

Abstract
Background
Axial spondyloarthritis (axSpA) comprises ankylosing spondylitis (radiographic axSpA) and non‐radiographic (nr‐)axSpA and is associated with psoriasis, uveitis and inflammatory bowel disease. Non‐steroidal anti‐inflammatory drugs (NSAIDs) are recommended as first‐line drug treatment.
Objectives
To determine the benefits and harms of NSAIDs in axSpA.
Search methods
We searched CENTRAL, MEDLINE and EMBASE to 18 June 2014.
Selection criteria
Randomised controlled trials (RCTs) or quasi‐RCTs of NSAIDs versus placebo or any comparator in adults with axSpA and observational cohort studies studying the long term effect (≥ six months) of NSAIDs on radiographic progression or adverse events (AEs). The main comparions were traditional or COX‐2 NSAIDs versus placebo. The major outcomes were pain, Bath Ankylosing Spondylitis Disease Activity Index (BASDAI), Bath Ankylosing Spondylitis Functional Index (BASFI), Bath Ankylosing Spondylitis Metrology Index (BASMI), radiographic progression, number of withdrawals due to AEs and number of serious AEs
Data collection and analysis
Two review authors independently selected trials for inclusion, assessed the risk of bias, extracted data and assessed the quality of evidence for major outcomes using GRADE.
Main results
We included 39 studies (35 RCTs, two quasi‐RCTs and two cohort studies); and 29 RCTs and two quasi‐RCTs (n = 4356) in quantitative analyses for the comparisons: traditional NSAIDs versus placebo, cyclo‐oxygenase‐2 (COX‐2) versus placebo, COX‐2 versus traditional NSAIDs, NSAIDs versus NSAIDs, naproxen versus other NSAIDs, low versus high dose. Most trials were at unclear risk of selection bias (n = 29), although blinding of participants and personnel was adequate in 24 trials. Twenty‐five trials had low risk of attrition bias and 29 trials had low risk of reporting bias. Risk of bias in both cohort studies was high for study participation, and low or unclear for all other criteria. No trials in the meta‐analyses assessed patients with nr‐axSpA.
Traditional NSAIDs were more beneficial than placebo at six weeks. High quality evidence (four trials, N=850) indicates better pain relief with NSAIDs (pain in control group ranged from 57 to 64 on a 100mm visual analogue scale (VAS) and was 16.5 points lower in the NSAID group (95% confidence interval (CI) ‐20.8 to ‐12.2), lower scores indicate less pain, NNT 4 (3 to 6)); moderate quality evidence (one trial, n = 190) indicates improved disease activity with NSAIDs (BASDAI in control group was 54.7 on a 100‐point scale and was 17.5 points lower in the NSAID group, 95% CI ‐23.1 to ‐11.8), lower scores indicate less disease activity, NNT 3 (2 to 4)); and high quality evidence (two trials, n = 356) indicates improved function with NSAIDs (BASFI in control group was 50.0 on a 100‐point scale and was 9.1 points lower in the NSAID group (95% CI ‐13.0 to ‐5.1), lower scores indicate better functioning, NNT 5 (3 to 8)). High (five trials, n = 1165) and moderate (three trials, n = 671) quality evidence (downgraded due to potential imprecision) indicates that withdrawals due to AEs and number of serious AEs did not differ significantly between placebo (52/1000 and 2/1000) and NSAID (39/1000 and 3/1000) groups after 12 weeks (risk ratio (RR) 0.75, 95% CI 0.46 to 1.21; and RR 1.69, 95% CI 0.36 to 7.97, respectively). BASMI and radiographic progression were not reported.
COX‐2 NSAIDS were also more efficacious than placebo at six weeks. High quality evidence (two trials, n = 349) indicates better pain relief with COX‐2 (pain in control group was 64 points and was 21.7 points lower in the COX‐2 group (95% CI ‐35.9 to ‐7.4), NNT 3 (2 to 24)); moderate quality evidence (one trial, n = 193) indicates improved disease activity with COX‐2 (BASDAI in control groups was 54.7 points and was 22 points lower in the COX‐2 group (95% CI ‐27.4 to ‐16.6), NNT 2 (1 to 3)); and high quality evidence (two trials, n = 349) showed improved function with COX‐2 (BASFI in control group was 50.0 points and was 13.4 points lower in the COX‐2 group (95% CI ‐17.4 to ‐9.5), NNT 3 (2 to 4)). Low and moderate quality evidence (three trials, n = 669) (downgraded due to potential imprecision and heterogeneity) indicates that withdrawals due to AEs and number of serious AEs did not differ significantly between placebo (11/1000 and 2/1000) and COX‐2 (24/1000 and 2/1000) groups after 12 weeks (RR 2.14, 95% CI 0.36 to 12.56; and RR 0.92, 95% CI 0.14 to 6.21, respectively). BASMI and radiographic progression were not reported.
There were no significant differences in benefits (pain on VAS: MD ‐2.62, 95% CI ‐10.99 to 5.75; three trials, n = 669) or harms (withdrawals due to AEs: RR 1.04, 95% CI 0.60 to 1.82; four trials, n = 995) between NSAID classes. While indomethacin use resulted in significantly more AEs (RR 1.25, 95% CI 1.06 to 1.48; 11 studies, n = 1135), and neurological AEs (RR 2.34, 95% CI 1.32 to 4.14; nine trials, n = 963) than other NSAIDs, these findings were not robust to sensitivity analyses. We found no important differences in harms between naproxen and other NSAIDs (three trials, n = 646), although other NSAIDs appeared more effective for relieving pain (MD 6.80, 95% CI 3.72 to 9.88; two trials, n = 232). We found no clear dose‐response effect on benefits or harms (five studies, n = 1136). Single studies suggest NSAIDs may be effective in retarding radiographic progression, especially in certain subgroups of patients, e.g. patients with high CRP, and that this may be best achieved by continuous rather than on‐demand use of NSAIDs.
Authors' conclusions
High to moderate quality evidence indicates that both traditional and COX‐2 NSAIDs are efficacious for treating axSpA, and moderate to low quality evidence indicates harms may not differ from placebo in the short term. Various NSAIDs are equally effective. Continuous NSAID use may reduce radiographic spinal progression, but this requires confirmation.
Plain language summary
Non‐steroidal anti‐inflammatory drugs (NSAIDs) for axial spondyloarthritis (axSpA)
In this Cochrane review of the effect of NSAIDs for people with axSpA (including ankylosing spondylitis and non‐radiographic (nr‐)axSpA), we included 39 studies with 4356 people (search up to 18 June 2014). One study looked at people with nr‐axSpA.
In people with axSpA:
Traditional and COX‐2 NSAIDs improve pain, disease activity and functioning (high quality evidence) and probably do not result in more withdrawals due to adverse events or serious adverse events compared with placebo in the short term (moderate quality evidence, as some outcomes suffered potential imprecision). We often do not have precise information about side effects, particularly for rare but serious side effects. Possible side effects may include gastrointestinal complaints. Rare complications may include gastrointestinal bleeding or problems with heart or blood vessels.
What is axSpA and what are NSAIDs?
AxSpA is a form of arthritis involving the joints of the pelvis or spine or both. It causes pain and stiffness in those regions and can result in deformities of the spine and poor functioning.
NSAIDs are commonly used to reduce pain and inflammation and are considered first‐line treatment for people with axSpA. COX‐2 NSAIDs are a subgroup of NSAIDs that potentially lead to less gastrointestinal complaints than traditional NSAIDs, although there is evidence that they may lead to other complications, like a higher risk of cardiovascular events.
What happens to people with axSpA taking NSAIDs after six weeks:
People who used a traditional NSAID rated their pain to be 16.5 points lower on a scale of 0 to 100 (lower score means less pain) (17% absolute improvement).
‐ People using a traditional NSAID rated their pain to be 44 points; people using placebo 60.5 points.
People who used a traditional NSAID rated their disease activity to be 17.5 points lower on a scale of 0 to 100 (lower score means less disease activity) (18% absolute improvement).
‐ People using a traditional NSAID rated their disease activity to be 37.2 points; people using placebo 54.7 points.
People who used a traditional NSAID rated their functioning to be 9.1 points lower on a scale of 0 to 100 (lower score means better functioning) (9% absolute improvement).
‐ People using a traditional NSAID rated their functioning to be 40.9 points; people using placebo 50.0 points.
Thirteen people less out of 1,000 stopped taking a traditional NSAID before the end of the study because of side effects (0% absolute difference).
‐ 39 people out of 1,000 receiving a traditional NSAID stopped, compared to 52 out of 1,000 receiving placebo.
One more person out of 1,000 had a serious adverse event while taking a traditional NSAID during the study (0% absolute difference).
‐ 3 people out of 1,000 receiving a traditional NSAID had a serious adverse event during the study, compared to 2 out of 1,000 receiving placebo.
People who used a COX‐2 NSAID rated their pain to be 21.7 points lower on a scale of 0 to 100 (22% absolute improvement).
‐ People using a COX‐2 NSAID rated their pain to be 42.3 points; people using placebo 64 points.
People who used a COX‐2 NSAID rated their disease activity to be 22 points lower on a scale of 0 to 100 (22% absolute improvement).
‐ People using a COX‐2 NSAID rated their disease activity to be 32.7 points; people using placebo 54.7 points.
People who used a selective COX‐2 NSAID rated their functioning to be 13.4 points lower on a scale of 0 to 100 (13% absolute improvement).
‐ People using a COX‐2 NSAID rated their functioning to be 36.6 points; people using placebo 50.0 points.
Thirteen people more out of 1,000 stopped taking a COX‐2 NSAID before the end of the study because of side effects (2% absolute difference).
‐ 24 people out of 1,000 receiving a COX‐2 NSAID stopped, compared to 11 out of 1,000 receiving placebo.
The same number of people had a serious adverse event while taking a COX‐2 NSAID or placebo during the study (0% absolute difference).
‐ 2 people out of 1,000 receiving a COX‐2 NSAID had a serious adverse event during the study, compared to 2 out of 1,000 receiving placebo.
Summary of findings
Background
Description of the condition
Spondyloarthritis (SpA) is an umbrella term that comprises ankylosing spondylitis (AS), psoriatic arthritis, arthritis/spondylitis with inflammatory bowel disease, and reactive arthritis (Amor 1990; Dougados 1991; van der Linden 1984). Patients with typical features of SpA that do not fulfil the criteria for one of these subgroups have also been incorporated in the SpA concept as undifferentiated SpA (Khan 1985; Khan 1990). Patients with SpA can also be distinguished according to their clinical presentation as patients with either predominantly peripheral (including peripheral arthritis, enthesitis and dactylitis) or axial (inflammation of the sacroiliac joints or the spine, or both) SpA (axSpa), with some overlap between these subtypes.
Patients with axSpA constitute a partly heterogeneous group of patients with specific clinical manifestations, such as spinal inflammation (Braun 2007). Sacroiliac joint involvement is considered the hallmark of the disease and structural consequences of sacroiliitis, visible on radiographs (using the modified New York criteria), are required for the classification of AS, a major subgroup of axSpA (van der Linden 1984). However, there is evidence from several studies that it often takes years from the onset of back pain until definite sacroiliitis on plain radiographs is detectable (Mau 1988; Oostveen 1999; Said‐Nahal 2000; Sampaio‐Barros 2001). This causes a diagnostic delay, on average six to eight years, as sacroiliitis on radiographs is a requirement for classification according to the modified New York criteria (Dougados 1995; Mau 1988; Rudwaleit 2005). Consequently, while these criteria perform well in patients with established disease, they lack sensitivity in early disease. The absence of radiographic sacroiliitis during the early stage of disease does not necessarily imply that inflammation is absent in the sacroiliac joints, as inflammation has been demonstrated on magnetic resonance imaging (MRI) in people with normal plain radiographs (Rudwaleit 2009a). Thus, the presence and absence of radiographic sacroiliitis in patients with SpA may represent different stages of one disease continuum (Rudwaleit 2005). Furthermore, the presence or absence of radiographic sacroiliitis does not affect the burden of disease (Rudwaleit 2004).
Classification criteria for axSpA have recently been developed by the Assessment of SpondyloArthritis International Society (ASAS) (Rudwaleit 2009a; Rudwaleit 2009b). According to these criteria, a patient with chronic back pain (≥ three months) and age at onset of < 45 years can be classified as having axSpA in the presence of sacroiliitis (either definite radiographic sacroiliitis or active inflammation of sacroiliac joints on MRI, which is highly suggestive of sacroiliitis associated with SpA) plus at least one typical SpA feature, or in the presence of HLA‐B27 plus at least two other SpA features (Rudwaleit 2009b). Using this set of criteria, patients can be classified as not having established radiographic changes in the sacroiliac joint, i.e. non‐radiographic axial SpA (nr‐axSpA), or as having developed radiographic changes in the sacroiliac joint, i.e. radiographic axSpA or AS. In Western European countries axSpA prevalence is between 0.3% and 2.5%. The prevalence rate of AS in Western countries is up to 0.53% (Stolwijk 2012).
Description of the intervention
Non‐steroidal anti‐inflammatory drugs (NSAIDs), including traditional NSAIDs and selective cyclo‐oxygenase (COX) inhibitors, are well‐established drugs commonly used to treat people with inflammatory conditions. The primary goal in the treatment of patients with AS is to maximize long‐term health‐related quality of life through control of symptoms and inflammation, to prevent progressive, structural damage, and to preserve or normalize function and social participation (Braun 2011). NSAIDs are recommended as first‐line drug treatment for patients with axSpA with pain and stiffness (Braun 2011). Current recommendations for the use of tumour necrosis factor (TNF) inhibitors in patients with axSpA recommend that patients should have had an adequate therapeutic trial of at least two NSAIDs, defined as at least two NSAIDs over a four‐week period in total at maximum recommended or tolerated anti‐inflammatory dose unless contraindicated (van der Heijde 2011). Continuous treatment with NSAIDs is preferred for patients with persistently active, symptomatic disease (Braun 2011).
Nevertheless, cardiovascular, gastrointestinal and renal risks should be taken into account when prescribing NSAIDs (Braun 2011). There is overwhelming evidence that these agents can lead to a variety of gastrointestinal toxicities by inhibition of mucosal prostaglandin production (Armstrong 1987; Fries 1991; Gabriel 1991; Griffin 1988; Langman 1994; MacDonald 1997; Stalnikowicz 1993). The increased risk of cardiovascular events was highlighted by the withdrawal of rofecoxib from the market due to findings that it may have increased the risk of myocardial infarction (Bresalier 2005). Several other NSAIDs have also been shown to be associated with an increased risk of cardiovascular disease (Kearney 2006). Recently, a network meta‐analysis on the cardiovascular safety of NSAIDs was published, which concluded that in comparison to placebo all studied NSAIDs posed an increased risk of cardiovascular morbidity and mortality (Trelle 2011). These harmful effects may occur through a variety of mechanisms, including an increase in blood pressure and peripheral oedema. NSAIDs have also consistently been associated with the development of congestive heart failure (Feenstra 2002). Furthermore, evidence exists that NSAIDs may produce either reversible or permanent renal toxicity and a variety of negative effects on electrolyte and water homeostasis (Murray 1993).
How the intervention might work
The primary site of action of NSAIDs is the enzyme COX that converts arachidonic acid into prostaglandins, which ‐ amongst other functions ‐ mediate inflammation and pain. Two forms of COX have been described, COX‐1 and COX‐2 (Vane 1998). COX‐1 is normally present in high concentration in platelets, vascular endothelial cells, stomach and kidney collecting tubules. It is responsible for the production of prostaglandins which are essential for maintenance of normal endocrine and renal function, gastric mucosal integrity and haemostasis. COX‐2 was first thought to be virtually undetectable in most tissues under physiological circumstances, and its activity only increased by inflammatory and mitogenic stimuli. However, the conventional distinctions between COX‐1 and COX‐2 (that prostaglandins important in physiological function are produced solely via COX‐1 and those that mediate local inflammation are produced solely via COX‐2) have been challenged by more recent evidence (Bertolini 2002). Most NSAIDs are non‐selective COX inhibitors, which means they inhibit both COX‐1 and COX‐2. A newer class of NSAIDs are the COX inhibitors, which selectively inhibit COX‐2.
It has recently been suggested that NSAIDs may also inhibit structural progression in the spine in patients with AS, especially in the group of patients with an elevated C‐reactive protein (CRP) (Poddubnyy 2012; Wanders 2005). This is in contrast to TNF inhibitors, which have been shown not to inhibit radiographic progression despite their ability to rapidly restore CRP levels and the erythrocyte sedimentation rate (ESR) to normal (van der Heijde 2008a; van der Heijde 2008b; van der Heijde 2009b). These findings suggest that there are inflammation‐independent mechanisms, sensitive to the effects of NSAIDs, which contribute to the process of syndesmophyte formation, the hallmark of structural progression in AS. A possible biological explanation for this is that COX‐2 is relevant in bone formation. This was concluded by a study in which both COX‐2 knock‐out mice and mice treated with COX‐2 inhibiting drugs showed reduced callus formation after a fracture, which seems to be attributable to suppression of osteoblasts (Zhang 2002). Furthermore, in an immuno‐histochemical analysis comparing synovial tissue samples obtained from patients with different forms of inflammatory arthritis (AS, osteoarthritis, rheumatoid arthritis and psoriatic arthritis), COX‐2 expression appeared to be highest in the samples from patients with AS (Siegle 1998). So if an upregulated level of COX‐2 in AS is indeed responsible for increased osteoblastic bone formation (in the form of syndesmophytes), inhibition of COX‐2 with NSAIDs might be a rational approach to prevent the occurrence of syndesmophytes. In contrast to the effects of NSAIDs seen on bone formation on radiographs, a six‐week open‐label study in patients with AS treated with etoricoxib found only a small effect on MRI‐detected lesions. However, other studies have consistently shown a substantial improvement of MRI lesions after anti‐TNF therapy (Braun 2006; Jarrett 2009).
Why it is important to do this review
There are several reasons why it is important to do this Cochrane review. The review will synthesise the existing data on the benefits and harms of NSAIDs in controlling disease activity, symptoms and radiographic progression in axSpA, and, for the first time, will also include nr‐axSpA. Although NSAIDs are recommended as first‐line therapy for axSpA, no systematic review has yet been undertaken on their effect considering outcomes relevant for clinical practice as recommended by ASAS (Sieper 2009). Although NSAIDs are recommended as first‐line therapy, they may have important side effects, so it is crucial to know whether the benefits offset the risks, especially because the therapy is often given for extended periods of time. Finally, this will be the first systematic review to examine the effect of NSAIDs on radiographic damage, an outcome that has been shown to be associated with impaired spinal mobility and function (Machado 2010; Machado 2011a). This review should provide clinicians with information to guide their decisions about NSAID therapy for patients with axSpA.
Objectives
To assess the benefit and harm of NSAIDs in controlling disease activity, symptoms and radiographic progression in patients with axSpA.
Methods
Criteria for considering studies for this review
Types of studies
We included all published randomised controlled trials (RCTs) and quasi‐RCTs (i.e. where allocation was not truly random). We included only trials that were published as full articles or were available as a full trial report. Extension phases and post‐hoc analyses of RCTs were also included to enable a comprehensive overview of the benefits and harms of NSAIDs.
As radiographic progression, as well as long term harmful effects, were unlikely to be assessed in short‐term RCTs of NSAIDs, we also included observational cohort studies to investigate the effect of NSAIDs on these outcomes. We assessed all included studies in an assessment of adverse events/harms of therapy with NSAIDs. In addition, cohort studies assessing the effect of NSAIDs on radiographic progression had to have a minimum duration of six months in order to be included. There were no restrictions on language of the paper.
Types of participants
We selected studies that included adults aged ≥ 18 years with a clinical diagnosis of axSpA, or patients fulfilling modified New York criteria or ASAS axial SpA criteria, including nr‐axSpA and AS. We included both disease subgroups. Studies containing patients with other diagnoses (e.g. trials that included participants based upon fulfilment of the European Spondyloarthropathy Study Group (ESSG) criteria or the Amor criteria (Amor 1990)) were only eligible if they presented results from patients with axSpA separately (Dougados 1991).
Types of interventions
We included studies that evaluated NSAIDs and all possible variations (dosage, intensity, mode of delivery, duration of delivery, timing of delivery, traditional and COX‐2 selective).
Comparators were:
Placebo;
No therapy;
Another NSAID;
Other pharmacological therapy;
Non‐pharmacological therapy;
Combination therapy;
Different doses, modes of delivery, frequency and duration.
Types of outcome measures
Primary outcomes
-
Benefits
Pain (as assessed by the mean change in pain score on a visual analogue scale (VAS) or numerical rating scale (NRS)); back pain was used but if not present in a study, overall pain was used;
-
Harms
Total number of withdrawals due to adverse events;
Disease activity as assessed by the mean improvement in Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) (Garrett 1994);
Physical function as assessed by the mean improvement in Bath Ankylosing Spondylitis Functional Index (BASFI) (Calin 1994);
Spinal mobility as assessed by the mean improvement in the Bath Ankylosing Spondylitis Metrology Index (BASMI) (Jenkinson 1994);
Radiographic progression as assessed by the mean change in the modified Stoke Ankylosing Spondylitis Spinal Score (mSASSS) (Wanders 2004);
Number of serious adverse events.
Secondary outcomes
-
Disease activity
Mean improvement in Ankylosing Spondylitis Disease Activity Index (ASDAS) (van der Heijde 2009a)
Mean improvement in patient's global assessment of disease activity
Mean improvement in fatigue (BASDAI question) (Garrett 1994)
Mean improvement in peripheral joint pain (BASDAI question) (Garrett 1994)
Mean improvement in tenderness of the joints (BASDAI question) (Garrett 1994)
Mean improvement in duration of morning stiffness (BASDAI question) (Garrett 1994)
Mean improvement in severity of morning stiffness (BASDAI question) (Garrett 1994)
Mean improvement in CRP
Mean improvement in ESR
Proportion of patients achieving ASDAS clinically important improvement (improvement ≥ 1.1 in ASDAS) (Machado 2011b)
Proportion of patients achieving ASDAS major improvement (improvement ≥ 2.0 in ASDAS) (Machado 2011b)
Proportion of patients achieving ASDAS inactive disease (ASDAS < 1.3) (Machado 2011b)
Proportion of patients achieving BASDAI 50 (improvement ≥ in BASDAI);
-
Fulfilment of response criteria
Proportion of responders according to ASAS20 (20% improvement in disease activity according to criteria of ASAS) (Anderson 2001)
Proportion of responders according to ASAS40 (40% improvement in disease activity according to criteria of ASAS) (Brandt 2004)
Proportion of responders according to ASAS 5/6 (20% improvement in disease activity according to criteria of ASAS) (Brandt 2004)
Proportion of patients achieving ASAS partial remission (Brandt 2004);
-
Spinal mobility
Mean improvement in lateral spinal flexion (Sieper 2009)
Mean improvement in chest expansion (Sieper 2009)
Mean improvement in tragus‐to‐wall distance (Sieper 2009)
Mean improvement in occiput‐to‐wall distance (Sieper 2009)
Mean improvement in cervical rotation (Sieper 2009)
Mean improvement in intermalleolar distance (Sieper 2009)
Mean improvement in 10 cm modified Schober's test (Sieper 2009);
Pain as assessed by the proportion of patients reporting pain relief of ≥ 50%;
-
Quality of life
Mean improvement in the Short‐Form 36 (SF‐36) (Ware 1992)
Mean improvement in the Ankylosing Spondylitis Quality of Life (ASQoL) score (Doward 2003);
-
Radiographic progression
The proportion of patients showing progression of at least two mSASSS units (Wanders 2004);
-
Adverse events
Number of (all) adverse events
Adverse events broken up by bodily system (e.g. gastrointestinal, cardiovascular, pulmonary).
We extracted all immediate (after up to two weeks of NSAID treatment), intermediate (up to and including six months of NSAID treatment) and longer‐term data (longer than six months of NSAID treatment).
Search methods for identification of studies
Electronic searches
One review author (LF) searched the following electronic bibliographical databases: MEDLINE (1946 to June 2014), EMBASE (1980 to June 2014), the Cochrane Central Register of Controlled Trials (CENTRAL) (the Cochrane Library (Issue 6, 2014) without language restrictions (Lefebvre 2011). We have provided the complete search strategies for the database searches in Appendix 1.
Searching other resources
In order to retrieve additional references, we pursued an additional search for systematic reviews in the Database of Abstracts of Review of Effects (DARE) and Health Technology Assessment (HTA) database. We also searched Scopus for conference proceedings, as well as two clinical trial registries for ongoing and recently finished studies (ClinicalTrials.gov (http://clinicaltrials.gov/) and the World Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP) search portal (http://apps.who.int/trialsearch/) for unpublished studies (Appendix 1). We screened the reference lists from included RCTs and other systematic reviews on the benefits and harms of NSAIDs for axSpA in order to identify all possible studies for this systematic review.
We also searched the websites of the regulatory agencies (e.g. the US Food and Drug Administration (FDA) MedWatch (http://www.fda.gov/Safety/MedWatch/default.htm), the European Medicines Evaluation Agency (http://www.ema.europa.eu), the Australian Adverse Drug Reactions Bulletin (http://www.tga.gov.au/safety/ews‐monitoring.htm), and the UK Medicines and Healthcare products Regulatory Agency pharmacovigilance and drug safety updates (http://www.mhra.gov.uk/Safetyinformation/index.htm) to identify any reported safety concerns.
Data collection and analysis
Selection of studies
Two review authors (FK, LvdB) independently assessed each title and abstract for suitability for inclusion. They decided independently of each other the eligibility of the article according to the pre‐determined selection criteria (Criteria for considering studies for this review). If there was any doubt, we retrieved and assessed the full‐text article. We resolved any disagreements between the review authors about the eligibility of the articles in a consensus meeting. In case of non‐consensus, a third review author (SR) decided if the study was eligible.
Data extraction and management
Two review authors (FK, LvdB) independently extracted data regarding study design (including funding source and number of centres), study duration, characteristics of study population, interventions, outcome measures and timing of outcome assessment, co‐interventions, benefits and adverse effect data, and losses to follow‐up by using a standardized data extraction form. We resolved any disagreements in data‐extraction by referring back to the original articles and by establishing consensus thereafter. If necessary, we consulted a third review author (SR).
We extracted the results (i.e. raw data: means and standard deviations (SDs) for continuous outcomes and number of events for dichotomous outcomes) for outcomes of interest in order to assess the benefIts and harms. For studies published in languages other than English, German, Portuguese, French, Spanish or Dutch, we consulted a native speaker or translator with content and methodological expertise.
Assessment of risk of bias in included studies
Two review authors (FK, LvdB) independently assessed the risk of bias of each included RCT (except for one trial that involved FK which was assessed by LvdB and SR) with regard to the following items: random sequence generation, allocation concealment, blinding of participants, care provider, and outcome assessor for each outcome measure (Types of outcome measures), incomplete outcome data, selective outcome reporting, and other sources of bias (including bias associated with cross‐over design of included studies if applicable (e.g. whether there was a carry‐over effect), baseline imbalance, co‐interventions and contamination), conforming to the methods recommended by Cochrane (Higgins 2011a). To determine the risk of bias of a study, for each criterion the presence of sufficient information and the likelihood of potential bias was evaluated. Each criterion is rated as "low risk of bias", "high risk of bias" or "unclear" (either lack of information or uncertainty over the potential for bias). In a consensus meeting we discussed and resolved any disagreements between the review authors. If consensus could not be reached, a third review author (SR) made the final decision.
Two review authors (FK, LvdB) independently assessed the risk of bias of each included observational study, with regard to the following items: study participation (i.e. representativeness of the study sample), study attrition, prognostic factor measurement, outcome measurement, confounding measurement and account, and analysis, as recommended in Hayden 2006. To determine the risk of bias of a study, for each criterion the presence of sufficient information and the likelihood of potential bias was evaluated. Each criterion is rated as "low risk of bias", "high risk of bias" or "unclear" (either lack of information or uncertainty over the potential for bias). In a consensus meeting, we resolved any disagreements between the review authors. If consensus could not be reached, a third author (SR) made the final decision.
Measures of treatment effect
We analysed the results of the studies using Cochrane's statistical software, Review Manager 2014. We only performed meta‐analysis if the data of the studies were clinically and statistically sufficiently homogeneous. We expressed the results as risk ratios (RRs) with corresponding 95% confidence intervals (CIs) for dichotomous data. A RR greater than 1.0 indicates a beneficial effect of NSAIDs (Deeks 2011).
For continuous data, we analysed results as mean differences (MDs) between the intervention and comparator group, with corresponding 95% CIs. The MD between treated group and control group was weighted by the inverse of the variance in the pooled treatment estimate. However, when different scales were used to measure the same conceptual outcome (e.g. functional status or pain), we calculated the standardized mean differences (SMDs) instead with corresponding 95% CIs. SMDs are calculated by dividing the MD by the SD, resulting in a unit‐less measure of treatment effect (Deeks 2011). SMD greater than zero indicate a beneficial effect in favour of NSAIDs for axSpA and we computed 95% CIs for the SMD. We interpreted the SMD as described by Higgins 2011b; i.e. a SMD of 0.2 was considered to indicate a small beneficial effect, 0.5 a medium effect, and 0.8 a large effect of NSAIDs for axSpA. SMDs were considered to indicate a clinically relevant effect if the SMD was > 0.5. Upon completion of the analysis, we translated the SMD back into a MD, on a scale of 0 to 10, which can be better appraised by clinicians.
For studies containing more than two intervention groups, making multiple pair‐wise comparisons between all possible pairs of intervention groups, we included the same group of participants only once in the meta‐analysis, or we split the group with the 'shared' intervention into two equally large groups to include two comparisons if deemed necessary. Whenever we had to decide between multiple dosages of a NSAID for studies containing more than two intervention groups, we used the proposed equivalent dose of 150 mg Diclofenac based on voting during the ASAS annual meeting (Dougados 2011).
Unit of analysis issues
Unit of analysis problems were not expected in this review. In the event that we identified crossover trials in which the reporting of continuous outcome data precluded paired analysis, we did not include these data in a meta‐analysis in order to avoid unit‐of‐analysis error. Where carry‐over effects were thought to exist, and where sufficient data existed, we included only data from the first period in the analysis (Higgins 2011b). Also, in studies of long duration, results may be presented for several periods of follow‐up. In that case we did not combine results from more than one time point for each study in a meta‐analysis to avoid unit‐of‐analysis error.
Dealing with missing data
Where important data are missing or incomplete, we planned to seek further information from the study authors.
In case individuals were missing from the reported results, we assumed the missing values to have a poor outcome. For dichotomous outcomes (e.g. number of withdrawals due to adverse events), we calculated the withdrawal rate using the number of patients randomised in the group as the denominator (worst case scenario).
For continuous outcomes (e.g. mean change in pain score), we calculated the MD or SMD based on the number of patients analysed at that time point. If the number of patients analysed was not presented for each time point, we used the number of randomised patients in each group at baseline.
Where possible, we computed missing SDs from other statistics such as standard errors, CIs or P values, according to the methods recommended in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011c). If SDs could not be calculated, they were imputed (e.g. from other studies in the meta‐analysis) (Higgins 2011b). If studies with final measurement data and change scores had to be combined using a SMD (e.g. because the studies used different scales), we calculated the final measurement data from the studies presenting change scores and imputed the SD for these final measurement data from the baseline SD from the same study.
Where data were presented graphically only, we extracted data from the graph when possible.
Assessment of heterogeneity
In this Cochrane review, we explored step‐by‐step the clinical and statistical heterogeneity between the studies. Firstly, we assessed studies for clinical homogeneity with respect to intervention groups, control groups, timing of outcome assessment and outcome measures. For any study judged as clinically homogeneous, we assessed statistical heterogeneity using the I² statistic (Deeks 2011), using the following as a rough guide for interpretation:
0 to 40%: might not be important;
30 to 60%: may represent moderate heterogeneity;
50 to 90%: may represent substantial heterogeneity;
75 to 100%: considerable heterogeneity.
In cases of considerable heterogeneity, we explored the data further, including subgroup analyses, in an attempt to explain the heterogeneity.
Assessment of reporting biases
In order to determine if reporting bias was present, we determined whether the protocol of the RCT was published before recruitment of patients of the study was started. For studies published after 1 July 2005, we screened the WHO ICTRP search portal (http://apps.who.int/trialsearch/). We evaluated whether selective reporting of outcomes was present (outcome reporting bias).
We compared the fixed‐effect model against the random‐effects model to assess the possible presence of small sample bias in the published literature (i.e. in which the intervention effect is more beneficial in smaller studies). In the presence of small sample bias, the random‐effects estimate of the intervention is more beneficial than the fixed‐effect estimate (Sterne 2011).
We further explored the potential for reporting bias by funnel plots if ≥ 10 studies were included.
We planned to add the unpublished trials in the Studies awaiting classification section, but we encountered none through our search strategy.
Data synthesis
We pooled the results of clinically and statistically homogeneous studies using the random‐effects model. We performed data analyses using Review Manager 2014 and produced forest plots for all analyses.
The main comparison of this review was NSAIDs versus placebo. However, many trials included both traditional and COX‐2 NSAIDs so we decided to assess the two NSAID classes separately.
We also considered the following comparisons in this review:
COX‐2 inhibitors versus traditional NSAIDs;
NSAIDs versus NSAIDs;
Naproxen versus other NSAIDs;
Low versus high dose NSAIDs; and
Continuous versus on‐demand NSAID use.
We included the comparison 'naproxen vs other NSAIDs' as a recent meta‐analysis of vascular and upper gastro‐intestinal effects of NSAIDs in various patients (prescribed mostly for rheumatoid arthritis or osteoarthritis, but also for prevention of colorectal adenomata or of Alzheimer's disease) found that naproxen was associated with less vascular (but increased upper gastro‐intestinal) risk than other NSAIDs (Bhala 2013).
'Summary of findings' table
We presented the main results of the review in 'Summary of findings' tables, which includes an overall grading of the evidence using the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach, and a summary of the available data on the main outcomes as described in the Cochrane Handbook for Systematic Reviews of Interventions (Schünemann 2011). We included the following outcomes in the 'Summary of findings' table:
Pain outcomes (mean change in pain score on a VAS or NRS);
Total number of withdrawals due to adverse events;
Mean improvement in BASDAI;
Mean improvement in BASFI;
Mean improvement in BASMI;
Radiographic progression;
Number of serious adverse events.
We illustrated data from the main comparison (NSAIDs vs placebo) in the main 'Summary of findings' table. Overall outcome data presented in the 'Summary of findings' tables were based on the longest time points measured in each study.
Grading of the evidence involved consideration of within‐study risk of bias, directness of evidence, heterogeneity, precision of effect estimates and risk of publication bias. However, other factors could affect the quality of evidence (e.g. it could be increased by a large magnitude of effect, plausible confounding and dose‐response gradients). Using this system, we graded the quality of the body of evidence as either high, moderate, low or very low (Atkins 2004).
In addition to the absolute and relative magnitude of effect provided in the 'Summary of findings' table, for dichotomous outcomes we calculated the number needed to treat to benefit (NNTB) or the number needed to treat to harm (NNTH) where appropriate from the control group event rate (unless the population event rate is known) and the RR using the Visual Rx calculator (Visual Rx 2008). We calculated the number needed to treat (NNT) for continuous outcomes using the Well's calculator software, which is based on the theory of Norman 2001 of determining the NNT based on achieving the minimal clinically important difference (MCID) for a particular outcome.
In the 'Summary of findings' table, we provided the absolute percent difference, the relative percent change from baseline, and the NNT (the NNT was provided only when the outcome showed a statistically significant difference).
For dichotomous outcomes, we calculated the absolute risk difference using the risk difference statistic in Review Manager 2014 and expressed the result as a percentage. For continuous outcomes, we calculated the absolute benefit as the improvement in the intervention group minus the improvement in the control group, in the original units.
We calculated the relative percent change for dichotomous data as the RR ‐ 1 and expressed it as a percentage. For continuous outcomes, the relative difference in the change from baseline was calculated as the absolute benefit divided by the baseline mean of the control group.
Subgroup analysis and investigation of heterogeneity
Where sufficient data was available, we conducted the following subgroup analyses to examine the influence of:
Gender (male vs female) on the effect of NSAIDs on all the outcomes;
Baseline radiographic damage (present vs absent) on the effect of NSAIDs on radiographic damage;
Baseline CRP (normal vs abnormal) on the effect of NSAIDs on radiographic damage;
Radiographic vs nr‐axSpA on the effect of NSAIDs on all the outcomes.
We selected these subgroups based on some evidence that these factors have prognostic value. Therefore, we wanted to assess whether this held true in this Cochrane review.
Sensitivity analysis
We conducted a sensitivity analysis to explore effect size differences and the robustness of conclusions. Where sufficient studies existed, we planned sensitivity analyses to assess the impact of any bias attributable to inadequate or unclear treatment allocation, blinding of patient/assessor and loss to follow‐up compared to studies without these study limitations ("low risk" vs "high risk" or "unclear").
Results
Description of studies
See Characteristics of included studies; Characteristics of excluded studies and Table 3 (Characteristics of included cohort studies) and Table 4 (Characteristics of included post‐hoc studies).
1. Characteristics of included cohort studies.
| Boersma 1976 | |
| Methods |
Design: Retrospective cohort study Number of centres: 1 Time point of assessments: Whenever radiological examination of the lumbar vertebral column was available. Length of follow‐up: Variable (up to 20 years) |
| Participants |
Inclusion criteria: Definite AS, with a radiological examination available. Exclusion criteria: NA Classification: New York criteria Continuous Phenylbutazone: Number of participants: 18 Number of completers: 18 Age: NA Male (%): NA Symptom duration: NA Disease duration: NA HLA‐B27 positive (%): NA Not continuous Phenylbutazone: Number of participants: 12 Number of completers: 12 Age: NA Male (%): NA Symptom duration: NA Disease duration: NA HLA‐B27 positive (%): NA No medication: Number of participants: 10 Number of completers: 10 Age: NA Male (%): NA Symptom duration: NA Disease duration: NA HLA‐B27 positive (%): NA |
| Interventions | Continuous Phenylbutazone vs not continuous Phenylbutazone vs no medication |
| Outcomes | Extracted outcomes: Unable to extract outcomes due to the method of presentation (individual data in graphs). |
| Notes | Funding source: Not reported |
| Risk of bias |
|
| Poddubnyy 2012 | |
| Methods |
Design: Post‐hoc analysis of prospective cohort study Number of centres: 13 Time point of assessments: BL, 2 years Length of follow‐up: 2 years |
| Participants |
Inclusion criteria (participants with AS): 1. Definite clinical diagnosis of axial SpA according to the local rheumatologist; 2. Duration of symptoms ≤ 10 years at the time of inclusion. Exclusion criteria (participants with AS): NA Classification (participants with AS): modified New York criteria Inclusion criteria (participants with nr‐axSpA): 1. Fulfilling ESSG criteria with minor modifications; 2. Duration of symptoms of ≤ 5 years. Exclusion criteria (participants with nr‐axSpA): NA Classification (participants with nr‐axSpA): ESSG criteria AS, low NSAIDs intake (NSAIDs index < 50): Number of participants: 64 Number of completers: 64 Age (mean (SD)): 36.2 (12.4) Male (%): 67 Symptom duration (mean (SD)): 5.0 (2.9) years Disease duration: NA HLA‐B27 positive (%): 86 AS, high NSAIDs intake (NSAIDs index ≥ 50): Number of participants: 24 Number of completers: 24 Age (mean (SD)): 38.7 (9.8) Male (%): 67 Symptom duration (mean (SD)): 5.5 (2.7) years Disease duration: NA HLA‐B27 positive (%): 79 nr‐axSpA, low NSAIDs intake (NSAIDs index < 50): Number of participants: 57 Number of completers: 57 Age (mean (SD)): 38.6 (9.3) Male (%): 32 Symptom duration (mean (SD)): 3.0 (2.2) years Disease duration: NA HLA‐B27 positive (%): 71 nr‐axSpA, high NSAIDs intake (NSAIDs index ≥50): Number of participants: 19 Number of completers: 19 Age (mean (SD)): 43.0 (9.6) Male (%): 32 Symptom duration (mean (SD)): 3.7 (2.1) years Disease duration: NA HLA‐B27 positive (%): 68 |
| Interventions | Low NSAIDs intake (NSAIDs index < 50) vs high NSAIDs intake (NSAIDs index ≥ 50) Co‐medication: No anti‐TNF therapy was allowed. |
| Outcomes | Extracted outcomes: mSASSS |
| Notes | Funding source: As part of the German competence network in rheumatology (Kompetenznetz Rheuma), GESPIC has been financially supported by the German Federal Ministry of Education and Research (Bundesministerium für Bildung und Forschung – BMBF), grant number: FKZ 01G19946. As funding by BMBF was reduced according to schedule in 2005 and stopped in 2007, complementary financial support has been obtained also from Abbott, Amgen, Centocor, Schering‐Plough and Wyeth. Since 2010, additional support has being obtained also from ANCYLOSS and ArthroMark projects funded by the German Federal Ministry of Education and Research. |
| Risk of bias |
|
2. Characteristics of included post‐hoc studies.
| Gossec 2005 | ||||||||||
|
Original study: Post hoc analysis of van der Heijde 2005. Comparison: Active drug (etoricoxib 90, etoricoxib 120 and naproxen 500) vs placebo. Analysis: Subgroup analysis in patients with and without chronic peripheral arthritis (defined as painful or swollen peripheral arthritis of > 4 weeks' duration, or a history of peripheral arthritis (anamnestic and based on medical chart), provided that the spine was the primary source of pain). Outcomes: BASDAI question on spine pain, patient global assessment of disease activity (BASFI), BASDAI question on peripheral pain, BASDAI questions on stiffness, BASDAI question on enthesopathy, ASAS 20 responders. | ||||||||||
| Baseline characteristics | ||||||||||
| Characteristic | Peripheral arthritis ‐ Yes | Peripheral arthritis ‐ No | ||||||||
| Number of participants | 115 | 186 | ||||||||
| Age (mean (SD)) | 43.8 (13.9) | 43.5 (10.4) | ||||||||
| "The two groups appeared to be well balanced, except for a higher percentage of concomitant DMARD and prior corticosteroid use in the group with peripheral arthritis." | ||||||||||
| Results | ||||||||||
| Outcome | Peripheral arthritis? | Treatment (N) | Baseline (mean (SD)) | Change from BL (mean (95% CI)) | P value | |||||
| Spine pain (VAS, 0 to 100, higher is worse) | Yes Yes No No |
Placebo (37) Active drug (117) Placebo (56) Active drug (175) |
78.7 (17.3) 77.6 (15.4) 76.2 (13.8) 77.7 (14.5) |
‐17.5 (‐24.7 to ‐10.3) ‐34.5 (‐38.6 to ‐30.4)* ‐10.0 (‐15.9 to ‐4.1) ‐42.5 (‐45.8 to ‐39.2)* |
*P < 0.05 vs placebo | |||||
| Peripheral pain (BASDAI) (VAS, 0 to 100, higher is worse) | Yes Yes No No |
Placebo (37) Active drug (117) Placebo (56) Active drug (175) |
61.8 (27.0) 61.2 (27.5) 45.4 (31.9) 43.5 (31.5) |
0.9 (‐5.9 to 7.6) ‐16.4 (‐20.3 to ‐12.6)* ‐5.5 (‐11.0 to ‐0.1) ‐26.6 (‐29.7 to ‐23.5)* |
*P < 0.05 vs placebo | |||||
| Patient global (VAS, to 100, higher is worse) | Yes Yes No No |
Placebo (37) Active drug (117) Placebo (56) Active drug (175) |
66.5 (21.8) 64.8 (23.3) 62.8 (20.5) 63.4 (20.3) |
‐3.3 (‐10.0 to 3.5) ‐22.0 (‐25.7 to ‐18.2)* ‐4.3 (‐9.7 to 1.2) ‐28.0 (‐31.1 to ‐24.9)* |
*P < 0.05 vs placebo | |||||
| BASFI (VAS, 0 to 100, higher is worse) | Yes Yes No No |
Placebo (37) Active drug (117) Placebo (56) Active drug (175) |
55.2 (29.8) 58.3 (23.8) 53.4 (25.2) 53.7 (23.3) |
‐3.5 (‐9.2 to 2.3) ‐14.9 (‐18.2 to ‐11.7)* ‐5.1 (‐9.8 to ‐0.4) ‐20.3 (‐22.9 to ‐17.6)* |
*P < 0.05 vs placebo | |||||
| Morning stiffness (duration + severity) (VAS, 0 to 100, higher is worse) | Yes Yes No No |
Placebo (37) Active drug (117) Placebo (56) Active drug (175) |
61.8 (26.0) 61.8 (25.4) 65.0 (21.4) 62.6 (23.7) |
‐5.7 (‐12.4 to 1.0) ‐24.4 (‐28.1 to ‐20.6)* ‐6.2 (‐11.6 to ‐0.7) ‐28.7 (‐31.8 to ‐25.6)* |
*P < 0.05 vs placebo | |||||
| Outcome | Peripheral arthritis? | Treatment (N) | % reaching ASAS 20 | Difference significant? | ||||||
| ASAS 20 | Yes Yes No No |
Placebo (37) Active drug (117) Placebo (56) Active drug (175) |
25% 61%* 25% 71%* |
*P = 0.001 vs placebo *P < 0.001 vs placebo |
||||||
| Discussion: "the combined active drug group provided significant clinical efficacy in AS patients with and without peripheral arthritis. The treatment responses that the authors observed…compared with placebo are in agreement with those seen in other trials...However, the magnitude of these responses was greater in patients without chronic peripheral arthritis or a history of peripheral arthritis. Although a significant difference in treatment effect among those with compared with those without peripheral arthritis was only seen for the primary end point of spinal pain, other end points demonstrated qualitatively similar differences, suggesting an overall difference in response between the two patient subgroups." | ||||||||||
| Kroon 2012 | ||||||||||
|
Original study: Post hoc analysis of Wanders 2005. Comparison: Continuous vs on‐demand NSAID treatment (ketoprofen and celecoxib). Analysis: Relevant subgroups were created by splitting ta‐CRP, ta‐ESR, ta‐BASDAI, ta‐AS‐ DAS‐CRP and ta‐ASDAS‐ESR at predefined values considered as elevated (for the acute phase reactants and representing high and low disease activity for the disease activity measures) ('low' vs 'high'). CRP levels > 5 mg/L and ESR > 12 mm/h were considered elevated; BASDAI > 4 and ASDAS > 2.1 were considered high. These subgroups were further split according to NSAID use (comparing continuous use with on‐demand use). Statistical interactions between subgroups of disease activity and mode of NSAID use, as well as their independent contributory effects, on radiographic progression were tested using multiple regression analysis and logistic regression analysis. Outcomes: BASDAI, inflammation (ESR and CRP), mSASSS, ASDAS‐ESR, ASDAS‐CRP. | ||||||||||
| Baseline characteristics | ||||||||||
| All patients | Patients with complete set of x‐rays | |||||||||
| Characteristic | Continuous use (N = 111) | On‐demand use (N = 103) | Continuous use (N = 76) | On‐demand use (N = 74) | ||||||
| Age (mean (SD) years) | 38.0 (10.7) | 40.1 (10.5) | 40.9 (9.8) | 37.9 (11.9) | ||||||
| Male (%) | 67 | 72 | 66 | 70 | ||||||
| Disease duration (mean (SD) years) | 11.9 (9.3) | 11.0 (9.4) | 13.0 (10.2) | 10.2 (9.3) | ||||||
| HLA‐B27 (pos. %) | 86 | 87 | 88 | 88 | ||||||
| "Between‐group differences at baseline were small and negligible… About 73% of the patients in both groups used celecoxib during the entire study period." | ||||||||||
| Results | ||||||||||
| Time‐averaged determinant | Outcome | Continuous treatment | On‐demand treatment | P value | ||||||
| CRP | High | dmSASSS (SD)* | 0.2 (1.6) (N = 52) | 1.7 (2.8) (N = 45) | 0.003 | |||||
| Nprog (%) | 7 (13%) (N = 52) | 17 (38%) (N = 45) | 0.011 | |||||||
| Low | dmSASSS (SD)* | 0.9 (1.8) (N = 21) | 0.8 (1.1) (N = 25) | 0.62 | ||||||
| Nprog (%) | 5 (24%) (N = 21) | 7 (28%) (N = 25) | 0.97 | |||||||
| ESR | High | dmSASSS (SD)* | 0.9 (1.6) (N = 37) | 2.0 (2.4) (N = 35) | 0.038 | |||||
| Nprog (%) | 8 (22%) (N = 37) | 17 (49%) (N = 35) | 0.031 | |||||||
| Low | dmSASSS (SD)* | 0.1 (1.8) (N = 35) | 0.7 (2.2) (N = 35) | 0.03 | ||||||
| Nprog (%) | 4 (11%) (N = 35) | 7 (20%) (N = 35) | 0.51 | |||||||
| BASDAI | High | dmSASSS (SD)* | 0.1 (1.1) (N = 18) | 1.1 (1.6) (N = 24) | 0.021 | |||||
| Nprog (%) | 1 (6%) (N = 18) | 7 (29%) (N = 24) | 0.126 | |||||||
| Low | dmSASSS (SD)* | 0.5 (1.8) (N = 58) | 1.6 (2.8) (N = 50) | 0.015 | ||||||
| Nprog (%) | 11 (19%) (N = 58) | 19 (38%) (N = 50) | 0.047 | |||||||
| ASDAS‐CRP | High | dmSASSS (SD)* | 0.4 (1.2) (N = 36) | 1.9 (2.7) (N = 40) | 0.005 | |||||
| Nprog (%) | 4 (11%) (N = 36) | 15 (38%) (N = 40) | 0.017 | |||||||
| Low | dmSASSS (SD)* | 0.4 (2.0) (N = 40) | 0.9 (2.1) (N = 34) | 0.11 | ||||||
| Nprog (%) | 8 (20%) (N = 40) | 11 (32%) (N = 34) | 0.35 | |||||||
| ASDAS‐ESR | High | dmSASSS (SD)* | 0.4 (1.3) (N = 30) | 1.8 (2.5) (N = 37) | 0.006 | |||||
| Nprog (%) | 3 (10%) (N = 30) | 15 (41%) (N = 37) | 0.012 | |||||||
| Low | dmSASSS (SD)* | 0.4 (1.9) (N = 46) | 1.1 (2.5) (N = 37) | 0.097 | ||||||
| Nprog (%) | 9 (20%) (N = 46) | 11 (30%) (N = 37) | 0.41 | |||||||
| * Mean (SD) value of δ modified stoke ankylosing spondylitis spine score (dmSASSS) in this (sub) group, defined as the difference between the modified Stoke Ankylosing Spondylitis Spine Score (mSASSS) at month 0 and month 24. Nprog is the number (percentage) of participants in this (sub) group with a progression score on the mSASSS of 2 or more. | ||||||||||
| Discussion: "continued inflammation in this study represented by ESR, CRP or the combined index ASAS‐ESR and ASDAS‐CRP plays an important role in radiographic progression. … this means we would be able to select patients who may benefit more from continuous use of NSAIDs with regards to radiographic progression… continuous use of NSAIDs can almost completely counteract the negative influence of high ESR on structural damage…The application of continuous therapy with NSAIDs in patients with elevated acute phase reactants may lead to an improved benefit to RR of these drugs, although it remains important to weigh the risk and benefit in individual patients". | ||||||||||
Results of the search
Through database searching we initially identified 7883 records (see Figure 1). No additional records were found through other sources. We assessed 177 full‐text articles for eligibility, of which we included 39 studies. We excluded 88 full‐text articles, for the reasons listed in the Excluded studies section. Finally, we included 31 studies in the quantitative analysis of this Cochrane review. In case the extracted data could not be used in the meta‐analysis, we reported these results in the Characteristics of included studies tables.
1.
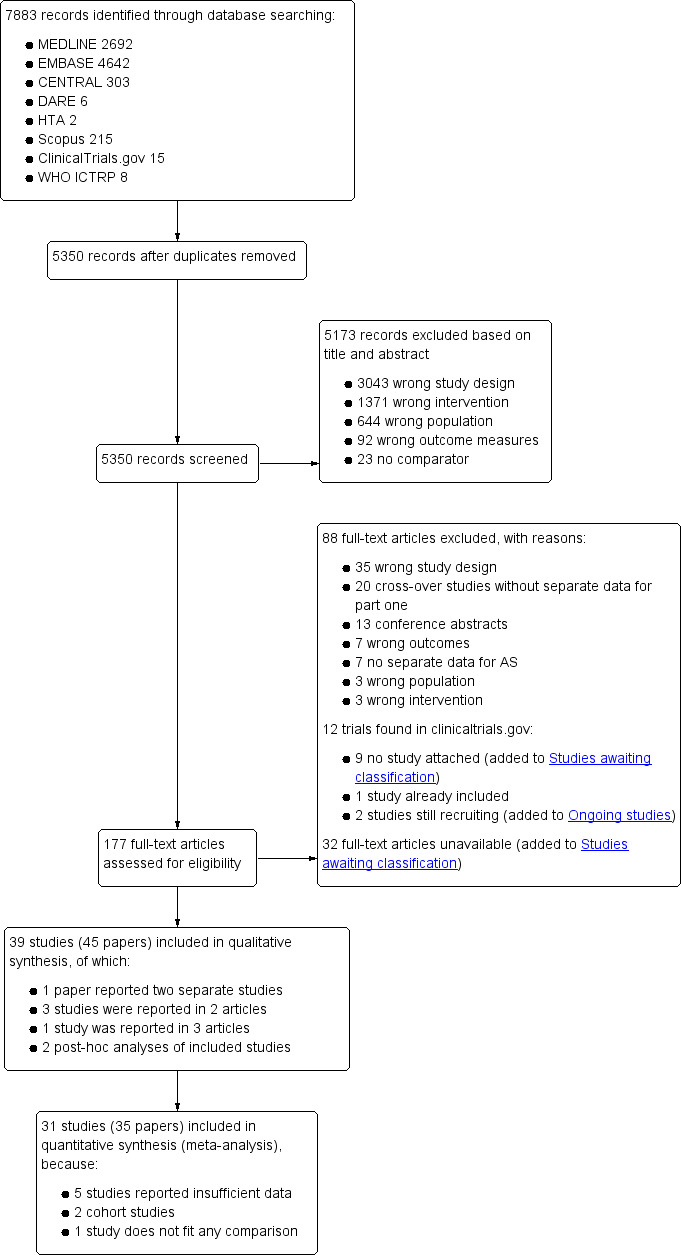
Study flow diagram.
