditions and often requires customized statistical analysis to evaluate the effects of the experimental variables. As a first step, Transit has some tools for evaluating variability of insertion counts across conditions. The analysis below focus on identifying genes that exhibit some statistically signficant differences in insertion counts across the panel of conditions. This is a useful starting point to begin to assess effects of the treatments (and similarities among them) based on the subsets of genes that respond.
In this section, we use an example from a study of iron utilization in mycobacteria [33]. The data includes 2-3 replicates each of an M. tuberculosis H37Rv transposon library grown in in-vitro conditions involving several different vehicles for iron delivery, focusing on various forms of heme and mycobactin (6 conditions). The 14 wig files for this example have already been consolidated into a combined wig file (iron_combined_wig4.txt), and a samples metadata file is used to encode which samples belong to which condition.
3.4.1. Genetic Interaction Analysis
A special case of multi-condition analysis that occurs frequently is when TnSeq libraries in a wild-type strain and a mutant strain (e.g. gene-knockout) are compared between two conditions (e.g. a stress and a control). Typically, one is looking for genes that are conditionally essentially in the stress condition, but one wants to factor out those genes with a similar response in the wild-type strain and focus on differences unique to the mutant. There are many variations of this scheme which also require a comparison of 4 TnSeq datasets, arranged as 2 × 2. Transit has a specialized method based on Bayesian analysis [34] to identify significantly interacting genes (that interact with the knockout in the context of the stress). An example of how to run this command is as follows, which takes 4 groups of comma-separated wig files as input:
> python3 ../../transit/src/transit.py GI
<wigs_for_strA_cond1> <wigs_for_strA_cond2> <wigs_for_strB_cond1> <wigs_for_strB_cond2>
<annotation .prot_table or GFF3> <output file>
In the output file, significant genes are categoried as ‘aggravating’, ‘alleviating’, or ‘suppressive’ interactions, depending on whether they exhibit increased or decreased insertion counts in the mutant compared to the wild-type in the stress condition.
3.4.2. ANOVA
ANOVA analysis in Transit can be used to identify genes that exhibit significant variability of insertion counts across multiple conditions. ANOVA is a traditional statistical method for determining whether there are differences in observations among several groups. This method can be run in Transit using the following command:
> transit anova --help
Usage: python3 transit.py anova <combined wig file> <samples_metadata file>
<annotation .prot_table> <output file> [Optional Arguments]
Optional Arguments:
-n <string> := Normalization method. Default: -n TTR
--include-conditions <cond1,…> := Comma-sep list of conds to use for analysis
--ignore-conditions <cond1,…> := Comma-sep list of conds to ignore
-iN <int> := Ignore TAs within given percentage of N terminus. Def: -iN 0
-iC <int> := Ignore TAs within given percentage of C terminus. Def: -iC 0
-PC <int> := pseudocounts to use for calculating LFC. Default: -PC 5
For example,
> transit anova iron_combined_wig4.txt iron_samples_metadata.txt
H37Rv.prot_table anova_iron.txt
The use of a combined_wig file makes it easy to work with a large number of datasets in this context. The samples metadata file is a spreadsheet (in tab-separated text format) prepared by users that contains information about each sample/dataset/wig file. The headers must contain ‘Id’, ‘Filename’, and ‘Condition’, where Id is a unique name for each sample, Filename is the name of its wig file (incorporated in the combined_wig file) and Condition is a symbolic name used to represent the treatment (typically represented by multiple replicates). The samples metadata file can contain additional information as well (e.g. time points, concentrations, batches, etc.). During the ANOVA analysis, the (TTR-normalized) insertion counts for the TA sites in each gene will be pooled into groups based on the condition labels, before computing the F-statistic and p-value. P-values are adjusted post-hoc by the Benjamini-Hochberg method. Flags --ignore-conditions and --include-conditions can be used to focus on the analysis on just a subset of desired conditions (provided as a comma-separated list). For example ‘--include-conditions HighFeMBT,Hemin,Hemoglobin’ or ‘--ignore-conditions LowFeMBT’.
The output of ANOVA analysis is a tab-separated spreadsheet which can be sorted by adjusted P-value. In this data, 181 genes were found to exhibit significant variability in insertion counts among the 6 iron-supplementation conditions. If a gene is determined to be significant by ANOVA, it only means that its counts vary in some condition (at least one) relative to the others, but does not indicate which one. Like traditional ANOVA analysis, post-hoc analyses must be employed to determine which conditions a gene is responding to, such as Tukey’s range-test (or honestly significant difference), which is based on pairwise comparisons between conditions [35].
