

ABSTRACT
The sensu stricto autophagy, macroautophagy, is considered to be both a metabolic process as well as a bona fide quality control process. The question as to how these two aspects of autophagy are coordinated and whether and why they overlap has implications for fundamental aspects, pathophysiological effects, and pharmacological manipulation of autophagy. At the top of the regulatory cascade controlling autophagy are master regulators of cellular metabolism, such as MTOR and AMPK, which render the system responsive to amino acid and glucose starvation. At the other end exists a variety of specific autophagy receptors, engaged in the selective removal of a diverse array of intracellular targets, from protein aggregates/condensates to whole organelles such as mitochondria, ER, peroxisomes, lysosomes and lipid droplets. Are the roles of autophagy in metabolism and quality control mutually exclusive, independent or interlocked? How are priorities established? What are the molecular links between both phenomena? This article will provide a starting point to formulate these questions, the responses to which should be taken into consideration in future autophagy-based interventions.
KEYWORDS: mitochondria, lysosomes, glycolysis, oxidative phosphorylation, sirtuin, mitophagy, NASH, Tor
Introduction
The present day view of the sensu stricto autophagy (often referred to as macroautophagy) is primarily based on the genetic analyses initially conducted in yeast strains that were deficient in a series of ATG (autophagy related) genes and that were found to be largely conserved in mammals [1]. However, despite similarities, including a the level of regulation of autophagy by the principal regulators of cellular metabolism, MTOR and AMPK [2,3], there are notable differences between mammalian and yeast systems regarding regulatory inputs and effector functions [4,5]. With respect to the mammalian system, which is the exclusive focus of this review, three sets of conclusions have transpired with important ramifications for the future of autophagy studies as they relate to pathway fundamentals, normative physiology and human diseases [4,5]: (i) the core autophagy orthologs (ATG) or paralogs engage in additional activities in mammalian cells; (ii) some of the principal findings regarding the mammalian ATG paralogs are difficult to reconcile with the linear pathway discovered in yeast; and (iii) the sensu stricto autophagy in mammalian cells engages a plethora of additional proteins outside of the core autophagy apparatus defined per the yeast paradigm. This brings us to the question of the evolutionarily conserved integration of the metabolic and quality control functions of autophagy as manifested in mammalian systems. We refer to metabolic autophagy as autophagy that primarily generates nutrients and supports general cellular growth vs. quality control (QC) autophagy that keeps the cytoplasm free of damaged or surplus organelles, aggregates and invading microbes. Here, we will discuss the differences and similarities of both types of autophagy.
Brief description of mammalian autophagy
There are numerous reviews summarizing the core autophagy pathway, and the reader is referred to schematics in such articles [1,6]. The ignition of autophagy involves phagophores sequestering cytoplasmic cargo, which in some cases is captured in a selective fashion via receptors that collect protein aggregates/condensates, damaged/surplus organelles or invading microbes (Figure 1). The resulting autophagosomes then fuse with lysosomes to digest or eliminate the captured material [4]. Of note, autophagy can be activated by metabolic cues [7–11] (Figure 1A) or triggered by cargo recognition via autophagic receptors [4,12,13] that actively stimulate autophagy [13–16] (Figure 1B), with one of the binding surfaces of mammalian Atg8 orthologs (mAtg8s), resulting in pairings such as LC3-interacting region:LIR-docking site (LIR:LDS; the most common) [12], ubiquitin-interacting motif:UIM-docking site (UIM:UDS) [17], or potentially other non-LDS surfaces, thus linking the cargo to autophagic membranes. Nevertheless, the ability to bind LC3 or its paralogs is not essential for initiation of autophagy [18] and this can be experimentally uncoupled [16].
Figure 1.
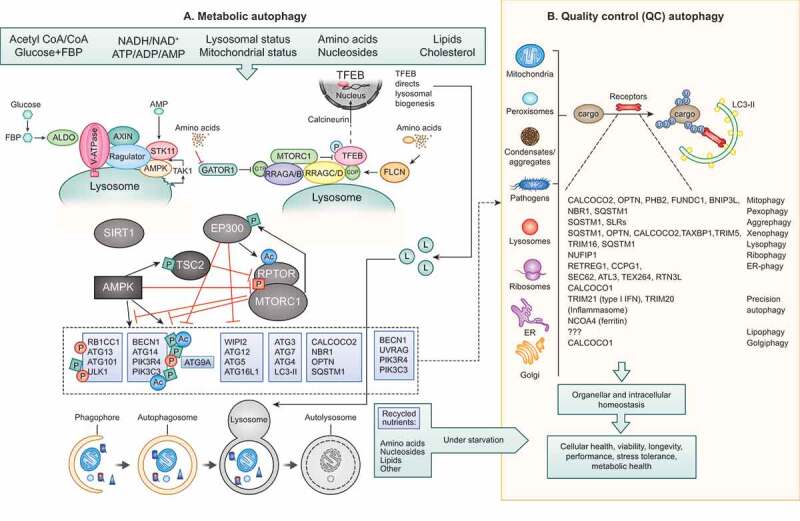
Metabolic autophagy vs QC autophagy. (A) Metabolic autophagy is a response to diminishing sources of indicated nutrients and energy. AMPK and MTOR are at the center of responses and both along with their regulatory circuitry are located on lysosomes. Note: canonical activation of AMPK in response to AMP occurs in the cytosol (dashed arrow), whereas its noncanonical activation occurs on lysosomes in response to glucose starvation via ALDO (aldolase)-V-ATPase-AXIN-STK11/LKB1 or in response to lysosomal damage via MAP3K7/TAK1. Regulatory circuitry is indicated. TFEB is phosphorylated by MTOR and retained in the cytosol, but when nutrients are sparse and MTOR inactive, TFEB translocates to the nucleus stimulating lysosomal biogenesis. The autophagy systems (see text for definition of different protein complexes) are under control of AMPK, EP300, SIRT1 and MTOR, thought to be responsible for conducting metabolic (“bulk”) autophagy that digests macromolecules upon fusion with lysosomes to recycle/supply nutrients during starvation. Dashed box and arrow in (A) indicate engagement of components of the basal autophagy apparatus with QC autophagy in (B). (B) Quality control (QC) autophagy removes a variety of specific cytoplasmic targets, with individual cargos depicted along with their “selective autophagy” names as well as the cargo receptors implicated in each case. Both metabolic and QC autophagy contribute to cellular health and fitness.
Autophagosome formation is controlled through assembly and interactions of a number of modules [1] (Figure 1A). The first autophagy-dedicated module is the ULK1-ULK2 kinase complex with RB1CC1/FIP200, ATG13 and ATG101 [19–21]. A second system is an ATG14-endowed class III phosphatidylinositol 3-kinase (PtdIns3K) complex that includes PIK3C3/VPS34 and BECN1 (beclin 1) [22]. There are additional modules including ATG9, WIPI1-WIPI2 and WDR45B/WIPI3-WDR45/WIPI4 proteins and ATG2 complexes [23–25]. ATG2 plays a role in lipid transfer between the donor membrane and the growing phagophore [26], whereas ATG9A appears to cause membrane bending and lipid scrambling [27,28]. Other proteins with lipid scramblase properties, VMP1 and TMEM41, may assist during generation of autophagic membranes [29]. The above modules interconnect during autophagy, e.g. RB1CC1 bridges the ULK1-ULK2 complex with the mAtg8 conjugation system by binding ATG16L1 [30–32], ATG16L1 and WIPI1-WIPI2 interact [33], and ATG13 likely connects the ULK1-ULK2 complex with ATG14-PIK3C3/VPS34 [34]. A special place in the autophagy system is reserved for the family of six principal mAtg8s (depending on the organism, 5–7): LC3A, LC3B, LC3C and GABARAP, GABARAPL1 and GABARAPL2. LC3B, one of the most commonly used autophagosomal membrane markers [35], however, is also found in a variety of other non-autophagic membranes [5,36,37]. The special, almost iconic, status of LC3 is reinforced by the ATG12–ATG5-ATG16L1 E3 ligase conjugation system [38], which lipidates LC3B and other mAtg8s. Whereas lipidation of proteins (e.g., myristylation, palmitoylation) per se is not a unique process, in the case of mAtg8s and autophagy, lipidation (in this case, covalent attachment of phosphatidylethanolamine) of mAtg8s has been proposed (but not proven) to be central to the nucleation and expansion of nascent autophagosomal membranes. Of note, lipidated mAtg8s bind to membranes other than autophagosomes [5,36,37,39], and mAtg8s play important regulatory roles [40] including the control of MTOR [41] and TFEB (transcription factor EB) [37,41], suggesting that mAtg8s have multiple functions beyond their proposed role in building autophagosomes.
