

Abstract
The spike (S) protein of SARS-CoV-2 plays a crucial role in cell entry, and the nucleocapsid (N) protein is highly conserved among human coronavirus homologs. For potentially broad effectiveness against both original virus and emerging variants, we developed Alphavirus-based self-amplifying mRNA (sa-mRNA) SARS-CoV-2 vaccines: an sa-mRNA S encoding a full-length S protein stabilized in a prefusion conformation and an sa-mRNA S-N co-expressing S and N proteins for the original virus. We show that these sa-mRNA SARS-CoV-2 vaccines raised potent neutralizing antibody responses in mice against not only the original virus but also the Alpha, Beta, Gamma, and Delta variants. sa-mRNA S vaccines against the Alpha and Beta variants also raised robust cross-reactive neutralizing antibody responses against their homologous viruses and heterologous variants. sa-mRNA S and sa-mRNA S-N vaccines elicited Th1-dominant, antigen-specific CD4+ T cell responses to S and N proteins and robust and broad CD8+ T cell responses to S protein. Hamsters immunized with either vaccine were fully protected from lung infection and showed significant reduction of viral load in upper respiratory tract. Our findings demonstrate that sa-mRNA SARS-CoV-2 vaccines are potent in animal models with potential to be highly effective against SARS-CoV-2 infection in humans.
Keywords: self-amplifying mRNA, SARS-CoV-2 vaccine, spike protein, nucleocapsid protein, cross-reactive immunogenicity, protection
Graphical abstract
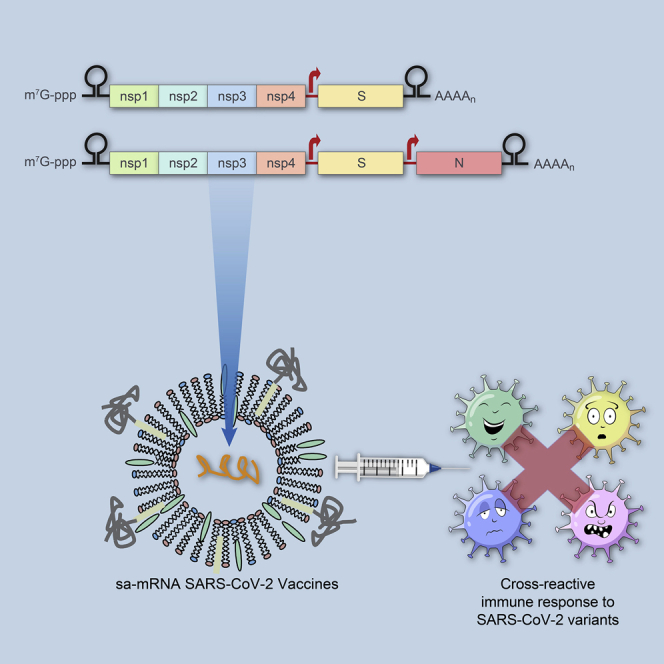
Self-amplifying mRNA (sa-mRNA) SARS-CoV-2 vaccines containing S protein or S and N proteins raised robust cross-reactive neutralizing antibody and T cell responses against variants in mice. Hamsters immunized with either vaccine were fully protected from lung infection with significantly reduced viral loads in upper respiratory tracts.
Introduction
SARS-CoV-2 belongs to the Betacoronavirus family and is closely related to several bat coronaviruses.1,2 A 1,273-amino acid viral surface homotrimeric glycoprotein, the spike (S) protein is the main target for neutralizing antibody response and has become the main immunogen for coronavirus vaccine development.3,4 Each S monomer comprises two subunits: the S1 subunit, which contains the receptor-binding domain that engages with the host cell receptor angiotensin-converting enzyme 2 (ACE2) for viral attachment, and the S2 subunit, which mediates fusion between viral and host cell membranes through a substantial structural rearrangement from a metastable, prefusion to a more stable, post-fusion conformation.5,6 Prefusion stabilization of SARS-CoV-1 S protein greatly increases the yield of the recombinant protein antigens and elicits high neutralizing antibody titers relative to its wild-type counterpart, which supports the superiority of the stabilized prefusion S protein as an immunogen.7
There is an inverse correlation between effective SARS-CoV-2-specific T cell responses and COVID-19 disease severity.8 The nucleocapsid (N) protein, a 419-amino acid viral internal protein that makes up the helical nucleocapsid and binds along the viral RNA genome, is more genetically conserved than the S protein and, in addition to S protein, is a prominent target for T cell responses in SARS-CoV-2-infected individuals.4 In COVID-19 convalescent patients, CD4+ T helper cell responses to the S protein were robust and correlated with anti-S antibody titers, and CD8+ T cells targeting S protein were detected.9 In addition, both N protein and anti-N antibody are co-dominant targets for CD4+ T cells; furthermore, N protein is strongly recognized by CD8+ T cells. These results suggest that N protein is worthy of study as an additional potential immunogen for vaccine development.
Alphavirus-based self-amplifying mRNA (sa-mRNA) encapsulated within a lipid nanoparticle (LNP) has been develope1d as a novel vaccine platform technology.10 sa-mRNA retains the genes encoding the Alphavirus RNA replication machinery, but those encoding the viral structural proteins are replaced with genes encoding antigens of interests, which are then abundantly expressed via subgenomic transcription-mediated amplifications of antigen-encoding mRNAs in the cell’s cytoplasm. The self-replication of sa-mRNA in a target cell greatly reduces the RNA dose relative to non-amplifying mRNA and expands the protein expression time in an individual cell.11 LNP encapsulation of sa-mRNA allows efficient and low-cytopathic delivery of RNA to cells without the complication of anti-vector immunity that can thwart repeated immunization as seen with DNA viral vector-based vaccines.12
sa-mRNA production starts from a cell-free enzymatic in vitro transcription reaction, followed first by a simple downstream purification and then by LNP formulation with synthetic lipids, a process that enables rapid and cost-effective vaccine manufacturing.10,13 The rapid production of potent sa-mRNA H7N9 influenza vaccine demonstrated the potential of the sa-mRNA platform in response to a pandemic.14 In additional to eliciting a robust antibody response, sa-mRNA vaccines expressing one or more conserved influenza antigens raised robust CD4+ T helper cells and CD8+ T cytotoxic cells.15 During the COVID-19 pandemic, sa-mRNA technology has been used to develop SARS-CoV-2 vaccines and further demonstrated its potential as an effective vaccine platform that addresses pandemic challenges.16, 17, 18
In this paper, we report a study of two sa-mRNA SARS-CoV-2 vaccine candidates: sa-mRNA S encoding the prefusion S protein and sa-mRNA S-N co-expressing S protein and N protein. We sought to determine whether these vaccines would raise neutralizing antibody titers against SARS-CoV-2 variants, increase T helper type 1 (Th1)-dominant antigen-specific CD4+ T cell responses, and elicit CD8+ T cell responses in mice. Furthermore, we also studied whether hamsters immunized with either sa-mRNA SARS-CoV-2 vaccine would be protected from a SARS-CoV-2 virus challenge.