To facilitate this post-hoc analysis, the ANOVA method in Transit also prints out LFCs for each gene in each condition. The LFCs can be used to look for genes that respond to specific conditions, e.g. by sorting on these columns to look for genes with the most enrichment or depletion in a given condition. Note that LFCs are computed relative to the mean insertion count for a gene across all conditions. Thus, there will almost always be some condition(s) with higher counts (representing more fitness for the mutant) and other condition(s) with lower counts (where disruption of gene has less fitness, and hence is relatively more essential) than average. Pseudocounts of 5 are used in calculating the LFCs (which helps reduce high-magnitude LFCs for genes with low counts, which are susceptible to noise), though this can be changed with the -PC flag.
The LFC columns in the ANOVA output can be colored as a heatmap in the spreadsheet (using Excel) to make the patterns of variation among the genes more clear. In addition, the ANOVA output file can be used to generate a heatmap (see Figure 1, adapted from [33]) that simultaneously clusters the significant genes and the conditions, which is especially useful for shedding light on the relationships among the conditions apparent in the data:
> transit heatmap -anova anova_iron.txt heatmap_iron.png
Similarly, the anova file can be used as input the corrplot command (with the ‘-anova’ flag suffixed) to show the similarities among the condition.
> transit corrplot anova_iron.txt heatmap_iron.png -anova
Importantly, the heatmap and corrplot analyses are based only on the significantly varying genes (Padj > 0.05, typically only a few hundred) in order to enhance the patterns, since otherwise they would be washed out by the rest of the genes in the genome, the majority of which do not exhibit significant variation.
Figure 1:
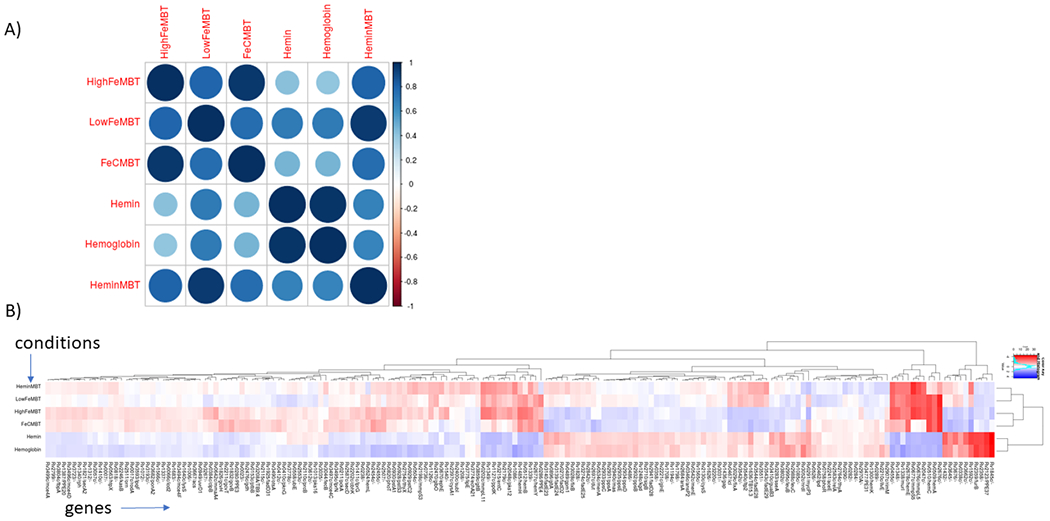
a) Correlation plot among iron-supplementation conditions based on significantly varying genes according to ANOVA. b) Heatmap of same data showing clustering of genes and conditions. Blue means more insertions than average (i.e. less essential), and red means less insertions than average (i.e. more essential).
3.4.3. Zero-Inflated Negative Binomial (ZINB)
An alternative method for identifying genes exhibiting variability of insertion counts across conditions is Zero-Inflated Negative Binomial (ZINB) regression, which has recently been added to Transit [36]. One of the limitations of ANOVA analysis is that it assumes the data are Normally distributed. However, transposon insertion counts clearly violate this assumption (for several reasons). The ZINB method is designed to be a better model for insertion count data by: a) representing the non-zero counts using a Negative Binomial distribution, and b) representing the saturation (TA sites with counts of 0) using a zero component in a mixture model. Thus, when analyzing the counts in a gene across multiple conditions, ZINB can detect variations in either the magnitudes in the insertion counts or the local level of saturation. This enables ZINB to be a little more sensitive than ANOVA, and empirical studies suggest ZINB can detect about 30-50% more significant genes.