Metabolic autophagy
Early in phylogeny, autophagy is employed by unicellular eukaryotes to mobilize energy reserves in conditions of nutrient deprivation and to catabolize macromolecules (RNA, proteins, complex lipids and carbohydrates) into small metabolites (nucleotides, amino acids, fatty acids and glycerol, simple sugars) that either can be used for energy metabolism or constitute building blocks for adaptive anabolic reactions [42]. This latter possibility comes into play when the adaptation to nutrient scarcity goes beyond the maintenance of vital bioenergetics and involves differentiation of cells (e.g., to generate spores by fungi or dauer forms by nematodes) [43,44], allowing them to resist harsh environmental conditions. Moreover, in evolutionary terms, it appears plausible that the ignition of the autophagic program has been coupled to other non-nutritional forms of cellular stress (such as hyperthermia, exposure to toxins, oxidative stress, or DNA damage) to rapidly mount rapid adaptive response requiring a combination of catabolic and anabolic reactions.
In mammalians, nutrient scarcity induced by absent or hypocaloric feeding causes a reduction of extracellular nutrients (such as glucose and amino acids) that is compensated by the generation of ketone bodies and a surge in free fatty acids generated by lipolysis [45]. However, the depletion of glucose and branched chain amino acids (isoleucine, leucine, valine) and the accompanying reduction of trophic hormones (such as insulin and IGF1; insulin like growth factor 1], which regulate the transport of glucose and amino acids on cell membranes) results in the activation of nutrient sensors such as AMPK, EP300, SIRT1 (sirtuin 1) and inactivation of MTOR (Figure 1A) with the consequent activation of “metabolic” autophagy in all major cell types of the body [46]. Adult mice that have undergone an inducible whole-body knockout of the perinatally essential autophagy gene Atg7 develop fatal hypoglycemia upon starvation [47], illustrating the importance of autophagy for adaptation to nutrient stress. In fed conditions, such a whole-body inactivation of autophagy results in the reduction of specific metabolites (such as arginine, cholesteryl sulfate, malate/fumarate, cystathionine, S-adenosyl-L-methionine and carnitine), supporting the role of autophagy in basal metabolism [48].
Experiments in a variety of model organisms including mice suggest that caloric restriction, intermittent fasting and endurance training mediate their positive effects on aging and age-associated diseases (such as diabetes) through the induction of metabolic autophagy [49,50]. Conversely, it appears plausible that obesity with its accompanying excess of nutrients (glucose, amino acids, fatty acids, cholesterol), and autophagy-inhibitory hormones (insulin, IGF1, DBI/ACBP [diazepam binding inhibitor, acyl CoA binding protein]) causes chronic suppression of autophagy [51,52]. Chronic autophagy inhibition may be (one of) the mechanism(s) through which obesity accelerates the manifestation of major time-dependent ailments including cardiovascular disease, cancer and neurodegeneration [53].
Quality control autophagy
QC autophagy removes specific targets in the cell whereas metabolic autophagy, i.e., autophagy activated by nutrient scarcity, leads to the degradation of a vast array of cytoplasmic targets, apparently without a true specificity for a particular type of cargo, meaning that any kind of cargo including portions of the cytosol, endoplasmic reticulum (ER) and mitochondria can be detected in autophagosomes (Figure 1B). This “nonselective” (or “bulk”) autophagy contrasts with “selective” autophagy or “QC” autophagy, where the same or a largely overlapping molecular machinery detects specific cargo. Such a selective autophagy is designated by a term comprising a prefix derived from the cargo (e.g., aggre-, reticulo-, ferritino-, glyco-,lipo-, mito-, nucleo-, ribo-, pexo-, viro-, xeno-) and the suffix “phagy” [4]. The latest addition to this repertoire is Golgi-phagy [54]. Such a specific cargo is often recognized by receptors containing LIR motifs (W/F/Y1x2x3L/I/V4) that directly bridge the cargo to mAtg8s present on nascent phagophores [12]. Recent in-depth advances have increased our understanding of the global cargo repertoire for soluble selective autophagy receptors [55], including how they physically act during autophagic capture of cargo such as aggregates/liquid droplets (condesates) [56]. One example of integral membrane selective autophagy receptors is the mitochondrial protein PHB2 (prohibitin 2) that is usually hidden within mitochondria, yet becomes accessible with its LIR when the outer mitochondrial membrane ruptures, resulting in mitophagy [57]. Other LIR-containing autophagy receptors possess a ubiquitin-binding domain empowering them to recognize cargo that has been “marked up” for autophagic destruction by ubiquitination. A prominent example for this mechanism is mitophagy that is initiated upon loss of the mitochondrial transmembrane potential, leading to the accumulation of the kinase PINK1, the consequent recruitment/activation of the cytosolic E3 ubiquitin ligase PRKN/Parkin, and the final ubiquitination of proteins at the mitochondrial surface recognized by selective autophagy receptors [58] (Figure 2A). Alternatively, specific targets (including proteins involved in interferon and inflammasome signaling, e.g. IRF3, NLRP3, and pro-CASP1 (caspase 1) [59]) can bind to TRIM (tripartite motif) family members (Figure 2A), which serve as adaptors that interact with GABARAPs to target such proteins for “precision autophagy” [13,59,60].
Figure 2.
