

ABSTRACT
Cholesterol overloading-induced damages on hepatocytes cause liver dysfunctions, which further damages cholesterol metabolism and results in visceral fat accumulation in patients with type 2 diabetes mellitus (T2DM). The sodium-glucose cotransporter 2 (SGLT2) inhibitor Dapagliflozin has been reported to regulate cholesterol levels in T2DM patients, but the underlying mechanisms have not been studied. In the present study, we initially established in vivo T2DM mice models, and our results showed that both free cholesterol (FC) and cholesteryl ester (CE) were accumulated, while the pro-proliferation associated genes were downregulated in T2DM mice liver tissues, which were reversed by Dapagliflozin co-treatment. Similarly, the mice primary hepatocytes were loaded with cholesterol to establish in vitro models, and we expectedly found that Dapagliflozin attenuated cholesterol-overloading induced cytotoxicity and cellular senescence in the hepatocytes. Then, we noticed that oxidative damages occurred in T2DM mice liver tissues and cholesterol treated hepatocytes, which could be suppressed by Dapagliflozin. Also, elimination of Reactive Oxygen Species (ROS) by N-acetyl-L-cysteine (NAC) recovered cellular functions of hepatocytes in vitro and in vivo. Furthermore, the potential underlying mechanisms were uncovered, and our data suggested that Dapagliflozin activated the anti-oxidant Nrf2/HO-1 pathway in mice hepatocytes, and silencing of Nrf2 abrogated the protective effects of Dapagliflozin on cholesterol-overloaded hepatocytes. Collectively, we concluded that Dapagliflozin recovered cholesterol metabolism functions in T2DM mice liver via activating the anti-oxidant Nrf2/HO-1 pathway, and our data supported that Dapagliflozin was a potential therapeutic drug to eliminate cholesterol-induced cytotoxicity during T2DM pathogenesis.
KEYWORDS: Type 2 diabetes mellitus, cholesterol-overloading stress, hepatocytes, Dapagliflozin, oxidative stress
Background
Type 2 diabetes mellitus (T2DM) is a serious chronic disease which brings huge health burden for human being worldwide [1,2], and recent data hint that visceral fat accumulation is an independent and important risk factor for T2DM [3,4]. To our knowledge, liver is the major organ to regulate fat deposition, metabolism and homeostasis [5,6], and T2DM has been reported to be closely associated with fatty liver, and hepatic ectopic fat accumulation contributes to insulin resistance and worse prognosis in T2DM patients [7–9]. According to the previous literatures, cholesterol is ectopically accumulated in the liver of T2DM patients [10], and data from different teams show that cholesterol-overloading induces hepatocyte death, which further cause liver dysfunctions [11–13]. For example, cholesterol overloading respectively activates the unfolded protein response (UPR) [12] and induces mitochondrial dynamic changes [13] to promote hepatocyte death. Cholesterol overloading-induced hepatocyte death impairs the normal functions of liver for cholesterol metabolism, which further aggravates fat accumulation and cell death in liver [11,13].
As previously described, cholesterol overloading induced oxidative damages in various types of cells, such as vascular endothelial cells [14], renal cells [15] and hepatocytes [13,16], and oxidative stress contributes to apoptotic cell death [17,18]. Specifically, high-cholesterol diet induces cytotoxicity in the hepatocytes with steatohepatitis [16] and liver injury [13] through triggering oxidative damages. Also, the published clinical data support that oxidative stress has close relationship with fatty liver index in T2DM patients [19,20], and elimination of cholesterol-induced oxidative damages in hepatocytes may be a novel strategy to recover normal cholesterol metabolism functions [21,22]. When the cells suffer from oxidative stress, the reduced glutathione (GSH) is transformed into the oxidized glutathione (GSSG) to exert its anti-oxidant effects, thus, GSH/GSSH ratio is considered as a pivotal indicator to evaluate oxidative stress [23,24]. Dapagliflozin is a sodium-glucose cotransporter 2 (SGLT2) inhibitor, which has been commonly used as therapeutic drug for T2DM treatment to decrease serum glucose levels [25–27]. Interestingly, clinical data from Kurinami et al. report that Dapagliflozin significantly reduces liver fat accumulation in patients with T2DM [28]. In addition, Dapagliflozin is reported to eliminate Reactive Oxygen Species (ROS) to exert its anti-oxidant effects and attenuate steatohepatitis and Parkinson’s Disease [29–31], but it is still unclear whether Dapagliflozin recover liver functions in T2DM patients by suppressing oxidative stress.
The anti-oxidant Nrf2/HO-1 pathway is critical for regulating oxidative stress in various diseases, such as lipopolysaccharide (LPS)-induced acute lung injury (ALI) [32], colitis [33], and neuroinflammation [34]. Interestingly, data from Jin et al. evidence that HO-1 activates the Nrf2/ERK pathway to alleviate cholesterol-induced oxidative damages in endothelial cells [35]. In addition, the PI3K/Akt pathway interplays with the Nrf2/HO-1 pathway, and recent literatures report that the PI3K/Akt pathway is capable of promoting the activation of the Nrf2 signaling pathway to exert its anti-oxidant effects [36–38]. In addition, both the PI3K/Akt pathway [39,40] and Nrf2/HO-1 pathway [29,41] can be activated by Dapagliflozin, for example, Dapagliflozin activates the PI3K/Akt signaling pathway to inhibit cell apoptosis in myocardial ischemia/reperfusion injury rats [39] and induce renal gluconeogenesis in T2DM mice models [40], and the Nrf2/HO-1 pathway can be activated by Dapagliflozin to ameliorate inflammatory bowel disease [41] and Parkinson’s disease [29]. Notably, Arab et al. evidence that Dapagliflozin concurrently targets both PI3K/Akt pathway and Nrf2/HO-1 pathway to attenuate neuronal injury in the rat models with Parkinson’s Diseases [29].
Taken together the above information, we aimed to investigate the academic issues as follows: (1) The therapeutic effects of Dapagliflozin on the cellular functions of hepatocytes loaded with cholesterol. (2) The regulating effects of Dapagliflozin on cholesterol-induced oxidative stress. (3) The interplays between the PI3K/Akt pathway and the Nrf2/HO-1 pathway. (4) The underlying mechanisms by which Dapagliflozin exerts its protective effects in cholesterol-overloaded hepatocytes.
Methods
In vivo mice models for T2DM
The male C57BL/6 mice were purchased and housed in a specific-pathogen-free (SPF) environment with 12 h light/dark cycle at 22°C, and according to the published experimental procedures [42–44], the T2DM mice models were established by feeding the C57BL/6 mice with high-fat diet combined with low-dose streptozotocin (STZ) injections. Specifically, the mice in normal group and T2DM group were respectively fed with normal diet (20% kcal fat, 20% kcal protein, 60% kcal carbohydrates) and high-fat diet (60% kcal fat, 20% kcal protein, 20% kcal carbohydrates) combined with low-dose STZ for 12 consecutive weeks, each group had 6 mice. In addition, dapagliflozin was provided through the water at 100 mg/kg doses for 12 weeks before anesthesia by phenobarbital sodium. The mice liver tissues were obtained by surgical resection and were stored in the refrigerator with −70°C conditions for further analysis. The animal experiments were conducted in accordance with the guidelines of the Ethics Committee Affiliated to Jingjiang People’s Hospital.