Included studies
We included a total of 39 studies. In terms of study design there were 35 RCTs (of which six were cross‐over trials (Ansell 1978; Jessop 1976; Lehtinen 1984; Muller‐Fassbender 1985; Simpson 1966; Sydnes 1981)), two were quasi‐RCTs (Caldwell 1986; Calin 1979) and two were cohort studies (Boersma 1976; Poddubnyy 2012).
Characteristics of the included RCTs and quasi‐RCTs
Trial design
The 37 included RCTs and quasi‐RCTs involved a total of 4908 participants (range 14 to 611, mean 133) and were published between 1966 and 2006. Twenty‐four studies (65%) were published before 1990 (Ansell 1978; Astorga 1987; Caldwell 1986; Calin 1979; Ebner 1983; Franssen 1986; Good 1977; Heinrichs 1985; Jessop 1976; Khan 1985; Lehtinen 1984; Lomen 1986 I; Lomen 1986 P; Mena 1977; Muller‐Fassbender 1985; Myklebust 1986; Nahir 1980; Nissilä 1978a; Nissilä 1978b; Rejholec 1980; Santo 1988; Simpson 1966; Sydnes 1981; Tannenbaum 1984). Most trials were published in English, except for one trial that was in German (Heinrichs 1985) and one that was in Norwegian (Myklebust 1986). The treatment duration ranged from one week to two years, with a median duration of 12 weeks. Four studies also had an extension phase (Dougados 1999; Franssen 1986; Tannenbaum 1984; van der Heijde 2005). Fifteen trials were multicentre (Batlle‐Gualda 1996; Caldwell 1986; Dougados 1999; Dougados 2001; Khan 1985; Lomen 1986 I; Lomen 1986 P; Myklebust 1986; Palferman 1991; Sieper 2008; Sydnes 1981; Tannenbaum 1984; van der Heijde 2005; Villa Alcázar 1996; Wanders 2005), seven single centre (Carcassi 1990; Jessop 1976; Lehtinen 1984; Nahir 1980; Nissilä 1978a; Nissilä 1978b; Simpson 1966) and in 15 no information was reported on the number of centres involved.
Relevant post‐hoc analyses of two included RCTs (van der Heijde 2005; Wanders 2005) were published in a separate paper. Gossec 2005 is a post‐hoc analysis of data from van der Heijde 2005, in which a subgroup analysis was performed in patients with and without chronic peripheral arthritis. Kroon 2012 is a post‐hoc analysis of data from Wanders 2005, in which the effect of continuous versus on‐demand NSAID treatment was analysed in subgroups of patients with high vs low CRP, ESR, BASDAI, ASDAS‐CRP and ASDAS‐ESR.
Trial participants
Sixteen studies used a flare design (i.e. including only patients with a pre‐defined increase in symptoms after discontinuation of their usual treatment) and 28 studies required a wash‐out period of variable length. In 16 studies no classification criteria were reported for the inclusion of patients, and in the other 21 studies variable classification criteria were used (six trials used the modified New York criteria (Barkhuizen 2006; Dougados 1999; Dougados 2001; Sieper 2008; van der Heijde 2005; Wanders 2005), eight used the New York criteria (Batlle‐Gualda 1996; Calin 1979; Carcassi 1990; Heinrichs 1985; Lehtinen 1984; Schwarzer 1990; Tannenbaum 1984; Villa Alcázar 1996), three used the Rome criteria (Good 1977; Mena 1977; Palferman 1991), two used the ARA criteria (Khan 1985; Sydnes 1981), one used the Bennet and Wood criteria from 1968 (Jessop 1976) and one used multiple classification criteria (Dougados 1994).
All studies recruited adult participants. The mean age of participants was reported in 26 studies and was 40.5 years (SD 11.1 years, range 18 to 78). Of all participants, 81% were male (reported in 36 studies). Sixteen studies reported the mean disease duration, which ranged from 5.9 to 14 years, with a mean disease duration of 9.7 years. Nine trials reported the percentage of participants that were HLA‐B27 positive, which was on average 88.7%. Thirty‐six trials included only patients with AS, while one trial included patients with AS as well as patients with rheumatoid arthritis (Myklebust 1986). For the latter trial, we have included only the results for the AS subset in this review. No studies were found that included patients classified as having nr‐axSpA.
Interventions
The most frequently studied drug was indomethacin (15 studies: Batlle‐Gualda 1996; Caldwell 1986; Calin 1979; Carcassi 1990; Ebner 1983; Good 1977; Khan 1985; Lehtinen 1984; Lomen 1986 I; Nissilä 1978a; Nissilä 1978b; Palferman 1991; Rejholec 1980; Sydnes 1981; Tannenbaum 1984). Other frequently studied NSAIDs were diclofenac (six studies: Heinrichs 1985; Khan 1985; Nahir 1980; Santo 1988; Schwarzer 1990; Sieper 2008), naproxen (five studies: Ansell 1978; Barkhuizen 2006; Myklebust 1986; Pasero 1994; van der Heijde 2005), phenylbutazone (five studies: Franssen 1986; Jessop 1976; Lomen 1986 P; Mena 1977; Simpson 1966), piroxicam (five studies: Astorga 1987; Dougados 1999; Myklebust 1986; Sydnes 1981; Tannenbaum 1984), celecoxib (four studies: Barkhuizen 2006; Dougados 2001; Sieper 2008; Wanders 2005), flurbiprofen (four studies: Good 1977; Lomen 1986 I; Lomen 1986 P; Mena 1977), aceclofenac (three studies, Batlle‐Gualda 1996; Pasero 1994; Villa Alcázar 1996), ketoprofen (three studies: Dougados 2001; Jessop 1976; Muller‐Fassbender 1985), tenoxicam (three studies: Astorga 1987; Schwarzer 1990; Villa Alcázar 1996), oxaprozin (two studies: Caldwell 1986; Santo 1988), proquazone (two studies: Nissilä 1978a; Nissilä 1978b) and sulindac (two studies: Calin 1979; Nahir 1980). Single trials studied the following NSAIDs: butacote (Ansell 1978), diflunisal (Franssen 1986), etoricoxib (van der Heijde 2005), flufenamic acid (Simpson 1966), meclofenamate sodium (Ebner 1983), meloxicam (Dougados 1999), nabumetone (Palferman 1991), pirazolac (Carcassi 1990), tiaprofenacid (Heinrichs 1985), tolfenamic acid (Rejholec 1980) and ximoprofen (Dougados 1994).
There were five trials that included a placebo‐group (Barkhuizen 2006; Dougados 1994; Dougados 1999; Dougados 2001; van der Heijde 2005). Eighteen trials provided information about concurrent Disease Modifying Anti‐Rheumatic Drug (DMARD) and analgesic therapy. Six trials reported the allowance of stable doses of DMARDs, such as gold, penicillamine, chloroquine, sulfasalazine, methotrexate or low dose steroids (Barkhuizen 2006; Caldwell 1986; Myklebust 1986; Sieper 2008; van der Heijde 2005; Wanders 2005). Eleven trials reported that rescue analgesics without anti‐inflammatory effects, such as paracetamol, were allowed (Barkhuizen 2006; Batlle‐Gualda 1996; Caldwell 1986; Dougados 1999; Dougados 2001; Franssen 1986; Jessop 1976; Lehtinen 1984; Sydnes 1981; Villa Alcázar 1996; Wanders 2005). Four studies explicitly reported that no other analgesics or anti‐inflammatory drugs were allowed besides the study drugs (Ebner 1983; Good 1977; Mena 1977; Muller‐Fassbender 1985). None of the participants in the included studies were receiving biological DMARDs.
Characteristics of the included cohort studies
We included two cohort studies in this review:
Boersma 1976 was a retrospective cohort study comparing the effects of continuous phenylbutazone versus intermittent phenylbutazone versus no medication on radiographic progression in 40 patients with definite AS according to the New York criteria. Duration of follow‐up was variable (up to 20 years). No information was provided by trial authors on patient characteristics such as age, gender, HLA‐B27‐positivity or symptom duration.
Poddubnyy 2012 was a post‐hoc analysis of a prospective cohort study comparing the effects of low NSAID intake versus high NSAID intake on radiographic progression in 164 patients with AS (according to the modified New York criteria) or nr‐axSpA (according to the ESSG criteria). Duration of follow‐up was two years. The mean age of participants was 39.1 years, 49.5% were males, 76% were HLA‐B27 positive and the mean symptom duration was 4.3 years.
Reported outcomes
Not all included studies reported all outcomes that we planned to extract (see Types of outcome measures). In the Additional tables section we specified which outcome data were available for each study in each comparison, with a "+" indicating that the data were available in a format that could be used in the meta‐analysis, and a "*" indicating that the data were available but could not be used in the meta‐analysis (e.g. because data were reported without a measure of variance (SD, SE or CI), the number of patients per treatment group was not reported, or the data were only reported graphically) (see Table 5; Table 6; Table 7; Table 8; Table 9; Table 10).
3. Available data for comparison 1 (traditional NSAID vs Placebo).
| Trial | Groups | Pain on VAS | Withdrawals due to adverse events | BASDAI | Patient's global assessment of disease activity | Duration of morning stiffness | CRP | ASAS 20 | ASAS partial remission | BASFI | Chest expansion | Schober's test | Pain relief ≥ 50% | Number of any adverse events | Number of serious adverse events | Adverse events per organ system |
| Barkhuizen 2006 | Naproxen 500 mg vs Placebo | * | + | * | * | * | * | + | * | + | + | + | ||||
| Dougados 1994 | Ximoprofen 30 mg vs Placebo | + | + | + | + | + | + | + | ||||||||
| Dougados 2001 | Ketoprofen 200 mg vs Placebo | + | + | + | + | + | + | + | + | + | + | + | + | |||
| Dougados 1999 | Meloxicam 15 mg vs Placebo | + | + | + | + | + | + | + | + | + | + | |||||
| Dougados 1999 | Piroxicam 20 mg vs Placebo | + | + | + | + | + | + | + | + | + | + | |||||
| van der Heijde 2005 | Naproxen 1000 mg vs Placebo | + | + | + | + | + | + | + | + | + | + | + | + |
+ Available data that was used in the meta‐analysis. * Available data that could not be used in the meta‐analysis. Additional information on all included trials can be found in the Characteristics of included studies.
4. Available data for comparison 2 (COX‐2 vs Placebo).
| Trial | Groups | Pain on VAS | Withdrawals due to adverse events | BASDAI | Patient's global assessment of disease activity | Duration of morning stiffness | CRP | ASAS 20 | ASAS partial remission | BASFI | Chest expansion | Schober's test | Pain relief ≥ 50% | Number of any adverse events | Number of serious adverse events | Adverse events per organ system |
| Barkhuizen 2006 | Celecoxib 400 mg vs Placebo | * | + | * | * | * | * | + | * | + | + | + | ||||
| Dougados 2001 | Celecoxib 200 mg vs Placebo | + | + | + | + | + | + | + | + | + | + | + | + | |||
| van der Heijde 2005 | Etoricoxib 90 mg vs Placebo | + | + | + | + | + | + | + | + | + | + | + | + |
+ Available data that was used in the meta‐analysis. * Available data that could not be used in the meta‐analysis. Additional information on all included trials can be found in the Characteristics of included studies.
5. Available data for comparison 3 (COX‐2 vs traditional NSAID).
| Trial | Groups | Pain on VAS | Withdrawals due to adverse events | BASDAI | Patient's global assessment of disease activity | Duration of morning stiffness | CRP | ASAS 20 | ASAS partial remission | BASFI | BASMI | Chest expansion | Schober's test | Pain relief ≥ 50% | Number of any adverse events | Number of serious adverse events | Adverse events per organ system |
| Barkhuizen 2006 | Celecoxib 400 mg vs Naproxen 500 mg | * | + | * | * | * | * | + | * | + | + | + | |||||
| Dougados 2001 | Celecoxib 200 mg vs Ketoprofen 200 mg | + | + | + | + | + | + | + | + | + | + | + | + | ||||
| Sieper 2008 | Celecoxib 400 mg vs Diclofenac 75 mg | + | + | + | + | + | + | + | + | + | + | + | |||||
| van der Heijde 2005 | Etoricoxib 90 mg vs Naproxen 1000 mg | + | + | + | + | + | + | + | + | + | + | + | + |
+ Available data that was used in the meta‐analysis. * Available data that could not be used in the meta‐analysis. Additional information on all included trials can be found in the Characteristics of included studies.
6. Available data for comparison 4 (NSAID vs NSAID).
| Trial | Groups | Pain on Likert scale | Pain on VAS | Withdrawals due to adverse events | Patient's global assessment of disease activity | Duration of morning stiffness | Severity of morning stiffness | CRP | ESR | Lateral spinal flexion | Chest expansion | Tragus‐to‐wall distance | Occiput‐to‐wall distance | Intermalleolar distance | Schober's test | Pain relief ≥ 50% | Number of any adverse events | Number of serious adverse events | Adverse events per organ system |
| Astorga 1987 | Piroxicam 20 mg vs Tenoxicam 20 mg | + | * | * | + | + | |||||||||||||
| Batlle‐Gualda 1996 | Aceclofenac 200 mg vs Indomethacin 100 mg | + | + | + | + | + | + | + | + | + | + | ||||||||
| Caldwell 1986 | Oxaprozin 1200 mg vs Indomethacin 50 to 150 mg | * | + | * | * | * | * | * | * | * | + | + | |||||||
| Calin 1979 | Sulindac 200 to 400 mg vs Indomethacin 75 to 150 mg | + | * | * | * | * | + | ||||||||||||
| Dougados 1999 | Piroxicam 20 mg vs Meloxicam 15 mg | + | + | + | + | + | + | + | + | * | * | ||||||||
| Ebner 1983 | Meclofanamate sodium 300 mg vs Indometacin 150 mg | * | + | * | * | * | * | + | + | ||||||||||
| Franssen 1986 | Diflunisal 100 mg vs Phenylbutazone 400 mg | + | + | + | + | * | * | + | |||||||||||
| Good 1977 | Flurbiprofen 150 to 200 mg vs Indomethacin 75 to 100 mg | * | + | * | * | * | * | * | * | + | + | ||||||||
| Heinrichs 1985 | Diclofenac 150 to 200 mg vs Tiaprofenacid 600 to 700 mg | * | + | * | * | * | * | + | |||||||||||
| Jessop 1976 | Ketoprofen 200 mg vs Phenylbutazone 300 mg | + | + | + | + | * | * | * | * | * | |||||||||
| Khan 1985 | Diclofenac 125 mg vs Indomethacin 125 mg | * | + | * | * | * | * | * | + | + | |||||||||
| Lomen 1986 I | Flurbiprofen 150 to 300 mg vs Indomethacin 75 to 150 mg | * | + | * | * | * | * | * | + | + | |||||||||
| Lomen 1986 P | Flurbiprofen 200 to 300 mg vs Phenylbutazone 300 to 500 mg | * | + | * | * | * | * | * | + | + | |||||||||
| Mena 1977 | Flurbiprofen 150 to 200 mg vs Phenylbutazone 300 to 400 mg | * | + | * | * | * | * | * | + | + | |||||||||
| Myklebust 1986 | Piroxicam 20 mg vs Naproxen 1000 mg | + | * | * | * | ||||||||||||||
| Nahir 1980 | Diclofenac 150 mg vs Sulindac 600 mg | + | + | + | + | + | + | + | + | ||||||||||
| Nissilä 1978a | Proquazone 900 mg vs Indomethacin 75 mg | + | + | + | |||||||||||||||
| Nissilä 1978b | Proquazone 900 mg vs Indomethacin 75 mg | + | + | + | |||||||||||||||
| Palferman 1991 | Nabumetone 2000 mg vs Indomethacin 150 mg | + | + | + | + | + | |||||||||||||
| Pasero 1994 | Aceclofenac 100 mg vs Naproxen 500 mg | * | + | * | + | + | + | ||||||||||||
| Rejholec 1980 | Tolfenamic acid 600 mg vs Indomethacin 75 mg | * | + | * | * | * | |||||||||||||
| Santo 1988 | Diclofenac 100 mg vs Oxaprozin 1200 mg | + | + | + | + | + | + | + | + | ||||||||||
| Schwarzer 1990 | Diclofenac 50 mg vs Tenoxicam 20 mg | + | + | + | + | + | |||||||||||||
| Simpson 1966 | Flufenamic acid 600 mg vs Phenylbutazone 300 mg | + | + | ||||||||||||||||
| Tannenbaum 1984 | Piroxicam 10 to 20 mg vs Indomethacin 75 to 125 mg | + | + | + | + | + | + | + | + | + | |||||||||
| Villa Alcázar 1996 | Aceclofenac 200 mg vs Tenoxicam 20 mg | + | + | * | * | * | * | * | + |
+ Available data that was used in the meta‐analysis. * Available data that could not be used in the meta‐analysis. Additional information on all included trials can be found in the Characteristics of included studies.
7. Available data for comparison 5 (Naproxen vs other NSAID).
| Trial | Groups | Pain on VAS | Withdrawals due to adverse events | BASDAI | Patient's global assessment of disease activity | Duration of morning stiffness | CRP | ASAS 20 | ASAS partial remission | BASFI | Chest expansion | Schober's test | Number of any adverse events | Number of serious adverse events | Adverse events per organ system |
| Barkhuizen 2006 | Naproxen 500 mg vs Celecoxib 400 mg | * | + | * | * | * | * | + | * | + | + | + | |||
| Myklebust 1986 | Naproxen 1000 mg vs Piroxicam 20 mg | + | * | * | * | ||||||||||
| Pasero 1994 | Naproxen 500 mg vs Aceclofenac 100 mg | * | + | * | + | + | + | ||||||||
| van der Heijde 2005 | Naproxen 1000 mg vs Etoricoxib 90 mg | + | + | + | + | + | + | + | + | + | + | + | + |
+ Available data that was used in the meta‐analysis. * Available data that could not be used in the meta‐analysis. Additional information on all included trials can be found in the Characteristics of included studies.
8. Available data for comparison 6 (low dose vs high dose NSAID).
| Trial | Groups | Pain on Likert scale | Pain on VAS | Withdrawals due to adverse events | BASDAI | Patient's global assessment of disease activity | Duration of morning stiffness | CRP | ASAS20 | ASAS partial remission | BASFI | BASMI | Chest expansion | Schober's test | Pain relief ≥ 50% | Number of any adverse events | Number of serious adverse events | Adverse events per organ system |
| Barkhuizen 2006 | Celecoxib 200 mg vs Celecoxib 400 mg | * | + | * | * | * | * | + | * | + | + | + | ||||||
| Dougados 1994 | Ximoprofen 5 mg vs Ximoprofen 30 mg | + | + | + | + | + | + | + | ||||||||||
| Dougados 1999 | Meloxicam 15 mg vs Meloxicam 22.5 mg | + | + | + | + | + | + | + | + | * | * | |||||||
| Sieper 2008 | Celecoxib 200 mg vs Celecoxib 400 mg | + | + | + | + | + | + | + | + | + | + | + | ||||||
| van der Heijde 2005 | Etoricoxib 90 mg vs Etoricoxib 120 mg | + | + | + | + | + | + | + | + | + | + | + | + |
+ Available data that was used in the meta‐analysis. * Available data that could not be used in the meta‐analysis. Additional information on all included trials can be found in the Characteristics of included studies.
Primary outcomes
Of the 31 studies that were included in the quantitative data‐analysis, 14 trials reported pain on a VAS (which could be used in the analyses of 11 studies) and 15 reported pain on a NRS (which could be used in the analyses of six studies). There were four trials that did not report pain as an outcome (Calin 1979; Nissilä 1978a; Nissilä 1978b; Palferman 1991).
The number of withdrawals due to adverse events was reported in 28 trials (and could be used in the analyses of 28 trials). There were three trials that did not report the number of withdrawals due to adverse events (Astorga 1987; Jessop 1976; Myklebust 1986).
Three studies reported the BASDAI (which could be used in analyses of two studies) (Barkhuizen 2006; Sieper 2008; van der Heijde 2005). BASFI was reported in four studies (and could be used in the analyses of three studies) (Barkhuizen 2006; Dougados 2001; Sieper 2008; van der Heijde 2005). BASMI was only reported by Sieper 2008, and radiographic progression was not reported in any of the studies included in the meta‐analysis. Six studies reported the number of serious adverse events (Barkhuizen 2006; Dougados 2001; Nahir 1980; Schwarzer 1990; Sieper 2008; van der Heijde 2005).
Secondary outcomes
The following secondary outcomes were reported by one or more of the studies included in the meta‐analysis: patient's global assessment of disease activity (9/31 studies, five included in analyses), duration of morning stiffness (22/31 studies, 10 included in analyses), severity of morning stiffness (2/31 studies, two included in analyses), CRP (4/31 studies, three included in analyses), ESR (7/31 studies, three included in analyses), proportion of responders according to ASAS20 (3/31 studies, three included in analyses), proportion of patients achieving ASAS partial remission (1/31 studies, one included in analyses), lateral spinal flexion (4/31 studies, one included in analyses), chest expansion (23/31 studies, seven included in analyses), tragus‐to‐wall distance (2/31 studies, one included in analyses), occiput‐to‐wall distance (11/31 studies, two included in analyses), intermalleolar distance (6/31 studies, 0 included in analyses), Schober's test (26/31 studies, 11 included in analyses), proportion of patients reporting pain relief of ≥ 50% (3/31 studies, three included in analyses), number of (any) adverse events (25/31 studies, 24 included in analyses) and adverse events broken up by bodily system (e.g. gastrointestinal, cardiovascular, neurological) (22/31 studies, 21 included in analyses).
The secondary outcomes not reported by any of the 31 studies included in the meta‐analysis were: ASDAS, fatigue, joint pain, tenderness of the joints, proportion of patients achieving ASDAS clinically important improvement (improvement ≥ 1.1 in ASDAS), proportion of patients achieving ASDAS major improvement (improvement ≥ 2.0 in ASDAS), proportion of patients achieving ASDAS inactive disease (ASDAS < 1.3), proportion of patients achieving BASDAI 50 (improvement ≥ in BASDAI), proportion of responders according to ASAS40, proportion of responders according to ASAS 5/6, cervical rotation, quality of life (assessed by SF‐36 or ASQoL score) and proportion of patients showing radiographic progression of at least two mSASSS units.
Excluded studies
Of the 177 full‐text papers that were assessed for eligibility, we excluded 88 full‐text articles for the following reasons: wrong study design (n = 35), cross‐over study without separate data for part one of the cross‐over (n = 20), conference abstract (n = 13), wrong outcomes (n = 7), no separate data available for patients with AS (n = 7), wrong population (n = 3) and wrong intervention (n = 3) (see Figure 1).
Furthermore, nine trials found in ClinicalTrials.gov did not provide any results, were not published and were thus added to Studies awaiting classification. One trial that was found in ClinicalTrials.gov was a trial that was already included and two trials were still recruiting participants. Thus we added these studies to the Ongoing studies section. Of 32 articles, no full‐text could be obtained after extensive searching, and we added these trials to the Studies awaiting classification. The most relevant excluded trials and the reasons for exclusion are listed in the Characteristics of excluded studies table.
Risk of bias in included studies
We assessed the risk of bias for each study (see Characteristics of included studies for RCTs and quasi‐RCTs, see Table 3 for observational studies). The results for RCTs and quasi‐RCTs are also summarised in Figure 2 and Figure 3.
2.
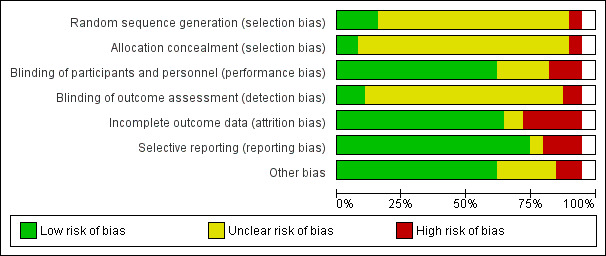
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.
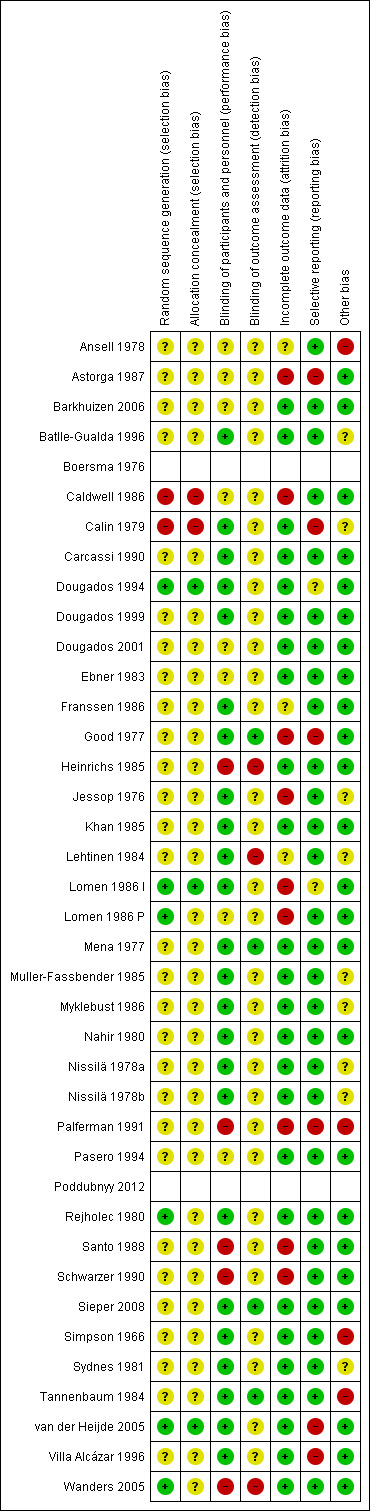
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
Three studies were assessed as being at low risk of selection bias, as they described adequate sequence generation and allocation concealment (Dougados 1994; Lomen 1986 I; van der Heijde 2005). Three trials described adequate sequence generation, but the risk of bias concerning allocation concealment remained unclear (Lomen 1986 P; Rejholec 1980; Wanders 2005). We assessed two trials as being at high risk of selection bias (Caldwell 1986; Calin 1979). In the remaining 29 trials the risk of selection bias remained unclear, based on both random sequence allocation and allocation concealment, mainly due to lack of information.
Blinding
Participants and personnel were adequately blinded in most trials and we judged 24 trials as being at low risk of performance bias. The risk of performance bias was assessed as being high in five trials (Heinrichs 1985; Palferman 1991; Santo 1988; Schwarzer 1990; Wanders 2005) due to inadequate blinding of participants or personnel, or both. In the remaining eight trials the risk of performance bias remained unclear (Ansell 1978; Astorga 1987; Barkhuizen 2006; Caldwell 1986; Dougados 2001; Ebner 1983; Lomen 1986 P; Pasero 1994).
We assessed the risk of detection bias as being low in four studies as they described an adequate blinding of outcome assessment (Good 1977; Mena 1977; Sieper 2008; Tannenbaum 1984). In two trials we judged the risk of detection bias to be high (Heinrichs 1985; Lehtinen 1984). One trial, Wanders 2005, adequately blinded the scoring of radiographs by the investigators. However, clinical outcomes were all assessed by self‐report by participants who could not have been blinded to the treatment regiment to which they were allocated. However, most studies did not clearly describe the blinding of outcome assessment, and thus the risk of detection bias was unclear in these 30 trials.
Incomplete outcome data
Twenty‐five trials were assessed as low risk of attrition bias. Nine trials had unexplained incomplete outcome data and were assessed as being at high risk of attrition bias (Astorga 1987; Caldwell 1986; Good 1977; Jessop 1976; Lomen 1986 I; Lomen 1986 P; Palferman 1991; Santo 1988; Schwarzer 1990). The risk of detection bias was unclear in three trials (Ansell 1978; Franssen 1986; Lehtinen 1984).
Selective reporting
We judged most studies as being at low risk of reporting bias, as these 29 trials did not selectively report outcomes. Six studies were assessed as being at high risk of reporting bias (Astorga 1987; Calin 1979; Good 1977; Palferman 1991; van der Heijde 2005; Villa Alcázar 1996). We assessed two studies as having an unclear risk of reporting bias, as the impact of minor outcomes that were not reported or the impact of not reporting outcomes at all time points was unclear, or both (Dougados 1994; Lomen 1986 I).
Other potential sources of bias
In four studies another potential source of bias was detected, including possible carry‐over effects in a cross‐over design (Ansell 1978), baseline imbalance between groups (Palferman 1991), no baseline information on participants characteristics (Simpson 1966), and significant differences in compliance to the studied drugs (Tannenbaum 1984). In nine studies we assessed the risk of an additional potential source of bias as unclear, as there was insufficient information provided by the study authors or insufficient evidence that an identified problem has introduced bias, or both. In the remaining 24 trials no other potential source of bias was identified.
Of the six studies that were included with a cross‐over design, only one documented that they did not find any carry‐over effects (Lehtinen 1984). In one study a carry‐over effect was present, and this study was thus assessed as having a high risk of 'other bias' (Ansell 1978). The remaining four cross‐over studies were judged as having an unclear risk of carry‐over effects (Jessop 1976; Muller‐Fassbender 1985; Simpson 1966; Sydnes 1981).
Risk of bias in observational studies
We judged both included observational studies as being at high risk of bias regarding study participation. We assessed the risk of bias due to study attrition and outcome measurement as low in both studies. In one study, Poddubnyy 2012, prognostic factor measurement was accurately performed. However in the other study, Boersma 1976, we judged the risk of bias due to inadequate prognostic factor measurement to be unclear. Furthermore, confounding measurement and account were judged to be unclear in one study, Boersma 1976, and introduced a high risk of bias in the other, Poddubnyy 2012. Finally analysis was appropriate in one study, Poddubnyy 2012, and thus at low risk of introducing bias. However, it was not reported in the other, Boersma 1976, so we assessed the risk of bias due to analysis as unclear in the last study.
Effects of interventions
Summary of findings for the main comparison. Traditional NSAIDs compared with placebo for axSpA.
| Traditional NSAIDs compared with placebo for axSpA (AS and nr‐axSpA) | ||||||
|
Patient or population: patients with axSpA (AS and nr‐axSpA) Settings: outpatient, hospital Intervention: traditional NSAID Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Traditional NSAID | |||||
|
Pain on VAS Scale from 0 to 100 mm (higher is worse) Follow‐up: 2 to 6 weeks |
The mean pain score in the control group was 61 points1 |
The mean pain scores in the intervention groups was 16.5 points lower (12.2 to 20.8 lower) | 850 (four studies) | ⊕⊕⊕⊕ high | Absolute percent difference: 17% lower (12% to 21% lower) Relative percent change from baseline: 21% lower (16% to 27% lower)2 NNT: 4 (3 to 6)3 |
|
|
Withdrawals due to adverse events Due to adverse events Follow‐up: 2 to 12 weeks |
52 per 10004 | 39 per 1,000 (24 to 63) | RR 0.75 (0.46 to 1.21) | 1165 (five studies) | ⊕⊕⊕⊕ high | Absolute percent difference: 0% more (3% less to 2% more) Relative percent difference from baseline: decrease 25% (54% decrease to 21% increase) |
|
BASDAI Scale from 0 to 100 (higher is worse) Follow‐up: 6 weeks |
The mean BASDAI in the control group was 54.7 points |
The mean BASDAI in the intervention group was 17.5 points lower (11.8 to 23.1 lower) | 190 (one study) | ⊕⊕⊕⊝ moderate5 | Absolute percent difference: 18% lower (12% to 23% lower) Relative percent change from baseline: 28% lower (19% to 37% lower)6 NNT: 3 (2 to 4)7 |
|
|
BASFI Scale from 0 to 100 (higher is worse) Follow‐up: 6 weeks |
The mean BASFI in the control groups was 50.0 points8 | The mean BASFI in the intervention groups was 9.1 points lower (5.1 to 13.0 lower) | 356 (two studies) | ⊕⊕⊕⊕ high | Absolute percent difference: 9% lower (5% to 13% lower) Relative percent change from baseline: 17% lower (9% to 24% lower)9 NNT: 5 (3 to 8)10 |
|
|
BASMI Scale from 0 to 10 (higher is worse) |
See comment | See comment | See comment | See comment | None of the trials included in this comparison reported BASMI. | |
|
Radiographic progression Mean change in mSASSS. Scale from 0 to 72 (higher is worse) |
See comment | See comment | See comment | See comment | None of the trials included in this comparison reported mSASSS. | |
|
Number of serious adverse events Follow‐up: 6 to 12 weeks |
2 per 100011 |
3 per 1,000 (1 to 16) |
RR 1.69 (0.36 to 7.97) | 671 (three studies) | ⊕⊕⊕⊝ moderate12 | Absolute percent difference: 0% more (1% less to 2% more) Relative percent change from baseline: increase 69% (64% decrease to 697% increase) |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). Abbreviations: CI: confidence interval; RR: risk ratio; NSAID: non‐steroidal anti‐inflammatory drug; BASDAI: Bath Ankylosing Spondylitis Disease Activity Index; BASFI: Bath Ankylosing Spondylitis Functional Index; BASMI: Bath Ankylosing Spondylitis Metrology Index; mSASSS: modified Stoke Ankylosing Spondylitis Spinal Score; VAS: Visual Analogue Scale. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1Assumed risk based on mean control group final values taken from Dougados 1994; van der Heijde 2005. 2Estimated relative changes based on mean (SD) pain on VAS in placebo group at baseline 77.22 (15.24) from van der Heijde 2005. 3Based on MCID of 15 points on a 0 to 100 point scale. 4Assumed risk based on the median risk in the control groups. 5Downgraded due to potential imprecision due to data available only from a single study (N = 190). 6Estimated relative changes based on mean (SD) BASDAI in placebo group at baseline 61.78 (18.70) from van der Heijde 2005. 7Based on MCID of 10 points on a 0 to 100 point scale. 8Assumed risk based on the control group final values from van der Heijde 2005. 9Estimated relative changes based on mean (SD) BASFI in placebo group at baseline 54.12 (26.99) from van der Heijde 2005. 10Based on MCID of 10 points on a 0 to 100 point scale. 11Assumed risk based on the mean risk in the control groups. 12Downgraded due to potential imprecision because the 95% CI includes 'no effect' and the upper confidence limit also crosses 'appreciable harm'.
Since the studies included in the analyses of this comparison were more high quality studies compared to the other included studies in the review, it was decided not to downgrade the evidence for study limitations (as assessed in the risk of bias), as the authors believe this did not importantly affect the quality of the evidence of this comparison.
Summary of findings 2. COX‐2 NSAIDs compared with placebo for axSpA.
| COX‐2 NSAIDs compared with placebo for axSpA (AS and nr‐axSpA) | ||||||
|
Patient or population: patients with axSpA (AS and nr‐axSpA) Settings: outpatient, hospital Intervention: COX‐2 NSAID Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | COX‐2 NSAID | |||||
|
Pain on VAS Scale from 0 to 100 mm (higher is worse) Follow‐up: 6 weeks |
The mean pain scores across control groups was 64 points1 |
The mean pain scores in the intervention groups was 21.7 points lower (7.4 to 35.9 lower) | 349 (two studies) | ⊕⊕⊕⊕ high | Absolute percent difference: 22% lower (7% to 36% lower) Relative percent change from baseline: 28% lower (10% to 47% lower)2 NNT: 3 (2 to 24)3 |
|
|
Withdrawals due to adverse events Follow‐up: 6 to 12 weeks |
11 per 10004 |
24 per 1,000 (4 to 142) |
RR 2.14 (0.36 to 12.56) | 669 (three studies) | ⊕⊕⊝⊝ low5 | Absolute percent difference: 2% more (2% less to 6% more) Relative percent difference from baseline: increase 114% (64% decrease to 1156% increase) |
|
BASDAI Scale from 0 to 100 (higher is worse) Follow‐up: 6 weeks |
The mean BASDAI in the control group was 54.7 points |
The mean BASDAI in the intervention group was 22 points lower (16.6 to 27.4 lower) | 193 (one study) | ⊕⊕⊕⊝ moderate6 | Absolute percent difference: 22% lower (17% to 27% lower) Relative percent change from baseline: 36% lower (27% to 44% lower)7 NNT: 2 (1 to 3)8 |
|
|
BASFI Scale from 0 to 100 (higher is worse) Follow‐up: 6 weeks |
The mean BASFI in the control groups was 50.0 points1 | The mean BASFI in the intervention groups was 13.4 points lower (9.5 to 17.4 lower) | 349 (two studies) | ⊕⊕⊕⊕ high | Absolute percent difference: 13% lower (9% to 17% lower) Relative percent change from baseline: 25% lower (18% to 32% lower)9 NNT: 3 (2 to 4)10 |
|
|
BASMI Scale from 0 to 10 (higher is worse) |
See comment | See comment | See comment | See comment | None of the trials included in this comparison reported BASMI. | |
|
Radiographic progression Mean change in mSASSS. Scale from 0 to 72 (higher is worse) |
See comment | See comment | See comment | See comment | None of the trials included in this comparison reported mSASSS. | |
|
Number of serious adverse events Follow‐up: 6 to 12 weeks |
2 per 10004 | 2 per 1000 (0 to 13) | RR 0.92 (0.14 to 6.21) | 669 (three studies) | ⊕⊕⊕⊝ moderate11 | Absolute percent difference: 0% more (1% less to 1% more) Relative percent change from baseline: decrease 8% (86% decrease to 512% increase) |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). Abbreviations: CI: confidence interval; COX: cyclo‐oxygenase; RR: risk ratio; NSAID: non‐steroidal anti‐inflammatory drug; BASDAI: Bath Ankylosing Spondylitis Disease Activity Index; BASFI: Bath Ankylosing Spondylitis Functional Index; BASMI: Bath Ankylosing Spondylitis Metrology Index; mSASSS: modified Stoke Ankylosing Spondylitis Spinal Score; VAS: Visual Analogue Scale. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1Assumed risk based on the control group final values from van der Heijde 2005. 2Estimated relative changes based on mean (SD) pain on VAS in placebo group at baseline 77.22 (15.24) from van der Heijde 2005. 3Based on MCID of 15 points on a 0 to 100 point scale. 4Assumed risk based on the mean risk in the control groups. 5Downgraded due to potential imprecision because the 95% CI includes 'no effect' and the upper confidence limit also crosses 'appreciable harm', as well as inconsistency in the results with large heterogeneity (I2=84%). 6Downgraded due to potential imprecision due to data available only from a single study (N = 193). 7Estimated relative changes based on mean (SD) BASDAI in placebo group at baseline 61.78 (18.70) from van der Heijde 2005. 8Based on MCID of 10 points on a 0 to 100 point scale. 9Estimated relative changes based on mean (SD) BASFI in placebo group at baseline 54.12 (26.99) from van der Heijde 2005. 10Based on MCID of 10 points on a 0 to 100 point scale. 11Downgraded due to potential imprecision because the 95% CI includes 'no effect' and the upper confidence limit also crosses 'appreciable harm'.
Since the studies included in the analyses of this comparison were more high quality studies compared to the other included studies in the review, it was decided not to downgrade the evidence for study limitations (as assessed in the risk of bias), as the authors believe this did not importantly affect the quality of the evidence of this comparison.
We only included 31 studies (out of the total 39 included trials) with 4356 participants and a median duration of 12 weeks (range two to 26 weeks) in quantitative data‐analysis. The eight remaining trials could not be included in the meta‐analysis due to the study design, as these were either not RCTs or not quasi‐RCTs (Boersma 1976; Poddubnyy 2012), or because the authors did not provide the number of participants per treatment arm (Ansell 1978; Carcassi 1990; Lehtinen 1984; Sydnes 1981), or there was no fitting comparison (Wanders 2005) or the outcomes were presented in a way that we could not extract quantitative data (Muller‐Fassbender 1985).
Of the 31 trials that could be included in the meta‐analysis, only 19 studies provided data for any of the efficacy variables. In the Characteristics of included studies table we provided information for each study on which outcomes were extracted and whether these outcomes were used in the quantitative data‐analysis. If an outcome could not be used in the quantitative data‐analysis, we reported any relevant information that was provided by the trial authors on that outcome. The eight studies that could not be included in the meta‐analysis are discussed separately under the appropriate comparison.
Comparison 1: Traditional NSAID versus placebo
In comparison 1 we included all studies that compared a traditional NSAID to placebo (five trials: Barkhuizen 2006; Dougados 1994; Dougados 1999; Dougados 2001; van der Heijde 2005). In Table 5 we listed the outcomes that were available for each study in this comparison.
Benefits
Five trials with 1165 participants and a duration of two to 12 weeks, showed a consistent significant effect favouring NSAIDs in all efficacy variables: pain on VAS (four trials, N = 850; MD ‐16.51, 95% CI ‐20.84 to ‐12.17 on a scale of 0 to 100 (higher is worse); Analysis 1.1); BASDAI (one trial, N = 190; MD ‐17.45, 95% CI ‐23.10 to ‐11.80 on a scale of 0 to 100 (higher is worse); Analysis 1.3); patient's global assessment of disease activity (three trials, N = 705; MD ‐17.75, 95% CI ‐24.39 to ‐11.10 on a scale of 0 to 100 (higher is worse); Analysis 1.4); duration of morning stiffness (four trials, N = 850; SMD ‐0.40, 95% CI ‐0.58 to ‐0.22; Analysis 1.5); CRP (two trials, N = 515; MD ‐3.37 mg/dL, 95% CI ‐6.11 to ‐0.62; Analysis 1.6); ASAS 20 (two trials, N = 503; RR 2.49, 95% CI 1.94 to 3.18; Analysis 1.7); BASFI (two trials, N = 356; MD ‐9.07, 95% CI ‐13.04 to ‐5.10 on a scale of 0 to 100 (higher is worse); Analysis 1.9); chest expansion (two trials, N = 515; MD 0.44 cm, 95% CI 0.20 to 0.68; Analysis 1.10); Schober's test (four trials, N = 850; MD 0.37 cm, 95% CI 0.18 to 0.57; Analysis 1.11) and pain relief ≥ 50% (three trials, N = 660; RR 2.27, 95% CI 1.77 to 2.91; Analysis 1.12). Only ASAS partial remission was not significantly different according to one trial with 190 participants (RR 2.88, 95% CI 0.80 to 10.30; Analysis 1.8). No studies reported data for a comparison between traditional NSAIDs and placebo with respect to our other major efficacy outcomes BASMI or radiographic progression (see Table 5). The outcome BASDAI was downgraded from high to moderate quality evidence because of potential imprecision as this outcome only included data from a single study (N = 190).
1.1. Analysis.
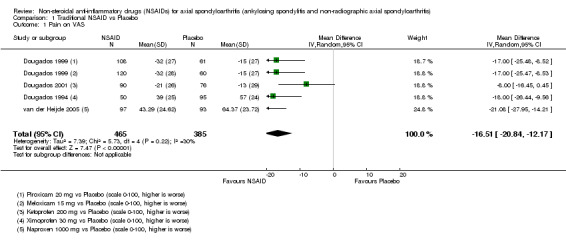
Comparison 1 Traditional NSAID vs Placebo, Outcome 1 Pain on VAS.
1.3. Analysis.
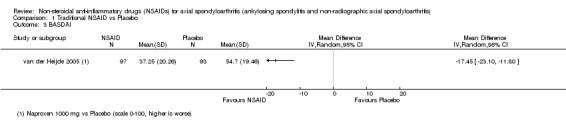
Comparison 1 Traditional NSAID vs Placebo, Outcome 3 BASDAI.
1.4. Analysis.
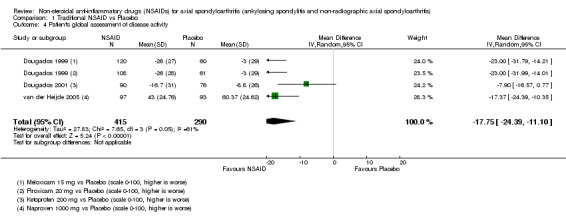
Comparison 1 Traditional NSAID vs Placebo, Outcome 4 Patient's global assessment of disease activity.
1.5. Analysis.
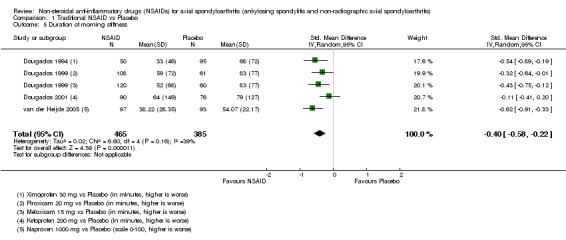
Comparison 1 Traditional NSAID vs Placebo, Outcome 5 Duration of morning stiffness.
1.6. Analysis.
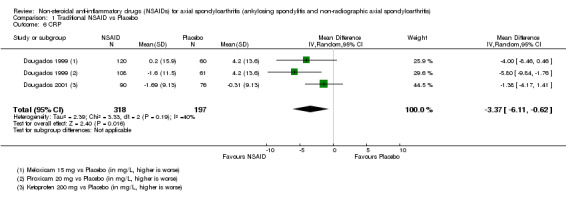
Comparison 1 Traditional NSAID vs Placebo, Outcome 6 CRP.
1.7. Analysis.
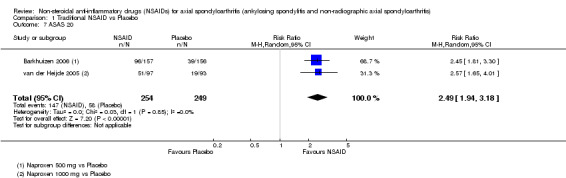
Comparison 1 Traditional NSAID vs Placebo, Outcome 7 ASAS 20.
1.9. Analysis.
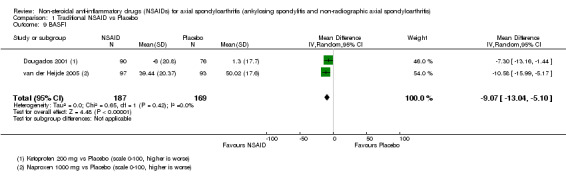
Comparison 1 Traditional NSAID vs Placebo, Outcome 9 BASFI.
1.10. Analysis.
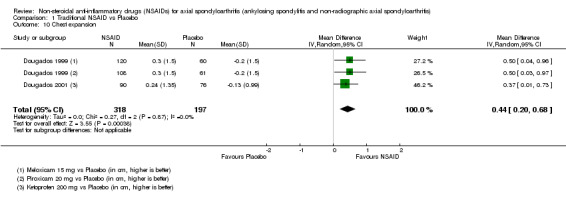
Comparison 1 Traditional NSAID vs Placebo, Outcome 10 Chest expansion.
1.11. Analysis.
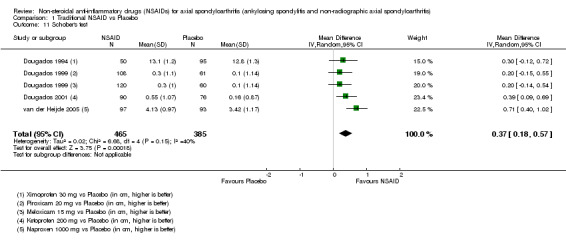
Comparison 1 Traditional NSAID vs Placebo, Outcome 11 Schober's test.
1.12. Analysis.
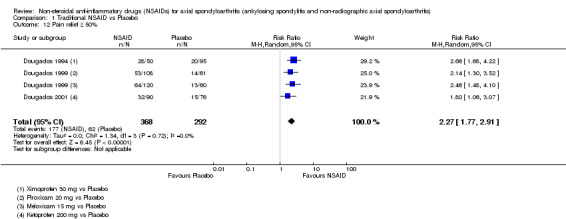
Comparison 1 Traditional NSAID vs Placebo, Outcome 12 Pain relief ≥ 50%.
1.8. Analysis.
Comparison 1 Traditional NSAID vs Placebo, Outcome 8 ASAS partial remission.
Harms
There was no difference in the number of withdrawals due to adverse events (five trials, N = 1165; RR 0.75, 95% CI 0.46 to 1.21; Analysis 1.2) or the number of any adverse events (RR 1.08, 95% 0.92 to 1.26; Analysis 1.13) or serious adverse events (RR 1.69, 95% CI 0.36 to 7.97; Analysis 1.14). However, when looking at the number of adverse events per organ system, five trials with 1289 participants showed that there were more gastro‐intestinal adverse events in patients taking NSAIDs compared to placebo (RR 1.92, 95% CI 1.41 to 2.61). Also, four studies with 1144 participants indicated that there were fewer neurological adverse events (including headache and dizziness) in the NSAID‐group compared with the placebo‐group (RR 0.44, 95% CI 0.24 to 0.82). In the other organ systems that were assessed (respiratory, hematological and dermatological) there was no difference in the number of adverse events (Analysis 1.15). The outcome serious adverse events was downgraded from high to moderate quality evidence because of potential imprecision as the 95% CI of this outcome includes 'no effect' and the upper confidence limit also crosses 'appreciable harm'.
1.2. Analysis.
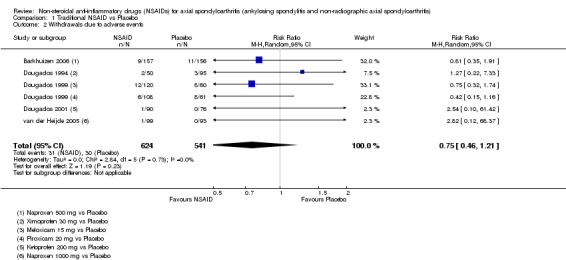
Comparison 1 Traditional NSAID vs Placebo, Outcome 2 Withdrawals due to adverse events.
1.13. Analysis.
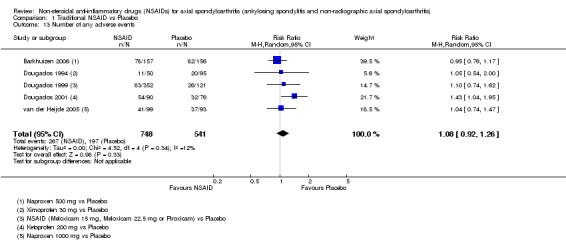
Comparison 1 Traditional NSAID vs Placebo, Outcome 13 Number of any adverse events.
1.14. Analysis.
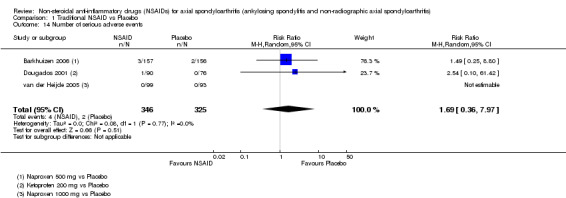
Comparison 1 Traditional NSAID vs Placebo, Outcome 14 Number of serious adverse events.
1.15. Analysis.
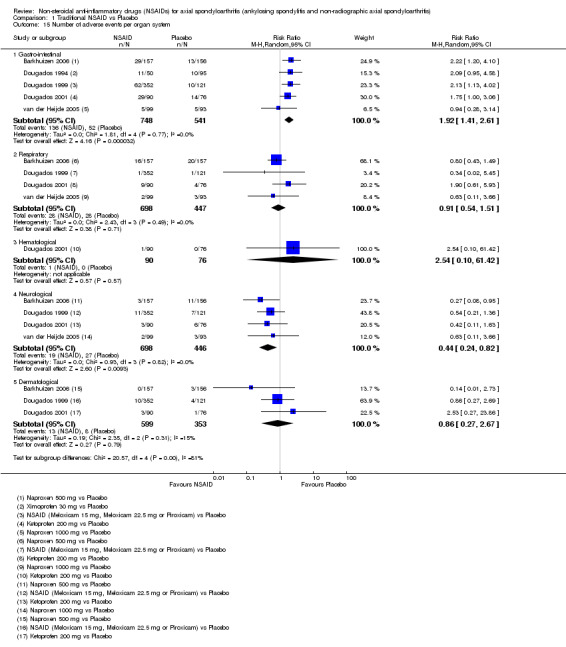
Comparison 1 Traditional NSAID vs Placebo, Outcome 15 Number of adverse events per organ system.
Extension phase of included trials
One study in this comparison, Dougados 1999, had an extension phase after the initial trial. Dougados 1999 had a double‐blind extension of the original six week trial (comparing piroxicam 20 mg to meloxicam 15 mg to meloxicam 22.5 mg and to placebo) up to 52 weeks, in which patients remained in the original treatment arm, showing comparable results as the six‐week trial for both benefits and harms.
Comparison 2: COX‐2 NSAIDs versus placebo
In comparison 2 we included all studies that compared a COX‐2 NSAID to placebo (three trials: Barkhuizen 2006; Dougados 2001; van der Heijde 2005). In Table 6 we listed the outcomes that were available for each study in this comparison.
