

Abstract
There is a growing need for public health and veterinary laboratories to perform whole genome sequencing (WGS) for monitoring antimicrobial resistance (AMR) and protecting the safety of people and animals. With the availability of smaller and more affordable sequencing platforms coupled with well-defined bioinformatic protocols, the technological capability to incorporate this technique for real-time surveillance and genomic epidemiology has greatly expanded. There is a need, however, to ensure that data are of high quality. The goal of this study was to assess the utility of a small benchtop sequencing platform using a multi-laboratory verification approach. Thirteen laboratories were provided the same equipment, reagents, protocols and bacterial reference strains. The Illumina DNA Prep and Nextera XT library preparation kits were compared, and 2×150 bp iSeq i100 chemistry was used for sequencing. Analyses comparing the sequences produced from this study with closed genomes from the provided strains were performed using open-source programs. A detailed, step-by-step protocol is publicly available via protocols.io (https://www.protocols.io/view/iseq-bacterial-wgs-protocol-bij8kcrw). The throughput for this method is approximately 4–6 bacterial isolates per sequencing run (20–26 Mb total load). The Illumina DNA Prep library preparation kit produced high-quality assemblies and nearly complete AMR gene annotations. The Prep method produced more consistent coverage compared to XT, and when coverage benchmarks were met, nearly all AMR, virulence and subtyping gene targets were correctly identified. Because it reduces the technical and financial barriers to generating WGS data, the iSeq platform is a viable option for small laboratories interested in genomic surveillance of microbial pathogens.
Keywords: sequencing quality, sequencing accessibility, One Health, foodborne illness
Data Summary
Bench protocol: published on the protocols.io platform: https://www.protocols.io/view/iseq-bacterial-wgs-protocol-bij8kcrw (dx.doi.org/10.17504/protocols.io.bij8kcrw)
Raw data: deposited in NCBI SRA under BioProject PRJNA726961.
Supporting data and computational protocols: provided on the GitHub platform: https://github.com/pkmitchell/iSeq_EC_LM_mlv
The authors confirm all supporting data, code and protocols have been provided within the article or through supplementary data files.
Impact Statement.
The paper describes an effort from a consortium of 13 veterinary laboratories in the USA working in partnership with the US FDA Veterinary Laboratory Investigation and Response and Genome Trakr networks to evaluate a small, affordable platform for bacterial whole genome sequencing. The goal of this work is to make sequencing technology more accessible to small laboratories while still ensuring a high standard of quality. In particular, more widespread deployment of sequencing in animal health laboratories is needed in order to fill critical surveillance gaps. We find that this equipment combined with a step-by-step protocol was easy to deploy in labs that had never done sequencing and provided high-quality genomes for pathogens of human health importance, with some limitations on throughput.
Introduction
Whole genome sequencing (WGS) data have become an integral component of public health investigations and clinical diagnostics. The integration of WGS into foodborne disease surveillance systems has allowed for more rapid and accurate outbreak detection and source attribution [1]. As a result, these data have contributed to high-profile US Food and Drug Administration (FDA) and Centers for Disease Control and Prevention (CDC) investigations of public health incidents such as from Salmonella in pig-ear dog treats causing multistate outbreaks in humans (https://www.cdc.gov/salmonella/pet-treats-07-19/index.html). As the costs of WGS analyses decrease and the accessibility to bioinformatic tools increases, their use in academic and state veterinary laboratories will continue to expand. To ensure that findings are reproducible and reliable, the accuracy of sequencing should be assessed when adopting new platforms.
The FDA has played a key role in increasing the use of WGS-based tools in the USA through the GenomeTrakr and Veterinary Laboratory Investigation and Response Network (Vet-LIRN) programmes, which provide equipment, reagents and training to food and public or animal health testing laboratories. Illumina chemistry is the most widely used in public health for bacterial WGS, but most of their platforms have a high startup cost (>$100000 per instrument) and require labour-intensive maintenance. Vet-LIRN began piloting the Illumina iSeq 100 sequencing platform in 2018, shortly after its release, in order to assess its suitability for laboratories with lower throughput needs. An initial deployment to eight pilot laboratories was expanded to 13 institutions in 2019. Illumina also released the Nextera Flex library preparation kit in 2019, renamed as DNA Prep in 2020, which simplified certain aspects of the procedure compared to the Nextera XT kit that had been used by laboratory networks supported by the FDA and CDC.
