

Abstract
In the present study, novel phthalimide derivatives 8(a-f) and 9(a-f) bearing a 1,2,3-triazole subunit were synthesized via CuAAC reactions and characterized by 1H, 13C NMR, HR-MS, and FT-IR analyses. To support the fight against SARS-CoV-2, in silico molecular docking studies were carried out to examine their interactions with the proteins of SARS-CoV-2 (Mpro and PLpro) and the protein-protein interactions (PPI) between the ACE2-spike (S1) in comparison with various inhibitors reported to be active by in vitro experiments. The ligand-protein stabilities of compounds 8a-Mpro, 8b-PLpro, and 9a-‘ACE2-S1’ showing the best binding energy and predicted inhibition constant values (Ki) were examined by molecular dynamics simulation studies. Finally, in silico ADMET properties of the target compounds were investigated using the Swiss ADME and ProTox-II web tools. According to in silico results, all phthalimide analogs may block the PPI between S1 and ACE2. The compounds may also inhibit the progression of the Mpro, and PLpro proteins of SARS-CoV-2. Additionally, it has been estimated that the compounds are suitable for oral administration and exhibit low levels of toxicity.
Keywords: Phthalimide; 1,2,3-triazole; SARS-CoV-2; ADMET; Molecular docking studies
Graphical abstract

1. Introduction
Since a new type of coronavirus disease, COVID-19 caused by SARS-CoV-2 virus, emerged in December 2019 in China [1], scientists have conducted many studies on the mechanism of action of the SARS-CoV-2 in the human body. The spike (S) protein of SARS-CoV-2 has been reported to promote virus entry into the host cell by binding with the human ACE2 protein [2]. The S protein comprises two subunits: S1 and S2. The S1 facilities the binding to ACE2 of the virus and it consists of the receptor-binding domain (RBD), the N-terminal domain (NTD), and the other several subdomains, while the S2 mediates virus-cell membrane fusion and initiates the infection [3], [4], [5]. The S1 binds to ACE2 via its RBD part [6,7]. Blocking of the protein-protein interactions (PPI) between the S1 and ACE2 can impair infection efficiency [8], therefore, this blockade has been considered as a possible drug target [9]. After the virus enters the host cell, viral replication and transcription are initiated by the other proteins of SARS-CoV-2 [10,11]. The main protease protein (Mpro or 3CLpro) of the virus is considered an attractive drug target because it is required in the processing and maturation of viral polyproteins [12]. The papain-like protease protein (PLpro), which is another viral protease of the virus, plays an important role in both the cleavage for the proliferation cycle of viral polyproteins and the disruption of host responses [13,14]. Therefore, the PLpro has been reported as an attractive drug target [15], as well. In silico approaches have been used to discover potential inhibitors of SARS-CoV-2 by scientists. The interactions of both commercially available antiviral drugs and some known bioactive compounds with the proteins (especially, Mpro and PLpro) of SARS-CoV-2 have been examined via in silico studies [15], [16], [17], [18]. Recently, the synthesis of some new compounds containing various bioactive moieties such as 1,2,3-triazole, Schiff base, piperidine, and norcantharimides have been reported by scientists and some of these compounds have been suggested as possible therapeutic agents for the COVID-19 via in silico studies [19], [20], [21]. In addition to in silico studies, it has been reported in vitro antiviral studies against SARS-CoV-2 [22].
Phthalimide is a core structure of some pharmaceutically active compounds (Fig. 1 ). The phthalimide framework is used as starting materials for the synthesis of the compounds, such as alkaloids, polymers, pesticides, and copolymers [23]. 4-amino phthalimide derivatives are known for their strong fluorescent properties [24,25]. Phthalimide derivatives are also known to have potent biological activities, such as anti-HIV, anticancer, anti-inflammatory, CK2 inhibitory, hypoglycemic, antioxidant anticonvulsant, antimalarial, antiviral, histone deacetylase inhibitory, cholinesterase inhibitory, and COX inhibitory [26], [27], [28], [29], [30], [31], [32].
Fig. 1.

Pharmaceutically active phthalimide and its analogs.
The 1,2,3-triazole ring constitutes the basic building block of many organic and inorganic molecules having various bioactive properties, such as antitubercular, antifungal, anticancer, anti-HIV, antibacterial, antimicrobial, anti-inflammatory, antiallergic, xanthine oxidase (XO) inhibitory, and IDO inhibitory [33], [34], [35], [36], [37], [38], [39] (Fig. 2 ). The 1,2,3-triazole ring is resistant to oxidation–reduction and hydrolysis reactions in acidic or basic media due to their high aromatic stability [35]. In recent years, the 1,2,3-triazole compounds have been used as a key motif in many fields, such as polymer chemistry, materials science, and drug discovery [40,41]. The primary and secondary amines, another bioactive subunits, have been reported to have favorable effects on the biological activities of some hybrid molecules [29,37,[42], [43], [44]].
Fig. 2.

Some biologically active 1,2,3-triazole compounds.
In the light of these studies, in the present study, novel hybrid phthalimide derivatives 8(a-f) and 9(a-f) bearing 1,2,3-triazole subunit having three active components (the phthalimide ring, various amines, and 1,2,3-triazole ring) were synthesized. To support the fight against SARS-CoV-2, in silico molecular modeling studies were carried out to examine their interactions with the PPI of ACE2-S1 and the proteins of SARS-CoV-2 (Mpro and PLpro) by comparison with various inhibitors reported to be active in vitro experiments.
2. Results and discussions
2.1. Synthesis
Novel hybrid phthalimide analogs bearing a 1,2,3-triazole framework were regioselectively synthesized via Cu(I)-catalyzed azide-alkyne cycloaddition (CuAAC) [34,36,45]. For this purpose, phthalimide was reacted with propargyl bromide in the presence of K2CO3 in acetonitrile at 75 °C to obtain compound 1 [46] (Scheme 1 ).
Scheme 1.