Isolation, culture, and treatment of the primary mice hepatocytes
Primary hepatocytes were isolated from C57BL/6 mice based on the experimental procedures in the previous work [45], and briefly, the fresh liver tissues were obtained, and the portal vein was carefully removed. Then, the tissues were washed by Hank’s buffer solution for 3 times, and digested by collagenase reagent (0.8 mg/ml) for 10 min with constant 5 ml/min collagenase perfusion at 37°C. Next, the isolated cells were obtained and were cultured in the 6-well plates in DMEM medium (Gibco, USA) containing 10% fetal bovine serum (Gibco, USA) and 100 U penicillin/streptomycin antibiotics in the incubator with 5% CO2 humidified atmosphere at 37°C. The dead cells were removed during cell culture process, and at 2 days post-culture, the hepatocytes were subjected to 60 μg/ml of dapagliflozin (Selleck, Houston, Texas, USA), loaded with 50 μg/ml of water-soluble cholesterol (Sigma-Aldrich, USA), treated with NAC (Sigma-Aldrich, USA) and LY294002 (Sigma-Aldrich, USA) according to our preliminary experiments (data not shown) and published literature [46].
Delivery of the vectors
Nrf2 and HO-1 silencing vectors were designed as previously reported [32–34] and were constructed by a commercial third-party company (Sangon Biotech, Shanghai, China), and the vectors were delivered into the mice hepatocytes by using the Lipofectamine 2000 transfection reagent (Invitrogen, CA, USA) in accordance with the manufacturer’s protocols. The vectors transfection efficiency was determined by Real-Time qPCR at 48 h post-delivery.
Measurement of cell proliferation and viability
The primary hepatocytes were cultured in the 96-well plates at the density of 1 × 105 cells per well, and the cells were subjected to different treatments and were cultured in the standard culture conditions (5% CO2 humidified air at 37°C) for 48 h with 12 h intervals. MTT assay and trypan blue staining assay were respectively performed to examine cell proliferation and viability. For the MTT assay, the cells were incubated with 5 mg/ml MTT reaction solution at the volume of 20 μl per well for 3 h at 37°C, the supernatants were carefully discarded and the formazan was resolved by using 150 μl DMSO solution. The plates were fully vortexed and a microplate reader (Molecular Devices, CA, USA) was employed to determine optical density at 490 nm absorbance. For trypan blue staining assay, the cells were stained with trypan blue dye for 15 min at 37°C, and a light microscope (ThermoFisher Scientific, USA) was employed to count the dead blue cell numbers to calculate cell viability.
Detection of cell apoptosis ratio
A commercial Apoptosis Detection Kit (KeyGEN, Nanjing, China) was used to measure cell apoptosis ratio in the primary hepatocytes. Specifically, the hepatocytes were cultured and subjected to differential treatments, and the cells were suspended by the 1× binding buffer in the flow cytometry (FCM) tubes, and were concurrently incubated with Annexin V-FITC (5 μl) and PI (5 μl) for 10 min at room temperature without light exposure. Finally, the FCM assay was performed to analyze the apoptotic cells, which were labeled with Annexin V-FITC and PI. The ratio of the Annexin V-FITC-positive and PI-positive cells were considered as apoptotic ratio in this study.
Examination of mRNA levels by real-time qPCR
Total RNA from mice tissues and primary hepatocytes was extracted by using the TRizol reagent (Invitrogen, USA), and RNA quality was controlled by 1% agarose gel electrophoresis. The cDNA synthesis kit (Takara, Japan) was used to reversely transcribe the mRNA into cDNA, and further SYBR Mixture (Takara, Japan) and Real-Time qPCR analysis (Applied Biosystems, USA) were used to quantify the expression levels of the associated genes. The primer sequences for Nrf2 (Forward: 5’- GTG GTT TAG GGC AGA AGG-3’, Reverse: 5’-TCT TTC TTA CTC TGC CTC TA-3’), HO-1 (Forward: 5’-TAC CGC TCC CGA ATG AAC AC-3’, Reverse: 5’-GTC ACG GGA GTG GAG TCT TG-3’), CCND1 (Forward: 5’-GAT GGC GAT CGT CCT GTC AT-3’, Reverse: 5’-ATG ACA GGA CGA TCG CCA TC-3’), CDK2 (Forward: 5’- CCA GGA GTT ACT TCT ATG CCT GA-3’, Reverse: 5’-TTC ATC CAG GGG AGG TAC AAC-3’), CDK4 (Forward: 5’- CTG GAC ACT GAG AGG GCA AT-3’, Reverse: 5’- TGG GAA GGA GAA GGA GAA GC-3’), CDK6 (Forward: 5’- GTG AAC CAG CCC AAG ATG AC-3’, Reverse: 5’-TGG AGG AAG ATG GAG AGC AC-3’), CDKN3 (Forward: 5’-GGA CTC CTG ACA TAG CCA GC-3’, Reverse: 5’- CTG TAT TGC CCC GGA TCC TC-3’) and GAPDH (Forward: 5’-AAG ACC CAG AAA TGA AC-3’, Reverse: 5’- TCT ACA CGA TAA CAA CCA-3’) were designed according to the previous publications [32–34,47,48], which were synthesized by Sangon Biotechnology (Shanghai, China). The relative expressions of the above genes at mRNA levels were normalized by using GAPDH mRNA as internal control.
Measurement of protein levels by Western blot analysis
The hepatocytes were lysed with RIPA lysis buffer (Beyotime, Shanghai, China), separated by 10% SDS-PAGE, and transferred onto the PVDF membranes (Millipore, USA). The membranes were sequentially blocked with 5% nonfat milk and probed with the primary antibodies against Nrf2 (1:1500, Abcam, UK), HO-1 (1:2000, Abcam, UK), p-PI3K (1:2000, Abcam, UK), PI3K (1:1500, Abcam, UK), p-Akt (1:1500, Abcam, UK), Akt (1:2000, Abcam, UK), Ki67 (1:2000, Abcam, UK), pRPS6 (1:1500, Abcam, UK) and GAPDH (1:2000, Abcam, UK) at 4°C overnight, and were subsequently probed with the secondary antibody (1:5000, Abcam, UK) for 1 h at room temperature. The protein bands were visualized by using an ECL immunoblotting reagent (Solarbio Life Sciences, USA) in keeping with the manufacturer protocol, and the Image J software was used to analyze gray values of the protein bands.
Evaluation of oxidative stress in vitro and in vivo
To determine the status of oxidative damages in vitro and in vivo, we respectively purchased the DHE (Dihydroethidium) staining assay kit (QCbio, Shanghai, China), MDA detection kit (Abcam, UK), GSH/GSSG ratio kit (Sigma-Aldrich, USA) to measure ROS generation, MDA levels and GSH/GSSH ratio in keeping with their corresponding instructions.
Cellular localization of Nrf2 was determined by immunofluorescent staining assay
Cells were fixed by 4% paraformaldehyde for 15 min, and were subsequently permeabilized by treating cells with TBS buffer containing 0.3% Triton X-100 for 1 h. The cells were then blocked with 5% BSA and were incubated with the primary Nrf2 antibody (1:200, Abcam, UK) at 4°C overnight, and the cells were then probed with the secondary antibody (1:1000, Invitrogen, USA) for 1 h at room temperature. Nucleus was stained by 4ʹ6-diamidino-2-phenylindole (DAPI) for 5 min. A fluorescence microscope (Olympus, Japan) was employed to observe the expression levels and localization of the Nrf2 protein.