The most common bacterial pathogens causing foodborne illness in people, Escherichia coli , Salmonella enterica , Campylobacter species and Listeria monocytogenes , are all considered zoonotic. Several large surveys of pets in the USA and UK have implicated raw meat diets as a major source of foodborne zoonoses [2–4]. E. coli is the most ubiquitous of these and can vary drastically in its pathogenicity, ranging from strains found in normal intestinal flora that are non-pathogenic to those causing significant intestinal or extra-intestinal infections. In animal health, it is one of the most common pathogens tested for antibiotic susceptibility [5] and an emerging concern for multi-drug resistance [6–8]. Because the methods for susceptibility testing are not standardized across animal species [9] and specific drug panels are used for animal testing that do not cover all antibiotics used in humans, it was not until these strains were sequenced that the severity of carbapenem resistance in dogs and cats in the USA was fully appreciated [10]. Having sequencing capacity for commensal and pathogenic Enterobacteriaceae as well as other Gram-negative zoonotic bacterial pathogens is critically needed. E. coli has a large and complex genome that makes it one of the most challenging foodborne bacteria for performing WGS, and therefore an ideal target for interlaboratory verification.
Salmonella enterica subspecies enterica is less common but also has the ability to infect a wide variety of animal species in addition to humans. Drug-resistant non-typhoidal Salmonella is considered a ‘serious’ antibiotic resistance threat by the CDC (https://www.cdc.gov/drugresistance/biggest-threats.html) and is a frequent cause of foodborne illness in people and animals. Salmonella serovar Dublin is an emerging concern due to its ability to cause systemic disease and to the high proportion of isolates capable of multidrug resistance in the USA [11, 12]. Salmonella species have a comparable genome size to E. coli , but are more clonal and consistent in terms of accessory gene content. Listeria species are Gram-positive species common in animals and in the environment, of which L. monocytogenes is the major species causing foodborne illness, as reviewed by Orsi and Wiedmann [13]. L. monocytogenes causes a foodborne illness in humans mainly through various contaminated foods including cheeses, sausages, raw meat, sandwiches, lettuce, raw mushrooms and seafood. People become infected with L. monocytogenes by either eating contaminated food or handling contaminated food including pet food. Sequencing of Gram-positives has different challenges in terms of propagation and DNA extraction, and these organisms should be represented in method development studies as well.
The use of WGS as a primary tool for antimicrobial resistance (AMR) monitoring in these organisms that are well represented in public databases is supported by over a decade of publications [14–16] from the National Antimicrobial Resistance Monitoring System (NARMS), which has begun integrating veterinary laboratory susceptibility testing into its reports. In 2017, Vet-LIRN initiated systematic representation of animal health strains in the NCBI Pathogen Browser (i.e. isolates from sick animals or veterinary necropsies) through its Antimicrobial Resistance Monitoring Programme [11], which provides important data for comparative medicine studies and surveillance. Although the collection of phenotypic antimicrobial susceptibility testing (AST) data is still critically important, WGS overcomes the lack of standardization in AST methods, thanks to public databases such as NCBI and NARMS. Using standardized metrics to assess performance is still needed, not only for new platforms but also for ongoing evaluation. This study aimed to evaluate the utility of a small benchtop sequencing platform to increase the accessibility of WGS to smaller laboratories in order to continue and expand these databases with high-quality sequencing data.
Methods
Participating laboratories and bacterial isolates
Eight laboratories were enrolled in 2018 for the S. enterica and library kit comparison phase of the project, and an additional five laboratories were added in 2019 for the E. coli and L. monocytogenes phase (Table 1). One lab did not complete all runs and was excluded from the analysis. Each site was provided with funding to purchase the iSeq instrument and reagents. For the S. enterica phase, laboratories were provided with a common set of four S. enterica samples as agar slants. Each lab completed two library preparations and sequencing runs, one using the Nextera XT protocol and the other using the Illumina DNA Prep protocol (Illumina).
Table 1.
Details of participating laboratories
|
List of collaborating laboratories |
|---|
|
Cornell University Animal Health Diagnostic Center Florida Dept. of Agriculture and Consumer Services Animal Disease Diagnostic Laboratory* Michigan State University Veterinary Diagnostic Laboratory Mississippi State University Veterinary Research and Diagnostic Laboratory* Ohio Dept. of Agriculture Animal Disease Diagnostic Laboratory Oklahoma State University Animal Disease Diagnostic Laboratory* Oregon State University Veterinary Diagnostic Laboratory* University of Georgia, Athens State Veterinary Diagnostic Laboratory University of Illinois Veterinary Diagnostic Laboratory University of Minnesota Veterinary Diagnostic Laboratory University of Missouri Veterinary Medical Diagnostic Laboratory University of Wisconsin-Madison Veterinary Diagnostic Laboratory* Virginia Tech Animal Laboratory Services |
*Joined during phase II (E. coli and Listeria) but not the Salmonella XT vs. Prep comparison.