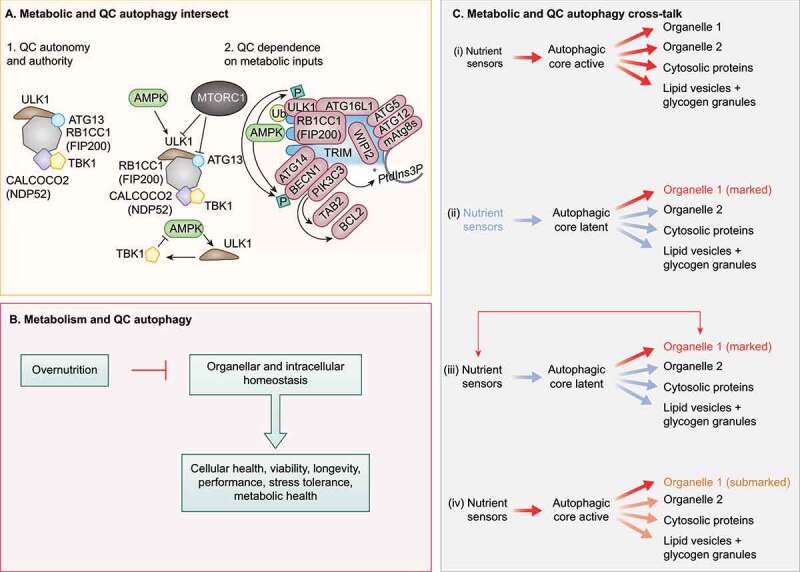
Crosstalk between QC and metabolic autophagy. (A) Subpanel 1. Mitophagy sponsored by CALCOCO2 can be experimentally rendered independent of upstream metabolic regulators AMPK and MTOR. Note the key contribution of the ULK1 complex, which here can be independent of its upstream regulators AMPK and MTOR, Note that CALCOCO2 remains linked to TBK1. Subpanel 2, left top. AMPK and MTOR normally control the ULK1 complex (ULK1, RB1CC1, ATG13, ATG101). Subpanel 2, left bottom. TBK1 may provide additional regulatory loops with AMPK and MTOR under physiological conditions. Subpanel 2, right. TRIMs are receptor-regulators of precision autophagy (a highly focused selective autophagy) remaining connected to the metabolic regulator AMPK. (B) Overnutrition inhibits metabolic autophagy and may compromise QC autophagy. (C) Proposed combinatorial relationships between metabolic and QC autophagy: (I) metabolic autophagy is activated by nutrient sensors acting on the autophagic core machinery and is nonselective; (ii) QC autophagy only concerns organelles (exemplified), proteins or other structures that have been marked for destruction with appropriate tags, following the detection of deficient quality (“marked”). (iii) Nutrient sensors may affect specific organelles to contribute to their marking (“marked”). (iv) When the core machinery of autophagy is activated downstream of nutrient sensors, the primary autophagic cargo may be constituted by partially deficient organelles that bear a few marking tags that usually would not lead to their destruction.
Selective autophagy is designed to ensure quality control of aged or damaged organelles, to remove potentially toxic protein aggregates, to mobilize specific nutrient sources (such as glycogen or lipid vesicles), to destroy intracellular pathogens or to avoid excessive inflammatory reactions. Thus, mutations affecting genes/proteins involved in selective autophagy, upstream of the common core machinery of autophagy are involved in a series of neurodegenerative and autoinflammatory diseases [4,61]. For example, genetic defects in mitophagy can lead to Parkinson disease because the accumulation of dysfunctional mitochondria elicits elements of inflammation and ultimately compromises the function and survival of vulnerable neurons [58].
Regulation of the core autophagy machinery by nutrient sensors
Two key regulators of cellular metabolism, MTOR, which is inhibited upon depletion of amino acids or growth factors [3] and AMPK, which is canonically activated when AMP levels increase at the expense of ATP in addition to other stimuli [2], directly regulate autophagy. In general, MTOR favors anabolic and biosynthetic processes, whereas AMPK favors catabolic, degradative pathways. To close and coordinate this metabolic regulatory loop, AMPK negatively controls MTOR via phosphorylation of its key regulator TSC2 and the MTOR complex 1 (MTORC1) component RPTOR/Raptor [2,3]. In principle, several key autophagy factors are subject to activating phosphorylation by AMPK and inhibitory phosphorylation by MTOR [2,3]. The ULK1-ULK2 kinase complex with RB1CC1, ATG13 and ATG101 is a conduit for both inhibitory phosphorylation by MTOR [19–21] and activating phosphorylation by AMPK [62]. A second key system activated by AMPK is the ATG14-endowed class III PtdIns3K complex that includes PIK3C3/VPS34 and BECN1 [22]. Within this complex, PIK3C3/VPS34 is directly phosphorylated by AMPK [63] and BECN1 is phosphorylated by ULK1 upon its activation by AMPK [64]. Thus, the key complexes setting autophagy in motion are under direct control by MTOR and AMPK, jumpstarting autophagy under starvation conditions [65] (Figure 1A).
Nonetheless, MTOR and AMPK are not the sole nutrient-sensing kinases, and other types of enzymes are involved. For example, the acetyl transferase EP300 senses the drop of cytosolic acetyl CoA concentrations secondary to the reduction of glycolysis, fatty acid oxidation or amino acid depletion [9], whereas deacetylases from the sirtuin family become activated when the NAD+ levels increase [66]. Inhibition of EP300 and/or activation of SIRT1 result in the deacetylation of multiple ATG proteins including but not limited to ATG5, ATG7, BECN1, LC3B and PIK3C3/VPS34, thus activating autophagy through a multipronged effect [9,67]. Moreover, there is a crosstalk between the two metabolism sensing autophagy inhibitors, MTORC1 and EP300, in the sense that MTOR phosphorylates EP300 to activate its acetyltransferase activity [68], whereas EP300 acetylates and activates the MTORC1 component RPTOR [69]. This intersection (Figure 1A) allows for the coordination among distinct types of biosensors that regulate the core pathway of autophagy by affecting the phosphorylation and acetylation of ATG proteins.
Regulation of autophagy by macro- and micronutrients
Autophagy has an important role in mobilizing latent macro- or micronutrients, for instance by facilitating the digestion of lipid vesicles, a process referred to as “lipophagy”, that ultimately allows for the mobilization of free fatty acids from complex lipids [70], glycogen through “glycophagy”, which generates free glucose from this storage molecule, and that of the iron-binding protein ferritin in a process logically denoted as “ferritinophagy” leading to the liberation of free iron [71]. Conversely, a number of macro- and micronutrients suppress autophagy.
As a general rule, energy-rich metabolites (including glucose, fatty acids and amino acids) enhance the cytosolic levels of acetyl coenzyme A (AcCoA). AcCoA is a central metabolite that is generated as a side product of glycolysis from pyruvate introduced into the tricarboxylic acid cycle in mitochondria, yielding citrate that is exported to the cytosol and then converted to AcCoA by ACLY [ATP citrate lyase], the catabolism of branched chain amino acids (through the intermediate branched chain α-keto acids), glutaminolysis (through the conversion of glutamine into glutamate and then the anaplerotic substrate α-ketoglutarate), or fatty acid oxidation (which directly yields AcCoA) [9]. Excessive cytosolic AcCoA then inhibits autophagy through the activation of acetyl transferases including EP300 (which can also be activated by phosphorylation mediated by AKT/protein kinase B downstream of receptors receiving trophic signals), inducing acetylation of key regulators of autophagy including several ATG gene products [72] as well as the MTORC1 component RPTOR [69]. More specifically, methionine can suppress autophagy through its metabolite S-adenosyl methionine (SAM), which then acts via BMT2/SAMTOR leading to activation of MTORC1 [73]. Furthermore, specific amino acids (such as arginine, glutamine and leucine) affect MTORC1 signaling through additional amino acid sensory systems (such as those involving SESN2, Leucyl-tRNA synthetase/LRS, CASTOR1, and SLC38A9) or complex pathways involving G protein coupled receptors and calcium signals [74].
Glucose reportedly favors HK2 (hexokinase 2) binding to MTORC1, thereby mediating autophagy suppression [75]. Moreover, the decrease of the intracellular glucose metabolite fructose-1,6-bisphosphate is sensed by ALDO (aldolase) to activate AMPK (independently of AMP) during glucose starvation [76], presumably through the inhibition of the endoplasmic reticulum (ER)-localized TRPV (transient receptor potential cation channel subfamily V) [77], the pharmacological inhibition of which also elevates NAD+ levels in aged muscles [77]. High NADH levels (or NADH:NAD+ ratios) reflecting plethoric bioenergetics may suppress SIRTs, favoring autophagy inhibition [78]. In contrast, hunger-associated metabolites such as ketone bodies and free fatty acids stimulate autophagy. Thus, the ketone body β-hydroxybutyric acid (BHB), which is overproduced in the context of hunger and ketogenic diet, induces autophagy through the transcriptional activation of FOXO1 [79]. Similarly, free fatty acids trigger autophagy [80]. In sharp contrast, trans-unsaturated fatty acids, which are contained in ultra-processed food items, repress autophagy induced by natural saturated fatty acids, perhaps explaining some aspects of their broad toxicity [81]. In sum, multiple metabolites regulate autophagy and regulators of autophagy to ensure that starvation-induced diminution of specific metabolites (such as glucose and amino acids) as well as starvation-elicited increases in other metabolites (ketone bodies and free fatty acids) stimulate autophagy.