Analysis and presentation of data
Data was collected and analyzed by using the SPSS 18.0 software and Prism GraphPad 8.0 software. Data from two groups were compared by using the student’s t-test, and the comparisons among different groups (>2) were analyzed by using the one-way ANOVA analysis methods. *P < 0.05 was deemed as statistical significance.
Results
Dapagliflozin recovered cellular functions in cholesterol-loaded hepatocytes in vivo and in vitro
The T2DM mice models were established by feeding C57BL/6 mice with high-fat diet combined with low-dose STZ treatments according to the previous publications [42,43], which were further subjected to Dapagliflozin treatments, and the mice liver tissues were collected for further analysis. The data in Figure 1(a,b) showed that both free cholesterol (FC) and cholesteryl ester (CE) tended to be enriched in T2DM mice tissues, in contrast with the normal mice. Also, our Real-Time qPCR analysis evidenced that the mRNA levels of the cell cycle-associated genes, including CCND1, CDK2, CDK4, CDK6, CDKN3, and SKP2, were all downregulated in T2DM mice (Figure 1(c)), suggesting that cholesterol was enriched, and cell cycle arrest was also induced in T2DM mice liver cells. Consistently, as previously described [45], the primary mice hepatocytes were isolated, purified and cultured in vitro, and the cells were loaded with cholesterol based on our preliminary experiments (data not shown) and published data. The MTT assay (Figure 1(d)) and trypan blue staining assay (Figure 1(e)) results showed that cholesterol suppressed cell proliferation and viability in a time-dependent manner. Similarly, mice hepatocytes were exposed to cholesterol-overloading treatments for 12 h, and the FCM assay results evidenced that cholesterol triggered apoptotic cell death in mice hepatocytes (figure 1(f)). Interestingly, by pre-treating T2DM mice and mice primary hepatocytes with Dapagliflozin, we surprisingly found that Dapagliflozin decreased the levels of FC (Figure 1(a)) and CE (Figure 1(b)), and upregulated the pro-proliferation associated genes (Figure 1(c)) in T2DM mice in vivo. Consistently, Dapagliflozin also recovered cell proliferation (Figure 1(d)) and viability (Figure 1(e)), and suppressed cell apoptosis (figure 1(f)) in cholesterol-loaded mice hepatocytes in vitro. In addition, cellular senescence is a process that occurs following genotoxic stimuli and induces permanent cell cycle arrest with a loss of cellular functions [49], and it has been shown that Dapagliflozin prevents the progression of diabetic kidney disease by inhibiting cellular senescence and oxidative stress via ketone-induced NRF2 activation [50], which were in consistent with our results that cholesterol overloading significantly decreased Ki67 levels, whereas upregulated pRPS6 and β-galactosidase levels to induce cellular senescence in the mice hepatocytes, which were reversed by co-treating cells with Dapagliflozin (Figure S1A-C), indicating that Dapagliflozin suppressed cholesterol overloading-induced cellular senescence in cholesterol-overloaded mice hepatocytes. The above results indicated the facts as follows: (1) cholesterol-induced hepatocyte apoptosis and liver injury was closely associated with T2DM. (2) Dapagliflozin was effective to recover normal cellular functions and suppress cellular senescence in cholesterol-loaded hepatocytes.
Figure 1.

Dapagliflozin ameliorated cholesterol-induced cytotoxicity in mice hepatocytes with T2DM. The T2DM mice models were established, and (a) CE and (b) FC tended to be enriched in the liver tissues collected from T2DM mice, which were decreased by Dapagliflozin co-treatment. (c) The expression levels of cell cycle-associated genes in mice liver tissues were screened by performing the Real-Time qPCR analysis. (d) MTT assay and (e) trypan blue staining assay were respectively conducted to examine the effects of Dapagliflozin on cell proliferation and viability in cholesterol-treated hepatocytes. (f) FCM assay was employed to determine the Annexin V-FITC and PI-positive apoptotic cell ratio in mice hepatocytes. We performed each experiment with at least three repetitions, and *P < 0.05 was confirmed as statistical significance.
Cholesterol-induced oxidative stress was eliminated by Dapagliflozin in T2DM mice and cholesterol treated hepatocytes
Oxidative stress can be induced by various environmental stimulus, which participates in the regulation of different diseases, including T2DM [13–16]. Also, cholesterol accumulation is reported to induce oxidative damages in hepatocytes [7–9], which were evidenced by our study in the following experiments. Specifically, by analyzing the mice tissues, we found that the mRNA levels of anti-oxidant genes, including Nrf2 (Figure 2(a)) and HO-1 (Figure 2(b)), were downregulated, and the GSH/GSSG ratio was also suppressed (Figure 2(c)) in T2DM mice liver tissues. The above in vivo results were supported by the following in vitro data, which showed that cholesterol treatment increased DHE-positive cell ratio (Figure 2(d)) and MDA levels (Figure 2(e)), inhibited Nrf2 (figure 2(f)) and HO-1 (Figure 2(g)) mRNA levels, and suppressed GSH/GSSG ratio (Figure 2(h)) in mice primary hepatocytes. Next, we evidenced that Dapagliflozin eliminated oxidative stress in vivo and in vitro (Figure 2(a-h)). As shown in Figure 2(a-c), Dapagliflozin was effective to increased mRNA levels of Nrf2 and HO-1, and GSH/GSSG ratio in T2DM mice liver tissues. In addition, the cellular experiments also evidenced that Dapagliflozin suppressed ROS generation (Figure 2(d)) and MDA levels (Figure 2(e)), increased Nrf2 (figure 2(f)) and HO-1 (Figure 2(g)) expressions, and upregulated GSH/GSSG ratio (Figure 2(h)) in the mice primary hepatocytes loaded with cholesterol. The above results hinted that: (1) oxidative stress was critical for cholesterol-induced liver injury. (2) Dapagliflozin could eliminate cholesterol-induced oxidative damages in mice hepatocytes.
Figure 2.