Synthesis of compounds 8(a-f) and 9(a-f). Reagents and conditions: (a) Propargyl bromide, K2CO3, acetonitrile, 75 °C, (b) Chloroacetyl chloride, NaHCO3, acetonitrile, 0 °C, (c) NaN3, dimethyl sulfoxide, r.t., (d) CuSO4•5H2O, sodium L-ascorbate, methanol:water (1:1), r.t.
On the other hand, corresponding organic azido compounds 6(a-f) [42,43,47] and 7(a-f) [48], [49], [50], [51], which are known in the literature, were synthesized by the reaction steps given in Scheme 1. For this, in the first step, 4(a-f) and 5(a-f) were synthesized from the reaction of various amines (secondary amines 2(a-f) and primary amines 3(a-f)) with chloroacetyl chloride in the presence of NaHCO3 in acetonitrile at 0 °C. In the second step, 2-chloro-N-substituted acetamide derivatives 4(a-f) and 5(a-f) were treated with NaN3 in dimethyl sulfoxide at room temperature and azido compounds 6(a-f) and 7(a-f) were obtained in good yields. Finally, compound 1 was treated with azido compounds 6(a-f) or 7(a-f) in the presence of CuSO4•5H2O [45] and sodium L-ascorbate in a methanol:water solvent system at room temperature, and the target compounds (8(a-f) and 9(a-f)) were obtained in good yields (Scheme 1).
2.2. Characterization
The structures of phthalimide-1,2,3-triazole hybrid compounds 8(a-f) and 9(a-f) are similar. Among these compounds, 8a and 9a were chosen as the model compounds. The 1H NMR spectrum of the compound 8a was recorded in CDCl3 and the 13C NMR spectrum of 8a was recorded in CDCl3/DMSO-d 6 solvent system at 40 °C (see Supplementary Information (SI), Figures S1 and S2). In the 1H NMR spectrum of compound 8a, the aromatic protons of the phthalimide moiety and the proton (=C—H*) in the triazole ring of 8a were observed together at δ 7.85–7.73 ppm range (m, 5H). The 1,4-disubstituted methylene protons of compound 8a were observed at δ 5.03 (s, 2H) ppm and 5.20 ppm (dd, J = 16, 6.4 Hz, 2H), respectively. While the methylene protons and tertiary proton (-CH) of the piperidine ring of 8a were observed at δ 4.9–1.09 ppm range (9H), the 4-methyl protons (-CH3) were observed at 0.95 ppm (d, J = 6 Hz, 3H). In the 13C NMR spectrum of 8a, the signals of the carbonyl carbons of the imide and amide moieties were observed at δ 172.17 (2C), 168.28 ppm. The aromatic carbons of the phthalimide moiety were observed at δ 139.12, 136.77, 129.92 ppm. The quaternary carbon and =C*-H carbons in the triazole ring of 8a were observed at 147.24 and 128.09 ppm, respectively. The 1,4-disubstituted methylene carbons attached to the triazole ring of 8a were detected at 47.49 and 55.81 ppm. The carbons of the piperidine ring of 8a were detected at 49.97 ppm and 39.04–26.45 ppm range. In the FT-IR spectrum of 8a (see SI, Figure S25), the υ(C—H) characteristic band of the triazole ring was observed at 3164 cm−1. The aromatic and aliphatic υ(C—H) stretching vibrations were observed at 2992–2864 cm−1 range. While the υ(C O) asymmetric and symmetric stretching vibrations of the imide ring were observed at 1768 and 1720 cm−1, υ(C O) stretching vibration of the amide moiety of 8a was observed at 1655 cm−1. The peak of the [M + H]+ ion in the mass spectrum of 8a (see SI, Figure S36) was determined at 368.17172 m/z (for C19H21N5O3). Regarding 9a, in the 1H NMR spectrum in CDCl3 of compound 9a (see SI, Figure S13), the NH proton in the amide group was observed at δ 6.61 ppm (s, 1H). The =C—H proton in the triazole ring of 9a was observed together with the aromatic protons of the phthalimide ring at δ 7.86–7.72 ppm range (m, 5H). While the aromatic protons of the benzyl group were observed at δ 7.30–7.19 ppm range (5H), the signals of its methylene protons were detected at δ 4.41 (d, J = 4.8 Hz, 2H) ppm. The 1,4-disubstituted methylene protons of 9a were detected at 4.99 ppm (s, 2H) and δ 5.05 ppm (s, 2H), respectively. When the 13C NMR spectrum recorded in CDCl3-DMSO-d6 at 40 °C (see SI, Figure S14) of 9a was examined, the carbonyl carbons of the phthalimide and acetamide units of 9a were observed at 167.62 ppm and 164.90 ppm, respectively. The 1,4-disubstituted methylene carbons of compound 9a were detected at δ 43.77 ppm and 53.15 ppm. The quaternary and =C*-H carbons in the triazole ring of 9a were observed at 143.84–123.52 ppm range (9C) together with the aromatic carbons of the phthalimide and benzyl units. The methylene carbon of the benzyl group was detected at δ 32.90 ppm. In the FT-IR spectrum of compounds 9a (see SI, Figure S31), υ(N—H) stretching band was determined at 3297 cm−1 and the υ(C—H) characteristic band of the triazole ring was observed at 3153 cm−1. The aromatic and aliphatic υ(C—H) stretching vibrations of 9a were detected at 3067–2957 cm−1 range. The υ(C O) asymmetric/symmetric stretching vibrations of the imide ring and υ(C O) stretching vibrations of the amide unit in 9a were observed at 1766 cm−1 (asym.), 1709 cm−1 (sym.), and 1651 cm−1, respectively. The peak of the [M + H]+ ion in the mass spectrum of 9a (see SI, Figure S42), was determined at 376.14039 m/z (for C20H17N5O3).
2.3. Molecular docking studies
Molecular docking studies play an important role in explaining the protein-ligand relationship [52], [53], [54] and prevent both in vitro and in vivo studies of molecules that are impossible to be effective, thus the studies provide great financial savings. In the present studies, the interactions of the target compounds with SARS-CoV-2 proteins were examined and their estimated Ki values were calculated via molecular docking studies. For this purpose. The interactions of the target compounds with the PPI of ACE2-S1, Mpro, and PLpro proteins were investigated. PDB files: 6M0J for the ACE2-S1, 7K40 for the Mpro, and 7CMD for the PLpro were used. Bojadzic et al. [9] have reported that methylene blue (MB) (IC50: 3.03 µM) inhibits the PPI of between the S1 and ACE2. Therefore, MB was used as a reference inhibitor/ligand. The target compounds and MB were docked on the PPI of ACE2-S1. According to the docking results, all target compounds showed better interactions than MB with the PPI. The binding energy values vary between −6.40 and −9.70 kcal/mol, while the estimated Ki values vary between 0.077 and 20.35 µM. The best binding energy and estimated Ki values of MB were −6.18 kcal/mol and 29.32 µM. Compound 9a in the target compounds exhibited the best binding energy (−9.70 kcal/mol) and estimated Ki value (0.077 µM) (Table 1 ). According to the docking results, MB interacted with the RDB of ACE2, while compound 9a interacted with both the RDB of ACE2 and S1. The phthalimide moiety of 9a formed a conventional hydrogen bond with Lys353 residue (3.00 Å) of the ACE2, its triazole ring formed a π-anion with Asp38 residue (3.92 Å) of the ACE2 protein. The phthalimide moiety of 9a also formed a π-cation interaction with Arg403 (3.91 Å) and a carbon-hydrogen bond with Tyr495 residue (2.97 Å) of the S1 protein. At the same time, while the carbonyl group of amide moiety of 9a formed a conventional hydrogen bond with Ser494 residue (2.07 Å) of the S1 protein, its benzyl group formed a π-alkyl interaction with Leu452 (5.28 Å) of the S1. 9a also formed van der Waals interactions with both the ACE2 and S1 proteins (Figs. 3 and 6).
Table 1.
The docking results of the target compounds and the inhibitors with the PPI of ACE2-S1, Mpro, and PLpro.
| Compounds | ACE2-S1 |
Mpro |
PLpro |
|||
|---|---|---|---|---|---|---|
| BE*(kcal/mol) | EIC*, Ki(µM) | BE*(kcal/mol) | EIC*, Ki(µM) | BE*(kcal/mol) | EIC*, Ki(µM) | |
| 8a | −8.16 | 1.05 | −8.76 | 0.379 | −8.80 | 0.353 |
| 8b | −8.05 | 1.26 | −7.81 | 1.89 | −8.87 | 0.315 |
| 8c | −8.69 | 0.428 | −7.82 | 1.85 | −8.64 | 0.463 |
| 8d | −6.40 | 20.35 | −7.12 | 6.06 | −7.31 | 4.30 |
| 8e | −6.80 | 10.39 | −7.73 | 2.15 | −8.57 | 0.522 |
| 8f | −7.26 | 4.79 | −7.67 | 2.39 | −7.35 | 4.08 |
| 9a | −9.70 | 0.077 | −8.40 | 0.699 | −8.54 | 0.546 |
| 9b | −7.02 | 7.15 | −7.03 | 7.03 | −8.40 | 0.699 |
| 9c | −7.55 | 2.91 | −7.03 | 6.97 | −8.33 | 0.789 |
| 9d | −6.80 | 10.45 | −7.14 | 5.82 | −8.04 | 1.28 |
| 9e | −7.03 | 7.04 | −7.39 | 3.81 | −7.70 | 2.27 |
| 9f | −8.33 | 0.789 | −7.47 | 3.35 | −8.07 | 1.21 |
| MB | −6.18 | 29.32 | −6.39 | 20.75 | −7.02 | 7.14 |
| Boceprevir | −6.24 | 26.49 | −10.04 | 0.043 | −7.51 | 3.10 |
| GRL0617 | −6.43 | 19.20 | −7.75 | 2.09 | −9.83 | 0.061 |
BE* = Binding Energy
EIC* = Estimated Inhibition Constant.
Fig. 3.

3D interaction diagrams of 9a (a) and MB (c) with the PPI of ACE2-S1. The best binding poses: b) 9a (red) and MB (blue).
Fig. 6.

2D ligand–receptor interaction diagrams: a) 8a-Mpro, b) Boceprevir-Mpro, c) 8b-PLpro, d) GRL0617-PLpro, e) 9a-‘ACE2-S1’, f) MB-‘ACE2-S1’.
The Mpro protein of SARS-CoV-2 are necessary for maturation, viral polyproteins processing [12], and it has been recognized as an important drug target. Therefore, all target compounds were docked on the active site of the Mpro protein (PDB ID: 7K40) containing boceprevir as a co-crystallized ligand. The docking mode of 7K40 was determined according to the location of boceprevir using a directed docking procedure. The validation study was carried out for boceprevir and the root mean square deviation (RMSD) value was 1.630 Å (see SI, Figure S49-a). This value shows to be valid for the docking procedure. Boceprevir was also used as a reference inhibitor/ligand, it has been reported to inhibit the Mpro protein (IC50: 8.0 µM) [12]. For all target compounds, the binding energy values vary between −7.03 and −8.76 kcal/mol, while the estimated Ki values vary between 7.03 and 0.379 µM. The best binding energy and estimated Ki values of boceprevir were −10.04 kcal/mol and 0.043 µM, respectively. 8a in the target compounds showed the best binding energy (−8.76 kcal/mol) with the active site of the Mpro. The estimated Ki value of 8a was 0.379 µ (Table 1). The other target compounds were predicted to show acceptable inhibition constants. When the interactions of 8a with the Mpro, which showed the best binding energy and inhibition constant values, were examined, it was observed that the amide carbonyl group of 8a formed two conventional hydrogen bonds with His41 (2.65 Å) and Cys145 (3.69 Å) residues of the Mpro. The phthalimide moiety of 8a formed a π-sigma interaction with Leu167 (3.64 Å) and a carbon-hydrogen bond with Gln189 (3.61 Å). Its triazole ring formed a π-alkyl with Met165 (5.36 Å) and unfavorable acceptor-acceptor interactions with Glu166 (2.96 Å). The 4-methyl piperidine moiety of compound 8a formed alkyl interactions with Pro52 (4.60 Å), Met49 (3.86 Å and 4.36 Å), Met165 (5.40 Å) and an alkyl interaction with His41 (4.66 Å) residue. 8a also formed van der Waals interactions (Figs. 4 and 6).
Fig. 4.