The usage of the ZINB command in Transit is as follows:
> transit zing --help
Usage: transit zinb <combined wig file> <samples_metadata file> <annotation .prot_table>
<output file> [Optional Arguments]
Optional Arguments:
-n <string> := Normalization method. Default: -n TTR
--condition := column name in samples_metadata to use. Default: “Condition”
--ignore-conditions <cond1,…> := Comma separated list of conditions to ignore
--include-conditions <cond1,…> := Comma separated list of conditions to include
-iN <float> := Ignore TAs within given percentage of the N terminus. Def: 5
-iC <float> := Ignore TAs within given percentage of the C terminus. Def: 5
-PC <N> := Pseudocounts used in calculating LFCs in output file. Default: -PC 5
--covars … := Comma-sep. list of variables (columns in metadata) to use as covariates
--interactions … := Comma-sep. list of variables to test for interations
--gene <ORF id or gene name> := Run method for just one gene and print model output.
For example,
> transit zinb iron_combined_wig4.txt iron_samples_metadata.txt
H37Rv.prot_table zinb_iron.txt
If there are just two conditions provided, then using ZINB analysis to identify genes with variable insertion counts is equivalent to detecting conditional essentiality. Hence ZINB can be viewed as an alternative to resampling in this limiting case, and anecdotal testing suggests that there is a great deal of overlap. ZINB can sometimes identify additional genes as conditionally essential where there is a difference in either local saturation or magnitudes of counts alone, even if the overall mean count between conditions is not significantly different, and hence not detected by resampling. (see [36] for a more thorough comparison of the differences in significant hits between ZINB and resampling on example datasets).
In the output file for ZINB, multiple columns of information are provided on the mean counts in each condition, LFCs (relative to the mean across all conditions), non-zero mean, and saturation. For convenience, the –gene flag can be used to run the analysis just on a single gene, and the Transit will print out the condition-dependent and condition-independent models, likelihoods, etc., as in this example:
> transit zinb iron_combined_wig4.txt iron_samples_metadata.txt
H37Rv.prot_table output.txt --gene glpK
The significance (p-value) of each gene is determined by a likelihood ratio test (LRT). First, a condition-dependent model is generated by fitting independent parameters for each condition (such as mean and dispersion of insertion counts in the Negative Binomial, and saturation as the mixing coefficient with the zero component). Then a condition-independent ZINB model is generated by fitting a common set of parameters based on the counts pooled across all conditions. Finally, a likelihood ratio test is performed to determine whether the increase in likelihood of the condition-dependent model is justified, given the increased number of parameters. A p-value is derived for each gene using a chi-square distribution, and then the p-values are adjusted post-hoc by the Benjamini-Hochberg procedure to control the overall FDR.
One of the advantages of the ZINB model is that it is implemented in a Generalized Linear Model (GLM) framework, which allows incorporation of experimental variables (attributes of the conditions) as covariates. One application of this idea is to factor our the effect of an attribute that is known to affect the main condition in a way that is not of interest. For example, suppose we are interested in identifying genes that exhibit variable responses to different antibiotics. Futher, suppose the samples were each cultured for varying amounts of time (e.g. 0, 1, or 2 weeks). It is natural to expect that there will variation in insertion counts between samples at different time-points, even if they are treated with the same drug. To evaluate the effect of the drug, it is desirable to subtract out any systematic effects on the insertion counts due to time (independent of drug). If a “Time” column is included in the samples metadata file, and a column with the header “Drug” encodes the drug treatment for each sample, then this may be achieved by using the --covars flag, as in this hypothetical example:
> transit zinb combined_wig.txt samples_metadata.txt H37Rv.prot_table
ZINB_cond_drug_covar_time.txt --condition Drug --covars Time
If there are multiple covariates, they can by specified using a comma-separated list (i.e. corresponding to separate columns in the metadata file). The --covars flag could also potentially be used to correct for batch effects (where the variation in insertion counts in some samples appear to be determined by the batch of the experiment or data collection).
A similar approach can be used to test for interactions of variables with the main condition. (see Note 15)
In the resulting output file, significant genes are those the exhibit some variablility among the drug treatments that is dependent on carbon source.
As with ANOVA, the ZINB output file can be used to make a heatmap showing the clustering of the significant genes and the the conditions:
> transit heatmap -zinb zinb_iron.txt zinb_iron_heatmap.png
4. Summary
Transit is designed to be a platform for statistical analysis of TnSeq data, with a focus on analysis of Himar1 transposon libraries (where insertions are restricted to TA dinucleotides). Some of the analytical methods can be applied to other transposons, like Tn5, though currently, they don’t work as robustly. Although Transit has a graphical interface (GUI), some of the more recent tools that have been added can only be invoked at the command line, which has been the focus of this paper. The Transit Pre-Processor (TPP) provides a way of processing raw sequencing data files and reducing the raw data to TA-site insertion counts in the form of .wig files. The ‘tnseq_stats’ command provides important summary statistics on datasets which is useful for diagnositics (i.e. identifying poor-quality datasets that might need to be excluded or re-collected). The analytical tools can be divided into 3 major tasks. First, individual datasets from an organism in a single (e.g. reference) condition can be used to identify essential genes using methods such as Gumbel analysis (gaps) or a Hidden Markov Model. Second, pairs of conditions can be compared to identify conditionally essential genes using resampling (a permutation test on mean counts). The recently added ability to apply resampling on datasets mapped to different genome sequences has proven useful for studying differences in gene essentiality between libraries made from different strains, such as clinical isolates. Several methods for pathway enrichment analysis have also been added to gain additional insight from functional similarities among conditionally essential genes.