Results
Design and production of SARS-CoV-2 vaccines
To generate SARS-CoV-2 vaccine candidates, a monocistronic vector was constructed encoding the full-length, codon-optimized S glycoprotein, based on the sequences from SARS-CoV-2/human/USA/WA-CDC-WA1/2020 (original virus), wherein the S1/S2 furin-like cleavage site, RRAR, was mutated to QQAA to stabilize the protein in a prefusion conformation (Figure 1A). A bicistronic vector was also generated encoding both this prefusion S protein and a full-length, codon-optimized N protein, driven by a duplicated subgenomic promoter, to elicit immune responses by both antigens. The corresponding sa-mRNAs (i.e., sa-mRNA S and sa-mRNA S-N) were synthesized and 5′-capped in vitro by enzymatic reactions. Then purified sa-mRNAs were encapsulated into LNPs composed of synthetic lipids and characterized for particle biophysical attributes (Table S1) and antigen expression in transfected baby hamster kidney (BHK) cells. Both flow cytometry (Figure 1B) and western blot (Figure 1C) confirmed the expression of S protein by LNP-formulated sa-mRNA S and both S and N proteins by LNP-formulated sa-mRNA S-N. These assays also showed that the expression level of S antigen was comparable between sa-mRNA S and sa-mRNA S-N (Figures 1C and 1D).
Figure 1.

Design and production of sa-mRNA S and sa-mRNA S-N vaccines
(A) Full-length SARS-CoV-2 prefusion S and full-length N protein sequences based on the SARS-CoV-2/human/USA/WA-CDC-WA1/2020 virus (original virus) were inserted into Alphavirus-based sa-mRNA with both S and N downstream of two distinct subgenomic promoters. sa-mRNA S or sa-mRNA S-N transfected baby hamster kidney (BHK) cells were analyzed by flow cytometry for S+ or S+N+ expressing cells (B) and western blotting for expression of S and N proteins (C). (D) Flow cytometry was also used to analyze relative level of S protein expression in S+ expressing cells.
Immune response in mice
To evaluate the antibody immune response by sa-mRNA vaccines in a preclinical animal model, female BALB/c mice were immunized at day 1 with either sa-mRNA S or sa-mRNA S-N at a dose of 1 μg RNA (Figure 2A). Half of the animals were boosted at day 22 with the same vaccines used for priming, and all animals were sacrificed at day 43. Serum was tested for antibodies neutralizing Vero E6 cell infection by homologous original virus and for antibodies inhibiting S protein binding to the ACE2 receptor. Both assays showed that immunization with one dose of either sa-mRNA S or sa-mRNA S-N generated neutralizing antibody titers. Microneutralization (MN) geometric mean titer (GMT) was 211 for sa-mRNA S and 98 for sa-mRNA S-N (Figure 2B). The GMT for ACE2-binding inhibition was 1,004 for sa-mRNA S and 941 for sa-mRNA S-N (Figure 2C). The boost dose increased MN GMT ≥10-fold to 2,774 for sa-mRNA S and 1,280 for sa-mRNA S-N. The GMTs for ACE2-binding inhibition were also increased ≥10-fold to 12,592 for sa-mRNA S and 10,791 for sa-mRNA S-N. With the same assay protocols, eight human COVID-19 convalescent plasma samples were shown to have GMTs of 199 for MN (Figure 2B) and 1,715 for ACE2-binding inhibition, comparable to the titers raised with a single dose but ∼10-fold lower than those raised with two doses of sa-mRNA S or sa-mRNA S-N.
Figure 2.

Neutralizing antibody response raised by one or two doses of sa-mRNA S and sa-mRNA S-N vaccines in BALB/c mice
(A) BALB/c female mice (n = 10) were immunized once (one dose) or twice (two doses, 3 weeks apart) with 1.0 μg of sa-mRNA S (blue) or sa-mRNA S-N (red). Serum samples collected at day 43 were tested in a live virus microneutralization (MN) assay for homologous original virus (B) or an assay on inhabitation of angiotensin-converting enzyme 2 (ACE2) binding by SARS-CoV-2 S ectodomain (Secto) protein with sequences from original virus (C). Each dot represents an individual serum sample, and the column represents the geometric mean for the group. Green dots and column (B) represent MN results for the reference convalescent plasma, eight convalescent plasma samples from CSL plasma, and a pooled plasma sample from the National Institute for Biological Standards and Control. The dotted line in each panel represents the lower limit of quantitation for the assay. Statistical analysis by two-way ANOVA with Tukey’s multiple comparison test was performed using GraphPad Prism 9.1.0. ∗p < 0.05. ∗∗p < 0.01. ∗∗∗p < 0.001. ∗∗∗∗p < 0.0001. ns, not significant (p ≥ 0.05).
To confirm the neutralizing antibody response, additional studies were performed with both BALB/c mice and C57BL/6J mice immunized at day 1 and day 22 with sa-mRNA S or sa-mRNA S-N at doses of 1.0 μg RNA or 0.01 μg RNA as well as MF59-adjuvanted recombinant S protein (Adj-Pro S) at dose of 1.0 μg protein. Tests for antibody responses confirmed a significant dose response by both sa-mRNA S and sa-mRNA S-N in both mouse species (Figures 3A and 3B). In BALB/c mice, MN GMTs against the homologous original virus were 197 for the 0.01 μg dose and 1,372 for the 1.0 μg dose of sa-mRNA S, and 149 for the 0.01 μg dose and 905 for the 1.0 μg dose of sa-mRNA S-N (Figure 3A). In C57BL/6J mice, MN GMT was 197 and 970 for the 0.01 and 1.0 μg doses of sa-mRNA S, respectively, and 98 and 735 for the 0.01 and 1.0 μg doses of sa-mRNA S-N, respectively (Figure 3B). The antibody responses with the 0.01 μg dose for both sa-mRNA S and sa-mRNA S-N were comparable to titer from human COVID-19 convalescent plasma samples (Figure 2B). Mice immunized with Adj-Pro S 1.0 μg exhibited MN GMTs of 6,756 in BALB/c mice and 4,159 in C57BL/6J mice. In addition, anti-N binding antibody was detected from sera samples raised by sa-mRNA S-N at 0.01 and 1.0 μg doses in both BALB/c (Figure S1A) and C57BL/6J (Figure S1B) mice.
Figure 3.