For the E. coli and L. monocytogenes phase, three runs were performed all using the Illumina DNA Prep protocol. These runs were designed to test performance closer to the maximum sequence output of the iSeq platform. For the first two sequencing runs, E. coli and L. monocytogenes libraries were pooled evenly as recommended by the manufacturer (equal volumes from each library). For the third, the same set of isolates were sequenced as in the second run but with pooling volume ratios adjusted to improve E. coli coverage by reducing the input volume for the two Listeria isolates by half. Labs that joined during this phase were first required to run the S. enterica panel from the first phase using the Illumina DNA Prep protocol (Illumina) and meet the quality control metrics defined by Timme et al. [17]. All S. enterica , E. coli and L. monocytogenes samples were previously sequenced in other research or surveillance efforts. Characteristics and NCBI accessions of these strains are provided in Tables 2 and S1 (available in the online version of this article). For S. enterica samples, Sequence Read Archive accessions are provided for their original sequencing runs. These were run through the same analysis pipeline as the test samples in order to generate reference annotations. For the E. coli and L. monocytogenes samples, GenBank accessions of previously generated closed genomes are provided as gene annotations were compared against these assemblies.
Table 2.
Summary information for the bacterial strains used in the study
|
Organism |
Sequence type |
Reference accession |
Set |
|---|---|---|---|
|
Salmonella enterica ser. Albany |
292 |
SRR3933152 |
S. enterica panel |
|
Salmonella enterica ser. Typhimurium |
19 |
SRR3933092 |
|
|
Salmonella enterica ser. Enteritidis |
11 |
SRR3933133 |
|
|
Salmonella enterica ser. I 4,[5],12:i:- |
19 |
SRR3933156 |
|
|
Escherichia coli O157:H7 |
11 |
GCF_016458895.1 |
E. coli and L. monocytogenes run 1 |
|
Escherichia coli O157:H7 |
11 |
GCF_016458935.1 |
|
|
Escherichia coli O157:H7 |
11 |
GCF_016458905.1 |
|
|
Escherichia coli O157:H7 |
11 |
GCF_016458945.1 |
|
|
3 |
GCA_004142705.2 |
||
|
7 |
GCA_003900515.2 |
||
|
Escherichia coli O9:H10 |
540 |
GCA_003792995.1 |
E. coli and L. monocytogenes runs 2 and 3 |
|
Escherichia coli O89:H9 |
10 |
GCA_003792995.1 |
|
|
Escherichia coli O83:H42 |
1485 |
GCA_003769125.2 |
|
|
Escherichia coli O117:H42 |
2207 |
GCA_008386435.1 |
|
|
554 |
GCA_004434345.3 |
||
|
6 |
GCA_004501425.3 |
Laboratory procedures
A detailed, step-by-step wet lab protocol including Excel worksheets with calculations tailored to each experiment was provided to participating laboratories and is now publically available on protocols.io (https://www.protocols.io/view/iseq-bacterial-wgs-protocol-bij8kcrw). Briefly, bacterial genomic DNA was extracted using the DNeasy Blood and Tissue Kit (Qiagen). The concentration of purified DNA was measured by Qubit fluorometry (Thermo Fisher). For labs that had internally verified an automated extraction method, they had the option of using that method as long as the DNA concentration was sufficient to have >100 ng in 20 µl. Barcoded sequencing libraries were prepared using either Nextera XT or Nextera Flex/DNA Prep library preparation kits (Illumina). Sequencing was performed using iSeq i100 2×150 bp chemistry (Illumina).