Benefits
Three studies with 669 participants and a duration of six to 12 weeks provided data for this comparison. A significant effect favouring COX‐2 NSAIDs over placebo was found for most efficacy outcomes: pain on VAS (two trials, N = 349; MD ‐21.68, 95% CI ‐35.94 to ‐7.42 on a scale of 0 to 100 (higher is worse); Analysis 2.1); BASDAI (one trial, N = 193; MD ‐22.00, 95% CI ‐27.44 to ‐16.56 on a scale of 0 to 100 (higher is worse); Analysis 2.3); patient's global assessment of disease activity (two trials, N = 349; MD ‐20.82, 95% CI ‐29.88 to ‐11.75 on a scale of 0 to 100 (higher is worse); Analysis 2.4); ASAS 20 (two trials, N = 510; RR 2.51, 95% CI 1.66 to 3.79; Analysis 2.7); ASAS partial remission (one trial, N = 193; RR 4.65, 95% CI 1.39 to 15.55; Analysis 2.8); BASFI (two trials, N = 349, MD ‐13.42, 95% CI ‐17.35 to ‐9.49 on a scale of 0 to 100 (higher is worse); Analysis 2.9); Schober's test (two trials, N = 349; MD 0.42 cm, 95% CI 0.21 to 0.63; Analysis 2.11); and pain relief ≥ 50% (one trial, N = 156; RR 2.41, 95% CI 1.45 to 4.00; Analysis 2.12). However, we found no significant difference between coxibs and placebo in the outcome duration of morning stiffness (two trials, N = 349; SMD ‐4.72, 95% CI ‐13.33 to 3.90; Analysis 2.5), CRP (one trial, N = 156; MD ‐2.17 mg/dL, 95% CI ‐5.39 to 1.05; Analysis 2.6) and chest expansion (one trial, N = 156; MD 0.40 cm, 95% CI ‐0.00 to 0.80; Analysis 2.10). No studies reported data for a comparison between COX‐2 NSAIDs and placebo with respect to our other major efficacy outcomes BASMI or radiographic progression (see Table 6). The outcome BASDAI was downgraded from high to moderate quality evidence because of potential imprecision as this outcome only included data from a single study (N = 193).
2.1. Analysis.
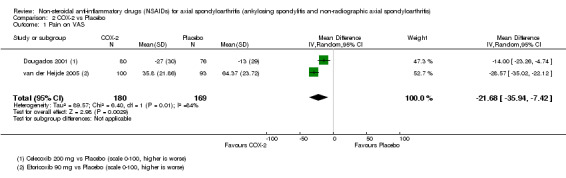
Comparison 2 COX‐2 vs Placebo, Outcome 1 Pain on VAS.
2.3. Analysis.
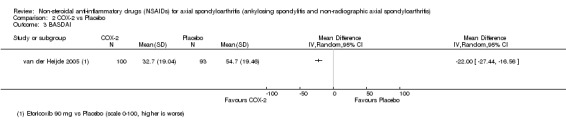
Comparison 2 COX‐2 vs Placebo, Outcome 3 BASDAI.
2.4. Analysis.
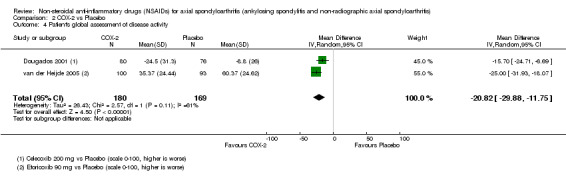
Comparison 2 COX‐2 vs Placebo, Outcome 4 Patient's global assessment of disease activity.
2.7. Analysis.
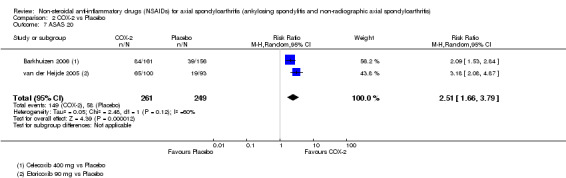
Comparison 2 COX‐2 vs Placebo, Outcome 7 ASAS 20.
2.8. Analysis.
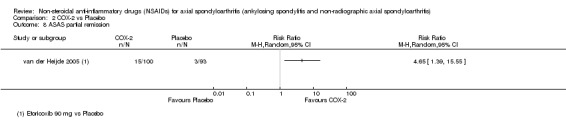
Comparison 2 COX‐2 vs Placebo, Outcome 8 ASAS partial remission.
2.9. Analysis.
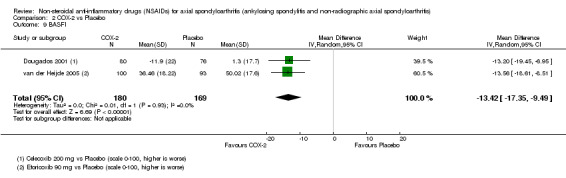
Comparison 2 COX‐2 vs Placebo, Outcome 9 BASFI.
2.11. Analysis.
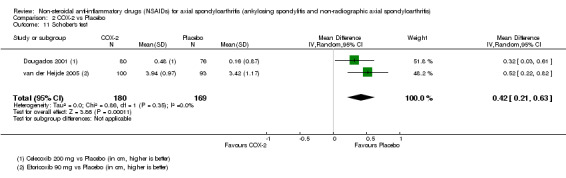
Comparison 2 COX‐2 vs Placebo, Outcome 11 Schober's test.
2.12. Analysis.
Comparison 2 COX‐2 vs Placebo, Outcome 12 Pain relief ≥ 50%.
2.5. Analysis.
Comparison 2 COX‐2 vs Placebo, Outcome 5 Duration of morning stiffness.
2.6. Analysis.
Comparison 2 COX‐2 vs Placebo, Outcome 6 CRP.
2.10. Analysis.
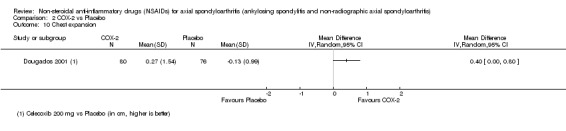
Comparison 2 COX‐2 vs Placebo, Outcome 10 Chest expansion.
Harms
Regarding harms we found similar results as in the comparison traditional NSAIDs versus placebo. The studies detected no difference in the number of withdrawals due to adverse events (three trials, N = 669; RR 2.14, 95% CI 0.36 to 12.56; Analysis 2.2) or the number of any adverse events (three trials, 669 participants; RR 1.22, 95% CI 0.93 to 1.62; Analysis 2.13) or any serious adverse events (three trials, 669 participants; RR 0.92, 95% CI 0.14 to 6.21; Analysis 2.14). However, when looking at the number of adverse events per organ system, we found also here that there were more gastro‐intestinal adverse events in patients taking coxibs compared to placebo (three studies, N = 669; RR 1.80, 95% CI 1.22 to 2.67). In the other organ systems that were assessed (respiratory, neurological and dermatological) there was no difference in the number of adverse events (Analysis 2.15). The outcome withdrawals due to adverse events was downgraded from high to low quality evidence because of potential imprecision (the 95% CI includes 'no effect' and the upper confidence limit also crosses 'appreciable harm') and large heterogeneity (I² statistic = 84%). The outcome serious adverse events was downgraded from high to moderate quality evidence because of potential imprecision as the 95% CI of this outcome includes 'no effect' and the upper confidence limit also crosses 'appreciable harm'.
2.2. Analysis.
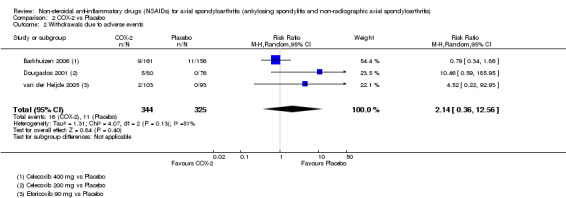
Comparison 2 COX‐2 vs Placebo, Outcome 2 Withdrawals due to adverse events.
2.13. Analysis.
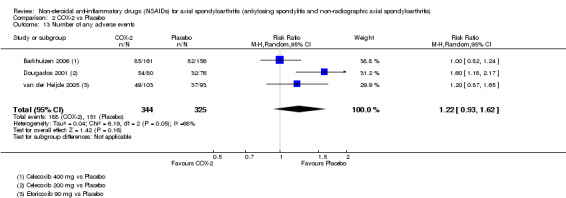
Comparison 2 COX‐2 vs Placebo, Outcome 13 Number of any adverse events.
2.14. Analysis.
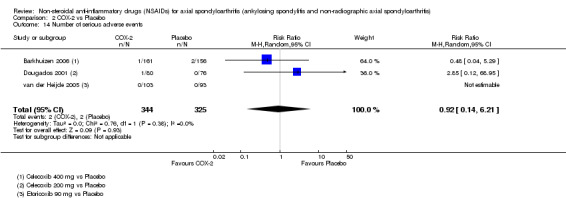
Comparison 2 COX‐2 vs Placebo, Outcome 14 Number of serious adverse events.
2.15. Analysis.
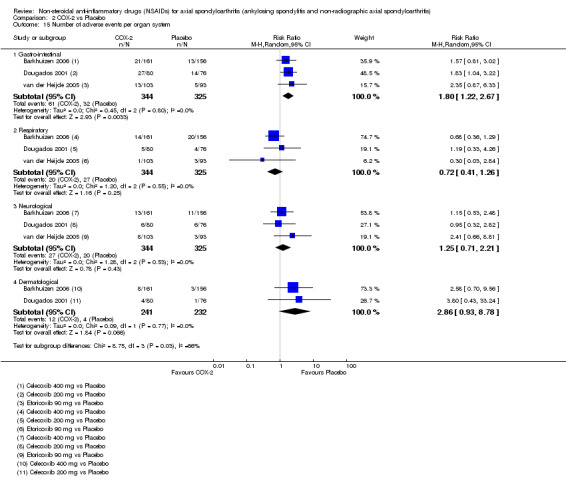
Comparison 2 COX‐2 vs Placebo, Outcome 15 Number of adverse events per organ system.
Comparison 3: COX‐2 NSAIDs versus traditional NSAIDs
In comparison 3 we included all studies that compared COX‐2 versus traditional NSAIDs (four trials: Barkhuizen 2006; Dougados 2001; Sieper 2008; van der Heijde 2005). In Table 7 we listed the outcomes that were available for each study in this comparison.
Benefits
In four studies with 995 participants, we found no difference in any of the efficacy measures between coxibs and traditional NSAIDs (pain on VAS (Analysis 3.1); BASDAI (Analysis 3.3); patient's global assessment of disease activity (Analysis 3.4); duration of morning stiffness (Analysis 3.5); CRP (Analysis 3.6); ASAS 20 (Analysis 3.7); ASAS partial remission (Analysis 3.8); BASFI (Analysis 3.9); BASMI (Analysis 3.10); chest expansion (Analysis 3.11); Schober's test (Analysis 3.12); and pain relief ≥ 50% (Analysis 3.13). However, several outcomes were only measured in one trial (ASAS partial remission (Analysis 3.8); BASMI (Analysis 3.10); chest expansion (Analysis 3.11); and pain relief ≥ 50% (Analysis 3.13). No studies reported data for a comparison between COX‐2 and traditional NSAIDs with respect to our other major efficacy outcome radiographic progression (see Table 7).
3.1. Analysis.
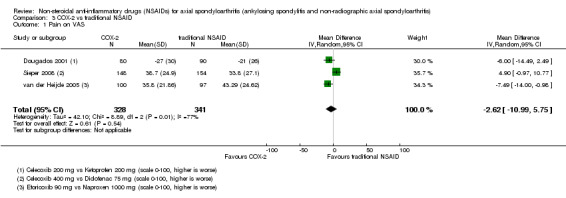
Comparison 3 COX‐2 vs traditional NSAID, Outcome 1 Pain on VAS.
3.3. Analysis.
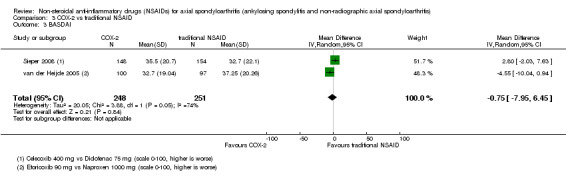
Comparison 3 COX‐2 vs traditional NSAID, Outcome 3 BASDAI.
3.4. Analysis.
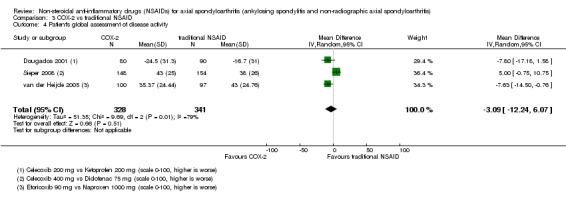
Comparison 3 COX‐2 vs traditional NSAID, Outcome 4 Patient's global assessment of disease activity.
3.5. Analysis.
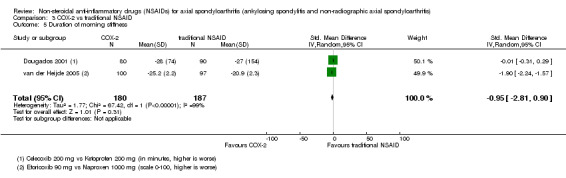
Comparison 3 COX‐2 vs traditional NSAID, Outcome 5 Duration of morning stiffness.
3.6. Analysis.
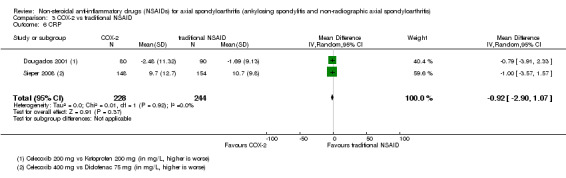
Comparison 3 COX‐2 vs traditional NSAID, Outcome 6 CRP.
3.7. Analysis.
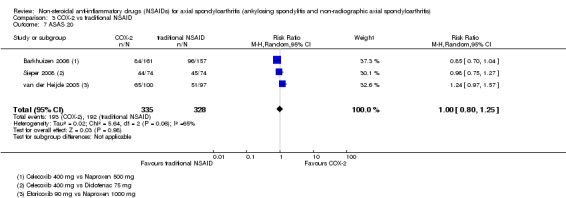
Comparison 3 COX‐2 vs traditional NSAID, Outcome 7 ASAS 20.
3.8. Analysis.
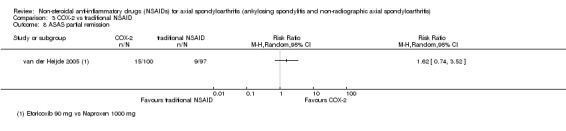
Comparison 3 COX‐2 vs traditional NSAID, Outcome 8 ASAS partial remission.
3.9. Analysis.
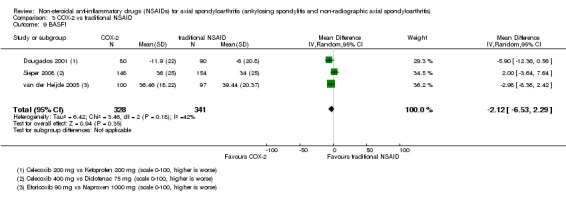
Comparison 3 COX‐2 vs traditional NSAID, Outcome 9 BASFI.
3.10. Analysis.
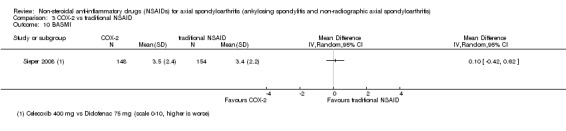
Comparison 3 COX‐2 vs traditional NSAID, Outcome 10 BASMI.
3.11. Analysis.
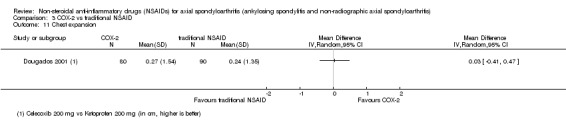
Comparison 3 COX‐2 vs traditional NSAID, Outcome 11 Chest expansion.
3.12. Analysis.
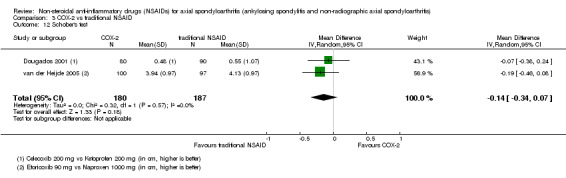
Comparison 3 COX‐2 vs traditional NSAID, Outcome 12 Schober's test.
3.13. Analysis.
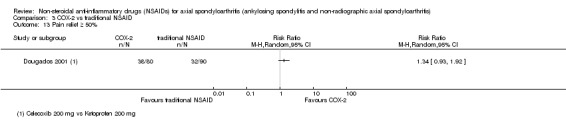
Comparison 3 COX‐2 vs traditional NSAID, Outcome 13 Pain relief ≥ 50%.
Harms
No differences between COX‐2 and traditional NSAIDs could be detected for any of the safety outcomes (including: withdrawals due to adverse events (Analysis 3.2); any adverse events (Analysis 3.14); serious adverse events (Analysis 3.15); and adverse events per organ system (cardiovascular, gastro‐intestinal, hepatic, respiratory, haematological, neurological or dermatological; Analysis 3.16).
3.2. Analysis.
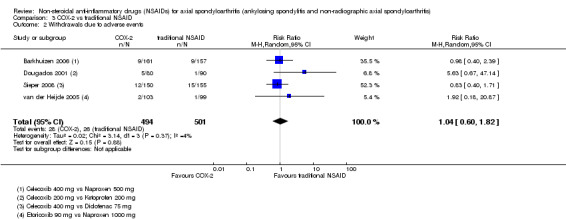
Comparison 3 COX‐2 vs traditional NSAID, Outcome 2 Withdrawals due to adverse events.
3.14. Analysis.
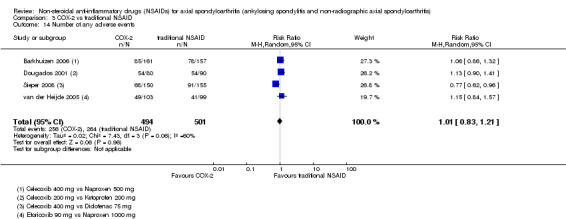
Comparison 3 COX‐2 vs traditional NSAID, Outcome 14 Number of any adverse events.
3.15. Analysis.
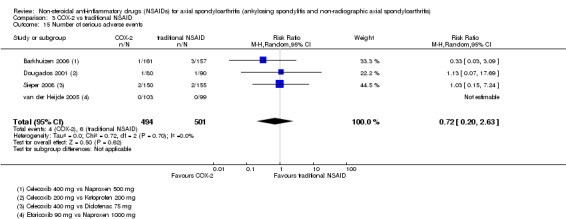
Comparison 3 COX‐2 vs traditional NSAID, Outcome 15 Number of serious adverse events.
3.16. Analysis.
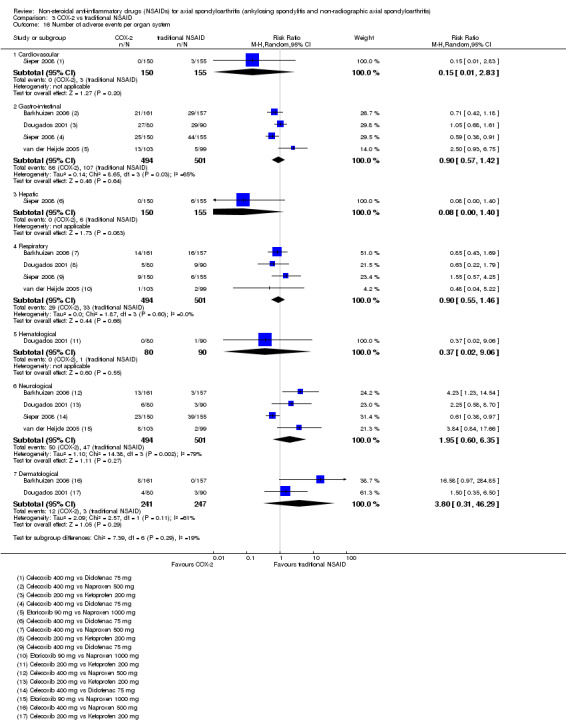
Comparison 3 COX‐2 vs traditional NSAID, Outcome 16 Number of adverse events per organ system.
Extension phase of included trials
One study in this comparison, van der Heijde 2005, had an extension phase after the initial trial. van der Heijde 2005 had a double‐blind extension of the original six week trial (comparing etoricoxib 90 mg to etoricoxib 120 mg to naproxen 1000 mg and to placebo) up to 52 weeks. The patients in the placebo‐group were reassigned to one of the three active treatment arms (1:1:1), and patients that were originally randomised into an active treatment arm continued the same study drug. Results of this extension phase showed that etoricoxib 90 mg and etoricoxib 120 mg were both more effective than naproxen 1000 mg, with a similar efficacy of both dosages of etoricoxib and comparable harms of both etoricoxib dosages and naproxen (see the Characteristics of included studies section for full report of results of extension phase).
Comparison 4: NSAIDs versus NSAIDs
In comparison 4 we included all trials that compared one NSAID to another (24 trials with 2076 participants) (Batlle‐Gualda 1996; Caldwell 1986; Calin 1979; Dougados 1999; Ebner 1983; Franssen 1986; Good 1977; Heinrichs 1985; Khan 1985; Lomen 1986 I; Lomen 1986 P; Mena 1977; Myklebust 1986; Nahir 1980; Nissilä 1978a; Nissilä 1978b; Palferman 1991; Pasero 1994; Rejholec 1980; Santo 1988; Schwarzer 1990; Simpson 1966; Tannenbaum 1984; Villa Alcázar 1996). The comparison includes many different NSAIDs and must therefore be interpreted with caution.
We performed a sensitivity analysis for all NSAIDs that were assessed by two or more trials in any outcome, to see whether one NSAID was consistently different from other NSAIDs with regards to benefits or harms. In Table 8 we listed the outcomes that were available for each study in this comparison.
Four additional trials could not be included in the quantitative data analyses as the numbers of participants per treatment arm were not specified (Ansell 1978; Carcassi 1990; Lehtinen 1984; Sydnes 1981), and we have summarised these trials after presentation of the pooled analyses below.
Benefits
Overall, no consistent difference in efficacy could be determined in any of the efficacy variables that were assessed (pain on Likert scale (Analysis 4.1); pain on VAS (Analysis 4.2); patient's global assessment of disease activity (Analysis 4.4); duration of morning stiffness (Analysis 4.5); severity of morning stiffness (Analysis 4.6); CRP (Analysis 4.7); ESR (Analysis 4.8); lateral spinal flexion (Analysis 4.9); chest expansion (Analysis 4.10); tragus‐to‐wall distance (Analysis 4.11); and pain relief ≥ 50%) (Analysis 4.14). In the outcome occiput‐to‐wall distance, two studies with 354 participants (Batlle‐Gualda 1996; Tannenbaum 1984) indicated that indomethacin performed significantly worse than another NSAID (aceclofenac and piroxicam) (MD ‐0.10 cm, 95% CI ‐0.12 to ‐0.08; Analysis 4.12). However, this result was completely determined by one trial, Batlle‐Gualda 1996. In the outcome Schober's test (Analysis 4.13) two trials with 412 participants (Batlle‐Gualda 1996; Pasero 1994) indicated that aceclofenac performed significantly better than another NSAID (indomethacin and naproxen) (MD 0.10 cm, 95% CI 0.08 to 0.12), but this result was again completely determined by Batlle‐Gualda 1996. In the same outcome, three studies with 396 participants (Batlle‐Gualda 1996; Palferman 1991; Tannenbaum 1984) indicated that indomethacin performed significantly worse than another NSAID (aceclofenac, nabumetone and piroxicam), which was again fully determined by Batlle‐Gualda 1996 (MD 0.10 cm, 95% CI 0.08 to 0.12). No studies reported data for a comparison between NSAIDs with respect to our other major efficacy outcomes BASDAI, BASFI, BASMI or radiographic progression (see Table 8).
4.1. Analysis.
Comparison 4 NSAID vs NSAID, Outcome 1 Pain on Likert scale.
4.2. Analysis.
Comparison 4 NSAID vs NSAID, Outcome 2 Pain on VAS.
4.4. Analysis.
Comparison 4 NSAID vs NSAID, Outcome 4 Patient's global assessment of disease activity.
4.5. Analysis.
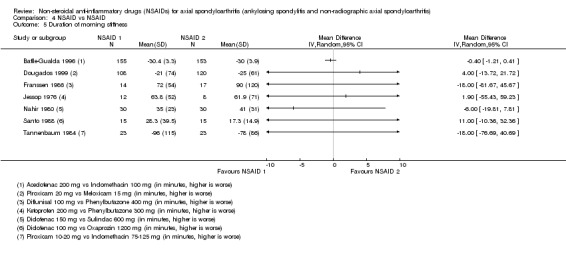
Comparison 4 NSAID vs NSAID, Outcome 5 Duration of morning stiffness.
4.6. Analysis.
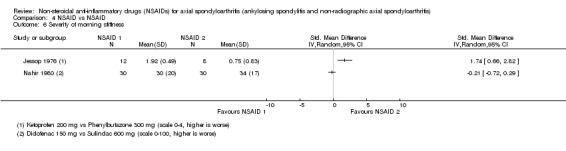
Comparison 4 NSAID vs NSAID, Outcome 6 Severity of morning stiffness.
4.7. Analysis.
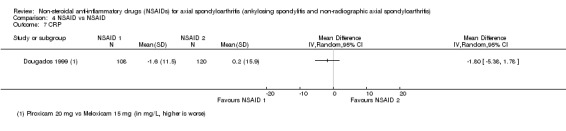
Comparison 4 NSAID vs NSAID, Outcome 7 CRP.
4.8. Analysis.
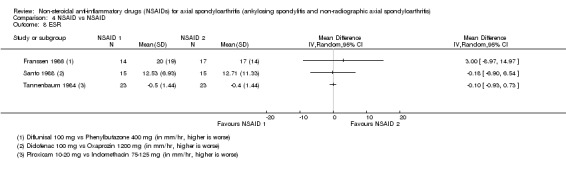
Comparison 4 NSAID vs NSAID, Outcome 8 ESR.
4.9. Analysis.
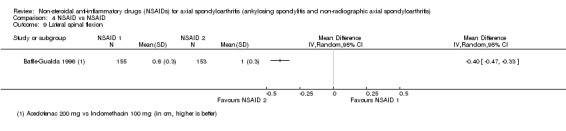
Comparison 4 NSAID vs NSAID, Outcome 9 Lateral spinal flexion.
4.10. Analysis.
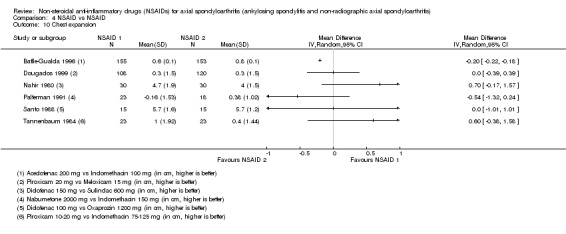
Comparison 4 NSAID vs NSAID, Outcome 10 Chest expansion.
4.11. Analysis.
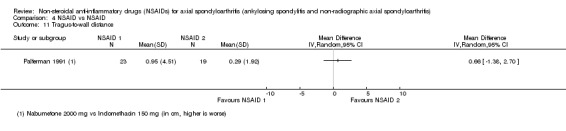
Comparison 4 NSAID vs NSAID, Outcome 11 Tragus‐to‐wall distance.
4.14. Analysis.
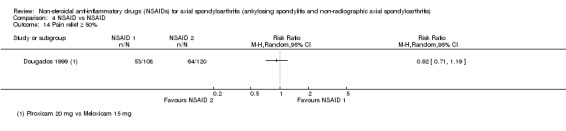
Comparison 4 NSAID vs NSAID, Outcome 14 Pain relief ≥ 50%.
4.12. Analysis.
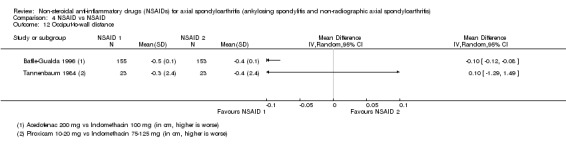
Comparison 4 NSAID vs NSAID, Outcome 12 Occiput‐to‐wall distance.
4.13. Analysis.
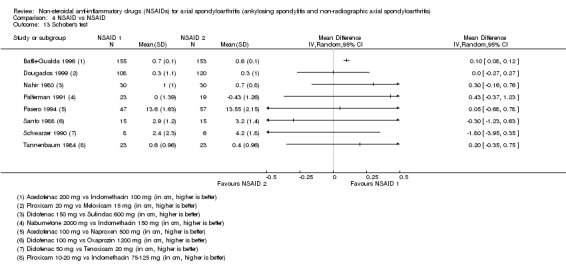
Comparison 4 NSAID vs NSAID, Outcome 13 Schober's test.
Harms
Twenty‐three studies with 2041 participants reported no difference in withdrawals due to adverse events in one of the NSAIDs assessed versus another (Analysis 4.3). Eleven studies with 1135 participants showed that indomethacin resulted in significantly more (any) adverse events than other NSAIDs (RR 1.25, 95% CI 1.06 to 1.48; Analysis 4.15; see Figure 4 for a forest plot of this analysis that only includes studies comparing indomethacin to another NSAID). Furthermore, two trials with 137 participants (Caldwell 1986; Santo 1988) indicated that oxaprozine had significantly less (any) adverse events than another NSAID (diclofenac and indomethacin) (RR 0.64, 95% CI 0.42 to 0.96), although this result was mainly based on Caldwell 1986 with an overall high risk of bias. In two trials with 86 participants no difference was detected in serious adverse events between the different NSAIDs (Analysis 4.16).
4.3. Analysis.
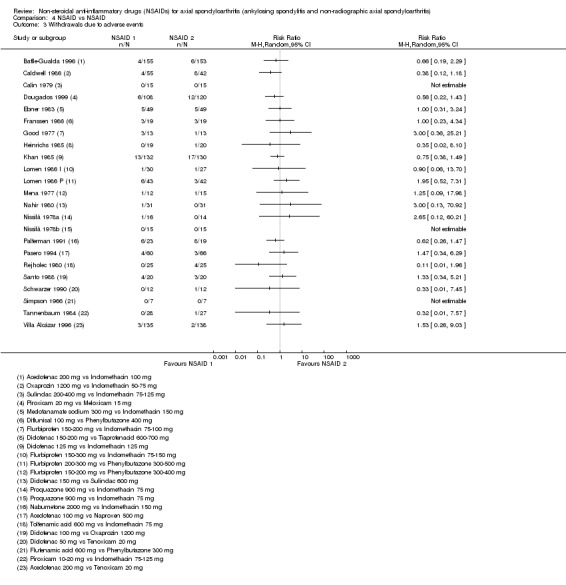
Comparison 4 NSAID vs NSAID, Outcome 3 Withdrawals due to adverse events.
4.15. Analysis.
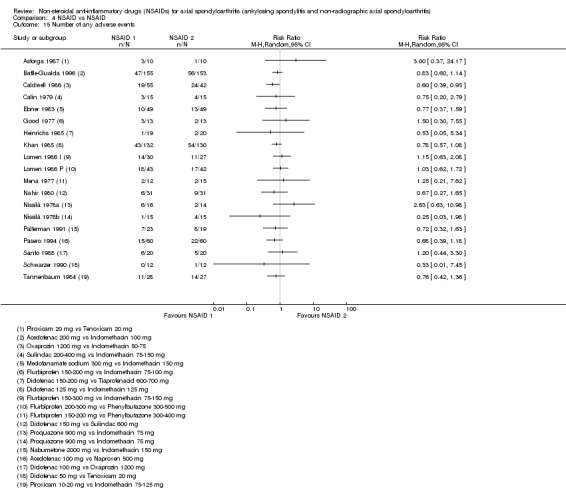
Comparison 4 NSAID vs NSAID, Outcome 15 Number of any adverse events.
4.
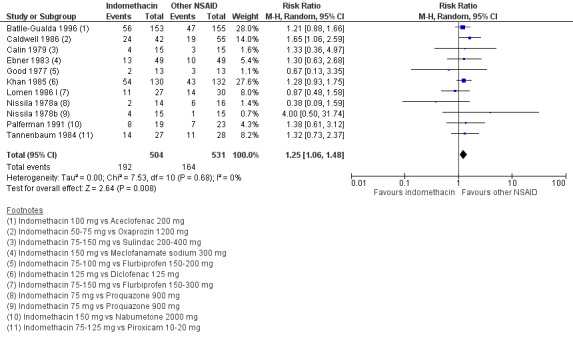
Forest plot of comparison 4 NSAID vs NSAID, outcome 4.15 Number of any adverse events, only studies with indomethacin.
4.16. Analysis.
Comparison 4 NSAID vs NSAID, Outcome 16 Number of serious adverse events.
When assessing adverse event per organ system, nine trials with 963 participants showed that indomethacin was associated with more neurological adverse events (like headache and dizziness) than another NSAID (RR 2.34, 95% CI 1.32 to 4.14; Analysis 4.17; see Figure 5 for a forest plot of this analysis that only includes studies comparing indomethacin to another NSAID). Two studies with 302 participants (Khan 1985; Santo 1988) indicated that diclofenac resulted in more neurological adverse events (like headache and dizziness) in comparison to another NSAID (indomethacin and oxaprozine) (RR 2.94, 95% CI 1.49 to 5.88), but again this result was completely determined by one study (Khan 1985). Adverse events in the other organ systems that were assessed (cardiovascular, gastro‐intestinal, hepatic, respiratory, hematological, renal and dermatological) were not more prevalent in one NSAID versus the others.
4.17. Analysis.
Comparison 4 NSAID vs NSAID, Outcome 17 Adverse events per organ system.
5.
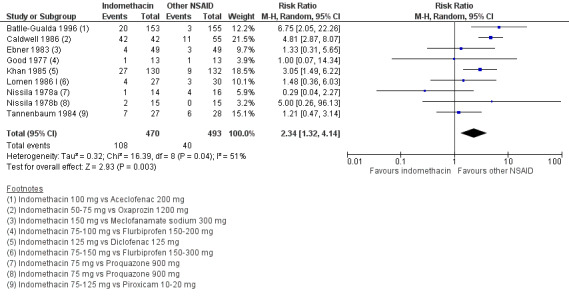
Forest plot of comparison 4 NSAID vs NSAID, outcome 4.17 Adverse events per organ system, only studies with indomethacin.
Summary of trials not included in the quantitative analyses
Ansell 1978 was a cross‐over trial with 25 participants comparing naproxen 750 mg to butacote 300 mg in treatment periods of four weeks. The trial authors reported that although they found no difference in benefits or harms, patients and physicians favoured butacote in their global assessments.
Carcassi 1990 was an RCT with 151 participants comparing pirazolac 300 to 600 mg to indomethacin 25 to 50 mg (treatment duration 12 weeks). The trial authors reported no between‐group differences in either benefits or harms.
Lehtinen 1984 was a cross‐over trial with 30 participants comparing indomethacin slow‐release tablets (50 mg) to indomethacin capsules (25 mg) in treatment periods of one week. The trial authors concluded that slow‐release tablets were as effective as capsules, but had fewer side‐effects, especially diarrhoea, epigastric pain and dizziness.
Sydnes 1981 was a cross‐over study with 93 participants comparing piroxicam 20 mg to indomethacin 75 mg in treatment periods of four weeks. The study authors concluded that piroxicam was more effective and better tolerated than indomethacin.
Extension phase of included studies
Three studies in this comparison (Dougados 1999; Franssen 1986; Tannenbaum 1984) had an extension phase after the initial trial. The extension phase of Franssen 1986 (comparing diflunisal 1000 mg to phenylbutazone 400 mg) and Tannenbaum 1984 (comparing piroxicam 10 to 20 mg to indomethacin 75 to 125 mg) were both open extensions up to 48 weeks duration, showing similar results in benefits and harms as the original trials. Dougados 1999 (extension phase is described in more detail above) found comparable results in the extension phase as in the six week trial for both benefits and harms of piroxicam and meloxicam (see Characteristics of included studies section for full report of results of extension phases).
Comparison 5: Naproxen versus other NSAIDs
In comparison 5 we included all studies that compared naproxen to other NSAIDs (three trials: Barkhuizen 2006; Pasero 1994; van der Heijde 2005). In Table 9 we listed the outcomes that were available for each study in this comparison.
Benefits
We included three studies with 646 participants in this comparison. Naproxen performed significantly worse than another NSAID in the outcomes pain on VAS (two trials, N = 232; MD 6.80, 95% CI 3.72 to 9.88 on a scale of 0 to 100 (higher is worse); Analysis 5.1) and patient's global assessment of disease activity (one trial, N = 197; MD 7.63, 95% CI 0.61 to 14.65 on a scale of 0 to 100 (higher is worse); Analysis 5.4). There was no difference between naproxen and another NSAID in the other efficacy variables (BASDAI (Analysis 5.3); duration of morning stiffness (Analysis 5.5); ASAS 20 (Analysis 5.6); ASAS partial remission (Analysis 5.7); BASFI (Analysis 5.8); and Schober's test (Analysis 5.9). No studies reported data for a comparison between naproxen and other NSAIDs with respect to our other primary efficacy outcomes BASMI or radiographic progression (see Table 9).
5.1. Analysis.
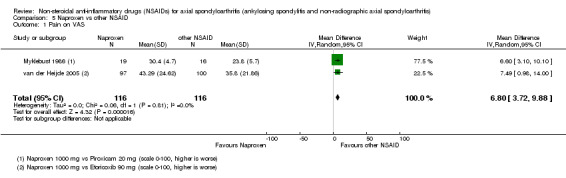
Comparison 5 Naproxen vs other NSAID, Outcome 1 Pain on VAS.
5.4. Analysis.
Comparison 5 Naproxen vs other NSAID, Outcome 4 Patient's global assessment of disease activity.
5.3. Analysis.
Comparison 5 Naproxen vs other NSAID, Outcome 3 BASDAI.
5.5. Analysis.
Comparison 5 Naproxen vs other NSAID, Outcome 5 Duration of morning stiffness.
5.6. Analysis.
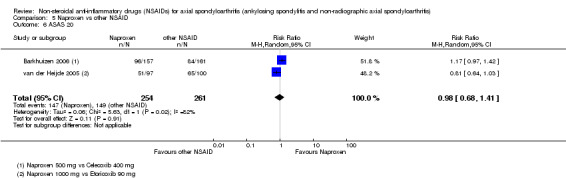
Comparison 5 Naproxen vs other NSAID, Outcome 6 ASAS 20.
5.7. Analysis.
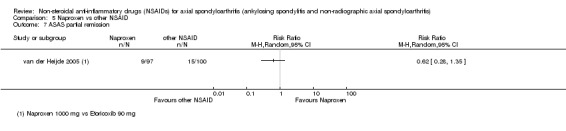
Comparison 5 Naproxen vs other NSAID, Outcome 7 ASAS partial remission.
5.8. Analysis.
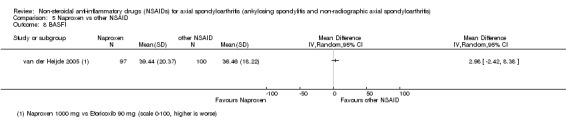
Comparison 5 Naproxen vs other NSAID, Outcome 8 BASFI.
5.9. Analysis.
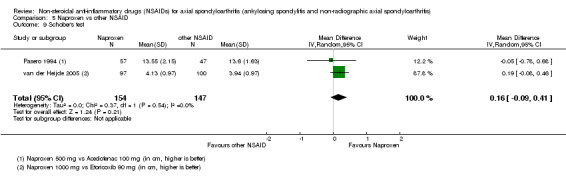
Comparison 5 Naproxen vs other NSAID, Outcome 9 Schober's test.
Harms
The number of withdrawals due to adverse events (Analysis 5.2), as well as the number of any adverse events (Analysis 5.10) or serious adverse events (Analysis 5.11) did not differ between naproxen and other NSAIDs that were assessed. However, the three included trials showed that naproxen resulted in significantly less neurological adverse events in comparison to other NSAIDs (RR 0.24, 95% CI 0.10 to 0.60; Analysis 5.12). No such difference was found in adverse events in the other organ systems that were assessed (gastro‐intestinal, hepatic, respiratory and dermatological; Analysis 5.12).
5.2. Analysis.
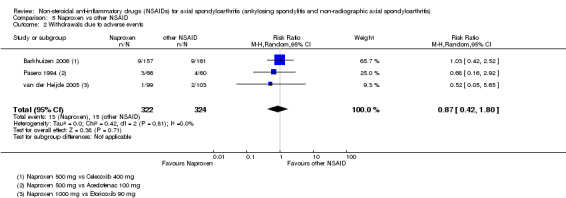
Comparison 5 Naproxen vs other NSAID, Outcome 2 Withdrawals due to adverse events.
5.10. Analysis.
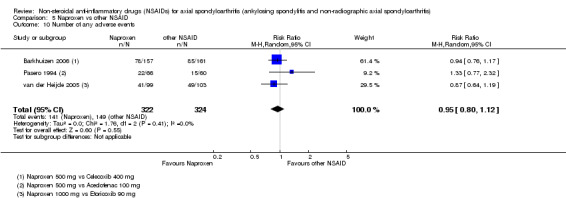
Comparison 5 Naproxen vs other NSAID, Outcome 10 Number of any adverse events.
5.11. Analysis.
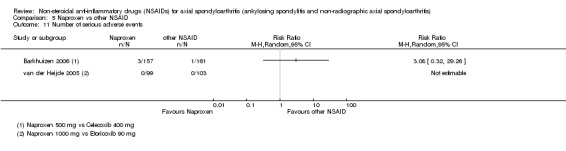
Comparison 5 Naproxen vs other NSAID, Outcome 11 Number of serious adverse events.
5.12. Analysis.
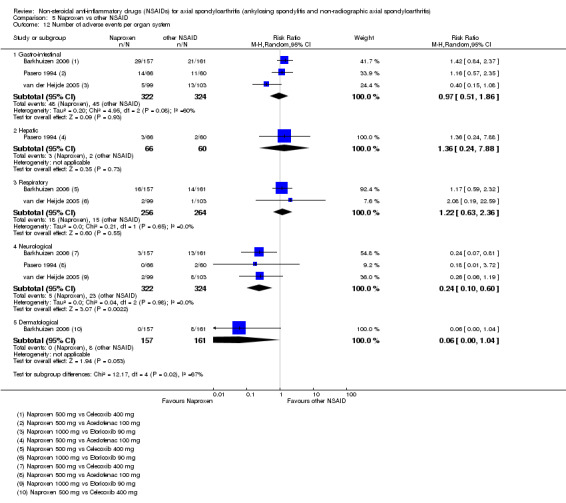
Comparison 5 Naproxen vs other NSAID, Outcome 12 Number of adverse events per organ system.
Extension phase of included trials
One study in this comparison had an extension phase after the initial trial as described earlier (van der Heijde 2005) in which etoricoxib 90 mg and etoricoxib 120 mg were both more effective than naproxen 1000 mg, with comparable harms of both etoricoxib dosages and naproxen (see the Characteristics of included studies table for the full report of results of extension phase).
Comparison 6: Low versus high dose NSAID
In comparison 6 we included all studies that compared low versus higher doses of an NSAID. There were five trials (1136 participants) (Barkhuizen 2006; Dougados 1994; Dougados 1999; Sieper 2008; van der Heijde 2005) and one post‐hoc analysis of a prospective cohort study (Poddubnyy 2012).
Similar to comparison 4, the trials included many different NSAIDs, therefore interpretation of pooled analyses was done with caution. In Table 10 we listed the outcomes that were available for each trial in this comparison.
We could not include one additional trial in pooled analyses as we could not extract suitable quantitative data (Muller‐Fassbender 1985).
Benefits
Based upon the trial data, we found no clear dose‐effect on any of the efficacy variables (pain on VAS (Analysis 6.1); BASDAI (Analysis 6.3); patient's global assessment of disease activity (Analysis 6.4); duration of morning stiffness (Analysis 6.5); CRP (Analysis 6.6); ASAS 20 (Analysis 6.7); ASAS partial remission (Analysis 6.8); BASFI (Analysis 6.9); BASMI (Analysis 6.10); chest expansion (Analysis 6.11); Schober's test (Analysis 6.12); and pain relief ≥ 50% (Analysis 6.13). No studies reported data for a comparison between different dosages of NSAIDs with respect to our other major efficacy outcome radiographic progression (see Table 10).
6.1. Analysis.
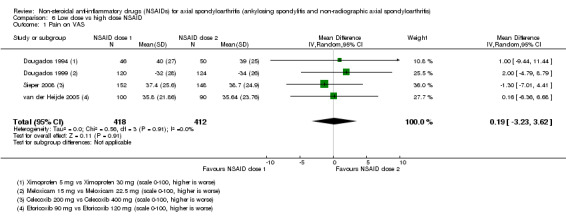
Comparison 6 Low dose vs high dose NSAID, Outcome 1 Pain on VAS.
6.3. Analysis.
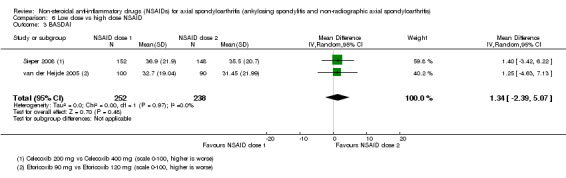
Comparison 6 Low dose vs high dose NSAID, Outcome 3 BASDAI.
6.4. Analysis.
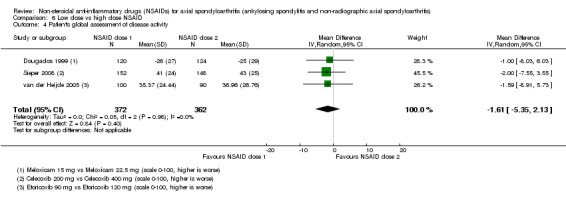
Comparison 6 Low dose vs high dose NSAID, Outcome 4 Patient's global assessment of disease activity.
6.5. Analysis.
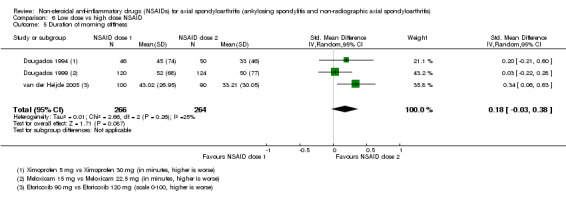
Comparison 6 Low dose vs high dose NSAID, Outcome 5 Duration of morning stiffness.
6.6. Analysis.
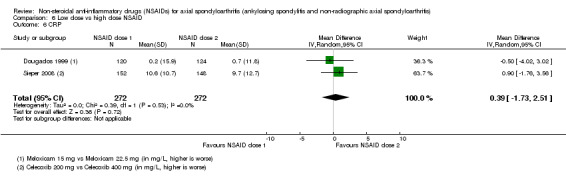
Comparison 6 Low dose vs high dose NSAID, Outcome 6 CRP.
6.7. Analysis.
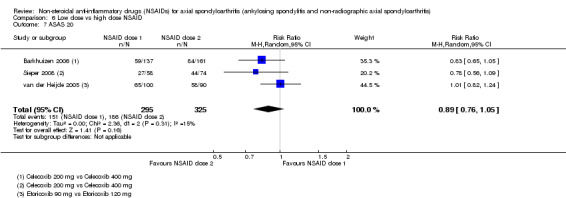
Comparison 6 Low dose vs high dose NSAID, Outcome 7 ASAS 20.
6.8. Analysis.
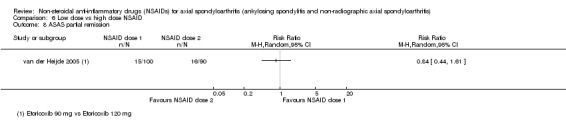
Comparison 6 Low dose vs high dose NSAID, Outcome 8 ASAS partial remission.
6.9. Analysis.
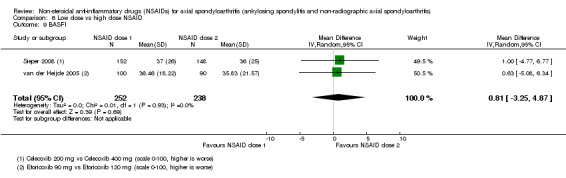
Comparison 6 Low dose vs high dose NSAID, Outcome 9 BASFI.
6.10. Analysis.
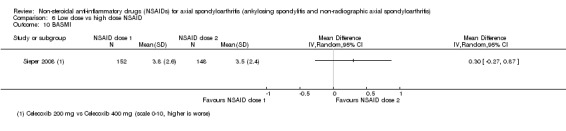
Comparison 6 Low dose vs high dose NSAID, Outcome 10 BASMI.
6.11. Analysis.
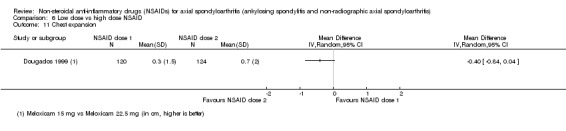
Comparison 6 Low dose vs high dose NSAID, Outcome 11 Chest expansion.
6.12. Analysis.
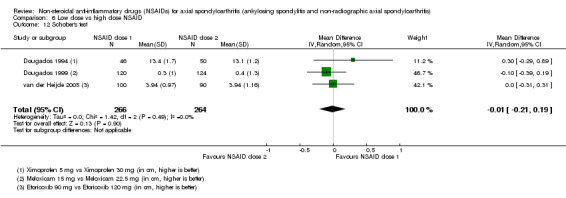
Comparison 6 Low dose vs high dose NSAID, Outcome 12 Schober's test.
6.13. Analysis.
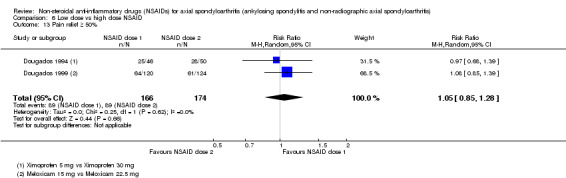
Comparison 6 Low dose vs high dose NSAID, Outcome 13 Pain relief ≥ 50%.
Muller‐Fassbender 1985 was a cross‐over trial with 39 participants comparing ketoprofen 150 mg two times a day to ketoprofen in a dosage of 100 mg three times a day. The results showed an improvement in both groups, although the global assessment of patient and physician were both in favour of ketoprofen 100 mg three times a day.
Poddubnyy 2012 was a post‐hoc analysis of a prospective cohort study including 164 participants with AS and axial SpA. They reported a mean change in mSASSS over two years in participants with AS and high NSAID intake of 0.02 ± 1.39 units versus 0.96 ± 2.78 units in participants with low NSAID intake. After adjustment for radiographic status at baseline, this difference was not significant (P = 0.22). Fewer participants with a high NSAID intake had worsening of mSASSS score by ≥ two units compared with those with a low NSAID intake (8.3% versus 21.9%, P = 0.142). After adjustment for factors independently associated with radiographic spinal progression, this resulted in OR 0.15 (95% CI 0.02 to 0.96; P = 0.045) for retarded radiographic progression in the spine with high NSAID intake. There was no clear and consistent difference between low versus high NSAID intake and radiographic progression in participants with nr‐axSpA (for full study description and results see Table 3).
Harms
Based upon the trial data, no clear dose‐effect with respect to harms (including withdrawals due to adverse events (Analysis 6.2); any adverse events (Analysis 6.14); serious adverse events (Analysis 6.15); and adverse events per organ system (cardiovascular, gastro‐intestinal, respiratory, neurological and dermatological; Analysis 6.16) could be detected.
6.2. Analysis.
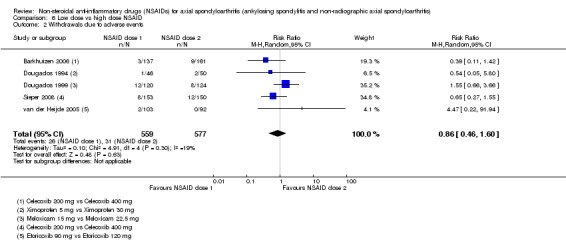
Comparison 6 Low dose vs high dose NSAID, Outcome 2 Withdrawals due to adverse events.
6.14. Analysis.
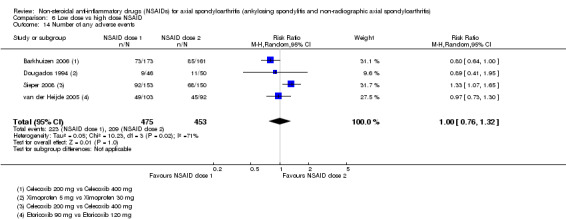
Comparison 6 Low dose vs high dose NSAID, Outcome 14 Number of any adverse events.
6.15. Analysis.
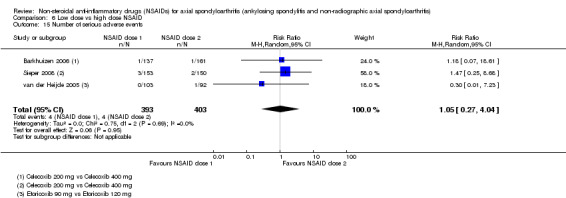
Comparison 6 Low dose vs high dose NSAID, Outcome 15 Number of serious adverse events.