Cross-reactive neutralizing antibody responses raised by sa-mRNA S and sa-mRNA S-N vaccines or Adj-Pro S vaccine against original virus in BALB/C and C57BL/6J mice
BALB/c or C57BL/6J female mice (n = 10 each) were immunized twice, 3 weeks apart, with 1.0 μg or 0.01 μg of sa-mRNA S (blue) or sa-mRNA S-N vaccines (red), or 1.0 μg Adj-Pro S vaccine (green). Serum samples were collected at day 43. Sera from BALB/C (A) or C57BL/6J (B) mice were tested in MN assay for the original virus. Sera from BALB/c mice raised by sa-mRNA S (C) and sa-mRNA S-N (D) were tested additionally in MN assay for the original virus (red) and the emerging Alpha (blue), Beta (green), Gamma (purple), and Delta (gray) variants. Each dot represents an individual serum sample, and the column represents the geometric mean for the group. The dotted line in each panel represents the lower limit of quantitation for the assay. Statistical analysis by two-way ANOVA with Tukey’s multiple comparison test was performed using GraphPad Prism 9.1.0. ∗p < 0.05. ∗∗p < 0.01. ∗∗∗p < 0.001. ∗∗∗∗p < 0.0001. ns, not significant (p ≥ 0.05).
To characterize Th1 versus Th2 type immune response generated by sa-mRNA S and sa-mRNA S-N, the S-specific immunoglobulin G1 (IgG1) and IgG2 subclasses were evaluated by enzyme-linked immunosorbent assay (ELISA). In BALB/c mice immunized with 1.0 μg sa-mRNA S or sa-mRNA S-N, the production of the two IgG subclasses was well balanced, and both correlated with MN titers (Figures S2A and S2C). Conversely, for Adj-Pro S, the IgG production was highly skewed toward IgG1. In C57BL/6J mice, IgG production was IgG1-dominant for sa-mRNA S and sa-mRNA S-N and IgG2-dominant for Adj-Pro S (Figure S2B). These findings are consistent with a Th1 type response generated by the sa-mRNA vaccines in mice.
During our study, new SARS-CoV-2 variants of concern, the Alpha, Beta, Gamma, and Delta variants, emerged and started to spread, with the potential to be more infectious and virulent than the earlier pandemic viruses.19, 20, 21, 22 To evaluate the cross-reactivity of antibodies generated with sa-mRNA vaccines for the original virus, MN assays were used to evaluate inhibition of viral infection by the heterologous SARS-CoV-2 Alpha, Beta, Gamma, and Delta variants as well as the original virus. For sa-mRNA S, MN GMTs were 211 and 1,114 for the 0.01 and 1.0 μg doses against the original virus, 453 and 1,810 against the Alpha variant, 130 and 905 against the Beta variant, 197 and 905 against the Gamma variant, and 113 and 299 against the Delta variant (Figure 3C). For sa-mRNA S-N, MN GMTs were 184 and 788 for the 0.01 and 1.0 μg doses, respectively, against the original virus and 98 and 394, respectively, against the Delta variant (Figure 3D). The GMTs against the variants raised by sa-mRNA S and sa-mRNA S-N were comparable or <2-fold lower than the GMTs against the homologous original virus.
To further evaluate the cross-neutralization potential of sa-mRNA S vaccines, sa-mRNA S vaccines with S protein sequences from original virus plus the D614G mutation (614G), from the Alpha variant, and from the Beta variant were generated using the same vaccine approach for the original virus. The MN assays showed that comparable and robust GMTs against the original virus (Figure 4A), Alpha variant (Figure 4B), Beta variant (Figure 4C), and Gamma variant (Figure 4D) were raised by sa-mRNA S vaccines against 614G, the Alpha and Beta variants, as well as the original virus in BALB/c mice. The GMTs against the Delta variant (Figure 4E) by these sa-mRNA S vaccines were 2- to 3-fold lower than the GMTs against their own homologous viruses.
Figure 4.

Cross-reactive neutralizing antibody responses raised by sa-mRNA S vaccines against original virus, 614G, and Alpha and Beta variants in BALB/c mice
BALB/c female mice (n = 10) were immunized twice, 3 weeks apart, with 1.0 μg or 0.01 μg of sa-mRNA S against the original virus (blue), 614G (red), Alpha variant (green), or Beta variant (purple). Serum samples were collected at day 43. Sera were tested in MN assay for the original virus (A), Alpha variant (B), Beta variant (C), Gamma variant (D), and Delta variant (E). Each dot represents an individual serum sample, and the column represents the geometric mean for the group. The dotted line in each panel represents the lower limit of quantitation for the assay. Statistical analysis by two-way ANOVA with Tukey’s multiple comparison test was performed using GraphPad Prism 9.1.0. ∗p < 0.05. ∗∗p < 0.01. ∗∗∗p < 0.001. ∗∗∗∗p < 0.0001. ns, not significant (p ≥ 0.05).
In addition to antibody response, T cell responses in BALB/c and C57BL/6J mice raised with sa-mRNA S and sa-mRNA S-N were evaluated. Spleens from BALB/c mice were collected on day 43 and tested for S-specific and N-specific CD4+ and CD8+ T cells. Splenocyte suspensions were cultured in the presence or absence of synthetic peptide pools representing full-length S or N protein, and cultures were analyzed for cell surface markers and intracellular cytokines. Immunization with sa-mRNA S or sa-mRNA S-N resulted in both CD4+ (Figures 5A and 5C) and CD8+ (Figures 5B and 5D) T cells reactive with epitopes in the S1 and S2 domains of S protein. In addition, sa-mRNA S-N induced N-reactive CD4+ T cells (Figure 5E), although no N-reactive CD8+ T cells were detected in BALB/c mice.
Figure 5.

Antigen-specific CD4+ and CD8+ T cells raised by sa-mRNA S and sa-mRNA S-N vaccines or Adj-Pro S vaccines in BALB/c mice
BALB/c female mice (n = 10) were immunized twice, 3 weeks apart, with 1.0 μg or 0.01 μg of sa-mRNA S or sa-mRNA S-N vaccines or with 1.0 μg Adj-Pro S vaccine. Spleens were collected on day 43 and pooled (five spleens/vaccine). Splenocytes were prepared, cultured in the presence or absence of antigen peptide mixtures, and analyzed by flow cytometry. The net (antigen-specific) percentage of cytokine-producing CD4+ and CD8+ T cells induced by each vaccine are shown for S1-specific CD4+ T cells (A), S1-specific CD8+ T cells (B), S2-specific CD4+ T cells (C), S2-specific CD8+ T cells (D), and N-specific CD4+ T cells (E). Contribution of various T helper subsets to the overall CD4 response was determined as follows: Th1, CD4+IFNγ+IL-5negIL-13neg; Th2, CD4+IFNγ−IL-5+/IL-13+; and Th0, CD4+IL-2+/TNFα+.