Data analysis
Once each sequencing run was complete, the participating laboratories shared their data with one of the two institutions performing analyses for the study (Cornell and Illinois). Participants then received a standardized report on their run performance. All analysis files used for this study are available at https://github.com/pkmitchell/iSeq_EC_LM_mlv. To evaluate the DNA Prep protocol relative to Nextera XT, data were compared between Salmonella runs 1 and 4. For labs that repeated run 1 due to missed benchmarks the best available replicate was chosen for the comparison. Reads from each run were quality checked using FastQC v.0.11.8 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and assembled using SKESA v.2.3.0 [18]. Assembly quality was checked using QUAST v.5.0.2 [19] and reads were mapped back to the corresponding assembly using BBMap v.38.58 to calculate coverage depth. Average nucleotide identity (ANI) to the correct species was confirmed for all samples. Serotypes were predicted using SISTR v.1.0.2 and AMR genes were identified using AMRFinderPlus v.3.0.12 [20]. Quality control thresholds were set at a minimum of 30× read depth, maximum contig count of 500 and minimum N50 of 10 kbp [17]. AMR genes were considered to be successfully detected if there was a match to the appropriate gene family, and the default parameters were used to determine if genes were complete (>90 %) or partial (50–90 %) matches to the reference in the AMRFinderPlus database. Differences in read and assembly quality metrics were compared using the Wilcoxon rank-sum test. All statistical analyses were run using R v.3.4.2 (https://www.R-project.org/).
The same assembly, quality control, sequence typing and AMR gene detection methods were followed for the E. coli and L. monocytogenes runs. Additionally, for the E. coli samples, ECtyper v.0.8.1 (https://github.com/phac-nml/ecoli_serotyping) was used to identify serotypes and ABRicate v.0.9.8 (https://github.com/tseemann/abricate) was used to search for sets of veterinary and zoonotic virulence factors (https://github.com/pkmitchell/VPECdb). Subtyping and gene predictions for each assembly were compared against those from previously generated closed genomes for each sample. Reads were also mapped against these closed genomes to identify SNPs using the CFSAN SNP pipeline [21]. Quality control thresholds for E. coli were set at a minimum estimated read coverage depth of 40×, maximum contig count of 600 and minimum N50 of 10 kbp. Thresholds for L. monocytogenes were 20× read depth, maximum contig count of 200 and minimum N50 of 10 kbp. To determine whether the pooling adjustment improved E. coli results, the proportion of samples meeting quality control and annotation benchmarks [correct multi-locus sequence type (MLST) profile and serotype called and all expected AMR and virulence gene sequences detected compared to closed reference genomes] was compared between runs 2 and 3 using Fisher’s exact test.
Results
Comparison of Nextera XT and DNA Prep on Salmonella isolates
All samples met read quality benchmarks for both the Prep and XT runs pooling four Salmonella isolates. However, read quality metrics were slightly higher for XT while the average read length was higher and the read length distribution was tighter for Prep (Table 3, Fig. 1). Assembly quality was consistently better using the Prep data, with significantly lower contig counts and higher N50 values (Table 3, Fig. 1). While the median read count and coverage depth did not differ between the two methods, their distributions were tighter using the Prep protocol.
Table 3.
Comparison of DNA Prep and Nextera XT
|
Prep Median (IQR) |
XT Median (IQR) |
P-value |
|
|---|---|---|---|
|
Read count (×105) |
9.18 (7.78–10.77) |
11.24 (8.19–13.03) |
0.059 |
|
Read length (bp) |
293.6 (292.3–294.8) |
276.9 (264.8–290.4) |
<0.001 |
|
Quality Score |
34.4 (34.3–34.9) |
35.3 (35.2–35.4) |
<0.001 |
|
Q30 % |
93.1 (92.4–94.7) |
95.9 (95–96.3) |
<0.001 |
|
Assembly length (Mbp) |
4.90 (4.83–4.95) |
4.87 (4.69–4.93) |
0.100 |
|
Contig count |
137.5 (107.8–161.8) |
257 (176.5–565) |
<0.001 |
|
N50 (kbp) |
95.71 (76.64–119.90) |
44.90 (18.03–63.82) |
<0.001 |
|
Coverage depth |
55.9 (46.4–65.6) |
65.3 (39.5–77.5) |
0.226 |
IQR, interquartile range.
Fig. 1.

Comparison of library preparation methods with four pooled Salmonella strains.
All expected AMR genes were detected in all samples from both methods, though there were five instances in which the correct gene family, but not the specific allele, were found in the XT assembly. Each of these non-specific matches were attributable to partial length matches and four of the five affected the same gene target. MLST profiles were successfully identified from all Prep assemblies but missed in 5/32 (15.6 %) XT assemblies. Of these five, one had a partial match for 1/7 alleles, one had a partial match for two alleles, and for the remaining three, the E. coli MLST scheme was selected as the best match rather than the S. enterica scheme. When the programme was manually forced to use the S. enterica scheme for these three, one matched to 2/7, another had partial matches to 4/7 and the third had partial matches to 5/7. The correct serotype was identified in all Prep assemblies and all but one XT assembly.