However, more recent developments in Transit have focused on supplying tools for analyzing larger collections of datasets from experiments involving multiple conditions. To keep things manageble when working with large collections of datasets, many of the tools in Transit have been extended to use ‘combined_wig’ files and accompanying metadata files that encode relevant information about the different conditions. A starting point for analyzing such complex experiments is to identify genes exhibiting statistically significant variability across the conditions, using ANOVA or ZINB analysis, and then to begin to cluster and assess genes based on the similarity of their patterns of count variations, utilizing correlation plots and heatmaps. ZINB can be used to perform more sophisticated analyses through the exploitation of variables relating the different conditions as covariates and/or interactions (including capturing the trend or dependence of insertion counts on quantitative variables such as time or concentration). Genetic interaction (GI) analysis can be used to evaluate experiments where two different experimental variables are evaluated, producing a 2x2=4-way comparison of conditions, and test for significant interactions (e.g. suppressive, alleviating, or aggravating), which is especially useful for identifying genes associated with phenotypic changes in a knock-out strain compared to a wild-type strain.
Transit continues to evolve and improve, especially through feedback and suggestions from users (send email to ioerger@cs.tamu.edu). In the future, we hope to add new statistical methods to support analysis of more complex experiments, improve integration with the GUI, and also extend and improve the analyses to TnSeq libraries made with other transposons, especially Tn5.
Acknowledgements
This work has been supported by NIH grants U19 AI107774 and P01 AI143575.
Footnotes
For example, TnSeq has been used to study genes required for metabolizing/utilizing specific nutrients such as cholesterol [24] or iron [33], tolerance of stress conditions [37, 38], genetic interactions with knocked-out genes in libraries made from null mutants [39–41], and virulence in animal models [4, 5, 9, 42, 43].
For Himar1, there is an apparent non-permissiveness of TA sites with a G at flanking positions +/−2 bp, with G or C at +/−3 [44], and also a general preference for insertions in more bendable regions of the DNA [45]). Similarly, only a generalized sequence preference has been identified for Tn5, but it is insufficient to make insertion locations predictable [46].
Some studies have observed large-scale chromosomal biases, where mean insertion counts exhibit a trend based on position on the chromosome, such as a gradation based on distance between the origin and termination of replication. Adjustments for such biases can be made through methods such as the LOESS correction [47], which equalizes the smoothed mean across the whole genome.
A common case of single-condition analysis might be assessing essential genes and pathways in a new bacterial species. A common case of pairwise analysis might be comparing essentiality in a stress condition, such as starvation, iron limitation, low pH, hypoxia, antibiotic exposure, or growth in an animal model, compared to a reference condition, such as growth on rich medium. However, recently, more complex TnSeq experiments are being conducted involving multiple experimental variables/treatments, such as varying antibiotic concentrations, varying durations (number of days or weeks in culture or in vivo), comparison of survival in different animal breeds/species/genotypes, or supplementation with various nutrients, making tools for task multi-condition analysis necessary to explore/characterize patterns of response over larger sets of experimental conditions.
Since the reads represent junctions between the transposon and chromosome, the first step is identifying reads (in read 1) with a prefix matching the terminus of the transposon, and the stripping this prefix off to map the genomic suffix. Many protocols introduce random shifts in the location of the prefix sequence within the reads, to reduce sequencing problems when all reads begin with the same nucleotides. By default, TPP searches for a prefix corresponding to Himar1, but other transposons can be accommodated by specifying the search sequence using the ‘-prefix’ flag. Illumina sequencers often provide pairs of reads, and while read 2 is not absolutely necessary for mapping reads (i.e. optional), Sassetti et al. have shown how to embed random nucleotide barcodes in read 2 that can be used by TPP to reduce raw read counts at each TA site to counts of unique DNA templates, which helps reduce noise due to PCR jackpotting effects [15]. Because of the lengths of the prefix in read 1 and barcodes (and surrounding constant regions and genomic regions in read 2), it is recommended to use a read length of at least 75 bp (i.e. 75x75 bp paired-end sequencing). Various flags can be used to adjust the methods and parameters for mapping reads, such as number of mismatches allowed or constraints on location of prefix.