3D interaction diagrams of 8a (a) and boceprevir (c) with the active site of Mpro. The best binding poses: b) 8a (pink) and boceprevir (dark green).
The interactions with the PLpro of the target compounds were also examined. The target compounds were docked on the active site of the PLpro protein. 7CMD PDB file containing 5-amino-2-methyl-N-[(1R)−1-naphthalen-1-ylethyl]benzamide (GRL0617) as a co-crystallized ligand was chosen for PLpro. The docking mode of 7CMD was determined according to the location of GRL0617 using a directed docking procedure. The RMSD value was 1.021 Å (see SI, Figure S49-b) for GRL0617. This value is valid for validation. At the same time, GRL0617 was a strong inhibitor against PLpro (IC50: 2.4 µM) [55] and was evaluated as a reference inhibitor/ligand. According to the docking results, the binding energy values of the target compounds vary between −7.31 and −8.87 kcal/mol, while its estimated Ki values vary between 4.30 and 0.315 µM. The best binding energy and estimated Ki values of GRL0617 were −9.83 kcal/mol and 0.061 µM, respectively. Compound 8b exhibited the best binding (−8.87 kcal/mol) and estimated Ki value (0.315 µM) in the target compounds (Table 1). When the results were examined in detail, the triazole ring of 8b formed a conventional hydrogen bond with Gln269 (2.02 Å), a π-π stacked with Tyr264 (3.69 Å). The triazole ring also formed π-anion and carbon-hydrogen bond interactions with Asp164 (4.25 Å and 3.77 Å, respectively). The phthalimide moiety formed two amide-π-stacked interactions with Gly163 (3.67 Å and 4.55 Å), π-anion with Glu167 (4.94 Å), π-alkyl with Leu162 (5.42 Å), and π-sigma interaction with Gln269 (3.64 Å). Its pyrrolidine ring formed an alkyl interaction with Arg166 (4.57 Å) (Figs. 5 and 6 ).
Fig. 5.

3D interaction diagrams of 8b (a) and GRL0617 (c) with the active site of PLpro. The best binding poses: b) 8b (blue) and GRL0617 (yellow).
In this study, the interactions of the respective inhibitors (Fig. 7 ) with all proteins were investigated at the same time. Both the interactions of MB with the Mpro and PLpro proteins and the interactions of boceprevir with the PPI of ACE2-S1 complex are weaker than the target compounds. Regarding GRL0617, while it showed weaker than the target compounds in the interactions with the PPI of the ACE2-S1, it showed the binding energy value close to the target compounds in the interactions with Mpro. Multi-targeting inhibitors can be beneficial for solving limited efficiencies, resistant profiles of an individual target. Therefore, the target compounds can be considered as effective inhibitors than the respective inhibitors against all three proteins (Table 1).
Fig. 7.

The structures of boceprevir, GRL0617, and MB.
2.4. Molecular dynamics simulation
Molecular docking studies were supported by molecular dynamics (MD) simulations for the complexes 8a-Mpro, 8b-PLpro, and 9a-the PPI of ACE2-S1 to check their stability and dynamic properties. Their Desmond simulation trajectories were analyzed. The RMSD, root mean square fluctuation (RMSF), and protein-ligand contacts were calculated from MD trajectory analysis. The RMSD plots are shown in Fig. 8 . The RMSD plot of the complex 8a-Mpro indicates that the complex reaches stability at 5 ns. From then, an average RMSD value of 0.8 Å for 8a-Mpro persists up to 50 ns. After that, the changes in the RMSD values remain within 0.5 Å during the simulation period, which is quite acceptable (Fig. 8a). The ligand fit to protein RMSD values fluctuates within an average of 1.0 Å after being stable. There was a decrease in the RMSD of ligand due to flip. It increases at 40 ns and became stable for the rest of the time. The RMSD plot of the complex 8b-PLpro indicates that the complex reaches stability at 10 ns. From then, an average RMSD value of 1.0 Å for 8b-PLpro persists up to 50 ns. The ligand fit to protein RMSD values fluctuates within 1.0 Å after being stable (Fig. 8b). The RMSD plot of the complex 9a-‘ACE2-S1’ indicates that the complex reaches stability at 5 ns. From then, an average RMSD value of 1.0 Å for 9a-‘ACE2-S1’ persists up to 50 ns. The ligand fit to protein RMSD values fluctuates within 1.0 Å after being stable. There was a slight increase in the ligand's RMSD at 15 ns but became stable again. These indicate that the ligand remains stably bound to the binding site of the receptor during the simulation period (Fig. 8c). Fig. 9 shows the residue-wise RMSF value of the protein bound to the ligand. The residues showing higher peaks correspond to loop regions, as identified from MD trajectories (Fig. 10 ), or N and C-terminal zones. The low RMSF values of binding site residues indicate the stability of ligand binding to the protein. Most of the important interactions of ligand–proteins determined with MD are hydrogen bonds and hydrophobic interactions. Gln189, Thr190, and Gln192 are the most important ones in terms of H-bonds for 8a-Mpro (Fig. 11 a and 11b). Tyr273 is the most important one in terms of H-bonds for 8b-PLpro (Fig. 11c and 11d). A: His34, A: Glu35, A: Asp38, A: Lys353, E: Ser494, and E: Gly496 are the most important ones in terms of H-bonds for 9a-‘ACE2-S1’ (Fig. 11e and 11f).
Fig. 8.

RMSD plots of the Cα atoms of protein and the ligand with time: a) 8a-Mpro, b) 8b-PLpro, c) 9a-‘ACE2-S1’.
Fig. 9.

RMSF plots of the proteins: a) Mpro, b) PLpro, c) ACE2-S1.
Fig. 10.

Protein Secondary Structure element distribution by residue index throughout the protein structure a) Mpro, b) PLpro, c) ACE2-S1. Red columns indicate alpha helices, and blue columns indicate beta-strands.
Fig. 11.