Whereas overnutrition inhibits autophagy, there are several possibilities to manipulate metabolism or metabolite sensors to stimulate autophagy beyond the well-established strategy to inhibit receptor tyrosine kinases and their downstream signal following the phosphoinositide 3-kinase/PI3K→AKT→MTORC1 pathway. Thus, agents that inhibit the generation of AcCoA (such as inhibitors of ACLY; e.g., hydroxycitrate), activate AcCoA activating enzymes (such as triethylenetetramine, which activates SAT1 [spermidine/spermine N1-acetyltransferase 1]), inhibit autophagy-inhibitory acetyl transferases (such as EP300, which is inhibited by spermidine as well as by anti-inflammatory agents such as aspirin and nordihydroguaiaretic acid) or activate autophagy-activating deacetylases (such as SIRT1, which is activated by resveratrol and NAD+ precursors such as nicotinamide) act as “caloric restriction mimetics” to induce autophagy and to mediate broad effects against obesity, metabolic syndrome and age-associated diseases [81–83].
Whether and how these aspects affect selective autophagy has not been investigated, but given the above considerations of the crosstalk between metabolic and QC autophagy this may open a window of opportunity to intervene in specific diseases. In theory, the homeostasis of one or a subset of selective autophagy targets could be ameliorated by nutriceuticals and pharmaceuticals targeting metabolic autophagy. This parallels the recognition of the utility and translational potential of intermediary metabolites in other processes, such as control of inflammation and immune activation [84–86], to which autophagy is connected [87].
Regulation of quality control autophagy by nutrient sensors
While there is no doubt that bulk autophagy can be jumpstarted by nutrient sensors, the question arises as to whether selective autophagy also falls under the “supervision” by MTOR, AMPK and (de-)acetylation reactions, or whether it can be triggered without these upstream cues. Recent studies support the notion that autophagy receptors have the authority to initiate autophagy as they recognize the cargo earmarked for removal [13–16]. But do they have the autonomy?
An elegant set of experiments has recently uncoupled ULK1 function in mitophagy from AMPK and MTOR inputs, by artificially localizing the RB1CC1-ULK1 complex to mitochondria and initiating autophagy independently of AMPK activation or MTOR inhibition [16]. This finding suggests that the ATG machinery may autonomously initiate autophagy (Figure 2A, situation 1). Nonetheless, in the aforementioned case, another kinase, TBK1, takes over the role of the AMPK-MTOR axis, by promoting autophagy initiation through formation of RB1CC1-ULK1 complexes [16,88]. TBK1, a kinase that has been associated with autophagy [89], is primarily known for mediating critical inflammatory processes (including in the context of COVID-19) [90]. Nevertheless, TBK1 is interlinked with metabolic regulators: TBK1 inhibits AMPK, whereas ULK1 downstream of AMPK activates TBK1, creating a loop between metabolic, inflammatory, and autophagy processes [91]. Thus, TBK1 [88], as well as related IKK kinases [92], contribute to metabolic (starvation-induced) autophagy and interconnect with AMPK. In conclusion, despite the demonstration of experimentally forced autonomy of mitophagy, it appears likely that under physiological circumstances the process is coupled to MTOR and/or AMPK inputs as well as to inflammatory inputs (Figure 2A, situation 2). A similar argument can be made for precision autophagy conducted by TRIMs [13,59] (Figure 2A, situation 2).
Lysophagy constitutes another example in which nutrient-sensitive kinases play a major role. When endosomal/lysosomal membranes are damaged, a form of QC autophagy termed lysophagy comes into action to isolate and remove damaged lysosomes that are tagged by ubiquitin [32]. Of special importance for the discussion here, lysosomes are the organelles where MTOR and the MTORC1 regulatory machinery (Ragulator and RRAG GTPases) reside [10]. Furthermore, lysosomes are the organelles where AMPK is noncanonically activated in response to inputs other than the canonical activation in the cytosol by AMP [93]. Two different systems on lysosomes activate AMPK in immediate response to glucose starvation [76,93] or upon lysosomal membrane permeabilizaton/damage [94,95] (Figure 1A). The former system also contributes to MTOR inactivation [96] under glucose-starvation conditions, critically important for perinatal/neonatal induction of autophagy to free gluconeogenic amino acids and stave off hypoglycemia at birth [97]. The latter system, acting in response to lysosomal damage, is a part of a larger protective response called membrane repair, removal and replacement (MERiT) which is coordinated by cytosolic lectins called LGALS/galectins that recognize membrane tears [94,95,98,99] . Ubiquitination [100] ESCRT machinery [101,102], and LGALS proteins [99] are required for repair and other processes. If damage persists, this leads to LGALS-sponsored inactivation of MTOR [94] and activation of AMPK [95], which then stimulate lysophagy [99,103]. Finally, the TFEB-directed lysosomal biogenesis program is turned on to replenish lysosomes [99,103]. TFEB is under negative control by MTOR [37], and the same process of MTOR inactivation and AMPK activation that leads to the assembly of ULK1 complexes and induction of lysophagy [103] triggers the translocation of TFEB from the cytoplasm to the nucleus, ultimately facilitating the transactivation of lysosome-relevant genes [99]. TFEB repositioning to the nucleus is reinforced by autophagy-independent functions of mAtg8s on MTOR and TFEB [41], by noncanonical lipidation of mAtg8’s elicited by lysosomal damage [37], and by mAtg8-dependent activation of TFEB phosphatases [37,41]. Metformin, a widely used anti-diabetic drug that reportedly activates AMPK [104], elicits lysosomal damage and induces MERiT processes that include autophagy [95], perhaps explaining its broad antiaging effects [105]. Thus, lysophagy exemplifies another type of QC autophagy that is inextricably linked to metabolic inputs and outputs and is under direct control by MTOR and AMPK.
There is also evidence that (de-)acetylation reactions can affect QC (organelle-specific) autophagy. Thus, BLOC1S1/GCN5L1-mediated acetylation and SIRT3-mediated deacetylation of mitochondrial proteins inhibit and induce mitophagy, respectively [106,107]. Moreover, the membrane transporter SLC33A1/AT-1, which translocates cytosolic acetyl-CoA in the ER (and hence is required for the acetylation of ER-resident proteins by NAT8/ATase2-NAT8B/ATase1 acetyltransferase) negatively regulates reticulophagy, and its transgenic overexpression in mice is pathogenic, causing a progerial-like syndrome [108]. Although there is no formal proof for this conjecture, it appears possible that a scarcity of cytosolic CoA, as is observed in conditions of nutrient depletion [9], may favor deacetylation reactions at mitochondria and within the ER to stimulate mitophagy and reticulophagy, respectively.
Transcriptional regulation of lysosomal biogenesis and autophagy in metabolism and quality control
Although autophagy can be turned on in a transcription-independent fashion, it requires the transactivation of multiple genes for its maintenance, through the action of a series of transcription factors [109]. However, transcriptional regulation of metabolic vs. QC autophagy has not been systematically addressed.