Generally speaking, the number of TA sites with low counts are most abundant, and sites with high counts are far less frequent. While it is not theoretically guaranteed, well-behaved datasets typically conform closer to a geometric distribution (though possibly with some excess dispersion, which could be modeled as a negative binomial). In contrast, lower-quality datasets often exhibit a significant skew away from this distribution. Skewed datasets are often more dominated by excessively high counts at a few sites, while the counts at the vast majority of remaining sites are quite low by comparison. This can cause problems with normalization; even though TTR is designed to be robust to a few outliers, it is only linear transformation and cannot correct for skew in the data. This can lead to an inflation of apparently differentially essential genes (artifacts) when skewed datasets are compared to other conditions.
The Transit GUI provides plots of the insertion-count distribution, including a QQ-plot (quantile-quantile plot) against an ideal geometric, to visually assess the degree of skew. The closer to a diagonal, the better. Aside from ‘skewness’ as a metric itself, the tnseq_stats table also report the Pickand’s tail index [48]. Anecdotal experience suggests that dataset with PTI > 1.0 (and maybe even PTI > 0.5) are potentially problematic.
The causes of skew are (currently) not well understood, though, anecdotally, it often seems to be associated with loss of diverity (e.g. in animal-passeged samples) over ‘over-selection’ of libraries. For example, culturing too long under a stress condition can amplify the effects of even small fitness differences.
> transit normalize G1 .wig G1_BGC. wig –n betageom
If multiple replicates are available, they can be provided to Gumbel via a comma-separated list, as shown above, and the data will be merged. The output file will contains values for each gene, such as number of TA sites, number of sites with insertion, longest run of sites without insertion, and finally a posterior probability (‘zbar’) and call (ES=essential, NE=non-ess, U=uncertain, S=too short to analyze). This command also has flags for explicitly trimming (ignoring) TA sites in termini of genes, etc.
Note that one can add other types of genes to a prot_table, such as tRNAs, rRNAs, and ncRNAs. All that is required it to include lines with the expected format, including start and end coordinates and strand. Furthermore, the same approach can be used to represent other types of loci, such as operons (spanning multiple genes) or intergenic regions, to be analyzed for essentiality. However, note that most intergenic regions and ncRNAs are relatively small compared to typical protein-coding regions, and hence they often have only a few (if any) TA sites, making essentiality analysis highly uncertain (except for the most highly saturated libraries [44]).
Because the HMM takes the magnitudes of insertions into account, it can be affected by large-scale varations in insertion counts across the genome. In the GUI, a plot of the smoothed mean insertion count can be generated. If there are noticable differences, the local deviations from the global mean can be subtracted out using the LOESS correction [17] (‘-l’ flag for the hmm command). While this is typically not necessary for Tn libraries in M. tuberculosis, chromosomal biases can be more severe in faster-replicating species, like Vibrio cholera [3] and E. coli [49].
First, the normalized insertion counts are pooled over all TA sites and replicates for both conditions. Then a null distribution is created by repeatedly drawing random two samples (of the same size as the original number of observations) from the pooled counts and calculating the difference of means 10,000 times. From this a two-tailed p-value is derived for the observed difference, and the p-values are adjusted post-hoc for multiple testing via the Benjamini-Hochberg correction [50] to limit the overall false discovery rate (FDR) to 5%.
This comparative approach to identifying conditionally essential genes is preferred over simply combining the results of individual analyses of each condition (e.g. Gumbel) and selecting genes that are called Essential in one condition but not the other, because the results of that approach could be influenced by which genes fall just above or below the significance cutoff. Resampling is a direct comparison that looks at differences in insertion counts. The resampling procedure is designed to be (somewhat) robust to noise (e.g. outliers, high variance among counts), and is appropriately less sensitive for smaller genes and more sensitive with more replicates.
For each dataset, the total insertions over all TA sites is tabulated, after removing the top and bottom 1% of sites (to reduce the influence of outliers). The sites are divided by this total, and then scaled-up so that the mean insertion count over the whole genome is 100. While the calculation is simple, it puts counts from different datasets on a comparable basis. Furthermore, it maintains a balance so that counts from less-saturated datasets are inflated proportionally, so that the mean insertion for most genes (averaged over multiple TA sites) stays about the same on average. This objective is important for the assumptions of resampling (see explanation in [23]). A way to verify that TTR normalization is doing the right thing is to make a scatter plot of the mean insertion counts for each gene between any two datasets; the data should scatter along the X=Y diagonal. Individual points can deviate from this line due to: experimental noise, sampling error for small genes with few TA sites, or biological differences (if the datasets represent distinct experimental conditions). But if there is a systematic deviation away from the X=Y line in this scatter plot (due to improper normalization), it would likely produce an excess of conditional essentials detected by resampling.