Protein-ligand contact histograms: a) 8a-Mpro, c) 8b-PLpro, e) 9a-‘ACE2-S1’ and 2D interaction diagrams: b) 8a-Mpro, d) 8b-PLpro, f) 9a-‘ACE2-S1’.
2.5. In silico ADMET studies
In silico ADME studies of compounds 8(a-f) and 9(a-f) were performed to predict their physicochemical descriptors using the Swiss ADME website [56]. Some of the ADME values of the compounds are given in Table 2 and the bioavailability radar images are shown in Figure S48 (see SI). The colored zone in the bioavailability radar image is the convenient physicochemical region for oral bioavailability [29] (the colored zone; Lipo (Lipophilicity):−0.7<XLOGP3<+5.0, size: 150<MW<500 g/mol, polar (polarity): 20Å2<TPSA<130Å2, insolu (Insolubility): 0<log S(ESOL)<6, insatu (insaturation): 0.25<Fraction Csp3<1, flex (flexibility): 0.25<num. rotatable bonds<9). According to this, the bioavailability radar images show that all compounds, except 8f and 9a, have acceptable physicochemical properties. These results mean that the compounds are suitable for oral bioavailability. Log S (ESOL) value is defined as in the water solubility and classes of solubility are listed as follows insoluble<−10<poorly<−6<moderately<−4<soluble<−2<very<0 <highly. According to this, while compounds 8c, 8d, 9b are very soluble in the water, the other compounds are soluble in the water. All compounds have high GI absorption and do not cross the blood-brain barrier (BBB). The BBB diffusion is a crucial parameter due to toxicity, therefore the target compounds may be considered as good drug candidates. While compounds 8(a-c), 9a, 9c, and 9e act as P- glycoprotein substrates, compounds 8d, 8e, 8f, 9b, and 9e do not act as P- glycoprotein substrates. All compounds obey the Lipinski, Ghose, Veber, Egan, and Muegge rules. In silico toxicity predictions of the target compounds were evaluated through the ProTox-II [57], [58], [59], which is a free website. The oral rat acute toxicity (LD50) values of the target compounds were predicted as 500 mg/kg, and the predicted toxicity classes of all were 4 (Table 2). These predicted results mean that the target compounds show low toxicity profiles.
Table 2.
Some predicted ADMET values of compounds 8(a-f) and 9(a-f).
| Some ADME properties | Compounds |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 8a | 8b | 8c | 8d | 8e | 8f | 9a | 9b | 9c | 9d | 9e | 9f | |
| Molecular weight (g/mol) | 367.40 | 339.35 | 355.35 | 313.31 | 341.36 | 397.47 | 375.38 | 313.31 | 327.34 | 341.36 | 327.34 | 341.36 |
| Fraction Csp3 | 0.42 | 0.35 | 0.35 | 0.27 | 0.35 | 0.48 | 0.15 | 0.27 | 0.31 | 0.35 | 0.31 | 0.35 |
| Num. rotatable bonds | 5 | 5 | 5 | 5 | 7 | 11 | 7 | 6 | 7 | 8 | 6 | 6 |
| Num. H-bond acceptors | 5 | 5 | 6 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 |
| Num. H-bond donors | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 1 |
| TPSA (Å2) | 88.40 | 88.40 | 97.63 | 88.40 | 88.40 | 88.40 | 97.19 | 97.19 | 97.19 | 97.19 | 97.19 | 97.19 |
| Log S (ESOL) | −2.79 | −2.13 | −1.68 | −1.70 | −2.17 | −3.33 | −2.87 | −1.75 | −2.09 | −2.31 | −2.09 | −2.28 |
| Consensus Log Po/w | 1.31 | 0.70 | 0.23 | 0.41 | 1.06 | 2.44 | 1.41 | 0.54 | 0.87 | 1.18 | 0.89 | 1.05 |
| GI absorption | High | High | High | High | High | High | High | High | High | High | High | High |
| BBB permeant | No | No | No | No | No | No | No | No | No | No | No | No |
| P-gp substrate | Yes | Yes | Yes | No | No | No | Yes | No | Yes | Yes | No | Yes |
| Log Kp (skin permeation) (cm/s) | −7.75 | −8.15 | −8.86 | −8.33 | −7.98 | −7.07 | −7.78 | −8.20 | −7.91 | −7.75 | −7.98 | −7.94 |
| Lipinski | Yes, 0 violation | Yes, 0 violation | Yes, 0 violation | Yes, 0 violation | Yes, 0 violation | Yes, 0 violation | Yes, 0 violation | Yes, 0 violation | Yes, 0 violation | Yes, 0 violation | Yes, 0 violation | Yes, 0 violation |
| Bioavailability Score | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 |
| PAINS | 0 alert | 0 alert | 0 alert | 0 alert | 0 alert | 0 alert | 0 alert | 0 alert | 0 alert | 0 alert | 0 alert | 0 alert |
| LD50 (mg/kg) | 500 | 500 | 500 | 500 | 500 | 500 | 500 | 500 | 500 | 500 | 500 | 500 |
| Predicted Toxicity Class | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 |
3. Materials and methods
All reagents and anhydrous solvents were purchased from Sigma-Aldrich, Alpha, Acros, and Merck and used without further purification. The 1H NMR and 13C NMR spectra were recorded on a Bruker (Avance III) 400 MHz NMRspectrometer. The FT-IR spectrum was recorded with a Perkin Elmer spectrophotometer using an ATR head in the range of 4000–600 cm−1. Melting point (M.p.) was measured with a Thermo Scientific melting point device. The mass spectra were recorded on an Agilent Technologies 6530 Accurate-Mass Q-TOF LC/MS.
3.1. Synthesis
Compounds 1, 4(a-f), 5(a-f), 6(a-f), and 7(a-f) were synthesized as mentioned in previous studies [42,43,[46], [47], [48], [49], [50], [51]].
General procedure for the preparation of 8a-f and 9a-f
To a stirred solution of compound 1 (1 mmol) in 20 mL of methanol were added 6(a-f) or 7(a-f) (1 mmol) and then, added solutions of sodium L-ascorbate (0.1 mmol) in 5 mL of H2O and CuSO4•5H2O (0.1 mmol) in 5 mL of H2O, consecutively, at room temperature. Until the consumption of compound 1, the mixture was stirred at room temperature for about 5 h. Then, to the mixture was added 20 mL of ethyl acetate, and extraction was performed with H2O (3 × 10 mL). The organic layer was dried over Na2SO4. The crude product was purified by washing with diethyl ether.
2-((1-(2-(4-methylpiperidin-1-yl)−2-oxoethyl)−1H-1,2,3-triazol-4-yl)methyl)isoindoline-1,3-dione (8a)

Yield: 85%, white crystals, M.p: 182–184 °C, 1H NMR (δ, ppm, 400 MHz, CDCl3): 7.85–7.82 (m, 3H), 7.75–7.73 (m, 2H), 5.20 (dd, J = 16, 6.4, Hz, 2H), 5.03 (s, 2H), 4.49 (d, J = 12.8 Hz, 1H), 3.83 (d, J = 12.8 Hz, 1H), 3.09 (t, J = 12.4 Hz, 1H), 2.63 (t, J = 12.4 Hz, 1H), 1.72 (t, J = 15.2 Hz, 3H), 1.09 (q, J = 11.2 Hz, 2H), 0.95 (d, J = 6 Hz, 3H), 13C NMR (δ, ppm, 100 MHz, CDCl3 + DMSO-d 6 (40 °C)): 172.17, 168.28, 147.24, 139.12, 136.77, 129.92, 128.09, 55.81, 49.97, 47.29, 39.04, 38.33, 37.91, 35.45, 26.45, FT-IR (cm−1):3164, 2992, 2956, 2921, 2864, 1768, 1720, 1655, 1607, 1555, 1466, 1426, HR-ESI-MS (C19H21N5O3): m/z 368.17172 [M + H]+ (calcd. 368.1615 [M + H]+).
2-((1-(2-oxo-2-(pyrrolidin-1-yl)ethyl)−1H-1,2,3-triazol-4-yl)methyl)isoindoline-1,3‑dione (8b)
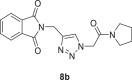
Yield: 80%, white crystals, M.p: 177–178 °C, 1H NMR (δ, ppm, 400 MHz, CDCl3): 7.87–7.85 (m, 3H), 7.73–7.72 (m, 2H) 5.12 (s, 2H), 5.03 (s, 2H), 3.55–3.48 (m, 4H), 2.07–2.00 (m, 2H), 1.93–1.87 (m, 2H), 13C NMR (δ, ppm, 100 MHz, CDCl3): 167.68, 163.03, 142.95, 134.08, 132.08, 124.57, 123.47, 51.73, 46.39, 46.31, 33.10, 26.13, 24.10, FT-IR (cm−1): 3132, 3085, 2975, 2942, 2882, 1768, 1714, 1662, 1611, 1554, 1444, 1428, HR-ESI-MS (C17H17N5O3): m/z 340.14049 [M + H]+ (calcd. [60] 340.1394 [M + H]+).
2-((1-(2-morpholino-2-oxoethyl)−1H-1,2,3-triazol-4-yl)methyl)isoindoline-1,3‑dione (8c)