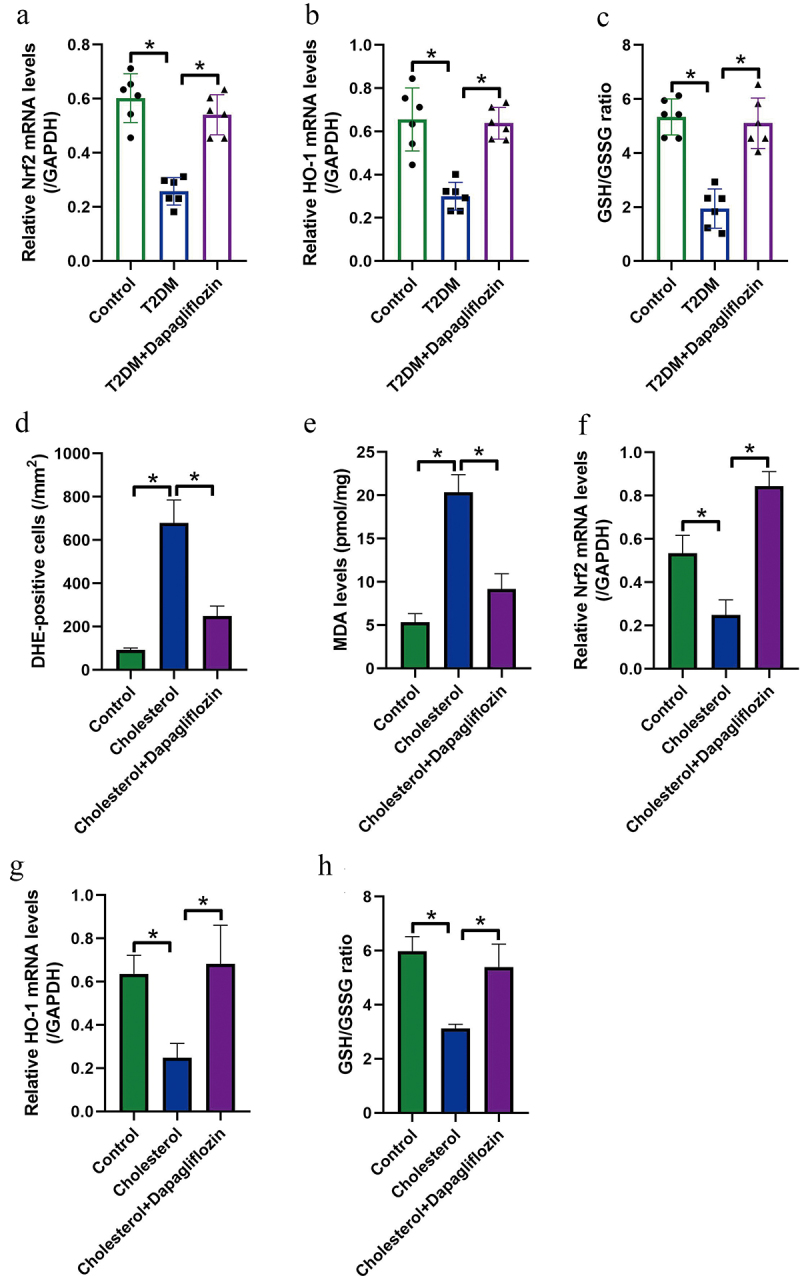
Dapagliflozin alleviated cholesterol overloading-induced oxidative damages in mice hepatocytes. The mRNA levels of (a) Nrf2 and (b) HO-1 in mice liver tissues were examined by conducting Real-Time qPCR analysis. (c) The GSH/GSSG ratio in mice liver was examined by using the corresponding commercial kit. (d) DHE staining assay was performed, and DHE-positive cells were counted under immunofluorescent microscope to evaluate ROS generation. (e) MDA levels in mice hepatocytes were evaluated. Real-Time qPCR was performed to determine (f) Nrf2 mRNA and (g) HO-1 mRNA levels in mice hepatocytes. (h) GSH/GSSG ratio in mice hepatocytes was examined. We performed each experiment with at least three repetitions, and *P < 0.05 was confirmed as statistical significance.
Elimination of ROS by its scavenger NAC rescued cell viability in mice primary hepatocytes loaded with cholesterol
As previously reported, oxidative stress contributes to cell death and is closely associated with cholesterol-induced cytotoxicity [18], and we next explored whether elimination of oxidative damages rescued cellular functions in cholesterol-treated hepatocytes. Thus, the mice hepatocytes were pre-treated with ROS scavenger NAC, and the MTT assay results in Figure 3(a) showed that NAC restored cell proliferation in mice hepatocytes loaded with cholesterol. Also, we performed trypan blue staining assay (Figure 3(b)), which evidenced that the inhibiting effects of cholesterol overloading on hepatocyte viability were also abrogated by NAC. Finally, the FCM assay was conducted to determine cell apoptosis, and as expected, cholesterol induced apoptotic cell death in mice hepatocytes, which were reversed by co-treating cells with NAC (Figure 3(c)). In general, the above data suggested that cholesterol-overloading promoted hepatocyte death in an oxidative stress-dependent manner.
Figure 3.
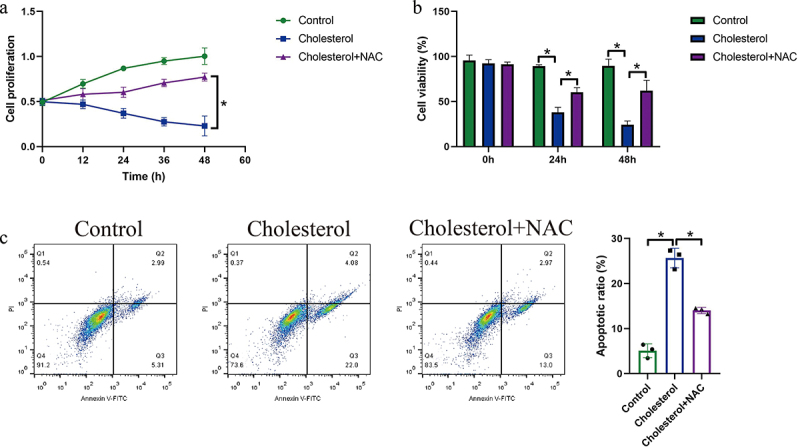
Elimination of ROS by NAC rescued cellular functions in cholesterol-treated mice hepatocytes. The mice hepatocytes were treated with cholesterol and NAC, and (a) MTT assay, (b) trypan blue staining assay, and (c) FCM assay were respectively performed to evaluate cell proliferation, viability and apoptosis. We performed each experiment with at least three repetitions, and *P < 0.05 was confirmed as statistical significance.
Dapagliflozin regulated the Nrf2/HO-1 pathway in mice hepatocytes through activating the PI3K/Akt pathway
Next, we investigated the potential underlying mechanisms by which Dapagliflozin suppressed cholesterol overloading-induced oxidative damages in mice hepatocytes. According to the published literatures, the anti-oxidant Nrf2/HO-1 pathway is crucial for eliminating ROS, which can also be activated by Dapagliflozin according to the available data from other teams [29,41]. Thus, through conducting the Western blot analysis, we evidenced that Dapagliflozin upregulated the expression levels of both Nrf2 and HO-1 in mice hepatocytes (Figure 4(a,b)). Consistently, as Nrf2 exerted its anti-oxidant effects by translocating to the nucleus to bind to the promoter region of antioxidant response element (ARE) for its transcription [51,52], thus, by performing the immunofluorescent staining assay, we noticed that Dapagliflozin not only promoted Nrf2 expressions, but accelerated the translocation of Nrf2 from cytoplasm to nucleus (Figure 4(c)). In addition, Dapagliflozin is reported to activate the PI3K/Akt pathway [39,40], which plays important role during Nrf2/HO-1 pathway activation [38], and our subsequent experiments evidenced that Dapagliflozin upregulated the levels of phosphorylated PI3K (p-PI3K) and Akt (p-Akt) to activate the PI3K/Akt pathway (Figure 4(d,f)), and LY294002 co-treatment significantly inactivated the PI3K/Akt pathway in the mice hepatocytes treated with Dapagliflozin (Figure S2A-C). As expected, inhibition of the PI3K/Akt pathway by its inhibitor LY294002 abrogated the promoting effects of Dapagliflozin on Nrf2/HO-1 pathway in mice hepatocytes (Figure 4(a-c)), indicating that Dapagliflozin activated the Nrf2/HO-1 pathway through modulating the PI3K/Akt pathway.