6.16. Analysis.
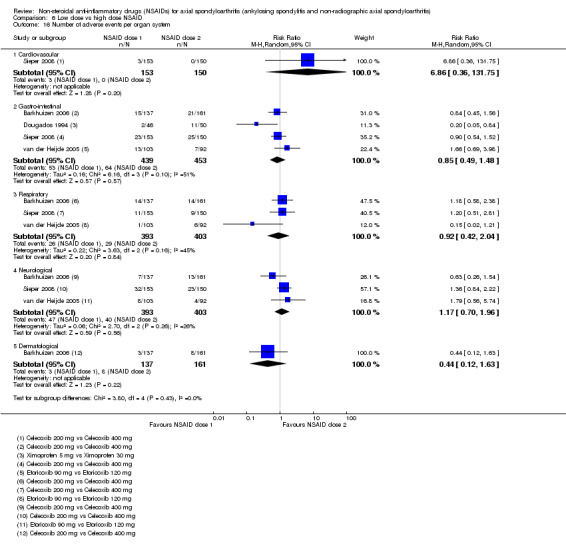
Comparison 6 Low dose vs high dose NSAID, Outcome 16 Number of adverse events per organ system.
Extension phase of included trials
As noted above, two trials in this comparison had an extension phase (Dougados 1999; van der Heijde 2005). Dougados 1999 found comparable results as the six‐week trial for both benefits and harms of meloxicam 15 and 22.5 mg. van der Heijde 2005 also found similar benefits and harms of both doses of etoricoxib (see Characteristics of included studies section for full report of results of extension phase).
Comparison 7: Continuous versus on‐demand NSAID use
One trial, Wanders 2005, with a post‐hoc subgroup analysis, Kroon 2012, and one retrospective cohort study, Boersma 1976, compared continuous versus on‐demand NSAID use. We could not pool data.
Wanders 2005 was an open‐label RCT with 214 participants (N = 111 in the continuous treatment group (N = 76 had complete set of radiographs); N = 103 in the on‐demand group (N = 74 had complete set of radiographs)). All patients started with celecoxib 400 mg, but were allowed to switch to another NSAID at their discretion. The trial had a duration of two years. Results of this trial indicated that the patients who were continuously treated with NSAIDs showed significantly less radiographic progression than the patients who took NSAIDs on‐demand (mean (SD) 0.4 (1.7) mSASSS units radiographic progression after two years in the continuous treatment group versus 1.5 (2.5) mSASSS units in the on‐demand group; number of patients with at least two mSASSS units radiographic progression after two years was 12 out of 76 in the continuous treatment group versus 26 out of 74 in the on‐demand treatment group), although the two treatment strategies had a similar effect on signs and symptoms (pain on VAS, BASDAI, patient's global assessment of disease activity, fatigue, duration of morning stiffness, severity of morning stiffness, CRP, ESR, BASFI, chest expansion, occiput‐to‐wall distance, Schober's test) and the number of adverse events in both arms was similar as well (withdrawals due to adverse events, number of serious adverse events, number of adverse events per organ system) (for full study description and results see table of Characteristics of included studies).
The post‐hoc subgroup analysis included 150 participants from the original study, and compared outcomes based on elevated/high versus normal/low CRP, ESR, BASDAI, ASDAS‐CRP and ASDAS‐ESR. Kroon 2012 found that the effect of slowing of radiographic progression in the spine with continuous NSAID use was more pronounced in subgroups with elevated/high CRP (mean (SD) mSASSS progression after two years 0.2 (16) in continuous NSAID group vs 1.7 (2.8) in on‐demand NSAID group (compared to 0.9 (1.8) vs 0.8 (1.1) in participants with normal/low CRP)), ESR (0.9 (1.6) vs 2.0 (2.4) (compared to 0.1 (1.8) vs 0.7 (2.2))), ASDAS‐CRP (0.4 (1.2) vs 1.9 (2.7) (compared to 0.4 (2.0) vs 0.9 (2.1))) and ASDAS‐ESR (0.4 (1.3) vs 1.8 (2.5) (compared to 0.4 (1.9) vs 1.1 (2.5)), but not BASDAI (0.1 (1.1) vs 1.1 (1.6) (compared to 0.5 (1.8) vs 1.6 (2.8))) (for full study description and results see Table 4).
Boersma 1976 was a retrospective cohort study (40 participants) comparing radiographic progression of the spine in participants with continuous NSAID (phenylbutazone) intake versus non‐continuous NSAID (phenylbutazone) intake versus no NSAID intake. The trial authors concluded that in early or relatively early stages of AS, continuous NSAID medication can completely or largely control ossification of the spine. However, we could not extract any quantitative data due to the method of presentation (individual data in graphs) (for full study description and results see Table 3).
Subgroup analyses
We had planned to conduct several subgroup analyses (see Subgroup analysis and investigation of heterogeneity), but due to unavailability of data we could not perform any of the pre‐specified subgroup analyses.
The prespecified main comparison of this review was NSAIDs vs placebo. However as many trials included both traditional and COX‐2 NSAIDs, we elected to present data for the two NSAID classes separately. While we also considered direct comparison of traditional to COX‐2 NSAIDs, for completeness we also performed a post‐hoc subgroup analysis for one of our main outcomes (pain on VAS; Analysis 7.1). This analysis shows that both NSAID classes are equally efficacious for pain relief (test for subgroup differences: Chi² test = 0.46, df = 1; P = 0.50; I² statistic = 0%).
7.1. Analysis.
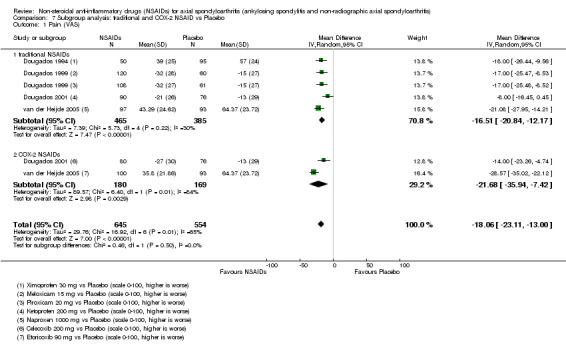
Comparison 7 Subgroup analysis: traditional and COX‐2 NSAID vs Placebo, Outcome 1 Pain (VAS).
We identified two studies that were post‐hoc subgroup analyses of included RCTs (Kroon 2012; Gossec 2005). Kroon 2012 addressed one of the subgroup analyses we had prespecified (e.g. effect of CRP on the effect of NSAIDs on radiographic damage). For full study description and results, see Comparison 7 and Table 4.
Gossec 2005 was a post‐hoc subgroup analysis of van der Heijde 2005 performed to determine whether peripheral arthritis is a treatment effect modifier of NSAID therapy. The authors found that receipt of either etoricoxib 90 mg or 120 mg or naproxen 1000 mg resulted in significantly greater improvement in spinal pain among participants without peripheral arthritis (change from baseline on a scale of 0 to 100 VAS (higher is worse) ‐42.5 (95% CI ‐45.8 to ‐39.2) compared with ‐34.5 (95% CI ‐38.6 to ‐30.4) in participants with peripheral arthritis (test for subgroup differences: strata interaction test, P = 0.005)). The authors also found non‐significant differences in other end points suggesting better outcomes in those without peripheral arthritis. For the full study description and results see Table 4.
Sensitivity analysis
We performed pre‐planned sensitivity analyses in all significant comparisons where sufficient studies existed to assess the impact of any selection bias, performance bias, detection bias or attrition bias compared to studies without these study limitations, in order to explore the robustness of our conclusions.
In all sensitivity analyses of efficacy measures, results were unchanged after excluding studies with high or unclear risk of bias (Analysis 1.1; Analysis 1.4; Analysis 1.5; Analysis 1.6; Analysis 1.7; Analysis 1.9; Analysis 1.10; Analysis 1.11; Analysis 1.12; Analysis 2.1; Analysis 2.4; Analysis 2.7; Analysis 2.9; Analysis 2.11; Analysis 5.1).
However, when excluding studies at high or unclear risk of bias in the comparisons of safety outcomes, none of the between‐group differences remained significant (Analysis 1.15; Analysis 2.15; Analysis 5.12). In comparison 1 (traditional NSAIDs versus placebo), after excluding the trials with an unclear risk of bias (Barkhuizen 2006; Dougados 1999; Dougados 2001), the two trials with a low risk of bias (Dougados 1994; van der Heijde 2005) showed that NSAIDs did not result in more or less gastro‐intestinal adverse events than placebo (RR 1.61, 95% CI 0.77 to 3.36). One trial at low risk of bias, van der Heijde 2005, also showed that NSAIDs did not result in more or less neurological adverse events than placebo (RR 0.63, 95% CI 0.11 to 3.66). Also, in comparison 2 (COX‐2 NSAIDs versus placebo), after excluding the studies at unclear risk of bias (Barkhuizen 2006; Dougados 2001), the only trial with a low risk of bias, van der Heijde 2005, showed that coxibs did not result in more or less gastro‐intestinal adverse events than placebo (RR 2.35, 95% CI 0.87 to 6.33). In comparison 4 (NSAIDs versus NSAIDs, only trials that compared indomethacin to another NSAID), after excluding the trials at unclear or high risk of bias (Caldwell 1986; Calin 1979; Ebner 1983; Good 1977; Palferman 1991), the six remaining trials with a low risk of bias (Batlle‐Gualda 1996; Khan 1985; Lomen 1986 I; Nissilä 1978a; Nissilä 1978b; Tannenbaum 1984) showed that indomethacin did not result in more or less adverse events (RR 0.84, 95% CI 0.69 to 1.03) or neurological adverse events (RR 0.48, 95% CI 0.23 to 1.01) than another NSAID. Finally, in comparison 6 (naproxen versus other NSAIDs), after excluding the studies with an unclear risk of bias (Barkhuizen 2006; Pasero 1994), the only trial with a low risk of bias, van der Heijde 2005, showed that naproxen did not result in more or less neurological adverse events than other NSAIDs (RR 0.26, 95% CI 0.06 to 1.19).
We also planned to perform a post‐hoc sensitivity analysis in all significant comparisons where sufficient studies existed to assess whether results could be influenced by the year of publication of the study. We planned to compare studies published before 1990 to studies published after 1990. However, none of the comparisons with significant results included studies older than 1990.
Assessment of reporting biases
We explored the potential for reporting bias by creating funnel plots for outcomes where at least 10 studies were available (i.e. Outcome 4.3: withdrawals due to adverse events; Outcome 4.15: number of any adverse events; Outcome 4.17.2: gastro‐intestinal adverse events; and Outcome 4.17.7: neurological adverse events), which were symmetrical without an indication for bias (we did not included the figures in this Cochrane review).
Discussion
Summary of main results
This systematic review assessed the benefits and harms of NSAIDs for patients with axSpA by employing rigorous and systematic methods of searching, appraising and synthesising the evidence. Overall we have presented outcome data of the two main comparisons (traditional NSAIDs versus placebo and COX‐2 NSAIDs versus placebo) in the Table 1 and Table 2.
Evidence of moderate to high quality based upon five trials with 1165 participants indicates that traditional NSAIDs are consistently more efficacious than placebo in the outcomes pain on VAS, BASDAI, patient's global assessment of disease activity, duration of morning stiffness, CRP, ASAS 20, BASFI, chest expansion, Schober's test and pain relief ≥ 50% (Table 1). Only ASAS partial remission was not significantly different between the two groups (one study, 190 participants, six weeks). Evidence of moderate to high quality indicates that traditional NSAIDs do not lead to significantly more withdrawals due to adverse events, or to a significant increase in the number of (serious or any) adverse events. However, in five trials (1289 participants) there were more gastro‐intestinal adverse events in patients taking NSAIDs compared to placebo, and in four trials (1144 participants) there were more neurological adverse events (including headache and dizziness) in the placebo‐group compared with the NSAID‐group. In the sensitivity analyses that we performed excluding studies at high or unclear risk of bias, there were no longer any between‐group differences with respect to harms.
Moderate to high quality evidence based upon three studies (669 participants) indicates that COX‐2 NSAIDs are consistently more efficacious than placebo in the outcomes pain on VAS, BASDAI, patient's global assessment of disease activity, ASAS 20, ASAS partial remission, BASFI, Schober's test and pain relief ≥ 50% (Table 2). No significant difference between coxibs and placebo was found in the outcomes duration of morning stiffness, CRP and chest expansion. Evidence of low to moderate quality shows similar results as in the comparison traditional NSAIDs versus placebo regarding harms. We found no difference in the number of withdrawals due to adverse events, or the number of (serious or any) adverse events. However, when looking at the number of adverse events per organ system, three studies (669 participants) reported more gastro‐intestinal adverse events in patients taking coxibs compared to placebo. Also here, in the sensitivity analyses that we performed excluding studies with a high or unclear risk of bias, there was no difference in safety outcomes between the interventions.
Evidence of moderate to high quality based upon four trials (995 participants) shows that there is no difference in any of the efficacy measures between coxibs and traditional NSAIDs (pain on VAS, BASDAI, patient's global assessment of disease activity, duration of morning stiffness, CRP, ASAS 20, ASAS partial remission, BASFI, BASMI, chest expansion, Schober's test and pain relief ≥ 50%). A post‐hoc subgroup analysis confirmed these findings. Similarly, for the safety outcomes (including withdrawals due to adverse events, any adverse events, serious adverse events and adverse events per organ system (cardiovascular, gastro‐intestinal, hepatic, respiratory, haematological, neurological or dermatological)) no difference between COX‐2 and traditional NSAIDs could be detected.
When comparing different NSAIDs to each other, no important difference in benefits could be determined (24 trials, 2076 participants). While indomethacin seemed to result in more adverse events (RR 1.25, 95% CI 1.06 to 1.48; 11 studies, N = 1135), in particular neurological adverse events like headache and dizziness (RR 2.34, 95% CI 1.32 to 4.14; 9 studies, N = 963), compared with other NSAIDs, this did not result in an increased rate of withdrawals and the differences were no longer significant when we excluded studies at high or unclear risk of bias.
We found no important differences in harms (withdrawals due to adverse events, number of any or serious adverse events or adverse events per organ system) between naproxen and other NSAIDs, although naproxen appeared less effective for relieving pain based upon two trials at low and unclear risk of bias.
We also compared low versus higher dose of the same NSAID, but in general we found no clear dose‐effect on benefits or harms in five studies (N = 1136), although one post‐hoc analysis of a prospective cohort study suggested that higher NSAID intake may retard radiographic progression.
We found a suggestion from one RCT (low risk of bias) and one retrospective cohort study that NSAIDs may be effective in retarding radiographic progression of the spine in axSpA, especially in certain subgroups of patients, e.g. patients with high CRP, and this may be best achieved by continuous rather than on‐demand use of NSAIDs.
We performed sensitivity analyses to explore the robustness of our conclusions, by assessing the impact of studies with high or unclear risk of bias in all significant comparisons. Results of all efficacy measures remained significant when excluding studies with high or unclear risk of bias. However, when excluding studies with a high or unclear risk of bias in the comparisons of harms, none of the results remained significant. This possibly means that there is no increased risk of (gastrointestinal) adverse events in patients taking NSAIDs for a short period of time, or that studies were underpowered to assess this outcome.
Overall completeness and applicability of evidence
Evidence of moderate to high quality for the main comparisons, as presented in the 'Summary of findings' tables, indicates that traditional and COX‐2 NSAIDs are consistently more efficacious than placebo, without a difference between the two classes of NSAIDs. Low to moderate quality evidence showed that there were not significantly more (serious or any) adverse events or withdrawals due to adverse events in patients taking traditional or COX‐2 NSAIDs in comparison to patients taking placebo. However, in both NSAID classes significantly more patients taking NSAIDs complained of gastrointestinal adverse events, although this result did not remain significant when excluding studies at high or unclear risk of bias.
When comparing the two NSAID classes to each other, we found no difference in harms. This last result is surprising, as it is commonly thought that the traditional non‐selective COX inhibitors (inhibiting both COX‐1 and COX‐2) result in more, mainly gastrointestinal, adverse events, than the selective COX inhibitors (selectively inhibiting COX‐2) (Chan 2010; Emery 1999; Silverstein 2000; Simon 1999). The fact that we found no difference in harms between traditional NSAIDs, coxibs and placebo could be due to the fact that most AS patients are relatively younger and 'healthier' (i.e. have fewer comorbidities) than patients with other rheumatic diseases (like rheumatoid arthritis and osteoarthritis). This is supported by the finding that biologicals also result in less adverse events in patients with AS, than in patients with other rheumatic diseases (Burmester 2013). Moreover, it is also possible that the studies in this review were underpowered to assess harms, in other words, were too small or the duration of the studies too short to find the differences in adverse events between the two NSAID classes. Overall, it is reassuring to find that both classes of NSAIDs can be prescribed relatively safely, at least in the short‐term.
We considered the benefits and harms of naproxen in comparison to other NSAIDs, because a recent meta‐analysis of vascular and upper gastro‐intestinal effects of NSAIDs in various patients (prescribed mostly for rheumatoid arthritis or osteoarthritis, but also for prevention of colorectal adenomata or of Alzheimer's disease) showed that naproxen was associated with less vascular (but increased upper gastro‐intestinal) risk than other NSAIDs (Bhala 2013). In this review, no important differences in harms (withdrawals due to adverse events, number of any or serious adverse events or adverse events per organ system) could be determined. However, we could not include many studies (n = 3) in this comparison, and therefore we could not confirm nor reject the results of Bhala 2013 about the harms of naproxen.
In general, we found no clear dose‐effect on benefits or harms in five studies (three out of five assessed to be at low risk of bias), although they only compared a few different doses of a few NSAIDs (celecoxib, etoricoxib, meloxicam and ximoprofen). These data suggest that it might be preferable to choose a lower NSAID dosage to minimize the risk of adverse events. However ASAS members, who are experts in the field, have agreed to use relatively high dosage of NSAIDs to treat patients with axSpA (150 mg diclofenac, or an equivalent dose of another NSAID) based upon their experience in clinical practice (Dougados 2011). Further robust data are needed to resolve this issue.
However we also found that NSAIDs may be effective in retarding radiographic progression of the spine in axSpA, especially in patients with high CRP and this may be best achieved by continuous rather than on‐demand use of NSAIDs. These findings are in keeping with a recent study that found that high disease activity leads to more structural damage in the spine (Ramiro 2014). It has also been shown that radiographic damage is associated with impaired spinal mobility and function (Machado 2010; Machado 2011a). These findings stress the importance of retarding the progression of structural damage in the spine, and taking NSAIDs for a longer period of time may be an effective way to do so.
We included several types of study designs to assess specific questions like long term harms and radiographic progression. However, quantitative data‐analysis is limited to RCTs and quasi‐RCTs. The consequence of this limitation is that we were unable to quantitatively assess the data from these long term studies, especially the outcomes that we were specifically interested in. Also, the duration of NSAID therapy in the RCTs and quasi‐RCTs included in the meta‐analysis was limited to up to 26 weeks, with a median treatment duration of 12 weeks. This limits the (quantitative) results of this Cochrane review to an assessment of short term benefits and harms of NSAIDs, rather than the long term assessment that might even be more important for clinical practice, especially in terms of harms. For a full safety profile in axSpA one still depends on studies in other rheumatic diseases or non‐comparative cohort studies.
One of the objectives of this review was to consider all outcomes relevant for clinical practice as recommended by ASAS (Sieper 2009). However, many studies did not report all outcomes that we planned to assess. None of the included studies reported the outcome radiographic progression, which is also due to the fact that the duration of the studies was not long enough to assess this outcome. Of the other main efficacy outcomes, most studies reported on pain (although four studies did not report pain as an outcome), Three studies reported BASDAI, four studies reported BASFI and one study reported BASMI. At least one of our main safety outcomes was reported by most studies, although three trials did not report the number of withdrawals due to adverse events, and the number of serious adverse events was only reported in six studies. Many of the other secondary outcomes in this review were not reported by any of the included studies (for a list see Reported outcomes in Description of studies). This lack of outcome reporting can partly be explained by the fact that the core set as recommended by ASAS has only been published in 2009, and most studies included in this review have been published many years earlier. Nevertheless, we expect that NSAIDs will also be efficacious when measured with these other, newer outcomes (like the ASAS response criteria, ASDAS, etcetera), as NSAIDs have proven to be efficacious in almost all other outcomes that were reported in this review.
As reported in the Description of studies, most studies were older studies, as 25 of 41 included studies (61%) were published before 1990. Many of the older studies were of a lower quality than the more recently published studies, and had a higher risk of bias. Also, many of the older studies reported outcomes in a manner that made it impossible to utilise these results in our meta‐analysis (e.g. only in graphs, without a measure of variance, without presenting the number of participants per treatment group). For example, the outcome 'duration of morning stiffness' was reported in 22 out of 31 studies included in the meta‐analysis, but the data from 12 studies could not be used in the analyses. Of these 12 studies that could not be used, 10 were published before 1990. To provide an insight for our readers in which data was available for which comparison, and which studies reported data that could not be included in the analyses, we designed Table 5, Table 6, Table 7, Table 8, Table 10, and Table 9. Another consequence of the fact that most studies were not published very recently, is we could not include data on patients diagnosed with nr‐axSpA in our review as we had planned. Although we expect that these data about the benefits and harms of NSAIDs in patients with radiographic axSpA (AS) will also apply to patients with nr‐axSpA, we cannot confirm this assumption in the current review and RCTs in this patient population are needed.
Quality of the evidence
For the first main comparison, traditional NSAIDs versus placebo (five trials, N = 1165), the overall quality of evidence was graded as moderate to high (Table 1). We downgraded the evidence because of potential imprecision of a few outcomes. For the second main comparison, COX‐2 NSAIDs versus placebo (three trials, N = 669), the overall quality of evidence was graded as moderate to high (Table 2), although we graded the evidence of one outcome (number of withdrawals due to adverse events) as low. We downgraded the evidence because of potential imprecision of a few outcomes, and inconsistency of the results of the outcome number of withdrawals due to adverse events.
We did not present the quality of the evidence of our other five comparisons (COX‐2 vs traditional NSAIDs, NSAIDs vs NSAIDs, naproxen vs other NSAIDs, low vs high dose NSAIDs and continuous vs on‐demand NSAID use) in 'Summary of findings' tables. The quality of the evidence of the studies in the comparison COX‐2 vs traditional NSAIDs was graded moderate to high. We downgraded the evidence because of potential imprecision of a few outcomes. Overall, the quality of the evidence of the studies in the other four comparisons varied from low to high. Evidence of the studies of these comparisons was downgraded mainly because of potential imprecision as well as inconsistency of the results of a few outcomes.
Potential biases in the review process
We believe that we identified all relevant published trials for inclusion in this review by devising a thorough search strategy, searching all major databases for relevant studies with no language restrictions applied. As described in the methods, two review authors assessed the studies for inclusion in the review, and a third review author served as an adjudicator to solve any discussions or discrepancies. A potential bias in the review process could have been that several included studies could not be included in the meta‐analysis (eight studies), e.g. due to the study design (as these were no controlled trials) or because the authors did not provide the number of participants per treatment group.
However, we tried to overcome this by publishing all results that we could not include in our quantitative data‐analysis in the table of Characteristics of included studies and in the review text under the appropriate comparison.
Another potential bias in the review process is that we could not locate 32 full texts eligible for inclusion in the review after screening their title and abstract, in spite of thorough searching in many libraries. We also identified nine trials in ClinicalTrials.gov that did not provide any results, were not published and were thus added to Studies awaiting classification, although it is unlikely that these studies are ever going to be published. We believe, however, that even if a few of these studies would have been eligible for final inclusion in the review, these studies would not have substantially changed our conclusions. We further explored the potential for reporting bias by creating funnel plots for outcomes where at least 10 studies were available, which were symmetrical without an indication for bias (figures not included in this review).
Two review authors independently assessed the trials for inclusion in the review, extracted data and assessed the risk of bias, and a third review author adjudicated whenever there was any discrepancy. Three review authors (FK, RL and DvdH) are authors of several trials included in this review (Dougados 2001; Gossec 2005; Kroon 2012; van der Heijde 2005; Wanders 2005). To avoid any bias, an independent review author assessed these papers for inclusion in this review. None of the review authors were involved in data extraction or 'Risk of bias' assessment of their own trials.
Agreements and disagreements with other studies or reviews
Previous systematic reviews that have investigated the effects of NSAIDs for SpA have reported broadly similar findings as our review, although all these reviews only included trials comparing NSAIDs to placebo (Escalas 2010; Van den Berg 2012; Zochling 2006). Safety concerns associated with both traditional NSAIDs and COX‐2 NSAIDs reported in previous systematic reviews (Van den Berg 2012; Zochling 2006), in short that NSAIDs cause an increased risk of gastrointestinal toxicity which is lower with coxibs but still considerable also in this class of NSAIDs, could not be confirmed in this Cochrane review.
Initially we found that significantly more patients taking traditional NSAIDs, as well as those taking COX‐2 NSAIDs, complained of gastrointestinal adverse events. However, after excluding studies with high or unclear risk of bias these results did not remain significant. This possibly means that there is no increased risk of (gastrointestinal) adverse events in patients taking NSAIDs for a short period of time. It is possible that the included studies were underpowered to assess harms, e.g. because the studies were too small or the duration of the studies too short. Previous systematic reviews, in axSpA and in other diseases, also indicate an increased risk of cardiovascular toxicity, most importantly in COX‐2 NSAIDs, which we could not confirm in this review (Kearney 2006; Trelle 2011; Zochling 2006). However, as highlighted earlier, the trials included in this review were all of short duration, and may have not picked up on the cardiovascular adverse events related with these drugs. Furthermore, also in this case, it is possible that the included trials were underpowered to assess (cardiovascular) adverse events.
Authors' conclusions
Implications for practice.
We found high quality evidence that in patients with axSpA, both traditional and COX‐2 NSAIDs are more efficacious than placebo. Moderate quality evidence indicates that harms are, in the short term, not different to placebo. All studied NSAIDs (including traditional versus COX‐2 NSAIDs) were found to be equally effective. The results of this review support current recommendations for treatment of patients with axSpA with NSAIDs as a first‐line drug (Braun 2011). We found no comparative long‐term studies evaluating harms therefore consideration could be given to findings regarding long‐term harms of NSAIDs in other rheumatic diseases. Based upon moderate quality evidence, continuous NSAID use may reduce radiographic spinal progression, but further rigorous long‐term studies that also consider harms are needed before definitive conclusions can be drawn. Until these data are available, the potential benefits of continuous NSAID use should be considered in comparison to the potential risks in individual patients.
Implications for research.
The short term benefits and harms of traditional and COX‐2 NSAIDs for treatment of SpA have been studied extensively. We believe that further studies investigating the short‐term effects of NSAIDs are not likely to substantially change the conclusions of this Cochrane review. However, we found no comparative studies assessing long‐term harms. Rigorous studies with adequate follow‐up are needed to establish the long‐term benefits and harms of NSAIDs in patients with SpA including establishing whether or not continuous use of NSAIDs is superior to on‐demand use for slowing radiographic progression. No trials to date have studied the benefits and harms of NSAIDs for patients with nr‐axSpA. Although we suspect that these may be similar to effects observed in people with radiographic SpA (AS), this requires verification.
Acknowledgements
None.
Appendices
Appendix 1. Search strategies
MEDLINE
1. exp Spondylitis/
2. (ankylos$ or spondyl$).tw.
3. SpA.tw.
4. (bekhterev$ or bechterew$).tw.
5. or/1‐4
6. exp Cyclooxygenase 2 Inhibitors/
7. cox 2 inhibitor$.tw.
8. cyclooxygenase 2 inhibitor$.tw.
9. cyclo‐oxygenase‐2 inhibitor$.tw.
10. (meloxicam or movalis or mobec or mobic or movicox or mobicox or parocin or uticox or etoricoxib or arcoxia or celecoxib or celebrex).tw.
11. coxib$.tw.
12. exp Anti‐Inflammatory Agents, Non‐Steroidal/
13. (Anti‐Inflammator$ or (Anti adj Inflammator$) or AntiInflammator$).tw.
14. nsaid$.tw.
15. Aceclofenac.tw.
16. Acetylsalicylic acid.tw.
17. acephen.tw.
18. Ampyrone.tw.
19. Amynopirin.tw.
20. Antipyrine.tw.
21. Apazone.tw.
22. Aspirin.tw.
23. Bufexamac.tw.
24. Clofazimine.tw.
25. Clonixin.tw.
26. Curcumin.tw.
27. Dexketoprofen.tw.
28. Dexibruprofen.tw.
29. Diclofenac.tw.
30. Diflunisal.tw.
31. Dipyrone.tw.
32. Epirizole.tw.
33. Etodolac.tw.
34. Fenbufen.tw.
35. Fenclofenac.tw.
36. Fenoprofen.tw.
37. Floctafenine.tw.
38. Flurbiprofen.tw.
39. Ibuprofen.tw.
40. Indomethacin.tw.
41. Ketoprofen.tw.
42. Ketorolac.tw.
43. Lederfen.tw.
44. Meclofenamic Acid.tw.
45. Mefenamic Acid.tw.
46. Mesalamine.tw.
47. Nabumetone.tw.
48. Naproxen.tw.
49. Niflumic Acid.tw.
50. Oxaprozin.tw.
51. Oxyphenbutazone.tw.
52. Phenazone.tw.
53. Phenylbutazone.tw.
54. Piroxicam.tw.
55. pirazolac.tw.
56. pirprofen.tw.
57. Ponstan.tw.
58. Prenazone.tw.
59. Salicylate$.tw.
60. Salsalate.tw.
61. Seractil.tw.
62. Sulfasalazine.tw.
63. Sulindac.tw.
64. Suprofen.tw.
65. Tenoxicam.tw.
66. Tiaprofenic acid.tw.
67. tolfenamic acid.tw.
68. Tolmetin.tw.
69. ximoprofen.tw.
70. or/6‐69
71. 5 and 70
72. (animals not (humans and animals)).sh.
73. 71 not 72
EMBASE
1. spondylitis/
2. (ankylos$ or spondyl$).tw.
3. SpA.tw.
4. (bekhterev$ or bechterew$).tw.
5. or/1‐4
6. exp cyclooxygenase 2 inhibitor/
7. cox 2 inhibitor$.tw.
8. cox2 inhibitor$.tw.
9. cyclooxygenase 2 inhibitor$.tw.
10. cyclo‐oxygenase‐2 inhibitor$.tw.
11. (meloxicam or movalis or mobec or mobic or movicox or mobicox or parocin or uticox or etoricoxib or arcoxia or celecoxib or celebrex).tw.
12. coxib$.tw.
13. exp nonsteroid antiinflammatory agent/
14. (Anti‐Inflammator$ or (Anti adj Inflammator$) or AntiInflammator$).tw.
15. nsaid$.tw.
16. Aceclofenac.tw.
17. Acetylsalicylic acid.tw.
18. acephen.tw.
19. Ampyrone.tw.
20. Amynopirin.tw.
21. Antipyrine.tw.
22. Apazone.tw.
23. Aspirin.tw.
24. Bufexamac.tw.
25. Clofazimine.tw.
26. Clonixin.tw.
27. Curcumin.tw.
28. Dexketoprofen.tw.
29. Dexibruprofen.tw.
30. Diclofenac.tw.
31. Diflunisal.tw.
32. Dipyrone.tw.
33. Epirizole.tw.
34. Etodolac.tw.
35. Fenbufen.tw.
36. Fenclofenac.tw.
37. Fenoprofen.tw.
38. Floctafenine.tw.
39. Flurbiprofen.tw.
40. Ibuprofen.tw.
41. Indomethacin.tw.
42. Ketoprofen.tw.
43. Ketorolac.tw.
44. Lederfen.tw.
45. Meclofenamic Acid.tw.
46. Mefenamic Acid.tw.
47. Mesalamine.tw.
48. Nabumetone.tw.
49. Naproxen.tw.
50. Niflumic Acid.tw.
51. Oxaprozin.tw.
52. Oxyphenbutazone.tw.
53. Phenazone.tw.
54. Phenylbutazone.tw.
55. Piroxicam.tw.
56. pirazolac.tw.
57. pirprofen.tw.
58. Ponstan.tw.
59. Prenazone.tw.
60. Salicylate$.tw.
61. Salsalate.tw.
62. Seractil.tw.
63. Sulfasalazine.tw.
64. Sulindac.tw.
65. Suprofen.tw.
66. Tenoxicam.tw.
67. Tiaprofenic acid.tw.
68. tolfenamic acid.tw.
69. Tolmetin.tw.
70. ximoprofen.tw.
71. or/6‐70
72. 5 and 71
73. (animal$ not human$).sh,hw.
74. 72 not 73
CENTRAL
#1 MeSH descriptor: [Spondylitis] explode all trees
#2 (ankylos* or spondyl*):ti,ab
#3 SpA:ti,ab
#4 (bekhterev* or bechterew*):ti,ab
#5 #1 or #2 or #3 or #4
#6 MeSH descriptor: [Cyclooxygenase 2 Inhibitors] explode all trees
#7 "cox 2 inhibitor*":ti,ab
#8 "cyclooxygenase 2 inhibitor*":ti,ab
#9 "cyclo‐oxygenase‐2 inhibitor*":ti,ab
#10 (meloxicam or movalis or mobec or mobic or movicox or mobicox or parocin or uticox or etoricoxib or arcoxia or celecoxib or celebrex):ti,ab
#11 coxib*:ti,ab
#12 MeSH descriptor: [Anti‐Inflammatory Agents, Non‐Steroidal] explode all trees
#13 Anti‐Inflammator*:ti,ab or Anti nextj Inflammator*:ti,ab or AntiInflammator*:ti,ab
#14 nsaid*:ti,ab
#15 Aceclofenac.:ti,ab
#16 "Acetylsalicylic acid":ti,ab
#17 acephen:ti,ab
#18 Ampyrone:ti,ab
#19 Amynopirin:ti,ab
#20 Antipyrine:ti,ab
#21 Apazone:ti,ab
#22 Aspirin:ti,ab
#23 Bufexamac:ti,ab
#24 Clofazimine:ti,ab
#25 Clonixin:ti,ab
#26 Curcumin:ti,ab
#27 Dexketoprofen:ti,ab
#28 Dexibruprofen:ti,ab
#29 Diclofenac:ti,ab
#30 Diflunisal:ti,ab
#31 Dipyrone:ti,ab
#32 Epirizole:ti,ab
#33 Etodolac:ti,ab
#34 Fenbufen:ti,ab
#35 Fenclofenac:ti,ab
#36 Fenoprofen:ti,ab
#37 Floctafenine:ti,ab
#38 Flurbiprofen:ti,ab
#39 Ibuprofen:ti,ab
#40 Indomethacin:ti,ab
#41 Ketoprofen:ti,ab
#42 Ketorolac:ti,ab
#43 Lederfen:ti,ab
#44 "Meclofenamic Acid":ti,ab
#45 "Mefenamic Acid":ti,ab
#46 Mesalamine:ti,ab
#47 Nabumetone:ti,ab
#48 Naproxen:ti,ab
#49 "Niflumic Acid":ti,ab
#50 Oxaprozin:ti,ab
#51 Oxyphenbutazone:ti,ab
#52 Phenazone:ti,ab
#53 Phenylbutazone:ti,ab
#54 Piroxicam:ti,ab
#55 pirazolac:ti,ab
#56 pirprofen:ti,ab
#57 Ponstan:ti,ab
#58 Prenazone:ti,ab
#59 Salicylate*:ti,ab
#60 Salsalate:ti,ab
#61 Seractil:ti,ab
#62 Sulfasalazine:ti,ab
#63 Sulindac:ti,ab
#64 Suprofen:ti,ab
#65 Tenoxicam:ti,ab
#66 "Tiaprofenic acid":ti,ab
#67 "tolfenamic acid":ti,ab
#68 Tolmetin:ti,ab
#69 ximoprofen:ti,ab
#70 #6 or #7 or #8 or #9 or #10 or #11 or #12 or #13 or #14 or #15 or #16 or #17 or #18 or #19 or #20 or #21 or #22 or #23 or #24 or #25 or #26 or #27 or #28 or #29 or #30 or #31 or #32 or #33 or #34 or #35 or #36 or #37 or #38 or #39 or #40 or #41 or #42 or #43 or #44 or #45 or #46 or #47 or #48 or #49 or #50 or #51 or #52 or #53 or #54 or #55 or #56 or #57 or #58 or #59 or #60 or #61 or #62 or #63 or #64 or #65 or #66 or #67 or #68 or #69
#71 #5 and #70
Scopus
#1 TITLE‐ABS‐KEY(ankylos* OR spondyl*)
#2 TITLE‐ABS‐KEY(spa)
#3 TITLE‐ABS‐KEY(bekhterev* OR bechterew*)
#4 #1 OR #2 OR #3
#5 TITLE‐ABS‐KEY("cox 2 inhibitor*" OR "cyclooxygenase 2 inhibitor*" OR "cyclo‐oxygenase‐2 inhibitor*")
#6 TITLE‐ABS‐KEY(meloxicam OR movalis OR mobec OR mobic OR movicox OR mobicox OR parocin OR uticox OR etoricoxib OR arcoxia OR celecoxib OR celebrex)
#7 TITLE‐ABS‐KEY(coxib*)
#8 TITLE‐ABS‐KEY(anti‐inflammator* OR "Anti Inflammator*" OR antiinflammator*)
#9 TITLE‐ABS‐KEY(nsaid*)
#10 TITLE‐ABS‐KEY(Aceclofenac or "Acetylsalicylic acid" or acephen or Ampyrone or Amynopirin or Antipyrine or Apazone or Aspirin or Bufexamac or Clofazimine or Clonixin or Curcumin or Dexketoprofen or Dexibruprofen or Diclofenac or Diflunisal or Dipyrone or Epirizole or Etodolac or Fenbufen or Fenclofenac or Fenoprofen or Floctafenine or Flurbiprofen or Ibuprofen or Indomethacin or Ketoprofen or Ketorolac or Lederfen or "Meclofenamic Acid" or "Mefenamic Acid" or Mesalamine or Nabumetone or Naproxen or "Niflumic Acid" or Oxaprozin or Oxyphenbutazone or Phenazone or Phenylbutazone or Piroxicam or pirazolac or pirprofen or Ponstan or Prenazone or Salicylate* or Salsalate or Seractil or Sulfasalazine or Sulindac or Suprofen or Tenoxicam or "Tiaprofenic acid" or "tolfenamic acid" or Tolmetin or ximoprofen)
#5 OR #6 OR #7 OR #8 OR #9 OR #10
#11 #4 AND #10 AND LIMIT‐TO(DOCTYPE, "cp"
ClinicalTrials.gov
"Spondylitis" in Condition
WHO ICTRP search portal
ankylos* OR spondyl* OR SpA OR bekhterev* OR bechterew* in Condition
Data and analyses
Comparison 1. Traditional NSAID vs Placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Pain on VAS | 4 | 850 | Mean Difference (IV, Random, 95% CI) | ‐16.51 [‐20.84, ‐12.17] |
| 2 Withdrawals due to adverse events | 5 | 1165 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.46, 1.21] |
| 3 BASDAI | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 4 Patient's global assessment of disease activity | 3 | 705 | Mean Difference (IV, Random, 95% CI) | ‐17.75 [‐24.39, ‐11.10] |
| 5 Duration of morning stiffness | 4 | 850 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.40 [‐0.58, ‐0.22] |
| 6 CRP | 2 | 515 | Mean Difference (IV, Random, 95% CI) | ‐3.37 [‐6.11, ‐0.62] |
| 7 ASAS 20 | 2 | 503 | Risk Ratio (M‐H, Random, 95% CI) | 2.49 [1.94, 3.18] |
| 8 ASAS partial remission | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9 BASFI | 2 | 356 | Mean Difference (IV, Random, 95% CI) | ‐9.07 [‐13.04, ‐5.10] |
| 10 Chest expansion | 2 | 515 | Mean Difference (IV, Random, 95% CI) | 0.44 [0.20, 0.68] |
| 11 Schober's test | 4 | 850 | Mean Difference (IV, Random, 95% CI) | 0.37 [0.18, 0.57] |
| 12 Pain relief ≥ 50% | 3 | 660 | Risk Ratio (M‐H, Random, 95% CI) | 2.27 [1.77, 2.91] |
| 13 Number of any adverse events | 5 | 1289 | Risk Ratio (M‐H, Random, 95% CI) | 1.08 [0.92, 1.26] |
| 14 Number of serious adverse events | 3 | 671 | Risk Ratio (M‐H, Random, 95% CI) | 1.69 [0.36, 7.97] |
| 15 Number of adverse events per organ system | 5 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 15.1 Gastro‐intestinal | 5 | 1289 | Risk Ratio (M‐H, Random, 95% CI) | 1.92 [1.41, 2.61] |
| 15.2 Respiratory | 4 | 1145 | Risk Ratio (M‐H, Random, 95% CI) | 0.91 [0.54, 1.51] |
| 15.3 Hematological | 1 | 166 | Risk Ratio (M‐H, Random, 95% CI) | 2.54 [0.10, 61.42] |
| 15.4 Neurological | 4 | 1144 | Risk Ratio (M‐H, Random, 95% CI) | 0.44 [0.24, 0.82] |
| 15.5 Dermatological | 3 | 952 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.27, 2.67] |
Comparison 2. COX‐2 vs Placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Pain on VAS | 2 | 349 | Mean Difference (IV, Random, 95% CI) | ‐21.68 [‐35.94, ‐7.42] |
| 2 Withdrawals due to adverse events | 3 | 669 | Risk Ratio (M‐H, Random, 95% CI) | 2.14 [0.36, 12.56] |
| 3 BASDAI | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 4 Patient's global assessment of disease activity | 2 | 349 | Mean Difference (IV, Random, 95% CI) | ‐20.82 [‐29.88, ‐11.75] |
| 5 Duration of morning stiffness | 2 | 349 | Std. Mean Difference (IV, Random, 95% CI) | ‐4.72 [‐13.33, 3.90] |
| 6 CRP | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 7 ASAS 20 | 2 | 510 | Risk Ratio (M‐H, Random, 95% CI) | 2.51 [1.66, 3.79] |
| 8 ASAS partial remission | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9 BASFI | 2 | 349 | Mean Difference (IV, Random, 95% CI) | ‐13.42 [‐17.35, ‐9.49] |
| 10 Chest expansion | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 11 Schober's test | 2 | 349 | Mean Difference (IV, Random, 95% CI) | 0.42 [0.21, 0.63] |
| 12 Pain relief ≥ 50% | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13 Number of any adverse events | 3 | 669 | Risk Ratio (M‐H, Random, 95% CI) | 1.22 [0.93, 1.62] |
| 14 Number of serious adverse events | 3 | 669 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.14, 6.21] |
| 15 Number of adverse events per organ system | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 15.1 Gastro‐intestinal | 3 | 669 | Risk Ratio (M‐H, Random, 95% CI) | 1.80 [1.22, 2.67] |
| 15.2 Respiratory | 3 | 669 | Risk Ratio (M‐H, Random, 95% CI) | 0.72 [0.41, 1.26] |
| 15.3 Neurological | 3 | 669 | Risk Ratio (M‐H, Random, 95% CI) | 1.25 [0.71, 2.21] |
| 15.4 Dermatological | 2 | 473 | Risk Ratio (M‐H, Random, 95% CI) | 2.86 [0.93, 8.78] |
Comparison 3. COX‐2 vs traditional NSAID.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Pain on VAS | 3 | 669 | Mean Difference (IV, Random, 95% CI) | ‐2.62 [‐10.99, 5.75] |
| 2 Withdrawals due to adverse events | 4 | 995 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.60, 1.82] |
| 3 BASDAI | 2 | 499 | Mean Difference (IV, Random, 95% CI) | ‐0.75 [‐7.95, 6.45] |
| 4 Patient's global assessment of disease activity | 3 | 669 | Mean Difference (IV, Random, 95% CI) | ‐3.09 [‐12.24, 6.07] |
| 5 Duration of morning stiffness | 2 | 367 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.95 [‐2.81, 0.90] |
| 6 CRP | 2 | 472 | Mean Difference (IV, Random, 95% CI) | ‐0.92 [‐2.90, 1.07] |
| 7 ASAS 20 | 3 | 663 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.80, 1.25] |
| 8 ASAS partial remission | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9 BASFI | 3 | 669 | Mean Difference (IV, Random, 95% CI) | ‐2.12 [‐6.53, 2.29] |
| 10 BASMI | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 11 Chest expansion | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 12 Schober's test | 2 | 367 | Mean Difference (IV, Random, 95% CI) | ‐0.14 [‐0.34, 0.07] |
| 13 Pain relief ≥ 50% | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 14 Number of any adverse events | 4 | 995 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.83, 1.21] |
| 15 Number of serious adverse events | 4 | 995 | Risk Ratio (M‐H, Random, 95% CI) | 0.72 [0.20, 2.63] |
| 16 Number of adverse events per organ system | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 16.1 Cardiovascular | 1 | 305 | Risk Ratio (M‐H, Random, 95% CI) | 0.15 [0.01, 2.83] |
| 16.2 Gastro‐intestinal | 4 | 995 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.57, 1.42] |
| 16.3 Hepatic | 1 | 305 | Risk Ratio (M‐H, Random, 95% CI) | 0.08 [0.00, 1.40] |
| 16.4 Respiratory | 4 | 995 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.55, 1.46] |
| 16.5 Hematological | 1 | 170 | Risk Ratio (M‐H, Random, 95% CI) | 0.37 [0.02, 9.06] |
| 16.6 Neurological | 4 | 995 | Risk Ratio (M‐H, Random, 95% CI) | 1.95 [0.60, 6.35] |
| 16.7 Dermatological | 2 | 488 | Risk Ratio (M‐H, Random, 95% CI) | 3.80 [0.31, 46.29] |
Comparison 4. NSAID vs NSAID.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Pain on Likert scale | 6 | Std. Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 2 Pain on VAS | 7 | Std. Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 3 Withdrawals due to adverse events | 23 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4 Patient's global assessment of disease activity | 2 | Std. Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 5 Duration of morning stiffness | 7 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 6 Severity of morning stiffness | 2 | Std. Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 7 CRP | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 8 ESR | 3 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 9 Lateral spinal flexion | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 10 Chest expansion | 6 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 11 Tragus‐to‐wall distance | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 12 Occiput‐to‐wall distance | 2 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 13 Schober's test | 8 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 14 Pain relief ≥ 50% | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 15 Number of any adverse events | 19 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 16 Number of serious adverse events | 2 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 17 Adverse events per organ system | 16 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 17.1 Cardiovascular | 6 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.2 Gastro‐intestinal | 16 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.3 Hepatic | 3 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.4 Respiratory | 3 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.5 Hematological | 2 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.6 Renal | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.7 Neurological | 14 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.8 Dermatological | 9 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 5. Naproxen vs other NSAID.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Pain on VAS | 2 | 232 | Mean Difference (IV, Random, 95% CI) | 6.80 [3.72, 9.88] |
| 2 Withdrawals due to adverse events | 3 | 646 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.42, 1.80] |
| 3 BASDAI | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 4 Patient's global assessment of disease activity | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 5 Duration of morning stiffness | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 6 ASAS 20 | 2 | 515 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.68, 1.41] |
| 7 ASAS partial remission | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8 BASFI | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 9 Schober's test | 2 | 301 | Mean Difference (IV, Random, 95% CI) | 0.16 [‐0.09, 0.41] |
| 10 Number of any adverse events | 3 | 646 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.80, 1.12] |
| 11 Number of serious adverse events | 2 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 12 Number of adverse events per organ system | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 12.1 Gastro‐intestinal | 3 | 646 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.51, 1.86] |
| 12.2 Hepatic | 1 | 126 | Risk Ratio (M‐H, Random, 95% CI) | 1.36 [0.24, 7.88] |
| 12.3 Respiratory | 2 | 520 | Risk Ratio (M‐H, Random, 95% CI) | 1.22 [0.63, 2.36] |
| 12.4 Neurological | 3 | 646 | Risk Ratio (M‐H, Random, 95% CI) | 0.24 [0.10, 0.60] |
| 12.5 Dermatological | 1 | 318 | Risk Ratio (M‐H, Random, 95% CI) | 0.06 [0.00, 1.04] |
Comparison 6. Low dose vs high dose NSAID.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Pain on VAS | 4 | 830 | Mean Difference (IV, Random, 95% CI) | 0.19 [‐3.23, 3.62] |
| 2 Withdrawals due to adverse events | 5 | 1136 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.46, 1.60] |
| 3 BASDAI | 2 | 490 | Mean Difference (IV, Random, 95% CI) | 1.34 [‐2.39, 5.07] |
| 4 Patient's global assessment of disease activity | 3 | 734 | Mean Difference (IV, Random, 95% CI) | ‐1.61 [‐5.35, 2.13] |
| 5 Duration of morning stiffness | 3 | 530 | Std. Mean Difference (IV, Random, 95% CI) | 0.18 [‐0.03, 0.38] |
| 6 CRP | 2 | 544 | Mean Difference (IV, Random, 95% CI) | 0.39 [‐1.73, 2.51] |
| 7 ASAS 20 | 3 | 620 | Risk Ratio (M‐H, Random, 95% CI) | 0.89 [0.76, 1.05] |
| 8 ASAS partial remission | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9 BASFI | 2 | 490 | Mean Difference (IV, Random, 95% CI) | 0.81 [‐3.25, 4.87] |
| 10 BASMI | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 11 Chest expansion | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 12 Schober's test | 3 | 530 | Mean Difference (IV, Random, 95% CI) | ‐0.01 [‐0.21, 0.19] |
| 13 Pain relief ≥ 50% | 2 | 340 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.85, 1.28] |
| 14 Number of any adverse events | 4 | 928 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.76, 1.32] |
| 15 Number of serious adverse events | 3 | 796 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.27, 4.04] |
| 16 Number of adverse events per organ system | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 16.1 Cardiovascular | 1 | 303 | Risk Ratio (M‐H, Random, 95% CI) | 6.86 [0.36, 131.75] |
| 16.2 Gastro‐intestinal | 4 | 892 | Risk Ratio (M‐H, Random, 95% CI) | 0.85 [0.49, 1.48] |
| 16.3 Respiratory | 3 | 796 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.42, 2.04] |
| 16.4 Neurological | 3 | 796 | Risk Ratio (M‐H, Random, 95% CI) | 1.17 [0.70, 1.96] |
| 16.5 Dermatological | 1 | 298 | Risk Ratio (M‐H, Random, 95% CI) | 0.44 [0.12, 1.63] |
Comparison 7. Subgroup analysis: traditional and COX‐2 NSAID vs Placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Pain (VAS) | 4 | 1199 | Mean Difference (IV, Random, 95% CI) | ‐18.06 [‐23.11, ‐13.00] |
| 1.1 traditional NSAIDs | 4 | 850 | Mean Difference (IV, Random, 95% CI) | ‐16.51 [‐20.84, ‐12.17] |
| 1.2 COX‐2 NSAIDs | 2 | 349 | Mean Difference (IV, Random, 95% CI) | ‐21.68 [‐35.94, ‐7.42] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Ansell 1978.
| Methods |
Design: Cross‐over study Number of centres: NA Treatment duration: Each treatment period was 4 weeks Flare design: Yes Wash‐out period: Yes (2 weeks, not between therapies) Time point of assessments: BL, 4, 8 weeks |
|
| Participants |
Inclusion criteria: Radiographic evidence of sacroiliitis of at least grade 2 and clinically active symptoms. Exclusion criteria:
Classification: NA All participants: Number of participants: 25 Number of completers: NA Age: range 25 to 69 Male (%): 92 Symptom duration: NA Disease duration: NA HLA‐B27 positive (%): NA |
|
| Interventions | Naproxen (750 mg) vs Butacote (300 mg) Co‐medication: NA |
|
| Outcomes |
Extracted outcomes: 1. Patient's global assessment of disease activity (BL not available, post‐treatment after 4 weeks (± SD)) (scale 0 to 3, higher is worse) Naproxen 750 mg: 1.71 ± 0.73 Butacote 300 mg: 1.27 ± 0.33 2. ESR (BL (± SD), change after 4 weeks (± SD)) (in mm/hr, higher is worse) Naproxen 750 mg: 26.31 ± 13.97, +2.17 ± 11.97 Butacote 300 mg: 28.82 ± 19.31, ‐3.54 ± 10.48 3. Tragus‐to‐wall distance (BL (± SD), change after 4 weeks (± SD)) (in cm, higher is worse, left side (no differences with right side)) Naproxen 750 mg: 15.07 ± 5.23, ‐0.16 ± 1.40 Butacote 300 mg: 15.23 ± 4.11, ‐1.14 ± 1.16 4. Schober's test (BL (± SD), change after 4 weeks (± SD)) (in cm, higher is better) Naproxen 750 mg: 3.25 ± 2.07, +0.42 ± 0.89 Butacote 300 mg: 3.34 ± 1.84, +0.90 ± 2.42 |
|
| Notes | Results are not included in the meta‐analysis, because the number of patients in each treatment group was not available. Available results are described in this table. Only results of first part of cross‐over trial are presented. Funding source: Geigy Pharmaceuticals provided Butacote and placebo. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "For analysis patients were split into two groups". No information was provided on sequence generation. |
| Allocation concealment (selection bias) | Unclear risk | "For analysis patients were split into two groups". No information was provided on allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "double‐blind, cross‐over study with double‐dummy technique". Probably done, but no further information was provided on blinding participants. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "double‐blind". Probably done, but no further information was provided on blinding of outcome assessor. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No information was provided on drop‐outs or missing data. |
| Selective reporting (reporting bias) | Low risk | All pre‐specified outcomes according to the methods section, are reported in the results section. |
| Other bias | High risk | Crossover design, possible carry‐over effect in Schober's test and ESR (not reported for other outcomes). |
Astorga 1987.