Yield: 90%, white crystals, M.p: 197–199 °C, 1H NMR (δ, ppm, 400 MHz, CDCl3): 7.87–7.85 (m, 2H), 7.81 (s, 1H), 7.74–7.72 (m, 2H), 5.21 (s, 2H), 5.03 (s, 2H), 3.70–3.69 (m, 4H), 3.64–3.61 (m, 2H), 3.57–3.55 (m, 2H), 13C NMR (δ, ppm, 100 MHz, CDCl3): 167.66, 163.47, 143.07, 134.13, 132.03, 124.63, 123.48, 66.58, 66.33, 50.81, 45.75, 42.52, 33.05, FT-IR (cm−1): 3135, 2989, 2964, 2928, 2900, 2853, 1775, 1717, 1664, 1611, 1552, 1470, 1457, 1436, 1416, HR-ESI-MS (C17H17N5O4): m/z 356.13663 [M + H]+ (calcd. 356.1343 [M + H]+).
2-(4-((1,3-dioxoisoindolin-2-yl)methyl)−1H-1,2,3-triazol-1-yl)-N,N-dimethylacetamide (8d)
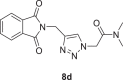
Yield:80%, white crystals, M.p: 210–212 °C, 1H NMR (δ, ppm, 400 MHz, DMSO‑d 6): 7.95 (s, 1H), 7.91–7.84 (m, 5H), 5.38 (s, 2H), 4.84 (s, 2H), 3.02 (s, 3H), 2.83 (s, 3H), 13C NMR (δ, ppm, 100 MHz, DMSO‑d 6): 167.84, 165.94, 142.32, 135.10, 132.01, 125.31, 123.71, 51.16, 36.32, 35.68, 33.39, FT-IR (cm−1): 3300, 3167, 2932, 1766, 1716, 1662, 1609, 1557, 1497, 1464, 1432, HR-ESI-MS (C15H15N5O3): m/z 314.12534 [M + H]+ (calcd. 314.1145 [M + H]+).
2-(4-((1,3-dioxoisoindolin-2-yl)methyl)−1H-1,2,3-triazol-1-yl)-N,N-diethylacetamide (8e)
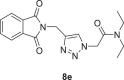
Yield: 90%, white crystals, M.p: 189–190 °C, 1H NMR (δ, ppm, 400 MHz, CDCl3): 7.87–7.85 (m, 3H), 7.74–7.71 (m, 2H), 5.19 (s, 2H), 5.03 (s, 2H), 3.41 (q, J = 7.2 Hz, 4H), 1.26 (t, J = 7.2 Hz, 3H), 1.14 (t, J = 7.2 Hz, 3H), 13C NMR (δ, ppm, 100 MHz, CDCl3): 167.64, 163.81, 142.90, 134.06, 132.07, 124.72, 123.44, 50.81, 41.90, 41.98, 33.12, 14.37, 12.85, FT-IR (cm−1): 3132, 3071, 2996, 2964, 2936, 2871, 1763, 1696, 1643, 1604, 1547, 1466, 1428, HR-ESI-MS (C17H19N5O3): m/z 342.15570 [M + H]+ (calcd. 342.1458 [M + H]+).
N,N-dibutyl-2-(4-((1,3-dioxoisoindolin-2-yl)methyl)−1H-1,2,3-triazol-1-yl)acetamide (8f)

Yield: 85%, white crystals, M.p: 120–122 °C, 1H NMR (δ, ppm, 400 MHz, CDCl3): 7.87.85 (m, 3H), 7.72–7.71 (m, 2H), 5.17 (s, 2H), 5.02 (s, 2H), 3.31 (dd, J = 10, 6.4 Hz, 4H), 1.63–1.47 (m, 4H), 1.41–1.24 (m, 4H), 0.96 (t, J = 7.2 Hz, 3H), 0.91 (t, J = 7.2 Hz, 3H), 13C NMR (δ, ppm, 100 MHz, CDCl3): 167.64, 164.14, 142.88, 134.06, 132.08, 124.73, 123.44, 50.81, 47.60, 46.49, 33.11, 31.17, 29.60, 20.19, 20.10, 13.79, FT-IR (cm−1): 3296, 3157, 3132, 2953, 2932, 2867, 1764, 1707, 1653, 1612, 1559, 1466, 1428, HR-ESI-MS (C21H27N5O3): m/z 398.21852 [M + H]+ (calcd. 398.2084 [M + H]+).
N-benzyl-2-(4-((1,3-dioxoisoindolin-2-yl)methyl)−1H-1,2,3-triazol-1-yl)acetamide (9a)
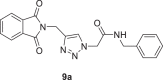
Yield: 90%, white crystals, M.p: 177–179 °C, 1H NMR (δ, ppm, 400 MHz, CDCl3): 7.86–7.84 (m, 3H), 7.74–7.72 (m, 2H), 7.30–7.24 (m, 3H), 7.19 (d, J = 7.2 Hz, 2H), 6.61 (s, 1H), 5.05 (s, 2H), 4.99 (s, 2H), 4.41 (d, J = 4.8 Hz, 2H), 13C NMR (δ, ppm, 100 MHz, CDCl3): 167.62, 164.90 (164.79), 143.84, 137.25, 137.13, 134.19, 131.97, 128.76, 128.71, 127.72, 123.52, 53.15, 43.77, 32.90, FT-IR (cm−1): 3297, 3153, 3067, 3035, 2957, 2103, 1766, 1709, 1651, 1607, 1556, 1465, 1451, 1427, HR-ESI-MS (C20H17N5O3): m/z 376.14039 [M + H]+ (calcd. 376.1365 [M + H]+).
2-(4-((1,3-dioxoisoindolin-2-yl)methyl)−1H-1,2,3-triazol-1-yl)-N-ethylacetamide (9b)
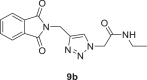
Yield: 95%, white crystals, M.p: 245–246 °C, 1H NMR (δ, ppm, 400 MHz, CDCl3): 7.90–7.88 (m, 3H), 7.77–7.75 (m, 2H), 6.11 (s, 1H), 5.02 (d, J = 8, 4H), 3.29 (p, J = 6.4 Hz, 2H), 1.12 (t, J = 7.2 Hz, 3H), 13C NMR (δ, ppm, 100 MHz, CDCl3-DMSO‑d 6 (40 °C): 172.19, 169.77, 147.40, 139.06, 136.77, 129.48, 128.08, 57.12, 39.07, 37.86, 19.32, FT-IR (cm−1): 3305, 3135, 3096, 3032, 2982, 2942, 2871, 1770, 1698, 1652, 1609, 1558, 1464, 1434, 1402, HR-ESI-MS (C15H15N5O3): m/z 314.12445 [M + H]+ (calcd. 314.1145 [M + H]+).
2-(4-((1,3-dioxoisoindolin-2-yl)methyl)−1H-1,2,3-triazol-1-yl)-N-propylacetamide (9c)
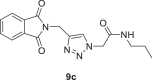
Yield: 90%, white crystals, mp: 171–173 °C, 1H NMR (δ, ppm, 400 MHz, CDCl3): 7.89–7.87 (m, 2H), 7.77–7.74 (m, 3H), 6.07 (s, 1H), 5.02 (d, J = 10.8 Hz, 4H), 3.20 (q, J = 6.8 Hz, 2H), 1.49 (h, J = 7.6 Hz, 2H), 0.85 (t, J = 7.2 Hz, 3H),13C NMR (δ, ppm, 100 MHz, CDCl3-DMSO‑d 6 (40 °C): 172.29, 169.85, 147.40, 138.97, 136.68, 129.33, 128.12, 57.22, 46.05, 37.75, 27.21, 16.20, FT-IR (cm−1): 3300, 3160, 3089, 2958, 2932, 2871, 1767, 1716, 1663, 1611, 1557, 1463, 1431, HR-ESI-MS (C16H17N5O3): m/z 328.14081 [M + H]+ (calcd. 328.1302 [M + H]+).
N‑butyl‑2-(4-((1,3-dioxoisoindolin-2-yl)methyl)−1H-1,2,3-triazol-1-yl)acetamide (9d)
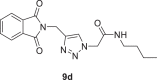
Yield: 92%, white crystals, M.p: 198–201 °C, 1H NMR (δ, ppm, 400 MHz, DMSO‑d6): 8.28 (t, J = 5.2 Hz, 1H), 8.02 (s, 1H), 7.92–7.85 (m, 4H), 5.01 (s, 2H), 4.84 (s, 2H), 3.06 (q, J = 6.8 Hz, 2H), 1.38 (p, J = 6.8 Hz, 2H), 1.27 (h, J = 6.8 Hz, 2H), 0.85 (t, J = 7.2 Hz, 3H), 13C NMR (δ, ppm, 100 MHz DMSO‑d6): 167.82, 165.54, 142.43, 135.06, 132.03, 125.17, 123.69, 52.08, 38.87, 33.36, 31.45, 19.96, 14.08, FT-IR (cm−1): 3285, 3153, 3103, 2960, 2935, 2864, 1766, 1725, 1718, 1663, 1611, 1561, 1463, 1429, HR-ESI-MS (C17H19N5O3): m/z 342.15644 [M + H]+ (calcd. 342.1458 [M + H]+).
2-(4-((1,3-dioxoisoindolin-2-yl)methyl)−1H-1,2,3-triazol-1-yl)-N-isopropylacetamide (9e)
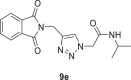
Yield: 96%, white crystals, M.p: 220–222 °C, 1H NMR (δ, ppm, 400 MHz, DMSO‑d6): 8.41 (d, J = 6.4 Hz, 1H), 8.02 (s, 1H), 7.91–7.85 (m, 4H), 5.00 (s, 2H), 4.83 (s, 2H), 3.80 (h, J = 6 Hz, 1H), 1.05 (dd, J = 6.4, 1.6 Hz, 6H), 13C NMR (δ, ppm, 100 MHz, DMSO‑d6): 167.83, 164.65, 142.36, 135.07, 132.02, 125.18, 123.69, 52.08, 41.35, 33.36, 22.70, FT-IR (cm−1): 3275, 3150, 3082, 2964, 2932, 2867, 1764, 1707, 1651, 1607, 1561, 1466, 1428, HR-ESI-MS (C16H17N5O3): m/z 328.14196 [M + H]+ (calcd. 328.1302 [M + H]+).
N-(tert‑butyl)−2-(4-((1,3-dioxoisoindolin-2-yl)methyl)−1H-1,2,3-triazol-1-yl)acetamide (9f)