CD4+ T cells elicited by sa-mRNA vaccines were mostly Th0 (interleukin [IL] 2+ and/or tumor necrosis factor alpha+ [TNFα+], interferon γ– [IFNγ–], IL5–, or IL13–) and Th1 (IFNγ+, IL5–, or IL13–) with few or no Th2 (IL5+ and/or IL13 + or IFNγ–). In contrast, S-specific CD4+ T cells in mice immunized with Adj-Pro S were mostly Th2 and Th0 with few or no Th1 cells (Figures 5A and 5C). These results aligned with the outcome of the IgG1 and IgG2 subclass analysis (Figure S2). Similar frequencies of S1- and S2-reactive CD4+ T cells were found. For CD8+ T cells, S1-reactive T cells dominated over S2-reactive T cells, with a broad cytokine phenotype, including triple, double, and single cytokine-producing CD8+ T cells (Figures 5B and 5D).
The T cell responses in C57BL/6J mice raised by sa-mRNA S and sa-mRNA S-N were evaluated with the same approaches, and the tests showed similar robust CD4+ and CD8+ T cell response patterns as in BALB/c mice (Figure S3). The T cell responses in BALB/c mice raised by the additional sa-mRNA S vaccines against 614G, Alpha, and Beta variants were also tested and shown with the robust S protein specific CD4+ and CD8+ T cells stimulated by these sa-mRNA vaccines (data not shown).
Protection from SARS-CoV-2 infection in hamsters
To evaluate the protective effect of immunization with sa-mRNA vaccines, hamsters were immunized with sa-mRNA S or sa-mRNA S-N at doses of 3 μg RNA or 0.3 μg RNA or Adj-Pro S at 5.0 μg on day 1 and day 22 (Figure 6A). All animals were challenged 28 days after the second immunization with SARS-CoV-2 original virus intranasally and sacrificed 4 days later, when infectious virus was measured in the lungs and nasal turbinates. MN assays on the sera collected before challenge showed GMTs as 557 and 485 against original virus, 368 and 211 against the Beta variant, and 61 and 53 against the Delta variant in hamsters by the 3.0 and 0.3 μg doses of sa-mRNA S, respectively, and were 485 and 243 against original virus, 368 and 211 against the Beta variant, and 160 and 139 against the Delta variant by 3.0 and 0.3 μg doses of sa-mRNA S-N, respectively (Figure 6B). The GMT with Adj-Pro S 5.0 μg was 422, 279, and 160 against original virus and the Beta and Delta variant, respectively.
Figure 6.

Cross-reactive neutralizing antibody response and challenge virus recovery in hamsters immunized with sa-mRNA S and sa-mRNA S-N vaccines or Adj-Pro S vaccine
(A) Female Syrian hamsters (n = 5) were immunized twice, 3 weeks apart, with 3.0 μg or 0.3 μg of sa-mRNA S (blue) or the sa-mRNA S-N (red) or with 5.0 μg Adj-Pro S (green). Hamsters were challenged 4 weeks after the second dose with live SARS-CoV-2 original virus at 100 50% tissue culture infectious dose (TCID50) per animal and sacrificed 4 days later. Lungs and nasal turbinates were collected for recovery of infectious virus. (B) Serum samples collected at time of challenge were tested in MN assay for original virus, Beta, and Delta variants. Each dot represents an individual serum sample, and the column represents the geometric mean for the group. The dotted line in each panel represents the lower limit of quantitation for the assay. Total virus recovery from lung (C) and nasal turbinates (D) expressed as TCID50/g of tissue. Each dot represents an individual sample, and the line the geometric mean for the group. The dotted line in each panel represents the lower limit of quantitation for the assay. Statistical analysis by two-way ANOVA with Tukey’s multiple comparison test was performed using GraphPad Prism 9.1.0. ∗p < 0.05. ∗∗p < 0.01. ∗∗∗p < 0.001. ∗∗∗∗p < 0.0001. ns, not significant (p ≥ 0.05).
To evaluate protection from viral infection, average virus recovery was compared between hamsters immunized with sa-mRNA S, sa-mRNA S-N, and Adj-Pro S and control hamsters immunized with phosphate-buffered saline (PBS). In the lung, the viral titer from control hamsters was 5,011,872 50% tissue culture infectious dose per gram (TCID50/g) and from vaccine-immunized hamsters was <20 TCID50/g, under the limit of quantitation of the assay and demonstrating full lower respiratory tract protection with all three vaccines (Figure 6C). In the upper respiratory tract (Figure 6D), virus recovery from nasal turbinates was 120,226,443 TCID50/g in control hamsters. In immunized hamsters, viral titers were reduced ∼104-fold, 1,995 and 9,120 TCID50/g for the 3.0 and 0.3 μg doses of sa-mRNA S, respectively, and 14,454 and 21,878 TCID50/g for the 3.0 and 0.3 μg doses of sa-mRNA S-N, respectively. The viral recovery from Adj-Pro S-immunized hamsters was reduced ∼102-fold, 1,000,000 TCID50/g, although the neutralizing antibody titers raised by Adj-Pro S were comparable to 0.3 μg of sa-mRNA S and sa-mRNA S-N (Figure 5B). Immunization also reduced the loss of body weight for hamsters caused by SARS-CoV-2 virus infection (Figure S4).
Discussion
The sa-mRNA technology described herein enables rapid vaccine production to meet the needs of pandemics such as COVID-19. sa-mRNA COVID-19 vaccines have been developed and advanced to preclinical or clinical stages, demonstrating the promise of the technology.16, 17, 18 As host cells produce antigen from the sa-mRNA, the proteins maintain the native, immunogenic, prefusion S protein conformation. This process places less stress on the protein, unlike the purification process normally required in standard protein vaccine manufacturing. The sa-mRNA S vaccine using the wild-type S protein sequence raised neutralizing antibody titers comparable to those of sa-mRNA S stabilized with additional prefusion mutations (Figure S5) that are essential for recombinant protein vaccine.5 This finding suggests that sa-mRNA vaccines can produce well-folded protein without modification to the antigen sequence, which itself may introduce non-natural epitopes.
In this study, a single dose at 1 μg or two doses at 0.01 μg each of either sa-mRNA SARS-CoV-2 vaccine (sa-mRNA S or sa-mRNA S-N) elicited antibody titers against the homologous original virus that were comparable to those from convalescent patient plasma. Neutralizing antibody responses were robust against not only homologous original virus but also the Alpha, Beta, Gamma, and Delta variants. In particular, we observed reduced but robust neutralizing antibody response, especially by sa-mRNA S-N vaccine, against the Delta virus, which differs antigenically from the original strain and has been resistant to neutralizing antibodies generated from convalescent plasma.19,21,22
In addition to neutralizing antibodies, sa-mRNA SARS-CoV-2 vaccines also elicited antigen-specific cellular-mediated immune responses. A Th1-dominant CD4+ cell response was elicited to the S and N proteins. The CD8+ T cell response to the S protein was significantly higher in mice immunized with sa-mRNA than with MF59-adjuvanted S protein. In the hamster challenge, sa-mRNA and adjuvanted protein vaccines raised comparable levels of neutralizing titers. Virus recovered in the upper respiratory tract was >100-fold lower in hamsters immunized with sa-mRNA vaccines than those receiving the adjuvanted protein vaccine. The CD8+ responses raised by sa-mRNA vaccines may have contributed to the superior protection we observed.
