

ABSTRACT
Macroautophagy/autophagy is an evolutionarily conserved catabolic pathway required to maintain cellular homeostasis. In cancer, the tumor cell-intrinsic effects of autophagy are highly context specific, which could promote cancer cell survival or induce programmed cell death. Here, we reveal that OLR1/LOX-1 (oxidized low density lipoprotein receptor 1), a scavenger receptor highly expressed in esophageal cancer cells, is involved in tumorigenesis by suppressing autophagic cell death. Mechanistically, OLR1 binding to RACK1 activates MAP2K/MEK-MAPK/ERK signaling leading to TFEB (transcription factor EB) being trapped outside the nucleus and inhibiting autophagy. In addition, we identify a polysaccharide which causes the degradation of OLR1 and suppresses this autophagic pathway to inhibit tumorigenesis. This study demonstrates novel molecular mechanisms underlying the tumor-suppressive effect of autophagy and provides therapeutic insight for esophageal cancer.
KEYWORDS: Cell death, ERK, fucoidan, RACK1, TFEB, tumorigenesis
Basal autophagy is a natural cellular process responsible for maintaining cell homeostasis by capturing and engulfing intracellular components including protein aggregates or organelles for delivery to the lysosome. The dysfunction of autophagy is involved in pathogenicity and progression in multiple diseases. The role of autophagy in cancer is very complex, which depends on different contexts including cancer type, driving oncogenes or tumor suppressor genes. OLR1/LOX-1 was first identified as a receptor of oxidized LDL (ox-LDL) expressed in endothelial cells for mediating uptake of ox-LDL. Under physiological conditions, vascular endothelial cells can promote lysosomal degradation of ox-LDL by activating autophagy and resist the damage induced by ox-LDL. When the accumulation of ox-LDL exceeds the capacity of cell metabolism excessive autophagy occurs in endothelial cells leading to cell death and atherosclerosis. Here, we describe a previously unrecognized autophagic modulation by OLR1 in esophageal cancer (EC). (Figure 1)
Figure 1.
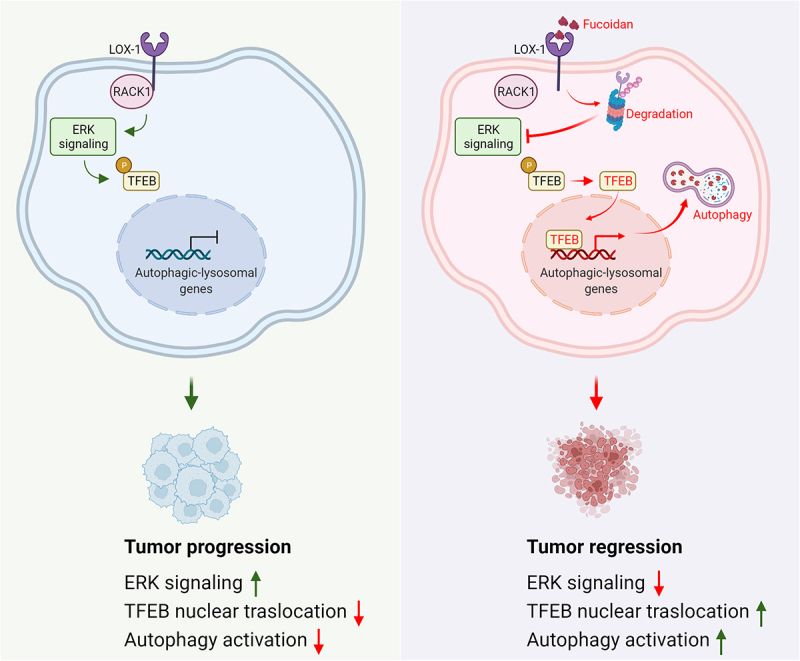
A model depicting roles of targeting LOX-1 in regulating autophagic program in esophageal cancer.
In our recent study [1], by analyzing the mRNA expression of OLR1 in multiple different kinds of tumors, we found OLR1 expression in EC is upregulated the most and predicts a poor prognosis in patients with EC. To know if OLR1 would function as an oncogene in EC development, we transfected siRNA targeting OLR1 into EC cell lines and found that knockdown of OLR1 specifically inhibits cell viability and colony formation. In addition, OLR1 deficiency dramatically restrains tumor development and prolongs the survival time of 4-nitroquinoline 1-oxide/4-NQO-induced EC mice, suggesting that OLR1 expressed in EC cells plays a critical role in supporting EC cell growth.