| Methods |
Design: RCT Number of centres: NA Treatment duration: 6 months Flare design: No Wash‐out period: No Time point of assessments: BL, 1, 2, 3, 4, 5 and 6 months |
|
| Participants |
Inclusion criteria: Active disease, as assessed in the following criteria: a) lumbar and stomach pain during the day and night but marked in the morning, b) marked morning lumbar stiffness, c) objective limitation of spinal movement, d) radiological signs characteristic of affected sacroiliac joints. Exclusion criteria: NA Classification: NA Tenoxicam: Number of participants: 10 Number of completers: 8 Age (mean (SD)): 47.8 (8.8) Male (%): 90 Symptom duration: NA Disease duration: NA HLA‐B27 positive (%): NA Piroxicam: Number of participants: 10 Number of completers: 9 Age (mean (SD)): 46.4 (7.9) Male (%): 80 Symptom duration: NA Disease duration: NA HLA‐B27 positive (%): NA |
|
| Interventions | Tenoxicam (20 mg) vs Piroxicam (20 mg) Co‐medication: NA |
|
| Outcomes |
Extracted outcomes:
|
|
| Notes | The outcomes duration of morning stiffness and ESR were also presented, but these data could not be used due to presentation in graphs from which the data could not be extracted. For the outcome pain on Likert scale individual patient data that were presented in the paper were combined for the meta‐analysis to a mean and SD. Funding source: Not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "were randomly assigned to treatment with either tenoxicam or piroxicam". Probably done, but no further information was provided on sequence generation. |
| Allocation concealment (selection bias) | Unclear risk | No information provided on allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "double‐blind study". Probably done, but no further information was provided on blinding of participants. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "double‐blind study". Probably done, but no further information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | No information provided on number of dropouts, however number of participants after 6 months doesn't add up to original number of included participants. |
| Selective reporting (reporting bias) | High risk | No pre‐specified outcomes defined in the methods section, however in discussion hand‐floor distance and Schober index are named (and not reported in results section). |
| Other bias | Low risk | No other bias was detected. |
Barkhuizen 2006.
| Methods |
Design: RCT Number of centres: NA Treatment duration: 12 weeks Flare design: Yes Wash‐out period: Yes (analgesics for 8 hours and anti‐inflammatory medication for 72 hours) Time point of assessments: Screening, BL, 1, 3, 6 and 12 weeks, at discontinuation |
|
| Participants |
Inclusion criteria: 1. Age 18 to 75; 2. AS with axial involvement; 3. Requiring daily treatment with NSAIDs during the previous 30 days; 4. Pain intensity ≥ 50mm on VAS, worsening by 30% after discontinuation therapy between pre‐inclusion visit and inclusion; 5. No analgesic use for at least 8 hours or anti‐inflammatory medication use for at least 72 hours prior to study start; 6. Negative pregnancy‐test at inclusion and on contraception throughout trial. Exclusion criteria: 1. Distal small‐joint synovitis; 2. Inflammatory enteropathy; 3. Extra‐articular signs; 4. Vertebral compression; 5. Needing to wear a corset during the trial; 6. Requiring physiotherapy or re‐education or manipulation during the trial; 7. Requiring concomitant use of muscle relaxants, hypnotics, anxiolytics, sedatives, tranquillizers, anticoagulants, ticlopidine, or lithium; 8. Use of antidepressants (unless taking stable dose for 2 weeks prior to inclusion); 9. Corticosteroids in 6 weeks prior to study start; 10. Receving MTX > 25 mg/week or anti‐TNF agents (SSZ only when taking stable dose for 60 days prior to screening); 11. History of gastroduodenal ulcer confirmed by endoscopy in 30 days prior to inclusion or with concurrent gastrointestinal bleeding; 12. Known hypersensitivity to analgesics, NSAIDs, celecoxib, COX‐2‐selective inhibitors, naproxen, lactose, sulfonamide, or APAP; 13. History of asthma, chronic disease that might interfere with study results or current/previous malignancy. Classification: modified New York criteria Celecoxib (200 mg): Number of participants: 137 Number of completers: 100 Age (mean (SD)): 43.9 (11.9) Male (%): 79 Symptom duration: NA Disease duration: NA HLA‐B27 positive (%): NA Celecoxib (400 mg): Number of participants: 161 Number of completers: 118 Age (mean (SD)): 45.1 (11.6) Male (%): 70 Symptom duration: NA Disease duration: NA HLA‐B27 positive (%): NA Naproxen (500 mg): Number of participants: 157 Number of completers: 118 Age (mean (SD)): 45.4 (12.6) Male (%): 75 Symptom duration: NA Disease duration: NA HLA‐B27 positive (%): NA Placebo: Number of participants: 156 Number of completers: 72 Age (mean (SD)): 43.8 (11.5) Male (%): 73 Symptom duration: NA Disease duration: NA HLA‐B27 positive (%): NA |
|
| Interventions | Celecoxib (200 mg) vs Celexocib (400 mg) vs Naproxen (500 mg) vs Placebo Co‐medication: Rescue APAP as needed (max 2000 mg/day), stable dose SSZ, MTX < 25 mg/week, or stable dose antidepressants, or both |
|
| Outcomes |
Extracted outcomes: 1. Withdrawals due to adverse events 2. BASDAI (mean change after 12 weeks) (scale 0 to 100, higher is worse) Celecoxib 200 mg: ‐15.4 (N = 137) Celecoxib 400 mg: ‐19.5 (N = 161) Naproxen 500 mg: ‐22.9 (N = 157) Placebo: ‐1.74 (N = 156) 3. Duration of morning stiffness (median change after 12 weeks) (in minutes, higher is worse) Celecoxib 200 mg: ‐5 (N = 137) (P < 0.05 all treatment groups versus placebo) Celecoxib 400 mg: ‐20 (N = 161) Naproxen 500 mg: ‐30 (N = 157) (P < 0.05 versus celecoxib 200 mg) Placebo: 0 (N = 156) 4. CRP (mean change after 12 weeks) (in mg/L, higher is worse) Celecoxib 200 mg: ‐2.46 (N = 137) (P < 0.05 all treatment groups versus placebo) Celecoxib 400 mg: ‐2.64 (N = 161) Naproxen 500 mg: ‐3.60 (N = 157) Placebo: +1.17 (N = 156) 5. ASAS 20 6. Number of any adverse events 7. Number of serious adverse events 8. Number of adverse events per organ system |
|
| Notes | The outcomes pain on VAS, patient's global assessment of disease activity and BASFI were also presented, but these data could not be used due to presentation in graphs from which the data could not be extracted. Outcomes that were not included in the meta‐analysis, because no measure of variance (SD, SE or CI) was reported for these outcomes: BASDAI, duration of morning stiffness, CRP. Available results are described in this table. In comparison 1 (NSAID vs NSAID), comparison 4 (COX‐2 vs Placebo), comparison 5 (COX‐2 vs traditional NSAID) and comparison 6 (Naproxen vs other NSAID) we chose to present data from Celecoxib 400 mg instead of Celecoxib 200 mg (see Measures of treatment effect for rationale). Funding source: Pfizer |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Patients were randomized to receive either…". Probably done, but no further information was provided. |
| Allocation concealment (selection bias) | Unclear risk | No information provided on allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "double‐blind, placebo‐controlled". Probably done, but no further information was provided on blinding of participants. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "double‐blind". Probably done, but no further information was provided on blinding of outcome assessor. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | "All efficacy analyses were performed on data from the intent‐to‐treat population cohort, defined as patients who were randomized to treatment and took at least one dose of study medication"; "In total, 408 (67%) patients completed the study: 72 (46%) in the placebo group, 100 (73%) in the celecoxib 200mg group, 188 (73%) in the celecoxib 400mg group, and 118 (75%) in the naproxen group. The most common reason for withdrawal was lack of efficacy, with a higher proportion of patients in the placebo group (38%) withdrawing for this reason than in the celecoxib 200 mg (18%) , celecoxib 400 mg (14%), or naproxen (11%) groups." |
| Selective reporting (reporting bias) | Low risk | All outcomes that would be reported according to the methods section, are reported in the results section. |
| Other bias | Low risk | Sufficient power for primary efficacy hypothesis (sample size calculations). No different co‐interventions between groups, other than "rescue" acetaminophen. |
Batlle‐Gualda 1996.
| Methods |
Design: RCT Number of centres: 18 Treatment duration: 3 months Flare design: NA Wash‐out period: Yes (1 week) Time point of assessments: Screening, BL, 15, 30, 60 and 90 days |
|
| Participants |
Inclusion criteria: 1. Age 20 to 50; 2. Active disease, defined by at least 2 of the following 3 criteria: a) morning stiffness ≥ 30 minutes, b) pain requiring daily treatment with NSAIDs, c) pain ≥ 40 mm on 100 mm VAS. Exclusion criteria: 1. Reiter's syndrome or any other type of arthritis; 2. Pregnancy or lactation; 3. Psoriasis, inflammatory bowel disease, Paget's disease, haemochromatosis, uncontrolled hypertension, renal (creatinine > 1.5 mg/dl) or hepatic disease; 4. Concomitant serious medical condition or expected survival time less than 2 years; 5. Myocardial infarction or stroke in the last 4 months, history of peptic ulceration or upper gastrointestinal bleeding; 6. History of angina or asthma associated with NSAIDs, or hypersensitivity to aspirin or other NSAIDs, 7. Use of SSZ, corticosteroids, or immunosuppressive drugs in the previous 3 months; 8. Concomittant use of oral anticoagulants, benzodiazepines, lithium, antidepressants, phenytoin, neuroleptics, diuretics, thyroxine, or probenecid; 9. Females with childbearing potential who were not using contraceptive measures; 10. Patients enrolled in any other clinical trial within the previous 3 months or who were applying for disability for any reason. Classification: New York criteria Aceclofenac (200 mg): Number of participants: 155 Number of completers: 127 Age (mean (SD)): 37.8 (7.9) Male (%): 90 Symptom duration (mean (SD)): 7.6 (7.2) years Disease duration: NA HLA‐B27 positive (%): NA Indomethacin (100 mg): Number of participants: 153 Number of completers: 126 Age (mean (SD)): 39.1 (7.6) Male (%): 82 Symptom duration (mean (SD)): 7.4 (7.6) years Disease duration: NA HLA‐B27 positive (%): NA |
|
| Interventions | Aceclofenac (200 mg) vs Indomethacin (100 mg) Co‐medication: APAP and antacid were allowed. Concurrent corticosteroid injection was not permitted. The participants received instructions to keep the same level of physical activity and physical therapy. |
|
| Outcomes |
Extracted outcomes:
|
|
| Notes | For Analysis 4.1 and Analysis 4.2 the SD was imputed from the BL (for rationale see Dealing with missing data). Funding source: Supported by Prodesfarma SA, Barcelona, Spain. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Patients were allocated randomly, in balanced groups of 4 within each center". Probably done, but no further information was provided on sequence generation. |
| Allocation concealment (selection bias) | Unclear risk | "Patients were allocated randomly, in balanced groups of 4 within each center". Probably done, but no further information was provided on allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "All the study tablets (aceclofenac, indomethacin, placebo) were identical. All medication was taken after meals." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "double blind trial". Probably done, but no further information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | "Of the 308 patients, 253 (82%) completed the full 3 months of the study (127 aceclofenac, 126 indomethacin). The reasons for early discontinuation are shown in Table 2. There were no significant differences between the groups."; "Two types of analysis of efficacy were performed: intention‐to‐treat and completers only. The results were similar, so the intention‐to‐treat results are reported." |
| Selective reporting (reporting bias) | Low risk | All pre‐specified outcomes according to the methods section, are reported in the results section. |
| Other bias | Unclear risk | Sufficient power for primary efficacy hypothesis (sample size calculations), similar compliance rate in both treatment groups, at BL more males in aceclofenac group but that has probably not introduced bias. |
Boersma 1976.
| Methods | Retrospective cohort study, see Table 3. | |
| Participants | ‐ | |
| Interventions | ‐ | |
| Outcomes | Results not included in meta‐analysis, but described in the text (Effects of interventions). | |
| Notes | ‐ | |
Caldwell 1986.
| Methods |
Design: CCT Number of centres: NA ("multicenter") Treatment duration: 6 months Flare design: Yes Wash‐out period: Yes (until flare) Time point of assessments: BL, 2 weeks, 4 weeks and 2, 3, 4, 5, 6 months |
|
| Participants |
Inclusion criteria: 1. Age 18 or over; 2. Diagnosis of AS for minimal 6 months; 3. At least 2 of the following: a) lumbar or dorsal lumbar junction pain and stiffness of over 3 months duration, b) major limitation of motion of lumbar spine in 3 directions (flexion‐extension, lateral bending and rotation), c) pain and stiffness in the thoracic region of over 3 months duration, d) nocturnal pain with morning stiffness or bilateral pain in buttocks, or both, or pain in either buttock; 4. Grade 2 or 3 bilateral sacroiliitis by the following X‐ray criteria: 0 = normal, 1 = suspicious, 2 = definitely abnormal, 3 = advanced abnormal; 5. HLA‐B27 positive; 6. ESR ≥ 18 mm/hr; 7. Rheumatoid factor titre ≤ 1/80; 8. Normal serum uric acid level (unless patient was receiving thiaxide diuretic therapy or had gouty arthritis); 9. Muscle spasm in the back and decreased range of motion of some part of the spine. Exclusion criteria: 1. Unable to walk 50 feet; 2. Receiving anticoagulant therapy; 3. Women of childbearing potential; 4. Patients with known allergies to aspirin or other NSAIDs, or both; 5. Coexisting gastrointestinal, inflammatory, malignant, or infectious diseases and renal or hepatic impairment. Classification: NA Oxaprozin (1200 mg): Number of participants: 55 Number of completers: 36 Indomethacin (50 to 150 mg): Number of participants: 42 Number of completers: 31 All participants: Age: 40 (range 19 to 70) Male (%): 84 Symptom duration: NA Disease duration: 11 (range 1 to 40) years HLA‐B27 positive (%): NA |
|
| Interventions | Oxaprozin (1200 mg) vs Indomethacin (50 to 150 mg) Co‐medication: Concomitant corticosteroid therapy (max 7.5 mg prednisone daily), pure analgesics having no anti‐inflammatory effect and medication for unrelated illnesses that had begun at least 3 months before study entry. |
|
| Outcomes |
Extracted outcomes: 1. Pain on Likert scale (BL, 2 weeks, post‐treatment after 6 months) (scale 0 to 4, higher is worse) Oxaprozin 1200 mg: 2.02 (N = 47), 1.62 (N = 47), 1.19 (N = 21) (P = not significant) Indomethacin 25 to 50 mg: 2.19 (N = 32), 1.38 (N = 32), 0.94 (N = 17) (P < 0.05 at 2 weeks, P < 0.001 post‐treatment) 2. Withdrawals due to adverse events 3. Patient's global assessment of disease activity (BL, 2 weeks, post‐treatment after 6 months) (scale 1 to 5, higher is worse) Oxaprozin 1200 mg: 3.09 (N = 47), 2.70 (N = 47), 2.18 (N = 22) (P < 0.05 at 2 weeks) Indomethacin 25 to 50 mg: 3.31 (N = 32), 2.47 (N = 32), 2.35 (N = 17) (P < 0.001 at 2 weeks, P < 0.01 post‐treatment) 4. Duration of morning stiffness (BL, 2 weeks, post‐treatment after 6 months) (median, in minutes, higher is worse) Oxaprozin 1200 mg: 120.0 (N = 48), 90.0 (N = 48), 15.0 (N = 22) (P < 0.001 post‐treatment) Indomethacin 25 to 50 mg: 120.0 (N = 32), 60.0 (N = 32), 20.0 (N = 17) (P < 0.01 at 2 weeks) 5. Lateral spinal flexion (BL, 2 weeks, post‐treatment after 6 months) (in cm, left‐sided, higher is better) Oxaprozin 1200 mg: 4.9 (N = 43), 5.8 (N = 43), 5.9 (N = 20) (P = not significant) Indomethacin 25 to 50 mg: 8.0 (N = 31), 8.2 (N = 31), 5.3 (N = 17) (P = not significant) 6. Chest expansion (BL, 2 weeks, post‐treatment after 6 months) (in cm, higher is better) Oxaprozin 1200 mg: 2.8 (N = 47), 3.3 (N = 47), 4.0 (N = 21) (P < 0.05 at 2 weeks, P < 0.01 post‐treatment) Indomethacin 25 to 50 mg: 2.8 (N = 30), 3.2 (N = 30), 3.2 (N = 16) (P = not significant) 7. Occiput‐to‐wall distance (BL, 2 weeks, post‐treatment after 6 months) (in cm, higher is worse) Oxaprozin 1200 mg: 5.2 (N = 43), 5.2 (N = 43), 7.8 (N = 17) (P = not significant) Indomethacin 25 to 50 mg: 3.7 (N = 29), 3.2 (N = 29), 2.2 (N = 15) (P = not significant) 8. Intermalleolar distance (BL, 2 weeks, post‐treatment after 6 months) (in cm, higher is better) Oxaprozin 1200 mg: 103.5 (N = 46), 102.2 (N = 46), 107.1 (N = 21) (P = not significant) Indomethacin 25 to 50 mg: 102.1 (N = 30), 105.5 (N = 30), 109.1 (N = 17) (P = not significant) 9. Schober's test (BL, 2 weeks, post‐treatment after 6 months) (in cm, higher is better) Oxaprozin 1200 mg: 4.7 (N = 43), 5.7 (N = 43), 5.3 (N = 20) (P = not significant) Indomethacin 25 to 50 mg: 6.5 (N = 30), 7.6 (N = 30), 4.8 (N = 17) (P = not significant) 10. Number of any adverse events 11. Number of adverse events per organ system |
|
| Notes | Outcomes that were not included in the meta‐analysis, because no measure of variance (SD, SE or CI) was reported for these outcomes: pain on Likert scale, patient's global assessment of disease activity, duration of morning stiffness, lateral spinal flexion, chest expansion, occiput‐to‐wall distance, intermalleolar distance, Schober's test. Available results are described in this table. For the outcome number of adverse events per organ system only the most frequently occurring adverse events were reported per organ system, adverse events judged as not drug‐related were excluded. Funding source: Not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | No comment is made by the authors anywhere that patients were randomised. |
| Allocation concealment (selection bias) | High risk | No comment is made by the authors anywhere that patients were randomised. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "double‐blind". Probably done, but no further information was provided on blinding of participants. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "double‐blind". Probably done, but no further information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | "Nineteen patients (35%) in the oxaprozin group and 11 (26%) in the indomethacin group discontinued treatment for drug‐related reasons including unsatisfactory response and/or adverse effects". In table 3 with results every outcome has another number of participants without explanation why the other patients did not provide data, also many patient data are not available for outcomes post‐treatment (up to 83% loss‐to‐follow‐up). |
| Selective reporting (reporting bias) | Low risk | All pre‐specified outcomes according to the methods section, are reported in the results section. |
| Other bias | Low risk | No other bias was detected. |
Calin 1979.
| Methods |
Design: CCT Number of centres: NA Treatment duration: 6 months Flare design: Yes Wash‐out period: Yes Time point of assessments: "on 8 occasions during the six‐month period" |
|
| Participants |
Inclusion criteria: HLA‐B27 positive, and fulfilling the New York criteria. Exclusion criteria: NA Classification: New York criteria Indomethacin (75 to 150 mg): Number of participants: 15 Number of completers: 15 Age (mean): 44.6 Male (%): 80 Symptom duration: NA Disease duration: NA HLA‐B27 positive (%): 100 Sulindac (200 to 400 mg): Number of participants: 15 Number of completers: 12 Age (mean): 32.7 Male (%): 80 Symptom duration: NA Disease duration: NA HLA‐B27 positive (%): 100 |
|
| Interventions | Indomethacin (75 to 150 mg) vs Sulindac (200 to 400 mg) Co‐medication: NA |
|
| Outcomes |
Extracted outcomes: 1. Withdrawals due to adverse events 2. Lateral spinal flexion (BL, post‐treatment after 6 months) (in cm, higher is better) Indomethacin 75 to 150 mg: 2.0 (N = 15), 3.8 (N = 15) Sulindac 200 to 400 mg: 3.1 (N = 15), 5.5 (N = 14) 3. Chest expansion (BL, post‐treatment after 6 months) (in cm, higher is better) Indomethacin 75 to 150 mg: 2.7 (N = 15), 3.7 (N = 15) Sulindac 200 to 400 mg: 3.1 (N = 15), 4.5 (N = 14) 4. Intermalleolar distance (BL, post‐treatment after 6 months) (in cm, higher is better) Indomethacin 75 to 150 mg: 89 (N = 15), 103 (N = 15) Sulindac 200 to 400 mg: 90 (N = 15), 114 (N = 14) 5. Schober's test (BL, post‐treatment after 6 months) (in cm, higher is better) Indomethacin 75 to 150 mg: 2.6 (N = 15), 3.9 (N = 15) Sulindac 200 to 400 mg: 2.8 (N = 15), 5.2 (N = 14) 6. Number of any adverse events |
|
| Notes | Outcomes that were not included in the meta‐analysis, because no measure of variance (SD, SE or CI) was reported for these outcomes: lateral spinal flexion, chest expansion, intermalleolar distance, Schober's test. Available results are described in this table. For the outcome lateral spinal flexion, "left lateral spinal flexion" was extracted ("right lateral spinal flexion" was also available, there was no difference between left and right). Funding source: Not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | No comment is made by authors that patients were randomised. |
| Allocation concealment (selection bias) | High risk | No comment is made by the authors that patients were randomised. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "…plus dummy sulindac tablets…plus dummy indomethacin tablets". Participants appear to have been blinded to treatment allocation. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "double‐blind". Probably done, but no further information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | "The three dropouts were unavailable for follow‐up for reasons not related to tolerance or efficacy." Although uneven number of dropouts in treatment groups (0 vs 3), there appears to be a low risk of attrition bias. |
| Selective reporting (reporting bias) | High risk | The following pre‐specified outcomes were not reported: pain during night and day, duration of morning stiffness, fatigue, global score by patient and physician, daily functioning, loss of lordosis and occiput‐to‐wall‐distance. |
| Other bias | Unclear risk | Baseline imbalance and administration of co‐medication cannot be ruled out. |
Carcassi 1990.
| Methods |
Design: RCT Number of centres: 1 Treatment duration: 12 weeks Flare design: No Wash‐out period: Yes (48 hours) Time point of assessments: BL, 2, 4, 8 and 12 weeks |
|
| Participants |
Inclusion criteria: 1. Out‐patients only; 2. Age 18 to 75; 3. If female, post‐menopausal or surgically sterile; 4. Definite diagnosis of AS; 5. Active disease as defined by the presence of spinal and/or sacroiliac pain with an increased sedimentation rate and/or active involvement of one or more peripheral joints and/or morning stiffness; 6. Onset after 16 years of age. Exclusion criteria: 1. Acute or active metabolic, neurological, infectious, endocrine, or auto‐immune disease giving rise to arthritis; 2. Evidence of clinically significant, uncontrolled heart, lung, kidney, liver, endocrinological, neurological, gastrointestinal, or hypertensive disease; 3. History of significant upper gastrointestinal bleeding or documented gastric/duodenal ulcer during 5 years prior to study entry; 4. Abnormal pre‐treatment laboratory values that were considered clinically significant but not due to AS if such values were believed to influence the safety evaluations; 5. History of blood dyscrasia, significant psychiatric disorder, drug abuse or alcoholism, allergy to NSAIDs or history of malignancy (unless patient was free of malignant disease for at least 1 year and required no active treatment); 6. Patients receiving steroids, intra‐articular injections, immunosuppressive therapy or investigational drugs during 8 weeks prior to enrolment; 7. If all NSAIDs or analgesics were not discontinued for at least 48 hours prior to enrolment and if patient took salicylates or other NSAIDs or anti‐coagulants during the study. Classification: New York criteria Pirazolac (300 to 600 mg): Age (mean): 36.8 (range 18 to 72) Disease duration: 119 months (range 5 to 480) Indomethacin (25 to 50 mg): Age (mean): 40 (range 20 to 62) Disease duration: 126 months (range 6 to 600) All participants: Number of participants: 151 Number of completers: 119 Male (%): 85 Symptom duration: NA HLA‐B27 positive (%): NA |
|
| Interventions | Pirazolac (300 to 600 mg) vs Indomethacin (25 to 50 mg) Co‐medication: Continuation of any prior physical therapy regimen was required, patients were allowed to take vitamins and medications for control of permitted conditions, antacids were permitted only on a non‐chronic basis |
|
| Outcomes |
Extracted outcomes: 1. Withdrawals due to adverse events (after 12 weeks) (in %) Pirazolac 300 to 600 mg: 20% Indomethacin 25 to 50 mg: 9% 2. Number of any adverse events (after 12 weeks) (""Only the adverse effects for which the severity and relationship to the study drug were known are included.") Pirazolac 300 to 600 mg: N = 14 Indomethacin 25 to 50 mg: N = 6 3. Number of adverse events per organ system (after 12 weeks) Pirazolac 300 to 600 mg: cardiovascular n = 1, gastro‐intestinal n = 6, neurologic n = 1, dermatologic n = 3 Indomethacin 25 to 50 mg: cardiovascular n = 0, gastro‐intestinal n = 3, neurologic n = 4, dermatologic n = 0 |
|
| Notes | Results are not included in the meta‐analysis, because the number of patients in each treatment group was not available. Available results are described in this table. The outcomes Schober's test, occiput‐to‐wall distance, chest expansion and duration of morning stiffness were also reported, but could not be used due to presentation in graphs from which the data could not be extracted. Funding source: Not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Each patient was randomly assigned to either". Probably done, but no further information was provided on sequence generation. |
| Allocation concealment (selection bias) | Unclear risk | No information provided on allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "All drugs were supplied in an identical capsule form." "Those patients who were randomized to pirazolac had received placebo at times corresponding to the second daily dose of indomethacin, so that all patients took medication on a t.i.d. basis". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "double‐blind". Probably done but no further information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | "There were no significant differences between the two treatment groups with regards to drop‐out rates (p=0.17)." |
| Selective reporting (reporting bias) | Low risk | All pre‐specified outcomes according to the methods section, are reported in the results section. |
| Other bias | Low risk | No other bias was detected. |
Dougados 1994.
| Methods |
Design: RCT Number of centres: NA Treatment duration: 2 weeks Flare design: Yes Wash‐out period: Yes (2 days) Time point of assessments: BL, 1 week, 2 weeks |
|
| Participants |
Inclusion criteria: 1. Having received NSAID daily for at least 1 month; 2. A 2‐day washout period for the concomitant NSAIDs; 3. A flare of the disease defined by the 2 following: pain evaluated on a VAS over 40, and increase in pain of at least 30% between screening and entry visit. Exclusion criteria: 1. Peripheral articular disease (at least 2 inflamed joints at screening visit); 2. Inflammatory bowel disease; 3. Serious concomitant medical illness; 4. Judged to be in functional class IV according to the Steinbrocker criteria. Classification: ESSG, Amor and modified New York criteria Ximoprofen (5 mg): Number of participants: 46 Number of completers: 41 Age (mean (SD)): 40 (10) Male (%): 63 Symptom duration: NA Disease duration (mean (SD)): 10 (8) years HLA‐B27 positive (%): 80 Ximoprofen (10 mg): Number of participants: 49 Number of completers: 46 Age (mean (SD)): 40 (10) Male (%): 71 Symptom duration: NA Disease duration (mean (SD)): 8 (7) years HLA‐B27 positive (%): 67 Ximoprofen (20 mg): Number of participants: 45 Number of completers: 41 Age (mean (SD)): 40 (13) Male (%): 62 Symptom duration: NA Disease duration (mean (SD)): 8 (8) years HLA‐B27 positive (%): 76 Ximoprofen (30 mg): Number of participants: 50 Number of completers: 44 Age (mean (SD)): 40 (12) Male (%): 76 Symptom duration: NA Disease duration (mean (SD)): 10 (8) years HLA‐B27 positive (%): 84 Placebo: Number of participants: 95 Number of completers: 71 Age (mean (SD)): 40 (11) Male (%): 68 Symptom duration: NA Disease duration (mean (SD)): 10 (8) years HLA‐B27 positive (%): 75 |
|
| Interventions | Ximoprofen (5 mg) vs Ximoprofen (10 mg) vs Ximoprofen (20 mg) vs Ximoprofen (30 mg) vs Placebo Co‐medication: NA |
|
| Outcomes |
Extracted outcomes:
|
|
| Notes | In comparison 2 (NSAID vs NSAID dose) we chose to compare the smallest to the highest dose (Ximoprofen 5 mg vs Ximoprofen 30 mg). In comparison 3 (NSAID vs Placebo) we chose to present data from the highest dose (Ximoprofen 30 mg). Funding source: Supported in part by Laboratories Jacques LOGEAIS. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "This randomization was performed centrally by using the computer system. The allocated drug for the recruited patient was then sent to the investigator's office." |
| Allocation concealment (selection bias) | Low risk | "This randomization was performed centrally by using the computer system. The allocated drug for the recruited patient was then sent to the investigator's office." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "All these capsules (placebo or Ximoprofen) were undistinguishable." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "double blind"; "Clinical assessment was made weekly by the same investigator for each patient." Comment: Probably done, but no further information provided on blinding of the outcome assessor. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | "During the trial 42 patients withdrew (24 in the placebo group, 25%: 21 because of inefficacy, 1 because of toxicity, and 2 because of both toxicity and inefficacy; 5 in the 5 mg Ximoprofen group, 11%: 4 because of inefficacy and 1 because of toxicity; 3 in the 10 mg Ximoprofen group, 6%: all because of inefficacy; 4 in the 20 mg Ximoprofen group, 9%: 2 because of inefficacy, 1 because of both inefficacy and toxicity and 1 for a reason unrelated to treatment; 6 in the 30 mg Ximoprofen group, 12%: 4 because of inefficacy and 2 because of toxicity." |
| Selective reporting (reporting bias) | Unclear risk | Patient and physicians assessment of disease activity was assessed (see methods section), but not reported. All other outcomes that were assessed, were reported. |
| Other bias | Low risk | Sufficient power for primary efficacy hypothesis (sample size calculations). No other bias detected. |
Dougados 1999.
| Methods |
Design: RCT Number of centres: NA ("different centres in four countries (Belgium, France, Germany, UK)") Treatment duration: 6 weeks (part I) and 52 week extension (part II) Flare design: Yes Wash‐out period: Yes (2 to 15 days) Time point of assessments: BL, 1, 3, 6, 13, 26, 39, 52 weeks |
|
| Participants |
Inclusion criteria: 1. Daily NSAID intake during the month preceding the selection visit; 2. A wash‐out period of NSAID of 2 to 15 days before the BL visit; 3. A flare of the disease at BL defined by both pain evaluated on a 100mm length VAS over 40mm and increase in pain of at least 30% between the screening and the BL visits. Exclusion criteria: 1. Peripheral articular disease defined by the presence at the screening visit of an active (painful or swollen) peripheral arthritis (excluding hip and shoulder); 2. Active inflammatory bowel disease; 3. Severe concomitant medical illness; 4. Patients who received corticosteroids during the previous month or any slow‐acting drug initiated, or both, or with an altered dose during the previous 6 months. Classification: modified New York criteria Piroxicam (20 mg): Number of participants: 108 Number of completers: 91 Age (mean (SD)): 44 (13) Male (%): 77 Symptom duration: NA Disease duration (mean (SD)): 12 (11) years HLA‐B27 positive (%): 84 Meloxicam (15 mg): Number of participants: 120 Number of completers: 99 Age (mean (SD)): 44 (12) Male (%): 79 Symptom duration: NA Disease duration (mean (SD)): 13 (9) years HLA‐B27 positive (%): 80 Meloxicam (22.5 mg): Number of participants: 124 Number of completers: 103 Age (mean (SD)): 42 (12) Male (%): 85 Symptom duration: NA Disease duration (mean (SD)): 12 (10) years HLA‐B27 positive (%): 91 Placebo: Number of participants: 121 Number of completers: 70 Age (mean (SD)): 40 (12) Male (%): 72 Symptom duration: NA Disease duration (mean (SD)): 12 (9) years HLA‐B27 positive (%): 90 |
|
| Interventions | Piroxicam (20 mg) vs Meloxicam (15 mg) vs Meloxicam (22.5 mg) vs Placebo Co‐medication: Paracetamol (500 mg) was used as an analgesic rescue during the study. |
|
| Outcomes |
Extracted outcomes: Six weeks results (included in meta‐analysis)
One year results (not included in meta‐analysis) 1. Pain on VAS (BL, mean change after 1 year (± SD)) (scale 0 to 100, in mm, higher is worse) Meloxicam 22.5 mg: 72 ± 14 (N = 124), ‐33 ± 27 (P < 0.05 vs placebo) Meloxicam 15 mg: 69 ± 18 (N = 120), ‐31 ± 30 (P < 0.05 vs placebo) Piroxicam 20 mg: 72 ± 15 (N = 108), ‐29 ± 28 (P < 0.05 vs placebo) Placebo: 72 ± 17 (N = 121), ‐11 ± 28 2. Withdrawals due to adverse events (post‐treatment after 1 year) Meloxicam 22.5 mg: n = 11 (total N = 124) Meloxicam 15 mg: n = 21 (total N = 120) Piroxicam 20 mg: n = 21 (total N = 108) Placebo: n = 10 (total N = 121) (P = 0.08 between different groups) 3. Patient's global assessment of disease activity (BL, mean change after 1 year (± SD)) (VAS 0 to 100, in mm, higher is worse) Meloxicam 22.5 mg: 65 ± 18 (N = 124), ‐26 ± 30 (P < 0.05 vs placebo) Meloxicam 15 mg: 62 ± 20 (N = 120), ‐25 ± 29 (P < 0.05 vs placebo) Piroxicam 20 mg: 65 ± 19 (N = 108), ‐21 ± 30 (P < 0.05 vs placebo) Placebo: 62 ± 20 (N = 121), +2 ± 31 4. Duration of morning stiffness (BL, mean change after 1 year (± SD) (in minutes, higher is worse)) Meloxicam 22.5 mg: 86 ± 77 (N = 124), ‐42 ± 77 (P = not significant) Meloxicam 15 mg: 77 ± 68 (N = 120), ‐23 ± 68 (P < 0.05 vs placebo) Piroxicam 20 mg: 80 ± 72 (N = 108), ‐26 ± 66 (P = not significant) Placebo: 88 ± 77 (N = 121), 2 ± 74 5. CRP (mean change after 1 year (± SD)) (in mg/L, higher is worse) Meloxicam 22.5 mg: ‐2.4 ± 15.7 (P < 0.05 vs placebo) Meloxicam 15 mg: ‐3.0 ± 16.1 (P < 0.05 vs placebo) Piroxicam 20 mg: 0.3 ± 17.0 (P = not significant) Placebo: 6.0 ± 14.2 6. BASFI (BL, mean change after 1 year (± SD)) (scale 0 to 10, higher is worse) Meloxicam 22.5 mg: 15 ± 6 (N = 124), ‐3.1 ± 7.0 (P < 0.05 vs placebo) Meloxicam 15 mg: 15 ± 7 (N = 120), ‐3.1 ± 7.2 (P < 0.05 vs placebo) Piroxicam 20 mg: 15 ± 6 (N = 108), ‐1.7 ± 7.4 (P < 0.05 vs placebo) Placebo: 16 ± 7 (N = 121), +1.5 ± 7.8 7. Chest expansion (BL, mean change after 1 year (± SD)) (in cm, higher is better) Meloxicam 22.5 mg: 3.5 ± 1.9 (N = 124), ‐0.7 ± 1.9 (P < 0.05 vs placebo) Meloxicam 15 mg: 3.8 ± 2.2 (N = 120), +0.3 ± 1.2 (P < 0.05 vs placebo) Piroxicam 20 mg: 3.5 ± 2.2 (N = 108), +0.5 ± 1.6 (P < 0.05 vs placebo) Placebo: 3.8 ± 2.2 (N = 121), ‐0.3 ± 1.6 8. Schober's test (BL, mean change after 1 year (± SD)) (in cm, higher is better) Meloxicam 22.5 mg: 12.7 ± 1.8 (N = 124), 0.4 ± 1.4 (P = not significant) Meloxicam 15 mg: 12.7 ± 1.5 (N = 120), 0.3 ± 1.2 (P = not significant) Piroxicam 20 mg: 12.8 ± 1.5 (N = 108), 0.3 ± 1.3 (P = not significant) Placebo: 12.8 ± 1.5 (N = 121), 0.1 ± 1.6 9. Pain relief ≥ 50% (percentage of responders after 1 year) (in %) Meloxicam 22.5 mg: 46% (total N = 124) (P < 0.0167 vs placebo) Meloxicam 15 mg: 50% (total N = 120) (P < 0.0167 vs placebo) Piroxicam 20 mg: 39% (total N = 108) (P < 0.0167 vs placebo) Placebo: 16% (total N = 121) 10. Number of any adverse events (post‐treatment after 1 year) Meloxicam 22.5 mg: n = 45 (total N = 124) Meloxicam 15 mg: n = 41 (total N = 120) Piroxicam 20 mg: n = 41 (total N = 108) Placebo: n = 32 (total N = 121) (P = not significant between different groups) |
|
| Notes | The outcomes number of any adverse events and number of adverse events per organ system were both only presented for the placebo‐group and the active‐NSAID‐group (meloxicam 15 mg, meloxicam 22.5 mg and piroxicam), so these data were only used in comparison 3 (NSAID vs Placebo). In comparison 1 (NSAID vs NSAID) and comparison 3 (NSAID vs Placebo) we chose to present data from Meloxicam 15 mg instead of Meloxicam 22.5 mg (see Measures of treatment effect for rationale). In comparison 3 (NSAID vs Placebo) we chose to present data from both Meloxicam 15 mg and Piroxicam 20 mg, by splitting the Placebo‐group into two groups and thus including two comparisons for this study (see Cochrane Handbook Chapter 16.5.4). For Analysis 4.2, Analysis 4.4, Analysis 6.5 and Analysis 1.5 the SD was imputed from the BL (for rationale see Dealing with missing data). Funding source: Supported in part by a grant from Boehringer Ingelheim Ltd. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "patients were randomly assigned to receive..." Probably done, but no further information provided on sequence generation. |
| Allocation concealment (selection bias) | Unclear risk | No information provided on allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Patients received two indistinguishable capsules every evening with a glass of water after food." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "double‐blind". Probably done but no further information provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | "The main reasons for discontinuation of the study drug were lack of efficacy and adverse events. A week 6, 35 patients withdrew (13, 6, 10 and 6 in placebo, piroxicam 20mg, meloxicam 15mg and meloxicam 22.5mg groups, respectively)". Similar number of dropouts in all treatment groups, with reasons provided and also similar. Intention‐to‐treat‐analysis presented (not different from completers analysis). |
| Selective reporting (reporting bias) | Low risk | All pre‐specified outcomes according to the methods section, are reported in the results section. |
| Other bias | Low risk | Sufficient power for primary efficacy hypothesis (sample size calculations). No other bias detected. |
Dougados 2001.
| Methods |
Design: RCT Number of centres: 76 Treatment duration: 6 weeks Flare design: Yes Wash‐out period: Yes (2 to 14 days) Time point of assessments: BL, 1, 3, 6 weeks |
|
| Participants |
Inclusion criteria: 1. Daily NSAID intake during the month preceding the screening visit; 2. NSAID washout period of 2 to 14 days before BL; 3. A flare of disease at BL, defined both by pain ≥ 40mm on a 100mm VAS and by an increase in pain of at least 30% between screening and baseline. Exclusion criteria: 1. Patients with peripheral articular disease, defined by the presence of active (with swelling) peripheral arthritis (excluding hip and shoulder) at screening; 2. Active inflammatory bowel disease; 3. Concomitant severe medical illness; 4. Corticosteroids during previous 6 weeks or any DMARDs with a change in dosage during previous 6 months, or both; 4. Peptic ulcer confirmed by endoscopy within 1 year preceding the screening visit. Classification: modified New York criteria Ketoprofen (100 mg): Number of participants: 90 Number of completers: 67 Age (mean (SD)): 38 (11) Male (%): 67 Symptom duration: NA Disease duration (mean (SD)): 11 (10) years HLA‐B27 positive (%): 89 Celecoxib (100 mg): Number of participants: 80 Number of completers: 54 Age (mean (SD)): 38 (11) Male (%): 70 Symptom duration: NA Disease duration (mean (SD)): 11 (9) years HLA‐B27 positive (%): 84 Placebo: Number of participants: 76 Number of completers: 44 Age (mean (SD)): 40 (11) Male (%): 71 Symptom duration: NA Disease duration (mean (SD)): 11 (9) years HLA‐B27 positive (%): 84 |
|
| Interventions | Ketoprofen (100 mg) vs Celecoxib (100 mg) vs Placebo Co‐medication: Acetaminophen (500 mg tablets, max 6 per day) was used as analgesic treatment during the study when needed. At the screening visit, concomitant therapies with gastrointestinal protective effects (misoprostol, proton pump inhibitors) were stopped when there was no history of gastroduodenal ulcers and were initiated or continued when there was a positive history of gastroduodenal ulcers, or both. |
|
| Outcomes |
Extracted outcomes:
|
|
| Notes | For Analysis 1.5 the SD was imputed from the BL (for rationale see Dealing with missing data). Funding source: Supported in part by a grant from Searle Ltd. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "patients were randomly assigned to receive..." Probably done, but no further information was provided on sequence generation. |
| Allocation concealment (selection bias) | Unclear risk | No information provided on allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "double‐blind". Probably done, but no further information was provided on blinding of participants. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "double‐blind". Probably done but no further information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Although a high number of participants dropped out, reasons are provided and equally divided amongst treatment groups. An intention‐to‐treat‐analysis was performed. |
| Selective reporting (reporting bias) | Low risk | All pre‐specified outcomes according to the methods section, are reported in the results section. |
| Other bias | Low risk | High compliance rate in all groups (> 95%). Sufficient power for primary efficacy hypothesis (sample size calculations). No other bias detected. |
Ebner 1983.
| Methods |
Design: RCT Number of centres: NA Treatment duration: 8 weeks Flare design: No Wash‐out period: Yes (1 week) Time point of assessments: BL, 1, 2, 3, 4, 6, 8 weeks |
|
| Participants |
Inclusion criteria: 1. Definite AS, defined as having 4 of the 5 following clinical criteria or one of these criteria and characteristic X‐ray findings: a) pain and stiffness in lumbar and dorsal lumbar junction existing for more than 3 months with no improvement at rest, b) pain and stiffness in thoracic region for more than 3 months, c) restricted mobility in lumbar vertebral column, d) restricted chest expansion, and e) clinical history or objective symptoms of an iritis sequels; 2. Active disease (marked pain in the vertebral column and at least one of the following: increased muscular tension in the back, restricted range of motion in any part of the vertebral column, an accelerated ESR). Exclusion criteria: 1. Treatment with corticosteroids or an experimental anti‐inflammatory drug within 4 weeks of onset of the study; 2. Treatment with gold or anti‐malarials within 3 months of onset of the study; 3. Concomitant treatment with anticoagulants; 4. Severe cardiorespiratory insufficiency; 5. Laboratory findings indicating hepatic or renal disease; 6. Signs of significant disease of the gastrointestinal tract; 7. Clinically significant eye disease, bone marrow suppression or vasculitis; 8. A history of severe allergic reactions either to indomethacin or to derivatives of anthralinic acid; 9. Hemoglobin < 10, hematocrit < 30, WBC < 4500/mm³. Classification: NA Meclofenamate sodium (300 mg): Number of participants: 49 Number of completers: 39 Age (median): 35 (range 16 to 67) Male (%): 90 Symptom duration: NA Disease duration (median): 8 (range 0.5 to 28) years HLA‐B27 positive (%): NA Indomethacin (150 mg): Number of participants: 49 Number of completers: 41 Age (median): 34 (range 20 to 70) Male (%): 83 Symptom duration: NA Disease duration (median): 7 (range 0.5 to 50) years HLA‐B27 positive (%): NA |
|
| Interventions | Meclofenamate sodium (300 mg) vs Indomethacin (150 mg) Co‐medication: Patients were requested to avoid taking any other analgesic or anti‐inflammatory drugs during the study. The physician could prescribe a muscle relaxant, if this was necessary. |
|
| Outcomes |
Extracted outcomes: 1. Pain on Likert scale (BL, percentage change from BL after treatment) (scale 0 to 4, higher is worse) Meclofenamate sodium 300 mg: 2.27 (N = 48), ‐53.3% (N = 48) Indomethacin 150 mg: 2.25 (N = 46), ‐58.7% (N = 46) (P = not significant between groups) 2. Withdrawals due to adverse events 3. Patient's global assessment of disease activity (BL, percentage change from BL after treatment) (scale 0 to 5, higher is worse) Meclofenamate sodium 300 mg: 2.58 (N = 48), ‐32.2% (N = 48) Indomethacin 150 mg: 2.57 (N = 46), ‐41.6% (N = 46) (P = not significant between groups) 4. Duration of morning stiffness (BL, percentage change from BL after treatment) (median in minutes, higher is worse) Meclofenamate sodium 300 mg: 25 (N = 48), ‐60.0% (N = 48) (P < 0.01 versus baseline) Indomethacin 150 mg: 30 (N = 46), ‐70.0% (N = 46) (P < 0.01 versus BL, P = not significant between groups) 5. Chest expansion (BL, percentage change from BL after treatment) (in cm, higher is better) Meclofenamate sodium 300 mg: 4.39 (N = 48), +22.1% (N = 48) (P < 0.01 versus BL) Indomethacin 150 mg: 4.13 (N = 46), +29.1% (N = 46) (P < 0.01 versus BL, P = not significant between groups) 6. Schober's test (BL, percentage change from BL after treatment) (in cm, higher is better) Meclofenamate sodium 300 mg: 2.23 (N = 48), +52.5% (N = 48) (P < 0.01 versus BL) Indomethacin 150 mg: 2.03 (N = 46), +51.2% (N = 46) (P < 0.01 versus BL, P = not significant between groups) 7. Number of any adverse events 8. Number of adverse events per organ system |
|
| Notes | Outcomes that were not included in the meta‐analysis, because no measure of variance (SD, SE or CI) was reported for these outcomes: pain on Likert scale, patient's global assessment of disease activity, duration of morning stiffness, chest expansion, Schober's test. Available results are described in this table. Funding source: Not reported. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "All patients in each participating center were randomly assigned to treatment with either…" Probably done, but no further information was provided on sequence generation. |
| Allocation concealment (selection bias) | Unclear risk | No information provided on allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "Following a single‐blind baseline period on placebo, patients received either meclofenamate sodium or indometacin for 8 weeks under parallel double‐blind conditions." Comment: Probably done, but no further information was provided on blinding of participants. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "double‐blind conditions" Probably done, but no further information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | "Patients were evaluated for efficacy if they received medication for more than one week and if premedication had been terminated as required in the protocol." "The rate of patients who withdrew from the study was similar in the 2 treatment groups." |
| Selective reporting (reporting bias) | Low risk | All pre‐specified outcomes according to the methods section, are reported in the results section. |
| Other bias | Low risk | No other bias detected. |
Franssen 1986.
| Methods |
Design: RCT Number of centres: NA Treatment duration: 12 weeks and 48 week extension Flare design: Yes Wash‐out period: Yes (2 weeks or until flare) Time point of assessments: BL, 2, 4, 6, 8, 10, 12 weeks |
|
| Participants |
Inclusion criteria: 1. Male; 2. Age 18 to 55; 3. Definite AS confirmed by two or more of the following symptoms: back pain, morning stiffness and progressive limitation of movement; 4. Flare‐up after wash‐out (defined as a worsening of the patient's condition in which pain and stiffness was an essential component requiring treatment). Exclusion criteria: 1. Active peptic ulcer or other significant internal or neurological disease; 2. Hypersensitivity or serious side effects while taking NSAIDs; 3. End‐stage and incapacitating disease (ARA functional class IV). Classification: NA Diflunisal (1000 mg): Number of participants: 19 Number of completers: 14 Phenylbutazone (400 mg): Number of participants: 19 Number of completers: 17 All participants: Age (mean): 37 (range 20 to 54) Male (%): 100 Symptom duration: NA Disease duration: NA HLA‐B27 positive (%): NA |
|
| Interventions | Diflunisal (1000 mg) vs Phenylbutazone (400 mg) Co‐medication: Patients received only one active compound. Any programme of physical therapy remained unchanged during the double‐blind period. No other drug intake was allowed during the study except 500 mg acetaminophen as a supplementary analgesic. |
|
| Outcomes |
Extracted outcomes: Twelve weeks results (included in the meta‐analysis)
Forty‐eight weeks results (not included in the meta‐analysis) 1. Pain on Likert scale (BL, post‐treatment after 48 weeks (± SD)) (scale 0 to 4, higher is worse) Diflunisal: 2.8 ± 0.6 (N = 19), 1.6 ± 0.9 (N = 14) (P < 0.01 vs BL) Phenylbutazone: 2.6 ± 0.7 (N = 19), 1.5 ± 1.2 (N = 17) (P < 0.01 vs BL) 2. Withdrawals due to adverse events (post‐treatment after 48 weeks) Diflunisal: n = 3 (total N = 19) Phenylbutazone: n = 3 (total N = 19) 3. Duration of morning stiffness (BL, post‐treatment after 48 weeks (± SD)) (in hours, higher is worse) Diflunisal: 2.1 ± 2.0 (N = 19), 1.4 ± 1.3 (N = 14) (P = not significant) Phenylbutazone: 2.7 ± 2.5 (N = 19), 2.0 ± 2.7 (N = 17) (P = not significant) 4. ESR (BL, post‐treatment after 48 weeks (± SD) (in mm/hr, higher is worse) Diflunisal: 31 ± 20 (N = 19), 20 ± 22 (N = 14) (P < 0.05 vs BL) Phenylbutazone: 19 ± 12 (N = 19), 17 ± 13 (N = 17) (P = not significant) |
|
| Notes | The outcomes chest expansion and Schober's test were also presented, but these data could not be used due to presentation in graphs from which the data could not be extracted. Funding source: Supported with a grant from Merck Sharp and Dohme/Chibret, Haarlem, The Netherlands. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "all patients were allocated at random to receive either…" Probably done, but no further information provided on sequence generation. |
| Allocation concealment (selection bias) | Unclear risk | "all patients were allocated at random to receive either…" Probably done, but no further information provided on allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Patients received only one active compound together with the corresponding placebo of the alternative drug." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "double blind"; "Clinical assessment was made weekly by the same investigator for each patient." Probably done, but no further information provided on blinding outcome assessor. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | A completers‐analysis is done, so possible attrition bias. |
| Selective reporting (reporting bias) | Low risk | All pre‐specified outcomes according to the methods section, are reported in the results section. |
| Other bias | Low risk | "The average patient compliance in the double‐blind period was 95±4% for DIF and 96±5% for PBZ..."; high compliance, no other bias detected. |
Good 1977.
| Methods |
Design: RCT Number of centres: NA Treatment duration: 6 weeks Flare design: Yes Wash‐out period: No Time point of assessments: BL, 3, 4, 6 weeks |
|
| Participants |
Inclusion criteria: 1. At least two Rome clinical criteria of the disease; 2. Exacerbation of disease, defined as a clear increase in spinal or sacro‐iliac pain and one or more of the following: muscle spasm in the back, decreased range of motion of some part of the spine, elevation of the ESR; 3. Abnormal or ankylosed sacroiliac joints by radiographic criteria. Exclusion criteria: 1. Age below 19 years; 2. Involvement of more than two peripheral joints not including the shoulders or hips; 3. Probability of pregnancy during the trial; 4. Hypersensitivity to the experimental drugs; 5. Other rheumatoid variants; 6. Positive rheumatoid factor; 7. Serious concomitant disease. Classification: Rome criteria Flurbiprofen (150 to 200 mg): Number of participants: 13 Number of completers: 9 Age: NA Male (%): 85 Symptom duration: NA Disease duration: NA HLA‐B27 positive (%): "HLA‐B27 antigen was tested in 8 patients and only one was negative" Indomethacin (75 to 100 mg): Number of participants: 13 Number of completers: 12 Age: NA Male (%): 85 Symptom duration: NA Disease duration: NA HLA‐B27 positive (%): "HLA‐B27 antigen was tested in 8 patients and only one was negative" |
|
| Interventions | Flurbiprofen (150 to 200 mg) vs Indomethacin (75 to 100 mg) Co‐medication: The use of any other analgesic or anti‐inflammatory drug was discouraged. |
|
| Outcomes |
Extracted outcomes: 1. Pain on Likert scale (BL, mean change after 6 weeks) (scale 0 to 4, higher is worse) Flurbiprofen 150 to 200 mg: 2.5 (N = 13), ‐0.7 (N = 9) (P = not significant versus BL) Indomethacin 75 to 100 mg: 2.6 (N = 13), ‐0.9 (N = 12) (P < 0.02 versus BL) 2. Withdrawals due to adverse events 3. Duration of morning stiffness (BL, mean change after 6 weeks) (in hours, higher is worse) Flurbiprofen 150 to 200 mg: 5.0 (N = 13), ‐4.2 (N = 9) (P = not significant versus BL) Indomethacin 75 to 100 mg: 4.2 (N = 13), ‐2.2 (N = 12) (P = not significant versus BL) 4. ESR (BL, mean change after 6 weeks) (in mm/hr, higher is worse) Flurbiprofen 150 to 200 mg: 32 (N = 13), +0.6 (N = 9) (P = not significant versus BL) Indomethacin 75 to 100 mg: 42 (N = 13), ‐1.5 (N = 12) (P = not significant versus BL) 5. Chest expansion (BL, mean change after 6 weeks) (in cm, higher is better) Flurbiprofen 150 to 200 mg: 3.9 (N = 13), +0.7 (N = 9) (P = not significant versus BL) Indomethacin 75 to 100 mg: 2.9 (N = 13), +0.8 (N = 12) (P < 0.05 versus BL) 6. Occiput‐to‐wall distance (BL, mean change after 6 weeks) (in cm, higher is worse) Flurbiprofen 150 to 200 mg: 5.9 (N = 13), ‐0.9 (N = 9) (P = not significant versus BL) Indomethacin 75 to 100 mg: 5.9 (N = 13), 0.3 (N = 12) (P = not significant versus BL) 7. Intermalleolar distance (BL, mean change after 6 weeks) (in cm, higher is better) Flurbiprofen 150 to 200 mg: 58.4 (N = 13), +7.1 (N = 9) (P = not significant versus BL) Indomethacin 75 to 100 mg: 50.4 (N = 13), +2.1 (N = 12) (P = not significant versus BL) 8. Schober's test (BL, mean change after 6 weeks) (in cm, higher is better) Flurbiprofen 150 to 200 mg: 12.2 (N = 13), +0.8 (N = 9) (P < 0.05 versus BL) Indomethacin 75 to 100 mg: 11.8 (N = 13), +0.5 (N = 12) (P = not significant versus BL) 9. Number of any adverse events 10. Number of adverse events per organ system |
|
| Notes | Outcomes that were not included in the meta‐analysis, because no measure of variance (SD, SE or CI) was reported for these outcomes: pain on Likert scale, duration of morning stiffness, ESR, chest expansion, occiput‐to‐wall distance, Schober's test. Available results are described in this table. Funding source: The Upjohn Company provided the grant to support this study. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Twenty‐six patients were randomly assigned to…" Probably done, but no further information was provided on sequence generation. |
| Allocation concealment (selection bias) | Unclear risk | No information was provided on allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "The drugs were available as identical‐looking capsules of 25mg indomethacin or 50mg flurbiprofen. The contents of the capsules were not known to the patient or the investigator." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "The contents of the capsules were not known to the patient or the investigator." |
| Incomplete outcome data (attrition bias) All outcomes | High risk | why some patients in the flurbiprofen‐group dropped out. Only completers‐analysis. Different number of dropouts between groups. |
| Selective reporting (reporting bias) | High risk | Same study is reported in two different papers, in which the provided information on adverse events differs. |
| Other bias | Low risk | No other bias detected. |
Heinrichs 1985.