A well-defined system links the master transcriptional regulator of oxidative stress response, NFE2L2/NRF2, with autophagy via one of the key selective autophagy receptors, SQSTM1/p62 and its bidning partner KEAP1 [110]. The class O forkhead box transcription factors (FOXO) including FOXO1, FOXO3 and FOXO4 become dephosphorylated upon inhibition of AKT and then translocate into the nucleus to transactivate autophagy-relevant genes [111]. A key regulator of lysosomal gene transcription, TFEB [112], a member of the MiT-TEF family of transcription factors, transduces inhibitory MTOR signaling to lysosomal biogenesis [113]] (Figure 1A) essential for both metabolic and QC autophagy. As introduced earlier, TFEB is phosphorylated by MTOR thus retaining TFEB in the cytoplasm, whereas the PPP3/calcineurin phosphatase PPP3CB dephosphorylates TFEB and promotes its translocation to the nucleus [114]. TFEB and other MiT-TEF factors close this regulatory loop by controlling expression of RRAGD (one of the 4 small GTPases RRAGA-RRAGB and RRAGC-RRAGD) [115]. RRAGC-RRAGD in a GTP bound state restricts MTOR suppression of TFEB [116]. When MTOR is inhibited, TFEB transactivates lysosomal and a small subset of pro-autophagic genes [112], but also activates multiple autophagy-independent metabolic functions such as cellular glucose uptake and the anabolism in exercising muscles, a function that apparently does not involve PPARGC1A/PGC1α [117]. Nevertheless, the lipid catabolism-stimulatory action of TFEB relies on PPARGC1A and synergizes with autophagy to protect against obesity and metabolic syndrome [118]. Similar considerations apply to the autophagy-modulatory action of several other transcription factors and epigenetic modulators, some of which may functionally interact with TFEB: FOXO1 and FOXO3, NFKB, E2F1, TP53/p53, NR1H4/FXR, CREB, ZKSCAN3, BRD4, CARM1, KAT8, etc. (for a recent comprehensive review, see Ref [119].).
Recent studies suggest that TFEB may play a direct role in QC autophagy through effects on the expression of at least one selective autophagy receptor [112]. It is also intriguing that MiT-TFE3 factors specifically induce RETREG1/FAM134B, a receptor for reticulophagy [120], and that TFEB is activated during mitophagy [121] and upon lysosomal damage [37,99,103].
Metabolic autophagy versus quality control autophagy and metabolism – answered and unanswered questions
Metabolic autophagy and QC autophagy exhibit a significant overlap in their regulation by nutrients and energy sensors, meaning that scarcity of nutrients can favor both phenomena. As discussed above, metabolic autophagy is triggered by the demand to retrieve energy and building blocks from intracellular macromolecules. Selective autophagy involves earmarking specific substrates to supply them for autophagic destruction, and this could be metabolic or QC. Thus, there is no fundamental opposition between both types of autophagy. For example, QC autophagy may be imperfect in its selectivity in thus far that “accidental” degradation of bystander cargo has been reported [122], albeit this remains to be fully explored. Moreover, it is conceivable that, in analogy to supply and demand that together regulate the market, both processes, i.e., metabolic and selective autophagy, crosstalk (Figure 2B,C). For example, when energy sensors are activated to induce metabolic autophagy, the sequestration of cargo may preferentially concern those cellular structures that exhibit signs of partial dysfunction (such as depolarized mitochondria that are surface-ubiquitinated due to the action of PRKN) [58] as well as protein aggregates rather than functional units (Figure 2C). Indeed, there is evidence that mitochondria that are eliminated during bulk autophagy are those with the lowest transmembrane potential and hence the poorest function [123]. Thus, even organelles with residual functions that are usually not subjected to specific autophagy may be preferentially eliminated by bulk autophagy activated in response to nutrient deprivation. This may explain why chronic or intermittent induction of autophagy by caloric restriction, caloric restriction mimetics or periodic fasting improves organismal fitness, delays aging, mediates cytoprotective effects and reduces the pace of neuro- or myodegenerative diseases [4].
Bulk autophagy, which may be particularly efficient in mobilizing nutrients, has been thought to be largely nonspecific. Nonetheless, from a teleological perspective, there should be mechanisms through which autophagy would mobilize specific nutrients when they are scarce, for instance fatty acids through lipophagy, glucose by autophagic digestion of glycogen granules, or amino acids by digestion of protein-rich organelles. Indeed, recent in-depth analyses suggest that nutritional turnover of cytoplasmic constituents may be guided by mechanisms that involve some degree of specificity and selectivity [65]. Thus, basic amino acids and nucleosides may be mobilized in a particularly efficient fashion via the degradation of ribosomes, potentially including ribophagy, which may be initiated by specific receptors including NUFIP1 [65]. However, other work indicates that adjustments in ribosomal content under acute nutrient stress, including their degradation, could be dominated by processes other than autophagy [124]. Whether cells may activate some sort of substrate-specific autophagy in response to metabolic signals, hormones and neurotransmitters remains to be determined. Moreover, specific neuroendocrine circuits might activate autophagy in an organ-specific fashion (e.g., in the liver for glycogenolysis, in white adipose tissue for lipolysis and in skeletal muscle for the digestion of protein and the provision of amino acids) to mobilize different metabolites, thus maintaining metabolic homeostasis. The comprehension of such – still hypothetical – cell-intrinsic or supracellular pathways might be extremely useful for designing specific interventions in metabolism, for instance in the context of diabetes, dyslipidemia or cachexia.
The subtle crosstalk between metabolic and QC autophagy may have a major impact on the evolution of human health [125]. For example, when extrapolating preclinical work obtained in mice, it appears obvious, yet remains to be confirmed, that the obesity-associated suppression of autophagy compromises cellular quality control, thereby reducing healthspan and lifespan [126]. This may have far-reaching consequences because obesity is not only the most prevalent pathological state in the world but also (one of) the most important risk factor(s) for developing nonalcoholic hepatosteatosis (NASH), cardiovascular disease, neurodegeneration and cancer [127]. It remains to be determined whether therapeutic stimulation of autophagy would be sufficient to reduce systemic inflammation, since the two are extensively linked [87], and to improve metabolic health or, more specifically, to prevent NASH, arteriosclerosis, cardiac failure, and Alzheimer disease. In cancer, autophagy plays complex and sometimes opposing, stage-dependent roles in carcinogenesis, progression and metastasis, and is complicated by cancer cell-autonomus, stromal, and immune contributions. In this context, it will be important to understand whether specific induction of metabolic autophagy (including for example glycophagy and lipophagy) or therapeutic stimulation of QC autophagy (including aggrephagy and mitophagy) will provide superior results against each of these age- and obesity-associated diseases.
Funding Statement
This work was supported by NIH grants [R37AI042999 and R01AI111935] and center grant P20GM121176 to V.D. GK is supported by the Ligue contre le Cancer (équipe labellisée); Agence National de la Recherche (ANR) – Projets blancs; AMMICa US23/CNRS UMS3655; Association pour la recherche sur le cancer (ARC); Association “Ruban Rose”; Cancéropôle Ile-de-France; Chancelerie des universités de Paris (Legs Poix), Fondation pour la Recherche Médicale (FRM); a donation by Elior; European Research Area Network on Cardiovascular Diseases (ERA-CVD, MINOTAUR); Gustave Roussy Odyssea, the European Union Horizon 2020 Project Oncobiome; Fondation Carrefour; High-end Foreign Expert Program in China (GDW20171100085), Institut National du Cancer (INCa); Inserm (HTE); Institut Universitaire de France; LeDucq Foundation; the LabEx Immuno-Oncology (ANR-18-IDEX-0001); the RHU Torino Lumière; the Seerave Foundation; the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); and the SIRIC Cancer Research and Personalized Medicine (CARPEM). This study contributes to the IdEx Université de Paris ANR-18-IDEX-0001.
Conflicts of interest
Guido Kroemer is a scientific co-founder of everImmune, Samsara Therapeutics and Therafast Bio.