In some cases, the treatment (e.g. a high-stress condition) might affect the essentiality (and fitness) of hundreds of genes. In other cases, biologically weak selection criteria might affect only a few (or perhaps no) genes. If the experimenter feels there are too few hits (e.g. compared to their expectations, possibly lacking genes known to be in affected pathways), then it might help to collect additional replicates, which can increase the sensitivity of detection. If, subjectively, too many significant genes are output, it might be a sign of problems with skewing of the data. The user can follow the QC guidelines above to possibly identify lower-quality datasets to try excluding, or they might consider applying BGC normalization (‘-n betageom’) to the datasets.
For example, a gene with a mean count of 1 in the first condition and 4 in the second would appear to have a fold-change of 4 and LFC of 2.0 (with PC=0). However, this is typically in the range of noise for TnSeq experiments. Considering that TTR normalization adjusts the mean insertion count of each sample to around 100, raising the pseudo-counts to a level of PC=5 would reduce this to log2(9/6) = 0.58, masking out differences below this level.
By loading the resampling output file into the GUI (bottom panel), a volcano plot can be generated (via ‘Choose Action’), showing a scatter plot of the P-values versus the LFC for each gene. Genes the become more essential (in the experimental condition over control) are on the left (negative LFC), and significant genes are above the dashed red line (p-value threshold adjusted for 5% FDR).
If the replicates in each conditions represent samples from multiple libraries, the sensitivity of resampling can sometimes be increased by performing separate random draws from the counts for each library and computing the difference of the means between conditions summed separately for each library. This can help reduce the variance in the difference in the mean insertion count between conditions by effectively subtracting out variability due to differences in abundance at each TA site between libraries. The transposon library each sample in the two conditions can be specified symbolically using a string of characters. For example ‘--exp_lib AABB --ctrl_lib AB’ would indicate that the first 2 experimental samples came from library A and the second two samples from library B; there can be different numbers of samples in the control condition, but they should use the same codes (in this case, one sample each from library A and B).
> transit zinb combined_wig.txt samples_metadata.txt H37Rv.prot_table
ZINB_cond_drug_interac_carbon.txt
--condition Drug --interactions CarbonSource
References
- 1.van Opijnen T, Camilli A (2013) Transposon insertion sequencing: a new tool for systems-level analysis of microorganisms. Nat Rev Microbiol 11(7):435–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barquist L, Boinett CJ, Cain AK (2013) Approaches to querying bacterial genomes with transposon-insertion sequencing. RNA Biol 10(7):1161–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chao M, S A, Davis B, Waldor M (2016) The design and analysis of transposon insertion sequencing experiments. Nat Rev Microbiol 14(2):119128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Langridge GC, Phan M, Turner D, Perkins T, Parts L, Haase J, Charles I, Maskell D, Peters S, Dougan G, et al. (2009) Simultaneous assay of every salmonella typhi gene using one million transposon mutants. Genome Research 19(12):2308–2316, URL http://www.ncbi.nlm.nih.gov/pubmed/19826075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gawronski JD, Wong SM, Giannoukos G, Ward DV, Akerley BJ (2009) Tracking insertion mutants within libraries by deep sequencing and a genome-wide screen for Haemophilus genes required in the lung. Proc Natl Acad Sci USA 106(38):16422–16427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goodman AL, McNulty NP, Zhao Y, Leip D, Mitra RD, Lozupone CA, Knight R, I GJ (2009) Identifying genetic determinants needed to establish a human gut symbiont in its habitat. Cell Host Microbe 6(3):279–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wetmore K, Price M, Waters R, Lamson J, He J, Hoover C, Blow M, Bristow J, Butland G, Arkin A, A D (2015) Rapid quantification of mutant fitness in diverse bacteria by sequencing randomly bar-coded transposons. mBio 6(3):e00306–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jensen P, Zhu Z, van Opijnen T (2017) Antibiotics disrupt coordination between transcriptional and phenotypic stress responses in pathogenic bacteria. Cell Reports 20(7):1705–1716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang YJ, Reddy MC, Ioerger TR, Rothchild AC, Dartois V, Schuster BM, Trauner A, Wallis D, Galaviz S, Huttenhower C, Sacchettini JC, Behar SM, J