Yield: 90%, white crystals, M.p: 216–218 °C, 1H NMR (δ, ppm, 400 MHz, CDCl3): 7.88–7.86 (m, 2H), 7.77 (s, 1H), 7.75–7.73 (m, 2H), 5.82 (s, NH), 5.03 (s, 2H), 4.91 (s, 2H), 1.32 (s, 9H), 13C NMR (δ, ppm, 100 MHz, CDCl3): 167.61, 163.82, 143.31, 134.15, 132.05, 124.42, 123.49, 53.75, 52.12, 33.02, 28.52, FT-IR (cm−1): 3288, 3160, 3089, 2996, 2960, 2864, 1769, 1728, 1718, 1670, 1614, 1562, 1456, 1431, HR-ESI-MS (C17H19N5O3): m/z 342.15595 [M + H]+ (calcd. 342.1458 [M + H]+)
3.3. Molecular docking studies
AutoDock 4.2 program was used to perform molecular docking studies [61]. 3D crystallographic structures of the ACE2-S1 complex, Mpro, and PLpro proteins were downloaded from the Protein Data Bank (www.rcsb.org) [62] (PDB ID:6M0J for the ACE2-S1 complex, PDB ID:7K40 for the Mpro, PDB ID:7CMD for the PLpro). All co-crystallized ligands and waters were deleted using Discovery Studio Visualizer 2021 [63]. The 2D structures of the compounds were drawn on ChemDraw Professional 16.0. The 2D structures of boceprevir, GRL0617, and MB were retrieved from the PubChem website [64]. The 2D structures of all compounds were minimized using the MMFF94s force field via Avogadro software [65]. In the docking analysis, the compounds were flexible, and the proteins were rigid. The grid boxes were created with the AutoDock 4.2 program. The grid box center values were arranged as X: −38.842 Å, Y:28.998 Å, Z: 1.222 Å for 6M0J; X: 10.151 Å, Y:−25.241 Å, Z: 21.899 Å for 7K40; X: −33.899 Å, Y:−11.293 Å, Z: −30.009 Å for 7CMD. The grid box sizes were set at 86 × 86 × 86 Å3 for 6M0J, 40 × 40 × 40 Å3 for 7K40, and 50 × 50 × 50 Å3 for 7CMD. The RMSD values for the validation were calculated via the Visual Molecular Dynamics program (VMD) [66]. The results of molecular docking were visualized with Discovery Studio Visualizer 2021 and VMD.
3.4. Molecular dynamics simulation
Molecular dynamics simulations were performed for 50 ns using Desmond [67], a Package of Schrödinger LLC (Schrödinger, LLC, New York, NY, 2014). The initial stage of protein and ligand complexes for molecular dynamics simulation were obtained from docking studies. Molecular Docking Studies provide a prediction of ligand binding status in static conditions. Simulations were carried out to predict the ligand binding status in the physiological environment. The protein-ligand complexes were preprocessed using Protein Preparation Wizard in Maestro, which also included optimization and minimization of the complexes. All systems were prepared by the System Builder tool. Solvent Model with an orthorhombic box was selected as TIP3P (Transferable Intermolecular Interaction Potential 3 Points). The OPLS_2005 force field [68,69] was used in the simulation. The models were made neutral by adding counter ions where needed. To mimic the physiological conditions, 0.15 M salt (NaCl) was added. The NPT [70,71] ensemble with 300 K temperature and 1 atm pressure were selected for complete simulation. The models were relaxed before the simulation. The trajectories were saved after every 100 ps for analysis, and the stability of simulations was evaluated by calculating the RMSD of the protein and ligand over time.
4. Conclusion
In summary, novel phthalimide analogs 8(a-f) and 9(a-f) bearing a 1,2,3-triazole framework were synthesized via CuAAC in good yields and characterized by various spectroscopic methods. The interactions with the proteins of SARS-CoV-2 (Mpro and PLpro) and the PPI between ACE2-S1 of the target compounds were examined via the molecular docking studies by comparison with MB, boceprevir, and GRL0617 inhibitors. All phthalimide analogs showed better interactions than MB in the interactions with the PPI of ACE2-S1 and compound 9a in the target compounds exhibited the best binding energy value (−9.70 kcal/mol) and estimated Ki value (0.077 µM). In the interactions with the Mpro protein, compound 8a showed the best binding energy (−8.76 kcal/mol) and estimated Ki value (0.379 µM). Regarding the interactions with the PLpro, compound 8b exhibited the best binding energy (−8.87 kcal/mol) and estimated Ki value (0.315 µM) in the target compounds. The ligand-protein stabilities of compounds 8a-Mpro, 8b-PLpro, and 9a-‘ACE2-S1’ showing the best binding energy and predicted Ki were investigated via the molecular dynamics simulation studies. The stabilities of 8a-Mpro, 8b-PLpro, and 9a-‘ACE2-S1’ support the docking results. Finally, in silico ADMET studies were carried out. According to the predicted results, the target compounds have acceptable pharmacokinetic profiles and are suitable for oral administration. The compounds were estimated to display low toxicity levels. According to the in silico results, the target compounds may prevent the entry of SARS-CoV-2 into the host-cell, and the progression of the Mpro and PLpro proteins. Therefore, the compounds can be evaluated in vitro and in vivo studies as potential inhibitors of SARS-CoV-2.
CRediT authorship contribution statement
Ayse Tan: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Visualization, Writing – original draft, Writing – review & editing.
Declaration of Competing Interest
The author declares no competing interests.
Acknowledgments
The author is grateful to Mus Alparslan University. 1H and 13C NMR analyses at Giresun University, HR-MS analyses at Ataturk University were performed with a fee.
Footnotes
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.molstruc.2022.132915.
Appendix. Supplementary materials
References
- 1.Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R. A novel coronavirus from patients with pneumonia in China. New Engl. J. Med. 2019:2020. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Narkhede R.R., Cheke R.S., Ambhore J.P., Dshinde S. The molecular docking study of potential drug candidates showing anti-COVID-19 activity by exploring of therapeutic targets of SARS-CoV-2. Eur. J. Med. Oncol. 2020;4(3):185–195. [Google Scholar]
- 3.Walls A.C., Park Y.-J., Tortorici M.A., Wall A., McGuire A.T., Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;181(2):281–292. doi: 10.1016/j.cell.2020.02.058. e286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wrapp D., Wang N., Corbett K.S., Goldsmith J.A., Hsieh C.-.L., Abiona O., Graham B.S., McLellan J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367(6483):1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li W., Zhang C., Sui J., Kuhn J.H., Moore M.J., Luo S., Wong S.K., Huang I.C., Xu K., Vasilieva N. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 2005;24(8):1634–1643. doi: 10.1038/sj.emboj.7600640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lan J., Ge J., Yu J., Shan S., Zhou H., Fan S., Zhang Q., Shi X., Wang Q., Zhang L. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581(7807):215–220. doi: 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- 7.Unni S., Aouti S., Thiyagarajan S., Padmanabhan B. Identification of a repurposed drug as an inhibitor of Spike protein of human coronavirus SARS-CoV-2 by computational methods. J. Biosci. 2020;45(1):1–20. doi: 10.1007/s12038-020-00102-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tai W., He L., Zhang X., Pu J., Voronin D., Jiang S., Zhou Y., Du L. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol. 2020;17(6):613–620. doi: 10.1038/s41423-020-0400-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bojadzic D., Alcazar O., Buchwald P. Methylene blue inhibits the SARS-CoV-2 spike–ACE2 protein-protein interaction–a mechanism that can contribute to its antiviral activity against COVID-19. Front. Pharmacol. 2021:2255. doi: 10.3389/fphar.2020.600372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Astuti I. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): an overview of viral structure and host response. Diab. Metab. Syndr. 2020;14(4):407–412. doi: 10.1016/j.dsx.2020.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mandal M., Chowdhury S.K., Khan A.A., Baildya N., Dutta T., Misra D., Ghosh N.N. Inhibitory efficacy of RNA virus drugs against SARS-CoV-2 proteins: an extensive study. J. Mol. Struct. 2021 doi: 10.1016/j.molstruc.2021.130152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fu L., Ye F., Feng Y., Yu F., Wang Q., Wu Y., Zhao C., Sun H., Huang B., Niu P. Both Boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nat. Commun. 2020;11(1):1–8. doi: 10.1038/s41467-020-18233-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Osipiuk J., Azizi S.-A., Dvorkin S., Endres M., Jedrzejczak R., Jones K.A., Kang S., Kathayat R.S., Kim Y., Lisnyak V.G. Structure of papain-like protease from SARS-CoV-2 and its complexes with non-covalent inhibitors. Nat. Commun. 2021;12(1):1–9. doi: 10.1038/s41467-021-21060-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Snijder E., Decroly E., Ziebuhr J. The nonstructural proteins directing coronavirus RNA synthesis and processing. Adv. Virus Res. 2016;96:59–126. doi: 10.1016/bs.aivir.2016.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sanachai K., Mahalapbutr P., Lee V.S., Rungrotmongkol T., Hannongbua S. Silico elucidation of potent inhibitors and rational drug design against SARS-CoV-2 Papain-like protease. J. Phys. Chem. B. 2021 doi: 10.1021/acs.jpcb.1c07060. [DOI] [PubMed] [Google Scholar]
- 16.Mandal M., Chowdhury S.K., Khan A.A., Baildya N., Dutta T., Misra D., Ghosh N.N. Inhibitory efficacy of RNA virus drugs against SARS-CoV-2 proteins: an extensive study. J. Mol. Struct. 2021;1234 doi: 10.1016/j.molstruc.2021.130152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stasiulewicz A., Maksymiuk A.W., Nguyen M.L., Bełza B., Sulkowska J.I. SARS-CoV-2 papain-like protease potential inhibitors—In silico quantitative assessment. Int. J. Mol. Sci. 2021;22(8):3957. doi: 10.3390/ijms22083957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holanda V.N., Lima E.M.d.A., Silva W.V.d., Maia R.T., Medeiros R.d.L., Ghosh A., Lima V.L.d.M., Figueiredo R.C.B.Q.d. Identification of 1, 2, 3-triazole-phthalimide derivatives as potential drugs against COVID-19: a virtual screening, docking and molecular dynamic study. J. Biomol. Struct. Dyn. 2021:1–19. doi: 10.1080/07391102.2020.1871073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Domínguez-Villa F.X., Durán-Iturbide N.A., Ávila-Zárraga J.G. Synthesis, molecular docking, and in silico ADME/Tox profiling studies of new 1-aryl-5-(3-azidopropyl) indol-4-ones: potential inhibitors of SARS CoV-2 main protease. Bioorg. Chem. 2021;106 doi: 10.1016/j.bioorg.2020.104497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Özkan H., Adem Ş. Synthesis, spectroscopic characterizations of novel norcantharimides, Their ADME properties and docking studies against COVID-19 Mpr°. ChemistrySelect. 2020;5(18):5422–5428. doi: 10.1002/slct.202001123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Asha R.N., Nayagam B.R.D., Bhuvanesh N. Synthesis, molecular docking, and in silico ADMET studies of 4-benzyl-1-(2, 4, 6-trimethyl-benzyl)-piperidine: potential Inhibitor of SARS-CoV2. Bioorg. Chem. 2021;112 doi: 10.1016/j.bioorg.2021.104967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seliem I.A., Panda S.S., Girgis A.S., Moatasim Y., Kandeil A., Mostafa A., Ali M.A., Nossier E.S., Rasslan F. Srour AM: new quinoline-triazole conjugates: synthesis, and antiviral properties against SARS-CoV-2. Bioorg. Chem. 2021;114 doi: 10.1016/j.bioorg.2021.105117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.da Silva M.R., Santos C.P. Standard molar enthalpies of formation and sublimation of N-phenylphthalimide. J. Therm. Anal. Calorim. 2007;87(1):21–25. [Google Scholar]
- 24.Tan A., Bozkurt E., Kishali N., Kara Y. A New and Convenient Synthesis of Amino-phthalimide (1H-Isoindole-1, 3 (2H)-dione) derivatives and their photoluminescent properties. Helv. Chim. Acta. 2014;97(8):1107–1114. [Google Scholar]
- 25.Tan A., Bozkurt E., Kara Y. Investigation of solvent effects on photophysical properties of new aminophthalimide derivatives-based on methanesulfonate. J. Fluoresc. 2017;27(3):981–992. doi: 10.1007/s10895-017-2033-2. [DOI] [PubMed] [Google Scholar]
- 26.Pan L., Li X., Gong C., Jin H., Qin B. Synthesis of N-substituted phthalimides and their antifungal activity against Alternaria solani and Botrytis cinerea. Microb. Pathog. 2016;95:186–192. doi: 10.1016/j.micpath.2016.04.012. [DOI] [PubMed] [Google Scholar]
- 27.Nayab P.S., Irfan M., Abid M., Pulaganti M., Nagaraju C., Chitta S.K. Experimental and molecular docking investigation on DNA interaction of N-substituted phthalimides: antibacterial, antioxidant and hemolytic activities. Luminescence. 2017;32(3):298–308. doi: 10.1002/bio.3178. [DOI] [PubMed] [Google Scholar]
- 28.Tan A., Yaglioglu A.S., Kishali N.H., Sahin E., Kara Y. Evaluation of cytotoxic potentials of some isoindole-1, 3-dione derivatives on HeLa, C6 and A549 cancer cell lines. Med. Chem. (Los Angeles) 2020;16(1):69–77. doi: 10.2174/1573406415666181206115638. [DOI] [PubMed] [Google Scholar]
- 29.Tan A., Almaz Z. Synthesis, investigation of the cholinesterase inhibitory activities and in silico studies of some novel N-substituted phthalimide derivatives. J. Iranian Chem. Soc. 2022:1–12. [Google Scholar]
- 30.Abdel-Hafez A.A.-M. Synthesis and anticonvulsant evaluation ofN-substituted-isoindolinedione derivatives. Arch. Pharm. Res. 2004;27(5):495–501. doi: 10.1007/BF02980121. [DOI] [PubMed] [Google Scholar]
- 31.LdM Lima, P Castro, Machado A.L., Fraga C.A.M., Lugnier C., De Moraes V.L.G., Barreiro E.J. Synthesis and anti-inflammatory activity of phthalimide derivatives, designed as new thalidomide analogues. Bioorg. Med. Chem. 2002;10(9):3067–3073. doi: 10.1016/s0968-0896(02)00152-9. [DOI] [PubMed] [Google Scholar]
- 32.Khedkar M.V., Khan S.R., Sawant D.N., Bagal D.B., Bhanage B.M. Palladium on carbon: an efficient, heterogeneous and reusable catalytic system for carbonylative synthesis of N-substituted phthalimides. Adv. Synth. Catal. 2011;353(18):3415–3422. [Google Scholar]
- 33.Aouad M.R., Almehmadi M.A., Rezki N., Al-blewi F.F., Messali M., Ali I. Design, click synthesis, anticancer screening and docking studies of novel benzothiazole-1, 2, 3-triazoles appended with some bioactive benzofused heterocycles. J. Mol. Struct. 2019;1188:153–164. [Google Scholar]
- 34.Tan A. Novel 1, 2, 3-triazole compounds: synthesis, In vitro xanthine oxidase inhibitory activity, and molecular docking studies. J. Mol. Struct. 2020;1211 [Google Scholar]
- 35.Haider S., Alam M.S., Hamid H. 1, 2, 3-Triazoles: scaffold with medicinal significance. Inflamm. Cell Signal. 2014;1:e95. [Google Scholar]
- 36.Ayşe T. Ksantin Oksidaz İnhibitörü Yeni 1, 2, 3-triazol Türevlerinin Sentezi, Karakterizasyonu ve Moleküler Doking Çalışmaları. Bitlis Eren. Üniversitesi. Fen. Bilimleri Dergisi. 2021;9(2):644–654. [Google Scholar]
- 37.Totobenazara J., Burke A.J. New click-chemistry methods for 1, 2, 3-triazoles synthesis: recent advances and applications. Tetrahedron Lett. 2015;56(22):2853–2859. [Google Scholar]
- 38.Poonia N., Kumar A., Kumar V., Yadav M., Lal K. Recent Progress in 1H-1, 2, 3-triazoles as potential antifungal agents. Curr. Top. Med. Chem. 2021;21(23):2109–2133. doi: 10.2174/1568026621666210913122828. [DOI] [PubMed] [Google Scholar]