| Methods |
Design: RCT Number of centres: NA Treatment duration: 3 weeks Flare design: No Wash‐out period: No Time point of assessments: BL, 1, 2, 3 weeks |
|
| Participants |
Inclusion criteria: Definite AS. Exclusion criteria: 1. Pregnancy; 2. Stomach or duodenal ulceration; 3. Other rheumatic disorders; 4. Patients who were not ambulant; 5. Anticoagulant therapy; 6. Hematologic changes; 7. Corticosteroid use in the last 6 weeks; 8. Hypersensitivity to tiaprofenacid, diclofenac or similar substances. Classification: New York criteria Tiaprofenacid (600 to 700 mg): Number of participants: 20 Number of completers: 19 Age (mean): 38.6 Male (%): NA Symptom duration: NA Disease duration (mean): 5 (range 0.1 to 13) years HLA‐B27 positive (%): NA Diclofenac (150 to 200 mg): Number of participants: 19 Number of completers: 19 Age (mean): 38.2 Male (%): NA Symptom duration: NA Disease duration (mean): 7 (range 0.1 to 10) years HLA‐B27 positive (%): NA |
|
| Interventions | Tiaprofenacid (600 to 700 mg) vs Diclofenac (150 to 200 mg) Co‐medication: All patients participated in some form of physiotherapy. |
|
| Outcomes |
Extracted outcomes: 1. Withdrawals due to adverse events 2. Occiput‐to‐wall distance (BL, post‐treatment after 3 weeks) (in cm, higher is worse) Tiaprofenacid 600 to 700 mg: 3.37 (N = 19), 2.47 (N = 19) Diclofenac 150 to 200 mg: 2.05 (N = 19), 0.68 (N = 19) 3. Schober's test (BL, post‐treatment after 3 weeks) (in cm, higher is better) Tiaprofenacid 600 to 700 mg: 1.89 (N = 19), 2.32 (N = 19) Diclofenac 150 to 200 mg: 2.52 (N = 19), 2.76 (N = 19) 4. Number of any adverse events |
|
| Notes | Outcomes that were not included in the meta‐analysis, because no measure of variance (SD, SE or CI) was reported for these outcomes: occiput‐to‐wall distance, Schober's test. Available results are described in this table. The outcomes pain on VAS, duration of morning stiffness and chest expansion were also presented, but these data could not be used due to presentation in graphs from which the data could not be extracted. Funding source: Not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Die Gruppenzuteilung erfolgte randomisiert." Probably done, but no further information provided on sequence generation. |
| Allocation concealment (selection bias) | Unclear risk | No information was provided on allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No information was provided on whether participants were blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | No information was provided on whether outcome assessor was blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | A completers analysis is performed, but only one participant dropped out due to an adverse event, so risk of bias is assessed as being low. |
| Selective reporting (reporting bias) | Low risk | All pre‐specified outcomes according to the methods section, are reported in the results section. |
| Other bias | Low risk | No other bias was detected. |
Jessop 1976.
| Methods |
Design: Cross‐over study Number of centres: 1 Treatment duration: each treatment period was 4 weeks Flare design: No Wash‐out period: No Time point of assessments: BL, 4, 8 weeks |
|
| Participants |
Inclusion criteria: AS for which patient was currently receiving treatment in an outpatient department. Exclusion criteria: 1. Renal, hepatic, or cardiac failure; 2. Severe dyspepsia or previous intolerance to phenylbutazone; 3. Sacro‐ileitis associated with ulcerative colitis, regional ileitis, Reiter's disease or psoriasis. Classification: Bennet and Wood 1968 Ketoprofen (200 mg) first: Number of participants: 15 Number of completers: 12 Age (mean): 46.0 (range 20 to 59) Male (%): 83 Symptom duration: NA Disease duration: 2 participants 1 to 5 years, and 10 participants > 5 years HLA‐B27 positive (%): NA Phenylbutazone (300 mg) first: Number of participants: 11 Number of completers: 8 Age (mean): 37.3 (range 28 to 54) Male (%): 88 Symptom duration: NA Disease duration: 1 participant 1 to 5 years, and 7 participants > 5 years HLA‐B27 positive (%): NA |
|
| Interventions | Ketoprofen (200 mg) first vs Phenylbutazone (300 mg) first Co‐medication: Only paracetamol tablets (500 mg) from a measured supply could be taken as a 'rescue' analgesic. Physiotherapy was allowed, provided it had been started at least 4 weeks prior to start of study and was allowed to continue unchanged throughout the trial period. |
|
| Outcomes |
Extracted outcomes: 1. Pain on Likert scale 2. Patient's global assessment of disease activity 3. Duration of morning stiffness 4. Severity of morning stiffness 5. Chest expansion (BL, post‐treatment after 4 weeks) (in cm, higher is better) Ketoprofen 200 mg: 4.2 (N = 9), 3.9 (N = 9) Phenylbutazone 300 mg: 4.5 (N = 7), 5.2 (N = 7) (P = not significant) 6. Tragus‐to‐wall distance (BL, post‐treatment after 4 weeks) (in cm, higher is worse) Ketoprofen 200 mg: 20.0 (N = 12), 19.6 (N = 12) Phenylbutazone 300 mg: 20.0 (N = 8), 20.1 (N = 8) (P = not significant) 7. Occiput‐to‐wall distance (BL, post‐treatment after 4 weeks) (in cm, higher is worse) Ketoprofen 200 mg: 8.0 (N = 12), 8.3 (N = 12) Phenylbutazone 300 mg: 8.8 (N = 8), 8.6 (N = 8) (P = not significant) 8. Intermalleolar distance (BL, post‐treatment after 4 weeks) (in cm, higher is better) Ketoprofen 200 mg: 85.2 (N = 12), 85.9 (N = 12) Phenylbutazone 300 mg: 102.0 (N = 8), 105.0 (N = 8) (P = not significant) 9. Schober's test (BL, post‐treatment after 4 weeks) (in cm, higher is better) Ketoprofen 200 mg: 2.0 (N = 12), 1.9 (N = 12) Phenylbutazone 300 mg: 2.4 (N = 6), 2.4 (N = 6) (P = not significant) |
|
| Notes | Outcomes that were not included in the meta‐analysis, because no measure of variance (SD, SE or CI) was reported for these outcomes: chest expansion, tragus‐to‐wall distance, occiput‐to‐wall distance, intermalleolar distance, Schober's test. Available results are described in this table. For the outcomes pain on Likert scale, patient global assessment of disease activity, duration of morning stiffness and severity of morning stiffness individual patient data that were presented in the paper were combined for the meta‐analysis to a mean and SD. For the outcome tragus‐to‐wall distance, "straight tragus‐to‐wall distance" was extracted, and not "turning to right" or "turning to left" (which was also presented in the paper). For the outcome occiput‐to‐wall distance, "greatest displacement tolerated" was extracted, and not "when pain first appears" (which was also presented in the paper. Presented demographics are those of the participants that completed the trial. Only results of first part of cross‐over trial are used in analysis. Funding source: May & Baker Ltd. provided the drugs and record cards used in this trial. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "The patients were randomly allocated..." Probably done, but no further information provided on sequence generation. |
| Allocation concealment (selection bias) | Unclear risk | No information provided on allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "in identical capsules". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "double‐blind". Probably done, but no further information provided on blinding outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Dropouts (6) were excluded from all analysis, no information provided on whether dropouts differed from analysed participants. Although number of dropouts per group was equal (2x3), the reason for dropout differed between treatment groups, possibly introducing bias. |
| Selective reporting (reporting bias) | Low risk | All pre‐specified outcomes according to the methods section, are reported in the results section. |
| Other bias | Unclear risk | Crossover design, no information provided on possible carry‐over effect. BL inequality cannot be assessed because demographic information was only provided on completers. |
Khan 1985.
| Methods |
Design: RCT Number of centres: 20 Treatment duration: 13 weeks Flare design: Yes Wash‐out period: Yes (2 days to 2 weeks) Time point of assessments: BL, 1, 2, 3, 4, 6, 10, 13 weeks |
|
| Participants |
Inclusion criteria: 1. Out‐patients aged 18 to 65; 2. Confirmed diagnosis of AS for which they had required therapy for at least 3 months. Exclusion criteria: Patients with coexisting rheumatic disease or significant medical problems. Classification: ARA function class I/II/III Diclofenac (125 mg): Number of participants: 132 (safety analysis)/118 (efficacy analysis) Number of completers: 93 Age (mean): 39 (range 19 to 64) Male (%): 86 Symptom duration: NA Disease duration: NA HLA‐B27 positive (%): NA Indomethacin (125 mg): Number of participants: 130 (safety analysis) / 106 (efficacy analysis) Number of completers: 81 Age (mean): 38 (range 19 to 64) Male (%): 80 Symptom duration: NA Disease duration: NA HLA‐B27 positive (%): NA |
|
| Interventions | Diclofenac (125 mg) vs Indomethacin (125 mg) Co‐medication: NA |
|
| Outcomes |
Extracted outcomes: 1. Pain on Likert scale (BL, post‐treatment after 13 weeks) (scale 0 to 4, higher is worse) Diclofenac 125 mg: 2.38 (N = 118), 0.93 (N = 93) (P < 0.001 versus BL) Indomethacin 125 mg: 2.45 (N = 106), 0.93 (N = 81) (P < 0.001 versus BL, P = not significant between groups) 2. Withdrawals due to adverse events 3. Patient's global assessment of disease activity (BL, post‐treatment after 13 weeks) (scale 0 to 10, higher is worse) Diclofenac 125 mg: 7.00 (N = 118), 3.56 (N = 93) (P < 0.001 versus BL) Indomethacin 125 mg: 7.01 (N = 106), 3.22 (N = 81) (P < 0.001 versus BL, P = not significant between groups) 4. Duration of morning stiffness (BL, post‐treatment after 13 weeks) (in hours, higher is worse) Diclofenac 125 mg: 6.38 (N = 118), 1.15 (N = 93) (P < 0.001 versus BL) Indomethacin 125 mg: 4.76 (N = 106), 0.99 (N = 81) (P < 0.001 versus BL, P = not significant between groups) 5. Chest expansion (BL, post‐treatment after 13 weeks) (in cm, higher is better) Diclofenac 125 mg: 2.57 (N = 118), 3.95 (N = 93) (P < 0.001 versus BL) Indomethacin 125 mg: 2.72 (N = 106), 4.28 (N = 81) (P < 0.001 versus BL, P = not significant between groups) 6. Occiput‐to‐wall distance (BL, post‐treatment after 13 weeks) (in cm, higher is worse) Diclofenac 125 mg: 6.19 (N = 118), 4.08 (N = 93) (P < 0.001 versus BL) Indomethacin 125 mg: 5.60 (N = 106), 4.06 (N = 81) (P < 0.001 versus BL, P = not significant between groups) 7. Schober's test (BL, post‐treatment after 13 weeks) (in cm, higher is better) Diclofenac 125 mg: 12.62 (N = 118), 13.70 (N = 93) (P < 0.001 versus BL) Indomethacin 125 mg: 12.53 (N = 106), 13.92 (N = 81) (P < 0.001 versus BL, P = not significant between groups) 8. Number of any adverse events 9. Number of adverse events per organ system |
|
| Notes | Outcomes that were not included in the meta‐analysis, because no measure of variance (SD, SE or CI) was reported for these outcomes: pain on Likert scale, patient's global assessment of disease activity, duration of morning stiffness, chest expansion, occiput‐to‐wall distance, Schober's test. Available results are described in this table. The results of the extension phase were not reported for this trial. Funding source: Supported by a grant from CIBA‐GEIGY Corporation. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Patients were randomized at each study center, using a blocking factor of one." Comment: Probably done, but no further information provided on sequence generation. |
| Allocation concealment (selection bias) | Unclear risk | No information was provided on allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "One group was instructed to take one 25 mg enteric coated diclofenac tablet and one placebo capsule TID, and the other was instructed to take one 25 mg indomethacin capsule and one placebo tablet TID." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "double‐blind". Probably done, but no further information provided on blinding of outcome assessor. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | "Data were analyzed employing the concept of "terminal visits"… This method ensured that each acceptable patient was represented at least once in the efficacy analysis." Similar dropout rates between groups. Analysis method with low risk of attrition bias. |
| Selective reporting (reporting bias) | Low risk | All pre‐specified outcomes according to the methods section, are reported in the results section. |
| Other bias | Low risk | No other bias detected. |
Lehtinen 1984.
| Methods |
Design: Cross‐over study Number of centres: 1 Treatment duration: Each treatment period was 1 week Flare design: No Wash‐out period: Yes (4 days; paracetamol or aspirin, or both, were allowed as rescue analgesic during this period) Time point of assessments: BL, 1, 2 weeks |
|
| Participants |
Inclusion criteria: 1. In‐patients with AS; 2. Sufficient disease activity to warrant anti‐inflammatory analgesic therapy. Exclusion criteria: 1. Patients with active peptic ulcer, asthmatic symptoms, hepatic or renal dysfunction; 2. Sensitivity to indomethacin or aspirin; 3. Pregnant women. Classification: New York criteria All participants: Number of participants: 30 Number of completers: 30 Age (median): 37 (range 27 to 56) Male (%): 83 Symptom duration: NA Disease duration (median): 12 years (range 5 months to 26 years) HLA‐B27 positive (%): NA |
|
| Interventions | Indomethacin (50 mg tablets) first vs Indomethacin (25 mg capsules) first Co‐medication: Rescue analgesics were permitted during the treatment periods. |
|
| Outcomes |
Extracted outcomes: 1. Withdrawals due to adverse events (after 1 week) Indomethacin 50 mg first: n = 0 Indomethacin 25 mg first: n = 3 2. Number of any adverse events (after 1 week) Indomethacin 50 mg first: n = 8 Indomethacin 25 mg first: n = 9 3. Number of adverse events per organ system (after 1 week) Indomethacin 50 mg first: gastro‐intestinal n = 5, neurological n = 8 Indomethacin 25 mg first: gastro‐intestinal n = 9, cardiovascular n = 9 |
|
| Notes | Results are not included in the meta‐analysis, because the number of patients in each treatment group was not available. Available results are described in this table. Only data on adverse events could be extracted, because other data was pooled from the first and second treatment period. Funding source: Not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "…the patients were randomized into two groups." Probably done, but no further information provided on sequence generation. |
| Allocation concealment (selection bias) | Unclear risk | No information provided on allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Owing to the difference in appearance of the preparations, a double‐dummy technique was used." |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | No information provided on whether outcome assessor was blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No clear information provided on the number of dropouts. However, there seem to be more withdrawals during treatment with indomethacin capsules in comparison to the tablets. |
| Selective reporting (reporting bias) | Low risk | All pre‐specified outcomes according to the methods section, are reported in the results section. |
| Other bias | Unclear risk | Crossover design, authors did not find any evidence for a cross‐over effect. For rescue analgesic in the washout period also aspirin was allowed; no indication of the height of the dose and if this might have influenced the results. |
Lomen 1986 I.
| Methods |
Design: RCT Number of centres: 8 Treatment duration: 26 weeks Flare design: No Wash‐out period: Yes (at least 48 hours) Time point of assessments: BL, 2, 4, 6, 10, 14, 18, 22, 26 weeks |
|
| Participants |
Inclusion criteria: 1. Age 18 to 60; 2. Definitive diagnosis of AS for which clinical and radiographic criteria include: a) pain and stiffness in the lumbar region for more than 3 months, b) major limitation of motion in the lumbar spine in all three planes, c) pain and stiffness in the thoracic region for more than 3 months, d) limitation of chest expansion, e) night pain, f) history or evidence of iritis or its sequelae, and g) bilateral sacroiliac disease on radiographic examination. Exclusion criteria: A serious adverse event during the first week of treatment. Classification: NA Flurbiprofen (150 to 300 mg): Number of participants: 30 Number of completers: 25 Age: NA Male (%): 87 Symptom duration: NA Disease duration: NA HLA‐B27 positive (%): NA Indomethacin (75 to 150 mg): Number of participants: 27 Number of completers: 22 Age: NA Male (%): 89 Symptom duration: NA Disease duration: NA HLA‐B27 positive (%): NA |
|
| Interventions | Flurbiprofen (150 to 300 mg) vs Indomethacin (75 to 150 mg) Co‐medication: All patients were given a daily diary card to record consumption of any non‐study drugs. |
|
| Outcomes |
Extracted outcomes: 1. Pain on Likert scale (mean change after 26 weeks) (scale 0 to 6, higher is worse) Flurbiprofen 150 to 300 mg: ‐1.9 (N = 24) Indomethacin 75 to 150 mg: ‐1.8 (N = 21) 2. Withdrawals due to adverse events 3. Duration of morning stiffness (mean change after 26 weeks) (in hours, higher is worse) Flurbiprofen 150 to 300 mg: ‐6.21 (N = 25) Indomethacin 75 to 150 mg: ‐5.73 (N = 20) 4. Chest expansion (mean change after 26 weeks) (in cm, higher is better) Flurbiprofen 150 to 300 mg: +0.86 (N = 25) Indomethacin 75 to 150 mg: +1.40 (N = 21) 5. Occiput‐to‐wall distance (mean change after 26 weeks) (in cm, higher is worse) Flurbiprofen 150 to 300 mg: ‐2.2 (N = 19) Indomethacin 75 to 150 mg: ‐3.1 (N = 15) 6. Intermalleolar distance (mean change after 26 weeks) (in cm, higher is better) Flurbiprofen 150 to 300 mg: ‐1.8 (N = 25) Indomethacin 75 to 150 mg: ‐5.4 (N = 21) 7. Schober's test (mean change after 26 weeks) (in cm, higher is better) Flurbiprofen 150 to 300 mg: +0.9 (N = 25) Indomethacin 75 to 150 mg: +0.8 (N = 21) 8. Number of any adverse events 9. Number of adverse events per organ system |
|
| Notes | Outcomes that were not included in the meta‐analysis, because no measure of variance (SD, SE or CI) was reported for these outcomes: pain on Likert scale, duration of morning stiffness, chest expansion, occiput‐to‐wall distance, intermalleolar distance, Schober's test. Available results are described in this table. BL results were not available for any of the outcomes. Funding source: Not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Assignment to one of the two treatment groups was in accordance with a standardized randomization scheme." |
| Allocation concealment (selection bias) | Low risk | "Treatment was double‐blind, with patients receiving…in identically appearing bottles with attached decoding labels." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Treatment was double‐blind, with patients receiving…in identically appearing bottles with attached decoding labels." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "double‐blind", Comment: probably done, but no further information provided on blinding of outcome assessor. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | "Patients were withdrawn before completing the first week if a serious adverse event occurred." "If symptoms did not subside after this time, the patient was withdrawn." "Patients who had normal values at baseline on any measure were excluded from the analyses." ; Comment: Several points in time at which the authors withdrew patients (how many?) for reasons with a high risk of attrition bias. Also unclear why some outcomes had less number of patients than those that completed the study. |
| Selective reporting (reporting bias) | Unclear risk | Pain during the day was assessed (see methods section), but not reported (see results section). All other outcomes that were assessed, were reported. |
| Other bias | Low risk | No other bias detected. |
Lomen 1986 P.
| Methods |
Design: RCT Number of centres: 11 Treatment duration: 26 weeks Flare design: No Wash‐out period: Yes (at least 48 hours) Time point of assessments: BL, 2, 4, 6, 10, 14, 18, 22, 26 weeks |
|
| Participants |
Inclusion criteria: 1. Age 18 to 60; 2. Definitive diagnosis of AS for which clinical and radiographic criteria include: a) pain and stiffness in the lumbar region for more than 3 months, b) major limitation of motion in the lumbar spine in all three planes, c) pain and stiffness in the thoracic region for more than 3 months, d) limitation of chest expansion, e) night pain, f) history or evidence of iritis or its sequelae, and g) bilateral sacroiliac disease on radiographic examination. Exclusion criteria: A serious adverse event or lack of efficacy during the first week of treatment. Classification: NA Flurbiprofen (200 to 300 mg): Number of participants: 43 Number of completers: 29 Age: NA Male (%): 95 Symptom duration: NA Disease duration: NA HLA‐B27 positive (%): NA Phenylbutazone (300 to 500 mg): Number of participants: 42 Number of completers: 32 Age: NA Male (%): 86 Symptom duration: NA Disease duration: NA HLA‐B27 positive (%): NA |
|
| Interventions | Flurbiprofen (200 to 300 mg) vs Phenylbutazone (300 to 500 mg) Co‐medication: All patients were given a daily diary card to record consumption of any non‐study drugs. |
|
| Outcomes |
Extracted outcomes: 1. Pain on Likert scale (mean change after 26 weeks) (scale 0 to 6, higher is worse) Flurbiprofen 200 to 300 mg: ‐1.5 (N = 27) Indomethacin 300 to 500 mg: ‐1.8 (N = 31) 2. Withdrawals due to adverse events 3. Duration of morning stiffness (mean change after 26 weeks) (in hours, higher is worse) Flurbiprofen 200 to 300 mg: ‐2.31 (N = 27) Indomethacin 300 to 500 mg: ‐3.12 (N = 31) 4. Chest expansion (mean change after 26 weeks) (in cm, higher is better) Flurbiprofen 200 to 300 mg: +0.80 (N = 26) Indomethacin 300 to 500 mg: +1.06 (N = 31) 5. Occiput‐to‐wall distance (mean change after 26 weeks) (in cm, higher is worse) Flurbiprofen 200 to 300 mg: ‐1.6 (N = 17) Indomethacin 300 to 500 mg: ‐0.6 (N = 14) 6. Intermalleolar distance (mean change after 26 weeks) (in cm, higher is better) Flurbiprofen 200 to 300 mg: ‐6.1 (N = 28) Indomethacin 300 to 500 mg: ‐10.0 (N = 31) 7. Schober's test (mean change after 26 weeks) (in cm, higher is better) Flurbiprofen 200 to 300 mg: +1.7 (N = 28) Indomethacin 300 to 500 mg: +1.0 (N = 31) 8. Number of any adverse events 9. Number of adverse events per organ system |
|
| Notes | Outcomes that were not included in the meta‐analysis, because no measure of variance (SD, SE or CI) was reported for these outcomes: pain on Likert scale, duration of morning stiffness, chest expansion, occiput‐to‐wall distance, intermalleolar distance, Schober's test. Available results are described in this table. BL results were not available for any of the outcomes. Funding source: Not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Assignment to flurbiprofen or phenylbutazone treatment groups was in accordance with a standardized randomization scheme..." |
| Allocation concealment (selection bias) | Unclear risk | No information provided on allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "treatment was double‐blind." Probably done, but no further information provided on blinding of participants. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "treatment was double‐blind." Probably done, but no further information provided on blinding of outcome assessor. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | "The study was stopped before completion of the first week if serious adverse reactions occurred, or after completion of the first week if a lack of efficacy could be definitively determined." "If, after two weeks of treatment with the low‐dose escalation package or after one week of treatment with the high‐dose escalation package, symptoms did not subside, the patient was withdrawn from the study." "Patients with normal values at baseline on any measure were excluded from the analysis of that measure." Several points in time at which the authors withdrew patients (how many?) for reasons with a high risk of attrition bias. Also unclear why some outcomes had less number of patients than those that completed the study. |
| Selective reporting (reporting bias) | Low risk | All pre‐specified outcomes according to the methods section, are reported in the results section. |
| Other bias | Low risk | No other bias detected. |
Mena 1977.
| Methods |
Design: RCT Number of centres: NA Treatment duration: 6 weeks Flare design: Yes Wash‐out period: No Time point of assessments: BL, 2, 4, 6 weeks |
|
| Participants |
Inclusion criteria: 1. At least two Rome clinical criteria of the disease; 2. Abnormal or ankylosed sacroiliac joints by radiographic criteria; 3. Suffering an exacerbation of their disease, defined as a clear increase in spinal or sacroiliac pain and one or more of the following: a) muscle spasm in the back, b) decreased range of motion of some part of the spine, c) elevation of the ESR. Exclusion criteria: 1. Age below 19 years; 2. Involvement of more than two peripheral joints not including the shoulders or hips; 3. Probability of pregnancy during the trial; 4. Hypersensitivity to the experimental drugs; 5. Other rheumatoid variants, positive rheumatoid factor or serious concomitant diseases. Classification: Rome criteria Flurbiprofen (150 to 200 mg): Number of participants: 12 Number of completers: 9 Age: NA Male (%): 75 Symptom duration: NA Disease duration: NA HLA‐B27 positive (%): "HLA‐B27 antigen was tested in 8 patients and only one was negative" Phenylbutazone (300 to 400 mg): Number of participants: 15 Number of completers: 13 Age: NA Male (%): 80 Symptom duration: NA Disease duration: NA HLA‐B27 positive (%): "HLA‐B27 antigen was tested in 8 patients and only one was negative" |
|
| Interventions | Flurbiprofen (150 to 200 mg) vs Phenylbutazone (300 to 400 mg) Co‐medication: Co‐medication was allowed, but the use of any other analgesia or anti‐inflammatory drug was discouraged. |
|
| Outcomes |
Extracted outcomes: 1. Pain on Likert scale (BL, mean change after 6 weeks) (scale 0 to 4, higher is worse) Flurbiprofen 150 to 200 mg: 2.3 (N = 12), ‐0.8 (N = 9) (P < 0.05 versus BL) Phenylbutazone 300 to 400 mg: 1.9 (N = 15), ‐0.7 (N = 13) (P < 0.05 versus BL) 2. Withdrawals due to adverse events 3. Duration of morning stiffness (BL, mean change after 6 weeks) (in hours, higher is worse) Flurbiprofen 150 to 200 mg: 4.6 (N = 12), ‐4.3 (N = 9) (P = not significant versus BL) Phenylbutazone 300 to 400 mg: 1.2 (N = 15), +0.5 (N = 13) (P = not significant versus BL) 4. ESR (BL, mean change after 6 weeks) (in mm/hr, higher is worse) Flurbiprofen 150 to 200 mg: 24.0 (N = 12), +5.6 (N = 9) (P = not significant versus BL) Phenylbutazone 300 to 400 mg: 28.0 (N = 15), ‐6.7 (N = 13) (P < 0.05 versus BL) 5. Chest expansion (BL, mean change after 6 weeks) (in cm, higher is better) Flurbiprofen 150 to 200 mg: 2.5 (N = 12), +0.5 (N = 9) (P < 0.05 versus BL) Phenylbutazone 300 to 400 mg: 3.2 (N = 15), +0.1 (N = 13) (P = not significant versus BL) 6. Occiput‐to‐wall distance (BL, mean change after 6 weeks) (in cm, higher is worse) Flurbiprofen 150 to 200 mg: 7.6 (N = 12), ‐1.1 (N = 9) (P < 0.05 versus BL) Phenylbutazone 300 to 400 mg: 6.6 (N = 15), ‐1.4 (N = 13) (P < 0.02 versus BL) 7. Schober's test (BL, mean change after 6 weeks) (in cm, higher is better) Flurbiprofen 150 to 200 mg: 12.3 (N = 12), +0.0 (N = 9) (P = not significant versus BL) Phenylbutazone 300 to 400 mg: 12.6 (N = 15), +0.5 (N = 13) (P < 0.05 versus BL) 8. Number of any adverse events 9. Number of adverse events per organ system |
|
| Notes | Outcomes that were not included in the meta‐analysis, because no measure of variance (SD, SE or CI) was reported for these outcomes: pain on Likert scale, duration of morning stiffness, ESR, occiput‐to‐wall distance, Schober's test. Available results are described in this table. Funding source: The Upjohn Company provided a grant to support this study. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Twelve patients were randomly assigned to a flurbiprofen group and 15 to a phenylbutazone group." No information was provided on sequence generation. |
| Allocation concealment (selection bias) | Unclear risk | No information provided on allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "The drugs were available in a blister package. Each blister contained one 50 mg tablet of flurbiprofen or one 100 mg tablet of phenylbutazone and a placebo tablet identical to the other drug." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "The contents of the tablets were not known to the patient or the investigator." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition is equal and low (< 25%) in both groups. |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the methods were reported. |
| Other bias | Low risk | No other bias was detected. |
Muller‐Fassbender 1985.
| Methods |
Design: Cross‐over study Number of centres: NA Treatment duration: each treatment period was 6 weeks Flare design: No Wash‐out period: No Time point of assessments: BL, 1, 2, 3, 4, 5, 6 weeks |
|
| Participants |
Inclusion criteria: None reported. Exclusion criteria: 1. Severe damage to the liver parenchyma or decreased kidney functioning; 2. History of peptic ulcer; 3. Pregnancy. Classification: NA Ketoprofen (2 times 150 mg): Number of participants: 20 Number of completers: NA Ketoprofen (3 times 100 mg): Number of participants: 19 Number of completers: NA All participants: Age (mean): 42.7 Male (%): 95 Symptom duration: NA Disease duration: NA HLA‐B27 positive (%): NA |
|
| Interventions | Ketoprofen (2 times 150 mg) vs Ketoprofen (3 times 100 mg) Co‐medication: No other anti‐inflammatory drugs were allowed. |
|
| Outcomes | Extracted outcomes: None | |
| Notes | The study was not included in the meta‐analysis, because no outcomes could be extracted. The outcomes pain on Likert scale, chest expansion and Schober's test were presented, but these data could not be used due to presentation in graphs from which the data could not be extracted. The authors state that they only used and reported the data from the first part of the cross‐over study, because they could not exclude a cross‐over effect in the second part of the cross‐over. Funding source: Not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information is provided on sequence generation. |
| Allocation concealment (selection bias) | Unclear risk | No information provided on allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Als behandlung A erhielten die Patienten morgens und abends je 1 kapsel mit 150mg Ketoprofen und mittags 1 Placebokapsel…" Patients were blinded from their allocated treatment and received a placebo capsule. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "In a randomized, double‐blind cross‐over study…" No further information is provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Outcome data from the first part of the cross‐over study was available from all participants. |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the methods are reported. |
| Other bias | Unclear risk | Unable to detect other causes of bias due to the way the data is presented. |
Myklebust 1986.
| Methods |
Design: RCT Number of centres: 9 Treatment duration: 12 weeks Flare design: No Wash‐out period: Yes (1 week) Time point of assessments: BL, 4, 8, 12 weeks |
|
| Participants |
Inclusion criteria: 1. Between 18 and 80 years of age, with Bechterew syndrome; 2. Disease duration of more than six months. Exclusion criteria: None reported. Classification: NA Naproxen (1000 mg): Number of participants: 21 Number of completers: NA Age (mean (SE)): 41.5 (2.3) Male (%): 57 Symptom duration: NA Disease duration: NA HLA‐B27 positive (%): NA Piroxicam (20 mg): Number of participants: 16 Number of completers: NA Age (mean (SE)): 41.7 (4.0) Male (%): 63 Symptom duration: NA Disease duration: NA HLA‐B27 positive (%): NA |
|
| Interventions | Naproxen (1000 mg) vs Piroxicam (20 mg) Co‐medication: Antimalarial drugs, penicillamines or gold were allowed if treatment had lasted more than 6 months, as well as low dose corticosteroids. |
|
| Outcomes |
Extracted outcomes: 1. Pain on VAS 2. Duration of morning stiffness (BL (median ± IQR), post‐treatment after 12 weeks (median ± IQR)) (in minutes, higher is worse) Naproxen 1000 mg: 210 ± 120 to 300 (N = 19), 90 ± 30 to 150 (N = 19) Piroxicam 20 mg: 120 ± 60 to 180 (N = 16), 60 ± 15 to 172.5 (N = 16) 3. Chest expansion (BL (± SE), percentage change after 12 weeks (± SE)) (in cm, higher is better) Naproxen 1000 mg: 4.9 ± 0.6 (N = 19), +28.8% ± 8.9% (N = 19) Piroxicam 20 mg: 4.1 ± 0.4 (N = 16), +22.2% ± 7.9% (N = 16) 4. Schober's test (BL (± SE)), percentage change after 12 weeks (± SE)) (in cm, higher is better) Naproxen 1000 mg: 3.0 ± 0.2 (N = 19), +13.1% ± 8.9% (N = 19) Piroxicam 20 mg: 2.6 ± 0.2 (N = 16), +59.2% ± 34.7% (N = 16) |
|
| Notes | Outcomes that were not included in the meta‐analysis, because they could not be extracted due to the presentation of these outcomes: duration of morning stiffness, chest expansion, Schober's test. Available results are described in this table. Adverse events were not reported separately for patients with rheumatoid arthritis and AS, and could therefore not be used. Funding source: Not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information was provided on sequence generation. |
| Allocation concealment (selection bias) | Unclear risk | No information was provided on allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | A double dummy technique was used for adequate blinding of participants. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No further information provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Overall dropout rates are low (n = 12, 11%) and mainly concerned participants with RA. |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the methods are reported. |
| Other bias | Unclear risk | BL characteristics imbalance cannot be determined. No information is provided on the statistical methods that were used. |
Nahir 1980.
| Methods |
Design: RCT Number of centres: 1 Treatment duration: 4 weeks Flare design: No Wash‐out period: Yes (7 days no anti‐inflammatory/analgesic medication) Time point of assessments: Start of wash‐out, BL, 7, 14, 28 days |
|
| Participants |
Inclusion criteria: 1. Radiographic evidence of sacroiliitis and clinically active disease; 2. Demonstrated spinal pain; 3. Decreased range of motion of some part of the spine; 4. Increased ESR. Exclusion criteria: 1. Hepatic, renal, or gastric disease; 2. Previous intolerance to indomethacin. Classification: NA Diclofenac (150 mg): Number of participants: 31 Number of completers: 30 Age (mean): 37 (range 26 to 58) Male (%): 97 Symptom duration: NA Disease duration: 32% 1 to 5 years, and 68% > 5 years HLA‐B27 positive (%): 77 Sulindac (600 mg): Number of participants: 31 Number of completers: 31 Age (mean): 37 (range 20 to 57) Male (%): 97 Symptom duration: NA Disease duration: 42% 1‐5 years, and 58% > 5 years HLA‐B27 positive (%): 90 |
|
| Interventions | Diclofenac (150 mg) vs Sulindac (600 mg) Co‐medication: Not reported |
|
| Outcomes |
Extracted outcomes:
|
|
| Notes | Funding source: Not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "…the patients were randomly assigned to diclofenac … or sulindac…" No information is provided on sequence generation. |
| Allocation concealment (selection bias) | Unclear risk | No information provided on allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "…the patients were randomly assigned to diclofenac 50 mg thrice daily plus sulindac placebo twice daily or sulindac 200 mg thrice daily plus diclofenac placebo thrice daily." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "A double‐blind between‐patient comparison…" No further information is provided on the blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Outcome data is available for > 95% of the study participants. |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the methods are reported. |
| Other bias | Low risk | No other bias was detected. |
Nissilä 1978a.
| Methods |
Design: RCT Number of centres: 1 Treatment duration: 3 weeks Flare design: No Wash‐out period: Yes (3 to 4 days) Time point of assessments: BL, 7, 14, 21 days |
|
| Participants |
Inclusion criteria: Definite diagnosis of AS. Exclusion criteria: 1. Previously known renal, liver or gastrointestinal disorders; 2. Intolerance to indomethacin. Classification: NA Proquazone (900 mg): Number of participants: 16 Number of completers: NA Age: NA Male (%): 88 Symptom duration: NA Disease duration: NA HLA‐B27 positive (%): NA Proquazone (900 mg): Number of participants: 14 Number of completers: NA Age: NA Male (%): 100 Symptom duration: NA Disease duration: NA HLA‐B27 positive (%): NA |
|
| Interventions | Proquazone (900 mg) vs Indomethacin (75 mg) Co‐medication: "An additional fourth capsule was taken 55 times (average 3.4 capsules/patient) in the proquazone group and 42 times (average 3.0 capsules/patient) in the indomethacin group." |
|
| Outcomes |
Extracted outcomes:
|
|
| Notes | Two separate studies (Nissilä 1978a; Nissilä 1978b) were reported in one paper. Funding source: Not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information is provided on sequence generation. |
| Allocation concealment (selection bias) | Unclear risk | No information provided on allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "The capsules used were identical in appearance." Study participants were adequately blinded to their allocated treatment. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "two separate 3‐week clinical, double‐blind, randomized studies…" No further information is provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | "A total of 56 patients out of 60 completed the study." Dropouts were low in both groups of both studies and equally distributed among all groups. |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the methods are reported. |
| Other bias | Unclear risk | "There was no significant difference between the groups regarding age, and duration of severity of the disease." No BL characteristics reported in the article, so BL characteristics imbalance cannot be determined other than this. |
Nissilä 1978b.
| Methods |
Design: RCT Number of centres: 1 Treatment duration: 3 weeks Flare design: No Wash‐out period: Yes (3 to 4 days) Time point of assessments: BL, 7, 14, 21 days |
|
| Participants |
Inclusion criteria: Definite diagnosis of AS. Exclusion criteria: 1. Previously known renal, liver or gastrointestinal disorders; 2. Intolerance to indomethacin. Classification: NA Proquazone (900 mg): Number of participants: 15 Number of completers: NA Age: NA Male (%): 93 Symptom duration: NA Disease duration: NA HLA‐B27 positive (%): NA Proquazone (900 mg): Number of participants: 15 Number of completers: NA Age: NA Male (%): 93 Symptom duration: NA Disease duration: NA HLA‐B27 positive (%): NA |
|
| Interventions | Proquazone (900 mg) vs Indomethacin (75 mg) Co‐medication: "An additional fourth capsule was taken 65 times (average 4.3 capsules/patient) in the proquazone group and 47 times (average 3.1 capsules/patient) in the indomethacin group." |
|
| Outcomes |
Extracted outcomes:
|
|
| Notes | Two separate studies (Nissilä 1978a; Nissilä 1978b) were reported in one paper. Funding source: Not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information is provided on sequence generation. |
| Allocation concealment (selection bias) | Unclear risk | No information provided on allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "The capsules used were identical in appearance." Study participants were adequately blinded to their allocated treatment. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "two separate 3‐week clinical, double‐blind, randomized studies…" No further information is provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | "A total of 56 patients out of 60 completed the study." Dropouts were low in both groups of both studies and equally distributed among all groups. |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the methods are reported. |
| Other bias | Unclear risk | "There was no significant difference between the groups regarding age, and duration of severity of the disease." No BL characteristics reported in the article, so BL characteristics imbalance cannot be determined other than this. |
Palferman 1991.
| Methods |
Design: RCT Number of centres: 2 Treatment duration: 3 months Flare design: No Wash‐out period: Yes (with Paracetamol as escape) Time point of assessments: BL, 1, 2, 3 months |
|
| Participants |
Inclusion criteria: 1. Over sixteen years of age; 2. Satisfied the Rome criteria for AS Exclusion criteria: 1. Pregnant; 2. Receiving concomitant treatment with hydantoins or sulphonamides; 3. Receiving prednisolone in excess of 7.5 mg daily; 4. History of indomethacin intolerance; 5. Renal or hepatic impairment. Classification: Rome criteria Nabumetone (2000 mg): Number of participants: 23 Number of completers: 14 Age (mean): 40 Male (%): 87 Symptom duration: NA Disease duration (mean): 13.7 years HLA‐B27 positive (%): NA Indomethacin (150 mg): Number of participants: 19 Number of completers: 10 Age (mean): 40 Male (%): 58 Symptom duration: NA Disease duration (mean): 14.64 years HLA‐B27 positive (%): NA |
|
| Interventions | Nabumetone (2000 mg) vs Indomethacin (150 mg) Co‐medication: Not reported |
|
| Outcomes |
Extracted outcomes:
|
|
| Notes | Funding source: Not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "…they were randomised to one of the two groups..." No information was provided on sequence generation. |
| Allocation concealment (selection bias) | Unclear risk | No information was provided on allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | "…to one of the two groups and treatment started with either indomethacin 50 mg tds or nabumetone 1 g bd." No actions were undertaken to blind participants from their allocated treatment. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | "If a patient withdrew from the study prior to month three, the data from the last assessment was used." "Eighteen patients withdrew early, that is, before the three month assessment…" Although actions were undertaken to impute for missing data, the level of missing data was considerable in both groups. |
| Selective reporting (reporting bias) | High risk | Spinal pain (on a 0 to 3 graded scale) was measured but not reported in the results. |
| Other bias | High risk | There was some BL imbalance that was not adjusted for in the analyses. |
Pasero 1994.
| Methods |
Design: RCT Number of centres: NA Treatment duration: 12 weeks Flare design: No Wash‐out period: No Time point of assessments: BL, 15 days, 4, 8, 12 weeks |
|
| Participants |
Inclusion criteria: Definite AS, defined as grade 2/3/4 of sacroiliitis confirmed on X‐ray and at least 2 of the following: a) lumbar or dorsal/lumbar junction pain and stiffness of over 3 months' duration, b) major limitation of motion of the lumbar spine in three directions (flexion/extension, lateral bending, rotation), c) pain and stiffness in thoracic region of over 3 months' duration, d) limited chest expansion, e) nocturnal pain with morning predominance and/or morning stiffness and/or pain in one or both buttocks. Exclusion criteria: 1. Other arthropathies, cardiovascular, neoplastic, gastro‐intestinal, or renal diseases; 2. Treated with drugs that could interfere with study drugs; 3. Women pregnant or lactating or receiving hormonal contraception; 4. Patients who in the eyes of the investigators would be unable to comply fully with the trial requirements. Classification: NA Aceclofenac (100 mg): Number of participants: 60 Number of completers: 47 Age (mean (SD)): 39.10 (7.93) Male (%): 50 Symptom duration: NA Disease duration (mean (SD)): 89.77 (74.22) months HLA‐B27 positive (%): NA Naproxen (500 mg): Number of participants: 66 Number of completers: 57 Age (mean (SD)): 38.50 (8.94) Male (%): 57 Symptom duration: NA Disease duration (mean (SD)): 85.82 (85.39) months HLA‐B27 positive (%): NA |
|
| Interventions | Aceclofenac (100 mg) vs Naproxen (500 mg) Co‐medication: NA |
|
| Outcomes |
Extracted outcomes:
|
|
| Notes | The outcome pain on VAS was also presented, but these data could not be used due to presentation in a graph from which the data could not be extracted. Funding source: Not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Sixty‐three patients were randomized to receive treatment with…" Probably done, but no further information was provided. |
| Allocation concealment (selection bias) | Unclear risk | No information was provided on allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "double‐blind". Probably done, but no further information was provided on blinding of participants. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "double‐blind". Probably done, but no further information was provided on blinding of outcome assessor. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Number of dropouts is low and equal in both groups (21.7% (13 patients) in the aceclofenac group and 13.6% (9 patients) in the naproxen group withdrew from the study (1 patient in the naproxen group because of improvement in symptoms)). |
| Selective reporting (reporting bias) | Low risk | All pre‐specified outcomes according to the methods section, are reported in the results section. |
| Other bias | Low risk | No other bias was detected. |
Poddubnyy 2012.
| Methods | Prospective cohort study, see Table 3. | |
| Participants | ‐ | |
| Interventions | ‐ | |
| Outcomes | Results not included in meta‐analysis, but described in the text (Effects of interventions). | |
| Notes | ‐ | |
Rejholec 1980.
| Methods |
Design: RCT Number of centres: NA Treatment duration: 6 months Flare design: No Wash‐out period: No Time point of assessments: BL, 1, 2, 3, 4, 5, 6 months |
|
| Participants |
Inclusion criteria: Diagnosis of AS verified clinically and radiographically. Exclusion criteria: None reported. Classification: NA Tolfenamic acid (600 mg): Number of participants: 25 Number of completers: 24 Age (mean (SD)): 38.6 (2.7) Male (%): 84 Symptom duration: NA Disease duration (mean (SD)): 13.9 (2.4) years HLA‐B27 positive (%): 100 Indomethacin (75 mg): Number of participants: 25 Number of completers: 21 Age (mean (SD)): 35.6 (2.7) Male (%): 88 Symptom duration: NA Disease duration (mean (SD)): 10.4 (2.1) years HLA‐B27 positive (%): 100 |
|
| Interventions | Tolfenamic acid (600 mg) vs Indomethacin (75 mg) Co‐medication: Not reported |
|
| Outcomes |
Extracted outcomes:
|
|
| Notes | The outcomes pain on Likert scale, ESR, chest expansion and Schober's test were also presented, but these data could not be used due to presentation in graphs from which the data could not be extracted. Funding source: Not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "The patients were sequentially given a study number; each study number had previously been assigned to either the tolfenamic acid or the indomethacin group by a person not directly involved in the trial, using a table of random numbers." |
| Allocation concealment (selection bias) | Unclear risk | No information provided on allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "The drugs were administered in gelatin capsules of identical appearance." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | "Four patients receiving indomethacin interrupted the 6‐month trial… In the tolfenamic acid group there was 1 discontinuation…" Outcome data is most likely available for > 84% of the study participants. |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the methods are reported. |
| Other bias | Low risk | No other bias was detected. |
Santo 1988.
| Methods |
Design: RCT Number of centres: NA Treatment duration: 6 weeks Flare design: No Wash‐out period: Yes (1 week, no analgesic) Time point of assessments: BL, 2, 6 weeks |
|
| Participants |
Inclusion criteria: 1. Over 18 years of age; 2. Presence of at least two of the following: a) lumbar or dorso‐lumbar pain and stiffness of more than 3 months' duration, b) severe limitation of motion of the lumbar spine in three directions (flexion‐extension, lateral flexion and rotation), c) pain and stiffness of the thoracic region of more than 3 months' duration, d) limitation of thoracic expansion (generally between 2 and 5 cm), e) night pain and morning stiffness of the gluteal regions; 3. Bilateral sacroiliitis (grades 2 or 3, according to radiological criteria); 4. Positive HLA‐B27 antigen; 5. Muscular contraction of the dorso‐lumbar region with limitation of motion of any segment of the vertebral column. Exclusion criteria: 1. Receiving anti‐coagulant therapy; 2. Pregnant women or those who might become pregnant during the study; 3. Allergic to aspirin or other NSAIDs; 4. Patients with gastro‐intestinal, infectious, cardiac, or malignant diseases; 5. Patients with hepatic or renal impairment. Classification: NA Oxaprozin (1200 mg): Number of participants: 20 Number of completers: 15 Age (mean (SD)): 36.6 (6.75) Male (%): 75 Symptom duration: NA Disease duration (mean (SD)): 11.2 (6.90) years HLA‐B27 positive (%): 100 Diclofenac (100 mg): Number of participants: 20 Number of completers: 15 Age (mean (SD)): 41.8 (13.04) Male (%): 75 Symptom duration: NA Disease duration (mean (SD)): 11.1 (9.78) years HLA‐B27 positive (%): 100 |
|
| Interventions | Oxaprozin (1200 mg) vs Diclofenac (100 mg) Co‐medication: Not reported |
|
| Outcomes |
Extracted outcomes:
|
|
| Notes | Funding source: Not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "The study was open, with patients assigned randomly to two groups of 20 patients each." No information provided on sequence generation. |
| Allocation concealment (selection bias) | Unclear risk | No information provided on allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No actions were undertaken to blind participants for their allocated treatment. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | "Of the 40 patients enrolled, 30 (15 in each treatment group) completed the clinical trial." Level of dropout is considerable (25%) in both groups. No actions were undertaken to impute in case of missing data. |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the methods are reported. |
| Other bias | Low risk | No other bias was detected. |
Schwarzer 1990.
| Methods |
Design: RCT Number of centres: NA Treatment duration: 12 weeks Flare design: Yes Wash‐out period: Yes (3 days, NSAIDs ceased) Time point of assessments: BL, 2, 4, 6, 8, 12 weeks |
|
| Participants |
Inclusion criteria: 1. Age between 16 and 65 years; 2. Diagnosis of definite or probable AS according to the New York criteria. Exclusion criteria: 1. Spinal arthritis showing active peripheral manifestations (articular or not); 2. Spinal arthritis secondary to an intestinal lesion or Behcet's syndrome; 3. Disc lesions in spinal arthritis; 4. Known intolerance to other NSAIDs; 5. Current treatment with anticoagulants; 6. Patients treated within the previous 2 months with radiotherapy, thorium, gold, immunosuppressives or steroids. Classification: New York criteria Tenoxicam (20 mg): Number of participants: 12 Number of completers: 6 Age (mean): 42 Male (%): 100 Symptom duration: NA Disease duration (mean): 9 years HLA‐B27 positive (%): NA Diclofenac (50 mg): Number of participants: 12 Number of completers: 8 Age (mean): 40 Male (%): 75 Symptom duration: NA Disease duration (mean): 7 years HLA‐B27 positive (%): NA |
|
| Interventions | Tenoxicam (20 mg) vs Diclofenac (50 mg) Co‐medication: Not reported |
|
| Outcomes |
Extracted outcomes:
|
|
| Notes | Funding source: Roche Products Pty. Limited, Dee Why, NSW Australia sponsored this study. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Twenty‐four patients were randomly allocated to the two treatments, 12 to each group." No information was provided on sequence generation. |
| Allocation concealment (selection bias) | Unclear risk | No information was provided on allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No actions were undertaken to blind participants for their allocated treatment. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | "Six of the 12 patients receiving tenoxicam withdrew from the study… .Four of the 12 patients receiving diclofenac withdrew from the study…" High level of withdrawals from the study from groups that were already very small at the start of the study. |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the methods are reported. |
| Other bias | Low risk | No other bias was detected. |
Sieper 2008.
| Methods |
Design: RCT Number of centres: 47 Treatment duration: 12 weeks Flare design: Yes Wash‐out period: Yes (2 to 14 days) Time point of assessments: BL, 1, 2, 6, 12 weeks |
|
| Participants |
Inclusion criteria: 1. Age 18 to 75 years; 2. Confirmed diagnosis of AS; 3. The presence of axial involvement; 4. No peripheral involvement; 5. The need for daily treatment with NSAIDs. Exclusion criteria: 1. Present or previous episodes of inflammatory bowel disease; 2. A history of upper gastrointestinal ulcers within the previous year and confirmed by endoscopy. Classification: modified New York criteria Celecoxib (200 mg): Number of participants: 153 Number of completers: 128 Age (mean): 44.9 Male (%): 69 Symptom duration: NA Disease duration: NA HLA‐B27 positive (%): NA Celecoxib (400 mg): Number of participants: 150 Number of completers: 122 Age (mean): 46.2 Male (%): 69 Symptom duration: NA Disease duration: NA HLA‐B27 positive (%): NA Diclofenac (75 mg): Number of participants: 155 Number of completers: 131 Age (mean): 43.4 Male (%): 70 Symptom duration: NA Disease duration: NA HLA‐B27 positive (%): NA |
|
| Interventions | Celecoxib (200 mg) vs Celecoxib (400 mg) vs Diclofenac (75 mg) Co‐medication: DMARDs in stable dosage, prednisolone < 10 mg/day, proton pump inhibitors. |
|
| Outcomes |
Extracted outcomes:
|
|
| Notes | In comparison 5 (COX‐2 vs traditional NSAID) we chose to present data from Celecoxib 400 mg instead of Celecoxib 200 mg (see Measures of treatment effect for rationale). Funding source: This study was sponsored by Pfizer Pharma GmbH, Germany. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "At baseline, eligible subjects were randomly assigned (ratio 1:1:1) to double‐dummy study medication…" No information provided on sequence generation. |
| Allocation concealment (selection bias) | Unclear risk | No information provided on allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "...to double‐dummy study medication (capsules of celecoxib, diclofenac, and matching placebo)…" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "… multicentre, randomised, controlled, double‐blind study…" Outcomes assessed by self‐report. Participants were blinded to their treatment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Outcome data is available for > 80% of all study participants. Last observation carried forward approach was used for the 'full analysis set'. |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the methods were reported. |
| Other bias | Low risk | No other bias was detected. |
Simpson 1966.