RE (2013) Tryptophan biosynthesis protects mycobacteria from CD4 T-cell-mediated killing. Cell 155(6):1296–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luo H, Lin Y, Gao F, Zhang CT, Zhang R (2014) DEG 10, an update of the Database of Essential Genes that includes both protein-coding genes and non-coding genomic elements. Nucleic Acids Research 42:D574–D580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rubin EJ, Akerley BJ, Novik VN, Lampe DJ, Husson RN, Mekalanos JJ (1999) In vivo transposition of mariner-based elements in enteric bacteria and mycobacteria. PNAS 96(4):1645–1650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sassetti CM, Boyd DH, Rubin EJ (2001) Comprehensive identification of conditionally essential genes in mycobacteria. PNAS 98(22):12712–12717, DOI 10.1073/pnas.231275498, URL http://www.pnas.org/content/98/22/12712.abstract, http://www.pnas.org/content/98/22/12712.full.pdf+html [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reznikoff WS (2003) Tn5 as a model for understanding DNA transposition. Molecular Microbiology 43(5):1199–1206 [DOI] [PubMed] [Google Scholar]
- 14.Lampe DJ, Churchill ME, Robertson HM (1996) A purified mariner transposase is sufficient to mediate transposition in vitro. the The European Molecular Biology Organization Journal 15(19):5470–5479 [PMC free article] [PubMed] [Google Scholar]
- 15.Long J, DeJesus M, Ward D, Baker R, Ioerger T, Sassetti C (2015) Identifying essential genes in Mycobacterium tuberculosis by global phenotypic profiling. In: Lu LJ (ed) Methods in Molecular Biology: Gene Essentiality, Springer, vol 1279, pp 79–95 [DOI] [PubMed] [Google Scholar]
- 16.DeJesus MA, Ioerger TR (2016) Normalization of transposon-mutant library sequencing datasets to improve identification of conditionally essential genes. J Bioinform Comput Biol p 1642004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zomer A, Burghout P, Bootsma HJ, Hermans PW, van Hijum SA (2012) ESSENTIALS: software for rapid analysis of high throughput transposon insertion sequencing data. PLoS ONE 7(8):e43012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Solaimanpour S, Sarmiento F, Mrazek J (2015) Tn-seq explorer: a tool for analysis of high-throughput sequencing data of transposon mutant libraries. PLoS ONE 10(5):e0126070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barquist L, Mayho M, Cummins C, Cain AK, Boinett CJ, Page AJ, Langridge GC, Quail MA, Keane JA, Parkhill J (2016) The tradis toolkit: sequencing and analysis for dense transposon mutant libraries. Bioinformatics 32(7):1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao L, Anderson MT, Wu W, T Mobley HL, Bachman MA (2017) TnseqDiff: identification of conditionally essential genes in transposon sequencing studies. BMC Bioinformatics 18(1):326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Opijnen T, Bodi KL, Camilli A (2009) Tn-seq: high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nat Methods 6(10):767–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pritchard JR, Chao MC, Abel S, Davis BM, Baranowski C, Zhang YJ, Rubin EJ, Waldor MK (2014) ARTIST: high-resolution genome-wide assessment of fitness using transposon-insertion sequencing. PLoS Genet 10(11):e1004782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DeJesus MA, Ambadipudi C, Baker R, Sassetti C, Ioerger TR (2015) TRANSIT–A Software Tool for Himar1 TnSeq Analysis. PLoS Comput Biol 11(10):e1004401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Griffin JE, Gawronski JD, DeJesus MA, Ioerger TR, Akerley BJ, Sassetti CM (2011) High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog 7(9):e1002251, DOI 10.1371/journal.ppat.1002251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25(14):1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DeJesus MA, Zhang YJ, Sassetti CM, Rubin EJ, Sacchettini JC, Ioerger TR (2013) Bayesian analysis of gene essentiality based on sequencing of transposon insertion libraries. Bioinformatics 29(6):695–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DeJesus MA, Ioerger TR (2013) A Hidden Markov Model for identifying essential and growth-defect regions in bacterial genomes from transposon insertion sequencing data. BMC Bioinformatics 14:303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cole ST, Brosch R, Parkhill J (1998) Deciphering the biology of mycobacterium tuberculosis from the complete genome sequence. Nature 393(6685):537–544, URL 10.1038/31159 [DOI] [PubMed] [Google Scholar]
- 29.Galperin MY, Makarova KS, Wolf YI, Koonin EV (2015) Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Research 43:D261–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Irizarry RA, Wang C, Zhou Y, Speed TP (2009) Gene set enrichment analysis made simple. Stat Methods Med Res 18(6):565–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. PNAS 102(43):15545–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grossmann S, Bauer S, Robinson PN, Vingron M (2007) Improved detection of overrepresentation of Gene-Ontology annotations with parent-child analysis. Bioinformatics 23(22):30243031. [DOI] [PubMed] [Google Scholar]