Figure 4.
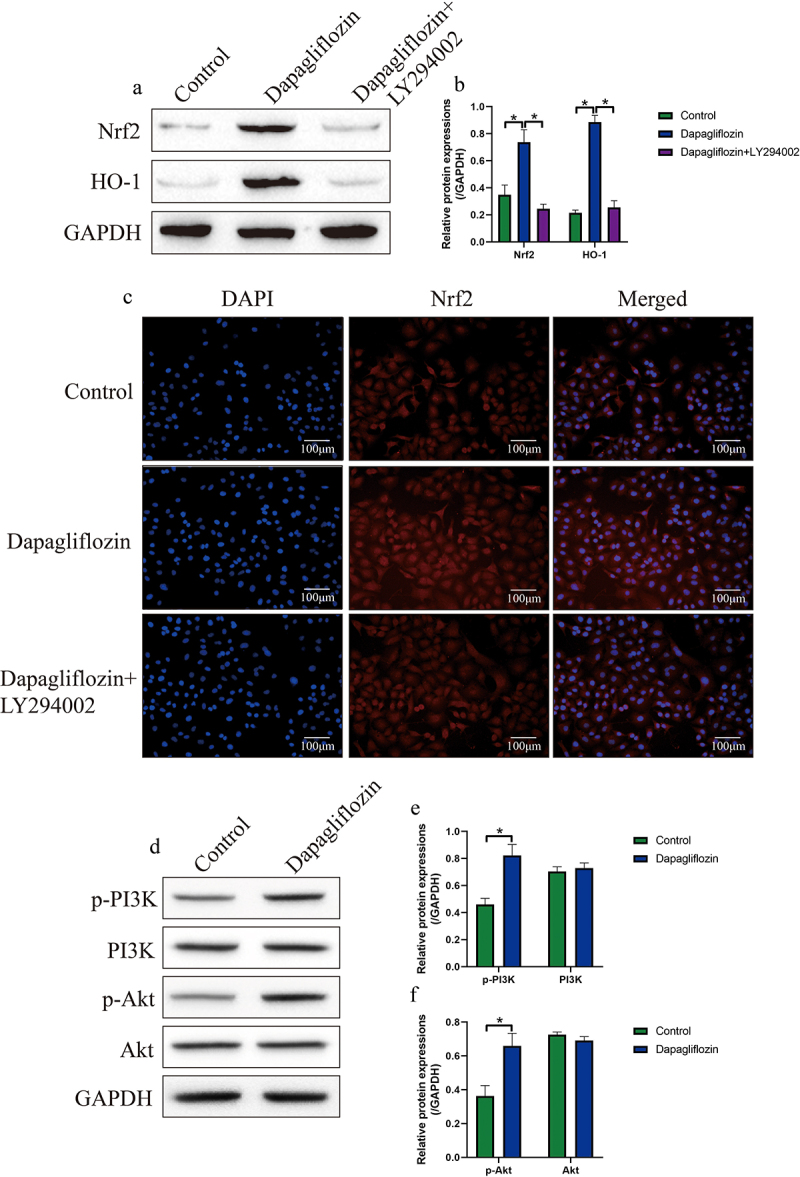
Dapagliflozin re-activated the anti-oxidant Nrf2/HO-1 pathway in cholesterol-treated mice hepatocytes through activating the PI3K/Akt pathway. (a, b) The effects of Dapagliflozin and LY294002 co-treatment on the expression levels of Nrf2 and HO-1 were determined by conducting Western blot analysis. (c) Immunofluorescent staining assay was performed to evaluate the expression levels and cellular localization of Nrf2 in mice hepatocytes. (d-f) Western blot analysis was used to measure the expression status of p-PI3K, PI3K, p-Akt and Akt in the hepatocytes. We performed each experiment with at least three repetitions, and *P < 0.05 was confirmed as statistical significance.
Genetically silencing of Nrf2 and HO-1 abolished the protective effects of Dapagliflozin on cholesterol-induced hepatocyte death
Since targeting oxidative stress is effective to recover cellular functions in cholesterol-overloaded mice hepatocytes, and the anti-oxidant Nrf2/HO-1 pathway can be modulated by Dapagliflozin in our work and previous data [36–38]. The following reversal experiments were conducted to investigate whether Dapagliflozin modulated cellular functions in cholesterol-treated hepatocytes through the Nrf2-HO-1 pathway. Therefore, the silencing vectors for Nrf2 and HO-1 were synthesized and respectively delivered into the mice hepatocytes for their downregulation (Figure 5(a,b)), and the MTT assay results in Figure 5(c) showed that the inhibiting effects of cholesterol on hepatocyte proliferation were attenuated by Dapagliflozin co-treatment, which were abrogated by silencing Nrf2 and HO-1. The above data were supported by the following data that genetically silencing of Nrf2 and HO-1 also suppressed cell viability in mice hepatocytes co-treated with Dapagliflozin and cholesterol (Figure 5(d)). Consistently, through performing the FCM assay, we found that the suppressing effects of Dapagliflozin on cholesterol-induced hepatocyte apoptosis were also abolished by downregulating Nrf2 and HO-1 (Figure 5(e,f)). Taken together the above data, we concluded that Dapagliflozin exerted its protective effects in cholesterol-loaded mice hepatocytes through activating the Nrf2/HO-1 pathway.
Figure 5.
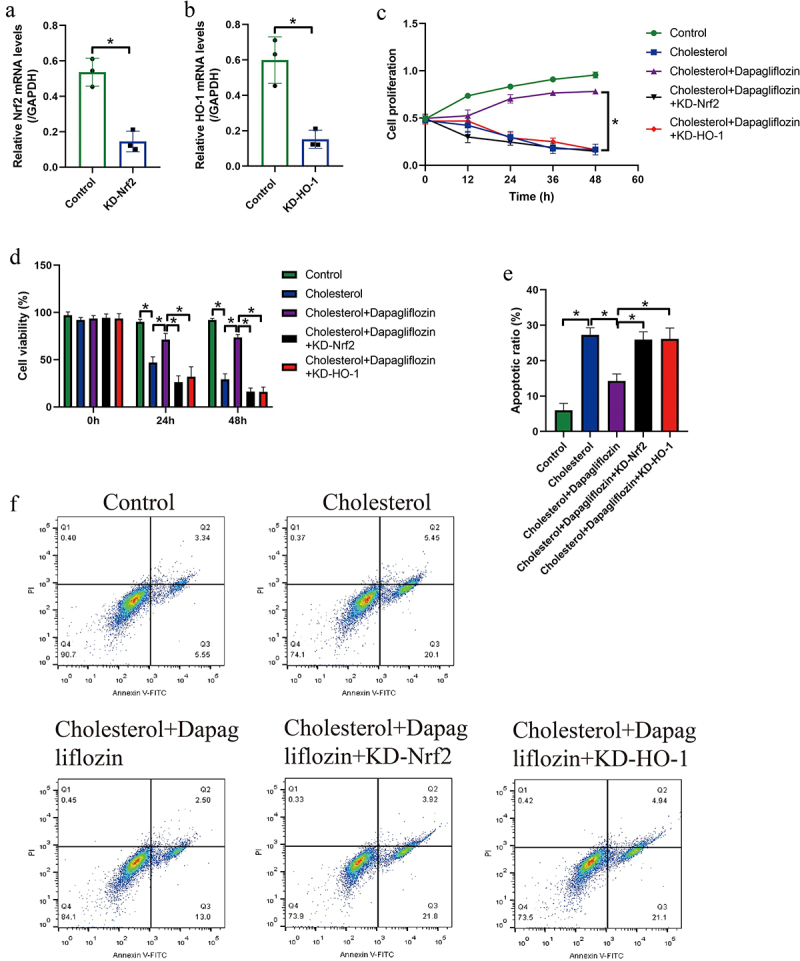
Genetically inactivation of the Nrf2/HO-1 pathway abrogated the protective effects of Dapagliflozin on cholesterol-treated mice hepatocytes. The silencing vectors for (a) Nrf2 and (b) HO-1 were respectively transfected into the hepatocytes, and the transfection efficiency was determined by Real-Time qPCR analysis. (c) MTT assay and (d) trypan blue staining assay were performed to examine cell proliferation and viability. (e, f) Cell apoptosis ratio in mice hepatocytes was examined by performing FCM assay. We performed each experiment with at least three repetitions, and *P < 0.05 was confirmed as statistical significance.