| Methods |
Design: Cross‐over study Number of centres: 1 Treatment duration: each treatment period was 4 weeks Flare design: No Wash‐out period: No Time pointof assessments: BL, 4, 8 weeks |
|
| Participants |
Inclusion criteria: 1. X‐ray evidence of involvement of sacroiliac joints, small joints of the hand were not involved; 2. Chest expansion had to be limited and morning stiffness present; 3. Latex test had to be negative; 4. Serum uric acid level not be in excess of 6.5 mg/100 mL; 5. Raised ESR. Exclusion criteria: None reported. Classification: NA Phenylbutazone (300 mg): Number of participants: 7 Number of completers: 6 Flufenamic acid (600 mg): Number of participants: 7 Number of completers: 7 All participants: Age (mean): males 35, females 51 Male (%): 79 Symptom duration: NA Disease duration: NA HLA‐B27 positive (%): NA |
|
| Interventions | Phenylbutazone (300 mg) vs Flufenamic acid (600 mg) Co‐medication: Not reported |
|
| Outcomes |
Extracted outcomes:
|
|
| Notes | For the outcome pain on Likert scale individual patient data that were presented in the paper were combined for the meta‐analysis to a mean and SD.Funding source: Not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "… and the order of administration was randomized." No information on sequence generation was provided. |
| Allocation concealment (selection bias) | Unclear risk | No information was provided on allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Matched capsules containing flufenamic acid 100 mg and phenylbutazone 50 mg were administered at the rate of six capsules per day." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information was provided on blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The number of missing observations were low (extracted from table 1). |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the methods were reported in the results. |
| Other bias | High risk | No information provided how statistical analyses were performed. Unable to determine BL characteristics imbalance. |
Sydnes 1981.
| Methods |
Design: Cross‐over study Number of centres: 13 Treatment duration: each treatment period was 4 weeks Flare design: No Wash‐out period: 7 days (placebo was given) Time point of assessments: BL, 5, 6, 10 weeks |
|
| Participants |
Inclusion criteria: 1. Patients of both sexes ages 18 to 70 years suffering from classical or definite AS; 2. Active disease requiring treatment with NSAIDs. Exclusion criteria: 1. A history of primary disease of less than 6 months duration; 2. AS associated with psoriasis; 3. Systemically or intra‐articularly administered corticosteroids in the preceding 3 months, or anticipated corticosteroid requirement during the course of the trial; 4. Pregnancy, lactating mothers; 5. Blood, liver or renal abnormalities unrelated to the primary disease; 6. Peptic ulceration or severe dyspepsia in preceding 12 months; 7. Known hypersensitivity to NSAID. Classification: American Rheumatism Association Piroxicam (20 mg): Number of participants: NA Number of completers: NA Age (mean (SD)): NA Male (%): NA Symptom duration: NA Disease duration (mean (SD)): NA HLA‐B27 positive (%): NA Indomethacin (75 mg): Number of participants: NA Number of completers: NA Age (mean (SD)): NA Male (%): NA Symptom duration: NA Disease duration (mean (SD)): NA HLA‐B27 positive (%): NA All participants: Number of participants: 93 Number of completers: 87 |
|
| Interventions | Piroxicam (20 mg) vs Indomethacin (75 mg) Co‐medication: Paracetamol |
|
| Outcomes |
Extracted outcomes: 1. Pain on VAS (BL (± SE), post‐treatment after 4 weeks (± SE)) (scale 0 to 10, higher is worse) Piroxicam 20 mg: 5.0 ± 0.4, 3.2 ± 0.4 Indomethacin 75 mg: 5.0 ± 0.3, 3.8 ± 0.4 2. Patient's global assessment of disease activity (BL (± SE), post‐treatment after 4 weeks (± SE)) (scale 1 to 5, higher is worse) Piroxicam 20 mg: 3.3 ± 0.1, 2.3 ± 0.1 Indomethacin 75 mg: 3.3 ± 0.1, 2.6 ± 0.1 3. Peripheral joint pain (BASDAI) (BL (± SE), post‐treatment after 4 weeks (± SE)) (scale 1 to 5, higher is worse) Piroxicam 20 mg: 3.6 ± 0.5, 2.5 ± 0.4 Indomethacin 75 mg: 3.7 ± 0.3, 3.2 ± 0.4 4. Duration of morning stiffness (BASDAI) (BL (± SE), post‐treatment after 4 weeks (± SE)) (in hours, higher is worse) Piroxicam 20 mg: 2.1 ± 0.3, 1.2 ± 0.3 Indomethacin 75 mg: 2.2 ± 0.2, 1.7 ± 0.2 5. Chest expansion (BL (± SE), post‐treatment after 4 weeks (± SE)) (in cm, higher is better) Piroxicam 20 mg: 4.2 ± 0.3, 4.7 ± 0.4 Indomethacin 75 mg: 4.4 ± 0.3, 4.7 ± 0.3 6. Schober's test (BL (± SE), post‐treatment after 4 weeks (± SE)) (in cm, higher is better) Piroxicam 20 mg: 2.6 ± 0.3, 2.9 ± 0.3 Indomethacin 75 mg: 2.2 ± 0.3, 2.6 ± 0.3 |
|
| Notes | No BL characteristics reported for this trial. Results are not included in the meta‐analysis, because the number of patients in each treatment group was not available. Available results are described in this table. Only results of first part of cross‐over trial are presented. Funding source: Not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "..and the order in which the drugs were given was randomised with a restriction to ensure a balance between treatments and orders." No information provided on method of sequence generation. |
| Allocation concealment (selection bias) | Unclear risk | No information provided on allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "The double‐blind design was maintained by supplementing two placebo tablets daily during the peroxicam period." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "Patients attended for assessment after one, five, six and ten weeks, as far as possible at the same hour of the day, and were seen by the same observer on each occasion." No further information is provided on the blinding of outcome assessors. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | "A total of 93 patients were included in the trial. Treatment was terminated in 6 patients for various reasons." Outcome data was available from a large proportion of the participants (94%). |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the methods are reported. |
| Other bias | Unclear risk | "piroxicam had a certain carry‐over effect as parameters did not reach baseline values after the wash‐out period. By contrast, corresponding values after indomethacin deteriorated to below pre‐treatment values." Carry‐over effect not relevant for the results extracted from this study because we only extracted data before cross‐over. Limited BL characteristics available to determine any BL imbalance. |
Tannenbaum 1984.
| Methods |
Design: RCT Number of centres: 4 Treatment duration: 12 weeks and 9 months open extension Flare design: Yes Wash‐out period: Yes (up to 7 days, placebo was provided) Time point of assessments: BL, 1, 2, 3, 7, 12, 9 months |
|
| Participants |
Inclusion criteria: 1. Between 18 and 65 years; 2. Active disease as evidenced by spinal or sacroiliac pain, or both, and one or more of the following: a) muscle spasm in the back, b) decreased range of motion of some part of the spine, c) increased sedimentation rate. Exclusion criteria: 1. Patients with other arthropathies or diseases closely related to AS, such as psoriatic spondylitis or spondylitis associated with inflammatory bowel disease; 2. Patients with active hematological, gastrointestinal, renal or hepatic disease and pregnant or nursing women. Classification: New York criteria Piroxicam (10 to 20 mg): Number of participants: 28 Number of completers: 23 Age (mean (SD)): 35.6 (1.3) Male (%): 75 Symptom duration: NA Disease duration (mean (SD)): 8.8 (1.4) years HLA‐B27 positive (%): 79 Indomethacin (75 to 125 mg): Number of participants: 27 Number of completers: 23 Age (mean (SD)): 34.0 (1.8) Male (%): 74 Symptom duration: NA Disease duration (mean (SD)): 9.7 (1.7) years HLA‐B27 positive (%): 81 |
|
| Interventions | Piroxicam (10 to 20 mg) vs Indomethacin (75 to 125 mg) Co‐medication: Not reported |
|
| Outcomes |
Extracted outcomes: Twelve‐weeks results (included in meta‐analysis):
Nine months open extension results (not included in meta‐analysis) Results were only described in the text. "This resulted in 27 piroxicam and 21 indomethacin treated subjects being followed for efficacy and safety for a prolonged period of up to 9 months. Efficacy was maintained in both treatment groups, but 3 patients experienced side effects (depression, constipation, and GI and CNS intolerance), which necessitated discontinuation of piroxicam therapy." |
|
| Notes | For Analysis 4.2 the SD was imputed from the BL (for rationale see Dealing with missing data). Funding source: Not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "The treatment distribution was randomized…" No information was provided on sequence generation |
| Allocation concealment (selection bias) | Unclear risk | No information was provided on allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "since the two drugs were not identical in appearance, the double‐dummy technique was used." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "… a blinded investigator performed all clinical assessments." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | "Twenty‐three patients in each group completed 12 weeks of treatment." 18% in piroxicam and 19% in indomethacin did not complete the trial. No differences between groups were found in the life table analysis. No imputation was done for missing data. |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the methods were reported. |
| Other bias | High risk | Compliance differed significantly between piroxicam (once daily, or placebo) and indomethacin (thrice daily, or placebo) |
van der Heijde 2005.
| Methods |
Design: RCT Number of centres: 43 Treatment duration: 6 weeks and 52 weeks extension Flare design: Yes Wash‐out period: Yes Time point of assessments: Screening, BL, 2, 4, 6 weeks, at discontinuation |
|
| Participants |
Inclusion criteria: 1. Age 18 or over; 2. Diagnosis AS at least 6 months prior to study; 3. History of positive therapeutic benefit with NSAIDs; 4. Use of NSAIDs at least 25 of previous 30 days at therapeutic dose level for at least 30 days prior to study; 5. Use of approved non‐study antirheumatic therapy at stable dose (MTX ≥3 months, SSZ ≥3 months, or other DMARD ≥6 months); 6. Satisfaction of flare criteria (≥ 40 mm worsening of spinal pain on 100 mm VAS and increase ≥ 30% (minimum 12 mm) compared with pain rating at screening visit after washout period for pre‐study NSAIDs). Exclusion criteria: 1. Concurrent rheumatic disease that could confound the evaluation of efficacy; 2. Acute peripheral articular disease (onset within 4 weeks prior to study of active (painful/swollen) peripheral arthritis); 3. Corticosteroid therapy within 1 month prior to screening visit; 4. Use of analgesic medications within 3 days of study entry and through week 6 (acetaminophen use was permitted prior to study entry); 5. Use of non‐study NSAID or selective COX‐2 inhibitor with the exception of low dose aspirin (≤ 100 mg). Classification: modified New York criteria Etoricoxib (90 mg): Number of participants: 103 Number of completers: 92 Age (mean (SD)): 43.1 (12.1) (range 20 to 74) Male (%): 73.8 Symptom duration: NA Disease duration: NA HLA‐B27 positive (%): NA Etoricoxib (120 mg): Number of participants: 92 Number of completers: 83 Age (mean (SD)): 42.5 (12.0) (range 20 to 78) Male (%): 78.3 Symptom duration: NA Disease duration: NA HLA‐B27 positive (%): NA Naproxen (1000 mg): Number of participants: 99 Number of completers: 78 Age (mean (SD)): 45.0 (11.4) (range 18 to 74) Male (%): 79.8 Symptom duration: NA Disease duration: NA HLA‐B27 positive (%): NA Placebo: Number of participants: 93 Number of completers: 48 Age (mean (SD)): 43.7 (12.1) (range 23 to 71) Male (%): 79.6 Symptom duration: NA Disease duration: NA HLA‐B27 positive (%): NA |
|
| Interventions | Etoricoxib (90 mg) vs Etoricoxib (120 mg) vs Naproxen (1000 mg) vs Placebo Co‐medication: Low‐dose aspirin (≤ 100 mg) or DMARDs at stable dose (MTX ≥ 3 months, SSZ ≥ 3 months, or other DMARD ≥ 6 months), or both. |
|
| Outcomes |
Extracted outcomes: Six weeks results (included in meta‐analysis)
Fifty‐two weeks active‐comparator‐controlled results (not included in meta‐analysis) 1. Pain on VAS (change after 1 year (least squares mean ± SEM) (scale 0 to 100, higher is worse) Etoricoxib 90 mg: ‐42.9 ± 2.2 (N = 100) (P < 0.05 vs naproxen) Etoricoxib 120 mg: ‐44.6 ± 2.4 (N = 90) (P < 0.01 vs naproxen) Naproxen 1000 mg: ‐35.4 ± 2.3 (N = 97) 2. Withdrawals due to adverse events (post‐treatment after 1 year) Etoricoxib 90 mg: 8 (total N = 103) Etoricoxib 120 mg: 4 (total N = 92) Naproxen 1000 mg: 6 (total N = 99) 3. BASDAI (change after 1 year (least squares mean ± SEM) (VAS 0 to 100, higher is worse) Etoricoxib 90 mg: ‐30.4 ± 1.9 (N = 100) (P < 0.05 vs naproxen) Etoricoxib 120 mg: ‐31.6 ± 2.0 (N = 90) (P < 0.05 vs naproxen) Naproxen 1000 mg: ‐24.5 ± 1.9 (N = 97) 4. Patient's global assessment of disease activity (change after 1 year (least squares mean ± SEM) (VAS 0 to 100, higher is worse) Etoricoxib 90 mg: ‐29.5 ± 2.2 (N = 100) (P < 0.05 vs naproxen) Etoricoxib 120 mg: ‐30.1 ± 2.3 (N = 90) (P < 0.05 vs naproxen) Naproxen 1000 mg: ‐22.6 ± 2.2 (N = 97) 5. Duration of morning stiffness (change after 1 year (least squares mean ± SEM) (VAS 0 to 100, higher is worse) Etoricoxib 90 mg: ‐28.8 ± 2.3 (‐34.5 minutes) (N = 100) (P < 0.05 vs naproxen) Etoricoxib 120 mg: ‐29.0 ± 2.4 (‐34.8 minutes) (N = 90) (P < 0.05 vs naproxen) Naproxen 1000 mg: ‐22.4 ± 2.3 (‐26.9 minutes) (N = 97) 6. BASFI (change after 1 year (least squares mean ± SEM) (VAS 0 to 100, higher is worse) Etoricoxib 90 mg: ‐21.7 ± 1.8 (N = 100) (P < 0.05 vs naproxen) Etoricoxib 120 mg: ‐22.4 ± 2.0 (N = 90) (P < 0.05 vs naproxen) Naproxen 1000 mg: ‐16.1 ± 1.9 (N = 97) 7. Schober's test (change after 1 year (least squares mean ± SEM) (in cm, higher is better)) Etoricoxib 90 mg: 0.56 ± 0.11 (N = 100) (P = not significant) Etoricoxib 120 mg: 0.70 ± 0.11 (N = 90) (P = not significant) Naproxen 1000 mg: 0.60 ± 0.11 (N = 97) 8. Number of any adverse events (post‐treatment after 1 year) Etoricoxib 90 mg: n = 76 (total N = 103) Etoricoxib 120 mg: n = 68 (total N = 92) Naproxen 1000 mg: n = 52 (total N = 99) 9. Number of serious adverse events (post‐treatment after 1 year) Etoricoxib 90 mg: n = 7 (total N = 103) Etoricoxib 120 mg: n = 6 (total N = 92) Naproxen 1000 mg: n = 6 (total N = 99) |
|
| Notes | In comparison 4 (COX‐2 vs Placebo), comparison 5 (COX‐2 vs traditional NSAID) and comparison 6 (Naproxen vs other NSAID) we chose to present data from Etoricoxib 90 mg instead of Etoricoxib 120 mg (see Measures of treatment effect for rationale). A post‐hoc analysis of this study was also included in the review (Gossec 2005), for detailed description see Table 4. Funding source: Merck. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "The patient was randomly allocated to a treatment sequence using a computer‐generated random‐allocation schedule." |
| Allocation concealment (selection bias) | Low risk | "The patient was randomly allocated to a treatment sequence using a computer‐generated random‐allocation schedule." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "double‐blind"; "patients received 3 bottles of study medication at randomization and at weeks 2 and 4. Each bottle contained active medication or matching placebo." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "double‐blind". Probably done, but no further information provided on blinding of outcome assessor. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | "The primary efficacy analyses were based on the modified intent‐to‐treat principle (i.e., inclusion of all patients in the analysis population for whom a baseline value and at least 1 postbaseline measurement were available) and per‐protocol approach"; "77.8% completed the first 6‐week period"; "of the 81 patients who discontinued part I of the study due to lack of efficacy after completing at least 2 weeks of treatment, 77 continued into part II"; "during the first 6 weeks, a significantly smaller percentage of patients discontinued treatment due to lack of efficacy in the 90 mg etoricoxib, 120 mg etoricoxib, and naproxen groups compared with placebo and in both etoricoxib groups compared with naproxen. No notable differences were observed among the treatment groups in discontinuation rates due to clinical AEs or laboratory AEs, or due to other reasons." |
| Selective reporting (reporting bias) | High risk | Occiput‐to‐wall distance and chest expansion was assessed (see methods section), but not reported (see results section). All other outcomes that were assessed, were reported. |
| Other bias | Low risk | High compliance rate in all groups (> 95%) over 52‐week‐course. Sufficient power for primary efficacy hypothesis (sample size calculations). No different co‐interventions between groups, other than "rescue" acetaminophen. |
Villa Alcázar 1996.
| Methods |
Design: RCT Number of centres: 16 Treatment duration: 3 months Flare design: No Wash‐out period: Yes (1 week, paracetamol was allowed) Time point of assessments: BL, 15 days, 30 days, 2 months, 3 months |
|
| Participants |
Inclusion criteria: 1. Outpatients of both sexes between 18 and 50 years of age with defined clinical and radiological AS by the New York criteria; 2. Morning stiffness lasting 30 minutes or longer; 3. Pain requiring medication with NSAIDs; 4. Visual analog pain scale (VAS) level of ≥ 40. Exclusion criteria: 1. Other spondyloarthropathies or psoriasis, Paget's disease of the bone, gout, haemochromatosis or arthritis of any etiology, or both; 2. Patients with history of peptic ulcers or digestive haemorrhage caused by NSAIDs; 3. Patients with hypersensitivity to either of the drugs under study; 4. Patients with any complaint with life expectancy of less than 2 years; 5. Significant pulmonary, cardiac, cerebrovascular, hepatic, or renal disease; 6. Pregnant women, nursing mothers, and women of child bearing potential; 7. Anticoagulant therapy or other treatments that could interfere with the drugs under study; 8. Treatment with sulfasalazine, steroids, or immunosuppressive drugs within the previous 3 months; 9. Concurrent pathologies or other circumstances that impeded the performance of trial controls; 10. Patients who had applied for invalid classification or had participated in other trials within 3 months. Classification: New York criteria Aceclofenac (200 mg): Number of participants: 135 Number of completers: 120 Age (mean (SD)): 37.4 (8.4) Male (%): 83 Symptom duration: NA Disease duration (mean (SD)): 6.3 (5.7) years HLA‐B27 positive (%): NA Tenoxicam (20 mg): Number of participants: 138 Number of completers: 115 Age (mean (SD)): 37.1 (8.1) Male (%): 77 Symptom duration: NA Disease duration (mean (SD)): 5.5 (5.4) years HLA‐B27 positive (%): NA |
|
| Interventions | Aceclofenac (200 mg) vs Tenoxicam (20 mg) Co‐medication: Paracetamol as emergency medication (500 mg). |
|
| Outcomes |
Extracted outcomes: 1. Pain on VAS 2. Withdrawals due to adverse events 3. Duration of morning stiffness (BL, mean change after 3 months) (in minutes, higher is worse) Aceclofenac 200 mg: 55.3 (N = 135), ‐31.4 (N = 135) (P < 0.01 versus BL) Tenoxicam 20 mg: 61.1 (N = 138), ‐38.9 (N = 138) (P < 0.01 versus BL) 4. Lateral spinal flexion (BL, mean change after 3 months) (in cm, higher is better) Aceclofenac 200 mg: 114.6 (N = 135), +7.7 (N = 135) (P < 0.02 versus BL) Tenoxicam 20 mg: 114.6 (N = 138), +14.5 (N = 138) (P < 0.01 versus BL) 5. Chest expansion (BL, mean change after 3 months) (in mm, higher is better) Aceclofenac 200 mg: 34.4 (N = 135), +5.1 (N = 135) (P < 0.01 versus BL) Tenoxicam 20 mg: 35.1 (N = 138), +7.8 (N = 138) (P < 0.01 versus BL) 6. Occiput‐to‐wall distance (BL, mean change after 3 months) (in mm, higher is worse) Aceclofenac 200 mg: 34.1 (N = 135), ‐6.2 (N = 135) (P < 0.01 versus BL) Tenoxicam 20 mg: 33.8 (N = 138), ‐3.8 (N = 138) (P < 0.05 versus BL) 7. Schober's test (BL, mean change after 3 months) (in mm, higher is better) Aceclofenac 200 mg: 47.0 (N = 135), +8.8 (N = 135) (P < 0.02 versus BL) Tenoxicam 20 mg: 48.1 (N = 138), +10.6 (N = 138) (P < 0.01 versus BL) 8. Number of any adverse events 9. Number of adverse events per organ system |
|
| Notes | Outcomes that were not included in the meta‐analysis, because no measure of variance (SD, SE or CI) was reported for these outcomes: duration of morning stiffness, lateral spinal flexion, chest expansion, occiput‐to‐wall distance, Schober's test. Available results are described in this table. Funding source: This study was sponsored by Prodesfarma SA, Barcelona, Spain. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "…patients were randomly assigned to receive.." No information was provided by the authors how randomization was done. |
| Allocation concealment (selection bias) | Unclear risk | No information was provided on allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "…study was double blind, so that all medications were identical in appearance." "Placebo tablets were matched to active drug tablets in the tenoxicam group." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "…a 3 month, multicenter, double blind, parallel study…" No further information is provided on the blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | "Fifteen (11%) patients in the aceclofenac group and 23 (17%) in the tenoxicam group abandoned the study." Outcome data is available for a large part of the study population. No imputation was done for missing data. |
| Selective reporting (reporting bias) | High risk | Semiquantitative pain scale (SPS) and laboratory test results (including CRP and ESR) are not reported. |
| Other bias | Low risk | No other bias was detected. |
Wanders 2005.
| Methods |
Design: RCT Number of centres: 76 Treatment duration: 2 years Flare design: Yes Wash‐out period: Yes (2 to 14 days, Paracetamol only) Time point of assessments: BL, 1, 4, 7, 10, 13, 16, 22, 24 months |
|
| Participants |
Inclusion criteria: 1. Daily NSAID intake during the month preceding the screening visit; 2. An NSAID washout period of 2 to 14 days before the BL visit; 3. A flare of the disease at BL, defined by absolute score for pain of ≥ 40 mm (100 mm VAS) and increase in pain of at least 30% between screening visit and BL visit. Exclusion criteria: 1. Patients with peripheral arthritis (presence of active synovitis with swelling of a peripheral joint at the screening visit; 2. Active inflammatory bowel disease; 3. Patients with severe concomitant medical illness; 4. Received corticosteroids in previous 6 weeks before start of the study; 5. Any DMARD with a change in dosage during previous 6 months; 6. Confirmed peptic ulcer by gastro‐duodenoscopy within the year preceding screening visit. Classification: modified New York criteria Celecoxib (400 mg) continuous: Number of participants: 111 Number of completers: 96 (68 completed the study while taking celecoxib, 28 patients completed the study while taking a different NSAID) Age (mean (SD)): 38.0 (10.7) Male (%): 67 Symptom duration: NA Disease duration (mean (SD)): 11.9 (9.3) years HLA‐B27 positive (%): 86 Celecoxib (400 mg) on‐demand: Number of participants: 103 Number of completers: 86 (67 completed the study while taking celecoxib, 19 patients completed the study while taking a different NSAID) Age (mean (SD)): 40.1 (10.5) Male (%): 72 Symptom duration: NA Disease duration (mean (SD)): 11.0 (9.4) years HLA‐B27 positive (%): 87 |
|
| Interventions | Celecoxib (400 mg) continuous vs Celecoxib (400 mg) on‐demand Co‐medication: Analgesics or DMARDs without changing dosage, or both. |
|
| Outcomes |
Extracted outcomes: 1. Pain on VAS (BL (± SD), post‐treatment after 2 years (± SD)) (scale 0 to 100, higher is worse) Celecoxib continuous: 50 ± 38, 36.41 ± 30.40 Celecoxib on‐demand: 54 ± 37, 39.05 ± 29.99 2. Withdrawals due to adverse events (post‐treatment after 2 years) Celecoxib continuous: n = 2 (total N = 111) Celecoxib on‐demand: n = 3 (total N = 103) 3. BASDAI (BL not available, post‐treatment after 2 years (± SD)) (scale 0 to 100, higher is worse) Celecoxib continuous: 28.88 ± 21.03 Celecoxib on‐demand: 32.40 ± 22.59 4. Patient's global assessment of disease activity (BL (± SD), post‐treatment after 2 years (± SD)) (scale 0 to 100, higher is worse) Celecoxib continuous: 43 ± 29, 34.73 ± 28.70 Celecoxib on‐demand: 47 ± 31, 40.05 ± 28.01 5. Fatigue (BL not available, post‐treatment after 2 years (± SD)) (scale 0 to 100, higher is worse) Celecoxib continuous: 37.54 ± 27.21 Celecoxib on‐demand: 37.75 ± 27.42 6. Duration of morning stiffness (BL not available, post‐treatment after 2 years (± SD)) (scale 0 to 100, higher is worse) Celecoxib continuous: 38.37 ± 52.48 Celecoxib on‐demand: 38.01 ± 46.65 7. Severity of morning stiffness (BL not available, post‐treatment after 2 years (± SD)) (scale 0 to 100, higher is worse) Celecoxib continuous: 27.51 ± 22.92 Celecoxib on‐demand: 33.07 ± 25.15 8. CRP (BL (± SD)), post‐treatment after 2 years (± SD)) (in mg/L, higher is worse) Celecoxib continuous: 14.7 ± 17.9, 12.56 ± 13.88 Celecoxib on‐demand: 12.7 ± 17.1, 11.98 ± 17.20 9. ESR (BL (± SD)), post‐treatment after 2 years (± SD)) (in mm/hr, higher is worse) Celecoxib continuous: 17.0 ± 13.8, 14.22 ± 12.11 Celecoxib on‐demand: 17.0 ± 16.7, 15.96 ± 14.49 10. BASFI (BL (± SD), post‐treatment after 2 years (± SD)) (scale 0 to 100, higher is worse) Celecoxib continuous: 33 ± 25, 28.49 ± 23.01 Celecoxib on‐demand: 38 ± 28, 31.76 ± 25.49 11. Chest expansion (BL (± SD), post‐treatment after 2 years (± SD)) (in cm, higher is better) Celecoxib continuous: 4.7 ± 2.3, 5.17 ± 2.49 Celecoxib on‐demand: 5.0 ± 2.3, 5.39 ± 2.20 12. Occiput‐to‐wall distance (BL not available, post‐treatment after 2 years (± SD)) (in cm, higher is worse) Celecoxib continuous: 3.56 ± 4.86 Celecoxib on‐demand: 2.63 ± 3.55 13. Schober's test (BL (± SD), post‐treatment after 2 years (± SD)) (in cm, higher is better) Celecoxib continuous: 3.2 ± 1.4, 3.10 ± 1.47 Celecoxib on‐demand: 3.2 ± 1.4, 3.19 ± 1.37 14. mSASSS (BL (± SD), post‐treatment after 2 years (± SD)) (in cm, higher is worse, n = only patients with a X‐ray) Celecoxib continuous: 7.9 ± 14.7 (N = 76), 8.28 ± 14.72 (N = 76) Celecoxib on‐demand: 9.3 ± 15.2 (N = 74), 10.75 ± 16.15 (N = 74) 15. Number of patients with at least 2 mSASSS units radiographic progression (post‐treatment after 2 years) (n = only patients with a X‐ray) Celecoxib continuous: n = 12 (total N = 76) Celecoxib on‐demand: n = 26 (total N = 74) 16. Number of serious adverse events (post‐treatment after 2 years) Celecoxib continuous: n = 22 (total N = 111) Celecoxib on‐demand: n = 16 (total N = 103) 17. Number of adverse events per organ system (post‐treatment after 2 years) Celecoxib continuous: cardiovascular 14, gastro‐intestinal 102, hepatic 3, respiratory 59, haematological 1, renal 1, neurologic 20, dermatological 14 (total N = 111) Celecoxib on‐demand: cardiovascular 11, gastro‐intestinal 75, hepatic 0, respiratory 61, haematological 3, renal 0, neurologic 18, dermatological 14 (total N = 103) |
|
| Notes | Not included in the meta‐analysis, because the study was not suitable for any of the comparisons in this review. Available results are reported in this table. A post‐hoc analysis of this study was also included in the review (Kroon 2012), for detailed description see Table 4. Funding source: Supported by an unrestricted grant from Pharmacia. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Randomization was performed using a computer‐generated randomization list". |
| Allocation concealment (selection bias) | Unclear risk | No information provided on allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Participants were not blinded because the treatment regimens differed. However, it questionable whether this introduced bias. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Only mSASSS scoring was blinded. Other outcomes were assessed by self‐report. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | "Missing values for variables assessing signs and symptoms were replaced by the last observation that was present, which was carried forward, provided that at least one value obtained while the patient was receiving treatment was available." "...imputation of missing data by different means did not influence the direction of the between‐group difference..." 86% in continuous treatment and 83% in on‐demand treatment completed trial. For radiographic progression only patients with a complete set of radiographs were used. The BL characteristics did not differ, indicating a non‐selective group of participants. |
| Selective reporting (reporting bias) | Low risk | All outcomes stated in the methods were reported. |
| Other bias | Low risk | Compliance with treatment regimen was high. No other biases were detected. |
Abbreviations: APAP = acetaminophen; ARA = American Rheumatism Association; AS = ankylosing spondylitis; ASAS = Assessment of SpondyloArthritis international Society; BASDAI = Bath Ankylosing Spondylitis Disease Activity Index; BASFI = Bath Ankylosing Spondylitis Functional Index; BASMI = Bath Ankylosing Spondylitis Metrology Index; BL = baseline; CCT = controlled clinical trial; CI = confidence interval; cm = centimetre; CNS = central nervous system; COX = cyclo‐oxygenase; CRP = C‐reactive protein; ESR = erythrocyte sedimentation rate; ESSG = European Spondylarthropathy Study Group; GI = gastrointestinal; HLA = human like antigen; mg = milligram; mm = millimetre; MTX = methotrexate; NA = not available; NSAID = non‐steroidal antiinflammatory drug; RCT = randomised controlled trial; SD = standard deviation; SE = standard error; SpA = spondylarthritis; SSZ = sulfasalazine; TNF = tumour necrosis factor; VAS = visual analogue scale; WBC = white blood cell count.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Armstrong 1984 | Cross‐over study without separate data of first part of the study. |
| Bird 1980 | Cross‐over study without separate data of first part of the study. |
| Burry 1980 | Cross‐over study without separate data of first part of the study. |
| Byron 1982 | Cross‐over study without separate data of first part of the study. |
| Calin 1974 | Cross‐over study without separate data of first part of the study. |
| Charlot 1982 | Wrong (presentation of) outcomes (only percent of improvement available). |
| Dougados 1989 | Cross‐over study without separate data of first part of the study. |
| Doury 1986 | Cross‐over study without separate data of first part of the study. |
| Esdaile 1982 | Cross‐over study without separate data of first part of the study. |
| Gibson 1980 | Cross‐over study without separate data of first part of the study. |
| Harkness 1977 | Cross‐over study without separate data of first part of the study. |
| Johnson 1992 | Cross‐over study without separate data of first part of the study. |
| Kennedy 1991 | Cross‐over study without separate data of first part of the study. |
| Kinsella 1967 | Cross‐over study without separate data of first part of the study. |
| Mayrhofer 1990 | Wrong (presentation of) outcomes (data presented categorical and in proportions). |
| Peloso 2011 | Wrong (presentation of) outcomes (post‐hoc analysis without relevant outcomes for this review). |
| Peter 1975 | Cross‐over study without separate data of first part of the study. |
| Sadowska‐Wroblewska 1980 | Wrong (presentation of) outcomes (data presented categorical and in proportions). |
| Schattenkirchner 1980 | No comparator. |
| Shipley 1980 | Cross‐over study without separate data of first part of the study. |
| Sieper 2014 | Wrong study design. |
| Simpson 1968 | Wrong (presentation of) outcomes (data presented categorical and in proportions). |
| Sturrock 1974 | Cross‐over study without separate data of first part of the study. |
| Thompson 1977 | Cross‐over study without separate data of first part of the study. |
| Van Gerwen 1978 | Cross‐over study without separate data of first part of the study. |
| Wasner 1981 | Cross‐over study without separate data of first part of the study. |
| Wordsworth 1980 | Cross‐over study without separate data of first part of the study. |
Characteristics of studies awaiting assessment [ordered by study ID]
1966.
| Methods | ‐ |
| Participants | ‐ |
| Interventions | Indomethacin |
| Outcomes | ‐ |
| Notes | Title: A nonsteroidal anti‐inflammatory agent. Indomethacin (indocin) We could not locate the full‐text article. |
Acqaviva 1983.
| Methods | ‐ |
| Participants | ‐ |
| Interventions | Bi‐Profenid |
| Outcomes | ‐ |
| Notes | Title: Clinical study of Bi‐Profenid in rheumatologic practice We could not locate the full‐text article. |
Aeidler 1975.
| Methods | ‐ |
| Participants | ‐ |
| Interventions | Naproxen vs indomethacin |
| Outcomes | ‐ |
| Notes | Title: Clinical results of a multicentral double‐blind examination of naproxen compared to indomethacin in chronic rheumatoid arthritis, ankylosing spondylitis, and osteoarthrosis We could not locate the full‐text article. |
Bachmann 1984.
| Methods | ‐ |
| Participants | ‐ |
| Interventions | Ibuprofen vs indomethacin |
| Outcomes | ‐ |
| Notes | Title: Comparison between sustained release formulations of ibuprofen and indomethacin in treatment of ankylosing spondylitis We could not locate the full‐text article. |
Baerwald 1999.
| Methods | ‐ |
| Participants | ‐ |
| Interventions | Oral hydrolytic enzymes vs indomethacin |
| Outcomes | ‐ |
| Notes | Title: Efficacy and tolerance of oral hydrolytic enzymes in ankylosing spondylitis as compared with indomethacin: A controlled double‐blind prospective clinical trial We could not locate the full‐text article. |
Becvar 1949.
| Methods | ‐ |
| Participants | ‐ |
| Interventions | Ethophenamate injections |
| Outcomes | ‐ |
| Notes | Title: Ethophenamate injections in treatment of acute painful conditions in rheumatic diseases We could not locate the full‐text article, unlikely to contain separate information for SpA. |
Beltrán Gutiérrez 1968.
| Methods | ‐ |
| Participants | ‐ |
| Interventions | Flufenamic acid vs oxyphenylbutazone |
| Outcomes | ‐ |
| Notes | Title: Double‐blind study using flufenamic acid (F.I. 440) and oxyphenylbutazone in rheumatoid arthritis and ankylosing spine We could not locate the full‐text article. |
Bernstein 1992.
| Methods | ‐ |
| Participants | ‐ |
| Interventions | Lornoxicam vs indomethacin |
| Outcomes | ‐ |
| Notes | Title: A comparison of the efficacy and tolerability of lornoxicam and indomethacin in ankylosing spondylitis We could not locate the full‐text article. |
Bird 1986.
| Methods | ‐ |
| Participants | ‐ |
| Interventions | Tenoxicam vs piroxicam |
| Outcomes | ‐ |
| Notes | Title: A parallel group comparison of tenoxicam and piroxicam in patients with ankylosing spondylitis We could not locate the full‐text article. |
Bocci 1972.
| Methods | ‐ |
| Participants | ‐ |
| Interventions | Indomethacin |
| Outcomes | ‐ |
| Notes | Title: Controlled clinical study of the therapeutic activity and side‐effects of a preparation of indomethacin in lactocomplex We could not locate the full‐text article. |
Burdeĭnĭ 1981.
| Methods | ‐ |
| Participants | ‐ |
| Interventions | Voltaren vs indomethacin |
| Outcomes | ‐ |
| Notes | Title: Double blind trial of voltaren and indomethacin in Bechterew's disease We could not locate the full‐text article. |
Dougados 2000.
| Methods | ‐ |
| Participants | ‐ |
| Interventions | Piroxicam vs meloxicam |
| Outcomes | ‐ |
| Notes | Title: Double‐blind, placebo‐controlled clinical study over 52 weeks on M. Bechterew patients with 20 mg Piroxicam, 15 mg and 22,5 mg Meloxicam We could not locate the full‐text article. |
Droste 1979.
| Methods | ‐ |
| Participants | ‐ |
| Interventions | Tolmetin |
| Outcomes | ‐ |
| Notes | Title: Tolmetin 400 mg capsules in treatment of ankylosing spondylitis We could not locate the full‐text article. |
Droste 1985.
| Methods | ‐ |
| Participants | ‐ |
| Interventions | Pirprofen |
| Outcomes | ‐ |
| Notes | Title: Pirprofen in the treatment of ankylosing spondylitis. A 3‐week open trial We could not locate the full‐text article. |
Dutu 1982.
| Methods | ‐ |
| Participants | ‐ |
| Interventions | Tolmetin vs phenylbutazone |
| Outcomes | ‐ |
| Notes | Title: Treatment with tolmetin in ankylosing spondylitis. Comparative cross‐study with phenylbutazone. We could not locate the full‐text article. |
Franke 1975.
| Methods | ‐ |
| Participants | ‐ |
| Interventions | Bumadizone‐calcium‐semihydrate (Eumotol) vs oxyphenbutazone |
| Outcomes | ‐ |
| Notes | Title: Bumadizone‐calcium‐semihydrate (Eumotol) and oxyphenbutazone in the treatment of ankylosing spondylitis. A clinical double‐blind study We could not locate the full‐text article, NSAID only available in a few countries. |
ICTRP.
| Methods | ‐ |
| Participants | ‐ |
| Interventions | Etoricoxib vs naproxen |
| Outcomes | ‐ |
| Notes | Title: Etoricoxib with naproxen in ankylosing spondylitis We could not locate the full‐text article. Clinical trial found in WHO ICTRP database. |
Jajic 1982.
| Methods | ‐ |
| Participants | ‐ |
| Interventions | Pirprofen vs indomethacin |
| Outcomes | ‐ |
| Notes | Title: Pirprofen, indomethacin and placebo in ankylosing spondylitis. Double‐blind comparison We could not locate the full‐text article. |
Leng Levy 1963.
| Methods | ‐ |
| Participants | ‐ |
| Interventions | Tanderil |
| Outcomes | ‐ |
| Notes | Title: Clinical Results of Tanderil Administration in Gout and Rheumatic Pelvispondylitis We could not locate the full‐text article, NSAID only available in a few countries. |
Maier‐Lenz 1981.
| Methods | ‐ |
| Participants | ‐ |
| Interventions | Exrheudon vs phenylbutazone |
| Outcomes | ‐ |
| Notes | Title: Results of a controlled double‐blind comparison of Exrheudon and phenylbutazone We could not locate the full‐text article. |
Mayrhofer 1991.
| Methods | ‐ |
| Participants | ‐ |
| Interventions | NSAIDs |
| Outcomes | ‐ |
| Notes | Title: The efficacy and tolerance of NSAIDs in patients with ankylosing spondylitis We could not locate the full‐text article. |
Mertz 1981.
| Methods | ‐ |
| Participants | ‐ |
| Interventions | Proquazone |
| Outcomes | ‐ |
| Notes | Title: Efficacy and tolerance of proquazone in ankylosing spondylarthritis We could not locate the full‐text article, NSAID not marketed for SpA anymore. |
Müller‐Fassbender 1979.
| Methods | ‐ |
| Participants | ‐ |
| Interventions | Tolmetin |
| Outcomes | ‐ |
| Notes | Title: Tolmetin treatment for ankylosing spondylitis We could not locate the full‐text article. |
Müller‐Fassbender 1981.
| Methods | ‐ |
| Participants | ‐ |
| Interventions | Tolmetin 800 vs 1200 mg |
| Outcomes | ‐ |
| Notes | Title: Treatment of ankylosing spondylitis with tolmetin in doses of 800 or 1200 mg We could not locate the full‐text article. |
NCT00367211.
| Methods | ‐ |
| Participants | ‐ |
| Interventions | PN 200 vs naproxen |
| Outcomes | ‐ |
| Notes | Title: Study to Evaluate the Incidence of Gastric Ulcers Following Administration of Either PN 200 or Naproxen in Subjects Who Are at Risk for Developing NSAID‐Associated Ulcers No trial publication could be located. Protocol on ClinicalTrials.gov. This study has been completed. |
NCT00715091.
| Methods | ‐ |
| Participants | ‐ |
| Interventions | Experimental intervention: continuous (daily) treatment with diclofenac cholestyramine 150 mg (Voltaren Resinate), divided into 75 mg Voltaren twice dailyControl intervention: treatment on‐demand (as needed) with diclofenac‐cholestyramine 75 to 150 mg (Voltaren Resinate) |
| Outcomes | ‐ |
| Notes | Title: Effects of Non‐Steroidal Anti‐Inflammatory Drugs (NSAIDs) on RAdiographic Damage in Ankylosing Spondylitis (ENRADAS) No trial publication could be located. Protocol on ClinicalTrials.gov. This study has been completed. |
NCT00766402.
| Methods | ‐ |
| Participants | ‐ |
| Interventions | Ultracet vs diclofenac |
| Outcomes | ‐ |
| Notes | Title: An Efficacy and Safety Study of Tramadol/Acetaminophen Versus Diclofenac in the Treatment of Pain in Participants With Ankylosing Spondylitis Receiving Stable Treatment of Disease Modifying Anti‐rheumatic Drugs (DMARDs) No trial publication could be located. Protocol on ClinicalTrials.gov. This study has been terminated. |
NCT01077843.
| Methods | ‐ |
| Participants | ‐ |
| Interventions | Etoricoxib and Other Anti‐inflammatory Therapies |
| Outcomes | ‐ |
| Notes | Title: Post‐authorization Safety Study of Etoricoxib and Other Anti‐inflammatory Therapies in European Patients With Ankylosing Spondylitis No trial publication could be located. Protocol on ClinicalTrials.gov. This study is ongoing, but not recruiting participants. |
NCT01091675.
| Methods | ‐ |
| Participants | ‐ |
| Interventions | Etoricoxib 90 mg |
| Outcomes | ‐ |
| Notes | Title: Assessment of the Response to Etoricoxib in Patients With Ankylosing Spondylitis (AS) and Inadequate Response to ≥2 NSAIDs No trial publication could be located. Protocol on ClinicalTrials.gov. This study has been completed. |
NCT01176682.
| Methods | ‐ |
| Participants | ‐ |
| Interventions | NSAID |
| Outcomes | ‐ |
| Notes | Title: Observational Study on Non‐steroid Anti‐inflammatory Drugs (NSAIDs) Treated Patients With Arthritic Disorder (EVIDENCE) No trial publication could be located. Protocol on ClinicalTrials.gov. This study has been completed. |
NCT01208207.
| Methods | ‐ |
| Participants | ‐ |
| Interventions | Etoricoxib vs naproxen |
| Outcomes | ‐ |
| Notes | Title: A Two‐Part 26‐Week Study of Etoricoxib as Treatment for Ankylosing Spondylitis (AS) No trial publication could be located. Protocol on ClinicalTrials.gov. This study has been completed. |
NCT01685424.
| Methods | ‐ |
| Participants | ‐ |
| Interventions | Etoricoxib |
| Outcomes | ‐ |
| Notes | Title: Etoricoxib Prescribing Patterns and Adverse Events of Interest in Primary Care in the United Kingdom No trial publication could be located. Protocol on ClinicalTrials.gov. This study is ongoing, but not recruiting participants. |
Orozco Medina 1983.
| Methods | ‐ |
| Participants | ‐ |
| Interventions | Indoprofen |
| Outcomes | ‐ |
| Notes | Title: Evaluation of efficacy and tolerance of oral indoprofen in patients with ankylosing spondilitis We could not locate the full‐text article, NSAID not marketed for SpA. |
Pattin 1990.
| Methods | ‐ |
| Participants | ‐ |
| Interventions | Etodolac vs piroxicam |
| Outcomes | ‐ |
| Notes | Title: Efficacy, safety and therapeutic benefit of etodolac (600 mg daily) versus piroxicam (20 mg daily) We could not locate the full‐text article. |
Reiter 1984.
| Methods | ‐ |
| Participants | ‐ |
| Interventions | Acemetacin vs piroxicam |
| Outcomes | ‐ |
| Notes | Title: Ankylosing spondylitis: comparison of acemetacin and piroxicam We could not locate the full‐text article. |
Renier 1982.
| Methods | ‐ |
| Participants | ‐ |
| Interventions | Pirprofen vs ketoprofen |
| Outcomes | ‐ |
| Notes | Title: Ankylosing spondylitis. Comparative trial of two non‐steroidal anti‐inflammatory agents: pirprofen and ketoprofen We could not locate the full‐text article. |
Schattenkirchner 1981.
| Methods | ‐ |
| Participants | ‐ |
| Interventions | Piroxicam |
| Outcomes | ‐ |
| Notes | Title: Long‐time results of Felden (piroxicam) in ankylosing spondylitis We could not locate the full‐text article. |
Schattenkirchner 1986.
| Methods | ‐ |
| Participants | ‐ |
| Interventions | Acemetacin vs diclofenac |
| Outcomes | ‐ |
| Notes | Title: Treatment of ankylosing spondylitis: Single‐blind crossover comparison of acemetacin and diclofenac We could not locate the full‐text article. |
Simon 1987.
| Methods | ‐ |
| Participants | ‐ |
| Interventions | Pirprofene vs phenylbutazone |
| Outcomes | ‐ |
| Notes | Title: A comparative study of pirprofene and phenylbutazone in the treatment of ankylosing spondylitis We could not locate the full‐text article. |
Stollenwerk 1985.
| Methods | ‐ |
| Participants | ‐ |
| Interventions | Piroxicam vs indomethacin |
| Outcomes | ‐ |
| Notes | Title: Comparative study of the treatment of ankylosing spondylitis with piroxicam suppositories and indomethacin suppositories in combination with indomethacin retard capsules We could not locate the full‐text article. |
Characteristics of ongoing studies [ordered by study ID]
ClinicalTrials.gov 2014a.
| Trial name or title | Delaying Ossification and Improving Inflammation of Celebrex Plus/or Enbrel Treatment on Active Ankylosing Spondylitis |
| Methods | ‐ |
| Participants | ‐ |
| Interventions | ‐ |
| Outcomes | ‐ |
| Starting date | ‐ |
| Contact information | ‐ |
| Notes | http://clinicaltrials.gov/ct2/show/NCT01934933?type=Intr&cond=Spondylitis&rcv_s=06%2F01%2F2013&rank=2 NCT01934933 |
ClinicalTrials.gov 2014b.
| Trial name or title | Treatment of Axial Spondyloarthritis With Reduced Doses of NSAIDs |
| Methods | ‐ |
| Participants | ‐ |
| Interventions | ‐ |
| Outcomes | ‐ |
| Starting date | ‐ |
| Contact information | ‐ |
| Notes | http://clinicaltrials.gov/ct2/show/NCT02089529?type=Intr&cond=Spondylitis&rcv_s=06%2F01%2F2013&rank=9 NCT02089529 |
Differences between protocol and review
We included extension phases and post‐hoc analyses of RCTs to enable a comprehensive overview of the benefits and harms of NSAIDs. This was pre‐planned, but not clearly specified in the protocol.
The prespecified main comparison of this review was NSAIDs versus placebo. However, because many trials included both traditional and COX‐2 NSAIDs we presented the results of traditional and COX‐2 NSAIDS versus placebo separately. We also performed a post‐hoc subgroup analysis of traditional versus COX‐2 NSAIDs for one of our main outcomes (pain on VAS).
The approach to handling unit of analysis issues as specified in the Methods ("We split the group with the 'shared' intervention into two equally large groups to include two comparisons if deemed necessary. Whenever we had to decide between multiple dosages of a NSAID for studies containing more than two intervention groups, we used the proposed equivalent dose of 150 mg Diclofenac based on voting during the ASAS annual meeting (Dougados 2011).") was pre‐planned, but not clearly specified in the protocol.
The approach to dealing with missing data as specified in the Methods ("If studies with final measurement data and change scores had to be combined using a SMD (e.g. because the studies used different scales), we calculated the final measurement data from the studies presenting change scores and imputed the SD for these final measurement data from the baseline SD from the same study.). Where data were presented graphically only, we extracted data from the graph when possible.") was pre‐planned, but not clearly specified in the protocol.
Contributions of authors
FK contributed to development of the protocol, study selection, data extraction and management, data analysis, interpretation of the data and writing of the review. LvdB contributed to development of the protocol, study selection, data extraction and management, data analysis, interpretation of the data and writing of the review. SR contributed to development of the protocol, study selection, data analysis, interpretation of the data and writing of the review. RL contributed to development of the protocol, interpretation of data and writing of the review. RB contributed to development of the protocol, interpretation of data and writing of the review. LF devised the search strategy, and executed and updated the search for the review. DvdH contributed to development of the protocol, interpretation of data and writing of the review.
Sources of support
Internal sources
No sources of support received, Other.
External sources
No sources of support received, Other.
Declarations of interest
FK was a principal investigator in Kroon 2012. RL was a principal investigator in Kroon 2012 and Wanders 2005. DvdH was a principal investigator in Dougados 2001, Gossec 2005, Kroon 2012, van der Heijde 2005 and Wanders 2005.