We next wanted to understand how OLR1 affects EC cell growth by assessing cell proliferation and cell death. Oddly, flow cytometry analysis demonstrated that OLR1 knockdown has no effects on cell cycle arrest, indicating the loss of OLR1-induced prevention of EC cell growth is not because of decreased cell proliferation but possibly because of accelerated cell death. Further by treating with different inhibitors, we found that only the autophagy inhibitor 3-methyladenine but not the apoptosis inhibitor ZVAD-FMK or the necrosis inhibitor necrostatin-1 prevent OLR1-deletion induced cell viability decline, suggesting that the activation of autophagy is involved in OLR1-knockdown induced EC cell death. By using transmission electron microscopy (TEM), we observed morphological characteristics of autophagy and further confirmed that autophagy is activated in the OLR1 knockdown cancer cells via detecting LC3/Atg8 and SQSTM1/p62 expression levels. As LC3-II accumulation is attributed to either increased autophagosome formation or impaired autophagosome-lysosome fusion, we next measured the rate of delivery of autophagosomes to lysosomes using an mCherry-GFP-LC3 reporter system and found autophagic flux is significantly increased in OLR1 knockdown EC cells. Thus, these findings suggest that the knockdown of OLR1 induces activation of autophagy that can contribute to EC suppression.
The expression of OLR1 is negatively associated with several autophagy-lysosome signature genes, which prompted us to explore whether TFEB, a master regulator of lysosomal biogenesis and autophagy, is involved in OLR1-knockdown induced autophagy activation. TFEB is phosphorylated and retained in the cytoplasm in an inactivated status, whereas it translocates into the nucleus after dephosphorylation to initiate gene transcription. Surprisingly, we found that OLR1 knockdown induces serine dephosphorylation of TFEB and increases its nuclear distribution in the EC cells. Remarkably, the activation of autophagy and EC cell death induced by OLR1 knockdown are prevented by a phosphomimetic TFEBS142D mutant. Therefore, these data demonstrate that knockdown of OLR1 induces autophagy-associated EC cell death through inducing dephosphorylation of TFEB S142 to promote TFEB nuclear localization.
OLR1, as a pattern recognition receptor, mainly induces intracellular signaling by a phosphorylation cascade that regulates the expression of a series of effector genes. For clarifying the molecular mechanism of OLR1 in EC, we conducted a phosphokinase antibody array and found that the MAPK1/ERK2-MAPK3/ERK1 (p-T202/Y204) level is significantly decreased in OLR1-knockdown EC cells. In parallel, constitutively activated MAPK1/ERK2 (caMAPK1) abolishes OLR1-deficiency induced TFEB nuclear-translocation and cell death, and pharmacological inhibition of MAPK1-MAPK3 impede OLR1-overexpression induced increase of tumor growth in vivo. As the cytoplasmic domain of OLR1 is very short with only 36 amino acid and lacks classical signal transduction motifs, we hypothesized that OLR1 might require a key cooperator for mediating downstream signaling. By mass spectroscopy, we identified an OLR1-binding partner, the scaffold protein RACK1 (receptor for activated C kinase 1). Importantly, knockdown of RACK1 blocks the activation of MAP2K/MEK-MAPK/ERK signaling, phosphorylation of TFEB, and inhibition of autophagy induced by OLR1 overexpression. Therefore, inhibitors of the MAP2K/MEK-MAPK/ERK pathway may serve as an attractive therapeutic strategy for OLR1-driven EC.
Considering the critical role of OLR1 in esophageal carcinogenesis, we sought to look for a strategy for inhibiting OLR1 signaling as a potential anticancer approach for EC therapy. Fucoidan, a sulfated polysaccharide from marine brown algae, directly binds to OLR1 and induces its proteasomal degradation. By administering fucoidan to the wild-type EC and OLR1 knockout EC model, we found that fucoidan dramatically inhibits EC growth in vitro and in vivo, and such suppressive effect is reduced in the condition of OLR1 deficiency. These results suggest that genetic ablation of OLR1 or the use of a bioactive polysaccharide targeting OLR1 would be promising interventions for EC.
In conclusion, our research uncovered that OLR1 coupled with the intracellular adaptor protein RACK1 promote RAS-MAP2K/MEK-MAPK/ERK signaling for TFEB-dependent autophagy inhibition and EC promotion (Figure 1). This route can be blocked by the sulfated polysaccharide fucoidan, which we confirmed as an intervention strategy to inhibit esophageal tumorigenesis via reducing the stability of OLR1 by ubiquitination-dependent degradation. OLR1 in human endothelial cells is upregulated by ox-LDL and induces NFKB (nuclear factor kappa B) activation for pro-inflammatory effects, which are very important for the tumor immune response. However, the crosstalk between cancer cells and the host immune system are very complicated and multifaceted, and further mechanistic study on immune functions regulated by OLR1 in EC is needed in the future.
Funding Statement
This work was supported by grants from the National Natural Science Fund 32071272, 31870800.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Reference
- [1].Li C, Liu F, Yang X, et al. Targeting lectin-like oxidized low-density lipoprotein receptor-1 triggers autophagic program in esophageal cancer. Cell Death Differ. 2021. doi: 10.1038/s41418-021-00884-y. [DOI] [PMC free article] [PubMed] [Google Scholar]