Discussion
Fatty liver is an independent risk factor which predicts worse prognosis in T2DM patients, and aberrant accumulation and metabolism of cholesterol contribute to hepatic ectopic fat accumulation, which contributes to insulin resistance and worse prognosis in T2DM patients [7–9]. Mechanistically, cholesterol overloading induces cytotoxicity in hepatocytes, which further damages the normal functions of cholesterol metabolism of liver, and accelerates the development of fatty liver, resulting in obesity [11–13]. Thus, it is feasible and urgent to search for therapeutic agents or drugs to reverse cholesterol-induced hepatotoxicity and recover normal liver functions. Dapagliflozin has been widely used for the treatment of T2DM through lowering the serum glucose levels [25–27], and interestingly, recent data suggest that Dapagliflozin also inhibits liver fat accumulation in patients with T2DM [28], and we evidenced that Dapagliflozin recovered cellular functions in mice hepatocytes loaded with cholesterol. Specifically, cholesterol overloading induced cell cycle arrest and apoptosis, and suppressed cell proliferation and viability in mice hepatocytes, which were reversed by Dapagliflozin co-treatments, implying that Dapagliflozin abrogated the cytotoxic effects of cholesterol overloading on hepatocytes, which were supported by the previous work [28].
Oxidative stress contributes the development of various diseases, including T2DM [13–16]. In recent studies, researchers notice that cholesterol induced cell death via triggering irreversible oxidative damages [13–16], which were supported by our results that cholesterol induced ROS generation in mice hepatocytes. As previously reported, oxidative stress promoted cell apoptosis and suppressed cell viability [18], and we found that elimination of oxidative stress by ROS scavenger NAC rescued cell proliferation and viability in mice hepatocytes loaded with cholesterol, suggesting that cholesterol-induced hepatotoxicity in an oxidative damage-dependent manner. Interestingly, Dapagliflozin is reported to regulate oxidative stress in various type of cells [29–31], and this study evidenced that Dapagliflozin effectively suppress cholesterol-induced oxidative stress in vitro and in vivo. In addition, oxidative stress is closely related with cellular senescence [49], and our data proved that Dapagliflozin was effective to suppress cholesterol-overloading induced cellular senescence in mice hepatocytes, which were supported by the previous work [50]. The above information indicated that Dapagliflozin eliminates ROS-induced damages to recover normal cellular functions in cholesterol-loaded hepatocytes.
Next, the underlying mechanisms by which Dapagliflozin exerts its protective effects on cholesterol-treated hepatocytes were investigated. According to the previous publications, the antioxidant Nrf2/HO-1 pathway is critical for regulating oxidative stress in various diseases [32–34], and this signal pathway can also be modulated by Dapagliflozin as previously described [29,41]. In consistent with the above literatures, we evidenced that Dapagliflozin upregulated Nrf2 and HO-1, and promoted Nrf2 protein translocation from cytoplasm to nucleus, resulting in the activation of the Nrf2/HO-1 pathway. In addition, the PI3K/Akt pathway has been reported to activate the Nrf2/HO-1 pathway [36–38], and Dapagliflozin also activate the PI3K/Akt pathway [39,40], which were validated by our results that Dapagliflozin activated the anti-oxidant Nrf2/HO-1 pathway through the PI3K/Akt pathway. Since Dapagliflozin is a sodium-glucose cotransporter 2 inhibitor to block glucose transportation [53], and transportation of glucose influences the activation of the PI3K/Akt pathway [54,55], which enlightens us to speculate that Dapagliflozin might activate the PI3K/Akt pathway through inhibiting glucose transportation. However, this issue is still needed to be verified in our future work. Finally, we evidenced that Dapagliflozin suppressed cholesterol-induced cell death in hepatocytes via regulating the Nrf2/HO-1 pathway, which were in consistent with the published data in other types of cells [39,40].
Conclusions
Taken together all the information, we summarized our conclusions as follows: (1) Dapagliflozin attenuated cholesterol overloading-induced cytotoxicity in mice hepatocytes with T2DM. (2) Cholesterol exerted its cytotoxic effects on the hepatocytes via triggering oxidative stress. (3) Dapagliflozin activated the anti-oxidant Nrf2/HO-1 pathway via modulating the PI3K/Akt pathway. (4) Dapagliflozin attenuated cholesterol overloading-induced hepatocyte death through activating the Nrf2/HO-1 pathway.
Supplementary Material
Funding Statement
The author(s) reported there is no funding associated with the work featured in this article.
Authors’ contributions
As the first author, Dr. Liu Yang designed and conducted the investigations of this work, and this author also drafted the initial version of the manuscript. As the co-authors, Dr. Dan Liu and Dr. Hongqin Yan helped to collect and analyze the data. Dr. Kaixia Chen is the corresponding author of this study, and provided conception and guidance. In addition, the corresponding author proofread the manuscript.
Availability of data and materials
All the involved data had been included in the manuscript, and the raw data and all the source files can be obtained from the corresponding author upon reasonable request.
Disclosure statement
No potential conflict of interest was reported by the authors.
Ethics approval and consent to participate
This study did not use human subjects, and the animal experiments were conducted in accordance with the guidelines of the Ethics Committee Affiliated to Jingjiang People’s Hospital. All the in vivo study on the animals were carried in accordance with the ARRIVE guidelines and were approved by the Ethics Committee Affiliated to Jingjiang People’s Hospital (Approval Number 2,019,082,703,001) in August 27th, 2019.