References
- [1].Mizushima N, Yoshimori T, Ohsumi Y.. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011. Nov;10(27):107–132. [DOI] [PubMed] [Google Scholar]
- [2].Herzig S, Shaw RJ.. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. 2018. Feb;19(2):121–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Liu GY, Sabatini DM. mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol. 2020. Apr;21(4):183–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell. 2019. Jan 10;176(1–2):11–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Galluzzi L, Green DR. Autophagy-independent functions of the autophagy machinery. Cell. 2019. Jun 13;177(7):1682–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Melia TJ, Lystad AH, Simonsen A. Autophagosome biogenesis: from membrane growth to closure. J Cell Biol. 2020. Jun 1;219(6):372-383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Noda T, Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem. 1998. Feb 13;273(7):3963–3966. [DOI] [PubMed] [Google Scholar]
- [8].Scott RC, Schuldiner O, Neufeld TP. Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev Cell. 2004. Aug;7(2):167–178. [DOI] [PubMed] [Google Scholar]
- [9].Marino G, Pietrocola F, Eisenberg T, et al. Regulation of autophagy by cytosolic acetyl-coenzyme A. Mol Cell. 2014. Mar 6;53(5):710–725. [DOI] [PubMed] [Google Scholar]
- [10].Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017. Mar 09;168(6):960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Garcia D, Shaw RJAMPK. Mechanisms of cellular energy sensing and restoration of metabolic balance. Mol Cell. 2017. Jun 15;66(6):789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins [Research Support, Non-U.S. Gov’t]. Autophagy. 2011. Mar;7(3):279–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kimura T, Mandell M, Deretic V. Precision autophagy directed by receptor regulators - emerging examples within the TRIM family. J Cell Sci. 2016. Mar 01;129(5):881–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Mandell MA, Jain A, Arko-Mensah J, et al. TRIM proteins regulate autophagy and can target autophagic substrates by direct recognition. Dev Cell. 2014. Aug 25;30(4):394–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lazarou M, Sliter DA, Kane LA, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015. Aug 20;524(7565):309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Vargas JNS, Wang C, Bunker E, et al. Spatiotemporal control of ULK1 Activation by NDP52 and TBK1 during selective autophagy. Mol Cell. 2019. Apr 18;74(2):347–362 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Marshall RS, Hua Z, Mali S, et al. ATG8-binding UIM proteins define a new class of autophagy adaptors and receptors. Cell. 2019. Apr 18;177(3):766–781 e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Nguyen TN, Padman BS, Usher J, et al. Atg8 family LC3/GABARAP proteins are crucial for autophagosome-lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J Cell Biol. 2016. Dec 19;215(6):857–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ganley IG, Lam du H, Wang J, et al. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009. May 1;284(18):12297–12305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Jung CH, Jun CB, Ro SH, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009. Apr;20(7):1992–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hosokawa N, Hara T, Kaizuka T, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009. Apr;20(7):1981–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Baskaran S, Carlson LA, Stjepanovic G, et al. Architecture and dynamics of the autophagic phosphatidylinositol 3-kinase complex. eLife. 2014;3. 10.7554/eLife.05115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Young AR, Chan EY, Hu XW, et al. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J Cell Sci. 2006. Sep 15;119(Pt 18):3888–3900. [DOI] [PubMed] [Google Scholar]
- [24].Velikkakath AK, Nishimura T, Oita E, et al. Mammalian Atg2 proteins are essential for autophagosome formation and important for regulation of size and distribution of lipid droplets [Research Support, Non-U.S. Gov’t]. Mol Biol Cell. 2012. Mar;23(5):896–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bakula D, Muller AJ, Zuleger T, et al. WIPI3 and WIPI4 beta-propellers are scaffolds for LKB1-AMPK-TSC signalling circuits in the control of autophagy. Nat Commun. 2017. May 31;8:15637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Valverde DP, Yu S, Boggavarapu V, et al. ATG2 transports lipids to promote autophagosome biogenesis. J Cell Biol. 2019. Jun 3;218(6):1787–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Guardia CM, Tan XF, Lian T, et al. Structure of human ATG9A, the only transmembrane protein of the core autophagy machinery. Cell Rep. 2020. Jun 30;31(13):107837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Maeda S, Yamamoto H, Kinch LN, et al. Structure, lipid scrambling activity and role in autophagosome formation of ATG9A. Nat Struct Mol Biol. 2020. Dec;27(12):1194–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ghanbarpour A, Valverde DP, Melia TJ, et al. A model for a partnership of lipid transfer proteins and scramblases in membrane expansion and organelle biogenesis. Proc National Acad Sci USA. 2021 Apr 20;118(16). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Nishimura T, Kaizuka T, Cadwell K, et al. FIP200 regulates targeting of Atg16L1 to the isolation membrane. EMBO Rep. 2013. Mar 1;14(3):284–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Gammoh N, Florey O, Overholtzer M, et al. Interaction between FIP200 and ATG16L1 distinguishes ULK1 complex-dependent and -independent autophagy. Nat Struct Mol Biol. 2013. Feb;20(2):144–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Fujita N, Morita E, Itoh T, et al. Recruitment of the autophagic machinery to endosomes during infection is mediated by ubiquitin [Research Support, Non-U.S. Gov’t]. J Cell Biol. 2013. Oct 14;203(1):115–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Dooley HC, Razi M, Polson HE, et al. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol Cell. 2014. Jul 17;55(2):238–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Jao CC, Ragusa MJ, Stanley RE, et al. A HORMA domain in Atg13 mediates PI 3-kinase recruitment in autophagy. Proc Natl Acad Sci U S A. 2013 Apr 2;110(14):5486–5491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000. Nov 1;19(21):5720–5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Heckmann BL, Teubner BJW, Tummers B, et al. LC3-Associated Endocytosis Facilitates beta-Amyloid Clearance and Mitigates Neurodegeneration in Murine Alzheimer’s Disease. Cell. 2019. Jul 25;178(3):536–551 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Nakamura S, Shigeyama S, Minami S, et al. LC3 lipidation is essential for TFEB activation during the lysosomal damage response to kidney injury. Nat Cell Biol. 2020. Oct;22(10):1252–1263. [DOI] [PubMed] [Google Scholar]
- [38].Mizushima N, Sugita H, Yoshimori T, et al. A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J Biol Chem. 1998. Dec 18;273(51):33889–33892. [DOI] [PubMed] [Google Scholar]
- [39].Lee C, Lamech L, Johns E, et al. Selective lysosome membrane turnover is induced by nutrient starvation. Dev Cell. 2020 Nov 55(3): 289-297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Alemu EA, Lamark T, Torgersen KM, et al. ATG8 family proteins act as scaffolds for assembly of the ULK complex: sequence requirements for LC3-interacting region (LIR) motifs [Research Support, Non-U.S. Gov’t]. J Biol Chem. 2012. Nov 16;287(47):39275–39290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kumar S, Jain A, Choi SW, et al. Mammalian Atg8 proteins and the autophagy factor IRGM control mTOR and TFEB at a regulatory node critical for responses to pathogens. Nat Cell Biol. 2020. Aug;22(8):973–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lahiri V, Hawkins WD, Klionsky DJ, et al. (Self-) Eat: autophagic mechanisms that modulate metabolism. Cell Metab. 2019. Apr 2;29(4):803–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Melendez A, Talloczy Z, Seaman M, et al. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science. 2003. Sep 5;301(5638):1387–1391. [DOI] [PubMed] [Google Scholar]