Strong anti-N antibodies (Figure S1) and CD4+ cells (Figure 5E) were raised with sa-mRNA S-N vaccines in the immunized mice, although no CD8+ cells were detected, likely due the limitation of the mouse models. In the hamster challenge study, no additional protection against the original viruses from N protein was observed when sa-mRNA S and sa-mRNA S-N responses were compared. However, microneutralization titers against the Delta variant were improved by sa-mRNA S-N. Additional studies are required to evaluate N protein as the additional SARS-CoV-2 vaccine antigen.
In conclusion, both sa-mRNA SARS-CoV-2 vaccines demonstrate robust humoral and cellular response in mice and conferred protection in a hamster challenge model. Relatively low doses elicited high levels of cross-neutralization against SARS-CoV-2 variants, suggesting that these vaccines warrant further evaluation in humans. Given the continued emergence and global spread of SARS-CoV-2 variants, along with the ever-present potential for novel pandemic pathogen emergence, this vaccine platform could serve a vital role in pandemic preparedness and response efforts.
Materials and methods
Generation of sa-mRNA
S and N coding DNA sequences (CDS) were codon optimized by GenScript based on amino acid sequences of SARS-CoV-2/human/USA/WA-CDC-WA1/2020 (GenBank MN985325.1), and S1/S2 cleavage sites of S protein were mutated to QQAA to stabilize S in the prefusion conformation for the sa-mRNA vaccines for the original virus. Two proline mutations at residues 986 and 987 were introduced by site-directed mutagenesis.5 For sa-mRNA S, S CDS was cloned after the subgenomic promoter (SGP) of Venezuelan equine encephalitis and Sindbis virus replicon chimera (VEE-SINV). For sa-mRNA S-N, N CDS was cloned after a minimal SGP, which is at the 3′ end of the S gene. For sa-mRNA S 614G (D614G mutation on the prefusion-stabilized WA S gene), sa-mRNA S Alpha variant (England/204820464/2020), and sa-mRNA S Beta variant (South Africa/KRISP-K005325/2020), the cloning approaches were the same as that for original virus.
RNA was prepared as previous reported.10 Briefly, the plasmid DNA template was linearized at the last base of 3′ end poly-A tail of the sa-mRNA sequence with BspQ1 (New England BioLabs) and purified by phenol-chloroform. Linearized DNA templates were enzymatically transcribed into RNA with T7 RNA polymerase (New England Biolabs, Ipswich, Massachusetts) using unmodified nucleosides, followed by digestion with Turbo DNAse (Life Technologies, Carlsbad, California) to remove template DNA, and capped using a Vaccinia capping system (New England BioLabs). RNA from transcription/capping reaction was purified by tangential flow filtration (TFF) and frozen at −80°C.
sa-mRNA/LNP formulation
RNA in citrate buffer was formulated into LNPs using a proprietary ionizable lipid, 1,2-diastearoylsn-glycero-3-phosphocholine (DSPC; Avanti Polar Lipid), 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (PEG-DMG 2000; NOF America Corporation, White Plains, New York), and cholesterol (Sigma-Aldrich, St. Louis, Missouri) dissolved in ethanol through a NanoAssemblr mixing instrument (Precision Nanosystems, Vancouver, Canada). The nanoparticles were buffer exchanged into Tris buffer with NaCl and sucrose by TFF, sterile filtered, and stored at −80°C.
Expression and purification of SARS-CoV-2 Secto and hACE2-Fc proteins
A construct for SARS-CoV-2 S protein (National Center for Biotechnology Information reference sequence YP_009724390.1) containing S ectodomain (Secto, residues 1–1,213) with a QQAA substitution at the S1/S2 cleavage site (residues 682–685), followed a C-terminal T4 fibritin trimerization motif and a strep tag, was codon optimized, synthesized, and cloned into the mammalian expression vector by GenScript. Two proline mutations at residues 986 and 987 were introduced by site-directed mutagenesis.5
The DNA plasmid was transfected into ExpiCHO cells (ThermoFisher Scientific, Waltham, Massachusetts) using Expifectamine, and on day 5 post transfection, cells were harvested and filtered using a 0.45-μm vacuum filtration system (Nalgene, ThermoFisher Scientific). To purify Secto, the supernatant containing secreted Secto was buffer exchanged by TFF and loaded onto Strep tactin column (GE Healthcare, Chicago, Illinois) using AKTA Pure (Cytiva, Hyclone Laboratories, Logan, Utah). Secto eluted from Strep column was concentrated and further purified by size exclusion chromatography with a superpose 6 10/300 GL column (Cytiva). For hACE2-Fc protein, the DNA plasmid containing codon-optimized hACE2 residues (1–615) with Fc was transfected into Expi293 (ThermoFisher) and purified with HiTrap Q HP column (Cytiva) using AKTA Pure. The protein was concentrated and further purified by size exclusion chromatography with superdex 200 10/300 GL (Cytiva).
Flow cytometry and western blot
BHK cells (American Type Culture Collection [ATCC], Manassas, Virginia) were cultured in Dulbecco’s modified Eagle’s medium [DMEM] (Gibco, ThermoFisher Scientific) containing 5% fetal bovine serum (Hyclone Laboratories) at 37°C and 5% CO2. LNP-formulated sa-mRNA S and sa-mRNA S-N were diluted in OptiMem (Life Technologies) and transfected into BHK cells. Transfections were incubated overnight at 37°C and 5% CO2.
Cells were collected, fixed, and permeabilized with Cytofix/Cytoperm (BD Biosciences, San Jose, California) and then stained with AF-647 conjugated rabbit anti-SARS-1-spike (ProSci, Poway, California) and/or AF-488 conjugated rabbit anti-SARS-1-NP antibodies (Sino Biological, Beijing, China). Frequencies of S- or N-protein-positive cells were enumerated by flow cytometry using a LSRFortessa flow cytometer (BD Biosciences) and analyzed using FlowJo software (BD Biosciences).