Supplementary material
Supplemental data for this article can be accessed here
References
- [1].Artasensi A, Pedretti A, and Vistoli G, et al. Type 2 diabetes mellitus: a review of multi-target drugs. Molecules. 2020;25(8 1987 doi: 10.3390/molecules25081987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Viigimaa M, Sachinidis A, Toumpourleka M, et al. Macrovascular complications of type 2 diabetes mellitus. Curr Vasc Pharmacol. 2020;18(2):110–116. [DOI] [PubMed] [Google Scholar]
- [3].Fuse K, Kadota A, Kondo K, et al. Liver fat accumulation assessed by computed tomography is an independent risk factor for diabetes mellitus in a population-based study: SESSA (Shiga Epidemiological Study of Subclinical Atherosclerosis). Diabetes Res Clin Pract. 2020;160:108002. [DOI] [PubMed] [Google Scholar]
- [4].Liu J, Fan D, Wang X, et al. Association of two novel adiposity indicators with visceral fat area in type 2 diabetic patients: novel adiposity indexes for type 2 diabetes. Medicine (Baltimore). 2020;99(19):e20046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Dewidar B, Kahl S, Pafili K, et al. Metabolic liver disease in diabetes - from mechanisms to clinical trials. Metabolism. 2020;111s:154299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lian CY, Zhai ZZ, Li ZF, et al. High fat diet-triggered non-alcoholic fatty liver disease: a review of proposed mechanisms. Chem Biol Interact. 2020;330:109199. [DOI] [PubMed] [Google Scholar]
- [7].Athyros VG, Polyzos SA, Kountouras J, et al. Non-alcoholic fatty liver disease treatment in patients with type 2 diabetes mellitus; new kids on the block. Curr Vasc Pharmacol. 2020;18(2):172–181. [DOI] [PubMed] [Google Scholar]
- [8].Bica C, Sandu C, Suceveanu AI, et al. Non-alcoholic fatty liver disease: a major challenge in type 2 diabetes mellitus (review). Exp Ther Med. 2020;20(3):2387–2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Daryabor G, Atashzar MR, Kabelitz D, et al. The effects of type 2 diabetes mellitus on organ metabolism and the immune system. Front Immunol. 2020;11:1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Orozco Morales JA, Medina Urrutia AX, Torres Tamayo M, et al. Effects of fatty liver on the size and composition of high-density lipoprotein cholesterol subpopulations in adolescents with type 2 diabetes mellitus. Pediatr Diabetes. 2020;21(7):1140–1149. [DOI] [PubMed] [Google Scholar]
- [11].Zhao ZH, Xin FZ, Zhou D, et al. Trimethylamine N-oxide attenuates high-fat high-cholesterol diet-induced steatohepatitis by reducing hepatic cholesterol overload in rats. World J Gastroenterol. 2019;25(20):2450–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Li Q, Liu Z, Guo J, et al. Cholesterol overloading leads to hepatic L02 cell damage through activation of the unfolded protein response. Int J Mol Med. 2009;24(4):459–464. [DOI] [PubMed] [Google Scholar]
- [13].Domínguez-Pérez M, Simoni-Nieves A, Rosales P, et al. Cholesterol burden in the liver induces mitochondrial dynamic changes and resistance to apoptosis. J Cell Physiol. 2019;234(5):7213–7223. [DOI] [PubMed] [Google Scholar]
- [14].Yamamoto K, Nogimori Y, Imamura H, et al. Shear stress activates mitochondrial oxidative phosphorylation by reducing plasma membrane cholesterol in vascular endothelial cells. Proc Natl Acad Sci U S A. 2020;117(52):33660–33667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sun Y, Ge X, Li X, et al. High-fat diet promotes renal injury by inducing oxidative stress and mitochondrial dysfunction. Cell Death Dis. 2020;11(10):914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wang X, Tanaka N, Hu X, et al. A high-cholesterol diet promotes steatohepatitis and liver tumorigenesis in HCV core gene transgenic mice. Arch Toxicol. 2019;93(6):1713–1725. [DOI] [PubMed] [Google Scholar]
- [17].Hou X, Yang S, Yin J.. Blocking the REDD1/TXNIP axis ameliorates LPS-induced vascular endothelial cell injury through repressing oxidative stress and apoptosis. Am J Physiol Cell Physiol. 2019;316(1):C104–c110. [DOI] [PubMed] [Google Scholar]
- [18].Yang M, Sun J, Stowe DF, et al. Knockout of VDAC1 in H9c2 cells promotes oxidative stress-induced cell apoptosis through decreased mitochondrial hexokinase II binding and enhanced glycolytic stress. Cell Physiol Biochem. 2020;54(5):853–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Klisic A, Isakovic A, Kocic G, et al. Relationship between oxidative stress, inflammation and dyslipidemia with fatty liver index in patients with type 2 diabetes mellitus. Exp Clin Endocrinol Diabetes. 2018;126(6):371–378. [DOI] [PubMed] [Google Scholar]
- [20].Sun P, Du Q, and Hao Q, et al. Correlations of liver function, oxidative stress and fatty liver severity with abnormal glucose metabolism in type 2 diabetes mellitus patients complicated with nonalcoholic fatty liver. Panminerva Med. 2021. 15 3 227–32 . [DOI] [PubMed] [Google Scholar]
- [21].Yuan S, Pan Y, Zhang Z, et al. Amelioration of the lipogenesis, oxidative stress and apoptosis of hepatocytes by a novel proteoglycan from ganoderma lucidum. Biol Pharm Bull. 2020;43(10):1542–1550. [DOI] [PubMed] [Google Scholar]
- [22].Zhou L, Tang J, Yang X, et al. Five constituents in psoralea corylifolia L. Attenuate palmitic acid-induced hepatocyte injury via inhibiting the protein kinase C-α/nicotinamide-adenine dinucleotide phosphate oxidase pathway. Front Pharmacol. 2019;10:1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Jiang C, Zhang C, Song J, et al. Cytidine-gold nanoclusters as peroxidase mimetic for colorimetric detection of glutathione (GSH), glutathione disulfide (GSSG) and glutathione reductase (GR). Spectrochim Acta A Mol Biomol Spectrosc. 2021;250:119316. [DOI] [PubMed] [Google Scholar]
- [24].Zhu Y, Wu J, Wang K, et al. Facile and sensitive measurement of GSH/GSSG in cells by surface-enhanced Raman spectroscopy. Talanta. 2021;224:121852. [DOI] [PubMed] [Google Scholar]
- [25].Dhillon S. Dapagliflozin: a review in type 2 diabetes. Drugs. 2019;79(10):1135–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kato ET, Silverman MG, Mosenzon O, et al. Effect of dapagliflozin on heart failure and mortality in type 2 diabetes mellitus. Circulation. 2019;139(22):2528–2536. [DOI] [PubMed] [Google Scholar]
- [27].Kelly MS, Lewis J, Huntsberry AM, et al. Efficacy and renal outcomes of SGLT2 inhibitors in patients with type 2 diabetes and chronic kidney disease. Postgrad Med. 2019;131(1):31–42. [DOI] [PubMed] [Google Scholar]
- [28].Kurinami N, Sugiyama S, Yoshida A, et al. Dapagliflozin significantly reduced liver fat accumulation associated with a decrease in abdominal subcutaneous fat in patients with inadequately controlled type 2 diabetes mellitus. Diabetes Res Clin Pract. 2018;142:254–263. [DOI] [PubMed] [Google Scholar]