New
References
References to studies included in this review
Ansell 1978 {published data only}
- Ansell BM. A comparative study of Butacote and naproxen in ankylosing spondylitis. Journal of International Medical Research 1977;5(Suppl 2):95. [PubMed] [Google Scholar]
- Ansell BM, Major G, Liyanage SP, Gumpel JM, Seifert MH, Mathews JA, et al. A comparative study of Butacote and Naprosyn in ankylosing spondylitis. Annals of the Rheumatic Diseases 1978;37(5):436‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Astorga 1987 {published data only}
- Astorga G. Double‐blind, parallel clinical trial of tenoxicam (Ro 12‐0068) versus piroxicam in patients with ankylosing spondylitis. European Journal of Rheumatology and Inflammation 1987;9(2):70‐3. [PubMed] [Google Scholar]
Barkhuizen 2006 {published data only}
- Barkhuizen A, Steinfeld S, Robbins J, West C, Coombs J, Zwillich S. Celecoxib is efficacious and well tolerated in treating signs and symptoms of ankylosing spondylitis. Journal of Rheumatology 2006;33(9):1805‐12. [PubMed] [Google Scholar]
Batlle‐Gualda 1996 {published data only}
- Batlle‐Gualda E, Figueroa M, Ivorra J, Raber A. The efficacy and tolerability of aceclofenac in the treatment of patients with ankylosing spondylitis: a multicenter controlled clinical trial. Aceclofenac Indomethacin Study Group. Journal of Rheumatology 1996;23(7):1200‐6. [PubMed] [Google Scholar]
Boersma 1976 {published data only}
- Boersma JW. Retardation of ossification of the lumbar vertebral column in ankylosing spondylitis by means of phenylbutazone. Scandinavian Journal of Rheumatology 1976;5(1):60‐4. [PubMed] [Google Scholar]
Caldwell 1986 {published data only}
- Caldwell JR, Altman RD, Burch FX, Calin A. Treatment of ankylosing spondylitis with oxaprozin: A comparison with indomethacin. Seminars in Arthritis and Rheumatism 1986;15(3 Suppl 2):95‐100. [Google Scholar]
Calin 1979 {published data only}
- Calin A, Britton M. Sulindac in ankylosing spondylitis. Double‐blind evaluation of sulindac and indomethacin. JAMA 1979;242(17):1885‐6. [PubMed] [Google Scholar]
Carcassi 1990 {published data only}
- Carcassi C, Nasa G, Perpignano G. A 12‐week double‐blind study of the efficacy, safety and tolerance of pirazolac b.i.d. compared with indomethacin t.i.d. in patients with ankylosing spondylitis. Drugs Under Experimental and Clinical Research 1990;16(1):29‐37. [PubMed] [Google Scholar]
Dougados 1994 {published data only}
- Dougados M, Nguyen M, Caporal R, Legeais J, Bouxin‐Sauzet A, Pellegri‐Guegnault B, et al. Ximoprofen in ankylosing spondylitis. A double blind placebo controlled dose ranging study. Scandinavian Journal of Rheumatology 1994;23(5):243‐8. [DOI] [PubMed] [Google Scholar]
Dougados 1999 {published data only}
- Dougados M, Gueguen A, Nakache JP, Velicitat P, Veys EM, Zeidler H, et al. Ankylosing spondylitis: what is the optimum duration of a clinical study? A one year versus a 6 weeks non‐steroidal anti‐inflammatory drug trial. Rheumatology 1999;38(3):235‐44. [DOI] [PubMed] [Google Scholar]
Dougados 2001 {published data only}
- Dougados M, Béhier JM, Jolchine I, Calin A, Heijde D, Olivieri I, et al. Efficacy of celecoxib, a cyclooxygenase 2‐specific inhibitor, in the treatment of ankylosing spondylitis: a six‐week controlled study with comparison against placebo and against a conventional nonsteroidal antiinflammatory drug. Arthritis and Rheumatism 2001;44(1):180‐5. [DOI] [PubMed] [Google Scholar]
Ebner 1983 {published data only}
- Ebner W, Poal Ballarin JM, Boussina I. Meclofenamate sodium in the treatment of ankylosing spondylitis. Report of a European double‐blind controlled multicenter study. Arzneimittel‐Forschung 1983;33(4A):660‐3. [PubMed] [Google Scholar]
Franssen 1986 {published data only}
- Franssen MJ, Gribnau FW, Putte LB. A comparison of diflunisal and phenylbutazone in the treatment of ankylosing spondylitis. Clinical Rheumatology 1986;5(2):210‐20. [DOI] [PubMed] [Google Scholar]
Good 1977 {published data only}
- Good A, Mena H. Treatment of ankylosing spondylitis with flurbiprofen and indomethacin. Current Medical Research and Opinion 1977;5(1):117‐21. [DOI] [PubMed] [Google Scholar]
- Mena HR, Good AE. Management of ankylosing spondylitis with flurbiprofen or indomethacin. Southern Medical Journal 1977;70(8):945‐7. [DOI] [PubMed] [Google Scholar]
Heinrichs 1985 {published data only}
- Heinrichs K, Kriech W. Treatment of spondylitis ankylosans: Controlled comparative study with tiaprofenic acid and diclofenac [Behandlung der Spondylitis Ankylosans: Kontrollierte Vergleichsstudie mit Tiaprofensaure und Diclofenac]. Medizinische Klinik 1985;80(21):597‐601. [Google Scholar]
Jessop 1976 {published data only}
- Jessop JD. Double‐blind study of ketoprofen and phenylbutazone in ankylosing spondylitis. Rheumatology and Rehabilitation 1976;Suppl:37‐42. [DOI] [PubMed] [Google Scholar]
Khan 1985 {published data only}
- Calabro JJ. Efficacy of diclofenac in ankylosing spondylitis. American Journal of Medicine 1986;80(4B):58‐63. [DOI] [PubMed] [Google Scholar]
- Khan MA. A double blind comparison of diclofenac and indomethacin in the treatment of ankylosing spondylitis. Journal of Rheumatology 1987;14(1):118‐23. [PubMed] [Google Scholar]
- Khan MA. Diclofenac in the treatment of ankylosing spondylitis: review of worldwide clinical experience and report of a double‐blind comparison with indomethacin. Seminars in Arthritis and Rheumatism 1985;15(2 Suppl 1):80‐4. [DOI] [PubMed] [Google Scholar]
Lehtinen 1984 {published data only}
- Lehtinen K, Kaarela K, Mäkisara P, Holttinen K, Gordin A. Tolerability and efficacy of a slow‐release indomethacin tablet in ankylosing spondylitis. British Journal of Rheumatology 1984;23(1):52‐6. [DOI] [PubMed] [Google Scholar]
Lomen 1986 I {published data only}
- Lomen PL, Turner LF, Lamborn KR, Brinn EL. Flurbiprofen in the treatment of ankylosing spondylitis. A comparison with indomethacin. American Journal of Medicine 1986;80(3A):127‐32. [DOI] [PubMed] [Google Scholar]
Lomen 1986 P {published data only}
- Lomen PL, Turner LF, Lamborn KR, Brinn EL, Sattler LP. Flurbiprofen in the treatment of ankylosing spondylitis. A comparison with phenylbutazone. American Journal of Medicine 1986;80(3A):120‐6. [DOI] [PubMed] [Google Scholar]
Mena 1977 {published data only}
- Mena HR, Willkens RF. Treatment of ankylosing spondylitis with flurbiprofen or phenylbutazone. European Journal of Clinical Pharmacology 1977;11(4):263‐6. [DOI] [PubMed] [Google Scholar]
Muller‐Fassbender 1985 {published data only}
- Muller‐Fassbender H. Ketoprofen in ankylosing spondylitis [Ketoprofen bei Morbus Bechterew]. Therapiewoche 1985;35(21):2619‐24. [Google Scholar]
Myklebust 1986 {published data only}
- Myklebust G. Comparison of naproxen and piroxicam in rheumatoid arthritis and Bechterew's syndrome. A double‐blind parallel multicenter study [Naproxen sammenlignet med piroxicam ved reumatoid artritt og Bechterews syndrom]. Tidsskrift for Den Norske Lægeforening 1986;106(17‐8):1485‐7. [PubMed] [Google Scholar]
Nahir 1980 {published data only}
- Nahir AM, Scharf Y. A comparative study of diclofenac and sulindac in ankylosing spondylitis. Rheumatology and Rehabilitation 1980;19(3):189‐98. [DOI] [PubMed] [Google Scholar]
Nissilä 1978a {published data only}
- Nissilä M, Kajander A. Proquazone (Biarison) and indomethacin (Indocid) in the treatment of ankylosing spondylitis. Two comparative, clinical, double‐blind studies. Scandinavian Journal of Rheumatology. Supplement 1978;21:36‐9. [DOI] [PubMed] [Google Scholar]
Nissilä 1978b {published data only}
- Nissilä M, Kajander A. Proquazone (Biarison) and indomethacin (Indocid) in the treatment of ankylosing spondylitis. Two comparative, clinical, double‐blind studies. Scandinavian Journal of Rheumatology. Supplement 1978;21:36‐9. [DOI] [PubMed] [Google Scholar]
Palferman 1991 {published data only}
- Palferman TG, Webley M. A comparative study of nabumetone and indomethacin in ankylosing spondylitis. European Journal of Rheumatology and Inflammation 1991;11(2):23‐9. [PubMed] [Google Scholar]
Pasero 1994 {published data only}
- Pasero G, Ruju G, Marcolongo R, Senesi M, Seni U, Mannoni A, et al. Aceclofenac versus naproxen in the treatment of ankylosing spondylitis: A double‐blind, controlled study. Current Therapeutic Research ‐ Clinical and Experimental 1994;55(7):833‐42. [Google Scholar]
Poddubnyy 2012 {published data only}
- Poddubnyy D, Haibel H, Listing J, Marker‐Hermann E, Zeidler H, Braun J, et al. NSAIDs retard radiographic spinal progression over two years in ankylosing spondylitis but not in non‐radiographic axial spondyloarthritis. Zeitschrift fur Rheumatologie 2011;70:102‐3. [Google Scholar]
- Poddubnyy D, Rudwaleit M, Haibel H, Listing J, Märker‐Hermann E, Zeidler H, et al. Effect of non‐steroidal anti‐inflammatory drugs on radiographic spinal progression in patients with axial spondyloarthritis: results from the German Spondyloarthritis Inception Cohort. Annals of the Rheumatic Diseases 2012;71(10):1616‐22. [DOI] [PubMed] [Google Scholar]
Rejholec 1980 {published data only}
- Rejholec V, Vapaatalo H, Tokola O, Gothoni G. Tolfenamic acid in ankylosing spondylarthritis: a double‐blind comparison to indomethacin. Scandinavian Journal of Rheumatology. Supplement 1980;36:1‐7. [PubMed] [Google Scholar]
Santo 1988 {published data only}
- Santo JE, Queiroz MV. Oxaprozin versus diclofenac sodium in the treatment of ankylosing spondylitis. Journal of International Medical Research 1988;16(2):150‐6. [DOI] [PubMed] [Google Scholar]
Schwarzer 1990 {published data only}
- Schwarzer AC, Cohen M, Arnold MH, Kelly D, McNaught P, Brooks PM. Tenoxicam compared with diclofenac in patients with ankylosing spondylitis. Current Medical Research and Opinion 1990;11(10):648‐53. [DOI] [PubMed] [Google Scholar]
Sieper 2008 {published data only}
- Sieper J, Klopsch T, Richter M, Kapelle A, Rudwaleit M, Schwank S, et al. Comparison of two different dosages of celecoxib with diclofenac for the treatment of active ankylosing spondylitis: results of a 12‐week randomised, double‐blind, controlled study. Annals of the Rheumatic Diseases 2008;67(3):323‐9. [DOI] [PubMed] [Google Scholar]
Simpson 1966 {published data only}
- Simpson MR, Simpson NR, Scott BO, Beatty DC. A controlled study of flufenamic acid in ankylosing spondylitis. A preliminary report. Annals of Physical Medicine 1966;Suppl:126‐8. [DOI] [PubMed] [Google Scholar]
Sydnes 1981 {published data only}
- Sydnes OA. Comparison of piroxicam with indomethacin in ankylosing spondylitis: a double‐blind crossover trial. British Journal of Clinical Practice 1981;35(1):40‐4. [PubMed] [Google Scholar]
Tannenbaum 1984 {published data only}
- Tannenbaum H, DeCoteau WE, Esdaile JM. A double blind multicenter trial comparing piroxicam and indomethacin in ankylosing spondylitis with long‐term follow‐up. Current Therapeutic Research ‐ Clinical and Experimental 1984;36(3):426‐35. [Google Scholar]
van der Heijde 2005 {published and unpublished data}
- Gossec L, Heijde D, Meilian A, Krupa DA, James MK, Cavanaugh PF, et al. Efficacy of cyclo‐oxygenase‐2 inhibition by etoricoxib and naproxen on the axial manifestations of ankylosing spondylitis in the presence of peripheral arthritis. Annals of the Rheumatic Diseases 2005;64(11):1563‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heijde D, Baraf HSB, Ramos‐Remus C, Calin A, Weaver AL, Schiff M, et al. Evaluation of the efficacy of etoricoxib in ankylosing spondylitis: results of a fifty‐two‐week, randomized, controlled study. Arthritis and Rheumatism 2005;52(4):1205‐15. [DOI] [PubMed] [Google Scholar]
Villa Alcázar 1996 {published data only}
- Villa Alcázar LF, Buergo M, Rico Lenza H, Montull Fruitós E. Aceclofenac is as safe and effective as tenoxicam in the treatment of ankylosing spondylitis: a 3 month multicenter comparative trial. Journal of Rheumatology 1996;23(7):1194‐9. [PubMed] [Google Scholar]
Wanders 2005 {published and unpublished data}
- Kroon F, Landewé R, Dougados M, Heijde D. Continuous NSAID use reverts the effects of inflammation on radiographic progression in patients with ankylosing spondylitis. Annals of the Rheumatic Diseases 2012;71(10):1623‐9. [DOI] [PubMed] [Google Scholar]
- Wanders A, Heijde D, Landewé R, Béhier JM, Calin A, Olivieri I, et al. Nonsteroidal antiinflammatory drugs reduce radiographic progression in patients with ankylosing spondylitis: a randomized clinical trial. Arthritis and Rheumatism 2005;52(6):1756‐65. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Armstrong 1984 {published data only}
- Amstrong RD, Laurent R, Panayi GS. A comparison of indoprofen and indomethacin in the treatment of ankylosing spondylitis. Pharmatherapeutica 1984;3(10):637‐41. [PubMed] [Google Scholar]
Bird 1980 {published data only}
- Bird HA, Rhind VM, Pickup ME, Wright V. A comparative study of benoxaprofen and indomethacin in ankylosing spondylitis. Journal of Rheumatology. Supplement 1980;6:139‐42. [PubMed] [Google Scholar]
Burry 1980 {published data only}
- Burry HC, Siebers R. A comparison of flurbiprofen with naproxen in ankylosing spondylitis. New Zealand Medical Journal 1980;92(670):309‐11. [PubMed] [Google Scholar]
Byron 1982 {published data only}
- Byron MA, Steele CE. A double‐blind crossover study comparing tolmetin sodium and naproxen in the treatment of ankylosing spondylitis. Current Medical Research and Opinion 1982;7:670‐6. [Google Scholar]
Calin 1974 {published data only}
- Calin A, Grahame R. Double‐bind cross‐over trial of flurbiprofen and phenylbutazone in ankylosing spondylitis. British Medical Journal 1974;4(5943):496‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Charlot 1982 {published data only}
- Charlot J, Villiaumey J. A comparative study of benoxaprofen and ketoprofen in ankylosing spondylitis. European Journal of Rheumatology and Inflammation 1982;5(2):277‐81. [PubMed] [Google Scholar]
Dougados 1989 {published data only}
- Dougados M, Caporal R, Doury P, Thiesce A, Pattin S, Laffez B, et al. A double blind crossover placebo controlled trial of ximoprofen in as. Journal of Rheumatology 1989;16(8):1167‐9. [PubMed] [Google Scholar]
Doury 1986 {published data only}
- Doury P, Roux H. Isoxicam vs ketoprofen in ankylosing spondylitis. British Journal of Clinical Pharmacology 1986;22(Suppl 2):157S‐60S. [DOI] [PMC free article] [PubMed] [Google Scholar]
Esdaile 1982 {published data only}
- Esdaile J, Rothwell R, MacLaughlin K, Percy J, Hawkins D. Double‐blind comparison of tolmetin sodium and indomethacin in ankylosing spondylitis. Journal of Rheumatology 1982;9(1):69‐74. [PubMed] [Google Scholar]
Gibson 1980 {published data only}
- Gibson T, Laurent R. Sulindac and indomethacin in the treatment of ankylosing spondylitis: a double‐blind cross‐over study. Rheumatology and Rehabilitation 1980;19(3):189‐92. [DOI] [PubMed] [Google Scholar]
Harkness 1977 {published data only}
- Harkness AJ, Burry HC, Grahame R. A trial of feprazone in ankylosing spondylitis. Rheumatology and Rehabilitation 1977;16(3):158‐61. [DOI] [PubMed] [Google Scholar]
Johnson 1992 {published data only}
- Johnsen V, Brun JG, Fjeld E, Hansen K, Sydnes OA, Ugstad MB. Morning stiffness and nightime pain in ankylosing spondylitis. A comparison between enteric‐coated and plain naproxen tablets. European Journal of Rheumatology & Inflammation 1992;12(2):37‐42. [PubMed] [Google Scholar]
Kennedy 1991 {published data only}
- Kennedy AC, Germain BF, Goldman AL, Vreede PD. A double‐blind, crossover comparison of ketoprofen and indomethacin in ankylosing spondylitis. Advances in Therapy 1991;8(3):148‐56. [Google Scholar]
Kinsella 1967 {published data only}
- Kinsella TD, MacKenzie KR, Kim SO, Johnson LG. Evaluation of indomethacin by a controlled, cross‐over technique in 30 patients with ankylosing spondylitis. Canadian Medical Association Journal 1967;96(22):1454‐9. [PMC free article] [PubMed] [Google Scholar]
Mayrhofer 1990 {published data only}
- Mayrhofer F, Broll H, Eberl R, Ebner W, Klein G, Rainer F, et al. Tenoxicam versus diclofenac in patients with ankylosing spondylitis. A double‐blind study. Drug Investigation 1990;2(Suppl 3):52‐3. [Google Scholar]
Peloso 2011 {published data only}
- Peloso PM, Gammaitoni A, Smugar SS, Wang H, Moore AR. Longitudinal numbers‐needed‐to‐treat (NNT) for achieving various levels of analgesic response and improvement with etoricoxib, naproxen, and placebo in ankylosing spondylitis. BMC Musculoskeletal Disorders 2011;12:165. [DOI] [PMC free article] [PubMed] [Google Scholar]
Peter 1975 {published data only}
- Peter E. A double blind comparison of naproxen and indomethacin on the after‐midnight‐pain of patients with morbus bechterew [Wirkungsvergleich von Naproxen und Indometacin im Doppelblindversuch beim Nachmitternachtsschmerz von Morbus Bechterew]. Arzneimittel‐Forschung 1975;25(2A):324‐5. [PubMed] [Google Scholar]
Sadowska‐Wroblewska 1980 {published data only}
- Sadowska‐Wroblewska M, Garwolinska H, Filipovicz‐Sosnowska A. Azapropazone versus indomethacin in a double blind test with patients with ankylosing spondylitis [Azapropazon Versus Indometacin im Doppelblindversuch bei Patienten mit Spondylitis ankylosans]. Zeitschrift für Rheumatologie 1980;39(11‐2):406‐11. [PubMed] [Google Scholar]
Schattenkirchner 1980 {published data only}
- Schattenkirchner M, Müller‐Fassbender H, Melzer H. An open long‐term study of piroxicam in ankylosing spondylitis patients [Eine offene Langzeitstudie mit Piroxicam bei Patienten mit Spondylitis ankylosans (M. Marie‐Strümpell, M.Bechterew)]. Aktuelle Rheumatologie 1980;5:38‐42. [Google Scholar]
Shipley 1980 {published data only}
- Shipley M, Berry H, Bloom B. A double‐blind cross‐over trial of indomethacin, fenoprofen and placebo in ankylosing spondylitis, with comments on patient assessment. Rheumatology and Rehabilitation 1980;19(2):122‐5. [DOI] [PubMed] [Google Scholar]
Sieper 2014 {published data only}
- Sieper J, Lenaerts J, Wollenhaupt J, Rudwaleit M, Mazurov VI, Myasoutova L, et al. Maintenance of biologic‐free remission with naproxen or no treatment in patients with early, active axial spondyloarthritis: results from a 6‐month, randomised, open‐label follow‐up study, INFAST Part 2. Annals of the Rheumatic Diseases 2014;73(1):108‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Simpson 1968 {published data only}
- Simpson MR, Simpson NR, Masheter HC. A controlled study of flufenamic acid in ankylosing spondylitis. A final report and follow‐up study. Annals of Physical Medicine 1968;9(6):229‐33. [DOI] [PubMed] [Google Scholar]
Sturrock 1974 {published data only}
- Sturrock RD, Hart FD. Double‐blind cross‐over comparison of indomethacin, flurbiprofen, and placebo in ankylosing spondylitis. Annals of the Rheumatic Diseases 1974;33(2):129‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
Thompson 1977 {published data only}
- Thompson M, Akyol MS. A controlled comparative trial of Butacote and fenclofenac in the treatment of ankylosing spondylitis. Journal of International Medical Research 1977;5(Suppl 2):96‐9. [PubMed] [Google Scholar]
Van Gerwen 1978 {published data only}
- Gerwen F, Korst JK, Gribnau FW. Double‐blind trial of naproxen and phenylbutazone in ankylosing spondylitis. Annals of the Rheumatic Diseases 1978;37(1):85‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wasner 1981 {published data only}
- Wasner C, Britton MC, Kraines RG, Kaye RL, Bobrove AM, Fries JF. Nonsteroidal anti‐inflammatory agents in rheumatoid arthritis and ankylosing spondylitis. JAMA 1981;246(19):2168‐72. [PubMed] [Google Scholar]
Wordsworth 1980 {published data only}
- Wordsworth BP, Ebringer RW, Coggins E, Smith S. A double‐blind cross‐over trial of fenoprofen and phenylbutazone in ankylosing spondylitis. Rheumatology and Rehabilitation 1980;19(4):260‐3. [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
1966 {published data only}
- Unknown. A nonsteroidal anti‐inflammatory agent. Indomethacin (indocin). JAMA 1966;196(7):651‐2. [DOI] [PubMed] [Google Scholar]
Acqaviva 1983 {published data only}
- Acqaviva B, Cayla J, Delcambre B, Goff P, Loyau G, Serratrice G, et al. Clinical study of Bi‐Profenid in rheumatologic practice [French]. La Semaine des Hôpitaux 1983;59(46):3229‐34. [PubMed] [Google Scholar]
Aeidler 1975 {published data only}
- Aeidler H. Clinical results of a multicentral double‐blind examination of naproxen compared to indomethacin in chronic rheumatoid arthritis, ankylosing spondylitis, and osteoarthrosis [German]. Arzneimittel‐Forschung 1975;25(2A):315‐8. [PubMed] [Google Scholar]
Bachmann 1984 {published data only}
- Bachmann F. Comparison between sustained release formulations of ibuprofen and indomethacin in treatment of ankylosing spondylitis [Zur Medikamentösen Therapie der Ankylosierenden Spondylitis mit Ibuprofen (Dolgit Retard) im Vergleich zu Indometacin Retard]. Therapiewoche 1984;34:4958‐64. [Google Scholar]
Baerwald 1999 {published data only}
- Baerwald CH, Willeke A, Lies S, Goebel KM, Engel HJ. Efficacy and tolerance of oral hydrolytic enzymes in ankylosing spondylitis as compared with indomethacin: A controlled double‐blind prospective clinical trial. Journal of Clinical Research 1999;2:17‐34. [Google Scholar]
Becvar 1949 {published data only}
- Becvar R, Pavelka K Jr. Ethophenamate injections in treatment of acute painful conditions in rheumatic diseases. Ceska Revmatologie 1994:79‐81. [Google Scholar]
Beltrán Gutiérrez 1968 {published data only}
- Beltrán Gutiérrez J, Fernández del Vallado P, Gijón Baños J. Double‐blind study using flufenamic acid (F.I. 440) and oxyphenylbutazone in rheumatoid arthritis and ankylosing spine. Revista Española de Reumatismo y Enfermedades Osteo‐Articulares 1968;12:219‐21. [PubMed] [Google Scholar]
Bernstein 1992 {published data only}
- Bernstein RM, Calin HJ, Ollier S, Calin A. A comparison of the efficacy and tolerability of lornoxicam and indomethacin in ankylosing spondylitis. European Journal of Rheumatology and Inflammation 1992;12:6‐13. [Google Scholar]
Bird 1986 {published data only}
- Bird HA, Gallez P, Astbury C, Looi D, Wright V. A parallel group comparison of tenoxicam and piroxicam in patients with ankylosing spondylitis. Pharmatherapeutica 1986;4(7):457‐62. [PubMed] [Google Scholar]
Bocci 1972 {published data only}
- Bocci U, Bignotti N, Luzi T, Pigani G, Santis AM. Controlled clinical study of the therapeutic activity and side‐effects of a preparation of indomethacin in lactocomplex [Italian]. Reumatismo 1972;24(1):1‐9. [PubMed] [Google Scholar]
Burdeĭnĭ 1981 {published data only}
- Burdeinyi AP. Double‐blind trial of the therapeutic efficacy of voltaren and indomethacin in Bechterew's disease. Terapevticheskii Arkhiv 1981;53(7):107‐11. [PubMed] [Google Scholar]
- Burdeĭnĭ AP. Double blind trial of voltaren and indomethacin in Bechterew's disease [Russian]. Terapevticheskiĭ Arkhiv 1981;53(7):107‐11. [PubMed] [Google Scholar]
Dougados 2000 {published data only}
- Dougados M, Calin A, Zeidler H. Double‐blind, placebo‐controlled clinical study over 52 weeks on M. Bechterew patients with 20 mg Piroxicam, 15 mg and 22,5 mg Meloxicam. Zeitschrift für Rheumatologie 2000;59(Suppl 3):49. [Google Scholar]
Droste 1979 {published data only}
- Droste U. Tolmetin 400 mg capsules in treatment of ankylosing spondylitis [Tolectin (Tolmetin) 400 Mg Kapseln in Der Behandlung Der Spondylitis Ankylosans]. Therapiewoche 1979;29:8901‐2. [Google Scholar]
Droste 1985 {published data only}
- Droste U. Pirprofen in the treatment of ankylosing spondylitis. A 3‐week open trial [German]. Fortschritte der Medizin 1985;103:173‐5. [PubMed] [Google Scholar]
Dutu 1982 {published data only}
- Dutu A, Bolosiu HD, Parasca I. Treatment with tolmetin in ankylosing spondylitis. Comparative cross‐study with phenylbutazone. Clujul Medical 1982;55:115‐8. [Google Scholar]
Franke 1975 {published data only}
- Franke M, Manz G. Bumadizone‐calcium‐semihydrate (Eumotol) and oxyphenbutazone in the treatment of ankylosing spondylitis. A clinical double‐blind study [German]. Die Medizinische Welt 1975;26(27‐8):1335‐40. [PubMed] [Google Scholar]
ICTRP {unpublished data only}
Jajic 1982 {published data only}
- Jajic I, Nekora A, Chadri HA. Pirprofen, indomethacin and placebo in ankylosing spondylitis. Double‐blind comparison [French]. La Nouvelle Presse Médicale 1982;11(33):2491‐3. [PubMed] [Google Scholar]
Leng Levy 1963 {published data only}
- Leng Levy J, David Chausse J, David Chausse F. Clinical results of tanderil administration in gout and rheumatic pelvispondylitis [French]. Journal de Médecine de Bordeaux et du Sud‐Ouest 1963;140:1679‐86. [PubMed] [Google Scholar]
Maier‐Lenz 1981 {published data only}
- Maier‐Lenz H, Ringwelski L. Results of a controlled double‐blind comparison of Exrheudon and phenylbutazone [Ergebnisse Einer Kontrollierten Doppelblindstudie mit Exrheudon Versus Phenylbutazon]. Therapiewoche 1981;31:1226‐34. [Google Scholar]
Mayrhofer 1991 {published data only}
- Mayrhofer H, Aglas F, Broll H, Eberl R, Ebner W, Klein G, et al. The efficacy and tolerance of NSAIDs in patients with ankylosing spondylitis. Therapiewoche Osterreich 1991;6:121‐31. [Google Scholar]
Mertz 1981 {published data only}
- Mertz DP. Efficacy and tolerance of proquazone in ankylosing spondylarthritis [Wirkung und Vertraglichkeit von Proquazon bei Spondylitis Ankylosans]. Therapiewoche 1981;31:3274‐8. [Google Scholar]
Müller‐Fassbender 1979 {published data only}
- Müller‐Fassbender H. Tolmetin treatment for ankylosing spondylitis [Behandlung der Spondylitis Ankylosans mit Tolectin® (Tolmetin)]. Therapiewoche 1979;29:8899‐900. [Google Scholar]
Müller‐Fassbender 1981 {published data only}
- Müller‐Fassbender H. Treatment of ankylosing spondylitis with tolmetin in doses of 800 or 1200 mg [Behandlung der Spondylitis Ankylosans mit Tolectin in Einer Dosierung von 800 mg Bzw, 1200 mg]. Verhandlungen der Deutschen Gesellschaft für Rheumatologie 1981;7:335‐7. [Google Scholar]
NCT00367211 {unpublished data only}
NCT00715091 {unpublished data only}
NCT00766402 {unpublished data only}
NCT01077843 {unpublished data only}
NCT01091675 {unpublished data only}
NCT01176682 {unpublished data only}
NCT01208207 {unpublished data only}
NCT01685424 {unpublished data only}
Orozco Medina 1983 {published data only}
- Orozco Medina JH. Evaluation of efficacy and tolerance of oral indoprofen in patients with ankylosing spondylitis [Valoracion de la eficacia y tolerancia de indoprofen oral en pacientes con espondilitis anquilosante]. Investigación Médica Internacional 1983;10(1):66‐9. [Google Scholar]
Pattin 1990 {published data only}
- Pattin S. Ankylosing spondylitis: efficacy, safety and therapeutic benefit of etodolac (600 mg daily) versus piroxicam (20 mg daily) [Spondylarthrite ankylosante: efficacité, tolérance et beneficé thérapeutique comparés de l'etodolac (600 mg/j) et du piroxicam (20 mg/j)]. Rhumatologie 1990;42(7A):207‐12. [Google Scholar]
Reiter 1984 {published data only}
- Reiter W, Pilger A. Ankylosing spondylitis: comparison of acemetacin and piroxicam. Therapiewoche 1984;34:7209‐15. [Google Scholar]
Renier 1982 {published data only}
- Renier JC, Fournier M, Loyau G, Roux H. Ankylosing spondylitis. Comparative trial of two non‐steroidal anti‐inflammatory agents: pirprofen and ketoprofen [French]. La Nouvelle Presse Médicale 1982;11(33):2494‐6. [PubMed] [Google Scholar]
Schattenkirchner 1981 {published data only}
- Schattenkirchner M. Long‐time results of Felden (piroxicam) in ankylosing spondylitis [Langzeiterfahrungen mit Felden (Piroxicam) bei Spondylitis Ankylosans (M. Bechterew)]. Therapiewoche 1981;31:2979‐81. [Google Scholar]
Schattenkirchner 1986 {published data only}
- Schattenkirchner M, Kellerer G, Kruger K. Treatment of ankylosing spondylitis: Single‐blind crossover comparison of acemetacin and diclofenac [Behandlung der Spondylitis Ankylosans: Einfachblinde Cross‐over‐Vergleichsstudie mit Acemetacin und Diclofenac]. Therapiewoche 1986;36:5276‐83. [Google Scholar]
Simon 1987 {published data only}
- Simon L, Bennet P. A comparative study of pirprofene and phenylbutazone in the treatment of ankylosing spondylitis [Etude comparative du pirprofene et de la phenylbutazone dans la spondylarthrite ankylosante]. Rheumatologie 1987;39(9):283‐7. [Google Scholar]
Stollenwerk 1985 {published data only}
- Stollenwerk R, Criegern T, Gierend M, Schilling F. Comparative study of the treatment of ankylosing spondylitis with piroxicam suppositories and indomethacin suppositories in combination with indomethacin retard capsules [Therapee der Spondylitis Ankylosans. Kurzzeitanwendung von Piroxicam‐Suppositorien oder Indometacin‐Suppositorien und ‐Retardkapseln]. Fortschritte der Medizin 1985;103(20):561‐5. [PubMed] [Google Scholar]
References to ongoing studies
ClinicalTrials.gov 2014a {unpublished data only}
- NCT01934933. Delaying Ossification and Improving Inflammation of Celebrex Plus/or Enbrel Treatment on Active Ankylosing Spondylitis. http://clinicaltrials.gov/ct2/show/NCT01934933?type=Intr&cond=Spondylitis&rcv_s=06%2F01%2F2013&rank=2 (accessed 18 June 2014).
ClinicalTrials.gov 2014b {unpublished data only}
- NCT02089529. Treatment of Axial Spondyloarthritis With Reduced Doses of NSAIDs. http://clinicaltrials.gov/ct2/show/NCT02089529?type=Intr&cond=Spondylitis&rcv_s=06%2F01%2F2013&rank=9 (accessed 18 June 2014).
Additional references
Amor 1990
- Amor B, Dougados M, Mijiyawa M. Criteria of the classification of spondyloarthropathies [French]. Revue du rhumatisme et des maladies ostéo‐articulaires 1990;57(2):85‐9. [PubMed] [Google Scholar]
Anderson 2001
- Anderson JJ, Baron G, Heijde D, Felson DT, Dougados M. Ankylosing spondylitis assessment group preliminary definition of short‐term improvement in ankylosing spondylitis. Arthritis and Rheumatism 2001;44(8):1876‐86. [DOI] [PubMed] [Google Scholar]
Armstrong 1987
- Armstrong CP, Blower AL. Non‐steroidal anti‐inflammatory drugs and life threatening complications of peptic ulceration. Gut 1987;28(5):527‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
Atkins 2004
- Atkins D, Best D, Briss PA, Eccles M, Falck‐Ytter Y, Flottorp S, et al. Grading quality of evidence and strength of recommendations. BMJ 2004;328(7454):1490‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bertolini 2002
- Bertolini A, Ottani A, Sandrini M. Selective COX‐2 inhibitors and dual acting anti‐inflammatory drugs: critical remarks. Current Medicinal Chemistry 2002;9(10):1033‐43. [DOI] [PubMed] [Google Scholar]
Bhala 2013
- Coxib and traditional NSAID Trialists' (CNT) Collaboration, Bhala N, Emberson J, Merhi A, Abramson S, Arber N, et al. Vascular and upper gastrointestinal effects of non‐steroidal anti‐inflammatory drugs: meta‐analyses of individual participant data from randomised trials. Lancet 2013;382(9894):769‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
Brandt 2004
- Brandt J, Listing J, Sieper J, Rudwaleit M, Heijde D, Braun J. Development and preselection of criteria for short term improvement after anti‐TNF alpha treatment in ankylosing spondylitis. Annals of the Rheumatic Diseases 2004;63(11):1438‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
Braun 2006
- Braun J, Landewé R, Hermann KG, Han J, Yan S, Williamson P, et al. Major reduction in spinal inflammation in patients with ankylosing spondylitis after treatment with infliximab: results of a multicenter, randomized, double‐blind, placebo‐controlled magnetic resonance imaging study. Arthritis and Rheumatism 2006;54(5):1646‐52. [DOI] [PubMed] [Google Scholar]
Braun 2007
- Braun J, Sieper J. Ankylosing spondylitis. Lancet 2007;369(9570):1379‐90. [DOI] [PubMed] [Google Scholar]
Braun 2011
- Braun J, Berg R, Baraliakos X, Boehm H, Burgos‐Vargoas R, Collantes‐Estevez E, et al. 2010 update of the ASAS/EULAR recommendations for the management of ankylosing spondylitis. Annals of the Rheumatic Diseases 2011;70(6):896‐904. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bresalier 2005
- Bresalier RS, Sandler RS, Quan H, Bolognese JA, Oxenius B, Horgan K, et al. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. New England Journal of Medicine 2005;352(11):1092‐102. [DOI] [PubMed] [Google Scholar]
Burmester 2013
- Burmester GR, Panaccione R, Gordon KB, McIlraith MJ, Lacerda APM. Adalimumab: long‐term safety in 23 458 patients from global clinical trials in rheumatoid arthritis, juvenile idiopathic arthritis, ankylosing spondylitis, psoriatic arthritis, psoriasis and Crohn's disease. Annals of the Rheumatic Diseases 2013;72(4):517‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Calin 1994
- Calin A, Garrett S, Whitelock H, Kennedy LG, O'Hea J, Mallorie P, et al. A new approach to defining functional ability in ankylosing spondylitis: the development of the Bath Ankylosing Spondylitis Functional Index. Journal of Rheumatology 1994;21(12):2281‐5. [PubMed] [Google Scholar]
Chan 2010
- Chan FK, Lanas A, Scheiman J, Berger MF, Nguyen H, Goldstein JL. Celecoxib versus omeprazole and diclofenac in patients with osteoarthritis and rheumatoid arthritis (CONDOR): a randomised trial. Lancet 2010;376(9736):173‐9. [DOI] [PubMed] [Google Scholar]
Deeks 2011
- Deeks JJ, Higgins JPT, Altman DG (editors). Chapter 9: Analysing data and undertaking meta‐analyses. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Dougados 1991
- Dougados M, Linden S, Juhlin R, Huitfeldt B, Amor B, Calin A, et al. The European Spondylarthropathy Study Group preliminary criteria for the classification of spondylarthropathy. Arthritis and Rheumatism 1991;34(10):1218‐27. [DOI] [PubMed] [Google Scholar]
Dougados 1995
- Dougados M. Diagnostic features of ankylosing spondylitis. British Journal of Rheumatology 1995;34(4):301‐3. [DOI] [PubMed] [Google Scholar]
Dougados 2011
- Dougados M, Paternotte S, Braun J, Burgos‐Vargas R, Maksymowych WP, Sieper J, et al. ASAS recommendations for collecting, analysing and reporting NSAID intake in clinical trials/ epidemiological studies in axial spondyloarthritis. Annals of the Rheumatic Diseases 2011;70(2):249–51. [DOI] [PubMed] [Google Scholar]
Doward 2003
- Doward LC, Spoorenberg A, Cook SA, Whalley D, Helliwell PS, Kay LJ, et al. Development of the ASQoL: a quality of life instrument specific to ankylosing spondylitis. Annals of the Rheumatic Diseases 2003;62(1):20‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Emery 1999
- Emery P, Zeidler H, Kvien TK, Guslandi M, Naudin R, Stead H, et al. Celecoxib versus diclofenac in long‐term management of rheumatoid arthritis: randomised double‐blind comparison. Lancet 1999;354(9196):2106‐11. [DOI] [PubMed] [Google Scholar]
Escalas 2010
- Escalas C, Trijau S, Dougados M. Evaluation of the treatment effect of NSAIDs/TNF blockers according to different domains in ankylosing spondylitis: results of a meta‐analysis. Rheumatology 2010;49(7):1317‐25. [DOI] [PubMed] [Google Scholar]
Feenstra 2002
- Feenstra J, Heerdink ER, Grobbee DE, Stricker BH. Association of nonsteroidal anti‐inflammatory drugs with first occurrence of heart failure and with relapsing heart failure: the Rotterdam Study. Archives of Internal Medicine 2002;162(3):265‐70. [DOI] [PubMed] [Google Scholar]
Fries 1991
- Fries JF. NSAID gastropathy: the second most deadly rheumatic disease? Epidemiology and risk appraisal. Journal of Rheumatology Supplement 1991;28:6‐10. [PubMed] [Google Scholar]
Gabriel 1991
- Gabriel SE, Jaakkimainen L, Bombardier C. Risk for serious gastrointestinal complications related to use of nonsteroidal anti‐inflammatory drugs. A meta‐analysis. Annals of Internal Medicine 1991;115(10):787‐96. [DOI] [PubMed] [Google Scholar]
Garrett 1994
- Garrett S, Jenkinson T, Kennedy LG, Whitelock H, Gaisford P, Calin A. A new approach to defining disease status in ankylosing spondylitis: the Bath Ankylosing Spondylitis Disease Activity Index. Journal of Rheumatology 1994;21(12):2286‐91. [PubMed] [Google Scholar]
Gossec 2005
- Gossec L, Heijde D, Meilian A, Krupa DA, James MK, Cavanaugh PF, et al. Efficacy of cyclo‐oxygenase‐2 inhibition by etoricoxib and naproxen on the axial manifestations of ankylosing spondylitis in the presence of peripheral arthritis. Annals of the Rheumatic Diseases 2005;64(11):1563‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Griffin 1988
- Griffin MR, Ray WA, Schaffner W. Nonsteroidal anti‐inflammatory drug use and death from peptic ulcer in elderly persons. Annals of Internal Medicine 1988;109(5):359‐63. [DOI] [PubMed] [Google Scholar]
Hayden 2006
- Hayden JA, Côté P, Bombardier C. Evaluation of the quality of prognosis studies in systematic reviews. Annals of Internal Medicine 2006;144(6):427‐37. [DOI] [PubMed] [Google Scholar]
Higgins 2011a
- Higgins JPT, Altman DG, Sterne JAC (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Higgins 2011b
- Higgins JPT, Deeks JJ, Altman DG (editors). Chapter 16: Special topics in statistics. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Higgins 2011c
- Higgins JPT, Deeks JJ (editors). Chapter 7: Selecting studies and collecting data. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Jarrett 2009
- Jarrett SJ, Sivera F, Cawkwell LS, Marzo‐Ortega H, McGonagle D, Hensor E, et al. MRI and clinical findings in patients with ankylosing spondylitis eligible for anti‐tumour necrosis factor therapy after a short course of etoricoxib. Annals of the Rheumatic Diseases 2009;68(9):1466‐9. [DOI] [PubMed] [Google Scholar]
Jenkinson 1994
- Jenkinson TR, Mallorie PA, Whitelock HC, Kennedy LG, Garrett SL, Calin A. Defining spinal mobility in ankylosing spondylitis (AS). The Bath AS Metrology Index. Journal of Rheumatology 1994;21(9):1694‐8. [PubMed] [Google Scholar]
Kearney 2006
- Kearney PM, Baigent C, Godwin J, Halls H, Emberson JR, Patrono C. Do selective cyclo‐oxygenase‐2 inhibitors and traditional non‐steroidal anti‐inflammatory drugs increase the risk of atherothrombosis? Meta‐analysis of randomised trials. BMJ 2006;332(7553):1302‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Khan 1985
- Khan MA, Linden SM, Kushner I, Valkenburg HA, Cats A. Spondylitic disease without radiologic evidence of sacroiliitis in relatives of HLA‐B27 positive ankylosing spondylitis patients. Arthritis and Rheumatism 1985;28(1):40‐3. [DOI] [PubMed] [Google Scholar]
Khan 1990
- Khan MA, Linden SM. A wider spectrum of spondyloarthropathies. Seminars in Arthritis and Rheumatism 1990;20(2):107‐13. [DOI] [PubMed] [Google Scholar]
Kroon 2012
- Kroon F, Landewé R, Dougados M, Heijde D. Continuous NSAID use reverts the effects of inflammation on radiographic progression in patients with ankylosing spondylitis. Annals of the Rheumatic Diseases 2012;71(10):1623‐9. [DOI] [PubMed] [Google Scholar]
Langman 1994
- Langman MJ, Weil J, Wainwright P, Lawson DH, Rawlins MD, Logan RF. Risks of bleeding peptic ulcer associated with individual non‐steroidal anti‐inflammatory drugs. Lancet 1994;343(8905):1075‐8. [DOI] [PubMed] [Google Scholar]
Lefebvre 2011
- Lefebvre C, Manheimer E, Glanville J. Chapter 6: Searching for studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
MacDonald 1997
- MacDonald TM, Morant SV, Robinson GC, Shield MJ, McGilchirst MM, Murray FE, et al. Association of upper gastrointestinal toxicity of non‐steroidal anti‐inflammatory drugs with continued exposure: cohort study. BMJ 1997;315(7119):1333‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Machado 2010
- Machado P, Landewé R, Braun J, Hermann KG, Baker D, Heijde D. Both structural damage and inflammation of the spine contribute to impairment of spinal mobility in patients with ankylosing spondylitis. Annals of the Rheumatic Diseases 2010;69(8):1465‐70. [DOI] [PubMed] [Google Scholar]
Machado 2011a
- Machado P, Landewé R, Braun J, Hermann KG, Baraliakos X, Baker D, et al. A stratified model for health outcomes in ankylosing spondylitis. Annals of the Rheumatic Diseases 2011;70(10):1758‐64. [DOI] [PubMed] [Google Scholar]
Machado 2011b
- Machado P, Landewé R, Lie E, Kvien TK, Braun J, Baker D, et al. Ankylosing Spondylitis Disease Activity Score (ASDAS): defining cut‐off values for disease activity states and improvement scores. Annals of the Rheumatic Diseases 2011;70(1):47‐53. [DOI] [PubMed] [Google Scholar]
Mau 1988
- Mau W, Zeidler H, Mau R, Majewski A, Freyschmidt J, Stangel W, et al. Clinical features and prognosis of patients with possible ankylosing spondylitis. Results of a 10‐year followup. Journal of Rheumatology 1988;15(7):1109‐14. [PubMed] [Google Scholar]
Murray 1993
- Murray MD, Brater DC. Renal toxicity of the nonsteroidal anti‐inflammatory drugs. Annual Review of Pharmacology and Toxicology 1993;33:435‐65. [DOI] [PubMed] [Google Scholar]
Norman 2001
- Norman GR, Sridhar FG, Guyatt GH, Walter SD. Relation of distribution‐ and anchor‐based approaches in interpretation of changes in health‐related quality of life. Medical Care 2001;39(10):1039‐47. [DOI] [PubMed] [Google Scholar]
Oostveen 1999
- Oostveen J, Prevo R, Boer J, Laar M. Early detection of sacroiliitis on magnetic resonance imaging and subsequent development of sacroiliitis on plain radiography. A prospective, longitudinal study. Journal of Rheumatology 1999;26(9):1953‐8. [PubMed] [Google Scholar]
Poddubnyy 2012
- Poddubnyy D, Rudwaleit M, Haibel H, Listing J, Märker‐Hermann E, Zeidler H, et al. Effect of non‐steroidal anti‐inflammatory drugs on radiographic spinal progression in patients with axial spondyloarthritis: results from the German Spondyloarthritis Inception Cohort. Annals of the Rheumatic Diseases 2012;71(10):1616‐22. [DOI] [PubMed] [Google Scholar]
Ramiro 2014
- Ramiro S, Heijde D, Tubergen A, Stolwijk C, Dougados M, Bosch F, et al. Higher disease activity leads to more structural damage in the spine in ankylosing spondylitis: 12‐year longitudinal data from the OASIS cohort. Annals of the Rheumatic Diseases 2014;73(8):1455‐61. [DOI: 10.1136/annrheumdis-2014-205178] [DOI] [PubMed] [Google Scholar]
Review Manager 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Rudwaleit 2004
- Rudwaleit M, Listing J, Märker‐Hermann E, Zeidler H, Braun J, Sieper J. The burden of disease in patients with ankylosing spondylitis (AS) and pre‐radiographic axial spondyloarthritis. Arthritis and Rheumatism 2004;50:S211. [Google Scholar]
Rudwaleit 2005
- Rudwaleit M, Khan MA, Sieper J. The challenge of diagnosis and classification in early ankylosing spondylitis: do we need new criteria?. Arthritis and Rheumatism 2005;52(4):1000‐8. [DOI] [PubMed] [Google Scholar]
Rudwaleit 2009a
- Rudwaleit M, Landewé R, Heijde D, Listing J, Brandt J, Braun J, et al. The development of Assessment of SpondyloArthritis international Society classification criteria for axial spondyloarthritis (part I): classification of paper patients by expert opinion including uncertainty appraisal. Annals of the Rheumatic Diseases 2009;68(6):770‐6. [DOI] [PubMed] [Google Scholar]
Rudwaleit 2009b
- Rudwaleit M, Heijde D, Landewé R, Listing J, Akkoc N, Brandt J, et al. The development of Assessment of SpondyloArthritis international Society classification criteria for axial spondyloarthritis (part II): validation and final selection. Annals of the Rheumatic Diseases 2009;68(6):777‐83. [DOI] [PubMed] [Google Scholar]
Said‐Nahal 2000
- Said‐Nahal R, Miceli‐Richard C, Berthelot JM, Duché A, Dernis‐Labous E, Blévec G, et al. The familial form of spondylarthropathy: a clinical study of 115 multiplex families. Groupe Francais d'Etude Genetique des Spondylarthropathies. Arthritis and Rheumatism 2000;43(6):1356‐65. [DOI] [PubMed] [Google Scholar]
Sampaio‐Barros 2001
- Sampaio‐Barros PD, Bertolo MB, Kraemer MH, Marques‐Neto JF, Samara AM. Undifferentiated spondyloarthropathies: a 2‐year follow‐up study. Clinical Rheumatology 2001;20(3):201‐6. [DOI] [PubMed] [Google Scholar]
Schünemann 2011
- Schünemann H, Oxman A, Higgins J, Vist GE, Glasziou P, Guyatt GH. Chapter 11: Presenting results and 'Summary of findings' tables. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Siegle 1998
- Siegle I, Klein T, Backman JT, Saal JG, Nüsing RM, Fritz P. Expression of cyclooxygenase 1 and cyclooxygenase 2 in human synovial tissue: differential elevation of cyclooxygenase 2 in inflammatory joint diseases. Arthritis and Rheumatism 1998;41(1):122‐9. [DOI] [PubMed] [Google Scholar]
Sieper 2009
- Sieper J, Rudwaleit M, Baraliakos X, Brandt J, Braun J, Burgos‐Vargas R, et al. The Assessment of SpondyloArthritis international Society (ASAS) handbook: a guide to assess spondyloarthritis. Annals of the Rheumatic Diseases 2009;68(Suppl 2):ii1‐44. [DOI] [PubMed] [Google Scholar]
Silverstein 2000
- Silverstein FE, Faich G, Goldstein JL, Simon LS, Pincus T, Whelton A, et al. Gastrointestinal toxicity with celecoxib vs nonsteroidal anti‐inflammatory drugs for osteoarthritis and rheumatoid arthritis: the CLASS study: A randomized controlled trial. Celecoxib Long‐term Arthritis Safety Study. JAMA 2000;284(10):1247‐55. [DOI] [PubMed] [Google Scholar]
Simon 1999
- Simon LS, Weaver AL, Graham DY, Kivitz AJ, Lipsky PE, Hubbard RC, et al. Anti‐inflammatory and upper gastrointestinal effects of celecoxib in rheumatoid arthritis: a randomized controlled trial. JAMA 1999;282(20):1921‐8. [DOI] [PubMed] [Google Scholar]
Stalnikowicz 1993
- Stalnikowicz R, Rachmilewitz D. NSAID‐induced gastroduodenal damage: is prevention needed? A review and metaanalysis. Journal of Clinical Gastroenterology 1993;17(3):238‐43. [DOI] [PubMed] [Google Scholar]
Sterne 2011
- Sterne JAC, Egger M, Moher D (editors). Chapter 10: Addressing reporting biases. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Stolwijk 2012
- Stolwijk C, Boonen A, Tubergen A, Reveille JD. Epidemiology of spondyloarthritis. Rheumatic Diseases Clinics of North America 2012;38(3):441‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
Trelle 2011
- Trelle S, Reichenbach S, Wandel S, Hildebrand P, Tschannen B, Villiger PM, et al. Cardiovascular safety of non‐steroidal anti‐inflammatory drugs: network meta‐analysis. BMJ 2011;342:c7086. [DOI] [PMC free article] [PubMed] [Google Scholar]
Van den Berg 2012
- Berg R, Baraliakos X, Braun J, Heijde D. First update of the current evidence for the management of ankylosing spondylitis with non‐pharmacological treatment and non‐biologic drugs: a systematic literature review for the ASAS/ EULAR management recommendations in ankylosing spondylitis. Rheumatology 2012;51(8):1388‐96. [DOI] [PubMed] [Google Scholar]
van der Heijde 2008a
- Heijde D, Landewé R, Baraliakos X, Houben H, Tubergen A, Willamson P, et al. Radiographic findings following two years of infliximab therapy in patients with ankylosing spondylitis. Arthritis and Rheumatism 2008;58(10):3063‐70. [DOI] [PubMed] [Google Scholar]
van der Heijde 2008b
- Heijde D, Landewé R, Einstein S, Ory P, Voss D, Ni L, et al. Radiographic progression of ankylosing spondylitis after up to two years of treatment with etanercept. Arthritis and Rheumatism 2008;58(5):1324‐31. [DOI] [PubMed] [Google Scholar]
van der Heijde 2009a
- Heijde D, Lie E, Kvien TK, Sieper J, Bosch F, Listing J, et al. ASDAS, a highly discriminatory ASAS‐endorsed disease activity score in patients with ankylosing spondylitis. Annals of the Rheumatic Diseases 2009;68(12):1811‐8. [DOI] [PubMed] [Google Scholar]
van der Heijde 2009b
- Heijde D, Salonen D, Weissman BN, Landewé R, Maksymowych WP, Kupper H, et al. Assessment of radiographic progression in the spines of patients with ankylosing spondylitis treated with adalimumab for up to 2 years. Arthritis Research & Therapy 2009;11(4):R127. [DOI] [PMC free article] [PubMed] [Google Scholar]
van der Heijde 2011
- Heijde D, Sieper J, Maksymowych WP, Dougados M, Burgos‐Vargas R, Landewé R, et al. 2010 Update of the international ASAS recommendations for the use of anti‐TNF agents in patients with axial spondyloarthritis. Annals of the Rheumatic Diseases 2011;70(6):905‐8. [DOI] [PubMed] [Google Scholar]
van der Linden 1984
- Linden S, Valkenburg HA, Cats A. Evaluation of diagnostic criteria for ankylosing spondylitis. A proposal for modification of the New York criteria. Arthritis and Rheumatism 1984;27(4):361‐8. [DOI] [PubMed] [Google Scholar]
Vane 1998
- Vane JR, Bakhle YS, Botting RM. Cyclooxygenases 1 and 2. Annual Review of Pharmacology and Toxicology 1998;38:97‐120. [DOI] [PubMed] [Google Scholar]
Visual Rx 2008 [Computer program]
- Dr Christopher Cates' EBM Web Site. Visual Rx. Version 3. Dr Christopher Cates' EBM Web Site, 2008.
Wanders 2004
- Wanders AJ, Landewé RB, Spoorenberg A, Dougados M, Linden S, Mielants H, et al. What is the most appropriate radiologic scoring method for ankylosing spondylitis? A comparison of the available methods based on the Outcome Measures in Rheumatology Clinical Trials filter. Arthritis and Rheumatism 2004;50(8):2622‐32. [DOI] [PubMed] [Google Scholar]
Wanders 2005
- Wanders A, Heijde D, Landewé R, Béhier JM, Calin A, Olivieri I, et al. Nonsteroidal antiinflammatory drugs reduce radiographic progression in patients with ankylosing spondylitis: a randomized clinical trial. Arthritis and Rheumatism 2005;52(6):1756‐65. [DOI] [PubMed] [Google Scholar]
Ware 1992
- Ware JE Jr, Sherbourne CD. The MOS 36‐item short‐form health survey (SF‐36). Conceptual framework and item selection. Medical Care 1992;30(6):473‐83. [PubMed] [Google Scholar]
Zhang 2002
- Zhang X, Schwarz EM, Young DA, Puzas JE, Rossier RN, O'Keefe RJ. Cyclooxygenase‐2 regulates mesenchymal cell differentiation into the osteoblast lineage and is critically involved in bone repair. Journal of Clinical Investigation 2002;109(11):1405‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zochling 2006
- Zochling J, Heijde D, Dougados M, Braun J. Current evidence for the management of ankylosing spondylitis: a systematic literature review for the ASAS/EULAR management recommendations in ankylosing spondylitis. Annals of the Rheumatic Diseases 2006;65(4):423–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to other published versions of this review
Kroon 2014
- Kroon FPB, Burg LRA, Ramiro S, Landewé RBM, Buchbinder R, Heijde D. Non‐steroidal anti‐inflammatory drugs (NSAIDs) for axial spondyloarthritis (ankylosing spondylitis and non‐radiographic axial spondyloarthritis). Cochrane Database of Systematic Reviews 2014, Issue 1. [DOI: 10.1002/14651858.CD010952] [DOI] [PMC free article] [PubMed] [Google Scholar]