Cell lysates were separated with 10% SDS-PAGE (Invitrogen, Carlsbad, California) and transferred onto a Nitrocellulose Membrane (Invitrogen). The membrane was probed with rabbit polyclonal antibody against SARS-CoV-2 S protein (ThermoFisher) and against N protein (VWR International, Radnor, Pennsylvania) and mouse monoclonal antibody against glyceraldehyde 3-phosphate dehydrogenase (GAPDH; GeneTex, Irvine, California). Proteins were detected with secondary anti-rabbit antibodies IRdye 800CW and IRdye 680RD (Li-COR Biosciences, Lincoln, Nebraska) and anti-mouse antibody IRDye800CW (Li-COR Biosciences) and visualized using Li-Cor Odyssey (Li-COR Biosciences).
Mouse immunogenicity studies
The mouse studies were conducted at Biomodels (Waltham, Massachusetts). Female BALB/c and C57BL/6J mice, 8–10 weeks old, maintained at Biomodels, Waltham, MA, were immunized (10 mice/group) with bilateral 50 μL intramuscular injections in the rear quadriceps on days 1 and 22. To evaluate antibody response, serum samples were obtained by retro-orbital sinus bleeds on day 21 and from bleed-outs of euthanized animals on day 43. To evaluate cell-mediated immunity, spleens were removed from each animal immediately after euthanasia.
Prior to bleeding, mice were anesthetized with 2.5% isoflurane. Mice were euthanized by exsanguination under anesthesia. Euthanasia was confirmed by cervical dislocation. All experiments were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 8023, revised 1978).
Propagation and titration of SARS-CoV-2 variants
SARS-CoV-2 isolates were provided by BEI Resources (Manassas, Virginia), USA-WA1/2020 (BEI RESOURCES/ATCC# NR-52281), England/204820464/2020 (Alpha variant, BEI RESOURCES/ATCC# NR-54000), South Africa/KRISP-K005325/2020 (Beta variant, BEI RESOURCES/ATCC# NR-54009), Japan/TY7-503/2021 (Gamma variant, BEI RESOURCES/ATCC# NR-54982), and USA/PHC658/2021 (Delta variant, BEI RESOURCES/ATCC# NR-55611).
Working stocks were propagated under biosafety level 3 (BSL3) conditions using the Vero E6 cells (Vero 76, clone E6, ATCC CRL-1586). Hundred times diluted seed stocks in fetal bovine serum (FBS)-free media, DMEM (Gibco), were added to cell monolayers in T225 flasks and incubated for 1 h at 37°C and 5% CO2. The viruses were removed and replaced with infective media, DMEM containing 2% of heat-inactivated FBS (HI-FBS, Gibco). The cells were incubated at 37°C and 5% CO2 and observed daily for the presence of cytopathic effect (CPE). Viruses were harvested when 70%–80% of the cells manifested CPE and were stored in cryovials at −80°C. Generated virus stocks were titrated by plaque assays using 0.6% microcrystalline cellulose (Sigma-Aldrich, St. Louis, Missouri) as described recently.23 Working stocks were characterized by obtaining a 50% tissue culture infective dose (TCID50) as described elsewhere.24 A series of 10-fold serial dilutions were transferred on 96-well cultured plates of Vero E6 cells, and cells were observed daily for a total of 4 days for the presence of CPE by means of an inverted optical microscope. The endpoint titers were calculated according to the Reed-Muench method based on eight replicates for titration.
Microneutralization assay
A CPE-based virus microneutralization test was performed in a 96-well format, using Vero-E6 cells. 2-fold serial dilutions of previously heat-treated sera (1 h at 56°C) were mixed with an equal volume of viral solution containing 100 TCID50 (diluted in FBS-free DMEM), then incubated for 1 h at 37°C and 5% CO2. Serum-virus mixture was transferred in quantities of 100 μL to each well of rows specific to the confluent cell monolayer and incubated for 1 additional hour at 37°C and 5% CO2. Another 100 μL of infective media (4% FBS supplemented DMEM) was added to each well, and plates were incubated for 4 days at 37°C in a humidified atmosphere with 5% CO2. Each series included positive-control wells containing cells with virus only and negative-control wells containing cells only. After 4 days, the plates were examined for the presence (no neutralization) or absence (neutralization) of CPE using an inverted microscope under the BSL3 practice (EVOS XL Core Cell Imaging System; Life Technology). The highest serum dilution that protected more than 50% of cells from CPE was taken as the microneutralization titer. After collecting data via microscope, the same cells were fixed (acetone: methanol) then stained with 1% of crystal violet (Electron Microscopy Sciences, Hatfield, Pennsylvania). Scanned plates were served for confirming microneutralization titers with the second readout method and for preserving data appropriately. For reference, multiple convalescent plasma samples provided by CSL Limited (Melbourne, Australia) were tested, as well as a pooled plasma sample from the National Institute for Biological Standards and Control (NIBSC, code # 20/150, part of the First World Health Organization International Reference Panel, NIBSC code # 20/268).
ACE2-binding inhibition assay
Inhibition of ACE2 binding was assayed using 384-well assay plates (Grenier Bio-One, Monroe, North Carolina), coated overnight with recombinant hACE2-Fc, washed three times with wash buffer (0.05% Tween 20 in PBS), and blocked with assay buffer (1% BSA and 0.1% Triton X-100 in PBS) for >30 min at room temperature (RT). Sera were diluted to a starting dilution of 1:10 with PBS and heat inactivated for 1 h at 56°C. In 384-well incubation plates (ThermoFisher Scientific), sera were serially diluted in assay buffer and incubated with equal volume of SARS-CoV-2 Secto in assay buffer. After 1 h at RT, blocking was removed from assay plates, and the serum-protein mixture was transferred to the assay plate and incubated for 1 h at RT. Plates were washed three times with wash buffer, and the binding of S protein to ACE2 was detected using horseradish peroxidase (HRP)-conjugated Anti-Strep-Tag antibodies (MilliporeSigma, Burlington, Massachusetts) for 1 h at RT. Plates were again washed three times with wash buffer, and the enzyme substrate was added (TMB substrate, Rockland Immunochemicals, Limerick, Pennsylvania) and incubated for 30 min at RT. The enzymatic reaction was stopped with stop solution (2N sulfuric acid, BDH) and the optical density (OD) at a wavelength of 405 nm was measured using a plate reader (NanoQuant Plate Infinite M200, Tecan Group, Männedorf, Switzerland). For each sample, OD values were plotted against the sample dilution using GraphPad Prism 9 software; a 4PL regression line was interpolated, and the half maximal effective concentration (EC50) was estimated. The inhibition titer was calculated as the inverse of EC50, i.e., the dilution of the sample that generated 50% reduction of OD.