- [44].Tekinay T, Wu MY, Otto GP, et al. Function of the Dictyostelium discoideum Atg1 kinase during autophagy and development. Eukaryot Cell. 2006. Oct;5(10):1797–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Pietrocola F, Demont Y, Castoldi F, et al. Metabolic effects of fasting on human and mouse blood in vivo. Autophagy. 2017. Mar 4;13(3):567–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Mizushima N, Yamamoto A, Matsui M, et al. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004. Mar;15(3):1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Karsli-Uzunbas G, Guo JY, Price S, et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 2014. Aug;4(8):914–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Poillet-Perez L, Xie X, Zhan L, et al. Autophagy maintains tumour growth through circulating arginine. Nature. 2018. Nov;563(7732):569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Morselli E, Maiuri MC, Markaki M, et al. Caloric restriction and resveratrol promote longevity through the Sirtuin-1-dependent induction of autophagy. Cell Death Dis. 2010;1:e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].He C, Bassik MC, Moresi V, et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature. 2012. Jan 26;481(7382):511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Galluzzi L, Pietrocola F, Levine B, et al. Metabolic control of autophagy [Review]. Cell. 2014. Dec 4;159(6):1263–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Bravo-San Pedro JM, Sica V, Martins I, et al. Acyl-CoA-binding protein is a lipogenic factor that triggers food intake and obesity. Cell Metab. 2019. Oct 1;30(4):754–767 e9. [DOI] [PubMed] [Google Scholar]
- [53].Lopez-Otin C, Galluzzi L, Freije JMP, et al. Metabolic control of longevity. Cell. 2016. Aug 11;166(4):802–821. [DOI] [PubMed] [Google Scholar]
- [54].Nthiga TM, Shrestha BK, Bruun JA, et al. Regulation of Golgi turnover by CALCOCO1-mediated selective autophagy. J Cell Biol. 2021. Jun 7;220(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Zellner S, Schifferer M, Behrends C. Systematically defining selective autophagy receptor-specific cargo using autophagosome content profiling. Molecular Cell. 2021. Jan28;88(6):1337-1354. [DOI] [PubMed] [Google Scholar]
- [56].Agudo-Canalejo J, Schultz SW, Chino H, et al. Wetting regulates autophagy of phase-separated compartments and the cytosol. Nature. 2021. Jan 20;591(7848):142-146. [DOI] [PubMed] [Google Scholar]
- [57].Wei Y, Chiang WC, Sumpter R Jr., et al. Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell. 2017. Jan 12;168(1–2):224–238 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Youle RJ. Mitochondria-striking a balance between host and endosymbiont. Science. 2019. Aug 16;365:6454. [DOI] [PubMed] [Google Scholar]
- [59].Kimura T, Jain A, Choi SW, et al. TRIM-mediated precision autophagy targets cytoplasmic regulators of innate immunity. J Cell Biol. 2015. Sep 14;210(6):973–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Di Rienzo M, Romagnoli A, Antonioli M, et al. TRIM proteins in autophagy: selective sensors in cell damage and innate immune responses. Cell Death Differ. 2020. Mar;27(3):887–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Fraiberg M, Elazar Z. Genetic defects of autophagy linked to disease. Prog Mol Biol Transl Sci. 2020;172:293–323. [DOI] [PubMed] [Google Scholar]
- [62].Kim J, Kundu M, Viollet B, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011. Feb;13(2):132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Kim J, Kim YC, Fang C, et al. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell. 2013. Jan 17;152(1–2):290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Russell RC, Tian Y, Yuan H, et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol. 2013. Jul;15(7):741–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Wyant GA, Abu-Remaileh M, Frenkel EM, et al. NUFIP1 is a ribosome receptor for starvation-induced ribophagy. Science. 2018. May 18;360(6390):751–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Hubbard BP, Sinclair DA. Small molecule SIRT1 activators for the treatment of aging and age-related diseases. Trends Pharmacol Sci. 2014. Mar;35(3):146–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Botti-Millet J, Nascimbeni AC, Dupont N, et al. Fine-tuning autophagy: from transcriptional to posttranslational regulation. Am J Physiol Cell Physiol. 2016. Sep 1;311(3):C351–62. [DOI] [PubMed] [Google Scholar]
- [68].Wan W, You Z, Xu Y, et al. mTORC1 phosphorylates acetyltransferase p300 to regulate autophagy and lipogenesis. Mol Cell. 2017. Oct 19;68(2):323–335 e6. [DOI] [PubMed] [Google Scholar]
- [69].Son SM, Park SJ, Stamatakou E, et al. Leucine regulates autophagy via acetylation of the mTORC1 component raptor. Nat Commun. 2020. Jun 19;11(1):3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Zechner R, Madeo F, Kratky D. Cytosolic lipolysis and lipophagy: two sides of the same coin. Nat Rev Mol Cell Biol. 2017. Nov;18(11):671–684. [DOI] [PubMed] [Google Scholar]
- [71].Tang M, Chen Z, Wu D, et al. Ferritinophagy/ferroptosis: iron-related newcomers in human diseases. J Cell Physiol. 2018. Dec;233(12):9179–9190. [DOI] [PubMed] [Google Scholar]
- [72].Figlia G, Willnow P, Teleman AA. Metabolites regulate cell signaling and growth via covalent modification of proteins. Dev Cell. 2020. Jul 20;54(2):156–170. [DOI] [PubMed] [Google Scholar]
- [73].Gu X, Orozco JM, Saxton RA, et al. SAMTOR is an S-adenosylmethionine sensor for the mTORC1 pathway. Science. 2017. Nov 10;358(6364):813–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Takahara T, Amemiya Y, Sugiyama R, et al. Amino acid-dependent control of mTORC1 signaling: a variety of regulatory modes. J Biomed Sci. 2020. Aug 17;27(1):87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Roberts DJ, Tan-Sah VP, Ding EY, et al . Hexokinase-II positively regulates glucose starvation-induced autophagy through TORC1 inhibition. Mol Cell. 2014. Feb 20;53(4):521–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Zhang CS, Hawley SA, Zong Y, et al. Fructose-1,6-bisphosphate and aldolase mediate glucose sensing by AMPK. Nature. 2017. Aug 3;548(7665):112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Li M, Zhang CS, Zong Y, et al. Transient receptor potential v channels are essential for glucose sensing by aldolase and AMPK. Cell metabolism. 2019. Jun 10;3:508-524 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Canto C, Menzies KJ, Auwerx J. NAD(+) metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab. 2015. Jul 7;22(1):31–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Miyauchi T, Uchida Y, Kadono K, et al. Up-regulation of FOXO1 and reduced inflammation by beta-hydroxybutyric acid are essential diet restriction benefits against liver injury. Proc Natl Acad Sci U S A. 2019 Jul 2;116(27):13533–13542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Niso-Santano M, Malik SA, Pietrocola F, et al. Unsaturated fatty acids induce non-canonical autophagy. EMBO J. 2015. Apr 15;34(8):1025–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Sauvat A, Chen G, Muller K, et al. Trans-fats inhibit autophagy induced by saturated fatty acids. EBioMedicine. 2018;30:261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Madeo F, Carmona-Gutierrez D, Hofer SJ, et al. Caloric restriction mimetics against age-associated disease: targets, mechanisms, and therapeutic potential. Cell Metab. 2019. Mar 5;29(3):592–610. [DOI] [PubMed] [Google Scholar]