- 39.Lal K., Poonia N., Rani P., Kumar A., Kumar A. Design, synthesis, antimicrobial evaluation and docking studies of urea-triazole-amide hybrids. J. Mol. Struct. 2020;1215 [Google Scholar]
- 40.Sajja Y., Vanguru S., Vulupala H.R., Bantu R., Yogeswari P., Sriram D. Nagarapu L: design, synthesis and in vitro anti-tuberculosis activity of benzo [6, 7] cyclohepta [1, 2-b] pyridine-1, 2, 3-triazole derivatives. Bioorg. Med. Chem. Lett. 2017;27(23):5119–5121. doi: 10.1016/j.bmcl.2017.10.071. [DOI] [PubMed] [Google Scholar]
- 41.Dong Y., Hu X., Duan C., Liu P., Liu S., Lan L., Chen D., Ying L., Su S., Gong X. A series of new medium-bandgap conjugated polymers based on naphtho [1, 2-c: 5, 6-c] bis (2-octyl-[1, 2, 3] triazole) for high-performance polymer solar cells. Adv. Mater. 2013;25(27):3683–3688. doi: 10.1002/adma.201301547. [DOI] [PubMed] [Google Scholar]
- 42.Aouad M.R. Click Synthesis and antimicrobial screening of novel isatin-1, 2, 3-triazoles with piperidine, morpholine, or piperazine moieties. Org. Prep. Proced. Int. 2017;49(3):216–227. [Google Scholar]
- 43.Deshmukh T.R., Khare S.P., Krishna V.S., Sriram D., Sangshetti J.N., Bhusnure O., Khedkar V.M., Shingate B.B. Design and synthesis of new aryloxy-linked dimeric 1, 2, 3-Triazoles via click chemistry approach: biological evaluation and molecular docking study. J. Heterocycl. Chem. 2019;56(8):2144–2162. [Google Scholar]
- 44.Shen Y., Sheng R., Zhang J., He Q., Yang B., Hu Y. 2-Phenoxy-indan-1-one derivatives as acetylcholinesterase inhibitors: a study on the importance of modifications at the side chain on the activity. Bioorg. Med. Chem. 2008;16(16):7646–7653. doi: 10.1016/j.bmc.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 45.Himo F., Lovell T., Hilgraf R., Rostovtsev V.V., Noodleman L., Sharpless K.B., Fokin V.V. Copper (I)-catalyzed synthesis of azoles. DFT study predicts unprecedented reactivity and intermediates. J. Am. Chem. Soc. 2005;127(1):210–216. doi: 10.1021/ja0471525. [DOI] [PubMed] [Google Scholar]
- 46.Phatak P.S., Bakale R.D., Dhumal S.T., Dahiwade L.K., Choudhari P.B., Siva Krishna V., Sriram D., Haval K.P. Synthesis, antitubercular evaluation and molecular docking studies of phthalimide bearing 1, 2, 3-triazoles. Synth. Commun. 2019;49(16):2017–2028. [Google Scholar]
- 47.Hou D.R., Hung M.-S., Liao C.-C., Lin C.-.C.: 1, 2, 3-Triazole derivatives as new cannabinoid-1 receptor antagonists. In.: Google Patents; 2012.
- 48.Lee D., Kim D., Lee S., Kim T., Kim J., Kim S., Liu K.-H., Lee S., Bae J.-S., Song K.-S. Efficient syntheses of 1, 2, 3-Triazoloamide derivatives using solid-and solution-phase synthetic approaches. Molecules. 2015;20(11):19984–20013. doi: 10.3390/molecules201119673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bunders C., Cavanagh J., Melander C. Flustramine inspired synthesis and biological evaluation of pyrroloindoline triazole amides as novel inhibitors of bacterial biofilms. Org. Biomol. Chem. 2011;9(15):5476–5481. doi: 10.1039/c1ob05605k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pokhodylo N., Savka R., Obushak M. Synthesis of (1 H-1, 2, 3-Triazol-1-yl) acetic Acid Derivatives. Russ. J. Org. Chem. 2020;56(8):1421–1431. [Google Scholar]
- 51.Dommerholt J., Van Rooijen O., Borrmann A., Guerra C.F., Bickelhaupt F.M., Van Delft F.L. Highly accelerated inverse electron-demand cycloaddition of electron-deficient azides with aliphatic cyclooctynes. Nat. Commun. 2014;5(1):1–7. doi: 10.1038/ncomms6378. [DOI] [PubMed] [Google Scholar]
- 52.Almaz Z., Oztekin A., Tan A., Ozdemir H. Biological evaluation and molecular docking studies of 4-aminobenzohydrazide derivatives as cholinesterase inhibitors. J. Mol. Struct. 2021;1244 [Google Scholar]
- 53.Tan A., Kizilkaya S., Kelestemur U., Akdemir A., Kara Y. The synthesis, anticancer activity, structure-activity relationships and molecular modelling studies of novel isoindole-1, 3 (2H)-dione compounds containing different functional groups. Anti-Cancer Agents Med. Chem. (Formerly Curr. Med. Chem.-Anti-Cancer Agents) 2020;20(11):1368–1378. doi: 10.2174/1871520620666200410080648. [DOI] [PubMed] [Google Scholar]
- 54.Tan A., Almaz Z. Synthesis, investigation of the cholinesterase inhibitory activities and in silico studies of some novel N-substituted phthalimide derivatives. J. Iranian Chem. Soc. 2022 [Google Scholar]
- 55.Freitas B.T., Durie I.A., Murray J., Longo J.E., Miller H.C., Crich D., Hogan R.J., Tripp R.A., Pegan S.D. Characterization and noncovalent inhibition of the deubiquitinase and deISGylase activity of SARS-CoV-2 papain-like protease. ACS Infect. Dis. 2020;6(8):2099–2109. doi: 10.1021/acsinfecdis.0c00168. [DOI] [PubMed] [Google Scholar]
- 56.Daina A., Michielin O., Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017;7(1):1–13. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Banerjee P., Eckert A.O., Schrey A.K., Preissner R. ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucl. Acids Res. 2018;46(W1):W257–W263. doi: 10.1093/nar/gky318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Banerjee P., Dehnbostel F.O., Preissner R. Prediction is a balancing Act: importance of sampling methods to balance sensitivity and specificity of predictive models based on imbalanced chemical data sets. Front. Chem. 2018;6:362. doi: 10.3389/fchem.2018.00362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Drwal M.N., Banerjee P., Dunkel M., Wettig M.R., Preissner R. ProTox: a web server for the in silico prediction of rodent oral toxicity. Nucl. Acids Res. 2014;42(W1):W53–W58. doi: 10.1093/nar/gku401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Patiny L., Borel A. ACS Publications; 2013. ChemCalc: a Building Block For Tomorrow's Chemical Infrastructure. [DOI] [PubMed] [Google Scholar]
- 61.Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30(16):2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Berman H., Westbrook J., Feng Z., Gilliland G., Bhat T., Weissig H., Shindyalov I., Bourne P.: The protein data Bank nucleic acids research, 28: 235–242. URL: www rcsb org Citation2000. [DOI] [PMC free article] [PubMed]
- 63.BIOVIA . BIOVIA Workbook; San Diego: 2021. Dassault Systèmes. Release; 2021. [Google Scholar]
- 64.Kim S., Chen J., Cheng T., Gindulyte A., He J., He S., Li Q., Shoemaker B.A., Thiessen P.A., Yu B. PubChem in 2021: new data content and improved web interfaces. Nucl. Acids Res. 2021;49(D1):D1388–D1395. doi: 10.1093/nar/gkaa971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hanwell M.D., Curtis D.E., Lonie D.C., Vandermeersch T., Zurek E., Hutchison G.R. Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J. Chem. Inf. 2012;4(1):17. doi: 10.1186/1758-2946-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14(1):33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 67.Bowers K.J., Chow D.E., Xu H., Dror R.O., Eastwood M.P., Gregersen B.A., Klepeis J.L., Kolossvary I., Moraes M.A., Sacerdoti F.D. SC'06: Proceedings of the 2006 ACM/IEEE Conference on Supercomputing. IEEE; 2006. Scalable algorithms for molecular dynamics simulations on commodity clusters. 43-43. [Google Scholar]
- 68.Jorgensen W.L., Maxwell D.S., Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996;118(45):11225–11236. [Google Scholar]
- 69.Kaminski G.A., Friesner R.A., Tirado-Rives J., Jorgensen W.L. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B. 2001;105(28):6474–6487. [Google Scholar]
- 70.Martyna G.J., Klein M.L., Tuckerman M. Nosé–Hoover chains: the canonical ensemble via continuous dynamics. J. Chem. Phys. 1992;97(4):2635–2643. [Google Scholar]
- 71.Martyna G.J., Tobias D.J., Klein M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994;101(5):4177–4189. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.