Assessment of with E. coli and L. monocytogenes with even pooling
The two sets of runs with equal pooling of four E. coli with two L . monocytogenes (all libraries made with DNA Prep) produced consistently high read quality (Fig. 2). However, of the 96 E. coli samples run by the 12 labs across these two runs, 25 (26.0 %) were below the 40× read depth target. Low-coverage samples were spread across labs, with 9/12 having at least one E. coli sample below the read depth target and 7/12 having multiple samples below target. One E. coli (1.0 %) and one L. monocytogenes (2.1 %) sample produced an assembly outside of the expected length. Among E. coli isolates, 13/96 (13.5 %) had >600 contigs and 10/96 (10.4 %) had an N50 <10 kbp. No E. coli samples that met the estimated coverage depth threshold failed to meet the N50 and contig count targets (Fig. 3). The correct MLST profile was called in 84/96 (87.5 %) E. coli samples, the correct O- and H-antigens were called in 94/96 (97.9 %), all expected AMR genes were detected in 84/96 (87.5 %) and all expected virulence factors were detected in 84/96 (87.5 %). An apparent sample swap in one run, wherein the sample identifiers were switched between two samples, accounted for two of the samples in which AMR genes and virulence factors were missed.
Fig. 2.
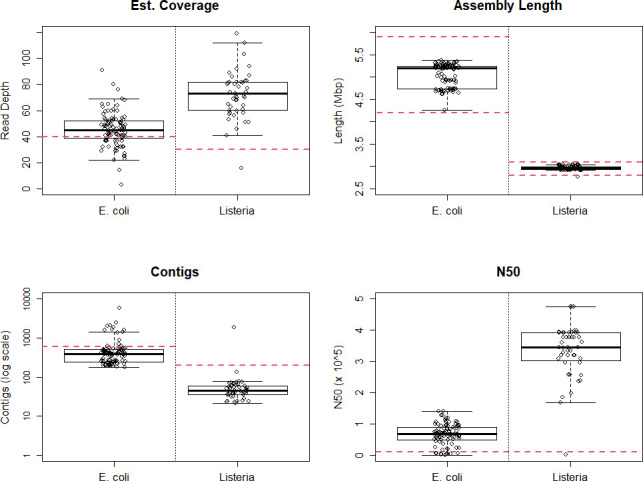
Box plots showing the distribution of quality metrics. The top and bottom of the box show the interquartile range and the midline shows the median. Red dashed lines show the target thresholds.
Fig. 3.

Assembly quality vs. coverage at higher loading. Dashed lines show the target thresholds for coverage depth, N50, and contig count.
Of 48 L . monocytogenes samples, one failed to sequence entirely and was excluded from further analysis and another missed the 20× read depth target, resulting in 46/48 (95.8 %) successfully meeting the read coverage target. Among the 47 L . monocytogenes isolates with any sequence data, one (2.1 %) was below the read depth target, had >200 contigs and had an N50<10 kbp. The correct MLST profile was called in 44/47 (93.6 %) L . monocytogenes samples and all expected AMR genes were detected in 45/47 (95.7 %), with two of the missed MLST calls and both sets of missed AMR genes resulting from another sample swap. The remaining missed MLST call was in the sample that missed the other quality benchmarks.
Comparison of equal volume pooling and adjustment by genome size
While the proportion of E. coli samples meeting quality benchmarks was higher after altering the pooling ratios to increase E. coli sequence output and improve coverage, these differences were not statistically significant (Fig. 4). With even pooling, 35/48 (72.9 %) met the coverage depth threshold, 42/48 (87.5 %) met the contig count threshold and 43/48 (89.6 %) met the N50 threshold. With adjusted pooling, 37/48 (77.1 %) met the coverage threshold, 46/48 (95.8 %) met the contig threshold and 46/48 (95.8 %) met the N50 threshold (Fisher’s exact test P-values: 0.82, 0.27 and 0.44 respectively). Similarly, changes in the proportions of samples in which the correct serotype (97.9 % vs. 100 %, P=1.00) and MLST profile (89.6 % vs. 93.8 %, P=0.20) were identified and all AMR genes (79.2 % vs. 89.6 %, P=0.26) and virulence genes (89.6 % vs. 91.7 %, P=1.00) were detected were not statistically significant.