- 33.Zhang L, Hendrickson RC, Meikle V, Lefkowitz EJ, Ioerger TR, Niederweis M (2020) Comprehensize analysis of iron utilization by Mycobacterium tuberculosis. PLoS Pathogens 16(3):e1008337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DeJesus MA, Nambi S, Smith CM, Baker RE, Sassetti CM, Ioerger TR (2017) Statistical analysis of genetic interactions in Tn-Seq data. Nucleic Acids Res 45(11):e93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tukey J (1949) omparing individual means in the analysis of variance. Biometrics 5(2):99–114 [PubMed] [Google Scholar]
- 36.Subramaniyam S, DeJesus MA, Zaveri A, Smith CM, Baker RE, Ehrt S, Schnappinger D, Sassetti CM, Ioerger TR (2019) Statistical analysis of variability in tnseq data across conditions using zero-inflated negative binomial regression. BMC Bioinformatics 20(1):603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu W, DeJesus MA, Rcker N, Engelhart CA, Wright MG, Healy C, Lin K, Wang R, Park SW, Ioerger TR, Schnappinger D, S E (2017) Chemical genetic interaction profiling reveals determinants of intrinsic antibiotic resistance in Mycobacterium tuberculosis. Antimicrobial Agents Chemotherapy 61(22):e01334–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matern WM, Rifat D, Bader JS, Karakousis PC (2018) Gene enrichment analysis reveals major regulators of Mycobacterium tuberculosis gene expression in two models of antibiotic tolerance. Front Microbiol 9:610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kieser KJ, Boutte CC, Kester JC, Baer CE, Barczak AK, Meniche X, Chao MC, Rego EH, Sassetti CM, Fortune SM, Rubin EJ (2015) Phosphorylation of the Peptidoglycan Synthase PonA1 Governs the Rate of Polar Elongation in Mycobacteria. PLoS Pathog 11(6):e1005010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nambi S, Long JE, Mishra BB, Baker R, Murphy KC, Olive AJ, Nguyen HP, Shaffer SA, Sassetti CM (2015) The Oxidative Stress Network of Mycobacterium tuberculosis Reveals Coordination between Radical Detoxification Systems. Cell Host Microbe 17(6):829–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baranowski C, Welsh MA, Sham LT, Eskandarian HA, Lim HC, Kieser KJ, Wagner JC, McKinney JD, Fantner GE, Ioerger TR, Walker S, Bernhardt TG, Rubin EJ, H RE (2018) Maturing Mycobacterium smegmatis peptidoglycan requires non-canonical crosslinks to maintain shape. Elife p e37516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fu Y, Waldor M, Mekalanos J (2013) Tn-seq analysis of vibrio cholerae intestinal colonization reveals a role for t6ss-mediated antibacterial activity in the host. Cell Host Microbe 14(6):652–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dragset MS, Ioerger TR, Loevenich M, Haug M, Sivakumar N, Marstad A, Cardona PJ, Klinkenberg G, Rubin EJ, Steigedal M, Flo TH (2019) Global assessment of Mycobacterium avium subsp. hominissuis genetic requirement for growth and virulence. mSystems; pp e00402–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.DeJesus MA, Gerrick ER, Xu W, Park SW, Long JE, Boutte CC, Rubin EJ, Schnappinger D, Ehrt S, Fortune SM, Sassetti CM, Ioerger TR (2017) Comprehensive Essentiality Analysis of the Mycobacterium tuberculosis Genome via Saturating Transposon Mutagenesis. MBio 8(1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lampe DJ, Grant TE, M RH (1998) Factors affecting transposition of the Himar1 mariner transposon in vitro. Genetics 149(1):179–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ason B, Reznikoff WS (2004) DNA sequence bias during Tn5 transposition. Journal of Molecular Biology 335:1213–1225 [DOI] [PubMed] [Google Scholar]
- 47.Chao MC, Pritchard JR, Zhang YJ, Rubin EJ, Livny J, Davis BM, Waldor MK (2013) High-resolution definition of the Vibrio cholerae essential gene set with hidden Markov model-based analyses of transposon-insertion sequencing data. Nucleic Acids Res 41(19):9033–9048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pickands J (1975) Statistical inference using extreme order statistics. Annals of Statistics 3:119–131 [Google Scholar]
- 49.Warr AR, Hubbard TP, Munera D, Blondel CJ, zur Wiesch PA, Abel S, Wang X, Davis BM, Waldor MK (2019) Transposon-insertion sequencing screens unveil requirements for ehec growth and intestinal colonization. PLoS Pathogens 15(8):e1007652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Benjamini Y, Yekutieli D (2005) False discovery rate controlling confidence intervals for selected parameters. Journal of the American Statistical Association 100(469):71–81 [Google Scholar]