ELISA for IgG to S protein or N protein
Sera were diluted to a starting dilution of 1:1,000 with PBS and heat inactivated for 1 h at 56°C. High-binding 384-well ELISA plates (Grenier Bio-One) were coated overnight with SARS-CoV-2 Secto at concentrations of 1 μg/mL in PBS or SARS-CoV-2 N protein (GeneTex cat#GTX135357). Plates were washed three times with wash buffer (0.05% Tween 20 in PBS) and blocked with assay buffer (1% BSA and 0.1% Triton X-100 in PBS) for 30 min at RT. The buffer was removed, and sera were serially diluted in assay buffer. After 1 h of incubation at RT, plates were washed three times with wash buffer, and the secondary detection antibody (HRP-conjugated goat anti-mouse IgG [H + L; Jackson ImmunoResearch], or HRP-conjugated goat anti-mouse immunoglobulin G1 [IgG1] and HRP-conjugated goat anti-mouse IgG2a [SouthernBiotech, Birmingham, Alabama]), diluted in assay buffer, was added, and the plates were incubated again for 1 h at RT. Plates were washed three more times with wash buffer, and the enzyme substrate was added (TMB substrate, Rockland Immunochemicals) and incubated for 30 min at RT. The enzymatic reaction was stopped with 50 μL/well of stop solution (2N sulfuric acid), and the OD at wavelength 405 nm was measured using a plate reader (NanoQuant Plate Infinite M200, Tecan Group Ltd.). For each sample, OD values were plotted against the sample dilution using GraphPad Prism 9 software; a 4PL regression line was interpolated; and the EC50 was estimated. The ELISA titer was calculated as the inverse of the EC50, i.e., the dilution of the sample that generated 50% reduction of OD.
Antigen-specific T cell detection
Five spleens per vaccine group were pooled together in gentleMACS C Tubes (Miltenyi Biotec, Bergisch Gladbach, Germany), and a spleen dissociation was run using a gentleMACS Dissociator (Miltenyi Biotec) in accordance with the manufacturer’s instructions to obtain single-cell suspensions for detection of antigen-specific T cells. Cells were plated at 2 × 106 per well in the presence of anti-CD28 (BD Biosciences), anti-CD107a BV421 (BioLegend, San Diego, California), and peptides. Peptide pools, consisting of a mix of peptides (15mers with 11-aa overlap) spanning the entire SARS-CoV-2 S and N protein sequence with >70% purity (JPT Peptide Technologies, Berlin, Germany) were used to detect S- or N-specific T cells. Unstimulated controls that had no peptide mixes were included. After 2 h, GolgiPlug (BD Biosciences) containing Brefeldin A was added for a total peptide stimulation of 6 h, and then cells were placed at 4°C overnight. The following day, cells were stained with a LIVE/DEAD fixable aqua dead cell stain kit (Invitrogen) for 30 min, washed, and incubated with Fc Block, anti-CD16/32 (BD Biosciences) for 10 min. Surface antigens were stained with anti-CD3 BV786 (BD Biosciences), anti-CD4 APC-H7 (BD Biosciences), anti-CD8 Alexa Fluor 700 (BD Biosciences), anti-CD44 BUV395 (BD Biosciences), and anti-CD278 BV711 (BioLegend) for an additional 30 min. Following surface staining, intracellular cytokines were detected by fixation for 20 min with Cytofix/Cytoperm (BD Biosciences), and then cells were stained with anti-interferon-γ (IFN-γ) PerCP-Cy5.5 (eBiosciences), anti-tumor necrosis factor (TNF) Alexa Fluor 488 (BD Biosciences), anti-IL-2 BV605 (BD Biosciences), anti-IL-5 APC (eBiosciences), and anti-IL-13 PE (eBiosciences) in perm buffer for 30 min. Cells were washed and analyzed on the BD LSRII flow cytometer (BD Biosciences). Data analysis was performed using the FlowJo software followed by the determination of the net percentage of antigen-specific T cells for either CD8 or CD4, a measure of the difference between stimulated and unstimulated cultures. Contribution of various T helper subsets to the overall CD4 response was determined as follows: Th1, CD4+IFNγ+IL-5negIL-13neg; Th2, CD4+IFNγ−IL-5+/IL-13+; and Th0, CD4+IL-2+/TNFα+. The 95% confidence limits for the percentage of antigen-specific cells were calculated using standard statistical methods.
Hamster challenge study
The hamster challenge study was approved by the Central Authority for Scientific Procedures on Animals (Centrale Commissie Dierproeven) and conducted in accordance with the European guidelines (EU directive on animal testing 86/609/EEC) and local Dutch legislation on animal experiments. The in-life phase took place at Viroclinics Biosciences BV, Viroclinics Xplore, Schaijk, the Netherlands. Female Syrian hamsters, 9–10 weeks old, were immunized twice, under isoflurane anesthesia, 3 weeks apart, at day 1 and 22, and challenged at day 50, 4 weeks after the second immunization, by intranasal infection with 100 TCID50 per dose of SARS-CoV-2 (BetaCoV/Munich/BavPat1/2020) in a total dose volume of 0.1 mL. Animals were followed for 4 days post challenge and sacrificed by cervical dislocation, under anesthesia, on day 54.
Detectable levels of replication-competent virus in lung and nasal turbinate tissues post challenge were analyzed. Quadruplicate, 10-fold serial dilutions were transferred to 96-well plates with Vero E6 cell culture monolayers and incubated for 1 h at 37°C. Cell monolayers were washed prior to incubation for 5 days at 37°C. Plates were then scored using the vitality marker WST8, and viral titers (Log10 TCID50/mL or per g) were calculated by the Spearman-Karber method.
Acknowledgments
We thank Viroclinics Xplore for performing hamster challenge study and protection analysis, and Seqirus Research members, Sukhmani Bedi, Harsh Patel, Yuhong Xie, Annette Ferrari, and Ghazal Hariri for technical support. Editorial support was provided by C. Gordon Beck and Amanda M. Justice and was funded by Seqirus, Inc.
Author contributions
Conceptualization, Y.W., G.P., P.S., G.R.O., and E.C.S.; Methodology, Y.W., G.P., C.C., C.L., N.M., G.R.O., and E.C.S.; Investigation, C.C., C.L., I.D., J.N., N.M., S.A., and G.P.; Writing – original draft, Y.W., G.P., C.C., C.L., N.M., S.A., G.R.O., and E.C.S.; Writing – review & editing: G.P., E.C.S., and Y.W.
Declaration of interests
All authors are employees of Seqirus Inc., which funded this work.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2022.03.013.