Fig. 4.

Box plots showing the distribution of quality metrics for equal volume vs. genome size adjusted pooling. The upper and lower bounds of the box show the interquartile range and the midline shows the median. Target thresholds are shown with dashed red lines.
Discussion
Our findings demonstrate that the iSeq platform can generate high-quality, accurate data for bacterial WGS, and it is a cost-effective addition to the AMR surveillance armamentarium. It seems ideally suited to small laboratories with low sample throughput, or as a supplement to other platforms to have on hand for time-sensitive situations. Although the options for run times are good (9.5–19 h), the cartridges have a minimum thawing time of 6 h, which is a drawback. Another major platform that is comparable in terms of throughput and cost is Oxford Nanopore Technologies, which has some attractive features such as a lower cost for equipment at less that $1000 per unit, longer reads up to 2 Mbases, and potential for realtime sequence data and to selectively sequence data. However, there is slightly lower accuracy in individual reads with this technology, and the newer basecalling algorithms require access to High Performance Computing Clusters or specialized GPU-based computers in order to keep up with the sequencing output. The flongle provides enough data at up to 2 Gb to sequence multiple bacterial genomes through barcoding and costs $90 per flowcell (compared to 1.2 Gb and $560 for iSeq), although they are currently available in packs of 12 with a shelflife of 3 months at room temperature (bulk pricing for eight iSeq kits is $495 each with longer cold storage). Combined with a ligation-based library preparation that requires 2 h and costs $100, this is a potential alternative. The Illumina DNA Prep library kit costs $48 per prep, if using the manufacturer-recommended volumes, and takes approximately 3.5 h. The larger MinION flow cell used on the MinION and GridION has the potential to sequence 10 Gbases or more of data at a cost of $900 for a single flow cell and as little as $500 per cell if purchased in larger quantities and would be similar to the cost of the iSeq cassettes with higher output. Overall, this is an alternative to the iSeq, but with some specific infrastructure requirements and slightly less refined bioinformatics. If a laboratory made WGS a routine part of their diagnostic workflow, the bulk purchase of library preparation kits and flow cells could make iSeq and Nanopore-based technologies similarly economically feasible. Similar studies would need to be performed in order to assess accuracy and utility for bacterial genomic epidemiology and AMR surveillance.
The level of bioinformatic skills needed to analyse and interpret the data from the iSeq platform is identical to that of the bigger Illumina platforms, but the lower number of samples being analysed lends itself well to cloud-based tools such as Galaxy (usegalaxy.org or galaxytrakr.org) and NCBI Pathogen Browser (www.ncbi.nlm.nih.gov/pathogens) that have point-and-click interfaces. The benchmarks used in this study were derived from standards set by the FDA GenomeTrakr Program [17], which are harmonized with NCBI and CDC PulseNet. Our findings suggest that labs starting out using this platform for bacteria should initially run no more than four isolates at a time, and with experience may be able to load 5–6 with smaller genomes. The DNA Prep library preparation method produced higher quality data than Nextera XT, and for pushing the throughput limits of this platform would be worth the slightly higher cost per prep. The Illumina DNA Prep library preparation method was designed to be more consistent and automation-friendly. The initial input and final pooling steps are much simplified compared to Nextera XT, particularly if using the manufacturer-recommended method of pooling equal volumes of purified libraries. We nevertheless attempted to scale the input volumes to improve assembly quality at the higher loading level. Adjustment of pooling volumes based on genome size and complexity gave modest improvement, which was not sufficient to reliably achieve benchmarks for all samples. Given that E. coli is similar in genome size to S. enterica but has a more complex and variable accessory genome, it is possible that a higher number of isolates could be loaded if E. coli were not included. Further refinement based on an individual lab’s typical case load would be expected. Pooling could also be adjusted by final DNA concentration, or by molarity, and different methods for checking the concentration prior to loading (e.g. quantitative PCR) could be explored.