- [83].Castoldi F, Hyvonen MT, Durand S, et al. Chemical activation of SAT1 corrects diet-induced metabolic syndrome. Cell Death Differ. 2020. Oct;27(10):2904–2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Hooftman A, Angiari S, Hester S, et al. The Immunomodulatory metabolite itaconate modifies NLRP3 and inhibits inflammasome activation. Cell Metab. 2020. Sep 1;32(3):468–478 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Mills EL, Ryan DG, Prag HA, et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature. 2018. Apr 5;556(7699):113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Ryan DG, Murphy MP, Frezza C, et al. Coupling Krebs cycle metabolites to signalling in immunity and cancer. Nat Metab. 2019. 1;Jan:16–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Deretic V. Autophagy in inflammation, infection, and immunometabolism. Immunity. 2021. Mar 9;54(3):437–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Kumar S, Gu Y, Abudu YP, et al. Phosphorylation of Syntaxin 17 by TBK1 controls autophagy initiation. Dev Cell. 2019. Apr 8;49(1):130–144 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Saitoh T, Fujita N, Hayashi T, et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc Natl Acad Sci U S A. 2009 Dec 8;106(49):20842–20846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Zhang Q, Bastard P, Liu Z, et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science. 2020. Sep 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Zhao P, Wong KI, Sun X, et al. TBK1 at the crossroads of inflammation and energy homeostasis in adipose tissue. Cell. 2018. Feb 8;172(4):731–743 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Criollo A, Niso-Santano M, Malik SA, et al. Inhibition of autophagy by TAB2 and TAB3. EMBO J. 2011. Dec 14;30(24):4908–4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Gonzalez A, Hall MN, Lin SC, et al. AMPK and TOR: the Yin and Yang of cellular nutrient sensing and growth control. Cell Metab. 2020. Mar 3;31(3):472–492. [DOI] [PubMed] [Google Scholar]
- [94].Jia J, Abudu YP, Claude-Taupin A, et al. Galectins control mTOR in response to endomembrane damage. Mol Cell. 2018. Apr 5;70(1):120–135 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Jia J, Bissa B, Brecht L, et al. AMPK, a regulator of metabolism and autophagy, is activated by lysosomal damage via a novel galectin-directed ubiquitin signal transduction system. Mol Cell. 2020. Jan 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Li M, Zhang CS, Feng JW, et al. Aldolase is a sensor for both low and high glucose, linking to AMPK and mTORC1. Cell Res 2020. Dec 21; 31:478-481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Efeyan A, Zoncu R, Chang S, et al. Regulation of mTORC1 by the Rag GTPases is necessary for neonatal autophagy and survival. Nature. 2013. Jan 31;493(7434):679–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Jia J, Claude-Taupin A, Gu Y, et al. MERIT, a cellular system coordinating lysosomal repair, removal and replacement. Autophagy. 2020. Aug;16(8):1539–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Jia J, Claude-Taupin A, Gu Y, et al. Galectin-3 Coordinates a Cellular System for Lysosomal Repair and Removal. Dev Cell. 2020. Jan 6;52(1):69–87 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Papadopoulos C, Kirchner P, Bug M, et al. VCP/p97 cooperates with YOD1, UBXD1 and PLAA to drive clearance of ruptured lysosomes by autophagy. EMBO J. 2017. Jan 17;36(2):135–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Skowyra ML, Schlesinger PH, Naismith TV, et al. Triggered recruitment of ESCRT machinery promotes endolysosomal repair. Science. 2018 Apr 6;360:6384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Radulovic M, Schink KO, Wenzel EM, et al. ESCRT-mediated lysosome repair precedes lysophagy and promotes cell survival. EMBO J. 2018. Nov 2;37:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Chauhan S, Kumar S, Jain A, et al. TRIMs and galectins globally cooperate and TRIM16 and galectin-3 co-direct autophagy in endomembrane damage homeostasis. Dev Cell. 2016. Oct 10;39(1):13–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].He L, Wondisford FE. Metformin action: concentrations matter. Cell Metab. 2015. Feb 3;21(2):159–162. [DOI] [PubMed] [Google Scholar]
- [105].Kulkarni AS, Gubbi S, Barzilai N. Benefits of metformin in attenuating the hallmarks of aging. Cell Metab. 2020. Jul 7;32(1):15–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Webster BR, Scott I, Han K, et al. Restricted mitochondrial protein acetylation initiates mitochondrial autophagy. J Cell Sci. 2013. Nov 1;126(Pt 21):4843–4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Scott I, Wang L, Wu K, et al. GCN5L1/BLOS1 links acetylation, organelle remodeling, and metabolism. Trends Cell Biol. 2018. May;28(5):346–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Peng Y, Shapiro SL, Banduseela VC, et al. Increased transport of acetyl-CoA into the endoplasmic reticulum causes a progeria-like phenotype. Aging Cell. 2018. Oct;17(5):e12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Pietrocola F, Izzo V, Niso-Santano M, et al. Regulation of autophagy by stress-responsive transcription factors. Semin Cancer Biol. 2013. Oct;23(5):310–322. [DOI] [PubMed] [Google Scholar]
- [110].Sanchez-Martin P, Komatsu M. Physiological stress response by selective autophagy. J Mol Biol. 2020. Jan 3;432(1):53–62. [DOI] [PubMed] [Google Scholar]
- [111].Webb AE, Brunet A. FOXO transcription factors: key regulators of cellular quality control. Trends Biochem Sci. 2014. Apr;39(4):159–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Settembre C, Di Malta C, Polito VA, et al. TFEB links autophagy to lysosomal biogenesis. Science.2011. Jun 17;332(6036):1429–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Ballabio A, Bonifacino JS. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat Rev Mol Cell Biol. 2020. Feb;21(2):101–118. [DOI] [PubMed] [Google Scholar]
- [114].Medina DL, Di Paola S, Peluso I, et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol. 2015. Mar;17(3):288–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Di Malta C, Siciliano D, Calcagni A, et al . Transcriptional activation of RagD GTPase controls mTORC1 and promotes cancer growth. Science. 2017. Jun 16;356(6343):1188–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Napolitano G, Di Malta C, Esposito A, et al . A substrate-specific mTORC1 pathway underlies Birt-Hogg-Dube syndrome. Nature. 2020. Sep;585(7826):597–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Mansueto G, Armani A, Viscomi C, et al. Transcription factor EB controls metabolic flexibility during exercise. Cell Metab. 2017. Jan 10;25(1):182–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Settembre C, De Cegli R, Mansueto G, et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol. 2013. Jun;15(6):647–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Di Malta C, Cinque L, Settembre C. Transcriptional regulation of autophagy: mechanisms and diseases. Front Cell Dev Biol. 2019;7:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Cinque L, De Leonibus C, Iavazzo M, et al. MiT/TFE factors control ER-phagy via transcriptional regulation of FAM134B. EMBO J. 2020. Sep 1;39(17):e105696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Nezich CL, Wang C, Fogel AI, et al. MiT/TFE transcription factors are activated during mitophagy downstream of Parkin and Atg5. J Cell Biol. 2015. Aug 3;210(3):435–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Nakamura S, Shigeyama S, Minami S, et al. LC3 lipidation is essential for TFEB activation during the lysosomal damage response to kidney injury. Nature Cell Bio. 2020. Sep 28;22:1252-1263. [DOI] [PubMed] [Google Scholar]
- [123].An H, Harper JW. Systematic analysis of ribophagy in human cells reveals bystander flux during selective autophagy. Nat Cell Biol. 2018. Feb;20(2):135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Gilkerson RW, De Vries RL, Lebot P, et al. Mitochondrial autophagy in cells with mtDNA mutations results from synergistic loss of transmembrane potential and mTORC1 inhibition. Hum Mol Genet. 2012. Mar 1;21(5):978–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].An H, Ordureau A, Korner M, et al. Systematic quantitative analysis of ribosome inventory during nutrient stress. Nature. 2020. Jul;583(7815):303–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Lopez-Otin C, Kroemer G. Hallmarks of health. Cell. 2021. Jan 7;184(1):33–63. [DOI] [PubMed] [Google Scholar]
- [127].Heymsfield SB, Wadden TA. Mechanisms, Pathophysiology, and. Management of obesity. N Engl J Med. 2017. Jan 19;376(3):254–266. [DOI] [PubMed] [Google Scholar]