Supplemental information
References
- 1.Lu R., Zhao X., Li J., Niu P., Yang B., Wu H., Wang W., Song H., Huang B., Zhu N., et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565–574. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu F., Zhao S., Yu B., Chen Y.M., Wang W., Song Z.G., Hu Y., Tao Z.W., Tian J.H., Pei Y.Y., et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Du L., He Y., Zhou Y., Liu S., Zheng B.J., Jiang S. The spike protein of SARS-CoV--a target for vaccine and therapeutic development. Nat. Rev. Microbiol. 2009;7:226–236. doi: 10.1038/nrmicro2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dai L., Gao G.F. Viral targets for vaccines against COVID-19. Nat. Rev. Immunol. 2021;21:73–82. doi: 10.1038/s41577-020-00480-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wrapp D., Wang N., Corbett K.S., Goldsmith J.A., Hsieh C.L., Abiona O., Graham B.S., McLellan J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367:1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cai Y., Zhang J., Xiao T., Peng H., Sterling S.M., Walsh R.M., Jr., Rawson S., Rits-Volloch S., Chen B. Distinct conformational states of SARS-CoV-2 spike protein. Science. 2020;369:1586–1592. doi: 10.1126/science.abd4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pallesen J., Wang N., Corbett K.S., Wrapp D., Kirchdoerfer R.N., Turner H.L., Cottrell C.A., Becker M.M., Wang L., Shi W., et al. Immunogenicity and structures of a rationally designed prefusion MERS-CoV spike antigen. Proc. Natl. Acad. Sci. U S A. 2017;114:E7348–E7357. doi: 10.1073/pnas.1707304114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sattler A., Angermair S., Stockmann H., Heim K.M., Khadzhynov D., Treskatsch S., Halleck F., Kreis M.E., Kotsch K. SARS-CoV-2-specific T cell responses and correlations with COVID-19 patient predisposition. J. Clin. Invest. 2020;130:6477–6489. doi: 10.1172/JCI140965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grifoni A., Weiskopf D., Ramirez S.I., Mateus J., Dan J.M., Moderbacher C.R., Rawlings S.A., Sutherland A., Premkumar L., Jadi R.S., et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in humans with COVID-19 disease and unexposed individuals. Cell. 2020;181:1489–1501. doi: 10.1016/j.cell.2020.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Geall A.J., Verma A., Otten G.R., Shaw C.A., Hekele A., Banerjee K., Cu Y., Beard C.W., Brito L.A., Krucker T., et al. Nonviral delivery of self-amplifying RNA vaccines. Proc. Natl. Acad. Sci. U S A. 2012;109:14604–14609. doi: 10.1073/pnas.1209367109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vogel A.B., Lambert L., Kinnear E., Busse D., Erbar S., Reuter K.C., Wicke L., Perkovic M., Beissert T., Haas H., et al. Self-amplifying RNA vaccines give equivalent protection against influenza to mrna vaccines but at much lower doses. Mol. Ther. 2018;26:446–455. doi: 10.1016/j.ymthe.2017.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huysmans H., Zhong Z., De Temmerman J., Mui B.L., Tam Y.K., Mc Cafferty S., Gitsels A., Vanrompay D., Sanders N.N. Expression kinetics and innate immune response after electroporation and LNP-mediated delivery of a self-amplifying mRNA in the skin. Mol. Ther. Nucleic Acids. 2019;17:867–878. doi: 10.1016/j.omtn.2019.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kis Z., Kontoravdi C., Shattock R., Shah N. Resources, production scales and time required for producing RNA vaccines for the global pandemic demand. Vaccines (Basel) 2020;9:3. doi: 10.3390/vaccines9010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hekele A., Bertholet S., Archer J., Gibson D.G., Palladino G., Brito L.A., Otten G.R., Brazzoli M., Buccato S., Bonci A., et al. Rapidly produced SAM® vaccine against H7N9 influenza is immunogenic in mice. Emerg. Microbes Infect. 2013;2:e52. doi: 10.1038/emi.2013.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Magini D., Giovani C., Mangiavacchi S., Maccari S., Cecchi R., Ulmer J.B., De Gregorio E., Geall A.J., Brazzoli M., Bertholet S. Self-amplifying mRNA vaccines expressing multiple conserved influenza antigens confer protection against homologous and heterosubtypic viral challenge. PLoS One. 2016;11:e0161193. doi: 10.1371/journal.pone.0161193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Alwis R., Gan E.S., Chen S., Leong Y.S., Tan H.C., Zhang S.L., Yau C., Low J.G.H., Kalimuddin S., Matsuda D., et al. A single dose of self-transcribing and replicating RNA-based SARS-CoV-2 vaccine produces protective adaptive immunity in mice. Mol. Ther. 2021;29:1970–1983. doi: 10.1016/j.ymthe.2021.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McKay P.F., Hu K., Blakney A.K., Samnuan K., Brown J.C., Penn R., Zhou J., Bouton C.R., Rogers P., Polra K., et al. Self-amplifying RNA SARS-CoV-2 lipid nanoparticle vaccine candidate induces high neutralizing antibody titers in mice. Nat. Commun. 2020;11:3523. doi: 10.1038/s41467-020-17409-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pollock K.M., Cheeseman H.M., Szubert A.J., Libri V., Boffito M., Owen D., Bern H., O'Hara J., McFarlane L.R., Lemm N.M., et al. Safety and immunogenicity of a self-amplifying RNA vaccine against COVID-19: COVAC1, a phase I, dose-ranging trial. EClinicalMedicine. 2022;44:101262. doi: 10.1016/j.eclinm.2021.101262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tao K., Tzou P.L., Nouhin J., Gupta R.K., de Oliveira T., Kosakovsky Pond S.L., Fera D., Shafer R.W. The biological and clinical significance of emerging SARS-CoV-2 variants. Nat. Rev. Genet. 2021;22:757–773. doi: 10.1038/s41576-021-00408-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cai Y., Zhang J., Xiao T., Lavine C.L., Rawson S., Peng H., Zhu H., Anand K., Tong P., Gautam A., et al. Structural basis for enhanced infectivity and immune evasion of SARS-CoV-2 variants. Science. 2021;373:642–648. doi: 10.1126/science.abi9745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gupta R.K. Will SARS-CoV-2 variants of concern affect the promise of vaccines? Nat. Rev. Immunol. 2021;21:340–341. doi: 10.1038/s41577-021-00556-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harvey W.T., Carabelli A.M., Jackson B., Gupta R.K., Thomson E.C., Harrison E.M., Ludden C., Reeve R., Rambaut A., Peacock S.J., Robertson D.L. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021;19:409–424. doi: 10.1038/s41579-021-00573-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jureka A.S., Silvas J.A., Basler C.F. Propagation, inactivation, and safety testing of SARS-CoV-2. Viruses. 2020;12:622. doi: 10.3390/v12060622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Manenti A., Maggetti M., Casa E., Martinuzzi D., Torelli A., Trombetta C.M., Marchi S., Montomoli E. Evaluation of SARS-CoV-2 neutralizing antibodies using a CPE-based colorimetric live virus micro-neutralization assay in human serum samples. J. Med. Virol. 2020;92:2096–2104. doi: 10.1002/jmv.25986. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.