Large platform WGS analysis fits best with a batch-wise approach for sample analysis, which is not ideal in a core hospital facility, or surveillance veterinary work with lower-throughput applications [22]. Overall, the iSeq platform seems to be a viable option for laboratories that need a sequencing option with low start-up and maintenance costs but at a somewhat higher per-sample cost. Regardless of platform, some specialized training is needed for both the wet and dry components of the workflow. The detailed protocol provided is intended to facilitate the technical hurdles on the wet lab side, and cloud-based analysis tools such as Galaxy (https://usegalaxy.org/) and NCBI Pathogen Browser (https://www.ncbi.nlm.nih.gov/pathogens) have greatly minimized the analytical barriers. Although not included in our study, iSeq seems ideal for amplicon-based applications that require much less sequencing power, for example influenza typing or clinical targeted metagenomics. Academic, food and/or diagnostic laboratories that have time-sensitive sequencing needs could benefit from this platform, but should also consider using other platforms if ultra-rapid results are needed. Ultimately, the time, cost and quality should be taken into consideration when choosing an next-generation sequencing platform. The capacity to perform advanced molecular characterization of bacterial isolates in a reliable manner is critical to our ability to monitor AMR across the One Health spectrum. Making this technology widely accessible will continue to have important impacts for both human and veterinary health.
Supplementary Data
Funding information
This study was funded (PAR-18-604) and performed in collaboration with the Food and Drug Administration’s Veterinary Laboratory Investigation and Response Network (FDA Vet-LIRN) under cooperative agreements 1U18FD006714-01 to L.B.G. and 1U18FD006866-01 to L.W. Additional funding for participating laboratories was as follows: 5U18FD006160-02, Florida Department of Agriculture and Consumer Services; 1U18 FD006444-01, Athens Veterinary Diagnostic Laboratory, The University of Georgia; 1U18 FD006593-01, Michigan State University, Veterinary Diagnostic Laboratory; 1U18 FD006568-01, University of Minnesota, Veterinary Diagnostic Laboratory; 1U18 FD006460-01, University of Missouri, Veterinary Medical Diagnostic Laboratory; 1U18 FD006866-01, 1U18 FD006714-01, Mississippi State University, Veterinary Diagnostic Laboratory; 1U18 FD006555-01, Ohio Animal Disease Diagnostic Laboratory; 1U18FD006671-01, Oklahoma Animal Disease Diagnostic Laboratory, Oklahoma State University; 1U18 FD006719-01, Oregon Veterinary Diagnostic Laboratory, Oregon State University; 1U18 FD006445-01, Virginia-Maryland College of Veterinary Medicine; 1U18 FD006859-01, University of Wisconsin-Madison Veterinary Diagnostic Laboratory.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Footnotes
Abbreviations: AMR, antimicrobial resistance; AST, antimicrobial susceptibility testing; CDC, Centers for Disease Control; FDA, Federal Drug Administration; MLST, multi-locus sequence typing; NARMS, National Antimicrobial Resistance Monitoring System; Vet-LIRN, Veterinary Laboratory Investigation and Response Network; WGS, whole genome sequencing.
All supporting data, code and protocols have been provided within the article or through supplementary data files. A supplementary table is available with the online version of this article.
References
- 1.Besser J, Carleton HA, Gerner-Smidt P, Lindsey RL, Trees E. Next-generation sequencing technologies and their application to the study and control of bacterial infections. Clin Microbiol Infect. 2018;24:335–341. doi: 10.1016/j.cmi.2017.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Davies RH, Lawes JR, Wales AD. Raw diets for dogs and cats: a review, with particular reference to microbiological hazards. J Small Anim Pract. 2019;60:329–339. doi: 10.1111/jsap.13000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nemser SM, Doran T, Grabenstein M, McConnell T, McGrath T, et al. Investigation of Listeria, Salmonella, and toxigenic Escherichia coli in various pet foods. Foodborne Pathog Dis. 2014;11:706–709. doi: 10.1089/fpd.2014.1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reimschuessel R, Grabenstein M, Guag J, Nemser SM, Song K, et al. Multilaboratory survey to evaluate salmonella prevalence in diarrheic and nondiarrheic dogs and cats in the United States between 2012 and 2014. J Clin Microbiol. 2017;55:1350–1368. doi: 10.1128/JCM.02137-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dargatz DA, Erdman MM, Harris B. A survey of methods used for antimicrobial susceptibility testing in veterinary diagnostic laboratories in the United States. J Vet Diagn Invest. 2017;29:669–675. doi: 10.1177/1040638717714505. [DOI] [PubMed] [Google Scholar]