- [29].Arab HH, Safar MM, Shahin NN. Targeting ROS-dependent AKT/GSK-3β/NF-κB and DJ-1/Nrf2 pathways by dapagliflozin attenuates neuronal injury and motor dysfunction in rotenone-induced parkinson’s disease rat model. ACS Chem Neurosci. 2021;12(4):689–703. [DOI] [PubMed] [Google Scholar]
- [30].Leng W, Wu M, Pan H, et al. The SGLT2 inhibitor dapagliflozin attenuates the activity of ROS-NLRP3 inflammasome axis in steatohepatitis with diabetes mellitus. Ann Transl Med. 2019;7(18):429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Uthman L, Homayr A, Juni RP, et al. Empagliflozin and dapagliflozin reduce ROS generation and restore NO bioavailability in tumor necrosis factor α-stimulated human coronary arterial endothelial cells. Cell Physiol Biochem. 2019;53(5):865–886. [DOI] [PubMed] [Google Scholar]
- [32].Huang CY, Deng JS, and Huang WC, et al. Attenuation of lipopolysaccharide-induced acute lung injury by hispolon in mice, through regulating the TLR4/PI3K/Akt/mTOR and keap1/Nrf2/HO-1 pathways, and suppressing oxidative stress-mediated ER stress-induced apoptosis and autophagy. Nutrients. 2020;12(6 1742 doi: 10.3390/nu12061742). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Mei Y, Wang Z, Zhang Y, et al. FA-97, a new synthetic caffeic acid phenethyl ester derivative, ameliorates DSS-induced colitis against oxidative stress by activating Nrf2/HO-1 pathway. Front Immunol. 2019;10:2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Yu J, Wang WN, Matei N, et al. Ezetimibe attenuates oxidative stress and neuroinflammation via the AMPK/Nrf2/TXNIP pathway after MCAO in rats. Oxid Med Cell Longev. 2020;2020:4717258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Jin X, Xu Z, Fan R, et al. HO‑1 alleviates cholesterol‑induced oxidative stress through activation of Nrf2/ERK and inhibition of PI3K/AKT pathways in endothelial cells. Mol Med Rep. 2017;16(3):3519–3527. [DOI] [PubMed] [Google Scholar]
- [36].Li ST, Dai Q, Zhang SX, et al. Ulinastatin attenuates LPS-induced inflammation in mouse macrophage RAW264.7 cells by inhibiting the JNK/NF-κB signaling pathway and activating the PI3K/Akt/Nrf2 pathway. Acta Pharmacol Sin. 2018;39(8):1294–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Sun X, Chen L, He Z. PI3K/Akt-Nrf2 and anti-inflammation effect of macrolides in chronic obstructive pulmonary disease. Curr Drug Metab. 2019;20(4):301–304. [DOI] [PubMed] [Google Scholar]
- [38].Zhuang Y, Wu H, Wang X, et al. Resveratrol attenuates oxidative stress-induced intestinal barrier injury through PI3K/Akt-mediated Nrf2 signaling pathway. Oxid Med Cell Longev. 2019;2019:7591840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Gong L, Wang X, Pan J, et al. The co-treatment of rosuvastatin with dapagliflozin synergistically inhibited apoptosis via activating the PI3K/AKt/mTOR signaling pathway in myocardial ischemia/reperfusion injury rats. Open Med (Wars). 2021;15(1):47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Sun X, Cao Z, Ma Y, et al. Resveratrol attenuates dapagliflozin-induced renal gluconeogenesis via activating the PI3K/Akt pathway and suppressing the FoxO1 pathway in type 2 diabetes. Food Funct. 2021;12(3):1207–1218. [DOI] [PubMed] [Google Scholar]
- [41].Arab HH, Al-Shorbagy MY, Saad MA. Activation of autophagy and suppression of apoptosis by dapagliflozin attenuates experimental inflammatory bowel disease in rats: targeting AMPK/mTOR, HMGB1/RAGE and Nrf2/HO-1 pathways. Chem Biol Interact. 2021;335:109368. [DOI] [PubMed] [Google Scholar]
- [42].Liu X, Zhang Y, Liang H, et al. Overexpression of microRNA-216a-3p accelerates the inflammatory response in cardiomyocytes in type 2 diabetes mellitus by targeting IFN-α2. Front Endocrinol (Lausanne). 2020;11:522340. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [43].Xu H, Du X, Xu J, et al. Pancreatic β cell microRNA-26a alleviates type 2 diabetes by improving peripheral insulin sensitivity and preserving β cell function. PLoS Biol. 2020;18(2):e3000603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Li XF, Zhang SH, Liu GF, et al. miR-363 alleviates detrusor fibrosis via the TGF-β1/Smad signaling pathway by targeting Col1a2 in rat models of STZ-induced T2DM. Mol Ther Nucleic Acids. 2020;22:1142–1153. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [45].Li S, Jin S, Chen W, et al. Mangiferin alleviates endoplasmic reticulum stress in acute liver injury by regulating the miR-20a/miR-101a-Nrf2 axis. J Biochem. 2020;168(4):365–374. [DOI] [PubMed] [Google Scholar]
- [46].Li L, Li Q, Huang W, et al. Dapagliflozin alleviates hepatic steatosis by restoring autophagy via the AMPK-mTOR pathway. Front Pharmacol. 2021;12:589273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Cui D, Bi J, Zhang ZN, et al. Organophosphorus flame retardant TDCPP-induced cytotoxicity and associated mechanisms in normal human skin keratinocytes. Sci Total Environ. 2020;726:138526. [DOI] [PubMed] [Google Scholar]
- [48].Sun M, Tan J, Wang M, et al. Inorganic arsenic-mediated upregulation of AS3MT promotes proliferation of nonsmall cell lung cancer cells by regulating cell cycle genes. Environ Toxicol. 2021;36(2):204–212. [DOI] [PubMed] [Google Scholar]
- [49].Campisi J, d’Adda DI, Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8(9):729–740. [DOI] [PubMed] [Google Scholar]
- [50].Kim MN, Moon JH, Cho YM. Sodium-glucose cotransporter-2 inhibition reduces cellular senescence in the diabetic kidney by promoting ketone body-induced NRF2 activation. Diabetes Obes Metab. 2021;23(11):2561–2571. [DOI] [PubMed] [Google Scholar]
- [51].Adelusi TI, Du L, Hao M, et al. Keap1/Nrf2/ARE signaling unfolds therapeutic targets for redox imbalanced-mediated diseases and diabetic nephropathy. Biomed Pharmacother. 2020;123:109732. [DOI] [PubMed] [Google Scholar]
- [52].Serafini MM, Catanzaro M, Fagiani F, et al. Modulation of keap1/Nrf2/ARE signaling pathway by curcuma- and garlic-derived hybrids. Front Pharmacol. 2019;10:1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Arow M, Waldman M, Yadin D, et al. Sodium-glucose cotransporter 2 inhibitor Dapagliflozin attenuates diabetic cardiomyopathy. Cardiovasc Diabetol. 2020;19(1):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Gao L, Yuan P, Zhang Q, et al. Taxifolin improves disorders of glucose metabolism and water-salt metabolism in kidney via PI3K/AKT signaling pathway in metabolic syndrome rats. Life Sci. 2020;263:118713. [DOI] [PubMed] [Google Scholar]
- [55].Xu L, Chen J, Jia L, et al. SLC1A3 promotes gastric cancer progression via the PI3K/AKT signalling pathway. J Cell Mol Med. 2020;24(24):14392–14404. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the involved data had been included in the manuscript, and the raw data and all the source files can be obtained from the corresponding author upon reasonable request.