- 6.Chen Y, Liu Z, Zhang Y, Zhang Z, Lei L, et al. Increasing prevalence of ESBL-producing multidrug resistance Escherichia coli from diseased pets in Beijing, China From 2012 to 2017. Front Microbiol. 2019;10:2852. doi: 10.3389/fmicb.2019.02852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hong JS, Song W, Jeong SH. Molecular characteristics of NDM-5-producing Escherichia coli from a cat and a dog in South Korea. Microb Drug Resist. 2020;26:1005–1008. doi: 10.1089/mdr.2019.0382. [DOI] [PubMed] [Google Scholar]
- 8.Tyson GH, Li C, Ceric O, Reimschuessel R, Cole S, et al. Complete genome sequence of a carbapenem-resistant Escherichia coli isolate with bla NDM-5 from a dog in the United States. Microbiol Resour Announc. 2019;8:e00872-19. doi: 10.1128/MRA.00872-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.CLSI Understanding Susceptibility Test Data as a Component of Antimicrobial Stewardship in Veterinary Settings. 1st ed. Wayne, PA: Clinical & Laboratory Standards Institute; 2019. [Google Scholar]
- 10.Cole SD, Peak L, Tyson GH, Reimschuessel R, Ceric O, et al. New Delhi Metallo-β-Lactamase-5-Producing Escherichia coli in companion animals, United States. Emerg Infect Dis. 2020;26:381–383. doi: 10.3201/eid2602.191221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ceric O, Tyson GH, Goodman LB, Mitchell PK, Zhang Y, et al. Enhancing the one health initiative by using whole genome sequencing to monitor antimicrobial resistance of animal pathogens: Vet-LIRN collaborative project with veterinary diagnostic laboratories in United States and Canada. BMC Vet Res. 2019;15:130. doi: 10.1186/s12917-019-1864-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hsu C-H, Li C, Hoffmann M, McDermott P, Abbott J, et al. Comparative genomic analysis of virulence, antimicrobial resistance, and plasmid profiles of salmonella dublin isolated from sick cattle, retail beef, and humans in the United States. Microb Drug Resist. 2019;25:1238–1249. doi: 10.1089/mdr.2019.0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Orsi RH, Wiedmann M. Characteristics and distribution of Listeria spp., including Listeria species newly described since 2009. Appl Microbiol Biotechnol. 2016;100:5273–5287. doi: 10.1007/s00253-016-7552-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tyson GH, McDermott PF, Li C, Chen Y, Tadesse DA, et al. WGS accurately predicts antimicrobial resistance in Escherichia coli . J Antimicrob Chemother. 2015;70:2763–2769. doi: 10.1093/jac/dkv186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McDermott PF, Tyson GH, Kabera C, Chen Y, Li C, et al. Whole-genome sequencing for detecting antimicrobial resistance in nontyphoidal salmonella. Antimicrob Agents Chemother. 2016;60:5515–5520. doi: 10.1128/AAC.01030-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.FDA NARMS Now: Integrated Data. Rockville, MD: US Department of Health and Human Services; [Google Scholar]
- 17.Timme RE, Wolfgang WJ, Balkey M, Venkata SLG, Randolph R, et al. Optimizing open data to support one health: best practices to ensure interoperability of genomic data from bacterial pathogens. One Health Outlook. 2020;2:20. doi: 10.1186/s42522-020-00026-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Souvorov A, Agarwala R, Lipman DJ. SKESA: strategic k-mer extension for scrupulous assemblies. Genome Biol. 2018;19:153. doi: 10.1186/s13059-018-1540-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29:1072–1075. doi: 10.1093/bioinformatics/btt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feldgarden M, Brover V, Haft DH, Prasad AB, Slotta DJ, et al. Validating the AMRFinder tool and resistance gene database by using antimicrobial resistance genotype-phenotype correlations in a collection of isolates. Antimicrob Agents Chemother. 2019;63:e00483-19. doi: 10.1128/AAC.00483-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davis S, Pettengill JB, Luo Y, Payne J, Shpuntoff A, et al. CFSAN SNP Pipeline: an automated method for constructing SNP matrices from next-generation sequence data. PeerJ Comput Sci. 2015;1:e20. doi: 10.7717/peerj-cs.20. [DOI] [Google Scholar]
- 22.Rossen JWA, Friedrich AW, Moran-Gilad J, ESCMID Study Group for Genomic and Molecular Diagnostics (ESGMD) Practical issues in implementing whole-genome-sequencing in routine diagnostic microbiology. Clin Microbiol Infect. 2018;24:355–360. doi: 10.1016/j.cmi.2017.11.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.